
Caspase-1-Independent IL-1 Release Mediates Blister Formation in Autoantibody-Induced Tissue Injury...
8
The Journal of Immunology Caspase-1–Independent IL-1 Release Mediates Blister Formation in Autoantibody-Induced Tissue Injury through Modulation of Endothelial Adhesion Molecules Hengameh Sadeghi,* Anike Lockmann, † Anna-Carina Hund, † Unni K. S. R. L. Samavedam,* ,‡ Elena Pipi,* ,‡ Katerina Vafia,* Eva Hauenschild, x Kathrin Kalies, x Hendri H. Pas, { Marcel F. Jonkman, { Hiroaki Iwata,* ,1 Andreas Recke,* ,‡ Michael P. Scho ¨n, † Detlef Zillikens,* ,‡ Enno Schmidt,* ,‡,2 and Ralf J. Ludwig* ,‡,2 Although reports documented aberrant cytokine expression in autoimmune bullous dermatoses (AIBDs), cytokine-targeting ther- apies have not been established in these disorders. We showed previously that IL-6 treatment protected against tissue destruction in experimental epidermolysis bullosa acquisita (EBA), an AIBD caused by autoantibodies to type VII collagen (COL7). The anti- inflammatory effects of IL-6 were mediated by induction of IL-1ra, and prophylactic IL-1ra administration prevented blistering. In this article, we demonstrate elevated serum concentrations of IL-1b in both mice with experimental EBA induced by injection of anti-COL7 IgG and in EBA patients. Increased IL-1a and IL-1b expression also was observed in the skin of anti-COL7 IgG- injected wild-type mice compared with the significantly less diseased IL-1R–deficient or wild-type mice treated with the IL-1R antagonist anakinra or anti–IL-1b. These findings suggested that IL-1 contributed to recruitment of inflammatory cells into the skin. Accordingly, the expression of ICAM-1 was decreased in IL-1R–deficient and anakinra-treated mice injected with anti- COL7. This effect appeared to be specifically attributable to IL-1 because anakinra blocked the upregulation of different endothelial adhesion molecules on IL-1–stimulated, but not on TNF-a–stimulated, cultured endothelial cells. Interestingly, injec- tion of caspase-1/11–deficient mice with anti-COL7 IgG led to the same extent of skin lesions as in wild-type mice. Collectively, our data suggest that IL-1, independently of caspase-1, contributes to the pathogenesis of EBA. Because anti–IL-1b in a prophylactic setting and anakinra in a quasi-therapeutic setting (i.e., when skin lesions had already developed) improved experimental EBA, IL-1 appears to be a potential therapeutic target for EBA and related AIBDs. The Journal of Immunology, 2015, 194: 000–000. A utoimmune bullous dermatoses (AIBDs) are character- ized by autoantibodies against structural proteins of the skin and mucosal tissues. Patients suffer from consid- erable morbidity and increased mortality. Based on the targeted Ags, AIBDs can be divided into pemphigus and pemphigoid diseases. In pemphigus disease, autoantibodies to desmosomal proteins directly cause blister formation, whereas in pemphigoid disease, blister formation requires the activation of immune mechanisms through the Fc portion of the autoantibodies (1, 2). Despite improved therapeutic options, the mortality of patients with pemphigus and pemphigoid remains high (3); this can be attributed, in part, to the extent of immunosuppressive therapy (4). Therefore, and because of the increasing incidence (3), there is a clear and unmet medical need for the development of effective and safe therapies for these patients. In other autoimmune and chronic inflammatory diseases, cytokine-modulating therapies have greatly improved the thera- peutic armamentarium (e.g., TNF-a inhibition in rheumatoid ar- thritis and psoriasis) (5). In AIBD, increased cytokine expression in several compartments, including serum, blister fluid, and skin, has been well documented (6). However, with the exception of two studies, one indicating the involvement of IL-1 and TNF-a in the pathogenesis of pemphigus vulgaris (7) and the other indi- cating an anti-inflammatory role for IL-6 in epidermolyis bullosa acquisita (EBA) (8), no functional data are available. Expression profiling identified IL-1ra as 1 of 33 gene products expressed differentially in the skin of BALB/c mice with experimental EBA. IL-1ra expression in diseased mice was closely linked to IL-1b (9), and its local and systemic expression were controlled by IL-6 (8). Furthermore, we recently observed increased IL-1a and IL-1b serum levels in C57BL/6 mice after the induction of experimental EBA induced by transfer of anti–type VII collagen (COL7) IgG. In addition, IL-1ra administration, which blocked IL-1 function, in a preventive experimental setting impaired skin blistering in a model of autoantibody transfer–induced EBA (8). Moreover, elevated levels of IL-1b, but not IL-1a, were detected in blister *Department of Dermatology, University of L€ ubeck, 23538 L€ ubeck, Germany; † Department of Dermatology, Venereology, and Allergology, University Medical Center, 37075 Go ¨ttingen, Germany; ‡ L€ ubeck Institute of Experimental Dermatology, University of L€ ubeck, 23538 L€ ubeck, Germany; x Institute of Anatomy, University of L€ ubeck, 23562 L€ ubeck, Germany; and { Center for Blistering Diseases, Department of Dermatology, University Medical Center Groningen, University of Groningen, 9700 RB Groningen, the Netherlands 1 Current address: Department of Dermatology, Hokkaido University Graduate School of Medicine, Sapporo, Japan. 2 E.S. and R.J.L. were equal contributors. Received for publication October 22, 2014. Accepted for publication February 15, 2015. This work was supported by the Deutsche Forschungsgemeinschaft (Excellence Cluster Inflammation at Interfaces Grant DFG EXC 306/2, Research Training Grant for Mod- ulation of Autoimmunity DFG GRK1727/1, and Grant DFG LU877/5-1), the Dr. Robert Pfleger Foundation, the B. Braun Foundation, the Focus Program on Autoimmunity of the University of L€ ubeck, and a research and development grant from Novartis. Address correspondence and reprint requests to Prof. Ralf J. Ludwig, L€ ubeck Insti- tute of Experimental Dermatogy, University of L€ ubeck, Ratzeburger Allee 160, D-23538 L€ ubeck, Germany. E-mail address: [email protected] Abbreviations used in this article: AIBD, autoimmune bullous dermatosis; AUC, area under the curve; COL7, type VII collagen; EBA, epidermolysis bullosa acquisita; IF, immunofluorescence; RT, room temperature; WT, wild-type. Copyright Ó 2015 by The American Association of Immunologists, Inc. 0022-1767/15/$25.00 www.jimmunol.org/cgi/doi/10.4049/jimmunol.1402688 Published March 20, 2015, doi:10.4049/jimmunol.1402688
Transcript of Caspase-1-Independent IL-1 Release Mediates Blister Formation in Autoantibody-Induced Tissue Injury...

The Journal of Immunology
Caspase-1–Independent IL-1 Release Mediates BlisterFormation in Autoantibody-Induced Tissue Injury throughModulation of Endothelial Adhesion Molecules
Hengameh Sadeghi,* Anike Lockmann,† Anna-Carina Hund,† Unni K. S. R. L. Samavedam,*,‡
Elena Pipi,*,‡ Katerina Vafia,* Eva Hauenschild,x Kathrin Kalies,x Hendri H. Pas,{
Marcel F. Jonkman,{ Hiroaki Iwata,*,1 Andreas Recke,*,‡ Michael P. Schon,†
Detlef Zillikens,*,‡ Enno Schmidt,*,‡,2 and Ralf J. Ludwig*,‡,2
Although reports documented aberrant cytokine expression in autoimmune bullous dermatoses (AIBDs), cytokine-targeting ther-apies have not been established in these disorders. We showed previously that IL-6 treatment protected against tissue destruction inexperimental epidermolysis bullosa acquisita (EBA), an AIBD caused by autoantibodies to type VII collagen (COL7). The anti-inflammatory effects of IL-6 were mediated by induction of IL-1ra, and prophylactic IL-1ra administration prevented blistering. Inthis article, we demonstrate elevated serum concentrations of IL-1b in both mice with experimental EBA induced by injection ofanti-COL7 IgG and in EBA patients. Increased IL-1a and IL-1b expression also was observed in the skin of anti-COL7 IgG-injected wild-type mice compared with the significantly less diseased IL-1R–deficient or wild-type mice treated with the IL-1Rantagonist anakinra or anti–IL-1b. These findings suggested that IL-1 contributed to recruitment of inflammatory cells into theskin. Accordingly, the expression of ICAM-1 was decreased in IL-1R–deficient and anakinra-treated mice injected with anti-COL7. This effect appeared to be specifically attributable to IL-1 because anakinra blocked the upregulation of differentendothelial adhesion molecules on IL-1–stimulated, but not on TNF-a–stimulated, cultured endothelial cells. Interestingly, injec-tion of caspase-1/11–deficient mice with anti-COL7 IgG led to the same extent of skin lesions as in wild-type mice. Collectively, ourdata suggest that IL-1, independently of caspase-1, contributes to the pathogenesis of EBA. Because anti–IL-1b in a prophylacticsetting and anakinra in a quasi-therapeutic setting (i.e., when skin lesions had already developed) improved experimental EBA,IL-1 appears to be a potential therapeutic target for EBA and related AIBDs. The Journal of Immunology, 2015, 194: 000–000.
A utoimmune bullous dermatoses (AIBDs) are character-ized by autoantibodies against structural proteins of theskin and mucosal tissues. Patients suffer from consid-
erable morbidity and increased mortality. Based on the targetedAgs, AIBDs can be divided into pemphigus and pemphigoiddiseases. In pemphigus disease, autoantibodies to desmosomalproteins directly cause blister formation, whereas in pemphigoid
disease, blister formation requires the activation of immunemechanisms through the Fc portion of the autoantibodies (1, 2).Despite improved therapeutic options, the mortality of patientswith pemphigus and pemphigoid remains high (3); this can beattributed, in part, to the extent of immunosuppressive therapy (4).Therefore, and because of the increasing incidence (3), there isa clear and unmet medical need for the development of effectiveand safe therapies for these patients.In other autoimmune and chronic inflammatory diseases,
cytokine-modulating therapies have greatly improved the thera-peutic armamentarium (e.g., TNF-a inhibition in rheumatoid ar-thritis and psoriasis) (5). In AIBD, increased cytokine expressionin several compartments, including serum, blister fluid, and skin,has been well documented (6). However, with the exception oftwo studies, one indicating the involvement of IL-1 and TNF-a inthe pathogenesis of pemphigus vulgaris (7) and the other indi-cating an anti-inflammatory role for IL-6 in epidermolyis bullosaacquisita (EBA) (8), no functional data are available. Expressionprofiling identified IL-1ra as 1 of 33 gene products expresseddifferentially in the skin of BALB/c mice with experimental EBA.IL-1ra expression in diseased mice was closely linked to IL-1b(9), and its local and systemic expression were controlled by IL-6(8). Furthermore, we recently observed increased IL-1a and IL-1bserum levels in C57BL/6 mice after the induction of experimentalEBA induced by transfer of anti–type VII collagen (COL7) IgG.In addition, IL-1ra administration, which blocked IL-1 function, ina preventive experimental setting impaired skin blistering ina model of autoantibody transfer–induced EBA (8). Moreover,elevated levels of IL-1b, but not IL-1a, were detected in blister
*Department of Dermatology, University of L€ubeck, 23538 L€ubeck, Germany;†Department of Dermatology, Venereology, and Allergology, University MedicalCenter, 37075 Gottingen, Germany; ‡L€ubeck Institute of Experimental Dermatology,University of L€ubeck, 23538 L€ubeck, Germany; xInstitute of Anatomy, University ofL€ubeck, 23562 L€ubeck, Germany; and {Center for Blistering Diseases, Departmentof Dermatology, University Medical Center Groningen, University of Groningen,9700 RB Groningen, the Netherlands1Current address: Department of Dermatology, Hokkaido University Graduate Schoolof Medicine, Sapporo, Japan.2E.S. and R.J.L. were equal contributors.
Received for publication October 22, 2014. Accepted for publication February 15,2015.
This work was supported by the Deutsche Forschungsgemeinschaft (Excellence ClusterInflammation at Interfaces Grant DFG EXC 306/2, Research Training Grant for Mod-ulation of Autoimmunity DFG GRK1727/1, and Grant DFG LU877/5-1), the Dr. RobertPfleger Foundation, the B. Braun Foundation, the Focus Program on Autoimmunity ofthe University of L€ubeck, and a research and development grant from Novartis.
Address correspondence and reprint requests to Prof. Ralf J. Ludwig, L€ubeck Insti-tute of Experimental Dermatogy, University of L€ubeck, Ratzeburger Allee 160,D-23538 L€ubeck, Germany. E-mail address: [email protected]
Abbreviations used in this article: AIBD, autoimmune bullous dermatosis; AUC, areaunder the curve; COL7, type VII collagen; EBA, epidermolysis bullosa acquisita; IF,immunofluorescence; RT, room temperature; WT, wild-type.
Copyright! 2015 by The American Association of Immunologists, Inc. 0022-1767/15/$25.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1402688
Published March 20, 2015, doi:10.4049/jimmunol.1402688

fluid from bullous pemphigoid patients, an AIBD caused byautoantibodies against type XVII collagen, and were correlatedwith the magnitude of skin lesions (10, 11).Given that some cytokines have potent anti-inflammatory ac-
tivities (12) or trigger the release of chemokine antagonists (13),we investigated the contribution of IL-1 to the prototypical AIBDEBA in which the autoimmune response is directed against COL7(14). Based on the observation of reduced skin blistering in au-toantibody transfer–induced EBA upon prophylactic anakinraapplication (8), we hypothesized that IL-1 would exert proin-flammatory activity in EBA. However, it remained unclear howIL-1 modulates blister formation in experimental EBA; whetherthis effect is mediated by IL-1a, IL-1b, or both; whether treatmentwith IL-1–modulating compounds can improve already estab-lished experimental EBA; how IL-1 expression is controlled; andwhether these findings are also applicable to EBA patients.We addressed these potentially clinically relevant questions using
several animal models of EBA, as well as a large collection of EBAsera. Given that blockade of IL-1 has therapeutic effects in establishedEBA, and there is increased IL-1 expression in EBApatients, blockadeof IL-1 might be a promising therapeutic option for EBA patients.
Materials and MethodsExperiments with human samples
For the determination of IL-1a and IL-1b concentrations, serum from EBApatients (n = 26) fulfilling the following criteria were used: presentationwith skin lesions resembling EBA; linear IgG and/or IgA deposition bydirect immunofluorescence (IF) microscopy; and an u-serrated pattern bydirect IF microscopy and/or detection of Abs against COL7. Serum fromblood donors (n = 52) served as a reference. All of the experiments usinghuman samples were approved by the local ethics committee (Universityof L€ubeck, L€ubeck, Germany) and were performed according to theDeclaration of Helsinki. Blood donors and patients provided their writteninformed consent prior to study participation.
Mice
SJL/J, C57BL/6, IL-1R-deficient (2/2), and caspase-1/11–deficient micewere obtained from Charles River Laboratories (Sulzfeld, Germany). Miceaged 6–8 wk were used for the experiments. The mice were kept in specificpathogen–free conditions and fed standard mouse chow and acidifieddrinking water ad libitum. All of the clinical examinations, biopsies, andblood collections were performed under anesthesia using i.p. administrationof a mixture of ketamine (100 mg/g) and xylazine (15 mg/g). The animalexperiments were approved by the local authority of the Animal Care andUse Committee (Kiel, Germany) and were performed by certified personnel.
Induction of experimental EBA
Rabbit anti-murine COL7 IgGwas prepared as previously described (15). Inbrief, rabbits were immunized with recombinant proteins of the NC1 do-main of murine COL7. IgG from immune and normal rabbit sera werepurified by affinity chromatography using protein G affinity. Passive-transfer studies into mice followed the published protocols (15, 16).Each experiment was performed twice, using different anti-COL7 IgGpreparations. Disease severity was expressed as the percentage of bodysurface area affected by skin lesions and was determined at three timepoints (days 4, 8, and 12). From these time points, the area under the curve(AUC) was calculated. Blood and tissue samples were collected on day 12.EBA was induced by immunization with an immunodominant fragmentlocated within murine COL7, as previously reported (17), with minormodifications described elsewhere (18). After immunization, the extent ofclinical EBA manifestation was determined weekly. If EBA skin lesionsaffected 2% or more of the body surface area of individual mice, they wereallocated to either anakinra or PBS (see below). The treatments were ad-ministered over a 2-wk period, and EBA severity (percentage of affectedbody surface area) was monitored weekly.
Determination of IL-1 levels in serum and skin
Serum from EBA patients and normal controls were analyzed for the ex-pression of IL-1a and IL-1b using Bio-Plex (Bioglobe, Hamburg, Ger-many). Serum concentrations of IL-1a and IL-1b were determined in mice
with experimental EBA and in normal rabbit IgG-injected controls at theindicated time points using Bio-Plex (Bioglobe). IL-1 expression in skinwas determined using immunohistochemistry. In brief, for both IL-1a andIL-1b staining, 6 mm-thick sections were incubated with the predilutedprimary Ab—goat anti-mouse IL-1a or goat anti-mouse IL-1b Ab, re-spectively (both R&D Systems, Wiesbaden, Germany)—at room temper-ature (RT) for 1 h. Nonspecific binding was blocked using 5% rabbitserum. Rabbit anti-goat Ab (Dako, Hamburg, Germany) was used assecondary Ab. Incubation with HistoGreen (LINARIS, Germany), coun-terstaining with hematoxylin, and dehydrating were performed beforemounting on slides. The intensity of IL-1 expression was scored semi-quantitatively by an observer who was unaware of the nature of thespecimen. The detection of ICAM-1 expression was performed similarly(i.e., Armenian hamster anti-mouse served as the primary Ab; BD Bio-sciences, Heidelberg, Germany). Rabbit anti-Armenian hamster (Abcam,Cambridge, U.K.) was used as a secondary Ab. For Gr-1 staining, rat anti-mouse Ly-6G (clone RB6-8C5) was used as primary Ab, and goat anti-ratIgG (both from Abcam) was used as secondary Ab.
RT-PCR
For RT-PCR analysis, lesional (n = 4) and nonlesional (n = 5) skin wasobtained from mice on day 6 after injection with rabbit anti-murine COL7IgG and normal rabbit IgG, respectively. RT-PCR of mouse skin wasperformed as described (19) using the following sets of primers: MLN51forward (59-CCAAGCCAGCCTTCATTCTTG-39) and MLN51 reverse(59-TAACGCTTAGCTCGACCACTCTG-39) and IL-1b forward (59-CTTCCAGGATGAGGACATGAG-39) and IL-1b reverse (59-CACAC-CAGCAGGTTATCATC-39).
Treatment protocols
Kineret (anakinra; Biovitrum, Stockholm, Sweden) was injected i.p. intomice at a dosage of either 100 or 200 mg/kg/d. PBS served as a control.Anakinra or PBS treatment was initiated on day 0 and maintainedthroughout the experiments in the Ab-transfer model. In immunization-induced EBA, anakinra was administered to mice when $2% of thebody surface area was affected by skin lesions; it was continued for a totalof 2 wk. Anti-IL-1b (clone ACZ885; Novartis, N€urnberg, Germany) wasinjected at 200 mg/mouse 1 d before and 2 and 4 d after the first anti-COL7IgG injection.
Western blotting with extracts of mouse skin
Ear specimens from mice were excised, immediately snap-frozen in liquidnitrogen, and stored at 280˚C until further analysis. To prepare totalprotein skin extracts, frozen ears were ground in liquid nitrogen and ho-mogenized in T-PER reagent (Thermo Scientific, Darmstadt, Germany;1:6, w/v), with the addition of protease inhibitor mixture set III (Calbio-chem, Darmstadt, Germany; 1:100, v/v). Tissue homogenates werecentrifuged at 10,000 3 g for 10 min at 4˚C to pellet debris, and thesupernatants were collected. Next, the samples were incubated with proteinA/G PLUS–Agarose beads (Santa Cruz Biotechnology, Heidelberg, Ger-many; 5:1, v/v) at 4˚C on a rotating device overnight. The beads werepelleted by centrifugation at 1000 3 g for 5 min at 4˚C, and the super-natants were collected and stored at 280˚C until use. The total proteinconcentration was determined according to the BCA method (ThermoScientific). Protein samples were separated by SDS-PAGE under reducingconditions on 15% polyacrylamide gel and transferred to a 0.2-mm poresize nitrocellulose membrane for Western blotting. The membranes wereblocked with 5% w/v skim milk powder in TBST for 1 h at RT. Beforeeach step, the membranes were washed five times for 10 min in TBST. Allof the Abs were diluted in blocking buffer. Goat polyclonal IgG againstmurine IL-1b (R&D Systems; cat. no. AF-401-NA) and mouse mono-clonal IgG against actin (Santa Cruz Biotechnology; cat. no. sc-47778)were used at 1:400 or 1:3000 dilution, respectively, overnight at 4˚C. HRP-labeled secondary Abs for goat (Dako; cat. no. P0449) and mouse (JacksonImmunoResearch, Suffolk, U.K.; cat. no. 715-035-150) were applied at1:20,000 and 1:50,000 dilution, respectively, for 1 h at RT and detectedusing Amersham ECL Prime reagent (GE Healthcare).
Culture and in vitro activation of human endothelial cells
HUVECs (PromoCell, Heidelberg, Germany) were cultured in EndothelialCell Growth Medium (PromoCell) at 37˚C in a humidified atmospherecontaining 5% CO2. Four hours prior to activation, the medium wasreplaced by Endothelial Cell Basal Medium (PromoCell) supplementedwith 10% FCS. To induce adhesion molecule expression, endothelial cellswere activated with recombinant human IL-1b (5 ng/ml) or TNF-a (25 ng/ml)for 4 h. These experiments were performed in the presence or absence
2 IL-1 IN EBA

of anakinra (5 mg/ml), which was added to the cell cultures 1 h beforetreatment. Nontreated cells served as negative controls.
Histology and IF microscopy
Murine specimens were subjected to H&E staining, according to a standardprotocol. The extent of leukocyte infiltration was scored semiquantitativelyin a blinded fashion, as described previously (20). Direct IF microscopy forthe detection of rabbit IgG and murine C3 in experimental EBA wasperformed on 6-mm cryosections, as described, using goat anti-rabbit IgG(Dako) and goat anti-murine C3 (MP Biomedicals, Kaysersberg, France),both labeled with FITC. IF microscopy on cultured HUVECs was per-formed as reported (21, 22). In brief, cells were fixed in 220˚C coldmethanol for 5 min and washed carefully with PBS before blocking with5% FCS in PBS for 1 h. Anti-VCAM Ab (clone STA; Immunotools,Friesoythe, Germany), anti-ICAM Ab (clone 15.2; FITC-labeled; Immu-notools), and anti–E-selectin Ab (clone 1-2B6; PE-labeled; Abs-online,Aachen, Germany) were diluted in PBS containing 2.5% FCS and in-cubated overnight before washing with PBS. Anti-VCAM reactivity wasvisualized by goat anti-mouse IgG conjugated with Alexa Fluor 488(Cell Signaling Technology, Danvers, MA). Mounting medium con-taining 1% DAPI was added to each culture slide. Stained cells wereanalyzed using an Axio Imager M1 microscope (Carl Zeiss Meditec,Jena, Germany). Standardized exposure times were used for all of theimages.
Statistics
The data were analyzed using SigmaPlot software, version 12 (SystatSoftware, Chicago, IL), and applied tests and 95% confidence intervals areindicated in the respective text and figure legends. For analysis of rank-scaled or semiquantitative data, we used ordinal logistic regression (polr)and mosaic plots, provided by GNU R open source statistical software(http://www.r-project.org), together with the “MASS” package. Ordinallogistic regression models were evaluated first by likelihood ratio testing.In the second step, models with a significant likelihood ratio testing wereevaluated further; p values are given for the difference between each groupand the controls. A p value , 0.05 was considered statistically significantafter appropriate adjustment for multiple testing.
ResultsIncreased IL-1a and IL-1b serum concentrations andcorrelations with disease severity in experimental and humanEBA
The induction of experimental EBA in BALB/c mice, by injectionof anti-COL7 IgG, led to increases in serum levels of IL-1b, but notIL-1a, compared with untreated control mice (Fig. 1A). The ex-tent of skin lesions correlated with IL-1b, but not with IL-1a,serum levels (Fig. 1B, 1C). Compared with healthy controls, 6-and 4-fold increases in IL-1a and IL-1b serum levels were foundin EBA patients (Fig. 1A). IL-1b mRNA expression also wassignificantly increased in lesional skin compared with healthymouse skin. Six days after the first IgG injection, IL-1b mRNAexpression was significantly increased in anti-COL7 IgG-injectedanimals compared with mice injected with normal rabbit IgG.Median relative (in relation to MLN51) IL-1b expression in miceinjected with normal rabbit IgG was 0.16 (0.12–0.57, 25/75percentiles), whereas it was 34.6 (21.1–194; p = 0.0159, Mann–Whitney rank-sum test) in lesional skin of mice injected with anti-COL7 IgG. Furthermore, we detected increased protein expressionof IL-1a and IL-1b in the skin of mice after EBA inductioncompared with IL-1R2/2 mice and anakinra-treated wild-type(WT) animals (Fig. 1D–F).
Induction of experimental EBA was impaired in mice witheither genetic or prophylactic pharmacologic inhibition of IL-1
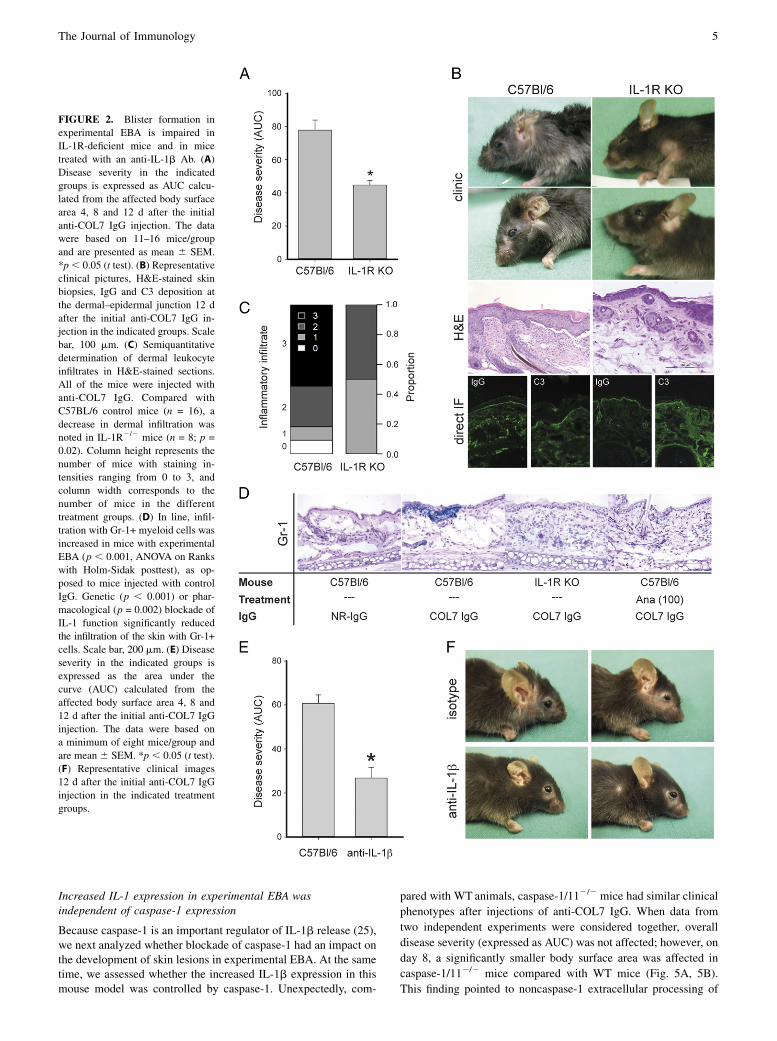
To test whether the increased local and/or systemic IL-1a andIL-1b levels were of functional relevance, IL-1 function wasinhibited. EBA was induced in IL-1R2/2 and WT C57BL/6 con-trol mice. Mice of both strains developed experimental EBA;however, in IL-1R2/2 mice, the overall extent of skin blistering
was reduced to 52% of that in control mice (p , 0.001; Fig. 2A,2B). Comparisons of clinical scores between the two strains at 4,8, and 12 d showed that the affected body surface area was sig-nificantly lower in IL-1R2/2 mice at all time points (day 4: 1.6 60.4% versus 0.6 6 0.2% [p = 0.008]; day 8: 7.4 6 0.7% versus5.2 6 0.7% [p = 0.04]; day 12: 20.7 6 1.8% versus 10.1 6 1.7%[p , 0.001] in WT and IL-1R2/2 mice, respectively). The clinicaleffects were accompanied by reduced dermal inflammatory infil-trates in IL-1R2/2 treated mice compared with controls (Fig. 2C).Staining for Gr-1+ cells revealed similar results, demonstratinga reduced expression of GR-1 in both IL-1R2/2 and anakinra-treated mice (Fig. 2D).Treatment with a function-blocking Ab directed against IL-1b
fully recapitulated and even exceeded the effects of total IL-1inhibition. Compared with C57BL/6 mice injected with isotype-control Abs, anti–IL-1b reduced blister formation to 44%(p = 0.001, Fig. 2E, 2F). Blockade of IL-1 function also was as-sociated with decreased expression of both IL-1a and IL-1b inmouse skin (Fig. 1). In contrast, levels of tissue-bound IgG and C3were similar in all of the mice (Fig. 2B), indicating that theclinical effects were not due to altered IgG metabolism and/oreffects on complement activation.
Therapeutic application of anakinra led to improvement ofimmunization-induced EBA
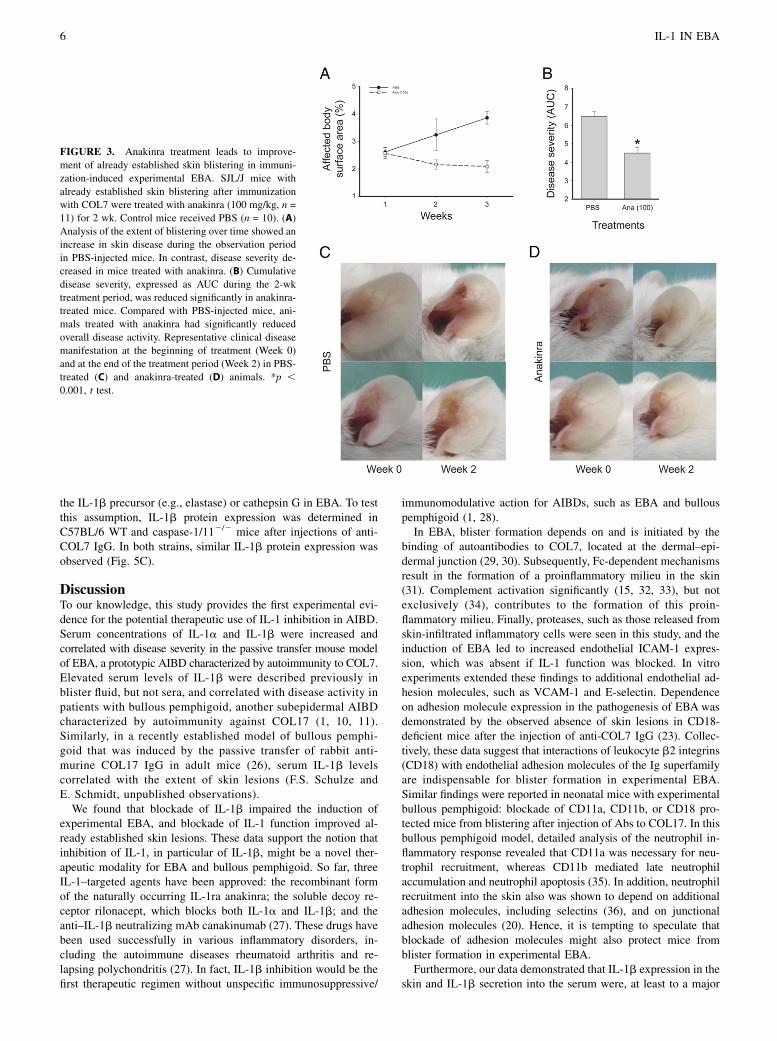
Following the prophylactic pharmacological inhibition of IL-1,an IL-1–targeting treatment was applied in a quasitherapeutic sett-ing (i.e., anakinra administration was initiated when mice alreadyhad established EBA skin lesions). For this purpose, theimmunization-induced EBA mouse model was used. At the ini-tiation of treatment, the clinical disease severity in mice ran-domly allocated to control and anakinra treatments was iden-tical (i.e., 2.6 6 0.2% affected body surface area). Clinical EBAmanifestations continuously progressed in PBS-injected mice(3.2 6 0.2% at week 1 and 3.9 6 0.1% at week 2), whereasit declined in anakinra-treated mice (2.1 6 0.2% at week 1 and2.0 6 0.2% at week 2, Fig. 3). Corresponding disease activityscores were significantly lower in the anakinra group comparedwith the PBS group (week 1, p = 0.004; week 2, p , 0.001;AUC, p , 0.001).
Inhibition of IL-1 function led to reduced expression ofendothelial adhesion molecules
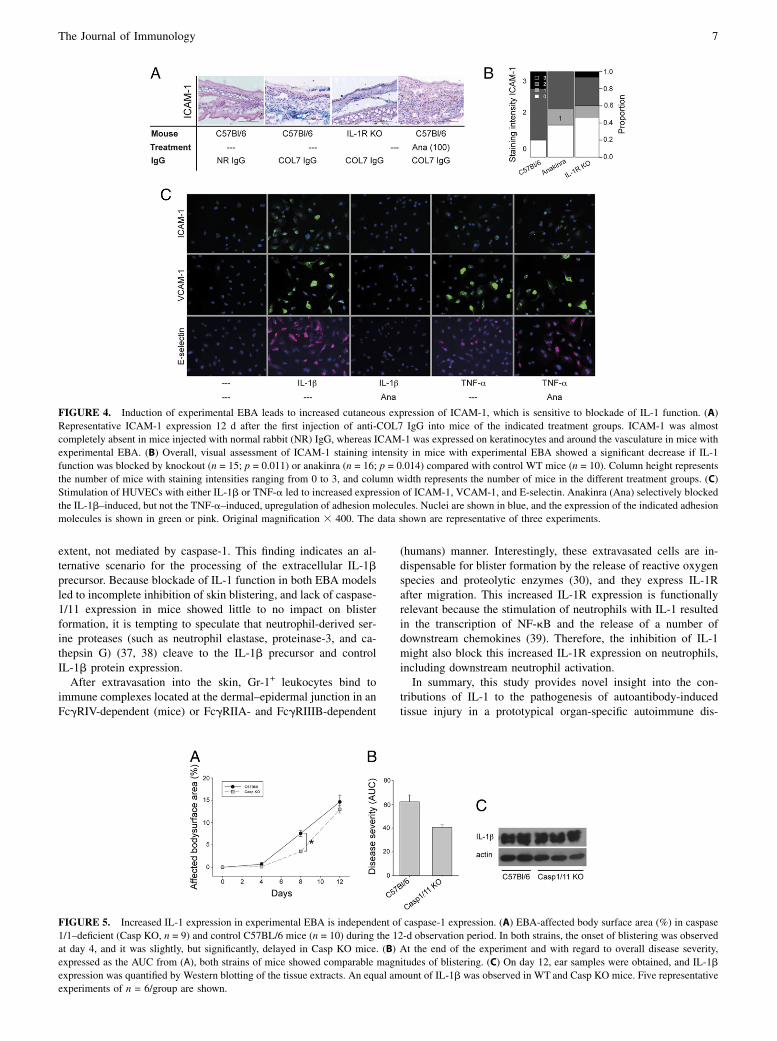
Because the induction of skin lesions in experimental EBA dependson CD18 (23) and because IL-1 regulates the expression of therespective endothelial ligands for LFA-1 (CD11a/CD18), Mac-1(CD11b/CD18), and integrin CD11c/CD18 (24), the expression ofICAM-1 (CD54) in the skin was evaluated in experimental EBA,with and without impaired IL-1 function. In mice with experi-mental EBA, ICAM-1 expression was detected both on endothe-lial cells and on basal keratinocytes (Fig. 4A, 4B). In contrast,ICAM-1 expression was almost absent in mice injected withnormal rabbit IgG (Fig. 4A). Similarly, blockade of IL-1 functionin mice injected with anti-COL7 IgG reduced ICAM-1 expression(Fig. 4A, 4B). To test whether this inhibitory effect of IL-1blockade also affected other endothelial adhesion molecules,such as VCAM-1 (CD106) and E-selectin (CD62E), the expres-sion of these molecules was assessed on HUVECs stimulated inthe absence or presence of anakinra. In line with the in vivofindings, IL-1 induced ICAM-1 expression, which could specifi-cally be inhibited by anakinra treatment (Fig. 4C). EndothelialVCAM-1 and E-selectin expression paralleled ICAM-1 expression(Fig. 4C).
The Journal of Immunology 3
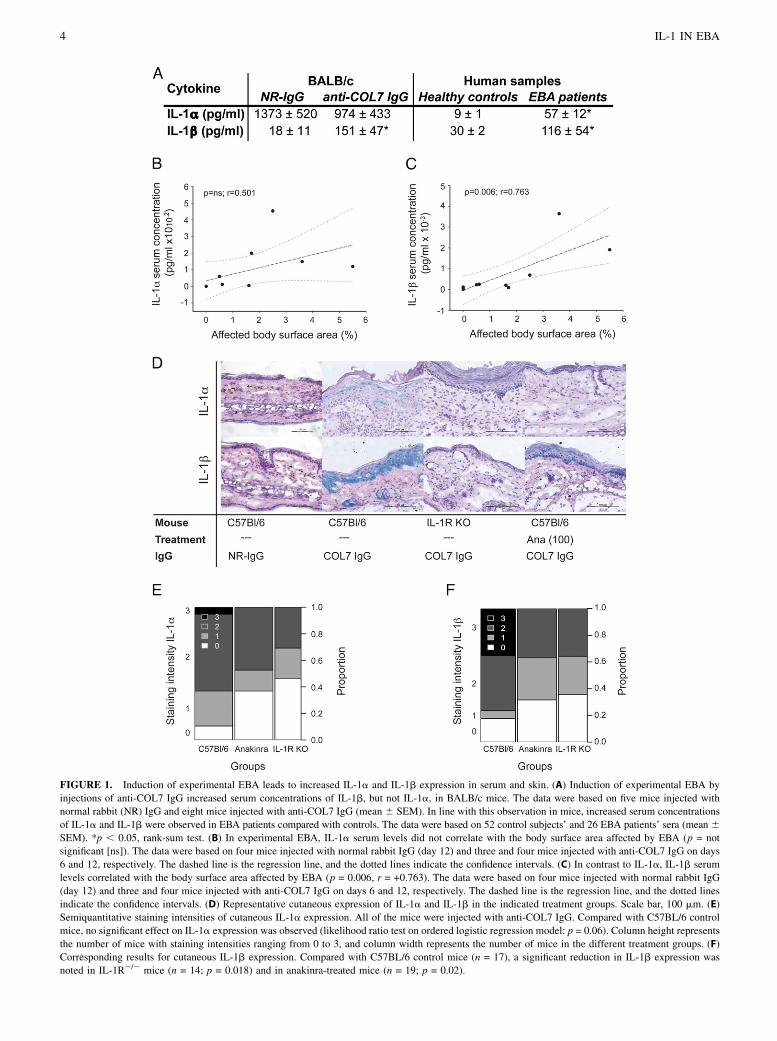
FIGURE 1. Induction of experimental EBA leads to increased IL-1a and IL-1b expression in serum and skin. (A) Induction of experimental EBA byinjections of anti-COL7 IgG increased serum concentrations of IL-1b, but not IL-1a, in BALB/c mice. The data were based on five mice injected withnormal rabbit (NR) IgG and eight mice injected with anti-COL7 IgG (mean 6 SEM). In line with this observation in mice, increased serum concentrationsof IL-1a and IL-1b were observed in EBA patients compared with controls. The data were based on 52 control subjects’ and 26 EBA patients’ sera (mean 6SEM). *p , 0.05, rank-sum test. (B) In experimental EBA, IL-1a serum levels did not correlate with the body surface area affected by EBA (p = notsignificant [ns]). The data were based on four mice injected with normal rabbit IgG (day 12) and three and four mice injected with anti-COL7 IgG on days6 and 12, respectively. The dashed line is the regression line, and the dotted lines indicate the confidence intervals. (C) In contrast to IL-1a, IL-1b serumlevels correlated with the body surface area affected by EBA (p = 0.006, r = +0.763). The data were based on four mice injected with normal rabbit IgG(day 12) and three and four mice injected with anti-COL7 IgG on days 6 and 12, respectively. The dashed line is the regression line, and the dotted linesindicate the confidence intervals. (D) Representative cutaneous expression of IL-1a and IL-1b in the indicated treatment groups. Scale bar, 100 mm. (E)Semiquantitative staining intensities of cutaneous IL-1a expression. All of the mice were injected with anti-COL7 IgG. Compared with C57BL/6 controlmice, no significant effect on IL-1a expression was observed (likelihood ratio test on ordered logistic regression model: p = 0.06). Column height representsthe number of mice with staining intensities ranging from 0 to 3, and column width represents the number of mice in the different treatment groups. (F)Corresponding results for cutaneous IL-1b expression. Compared with C57BL/6 control mice (n = 17), a significant reduction in IL-1b expression wasnoted in IL-1R2/2 mice (n = 14; p = 0.018) and in anakinra-treated mice (n = 19; p = 0.02).
4 IL-1 IN EBA
Increased IL-1 expression in experimental EBA wasindependent of caspase-1 expression
Because caspase-1 is an important regulator of IL-1b release (25),we next analyzed whether blockade of caspase-1 had an impact onthe development of skin lesions in experimental EBA. At the sametime, we assessed whether the increased IL-1b expression in thismouse model was controlled by caspase-1. Unexpectedly, com-
pared with WT animals, caspase-1/112/2 mice had similar clinicalphenotypes after injections of anti-COL7 IgG. When data fromtwo independent experiments were considered together, overalldisease severity (expressed as AUC) was not affected; however, onday 8, a significantly smaller body surface area was affected incaspase-1/112/2 mice compared with WT mice (Fig. 5A, 5B).This finding pointed to noncaspase-1 extracellular processing of
FIGURE 2. Blister formation inexperimental EBA is impaired inIL-1R-deficient mice and in micetreated with an anti-IL-1b Ab. (A)Disease severity in the indicatedgroups is expressed as AUC calcu-lated from the affected body surfacearea 4, 8 and 12 d after the initialanti-COL7 IgG injection. The datawere based on 11–16 mice/groupand are presented as mean 6 SEM.*p, 0.05 (t test). (B) Representativeclinical pictures, H&E-stained skinbiopsies, IgG and C3 deposition atthe dermal–epidermal junction 12 dafter the initial anti-COL7 IgG in-jection in the indicated groups. Scalebar, 100 mm. (C) Semiquantitativedetermination of dermal leukocyteinfiltrates in H&E-stained sections.All of the mice were injected withanti-COL7 IgG. Compared withC57BL/6 control mice (n = 16), adecrease in dermal infiltration wasnoted in IL-1R2/2 mice (n = 8; p =0.02). Column height represents thenumber of mice with staining in-tensities ranging from 0 to 3, andcolumn width corresponds to thenumber of mice in the differenttreatment groups. (D) In line, infil-tration with Gr-1+ myeloid cells wasincreased in mice with experimentalEBA (p , 0.001, ANOVA on Rankswith Holm-Sidak posttest), as op-posed to mice injected with controlIgG. Genetic (p , 0.001) or phar-macological (p = 0.002) blockade ofIL-1 function significantly reducedthe infiltration of the skin with Gr-1+cells. Scale bar, 200 mm. (E) Diseaseseverity in the indicated groups isexpressed as the area under thecurve (AUC) calculated from theaffected body surface area 4, 8 and12 d after the initial anti-COL7 IgGinjection. The data were based ona minimum of eight mice/group andare mean 6 SEM. *p , 0.05 (t test).(F) Representative clinical images12 d after the initial anti-COL7 IgGinjection in the indicated treatmentgroups.
The Journal of Immunology 5
the IL-1b precursor (e.g., elastase) or cathepsin G in EBA. To testthis assumption, IL-1b protein expression was determined inC57BL/6 WT and caspase-1/112/2 mice after injections of anti-COL7 IgG. In both strains, similar IL-1b protein expression wasobserved (Fig. 5C).
DiscussionTo our knowledge, this study provides the first experimental evi-dence for the potential therapeutic use of IL-1 inhibition in AIBD.Serum concentrations of IL-1a and IL-1b were increased andcorrelated with disease severity in the passive transfer mouse modelof EBA, a prototypic AIBD characterized by autoimmunity to COL7.Elevated serum levels of IL-1b were described previously inblister fluid, but not sera, and correlated with disease activity inpatients with bullous pemphigoid, another subepidermal AIBDcharacterized by autoimmunity against COL17 (1, 10, 11).Similarly, in a recently established model of bullous pemphi-goid that was induced by the passive transfer of rabbit anti-murine COL17 IgG in adult mice (26), serum IL-1b levelscorrelated with the extent of skin lesions (F.S. Schulze andE. Schmidt, unpublished observations).We found that blockade of IL-1b impaired the induction of
experimental EBA, and blockade of IL-1 function improved al-ready established skin lesions. These data support the notion thatinhibition of IL-1, in particular of IL-1b, might be a novel ther-apeutic modality for EBA and bullous pemphigoid. So far, threeIL-1–targeted agents have been approved: the recombinant formof the naturally occurring IL-1ra anakinra; the soluble decoy re-ceptor rilonacept, which blocks both IL-1a and IL-1b; and theanti–IL-1b neutralizing mAb canakinumab (27). These drugs havebeen used successfully in various inflammatory disorders, in-cluding the autoimmune diseases rheumatoid arthritis and re-lapsing polychondritis (27). In fact, IL-1b inhibition would be thefirst therapeutic regimen without unspecific immunosuppressive/
immunomodulative action for AIBDs, such as EBA and bullouspemphigoid (1, 28).In EBA, blister formation depends on and is initiated by the
binding of autoantibodies to COL7, located at the dermal–epi-dermal junction (29, 30). Subsequently, Fc-dependent mechanismsresult in the formation of a proinflammatory milieu in the skin(31). Complement activation significantly (15, 32, 33), but notexclusively (34), contributes to the formation of this proin-flammatory milieu. Finally, proteases, such as those released fromskin-infiltrated inflammatory cells were seen in this study, and theinduction of EBA led to increased endothelial ICAM-1 expres-sion, which was absent if IL-1 function was blocked. In vitroexperiments extended these findings to additional endothelial ad-hesion molecules, such as VCAM-1 and E-selectin. Dependenceon adhesion molecule expression in the pathogenesis of EBA wasdemonstrated by the observed absence of skin lesions in CD18-deficient mice after the injection of anti-COL7 IgG (23). Collec-tively, these data suggest that interactions of leukocyte b2 integrins(CD18) with endothelial adhesion molecules of the Ig superfamilyare indispensable for blister formation in experimental EBA.Similar findings were reported in neonatal mice with experimentalbullous pemphigoid: blockade of CD11a, CD11b, or CD18 pro-tected mice from blistering after injection of Abs to COL17. In thisbullous pemphigoid model, detailed analysis of the neutrophil in-flammatory response revealed that CD11a was necessary for neu-trophil recruitment, whereas CD11b mediated late neutrophilaccumulation and neutrophil apoptosis (35). In addition, neutrophilrecruitment into the skin also was shown to depend on additionaladhesion molecules, including selectins (36), and on junctionaladhesion molecules (20). Hence, it is tempting to speculate thatblockade of adhesion molecules might also protect mice fromblister formation in experimental EBA.Furthermore, our data demonstrated that IL-1b expression in the
skin and IL-1b secretion into the serum were, at least to a major
FIGURE 3. Anakinra treatment leads to improve-ment of already established skin blistering in immuni-zation-induced experimental EBA. SJL/J mice withalready established skin blistering after immunizationwith COL7 were treated with anakinra (100 mg/kg, n =11) for 2 wk. Control mice received PBS (n = 10). (A)Analysis of the extent of blistering over time showed anincrease in skin disease during the observation periodin PBS-injected mice. In contrast, disease severity de-creased in mice treated with anakinra. (B) Cumulativedisease severity, expressed as AUC during the 2-wktreatment period, was reduced significantly in anakinra-treated mice. Compared with PBS-injected mice, ani-mals treated with anakinra had significantly reducedoverall disease activity. Representative clinical diseasemanifestation at the beginning of treatment (Week 0)and at the end of the treatment period (Week 2) in PBS-treated (C) and anakinra-treated (D) animals. *p ,0.001, t test.
6 IL-1 IN EBA
extent, not mediated by caspase-1. This finding indicates an al-ternative scenario for the processing of the extracellular IL-1bprecursor. Because blockade of IL-1 function in both EBA modelsled to incomplete inhibition of skin blistering, and lack of caspase-1/11 expression in mice showed little to no impact on blisterformation, it is tempting to speculate that neutrophil-derived ser-ine proteases (such as neutrophil elastase, proteinase-3, and ca-thepsin G) (37, 38) cleave to the IL-1b precursor and controlIL-1b protein expression.After extravasation into the skin, Gr-1+ leukocytes bind to
immune complexes located at the dermal–epidermal junction in anFcgRIV-dependent (mice) or FcgRIIA- and FcgRIIIB-dependent
(humans) manner. Interestingly, these extravasated cells are in-dispensable for blister formation by the release of reactive oxygenspecies and proteolytic enzymes (30), and they express IL-1Rafter migration. This increased IL-1R expression is functionallyrelevant because the stimulation of neutrophils with IL-1 resultedin the transcription of NF-kB and the release of a number ofdownstream chemokines (39). Therefore, the inhibition of IL-1might also block this increased IL-1R expression on neutrophils,including downstream neutrophil activation.In summary, this study provides novel insight into the con-
tributions of IL-1 to the pathogenesis of autoantibody-inducedtissue injury in a prototypical organ-specific autoimmune dis-
FIGURE 4. Induction of experimental EBA leads to increased cutaneous expression of ICAM-1, which is sensitive to blockade of IL-1 function. (A)Representative ICAM-1 expression 12 d after the first injection of anti-COL7 IgG into mice of the indicated treatment groups. ICAM-1 was almostcompletely absent in mice injected with normal rabbit (NR) IgG, whereas ICAM-1 was expressed on keratinocytes and around the vasculature in mice withexperimental EBA. (B) Overall, visual assessment of ICAM-1 staining intensity in mice with experimental EBA showed a significant decrease if IL-1function was blocked by knockout (n = 15; p = 0.011) or anakinra (n = 16; p = 0.014) compared with control WT mice (n = 10). Column height representsthe number of mice with staining intensities ranging from 0 to 3, and column width represents the number of mice in the different treatment groups. (C)Stimulation of HUVECs with either IL-1b or TNF-a led to increased expression of ICAM-1, VCAM-1, and E-selectin. Anakinra (Ana) selectively blockedthe IL-1b–induced, but not the TNF-a–induced, upregulation of adhesion molecules. Nuclei are shown in blue, and the expression of the indicated adhesionmolecules is shown in green or pink. Original magnification 3 400. The data shown are representative of three experiments.
FIGURE 5. Increased IL-1 expression in experimental EBA is independent of caspase-1 expression. (A) EBA-affected body surface area (%) in caspase1/1–deficient (Casp KO, n = 9) and control C57BL/6 mice (n = 10) during the 12-d observation period. In both strains, the onset of blistering was observedat day 4, and it was slightly, but significantly, delayed in Casp KO mice. (B) At the end of the experiment and with regard to overall disease severity,expressed as the AUC from (A), both strains of mice showed comparable magnitudes of blistering. (C) On day 12, ear samples were obtained, and IL-1bexpression was quantified by Western blotting of the tissue extracts. An equal amount of IL-1b was observed in WT and Casp KO mice. Five representativeexperiments of n = 6/group are shown.
The Journal of Immunology 7

ease. We showed a proinflammatory contribution of IL-1, whichwas independent of caspase-1 expression. This proinflammatoryeffect of IL-1 was associated with the induction of endothelialadhesion molecule expression. This proinflammatory role for IL-1in experimental EBA could be considered when therapeuticdecisions must be made in patients with treatment-refractory and/orrelapsing EBA and other subepidermal autoimmune skin blisteringdiseases with a similar pathogenesis, such as bullous pemphigoid.
AcknowledgmentsThe anti–IL-1b Ab was kindly provided by Novartis (Basel, Switzerland).
DisclosuresThe authors have no financial conflicts of interest.
References1. Schmidt, E., and D. Zillikens. 2013. Pemphigoid diseases. Lancet 381: 320–332.2. Stanley, J. R., and M. Amagai. 2006. Pemphigus, bullous impetigo, and the
staphylococcal scalded-skin syndrome. N. Engl. J. Med. 355: 1800–1810.3. Langan, S. M., L. Smeeth, R. Hubbard, K. M. Fleming, C. J. Smith, and J. West.
2008. Bullous pemphigoid and pemphigus vulgaris—incidence and mortality inthe UK: population based cohort study. BMJ 337: a180.
4. Joly, P., J. C. Roujeau, J. Benichou, C. Picard, B. Dreno, E. Delaporte,L. Vaillant, M. D’Incan, P. Plantin, C. Bedane, et al; Bullous Diseases FrenchStudy Group. 2002. A comparison of oral and topical corticosteroids in patientswith bullous pemphigoid. N. Engl. J. Med. 346: 321–327.
5. Schon, M. P., and W. H. Boehncke. 2005. Psoriasis. N. Engl. J. Med. 352: 1899–1912.
6. Ludwig, R. J., and E. Schmidt. 2009. Cytokines in autoimmune bullous skindiseases. Epiphenomena or contribution to pathogenesis? G. Ital. Dermatol.Venereol. 144: 339–349.
7. Feliciani, C., P. Toto, P. Amerio, S. M. Pour, G. Coscione, G. Shivji, B. Wang,and D. N. Sauder. 2000. In vitro and in vivo expression of interleukin-1alpha andtumor necrosis factor-alpha mRNA in pemphigus vulgaris: interleukin-1alphaand tumor necrosis factor-alpha are involved in acantholysis. J. Invest. Dermatol.114: 71–77.
8. Samavedam, U. K., K. Kalies, J. Scheller, H. Sadeghi, Y. Gupta, M. F. Jonkman,E. Schmidt, J. Westermann, D. Zillikens, S. Rose-John, and R. J. Ludwig. 2013.Recombinant IL-6 treatment protects mice from organ specific autoimmunedisease by IL-6 classical signalling-dependent IL-1ra induction. J. Autoimmun.40: 74–85.
9. Kasperkiewicz, M., F. Nimmerjahn, S. Wende, M. Hirose, H. Iwata,M. F. Jonkman, U. Samavedam, Y. Gupta, S. Moller, E. Rentz, et al. 2012.Genetic identification and functional validation of FcgRIV as key molecule inautoantibody-induced tissue injury. J. Pathol. 228: 8–19.
10. Ameglio, F., L. D’Auria, C. Bonifati, C. Ferraro, A. Mastroianni, andB. Giacalone. 1998. Cytokine pattern in blister fluid and serum of patients withbullous pemphigoid: relationships with disease intensity. Br. J. Dermatol. 138:611–614.
11. Schmidt, E., A. Mittnacht, H. Schomig, R. Dummer, E. B. Brocker, andD. Zillikens. 1996. Detection of IL-1 alpha, IL-1 beta and IL-1 receptor an-tagonist in blister fluid of bullous pemphigoid. J. Dermatol. Sci. 11: 142–147.
12. Scheller, J., A. Chalaris, D. Schmidt-Arras, and S. Rose-John. 2011. The pro-and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Bio-phys. Acta 1813: 878–888.
13. Gabay, C., C. Lamacchia, and G. Palmer. 2010. IL-1 pathways in inflammationand human diseases. Nat Rev Rheumatol 6: 232–241.
14. Chen, M., G. H. Kim, L. Prakash, and D. T. Woodley. 2012. Epidermolysisbullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity 45:91–101.
15. Sitaru, C., S. Mihai, C. Otto, M. T. Chiriac, I. Hausser, B. Dotterweich, H. Saito,C. Rose, A. Ishiko, and D. Zillikens. 2005. Induction of dermal-epidermalseparation in mice by passive transfer of antibodies specific to type VII colla-gen. J. Clin. Invest. 115: 870–878.
16. Ishii, N., A. Recke, S. Mihai, M. Hirose, T. Hashimoto, D. Zillikens, andR. J. Ludwig. 2011. Autoantibody-induced intestinal inflammation and weightloss in experimental epidermolysis bullosa acquisita. J. Pathol. 224: 234–244.
17. Kasperkiewicz, M., R. M€uller, R. Manz, M. Magens, C. M. Hammers, C. Somlai,J. Westermann, E. Schmidt, D. Zillikens, R. J. Ludwig, and A. Orosz. 2011.Heat-shock protein 90 inhibition in autoimmunity to type VII collagen: evidencethat nonmalignant plasma cells are not therapeutic targets. Blood 117: 6135–6142.
18. Iwata, H., K. Bieber, B. Tiburzy, N. Chrobok, K. Kalies, A. Shimizu,S. Leineweber, A. Ishiko, A. Vorobyev, D. Zillikens, et al. 2013. B cells, den-dritic cells, and macrophages are required to induce an autoreactive CD4 helperT cell response in experimental epidermolysis bullosa acquisita. J. Immunol.191: 2978–2988.
19. Kalies, K., P. Konig, Y. M. Zhang, M. Deierling, J. Barthelmann, C. Stamm, andJ. Westermann. 2008. Nonoverlapping expression of IL10, IL12p40, and IFN-gamma mRNA in the marginal zone and T cell zone of the spleen after antigenicstimulation. J. Immunol. 180: 5457–5465.
20. Ludwig, R. J., T. M. Zollner, S. Santoso, K. Hardt, J. Gille, H. Baatz,P. S. Johann, J. Pfeffer, H. H. Radeke, M. P. Schon, et al. 2005. Junctionaladhesion molecules (JAM)-B and -C contribute to leukocyte extravasationto the skin and mediate cutaneous inflammation. J. Invest. Dermatol. 125:969–976.
21. Wallbrecht, K., N. Drick, A. C. Hund, and M. P. Schon. 2011. Downregulation ofendothelial adhesion molecules by dimethylfumarate, but not mono-methylfumarate, and impairment of dynamic lymphocyte-endothelial cellinteractions. Exp. Dermatol. 20: 980–985.
22. Oostingh, G. J., M. Pozgajova, R. J. Ludwig, T. Krahn, W. H. Boehncke,B. Nieswandt, and M. P. Schon. 2007. Diminished thrombus formation and al-leviation of myocardial infarction and reperfusion injury through antibody- orsmall-molecule-mediated inhibition of selectin-dependent platelet functions.Haematologica 92: 502–512.
23. Chiriac, M. T., J. Roesler, A. Sindrilaru, K. Scharffetter-Kochanek, D. Zillikens,and C. Sitaru. 2007. NADPH oxidase is required for neutrophil-dependentautoantibody-induced tissue damage. J. Pathol. 212: 56–65.
24. Schon, M. P., and R. J. Ludwig. 2005. Lymphocyte trafficking to inflamed skin—molecular mechanisms and implications for therapeutic target molecules. ExpertOpin. Ther. Targets 9: 225–243.
25. Dinarello, C. A. 2011. Interleukin-1 in the pathogenesis and treatment of in-flammatory diseases. Blood 117: 3720–3732.
26. Schulze, F. S., T. Beckmann, F. Nimmerjahn, A. Ishiko, M. Collin, J. Kohl,S. Goletz, D. Zillikens, R. J. Ludwig, and E. Schmidt. 2014. Fcg rεxεptοrs IIIand IV mediate tissue destruction in a novel adult mouse model of bullouspemphigoid. Am. J. Pathol. 184: 2185–2196.
27. Dinarello, C. A., A. Simon, and J. W. van der Meer. 2012. Treating inflammationby blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Dis-cov. 11: 633–652.
28. Kirtschig, G., P. Middleton, C. Bennett, D. F. Murrell, F. Wojnarowska, andN. P. Khumalo. 2010. Interventions for bullous pemphigoid. Cochrane DatabaseSyst. Rev. (10):CD002292.
29. Woodley, D. T., R. E. Burgeson, G. Lunstrum, L. Bruckner-Tuderman,M. J. Reese, and R. A. Briggaman. 1988. Epidermolysis bullosa acquisita an-tigen is the globular carboxyl terminus of type VII procollagen. J. Clin. Invest.81: 683–687.
30. Ludwig, R. J., K. Kalies, J. Kohl, D. Zillikens, and E. Schmidt. 2013. Emergingtreatments for pemphigoid diseases. Trends Mol. Med. 19: 501–512.
31. Hirose, M., K. Vafia, K. Kalies, S. Groth, J. Westermann, D. Zillikens,R. J. Ludwig, M. Collin, and E. Schmidt. 2012. Enzymatic autoantibody glycanhydrolysis alleviates autoimmunity against type VII collagen. J. Autoimmun. 39:304–314.
32. Mihai, S., M. T. Chiriac, K. Takahashi, J. M. Thurman, V. M. Holers,D. Zillikens, M. Botto, and C. Sitaru. 2007. The alternative pathway of com-plement activation is critical for blister induction in experimental epidermolysisbullosa acquisita. J. Immunol. 178: 6514–6521.
33. Karsten, C. M., M. K. Pandey, J. Figge, R. Kilchenstein, P. R. Taylor, M. Rosas,J. U. McDonald, S. J. Orr, M. Berger, D. Petzold, et al. 2012. Anti-inflammatoryactivity of IgG1 mediated by Fc galactosylation and association of FcgRIIB anddectin-1. Nat. Med. 18: 1401–1406.
34. Ludwig, R. J., S. M€uller, Ad. Marques, A. Recke, E. Schmidt, D. Zillikens,S. Moller, and S. M. Ibrahim. 2012. Identification of quantitative trait loci inexperimental epidermolysis bullosa acquisita. J. Invest. Dermatol. 132: 1409–1415.
35. Liu, Z., M. Zhao, N. Li, L. A. Diaz, and T. N. Mayadas. 2006. Differential rolesfor beta2 integrins in experimental autoimmune bullous pemphigoid. Blood 107:1063–1069.
36. Munro, J. M., J. S. Pober, and R. S. Cotran. 1991. Recruitment of neutrophilsin the local endotoxin response: association with de novo endothelial ex-pression of endothelial leukocyte adhesion molecule-1. Lab. Invest. 64: 295–299.
37. Guma, M., L. Ronacher, R. Liu-Bryan, S. Takai, M. Karin, and M. Corr. 2009.Caspase 1-independent activation of interleukin-1beta in neutrophil-predominantinflammation. Arthritis Rheum. 60: 3642–3650.
38. Liu, Z., S. D. Shapiro, X. Zhou, S. S. Twining, R. M. Senior, G. J. Giudice,J. A. Fairley, and L. A. Diaz. 2000. A critical role for neutrophil elastase inexperimental bullous pemphigoid. J. Clin. Invest. 105: 113–123.
39. Paulsson, J. M., A. Moshfegh, E. Dadfar, C. Held, S. H. Jacobson, andJ. Lundahl. 2012. In-vivo extravasation induces the expression of interleukin 1receptor type 1 in human neutrophils. Clin. Exp. Immunol. 168: 105–112.
8 IL-1 IN EBA




















