
Molecular mechanisms of neuroprotection by two natural antioxidant polyphenols
Upload
independentCategory
view
4download
0
84
Ann. N.Y. Acad. Sci. 1053: 84–96 (2005). © 2005 New York Academy of Sciences.doi: 10.1196/annals.1344.008
Neuroprotection by NGF in the PC12 In Vitro OGD Model
Involvement of Mitogen-Activated Protein Kinases and Gene Expression
RINAT TABAKMAN,a HAO JIANG,b IRIS SHAHAR,c HADAR ARIEN-ZAKAY,a ROBERT A. LEVINE,b AND PHILIP LAZAROVICIa
aDepartment of Pharmacology and Experimental Therapeutics, School of Pharmacy, Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem 91120, IsraelbWilliam T. Gossett Neurology Laboratories, Henry Ford Health System, Detroit, Michigan 48202, USAcFunctional Genomics Unit, Sheba Medical Center, Tel Hashomer 52621, Israel
ABSTRACT: Neurodegenerative disorders and chronic disability due to strokein the brain or spinal cord afflict a large sector of the population. To investigatethe mechanism involved in ischemic stroke and to develop neuroprotectivedrugs/therapies, in vivo and in vitro, pharmacological models are needed. Toinvestigate the cellular and molecular neuroprotective mechanisms of nervegrowth factor (NGF), a member of the nervous system neurotrophin family ofgrowth factors, under ischemia, we used an oxygen-glucose-deprivation (OGD)device and pheochromocytoma PC12 cells exposed to a paradigm of ischemicinsult. Pretreatment of the cultures with 50 ng/mL of NGF, 18 h prior to OGDinsult, conferred 30% of neuroprotection. Time-course experiments showedmarked activation of the ERK, JNK, and p-38 MAPK isoforms during the OGDphase, but not during OGD reperfusion. Pretreatment of the cultures with50 ng/mL of NGF, 18 h prior to OGD insult, resulted in 50% attenuation ofOGD-induced activation of JNK 1, and 20% and 50% attenuation of OGD-induced activation of p-38 � and �, respectively. The effect of NGF on geneexpression in the PC12 ischemic model using Affymatrix Rat DNA–Microarraytechnology indicates that only 6% of the genes are differentially regulated(induced/suppressed) by OGD insult and/or NGF. These findings support thenotion that pretreatment with NGF confers neuroprotection from OGD insult,a phenomenon coincidentally related to differential inhibition of MAPK stresskinase isoforms and differential gene expression. This ischemic model may beuseful to investigate molecular mechanisms of OGD-induced neurotoxicity andNGF-induced neuroprotection, and to generate novel therapeutic concepts forstroke treatment.
KEYWORDS: NGF; PC12; ischemia; neuroprotection; MAPK; gene expression
Address for correspondence: Prof. Philip Lazarovici, Ph.D., Department of Pharmacology andExperimental Therapeutics, School of Pharmacy, Faculty of Medicine, The Hebrew University ofJerusalem, Jerusalem 91120, Israel. Voice: +972-2-6758729, +972-2-6758767; fax: +972-2-6757490.

85TABAKMAN et al.: NEUROPROTECTION BY NGF
STROKE AND ISCHEMIA
Stroke refers to the neurological condition that develops when a part of the wholebrain is deprived of oxygen and glucose. In 70–80% of the cases, the precipitatingcause is a blood clot that blocks the supply of oxygenated blood to a region of thebrain, a situation termed ischemic stroke.
The damage caused to the neurons during ischemia is due to a reduction in oxy-gen and glucose supply—that is, oxygen and glucose deprivation (OGD).1,2 Insultsto the brain that interrupt its blood supply, as in ischemia, or its oxygen supply, as inhypoxia (>1% O2) and anoxia (0% O2), combined with a massive reduction inglucose supply (hypoglycemia), lead to rapid neuronal death.3,4 The precise mecha-nism of neuronal cell death is not clear, but it is believed that apoptosis is one of themechanisms involved in ischemic cell death.5
In Vitro Ischemic Models
In vitro models of cerebral ischemia may yield information, not possible in vivo,at a cellular and molecular level on the pharmacological properties of new drugs.The majority of in vitro cellular models, for ischemic investigation, include excita-tory neurons. Very few attempts have been made to investigate the effect of ischemiaon dopaminergic neurons. This aspect is very important since dopaminergic neuronsdegenerate during intrauterine embryo asphyxia and in brain substantia nigra,inducing the pathological syndrome named status marmoratus.6
PC12 Cells: A Neuronal Dopaminergic In Vitro Modelfor Investigating Ischemic Insults
Pheochromocytoma PC12 cells are a common dopaminergic neuronal model7 forin vitro studies of cell death. PC12 cells are small, round, catecholamine-containingcells, with a doubling time of 48–96 h, that grow in regular serum-supplementedmedium (FIG. 1). PC12 cells were cloned from a solid pheochromocytoma tumorpassaged subcutaneously in New England Deaconess Hospital Strain white rats.8
Upon treatment with nerve growth factor (NGF), PC12 cells stop dividing andinitiate the differentiation process expressed by electrical excitability and anincreased expression of cholinergic receptors and other differentiation markers.7
Unlike the adrenal medulla chromaffin cells, in which norepinephrine is the majorcatecholamine, the PC12 cells contain primarily dopamine (FIG. 1C, orange color).The enzymes required for the synthesis of catecholamines are also abundant in PC12cells (FIG. 1D, green color).
PC12 cells serve as the prime dopaminergic cellular tool in molecular neuro-science for investigating the NGF mechanism of action,9 under different insults.PC12 cells have served as cellular models of serum starvation,10 NGF depriva-tion,11,12 excitotoxicity,13 cytotoxicity,14 Parkinson’s disease,15 Alzheimer’s dis-ease,16 and ischemia.17–19 These models are also useful systems for exploringneuroprotective drugs.20,21
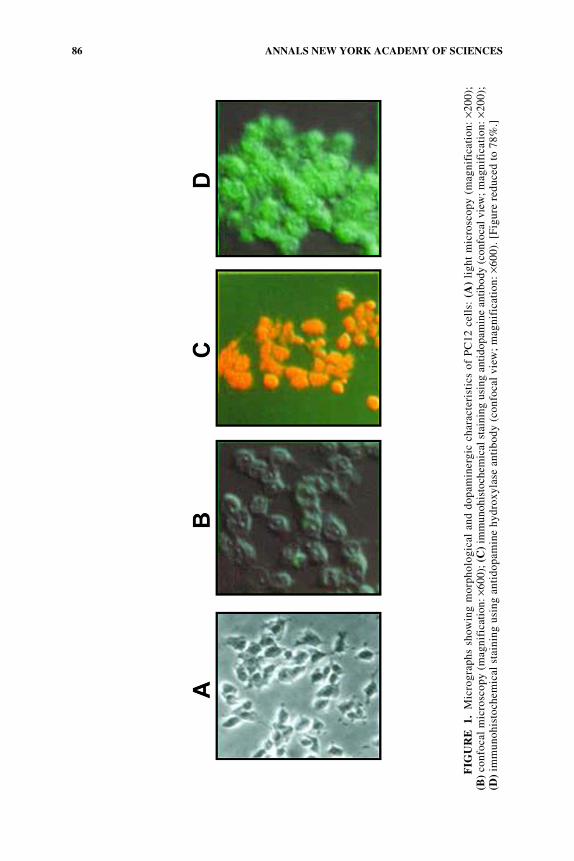
86 ANNALS NEW YORK ACADEMY OF SCIENCES
FIG
UR
E1.
Mic
rogr
aphs
sho
win
g m
orph
olog
ical
and
dop
amin
ergi
c ch
arac
teri
stic
s of
PC
12 c
ells
: (A
) li
ght
mic
rosc
opy
(mag
nifi
cati
on:
×200
);(B
) co
nfoc
al m
icro
scop
y (m
agni
fica
tion
: ×60
0); (
C)
imm
unoh
isto
chem
ical
sta
inin
g us
ing
anti
dopa
min
e an
tibo
dy (
conf
ocal
vie
w; m
agni
fica
tion
: ×2
00);
(D)
imm
unoh
isto
chem
ical
sta
inin
g us
ing
anti
dopa
min
e hy
drox
ylas
e an
tibo
dy (
conf
ocal
vie
w;
mag
nifi
cati
on:
×600
). [
Fig
ure
redu
ced
to 7
8%.]

87TABAKMAN et al.: NEUROPROTECTION BY NGF
PC12 Ischemic Model
Using an ischemic device prepared in our laboratory (FIG. 2) and PC12 cultures,we developed an in vitro model based on combined oxygen and glucose deprivation(OGD) insult, followed by reoxygenation, mimicking the pathological conditions ofischemia.19 In this model, the PC12 cells released large amounts of prostaglandinPGE2, lowered the ATP level, and underwent cell death, in part by apoptoticmechanism, measured by different cell death parameters.19,20,22
NERVE GROWTH FACTOR
NGF is the prototype of the neurotrophin family of growth factor molecules.23–25
NGF regulates the growth, development, and plasticity of selective neuronal popu-lations in the nervous system.26–30 It acts through binding and activating specific cellsurface receptors named trkA and p-75.31,32 TrkA is a receptor that stimulates several
FIGURE 2. Ischemic (oxygen-glucose-deprivation) device and protocol. (a) Ischemicsystem: (1) gas tank (N2/CO2 95%:5%); (2) cylinder containing water; (3) water bath withheating system and pump. (b) Ischemic chambers and oxygen detection: (4) oxygen monitorand oxygen sensor; (5) ischemic tissue culture chambers; arrow: flow of N2/CO2 throughTeflon tube (6) connecting in series the two chambers of the ischemic device (5). (c) Photo-graph of an open tissue culture ischemic chamber housing the petri dishes containing thePC12 cells (7) and tightly closed by metal screws (8) to seal the chambers. (d) Ischemia andnormoxia protocols.
88 ANNALS NEW YORK ACADEMY OF SCIENCES
signal transduction pathways, such as stimulation of phosphatidyl-inositol 3′-kinase,phospholipase C-γ, and mitogen-activated protein kinases (MAPKs).32
Neuronal NGF expression in vivo is markedly upregulated by seizures, forebrainischemia, marked hypoglycemia, and tissue injury.33–35 Studies in vivo and in vitroindicate that cerebral insults influence NGF gene expression via excitatory aminoacid receptors as well as through other pathways.34 The functional consequences ofthe increased production of neurotrophic factors following brain insults are not clear,but are supposed to represent intrinsic neuroprotective signals.35,36 Among its dif-ferent functions, NGF signaling appears to play important roles in response to injuryor disease that subserve neuroprotection and neural repair.37
NGF-Induced Neuroprotection in the PC12 Ischemic Modeland Implication to Neurodegeneration
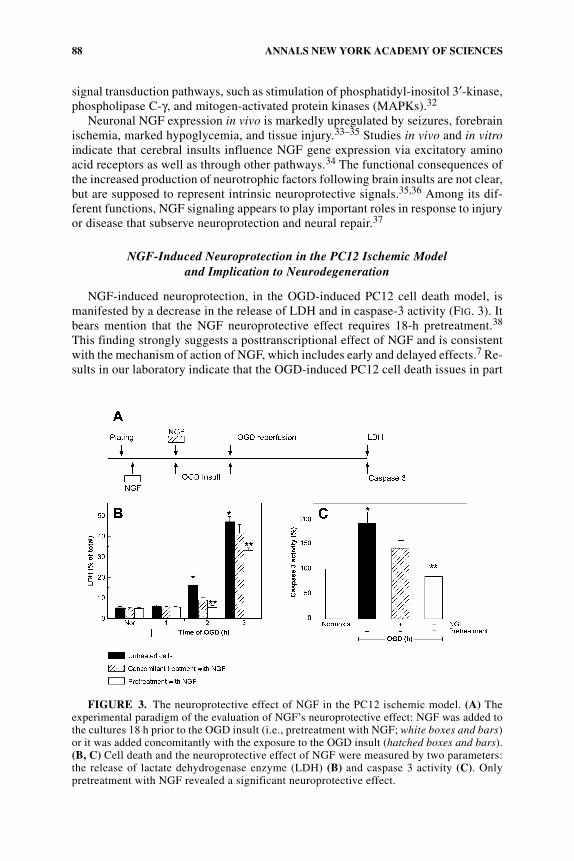
NGF-induced neuroprotection, in the OGD-induced PC12 cell death model, ismanifested by a decrease in the release of LDH and in caspase-3 activity (FIG. 3). Itbears mention that the NGF neuroprotective effect requires 18-h pretreatment.38
This finding strongly suggests a posttranscriptional effect of NGF and is consistentwith the mechanism of action of NGF, which includes early and delayed effects.7 Re-sults in our laboratory indicate that the OGD-induced PC12 cell death issues in part
FIGURE 3. The neuroprotective effect of NGF in the PC12 ischemic model. (A) Theexperimental paradigm of the evaluation of NGF’s neuroprotective effect: NGF was added tothe cultures 18 h prior to the OGD insult (i.e., pretreatment with NGF; white boxes and bars)or it was added concomitantly with the exposure to the OGD insult (hatched boxes and bars).(B, C) Cell death and the neuroprotective effect of NGF were measured by two parameters:the release of lactate dehydrogenase enzyme (LDH) (B) and caspase 3 activity (C). Onlypretreatment with NGF revealed a significant neuroprotective effect.

89TABAKMAN et al.: NEUROPROTECTION BY NGF
due to apoptosis.22 Therefore, it is tempting to suggest that the neuroprotective effectof NGF is related to a reduction in OGD-induced apoptosis. NGF-induced neuropro-tection, in the OGD model, supports the role of NGF as a neuroprotective agent inthe nervous system as previously documented in a variety of in vitro neuronalinsults.10,17,18,39–45 In recent neurological studies, a correlation was found betweendepletion and/or reduction in the central nervous system neurotrophin level and theoccurrence of neurodegeneration. It has been also shown that, in different neuro-degenerative disease models, treatment with neurotrophins like BDNF and NGF mayreduce the severity and death of the neurons. Thus, we hypothesize that controlledincrease in brain NGF level may be beneficial in ameliorating certain neurodegener-ative diseases such as Alzheimer’s disease. The fact that NGF’s neuroprotectiveeffect requires 18-h pretreatment emphasizes its essential endogenous, maintenance,neuroprotective role in the CNS. Also, we would like to emphasize that, during theischemic insult, part of the neurons are not dead, but damaged. In our opinion, anearly and long exposure of these neurons to NGF treatment may delay or rescue theirapoptotic cell death. To date, the major obstacle in therapeutic use of NGF for stroke,Alzheimer’s disease, and other neurodegenerative disorders is the inability of recom-binant human NGF to permeate through the blood-brain barrier. The development ofnovel delivery technologies for proteins such as NGF will provide the way for thisimportant neurotrophin to the clinic.
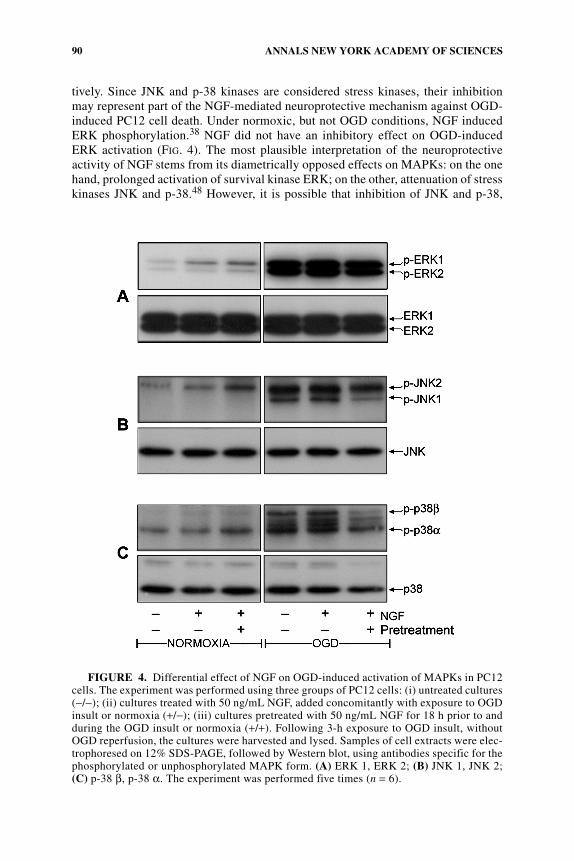
OGD-Induced Activation of MAPK Isoforms in the PC12 Ischemic Model
In view of the critical role of the MAPK pathways in the pathophysiologicalresponse of neurons to environmental stress, including ischemic injury,46,47 wemeasured MAPK isoform activity during the OGD paradigm. At the end of the 3-hinsult, there was a 12- and 25-fold increase in the activation of ERK 1 and ERK 2,respectively,38 dropping to the control level after 30 min of OGD reperfusion. Duringthe OGD insult, maximal 10-fold and 8-fold JNK 1 and JNK 2 activity, respectively,was observed 3 h from initiation of the insult, declining to a level similar to thatunder normoxia after an additional 2 h. Similar findings were observed for p-38enzymes (FIG. 4).
Our study shows that insult-induced activation of MAPKs occurs mainly duringthe OGD phase. A comparison between the kinetics of OGD-induced cell death38
and the kinetics of OGD-induced MAPK activation38 indicates that there is a similartime course, suggesting a coincidental relationship.
These findings support the emerging concept that MAPKs are activated duringischemia, stress, and inflammation,46 and may be crucial cross points in the neuro-toxicity/neuroprotection process.
NGF Attenuation of MAPK Activity during OGD-Induced PC12 Cell Death
The novel finding in our study is that NGF, aside from conferring neuroprotection,also differentially attenuates the phosphorylation of OGD-induced JNK 1, p-38 β,and p-38 α.38 The neuroprotective effect exerted by NGF after OGD was coinciden-tally related to 50%, 20%, and 50% inhibition of JNK 1, p-38 α, and p-38 β, respec-
90 ANNALS NEW YORK ACADEMY OF SCIENCES
tively. Since JNK and p-38 kinases are considered stress kinases, their inhibitionmay represent part of the NGF-mediated neuroprotective mechanism against OGD-induced PC12 cell death. Under normoxic, but not OGD conditions, NGF inducedERK phosphorylation.38 NGF did not have an inhibitory effect on OGD-inducedERK activation (FIG. 4). The most plausible interpretation of the neuroprotectiveactivity of NGF stems from its diametrically opposed effects on MAPKs: on the onehand, prolonged activation of survival kinase ERK; on the other, attenuation of stresskinases JNK and p-38.48 However, it is possible that inhibition of JNK and p-38,
FIGURE 4. Differential effect of NGF on OGD-induced activation of MAPKs in PC12cells. The experiment was performed using three groups of PC12 cells: (i) untreated cultures(−/−); (ii) cultures treated with 50 ng/mL NGF, added concomitantly with exposure to OGDinsult or normoxia (+/−); (iii) cultures pretreated with 50 ng/mL NGF for 18 h prior to andduring the OGD insult or normoxia (+/+). Following 3-h exposure to OGD insult, withoutOGD reperfusion, the cultures were harvested and lysed. Samples of cell extracts were elec-trophoresed on 12% SDS-PAGE, followed by Western blot, using antibodies specific for thephosphorylated or unphosphorylated MAPK form. (A) ERK 1, ERK 2; (B) JNK 1, JNK 2;(C) p-38 β, p-38 α. The experiment was performed five times (n = 6).
91TABAKMAN et al.: NEUROPROTECTION BY NGF
FIG
UR
E5.
Gen
e ex
pres
sion
com
pari
son
betw
een
OG
D a
nd n
orm
oxia
in
the
PC
12 i
sche
mic
mod
el.
Out
of
1476
gen
es t
hat
wer
e in
duce
d an
d80
8 ge
nes
that
wer
e su
ppre
ssed
, onl
y 36
5 or
483
gen
es h
ad a
2-f
old
indu
ctio
n or
sup
pres
sion
, res
pect
ivel
y. T
hese
gen
es w
ere
anal
yzed
, usi
ng D
avid
anal
ysis
(ht
tp:/
/dav
id.n
iaid
.nih
.gov
/dav
id/e
ase.
htm
), a
nd p
rese
nted
in
a pi
e ch
art
acco
rdin
g to
cel
lula
r fu
ncti
on.
92 ANNALS NEW YORK ACADEMY OF SCIENCES
upon NGF treatment, sustained the OGD-induced activation of ERK 1 and ERK 2by feedback regulation of the MAPK cascade.49
Pretreatment with NGF may be beneficial in neuronal ischemia because of itsability to attenuate stress kinase activity believed to cause OGD-induced cell death.
Gene Expression in the PC12 Ischemic Model
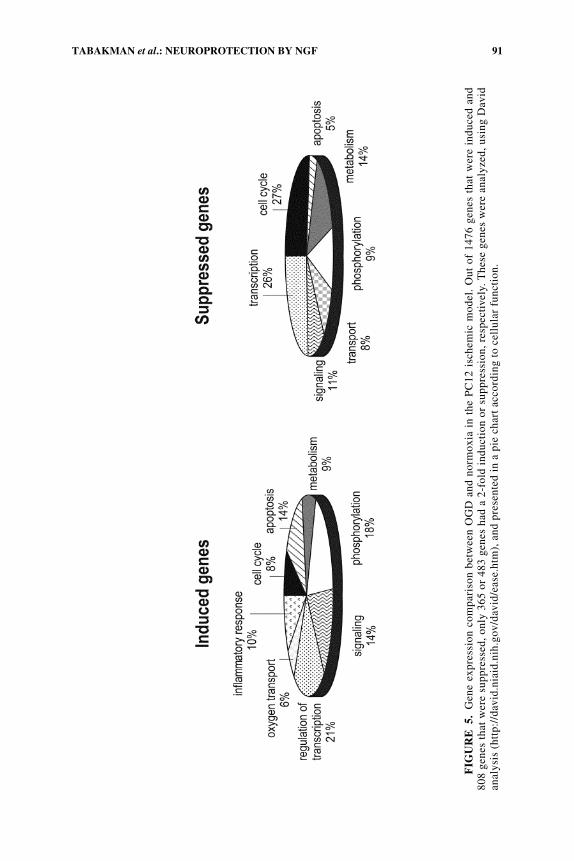
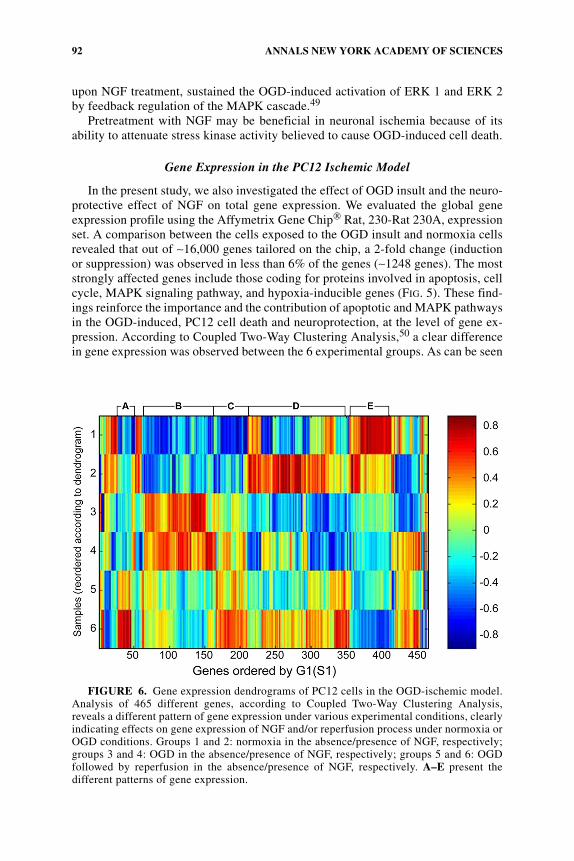
In the present study, we also investigated the effect of OGD insult and the neuro-protective effect of NGF on total gene expression. We evaluated the global geneexpression profile using the Affymetrix Gene Chip® Rat, 230-Rat 230A, expressionset. A comparison between the cells exposed to the OGD insult and normoxia cellsrevealed that out of ∼16,000 genes tailored on the chip, a 2-fold change (inductionor suppression) was observed in less than 6% of the genes (∼1248 genes). The moststrongly affected genes include those coding for proteins involved in apoptosis, cellcycle, MAPK signaling pathway, and hypoxia-inducible genes (FIG. 5). These find-ings reinforce the importance and the contribution of apoptotic and MAPK pathwaysin the OGD-induced, PC12 cell death and neuroprotection, at the level of gene ex-pression. According to Coupled Two-Way Clustering Analysis,50 a clear differencein gene expression was observed between the 6 experimental groups. As can be seen
FIGURE 6. Gene expression dendrograms of PC12 cells in the OGD-ischemic model.Analysis of 465 different genes, according to Coupled Two-Way Clustering Analysis,reveals a different pattern of gene expression under various experimental conditions, clearlyindicating effects on gene expression of NGF and/or reperfusion process under normoxia orOGD conditions. Groups 1 and 2: normoxia in the absence/presence of NGF, respectively;groups 3 and 4: OGD in the absence/presence of NGF, respectively; groups 5 and 6: OGDfollowed by reperfusion in the absence/presence of NGF, respectively. A–E present thedifferent patterns of gene expression.

93TABAKMAN et al.: NEUROPROTECTION BY NGF
in FIGURE 6, analysis of 465 different genes reveals a different pattern of gene ex-pression under various experimental conditions, clearly indicating the effects ongene expression of NGF and/or reperfusion process under normoxia or OGD condi-tions. Precise identification of the 465 genes is under way. These data will providenovel targets for neuroprotection in the ischemia-reperfusion process and resemblegene expression in the hypocampal neurons exposed to 3-h hypoxia51 and in the ratocclusion ischemic model.52
The Effect of NGF on Gene Expression in the PC12 Ischemic Model
We also examined the effect of NGF on gene expression under different experi-mental conditions: (i) under normoxic conditions, NGF affected 2284 genes, out ofwhich a 2-fold induction was observed in 365 genes and suppression of 483 genes,compared to nontreated cells; (ii) under OGD insult, NGF affected 2396 genes, outof which a 2-fold induction was observed in 345 genes and suppression of 258 genes,compared to nontreated cells; (iii) under OGD insult followed by reperfusion, NGFaffected 1884 genes, out of which a 2-fold induction was observed in 130 genes andsuppression of 128 genes, compared to nontreated cells (data not shown). These re-sults indicate a differential effect of NGF on the PC12 transcriptome under normoxiaand ischemic conditions. Evaluation of the specific genes and signal transductionpathways affected by ischemic insult may provide a new diagnostic tool for evaluationof the severity of the ischemic damage and may shed light on future therapeuticapproaches/targets.
Neuroprotection: A Balanced Process between Cell Death and Cell Survival
In neuronal models such as PC12 cells, the relationship between survival and celldeath is determined by the balance between the activated pro- and antiapoptoticsignaling proteins (the apoptotic thermostat),53 regulated in part by neurotrophinssuch as NGF.54
FIGURE 7. OGD-induced neurotoxicity and drug-induced neuroprotection: two antago-nistic processes in the PC12 ischemic model. Schematic presentation of the neuroprotectionconcept as a balanced neuropharmacological process between cell death and cell survivalpathways. White arrows indicate the neuroprotective processes; black arrows indicate OGD-induced neurotoxicity. Addition of neuroprotective compounds such as NGF antagonizes theneurotoxicity process and shifts the balance towards survival of the neurons. There is a dualeffect on gene expression under both neurotoxic and neuroprotective processes: part of thegenes are being induced, while others are being suppressed.

94 ANNALS NEW YORK ACADEMY OF SCIENCES
Our findings support the concept of a fine balance regulation (FIG. 7) between theneuron survival and death pathways: a differential inhibitory effect of NGF on theproapoptotic genes, while upregulating of the antiapoptotic genes (data not shown).
The most plausible interpretation of the neuroprotective activity of NGF stemsfrom its diametrically opposed effects on MAPKs: on the one hand, prolonged acti-vation of survival kinase ERK; on the other, attenuation of stress kinases JNK andp-38.48 In turn, these kinases, by phosphorylation of their transcription factorsubstrates, differentially regulate gene expression.49
Further investigation of the PC12 cell ischemic model, exploiting the OGDparadigm and specific inhibitors of signal transduction targets coupled with DNA-microarray analysis, may shed light on the differential contribution of the pro- andantiapoptotic proteins during ischemic insult and the neuroprotection conferred byNGF.
REFERENCES
1. SIESJO, B.K. 1992. Pathophysiology and treatment of focal cerebral ischemia. Part I:Pathophysiology. J. Neurosurg. 77: 169–184.
2. SIESJO, B.K. 1992. Pathophysiology and treatment of focal cerebral ischemia. Part II:Mechanisms of damage and treatment. J. Neurosurg. 77: 337–354.
3. LUO, X., G.N. LAMBROU, J.A. SAHEL & D. HICKS. 2001. Hypoglycemia induces generalneuronal death, whereas hypoxia and glutamate transport blockade lead to selectiveretinal ganglion cell death in vitro. Invest. Ophthalmol. Vis. Sci. 42: 2695–2705.
4. PEDERSEN, J.Z., G. BERNARDI, D. CENTONZE et al. 1998. Hypoglycemia, hypoxia, andischemia in a corticostriatal slice preparation: electrophysiologic changes and ascorbylradical formation. J. Cereb. Blood Flow Metab. 18: 868–875.
5. MATTSON, M.P., W. DUAN, W.A. PEDERSEN & C. CULMSEE. 2001. Neurodegenerativedisorders and ischemic brain diseases. Apoptosis 6: 69–81.
6. BERGER, R. & Y. GARNIER. 2000. Perinatal brain injury. J. Perinat. Med. 28: 261–285.7. FUJITA, K., P. LAZAROVICI & G. GUROFF. 1989. Regulation of the differentiation of
PC12 pheochromocytoma cells: regulation of differentiation in eukaryotic cells.Environ. Health Perspect. 80: 127–142.
8. GREENE, L.A. & A.S. TISCHLER. 1976. Establishment of a noradrenergic clonal line ofrat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc.Natl. Acad. Sci. USA 73: 2424–2428.
9. VAUDRY, D., P.J. STORK, P. LAZAROVICI & L.E. EIDEN. 2002. Signaling pathways forPC12 cell differentiation: making the right connections. Science 296: 1648–1649.
10. RUKENSTEIN, A., R.E. RYDEL & L.A. GREENE. 1991. Multiple agents rescue PC12 cellsfrom serum-free cell death by translation- and transcription-independent mechanisms.J. Neurosci. 11: 2552–2563.
11. BATISTATOU, A. & L.A. GREENE. 1991. Aurintricarboxylic acid rescues PC12 cells andsympathetic neurons from cell death caused by nerve growth factor deprivation:correlation with suppression of endonuclease activity. J. Cell Biol. 115: 461–471.
12. PARK, D.S., E.J. MORRIS, L. STEFANIS et al. 1998. Multiple pathways of neuronal deathinduced by DNA-damaging agents, NGF deprivation, and oxidative stress. J. Neurosci.18: 830–840.
13. MENEI, P., J.M. PEAN, V. NERRIERE-DAGUIN et al. 2000. Intracerebral implantation ofNGF-releasing biodegradable microspheres protects striatum against excitotoxicdamage. Exp. Neurol. 161: 259–272.
14. FISCHER, S.J., J.L. PODRATZ & A.J. WINDEBANK. 2001. Nerve growth factor rescue ofcisplatin neurotoxicity is mediated through the high affinity receptor: studies inPC12 cells and p75 null mouse dorsal root ganglia. Neurosci. Lett. 308: 1–4.
15. SHIMOKE, K. & H. CHIBA. 2001. Nerve growth factor prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced cell death via the Akt pathway by suppressing

95TABAKMAN et al.: NEUROPROTECTION BY NGF
caspase-3-like activity using PC12 cells: relevance to therapeutical application forParkinson’s disease. J. Neurosci. Res. 63: 402–409.
16. TROY, C.M., S.A. RABACCHI, Z. XU et al. 2001. Beta-amyloid-induced neuronal apoptosisrequires c-Jun N-terminal kinase activation. J. Neurochem. 77: 157–164.
17. PAN, Z., D. SAMPATH, G. JACKSON et al. 1997. Nerve growth factor and oxidative stressin the nervous system. In Brain Plasticity, pp. 173–193. Plenum. New York.
18. PAN, Z., D. SAMPATH, G. JACKSON et al. 1997. Nerve growth factor and oxidative stressin the nervous system. Adv. Exp. Med. Biol. 429: 173–193.
19. TABAKMAN, R., P. LAZAROVICI & R. KOHEN. 2002. Neuroprotective effects of carnosine andhomocarnosine on pheochromocytoma PC12 cells exposed to ischemia. J. Neurosci.Res. 68: 463–469.
20. ABU-RAYA, S., V. TREMBOVLER, E. SHOHAMI & P. LAZAROVICI. 1993. A tissue cultureischemic device to study eicosanoid release by pheochromocytoma PC12 cultures. J.Neurosci. Methods 50: 197–203.
21. ABU-RAYA, S., E. BLAUGRUND, V. TREMBOVLER et al. 1999. Rasagiline, a monoamineoxidase-B inhibitor, protects NGF-differentiated PC12 cells against oxygen-glucosedeprivation. J. Neurosci. Res. 5: 456–463.
22. TABAKMAN, R., H. JIANG, R.A. LEVINE et al. 2004. Apoptotic characteristics of celldeath and the neuroprotective effect of homocarnosine on pheochromocytoma PC12cells exposed to ischemia. J. Neurosci. Res. 75: 499–507.
23. BUTTE, M.J., P.K. HWANG, W.C. MOBLEY & R.J. FLETTERICK. 1998. Crystal structure ofneurotrophin-3 homodimer shows distinct regions are used to bind its receptors.Biochemistry 37: 16846–16852.
24. IBANEZ, C.F. 1994. Structure-function relationships in the neurotrophin family. J.Neurobiol. 25: 1349–1361.
25. ROBINSON, R.C., C. RADZIEJEWSKI, G. SPRAGGON et al. 1999. The structures of theneurotrophin 4 homodimer and the brain-derived neurotrophic factor/neurotrophin 4heterodimer reveal a common Trk-binding site. Protein Sci. 8: 2589–2597.
26. HUANG, E.J. & L.F. REICHARDT. 2001. Neurotrophins: roles in neuronal developmentand function. Annu. Rev. Neurosci. 24: 677–736.
27. REICHARDT, L.F. & I. FARINAS. 1997. Neurotrophic factors and their receptors: roles inneuronal development and function. In Molecular and Cellular Approaches toNeuronal Development, pp. 220–263. Oxford University Press. London/New York.
28. SCHINDER, A.F. & M. POO. 2000. The neurotrophin hypothesis for synaptic plasticity.Trends Neurosci. 23: 639–645.
29. THOENEN, H. 2000. Neurotrophins and activity-dependent plasticity. Prog. Brain Res.128: 183–191.
30. TABAKMAN, R., S. LECHT, S. SEPHANOVA et al. 2004. Interactions between the cells ofthe immune and nervous system: neurotrophins as neuroprotection mediators in CNSinjury. Prog. Brain Res. 146: 387–401.
31. BARBACID, M. 1994. The Trk family of neurotrophin receptors. J. Neurobiol. 25:1386–1403.
32. KAPLAN, D.R. & F.D. MILLER. 1997. Signal transduction by the neurotrophin receptors.Curr. Opin. Cell Biol. 9: 213–221.
33. GALL, C.M. & P.J. ISACKSON. 1989. Limbic seizures increase neuronal production ofmessenger RNA for nerve growth factor. Science 245: 758–761.
34. LINDVALL, O., Z. KOKAIA, J. BENGZON et al. 1994. Neurotrophins and brain insults.Trends Neurosci. 17: 490–496.
35. ZAFRA, F., E. CASTREN, H. THOENEN & D. LINDHOLM. 1991. Interplay betweenglutamate and gamma-aminobutyric acid transmitter systems in the physiologicalregulation of brain-derived neurotrophic factor and nerve growth factor synthesis inhippocampal neurons. Proc. Natl. Acad. Sci. USA 88: 10037–10041.
36. TOKUMINE, J., O. KAKINOHANA, D. CIZKOVA et al. 2003. Changes in spinal GDNF,BDNF, and NT-3 expression after transient spinal cord ischemia in the rat. J. Neurosci.Res. 74: 552–561.
37. SOFRONIEW, M.V., C.L. HOWE & W.C. MOBLEY. 2001. Nerve growth factor signaling,neuroprotection, and neural repair. Annu. Rev. Neurosci. 24: 1217–1281.

96 ANNALS NEW YORK ACADEMY OF SCIENCES
38. TABAKMAN, R., H. JIANG, E. SCHAEFER et al. 2004. Nerve growth factor pretreatmentattenuates oxygen and glucose deprivation–induced c-Jun amino-terminal kinase 1and stress-activated kinases p38alpha and p38beta activation and confers neuro-protection in the pheochromocytoma PC12 model. J. Mol. Neurosci. 22: 237–250.
39. BONIECE, I.R. & J.A. WAGNER. 1995. NGF protects PC12 cells against ischemia by amechanism that requires the N-kinase. J. Neurosci. Res. 40: 1–9.
40. HAVIV, R. & R. STEIN. 1999. Nerve growth factor inhibits apoptosis induced by tumornecrosis factor in PC12 cells. J. Neurosci. Res. 55: 269–277.
41. ENOKIDO, Y. & H. HATANAKA. 1990. High oxygen atmosphere for neuronal cell culturewith nerve growth factor. II. Survival and growth of clonal rat pheochromocytomaPC12h cells. Brain Res. 536: 23–29.
42. SATOH, T., T. YAMAGATA, Y. ISHIKAWA et al. 1999. Regulation of reactive oxygen speciesby nerve growth factor but not Bcl-2 as a novel mechanism of protection of PC12cells from superoxide anion–induced death. J. Biochem. (Tokyo) 125: 952–959.
43. DESHMUKH, M. & E.M. JOHNSON. 1997. Programmed cell death in neurons: focus on thepathway of nerve growth factor deprivation–induced death of sympathetic neurons.Mol. Pharmacol. 51: 897–906.
44. OHYASHIKI, T., E. SATOH, M. OKADA et al. 2002. Nerve growth factor protects againstaluminum-mediated cell death. Toxicology 176: 195–207.
45. ITANO, Y., Y. KITAMURA & Y. NOMURA. 1994. 1-Methyl-4-phenylpyridinium (MPP+)–induced cell death in PC12 cells: inhibitory effects of several drugs. Neurochem. Int.25: 419–424.
46. KYRIAKIS, J.M. & J. AVRUCH. 2001. Mammalian mitogen-activated protein kinase signaltransduction pathways activated by stress and inflammation. Physiol. Rev. 81: 807–869.
47. IRVING, E.A. & M. BAMFORD. 2002. Role of mitogen- and stress-activated kinases inischemic injury. J. Cereb. Blood Flow Metab. 22: 631–647.
48. XIA, Z., M. DICKENS, J. RAINGEAUD et al. 1995. Opposing effects of ERK and JNK-p38MAP kinases on apoptosis. Science 270: 1326–1331.
49. BRIGHTMAN, F.A. & D.A. FELL. 2000. Differential feedback regulation of the MAPKcascade underlies the quantitative differences in EGF and NGF signalling in PC12cells. FEBS Lett. 482: 169–174.
50. GETZ, G., E. LEVINE & E. DOMANY. 2000. Coupled Two-Way Clustering Analysis ofgene microarray data. Proc. Natl. Acad. Sci. USA 97: 12079–12084.
51. JIN, K., X.O. MAO, M.W. ESHOO et al. 2002. cDNA microarray analysis of changes in geneexpression induced by neuronal hypoxia in vitro. Neurochem. Res. 27: 1105–1112.
52. SORIANO, M.A., M. TESSIER, U. CERTA & R. GILL. 2000. Parallel gene expressionmonitoring using oligonucleotide probe arrays of multiple transcripts with an animalmodel of focal ischemia. J. Cereb. Blood Flow Metab. 20: 1045–1055.
53. CHAUM, E. 2003. Retinal neuroprotection by growth factors: a mechanistic perspective.J. Cell. Biochem. 88: 57–75.
54. MILLER, F.D. & D.R. KAPLAN. 2001. Neurotrophin signalling pathways regulating neuronalapoptosis. Cell. Mol. Life Sci. 58: 1045–1053.
Copyright © 2022 FDOKUMEN




















