
Alternative ERK5 regulation by phosphorylation during the cell cycle
9
Alternative ERK5 regulation by phosphorylation during the cell cycle Francisco A. Iñesta-Vaquera a , David G. Campbell b , Cathy Tournier c , Nestor Gómez d , Jose M. Lizcano d , A. Cuenda a, ⁎ a Departamento de Inmunología y Oncología, Centro Nacional de Biotecnología/CSIC, Campus de Cantoblanco, E-28049 Madrid, Spain b MRC Protein Phosphorylation Unit, Sir James Black Building, School of Life Sciences, University of Dundee, Dundee DD1 5EH, UK c Faculty of Life Sciences, University of Manchester, Michael Smith Building, Oxford Road, Manchester M13 9PT, UK d Institut de Neurociències i Departament de Bioquímica i Biologia Molecular, Universitat Autònoma de Barcelona, E-08193 Barcelona, Spain abstract article info Article history: Received 12 March 2010 Received in revised form 6 July 2010 Accepted 13 July 2010 Available online 25 July 2010 Keywords: Extracellular signal-regulated protein kinase 5 (ERK5) MEK5 Phosphorylation site Mitosis ERK5 is a member of the mitogen-activated protein kinase (MAPK) family that, after stimulation, is activated selectively by dual phosphorylation in the TEY motif by MAPK kinase 5 (MEK5). ERK5 plays an important role in regulating cell proliferation, survival, differentiation and stress response. Moreover, it is involved in G2/M progression and timely mitotic entry. ERK5 is phosphorylated during mitosis, but the molecular mechanism by which it is regulated during this phase is still unclear. Here we show that although ERK5 is phosphorylated in mitosis, this does not occur on the activation motif (TEY), but at its C-terminal half. We have identified five sites of ERK5 phosphorylation in mitosis, two of them unknown. Furthermore, we demonstrate that ERK5 phosphorylation in mitosis is not MEK5-dependent, but rather, cyclin-dependent kinase (CDK)-dependent. Using a mutagenesis approach, we analysed the importance of the phosphorylated residues in ERK5 function; our evidence show that phosphorylation in mitosis of the residues identified inhibits ERK5 activity and regulates ERK5 shuttling from cytoplasm to the nucleus. These results reveal a previously unreported form of ERK5 regulation by phosphorylation and establish a link between CDK and ERK5 pathways during mitosis, which could be crucial for the correct progression of the cell cycle. © 2010 Elsevier Inc. All rights reserved. 1. Introduction Extracellular signal-regulated kinase (ERK) 5 is a member of the mitogen-activated protein kinase (MAPK) family; it contains a TEY motif in its activation loop, similar to ERK1/2 [1,2]. After stimulation, ERK5 is activated selectively by dual phosphorylation on Thr 218 and Tyr 220 in the TEY motif by its only upstream kinase, MEK5, a member of the MEK (extracellular signal-regulated kinase kinase) family [3]. ERK5 is activated in cells by stress, but also by serum, growth factors and cytokines [4–7]. A few downstream targets of ERK5 have been identified, including the myocyte enhancer factor-2 (MEF2) tran- scription factor family members MEF2A, MEF2C and MEF2D, and other transcription factors such as SAP1, cMyc and cAMP response element-binding (CREB) (review in [8]). ERK5 is twice the size of other MAPK family members and hence, the largest kinase within its group. Unlike the classical MAPK, ERK5 has two intrinsic functions, as it has two distinct functional domains. The first is a catalytic N-terminal domain, which is very similar to that of ERK1/2 and is responsible for conventional protein kinase activity; the second is a unique C-terminal half, assumed to be responsible for transcriptional transactivation activity [8–10]. Interaction between the C- and N-terminal domains has an important role in regulating ERK5 localization and kinase activity [9,11,12]. The precise function of ERK5 is not clear, although genetic studies show that ERK5-deficient mice are embryonic lethal; they die around embryonic day E10.5 due to cardiovascular defects and angiogenesis failure in embryonic and extra-embryonic tissues [13–16]. Activation of the ERK5 pathway regulates tumour angiogenesis [17]; it is also associated with highly aggressive forms of breast and prostate cancers [18–20] and with cell proliferation in hepatocellular carcinomas [21]. Studies with cultured cells show ERK5 involvement in neuron survival, muscle cell differentiation, and cell proliferation and transformation [6,22–24]. ERK5 has also been implicated in cell cycle progression, and expression of an ERK5 dominant negative form blocks epithelial growth factor (EGF)-induced cell proliferation and prevents cells from entering the cell cycle S phase [6]. In addition, ERK5 is reported to be phosphorylated in mitosis [25,26]. ERK5 must be activated for G2/M progression and timely mitotic entry; this function requires ERK5 activation of nuclear factor κB (NFκB) through ribosomal S6 kinase2 (RSK2)-mediated phosphorylation [25]. ERK5 also acts as a survival factor in mitosis, phosphorylating and inactivating the apoptotic protein Bim [26]. Cellular Signalling 22 (2010) 1829–1837 Abbreviations: CDK, cyclin-dependent kinase; DMEM, Dulbecco's modified Eagle's medium; EGF, epithelial growth factor; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; MEF2, myocyte enhancer factor-2; MEK, MAP kinase-activated protein kinase kinase; MBP, myelin basic protein; NLS, nuclear localisation signal; NES, nuclear export signal; WT, wild type. ⁎ Corresponding author. Tel.: + 34 91 5855451; fax: + 34 91 3720493. E-mail address: [email protected] (A. Cuenda). 0898-6568/$ – see front matter © 2010 Elsevier Inc. All rights reserved. doi:10.1016/j.cellsig.2010.07.010 Contents lists available at ScienceDirect Cellular Signalling journal homepage: www.elsevier.com/locate/cellsig
Transcript of Alternative ERK5 regulation by phosphorylation during the cell cycle

Cellular Signalling 22 (2010) 1829–1837
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r.com/ locate /ce l l s ig
Alternative ERK5 regulation by phosphorylation during the cell cycle
Francisco A. Iñesta-Vaquera a, David G. Campbell b, Cathy Tournier c, Nestor Gómez d,Jose M. Lizcano d, A. Cuenda a,⁎a Departamento de Inmunología y Oncología, Centro Nacional de Biotecnología/CSIC, Campus de Cantoblanco, E-28049 Madrid, Spainb MRC Protein Phosphorylation Unit, Sir James Black Building, School of Life Sciences, University of Dundee, Dundee DD1 5EH, UKc Faculty of Life Sciences, University of Manchester, Michael Smith Building, Oxford Road, Manchester M13 9PT, UKd Institut de Neurociències i Departament de Bioquímica i Biologia Molecular, Universitat Autònoma de Barcelona, E-08193 Barcelona, Spain
Abbreviations: CDK, cyclin-dependent kinase; DMEMmedium; EGF, epithelial growth factor; ERK, extracelMAPK, mitogen-activated protein kinase; MEF2, myocytekinase-activated protein kinase kinase; MBP, myelinlocalisation signal; NES, nuclear export signal; WT, wild⁎ Corresponding author. Tel.: + 34 91 5855451; fax:
E-mail address: [email protected] (A. Cuenda).
0898-6568/$ – see front matter © 2010 Elsevier Inc. Aldoi:10.1016/j.cellsig.2010.07.010
a b s t r a c t
a r t i c l e i n f oArticle history:Received 12 March 2010Received in revised form 6 July 2010Accepted 13 July 2010Available online 25 July 2010
Keywords:Extracellular signal-regulated protein kinase5 (ERK5)MEK5Phosphorylation siteMitosis
ERK5 is a member of the mitogen-activated protein kinase (MAPK) family that, after stimulation, is activatedselectively by dual phosphorylation in the TEY motif by MAPK kinase 5 (MEK5). ERK5 plays an importantrole in regulating cell proliferation, survival, differentiation and stress response. Moreover, it is involved inG2/M progression and timely mitotic entry. ERK5 is phosphorylated during mitosis, but the molecularmechanism by which it is regulated during this phase is still unclear. Here we show that although ERK5 isphosphorylated in mitosis, this does not occur on the activation motif (TEY), but at its C-terminal half. Wehave identified five sites of ERK5 phosphorylation in mitosis, two of them unknown. Furthermore, wedemonstrate that ERK5 phosphorylation in mitosis is not MEK5-dependent, but rather, cyclin-dependentkinase (CDK)-dependent. Using a mutagenesis approach, we analysed the importance of the phosphorylatedresidues in ERK5 function; our evidence show that phosphorylation in mitosis of the residues identifiedinhibits ERK5 activity and regulates ERK5 shuttling from cytoplasm to the nucleus. These results reveal apreviously unreported form of ERK5 regulation by phosphorylation and establish a link between CDK andERK5 pathways during mitosis, which could be crucial for the correct progression of the cell cycle.
, Dulbecco's modified Eagle'slular signal-regulated kinase;enhancer factor-2; MEK, MAPbasic protein; NLS, nucleartype.+ 34 91 3720493.
l rights reserved.
© 2010 Elsevier Inc. All rights reserved.
1. Introduction
Extracellular signal-regulated kinase (ERK) 5 is a member of themitogen-activated protein kinase (MAPK) family; it contains a TEYmotif in its activation loop, similar to ERK1/2 [1,2]. After stimulation,ERK5 is activated selectively by dual phosphorylation on Thr218 andTyr220 in the TEY motif by its only upstream kinase, MEK5, a memberof the MEK (extracellular signal-regulated kinase kinase) family [3].ERK5 is activated in cells by stress, but also by serum, growth factorsand cytokines [4–7]. A few downstream targets of ERK5 have beenidentified, including the myocyte enhancer factor-2 (MEF2) tran-scription factor family members MEF2A, MEF2C and MEF2D, andother transcription factors such as SAP1, cMyc and cAMP responseelement-binding (CREB) (review in [8]).
ERK5 is twice the size of other MAPK family members and hence,the largest kinase within its group. Unlike the classical MAPK, ERK5has two intrinsic functions, as it has two distinct functional domains.
The first is a catalytic N-terminal domain, which is very similar to thatof ERK1/2 and is responsible for conventional protein kinase activity;the second is a unique C-terminal half, assumed to be responsible fortranscriptional transactivation activity [8–10]. Interaction betweenthe C- and N-terminal domains has an important role in regulatingERK5 localization and kinase activity [9,11,12].
The precise function of ERK5 is not clear, although genetic studiesshow that ERK5-deficient mice are embryonic lethal; they die aroundembryonic day E10.5 due to cardiovascular defects and angiogenesisfailure in embryonic and extra-embryonic tissues [13–16]. Activationof the ERK5 pathway regulates tumour angiogenesis [17]; it is alsoassociatedwith highly aggressive forms of breast and prostate cancers[18–20] and with cell proliferation in hepatocellular carcinomas [21].Studies with cultured cells show ERK5 involvement in neuronsurvival, muscle cell differentiation, and cell proliferation andtransformation [6,22–24]. ERK5 has also been implicated in cellcycle progression, and expression of an ERK5 dominant negative formblocks epithelial growth factor (EGF)-induced cell proliferation andprevents cells from entering the cell cycle S phase [6]. In addition,ERK5 is reported to be phosphorylated in mitosis [25,26]. ERK5 mustbe activated for G2/M progression and timely mitotic entry; thisfunction requires ERK5 activation of nuclear factor κB (NFκB) throughribosomal S6 kinase2 (RSK2)-mediated phosphorylation [25]. ERK5also acts as a survival factor in mitosis, phosphorylating andinactivating the apoptotic protein Bim [26].

1830 F.A. Iñesta-Vaquera et al. / Cellular Signalling 22 (2010) 1829–1837
Despite these findings, themolecularmechanism bywhich ERK5 isregulated during mitosis is unclear. Here we addressed this question,and our data confirm that ERK5 is phosphorylated in a cell cycle-dependent manner, with maximal phosphorylation at mitosis. UsingHeLa, HEK293 cells and mouse embryonic fibroblasts, we show thatERK5 is not phosphorylated on the activating sites T218 and Y220 in theTEY motif during mitosis. Our data also indicate that ERK5 is notphosphorylated by MEK5, but is regulated by Cyclin-dependentkinase (CDK) pathways. We identify five residues in the ERK5 C-terminal domain that are phosphorylated in mitosis; some arepreviously unreported phosphoacceptor sites. Using a mutagenesisapproach, we analysed the importance of these residues in ERK5function, and show evidence that ERK5 activity and subcellularlocalization are regulated during mitosis by MEK5-independentphosphorylation.
2. Materials and methods
2.1. Materials
Protein G-Sepharose and 32P γ-ATP were purchased fromAmersham Pharmacia Biotech (GE Healthcare); protease inhibitorcocktail tablets and Taq polymerase were from Roche (East Sussex,UK). Precast polyacrylamide gels, running and transfer buffer, CoomassieBlue and EGF were from Invitrogen (Paisley, UK), GSH-Sepharose wasfromGE Healthcare (Piscataway, NJ) and nocodazole from Sigma-Aldrich(St Louis, MO). Roscovitine was obtained from Calbiochem (Nottingham,UK) and PD184352 was made by custom synthesis [27]. Other chemicalswere of the highest purity available and purchased from Merck (Poole,UK) or Sigma-Aldrich.
2.2. Antibodies
Antibodies to ERK1/ERK2 and phospho-ERK1/ERK2 (Thr202/Tyr204) were from New England Biolabs (Ipswich, MA), anti-GSTand -ERK5 antibody were from the Division of Signal TransductionTherapy (Dundee, UK) and anti-phospho ERK5 (P-ERK5(T218EY220))was from Biosource (Invitrogen). Cyclin B1 was purchased from BDPharmingen (San Jose, CA).
Secondary Alexa-Fluor 488/Alexa-Fluor 594-conjugated donkeyanti-sheep antibodies were from Molecular Probes (Invitrogen),peroxidase-conjugated rabbit anti-sheep IgG, goat anti-rabbit andrabbit anti-mouse IgG antibodies were from Perbio Science UK(Tattenhall, UK).
2.3. Plasmids and proteins
pEBG2t plasmids encoding GST alone, GST-ERK5(WT) (humanERK5wild-type) or GST-ERK5(AEF) (ERK5 mutated at Thr218/Glu/Tyr220 toAla218/Glu/Phe220) were a generous gift of Dr. S. Arthur (MRCPhosphorylation Unit, Dundee, UK). GST-ATF2, GST-Elk1, GST-cMyc,GST-MEF2A and 2C were a gift of Dr. P. Cohen (MRC PhosphorylationUnit, Dundee, UK).
2.4. Generation of ERK5 mutants
pEBG2t ERK5(WT)wasmutated at the possiblemitotic phosphory-lation (Ser567, Ser720, Ser731, Thr733 and Ser803) sites using theQuickChange XL Site-Directed Mutagenesis kit (Stratagene, La Jolla,CA), using PfuTurbo DNA polymerase according to manufacturer'sinstructions.
2.5. Cell culture, transfection and lysis
Human embryonic kidney (HEK)293 cells and HeLa cells werecultured in DMEM supplemented with 10% (v/v) foetal bovine serum
(FBS), 2 mM L-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomy-cin.Mouseembryonicfibroblasts (MEF)were cultured inDMEMwith10%(v/v) FBS, 2 mM L-glutamine, 0.1 mM 2-mercaptoethanol, 100 μMnonessential amino acids, 100 U/ml penicillin and 0.1 mg/ml streptomy-cin. For transfection experiments, each 10 cm dish of HeLa cell wastransfected with 10–20 μg polyethyleneimine (Polysciences; Eppelheim,Germany) and 5–10 μg plasmid DNA as described [28]. Cellswere stimulated with sorbitol or synchronised with nocodazole 24–36 hpost-transfection, then lysed in lysisbuffer (50 mMTris–HClpH7.5, 1 mMEGTA, 1 mM EDTA, 1 mM sodium orthovanadate, 10 mM sodiumfluoride, 50 mM sodium β-glycerophosphate, 5 mM pyrophosphate,0.27 M sucrose, 0.1 mM phenylmethylsulphonyl fluoride, 1% (v/v) TritonX-100) plus 0.1% (v/v) 2-mercaptoethanol, 1 mM benzamidine and0.1 mM PMSF. Lysates were centrifuged (13,000×g, 15 min, 4 °C),supernatants removed, snap-frozen in liquid nitrogen and storedat −80 °C. Protein concentration was determined by the Bradfordmethod.
2.6. Immunofluorescence staining
HeLa cells were cultured on 22 mm-diameter coverslips. Transienttransfections were performed as indicated previously. Cells were fixedin 4% paraformaldehyde (10 min, room temperature (RT)), blocked in5% bovine serum albumin (BSA) in PBS (30 min, RT), then incubatedwith primary antibody (2 h, RT) in a humidified chamber. GST-ERK5was immunostained using 0.5 μg/ml anti-GST antibody; secondaryantibodywas used at 1:250 in PBS (1 h) in a dark humidified chamber.DNA was stained with DAPI (1 mM, 10 min, RT). Mounting mediumwas 50% glycerol in PBS. Epifluorescence microscopy was performedusing an Olympus FV1000 inverted microscope equipped withPlanApo x63 1.4NA oil immersion objectives. Images were assembledusing Adobe Photoshop.
2.7. Cell cycle synchronization
HeLa cells were arrested in G2/M by adding 50 ng/ml nocodazole(12 h). Mitotic cells were dislodged by gently tapping the plates, thenplaced in sterile tubes for lysis and analysis.
Cells were synchronised at the G1/S phase using a standarddouble-thymidine block-and-release protocol. Briefly, cells wereincubated with 2 mM thymidine (16 h), washed twice with PBS,incubated for 8 h in complete medium, followed by a second 14 hthymidine block. Cells were then released into complete medium forthe times indicated. Cell cycle arrest was confirmed by FACS.
2.8. Flow cytometry analysis (FACS)
Cells were trypsinised, resuspended in ice-cold 70% ethanol, andfixed (4 °C, 30 min) and stored (−20 °C). Cells were then resus-pended in PBS, centrifuged (1000×g, 4 °C, 10 min), resuspended in1 ml Beckman Coulter DNAprep and incubated (37 °C, 30 min). Cellswere then analysed on a Cytomics FC500 cytometer (BeckmanCoulter).
2.9. Western blotting
Protein samples were subjected to SDS-PAGE and transferred tonitrocellulose. Membranes were blocked (30 min) in 50 mM Tris/HCl(pH 7.5), 0.15 M NaCl, 0.5% (v/v) Tween (TBST buffer) containing 10%(w/v) skimmed milk powder. When phospho-ERK5 antibody wasused TBST buffer contained 5% (w/v) skimmed milk powder.Membranes were then incubated in TBST buffer with 10% skimmedmilk powder and 0.5–1 μg/ml antibody (2 h, RT or overnight at 4 °C),followed by the appropriate horseradish peroxidase-conjugatedsecondary antibodies and enhanced chemiluminescence reagent(Amersham Pharmacia Biotech).
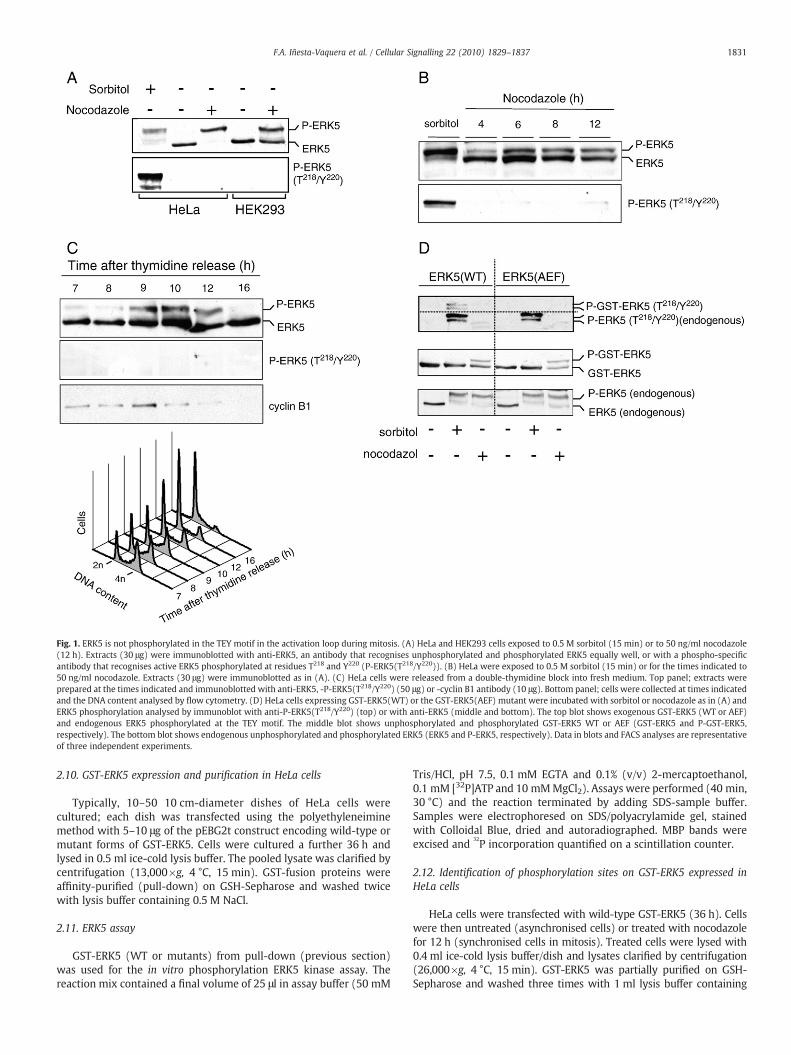
Fig. 1. ERK5 is not phosphorylated in the TEY motif in the activation loop during mitosis. (A) HeLa and HEK293 cells exposed to 0.5 M sorbitol (15 min) or to 50 ng/ml nocodazole(12 h). Extracts (30 μg) were immunoblotted with anti-ERK5, an antibody that recognises unphosphorylated and phosphorylated ERK5 equally well, or with a phospho-specificantibody that recognises active ERK5 phosphorylated at residues T218 and Y220 (P-ERK5(T218/Y220)). (B) HeLa were exposed to 0.5 M sorbitol (15 min) or for the times indicated to50 ng/ml nocodazole. Extracts (30 μg) were immunoblotted as in (A). (C) HeLa cells were released from a double-thymidine block into fresh medium. Top panel; extracts wereprepared at the times indicated and immunoblotted with anti-ERK5, -P-ERK5(T218/Y220) (50 μg) or -cyclin B1 antibody (10 μg). Bottom panel; cells were collected at times indicatedand the DNA content analysed by flow cytometry. (D) HeLa cells expressing GST-ERK5(WT) or the GST-ERK5(AEF) mutant were incubated with sorbitol or nocodazole as in (A) andERK5 phosphorylation analysed by immunoblot with anti-P-ERK5(T218/Y220) (top) or with anti-ERK5 (middle and bottom). The top blot shows exogenous GST-ERK5 (WT or AEF)and endogenous ERK5 phosphorylated at the TEY motif. The middle blot shows unphosphorylated and phosphorylated GST-ERK5 WT or AEF (GST-ERK5 and P-GST-ERK5,respectively). The bottom blot shows endogenous unphosphorylated and phosphorylated ERK5 (ERK5 and P-ERK5, respectively). Data in blots and FACS analyses are representativeof three independent experiments.
1831F.A. Iñesta-Vaquera et al. / Cellular Signalling 22 (2010) 1829–1837
2.10. GST-ERK5 expression and purification in HeLa cells
Typically, 10–50 10 cm-diameter dishes of HeLa cells werecultured; each dish was transfected using the polyethyleneiminemethod with 5–10 μg of the pEBG2t construct encoding wild-type ormutant forms of GST-ERK5. Cells were cultured a further 36 h andlysed in 0.5 ml ice-cold lysis buffer. The pooled lysate was clarified bycentrifugation (13,000×g, 4 °C, 15 min). GST-fusion proteins wereaffinity-purified (pull-down) on GSH-Sepharose and washed twicewith lysis buffer containing 0.5 M NaCl.
2.11. ERK5 assay
GST-ERK5 (WT or mutants) from pull-down (previous section)was used for the in vitro phosphorylation ERK5 kinase assay. Thereaction mix contained a final volume of 25 μl in assay buffer (50 mM
Tris/HCl, pH 7.5, 0.1 mM EGTA and 0.1% (v/v) 2-mercaptoethanol,0.1 mM [32P]ATP and 10 mMMgCl2). Assays were performed (40 min,30 °C) and the reaction terminated by adding SDS-sample buffer.Samples were electrophoresed on SDS/polyacrylamide gel, stainedwith Colloidal Blue, dried and autoradiographed. MBP bands wereexcised and
32P incorporation quantified on a scintillation counter.
2.12. Identification of phosphorylation sites on GST-ERK5 expressed inHeLa cells
HeLa cells were transfected with wild-type GST-ERK5 (36 h). Cellswere then untreated (asynchronised cells) or treated with nocodazolefor 12 h (synchronised cells in mitosis). Treated cells were lysed with0.4 ml ice-cold lysis buffer/dish and lysates clarified by centrifugation(26,000×g, 4 °C, 15 min). GST-ERK5 was partially purified on GSH-Sepharose and washed three times with 1 ml lysis buffer containing
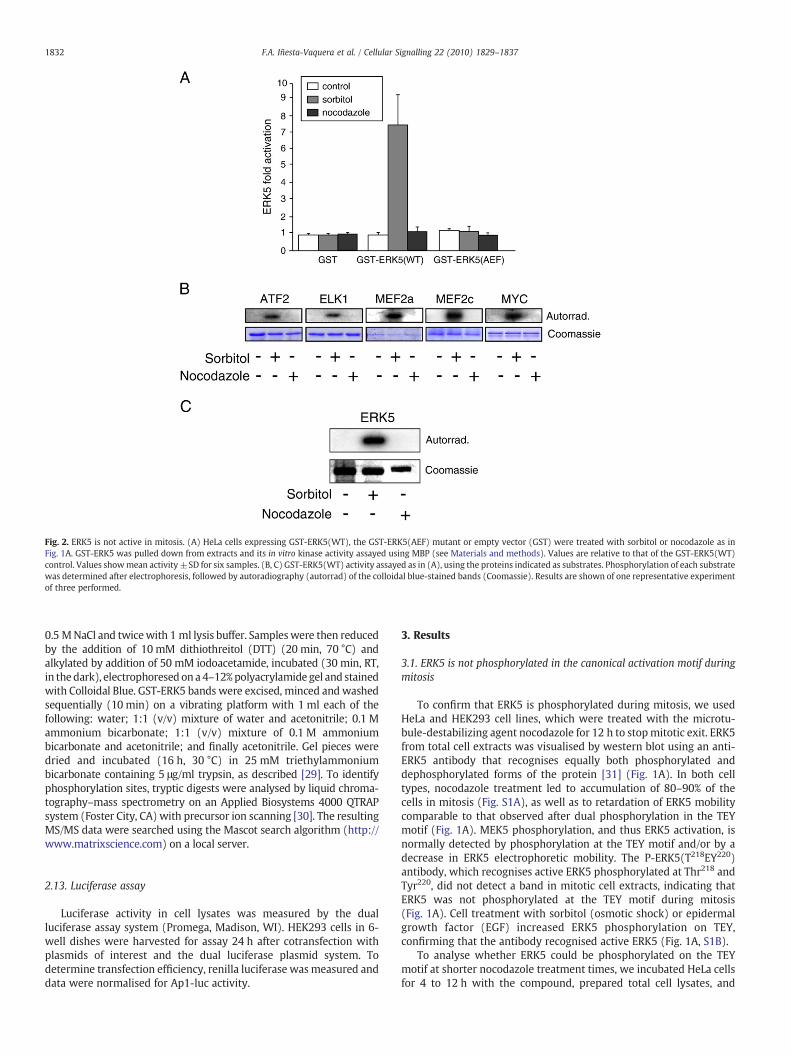
Fig. 2. ERK5 is not active in mitosis. (A) HeLa cells expressing GST-ERK5(WT), the GST-ERK5(AEF) mutant or empty vector (GST) were treated with sorbitol or nocodazole as inFig. 1A. GST-ERK5 was pulled down from extracts and its in vitro kinase activity assayed using MBP (see Materials and methods). Values are relative to that of the GST-ERK5(WT)control. Values showmean activity ±SD for six samples. (B, C) GST-ERK5(WT) activity assayed as in (A), using the proteins indicated as substrates. Phosphorylation of each substratewas determined after electrophoresis, followed by autoradiography (autorrad) of the colloidal blue-stained bands (Coomassie). Results are shown of one representative experimentof three performed.
1832 F.A. Iñesta-Vaquera et al. / Cellular Signalling 22 (2010) 1829–1837
0.5 MNaCl and twicewith 1 ml lysis buffer. Sampleswere then reducedby the addition of 10 mM dithiothreitol (DTT) (20 min, 70 °C) andalkylated by addition of 50 mM iodoacetamide, incubated (30 min, RT,in thedark), electrophoresed on a4–12%polyacrylamide gel and stainedwith Colloidal Blue. GST-ERK5 bands were excised, minced and washedsequentially (10 min) on a vibrating platform with 1 ml each of thefollowing: water; 1:1 (v/v) mixture of water and acetonitrile; 0.1 Mammonium bicarbonate; 1:1 (v/v) mixture of 0.1 M ammoniumbicarbonate and acetonitrile; and finally acetonitrile. Gel pieces weredried and incubated (16 h, 30 °C) in 25 mM triethylammoniumbicarbonate containing 5 μg/ml trypsin, as described [29]. To identifyphosphorylation sites, tryptic digests were analysed by liquid chroma-tography–mass spectrometry on an Applied Biosystems 4000 QTRAPsystem (Foster City, CA) with precursor ion scanning [30]. The resultingMS/MS data were searched using the Mascot search algorithm (http://www.matrixscience.com) on a local server.
2.13. Luciferase assay
Luciferase activity in cell lysates was measured by the dualluciferase assay system (Promega, Madison, WI). HEK293 cells in 6-well dishes were harvested for assay 24 h after cotransfection withplasmids of interest and the dual luciferase plasmid system. Todetermine transfection efficiency, renilla luciferase wasmeasured anddata were normalised for Ap1-luc activity.
3. Results
3.1. ERK5 is not phosphorylated in the canonical activation motif duringmitosis
To confirm that ERK5 is phosphorylated during mitosis, we usedHeLa and HEK293 cell lines, which were treated with the microtu-bule-destabilizing agent nocodazole for 12 h to stopmitotic exit. ERK5from total cell extracts was visualised by western blot using an anti-ERK5 antibody that recognises equally both phosphorylated anddephosphorylated forms of the protein [31] (Fig. 1A). In both celltypes, nocodazole treatment led to accumulation of 80–90% of thecells in mitosis (Fig. S1A), as well as to retardation of ERK5 mobilitycomparable to that observed after dual phosphorylation in the TEYmotif (Fig. 1A). MEK5 phosphorylation, and thus ERK5 activation, isnormally detected by phosphorylation at the TEY motif and/or by adecrease in ERK5 electrophoretic mobility. The P-ERK5(T218EY220)antibody, which recognises active ERK5 phosphorylated at Thr218 andTyr220, did not detect a band in mitotic cell extracts, indicating thatERK5 was not phosphorylated at the TEY motif during mitosis(Fig. 1A). Cell treatment with sorbitol (osmotic shock) or epidermalgrowth factor (EGF) increased ERK5 phosphorylation on TEY,confirming that the antibody recognised active ERK5 (Fig. 1A, S1B).
To analyse whether ERK5 could be phosphorylated on the TEYmotif at shorter nocodazole treatment times, we incubated HeLa cellsfor 4 to 12 h with the compound, prepared total cell lysates, and

Fig. 3. ERK5 phosphorylation in mitosis is independent of MEK5. (A)Wild-type (MEK5+/+)orMEK5-deficient (MEK5−/−)MEFwere treatedwithsorbitol ornocodazole as in Fig. 1A, andendogenous ERK5 phosphorylation was analysed by immunoblot with anti-ERK5,which recognises unphosphorylated and phosphorylated ERK5 equally well or with thephospho-specific antibody P-ERK5(T218/Y220). (B) Wild type or MEK5−/− MEF expressingGST-ERK5(WT) or GST-ERK5(AEF) were treated with sorbitol or nocodazole, and GST-ERK5phosphorylation was analysed by immunoblot as in (A). (C) HeLa cells were treated with50 ng/ml nocodazole (12 h) and then incubated alone or with 20 μM PD184352 or 50 μMroscovitine (3 h). ERK5phosphorylationwas analysedby immunoblot as in (A). All results arefrom one representative experiment of three performed.
1833F.A. Iñesta-Vaquera et al. / Cellular Signalling 22 (2010) 1829–1837
examined ERK5 band shift and phosphorylation at the TEYmotif. After4 h of nocodazole treatment, ERK5 underwent a small band shift thatlater increased; however, ERK5 was not phosphorylated in theactivation motif at any time tested (Fig. 1B, S1C). To confirm thatERK5 band shift is not accompanied by TEY motif phosphorylationduring the cell cycle, HeLa cells were synchronised in G1/S by double-thymidine block, then released in fresh medium and cell extractsprepared at different time points. Flow cytometry analysis (FACS)showed that the cells cycled synchronously between S phase, mitosisand G1 (Fig. 1C, bottom). Themitotic windowwas between 9 and 12 hpost-release, as shown by cyclin B1 expression (Fig. 1C, top). TotalERK5 protein was detected at all cell cycle stages, and underwent amarked decrease in electrophoretic mobility during mitosis, but wasnot phosphorylated at the TEY motif (Fig. 1C, top). These results showthat the increase in endogenous ERK5 phosphorylation during mitosisis not caused by phosphorylation in the TEY activation motif.
To verify our finding that ERK5 was not phosphorylated in the TEYmotif during mitosis, we transfected HeLa cells with cDNA coding forwild-type ERK5(WT) or for ERK5(AEF), an unphosphorylable mutantin which residues T218 and Y220 have been replaced by A218 and F220,respectively. Endogenous and overexpressed ERK5(WT) were phos-phorylated in the activating site in response to sorbitol, but not tonocodazole; as predicted, the ERK5(AEF) mutant was not phosphor-ylated in either condition (Fig. 1D). Sorbitol-induced phosphorylationof exogenous ERK5(WT) was not accompanied by a detectable band
shift (Fig. 1D, centre) and was weaker than that of endogenous ERK5(Fig. 1D, top). All ERK5 forms, overexpressed (ERK5(WT) and ERK5(AEF)) and endogenous, underwent a clear mobility shift afternocodazole treatment. These data confirm that ERK5 is not phos-phorylated in the TEY activation motif and indicate its possiblephosphorylation on other residue(s) during mitosis.
3.2. ERK5 catalytic function is not activated during mitosis
We examined whether ERK5 was active during mitosis, assuggested [25,26]. As the activity of endogenous ERK5 was notdetectable with available tools, we overexpressed ERK5(WT) or ERK5(AEF) in HeLa cells and determined their activity in in vitro kinaseassays using myelin basic protein (MBP) as substrate (Fig. 2A). Cellexposure to nocodazole did not cause ERK5(WT) or (AEF) activation,whereas sorbitol treatment led to an 7- to 9-fold ERK5(WT) activityincrease but did not activate ERK5(AEF) (Fig. 2A).
To test whether ERK5 phosphorylation during mitosis affects itssubstrate specificity, and since ERK5 also phosphorylates otherproteins such as MEF2A, MEF2C, Elk1, ATF2 and cMyc (review in[8]), we measured ERK5(WT) activity in cells treated with sorbitol ornocodazole, using these proteins as substrate. All transcription factorswere phosphorylated by sorbitol-activated ERK5, but not by ERK5from nocodazole-treated cells (Fig. 2B). In addition, it was shown thatan important cell substrate of ERK5 is ERK5 itself, since activated ERK5autophosphorylates on a number of residues [31]. As ERK5 autopho-sphorylation has been used in the past to monitor ERK5 activity, weanalysed this autophosphorylation in our experimental conditions,and observed that ERK5 autophosphorylated in response to sorbitolbut not to nocodazole (Fig. 1C). Using a number of substrates, ourresults thus show that ERK5 is activated by hyperosmotic stress, but itis not active during mitosis.
3.3. ERK5 is not phosphorylated by MEK5 in mitosis
MEK5 is the upstream kinase that specifically phosphorylatesERK5 at the T218EY220 motif, leading to its activation [6,31,32]. Toexamine the role ofMEK5 in ERK5 phosphorylation duringmitosis, weanalysed ERK5 activation/phosphorylation in response to osmoticshock and nocodazole in mouse embryonic fibroblasts (MEF) fromwild-type embryos (MEK5+/+) or embryos that did not expressMEK5(MEK5−/−) [33]. Sorbitol treatment led to ERK5 phosphorylation atthe TEY motif, and band shift in MEK5+/+ but not in MEK5−/− cells,whereas nocodazole treatment decreased the electrophoretic mobil-ity of endogenous ERK5 in MEK5+/+ and MEK5−/− equally (Fig. 3A).ERK5 was expressed at similar levels in both cell types. In addition,overexpressed ERK5(WT) and ERK5(AEF) showed a band shift duringmitosis in MEK5+/+ and MEK5−/− cells (Fig. 3B). These data indicatethat ERK5 is not phosphorylated by MEK5 in mitosis and suggest thatit might be phosphorylated by another kinase.
As ERK5 phosphorylation was not regulated in mitosis by its onlyreported kinase, MEK5, we tested CDK as obvious candidate kinasesthat could be involved in ERK5 phosphorylation duringM phase, sinceCDK are essential for driving each cell cycle phase [34]. To studywhether ERK5 were regulated by these kinases during mitosis, wearrested HeLa cells in mitosis. Cells were treated for 3 h with theMKK1/2/5 inhibitor PD184352 and/or the CDK-specific inhibitorroscovitine [35], followed by endogenous ERK5 band shift analysisby western blotting. The slowest migrating form of ERK5 severelydecreased after treatment with roscovitine, but not with PD184352(Fig. 3C), although PD184352 inhibited ERK5 and ERK1/2 phosphor-ylation in response to sorbitol (Fig. S2). These data demonstrate thatMEK5 does not mediate ERK5 phosphorylation in M phase, andindicate a role for CDK pathways in ERK5 regulation during mitosis.

1834 F.A. Iñesta-Vaquera et al. / Cellular Signalling 22 (2010) 1829–1837
3.4. Identification of ERK5 residues phosphorylated in mitosis
As these results suggest that ERK5 is phosphorylated in unknownresidue(s) during mitosis, we used phosphopeptide mapping toidentify phosphorylation sites. Human ERK5, expressed in asynchro-nised HeLa cells or synchronised inmitosis (Fig. 4A), was digested andthe resulting phosphopeptides analysed by liquid chromatography–mass spectrometry (LC–MS). Total ion current for each sampleshowed that approximately equal amounts of asynchronised andsynchronised sample had been applied to the LC–MS system (notshown). The mass spectrometric analysis revealed that ERK5 wasphosphorylated on either Ser731/Thr733 in asynchronised cells(peptide 1) and on at least three additional sites, Ser567, Ser720and Ser803 (peptides 2, 4, and 5) when cells were synchronised inmitosis. Phosphorylation of an ERK5 tryptic peptide (peptide 3)containing Ser731 and Thr733 was also observed in mitotic cells, butwe were unable to establish the residue phosphorylated (Fig. 4B).Analysis of extracted ion chromatograms for peptide 3 nonethelessshowed a N10-fold increase in abundance of the relevant ionsbetween the asynchronised and synchronised samples, whereaspeptides 2 and 4 showed a ≥80-fold increase. Peptide 5 was foundonly in synchronised samples (not shown). All of these phosphory-lation sites are located within the ERK5 C-terminal region (Fig. 4C).
To further examine ERK5 phosphorylation and to confirm that thephosphorylation sites responsible for ERK5 band shift are amongthose identified, we generated ERK5mutants in which all the residuesonly found in cells synchronised in mitosis (Ser567, Ser720 and
Fig. 4. Identification of ERK5 phosphorylation sites in mitosis. (A) HeLa cells were transfected wor treatedwith 50 ng/ml nocodazole (12 h), ERK5 partially purified on glutathione-Sepharose, andGST-ERK5 is indicated(arrow). (B)GST-ERK5wasexcised fromthegel, trypsindigestedandanalysespectra (seeMethods). The peaks identified are listed in the table, withmore certain phosphorylatasterisk (S* or T*). Similar resultswere obtained in two experiments. (C) Schematic representationsignal (NLS) and the localisationof thephosphorylation sites identified. Phosphorylationof Thr733HeLa cells were transfected with ERK5(WT), GST-ERK5(5A) (a mutant in which all phosphorylatphosphorylation sites identified in mitosis were replaced with Glu), GST-ERK5(3A) or GST-ERK5(
Ser803) were mutated to Glu (3E), to mimic phosphorylation byintroducing a negative charge, or to Ala (3A) to prevent phosphor-ylation. Moreover, we also generated mutants in which all phosphor-ylation sites in both asynchronised and synchronised cells werechanged to Glu (5E) or to Ala (5A). We then expressed these ERK5mutants in HeLa cells and blocked them in mitosis to examine theelectrophoretic band shift. Only the mutation of all identified residuesto Glu or Ala abolished the ERK5 band shift caused by nocodazoletreatment (Fig. 4D). These results indicate that ERK5 is phosphory-lated at Ser567, Ser720, Ser731, Thr733 and Ser803 in mitosis,although there could be additional phosphorylation sites.
3.5. Role of the identified phosphorylation sites in regulating ERK5subcellular localization
We then tested whether ERK5 phosphorylation on the mitoticphosphosites had functional effects. We first verified ERK5 localiza-tion in HeLa cells, as it was reported that ERK5 could shuttle fromcytoplasm to the nucleus, depending on its phosphorylation state [11].Thus when ERK5 undergoes phosphorylation, the intramolecularassociation between the N- and C-terminal halves is impaired, causingnuclear import of ERK5 [11]. Subcellular localization of exogenousERK5(WT), ERK5(5A) or ERK5(5E) was examined by confocalfluorescence microscopy in HeLa cells. ERK5(WT) and ERK5(5A)were distributed throughout the cytoplasm and excluded from thenucleus, while the ERK5(5E) mutant was localised mainly in thenucleus (Fig. 5); this indicates that ERK5 mitotic phosphorylation in
ith a construct expressing human GST-ERK5. At 36 h post-transfection, cells were untreatedresolved in SDS-PAGE. The gel was stainedwith Coomassie blue; the band corresponding todbyLC–MS.Thephosphorylated residueswere identifiedbymanual inspectionof theMS/MSed residues identified in bold (pS) and residueswith less certain assignmentsmarkedwith anof ERK5 showing the activationmotif T218EY220 in the kinase domain, the nuclear localisationcouldnotbeconfirmedbyMS/MS, but is included in the schemeandmarkedwithasterisk. (D)ion sites identified in mitosis were replaced with Ala), GST-ERK5(5E) (a mutant in which all3E), nocodazole-treated, and total ERK5 analysed by immunoblot as in Fig. 1A.

Fig. 5. Phosphorylation of residues identified in mitosis affects ERK5 subcellularlocalisation. ERK5 Wild-type (WT) or the indicated GST-ERK5 mutants were expressedin HeLa cells, fixed in paraformaldehyde, and GST-protein immunolocalisation (green)was analysed by fluorescence microscopy using anti-GST antibody. Nuclei stained withDAPI (blue).
1835F.A. Iñesta-Vaquera et al. / Cellular Signalling 22 (2010) 1829–1837
the C terminus controls its subcellular localization, possibly bydisrupting the ERK5 N- and C-terminal interaction.
3.6. Role of identified phosphorylation sites in regulating ERK5 activationand/or activity
We next analysed the effect of mutation of these phosphosites onERK5 activation in response to hyperosmotic stress. In Hela cells, weexpressed ERK5(WT), ERK5(5A) or ERK5(5E), and ERK5(AEF) ascontrol, and tested ERK5 activation using the P-ERK5(T218EY220)antibody. Sorbitol treatment caused phosphorylation on the TEYmotifof endogenous ERK5 and of ERK5(WT), ERK5(5A) and ERK5(5E), butnot ERK5(AEF) (Fig. 6A). When we examined ERK5 kinase activity,however, we found an 80% decrease for both ERK5(5A) and ERK5(5E)mutants when using MBP as substrate (Fig. 6B), indicating that thesesites are essential for regulation of ERK5 activity, independently ofphosphorylation in the TEY motif.
As ERK5 transactivates activator protein-1 (AP-1) promoteractivity [36], we examined whether phosphorylation at the identifiedC-terminal residues is necessary for AP-1 activation. We expressedERK5WT or the ERK5mutants (5A) or (5E) in HEK293 cells and used aluciferase-based reporter assay to measure AP-1 transcriptionalactivity. ERK5(WT) expression enhanced AP-1 activity by ~2.5-foldover that of controls (Fig. 6C). Expression of ERK5(5A) enhanced AP-1activity, showing an increase similar to the ERK5(WT); ERK5(5E) didnot lead to enhancement of AP-1 transcriptional activity (Fig. 6C).
All together, our results demonstrate that the phosphorylationsites identified in mitosis do not regulate ERK5 phosphorylation in theTEY motif, but might have an inhibitory effect on ERK5 kinase andtranscriptional activities.
4. Discussion
To better understand the molecular mechanism by which ERK5 isregulated during G2 to M phase transition, we examined itsphosphorylation in different cell types. We found that ERK5 isphosphorylated at its C-terminal tail during mitosis, but not on theactivating sites T218 and Y220 in the TEYmotif, whose phosphorylationis necessary for activation [31]. We then measured ERK5 activityduring mitosis and, contrary to reports from other groups [21,25,26],found that ERK5 is not active. This divergence could be due todifferences in the process by which ERK5 was partially purified fromcell extracts. After ERK5 precipitation and prior to assaying its activity,we used a high salt wash to eliminate possible contaminating kinasesthat could co-purify with ERK5, followed by a low salt wash (seeMaterials and methods). When the high salt wash was not included,we also detected ERK5 activity in the pellets from nocodazole-treatedcell extracts (Fig. S3), indicating one ormore contaminating kinases. Itis worth noting that in previous studies, ERK5 phosphorylation in theactivating TEY motif was not analysed and no high salt buffer wasused to wash the immunoprecipitated ERK5 before the activity assay[25,26].
Using mass spectrometry, we also report the identity of five ERK5residues that are phosphorylated during mitosis, Ser567, Ser720, Ser731,Thr733 and Ser803, all located in the C-terminal tail. We nonethelessconsider it likely that there are other phosphorylated sites near the C-terminal region. These putative sites proved difficult to identify, as theERK5 C terminus has an unusual amino acid sequence virtually devoid ofcleavage sites for trypsin or other proteinases; this gives rise to very largepeptides, impeding identification of phosphorylation sites. The generationof phospho-specific antibodies that recognise the sites identified herewillbe needed to determine whether ERK5 is phosphorylated in mitosis; thismay well be the case, as mutation of all the sites we identified wasaccompanied by a decrease in ERK5 electrophoretic mobility. The Ser731and Thr733 siteswere previously shown to be ERK5 autophosphorylationsites in vitro and also in ERK5-overexpressing cells [12,31]. Here we show
evidence supporting Ser731 and Thr733 phosphorylation during the Mphase, as mutation of these residues to Ala or Glu abolished ERK5 bandshift inmitosis. Althoughwe found phosphorylated Ser731 and Thr733 inboth asynchronous and synchronous cells in mitosis, phosphorylation ofthese sites increased by N10-fold in mitotic samples. Given thatapproximately 20% of asynchronous HeLa cells are in mitosis (Fig. S1A),thephospho-Ser731andphospho-Thr733 identified couldbe fromcells inM phase. Our data coincide with a recent study examining whole proteinphosphorylation in HeLa cells arrested in G1 and mitotic phases [37]reported that Ser731 andThr733, aswell as Ser720, are phosphorylated inERK5 during mitosis. We also identified two unreported ERK5 phosphor-ylation sites, Ser567 and Ser803.
Previous work showed that ERK5 autophosphorylation in differentresidues within the C-terminal tail enhances ERK5 kinase andtranscriptional activity [9,12,31,36]. The C-terminal half of ERK5 isunique among MAPK family members, as it contains a potenttranscriptional activation domain [38–40]. Moreover, activated ERK5undergoes autophosphorylation on its C-terminal half, necessary formaximal activation of ERK5 transcriptional activation [12]. Here weshow that phosphorylation in mitosis of the sites identified does notincrease ERK5 activity, but inhibits both functions, the kinase and thetranscriptional activities, although it does not regulate the phosphor-ylation of ERK5 TEY motif by MEK5. Our results illustrate thecomplexity of the regulation of ERK5 activity by phosphorylation atits C-terminal. Mutation of all mitotic residues to Ala (ERK5(5A))inhibits ERK5 kinase but not transcriptional activity, suggesting thatphosphorylation of one or more of these sites is needed for maximalkinase activity, and indicating dissimilar regulation of the two ERK5

Fig. 6. ERK5 phosphorylation in mitosis inhibits its kinase and transcriptional activities. (A) HeLa cells expressing GST-ERK5(WT), GST-ERK5(AEF), GST-ERK5(5A) or GST-ERK5(5E)mutants were incubated with sorbitol as in Fig. 1A, and ERK5 phosphorylation analysed by immunoblot with anti-P-ERK5(T218/Y220) (top) or anti-ERK5 (bottom). The top blot showsexogenous GST-ERK5 (WT or mutant) and endogenous ERK5 phosphorylated at the TEY motif. The bottom blot shows unphosphorylated and phosphorylated GST-ERK5WT or mutants(GST-ERK5 and P-GST-ERK5, respectively), as well as endogenous unphosphorylated and phosphorylated ERK5 (ERK5 and P-ERK5, respectively). Data for blots and FACS analyses arerepresentativeof two independent experiments. (B)HeLa cells expressingGST-ERK5WT(WT)or the indicatedmutants as in (A)were exposed to sorbitol andGST-ERK5kinase activitywasquantified using MBP as substrate as in Fig. 2. Results show mean activity±SD for four samples. (C) The indicated GST-ERK5 constructs were cotransfected with pAP1-Luc and renillaluciferase in HEK293 cells (24 h), cells were harvested, and lysates tested in the luciferase assay (see Materials and Methods). Results show mean activity ±SD for nine samples.
1836 F.A. Iñesta-Vaquera et al. / Cellular Signalling 22 (2010) 1829–1837
functions. Mutation of all ERK5 mitotic residues to Glu (ERK5(5E)),which mimics phosphorylation, inhibited both ERK5 activities,probably by causing a conformational change that impedes theERK5 interaction with its substrates but not with its activator MEK5.Indeed, the mitotic sites identified in this study lie in the region ofinteraction with transcription factors, but not that of interaction withMEK5 [40,41]. Other possibility is that ERK5(5E) could bind itssubstrates more tightly than ERK5(WT), this could results in asequestration of the substrate, which may hamper its activity. To thisregards we have obtained evidence showing that mutation of themitotic residues to Glu increases ERK5 binding to MEF2C compare toeither ERK5(WT) or ERK5(5A) (Fig. S4).
Howphosphorylation inmitosis of the ERK5 residues identified hereinhibits ERK5 kinase and transcriptional activation activity, and what isits biological relevance, must be elucidated in future studies. Wenonetheless show that phosphorylation at the ERK5 C-terminal domainis implicated in the regulation of its nuclear localisation. ERK5(WT) and
ERK5(5A) were distributed throughout the cytoplasm and excludedfrom the nucleus, whereas the ERK5(5E)mutant localisedmainly to thenucleus. This suggests that phosphorylation in mitosis at the ERK5 C-terminal residues controls its subcellular localisation through aconformational change. ERK5 has a nuclear localisation signal (NLS) aswell as a nuclear export signal (NES) in its C-terminal region [9,39,42].The balance between nuclear import and export signals determines thesubcellular localisation of ERK5 [9,11]. ERK5 is suggested to have both aclosed and an open conformation. In steady-state conditions inquiescent cells, the majority of ERK5 is assumed to be in a closedconformation in which the intramolecular interaction between the N-and C-terminal halves of ERK5 masks the NLS and exposes the NES,resulting in cytosolic localisation of ERK5. After stimulation, ERK5 isactivated by phosphorylation and the intramolecular associationbetween the N- and C-terminal halves is dissociated, disrupting NESactivity. ERK5 then translocates to the nucleus, as the NLS is constantlyactive [9–11]. Based on this model, any factor able to stabilise the open

1837F.A. Iñesta-Vaquera et al. / Cellular Signalling 22 (2010) 1829–1837
ERK5 conformation could trigger nuclear import of non-activated ERK5;it is thus possible that phosphorylation of the ERK5 C-terminal halfduring mitosis leads to an open conformation.
Previous studies showed translocation of inactive ERK5 fromcytoplasm to the nucleus in various cancer cell lines [42,43]. Inaddition to the ERK5 overexpression found in some forms of breastand prostate cancers [18–20], phosphorylated ERK5 is found in breastcancer patient samples [20]. It will be of interest to determinewhether the ERK5 residues identified here are also phosphorylated intumours, since their fine regulation might be essential for eukaryoticcellular processes such as cell cycle progression. We found that ERK5is not phosphorylated during mitosis by MEK5, so far its onlydescribed upstream kinase, but it is phosphorylated by CDK pathways.Although ERK5 is not phosphorylated directly by CDK in vitro (notshown), it is indirectly regulated by these kinases in vivo, since celltreatment with the specific CDK inhibitor roscovitine blocked ERK5band shift with an IC50 similar to the previously described in differenthuman tumour cell lines [44] (Fig. S5). CDK are the kinasesresponsible for driving the cell cycle and their misregulation inducesunscheduled cell proliferation [34], supporting a possible role forERK5 phosphorylation during mitosis and correct cell proliferation.
5. Conclusion
Our results demonstrate that ERK5 is not active in mitosis,although it is phosphorylated on five residues in the C-terminal half.We also show evidence that ERK5 activity and, maybe its structuralconformation, are regulated during mitosis by phosphorylation that isnot MEK5-dependent but is CDK-dependent. These observationsestablish a link between CDK and the ERK5 pathways during mitosis,which could be crucial for correct cell cycle progression.
Acknowledgments
We thank Drs Simon Arthur and Philip Cohen for reagents, theprotein production and antibody purification teams (Division ofSignal Transduction Therapy, University of Dundee), coordinated byDr. H. McLauchlan and J. Hastie, for generation and purification ofantibodies, and Catherine Mark for editorial support. We also thankEdgar Josue Rodriguez and Dr. Angel Nebreda (CNIO, Spain) for helpwith CDK assay. FAIV was supported by an FPU fellowship from theSpanish Ministry of Education. The work in the author's laboratory issupported by the Spanish Ministerio de Educación y Ciencia (MEC)Spain (BFU2007-67577).
Appendix A. Supplementery data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.cellsig.2010.07.010.
References
[1] J.D. Lee, R.J. Ulevitch, J. Han, Biochem. Biophys. Res. Commun. 213 (2) (1995) 715.[2] G. Zhou, Z.Q. Bao, J.E. Dixon, J. Biol. Chem. 270 (21) (1995) 12665.[3] J.M. English, C.A. Vanderbilt, S. Xu, S. Marcus, M.H. Cobb, J. Biol. Chem. 270 (48)
(1995) 28897.
[4] J. Abe, M. Kusuhara, R.J. Ulevitch, B.C. Berk, J.D. Lee, J. Biol. Chem. 271 (28) (1996)16586.
[5] X. Carvajal-Vergara, S. Tabera, J.C. Montero, A. Esparis-Ogando, R. Lopez-Perez, G.Mateo, N. Gutierrez, M. Parmo-Cabanas, J. Teixido, J.F. San Miguel, A. Pandiella,Blood 105 (11) (2005) 4492.
[6] Y. Kato, R.I. Tapping, S. Huang, M.H. Watson, R.J. Ulevitch, J.D. Lee, Nature 395(6703) (1998) 713.
[7] R.L. Nicol, N. Frey, G. Pearson, M. Cobb, J. Richardson, E.N. Olson, EMBO J. 20 (11)(2001) 2757.
[8] X. Wang, C. Tournier, Cell. Signal. 18 (6) (2006) 753.[9] M. Buschbeck, A. Ullrich, J. Biol. Chem. 280 (4) (2005) 2659.
[10] S. Nishimoto, E. Nishida, EMBO Rep. 7 (8) (2006) 782.[11] K. Kondoh, K. Terasawa, H. Morimoto, E. Nishida, Mol. Cell. Biol. 26 (5) (2006)
1679.[12] H. Morimoto, K. Kondoh, S. Nishimoto, K. Terasawa, E. Nishida, J. Biol. Chem. 282
(49) (2007) 35449.[13] M. Hayashi, J.D. Lee, J. Mol. Med. 82 (12) (2004) 800.[14] C.P. Regan, W. Li, D.M. Boucher, S. Spatz, M.S. Su, K. Kuida, Proc. Natl Acad. Sci. USA
99 (14) (2002) 9248.[15] S.J. Sohn, B.K. Sarvis, D. Cado, A. Winoto, J. Biol. Chem. 277 (45) (2002) 43344.[16] L. Yan, J. Carr, P.R. Ashby, V. Murry-Tait, C. Thompson, J.S. Arthur, BMC Dev. Biol. 3
(2003) 11.[17] M. Hayashi, C. Fearns, B. Eliceiri, Y. Yang, J.D. Lee, Cancer Res. 65 (17) (2005) 7699.[18] A. Esparis-Ogando, E. Diaz-Rodriguez, J.C. Montero, L. Yuste, P. Crespo, A.
Pandiella, Mol. Cell. Biol. 22 (1) (2002) 270.[19] P.B. Mehta, B.L. Jenkins, L. McCarthy, L. Thilak, C.N. Robson, D.E. Neal, H.Y. Leung,
Oncogene 22 (9) (2003) 1381.[20] J.C. Montero, A. Ocana, M. Abad, M.J. Ortiz-Ruiz, A. Pandiella, A. Esparis-Ogando,
PLoS ONE 4 (5) (2009) e5565.[21] K. Zen, K. Yasui, T. Nakajima, Y. Zen, K. Zen, Y. Gen, H. Mitsuyoshi, M. Minami, S.
Mitsufuji, S. Tanaka, Y. Itoh, Y. Nakanuma, M. Taniwaki, S. Arii, T. Okanoue, T.Yoshikawa, Genes Chromosom. Cancer 48 (2) (2009) 109.
[22] L. Liu, J.E. Cavanaugh, Y. Wang, H. Sakagami, Z. Mao, Z. Xia, Proc. Natl Acad. Sci.USA 100 (14) (2003) 8532.
[23] A. Shalizi, M. Lehtinen, B. Gaudilliere, N. Donovan, J. Han, Y. Konishi, A. Bonni, J.Neurosci. 23 (19) (2003) 7326.
[24] F.L. Watson, H.M. Heerssen, A. Bhattacharyya, L. Klesse, M.Z. Lin, R.A. Segal, Nat.Neurosci. 4 (10) (2001) 981.
[25] K. Cude, Y. Wang, H.J. Choi, S.L. Hsuan, H. Zhang, C.Y. Wang, Z. Xia, J. Cell Biol. 177(2) (2007) 253.
[26] A. Girio, J.C. Montero, A. Pandiella, S. Chatterjee, Cell. Signal. 19 (9) (2007) 1964.[27] Y. Kuma, G. Sabio, J. Bain, N. Shpiro, R. Marquez, A. Cuenda, J. Biol. Chem. 280 (20)
(2005) 19472.[28] F.A. Inesta-Vaquera, F. Centeno, P. del Reino, G. Sabio, M. Peggie, A. Cuenda, FEBS J.
276 (2) (2009) 387.[29] Y.L. Woods, G. Rena, N. Morrice, A. Barthel, W. Becker, S. Guo, T.G. Unterman, P.
Cohen, Biochem. J. 355 (Pt 3) (2001) 597.[30] B.L. Williamson, J. Marchese, N.A. Morrice, Mol. Cell. Proteomics 5 (2) (2006) 337.[31] N. Mody, D.G. Campbell, N. Morrice, M. Peggie, P. Cohen, Biochem. J. 372 (Pt 2)
(2003) 567.[32] S. Kamakura, T. Moriguchi, E. Nishida, J. Biol. Chem. 274 (37) (1999) 26563.[33] X. Wang, A.J. Merritt, J. Seyfried, C. Guo, E.S. Papadakis, K.G. Finegan, M. Kayahara,
J. Dixon, R.P. Boot-Handford, E.J. Cartwright, U. Mayer, C. Tournier, Mol. Cell. Biol.25 (1) (2005) 336.
[34] M. Malumbres, M. Barbacid, Nat. Rev. Cancer 9 (3) (2009) 153.[35] J. Bain, L. Plater, M. Elliott, N. Shpiro, C.J. Hastie, H. McLauchlan, I. Klevernic, J.S.
Arthur, D.R. Alessi, P. Cohen, Biochem. J. 408 (3) (2007) 297.[36] K. Terasawa, K. Okazaki, E. Nishida, Genes Cells 8 (3) (2003) 263.[37] N. Dephoure, C. Zhou, J. Villen, S.A. Beausoleil, C.E. Bakalarski, S.J. Elledge, S.P. Gygi,
Proc. Natl Acad. Sci. USA 105 (31) (2008) 10762.[38] M. Akaike, W. Che, N.L. Marmarosh, S. Ohta, M. Osawa, B. Ding, B.C. Berk, C. Yan, J.
Abe, Mol. Cell. Biol. 24 (19) (2004) 8691.[39] C. Yan, H. Luo, J.D. Lee, J. Abe, B.C. Berk, J. Biol. Chem. 276 (14) (2001) 10870.[40] H.G. Kasler, J. Victoria, O. Duramad, A.Winoto, Mol. Cell. Biol. 20 (22) (2000) 8382.[41] O.L. Roberts, K. Holmes, J. Muller, D.A. Cross, M.J. Cross, Biochem. Soc. Trans. 37 (Pt
6) (2009) 1254.[42] Z. Raviv, E. Kalie, R. Seger, J. Cell Sci. 117 (Pt 9) (2004) 1773.[43] J. Borges, A. Pandiella, A. Esparis-Ogando, Cell. Signal. 19 (7) (2007) 1473.[44] L. Meijer, A. Borgne, O. Mulner, J.P.J. Chong, J.J. Blow, N. Inagaki, M. Inagaki, J.-G.
Delcros, J.-P. Moulinoux, Eur. J. Biochem. 243 (1–2) (1997) 527.




















