
Translation Inhibition in Apoptosis. CASPASE-DEPENDENT PKR ACTIVATION AND eIF2-alpha PHOSPHORYLATION
10
Translation Inhibition in Apoptosis CASPASE-DEPENDENT PKR ACTIVATION AND eIF2- PHOSPHORYLATION* Received for publication, April 25, 2001, and in revised form, August 17, 2001 Published, JBC Papers in Press, September 12, 2001, DOI 10.1074/jbc.M103674200 Xavier Saelens‡, Michael Kalai‡, and Peter Vandenabeele§ From the Department of Molecular Biology, Unit of Molecular Signaling and Cell Death, Flanders Interuniversity Institute for Biotechnology and Ghent University, 9000 Ghent, Belgium The protein kinase PKR is a major player in the cel- lular antiviral response, acting mainly by phosphoryla- tion of the -subunit of the eukaryotic translation initi- ation factor 2 (eIF2-) to block de novo protein synthesis. PKR activation requires binding of double-stranded RNA or PACT/RAX proteins to its regulatory domain. Since several reports have demonstrated that transla- tion is inhibited in apoptosis, we investigated whether PKR and eIF2- phosphorylation contribute to this process. We show that PKR is proteolysed and that eIF2- is phosphorylated at the early stages of apoptosis induced by various stimuli. Both events coincide with the onset of caspase activity and are prevented by caspase inhibitors. Using site-directed mutagenesis we show that PKR is specifically proteolysed at Asp 251 dur- ing cellular apoptosis. This site is cleaved in vitro by recombinant caspase-3, caspase-7, and caspase-8 and not by the proinflammatory caspase-1 and caspase-11. The released kinase domain efficiently phosphorylates eIF2- at the cognate Ser 51 residue, and its overexpres- sion in mammalian cells impairs the translation of its own mRNA and of reporter mRNAs. Our results demon- strate a new and caspase-dependent activation mode for PKR, leading to eIF2- phosphorylation and translation inhibition in apoptosis. Protein synthesis is essential to sustain life. Inhibition of translation, for example by treatment with cycloheximide (CHX), 1 often enhances the induction of apoptosis by different stimuli (1). This sensitization of apoptosis by CHX has been explained by the inhibition of translation-dependent antiapo- ptotic mechanisms (2). Another known cytotoxic synergy, both in vitro and in vivo, is the combined addition of tumor necrosis factor (TNF) and interferon (IFN) (3–5). IFNs are pleiotropic cytokines with antiviral and antiproliferative properties (6). One of the best characterized proteins induced by IFNs is the dsRNA-activated protein kinase (PKR). PKR is intimately linked with the cellular IFN-inducible antiviral response. Its activation results in host cell protein synthesis shutoff, a pow- erful countermeasure of the cell against viral infection (7–9). Not surprisingly, many viruses, which are a prime source of dsRNA, encode PKR-inhibiting molecules. Down-regulation of PKR expression by antisense mRNA in U937 cells protects these cells against TNF-induced apoptosis (10), and overexpression of the cellular PKR inhibitor-p58 IPK renders cells resistant to dsRNA- and TNF-induced apoptosis (11). Thus PKR 0/0 mouse embryonic fibroblasts are resistant to TNF-induced apoptosis (12). Conversely, induction of apoptosis by TNF increases phosphorylation of the -subunit of the het- erotrimeric eukaryotic translation initiation factor (eIF) 2 (eIF2-) (13). The mechanism linking PKR activation to the TNF-signaling pathway leading to programmed cell death is still unclear. PKR is a serine-threonine kinase consisting of two function- ally distinct domains: an amino-terminal regulatory domain and a carboxyl-terminal catalytic domain (14). The regulatory domain consists of two dsRNA-binding motifs, followed by a spacer. Binding of dsRNA exposes the catalytic site and in- duces dimerization of PKR. Dimerization allows autophospho- rylation, rendering PKR active (15–19). Once the enzyme is activated, dsRNA is dispensable for further substrate phospho- rylation. Recently, also dsRNA-independent protein activators of PKR, such as PACT (human) and RAX (mouse), have been identified (20, 21). Activated PKR can phosphorylate eIF2-. eIF2 forms a ter- nary complex with the methionyl initiator tRNA (Met-tRNA i - Met ) and GTP. This complex is essential for recruiting Met- tRNA i Met to the 40 S ribosomal subunit and initiating mRNA translation. After delivery of Met-tRNA i Met at the proper AUG initiation codon of an mRNA, eIF2-bound GTP is hydrolyzed to GDP. Subsequent loading of eIF2 with Met-tRNA i Met requires exchange of eIF2-bound GDP by GTP, a reaction catalyzed by the guanine nucleotide exchange factor eIF2B. Phosphoryla- tion of eIF2- converts eIF2 to a competitive inhibitor of eIF2B, resulting in a general protein synthesis shutdown (22, 23). Phosphorylation of only a fraction of the cellular eIF2- is sufficient to severely impair translation initiation, because eIF2B is a limiting component of the cellular translation ma- chinery (24, 25). Apoptosis involves ordered activation of cysteinyl aspartate- specific proteinases (caspases), giving rise to cleavage of a particular subset of the cellular proteome (26). These cleavage * This work was supported in part by the Interuniversitaire Attrac- tiepolen, the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen (Grants 31.5189.00 and 3G.0006.01), and the EC-RTD (Grant QLRT- 1999-00739). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. ‡ These authors contributed equally to this work. § To whom correspondence should be addressed: Dept. of Molecular Biology, K. L. Ledeganckstraat 35, B-9000 Ghent, Belgium. Tel.: 32-9- 264-51-31; Fax: 32-9-264-53-48; E-mail: [email protected]. ac.be. 1 The abbreviations used are: CHX, cycloheximide; Ac-DEVD-amc, acetyl-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-aminomethylcoumarin; eIF, eukaryotic translation initiation factor; GFP, green fluorescent protein; EGFP, enhanced GFP; GPD, glyceraldehyde-3-phosphate dehydrogen- ase; IRES, internal ribosome entry site; KDcasp, caspase-generated kinase domain; PAGE, polyacrylamide gel electrophoresis; PARP, poly- (ADP-ribose) polymerase; ds, double-stranded; PKR, dsRNA-activated protein kinase; PMSF, phenylmethylsulfonyl fluoride; STS, staurospo- rine; TNF, tumor necrosis factor; IFN, interferon; wt, wild-type; zDEVD-cmk, benzyloxycarbonyl-Asp-Glu-Val-Asp-chloromethylketone; zVAD-fmk, benzyloxycarbonyl-Val-Ala-DL-Asp(OMe)-fluoromethylke- tone; PBS, phosphate-buffered saline. THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 276, No. 45, Issue of November 9, pp. 41620 –41628, 2001 © 2001 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. This paper is available on line at http://www.jbc.org 41620 by guest on December 2, 2015 http://www.jbc.org/ Downloaded from
Transcript of Translation Inhibition in Apoptosis. CASPASE-DEPENDENT PKR ACTIVATION AND eIF2-alpha PHOSPHORYLATION

Translation Inhibition in ApoptosisCASPASE-DEPENDENT PKR ACTIVATION AND eIF2-� PHOSPHORYLATION*
Received for publication, April 25, 2001, and in revised form, August 17, 2001Published, JBC Papers in Press, September 12, 2001, DOI 10.1074/jbc.M103674200
Xavier Saelens‡, Michael Kalai‡, and Peter Vandenabeele§
From the Department of Molecular Biology, Unit of Molecular Signaling and Cell Death, Flanders InteruniversityInstitute for Biotechnology and Ghent University, 9000 Ghent, Belgium
The protein kinase PKR is a major player in the cel-lular antiviral response, acting mainly by phosphoryla-tion of the �-subunit of the eukaryotic translation initi-ation factor 2 (eIF2-�) to block de novo protein synthesis.PKR activation requires binding of double-strandedRNA or PACT/RAX proteins to its regulatory domain.Since several reports have demonstrated that transla-tion is inhibited in apoptosis, we investigated whetherPKR and eIF2-� phosphorylation contribute to thisprocess. We show that PKR is proteolysed and thateIF2-� is phosphorylated at the early stages of apoptosisinduced by various stimuli. Both events coincide withthe onset of caspase activity and are prevented bycaspase inhibitors. Using site-directed mutagenesis weshow that PKR is specifically proteolysed at Asp251 dur-ing cellular apoptosis. This site is cleaved in vitro byrecombinant caspase-3, caspase-7, and caspase-8 andnot by the proinflammatory caspase-1 and caspase-11.The released kinase domain efficiently phosphorylateseIF2-� at the cognate Ser51 residue, and its overexpres-sion in mammalian cells impairs the translation of itsown mRNA and of reporter mRNAs. Our results demon-strate a new and caspase-dependent activation mode forPKR, leading to eIF2-� phosphorylation and translationinhibition in apoptosis.
Protein synthesis is essential to sustain life. Inhibition oftranslation, for example by treatment with cycloheximide(CHX),1 often enhances the induction of apoptosis by differentstimuli (1). This sensitization of apoptosis by CHX has beenexplained by the inhibition of translation-dependent antiapo-
ptotic mechanisms (2). Another known cytotoxic synergy, bothin vitro and in vivo, is the combined addition of tumor necrosisfactor (TNF) and interferon (IFN) (3–5). IFNs are pleiotropiccytokines with antiviral and antiproliferative properties (6).One of the best characterized proteins induced by IFNs is thedsRNA-activated protein kinase (PKR). PKR is intimatelylinked with the cellular IFN-inducible antiviral response. Itsactivation results in host cell protein synthesis shutoff, a pow-erful countermeasure of the cell against viral infection (7–9).Not surprisingly, many viruses, which are a prime source ofdsRNA, encode PKR-inhibiting molecules.
Down-regulation of PKR expression by antisense mRNA inU937 cells protects these cells against TNF-induced apoptosis(10), and overexpression of the cellular PKR inhibitor-p58IPK
renders cells resistant to dsRNA- and TNF-induced apoptosis(11). Thus PKR0/0 mouse embryonic fibroblasts are resistant toTNF-induced apoptosis (12). Conversely, induction of apoptosisby TNF increases phosphorylation of the �-subunit of the het-erotrimeric eukaryotic translation initiation factor (eIF) 2(eIF2-�) (13). The mechanism linking PKR activation to theTNF-signaling pathway leading to programmed cell death isstill unclear.
PKR is a serine-threonine kinase consisting of two function-ally distinct domains: an amino-terminal regulatory domainand a carboxyl-terminal catalytic domain (14). The regulatorydomain consists of two dsRNA-binding motifs, followed by aspacer. Binding of dsRNA exposes the catalytic site and in-duces dimerization of PKR. Dimerization allows autophospho-rylation, rendering PKR active (15–19). Once the enzyme isactivated, dsRNA is dispensable for further substrate phospho-rylation. Recently, also dsRNA-independent protein activatorsof PKR, such as PACT (human) and RAX (mouse), have beenidentified (20, 21).
Activated PKR can phosphorylate eIF2-�. eIF2 forms a ter-nary complex with the methionyl initiator tRNA (Met-tRNAi-Met) and GTP. This complex is essential for recruiting Met-tRNAi
Met to the 40 S ribosomal subunit and initiating mRNAtranslation. After delivery of Met-tRNAi
Met at the proper AUGinitiation codon of an mRNA, eIF2-bound GTP is hydrolyzed toGDP. Subsequent loading of eIF2 with Met-tRNAi
Met requiresexchange of eIF2-bound GDP by GTP, a reaction catalyzed bythe guanine nucleotide exchange factor eIF2B. Phosphoryla-tion of eIF2-� converts eIF2 to a competitive inhibitor of eIF2B,resulting in a general protein synthesis shutdown (22, 23).Phosphorylation of only a fraction of the cellular eIF2-� issufficient to severely impair translation initiation, becauseeIF2B is a limiting component of the cellular translation ma-chinery (24, 25).
Apoptosis involves ordered activation of cysteinyl aspartate-specific proteinases (caspases), giving rise to cleavage of aparticular subset of the cellular proteome (26). These cleavage
* This work was supported in part by the Interuniversitaire Attrac-tiepolen, the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen(Grants 31.5189.00 and 3G.0006.01), and the EC-RTD (Grant QLRT-1999-00739). The costs of publication of this article were defrayed inpart by the payment of page charges. This article must therefore behereby marked “advertisement” in accordance with 18 U.S.C. Section1734 solely to indicate this fact.
‡ These authors contributed equally to this work.§ To whom correspondence should be addressed: Dept. of Molecular
Biology, K. L. Ledeganckstraat 35, B-9000 Ghent, Belgium. Tel.: 32-9-264-51-31; Fax: 32-9-264-53-48; E-mail: [email protected].
1 The abbreviations used are: CHX, cycloheximide; Ac-DEVD-amc,acetyl-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-aminomethylcoumarin; eIF,eukaryotic translation initiation factor; GFP, green fluorescent protein;EGFP, enhanced GFP; GPD, glyceraldehyde-3-phosphate dehydrogen-ase; IRES, internal ribosome entry site; KDcasp, caspase-generatedkinase domain; PAGE, polyacrylamide gel electrophoresis; PARP, poly-(ADP-ribose) polymerase; ds, double-stranded; PKR, dsRNA-activatedprotein kinase; PMSF, phenylmethylsulfonyl fluoride; STS, staurospo-rine; TNF, tumor necrosis factor; IFN, interferon; wt, wild-type;zDEVD-cmk, benzyloxycarbonyl-Asp-Glu-Val-Asp-chloromethylketone;zVAD-fmk, benzyloxycarbonyl-Val-Ala-DL-Asp(OMe)-fluoromethylke-tone; PBS, phosphate-buffered saline.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 276, No. 45, Issue of November 9, pp. 41620–41628, 2001© 2001 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org41620
by guest on Decem
ber 2, 2015http://w
ww
.jbc.org/D
ownloaded from

events most often lead to loss of function of the target protein.In a number of cases, however, the substrate becomes activatedafter cleavage due to removal of regulatory domains (27).
Since PKR activation by an unknown mechanism has beendescribed downstream of TNF-induced apoptosis (10, 12, 13),we searched for a possible mechanism of PKR activation andconsequent phosphorylation of eIF2-� during extrinsic apo-ptosis by death domain receptor aggregation and during apo-ptosis by staurosporine (STS), a broad-spectrum kinase inhib-itor. In both apoptotic pathways phosphorylation of eIF2-�, aswell as proteolysis and activation of PKR, occur in a caspase-dependent manner, illustrating the existence of a novel, caspase-dependent mode of PKR activation.
MATERIALS AND METHODS
Cell Culture and Apoptosis Assays—Jurkat T cells and promonocyticU937 cells were cultured in RPMI supplemented with 10% fetal bovineserum, 1 mM L-glutamine, 100 units/ml penicillin, and 100 �g/ml strep-tomycin. Jurkat T cells were transfected with DMRIE-C reagent ac-cording to the manufacturer’s instructions (Life Technologies, Paisley,UK). Apoptosis was induced in Jurkat T cells by treatment with 100ng/ml CH-11 anti-Fas antibody (BioCheck, Burlingame, CA) or withSTS (Sigma). In U937 cells, apoptosis was induced with 5,000 IU/mlTNF in the presence of 10 �g/ml CHX. Inhibition of caspases wasobtained by preincubating cells for 30 min with 50 �M benzyloxycarbonyl-Val-Ala-DL-Asp(OMe)-fluoromethylketone (zVAD-fmk) or benzyloxy-carbonyl-Asp-Glu-Val-Asp-chloromethylketone (zDEVD-cmk) (Bachem,Bubendorf, Switzerland) before addition of an apoptotic stimulus. Celldeath was monitored by trypan blue exclusion.
At different time points after addition of anti-Fas antibody, cellswere collected by centrifugation, washed twice with ice-cold phosphate-buffered saline (PBS), and lysed on ice with cell-free system buffercontaining 220 mM mannitol, 68 mM sucrose, 2 mM NaCl, 2.5 mM
KH2PO4, 10 mM Hepes, pH 7.4, 1 mM aprotinin, 1 mM leupeptin, and100 �M phenylmethylsulfonyl fluoride (PMSF), supplemented with0.05% Nonidet-P40 and 1 mM oxidized glutathione. Supernatant (25 �gof 14,000 � g) was analyzed for caspase activity at 37 °C in the presenceof 50 �M of acetyl-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-aminomethyl-coumarin (Ac-DEVD-amc) (Peptide Institute, Osaka, Japan) as de-scribed previously (28). The relative caspase activity was defined as theratio of maximal Ac-DEVD-amc cleavage activity in test samples versusblank samples.
Metabolic Labeling of Proteins—Jurkat T cells (5 � 105/ml) weretreated with anti-Fas antibody or were left untreated. At selected timepoints, 1.5 � 106 cells were transferred to six-well plates and labeled for30 min with 10 �Ci of Trans35S-label (ICN Biomedicals, Irvine, CA).Cells were washed twice with ice-cold PBS and lysed on ice with lysisbuffer containing 20 mM Tris-HCl, pH 7.5, 50 mM KCl, 5 mM MgCl2, 400mM NaCl, 2 mM dithiothreitol, 20% glycerol, 1% Triton X-100, 1 mM
aprotinin, 1 mM leupeptin, and 100 �M PMSF. Supernatant fraction (30�g of protein of 14,000 � g) was resolved on SDS-polyacrylamide gelelectrophoresis (PAGE) and visualized by Coomassie Brilliant Bluestaining. The same gel was used for autoradiography.
Immunoblot Analysis—Protein extracts from cells (106 cells/sample)were prepared by lysis with sample buffer containing 6% SDS, 1.4 M
�-mercaptoethanol, 20% glycerol, 0.01% w/v bromphenol blue, and 125mM Tris-HCl, pH 6.8. Samples were boiled for 10 min, separated in12.5% SDS-PAGE gels, and transferred to nitrocellulose. Blocking, an-tibody incubation steps, and washing of the membrane were performedin Tris-buffered saline Tween containing 10 mM Tris-HCl, pH 8.0, 150mM NaCl, and 0.05% Tween 20, supplemented with 3% skimmed milk.Antibodies specific for the regulatory and kinase domain of human PKRwere purchased from Transduction Laboratories (Lexington, KY) andSanta Cruz Biotechnology (Santa Cruz, CA), respectively. Antibodiesagainst phosphorylated and unphosphorylated eIF2-� were obtainedfrom Research Genetics (Huntsville, AL) and Santa Cruz Biotechnol-ogy. Antibodies to human caspase-3, eIF4G, and poly(ADP-ribose)polymerase (PARP) were supplied by BIOSOURCE International(Camarillo, CA), Transduction Laboratories, and Biomol Research Lab-oratories (Plymouth Meeting, PA), respectively. Membranes were incu-bated with horseradish peroxidase-conjugated secondary antibodies tomouse and rabbit immunoglobulin (Amersham Pharmacia Biotech,Rainham, UK), as well as to goat antibody (Santa Cruz Biotechnology).Proteins were visualized using a chemiluminescence substrate (Du-Pont), and blots were exposed to film (Amersham Pharmacia Biotech).Signals were quantified by densitometric analysis (except for eIF2-�)and captured by a Lumi-Imager work station (Roche Molecular Bio-chemicals, Basel, Switzerland).
Plasmid Constructs—Plasmid constructs were made using currentmolecular biology techniques. Human PKR cDNA cloned in-frame withNH2-terminal His tag in pET15b was a kind gift from Dr. B. Williams(The Cleveland Clinic Foundation, Cleveland, OH). cDNA was trans-ferred to a pCDM8 vector (Invitrogen, San Diego, CA) with and withoutNH2-terminal His tag to obtain pCDM8-His-PKR and pCDM8-PKR,respectively. PKR-K296R, PKRD251N, and PKRD251NK296R mutants(Asp251 to Asn251) were generated by overlap polymerase chain reaction
FIG. 1. Inhibition of de novo protein synthesis and eIF2-� phosphorylation during Fas-mediated apoptosis in Jurkat T cells. A,apoptotic morphology of anti-Fas-treated cells as seen by light microscopy. B, caspase inhibitors delay Fas-mediated apoptosis. Cells werepretreated for 30 min with 50 �M zVAD-fmk or zDEVD-cmk, before an anti-Fas challenge. Cell death monitored by trypan blue exclusion isrepresented as means � S.D. from three independent experiments. C, inhibition of de novo protein synthesis. Cells were pulse-labeled for 30 minwith a mixture of [35S]L-methionine/cysteine, and cytosolic extracts were prepared. Equal amounts of total protein were separated by SDS-PAGE.Gels were stained with Coomassie Blue (left) and autoradiographed (right). D, eIF2-� phosphorylation during Fas-mediated apoptosis. Control (left)and anti-Fas-treated (right) cell lysates were analyzed by immunoblot with antibodies specific for Ser51-phosphorylated eIF2-� (eIF2-�P) and totaleIF2-� (eIF2-�T). The immunoreactive signal of intact eIF2-� was quantified densitometrically and the ratio eIF2-�P/eIF2-�T, normalized for 0 h,was determined. Arrowheads indicate cleavage products of eIF2-�.
Caspase-dependent PKR Activation 41621
by guest on Decem
ber 2, 2015http://w
ww
.jbc.org/D
ownloaded from

and cloned into pEF6/Myc-His C (Invitrogen) in-frame with a COOH-terminal Myc tag. pEF6A-E-tag-PKR-caspase-generated kinase domain(KDcasp) and pEF6A-E-tag-PKR-KDcasp K296R were obtained bytransferring cDNA coding for PKR and PKR K296R starting fromMet251, in-frame with an NH2-terminal E-tag (amino acid sequenceMGAPVPYPDPLEPRAAA), and inserted into a pEF6A/Myc-His A vec-tor (Invitrogen). Human eIF2-� cDNA was obtained from HeLa cellstotal RNA by reverse transcriptase-polymerase chain reaction andcloned into pEF6/Myc-His A in-frame with COOH-terminal tags to yieldpEF6-eIF2-�-Myc-His. Mutant eIF2-� S51A was obtained by overlappolymerase chain reaction. All introduced mutations and tag fusionswere confirmed by sequencing on an ABI373A sequencer (Applied Bio-systems, Foster City, CA).
In Vitro Cleavage Reactions—pCDM8-based PKR, amino-terminalHis-tagged PKR, and PKRD251N vectors (1 �g) were used as templatefor in vitro coupled transcription translation in a reticulocyte lysatesystem (Promega Biotec, Madison, WI). Translation reaction (2 �l) wasincubated with 200 nM recombinant-urified caspases (28) in a totalvolume of 25 �l of cell-free system buffer for 1.5 h at 37 °C. Theresulting cleavage products were analyzed on SDS-PAGE gels andautoradiography.
Kinase Assays—HEK293T were transiently transfected, using cal-cium phosphate precipitation, in 100-mm culture dishes with 5 �g ofdifferent PKR- and eIF2-�-encoding constructs. PKR, PKRK296R,eIF2-�, and eIF2-�S51A constructs were Myc-tagged; PKR-KD andPKR-KDK296R constructs were E-tagged. 36 h after transfection, cellswere harvested and washed twice with ice-cold PBS. Cell lysis, immu-noprecipitation, and kinase reaction were performed as described pre-viously (20). Briefly, tagged proteins were isolated from 500 �g of celllysate by overnight immunoprecipitation at 4 °C with anti-c-myc (RocheMolecular Biochemicals) or E-tag antibody (Amersham Pharmacia Bio-tech), followed by addition of protein A Trisacryl (Pierce). Immunecomplexes were washed twice with lysis buffer and twice with high saltbuffer containing 20 mM Tris-HCl, pH 7.5, 50 mM KCl, 400 mM NaCl, 1mM EDTA, 1 mM dithiothreitol, 20% glycerol, 1% Triton X-100, 1 mM
aprotinin, 1 mM leupeptin, and 100 �M PMSF. After the last wash,immune complexes were resuspended in 70 �l of kinase buffer contain-ing 20 mM Tris-HCl, pH 7.5, 50 mM KCl, 2 mM MgCl2, 2 mM MnCl2, 5%glycerol, and 100 �M PMSF. Kinase reactions were set up by mixing 10�l of the respective immune complexes with 5 �Ci of [�-32P]ATP (3,000Ci/mmol) in 25 �l total volume. Reactions were allowed for 15 min at30 °C and were analyzed by SDS-PAGE and autoradiography. A reac-tion without ATP added was set up in parallel and analyzed by Westernblot to estimate the input.
Translation Inhibition Assays—Transfections of HEK293T cells weredone using calcium phosphate precipitation in six-well plates with 300ng of the respective pEFPKR vector. To assay for translation inhibitionof PKR and PKR-KDcasp, cells from three independent transfectionswere pooled 36 h after transfection, washed in PBS, and devided in two.One half was used for Western blot analysis of expressed proteins, theother half was used for Northern blot analysis of total RNA isolatedwith an RNeasy kit (Qiagen, Chatsworth, CA). 10 �g of total RNA wasdenatured in glyoxal and Me2SO (29), separated in a phosphate-buff-ered 1% agarose gel, and transferred onto a nylon membrane. Filterswere hybridized with an [�-32P]dCTP-labeled PKR-KDcasp cDNA frag-ment, stripped, and rehybridized with a glyceraldehyde-3-phosphatedehydrogenase (GPD) probe. Western blot signals were quantitatedwith a Lumi-Imager, normalized for intensity of anti-�-actin. Northernblot signals were quantified with a Lumi-Imager, normalized for inten-sity of GPD.
The effect of PKR and PKR-KDcasp expression on translation of areporter mRNA was assayed by co-transfecting 300 ng of the respectivePKR constructs and 50 ng of a modified pEGFP-N1 plasmid (CLONTECH),encoding green fluorescent protein (GFP) with a nuclear localization sig-nal. 48 h after transfection, GFP expression was monitored by Westernblot with an anti-GFP antibody (CLONTECH). Western blot results wererepeated seven times with similar results. Northern blot analysis wasdone on total RNA from cells pooled from four independent transfectionsand probed with a BamHI-NotI EGFP fragment. Normalization of signalintensities was as described above. In addition, increasing amounts ofpEFPKR-KDcasp and pEFPKR-K296R plasmid were co-transfected with100 ng of pCAGGS encoding A/Victoria/3/75 influenza virus hemaggluti-nin (30). 48 h after transfection, cell lysates were assayed for expression ofhemagglutinin and PKR-KDcasp.
RESULTS
eIF2-� Is Phosphorylated during Apoptosis—Stimulation ofthe death domain-containing Fas receptor in Jurkat cells wasused as a prototype apoptotic model to assess if inhibition oftranslation occurs during apoptosis. Light microscopy exami-nation confirmed that anti-Fas-treated Jurkat cells rapidlydied from apoptosis, as exemplified by cell shrinkage and mem-brane blebbing (Fig. 1A). Cytotoxicity was caspase-dependent,since zVAD-fmk and zDEVD-cmk significantly delayed celldeath (Fig. 1B). Pulse labeling of cells with radiolabeled me-thionine and cysteine revealed that de novo protein synthesiswas inhibited starting from the third hour of anti-Fas treat-ment (Fig. 1C).
In eukaryotic cells, protein synthesis is tightly controlled atthe level of initiation (22). The heterotrimeric eukaryotic trans-lation initiation factor eIF2 recruits the initiator Met-tRNAi
Met
to the ribosome. This recruitment is severely impaired by phos-phorylation of the regulatory �-subunit of eIF2, viz. eIF2-�.Therefore, we monitored the degree of eIF2-� phosphorylationin the course of Fas-mediated apoptosis by Western blottingusing antibodies specific for phosphorylated and nonphospho-rylated eIF2-� (31). Increased eIF2-� phosphorylation was de-tected 3–4 h after anti-Fas treatment (Fig. 1D). Other groupsreported on the caspase-mediated cleavage of eIF2-� and sug-gested that it contributes to the shutdown of protein synthesisin apoptosis (32, 33). With both eIF2-�-specific antibodies weobserved minor proteolysis of the 35-kDa eIF2-� into a 32-kDafragment, coinciding with an increase in eIF2-� phosphoryla-tion (Fig. 1D). These proteolytic and phosphorylation eventsoccurred while the majority of the cells still had intact mem-branes, as demonstrated by trypan blue exclusion. This meansthat the events documented occur at least in the executioner
FIG. 2. Phosphorylation of eIF2-� coincides with and dependson caspase activation during anti-Fas-induced apoptosis inJurkat T cells. A, caspase activity and eIF2-� phosphorylation concurduring apoptosis. Control cells (open symbols) and anti-Fas-treated cells(filled symbols) were assayed for rate of Ac-DEVD-amc cleavage activity(circles). The same samples were used for immunoblotting with anti-bodies specific for Ser51-phosphorylated eIF2-� (eIF2-�P) and totaleIF2-� (eIF2-�T). Following quantification, the normalized ratio eIF2-�P/eIF2-�T was plotted as a function of time after anti-Fas treatment.Only the band intensity of intact eIF2-� was taken into account(squares). B, inhibition of eIF2-� phosphorylation by caspase inhibitors.Lysates were made from control and anti-Fas-treated cells, with orwithout pretreatment for 30 min with 50 �M of zVAD-fmk or zDEVD-cmk, and analyzed by immunoblotting with antibodies. Band intensi-ties were quantified by densitometry.
Caspase-dependent PKR Activation41622
by guest on Decem
ber 2, 2015http://w
ww
.jbc.org/D
ownloaded from
phase of the apoptotic process (Fig. 1B).eIF2-� Phosphorylation Is Caspase-dependent—To explore
the relationship between proteolytic activation of caspases andphosphorylation of eIF2-�, we compared the time kinetics ofthe enzymatic activity of caspases in cytosolic extracts of anti-Fas-triggered cells with the ratio of phosphorylated versus totaleIF2-� (Fig. 2A). The data demonstrate that inhibition of newprotein synthesis and eIF2-� phosphorylation are both co-lin-ear with DEVDase activity (Figs. 1C and 2A). Furthermore,treatment of Jurkat cells with zVAD-fmk or zDEVD-cmk priorto anti-Fas exposure abolished phosphorylation of eIF2-� aswell as caspase-3 maturation (Fig. 2B). These results suggestthat phosphorylation of eIF2-� coincides with and depends onactivation of caspases.
PKR Cleavage and eIF2-� Phosphorylation Concur duringApoptosis—A conceivable molecular mechanism to explain the
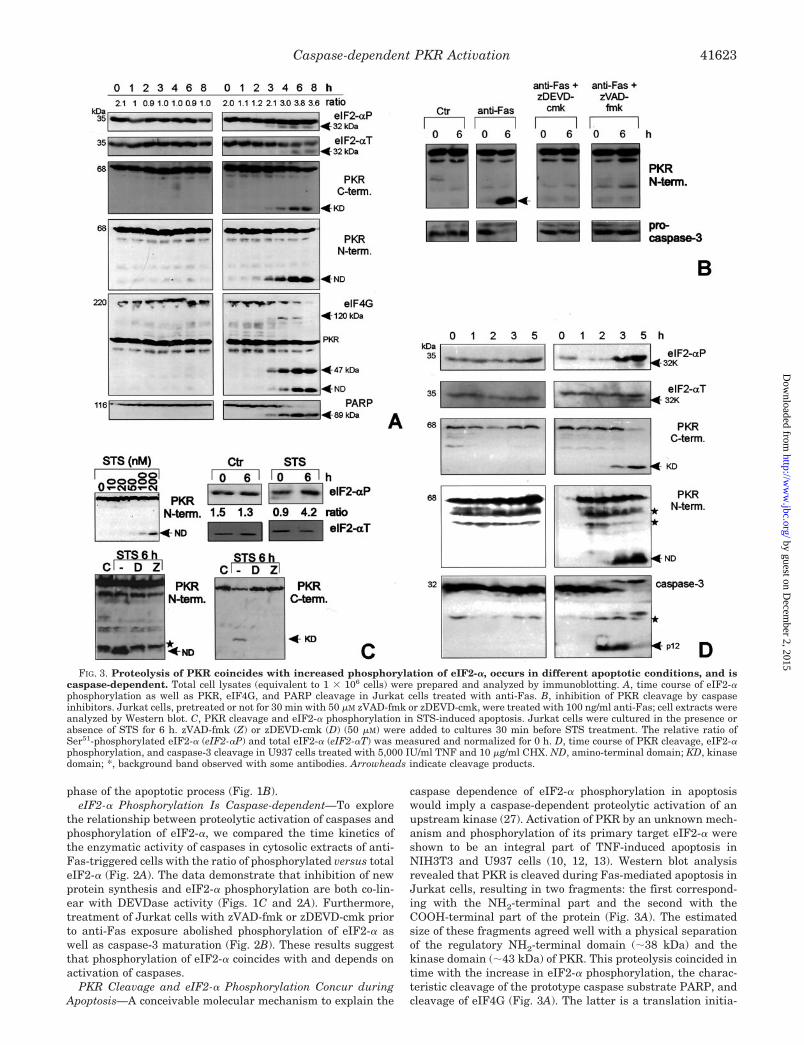
caspase dependence of eIF2-� phosphorylation in apoptosiswould imply a caspase-dependent proteolytic activation of anupstream kinase (27). Activation of PKR by an unknown mech-anism and phosphorylation of its primary target eIF2-� wereshown to be an integral part of TNF-induced apoptosis inNIH3T3 and U937 cells (10, 12, 13). Western blot analysisrevealed that PKR is cleaved during Fas-mediated apoptosis inJurkat cells, resulting in two fragments: the first correspond-ing with the NH2-terminal part and the second with theCOOH-terminal part of the protein (Fig. 3A). The estimatedsize of these fragments agreed well with a physical separationof the regulatory NH2-terminal domain (�38 kDa) and thekinase domain (�43 kDa) of PKR. This proteolysis coincided intime with the increase in eIF2-� phosphorylation, the charac-teristic cleavage of the prototype caspase substrate PARP, andcleavage of eIF4G (Fig. 3A). The latter is a translation initia-
FIG. 3. Proteolysis of PKR coincides with increased phosphorylation of eIF2-�, occurs in different apoptotic conditions, and iscaspase-dependent. Total cell lysates (equivalent to 1 � 106 cells) were prepared and analyzed by immunoblotting. A, time course of eIF2-�phosphorylation as well as PKR, eIF4G, and PARP cleavage in Jurkat cells treated with anti-Fas. B, inhibition of PKR cleavage by caspaseinhibitors. Jurkat cells, pretreated or not for 30 min with 50 �M zVAD-fmk or zDEVD-cmk, were treated with 100 ng/ml anti-Fas; cell extracts wereanalyzed by Western blot. C, PKR cleavage and eIF2-� phosphorylation in STS-induced apoptosis. Jurkat cells were cultured in the presence orabsence of STS for 6 h. zVAD-fmk (Z) or zDEVD-cmk (D) (50 �M) were added to cultures 30 min before STS treatment. The relative ratio ofSer51-phosphorylated eIF2-� (eIF2-�P) and total eIF2-� (eIF2-�T) was measured and normalized for 0 h. D, time course of PKR cleavage, eIF2-�phosphorylation, and caspase-3 cleavage in U937 cells treated with 5,000 IU/ml TNF and 10 �g/ml CHX. ND, amino-terminal domain; KD, kinasedomain; *, background band observed with some antibodies. Arrowheads indicate cleavage products.
Caspase-dependent PKR Activation 41623
by guest on Decem
ber 2, 2015http://w
ww
.jbc.org/D
ownloaded from

tion factor essential for mRNA recruitment to the ribosome. Itscleavage by caspases was suggested to be involved in the inhi-bition of cellular translation in apoptosis (34, 35). Treatmentwith the caspase inhibitors zVAD-fmk or zDEVD-cmk prior toanti-Fas exposure suppressed proteolysis of PKR and proteo-lytic maturation of caspase-3, suggesting that caspases led tocleavage of PKR (Fig. 3B). Similar results were obtained inapoptotic cell death mediated by STS, a potent Ser/Thr proteinkinase inhibitor that does not affect the eIF2-� kinase activityof PKR (Fig. 3C) (36), as well as in promonocytic U937 cellstreated with TNF (Fig. 3D). These results demonstrate thatPKR is proteolysed in apoptosis induced by death domain re-ceptor aggregation and in apoptotic cell death initiated by STS.PKR fragmentation in these apoptotic conditions coincides withphosphorylation of eIF2-�.
Identification of the Caspase Cleavage Site in PKR—To iden-tify the nature of the caspase(s) implicated and to demonstratethat PKR is a direct substrate for caspases, a panel of purifiedrecombinant caspases (28) was coincubated with [35S]methi-onine-labeled PKR. Among the six proteases tested, the apo-ptotic caspase-3, �7, and �8, and to some extent also caspase-6,could cleave PKR, whereas the inflammatory caspase-1 and �11did not (Fig. 4A). Cleavage of PKR in apoptotic cells resulted intwo peptide fragments of different sizes (�38 kDa for the NH2
terminus and 43 kDa for the COOH terminus). But the in vitro
FIG. 4. Identification of Asp251 in PKR as caspase cleavage site.In vitro translated 35S-labeled PKR was incubated with purifiedcaspases for 1 h at 37 °C. Reaction products were separated by SDS-PAGE; gels were dried and exposed to film (A, B, and D). Arrowheadsindicate cleavage products. A, recombinant purified caspases cleaveamino-terminal His-tagged PKR. B, wt and His-tagged PKR cleaved bycaspase-7. C, representation of PKR wt and D251N mutant. BD3, thirdbasic domain. Catalytic Lys296 is represented by a diamond. D, PKRsite-specific mutant D251N is resistant to cleavage by caspase-3, �7,and �8. E, PKRD251N mutant is not cleaved during Fas-mediatedapoptosis. Jurkat T cells were transiently transfected withpEFPKRK296R or pEFPKRK296RD251N, and treated with or withoutanti-Fas (500 ng/ml) for 4 h; cell extracts were subjected to Westernblotting with anti-Myc antibody. An aliquot of each lysate was assayedfor rate of Ac-DEVD-amc cleavage activity.
FIG. 5. Caspase-generated PKR-KDcasp inhibits its own trans-lation. A, representation of PKR and the kinase domain of PKR (PKR-KDcasp) generated by caspase cleavage. BD3, third basic domain; dia-mond, catalytic Lys296; *, phosphorylation sites. B, expression vectorsfor PKR, PKRK296R, PKR-Kdcasp, and PKR-KDcaspK296R weretransfected in HEK293T cells. 36 h after transfection, cells were col-lected, and half of the cell pellet was used to monitor the expression ofPKR (PKR) and KDcasp (KD) with anti-Myc and anti-E-tag, respec-tively, or �-actin by Western blot. Total RNA was isolated from theother half of the cell pellet and analyzed by Northern blot (N) probedwith a PKR-KD fragment or GPD.
Caspase-dependent PKR Activation41624
by guest on Decem
ber 2, 2015http://w
ww
.jbc.org/D
ownloaded from

cleavage assay seemingly yielded two closely running fragmentsof �40 kDa. This discrepancy with apoptotic cell lysates can beattributed to the NH2-terminal polyhistidine tag present in thein vitro translated PKR. Indeed, caspase-7 proteolysis performedon in vitro translated PKR without His tag revealed doublebands, resembling the fragments observed in apoptotic cell ly-sates (Fig. 4B). Analysis of the primary amino acid sequence ofPKR suggested Asp248-Leu-Pro-Asp251 as a potential caspasecleavage site that would generate the fragments observed duringapoptosis. Therefore, Asp251 was mutated to Asn, and the result-ing protein was tested for susceptibility to cleavage by caspase-3,�7, and �8 (Fig. 4, C and D). The PKR-D251N mutant wascompletely resistant to proteolysis by caspase-3 and �8, althoughsome minor proteolysis still occurred, mostly with caspase-7, butwith a different cleavage pattern. These in vitro data are consist-ent with the results obtained in apoptotic cells and show thatPKR is a potential substrate for caspase-3, �7, and �8. Toconfirm that Asp251 was the PKR cleavage site used in vivoduring apoptosis, we transfected Jurkat T cells with PKRK296R,PKRK296R, and D251N double-mutant cDNAs. 4 h after anti-Fas treatment, the tagged COOH-terminal PKR fragment wasdetected in cells transfected with the PKR construct containingthe wild-type (wt) caspase target site, but not in cells expressingthe D251N mutant (Fig. 4E). This demonstrates that PKR iscleaved by caspases at Asp251 during apoptotic cell death.
The Caspase-generated PKR Kinase Domain Is Active—Sev-eral structure-function studies have shown that the isolatedkinase domain of PKR can phosphorylate eIF2-� and inhibittranslation in vivo (17, 37, 38). Since these reports involved akinase domain different from the one released by caspases (Fig.5A), we ectopically expressed wt and catalytically inactiveK296R mutants of PKR and PKR-KDcasp in HEK293T cells,after which the autophosphorylation and eIF2-� phosphoryla-tion properties were assessed. Western blot analysis revealedthat the expression of wt PKR and PKR-KDcasp in transienttransfection experiments was lower than that of the PKR-K296R and PKR-KDcasp-K296R mutants (Fig. 5B). Northernblot analysis demonstrated that the difference in protein ex-pression level of wt versus K296R mutant PKR and PKR-
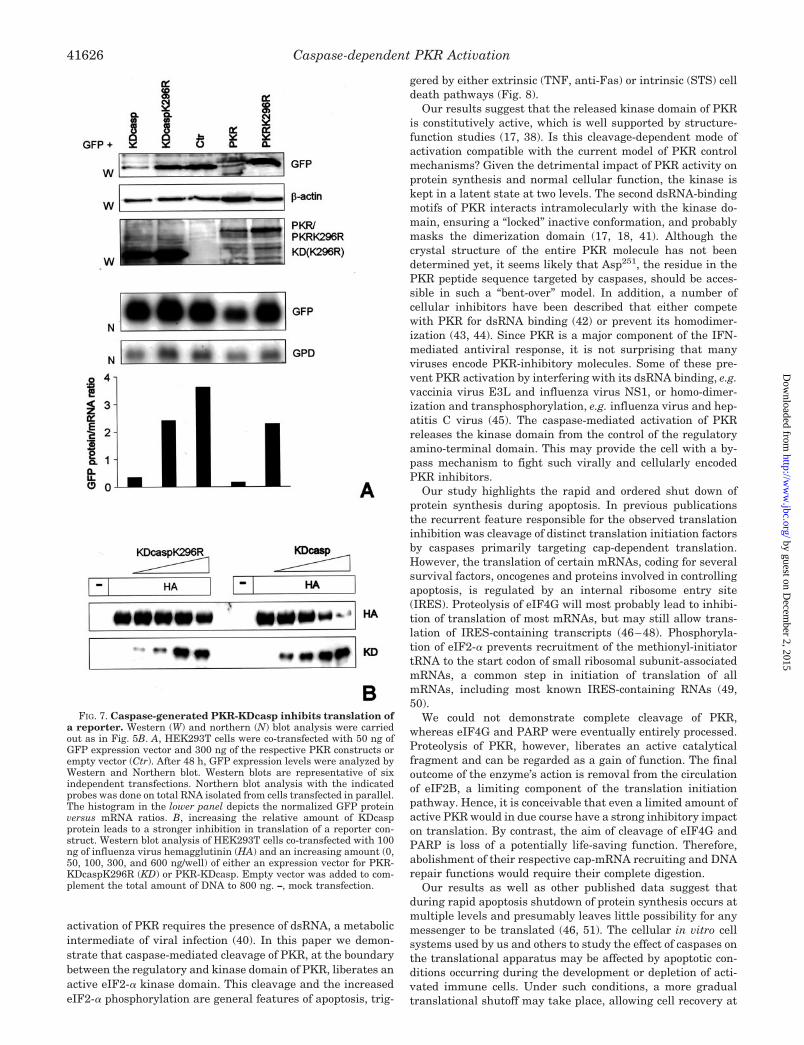
KDcasp was due to a lower translation efficiency, suggestingthat PKR and PKR-KDcasp can inhibit their own translation.
To further characterize the activity of PKR-KDcasp, an invitro kinase assay was performed. Tagged PKR and mutantderivatives were immunopurified from transfected HEK293Tlysates and incubated with [�-32P]ATP. Strong autophospho-rylation was observed with full-size wt PKR, whereas auto-phosphorylation of PKR-KDcasp was almost below detectionlevel (Fig. 6A). However, examination of the ability of PKR-KDcasp to phosphorylate eIF2-� demonstrated that caspase-generated PKR-KDcasp is fully capable to phosphorylateeIF2-� and to the same extent as full-size PKR (Fig. 6B). Thiskinase activity was dependent on the presence of Lys296, whichis directly involved in phosphate transfer, since the PKR-KD-casp-K296R mutant did not phosphorylate eIF2-� (Fig. 6B). Toverify that eIF2-� was phosphorylated at the orthodox phos-phorylation site (39), Ser51 was substituted by Ala by site-specific mutagenesis, generating eIF2-� S51A. Indeed, PKRand PKR-KDcasp failed to phosphorylate the eIF2-� S51A mu-tant, demonstrating that Ser51 is the target for transphospho-rylation by both PKR forms (Fig. 6C).
To assess whether the translation inhibition of overex-pressed PKR-KDcasp could be extended to other mRNAs, weco-expressed PKR-KDcasp with GFP and assayed its expres-sion in Western blot. Co-expression of PKR-KDcasp or PKRwith GFP decreased the expression level of the latter at least10-fold (Fig. 7A). Northern blot analysis of GFP mRNA levelsrevealed that the lower expression level was due to impairedtranslation. Similar results were obtained using influenza Avirus hemagglutinin as a reporter and increasing amounts ofPKR-KDcasp. These experiments demonstrate that an increasein relative amount of PKR-KDcasp inhibits hemagglutinin ex-pression (Fig. 7B). We conclude that the PKR-KDcasp fragmentreleased by caspases during apoptosis is active both in vitroand in vivo.
DISCUSSION
The antiviral protein kinase PKR is expressed in many cells,where it is present in a dormant state. The typical mode of
FIG. 6. Caspase-generated PKR-KDcasp is active. Expression vectors for PKR, PKRK296R, PKR-KDcasp, PKR-KDcaspK296R, eIF2-�, andeIF2-�S51A were transfected in HEK293T cells. After 36 h, cells were lysed, and expressed proteins were analyzed by immunoprecipitation withanti-Myc (PKR and eIF2-�) or anti-E-tag antibodies (PKR-KDcasp constructs). Immunoprecipitated PKR and PKR fragments were assayed forautophosphorylation and eIF2-� phosphorylation in the presence of [�-32P]ATP, followed by SDS-PAGE and autoradiography (R), or set up in aparallel “cold” reaction and analyzed by immunoblotting using antibodies (W). A, immunopurified PKR-KDcasp has low autophosphorylationcapacity. Middle and lower panels show low (20 min) and high (72 h) exposure of autophosphorylation assay, respectively. B, PKR-KDcaspphosphorylates eIF2-� as efficiently as PKR. PKR-KDcasp, PKR-KDcaspK296R, and PKR were incubated with eIF2-� in the presence of[�-32P]ATP. C, eIF2-� is phosphorylated at Ser51 by PKR-KDcasp. eIF2-� and eIF2-�S51A mutants were assayed as substrates for PKR andPKR-KDcasp kinases.
Caspase-dependent PKR Activation 41625
by guest on Decem
ber 2, 2015http://w
ww
.jbc.org/D
ownloaded from
activation of PKR requires the presence of dsRNA, a metabolicintermediate of viral infection (40). In this paper we demon-strate that caspase-mediated cleavage of PKR, at the boundarybetween the regulatory and kinase domain of PKR, liberates anactive eIF2-� kinase domain. This cleavage and the increasedeIF2-� phosphorylation are general features of apoptosis, trig-
gered by either extrinsic (TNF, anti-Fas) or intrinsic (STS) celldeath pathways (Fig. 8).
Our results suggest that the released kinase domain of PKRis constitutively active, which is well supported by structure-function studies (17, 38). Is this cleavage-dependent mode ofactivation compatible with the current model of PKR controlmechanisms? Given the detrimental impact of PKR activity onprotein synthesis and normal cellular function, the kinase iskept in a latent state at two levels. The second dsRNA-bindingmotifs of PKR interacts intramolecularly with the kinase do-main, ensuring a “locked” inactive conformation, and probablymasks the dimerization domain (17, 18, 41). Although thecrystal structure of the entire PKR molecule has not beendetermined yet, it seems likely that Asp251, the residue in thePKR peptide sequence targeted by caspases, should be acces-sible in such a “bent-over” model. In addition, a number ofcellular inhibitors have been described that either competewith PKR for dsRNA binding (42) or prevent its homodimer-ization (43, 44). Since PKR is a major component of the IFN-mediated antiviral response, it is not surprising that manyviruses encode PKR-inhibitory molecules. Some of these pre-vent PKR activation by interfering with its dsRNA binding, e.g.vaccinia virus E3L and influenza virus NS1, or homo-dimer-ization and transphosphorylation, e.g. influenza virus and hep-atitis C virus (45). The caspase-mediated activation of PKRreleases the kinase domain from the control of the regulatoryamino-terminal domain. This may provide the cell with a by-pass mechanism to fight such virally and cellularly encodedPKR inhibitors.
Our study highlights the rapid and ordered shut down ofprotein synthesis during apoptosis. In previous publicationsthe recurrent feature responsible for the observed translationinhibition was cleavage of distinct translation initiation factorsby caspases primarily targeting cap-dependent translation.However, the translation of certain mRNAs, coding for severalsurvival factors, oncogenes and proteins involved in controllingapoptosis, is regulated by an internal ribosome entry site(IRES). Proteolysis of eIF4G will most probably lead to inhibi-tion of translation of most mRNAs, but may still allow trans-lation of IRES-containing transcripts (46–48). Phosphoryla-tion of eIF2-� prevents recruitment of the methionyl-initiatortRNA to the start codon of small ribosomal subunit-associatedmRNAs, a common step in initiation of translation of allmRNAs, including most known IRES-containing RNAs (49,50).
We could not demonstrate complete cleavage of PKR,whereas eIF4G and PARP were eventually entirely processed.Proteolysis of PKR, however, liberates an active catalyticalfragment and can be regarded as a gain of function. The finaloutcome of the enzyme’s action is removal from the circulationof eIF2B, a limiting component of the translation initiationpathway. Hence, it is conceivable that even a limited amount ofactive PKR would in due course have a strong inhibitory impacton translation. By contrast, the aim of cleavage of eIF4G andPARP is loss of a potentially life-saving function. Therefore,abolishment of their respective cap-mRNA recruiting and DNArepair functions would require their complete digestion.
Our results as well as other published data suggest thatduring rapid apoptosis shutdown of protein synthesis occurs atmultiple levels and presumably leaves little possibility for anymessenger to be translated (46, 51). The cellular in vitro cellsystems used by us and others to study the effect of caspases onthe translational apparatus may be affected by apoptotic con-ditions occurring during the development or depletion of acti-vated immune cells. Under such conditions, a more gradualtranslational shutoff may take place, allowing cell recovery at
FIG. 7. Caspase-generated PKR-KDcasp inhibits translation ofa reporter. Western (W) and northern (N) blot analysis were carriedout as in Fig. 5B. A, HEK293T cells were co-transfected with 50 ng ofGFP expression vector and 300 ng of the respective PKR constructs orempty vector (Ctr). After 48 h, GFP expression levels were analyzed byWestern and Northern blot. Western blots are representative of sixindependent transfections. Northern blot analysis with the indicatedprobes was done on total RNA isolated from cells transfected in parallel.The histogram in the lower panel depicts the normalized GFP proteinversus mRNA ratios. B, increasing the relative amount of KDcaspprotein leads to a stronger inhibition in translation of a reporter con-struct. Western blot analysis of HEK293T cells co-transfected with 100ng of influenza virus hemagglutinin (HA) and an increasing amount (0,50, 100, 300, and 600 ng/well) of either an expression vector for PKR-KDcaspK296R (KD) or PKR-KDcasp. Empty vector was added to com-plement the total amount of DNA to 800 ng. –, mock transfection.
Caspase-dependent PKR Activation41626
by guest on Decem
ber 2, 2015http://w
ww
.jbc.org/D
ownloaded from

different levels of the cell death pathway. Such a rescue path-way could be mediated by an IRES-controlled expression ofantiapoptotic molecules, e.g. XIAP, c-Myc, eIF4G, Hsp70, andBiP (47). Translational impairment due to increased eIF2-�phosphorylation has been reported to preferentially allowtranslation of the proapoptotic molecules Bax and Fas (52). Onthe other hand, short-lived antiapoptotic proteins (such ascFLIP) were shown to be quickly lost by translation inhibition,unclosing the default cell death pathway (2). This suggests thatcontrol of translation plays a major role in the programmeddeath process.
In view of the paradigm that apoptosis is a clean way for acell to die, inhibition of de novo protein synthesis during theapoptotic process may be important to prevent inadvertentsynthesis of proinflammatory molecules (and of any other pro-teins), once the dying cell has been phagocytosed (53, 54).
Acknowledgments—We thank Dr. B. Williams (The Cleveland ClinicFoundation, Cleveland, OH) for kindly providing PKR cDNA and A.Meeus, W. Burm, and E. Van Damme for technical assistance.
REFERENCES
1. Polunovsky, V. A., Wendt, C. H., Ingbar, D. H., Peterson, M. S., and Bitterman,P. B. (1994) Exp. Cell Res. 214, 584–594
2. Fulda, S., Meyer, E., and Debatin, K. M. (2000) Cancer Res. 60, 3947–39563. Lejeune, F. J., Ruegg, C., and Lienard, D. (1998) Curr. Opin. Immunol. 10,
573–5804. Tsujimoto, M., Yip, Y. K., and Vilcek, J. (1986) J. Immunol. 136, 2441–24445. Fransen, L., Van der Heyden, J., Ruysschaert, R., and Fiers, W. (1986) Eur. J.
Cancer Clin. Oncol. 22, 419–4266. Stark, G. R., Kerr, I. M., Williams, B. R., Silverman, R. H., and Schreiber, R. D.
(1998) Annu. Rev. Biochem. 67, 227–2647. Katze, M. G. (1995) Trends Microbiol. 3, 75–788. Kaufman, R. J. (1999) Proc. Natl. Acad. Sci. U. S. A. 96, 11693–116959. Balachandran, S., Roberts, P. C., Brown, L. E., Truong, H., Pattnaik, A. K.,
Archer, D. R., and Barber, G. N. (2000) Immunity 13, 129–14110. Yeung, M. C., Liu, J., and Lau, A. S. (1996) Proc. Natl. Acad. Sci. U. S. A. 93,
12451–1245511. Tang, N. M., Korth, M. J., Gale, M., Jr., Wambach, M., Der, S. D., Bandyopadhyay,
S. K., Williams, B. R., and Katze, M. G. (1999) Mol. Cell. Biol. 19, 4757–476512. Der, S. D., Yang, Y. L., Weissmann, C., and Williams, B. R. (1997) Proc. Natl.
Acad. Sci. U. S. A. 94, 3279–328313. Srivastava, S. P., Kumar, K. U., and Kaufman, R. J. (1998) J. Biol. Chem. 273,
2416–242314. Meurs, E., Chong, K., Galabru, J., Thomas, N. S., Kerr, I. M., Williams, B. R.,
and Hovanessian, A. G. (1990) Cell 62, 379–39015. Galabru, J., and Hovanessian, A. (1987) J. Biol. Chem. 262, 15538–1554416. Manche, L., Green, S. R., Schmedt, C., and Mathews, M. B. (1992) Mol. Cell.
Biol. 12, 5238–524817. Wu, S., and Kaufman, R. J. (1997) J. Biol. Chem. 272, 1291–129618. Nanduri, S., Rahman, F., Williams, B. R., and Qin, J. (2000) EMBO J. 19,
5567–557419. Ung, T. L., Cao, C., Lu, J., Ozato, K., and Dever, T. E. (2001) EMBO J. 20,
3728–373720. Patel, R. C., and Sen, G. C. (1998) EMBO J. 17, 4379–439021. Ito, T., Yang, M., and May, W. S. (1999) J. Biol. Chem. 274, 15427–1543222. Gray, N. K., and Wickens, M. (1998) Annu. Rev. Cell Dev. Biol. 14, 399–45823. Dever, T. E., Feng, L., Wek, R. C., Cigan, A. M., Donahue, T. F., and Hinnebusch,
A. G. (1992) Cell 68, 585–59624. Safer, B. (1983) Cell 33, 7–825. Oldfield, S., Jones, B. L., Tanton, D., and Proud, C. G. (1994) Eur. J. Biochem.
221, 399–41026. Hengartner, M. O. (2000) Nature 407, 770–77627. Utz, P. J., and Anderson, P. (2000) Cell Death Differ. 7, 589–60228. Van de Craen, M., Declercq, W., Van den brande, I., Fiers, W., and Vandenabeele,
P. (1999) Cell Death Differ. 6, 1117–112429. McMaster, G. K., and Carmichael, G. G. (1977) Proc. Natl. Acad. Sci. U. S. A.
74, 4835–483830. Min Jou, W., Verhoeyen, M., Devos, R., Saman, E., Fang, R., Huylebroeck, D.,
Fiers, W., Threlfall, G., Barber, C., Carey, N., and Emtage, S. (1980) Cell 19,683–696
31. DeGracia, D. J., Sullivan, J. M., Neumar, R. W., Alousi, S. S., Hikade, K. R.,Pittman, J. E., White, B. C., Rafols, J. A., and Krause, G. S. (1997) J. Cereb.Blood Flow Metab. 17, 1291–1302
32. Marissen, W. E., Guo, Y., Thomas, A. A., Matts, R. L., and Lloyd, R. E. (2000)J. Biol. Chem. 275, 9314–9323
33. Satoh, S., Hijikata, M., Handa, H., and Shimotohno, K. (1999) Biochem. J. 342,65–70
34. Morley, S. J., McKendrick, L., and Bushell, M. (1998) FEBS Lett. 438, 41–4835. Marissen, W. E., and Lloyd, R. E. (1998) Mol. Cell. Biol. 18, 7565–757436. Martin de la Vega, C., Garcia, A., Martin, M. E., Alcazar, A., Marin, O.,
Quevedo, C., and Salinas, M. (1999) Cell. Signal. 11, 399–40437. Wu, S., and Kaufman, R. J. (1996) J. Biol. Chem. 271, 1756–176338. Zhu, S., Romano, P. R., and Wek, R. C. (1997) J. Biol. Chem. 272, 14434–1444139. Kaufman, R. J., Davies, M. V., Pathak, V. K., and Hershey, J. W. (1989) Mol.
Cell. Biol. 9, 946–95840. Williams, B. R. (1999) Oncogene 18, 6112–612041. Sharp, T. V., Moonan, F., Romashko, A., Joshi, B., Barber, G. N., and Jagus, R.
(1998) Virology 250, 302–315
FIG. 8. Representation of signalingevents leading to inhibition of transla-tion initiation in apoptosis. Caspases,activated through the extrinsic (e.g. TNFreceptor or Fas) or intrinsic (mitochondri-al) apoptotic pathway, cleave PKR, liberat-ing its constitutively active eIF2-� kinasedomain, which leads to inhibition of trans-lation. In parallel, caspases target struc-tural components of the translation initia-tion machinery, including the scaffoldeIF4G, the p35 subunit of eIF3, eIF4B, andeIF4E-BP1, as well as eIF2-�.
Caspase-dependent PKR Activation 41627
by guest on Decem
ber 2, 2015http://w
ww
.jbc.org/D
ownloaded from

42. Kumar, K. U., Srivastava, S. P., and Kaufman, R. J. (1999) Mol. Cell. Biol. 19,1116–1125
43. Cosentino, G. P., Venkatesan, S., Serluca, F. C., Green, S. R., Mathews, M. B.,and Sonenberg, N. (1995) Proc. Natl. Acad. Sci. U. S. A. 92, 9445–9449
44. Tan, S. L., Gale, M. J., Jr., and Katze, M. G. (1998) Mol. Cell. Biol. 18,2431–2443
45. Gale, M., Jr., and Katze, M. G. (1998) Pharmacol. Ther. 78, 29–4646. Clemens, M. J., Bushell, M., Jeffrey, I. W., Pain, V. M., and Morley, S. J. (2000)
Cell Death Differ. 7, 603–61547. Holcik, M., Sonenberg, N., and Korneluk, R. G. (2000) Trends Genet. 16,
469–47348. Henis-Korenblit, S., Strumpf, N. L., Goldstaub, D., and Kimchi, A. (2000) Mol.
Cell. Biol. 20, 496–50649. Hershey, J. W. B., and Merrick, W. C. (2000) in Translational Control and
Gene Expression (Sonenberg, N., Hershey, J. W. B., and Mathews, M. B.,eds) pp. 33–88, Cold Spring Harbor Laboratory Press, Cold Spring Harbor,NY
50. McCarthy, J. E. (2000) Curr. Biol. 10, R715–R71751. Nadano, D., and Sato, T. A. (2000) J. Biol. Chem. 275, 13967–1397352. Balachandran, S., Kim, C. N., Yeh, W. C., Mak, T. W., Bhalla, K., and Barber,
G. N. (1998) EMBO J. 17, 6888–690253. Savill, J., Fadok, V., Henson, P., and Haslett, C. (1993) Immunol. Today 14,
131–13654. Savill, J., and Fadok, V. (2000) Nature 407, 784–788
Caspase-dependent PKR Activation41628
by guest on Decem
ber 2, 2015http://w
ww
.jbc.org/D
ownloaded from

VandenabeeleXavier Saelens, Michael Kalai and Peter PHOSPHORYLATION
αACTIVATION AND eIF2-CASPASE-DEPENDENT PKR Translation Inhibition in Apoptosis:TRANSDUCTION:MECHANISMS OF SIGNAL
doi: 10.1074/jbc.M103674200 originally published online September 12, 20012001, 276:41620-41628.J. Biol. Chem.
10.1074/jbc.M103674200Access the most updated version of this article at doi:
.JBC Affinity SitesFind articles, minireviews, Reflections and Classics on similar topics on the
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/276/45/41620.full.html#ref-list-1
This article cites 52 references, 27 of which can be accessed free at
by guest on Decem
ber 2, 2015http://w
ww
.jbc.org/D
ownloaded from




















