
7-Bromoindirubin-3′-oxime induces caspase-independent cell death
Upload
independentCategory
view
0download
0
ORIGINAL PAPER
Calpain and caspase processing of caspase-12 contribute to the ERstress-induced cell death pathway in differentiated PC12 cells
Juan A. Martinez • Zhiqun Zhang •
Stanislav I. Svetlov • Ronald L. Hayes •
Kevin K. Wang • Stephen F. Larner
� Springer Science+Business Media, LLC 2010
Abstract Neuronal cell death after traumatic brain injury,
Alzheimer’s disease and ischemic stroke may in part be
mediated through endoplasmic reticulum (ER) stress and
unfolded protein response (UPR). UPR results in induction
of molecular chaperone GRP78 and the ER-resident cas-
pase-12, whose activation has been proposed to be medi-
ated by calpain and caspase processing, although their
relative contribution remains unclear. In this study we
induced ER stress with thapsigargin (TG), and determined
the activation profile of calpain-2, caspase-3, caspase-7,
and caspase-12 by analyses of protein levels, correspond-
ing substrates and breakdown products (BDP). Specific
calpain and caspase activity was assessed by analysis of
aII-spectrin BDP of 145 kDa (SBDP145), BDP of 150 kDa
(SBDP150) and BDP of 120 kDa (SBDP120). Decrease in
pro-calpain-2 protein and increased SBDP145 levels by 3 h
after TG treatment indicated early calpain activity. Active
caspase-7 (p20) increase occurred after 8 h, followed by
concomitant up-regulation of active caspase-3 and SBDP120
after 24 h. In vitro digestion experiments supported that
SBDP120 was exclusively generated by active caspase-3 and
validated that kinectin and co-chaperone p23 were calpain
and caspase-7 substrates, respectively. Pro-caspase-12 pro-
tein processing by the specific action of calpain and caspase-
3/7 was observed in a time-dependent manner. N-terminal
pro-domain processing of pro-caspase-12 by calpain
generated a 38 kDa fragment, while caspase-3/7 generated a
35 kDa fragment. Antibody developed specifically against
the caspase-3/7 C-terminal cleavage site D341 detected the
presence of large subunit (p20) containing 23 kDa fragment
that increased after 24 h of TG treatment. Significant cas-
pase-12 enzyme activity was only detected after 24 h of TG
treatment and was completely inhibited by caspase 3/7
inhibitor DEVD-fmk and partially by calpain inhibitor SNJ-
1945. ER-stress-induced cell death pathway in TG-treated
PC12 cells was characterized by up-regulation of GRP-78
and processing and activation of caspase-12 by the orches-
trated proteolytic activity of calpain-2 and caspase-3/7.
Keywords ER stress � Thapsigargin � Unfolded protein
response � Calpain � Caspase � PC12 cells
Abbreviations
ER Endoplasmic reticulum
UPR Unfolded protein response
GRP-78 Glucose responsive protein of 78 kDa
TG Thapsigargin
BDP Breakdown product
TBI Traumatic brain injury
Introduction
The endoplasmic reticulum (ER) specialized functions
include protein translation, folding and transport of pro-
teins for local cellular use (e.g. transmembrane recep-
tors and other integral membrane proteins), or export
(e.g., secreted digestive enzymes), calcium sequestration
for intracellular calcium homeostasis, post-translational
J. A. Martinez (&) � Z. Zhang � S. I. Svetlov �R. L. Hayes � K. K. Wang � S. F. Larner (&)
Center of Innovative Research, Banyan Biomarkers Inc,
12085 Research Drive, Alachua, FL 32615, USA
e-mail: [email protected]
S. F. Larner
e-mail: [email protected]
123
Apoptosis
DOI 10.1007/s10495-010-0526-4

modification and quality control of proteins, and the pro-
duction and storage of glycogen, steroids, and other mac-
romolecules [1]. Physiological and pathological conditions
that disrupt protein folding and/or cause loss of calcium
homeostasis leading to ER dysfunction include glycosyla-
tion deficiency [2], elevated protein synthesis and secretion
[3], glucose deprivation [4], neurodegenerative diseases [5]
and traumatic brain injury (TBI) [6, 7]. TBI leads to the ER
accumulation of misfolded proteins that can cause ER
stress [6]. Cells react to ER stress by activation of the
unfolded protein response (UPR) pathway which activates
signal transduction reactions in both the nucleus and Golgi
apparatus in a coordinated manner to cope with cellular
demand [8]. The UPR enables the cell to reduce the ER
unfolded protein load by translational attenuation, by pro-
moting protein folding through induction of molecular
chaperones and foldases, and by increasing degradation of
unfolded and misfolded ER proteins. However, if ER
homeostasis is not restored in a timely manner its func-
tional capacity is rapidly exceeded and apoptosis is
induced.
The mammalian UPR pathway is mediated primarily
through the ER transmembrane proteins, ATF6, IRE1 and
PERK [8]. Under normal conditions, the luminal domains
of these sensor proteins are occupied by GRP-78/BiP
(glucose responsive protein of 78 kDa/immunoglobulin
binding protein), an ER-resident belonging to the heat-
shock 70 (HSP70) family. Induction of ER stress causes
accumulation of unfolded proteins that have high affinity
for GRP-78 causing its release from these sensor proteins
and thus up-regulating the UPR signal. Although the
cooperative effect of these three sensors serves to protect
the cell until ER function is restored, they also contribute
to the induction of apoptosis if ER homeostasis is not
restored.
The ER-resident caspase-12 has been shown to mediate
apoptosis signaling induced by ER stress [9]. Both cal-
pain(s) and caspase(s) have been proposed to mediate
processing and activation of caspase-12 after induction of
the unfolded protein response and ER stress. Calpain-
deficient mouse embryonic fibroblasts (MEFs) have been
shown to have decreased ER stress-induced activation of
caspase-12 and are resistant to ER stress-induced apoptosis
[10]. Increased cytoplasmic Ca2? levels caused by thapsi-
gargin in primary MEFs appear to lead to the accumulation
and activation of calpain at the ER membrane where it
presumably activates caspase-12. Another protease, cas-
pase-7, has also been implicated in the activation of cas-
pase-12 in response to ER stress [11] and has been reported
to translocate from the cytosol to the ER to interact with
caspase-12 leading to its activation [10, 12].
Although caspase-12 is activated during the ER stress-
induced UPR, the involvement of caspase-12 in ER stress-
induced apoptosis remains controversial. Initial reports on
caspase-12 knockout mice showed increased resistance to
ER stress-induced apoptosis [13], but later studies sug-
gested caspase-12 had a more prominent role during the
inflammatory response [14]. It has also been suggested that
ER-stress induced apoptosis is primarily mediated by cal-
pains and not caspases [15]. The aim of this study was to
determine the relative contribution of calpains and caspases
to caspase-12 activation and the ER stress-induced cell
death pathway in vitro before in vivo TBI studies. A
common mechanism associated with TBI and neurotoxic
shock is the transient loss of calcium homeostasis. To
mimic this loss, thapsigargin was used to induce ER stress
in a neuronal-like cell model using pheochromocytoma cell
line (PC12), differentiated with nerve growth factor (NGF).
Our hypothesis is that the processing of caspase-12 by both
calpains and caspases, are involved in the activation of
caspase-12 and this activation contributes to the ER stress-
induced cell death pathway.
Materials and methods
Rat pheochromocytoma (PC12) cell culture
Rat PC12 YH cell cultures were grown on modified poly-
styrene T-75 tissue culture flasks (Cell-T plus, Sarstedt,
North Carolina, USA) in B-27 supplemented Neurobasal
medium with 10% horse serum, 5% fetal bovine serum,
2 mM glutamine, 1% Fungizone, 100 units/ml penicillin
and 100 lg/ml streptomycin (Gibco/Invitrogen, Carlsbad,
CA, USA). Cultures were kept at 37�C in a humidified 5%
CO2 incubator. Medium was replaced every 3 days and
cells were passaged by standard trypsinization (0.05%
Trypsin-EDTA) (Gibco/Invitrogen, Carlsbad, CA, USA)
upon reaching confluency at a 1:3 split ratio. For experi-
ments, 3 9 104 cells were seeded onto 6-well tissue culture
clusters (Nunclone, NUNC, Denmark) and grown for 48 h.
PC12 cells were differentiated by incubating in low serum
Neurobasal medium (2% horse serum, 1% fetal bovine
serum) with 50 ng/ml nerve growth factor (NGF) (2.5S
subunit, EMD Biosciences, Gibbstown, NJ, USA) and
glutamine/penicillin/streptomycin/amphotericin B as indi-
cated above, for 4–5 days. Cell differentiation was defined
by[80% cells having two or more neurites of any length as
determined by light microscopy of two randomly selected
visual fields using a Zeiss Axiovert 135 inverted micro-
scope. Media was then removed, cells washed with phos-
phate buffered saline (PBS) and experimental conditions
were carried out in serum-free Neurobasal medium with
NGF and supplements mentioned above but with reduced
(1/3) antibiotic/antimycotic content.
Apoptosis
123

Cell death assessment by lactate dehydrogenase (LDH)
assay
Cell death was determined by measuring LDH released
into culture medium using Promega’s CytoTox 96TM
(Madison, WI, USA) assay kit following manufacturer’s
protocol. Briefly, a 50 ll conditioned-medium aliquot was
collected from each sample and transferred to a 96-well
plate. A background control (medium alone) and a maxi-
mum positive control (total cell lysate) were measured to
establish assay range. After adding assay reagents, plates
were incubated for 30 min in the dark at room temperature.
Reaction was then stopped and plates read at 490 nm in a
SpectraMax Plus 384 microplate reader (Molecular Devi-
ces, Ontario, CA).
PC12 cell lysate sample preparation
ER stress was induced in differentiated PC12 cells by
exposure to 1 lM thapsigargin (Sigma Chemical Co, St.
Louis, MO, USA) at various time points (3, 8, 16, 24 h)
applying a reverse time course protocol for simultaneous
cell sampling, in the absence or presence of calpain
inhibitor (SNJ-1945) [16] or caspase inhibitor DEVD-fmk
(R&D Systems, Minneapolis, MN, USA) [17]. Cells were
washed with phosphate buffered saline (PBS) prior to
collection. Total cell lysate was collected by direct in-well
lysis by adding 600 ll of Triton lysis buffer [20 mM Tris-
HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA,
1% Triton X-100, 1 mM NaF, 1 mM Na3VO4, 1 mM DTT
and protease inhibitor cocktail (Roche, Indianapolis, IN,
USA)]. Plates were incubated for 90 min at 4�C in orbital
shaker. Cell lysate was transferred to microcentrifuge tube
and sonicated on ice for 3 sec (Fisher Sonic Dismembrator
60, Fisher Scientific, Pittsburgh, PA, USA) to shear any
remaining DNA. Samples were then centrifuged at
15,0009g for 10 min at 4�C to clarify crude lysate. Super-
natant was snap frozen on dry ice and stored at -80�C. Sub
cellular and size fractionation of PC12 lysate was collected
by scraping cells into 300 ll of cell resuspension buffer
(CRB) (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 2 mM
EGTA, 0.33 M sucrose, 1 mM DTT and protease inhibitor
cocktail). Cell suspension was mechanically disrupted by
passing through a 27G syringe needle (1 cc) on ice. The
suspension was then centrifuged at 8009g for 10 min at 4�C
to pellet membrane/nuclear fraction. Pellet was snap frozen
for future analysis of nuclear proteins and supernatant was
centrifuged at 12,0009g for 20 min at 4�C to pellet the
mitochondria. Supernatant (cytosol/ER fraction) was trans-
ferred to 1.5 ml microcentrifuge tube and snap frozen on dry-
ice. Sample preparation for immunodetection of cleaved
caspase-12 fragments required further processing by
enrichment of proteins in the 10–100 kDa range from either
total protein or cytoplasmic fractions using microconcen-
trators with 100 and 10 kDa molecular weight cutoff mem-
branes (Microcon-100, Microcon-10, Amicon, Bellerica,
MA, USA).
Caspase cleaved caspase-12 antibody development
The peptide sequence H2N–C-C9 linker-SITKAHVETD-OH
corresponding to amino acids 332–341 of murine caspase-
12, where D341 is a caspase-7 cleavage site, was synthesized
and purified by Proteos Inc. (Kalamazoo, MI), conjugated to
keyhole limpet hemocyanin (KLH) and injected into rabbit
host (Pocono Rabbit Farm and Lab, Canadensis, PA) with
subsequent booster injections in a 72-day antibody produc-
tion protocol. Antiserum was purified by peptide affinity
chromatography using disulfide linkage of peptide to agarose
resin (SulfoLink, Pierce/Thermo Scientific, Rockford, IL,
USA) following manufacturer protocol (2 ml columns). A
1:1 PBS-diluted antiserum solution was loaded onto column,
washed with 500 mM NaCl-containing PBS (pH 7.4) fol-
lowed by 5 column volume washes of PBS. The bound
IgG was eluted with 2 ml of 0.1 M glycine (pH 1.9) and
immediately neutralized with 100 ll 1 M Tris base (pH 9).
The eluted IgG’s were dialyzed against PBS (pH 7.4),
concentrated 5-fold with centrifugal microconcentrators
(Microcon-10, Amicon/Millipore, Bellerica, MA, USA) and
stored at -20�C in 50% glycerol.
Quantitative Western Blot Analysis (qWB)
The protein concentration of the PC12 lysates was deter-
mined by a modified Lowry method (DC Protein Assay Kit,
Bio-Rad Laboratories, Hercules, CA, USA) using bovine
serum albumin (BSA) standard curve reference in the
microplate format. Equal protein amounts were loaded onto
4–20% Tris/glycine gel (Invitrogen Life Technologies,
Carlsbad, CA, USA) for sodium dodecyl sulfate-poly-
acrylamide electrophoresis (SDS-PAGE). Samples were
then transferred onto a polyvinylidene fluoride (PVDF)
membrane (Bio-Rad Laboratories, Hercules, CA, USA) by
the semidry method in transfer buffer (39 mM glycine,
48 mM Tris and 5% methanol) at 20 V for 90 min at room
temperature (RT). Membranes were blocked with 5% non-
fat milk in Tween-20-containing Tris-buffered saline
(TBST) (20 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.05%
Tween-20) for 1 h at RT and then incubated with the pri-
mary antibody in 5% non-fat milk in TBST or Signal
BoostTM (EMD Biosciences, San Diego, CA, USA) at 4�C
overnight. After washing in TBST, the membranes were
then incubated for one hour at RT with corresponding
Apoptosis
123

peroxidase (1:20,000–1:40,000) or alkaline phosphatase
(1:5,000–1:10,000) conjugated secondary antibodies (EMD
Biosciences, San Diego, CA, USA). Alkaline phosphatase
conjugates were visualized using 5-bromo-4-chloro-
3-indolylphosphate/nitroblue tetrazolium (BCIP/NBT)
phosphatase substrate (Kirkegaard & Perry Laboratories,
Gaithersburg, MD, USA). Blots were scanned and analyzed
with NIH ImageJ software. Peroxidase conjugates were
visualized by chemiluminescent detection with SuperSig-
nalTM West Femto substrate (Pierce/Thermo Scientific,
Rockford, Il, USA) in a Kodak GL2200 CCD-based imag-
ing system and analyzed by Kodak Molecular Imaging
Software (Carestream Health, New Haven, CT, USA). For
multiple antigen probing, blots were stripped and reprobed
using Restore Plus reagent (Pierce/Thermo Scientific,
Rockford, Il, USA) following manufacturer specifications.
Graphical representation and statistics were performed with
Prism v.5 software (GraphPad, La Jolla, CA, USA).
Primary antibodies included: (1) mouse monoclonal anti-
a-fodrin (BioMol Intl, Plymouth Meeting, PA, USA; 1:5,000),
(2) caspase-12 (rabbit polyclonal, 1:1,000), (3) anti-active
caspase-7 (p20) (rabbit polyclonal, 1:1,000), (4) anti-cleaved
caspase-3 (Asp175) (rabbit polyclonal, 1:1,000), (5) anti-Grp-
78 (rabbit monoclonal, 1:2000), (#2–#5 from Cell Signaling,
Danvers, MA, USA), (6) pro-calpain-2 (in-house, rabbit
polyclonal, 1:500), (7) mouse monoclonal anti-p23 (Affinity
Bioreagents Ltd., Plymouth Meeting, PA, USA; 1:2,000), (8)
anti-kinectin-1 (rabbit polyclonal, Santa Cruz Biotechnology,
CA, USA 1:1000), (9) mouse monoclonal anti-b-actin (Sigma
Chemical Co, St. Louis, MO, USA, 1:10,000), (10) cleaved-
caspase-12 at D341 (C12D341, in-house, rabbit polyclonal,
1:300), (11) caspase-12 small fragment (rabbit polyclonal,
1 lg/ml, Anaspec, Fremont, CA, USA).
In vitro calpain and caspase substrate digestions
Differentiated PC12 cells were lysed in Triton X-100 lysis
buffer as previously described but without protease inhibi-
tors. For caspase enzyme digestions, 50 ll of cell lysate
(200 lg total protein) was incubated with (1) 20 U of
recombinant human caspase-3 (Chemicon/Millipore, Teme-
cula, CA, USA), or (2) 20 U of recombinant human caspase-
7 (EMD Biosciences, San Diego, CA, USA) in enzyme
buffer (EB) (100 mM Tris-HCl, pH 7.4 and 20 mM DTT) at
37�C for 4 h in rocking platform. Calpain digestion of PC12
lysate was performed by incubating 50 ll cell lysate (200 lg
total protein) with 1 lg recombinant rat calpain-2 (EMD
Biosciences, San Diego, CA, USA) at RT for 30 min in
rocking platform in EB with 10 mM CaCl2. Undigested
lysate served as control (30 min for calpain, 4 h for caspase-
3 and -7). After digestion samples were snap frozen in dry ice
and stored at -80�C until used.
Caspase-12 enzyme activity assay
Enzyme assay is based on the cleavage of the caspase-12-
specific substrate ATAD-AFC (MBL International, Woburn,
MA, USA). Total cell lysates from 8 and 24 h TG-treated
cultures were collected as previously described but in the
absence of protease and phosphatase inhibitors. Reactions
were carried in 100 ll volume with 5 lM substrate, enzyme
buffer, and 50 lg total protein sample from PC12 lysate in
duplicates. After 3 h of dark incubation at 37�C with shaking
in microplate incubator/shaker (Amersham, UK), fluores-
cence emission was measured using a SpectraMax Gemini
EM (Molecular Devices, Ontario, CA) set at Ex-400/Em-
505. Results were analyzed using SoftMax 5.0 software and
statistical analysis performed in Prism v5.0 (GraphPad, La
Jolla, CA).
Results
Induction of the unfolded protein response (UPR)
by thapsigargin precedes cytotoxicity in differentiated
PC12 cells
ER stress was induced in differentiated PC12 cells by
exposure to 1 lM thapsigargin (TG). Morphological
assessment during initial long-term time course studies
showed that[90% cell death occurred after 48 h and about
50% cell death was observed after 32 h (data not shown).
During the 24-h time course of TG treatment cellular mor-
phology appeared unremarkable under the light microscope.
Cell death analysis by LDH released into medium indicated a
significant level of cytotoxicity after 24 h of TG-treatment
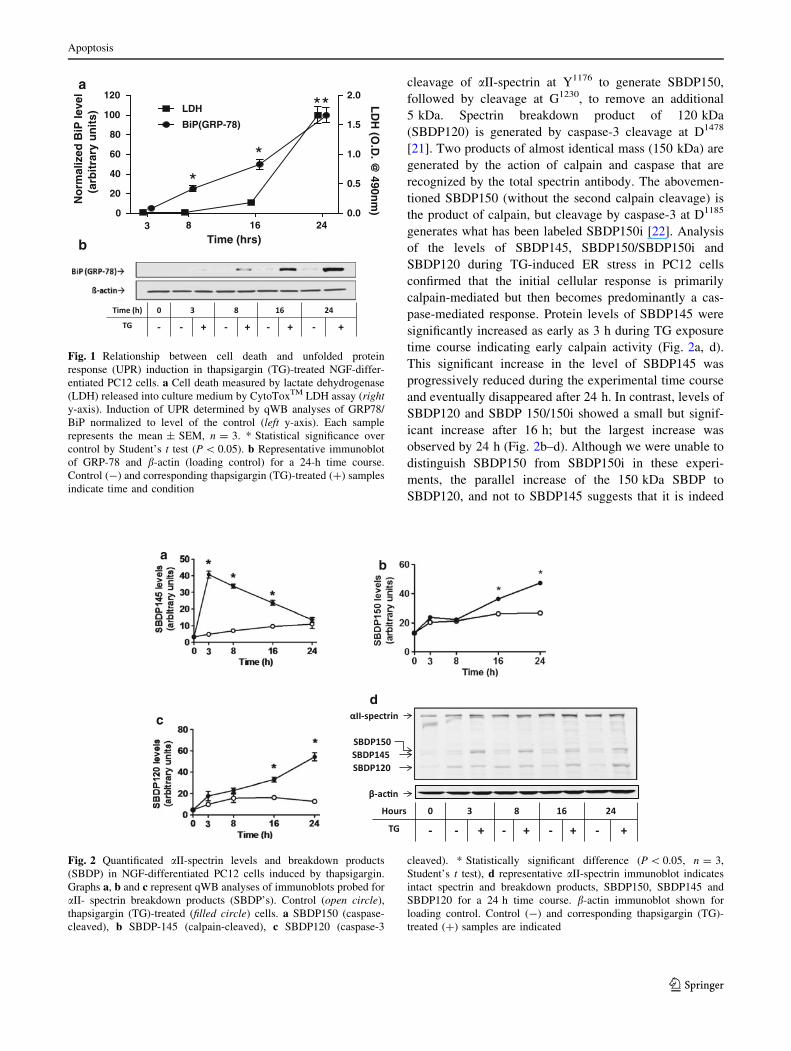
(Fig. 1a). No significant release of LDH was observed at
earlier time points during the 24-h time course. However,
TG-induced stress activated the UPR after 8 h as evidenced
by an increase in protein levels of GRP-78 as determined by
qWB analysis (Fig. 1a, b). There was no significant increase
in the level of GRP-78, before 8 h but after that the levels
began to almost double every 8 h, with peak levels recorded
at the end of the 24-h time course. Results suggest that the
UPR precedes ER stress-induced cell death in PC12 cells.
Calpain- and caspase-3-mediated aII-spectrin
breakdown products induced by TG in PC12 cells
indicate calpain activity precedes caspase-3
We examined the processing of aII-spectrin during TG-
induced ER stress in PC12 cells to determine the level of
calpain and caspase activity. Distinct breakdown products
of aII-spectrin are generated as the result of the action of
calpain and caspase-3 [18–20]. Spectrin breakdown prod-
uct of 145 kDa (SBDP145) results from sequential calpain
Apoptosis
123
cleavage of aII-spectrin at Y1176 to generate SBDP150,
followed by cleavage at G1230, to remove an additional
5 kDa. Spectrin breakdown product of 120 kDa
(SBDP120) is generated by caspase-3 cleavage at D1478
[21]. Two products of almost identical mass (150 kDa) are
generated by the action of calpain and caspase that are
recognized by the total spectrin antibody. The abovemen-
tioned SBDP150 (without the second calpain cleavage) is
the product of calpain, but cleavage by caspase-3 at D1185
generates what has been labeled SBDP150i [22]. Analysis
of the levels of SBDP145, SBDP150/SBDP150i and
SBDP120 during TG-induced ER stress in PC12 cells
confirmed that the initial cellular response is primarily
calpain-mediated but then becomes predominantly a cas-
pase-mediated response. Protein levels of SBDP145 were
significantly increased as early as 3 h during TG exposure
time course indicating early calpain activity (Fig. 2a, d).
This significant increase in the level of SBDP145 was
progressively reduced during the experimental time course
and eventually disappeared after 24 h. In contrast, levels of
SBDP120 and SBDP 150/150i showed a small but signif-
icant increase after 16 h; but the largest increase was
observed by 24 h (Fig. 2b–d). Although we were unable to
distinguish SBDP150 from SBDP150i in these experi-
ments, the parallel increase of the 150 kDa SBDP to
SBDP120, and not to SBDP145 suggests that it is indeed
8 16 240
20
40
60
80
100
120
0.0
0.5
1.0
1.5
2.0LDH
BiP(GRP-78)
3
**
**
Time (hrs)
No
rmal
ized
BiP
leve
l(a
rbit
rary
un
its)
LD
H (O
.D. @
490nm
)
b
a
Fig. 1 Relationship between cell death and unfolded protein
response (UPR) induction in thapsigargin (TG)-treated NGF-differ-
entiated PC12 cells. a Cell death measured by lactate dehydrogenase
(LDH) released into culture medium by CytoToxTM LDH assay (righty-axis). Induction of UPR determined by qWB analyses of GRP78/
BiP normalized to level of the control (left y-axis). Each sample
represents the mean ± SEM, n = 3. * Statistical significance over
control by Student’s t test (P \ 0.05). b Representative immunoblot
of GRP-78 and b-actin (loading control) for a 24-h time course.
Control (-) and corresponding thapsigargin (TG)-treated (?) samples
indicate time and condition
d
c
ab
Fig. 2 Quantificated aII-spectrin levels and breakdown products
(SBDP) in NGF-differentiated PC12 cells induced by thapsigargin.
Graphs a, b and c represent qWB analyses of immunoblots probed for
aII- spectrin breakdown products (SBDP’s). Control (open circle),
thapsigargin (TG)-treated (filled circle) cells. a SBDP150 (caspase-
cleaved), b SBDP-145 (calpain-cleaved), c SBDP120 (caspase-3
cleaved). * Statistically significant difference (P \ 0.05, n = 3,
Student’s t test), d representative aII-spectrin immunoblot indicates
intact spectrin and breakdown products, SBDP150, SBDP145 and
SBDP120 for a 24 h time course. b-actin immunoblot shown for
loading control. Control (-) and corresponding thapsigargin (TG)-
treated (?) samples are indicated
Apoptosis
123
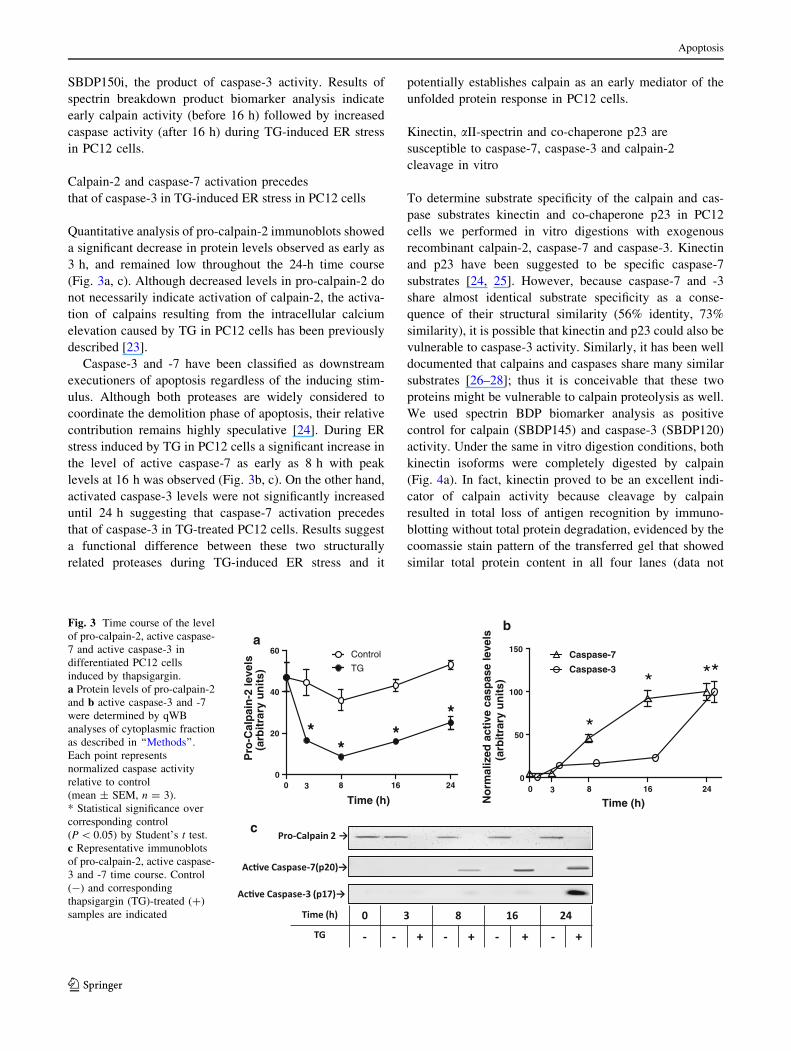
SBDP150i, the product of caspase-3 activity. Results of
spectrin breakdown product biomarker analysis indicate
early calpain activity (before 16 h) followed by increased
caspase activity (after 16 h) during TG-induced ER stress
in PC12 cells.
Calpain-2 and caspase-7 activation precedes
that of caspase-3 in TG-induced ER stress in PC12 cells
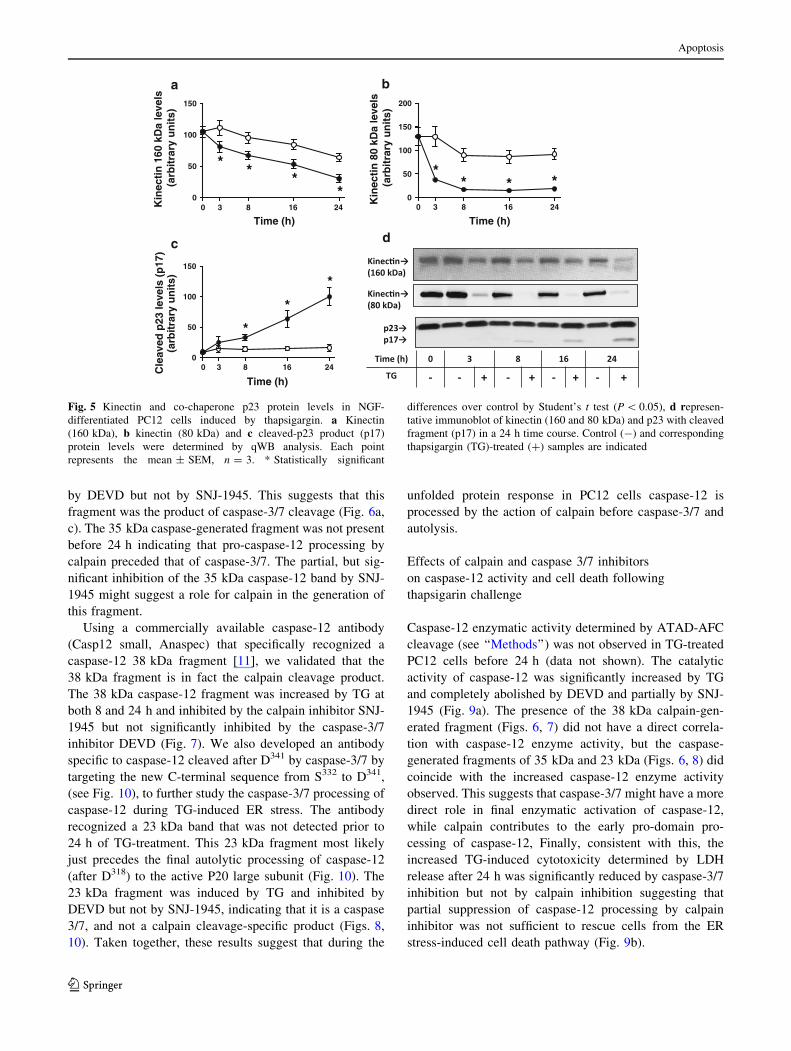
Quantitative analysis of pro-calpain-2 immunoblots showed
a significant decrease in protein levels observed as early as
3 h, and remained low throughout the 24-h time course
(Fig. 3a, c). Although decreased levels in pro-calpain-2 do
not necessarily indicate activation of calpain-2, the activa-
tion of calpains resulting from the intracellular calcium
elevation caused by TG in PC12 cells has been previously
described [23].
Caspase-3 and -7 have been classified as downstream
executioners of apoptosis regardless of the inducing stim-
ulus. Although both proteases are widely considered to
coordinate the demolition phase of apoptosis, their relative
contribution remains highly speculative [24]. During ER
stress induced by TG in PC12 cells a significant increase in
the level of active caspase-7 as early as 8 h with peak
levels at 16 h was observed (Fig. 3b, c). On the other hand,
activated caspase-3 levels were not significantly increased
until 24 h suggesting that caspase-7 activation precedes
that of caspase-3 in TG-treated PC12 cells. Results suggest
a functional difference between these two structurally
related proteases during TG-induced ER stress and it
potentially establishes calpain as an early mediator of the
unfolded protein response in PC12 cells.
Kinectin, aII-spectrin and co-chaperone p23 are
susceptible to caspase-7, caspase-3 and calpain-2
cleavage in vitro
To determine substrate specificity of the calpain and cas-
pase substrates kinectin and co-chaperone p23 in PC12
cells we performed in vitro digestions with exogenous
recombinant calpain-2, caspase-7 and caspase-3. Kinectin
and p23 have been suggested to be specific caspase-7
substrates [24, 25]. However, because caspase-7 and -3
share almost identical substrate specificity as a conse-
quence of their structural similarity (56% identity, 73%
similarity), it is possible that kinectin and p23 could also be
vulnerable to caspase-3 activity. Similarly, it has been well
documented that calpains and caspases share many similar
substrates [26–28]; thus it is conceivable that these two
proteins might be vulnerable to calpain proteolysis as well.
We used spectrin BDP biomarker analysis as positive
control for calpain (SBDP145) and caspase-3 (SBDP120)
activity. Under the same in vitro digestion conditions, both
kinectin isoforms were completely digested by calpain
(Fig. 4a). In fact, kinectin proved to be an excellent indi-
cator of calpain activity because cleavage by calpain
resulted in total loss of antigen recognition by immuno-
blotting without total protein degradation, evidenced by the
coomassie stain pattern of the transferred gel that showed
similar total protein content in all four lanes (data not
0 8 16 240
50
100
150Caspase-7
Caspase-3
3
*
* **
Time (h)No
rmal
ized
act
ive
casp
ase
leve
ls(a
rbit
rary
un
its)
ab
0 8 16 240
20
40
60
*
3
**
*
Control
TG
Time (h)
Pro
-Cal
pai
n-2
leve
ls(a
rbit
rary
un
its)
c
Fig. 3 Time course of the level
of pro-calpain-2, active caspase-
7 and active caspase-3 in
differentiated PC12 cells
induced by thapsigargin.
a Protein levels of pro-calpain-2
and b active caspase-3 and -7
were determined by qWB
analyses of cytoplasmic fraction
as described in ‘‘Methods’’.
Each point represents
normalized caspase activity
relative to control
(mean ± SEM, n = 3).
* Statistical significance over
corresponding control
(P \ 0.05) by Student’s t test.
c Representative immunoblots
of pro-calpain-2, active caspase-
3 and -7 time course. Control
(-) and corresponding
thapsigargin (TG)-treated (?)
samples are indicated
Apoptosis
123

shown). Full length kinectin (160 kDa) digestion by cas-
pase-7 and -3 showed a BDP of 120 kDa. This indicates
that the 160 kDa form of kinectin was susceptible to both
caspase-7 and -3 cleavage. But interestingly, the short form
of kinetin (80 kDa) was degraded to a truncated form of
60 kDa by caspase-3, but not caspase-7. We confirmed
activity of calpain and caspase-3 by spectrin BDP bio-
marker analysis of SBDP145, SBDP150i (see * in Fig. 4b)
and SBDP120 as we have reported previously [29].
Co-chaperone p23 was not susceptible to calpain cleavage
and was preferentially, although not exclusively, cleaved by
caspase-7 compared to caspase-3 (Fig. 4c). This demon-
strates that kinectin was a substrate preferentially processed
by calpain-2 and differentially processed by caspase-7 and -
3. Although kinectin could be used to determine activity of
the abovementioned proteases, its sensitivity to calpain-
mediated proteolysis makes it potentially useful for assess-
ment of calpain activity. Similarly, p23 was shown to be
preferentially processed by caspase-7 to generate p17 and
was not a calpain substrate, thus p17 could serve as indicator
of caspase-7 activity. The fact that SBDP120 could be pro-
duced only by the action of caspase-3 and not caspase-7
provides a unique marker to distinguish activity between
these two related proteases.
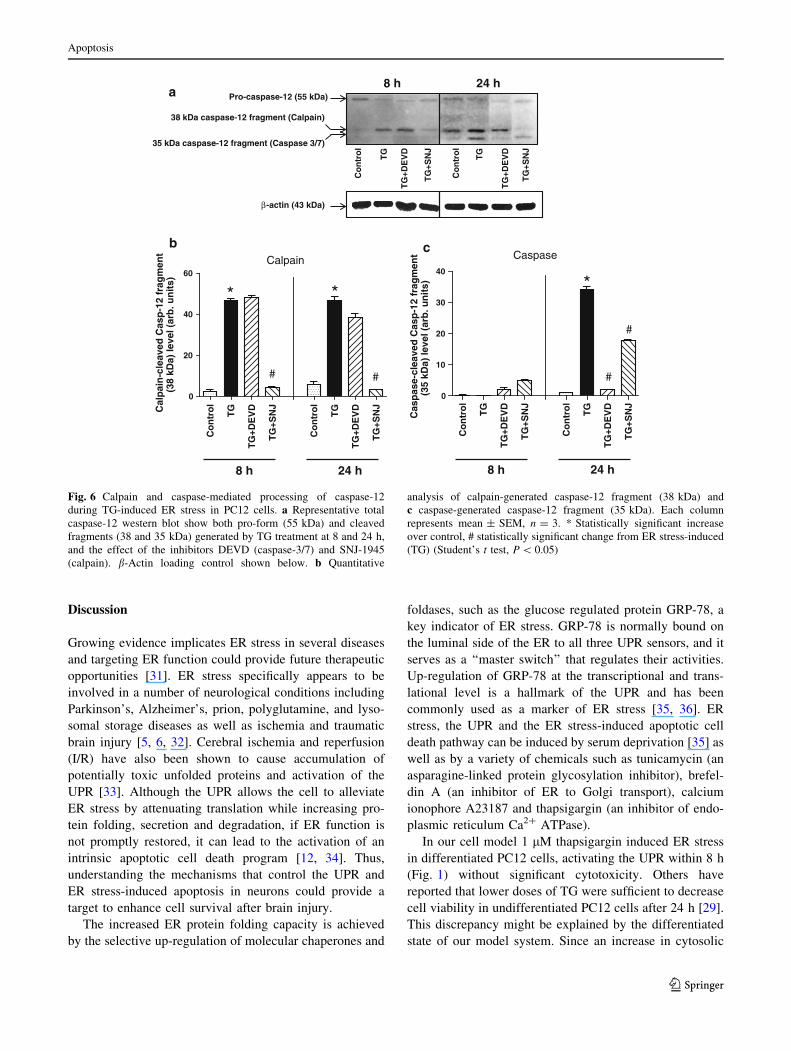
Temporal processing of kinectin and p23 support
that calpain activation precedes that of caspase-7
and -3 in TG-treated PC12 cells
Kinectin, the molecular motor kinesin receptor in ER,
although previously identified as a substrate for caspase-7
[25], as noted above, it was also a caspase-3, caspase-7 and
calpain substrate (Fig. 4a) and we decided to determine its
processing during TG-induced ER stress in PC12 cells.
Similarly, the processing of co-chaperone p23 protein, a
member of the heat shock protein 90 family (Hsp90),
shown to be a substrate with higher affinity for caspase-7
than caspase-3 [24], was examined. Analysis of the integ-
rity of these substrates during TG-induced ER stress in
PC12 cells showed different temporal profiles. Both the
native 160 kDa protein and the more abundant short form
of kinectin (80 kDa band) were immunodetected. It has
been previously reported that both are present in brain
tissue lysate [30]. Analysis of both immunoreactive species
showed a homologous temporal processing profile during
TG-induced ER stress. No breakdown products were
detected for either kinectin isoforms, but we did observe a
significant decrease in the protein level for both 160 kDa
and 80 kDa forms after 3 h of TG treatment (Fig. 5a, b, d).
The difference in the decreased level of the 80 kDa
kinectin band was more pronounced than that of the
160 kDa kinectin form. On the other hand, western blot
analysis of co-chaperone p23 showed a breakdown product
of 17 kDa (p17) that was significantly increased after 8 h
(Fig. 5c, d), indicating caspase-7 activity. Results of cal-
pain and caspase substrate processing during TG-induced
ER stress in PC12 cells again indicate that calpain activity
precedes that of caspase.
Processing and activation of caspase-12 by calpain
and caspase during ER stress produce distinct calpain
and caspase cleavage products
In TG-induced ER stress PC12 cells differential processing
of pro-caspase-12 by the action of calpain and caspase-3/7
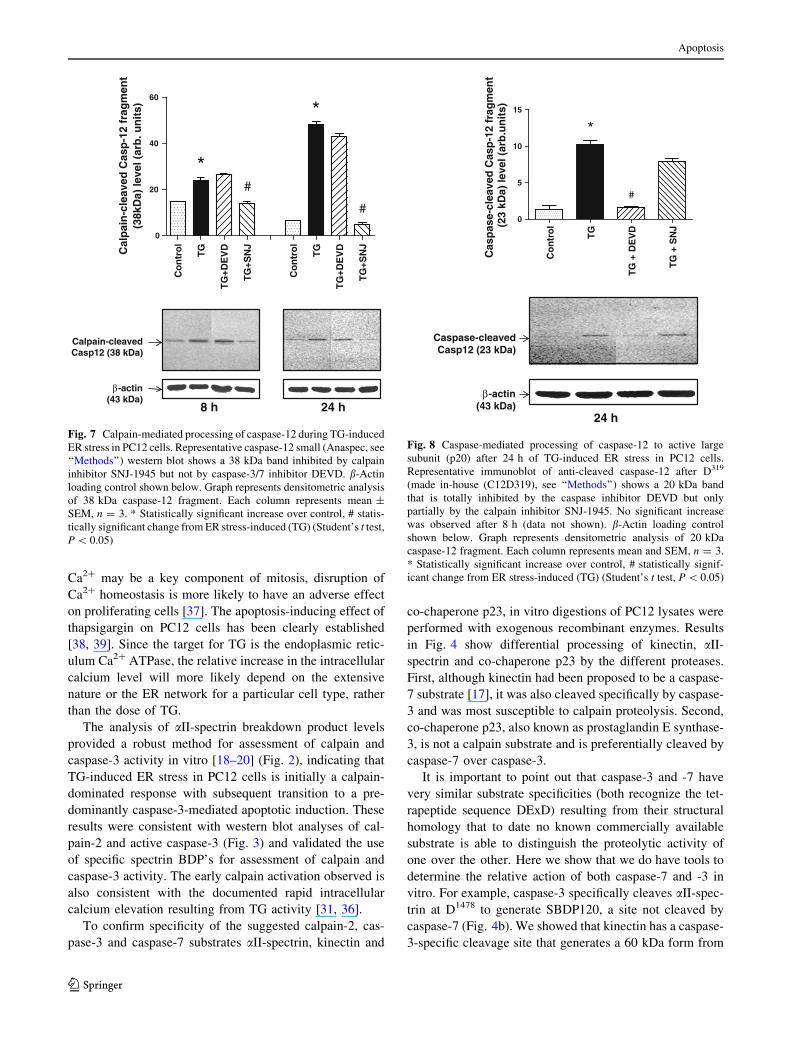
was observed. After 8 h, TG produced a 38 kDa fragment
that was inhibitable by the calpain inhibitor SNJ-1945, but
not by the caspase-3/7 inhibitor DEVD. This suggests this
38 kDa fragment was the product of calpain-cleavage
(Fig. 6a, b). This 38 kDa calpain product was also observed
at 24 h but only with 24 h of TG-treatment did we began to
observe a 35 kDa fragment that was completely inhibited
a
b
c
Fig. 4 Calpain and caspase in vitro digestion of kinectin and
co-chaperone p23 in NGF-differentiated PC12 cells. PC12 lysate
was digested with active calpain-2, caspase-3 and caspase-7 (see
‘‘Methods’’). a Kinectin immunoblot shows 160 and 80 kDa bands.
Kinectin was completely digested by calpain-2. Both caspase-3 and -7
cleaved the 160 kDa kinectin form to generate a 120 kDa band, but
only caspase-3 cleaved the 80 kDa kinectin form to generate a
60 kDa band. b Immunodetection of intact aII-spectrin and break-
down products SBDP145 and SBDP120 confirmed calpain and
caspase-3 activity, respectively. Caspase-7 and -3 processing of aII-
spectrin produced a SBDP band of 150 kDa (*SBDP-150i). c Co-
chaperone p23 immunoblot did not show degradation by calpain-2,
however it showed almost complete digestion to p17 with caspase-7,
but was minimally digested by caspase-3
Apoptosis
123
by DEVD but not by SNJ-1945. This suggests that this
fragment was the product of caspase-3/7 cleavage (Fig. 6a,
c). The 35 kDa caspase-generated fragment was not present
before 24 h indicating that pro-caspase-12 processing by
calpain preceded that of caspase-3/7. The partial, but sig-
nificant inhibition of the 35 kDa caspase-12 band by SNJ-
1945 might suggest a role for calpain in the generation of
this fragment.
Using a commercially available caspase-12 antibody
(Casp12 small, Anaspec) that specifically recognized a
caspase-12 38 kDa fragment [11], we validated that the
38 kDa fragment is in fact the calpain cleavage product.
The 38 kDa caspase-12 fragment was increased by TG at
both 8 and 24 h and inhibited by the calpain inhibitor SNJ-
1945 but not significantly inhibited by the caspase-3/7
inhibitor DEVD (Fig. 7). We also developed an antibody
specific to caspase-12 cleaved after D341 by caspase-3/7 by
targeting the new C-terminal sequence from S332 to D341,
(see Fig. 10), to further study the caspase-3/7 processing of
caspase-12 during TG-induced ER stress. The antibody
recognized a 23 kDa band that was not detected prior to
24 h of TG-treatment. This 23 kDa fragment most likely
just precedes the final autolytic processing of caspase-12
(after D318) to the active P20 large subunit (Fig. 10). The
23 kDa fragment was induced by TG and inhibited by
DEVD but not by SNJ-1945, indicating that it is a caspase
3/7, and not a calpain cleavage-specific product (Figs. 8,
10). Taken together, these results suggest that during the
unfolded protein response in PC12 cells caspase-12 is
processed by the action of calpain before caspase-3/7 and
autolysis.
Effects of calpain and caspase 3/7 inhibitors
on caspase-12 activity and cell death following
thapsigarin challenge
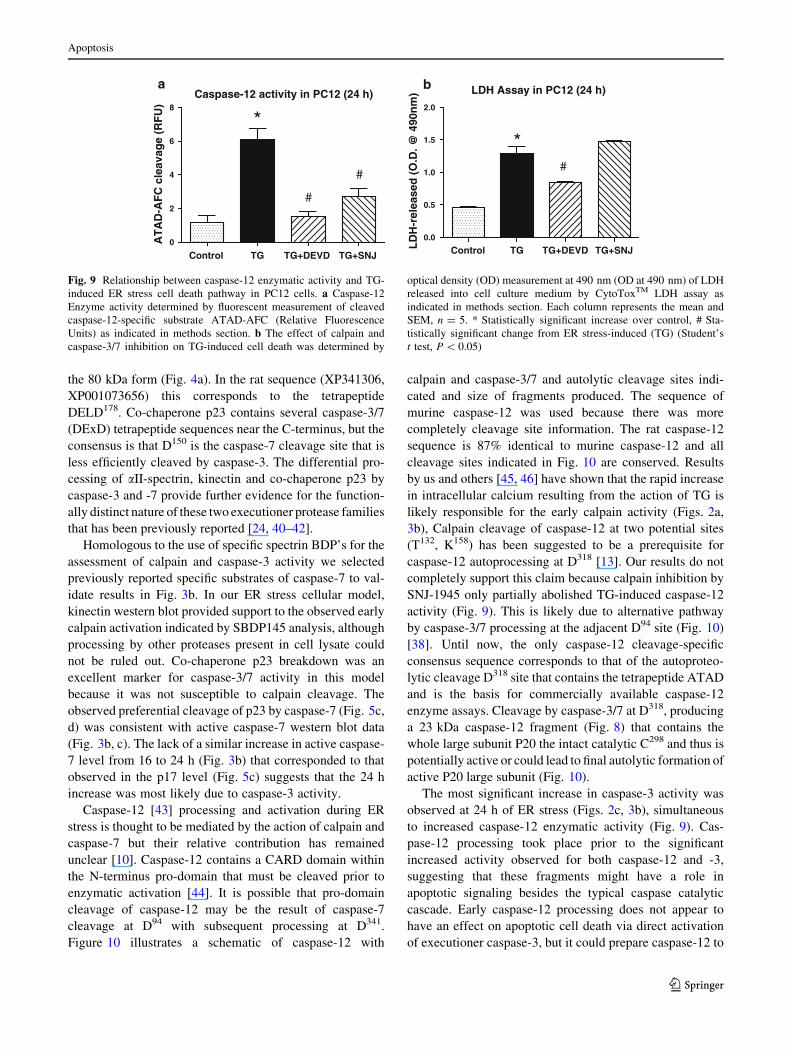
Caspase-12 enzymatic activity determined by ATAD-AFC
cleavage (see ‘‘Methods’’) was not observed in TG-treated
PC12 cells before 24 h (data not shown). The catalytic
activity of caspase-12 was significantly increased by TG
and completely abolished by DEVD and partially by SNJ-
1945 (Fig. 9a). The presence of the 38 kDa calpain-gen-
erated fragment (Figs. 6, 7) did not have a direct correla-
tion with caspase-12 enzyme activity, but the caspase-
generated fragments of 35 kDa and 23 kDa (Figs. 6, 8) did
coincide with the increased caspase-12 enzyme activity
observed. This suggests that caspase-3/7 might have a more
direct role in final enzymatic activation of caspase-12,
while calpain contributes to the early pro-domain pro-
cessing of caspase-12, Finally, consistent with this, the
increased TG-induced cytotoxicity determined by LDH
release after 24 h was significantly reduced by caspase-3/7
inhibition but not by calpain inhibition suggesting that
partial suppression of caspase-12 processing by calpain
inhibitor was not sufficient to rescue cells from the ER
stress-induced cell death pathway (Fig. 9b).
0 8 16 240
50
100
150
*
*
*
3
Time (h)
Cle
aved
p23
leve
ls (
p17
)(a
rbit
rary
un
its)
a
0 8 16 240
50
100
150
* * * *3
*
Time (h)
Kin
ecti
n 1
60 k
Da
leve
ls (
arb
itra
ry u
nit
s)
b
d
0 8 16 240
50
100
150
200
** * *
3
Time (h)
Kin
ecti
n 8
0 kD
a le
vels
(ar
bit
rary
un
its)
c
Fig. 5 Kinectin and co-chaperone p23 protein levels in NGF-
differentiated PC12 cells induced by thapsigargin. a Kinectin
(160 kDa), b kinectin (80 kDa) and c cleaved-p23 product (p17)
protein levels were determined by qWB analysis. Each point
represents the mean ± SEM, n = 3. * Statistically significant
differences over control by Student’s t test (P \ 0.05), d represen-
tative immunoblot of kinectin (160 and 80 kDa) and p23 with cleaved
fragment (p17) in a 24 h time course. Control (-) and corresponding
thapsigargin (TG)-treated (?) samples are indicated
Apoptosis
123
Discussion
Growing evidence implicates ER stress in several diseases
and targeting ER function could provide future therapeutic
opportunities [31]. ER stress specifically appears to be
involved in a number of neurological conditions including
Parkinson’s, Alzheimer’s, prion, polyglutamine, and lyso-
somal storage diseases as well as ischemia and traumatic
brain injury [5, 6, 32]. Cerebral ischemia and reperfusion
(I/R) have also been shown to cause accumulation of
potentially toxic unfolded proteins and activation of the
UPR [33]. Although the UPR allows the cell to alleviate
ER stress by attenuating translation while increasing pro-
tein folding, secretion and degradation, if ER function is
not promptly restored, it can lead to the activation of an
intrinsic apoptotic cell death program [12, 34]. Thus,
understanding the mechanisms that control the UPR and
ER stress-induced apoptosis in neurons could provide a
target to enhance cell survival after brain injury.
The increased ER protein folding capacity is achieved
by the selective up-regulation of molecular chaperones and
foldases, such as the glucose regulated protein GRP-78, a
key indicator of ER stress. GRP-78 is normally bound on
the luminal side of the ER to all three UPR sensors, and it
serves as a ‘‘master switch’’ that regulates their activities.
Up-regulation of GRP-78 at the transcriptional and trans-
lational level is a hallmark of the UPR and has been
commonly used as a marker of ER stress [35, 36]. ER
stress, the UPR and the ER stress-induced apoptotic cell
death pathway can be induced by serum deprivation [35] as
well as by a variety of chemicals such as tunicamycin (an
asparagine-linked protein glycosylation inhibitor), brefel-
din A (an inhibitor of ER to Golgi transport), calcium
ionophore A23187 and thapsigargin (an inhibitor of endo-
plasmic reticulum Ca2? ATPase).
In our cell model 1 lM thapsigargin induced ER stress
in differentiated PC12 cells, activating the UPR within 8 h
(Fig. 1) without significant cytotoxicity. Others have
reported that lower doses of TG were sufficient to decrease
cell viability in undifferentiated PC12 cells after 24 h [29].
This discrepancy might be explained by the differentiated
state of our model system. Since an increase in cytosolic
8 h 24 hPro-caspase-12 (55 kDa)
38 kDa caspase-12 fragment (Calpain)
35 kDa caspase-12 fragment (Caspase 3/7)
Co
ntr
ol
TG
TG
+DE
VD
TG
+SN
J
Co
ntr
ol
TG
TG
+DE
VD
TG
+SN
J
a
Co
ntr
ol
TG
TG
+DE
VD
TG
+SN
J
Co
ntr
ol
TG
TG
+DE
VD
TG
+SN
J
0
20
40
60
*
#
*
#
Cal
pai
n-c
leav
ed C
asp
-12
frag
men
t(3
8 kD
a) le
vel (
arb
. un
its)
8 h 24 hC
on
tro
l
TG
TG
+DE
VD
TG
+SN
J
Co
ntr
ol
TG
TG
+DE
VD
TG
+SN
J
0
10
20
30
40
*
#
#
Cas
pas
e-cl
eave
d C
asp
-12
frag
men
t(3
5 kD
a) le
vel (
arb
. un
its)
8 h 24 h
b cCalpain Caspase
β-actin (43 kDa)
Fig. 6 Calpain and caspase-mediated processing of caspase-12
during TG-induced ER stress in PC12 cells. a Representative total
caspase-12 western blot show both pro-form (55 kDa) and cleaved
fragments (38 and 35 kDa) generated by TG treatment at 8 and 24 h,
and the effect of the inhibitors DEVD (caspase-3/7) and SNJ-1945
(calpain). b-Actin loading control shown below. b Quantitative
analysis of calpain-generated caspase-12 fragment (38 kDa) and
c caspase-generated caspase-12 fragment (35 kDa). Each column
represents mean ± SEM, n = 3. * Statistically significant increase
over control, # statistically significant change from ER stress-induced
(TG) (Student’s t test, P \ 0.05)
Apoptosis
123
Ca2? may be a key component of mitosis, disruption of
Ca2? homeostasis is more likely to have an adverse effect
on proliferating cells [37]. The apoptosis-inducing effect of
thapsigargin on PC12 cells has been clearly established
[38, 39]. Since the target for TG is the endoplasmic retic-
ulum Ca2? ATPase, the relative increase in the intracellular
calcium level will more likely depend on the extensive
nature or the ER network for a particular cell type, rather
than the dose of TG.
The analysis of aII-spectrin breakdown product levels
provided a robust method for assessment of calpain and
caspase-3 activity in vitro [18–20] (Fig. 2), indicating that
TG-induced ER stress in PC12 cells is initially a calpain-
dominated response with subsequent transition to a pre-
dominantly caspase-3-mediated apoptotic induction. These
results were consistent with western blot analyses of cal-
pain-2 and active caspase-3 (Fig. 3) and validated the use
of specific spectrin BDP’s for assessment of calpain and
caspase-3 activity. The early calpain activation observed is
also consistent with the documented rapid intracellular
calcium elevation resulting from TG activity [31, 36].
To confirm specificity of the suggested calpain-2, cas-
pase-3 and caspase-7 substrates aII-spectrin, kinectin and
co-chaperone p23, in vitro digestions of PC12 lysates were
performed with exogenous recombinant enzymes. Results
in Fig. 4 show differential processing of kinectin, aII-
spectrin and co-chaperone p23 by the different proteases.
First, although kinectin had been proposed to be a caspase-
7 substrate [17], it was also cleaved specifically by caspase-
3 and was most susceptible to calpain proteolysis. Second,
co-chaperone p23, also known as prostaglandin E synthase-
3, is not a calpain substrate and is preferentially cleaved by
caspase-7 over caspase-3.
It is important to point out that caspase-3 and -7 have
very similar substrate specificities (both recognize the tet-
rapeptide sequence DExD) resulting from their structural
homology that to date no known commercially available
substrate is able to distinguish the proteolytic activity of
one over the other. Here we show that we do have tools to
determine the relative action of both caspase-7 and -3 in
vitro. For example, caspase-3 specifically cleaves aII-spec-
trin at D1478 to generate SBDP120, a site not cleaved by
caspase-7 (Fig. 4b). We showed that kinectin has a caspase-
3-specific cleavage site that generates a 60 kDa form from
Co
ntr
ol
TG
TG
+DE
VD
TG
+SN
J
Co
ntr
ol
TG
TG
+DE
VD
TG
+SN
J
0
20
40
60
*
*
#
#
Cal
pai
n-c
leav
ed C
asp
-12
frag
men
t(3
8kD
a) le
vel (
arb
. un
its)
Calpain-cleaved Casp12 (38 kDa)
8 h 24 h
β-actin(43 kDa)
Fig. 7 Calpain-mediated processing of caspase-12 during TG-induced
ER stress in PC12 cells. Representative caspase-12 small (Anaspec, see
‘‘Methods’’) western blot shows a 38 kDa band inhibited by calpain
inhibitor SNJ-1945 but not by caspase-3/7 inhibitor DEVD. b-Actin
loading control shown below. Graph represents densitometric analysis
of 38 kDa caspase-12 fragment. Each column represents mean ±
SEM, n = 3. * Statistically significant increase over control, # statis-
tically significant change from ER stress-induced (TG) (Student’s t test,
P \ 0.05)C
on
tro
l
TG
TG
+ D
EV
D
TG
+ S
NJ
0
5
10
15
*
#
Cas
pas
e-cl
eave
d C
asp
-12
frag
men
t (
23 k
Da)
leve
l (ar
b.u
nit
s)
Caspase-cleaved Casp12 (23 kDa)
24 h
β-actin(43 kDa)
Fig. 8 Caspase-mediated processing of caspase-12 to active large
subunit (p20) after 24 h of TG-induced ER stress in PC12 cells.
Representative immunoblot of anti-cleaved caspase-12 after D319
(made in-house (C12D319), see ‘‘Methods’’) shows a 20 kDa band
that is totally inhibited by the caspase inhibitor DEVD but only
partially by the calpain inhibitor SNJ-1945. No significant increase
was observed after 8 h (data not shown). b-Actin loading control
shown below. Graph represents densitometric analysis of 20 kDa
caspase-12 fragment. Each column represents mean and SEM, n = 3.
* Statistically significant increase over control, # statistically signif-
icant change from ER stress-induced (TG) (Student’s t test, P \ 0.05)
Apoptosis
123
the 80 kDa form (Fig. 4a). In the rat sequence (XP341306,
XP001073656) this corresponds to the tetrapeptide
DELD178. Co-chaperone p23 contains several caspase-3/7
(DExD) tetrapeptide sequences near the C-terminus, but the
consensus is that D150 is the caspase-7 cleavage site that is
less efficiently cleaved by caspase-3. The differential pro-
cessing of aII-spectrin, kinectin and co-chaperone p23 by
caspase-3 and -7 provide further evidence for the function-
ally distinct nature of these two executioner protease families
that has been previously reported [24, 40–42].
Homologous to the use of specific spectrin BDP’s for the
assessment of calpain and caspase-3 activity we selected
previously reported specific substrates of caspase-7 to val-
idate results in Fig. 3b. In our ER stress cellular model,
kinectin western blot provided support to the observed early
calpain activation indicated by SBDP145 analysis, although
processing by other proteases present in cell lysate could
not be ruled out. Co-chaperone p23 breakdown was an
excellent marker for caspase-3/7 activity in this model
because it was not susceptible to calpain cleavage. The
observed preferential cleavage of p23 by caspase-7 (Fig. 5c,
d) was consistent with active caspase-7 western blot data
(Fig. 3b, c). The lack of a similar increase in active caspase-
7 level from 16 to 24 h (Fig. 3b) that corresponded to that
observed in the p17 level (Fig. 5c) suggests that the 24 h
increase was most likely due to caspase-3 activity.
Caspase-12 [43] processing and activation during ER
stress is thought to be mediated by the action of calpain and
caspase-7 but their relative contribution has remained
unclear [10]. Caspase-12 contains a CARD domain within
the N-terminus pro-domain that must be cleaved prior to
enzymatic activation [44]. It is possible that pro-domain
cleavage of caspase-12 may be the result of caspase-7
cleavage at D94 with subsequent processing at D341.
Figure 10 illustrates a schematic of caspase-12 with
calpain and caspase-3/7 and autolytic cleavage sites indi-
cated and size of fragments produced. The sequence of
murine caspase-12 was used because there was more
completely cleavage site information. The rat caspase-12
sequence is 87% identical to murine caspase-12 and all
cleavage sites indicated in Fig. 10 are conserved. Results
by us and others [45, 46] have shown that the rapid increase
in intracellular calcium resulting from the action of TG is
likely responsible for the early calpain activity (Figs. 2a,
3b), Calpain cleavage of caspase-12 at two potential sites
(T132, K158) has been suggested to be a prerequisite for
caspase-12 autoprocessing at D318 [13]. Our results do not
completely support this claim because calpain inhibition by
SNJ-1945 only partially abolished TG-induced caspase-12
activity (Fig. 9). This is likely due to alternative pathway
by caspase-3/7 processing at the adjacent D94 site (Fig. 10)
[38]. Until now, the only caspase-12 cleavage-specific
consensus sequence corresponds to that of the autoproteo-
lytic cleavage D318 site that contains the tetrapeptide ATAD
and is the basis for commercially available caspase-12
enzyme assays. Cleavage by caspase-3/7 at D318, producing
a 23 kDa caspase-12 fragment (Fig. 8) that contains the
whole large subunit P20 the intact catalytic C298 and thus is
potentially active or could lead to final autolytic formation of
active P20 large subunit (Fig. 10).
The most significant increase in caspase-3 activity was
observed at 24 h of ER stress (Figs. 2c, 3b), simultaneous
to increased caspase-12 enzymatic activity (Fig. 9). Cas-
pase-12 processing took place prior to the significant
increased activity observed for both caspase-12 and -3,
suggesting that these fragments might have a role in
apoptotic signaling besides the typical caspase catalytic
cascade. Early caspase-12 processing does not appear to
have an effect on apoptotic cell death via direct activation
of executioner caspase-3, but it could prepare caspase-12 to
Caspase-12 activity in PC12 (24 h)
Control TG TG+DEVD TG+SNJ0
2
4
6
8
*
#
#A
TA
D-A
FC
cle
avag
e (R
FU
)
LDH Assay in PC12 (24 h)
Control TG TG+DEVD TG+SNJ0.0
0.5
1.0
1.5
2.0
*#
LD
H-r
elea
sed
(O
.D. @
490
nm
)
a b
Fig. 9 Relationship between caspase-12 enzymatic activity and TG-
induced ER stress cell death pathway in PC12 cells. a Caspase-12
Enzyme activity determined by fluorescent measurement of cleaved
caspase-12-specific substrate ATAD-AFC (Relative Fluorescence
Units) as indicated in methods section. b The effect of calpain and
caspase-3/7 inhibition on TG-induced cell death was determined by
optical density (OD) measurement at 490 nm (OD at 490 nm) of LDH
released into cell culture medium by CytoToxTM LDH assay as
indicated in methods section. Each column represents the mean and
SEM, n = 5. * Statistically significant increase over control, # Sta-
tistically significant change from ER stress-induced (TG) (Student’s
t test, P \ 0.05)
Apoptosis
123

participate in activation of caspase-3 via caspase-9 in
presence or absence of apoptosome complex formation [12,
47]. Studies using the caspase-12 inhibitor z-ATAD-fmk
indicate a reduction in both ER stress induced apoptosis,
active caspase-9 and active caspase-3 in PC12 cells [48]. It
has been suggested that a 36 kDa caspase-12 fragment
detected by a caspase-12 cleaved at D341 antibody trans-
locates to nucleus during ER stress [49].
Taken the literature data and our results together, our ER
stress caspase-12 activation model suggests that (i) cas-
pase-12 is initially processed by calpain, removing the
CARD-containing pro-domain producing a 38 kDa frag-
ment; (ii) if calpain is not available, caspase-3/7 can also
remove the prodomain; (iii) Caspase-3/7 will also proceed
to release the p10 small subunit by C-terminal cleavage (a
critical step in the process), followed by (iv) the subsequent
caspase-3/7 processing to generate the large subunit con-
taining 23 kDa fragment, (v) which is further autolytically
processed to generate the fully active caspase-12 large
subunit(p20) (Fig. 10). It is possible that active caspase-12
can initiate a positive feedback loop that activates caspase-
3 through caspase-9 activation and thus potentiating
apoptosis induction. Both activation of caspase-12 by
caspase-3 and activation of caspase-9 by caspase-12 have
been previously established [12, 34, 39, 47]. Finally,
inhibition of caspase-3/7 activity caused a larger decrease
in caspase-12 activation than calpain inhibition and a more
robust rescue against thapsigarin-induced PC12 cell death
(Fig. 9). In conclusion, although both calpain and caspase
are involved in the processing of caspase-12 during ER
stress, our results indicate that caspase-3/7 activity is more
critical than calpain for caspase-12 enzyme activation and
the overall activation of ER stress-induced cell death
pathway.
Acknowledgements This work was supported by NIH Grant
R01NS051431 to R. L. H. Dr. K. K. W. Wang and Dr. R. L. Hayes
hold equity in Banyan Biomarkers, Inc. (Alachua, FL), a company
commercializing the brain injury biomarker technology.
References
1. Ellgaard L, Helenius A (2003) Quality control in the endoplasmic
reticulum. Nat Rev Mol Cell Biol 4:181–191
2. Freeze HH (2007) Congenital disorders of glycosylation: CDG-I,
CDG-II, and beyond. Curr Mol Med 7:389–396
3. Zhang K, Kaufman RJ (2006) The unfolded protein response: a
stress signaling pathway critical for health and disease. Neurol-
ogy 66:S102–S109
4. Zhao L, Ackerman SL (2006) Endoplasmic reticulum stress in
health and disease. Curr Opin Cell Biol 18:444–452
Pro-domain/CARD Large Subunit (p20) Small Subunit (p10)D94 T132 K158 D318 D341 4191 C298
~55 kDa
Caspase-3/7 cleaved D94G only (~46 kDa, pre-active)
Calpain cleaved T132A, K158T (~38 kDa, pre-active)
Caspase-3/7 cleaved D94G, D341F (~35 kDa, active)
Calpain T132A, K158T and Caspase-3/7 cleaved D341F (~23 kDa, active)
Accession# NP_033938 419 aa caspase 12 [Mus musculus].
1 MAARRTHERD PIYKIKGLAK DMLDGVFDDL VEKNVLNGDE LLKIGESASF ILNKAENLVE 61 NFLEKTDMAG KIFAGHIANS QEQLSLQFSN DEDD*GPQKICTPSSPSESKR KVEDDEMEVN 121 AGLAHESHLM LT*APHGLQSS EVQDTLKLCP RDQFCKIK*TE RAKEIYPVME KEGRTRLALI181 ICNKKFDYLF DRDNADTDIL NMQELLENLG YSVVLKENLT AQEMETELMQ FAGRPEHQSS 241 DSTFLVFMSH GILEGICGVK HRNKKPDVLH DDTIFKIFNN SNCRSLRNKP KILIMQACRG 301 RYNGTIWVST NKGIATADTD EERVLSCKWN NSITKAHVETD*FIAFKSSTP HNISWKVGKT 361 GSLFISKLID CFKKYCWCYH LEEIFRKVQH SFEVPGELTQ MPTIERVSMT RYFYLFPGN //
Pro-caspase-12
Sizes indicated correspond to molecular weight estimated from western blots
Active Cys Autolytic site
Fig. 10 Schematic diagram of the calpain, caspase and autoproteo-
lytic cleavages of murine caspase-12 and sequence map. Pro-caspase-
12 is a 419 aa protein with calculated molecular weight of 49 kDa but
appears higher in western blots (about 55 kDa). N-terminus pro-
domain contains caspase-recruitment domain (CARD). Major
reported cleavages by calpain are after T132 and K158 (underlined),
whereas caspase 3/7 cleavages are at after D94 and D341. Active
cysteine (C) residue at C298 and caspase-12 autolytic site after D318
are also indicated [using murine caspase-12 amino acid sequence (see
lower panel)]. Thus, both calpain and caspase 3/7 initial cleavages
release the pro-domain. This is most likely followed by the release of
P10 small subunit by caspase 3/7, that generates the 23 kDa active
fragment that subsequently undergoes autolytic cleavage to release
the p20 subunit. In Figs. 6 and 7, we studied the calpain-cleaved
38 kDa fragment, in Figs. 6 and 8, the caspase-3/7-cleaved 35 kDa
fragment and the pre-autolytic processing 23 kDa active fragment,
respectively, which are also schematically shown here. Fragment
sizes indicated correspond to molecular weight estimated from
western blots. See text for more details. Underlined in caspase-12
sequence are peptides used to generate cleavage-specific antibodies,
C12D341 was used in Fig. 8
Apoptosis
123

5. Lindholm D, Wootz H, Korhonen L (2006) ER stress and neu-
rodegenerative diseases. Cell Death Differ 13:385–392
6. Larner SF, Hayes RL, Wang KK (2006) Unfolded protein
response after neurotrauma. J Neurotrauma 23:807–829
7. Truettner JS, Hu B, Alonso OF, Bramlett HM, Kokame K, Die-
trich WD (2007) Subcellular stress response after traumatic brain
injury. J Neurotrauma 24:599–612
8. Mori K (2000) Tripartite management of unfolded proteins in the
endoplasmic reticulum. Cell 101:451–454
9. Momoi T (2004) Caspases involved in ER stress-mediated cell
death. J Chem Neuroanat 28:101–105
10. Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, Greer PA (2006)
Ubiquitous calpains promote caspase-12 and JNK activation
during endoplasmic reticulum stress-induced apoptosis. J Biol
Chem 281:16016–16024
11. Masud A, Mohapatra A, Lakhani SA, Ferrandino A, Hakem R,
Flavell RA (2007) Endoplasmic reticulum stress-induced death of
mouse embryonic fibroblasts requires the intrinsic pathway of
apoptosis. J Biol Chem 282:14132–14139
12. Rao RV, Hermel E, Castro-Obregon S et al (2001) Coupling
endoplasmic reticulum stress to the cell death program. Mecha-
nism of caspase activation. J Biol Chem 276:33869–33874
13. Nakagawa T, Yuan J (2000) Cross-talk between two cysteine
protease families. Activation of caspase-12 by calpain in apop-
tosis. J Cell Biol 150:887–894
14. Saleh M, Vaillancourt JP, Graham RK et al (2004) Differential
modulation of endotoxin responsiveness by human caspase-12
polymorphisms. Nature 429:75–79
15. Sanges D, Marigo V (2006) Cross-talk between two apoptotic
pathways activated by endoplasmic reticulum stress: differen-
tial contribution of caspase-12 and AIF. Apoptosis 11:
1629–1641
16. Shirasaki Y, Miyashita H, Yamaguchi M, Inoue J, Nakamura M
(2005) Exploration of orally available calpain inhibitors: peptidyl
alpha-ketoamides containing an amphiphile at P3 site. Bioorg
Med Chem 13:4473–4484
17. Cryns V, Yuan J (1998) Proteases to die for. Genes Dev
12:1551–1570
18. Nath R, Raser KJ, Stafford D et al (1996) Non-erythroid
alpha-spectrin breakdown by calpain and interleukin 1 beta-
converting-enzyme-like protease(s) in apoptotic cells: contrib-
utory roles of both protease families in neuronal apoptosis.
Biochem J 319(Pt 3):683–690
19. Newcomb JK, Kampfl A, Posmantur RM et al (1997) Immu-
nohistochemical study of calpain-mediated breakdown products
to alpha-spectrin following controlled cortical impact injury in
the rat. J Neurotrauma 14:369–383
20. Newcomb-Fernandez JK, Zhao X, Pike BR et al (2001) Con-
current assessment of calpain and caspase-3 activation after
oxygen-glucose deprivation in primary septo-hippocampal cul-
tures. J Cereb Blood Flow Metab 21:1281–1294
21. Nath R, Huggins M, Glantz SB et al (2000) Development and
characterization of antibodies specific to caspase-3-produced
alpha II-spectrin 120 kDa breakdown product: marker for
neuronal apoptosis. Neurochem Int 37:351–361
22. Zhang Z, Larner SF, Liu MC, Zheng W, Hayes RL, Wang KK
(2009) Multiple alphaII-spectrin breakdown products distinguish
calpain and caspase dominated necrotic and apoptotic cell death
pathways. Apoptosis 14(11):1289–1298
23. Harriman JF, Liu XL, Aleo MD, Machaca K, Schnellmann RG
(2002) Endoplasmic reticulum Ca(2?) signaling and calpains
mediate renal cell death. Cell Death Differ 9:734–741
24. Walsh JG, Cullen SP, Sheridan C, Luthi AU, Gerner C, Martin SJ
(2008) Executioner caspase-3 and caspase-7 are functionally
distinct proteases. Proc Natl Acad Sci USA 105:12815–12819
25. Machleidt T, Geller P, Schwandner R, Scherer G, Kronke M
(1998) Caspase 7-induced cleavage of kinectin in apoptotic cells.
FEBS Lett 436:51–54
26. Chan SL, Mattson MP (1999) Caspase and calpain substrates:
roles in synaptic plasticity and cell death. J Neurosci Res
58:167–190
27. Wang KK (2000) Calpain and caspase: can you tell the differ-
ence? Trends Neurosci 23:20–26
28. Harwood SM, Yaqoob MM, Allen DA (2005) Caspase and cal-
pain function in cell death: bridging the gap between apoptosis
and necrosis. Ann Clin Biochem 42:415–431
29. Wang KK, Posmantur R, Nath R et al (1998) Simultaneous
degradation of alphaII- and betaII-spectrin by caspase 3 (CPP32)
in apoptotic cells. J Biol Chem 273:22490–22497
30. Tran H, Pankov R, Tran SD, Hampton B, Burgess WH, Yamada
KM (2002) Integrin clustering induces kinectin accumulation.
J Cell Sci 115:2031–2040
31. Kim I, Xu W, Reed JC (2008) Cell death and endoplasmic
reticulum stress: disease relevance and therapeutic opportunities.
Nat Rev Drug Discov 7:1013–1030
32. Yoshida H (2007) ER stress and diseases. FEBS J 274:630–658
33. Truettner JS, Hu K, Liu CL, Dietrich WD, Hu B (2009) Sub-
cellular stress response and induction of molecular chaperones
and folding proteins after transient global ischemia in rats. Brain
Res 1249:9–18
34. Rao RV, Castro-Obregon S, Frankowski H et al (2002) Coupling
endoplasmic reticulum stress to the cell death program. An Apaf-
1-independent intrinsic pathway. J Biol Chem 277:21836–21842
35. Voccoli V, Mazzoni F, Garcia-Gil M, Colombaioni L (2007)
Serum-withdrawal-dependent apoptosis of hippocampal neuro-
blasts involves Ca?? release by endoplasmic reticulum and
caspase-12 activation. Brain Res 1147:1–11
36. Muruganandan S, Cribb AE (2006) Calpain-induced endoplasmic
reticulum stress and cell death following cytotoxic damage to
renal cells. Toxicol Sci 94:118–128
37. Hiroi T, Wei H, Hough C, Leeds P, Chuang DM (2005) Pro-
tracted lithium treatment protects against the ER stress elicited by
thapsigargin in rat PC12 cells: roles of intracellular calcium,
GRP78 and Bcl-2. Pharmacogenomics J 5:102–111
38. Shimoke K, Kishi S, Utsumi T et al (2005) NGF-induced phos-
phatidylinositol 3-kinase signaling pathway prevents thapsigar-
gin-triggered ER stress-mediated apoptosis in PC12 cells.
Neurosci Lett 389:124–128
39. Lee W, Kim DH, Boo JH, Kim YH, Park IS, Mook-Jung I (2005)
ER stress-induced caspase-12 activation is inhibited by PKC in
neuronal cells. Apoptosis 10:407–415
40. Korfali N, Ruchaud S, Loegering D et al (2004) Caspase-7 gene
disruption reveals an involvement of the enzyme during the early
stages of apoptosis. J Biol Chem 279:1030–1039
41. Jang M, Park BC, Lee AY et al (2007) Caspase-7 mediated
cleavage of proteasome subunits during apoptosis. Biochem
Biophys Res Commun 363:388–394
42. Stegh AH, Kesari S, Mahoney JE et al (2008) Bcl2L12-mediated
inhibition of effector caspase-3 and caspase-7 via distinct
mechanisms in glioblastoma. Proc Natl Acad Sci USA 105:
10703–10708
43. Van de Craen M, Vandenabeele P, Declercq W et al (1997)
Characterization of seven murine caspase family members. FEBS
Lett 403:61–69
44. Hoppe V, Hoppe J (2004) Mutations dislocate caspase-12 from
the endoplasmic reticulum to the cytosol. FEBS Lett 576:
277–283
45. Korge P, Weiss JN (1999) Thapsigargin directly induces the
mitochondrial permeability transition. Eur J Biochem 265:
273–280
Apoptosis
123

46. Baumgartner HK, Gerasimenko JV, Thorne C et al (2009) Cal-
cium elevation in mitochondria is the main Ca2? requirement for
mitochondrial permeability transition pore (mPTP) opening.
J Biol Chem 284:20796–20803
47. Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko
Y (2002) An endoplasmic reticulum stress-specific caspase cas-
cade in apoptosis. Cytochrome c-independent activation of cas-
pase-9 by caspase-12. J Biol Chem 277:34287–34294
48. Mao W, Iwai C, Keng PC, Vulapalli R, Liang CS (2006) Nor-
epinephrine-induced oxidative stress causes PC-12 cell apoptosis
by both endoplasmic reticulum stress and mitochondrial intrinsic
pathway: inhibition of phosphatidylinositol 3-kinase survival
pathway. Am J Physiol Cell Physiol 290:C1373–C1384
49. Fujita E, Kouroku Y, Jimbo A, Isoai A, Maruyama K, Momoi T
(2002) Caspase-12 processing and fragment translocation into
nuclei of tunicamycin-treated cells. Cell Death Differ
9:1108–1114
Apoptosis
123
Copyright © 2022 FDOKUMEN




















