
Erythropoietin Enhances Long-Term Neuroprotection and Neurogenesis in Neonatal Stroke
138
Transcript of Erythropoietin Enhances Long-Term Neuroprotection and Neurogenesis in Neonatal Stroke


Basel • Freiburg • Paris • London • New York • Bangalore • Bangkok • Singapore • Tokyo • Sydney
Bridging Bench to Bedside in Fetal and Neonatal Brain Injury
Guest Editors
Sidhartha Tan, Evanston, Ill.Steven W. Levison, Newark, N.J.
67 figures, 16 in color, and 8 tables, 2007
All papers have undergone the Journal’s usual peer review

S. KargerMedical and Scientifi c PublishersBasel • Freiburg • Paris • LondonNew York • Bangalore • BangkokSingapore • Tokyo • Sydney
DisclaimerTh e statements, options and data contained in this publication are solely those of the individual authors and contributors and not of the publisher and the editor(s). Th e appearance of advertisements in the journal is not a warranty, endorsement, or approval of the products or services advertised or of their eff ectiveness, quality or safety. Th e publisher and the editor(s) disclaim responsibility for any injury to persons or property resulting from any ideas, methods, instructions or products referred to in the content or advertisements.
Drug DosageTh e authors and the publisher have exerted every eff ort to en-sure that drug selection and dosage set forth in this text are in accord with current recommendations and practice at the time of publication. However, in view of ongoing research, changes in government regulations, and the constant fl ow of informa-tion relating to drug therapy and drug reactions, the reader is urged to check the package insert for each drug for any change in indications and dosage and for added warnings and precau-tions. Th is is particularly important when the recommended agent is a new and/or infrequently employed drug.
All rights reserved.No part of this publication may be translated into other languages, reproduced or utilized in any form or by any means, electronic or mechanical, including photocopying, recording, microcopying, or by any information storage and retrieval system, without permission in writing from the publisher or, in the case of photocopying, direct payment of a specifi ed fee to the Copyright Clearance Center (see ‘General Information’).
© Copyright 2007 by S. Karger AG,P.O. Box, CH–4009 Basel (Switzerland)Printed in Switzerland on acid-free paper byReinhardt Druck, BaselISBN 978–3–8055–8319–2
Fax +41 61 306 12 34E-Mail [email protected]

Vol. 29, No. 4–5, 2007
Contents
Fax +41 61 306 12 34E-Mail [email protected]
© 2007 S. Karger AG, Basel
Access to full text and tables of contents, including tentative ones for forthcoming issues: www.karger.com/dne_issues
279 Preface
Levison, S.W. (Newark, N.J.) ; Tan, S. (Evanston, Ill.)
280 Perinatal Brain Damage Causation
Dammann, O. (Hannover/Boston, Mass.); Leviton, A. (Boston, Mass.)
289 Serial Diffusion Tensor Imaging Detects White Matter Changes That Correlate with Motor Outcome in Premature Infants
Drobyshevsky, A.; Bregman, J.; Storey, P.; Meyer, J.; Prasad, P.V.; Derrick, M.; MacKendrick, W.; Tan, S. (Evanston, Ill.)
302 Delayed IGF-1 Administration Rescues Oligodendrocyte Progenitors from Glutamate-Induced Cell Death and Hypoxic-Ischemic Brain Damage
Wood, T.L. (Newark, N.J./Hershey, Pa.); Loladze, V. (Hershey, Pa.); Altieri, S.; Gangoli, N.; Levison, S.W. (Newark, N.J.); Brywe, K.G.; Mallard, C.; Hagberg, H. (Gothenburg)
311 Prenatal Cord Clamping in Newborn Macaca nemestrina: A Model of Perinatal Asphyxia
Juul, S.E.; Aylward, E.; Richards, T.; McPherson, R.J.; Kuratani, J.; Burbacher, T.M. (Seattle, Wash.)
321 Erythropoietin Enhances Long-Term Neuroprotection and Neurogenesis in Neonatal Stroke
Gonzalez, F.F.; McQuillen, P. (San Francisco, Calif.); Mu, D. (San Francisco, Calif./Chengdu); Chang, Y. (San Francisco, Calif./Seoul); Wendland, M.; Vexler, Z.; Ferriero, D.M. (San Francisco, Calif.)
331 Perinatal Hypoxic/Ischemic Brain Injury Induces Persistent Production of Striatal Neurons from Subventricular Zone Progenitors
Yang, Z. (Shanghai); Levison, S.W. (Newark, N.J.)
341 Uteroplacental Inflammation Results in Blood Brain Barrier Breakdown, Increased Activated Caspase 3 and Lipid Peroxidation in the Late Gestation Ovine Fetal Cerebellum
Hutton, L.C.; Castillo-Melendez, M.; Walker, D.W. (Melbourne)
355 Identification of POSH2, a Novel Homologue of the c-Jun N-Terminal Kinase Scaffold Protein POSH
Wilhelm, M.; Kukekov, N.V.; Xu, Z.; Greene, L.A. (New York, N.Y.)
363 Pomegranate Polyphenols and Resveratrol Protect the Neonatal Brain against Hypoxic-Ischemic Injury
West, T.; Atzeva, M.; Holtzman, D.M. (St. Louis, Mo.)
373 Mast Cell Stabilization Limits Hypoxic-Ischemic Brain Damage in the Immature Rat
Jin, Y.; Silverman, A.-J.; Vannucci, S.J. (New York, N.Y.)
385 Gender-Dependent Pathways of Hypoxia-Ischemia-Induced Cell Death and Neuroprotection in the Immature P3 Rat
Nijboer, C.H.A.; Kavelaars, A.; van Bel, F.; Heijnen, C.J.; Groenendaal, F. (Utrecht)
393 Delayed Peripheral Administration of a GPE Analogue Induces Astrogliosis and Angiogenesis and Reduces Inflammation and Brain Injury following Hypoxia-Ischemia in the Neonatal Rat
Svedin, P. (Göteborg); Guan, J.; Mathai, S.; Zhang, R. (Auckland); Wang, X. (Göteborg); Gustavsson, M. (Auckland); Hagberg, H.; Mallard, C. (Göteborg)
403 Antioxidant Status Alters Levels of Fas-Associated Death Domain-Like IL-1B-Converting Enzyme Inhibitory Protein following Neonatal Hypoxia-Ischemia
Payton, K.S.E. (Baltimore, Md.); Sheldon, R.A. (San Francisco, Calif.); Mack, D.W. (Baltimore, Md.); Zhu, C.; Blomgren, K. (Göteborg); Ferriero, D.M. (San Francisco, Calif.); Northington, F.J. (Baltimore, Md.)
412 Author Index
412 Subject Index

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:279 DOI: 10.1159/000105468
Preface
Steven W. Levisona Sidhartha Tanb
a Department of Neurology and Neurosciences, UMDNJ – New Jersey Medical School, Newark, N.J. , and b Department of Pediatrics, Northwestern University, Evanston, Ill., USA
At the end of the conference, there was uniform agree-ment that this had been yet another extremely exciting and enlightening meeting, and it was unanimously de-cided to schedule a Sixth Conference on Developmental Brain Injury for the spring of June 2008 to be held outside of Paris, France. Drs. Susan Vannucci, Donna Ferriero, Pierre Gressens, Henrik Hagberg, and Steven Levison of-fered to organize the next meeting.
A general call for articles for this special issue was an-nounced at the 2006 conference, which resulted in many submissions. After rigorous peer review, 11 articles were selected for publication in this special issue. Several pa-pers in this issue report preclinical tests of new therapies that prevent injury to the developing brain including erythropoietin, insulin-like growth factor-1 peptides, and antioxidants. New insights into potential use of neu-ral stem cells to promote repair are also reported. Infor-mation obtained from MRI on human infants, combined with data from several animal models, highlights the ef-fects of interaction of cerebral hypoxia-ischemia, excito-toxicity, oxidative stress, seizures, and inflammation on the outcome of developmental brain injury and ultimate neurological outcome. This special issue will be of inter-est to both clinicians and basic scientists, who are inter-ested in the developing nervous system, its vulnerability to various pathophysiological processes, and the eventu-al consequences for neurological development. We hope the reader will find these articles both as stimulating and exciting as we have as editors.
The immature brain is vulnerable to prenatal and postnatal stresses, which may produce brain damage leading to neurological dysfunction in survivors. This special issue of Developmental Neuroscience presents new insights into the detection, pathophysiology and treat-ment of developmental brain injuries.
This volume represents the proceedings of the Fifth Hershey Conference on Developmental Brain Injury, which was held at the Doral Forrestal Conference Center and Spa, Princeton, N.J., May 31st to June 3rd 2006. Drs. Susan Vannucci, Donna Ferriero, Henrik Hagberg and Steven Levison organized the 2006 conference following four previously successful conferences. As in previous years, this conference was an international meeting (with approximately 104 participants) that was highly inter-active. Twenty-five participants from 9 countries, includ-ing the USA, presented plenary talks, with 51 additional poster presentations. To date, this was the best attended of the Hershey Conferences. The conference brought to-gether clinicians, basic scientists, fellows and graduate students in a relaxed setting to share their discoveries and thoughts with the goal of understanding the mechanisms that lead to perinatal brain injury, mechanisms of plastic-ity and repair, and progress in identifying new treatments and interventions. The conference was funded largely by the National Institute of Neurological Disorders and Stroke (1R13 NS 43136) with an additional contribution from the National Institute of Child Health and Human Development, Office of Rare Diseases, Morgan Stanley Children’s Hospital of New York and Olympic Corpora-tion.
Steven W. Levison, PhD Laboratory for Regenerative Neurobiology, Department of Neurology and Neuroscience and NJMS-UH Cancer Center, UMDNJ-New Jersey Medical School 205 South Orange Avenue, H-1226, Newark, NJ 07103 (USA) Tel. +1 973 972 5162, Fax +1 973 972 2668, E-Mail [email protected]
© 2007 S. Karger AG, Basel0378–5866/07/0295–0279$23.50/0
Accessible online at:www.karger.com/dne

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:280–288 DOI: 10.1159/000105469
Perinatal Brain Damage Causation
Olaf Dammann
a, b, c Alan Leviton
b
a Perinatal Infectious Disease Epidemiology Unit, OE 6415, Hannover Medical School, Hannover , Germany;
b Neuroepidemiology Unit, Children’s Hospital, and c
Division of Newborn Medicine, Floating Hospital forChildren at Tufts-New England Medical Center, Boston, Mass. , USA
measures, detailed knowledge about pathogenesis and etiology is paramount. However, before candidate causes can be identified, the causative framework, i.e. the con-cept of PBD causation in general, deserves consider-ation.
In this article, we do not offer an exhaustive overview of the various clinical and imaging characteristics of PBD, which is provided in several recent reviews [1–3] . Instead, we expand upon recent reviews of PBD etiology and pathogenesis [4–6] with an eye on causal inference, writing from the perinatal neuroepidemiologists’ per-spective.
Causal Inference and a Neopragmatic View of
Etiology Research
Modern etiology research in humans needs to be based on sound empirical methods [7] . This type of research relies on the observation of associations between expo-sures and outcomes, usually in humans, with the goal of identifying preventable causes of disease. On the other hand, we are all too familiar with the notion that ‘asso-ciation can never prove causation’. This notion has two components in the present context – the general problem of proof by observation, and the specific problem of how to define causes in empirical etiology research.
The general problem has been formulated by David Hume (1711–1776), who claimed that multiple observa-tions of similar co-occurrences do not justify any (induc-tive) conclusions beyond just these observations. This concept was extended by Sir Karl Popper (1902–1994)
Key Words
Brain damage � Fetus � Immature brain � Neonatal brain injury � Perinatal brain � Causation � Causal inference
Abstract
The search for causes of perinatal brain damage needs a sol-id theoretical foundation. Current theory apparently does not offer a unanimously accepted view of what constitutes a cause, and how it can be identified. We discuss nine poten-tial theoretical misconceptions: (1) too narrow a view of what is a cause (causal production vs. facilitation), (2) extrapolat-ing from possibility to fact (potential vs. factual causation), (3) if X, then invariably Y (determinism vs. probabilism), (4) co-occurrence in individuals vs. association in populations, (5) one cause is all that is needed (single cause attribution vs. multicausal constellations), (6) drawing causal inferences from very small numbers of observations (the tendency to generalize), (7) unstated causal inferences, (8) ignoring het-erogeneity, and (9) failing to consider alternative explana-tions for what is observed. We hope that our critical discus-sion will contribute to fruitful research and help reduce the burden of perinatal brain damage.
Copyright © 2007 S. Karger AG, Basel
Introduction
The heterogeneous spectrum of perinatal brain dam-age (PBD) in term and preterm infants warrants a de-tailed consideration of the equally heterogeneous spec-trum of causes of PBD. For identification of preventive
Received: April 6, 2006 Accepted after revision: September 26, 2006
Olaf Dammann, Dr. med., S.M. Director of Clinical Research, Division of Newborn MedicineDepartment of Pediatrics, Tufts-New England Medical Center750 Washington St., Boston, MA 02111 (USA)Tel. +1 617 636 0240, Fax +1 617 636 8943, E-Mail [email protected]
© 2007 S. Karger AG, Basel
Accessible online at:www.karger.com/dne

Perinatal Brain Damage Causation Dev Neurosci 2007;29:280–288 281
who offered his ‘falsificationist’ approach, which states that no number of similar observations can prove a hy-pothesis, while just one dissimilar observation can refute the conjecture – a theory perceived and discussed in both, statistics [8] and epidemiology [9–11] . An overview of the more recent philosophical discourse on causality [12, 13] is far beyond the scope of this paper.
The specific problem, i.e. how to conceptualize (and compute) ‘causation’, is far from simple and has produced a vast theoretical literature in the fields of epidemiology [14–18] and computer science [19, 20] . According to what is probably the most widely-known view in the epidemi-ologic literature [18, 21] , we should move from looking at individual component causes of disease, i.e. pieces of the pie, towards the appreciation of causal constellations, i.e. the entire pie. Only when the pie is complete will disease follow. This view might be considered inherently deter-ministic, because it holds that whenever the causal con-stellation is complete, disease follows inevitably . Howev-er, even in light of considerable progress in molecular epidemiology [22] , and even if we supplement epidemiol-ogy with appropriate experimental work, we are unlikely to discover the entire causal constellation in humans just by observation. ‘Even the most careful and detailed mech-anistic dissection of individual events cannot provide more than associations, albeit at a finer level’ [18] .
Thus, in practice, all etiology research can provide seems to be restricted to an approximation of ‘real’ causes via the identification of risk factors. In theory, research-ers may wish to fix the deterministic pie problem above by ‘simply think(ing) of the components as contributing together to the probability of the effect, rather than being sufficient for it’ [17] .
Could it thus be that finding ‘real’ causes of disease will remain elusive, because the question is ill posed? In-deed, some current thinking suggests we should stop ‘de-scribing (human inquiry) as an attempt to correspond to the intrinsic nature of reality (and start) describing it as an attempt to serve transitory purposes and solve transi-tory problems’ [23] . The major proponent of what is sometimes called ‘neopragmatism’ is Richard Rorty, self-declared ‘anti-dualist’, who suggests that Platonic-Carte-sian notions of ‘the absolute and the relative, the found and the made, object and subject, nature and convention, reality and appearance’ should be replaced with ‘new ways of speaking’. Rorty suggests we should ‘develop tools which will enable (humans) to enjoy more pleasure and less pain’. Along these very lines, could not we just stop taking ‘causal relationships to be the fundamental building blocks both of physical reality and of human
understanding of that reality’ [ 19 , p. xiii/xiv] and start ac-cepting observed associations, supplemented with some experimental evidence, as a sufficient starting point for change? Even if this was just changing language instead of solving a pressing philosophical problem, would not the ends (improved public health) justify the means (ig-noring Hume, plus dropping the dualistic worldview)?
In essence, we suggest that it does not matter whether we call an observed link between exposure and outcome ‘causation’ or ‘association’. What does matter is that iden-tifying risk factors for disease, and then proactively re-moving/reducing the identified risk factors from popula-tions can improve the human condition.
Clinical investigators are reluctant to expose human subjects to interventions that do not have a sound basic science rationale. To aid in the process of selecting risk factors for avoidance/modification, in the final section of this paper we offer criteria for what is worthy of evalua-tion. These should not be viewed as criteria for causes of disease.
Causal Concepts in PBD
Consider two scenarios. In the first, short-term intra-partum and neonatal insults are considered sufficient to damage the neonatal brain [24–26] . In the second, long-standing antenatal exposures are recognized as poten-tially changing the milieu in the fetal central nervous sys-tem, thereby playing a causative role in PBD [27–32] . In both scenarios, however, it might be helpful to integrate endogenous (e.g. maturation-related anatomic and/or physiologic/genetic) factors with exogenous (e.g. insult/milieu-related) phenomena, such as energy failure and/or inflammation [33] .
Moreover, the two scenarios differ with regard to what qualifies as an insult or harmful milieu. While matura-tional factors (for which gestational age is a surrogate [34] ) clearly contribute to PBD occurrence, circulatory and inflammatory risk factors are currently among the most frequently studied. Unfortunately, this recent devel-opment is frequently misinterpreted as an attempt to re-place one ‘old’ cause with a ‘new’ one. This perception, however, neglects the potential advantages of multiple cause appreciation, most importantly: an increase in the number of potential prevention opportunities.
A misperception of what constitutes a causal rela-tionship appears to contribute to this misunderstand-ing. Although the issues discussed in this paper apply to a large extent to disease causation in general, our area of

Dammann /Leviton
Dev Neurosci 2007;29:280–288282
concern is PBD causation. In particular, we are inter-ested in white matter damage (WMD), the term we use for focal and diffuse structural damage to the paraven-tricular white matter identified mainly in preterm new-borns [35] .
Nine Misconceptions in PBD Causation Thinking
We wrote this essay to raise awareness about issues that epidemiologists take for granted but others might not appreciate. We very much want to avoid embarrass-ing anyone who might have expressed any of these mis-conceptions. Thus, we have minimized identifying pa-pers that exemplify the very conceptual lapses we want the reader to recognize and avoid.
In the following sections, we expand on each of these nine issues. Because replacing some of what we consider misconceptions with new perspectives can be difficult, we created the table ( table 1 ) to aid in this process. We end this paper by offering some conclusions based on our dis-cussion.
Causal Production vs. Causal Facilitation We consider too simplistic the view that some factor is
a cause only if it produces disease. A more appropriate view might be that a factor is considered a cause when it contributes to disease occurrence, either by producing it or by facilitating its production by other causes.
Some exposures by themselves do not influence the risk of a disease. In the presence of other exposures, how-ever, they enhance the occurrence of disease. More com-monly, the exposure has a small influence in isolation, but in the presence of another exposure, has a greater than additive effect. This concept of ‘sensitization’ [36] is exemplified by the observation that the extent of cerebral infarction in 7-day old rats is greater following the com-bination of a low dose of endotoxin and a short period of hypoxia-ischemia than by either exposure alone [37] . Similarly, exposure to proinflammatory cytokines in-creases ibotenate-iduced excitotoxic cortical and WMD in a murine preterm model [38] .
On the epidemiologic level, this phenomenon is called ‘effect modification’, and under particular circumstances we may face effect modifiers that are themselves not pro-
Table 1. Nine conceptual problems in causation thinking, common assumptions associated with these, and proposals how to avoid causal misattribution
No. Conceptual problem Assumption Proposal
1 Causal production vs. causalfacilitation
X is cause of Y only if X produces Y X is cause of Y if X contributes to the occur-rence of Y, even if other exposures are required
2 Potential vs. factual causation(extrapolating from possibility to fact)
If X can cause Y (e.g. in experiments), then X does cause Y in real life
If X can cause Y, then X might cause Y
3 Determinism vs. probabilism If X then always Y If X then Y more frequently than if not X
4 Co-occurrence vs. association If X and Y co-occur, then X mustbe a cause of Y
If X and Y occur together more frequently than expected by chance, X might be a cause of Y
5 Single cause attribution vs. multicausal constellations
Y is caused only by X Y has many causes, including X, even in any individual
6 Generalization If X is a cause of Y in one instant,X is always a cause of Y
If X is a cause of Y in one instant, X can be a cause of Y
7 Unstated causal inferences If X is severe, it causes Y;if less severe, it causes Z
Find support before accepting
8 Ignoring heterogeneity If X1 is similar to X2, they can be combined as one
If X1 is similar to X2, evaluate similarity before combining
9 Failure to consider alternatives What I see is true Consider that what is seen can be explained away and is not what it appears to be
In this table, X is an exposure or characteristic and Y and Z are disorders of interest.

Perinatal Brain Damage Causation Dev Neurosci 2007;29:280–288 283
ducers of disease. For example, in one of our recent stud-ies we looked at the risk of WMD after exposure to hypo-carbia [39] . In these analyses, maternal antibiotic therapy was associated with neither hypocarbia nor WMD. How-ever, among children whose mothers had received antibi-otic treatment, the adjusted odds ratio for hypocarbia predicting WMD was 0.9 (95% confidence interval 0.3–2.9), while it was 2.7 (1.1–6.6) among infants whose moth-ers had not received antibiotic therapy. Clearly, although not associated with WMD in the first place, maternal an-tibiotic therapy was associated with WMD occurrence by virtue of conveying information about modifying the ef-fect of hypocarbia (or its antecedents or correlates) on WMD risk.
One (neopragmatic) way to avoid the failure to recog-nize causal facilitation is to enhance our etiologic lexicon by using terms such as ‘contributor’ or ‘risk factor’ a bit more frequently. Then we can talk about phenomena that contribute to disease, and not have confusion about what constitutes a cause.
Potential vs. Factual Cause We sometimes fall into the trap of what might be called
‘extrapolation from possibility to fact’. By this we mean that some tend to think that if X can cause Y in the ex-perimental setting, X does cause Y in real-life situations. The experimental evidence from perinatal neuroscience that some exposures can result in WMD is large and con-tinues to grow [4] . Such experimental evidence, however, shows only that these factors are candidate causes of WMD. We should not assume they are causes in hu-mans.
Along these lines, the current assumption that WMD in immature human newborns is caused by hypoxia-isch-emia is an extrapolation error, but not from possibility to fact, but from missing data. In essence, the inferential er-ror has no basis in fact. In the article that coined the term ‘periventricular leukomalacia’, the authors considered the entity ‘a neonatal form of anoxic encephalopathy’ [40] . However, they did not offer any supporting evidence why anoxia should be the cause for the damage observed. Despite this lack of data, an article published in 2002 cit-ed this paper this way, ‘[Banker and Larroche] found ad-ditional pathological changes within the lungs of all in-fants examined and noted that the majority had experi-enced a period of apnea or cardiac arrest requiring resuscitation. Anoxia was recorded as present without ex-ception in every infant in their series’ [25] . Although anoxia may have been recorded, it was not measured at all. The authors assumed anoxia was present because the
infants had had trouble breathing or had experienced cardiac arrest. It might be of interest to clinicians respon-sible for keeping oxygen saturation values of preterm in-fants in the ‘acceptable’ range that multiple experimental studies support the claim that hypoxia without ischemia does not cause brain damage [41–43] and that ventilated preterm newborns are ‘able to maintain adequate cere-bral perfusion at a MABP in the range of 23.7–39.3 mm Hg’ [44] .
Until today, the anoxia/asphyxia/hypoxia-ischemia complex of putative causes has its accustomed place in the introductions of scientific articles and textbooks on pre-term brain damage (see below, section ‘Single Cause At-tribution vs. Multicausal Constellations’), as well as in the courtroom. This is mainly because in response to such initial ‘observations’ of anoxia, a very successful rat mod-el of hypoxia-ischemia has been developed [43, 45] , mod-ifications of which are probably among the most com-monly used models of PBD. One should not cease asking the question how relevant hypoxia-ischemia is among term, and even more among preterm infants, for causa-tion of PBD and other neonatal disorders such as necro-tizing enterocolitis [46] .
In sum, the evidence in the literature that hypoxia-ischemia can indeed cause brain lesions is vast [26] . How-ever, the inference that brain damage in human new-borns must therefore be hypoxic-ischemic in origin is not well supported by solid observational evidence [47] .
Determinism vs. Probabilism Most people think of causation as an ‘if, then’ sequence
of events. The generalized example is ‘if X, then Y’. Now, the backbone of determinist thinking is the assumption that ‘if X, then always Y’. For some phenomena, such as those well-described by natural laws, the determinist view might indeed apply. However, the applicability of natural laws to disease processes might be less common than frequently assumed.
The generalized form for the probabilistic approach to causation [48, 49] is ‘if X, then the probability of Y is in-creased’. Our inference is that when we see X, we think that the occurrence of Y is more likely than if we do not see X.
Infection with HIV is considered the cause of AIDS [50] . However, not all HIV-infections lead to the rapid development of the full-blown clinical syndrome called AIDS, as illustrated by the existence of highly exposed, persistently seronegative individuals and HIV-1-infected long-term nonprogressors [51] . Simply put, it is not yet known whether some of these individuals will ever de-

Dammann /Leviton
Dev Neurosci 2007;29:280–288284
velop signs of AIDS. To elucidate the protective charac-teristics among these few cases will greatly improve our understanding of AIDS pathogenesis and causation.
Along these same lines, some investigators might put too much deterministic emphasis on what they expect to follow a certain exposure. In one recent study, for example, investigators noted that almost half of their cases of WMD ‘occurred unexpectedly in infants who did not appear to have an obvious cause, as they had not experienced a severe hemodynamic event’ [52] . Apparently, some favor certain-ty over likelihood. We are confident that based on our dis-cussion, at least some determinists will probably consider becoming determined probabilists (pun intended).
Co-Occurrence in Individuals vs. Association in Populations Some tend to rely on anecdotal evidence even in an era
when information about etiology comes from data de-rived from research in large groups of subjects. We, on the other hand, suggest that the evidence-based perspec-tive [53] should not only be applied to therapy studies, but also to etiology research [54] .
The emphasis on the individual is typically taken by clinicians interested in causation issues in individual pa-tients. For example, a physician would never have the slightest doubt about the etiology of lung cancer in a heavy smoker, but would always wonder about its patho-genesis in a lifelong nonsmoker.
WMD is often seen in the sickest preterm newborns. In the same infants, the blood pressure tends to be on the low side. Does this mean that a low systemic blood pres-sure must be a cause for WMD? Indeed, the majority of studies of observational studies have not shown an asso-ciation between measures of systemic blood flow and WMD [for an overview, see appendix of reference 55 ].
The concepts of co-occurrence and association are re-lated, but by no means identical. Hybrid papers that mix the small- and large-sample perspectives have the poten-tial to be confusing.
Association does not prove causation, as recently shown for the association between changes in regional stork populations and birth rates [56] . Similarly, the harmful effects of smoking are not supported by the ob-servation that all smokers die, but by the observation that smoking increases the likelihood of lung cancer and/or death at an earlier age above what we know from popula-tions of nonsmokers.
In keeping with this view, finding so-called ‘proin-flammatory’ cytokines in areas of neonatal WMD does not mean that they contributed to the damage. Their pres-
ence might also indicate that they are part of other re-sponses to an injurious stimulus. For example, they might play a neuroprotective role, contribute to diminishing the inflammation, or participate in repair [57, 58] .
Single Cause Attribution vs. Multicausal Constellations Imagine an elderly lady being hit by a bus. She breaks
her leg. Although this has shown that bus accidents can cause broken legs, to what extent does this support the hypothesis that bus accidents are a frequent, the main, the only cause of broken legs? How much of the accident can be attributed to the elderly lady (did she jay walk?), the driver, poor lighting, fog, slippery road conditions, me-chanical failure, another driver who caused the bus driv-er to swerve?
Attributing an outcome to one antecedent has been labeled ‘single cause attribution’ [59] . This is most often exemplified by what we call the ‘individual perspective’ in the previous section. One picks a case of PBD, search-es for some event deemed an appropriate insult, and at-tributes causal properties to this event.
The errors of single cause attribution are also be made by academicians and investigators. The following are ex-amples.
A popular textbook of neonatal neurology places the content dealing with WMD causation in a chapter labeled ‘Hypoxic-Ischemic Encephalopathy: Neuropathology and Pathogenesis’. The author tips his hat to other pos-sible etiologies, but makes abundantly clear that, in his eyes, his placement of the material is appropriate.
Hypoxia-ischemia (oxygen-glucose deprivation) causes brain damage in the laboratory. So do excitotoxins and inflammogens. Why choose hypoxia-ischemia as THE single cause, especially when this position is NOT supported by studies of humans?
Reports of clinical trials of head cooling for the neo-natal disorder characterized by obtundation and brain-stem dysfunction labeled the disorder hypoxic-ischemic encephalopathy. Sometimes this disorder has no obvious antecedent. Other times, epidemiologic studies show that it is associated with fetal/maternal inflammation. The disorder’s label reflects the simplistic thinking that the existence of the disorder means that there must have been unseen/undocumented hypoxia-ischemia. Plaintiffs’ at-torneys tell the jury that the doctors caring for the unre-sponsive baby diagnosed her condition as hypoxic-isch-emic encephalopathy, documenting that the obstetrician caused this child’s damaged condition. And sometimes these attorneys win.

Perinatal Brain Damage Causation Dev Neurosci 2007;29:280–288 285
Why cannot we attach the name ‘newborn encepha-lopathy’ to neonatal unresponsiveness accompanied by brainstem dysfunction and/or seizures? Doing so would avoid the fallacy of single cause attribution.
Extrapolating from the observation that ischemia causes brain damage in the laboratory to the belief that hypoxia-ischemia is the single cause of PBD in humans invokes both the misconception possibility to fact and this misconception (single cause attribution). The inter-relatedness of the misconceptions needs to be kept in mind when either one misconception is identified. Might another unstated misconception have been invoked?
Despite early [21] and widespread [60] recognition that all diseases have multiple causes, the concept of sin-gle-cause attribution appears to be widely accepted. We often see publications that include lists of cases and their putative causes, without showing any data on controls. It even made its way into the language of imaging colleagues who describe what they identify on ultrasound pictures as ‘ischemic lesions’ [see 61 for discussion and critique of this concept]. We raise the possibility that names of enti-ties/lesions that imply a known causation when the evi-dence is not convincing can impede progress identifying the myriad contributors to PBD causation.
Although we have attempted to avoid offending any-one by not identifying papers that exemplified one of the misconceptions we address, we consider it appropriate to use our own work to illustrate how single-cause attribu-tion was applied to a group of preterm children with ce-rebral palsy, the clinical neurodevelopmental symptom strongly associated with WMD [62] . The presence of cer-tain characteristics in these individual children’s histo-ries (e.g. cord prolapse) was assumed a ‘definite explana-tion’ of the child’s cerebral palsy, that of others (such as ‘severe postnatal asphyxia’) a ‘possible explanation’. In-terestingly, this way of attributing single causes yielded 32% explained, 32% possibly explained, and 36% not ex-plained cases. This result is not very different from what would be expected if one of three possible outcomes had been attributed by chance (1 of 3 = 33.3%).
A sophisticated extension of the single cause attribu-tion concept is the etiological pathway concept [63, 64] . This theory suggests that in individual infants, several different (but distinct) pathways can culminate in brain damage. The clear advantage of this concept is that not all cases of PBD are assumed equal with regard to causal mechanisms. A much more realistic way is to look at dis-ease causation from the probabilistic perspective, which holds that ‘the presence of X increases the likelihood of the occurrence of Y’ (see ‘Determinism vs. Probabilism’
above). However, this concept may be of limited use in individual patients. We advise against offering numbers and percents to parents, either in matters of causation or prognosis.
Nevertheless, the probabilistic approach offers enor-mous advantages in etiologic research and causal infer-ence. Moreover, it paves the way for what is now consid-ered the standard of sophisticated etiologic research, i.e. elaborate multivariable data analysis. Only this approach allows for the modeling of a multicausal network, the closest theoretical framework for the study of the multi-etiologic natural history of diseases.
Generalization Sometimes, we are sure we have found the cause of
PBD in a single individual newborn. A catastrophic situ-ation such as placental abruption is often considered highly suggestive of being the initiator of perfusion dis-turbances and associated energy failure, leading to PBD [65] . It is eminently conceivable that even in such a sup-posedly clear-cut situation, the origin of the observed brain damage might actually be what led to the catastro-phe, e.g. infection/inflammation [66–68] , not the catas-trophe itself (placental abruption). Thus, we consider it reasonable to argue that the mere observation of a rather suggestive co-occurrence (see ‘Co-Occurrence in Indi-viduals vs. Association in Populations’ above) of a plau-sible cause (here: abruption) and effect (here: PBD) in single clinical instances should not trigger the clinician’s, judge’s, lawyer’s, or jury member’s generalization that in all babies with placental abruption any co-occurring PBD must be due to the abruption.
Unstated Causal Inferences Amniotic fluid embolism results in profound and pro-
longed maternal hypotension, which interferes with the placenta’s ability to provide the fetus with oxygen. If the mother’s circulation is not improved, she will die. If the fetus cannot be delivered in time, the fetus, too, will die. If the fetus is delivered alive, however, brain damage may have occurred.
In light of this scenario, it seems eminently reasonable to invoke the ‘continuum of casualty’ hypothesis, which as applied to the brain, states that the same insult, if se-vere kills, but if less severe kills not the entire individual, but only the most vulnerable cells, assumed to be neurons [69] .
Although on one level this hypothesis is plausible, it does not apply to less catastrophic situations, for example, when the term infant with newborn encephalopathy is

Dammann /Leviton
Dev Neurosci 2007;29:280–288286
presumed to have suffered an intrapartum insult despite the mother having no documented physiologic distur-bance. The inference that low blood pressure in a sick infant born months before term who is presumed to have impaired cerebral blood flow regulation leads to WMD can be viewed as another example of inappropriately ap-plying the continuum of casualty hypothesis. The infer-ential error tends to be made by those who are not aware that they are invoking the hypothesis [70] . We encourage the explicit statement of all causal inferences.
Unstated Assumption that What Might Be Heterogeneous Is Homogeneous In PBD, this inferential error is exemplified by papers
that list grade III/IV intraventricular hemorrhage (IVH) as an outcome or a covariate [71] . In such situations, a hemorrhage that fills the lateral ventricle (IVH) is equat-ed with a large echodensity adjoining the lateral ventricle. An extension of this assumption is the assumption that WMD and IVH are etiologically equivalent. We are not arguing against the possibility that IVH is an antecedent or correlate of WMD damage. Rather, they just are not identical, nor should each be presumed to have exactly the same risk profile as the other.
Use of the entity called ‘grade III/IV IVH’ assumes that grade III IVH is the same as grade IV in terms of eti-ology, pathophysiology or consequences, depending on the setting in which it is used. Since what is called grade IV IVH might not be primarily a hemorrhage, continued use of the term perpetuates many errors and promotes inferential errors [72] .
When the borders of the lateral ventricle are lost and the ventricle contents cannot be distinguished from the surrounding echodense white matter, even highly com-petent sonologists might not be able to tell where is the boundary between hemocephalus and adjacent normal or damaged white matter. This quandary is real. Limiting use of the ‘grade III/IV IVH’ to these situations is inap-propriate. Failure to recognize the misconception leads to inferential errors.
Our main point is that progress in our thinking about causation will not come until we define exposures and characteristics as homogeneously as possible and seek the antecedents of the most homogeneous outcome possi-ble.
Failure to Consider Alternative Explanations In 1650, Oliver Cromwell wrote, ‘I beseech you, in the
bowels of Christ, think it possible you may be mistaken’ [73] . The last seven words of this quote constitute the
Cromwellian challenge taught to students of epidemiol-ogy. Unfortunately, some see a paper or a table of data that fits their favorite hypothesis and accept it without any reservation. We suggest that all findings and publications be interpreted with the Cromwellian challenge in mind.
Conclusions
Research in the field of PBD is either observational or experimental [for an overview, see 4 ]. By definition, ob-servational studies can only study association, but never prove causation. Experimental studies can show that an exposure ‘can’ produce an outcome, but this is never a ‘must’ outside the experimental world. Apparently, caus-al inference needs support from both observation and ex-periment, from both epidemiology and laboratory re-search [5] . Thus, we propose that the more of the follow-ing criteria are fulfilled, the stronger the support for the contention that some risk factor for PBD might be a ‘caus-al’ factor: • the factor precedes PBD (however, even if it does, how
do we distinguish ‘post hoc’ from ‘propter hoc’?); • the factor can produce PBD in the experimental set-
ting (still, a clear definition of ‘can’ should be sought in future causality research);
• it is (statistically) associated with PBD in (well-pow-ered) observational studies;
• its absence from populations reduces the prevalence of PBD compared to populations with the factor present, e.g. in clinical trials [5] . We do not suggest that our discussion and proposed
criteria solve any theoretical issue in causation/causality research. Rather, we hope they can serve as guiding lights for clinicians and public health workers in the dark wil-derness of causal inference and can contribute to fruitful research in this area, designed to reduce the individual and societal burden of PBD.
Acknowledgements
The authors appreciate Professor Uwe Meixner’s commentson their manuscript. While working on the paper, they were supported by grants from the National Institutes of Health (NS040069) and the Wilhelm Hirte Stiftung, Hannover, Ger-many.

Perinatal Brain Damage Causation Dev Neurosci 2007;29:280–288 287
References
1 De Vries LS, Groenendaal F: Neuroimaging in the preterm infant. Ment Retard Dev Dis-abil Res Rev 2002; 8: 273–280.
2 Ferriero DM: Neonatal brain injury. N Engl J Med 2004; 351: 1985–1995.
3 Rutherford M, Ward P, Allsop J, Malamaten-tiou C, Counsell S: Magnetic resonance im-aging in neonatal encephalopathy. Early Hum Dev 2005; 81: 13–25.
4 Hagberg H, Peebles D, Mallard C: Models of white matter injury: comparison of infec-tious, hypoxic-ischemic, and excitotoxic in-sults. Ment Retard Dev Disabil Res Rev 2002;
8: 30–38. 5 Dammann O, Leviton A: Inflammatory
brain damage in preterm newborns – dry numbers, wet lab, and causal inference. Ear-ly Hum Dev 2004; 79: 1–15.
6 Rees S, Inder T: Fetal and neonatal origins of altered brain development. Early Hum Dev 2005; 81: 753–761.
7 Rothman KJ, Greenland S: Modern Epide-miology, ed 2. Philadelphia, Lippincott-Ra-ven, 1998.
8 Dawid AP: Probability, causality and the em-pirical world: a Bayes-de Finetti-Popper-Borel synthesis. Stat Sci 2004; 19: 44–57.
9 Buck C: Popper’s philosophy for epidemiolo-gists. Int J Epidemiol 1975; 4: 159–168.
10 Maclure M: Popperian refutation in epide-miology. Am J Epidemiol 1985; 121: 343–350.
11 Greenland S: Induction versus Popper: sub-stance versus semantics. Int J Epidemiol 1998; 27: 543–548.
12 Sosa E, Tooley M (eds): Causation. Oxford, Oxford University Press, 1993.
13 Meixner U: Theorie der Kausalität. Pader-born, Mentis, 2001.
14 Hill AB: The environment and disease: as-sociation or causation? Proc R Soc Med 1965;
58: 295–300. 15 Susser MW: Causal Thinking in the Health
Sciences – Concepts and Strategies of Epide-miology. New York, Oxford University Press, 1973.
16 Susser M: What is a cause and how do we know one? A grammar for pragmatic epide-miology. Am J Epidemiol 1991; 133: 635–648.
17 Parascandola M, Weed DL: Causation in ep-idemiology. J Epidemiol Community Health 2001; 55: 905–912.
18 Rothman KJ, Greenland S: Causation and causal inference; in Rothman KJ, Greenland S (eds): Modern Epidemiology. Philadelphia, Lippincott Williams & Wilkins, 1998, pp 7–28.
19 Pearl J: Causality. Cambridge, Cambridge University Press, 2000.
20 Glymour C, Cooper GF (eds): Computation, Causation, and Discovery. Menlo Park/Cambridge, AAAI Press/MIT Press, 1999.
21 Rothman KJ: Causes. Am J Epidemiol 1976;
104: 87–92. 22 Boffetta P: Molecular epidemiology: a tool
for understanding mechanisms of disease. Eur J Surg Suppl 2002: 62–69.
23 Rorty R: Philosophy and Social Hope. Lon-don, Penguin, 1999, pp xvi–xxxii.
24 du Plessis AJ, Volpe JJ: Perinatal brain injury in the preterm and term newborn. Curr Opin Neurol 2002; 15: 151–157.
25 Rezaie P, Dean A: Periventricular leukoma-lacia, inflammation and white matter lesions within the developing nervous system. Neu-ropathology 2002; 22: 106–132.
26 Vannucci SJ, Hagberg H: Hypoxia-ischemia in the immature brain. J Exp Biol 2004; 207:
3149–3154. 27 Dammann O, Leviton A: Role of the fetus in
perinatal infection and neonatal brain dam-age. Curr Opin Pediatr 2000; 12: 99–104.
28 Kadhim H, Sebire G: Immune mechanisms in the pathogenesis of cerebral palsy: impli-cation of proinflammatory cytokines and T lymphocytes. Eur J Paediatr Neurol 2002; 6:
139–142. 29 Bracci R, Buonocore G: Chorioamnionitis: a
risk factor for fetal and neonatal morbidity. Biol Neonate 2003; 83: 85–96.
30 Hagberg H, Wennerholm UB, Savman K: Se-quelae of chorioamnionitis. Curr Opin In-fect Dis 2002; 15: 301–306.
31 Edwards AD: New approaches to brain in-jury in preterm infants. Dev Neurosci 2003;
24: 352–354. 32 Garnier Y, Coumans AB, Jensen A, Hasaart
TH, Berger R: Infection-related perinatal brain injury: the pathogenic role of impaired fetal cardiovascular control. J Soc Gynecol Investig 2003; 10: 450–459.
33 Dammann O, Leviton A: Brain damage in preterm newborns: might enhancement of developmentally-regulated endogenous pro-tection open a door for prevention? Pediat-rics 1999; 104: 541–550.
34 Leviton A, Blair E, Dammann O, Allred EN: The wealth of information conveyed by ges-tational age. J Pediatr 2005; 146: 123–127.
35 Leviton A, Paneth N: White matter damage in preterm newborns – an epidemiologic perspective. Early Hum Dev 1990; 24: 1–22.
36 Hagberg H, Dammann O, Mallard C, Levi-ton A: Preconditioning and the developing brain. Semin Perinatol 2004; 28: 389–395.
37 Eklind S, Mallard C, Leverin AL, Gilland E, Blomgren K, Mattsby-Baltzer I, Hagberg H: Bacterial endotoxin sensitizes the immature brain to hypoxic – ischaemic injury. Eur J Neurosci 2001; 13: 1101–1106.
38 Dommergues MA, Patkai J, Renauld JC, Ev-rard P, Gressens P: Proinflammatory cyto-kines and interleukin-9 exacerbate excito-toxic lesions of the newborn murine neopallium. Ann Neurol 2000; 47: 54–63.
39 Dammann O, Allred EN, Kuban KCK, Van Marter LJ, Pagano M, Leviton A, for the De-velopmental Epidemiology Network: Hypo-carbia during the first 24 postnatal hours and periventricular echolucencies in new-borns ! 28 weeks gestation. Pediatr Res 2001;
49: 388–393. 40 Banker BQ, Larroche JC: Periventricular
leukomalacia of infancy. Arch Neurol 1962;
7: 386–410. 41 Levine S: Anoxic-ischemic encephalopathy
in rats. Am J Pathol 1960; 36: 1–17. 42 Levy DE, Brierley JB, Silverman DG, Plum F:
Brief hypoxia-ischemia initially damages ce-rebral neurons. Arch Neurol 1975; 32: 450–456.
43 Rice J, Vannucci RC, Brierley JB: The influ-ence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 1981; 9:
131–141. 44 Tyszczuk L, Meek J, Elwell C, Wyatt JS: Ce-
rebral blood flow is independent of mean ar-terial blood pressure in preterm infants un-dergoing intensive care. Pediatrics 1998; 102:
337–341. 45 Hagberg H, Bona E, Gilland E, Puka-Sund-
vall M: Hypoxia-ischaemia model in the 7-day-old rat: possibilities and shortcomings. Acta Paediatr Suppl 1997; 422: 85–88.
46 Neu J: The ‘myth’ of asphyxia and hypoxia-ischemia as primary causes of necrotizing enterocolitis. Biol Neonate 2005; 87: 97–98.
47 Greisen G, Borch K: White matter injury in the preterm neonate: the role of perfusion. Dev Neurosci 2001; 23: 209–212.
48 Eells E: Probabilistic Causality. Cambridge, Cambridge University Press, 1991.
49 Hitchcock C: A generalized probabilistic theory of causal relevance. Synthese 1993; 97:
335–364. 50 Gallo RC, Montagnier L: The discovery of
HIV as the cause of AIDS. N Engl J Med 2003; 349: 2283–2285.
51 Beattie T, Rowland-Jones S, Kaul R: HIV-1 and AIDS: what are protective immune re-sponses? J HIV Ther 2002; 7: 35–39.
52 Batton DG, Kirtley X, Swails T: Unexpected versus anticipated cystic periventricular leu-komalacia. Am J Perinatol 2003; 20: 33–40.
53 Evidence-Based Medicine Working Group: Evidence-based medicine. A new approach to teaching the practice of medicine. JAMA 1993; 268: 2440–2445.
54 Dammann O: Evidence-based child neurol-ogy. Dev Med Child Neurol 2006; 48: 622–624.
55 Dammann O, Allred EN, Kuban KC, Van Marter LJ, Pagano M, Sanocka U, Leviton A: Systemic hypotension and white-matter damage in preterm infants. Dev Med Child Neurol 2002; 44: 82–90.
56 Hofer T, Przyrembel H, Verleger S: New evi-dence for the theory of the stork. Paediatr Perinat Epidemiol 2004; 18: 88–92.

Dammann /Leviton
Dev Neurosci 2007;29:280–288288
57 Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP: TNF alpha pro-motes proliferation of oligodendrocyte pro-genitors and remyelination. Nat Neurosci 2001; 4: 1116–1122.
58 Gupta S: A decision between life and death during TNF-alpha-induced signaling. J Clin Immunol 2002; 22: 185–194.
59 Leviton A: Single-cause attribution. Dev Med Child Neurol 1987; 29: 805–807.
60 Barnetche T, Gourraud PA, Cambon-Thom-sen A: Strategies in analysis of the genetic component of multifactorial diseases; bio-statistical aspects. Transpl Immunol 2005;
14: 255–266. 61 Kuban KC: White-matter disease of prema-
turity, periventricular leukomalacia, and ischemic lesions. Dev Med Child Neurol 1998; 40: 571–573.
62 Veelken N, Schopf M, Dammann O, Schulte FJ: Etiological classification of cerebral palsy in very low birthweight infants. Neuropedi-atrics 1993; 24: 74–76.
63 Blair E, Stanley F: Aetiological pathways to spastic cerebral palsy. Paediatr Perinat Epi-demiol 1993; 7: 302–317.
64 Blair E, Stanley F: When can cerebral palsy be prevented? The generation of causal hy-potheses by multivariate analysis of a case-control study. Paediatr Perinat Epidemiol 1993; 7: 272–301.
65 Gibbs JM, Weindling AM: Neonatal intra-cranial lesions following placental abrupti-on. Eur J Pediatr 1994; 153: 195–197.
66 Darby MJ, Caritis SN, Shen-Schwarz S: Pla-cental abruption in the preterm gestation: an association with chorioamnionitis. Obstet Gynecol 1989; 74: 88–92.
67 Kramer MS, Usher RH, Pollack R, Boyd M, Usher S: Etiologic determinants of abruptio placentae. Obstet Gynecol 1997; 89: 221–226.
68 Rana A, Sawhney H, Gopalan S, Panigrahi D, Nijhawan R: Abruptio placentae and chorio-amnionitis-microbiological and histologic correlation. Acta Obstet Gynecol Scand 1999; 78: 363–366.
69 Lilienfeld AM, Parkhurst E: A study of the association of factors of pregnancy and par-turition with the development of cerebral palsy: preliminary report. Am J Hyg 1951; 53:
262–282. 70 Ounsted M: Causes, continua and other con-
cepts. I. The ‘continuum of reproductive ca-sualty’. Paediatr Perinat Epidemiol 1987; 1:
4–7. 71 Papile LA, Burstein J, Burstein R, Koffler H:
Incidence and evolution of subependymal and intraventricular hemorrhage: a study of infants with birth weights less than 1,500 gm. J Pediatr 1978; 92: 529–534.
72 Paneth N: Classifying brain damage in pre-terms. J Pediatr 1999; 134: 527–529.
73 The Oxford Dictionary of Quotations. Ox-ford, Oxford University Press, 1979, p 169.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:289–301 DOI: 10.1159/000105470
Serial Diffusion Tensor Imaging DetectsWhite Matter Changes That Correlate withMotor Outcome in Premature Infants
Alexander Drobyshevsky
a Joanne Bregman
a Pippa Storey
b Joel Meyer
b
P.V. Prasad
b Matthew Derrick
a William MacKendrick
a Sidhartha Tan
a
Departments of a Pediatrics, and b
Radiology, Evanston Northwestern Healthcare, Evanston, Ill. , USA
white matter. A low value of FA at 30 weeks and a higher change of FA predicted less favorable motor outcome at 2 years and suggests that early subtle white matter injury can be detected in premature infants even without obvious signs of injury. Copyright © 2007 S. Karger AG, Basel
Introduction
Survival rates of extremely low birth weight infants have been improving in the past decades, but the inci-dence of neurodevelopmental disabilities in the survivors [Wilson-Costello et al., 2005] has not declined. This in-cludes cerebral palsy, as well as cognitive and psychomo-tor delays. In the US, around 20–22% of infants with birth weight less than 1,000 g may manifest cognitive or psy-chomotor delay at 20 months corrected age on a stan-dardized test of infant development, such as the Bayley Scales of Infant Development (BSID). Given this high risk for disability, there is a desire to improve our capacity for early diagnosis of brain injury in premature neonates [Miller et al., 2005]. Most frequently, serial cranial ultra-sounds (CUS) are used to diagnose brain injury and in-creasingly more sophisticated neuroimaging modalities including magnetic resonance imaging (MRI) are being
Key Words
Intraventricular hemorrhage � Premature infant � Hypoxia-ischemia � Brain development
Abstract
The objective of the study was to assess predictive value of serial diffusion tensor MRI (DTI) for the white matter injury and neurodevelopmental outcome in a cohort of premature infants. Twenty-four infants less than 32 weeks’ gestation were stratified to a control group (n = 11), mild brain injury with grades 1–2 of intraventricular hemorrhage (n = 6) and severe brain injury with grades 3–4 intraventricular hemor-rhage (n = 4). Serial DTI studies were performed at around 30 and 36 weeks’ gestation. Fractional anisotropy (FA) and apparent diffusion coefficient were calculated. Twelve in-fants were followed up for developmental outcome. Devel-opmental testing was performed with the Bayley Scales of Infant Development to obtain psychomotor index (Perfor-mance Developmental Index). Apparent diffusion coeffi-cient was higher in the severe injury group at the second MRI in the central and occipital white matter, and corona radiata; FA was lower in optic radiation compared to controls. Per-formance Developmental Indexscore correlated with FA on the scan taken at the 30th week and inversely with the change of FA between scans in internal capsule and occipital
Received: September 15, 2006 Accepted after revision: February 16, 2007
A. Drobyshevsky Department of Pediatrics Evanston Northwestern Healthcare Research Institute , 2650 Ridge Ave.Evanston, IL 60201 (USA) Tel. +1 847 570 2353, Fax +1 847 570 0231, E-Mail [email protected]
© 2007 S. Karger AG, Basel
Accessible online at:www.karger.com/dne

Drobyshevsky /Bregman /Storey /Meyer /Prasad /Derrick /MacKendrick /Tan
Dev Neurosci 2007;29:289–301290
used [Roelants-van Rijn et al., 2001; Inder et al., 2003]. Neonatologists often base their prognostication on the severity of the intraventricular hemorrhage (IVH) de-tected by CUS in the 1st week of life as well as evidence of periventricular leucomalacia (PVL) in any CUS through-out the hospital stay. Numerous epidemiological studies have shown that severe grades of IVH or PVL are associ-ated with poor developmental outcome [Volpe, 2001]. PVL is a form of white matter injury (WMI) character-ized by multiple small cystic lesions in the periventricular white matter [Volpe, 2001]. Another manifestation of WMI, less well detected by CUS, is a diffuse pattern of injury [Counsell et al., 2003].
Diffusion tensor imaging (DTI) with MR provides quantitative indexes of water diffusion in brain tissue, reflecting cellular and organelle density (apparent diffu-sion coefficient – ADC) and spatial organization of un-derlying structure (fractional anisotropy – FA). Studies using DTI have not only characterized normal brain mat-uration in developing white matter as white matter fiber tracts organize (a progressive increase of the FA and de-crease of ADC) [Neil et al., 1998], but deviations from this normal trend are considered diagnostic of perinatal WMI [Huppi and Inder, 2001; Miller et al., 2002].
We hypothesized that early, subtle WMI can be de-tected on serial DTI in premature neonates who have nor-mal conventional T 1 and T 2 scans and that this WMI causes later developmental deficits.
Methods
The initial MRI study and the follow-up study were approved by the Institutional Review Board of the Evanston Northwestern Healthcare Research Institute. Informed consent was obtained from the parents upon agreement to participate in each phase of the study. The diagram of the study protocol and subject workup is presented in figure 1 .
Subjects All premature newborns ! 32 weeks’ gestation born at the
Evanston Hospital and cared for in the Infant Special Care Unit (ISCU) were eligible for enrollment in the study if they were with-out major congenital anomalies, had undergone a cranial ultra-sound in the 1st week of life, and were deemed stable by the at-tending neonatologist. Out of 140 infants who met enrollment criteria during the enrollment period between July 2002, and Jan-uary 2004, 15 were determined too ill to participate. Parents of 7 patients were not approached because of language barrier or be-cause they were not present. Parents of 19 infants were not ap-proached when scanner hardware or logistical problems occurred. Out of those who were approached for consent, 32 parents de-clined and 43 did not respond during the 2-week enrollment pe-
riod. Mostly, the reason for refusing was because of absence of tangible benefits. Finally, 24 parents consented for the study. The infants enrolled in the study had rates of respiratory, infectious and cardiovascular complications that were similar to those seen in the general nursery population ! 32 weeks of gestation. There were 5 sets of twins, the rest singleton births. The subjects under-went MRI scans at 10–14 days of life, (approximately 30 weeks’ gestation) and again around 36 weeks’ postconceptional age or before discharge. Only one baby, the youngest study subject (24.1 weeks’ gestation at birth) required ventilator support during the first MRI study. Infants were transported to the MRI scanner in a thermo-controlled transport Isolette, accompanied by an ISCU-registered nurse and when indicated, an ISCU respiratory thera-pist. The infant’s head was softly restrained with fabric straps and beanbags. During the MRI procedure, a thermo-neutral environ-ment was maintained with warming blankets. Neonatal ear muffs were used to block out MRI noise. During MRI examination, the patient was continuously monitored by a pulse oximeter and closely observed by the neonatal intensive care nurse and respira-tory therapist if artificial ventilation was being provided. Imaging was done immediately after the infant was fed, so as to induce drowsiness and thus reduce head motion. No sedation was used in this study.
Magnetic Resonance Imaging The infant’s head was placed in a quadrature extremity coil
that was used for signal transmission and reception. The scanner parameters for the imaging protocol were as follows: T 1 -weighted imaging: spin echo with TE/TR = 14 ms/500 ms, NEX = 2; T 2 -weighted imaging: fast spin echo with TE/TR = 100 ms/3,650 ms, echo train = 8, NEX = 2; FLAIR imaging: spin echo with TE/TI/TR = 140 ms/2,200 ms/10,000 ms, NEX = 1; DTI: dual spin-echo EPI with bipolar diffusion gradients and b = 1,000 s/mm 2 , applied in six noncollinear directions, NEX = 8. Fourteen contiguous 5-mm axial slices were obtained for each sequence with a 15-cm FOV, covering the whole brain. Images were processed offline with in-house software written using Matlab 7.0.4 (MathWorks, Natick, Mass., USA). Parametric maps of diffusion indexes were calculated for ADC, FA [Basser et al., 1994]. Images were visually inspected and poor quality images due to excessive subjects’ mo-tion were excluded prior to analysis.
Assignment of Groups Ultrasound images were read by other pediatric radiologists,
not involved in the study, and these results were available to the clinical team taking care of the patient. Grading of IVH was done on the initial ultrasound. Data from 3 infants were excluded from the MRI protocol due to poor image quality secondary to exces-sive head motion as determined by 2 investigators. Using reports of ultrasound examinations, T 1 -, T 2 -weighted and FLAIR images, the remaining 21 subjects were assigned into 3 groups by the par-ticipating neuroradiologist (J.M.) who was blinded to the diffu-sion data:
(1) Controls without any abnormalities on cranial ultrasound and MRI (n = 11)
(2) Mild brain injury with IVH grades 1 or 2 (n = 6; 3 patients in each grade)
( 3) Severe brain injury (n = 4; 1 patient with multicystic PVL, 2 patients with IVH grade 3 and 1 with grade 4; all IVH patients in the group had ventriculomegaly) .

Serial DTI in White Matter and Motor Outcome in Premature Infants
Dev Neurosci 2007;29:289–301 291
In patients with IVH, no other abnormalities were detected on T 1 -, T 2 -weighted and FLAIR images.
Image Processing Regions of interest (ROIs) were positioned for each individual
by an investigator (A.D.) who was blinded to the grouping, as shown in figure 2. The nineteen ROIs covered major white matter (projection fibers, radiations, commissures, basal ganglia and gray matter structures in different lobes and calcarine cortex. Po-lygonal ROIs were placed on white matter tracts, using direction-ally encoded color FA-weighted map with the principal diffusiv-ity vector to identify the course of the fibers. Standard-size circu-lar 5-mm radius ROIs were used on T 2 -weighted images to sample thalamus and white matter regions in different lobes at the level of basal ganglia (not including the optic radiation) and centrum semiovale. In order to minimize partial volume effect, polygonal ROIs were also used to sample cortex in different lobes and calca-rine cortex. None of the positioned ROIs were in close proximity to the sites of hemorrhage to be affected by local magnetic field inhomogeneity.
Values for each selected structure were calculated as a sum for the ROIs from the left and right side, weighted by number of vox-els in the ROIs. Since polygonal ROIs were placed manually, we did not attempt to analyze ROIs from each side separately to avoid bias caused by asymmetry in IVH.
Standardizing Times of MRI Scans Since the MRI scans were obtained were obtained at different
ages, we attempted to standardize the data to two ages. Diffusion indices from the control group for each ROI were used to calculate the linear change in each ROI to age. The regression equations for each ROI were then used to extrapolate all the first scans to 30 weeks and all the second scans to 36 weeks of age. This is based on the assumption that diffusion indexes change linearly in the brain between 30 and 40 weeks [Partridge et al., 2004]. This was confirmed by the changes with age in our control group showing a linear fit of diffusion indices with age, exemplified by the scatter plot of FA in internal capsule ( fig. 3 a), parietal white matter ( fig. 3 b) and parietal cortex ( fig. 3 c). In the brain injury groups, the slopes were the similar or less, but without any significant re-lationship with age in FA. We used the same slopes, derived from the controls, to standardize our ages for the brain injury groups, a strategy that overestimates FA change a little for both scans.
Developmental Testing One infant who had IVH grade IV died during the hospital
course. The remaining 20 infants who had at least one good qual-ity DTI study were discharged home. Twelve (controls = 9, mild injury = 1, severe injury = 2) returned for neurobehavioral testing at the ENH ISCU Developmental Clinic at Evanston Hospital. Ten of those infants had 2 MRI exams done during their ISCU stay
Premature newborns
Consent
First MRI within 10–14 days
Second MRI at 36 weeks/discharge
Neuroradiologist assessment
Discharge
Analysis of MRI
Bayley’s Scales of Infant Developmentof 18–24 Month
24
21
3
motion artefacts,excluded
1 died
Control
11
9
Mild
6
1
Severe
4
2
Fig. 1. Diagram of the study protocol and workup of patients.

Drobyshevsky /Bregman /Storey /Meyer /Prasad /Derrick /MacKendrick /Tan
Dev Neurosci 2007;29:289–301292
(controls = 8, mild injury = 1, severe injury = 1). Reasons for at-trition included moving out of state, lack of insurance, transpor-tation issues, or were lost to follow-up. The 12 infants were evalu-ated between 18 and 24 months corrected age (for prematurity). The parents of these infants were Caucasians, were all married and had the same type of insurance. We did not collect any data on the socioeconomic status of parents.
At the clinic visit, a standardized developmental assessment, using the BSID-II [Bayley, 1993] was completed by a clinical psy-chologist (J.B.) or infant development specialist experienced and trained in administration of the BSID. Assessment of tone, hear-ing and vision status, was also obtained. The developmental as-sessment was done blinded to the results of the MRI scans.
Statistical Analysis of MRI Data Group averages for the first and the second MRI studies were
calculated for each ROI, as well as pairwise difference between the studies. Data are presented as mean 8 standard error of mean. Absolute changes in diffusion indices between studies and the rate of changes per week were compared using paired t test. Com-parison between groups was done by repeated measures ANOVA and Tukey’s post-hoc comparison procedure. To control overall alpha level due to multiple ROI comparison, Bonferroni correc-tion was applied to adjust alpha level for ANOVA of individual ROIs. Correlation analysis was done using Pearson and Spearman correlation. A value of p ! 0.05 was considered significant.
Results
Subjects and Pattern of WMI on Conventional Imaging The groups, defined by the severity of brain injury,
were not significantly different in birth weight and/or gestational age at birth, or at the first and the second MRI examination ( table 1 ). One infant had grade 1 IVH on the first study and multicystic PVL on the second study.
Fig. 2. Placement of ROIs on T 2 -weighted images with no diffu-sion weighting ( a , c ) and directionally encoded FA map ( b , d ). a , b Level of ventricles: frontal (1), occipital (2), temporal (3) white matter; parietal (4), occipital (5), temporal (6), frontal (7) gray matter; thalamus (8), putamen (9), caudate nucleus (10); white matter tracts – genu (11) and splenium (12) corpus callosum, op-tic radiation (13), posterior (14) and anterior (15) limbs of internal capsule. c , d Level of centrum semiovale: frontal (16), central (17) and parietal (18) white matter, corona radiata (19).
280.30
a
0.35
0.40
0.45
0.50
FA
0.55
0.60
30 32 34 36 38b
0.04
0.08
0.12
0.16
0.20
0.24
28 30 32Gestational age (weeks)
34 36 38c
0.10
0.15
0.20
0.25
0.30
0.35
0.40
28 30 32 34 36 38
Fig. 3. Diffusion indices experienced variable maturational changes for different regions in white and gray mat-ter. FA in control subjects linearly increased with age, as shown for internal capsule, posterior limb ( a ; slope 8.03 ! 10 –3 , R 2 = 0.40, p ! 0.01) and parietal white matter ( b ; slope 5.90 ! 10 –3 , R 2 = 0.29, p ! 0.01). c In con-trast, FA in parietal cortex was negatively correlated with age (slope –14.2 ! 10 –3 , R 2 = 0.34, p ! 0.001).

Serial DTI in White Matter and Motor Outcome in Premature Infants
Dev Neurosci 2007;29:289–301 293
Three infants from the severe injury group had small fo-cal hyperintensities on T 1 and corresponding hypointen-sities on T 2 -weighted images, that can be attributed to hemorrhage, within 1 cm but and not directly connected to ventricles. No other diffuse or focal WMI was detected in the parenchyma on conventional T 1 or T 2 images.
White Matter Maturation Assessed by DTI As expected, in the control group of infants, FA was
found to increase and ADC to decrease with age signifi-cantly for white matter regions and tracts. The opposite changes occurred for the cortical gray matter. The differ-ence in diffusion indices between the 1st study, at 30.3 8 0.4 weeks, and 2nd study, at 35.7 8 0.3 weeks (with 37.4 8 4.3 days between studies) can be visually appreciated on the maps of FA and ADC in figure 4 . Differences in values can be seen as differences in color based on the color maps.
The highest values of FA and lowest values for ADC were observed in compact white matter tracts, such as corpus callosum and internal capsule. In control prema-ture infants, the ADC of the overall white matter regions was higher than that of the gray matter for both studies ( fig. 5 a), and dropped between studies by 6.7% for the white matter and by 6.3% for the cortical gray matter, which corresponds to –1.5% and –1.4% change per week, respectively. ADC change ranged from –1 to –4% per
week in various regions, with the highest rate for the pos-terior limb of the internal capsule (–2.9 8 1.0%).
In contrast, values of FA increased between studies in the white matter by 8.9% and decreased in the cortical gray matter by 9.7%, which corresponds to 2.0% and 2.1% changes per week, respectively ( fig. 5 b). The rate of change was different for individual white matter ROIs, varying from 1.0 to 4.5% increase in FA per week, with the fastest rates in the frontal white matter (3.7 8 1.1%) followed by temporal white matter (2.7 8 1.6%) and the posterior limb of internal capsule (2.0 8 0.4%).
While the FA significantly decreased in the cortical gray matter, it had a trend to increase in the deep gray matter ROIs (caudate, lentiform and thalamus; table 2 ).
Regional Differences in Controls and Groups with Brain Injury by DTI Comparing the control group with groups with brain
injury for the selected ROIs, a significant difference was found in the white matter regions and tracts only in the second study ( tables 2 , 3 ). The ADC of the second study was higher for central, occipital white matter, corona ra-diata, and FA was lower in optic radiation in infants with severe brain injury compared to controls. The ADC of the second study was lower for central and occipital white matter in infants with mild brain injury compared to controls. There were no significant differences between controls and injury groups observed in the rest of select-
Table 1. Demographic data of the groups with different degree of brain injury
All Normal Mild Severe p value
Subjects 21 11 6 4Weight at birth, g 1,243870
(640–1,716)1,351880(995–1,689)
1,0698139(640–1,486)
1,2078195(766–1,716)
0.22
Postconceptional age at birth, weeks 28.780.40(0.4–30.9)
29.380.41(27.0–30.9)
27.981.03(24.1–30.9)
28.480.82(27.0–30.4)
0.33
Weight at first MRI exam, g 1,247874(605–1,696)
1,389870(1,020–1,620)
1,046870(605–1,615)
1,156870(750–1,696)
0.11
Age at first MRI exam,weeks after conception
30.480.41(25.9–32.9)
31.080.38(29.0–32.4)
29.681.08(25.9–32.9)
30.180.92(28.6–32.3)
0.32
Weight at second MRI exam, g 2,066870(1,570–2,590)
2,123870(1,830–2,590)
1,958870(1,570–2,370)
2,081870(2,046–2,115)
0.60
Age at second MRI exam,weeks after conception
35.780.21(34.1–37.1)
35.680.34(34.1–37.1)
35.980.37(34.9–37.0)
35.680.25(35.3–36.0)
0.18
Interval between MRI exams, days 3783(14–67)
3284(14–48)
4885(30–67)
2981(28–30)
0.07
Data are presented as mean 8 standard error and range; p values correspond to the single factor ANOVA test for equal mean in groups.

Drobyshevsky /Bregman /Storey /Meyer /Prasad /Derrick /MacKendrick /Tan
Dev Neurosci 2007;29:289–301294
0.5
ADC (×103 mm2/s) FA
1.5 2.5 0 0.5 1
Fig. 4. Representative T 2 -weighted ( a , e ), ADC ( b , f ), FA ( c , g ) and directionally encoded FA maps ( d , h ) of an infant, classified as normal, on 28 (top row) and 37 weeks (bottom row). Color map bars, representing pseudo-color mapping for ADC and FA values are in between corresponding images. Color schemes, representing en-coding of the predominant diffusivity directions are in the right bottom corners of d and h . Red is left-right, green is up-down and blue is perpendicular to the plane of the image. Note decreasing ADC of the gray and white matter structures and increasing anisotropy of white matter structures from the first to the second study.
AD
C ×
10–3
mm
2 /s
Frac
tion
al a
nis
otro
py
White matter1.0
a b
1.2
1.4
1.6
0.10
0.15
0.20
0.25
0.30
Gray matter White matter Gray matter
*
*
*
1st study2nd study
Fig. 5. a ADC values decreased in the white and cortical gray matter in premature in-fants from the time of the 1st study ( � 30 weeks) to the 2nd study at term-equivalent age ( � 36 weeks). b FA increased in the white matter and decreased in the gray matter between the studies. * p ! 0.05.

Serial DTI in White Matter and Motor Outcome in Premature Infants
Dev Neurosci 2007;29:289–301 295
ROI name Braininjurygroup
30 weeks 36 weeks
FA p ADC p FA p ADC p
White Internal capsule 0 0.3780.01 1.1880.03 0.4380.01 1.080.02matter posterior limb 1 0.3780.01 0.96 1.180.04 0.28 0.4380.01 0.94 0.9880.03 0.94
2 0.3780.03 0.96 1.1580.04 0.86 0.480.03 0.58 1.0480.13 0.75Internal capsule 0 0.2680.01 1.2780.04 0.2980.01 1.0380.02anterior limb 1 0.2880.01 0.46 1.1980.06 0.40 0.3280.01 0.17 0.9580.05 0.27
2 0.2880.01 0.25 1.2280.03 0.73 0.2780.01 0.80 1.0780.1 0.87Optic radiation 0 0.380.01 1.4680.06 0.3680.01 1.1480.03
1 0.2980.02 0.83 1.3380.04 0.30 0.4180.04 0.07 1.1480.1 1.002 0.380.02 0.83 1.4280.04 0.91 0.3180.03* 0.05 1.2680.04 0.59
Corona radiata 0 0.280.02 1.5580.05 0.2680.02 1.1480.061 0.2380.03 0.73 1.3380.07 0.08 0.3280.03 0.22 1.0380.08 0.172 0.2480.03 0.57 1.680.14 0.87 0.2580.05 0.99 1.3980.03* 0.01
Corpus callosum 0 0.4380.01 1.2980.04 0.4580.01 1.2380.04genu 1 0.4780.01 0.14 1.1380.04 0.05 0.4380.02 0.57 1.2180.05 0.94
2 0.4780.02 0.11 1.2880.05 0.99 0.4980.04 0.34 1.2580.02 0.98Corpus callosum 0 0.4580.01 1.3580.04 0.5380.02 1.1580.03splenum 1 0.4780.02 0.55 1.2480.06 0.28 0.5480.02 0.97 1.0680.03 0.17
2 0.4680.02 0.90 1.3180.04 0.87 0.5880.02 0.44 1.2480.02 0.49Frontal WM 0 0.1380.01 1.5580.04 0.1580.01 1.3680.04(level c. semiovale) 1 0.1480.01 0.89 1.4480.05 0.28 0.1780.01 0.41 1.1780.07 0.08
2 0.1280.01 0.83 1.5680.05 0.99 0.1880.01 0.42 1.3880.01 0.99Frontal WM 0 0.1480.0 1.5680.05 0.1580.01 1.4880.04(level ventricles) 1 0.1680.02 0.37 1.4980.06 0.60 0.1480.01 0.99 1.3280.04 0.07
2 0.1380.01 0.88 1.6180.03 0.76 0.1380.01 0.79 1.5380.1 0.83Central WM 0 0.1380.01 1.5380.04 0.1680.01 1.380.02(level c. semiovale) 1 0.1380.01 0.97 1.5180.06 0.92 0.1780.01 0.77 1.1580.06* 0.02
2 0.1380.01 0.95 1.5580.04 0.97 0.1580.02 0.96 1.3780.01* 0.04Parietal WM 0 0.1180.01 1.6680.04 0.1780.01 1.4280.03(level c. semiovale) 1 0.1180.01 0.85 1.6680.08 1.00 0.1980.02 0.37 1.1880.06* 0.04
2 0.180.01 0.77 1.6780.08 0.99 0.1780.01 1.00 1.4980.04 0.72Occipital WM 0 0.1480.01 1.5980.05 0.1680.01 1.3780.04(level ventricles) 1 0.1480.01 0.97 1.5980.06 1.00 0.1780.01 0.85 1.2180.04* 0.05
2 0.1380.01 0.86 1.6180.04 0.94 0.1480.02 0.52 1.5380.04* 0.04Temporal WM 0 0.1580.01 1.4980.05 0.2180.01 1.2280.03(level ventricles) 1 0.1480.01 0.97 1.4280.04 0.58 0.2180.01 0.95 1.1480.06 0.28
2 0.1680.0 0.81 1.4880.07 1.00 0.1980.02 0.76 1.2180.01 0.97
Gray Frontal GM 0 0.2780.03 0.86 1.2480.05 0.46 0.1680.02 0.84 1.2980.03 0.94matter 1 0.2980.03 1.00 1.1580.05 0.92 0.1880.03 0.60 1.3180.05 0.90
2 0.2780.03 1.2780.04 0.2180.03 1.2680.09Parietal GM 0 0.2580.02 1.1480.04 0.1180.02 1.280.04
1 0.3180.02 0.19 1.0980.04 0.72 0.0980.02 0.94 1.280.05 1.002 0.2780.03 0.82 1.2280.01 0.50 0.1580.0 0.71 1.2180.11 0.99
Perirolandic GM 0 0.1980.01 1.1780.04 0.1480.1 0.9680.021 0.1780.01 0.82 1.1280.05 0.70 0.1480.01 1.00 0.980.03 0.352 0.1780.02 0.64 1.1580.03 0.94 0.1480.01 0.93 1.0780.06 0.12
Temporal GM 0 0.1980.01 1.2480.06 0.1380.01 1.1380.051 0.2480.02 0.17 1.1580.06 0.59 0.1180.01 0.74 1.0780.06 0.712 0.1980.03 0.99 1.2580.03 1.00 0.1280.02 0.98 1.1180.08 0.97
Caudate nucleus 0 0.1480.03 1.2980.05 0.1380.01 1.0680.031 0.1280.0 0.81 1.2880.08 1.00 0.1480.01 0.86 0.9880.04 0.252 0.0980.01 0.47 1.3580.08 0.83 0.1280.01 0.85 1.180.08 0.85
Lentiform nucleus 0 0.1380.01 1.2780.04 0.1380.01 0.9280.021 0.1280.01 0.92 1.280.04 0.55 0.1480.01 0.79 0.8580.05 0.182 0.180.0 0.32 1.2880.05 0.99 0.1380.01 0.99 1.0080.08 0.41
Thalamus 0 0.1680.02 1.1580.04 0.280.01 0.9180.021 0.1680.01 0.96 1.180.05 0.71 0.1980.02 0.99 0.8480.04 0.222 0.1480.01 0.90 1.1280.03 0.88 0.1980.02 0.92 0.9980.12 0.45
Values of FA and ADC (!10–3 mm2/s) are interpolated to the standardized time points of the first study (30 weeks) and the second study (36 weeks). Brain injury groups are: control group (0), mild injury (1), severe injury (2).
* p < 0.05 on post-hoc comparison when there is significant difference between groups in ANOVA.
Table 2. Diffusion indices in ROIs at first and second DTI examination (means 8 standard error of means)

Drobyshevsky /Bregman /Storey /Meyer /Prasad /Derrick /MacKendrick /Tan
Dev Neurosci 2007;29:289–301296
Table 3. Diffusion indices in ROIs at first and second DTI examination (means 8 standard error of means)
ROI name Braininjurygroup
First MRI study Second MRI study
FA p ADC p FA p ADC p
Internal capsule, 0 0.3880.01 1.1580.04 0.4280.01 1.0180.02posterior limb 1 0.3780.01 0.92 1.1180.05 0.78 0.4380.01 0.88 0.9880.02 0.81
2 0.3780.03 1.00 1.1580.05 1.00 0.4080.03 0.63 1.0580.13 0.76Internal capsule, 0 0.2780.01 1.2580.04 0.2780.01 1.1480.03anterior limb 1 0.2880.01 0.71 1.2080.06 0.76 0.3080.01 0.27 1.1180.03 0.75
2 0.2880.01 0.36 1.2280.04 0.91 0.2680.01 0.82 1.1780.11 0.95Optic radiation 0 0.3180.01 1.4380.07 0.3380.01 1.2980.04
1 0.2980.02 0.51 1.3580.04 0.61 0.4080.04 0.08 1.3680.10 0.742 0.3080.01 0.99 1.4280.04 0.99 0.2980.03 0.61 1.3980.05 0.73
Corona radiata 0 0.2180.02 1.5180.05 0.2380.02 1.3380.061 0.2380.03 0.82 1.3480.05 0.16 0.2880.02 0.16 1.1180.04* 0.032 0.2480.03 0.61 1.6080.11 0.64 0.2380.05 0.64 1.5680.00* 0.05
Corpus callosum, 0 0.4380.01 1.2980.04 0.4580.01 1.2680.04genu 1 0.4780.01 0.15 1.1380.04 0.06 0.4280.02 0.54 1.2580.05 0.98
2 0.4780.02 0.12 1.2880.05 0.99 0.4980.03 0.34 1.2780.02 0.99Corpus callosum, 0 0.4580.01 1.3380.05 0.5080.02 1.2480.04splenum 1 0.4780.02 0.84 1.2580.07 0.50 0.4980.02 0.94 1.1880.03 0.49
2 0.4680.03 0.99 1.3180.05 0.95 0.5580.02 0.39 1.3180.00 0.61Frontal WM 0 0.1380.01 1.5580.05 0.1380.01 1.5080.05(level c. semiovale) 1 0.1380.01 0.98 1.4580.03 0.35 0.1480.01 0.66 1.3680.05 0.20
2 0.1380.01 0.94 1.4680.03 0.54 0.1680.02 0.31 1.4980.00 1.00Frontal WM 0 0.1380.00 1.5780.05 0.1480.01 1.5480.04(level ventricles) 1 0.1580.01 0.43 1.5080.05 0.56 0.1480.01 0.97 1.4180.03 0.14
2 0.1780.03 0.11 1.5580.04 0.98 0.1380.01 0.80 1.5980.10 0.87Central WM 0 0.1380.00 1.5380.05 0.1480.01 1.4480.02(level c. semiovale) 1 0.1280.01 0.64 1.4980.05 0.79 0.1580.01 0.94 1.3480.03* 0.05
2 0.1480.01 0.85 1.5580.04 0.98 0.1480.01 0.98 1.4880.03 0.66Parietal WM 0 0.1180.01 1.6680.06 0.1480.01 1.5680.04(level c. semiovale) 1 0.1180.01 0.93 1.6380.05 0.92 0.1580.01 0.67 1.3880.03* 0.02
2 0.1180.01 0.99 1.7080.04 0.92 0.1480.01 0.97 1.6180.06 0.84Occipital WM 0 0.1580.01 1.5780.05 0.1580.01 1.5080.04(level ventricles) 1 0.1380.01 0.64 1.5480.06 0.91 0.1580.01 0.99 1.3980.02 0.19
2 0.1280.01 0.38 1.6780.07 0.58 0.1380.02 0.54 1.6580.05 0.22Temporal WM 0 0.1580.01 1.4680.05 0.1780.01 1.3680.03(level ventricles) 1 0.1480.01 0.66 1.4380.05 0.91 0.1780.01 0.84 1.3380.03 0.78
2 0.1680.00 0.98 1.4880.07 0.97 0.1680.01 0.84 1.3380.00 0.87Frontal GM 0 0.2780.02 1.2480.04 0.2180.02 1.2880.03
1 0.2880.03 0.97 1.1780.03 0.50 0.2680.04 0.55 1.3080.05 0.962 0.3280.03 0.37 1.1580.07 0.38 0.2680.03 0.70 1.2580.08 0.91
Parietal GM 0 0.2580.02 1.1680.04 0.1880.02 1.1880.041 0.2780.03 0.84 1.1280.06 0.82 0.1980.02 0.85 1.1780.05 0.982 0.3280.02 0.16 1.1080.04 0.71 0.2180.01 0.76 1.1980.10 0.99
Perirolandic GM 0 0.1880.01 1.1580.04 0.1580.01 1.0680.021 0.1780.01 0.72 1.1480.06 1.00 0.1680.01 0.89 1.0580.02 0.942 0.1780.00 0.77 1.1680.07 0.99 0.1680.01 0.97 1.1680.07 0.17
Temporal GM 0 0.2080.01 1.2180.04 0.1780.02 1.1980.061 0.2280.03 0.76 1.1980.05 0.92 0.1680.01 1.00 1.1580.06 0.892 0.2480.03 0.48 1.1580.05 0.67 0.1680.03 0.95 1.1680.09 0.96
Caudate nucleus 0 0.1180.01 1.3080.05 0.1280.01 1.1880.031 0.1280.01 0.34 1.2580.08 0.84 0.1380.01 0.97 1.1580.02 0.782 0.1180.00 0.79 1.3780.09 0.82 0.1180.01 0.86 1.2180.10 0.95
Lentiform nucleus 0 0.1280.00 1.2580.04 0.1280.01 1.0980.031 0.1280.01 1.00 1.2480.07 0.99 0.1380.01 0.87 1.0880.01 0.972 0.1280.01 0.84 1.2380.07 0.97 0.1280.01 1.00 1.1580.10 0.59
Thalamus 0 0.1580.00 1.1380.04 0.1780.01 1.0380.021 0.1480.01 0.90 1.1180.06 0.94 0.1680.01 0.74 1.0080.02 0.752 0.1780.02 0.46 1.1280.07 0.99 0.1780.02 0.95 1.0980.14 0.56
Values FA and ADC (!10–3 mm2/s) are presented as average for the first and second MRI study without interpolation to the standardized time points as in table 2.
* p < 0.05 on post-hoc comparison when there is significant difference between groups in ANOVA.

Serial DTI in White Matter and Motor Outcome in Premature Infants
Dev Neurosci 2007;29:289–301 297
Table 4. Change in diffusion indices in ROIs between first and second DTI examinations normalized for 30 and 36 weeks (means 8 standard error of means)
ROI name Braininjurygroup
delta FA p delta ADC p delta FA/week p delta ADC/week p
White Internal capsule, 0 0.03780.011* 0.03 –0.12680.043* 0.02 0.00880.002* 0.01 –0.02680.006* 0.01matter posterior limb 1 0.06180.011* 0.02 –0.13580.066 0.11 0.01080.003* 0.03 –0.01880.007 0.06
2 0.04580.022 0.87 –0.04780.045 0.48 0.01180.005 0.26 –0.01380.012 0.49Internal capsule, 0 0.01480.018 1 –0.03280.055 0.58 0.00680.005 0.21 –0.00680.010 0.55anterior limb 1 –0.04380.021 0.35 0.11080.077 0.23 –0.00780.004 0.16 0.02080.017 0.29
2 –0.00880.007 1 –0.03480.041 0.56 –0.00280.002 0.45 –0.00980.011 0.55Optic radiation 0 0.02980.011 0.08 –0.06480.014 0.20 0.00680.002* 0.01 –0.02480.014 0.13
1 0.10680.049 0.30 –0.09380.018 0.78 0.01480.005 0.06 0.00280.008 0.812 0.00480.027 1 –0.02580.022 0.36 0.00180.007 0.94 –0.00480.002 0.34
Corona radiata 0 0.01780.021 1 –0.10880.048 0.08 0.00180.004 0.56 –0.02280.008 0.051 –0.00480.012 12 0.01080.030 1 –0.10080.064 0.36 0.00280.007 0.84 –0.02680.018 0.38
Corpus callosum, 0 0.04180.012* 0.03 –0.07680.040 0.09 0.00980.003* 0.01 –0.01380.013 0.33genu 1 0.02580.034 1 –0.08480.056 0.21 0.00680.006 0.42 –0.01280.008 0.21
2 0.05080.027 0.96 0.04480.083 0.69 0.01280.006 0.29 0.01380.022 0.67Corpus callosum, 0 0.02080.020 0.99 –0.16580.088 0.10 0.00480.004 0.37 –0.03380.020 0.14splenum 1 0.03780.032 0.91 –0.21380.075* 0.05 0.00680.004 0.27 –0.02980.008* 0.02
2 0.02380.014 1.07 –0.17880.127 0.39 0.00680.004 0.38 –0.04780.035 0.41Frontal WM 0 0.01680.015 0.95 –0.10580.051 0.07 0.00280.003 0.48 –0.01880.009 0.06(level c. semiovale) 1 0.00580.008 1 –0.12880.105 0.29 0.00180.001 0.59 –0.01780.013 0.27
2 0.00180.011 1 –0.07280.003* 0.22 0.00080.003 0.99 –0.01880.002 0.07Frontal WM 0 0.04880.016 0.05 –0.03380.045 0.48 –0.01080.004 0.06 0.01380.012 0.33(level ventricles) 1 0.03380.010 0.09 –0.01280.016 0.05 0.00580.002 0.05 –0.02080.004 0.07
2 –0.09280.057 1 0.00380.193 0.99 –0.02480.016 0.37 0.00380.048 0.97Central WM 0 0.01380.011 0.91 –0.01980.052 0.73 0.00580.005 0.29 0.00080.010 0.99(level c. semiovale) 1 –0.01980.015 0.81 –0.07880.089 0.43 –0.00280.002 0.44 –0.00980.012 0.50
2 –0.07380.070 1 –0.02880.146 0.88 –0.02080.019 0.49 –0.01080.037 0.84Parietal WM 0 0.00780.014 1 –0.08880.030* 0.02 0.00080.004 0.95 –0.01980.005* 0.01(level c. semiovale) 1 0.02280.018 0.85 –0.10680.089 0.30 0.00380.002 0.26 –0.01180.011 0.39
2 –0.04080.014 0.63 –0.01180.042 0.84 –0.01080.004 0.24 –0.00480.011 0.80Occipital WM 0 0.01880.014 0.68 –0.09480.068 0.20 0.00280.003 0.59 –0.01780.012 0.20(level ventricles) 1 0.02880.012 0.25 –0.11580.086 0.25 0.00480.002 0.06 –0.01180.012 0.42
2 0.00180.017 1 –0.15280.064 0.253 0.00080.004 0.99 –0.03780.013 0.22Temporal WM 0 0.03280.017 0.27 0.13580.084 0.15 0.01180.006 0.11 0.04980.030 0.14(level ventricles) 1 0.01880.013 0.70 –0.12280.051 0.078 0.00480.003 0.22 –0.02080.009 0.08
2 –0.00280.041 2.93 0.32780.161 0.29 –0.00180.010 0.93 0.08580.046 0.32
Gray Frontal GM 0 –0.02380.018 0.68 –0.06980.045 0.16 –0.00680.004 0.12 –0.01280.007 0.15matter 1 –0.01480.012 1.02 –0.12280.071 0.16 –0.00280.002 0.32 –0.01580.007 0.10
2 –0.00480.018 1 –0.01680.034 0.71 –0.00180.005 0.82 –0.00380.008 0.75Parietal GM 0 –0.02980.025 0.86 –0.02280.053 0.68 –0.00380.008 0.69 –0.00780.012 0.57
1 –0.06780.033 0.33 –0.05580.046 0.30 –0.00880.004 0.10 –0.00780.007 0.372 –0.06080.073 1 –0.05580.086 0.63 –0.01680.020 0.56 –0.01580.023 0.62
Perirolandic GM 0 0.02580.012 0.18 –0.08480.045* 0.01 0.00680.003 0.12 –0.01680.008 0.081 0.01580.016 1.23 –0.13280.063 0.10 0.00180.003 0.66 –0.01980.008 0.072 0.02280.045 2.11 –0.02780.043 0.64 0.00580.011 0.73 –0.00680.010 0.66
Temporal GM 0 0.02880.014 0.25 –0.07980.072 0.30 0.00680.004 0.13 –0.01480.017 0.441 0.04280.007* 0.02 –0.25480.079* 0.03 0.00680.001* 0.01 –0.03380.010* 0.032 0.04080.022 0.97 –0.10580.019 0.11 0.01080.005 0.30 –0.02780.007 0.15
Caudate nucleus 0 0.00880.010 1.28 –0.14780.035* 0.01 0.00180.002 0.61 –0.03280.005* 0.011 0.01280.012 1.14 –0.19180.088 0.09 0.00280.002 0.31 –0.02580.010 0.0712 0.00380.011 2.56 –0.03780.017 0.27 0.00080.003 0.90 –0.01080.005 0.30
Lentiform nucleus 0 –0.07680.028 0.08 0.02180.039 0.61 –0.01480.007 0.10 0.00680.012 0.641 –0.09380.035 0.17 0.02780.079 0.75 –0.01380.005* 0.05 0.00480.010 0.682 –0.08680.022 0.49 0.01380.108 0.92 –0.02180.004 0.12 0.00180.027 0.97
Thalamus 0 0.00180.016 2.80 –0.06180.066 0.39 –0.00480.004 0.34 –0.00380.014 0.821 0.02180.012 0.45 –0.17380.061* 0.05 0.00380.002 0.21 –0.02280.008* 0.042 0.00080.040 2.99 0.01480.056 0.84 –0.00180.010 0.96 0.00580.014 0.80
Changes in FA and ADC (!10–3 mm2/s) are presented as an absolute pairwise difference between the first and the second MRI study (not normal-ized) and as a rate of change per week.
* p < 0.05 on post-hoc comparison when there is significant difference between the first and the second study, paired t test.

Drobyshevsky /Bregman /Storey /Meyer /Prasad /Derrick /MacKendrick /Tan
Dev Neurosci 2007;29:289–301298
ed white and gray matter ROIs. No significant differenc-es were found between groups in the rate of change of the diffusion indices.
The absolute differences in diffusion indices between studies and rate of changes are presented for each ROI in table 4 . FA significantly increased between studies in the posterior limb of internal capsule, optic radiation, corpus callosum genu, parietal white matter in controls, and in the posterior limb of internal capsule, temporal gray mat-ter in the mild injury group. ADC significantly decreased between studies in the posterior limb of internal capsule, parietal white matter, caudate nucleus in controls; in the posterior limb of internal capsule, corpus callosum sple-num, temporal GM an thalamus in the mild injury group, and frontal white matter in the severe injury group. The differences in diffusion indices in the other ROIs were not significant.
Analysis of Developmental Data All 12 subjects of the follow-up study had normal hear-
ing and vision, documented by ABR screening prior to ISCU discharge, and parental report of ophthalmologic evaluations after discharge. Five out of 8 control infants and 1 infant with IVH grade 3 had hypotonia on exami-nation. Two infants, one with IVH grade 1 (diagnosed with PVL on the second MRI) and one with IVH grade 3, were diagnosed with cerebral palsy on examination. The second infant with IVH grade 1 had a normal follow-up ultrasound examination. FA of posterior limb of in-ternal capsule on the first MRI study in infants who de-veloped hypotonia was not different (0.36 8 0.01) than infants with normal tone (0.37 8 0.00), and there was a trend toward a higher FA on the second MRI (0.42 8 0.01 vs. 0.39 8 0.01, respectively). We could not do a statisti-cal comparison due to the small number of subjects.
There was a significant correlation between FA in the posterior limb of the internal capsule on the scan per-formed at 30 weeks and the Performance Developmental Index (PDI) at 24 months’ corrected age ( fig. 6 a, r = 0.55, p ! 0.05). FA on the second study at 36 weeks did not show a significant correlation with PDI ( fig. 6 b). How-ever, a low PDI was significantly correlated with a higher
r = 0.18, NS
r = 0.63, p < 0.02
ControlsMild injurySevere injury
CP
CP
FA0.25 0.30 0.35 0.40 0.45 0.50
40
50
60
70
80
90
100
110
a
b
c
Bayl
ey’s
PD
I
400 2 4 6 8 10
Delta FA per week (× 10–3)
12 14 16 18
50
60
70
80
90
100
110
Bayl
ey’s
PD
I40
FA0.25
CP CPr = 0.55, p < 0.05
0.30 0.35 0.40 0.45 0.50
50
60
70
80
90
100
110
Bayl
ey’s
PD
I
Fig. 6. Developmental testing was performed between 18 and 24 months’ corrected age with the BSID-II. a Significant correlation was found between a low psychomotor scale index (PDI) at 24 months and low FA in the posterior limb of the internal capsule at 30 weeks. b By the time of the second MRI study at term (36th week), correlation between FA in internal capsule and PDA was not significant. c Significant negative correlation was observed in internal capsule between PDA and absolute change of FA per week between studies. Patients were suspected to have cerebral palsy (CP) by the developmental psychologist and neonatologist, and the diagnoses were confirmed by a neurologist or a rehabilitation specialist elsewhere. A systematic neurological examination was not provided to the subjects of the study.

Serial DTI in White Matter and Motor Outcome in Premature Infants
Dev Neurosci 2007;29:289–301 299
change of FA per week between MRI studies in the inter-nal capsule ( fig. 6 c, r = –0.63, p ! 0.02), and occipital white matter (r = –0.59, p ! 0.05). The relationship re-mained significant even after removal of the data point of the patient with CP. The correlation between Bayley PDI and absolute delta FA between MRI studies was sig-nificant by Spearman’s rho (p ! 0.05).
There was no correlation between FA and Bayley Men-tal Developmental Index at either age.
Discussion
This study shows that two serial MRI scans performed in the postnatal period are useful for predicting psycho-motor developmental disabilities at 2 years of age. The first MRI scan in the first 10–14 days showed that low FA in the internal capsule was correlated with lower PDI scores. The implication is that evidence of WMI in pro-jection fibers was present in the latter part of the 2nd week. It is possible that the actual onset of insult may have been earlier, either in the immediate perinatal or antena-tal periods.
The study also showed that an increased rate of FA increase between the two serial scans significantly cor-related with a lower 24-month PDI. This surprising re-sult could possibly be explained by the study population. Patients who had serial scans consisted mostly of ‘healthy’ premature infants as the PDI was 1 70 in 80% of this cohort. One explanation is that WMI occurred at an early time point in life, resulting in decreased FA on the first scan, but this population, being relatively ‘healthy’, recovered to ‘normal’ levels by the second scan. However, our findings suggest that this recovery process, while accounting for a higher rate of change of FA, did not obviate the neurodevelopmental impact of the initial WMI. Interestingly, one of the subjects who developed cerebral palsy showed the maximum FA change between the two scans. However, a closer exam-ination reveals that the first FA was very low and the second FA was still among the lowest in the population. This inverse correlation of delta FA and PDI will need to be corroborated by larger multicentric studies. Our study suggests that early WMI can be detected in pre-mature infants even without obvious signs of injury. An early MRI study may not always be feasible due to the instability of premature infants but is of higher diagnos-tic value than one at discharge. Two serial scans even if done beyond the unstable period may have an even stronger predictive value.
Using diffuse hyperintensities on T 2 -weighted scans as a criteria, studies have reported the rate of WMI as high as 80% of infants [Dyet et al., 2006]. Using a differ-ent criteria of focal hyperintensities on T 1 -weighted im-ages with absence of corresponding hypointensities on T 2 -weighted images, other investigators have reported as much as 56% incidence of WMI [Miller et al., 2003]. This finding was elegantly shown to be a result of gliosis and not hemorrhage [Schouman-Claeys et al., 1993]. Al-though not a primary endpoint variable, there were not many changes suggestive of WMI determined by con-ventional T 1 - and T 2 - weighted images as reported previ-ously [Counsell et al., 2003; Miller et al., 2005; Dyet et al., 2006]. We did not identify either pattern of WMI in our data. We can only speculate as to the cause of such discrepancies in the studies on different patient popula-tions. (1) It is possible that there was some sampling bias but this is not probable because the enrollment criteria in our study were similar to previous studies. (2) We used 2-D spin echo sequence and did not use high-resolution gradient echo 3-D SPGR sequence for T 1 -weighted imag-ing [Miller et al., 2005], which may improve the sensitiv-ity of MRI and consequently detection of very small le-sions such as those less than 2 mm in size. (3) There has been no consensus on the exact definition of WMI be-cause different criteria of diffuse hyperintensities on T 2 -weighted scans [Dyet et al., 2006], or focal hemorrhages [Maalouf et al., 2001; Dyet et al., 2006] or focal hyperin-tensities on T 1 -weighted images attributed to gliosis have been used by various investigators. Assessment of T 2 -weighted and FLAIR but not T 1 -weighted images is rou-tinely performed at our institution. No diffuse hyperin-tensities were observed on T 2 -weighted images. On T 1 -weighted images, only 14% showed focal hemorrhages but no focal lesions suggestive of gliosis. (4) A probable reason is the difference in the neonatal populations stud-ied, specifically in the rate of inflammatory processes between our neonatal population and the national aver-age, as gliosis often is associated with an inflammatory process. The nosocomial infection rate in our very low birthweight population was 11% for 2003–2005, approx-imately half that reported in other series [Stoll et al., 2002; Vermont Oxford Network, 2005]. Over the same time period, our rate of chronic lung disease was 16%, again significantly less than that reported elsewhere [Eh-renkranz et al., 2005; Vermont Oxford Network, 2005]. It is possible then that inflammatory triggers for brain inflammation were lower than the experience for similar populations at other centers, resulting in an absence of finding of ‘gliosis’ on MRI.

Drobyshevsky /Bregman /Storey /Meyer /Prasad /Derrick /MacKendrick /Tan
Dev Neurosci 2007;29:289–301300
References
Basser PJ, Mattiello J, et al. (1994): Estimation of the effective self-diffusion tensor from the NMR spin echo. J Magn Reson B 103: 247–254.
Bayley N (1993): Bayley Scales of Infant Develop-ment. San Antonio, Psychological Corpora-tion.
Berman JI, Mukherjee P, et al. (2005): Quantita-tive diffusion tensor MRI fiber tractography of sensorimotor white matter development in premature infants. Neuroimage 27: 862–871.
Counsell SJ, Allsop JM, et al. (2003): Diffusion-weighted imaging of the brain in preterm in-fants with focal and diffuse white matter ab-normality. Pediatrics 112: 1–7.
Dyet LE, Kennea N, et al. (2006): Natural history of brain lesions in extremely preterm infants studied with serial magnetic resonance im-aging from birth and neurodevelopmental assessment. Pediatrics 118: 536–548.
Ehrenkranz RA, Walsh MC, et al. (2005): Vali-dation of the National Institutes of Health consensus definition of bronchopulmonary dysplasia. Pediatrics 116: 1353–1360.
Huppi PS, Inder TE (2001): Magnetic resonance techniques in the evaluation of the perinatal brain: recent advances and future directions. Semin Neonatol 6: 195–210.
Huppi PS, Maier SE, et al. (1998): Microstruc-tural development of human newborn cere-bral white matter assessed in vivo by diffu-sion tensor magnetic resonance imaging. Pediatr Res 44: 584–590.
Inder TE, Volpe JJ (2000): Mechanisms of peri-natal brain injury. Semin Neonatol 5: 3–16.
Inder TE, Wells SJ, et al. (2003): Defining the na-ture of the cerebral abnormalities in the pre-mature infant: a qualitative magnetic reso-nance imaging study. J Pediatr 143: 171–179.
Maalouf EF, Duggan PJ, et al. (2001): Compari-son of findings on cranial ultrasound and magnetic resonance imaging in preterm in-fants. Pediatrics 107: 719–727.
Miller SP, Cozzio CC, et al. (2003): Comparing the diagnosis of white matter injury in pre-mature newborns with serial MR imaging and transfontanel ultrasonography findings. AJNR Am J Neuroradiol 24: 1661–1669.
Miller SP, Ferriero DM, et al. (2005): Early brain injury in premature newborns detected with magnetic resonance imaging is associated with adverse early neurodevelopmental out-come. J Pediatr 147: 609–616.
Miller SP, Vigneron DB, et al. (2002): Serial quantitative diffusion tensor MRI of the pre-mature brain: development in newborns with and without injury. J Magn Reson Im-aging 16: 621–632.
Mukherjee P, Miller JH, et al. (2001): Normal brain maturation during childhood: devel-opmental trends characterized with diffu-sion-tensor MR imaging. Radiology 221:
349–358.
There were several limitations of the study, similar to those performed on neonatal and infant subjects: rela-tively large slice thickness, ROI may include other cere-bral tissue types due to partial volume effect, small num-ber of infants enrolled, inability to do subgroup analysis for twins, low follow-up rate, correction model for differ-ing GA at scan time.
We could not form any conclusions about higher grades of IVH being a substantial risk factor for WMI as shown previously [Miller et al., 2005]. As might be an-ticipated, there was no significant difference between the controls and the infants with grade IVH ̂ 2 in the mild brain injury group. Grade 1 or 2 IVH is a frequent com-plication in the premature infants, but the neurodevelop-mental prognosis for this group of patients is generally good [Volpe, 2001]. We also did not find any difference for FA in the posterior limb of the internal capsule in the group of infants with severe brain injury, although the infant with PVL did have lower values of FA. Internal capsule abnormalities have been reported in numerous studies in premature infants with WMI [Huppi et al., 1998; Inder and Volpe, 2000].
The absence of significant differences with the higher IVH group can be explained by a type II error due to such a small number of infants in our study with severe brain injury. This study corroborated the normal maturational pattern in the postnatal period for the major diffusion indices as shown by others [Mukherjee et al., 2001; Neil
et al., 2002; Berman et al., 2005], i.e. a decrease with age for ADC in white and gray matter, and an increase for FA in white matter and decrease for FA in the gray matter. The fastest rate of change for FA was observed in the fron-tal white matter region, although the previously reported fastest region, the corticospinal tracts [Partridge et al., 2004], was a close second. Among infants with grade 6 2 IVH, we also found significant differences in diffusion indices, suggesting WMI, in white matter regions and tracts adjacent or close to the ventricles, such as occipital white matter and optic radiation.
In summary, MRI can be an important tool to iden-tify early, subtle WMI, which could be used to prognos-ticate outcome and design earlier, more appropriate ther-apies for identified infants. Though a larger definitive study would be desirable before drawing conclusions, our study suggests that two MRI scans are probably better than one. If only one scan is done, an early MRI scan may be more useful than one done at term.
Acknowledgements
This study was supported by NIH NS 043285 and NS 41476 (S.T.) grants. The authors wish to thank Ilene Susan Wolf, clinical research coordinator, for her help throughout the study and Nora Manago, infant development specialist, for her help in the devel-opmental assessment.

Serial DTI in White Matter and Motor Outcome in Premature Infants
Dev Neurosci 2007;29:289–301 301
Neil J, Miller J, et al. (2002): Diffusion tensor im-aging of normal and injured developing hu-man brain – a technical review. NMR Bio-med 15: 543–552.
Neil J, Shiran S, et al. (1998): Normal brain in human newborns: apparent diffusion coef-ficient and diffusion anisotropy measured by using diffusion tensor MR imaging. Radi-ology 209: 57–66.
Partridge SC, Mukherjee P, et al. (2004): Diffu-sion tensor imaging: serial quantitation of white matter tract maturity in premature newborns. Neuroimage 22: 1302–1314.
Roelants-van Rijn AM, Groenendaal F, et al. (2001): Parenchymal brain injury in the pre-term infant: comparison of cranial ultra-sound, MRI and neurodevelopmental out-come. Neuropediatrics 32: 80–89.
Schouman-Claeys EM, Henry-Feugeas C, et al. (1993): Periventricular leukomalacia: corre-lation between MR imaging and autopsy findings during the first 2 months of life. Ra-diology 189: 59–64.
Stoll BJ, Hansen N, et al. (2002): Late-onset sep-sis in very low birth weight neonates: the ex-perience of the NICHD Neonatal Research Network. Pediatrics 110: 285–291.
Vermont Oxford Network (2005): Database Summary. Burlington, Vermont Oxford Network.
Volpe JJ (2001): Neurology of the Newborn. Phil-adelphia, W.B. Saunders.
Wilson-Costello D, Friedman H, et al. (2005): Improved survival rates with increased neu-rodevelopmental disability for extremely low birth weight infants in the 1990s. Pedi-atrics 115: 997–1003.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:302–310 DOI: 10.1159/000105471
Delayed IGF-1 Administration RescuesOligodendrocyte Progenitorsfrom Glutamate-Induced Cell Death andHypoxic-Ischemic Brain Damage
Teresa L. Wood
a, b Vaho Loladze
b Stefanie Altieri
a Nitish Gangoli
a
Steven W. Levison
a Katarina G. Brywe
c, d Carina Mallard
c Henrik Hagberg
c, d
a Department of Neurology and Neuroscience, New Jersey Medical School, University of Medicine and Dentistry
New Jersey, Newark, N.J. , b Department of Neural and Behavioral Sciences, Penn State College of Medicine,
Hershey, Pa. , USA; Departments of c Neuroscience and Physiology, and d
Obstetrics and Gynecology,Sahlgrenska Academy, Gothenburg , Sweden
Introduction
White matter damage occurs after ischemia in both the adult and the immature brain. However, in contrast to adult stroke where the predominant pathology is neu-ronal cell death, many of the neurological problems of the infant subsequent to hypoxia-ischemia (HI) are attrib-uted to white matter damage [Volpe, 2001a, b]. White matter damage in perinatal HI leads to periventricular leukomalacia, which is the neuropathology classically as-sociated with brain injuries in the premature infant [Vol-pe, 2001a]. As in the adult, glutamate is elevated in the immature rat brain including in the white matter after HI and is thought to have a primary role in the subsequent damage [Benveniste et al., 1984; Hagberg et al., 1987; An-dine et al., 1991; Silverstein et al., 1991]. Similarly, gluta-mate is elevated in the cerebral spinal fluid in term in-fants after perinatal HI [Hagberg et al., 1993]. Recently, Back et al. [2007] demonstrated that HI causes release of glutamate preferentially from axons and oligodendroglia in the perinatal brain. Multiple lines of evidence support the hypothesis that the AMPA/kainate GluR receptors are primarily responsible for glutamate-mediated death of oligodendrocyte progenitors (OPs) and immature oli-godendrocytes both in vitro [Yoshioka et al., 1995; Mat-
Key Words
Insulin-like growth factor � Trophic factors � Periventricular leukomalacia � White matter damage � Excitotoxicity � Apoptosis � Myelin
Abstract
We previously demonstrated that IGF-1 blocks glutamate-mediated death of late oligodendrocyte progenitors (OPs) by preventing Bax translocation, mitochondrial cytochrome c release and cleavage of caspases 9 and 3. Here, we demon-strate that IGF-1 prevents caspase 3 activation in late OPs when administered up to 16 h following exposure to gluta-mate. Moreover, late addition of IGF-1 to OPs previouslyexposed to toxic levels of glutamate promotes oligoden-drocyte maturation as measured by myelin basic protein ex-pression. We also demonstrate that intraventricularly admin-istered IGF-1 retains OPs in the perinatal white matter after hypoxia-ischemia when given after insult. These results sug-gest that delayed administration of IGF-1 will rescue OPs in the immature white matter and promote myelination fol-lowing hypoxia-ischemia. Copyright © 2007 S. Karger AG, Basel
Received: January 6, 2007 Accepted after revision: March 28, 2007
Teresa L. Wood, PhD Department of Neurology and Neuroscience, New Jersey Medical School/UMDNJ UH-Cancer Center H1200, 205 South Orange Avenue Newark, NJ 07103 (USA) Tel. +1 973 972 6529, Fax +1 973 972 0008, E-Mail [email protected]
© 2007 S. Karger AG, Basel
Accessible online at:www.karger.com/dne

IGF-1 Protection of Oligodendroglia Dev Neurosci 2007;29:302–310 303
ute et al., 1997; Sanchez-Gomez and Matute 1999; Alber-di et al., 2002] and in vivo [Follett et al., 2000].
The differential vulnerability of oligodendroglia as they progressively differentiate has led to the view that the immature progenitors are significantly more vulner-able than mature oligodendrocytes. In particular, the late progenitor (late OP) is the most sensitive developmental stage to kainate- and glutamate-mediated death [Mc-Donald et al., 1998b; Fern and Moller, 2000] and to oxi-dative stress induced by glutathione depletion [Back et al., 1998]. Follett et al. [2000] reported a similar age-depen-dent vulnerability to white matter lesions after intracere-bral injections of AMPA into the pericallosal white mat-ter. These data have led to the proposal that the late OP is intrinsically vulnerable to hypoxic/ischemic insult [Vol-pe, 1997]. Indeed, the age of highest incidence of white matter damage leading to periventricular leukomalacia in the premature infant directly correlates with the pre-dominance of late OPs in the immature brain [Back et al., 2001; Riddle et al., 2006].
In contrast to the classic NMDA-mediated excitotoxic death of neurons that occurs within a few hours, death of oligodendroglia after HI in the perinatal brain occurs over 24–48 h [Follett et al., 2000; Ness et al., 2001]. This sug-gests that the initial elevation of glutamate initiates a slow death cascade within the glial cells. Until recently, little was known about the mechanisms by which perinatal HI and glutamate promote death of oligodendroglia or of the ability of trophic factors to prevent this death. Previous in vitro studies suggested that maximal survival of OPs un-der normal culture conditions could be achieved using a combination of the IGF, neurotrophin and interleukin-6 (IL-6)-like families [Barres et al., 1993]. Several of these factors have been further tested for their ability to protect oligodendroglia against damaging or toxic agents. IGF-1 and CNTF (a member of the IL-6 type cytokines) protect immature oligodendrocytes from death induced by tumor necrosis factor- � [Louis et al., 1993; D’Souza et al., 1996; Ye and D’Ercole, 1999]. Prior to our studies, there was one report suggesting that NT-3 partially inhibited glutamate excitotoxicity of oligodendroglia [Kavanaugh et al., 2000]. Our studies demonstrated that IGF-1 and NT-3, but not CNTF, protect late OP cells from high levels of glutamate through 24 h in vitro [Ness and Wood, 2002]. However, only IGF-1 protects the late OP cells from glutamate toxic-ity longer than 24 h [Ness and Wood, 2002]. More recent-ly, we determined that IGF-1 blocks glutamate-mediated death in the late OPs by preventing Bax translocation, mi-tochondrial cytochrome c release and cleavage of caspases 9 and 3 [Ness et al., 2004]. However, IGF-1 has no effect on
initial calcium influx into the late OPs after exposure to glutamate nor does it promote recovery of intracellular cal-cium levels [Ness et al., 2004] as has been reported for IGF-1 trophic actions in neurons [Cheng and Mattson, 1991, 1992, 1994; Cheng et al., 1993; Mattson and Cheng, 1993; Mattson et al., 1993].
There are limited data concerning the ability of IGF-1 to prevent white matter damage and OP death in vivo fol-lowing HI in the immature brain. Recently, Lin et al. [2005] demonstrated that IGF-1 given prior to the insult partially rescues OPs and myelin basic protein (MBP) expression following bilateral HI in the perinatal rat brain. However, there is no information as to the effectiveness of IGF-1 to rescue white matter OPs if given after the insult. The goal of the studies presented here was to further understand the timing and mechanisms for IGF-1 protection and rescue of the late OPs from insult both in vitro and in vivo.
Materials and Methods
Minimal essential media (MEM), fetal bovine serum (FBS), and trypsin were purchased from Gibco/Invitrogen (Carlsbad, Ca-lif., USA). Cell culture supplements and glutamate were purchased from Sigma Chemical Company (St. Louis, Mo., USA). Recombi-nant rat IGF-1 was purchased from Upstate Biochemicals (Lake Placid, N.Y., USA). Recombinant human fibroblast growth factor-2 (FGF-2) was purchased from R&D Systems (Minneapolis, Minn., USA). Rabbit polyclonal antibodies to activated caspase 3 and Olig 2 were obtained from Cell Signaling Technologies (No. 9661; Dan-vers, Mass., USA) and Chemicon International (Te mecula, Calif., USA), respectively. Antibodies to MBP and to � -actin were ob-tained from Chemicon International (rat monoclonal to MBP a.a. 82–87) and from Sigma-Aldrich Corp. (mouse monoclonal; St. Louis, Mo., USA), respectively. Goat anti-rabbit and mouse horse-radish peroxidase-conjugated secondary antibodies were pur-chased from Jackson Laboratories (West Grove, Pa., USA).
Cell Culture and Treatment Conditions Newborn Sprague Dawley rat forebrain cortices were enzy-
matically digested with trypsin and DNase I and then mechani-cally dissociated and plated in MEM containing 10% FBS with antibiotics as previously described [Levison and McCarthy, 1991]. The mixed glial cells were grown in T75 flasks until they were confluent (10–14 days). Microglia were separated from the cul-tures by shaking the flasks on a rotary shaker for 1.5 h at 260 rpm. OP cells were isolated following an additional 18-hour shake as previously described [McCarthy and de Vellis, 1980]. OP cells were seeded into poly- D -lysine-coated T75 flasks at a density of 1.5 ! 10 4 /cm 2 in a chemically defined medium, N2S, composed of: (1) 66% N2B2 media [DMEM:F12 (Gibco, Grand Island, N.Y., USA; containing 15 m M HEPES, 2 m M glutamine) supplemented with 0.66 mg/ml BSA, 10 ng/ml d-biotin, 5 � g/ml insulin, 20 n M progesterone, 100 � M putrescine, 5 ng/ml selenium, 50 � g/ml apo-transferrin, 100 U/ml penicillin, and 100 � g/ml streptomy-cin], (2) 34% B104-conditioned medium (N2B2 preconditioned

Wood /Loladze /Altieri /Gangoli /Levison /Brywe /Mallard /Hagberg
Dev Neurosci 2007;29:302–310304
by B104 neuroblastoma cells), (3) 5 ng/ml FGF-2, and (4) 0.5% FBS. OP cultures were amplified for 4–10 days and passaged once using papain [Young and Levison, 1997] prior to performing ex-periments. The lineage stages of oligodendrocyte differentiation have been extensively characterized in vitro using stage-specific monoclonal antibodies as markers [Ranscht et al., 1982; Bansal et al., 1989; Bansal and Pfeiffer, 1992, 1997; McMorris and McKin-non, 1996; Orentas and Miller, 1999; Baumann and Pham-Dinh, 2001]. The early OP is bipolar, mitotic and can be identified by expression of glanglioside antigens (detected by immunostaining with either A2B5 or R24 antibodies). Using these isolation proce-dures, we consistently obtain greater than 95% purity of early OP cells (A2B5+/O4–) as previously reported [Jiang et al., 2001].
As the early OP begins to differentiate, it progresses through an intermediate progenitor stage that is still mitotic but multipolar. Late OPs can be identified by the surface antigen POA (detected by immunostaining with the O4 antibody). Under differentiation conditions, the late OP differentiates into a postmitotic immature oligodendrocyte that can be identified by immu nostaining for ga-lactocerebroside (GalC, detected by the O1 or Ranscht antibodies). Ultimately, the immature oligodendrocyte will further mature into a fully differentiated oligodendrocyte characterized by pro-duction of myelin proteins including MBP and proteolipid protein. To obtain highly enriched cultures of late OPs for experiments, early OP cells were replated at a density of 4.0 ! 10 4 cells/cm 2 onto poly- D -lysine-coated 60-mm dishes in N2B2 with 0.5% FBS and 10 ng/ml FGF-2 for 48 h. Under these conditions, we obtain cul-tures that contain 90–95% O4+ late OPs, 6–8% R24+/O4– OPs, 1–2% Ranscht+ immature OLs, ! 2% GFAP+ astrocytes and ! 0.01% OX42+ microglia [Ness and Wood, 2002]. For glutamate and tro-phic factor treatments, a hormone-supplemented medium was used that was identical to the N2B2 medium with the absence of insulin. Treatment media did not contain FGF-2. Cells were treat-ed for various time points with 500 � M glutamate and/or IGF-1 (10 ng/ml), and media were replaced every 18 h.
Western Blotting Following treatments, OP cells were washed in ice-cold PBS
and total cell lysates were isolated in SDS sample buffer (62.5 m M Tris-HCl, 2% SDS, 10% glycerol, 50 m M DTT, 1/100 protease in-hibitor cocktail (Sigma), 1 m M Na 3 VO 4 , and 1 m M NaF) and son-icated for 10–15 s on ice. Protein assays were performed to deter-mine protein concentrations (BioRad, Hercules, Calif., USA). 15–25 � g of protein was heated to 100 ° C for 5 min, cooled, and separated on 10, 12 or 3–8% mini-SDS polyacrylamide gels (Invi-trogen). Proteins were then electrotransferred to nitrocellulose membranes. Membrane blocking and primary and secondary an-tibody incubations were done in 5% milk (or 5% BSA for some antibodies) in TBS 0.05% (or 0.1%) Tween-20. Protein was visual-ized using an enhanced chemiluminescence system (New Eng-land Nuclear, Boston, Mass., USA) following incubation with ap-propriate horseradish peroxidase-conjugated secondary antibod-ies. Images were digitized and quantified using NIH Image 1.62.
HI Model and Olig2 Immunocytochemistry The left common carotid artery of PND7 outbred Wistar rats
was ligated under anesthesia. Following recovery, the pups were exposed to 60 min of hypoxia in a humidified chamber at 36 ° C with 7.77 8 0.01% oxygen in nitrogen. Immediately after HI, pups were treated with human recombinant IGF-1 (50 � g; Genentech,
Inc.) or vehicle (NaCl 0.9 mg/ml) i.c.v. and then this dose was re-peated once daily for 2 days as described previously [Brywe et al., 2005]. The dose of IGF-1 was chosen based on our previous stud-ies in 7-day-old rats where this dose showed neuroprotection [Brywe et al., 2005] and on similar studies in adult rats [Guan, 1993] where this dose showed the most significant neuroprotec-tion without affecting systemic glucose concentrations or cortical temperature. The injection procedure was well tolerated by the pups and there was no mortality. Animals were perfused at PND10 and the brains removed from the skull and immersion-fixed at 4 ° C for 24 h, dehydrated and embedded in paraffin.
Coronal brain sections (5 � m) were cut from paraffin blocks, deparaffinized and dehydrated through a graded series of xylenes and ethanol. For analysis of Olig2 expression, sections were ana-lyzed at Bregma –0.3 to –0.9 from all brains. After incubation in proteinase K (10 � g/ml; Sigma) in 10 m M Tris, pH 8.0 for 15 min, antigen retrieval was performed in 10 m M sodium citrate buffer using the pressure cooker method. Sections were incubated with a rabbit polyclonal antibody to Olig2 (1: 100) overnight at 4 ° C fol-lowed by incubation with a goat anti-rabbit IgG alkaline phospha-tase (1: 250; Southern Biotech, Birmingham, Ala., USA). Immu-noreactivity was detected with VectorRed Alkaline Phosphatase Substrate Kit I (Vector Laboratories, Burlingame, Calif., USA) so-lution for 30 min at room temperature in the dark. Sections were incubated with 4 � ,6 � -diamidino-2-phenylindole (1: 2,500; Sigma) for 4 min at room temperature in the dark to detect nuclei. Sec-tions were then dehydrated, and mounted in CytosealXYL (Rich-ard-Allan Scientific, Kalamazoo, Mich., USA).
Quantification and Statistical Analyses Photomicrographic images were captured by an individual
blinded to the experimental conditions using a Sensys camera mounted on an Olympus Provis AX70 microscope at 40 ! mag-nification. DAPI and rhodamine images were captured on each of three, adjacent fields along the white matter tracts (lateral to the subventricular zone) on both left and right hemispheres of sec-tions from IGF-1- (n = 6) or saline-treated (n = 6) animals. Cell counts were performed using a grid overlay. Differences between treatment groups were determined by Student’s t test.
Results
Transient Exposure to Glutamate Results in Delayed Death of Late OPs Our previous studies demonstrated that exposing late
OPs to glutamate in the absence of either IGF-1 or insulin induced cell death beginning at 22–24 h [Ness and Wood, 2002; Ness et al., 2004]. Glutamate-mediated death of the late OPs is initiated by calcium influx through AMPA/kainate receptors; however, Bax translocation to the mi-tochondria was not evident until at least 20 h following glutamate treatment [Ness et al., 2004]. It was not clear from these studies whether continuous exposure to glu-tamate is required to kill the late OPs. Since we do not use a glutamate receptor desensitizing blocker, we predicted

IGF-1 Protection of Oligodendroglia Dev Neurosci 2007;29:302–310 305
that continuous glutamate exposure is not required for death of the cells. In order to determine whether a tran-sient exposure to glutamate is sufficient to kill the late OPs, we tested whether ̂ 1 h exposure to glutamate is sufficient to induce apoptosis at 24 h. In these experi-ments, we used cultures enriched for the O4+/GalC– stage of the late OP. The late OPs were exposed to 500 � M glu-tamate for 1 or 24 h and then the media were replaced with glutamate-free media. Levels of active caspase 3 were determined by Western immunoblotting at 24 and 48 h ( fig. 1 ). Confirming our hypothesis, 1-hour exposure to glutamate was sufficient to activate caspase 3 in the late OPs, and this short exposure was as effective as continu-ous exposure to glutamate for 24 h. Adding IGF-1 to the
medium immediately after stimulating the cells with glu-tamate almost completely attenuated caspase 3 activation at both 24 h and 48 h of recovery ( fig. 1 b). Exposure to glutamate for 15 min similarly activated caspase 3 at 24 h ( fig. 1 a). These data suggest that a transient exposure to glutamate causes the delayed death of OPs in the absence of a receptor densensitizing agent and of IGF-1R signal-ing (due to either IGF-1 or micromolar levels of insulin). These results suggest that the initial calcium influx from glutamate exposure initiates a cascade of intracellular events leading to cell death.
Late Addition of IGF-1 Can PreventGlutamate-Mediated OP Death The delayed death of the OPs following exposure to
glutamate suggests the possibility of interfering with the death cascade during the 24-hour time period prior to caspase activation. To determine whether delayed addi-tion of IGF-1 could prevent glutamate-mediated apopto-sis of the late OPs, we performed experiments where the late OPs were exposed to glutamate for 12 or 16 h prior to addition of IGF-1 ( fig. 2 ). After this initial period of glu-tamate exposure, cells were maintained in medium con-taining either IGF-1 alone or glutamate plus IGF-1. Strik-ingly, adding IGF-1 almost completely attenuated the cleavage of caspase 3 at both 12 and 16 h after glutamate exposure ( fig. 2 ).
Late Addition of IGF-1 after Glutamate Exposure Promotes Oligodendrocyte Differentiation While inhibiting caspase 3 activation correlates well
with cell survival in the short term, there are noncaspase-mediated cell death cascades that might still be initiated by glutamate that would compromise the survival and differentiation of OP cells. To determine whether IGF-1 can completely enhance OP cell viability, we tested wheth-er exposure to glutamate followed by recovery in medium that would stimulate the IGF-1R would allow the late OPs to survive and then to differentiate. Late OP cells were exposed to 18 h of glutamate in the absence of IGF-1 or insulin and then transferred to medium containing glu-tamate and/or IGF-1. After 12 h, cells were transferred to differentiation media for 3 days (that contained supra-physiological levels of insulin and thyroid hormone). Dif-ferentiation into immature oligodendrocytes was subse-quently determined using morphological criteria and the expression of MBP. Cells exposed to glutamate and de-prived of IGF-1R stimulation had weak expression of MBP compared to control cells, whereas cells exposed to IGF-1 18 h after exposure to glutamate expressed levels of
15 minGlu
Casp-3
�-Actin
a
-----Glu
-----I
1 hGlu
24 hG/I
0b IGF Glu 1 h Glu 1 h Glu + IGF
Cle
aved
cas
pas
e 3
10
20
30
40
50
60
70
***
**
**24 h48 h
Fig. 1. Transient ( ̂ 1-hour) exposure to glutamate is sufficient to activate caspase 3. Primary cultures of late OPs were exposed to IGF-1 (10 ng/ml), or glutamate (500 � M ) for 24 h (Glu) or to glu-tamate for 15 min or 1 h only and then switched either to control media (1 h Glu) or to media containing IGF-1 (1 h Glu + IGF) for an additional 23 h. Levels of cleaved caspase 3 were analyzed by Western immunoblotting at 24 and 48 h. a Representative West-ern immunoblot showing cleaved caspase 3 and � -actin on the same blot. b Band intensities were quantified using NIH image and normalized to levels of � -actin. * * p ! 0.001 vs. IGF-1 or 1 h Glu + IGF-1; * p ! 0.04 vs. IGF-1 or 1 h Glu + IGF-1.

Wood /Loladze /Altieri /Gangoli /Levison /Brywe /Mallard /Hagberg
Dev Neurosci 2007;29:302–310306
MBP that were higher than the glutamate only-stimu-lated group, and which were almost equivalent to cells in complete medium that were not exposed to glutamate ( fig. 3 ). Analysis of MBP levels from duplicates of the 30-hour experiment shown in figure 3 showed that MBP lev-els in the glutamate only treatment were 50–60% of those in the group treated for 18 h with glutamate and then switched to IGF-1-containing media for 12 h prior todifferentiation. An additional experiment performed through 36 h with 24 h glutamate plus 12 h of IGF-1 showed similar results: MBP protein levels in the gluta-mate alone condition were 30–40% of MBP levels in the conditions switched to IGF-containing media. Interest-ingly, while there was significant death of cells in the cul-tures exposed to glutamate alone for 30 h until the dif-ferentiation treatment, the remaining cells did go on to express MBP; however, at levels lower than levels in the IGF-1 rescue conditions. Our results suggest that the ex-
posure to glutamate does not irreversibly alter OP viabil-ity or their capacity to differentiate if they are subse-quently stimulated with IGF-1 or high insulin-contain-ing media. Moreover, these cells can go on to produce myelin proteins. These results support the hypothesis that the cascade of events initiated by glutamate-induced calcium influx is reversible within a few hours prior to Bax translocation and cytochrome c release.
IGF-1 Administration after HI in the Perinatal Brain Retains Normal OP Numbers The in vitro studies described above demonstrate that
IGF-1 can rescue OPs subsequent to glutamate exposure. While numerous studies have investigated neuronal death and protection following HI in the perinatal brain, few studies have evaluated the vulnerability and rescue of immature oligodendroglia after HI. To test whether ad-dition of IGF-1 after HI can rescue oligodendroglia in
Casp-3
a
�-Actin
Glu Glu GluG/I28 h
G/I
I
I
Glu Glu GluG/I32 h
G/I
I
I
240
b
20
40
Cle
aved
cas
pas
e 3
(�-a
ctin
ad
j.)
60
IGF Glu + IGF 12 h
I G/I I G/I I G/I I G/I
16 h 8 h
16 h Glu12 h Glu
16 h
80
100
120
28 32 24 28 32 24
Total treatment time (h)
28 32 24 24 24 2428 28 3232
Glu Fig. 2. IGF-1 can rescue late OPs after 12- or 16-hour exposure to glutamate. Prima-ry late OPs were treated with IGF-1, gluta-mate or glutamate + IGF-1 for the times indicated above the immunoblots ( a ) or along the x-axis ( b ). Isolated protein was analyzed for cleaved caspase 3 by Western immunoblotting and normalized to � -ac-tin as for the previous figure. Some cells were treated for 12 or 16 h with glutamate and then switched to media containing ei-ther IGF-1 (I) or Glu + IGF-1 (G/I) for 8, 12 or 16 h as indicated. a Representative West-ern immunoblots showing cleaved caspase 3 and � -actin from the same blots from the 28- or 32-hour experiments. b Quantifica-tion of active caspase 3 after normalization to � -actin. Total treatment times are indi-cated on the x-axis and coded as 24 h (light gray bars), 28 h (dark gray bars), and 32 h (black bars).

IGF-1 Protection of Oligodendroglia Dev Neurosci 2007;29:302–310 307
vivo, we analyzed PND10 rat brains 72 h followingHI. Differentiation of oligodendrocytes begins between PNDs 10–14 in rats; thus, the oligodendroglia at PND 7–10 are predominantly late OPs. Olig2 is a transcription factor whose expression identifies cells of the oligoden-droglia lineage. Therefore, we quantified the number of Olig2+ cells in the ipsilateral white matter following ad-ministration of either IGF-1 (50 � g) or saline i.c.v. after HI. A prior analysis of damage and neuronal protection by IGF-1 in these brains revealed that IGF-1 reduced neu-ronal damage by 40% overall with significant neuropro-tection observed in the cortex, hippocampus, and stria-tum [Brywe et al., 2005]. We analyzed sections from these same brains to evaluate the extent to which IGF-1 could protect the white matter and resident immature oligo-dendroglia ( fig. 4 ). Cell counts performed by an investi-gator blinded to the conditions revealed that the total number of cells in the white matter was reduced to 78% in the HI vs. contralateral hemisphere ( fig. 4 a, b, e; p = 0.02). The number of Olig2+ cells in white matter of the ipsilateral hemisphere was reduced to 65% of the contra-lateral hemisphere ( fig. 4 a, b, e; p = 0.04). Administration of IGF-1 completely rescued the loss of Olig2+ cells in white matter ( fig. 4 c–e; p = 0.04). Interestingly, while there was a trend towards rescue of total white matter
cells with IGF-1 administration, this did not reach statis-tical significance suggesting that IGF-1 preferentially res-cued the Olig2+ cell population ( fig. 4 e).
Discussion
In previous studies, we demonstrated that death of late OPs from glutamate in vitro involves Bax translocation and a mitochondrial apoptotic pathway that occurs 20–24 h following exposure to glutamate. Here, we demonstrate that 1-hour exposure to glutamate is sufficient to initiate death of the late OPs in the same time frame. Moreover, we found that IGF-1 interferes with the glutamate-in-duced death pathway in the late OPs when administration is delayed as much as 16–18 h following glutamate expo-sure. Moreover, these cells can proceed to differentiate and to produce myelin proteins, suggesting that the cells are not irreversibly committed to die until near the time when Bax translocation occurs. We also demonstrate that, when administered following the insult, IGF-1 protects white matter OPs from HI in vivo in the perinatal brain.
Multiple investigators have used cell culture paradigms to characterize the molecular mechanisms of glutamate toxicity on oligodendrocyte lineage cells. There are sig-nificant differences in the approaches taken in these in vi-tro investigations and, thus, in the results obtained and conclusions drawn. The major differences that are critical for interpreting the data and generalizing to the in vivo conditions are the stage of cells utilized, the method for glutamate treatment and the culture conditions. Of note is that the majority of the in vitro systems used to study glu-tamate toxicity in the oligodendroglial lineage have used cells that are positive for galactocerebroside (GalC+), a marker for postmitotic immature or mature oligodendro-cytes [Yoshioka et al., 1995; Matute et al., 1997; McDonald et al., 1998a; Sanchez-Gomez and Matute 1999; Alberdi et al., 2002; Sanchez-Gomez et al., 2003]. As discussed previ-ously, increasing evidence suggests that the most vulner-able stage of the lineage in rodent, sheep and human white matter is the O4+/GalC– late OP. Thus, we developed a cul-ture system to enrich for the late OP cell [Ness and Wood, 2002]. Under conditions that promote progression to the late OP stage but prevent further differentiation, we gener-ate cultures that are greater than 90–95% O4+ late OPs with only 1–2% GalC+ cells (detected by O1 or Ranscht antibodies), 6–8% early progenitors (A2B5+/O4–), ! 2% as-trocytes, and ! 0.01% microglia [Ness and Wood, 2002].
A second distinguishing difference between our in vi-tro system vs. other paradigms is the absence of the recep-
– 21.5MBP
18 h Glu + 12 h Tx
N2B2Ctl Glu
/Ctl
Glu/G
lu
Glu/G
lu +
IGF
Glu/IG
F
�-Actin
– 18.5
Fig. 3. Late addition of IGF-1 to OPs after glutamate exposure res-cues myelin protein expression following differentiation. Prima-ry late OPs were treated for 30 h with full media containing high insulin (N2B2), control media without insulin (Ctl). Additional cultures were treated with glutamate for 18 h followed by an ad-ditional 12 h in glutamate alone (Glu/Glu), glutamate plus IGF-1 (Glu/Glu + IGF) or IGF-1 alone (Glu/IGF). After the 30 h of treat-ment, all cultures were switched to differentiation media for 72 h. Isolated protein was analyzed for expression of MBP or � -actin by Western immunoblotting. Blot is representative of 3 indepen-dent experiments with each condition per experiment analyzed in duplicate.

Wood /Loladze /Altieri /Gangoli /Levison /Brywe /Mallard /Hagberg
Dev Neurosci 2007;29:302–310308
tor-desensitizing blocker, cyclothiazide, which inhibits desensitization at AMPA-preferring receptors [Yoshioka et al., 1995; Matute et al., 1997; Matute, 1998; McDonald et al., 1998a; Sanchez-Gomez and Matute, 1999; Li and Stys, 2000; Alberdi et al., 2002; Sanchez-Gomez et al., 2003]. In addition, other laboratories use culture media containing micromolar levels of insulin, which activate the IGF-1R. In contrast, the insulin levels in our system are absent or physiological (nanomolar range), which do not activate the IGF-1R. This modification to the culture medium formulation is critical since our previous studies demonstrated that stimulating the IGF-1R antagonizes
glutamate-stimulated OP death through sustained acti-vation of Akt and downstream survival pathways [Ness and Wood, 2002]. The required addition of AMPA recep-tor-desensitizing agents used in other studies to obtain significant glial cell death is likely a direct consequence of including high insulin in the media as well as to using the postprogenitor stage of immature oligodendrocytes. Without the AMPA-R desensitizing blocker, we observe death of the late OPs after 20–24 h, a time frame consis-tent with the peak of active caspase 3 in the perinatal HI brain and with the incidence of ISEL+ cells in the white matter in the p7 rat brain after HI [Han and Holtzman,
120
100
80
60
40
20
0e DAPI
*
***
Ave
rag
e ce
ll n
umb
er/a
rea
Olig2
CtlCtl IGFHIHI IGF
Fig. 4. IGF-1 administration after HI in the perinatal brain prevents loss of Olig2+ cells in white matter. a–d Olig2 immunostain-ing in corpus callosum from PND10 rat brains 72 h after exposure to HI ( a , c ) or hypoxia alone ( b , d ) and treatment with either IGF-1 ( c , d ) or saline ( a , b ). DAPI (blue) was used to detect all nuclei. Dou-ble-positive cells appear as pink since the Olig2+ transcription factor is present in the nucleus. e Graph showing average number of total DAPI+ cells or Olig2+ cells per field (average of 3 fields per hemi-sphere, n = 6 for each condition). * p = 0.02 vs. Ctl for DAPI counts and p = 0.04 vs. Ctl for Olig2 counts; * * p = 0.04 vs. HI.

IGF-1 Protection of Oligodendroglia Dev Neurosci 2007;29:302–310 309
2000; Ness et al., 2001]. HI in the perinatal rat brain also reduces expression of IGF-1 within the first 24 h [Lee et al., 1996]. Taken together, the absence of a desensitizing blocker and reduced IGF-1R signaling as employed in our in vitro studies are likely to better model the in vivo state of the white matter after perinatal HI.
A critical finding of these studies is that administering IGF-1 following HI completely rescues loss of OPs in the white matter of the neonatal rat brain. Moreover, our data suggest that IGF-1 preferentially rescues oligodendroglia vs. other white matter glia after HI in the immature brain. A recent study demonstrated that IGF-1 partially rescued O4+ late OPs and MBP expression following bilateral HI in the perinatal rat brain [Lin et al., 2005]. However, in this study, IGF-1 was administered as a single dose prior to the HI insult, which may account for the partial rescue of OPs in this study. Consistent with our results, postinsult ad-ministration of IGF-1 following bilateral artery occlusion in the near-term sheep prevented loss of proteolipid pro-tein-positive cells in the white matter [Cao et al., 2003]. While IGF-1 is a poor mitogen for OPs in vitro, our previ-ous studies demonstrate that it significantly enhances OP proliferation induced by mitogens such as FGF-2 and PDGF [Jiang et al., 2001; Frederick and Wood 2004]. HI in the immature brain results in proliferation of stem/pro-
genitor populations [Back et al., 2002; Felling et al., 2006; Yang and Levison, 2006]. Thus, it is possible that rescue of the OP cells in our study was due in part to an increase in proliferation of the OPs that was augmented by infusion of IGF-1. However, our previous studies demonstrated that excitotoxic death of late OPs in vitro occurs over a pro-tracted time course [Ness and Wood, 2002] similar to the time course observed for death of OPs following HI in vivo in the neonatal rat brain [Ness et al., 2001]. Moreover, the in vitro results presented here suggest that IGF-1 treatment is effective in blocking excitotoxic death when adminis-tered just prior to the time when we observe initiation of mitochondrial-mediated apoptosis in the late OPs [Ness et al., 2004]. Thus, the results of these studies contribute to the viewpoint that administering IGF-1 will prevent white matter damage and promote myelination in the immature brain even if given several hours after insult.
Acknowledgements
The authors thank Bill Tyler for technical advice and helpful comments. This work was supported by Public Health Service grants from the National Institute of Neurological Disorders and Stroke, NS37560 and NS50742 awarded to T.L.W.
References
Alberdi E, Sanchez-Gomez M, Marino A, Ma-tute C (2002): Ca 2+ influx through AMPA or kainate receptors alone is sufficient to initi-ate excitotoxicity in cultured oligodendro-cytes. Neurobiol Dis 9: 234–243.
Andine P, Sandberg M, Bagenholm R, Lehmann A, Hagberg H (1991): Intra- and extracellular changes of amino acids in the cerebral cortex of the neonatal rat during hypoxic-ischemia. Brain Res Dev Brain Res 64: 115–120.
Back SA, Craig A, Kayton RJ, Luo NL, Meshul CK, Allcock N, Fern R (2007): Hypoxia-isch-emia preferentially triggers glutamate deple-tion from oligodendroglia and axons in peri-natal cerebral white matter. J Cereb Blood Flow Metab 27: 334–347.
Back S, Gan X, Li Y, Rosenberg P, Volpe J (1998): Maturation-dependant vulnerability of oli-godendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci 18: 6241–6253.
Back SA, Han BH, Luo NL, Chricton CA, Xan-thoudakis S, Tam J, Arvin KL, Holtzman DM (2002): Selective vulnerability of late ol-igodendrocyte progenitors to hypoxia-isch-emia. J Neurosci 22: 455–463.
Back SA, Luo NL, Borenstein NS, Levine JM, Volpe J, Kinney HC (2001): Late oligoden-drocyte progenitors coincide with the devel-opmental window of vulnerability for hu-man perinatal white matter injury. J Neurosci 21: 1302–1312.
Bansal R, Pfeiffer S (1992): Novel stages in the oligodendrocyte lineage defined by reactiv-ity of progenitors with R-mAb prior to O1 galactocerebroside. J Neurosci Res 32: 309–316.
Bansal R, Pfeiffer SE (1997): Regulation of oligo-dendrocyte differentiation by fibroblast growth factors. Adv Exp Med Biol 429: 69–77.
Bansal R, Warrington A, Gard A, Ranscht B, Pfei-ffer S (1989): Multiple and novel specificities of monoclonal antibodies O1, O4, and R-mAB used in the analysis of oligodendrocyte development. J Neurosci Res 24: 548–557.
Barres B, Schmid R, Sendtner M, Raff M (1993): Multiple extracellular signals are required for long-term oligodendrocyte survival. De-velopment 118: 283–295.
Baumann N, Pham-Dinh D (2001): Biology of oligodendrocyte and myelin in the mamma-lian central nervous system. Physiol Rev 81:
871–927.
Benveniste H, Drejer J, Schousboe A, Diemer NH (1984): Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral micro-dialysis. J Neurochem 43: 1369–1374.
Brywe KG, Mallard C, Gustavsson M, Hedtjarn M, Leverin AL, Wang X, Blomgren K, Is-gaard J, Hagberg H (2005): IGF-I neuropro-tection in the immature brain after hypoxia-ischemia, involvement of Akt and GSK3beta? Eur J Neurosci 21: 1489–1502.
Cao Y, Gunn AJ, Bennet L, Wu D, George S, Gluckman PD, Shao XM, Guan J (2003): In-sulin-like growth factor (IGF)-1 suppresses oligodendrocyte caspase-3 activation and increases glial proliferation after ischemia in near-term fetal sheep. J Cereb Blood Flow Metab 23: 739–747.
Cheng B, Mattson MP (1991): NGF and bFGF protect rat hippocampal and human cortical neurons against hypoglycemic damage by stabilizing calcium homeostasis. Neuron 7:
1031–1041. Cheng B, Mattson MP (1992): IGF-I and IGF-II
protect cultured hippocampal and septal neurons against calcium-mediated hypogly-cemic damage. J Neurosci 12: 1558–1566.

Wood /Loladze /Altieri /Gangoli /Levison /Brywe /Mallard /Hagberg
Dev Neurosci 2007;29:302–310310
Cheng B, Mattson MP (1994): NT-3 and BDNF protect CNS neurons against metabolic/ex-citotoxic insults. Brain Res 640: 56–67.
Cheng B, McMahon DG, Mattson MP (1993): Modulation of calcium current, intracellular calcium levels and cell survival by glucose deprivation and growth factors in hippo-campal neurons. Brain Res 607: 275–285.
D’Souza S, Alinauskas K, Antel J (1996): Ciliary neurotrophic factor selectively protects hu-man oligodendrocytes from tumor necrosis factor-mediated injury. J Neurosci Res 43:
289–298. Felling RJ, Snyder MJ, Romanko MJ, Rothstein
RP, Ziegler AN, Yang Z, Givogri MI, Bongar-zone ER, Levison SW (2006): Neural stem/progenitor cells participate in the regenera-tive response to perinatal hypoxia/ischemia. J Neurosci 26: 4359–4369.
Fern R, Moller T (2000): Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci 20: 34–42.
Follett PL, Paul A, Rosenberg PA, Volpe JJ, Jen-sen FE (2000): NBQX attenuates excitotoxic injury in developing white matter. J Neurosci 20: 9235–9241.
Frederick TJ, Wood TL (2004): IGF-I and FGF-2 coordinately enhance cyclin D1 and cyclin E-cdk2 association and activity to promote G(1) progression in oligodendrocyte pro-genitor cells. Mol Cell Neurosci 25: 480–492.
Guan J, Williams C, Gunning M, Mallard C,Gluckman P (1993): The effects of IGF-I treatment after hypoxic-ischemic brain in-jury in adult rats. J Cereb Blood Flow Metab 13:609–616.
Hagberg H, Andersson P, Kjellmer I, Thiringer K, Thordstein M (1987): Extracellular over-flow of glutamate, aspartate, GABA and tau-rine in the cortex and basal ganglia of fetal lambs during hypoxia-ischemia. Neurosci Lett 78: 311–317.
Hagberg H, Thornberg E, Blennow M, Kjellmer I, Lagercrantz H, Thiringer K, Hamberger A, Sandberg M (1993): Excitatory amino acids in the cerebrospinal f luid of asphyxiated in-fants: relationship to hypoxic-ischemic en-cephalopathy. Acta Paediatr 82: 925–929.
Han BH, Holtzman DM (2000): BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neuro-sci 20: 5775–5781.
Jiang F, Frederick TJ, Wood TL (2001): IGF-I synergizes with FGF-2 to stimulate oligo-dendrocyte progenitor entry into the cell cy-cle. Dev Biol 232: 414–423.
Kavanaugh B, Beesley J, Itoh T, Itoh A, Grinspan J, Pleasure D (2000): Neurotrophin-3 (NT-3) diminishes susceptibility of the oligoden-droglial lineage to AMPA glutamate recep-tor-mediated excitotoxicity. J Neurosci Res 60: 725–732.
Lee WH, Wang GM, Seaman LB, Vannucci SJ (1996): Coordinate IGF-I and IGFBP5 gene expression in perinatal rat brain after hy-poxia-ischemia. J Cereb Blood Flow Metab 16: 227–236.
Levison S, McCarthy K (1991): Astroglia in cul-ture; in Banker G, Goslin K (eds): Culturing Nerve Cells. Cambridge, MIT press, vol 1, pp 309–336.
Li S, Stys PK (2000): Mechanisms of ionotropic glutamate receptor-mediated excitotoxicity in isolated spinal cord white matter. J Neuro-sci 20: 1190–1198.
Lin S, Fan LW, Pang Y, Rhodes PG, Mitchell HJ, Cai Z (2005): IGF-1 protects oligodendro-cyte progenitor cells and improves neurolog-ical functions following cerebral hypoxia-ischemia in the neonatal rat. Brain Res 1063:
15–26. Louis JC, Magal E, Takayama S, Varon S
(1993): CNTF protection of oligodendro-cytes against natural and tumor necrosis fac-tor-induced death. Science 259: 689–692.
Mattson MP, Cheng B (1993): Growth factors protect neurons against excitotoxic/isch-emic damage by stabilizing calcium homeo-stasis. Stroke 24:I136–I140; discussion I144–I135.
Mattson MP, Zhang Y, Bose S (1993): Growth factors prevent mitochondrial dysfunction, loss of calcium homeostasis, and cell injury, but not ATP depletion in hippocampal neu-rons deprived of glucose. Exp Neurol 121: 1–13.
Matute C (1998): Characteristics of acute and chronic kainate excitotoxic damage to the optic nerve. Proc Nat Acad Sci USA 95:
10229–10234. Matute C, Sanchez-Gomez M, Martinez-Millan
L, Miledi R (1997): Glutamate receptor-me-diated toxicity in optic nerve oligodendro-cytes. Proc Nat Acad Sci USA 94: 8830–8835.
McCarthy K, de Vellis J (1980): Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85: 890–902.
McDonald J, Althomsons S, Hyrc K, Choi D, Goldberg M (1998a): Oligodendrocytes from forebrain are highly vulnerable to AKPA/kainate receptor-mediated excitotoxicity. Nat Med 4: 291–297.
McDonald JW, Levine JM, Qu Y (1998b): Mul-tiple classes of the oligodendrocyte lineage are highly vulnerable to excitotoxicity. Neu-roreport 9: 2757–2762.
McMorris F, McKinnon R (1996): Regulation of oligodendrocyte development and CNS my-elination by growth factors: prospects for therapy of demyelinating disease. Brain Pathol 6: 313–329.
Ness JK, Romanko MJ, Rothstein RP, Wood TL, Levison SW (2001): Perinatal hypoxia-isch-emia induces apoptotic and excitotoxic death of periventricular white matter oligodendro-cyte progenitors. Dev Neurosci 23: 203–208.
Ness JK, Scaduto RC Jr, Wood TL (2004): IGF-I prevents glutamate-mediated bax transloca-tion and cytochrome C release in O4+ oligo-dendrocyte progenitors. Glia 46: 183–194.
Ness JK, Wood TL (2002): Insulin-like growth factor I, but not neurotrophin-3, sustains Akt activation and provides long-term pro-tection of immature oligodendrocytes from glutamate-mediated apoptosis. Mol Cell Neurosci 20: 476–488.
Orentas DM, Miller RH (1999): Regulation of oligodendrocyte development. Mol Neuro-biol 18: 247–259.
Ranscht B, Clapshaw P, Price J, Noble M, Seifert W (1982): Development of oligodendrocytes and Schwann cells studied with a nomoclo-nal antibody against galactocerebroside. Proc Nat Acad Sci USA 79: 2709–2713.
Riddle A, Luo NL, Manese M, Beardsley DJ, Green L, Rorvik DA, Kelly KA, Barlow CH, Kelly JJ, Hohimer AR, Back SA (2006): Spa-tial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow pre-dicts fetal ovine periventricular white matter injury. J Neurosci 26: 3045–3055.
Sanchez-Gomez MV, Alberdi E, Ibarretxe G, Torre I, Matute C (2003): Caspase-depen-dent and caspase-independent oligodendro-cyte death mediated by AMPA and kainate receptors. J Neurosci 23: 9519–9528.
Sanchez-Gomez M, Matute C (1999): AMPA and kainate receptors each mediate excitotoxici-ty in oligodendroglial cultures. Neurobiol Dis 6: 475–485.
Silverstein FS, Naik B, Simpson J (1991): Hypox-ia-ischemia stimulates hippocampal gluta-mate efflux in perinatal rat brain: an in vivo microdialysis study. Pediatr Res 30: 587–590.
Volpe J (1997): Brain injury in the prematureinfant – from pathogenesis to prevention. Brain Dev 19: 519–534.
Volpe J (2001a): Neurobiology of periventricular leukomalacia in the premature infant. Pedi-atr Res 50: 553–562.
Volpe J (2001b): Perinatal brain injury: from pathogenesis to neuroprotection. Ment Re-tard Dev Disabil Res Rev 7: 56–64.
Yang Z, Levison SW (2006): Hypoxia/ischemia expands the regenerative capacity of progen-itors in the perinatal subventricular zone. Neuroscience 139: 555–564.
Ye P, D’Ercole AJ (1999): Insulin-like growth fac-tor I protects oligodendrocytes from tumor necrosis factor-alpha-induced injury. Endo-crinology 140: 3063–3072.
Yoshioka A, Hardy M, Younkin D, Grinspan J, Stern JL, Pleasure D (1995): Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors mediate excitotoxicity in the oligodendroglial lineage. J Neurochem 64: 2442–2448.
Young G, Levison S (1997): An improved meth-od for propagating oligodendrocyte progen-itors in vitro. J Neurosci Methods 77: 163–168.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:311–320 DOI: 10.1159/000105472
Prenatal Cord Clamping in Newborn Macaca nemestrina: A Model of Perinatal Asphyxia
Sandra E. Juul Elizabeth Aylward Todd Richards Ronald J. McPherson
John Kuratani Thomas M. Burbacher
University of Washington, Seattle, Wash. , USA
emic injury in M. nemestrina newborns. This model, which combines structural, biochemical, and behavioral assess-ments over time can be used to assess the safety and effi-cacy of neuroprotective strategies.
Copyright © 2007 S. Karger AG, Basel
Introduction
One common cause of brain injury in children is peri-natal asphyxia, defined as a critical and severe lack of oxygen to the fetus during labor and delivery. This occurs in 2–4 of every 1,000 live-born term infants, and more frequently in preterm births [1] . Between 20–50% of term asphyxiated newborns die during the newborn period, accounting for 23% of neonatal deaths worldwide [2] . Up to 60% of the survivors who sustain an hypoxic-ischemic encephalopathy are left with severe and life-long neuro-developmental handicaps that include mental retarda-tion, cerebral palsy, seizures and learning disabilities [3, 4] . Treatment strategies that reliably lessen the harmful effects of hypoxia on subsequent neurodevelopment are limited, and not fully effective [5] . There is, however, rea-son to believe that effective treatments can be developed because, while acute hypoxia-ischemia may cause some immediate neuronal death, much of the damage to the neonatal brain occurs in the hours following the acute incident [6, 7] . A cascade of biochemical events during
Key Words
Hypoxia-ischemia � Neurodevelopment � Erythropoietin
Abstract
Our objective was to establish a nonhuman primate model of perinatal asphyxia appropriate for preclinical evaluation of neuroprotective treatment strategies under conditions that closely resemble human neonatal emergencies, and to begin testing the safety and efficacy of erythropoietin neu-roprotective treatment. Prior to delivery by hysterotomy, the umbilical cords of near term Macaca nemestrina (n = 8) were clamped for times ranging between 12 and 15 min. Animals received erythropoietin (5,000 U/kg/dose ! 2 i.v., n = 3), or vehicle (n = 5) after resuscitation. We assessed physiologic parameters, continuous electroencephalogram, magnetic resonance imaging/spectroscopy, safety parameters and behavior. Animals were euthanized at 4 months of age. Mean birth weight was 507 8 62 g. Initial arterial pH ranged from 6.75 to 7.12, with base deficits of 17–25 mEq. Animals were flaccid at birth, with attenuated electroencephalograms, and seizures occurred in 3 of 8 animals. We demonstrated magnetic resonance imaging/spectroscopy changes consis-tent with hypoxia (elevated lactate levels were present in some animals), significant motor and behavioral abnormali-ties (particularly with 15 min of cord clamping), and evidence of gliosis at the time of death. We have established a repro-ducible model of moderate to severe perinatal hypoxic-isch-
Received: June 6, 2006 Accepted after revision: September 8, 2006
Sandra E. Juul, MD, PhD University of Washington, Department of Pediatrics, Division of Neonatology 1959 NE Pacific St. HSB RR542D, UW Box 356320 Seattle, WA 98195-6320 (USA) Tel. +1 206 221 6814, Fax +1 206 543 8926, E-Mail [email protected]
© 2007 S. Karger AG, Basel
Accessible online at:www.karger.com/dne

Juul/Aylward/Richards/McPherson/Kuratani/Burbacher
Dev Neurosci 2007;29:311–320312
this post-event period culminate in selective neuronal death [8] . Thus an intervention during this critical period might reduce the severity of brain injury [7, 9] .
Recombinant erythropoietin (Epo) has been estab-lished as a credible neuroprotective agent, improving short- and long-term neurologic outcomes following brain injury, in both neonatal and adult animal models [10] . Unlike other agents that have shown promise of neu-roprotection in rodent models, but which failed in human translational trials, Epo has shown clinical benefit in a randomized controlled trial of adults with middle cere-bral artery stroke [11] . The neuroprotective effects of Epo range from 50 to 75% because studies vary with respect to the animal model, mechanism of injury, targeted in-jury site (central or peripheral nervous system), dose of Epo, route of administration, and frequency of dosing [12–14] . Protective effects have been reported with doses ranging from 1,000 to 30,000 U/kg/dose, a range well above those used to promote erythropoiesis. Thus, the safety of these higher doses must be established in neo-nates before the clinical application of this therapy.
Rodent models have been critical to the development of new therapeutic approaches to many human illnesses. Although rodents are commonly used, there are substan-tial morphological and functional differences between developing human and rodent brains. Species differences such as the comparative proportion of white matter lim-it our capacity to translate neuroprotective strategies from rodents to humans. We are confident that the non-human primate can be valuable as an intermediate ani-mal model, because of the close phylogenetic relationship with humans.
The objectives for this pilot study were to (1) create a reproducible model of moderate to severe perinatal as-phyxia that allows for monitoring of biochemical, func-tional and structural data; (2) lay the groundwork for de-termining whether Epo will diminish the structural and functional sequelae associated with perinatal asphyxia, and (3) determine whether neonatal Epo treatment is well tolerated.
We have developed a nonhuman primate model of perinatal asphyxia, incorporating evaluation of brain structure (magnetic resonance imaging, MRI, studies with spectroscopy, MRS, gross anatomy and immuno-histochemistry), function (electroencephalogram, EEG, aEEG) and neurodevelopment. This project employed an interdisciplinary team which included members of the Washington National Primate Center and Infant Primate Research Laboratory (IPRL), Center on Human Develop-ment and Disability, Diagnostic Imaging Science Center,
and the Departments of Neurology, and Pediatrics. This model is ideal for preclinical evaluation of the safety and efficacy of a combined neuroprotective treatment strat-egy under conditions that closely resemble human neo-natal emergencies.
Methods
All experimental protocols were approved by the Animal Care and Use Committees at the University of Washington in accor-dance with U.S. NIH guidelines.
Animal Care and Procedures Pregnant dams were anesthetized with Isoflurane at 165 8 1
day of gestation (term = 172 days). A midline incision was made, the uterus isolated and supported with sterile towels and kept moist with warmed saline. The umbilical cord was exteriorized, while keeping the amniotic fluid and the fetus in the womb. The cord was clamped for 12–15 min, during which time the fetal heart rate was monitored by femoral pulse Doppler. The fetus was then delivered, dried, weighed, and resuscitated according to NRP guidelines. Re-suscitations included intubation, manual ventilation, and cardio-tonic support as needed [15] . Apgar scores were assigned. A 2.5 Fr. umbilical arterial catheter was placed immediately after delivery to allow for blood access and fluid administration. A pulse oximeter, indwelling rectal thermometer, and EEG electrodes were placed. A covered heating pad and radiant warmer were used initially to pro-vide thermal support during stabilization, and then animals were moved into a neutral-thermal isolette.
During the first 24 h of life, animals were maintained exclu-sively on parenteral fluids (10% dextrose at 150 ml/kg/day to start). Serum electrolytes and glucose levels were checked at 12 and 24 h, and fluid content and volume adjusted as needed. Feed-ings began when the abdominal exam was normal, bowel tones were present, and the infant had stooled (24–48 h). Heparinized maternal blood (collected at the time of delivery) was transfused when the phlebotomized blood volume reached 10 ml. Physical growth parameters (weight, crown-rump length, head circumfer-ence, head width, and head length) were measured every 2 weeks throughout life [16, 17] . Animals were sedated with ketamine, then euthanized with an overdose of i.v. sodium pentobarbital at 4 months of life.
Study Drug Animals received either saline (n = 5) or 5,000 U/kg/dose Epo
(n = 3) at t = 0 (range 5–24 min of life), then 24 h later. Plasma Epo concentrations were determined at time 0 (baseline), 30 min, 3,6, 12, 24, 72 and 168 h. Epo concentrations were determined by ELISA (R&D System, Minneapolis, Minn., USA).
Behavioral Measurements Feeding and housing milestones (age at self-feeding, age re-
moved from incubator) were assessed using a standard husband-ry schedule developed for infant monkeys [18] . A behavioral as-sessment procedure modeled after the Brazelton Neonatal Behav-ioral Assessment Scale used with human infants [19] was used to evaluate newborn reflexes (e.g. grasping and clasping) and behav-ioral responses (e.g. visual and auditory orientation and follow-

Primate Model of Perinatal Asphyxia Dev Neurosci 2007;29:311–320 313
ing, startle response) [18] . Neonates were tested 5 days a week for the first 20 days. These tests are sensitive measures of neonatal capacity [18, 20] . Object permanence, a measure of early spatial memory [25, 26] , was assessed beginning at 14 days of age [18] . The Fagan Test of Infant Intelligence, a visual recognition mem-ory test based on the familiarization-novelty paradigm [27] and adapted for monkeys [28] was used to assess object memory. Rec-ognition ability correlates with performance on standardized childhood intelligence tests [29, 30] . All infants were screened for deficits in visual acuity prior to the Fagan Test using a forced-choice preferential looking technique [21–24] . Normative data were derived from concurrent nursery controls delivered vagi-nally (n = 26).
Continuous EEG EEG monitoring was performed for the first 48 h of life, begin-
ning by 1 h of life. We recorded EEG using either conventional multichannel EEG (Grass-Telefactor Comet/Twin TM ), or single-channel amplitude-integrated EEG (aEEG/CFM, Olympic Medi-cal CFM 6000).
Magnetic Resonance Imaging To noninvasively evaluate brain structure, animals had 2
brain MRI/MRS done: the first at 48–72 h of life, and the second at 2 months of age. Intubated infants anesthetized with Isoflu-rane were placed supine in a custom MR-compatible holding de-vice to control position and prevent motion. Studies were ob-tained on a 1.5-tesla GE Signa scanner using a phased-array coil. MR imaging sequences included 2-D T 2 (TR/TE 4s/128ms), 2-D FLAIR (TR/TI/TE 10 s/2.2 s/136 ms), and 3-D T 1 SPGR (TR/TE/ � 44 ms/28 ms/35°) with and without magnetic transfer. Vol-umetric measurement of total brain volume, thalamus and cer-ebellum were done using the software MEASURE [31–33] . Single voxel MR spectroscopy was measured on the basal ganglia and thalami using the point-resolved spectroscopy pulse sequence and the proton brain exam technique [34] . Data were acquired using the following parameters: FID size 2,048 complex points; spectral width 2,000 Hz; spectral frequency of 63.8 MHz; TR/TE 2,000/30 ms. The spectral voxel size was 15 ! 15 ! 15 mm which is 3.375 cm 3 volume. For this MRS voxel, there was minimal con-tamination from CSF because in the infant primate, the CSF spaces are extremely small in the region of interest that was scanned with MRS as determined by T 2 -weighted high-resolu-tion MRI scans. A water spectrum of the same region was also acquired (without water suppression) for quantitation used in the LC model software. Data were processed using the Octane Sili-con Graphics workstation and Provencher’s LC model spectros-copy software. The detectable chemicals are choline, creatine, glutamine (GLN), glutamate (GLU; but these two chemicals can-not be separately determined and are combined together to give a GLX level), inositol, lactate (Lac), N-acetyl aspartate (NAA), scyllo-inositol, and taurine. MRS is curve fitted and analyzed for abnormal peaks [35] .
Laboratory Assessments Arterial blood gases were drawn immediately after umbilical
line placement and at 1, 3, 6, and 12 h of life, and as clinically in-dicated. Safety assessments included complete blood counts, se-rum electrolytes, liver and kidney function tests.
Immunohistochemistry Brains were perfusion fixed with 4% paraformaldehyde, par-
affin embedded and sectioned (5 � m). Brains from the 4 animals that received 15 min of cord clamping were evaluated. Sections of hippocampus, cerebellum, thalamus, caudate/putamen, water-shed region frontal cortex, and visual cortex were stained with HE, Luxol fast blue/cresyl violet, and underwent immunohisto-chemistry. Primary antibodies used were: Fluoro-Jade B (Chemi-con), goat anti-GFAP (Santa Cruz SC6170).
Results
Seven of 8 animals were male. Mean birth weight was 507 8 62 g. The duration of the umbilical cord clamp time started at 12 min, and as the pilot project progressed, was increased to 15 min. Details of each animal’s resus-citation and first hours of life are shown in table 1 . Two animals had 12 min of cord clamping, 2 had 14 min, and 4 were clamped for 15 min prior to birth. All animals were intubated by 2 min of life, and received positive pressure ventilation, followed by free-flow oxygen for the first 24 h of life. Duration of positive pressure ventilation is shown in table 1 . Seven animals received epinephrine for initial bradycardia. Three animals received bicarbon-ate for persistent metabolic acidosis. One (animal 3) suf-fered a spinal cord injury after a cisternal tap to collect CSF for Epo measurements. It was euthanized on day 3 of life. Necropsy findings confirmed the spinal cord in-jury, and also documented acute cerebral swelling, rar-efaction of white matter, and gliosis.
The relationship between duration of cord clamping with initial pH, base deficit, and 20 min Apgar scores are shown in figure 1 . The duration of cord clamping clearly affected the early biochemical and physiologic state of the delivered animal, and correlated best with the 20 min Ap-gar score (r = 0.892, p ! 0.01). It is notable that the initial acidosis, while severe, began to clear quickly: the magni-tude of the initial base deficit correlated (r = 0.83) with the number of minutes after delivery the gas was obtained, (range 4–22 min after delivery, p ! 0.01). For the 4 animals that sustained 15 min of cord clamping, the initial mean pH was 6.86 8 0.03, with a mean base deficit of 23.2 8 1.9 mEq/l. The mean 5, 10 and 20 min Apgar scores were 1.7 8 0.6, 2.3 8 0.6 and 3.7 8 0.6, respectively, indicat-ing moderate to severe hypoxic-ischemic injury, with small variability between subjects. Two animals clamped for 15 min received Epo (5,000 U/kg ! 2 doses 24 h apart), and 2 received saline. First spontaneous breath in the 15 min group occurred at 24 8 11 min, with regular spon-taneous respirations established at 4.1 8 1.3 h.

Juul/Aylward/Richards/McPherson/Kuratani/Burbacher
Dev Neurosci 2007;29:311–320314
The neurologic exam of all infants was abnormal ini-tially, with marked hypotonia, but the length of time it took animals to begin spontaneous movements varied with the clamp time. Myoclonic activity was present in all animals, and subclinical seizures occurred in 3 of 8 animals within the first 30 h of life. One animal received phenobarbital for recurrent clinically apparent seizure activity. Both conventional and aEEG were consistent in showing severely attenuated brain activity for the first 3 h of life in all animals. Figure 2 shows conventional EEG tracings on the left, compared with CFM tracings on the right. Both methods of CNS monitoring show severe at-tenuation of brain activity during the first hours of life, with slow improvement. By 24 h of life, the baseline activ-ity had increased, but burst suppression was still ongoing in most animals, particularly those with more prolonged cord clamping.
Seven of 8 animals had the early MRI done, and due to one death, only 7 animals had a 2 month scan. Five animals had usable MRS data from both time points. Fig-ure 3 shows an example of an MRI/MRS completed at48 h of life. This saline-treated animal received 15 min of cord clamping, and went on to develop significant cere-bral palsy, with flexion contractures at the hips, knees, and hands. Figure 3 a shows the T 1 -weighted scan, fig-ure 3 b the T 2 -weighted scan, and figure 3 c shows a sam-ple output from the LC model software. Within the spec-trum, the black line is the real proton MR spectrum from
the monkey brain and the red line is the model fit. Sev-eral of the chemicals are labeled on the spectrum. Note that there are abnormally high Lac and low NAA levels in this brain. An elevation of the myoinositol peak was also noted in some animals on their early MRS. Table 2 shows the MRS findings from the 4 animals that sus-tained 15 min of cord clamping, and 1 that had 12 min of clamping. As shown in table 2 , there was an increase in
Table 2. MR spectroscopy results (mM concentration)
Clamp time, min 12 15 15 15 15Study drug Epo Epo Epo Veh VehNAA
48 h 6 4.2 6 5.6 5.32 months 7.8 6.7 7.8 7.6 6.8
GLU + GLN48 h 8 10.9 11 13 10.42 months 11.3 15.2 16.6 15.5 9
Lactate48 h 0.15 0.8 0.16 9.5 0.222 months 0.55 1.14 1.17 0.212 0.75
Creatine48 h 4.63 3.6 4.9 3.6 4.52 months 7.4 8 6.2 7.6 6.1
Choline48 h 1.5 1.1 1.4 1.4 1.42 months 1.4 1.9 1.2 1.7 1.4
Table 1. Resuscitation characteristics
Clamp time, min 12 12 14 14 15 15 15 15
Birth weight, g 600 520 400 500 540 540 440 520Average weight gain/day, g 4.4 3.9 – 5.1 4.2 1.7 2.7 4.8
Sex male male female male male male male male
Initial pH 6.88 7.02 6.75 7.12 6.85 6.82 6.89 6.86
Initial base deficit, mEq/l 22 19 24 17 22 21 25 24
Apgar scores1 min 0 1 1 1 1 1 1 15 min 2 1 2 3 3 1 2 2
10 min 3 6 2 5 3 2 3 220 min 9 9 3 6 5 3 4 4
Study drug Veh Epo Veh Veh Veh Veh Epo Epo
Respiratory effort1st breath, min 11 5 80 30 16 36 31 13Regular respiration, h 0.5 1 3.0 1.0 3.0 5.5 3.5 0.3Extubated, h 1.5 1 3.5 1.5 3.5 6.0 4.0 0.5

Primate Model of Perinatal Asphyxia Dev Neurosci 2007;29:311–320 315
NAA and creatine in all animals from 48 h to 2 months, which may be related to brain development and matura-tion in infancy. There was a greater increase (from 48 h to 2 months) in GLU + GLN levels in the Epo group com-pared with the Veh group, but there were too few subjects to make any assessment of treatment effect.
Structural MRI enabled total and regional brain vol-ume measurements. Brain volumes from 2 days were
compared with 2 months of age. All brains grew over the 2-month period, with mean volume of 64.2 8 6.9 cm 3 at 2 days, and 82.9 8 12.1 cm 3 at 2 months. Table 3 gives specific brain volume measurements at 48 h and 2 months. There were no differences between initial or final brain/body ratios (volume/weight) by group (saline vs. Epo treated). There was also no correlation between clamp time and overall brain growth, nor with volume of spe-cific brain compartments.
Feeding and housing milestones were delayed for all of the infants compared with controls. Self feeding behavior was achieved by day 11 8 1 in control animals. Of the 4 animals with 15-min clamp times, one vehicle-treated animal achieved self-feeding at day 78, while the other never did. The 2 Epo-treated animals were able to self-feed by days 24 and 31 of life, and were therefore able to live independently as they matured. Asphyxiated infants also required assistance with temperature control for lon-ger than concurrent controls, with vehicle-treated ani-mals exiting their isolette at 25 and 81 days, Epo animals by 15 and 25 days, and controls by day 5 8 1. Neurode-velopmental evaluation was done using a previously vali-dated battery of behavioral tests throughout their 4 months of life. All animals had marked hypotonia after birth, and in some, this was followed by progressive hy-pertonia of upper and/or lower extremities, head lag, and delay in their early skills as measured by modified Bra-zelton testing [19] . Animals clamped for 12 min showed minimal (vehicle) to no spasticity (Epo); the surviving animal that sustained 14 min of clamping showed mild-moderate spasticity (vehicle), and of the 4 animals clamped for 15 min, the 2 had severe motor impairment (vehicle), while the 2 showed only mild motor impair-ment (Epo treated). Increased tone was detectable by 48 h of life. Figure 4 shows examples of normal positioning of the hands and feet from a concurrent control com-pared with the progressively severe contractures present
1
2
3
4
5
6
7
8
9
10
11 12 13 14 15 16
Clamp time (min)
20
min
Ap
ga
r
c
16
6.65
17
6.70
18
6.75
19
6.80
20
6.85
21
6.90
22
6.95
23
7.00
24
7.10
26
7.15
Init
ialb
ase
de
fici
tIn
itia
l pH
b
a
25
7.05
Fig. 1. Initial resuscitation. a Initial pH as a function of time of cord clamping. b Initial base deficit as a function of clamp time. c Relationship between clamp time and the 20 min Apgar score.
Table 3. Brain volume measurements over time
Clamp time, min 12 14 15 15 15 15Study drug Epo Veh Veh Veh Epo EpoTotal brain volume, cm3
48 h 63.4 62.5 72.3 70.1 52.7 64.12 months 87.8 74.9 97.2 93.1 64.9 79.3
Cerebellar volume, cm3
48 h 3.5 2.8 3.6 5.0 3.3 3.12 months 6.3 4.7 7.8 6.9 4.1 6.0
Thalamus volume, cm3 1.5 1.0 1.3 1.4 1.1 1.8

Juul/Aylward/Richards/McPherson/Kuratani/Burbacher
Dev Neurosci 2007;29:311–320316
in the brain-injured animals. The saline-treated animals developed severe contractures ( fig. 4 d), while Epo-treat-ed animals did not ( fig. 4 b, c). All animals also displayed delays in behavioral responsivity when compared with concurrent control animals born at term with no expo-sure to hypoxia-ischemia, and these deficits were most apparent in animals that underwent 15 min of cord clamping. Optimal behavioral responses on items such as visual and auditory orientation and follow and startle and habituation to auditory stimuli were observed for con-
trols by 3 8 1 days of age. Of the 4 animals clamped for 15 min, vehicle-treated animals displayed optimal re-sponses by day 8 and 13 and Epo animals by day 4 and 5. Visual acuity screening showed normal vision for all of the animals. Spatial memory as measured by the day of age to display object permanence was normal (control value = 77 8 19 days) in one of the two 15-min clamped Epo animals (61 days of age), while the other Epo animal did not display object permanence at the end of testing (135 days). Similar results were observed for the vehicle-
Fig. 2. EEG and aEEG tracings. a–c Conventional EEG tracings. The vertical lines are at 1-second intervals. Low-frequency fil-ter = 0.3 Hz. High-frequency filter = 35 Hz. a Severely attenuated cortical activity at 1.5 h of life from an animal that received 14 min of cord clamping. Note the tracings in each channel are nearly flat. b Improved background activity in this animal at 6.5 h of life. A seizure evolves in the left frontal region (Fp1) indicated by the re-petitive sharp discharges (arrows). c Normal-amplitude EEG from another animal at 24 h of life. d–f CFM tracings from an animal whose cord was clamped for 15 min. Note that the time
frame of these tracings is different, where one CFM panel repre-sents 3 h of recording. d Activity recorded at hours 2–5 of life shows severely depressed baseline (the tracings are relatively low on the graph) with a great deal of burst suppression (intermittent, broad vertical lines). e Hours 5–7 of life. The baseline slowly rises on the graph, correlating with the animal beginning to wake up and move spontaneously during this epoch. f Twenty-four hours of life. The baseline activity had increased further, but burst sup-pression was still ongoing, as indicated by the intermittent verti-cal lines.

Primate Model of Perinatal Asphyxia Dev Neurosci 2007;29:311–320 317
treated animals, one displaying object permanence at 86 days with the other never reaching criterion. Object memory as measured by the percent novelty preference on the Fagan Test was normal (control value = 57 8 7%) in the two 15-min clamped Epo animals (53 and 68%), while one of the vehicle-treated animals displayed a nov-
elty preference (61%) and the other did not (44%). There was no difference in weight gain by treatment group (440 8 206 g over 120 days vs. 455 8 126 g saline vs. Epo group), although average weight gain in all asphyxiated infants was less than for concurrent controls.
Fig. 3. MRI and MRS. An example of an MRI/MRS completed at 48 h of life. This vehicle-treated animal received 15 min of cord clamping, and went on to develop significant cerebral palsy, with flexion contractures at the hips, knees, and hands. a T 1 -weighted scan. b T 2 -weighted scan. c Sample output from the LC model software. Within the spectrum, the black line is the real proton
MR spectrum from the monkey brain and the red line is the mod-el fit. Several of the chemicals are labeled on the spectrum. Note that there are abnormally high Lac levels in this brain and also abnormally low NAA. This spectrum was acquired at 1.5 T with the point-resolved spectroscopy pulse sequence and with param-eters TR/TE 2,000/30 ms.
Fig. 4. Contractures. a A normal 11-day-old animal with no contractures (play group control). Note the open positioning of the hands and feet. b An 8-day-old Epo-treated animal with minor contractures of the feet. c A close-up of the feet of another Epo-treated animal with fixed toe con-tractures. d A saline-treated animal at 2 months of age with severe contractures at the hips, knees and elbows. These contrac-tures remained stable until the time of death at 4 months.

Juul/Aylward/Richards/McPherson/Kuratani/Burbacher
Dev Neurosci 2007;29:311–320318
Serum Epo concentrations were measured at timed in-tervals. In saline-treated animals, serum Epo concentra-tions peaked at 6 h, but remained surprisingly low ( ! 25 mU/ml), given the hypoxic insult. In contrast, those ani-mals receiving i.v. Epo had serum Epo concentrations up to 74,590 mU/ml at 0.5 h after injection ( fig. 5 ). The sec-ond dose of Epo given 24 h after birth did not result in a peak Epo concentration as high as the initial dose. CSF Epo concentrations were measured from 3 saline-treated animals. Concentrations ranged from 3.18 to 5.8 mU/ml. This procedure was discontinued after one animal sus-tained a spinal cord injury.
The asphyxial insult caused a transient rise in GGT, ALT and AST in all animals, which resolved by day 2 of life. Renal function was also transiently impaired, with mean serum creatinine of 1.4 8 0.2 at 48 h, resolving by 1 week of life. No long-term hematologic effects were not-ed. Two saline-treated animals had significant feeding problems consistent with ischemic gut injury (gastric re-siduals and bloody stools) and required i.v. nutritional and fluid support for several days. There were no evident negative side effects from high-dose Epo treatment.
Immunohistochemical staining of the 4 animals that sustained 15 min of cord clamping was done (data not shown). We were able to demonstrate increased GFAP immunoreactivity in the thalamus (gliosis), as well as neurodegeneration of cortical neurons as denoted by Flu-oro Jade B staining. No positive staining for macrophages (CD68) was noted. Comparisons to control brains were
not available, as the pilot study was designed to develop a working model of perinatal asphyxia, not to compare the histology of normal vs. injured animals. As such, ex-tensive immunohistochemical studies of these tissues are beyond the scope of this paper.
Discussion
Our goal was to develop a nonhuman primate model of perinatal asphyxia, which can be used for testing neu-roprotective strategies deemed promising in studies using lower mammals. Animal models of neonatal hypoxic-ischemic brain injury are essential for the development and testing of novel therapeutic approaches. Rodent mod-els are most commonly used because they are inexpensive, easy to work with, and often respond to pharmacotherapy in a manner similar to humans. Models of perinatal hy-poxic brain injury include the application of prolonged hypoxia alone [36, 37] , transient middle cerebral artery occlusion [38, 39] , and unilateral ligation of the common carotid artery followed by exposure of the suckling rat to hypoxia [40] . There are, however, substantial differences between human and rodent anatomy and physiology. In addition, the small circulating blood volume in these an-imals limits the ability to do sequential blood sampling, and rodent behavioral testing is limited. These features have made the translation of neuroprotective strategies from rodents to humans largely unsuccessful. To over-come these limitations, larger mammals including rab-bits, dogs, piglets, sheep, and nonhuman primates have been used to model perinatal asphyxia [41–46] . The non-human primate is valuable because complex behavior more comparable to that of humans can be tested. Such neurodevelopmental assessments have been developed and validated in the Infant Primate Laboratory of the Uni-versity of Washington Primate Center [47] . Ideally, thera-pies should be founded on data derived from both in vitro and in vivo approaches, with nonhuman primates used as the penultimate preclinical step [46, 48, 49] .
We sought to establish a degree of perinatal asphyxia such that our animals would meet the entry criteria used in recent studies of hypothermia, thereby modeling the clinical population targeted for neuroprotection [3, 4] . Entry criteria for the Cool Cap Trial included: (1) a 10 min Apgar score of ! 6, pH ! 7, BE 1 –16 mEq/l, or ongoing resuscitation at 10 min; (2) a clinical exam showing mod-erate to severe encephalopathy, and (3) abnormal aEEG findings for a minimum of 20 min, or seizures, with sim-ilar criteria used for the recent NICHD study [3, 4] . In
0
10,000
20,000
30,000
40,000
50,000
60,000
70,000
80,000
0
Time (h)
Ep
o(m
U/m
l)
3 6 9 12 15 18 21 24 48 72 96
Fig. 5. Serum Epo concentrations. Serial serum Epo concentra-tions were measured at timed intervals after the study drug dos-ing (0, 0.5, 1, 3, 6 12, 24, 48 and 96 h). Epo dosing (5,000 U/kg i.v.) occurred at birth and 24 h of life (n = 3).

Primate Model of Perinatal Asphyxia Dev Neurosci 2007;29:311–320 319
addition, we sought to model the neurodevelopmental deficits common to survivors of perinatal asphyxia. We began this pilot study by clamping the umbilical cord for 12 min, but found that the overall injury was insufficient to meet the above criteria consistently. We cautiously in-creased the duration of cord clamping to 14, and then 15 min. At 15 min, we found consistent, reproducible injury, with physiologic, biochemical, and electroencephalo-graphic findings that met study entry criteria. As our clamp time increased, animals showed varying degrees of muscle spasticity and cognitive delay, ranging from mild to severe. We did not, however, detect any atrophy or cyst formation by MRI at 2 months on T 1 , T 2 or FLAIR. This may be due to the relatively short interval between initial and follow-up scans (2 months). The lack of nor-mal, nonasphyxiated controls also made it difficult to as-sess possible ischemic-related changes across different anatomic locations. MRI/S detected an elevation of the myoinositol peak in some animals that has been described in association with hypoxic-ischemic brain injury in hu-man infants, as well as the low NAA, and elevated Lac demonstrated in figure 3 [50, 51] . The increase in NAA noted from 48 h to 2 months was consistent with normal neurodevelopment [52] . This pilot study was not powered to detect treatment differences.
We successfully documented many of the clinical findings often associated with significant perinatal hy-
poxia-ischemia in human infants: encephalopathy by physical exam and aEEG findings, abnormal EEG, pro-gressive spasticity, cognitive delay, transient somatic or-gan dysfunction (kidney and liver biochemical dysfunc-tion, feeding intolerance), and neuronal degeneration with gliosis on immunohistochemistry. Three of the 8 animals were treated with Epo. There were too few sub-jects to make any assessment of treatment effect; how-ever, no treatment complications were associated with high-dose Epo treatment.
In conclusion, these preliminary experiments demon-strate that 15 min of cord occlusion prior to delivery re-sults in a reproducible nonlethal hypoxic-ischemic injury in Macaca nemestrina newborns. We submit that this is an ideal model of perinatal hypoxia-ischemia which can be used for the preclinical testing of promising neuropro-tective strategies.
Acknowledgements
This study was supported by NIH grants RR00166 and P30 HD02274 through the Washington Regional Primate Research Center and the National Institute of Child Health and Human Development. We thank Jeff Stephenson for his expertise in MRI, Dr. Eric Demers and Infant Primate Lab staff for all their help with the care of these animals.
References
1 Vannucci RC: Hypoxia-ischemia: clinical aspects; in Fanaroff AA (ed): Neonatal-Peri-natal Medicine IV. Philadelphia, Mosby-Yearbook, 1997, pp 877–891.
2 Lawn JE, Cousens S, Zupan J: 4 million neo-natal deaths: When? Where? Why? Lancet 2005; 365: 891–900.
3 Gluckman PD, Wyatt JS, Azzopardi D, Bal-lard R, Edwards AD, Ferriero DM, Polin RA, Robertson CM, Thoresen M, Whitelaw A, Gunn AJ: Selective head cooling with mild systemic hypothermia after neonatal en-cephalopathy: multicentre randomised trial. Lancet 2005; 365: 663–670.
4 Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, Fan-aroff AA, Poole WK, Wright LL, Higgins RD, Finer NN, Carlo WA, Duara S, Oh W, Cotten CM, Stevenson DK, Stoll BJ, Lemons JA, Guillet R, Jobe AH: Whole-body hypo-thermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med 2005; 353:
1574–1584.
5 Whitelaw A, Thoresen M: Clinical trials of treatments after perinatal asphyxia. Curr Opin Pediatr 2002; 14: 664–668.
6 du Plessis AJ, Johnston MV: Hypoxic-isch-emic brain injury in the newborn: cellular mechanisms and potential strategies for neuroprotection. Clin Perinatol 1997; 24:
627–654. 7 Vannucci RC, Perlman JM: Interventions for
perinatal hypoxic-ischemic encephalopathy. Pediatrics 1997; 100: 1004–1014.
8 Vannucci RC: Experimental biology of cere-bral hypoxia-ischemia: relation to perinatal brain damage. Pediatr Res 1990; 27: 317–326.
9 Groenendaal F, de Vries LS: Selection of ba-bies for intervention after birth asphyxia. Semin Neonatol 2000; 5: 17–32.
10 Juul S: Erythropoietin in the central nervous system, and its use to prevent hypoxic-isch-emic brain damage. Acta Paediatr Suppl 2002; 91: 36–42.
11 Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M, Rustenbeck HH, Breiter N, Jacob S, Knerlich F, Bohn M, Poser W, Ruther E, Kochen M, Gefeller O, Gleiter C, Wessel TC, De Ryck M, Itri L, Prange H, Cerami A, Brines M, Siren AL: Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med 2002; 8:
495–505. 12 Brines ML, Ghezzi P, Keenan S, Agnello D,
de Lanerolle NC, Cerami C, Itri LM, Cerami A: Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 2000; 97:
10526–10531. 13 Kumral A, Ozer E, Yilmaz O, Akhisaroglu
M, Gokmen N, Duman N, Ulukus C, Genc S, Ozkan H: Neuroprotective effect of erythro-poietin on hypoxic-ischemic brain injury in neonatal rats. Biol Neonate 2003; 83: 224–228.

Juul/Aylward/Richards/McPherson/Kuratani/Burbacher
Dev Neurosci 2007;29:311–320320
14 Demers EJ, McPherson RJ, Juul SE: Erythro-poietin protects dopaminergic neurons and improves neurobehavioral outcomes in juve-nile rats after neonatal hypoxia-ischemia. Pediatr Res 2005; 58: 297–301.
15 Jackson JC, Truog WE, Standaert TA, Mur-phy JH, Juul SE, Chi EY, Hildebrandt J, Hod-son WA: Reduction in lung injury after com-bined surfactant and high-frequency venti-lation. Am J Respir Crit Care Med 1994; 150:
534–539. 16 Ruppenthal GC: Weight gain and intake re-
quirements in nursery reared macaques; in Meehan TP, Allen ME (eds): Proceedings of the 4th Annual Dr. Scholl Conference on the Nutrition of Captive Wild Animals. Chica-go, Lincoln Park Zoological Society, 1989, pp 20–35.
17 Sackett GP, Ruppenthal GC: Growth of nurs-ery-raised Macaca nemestrina infants: Ef-fects of feeding schedules, sex, and birth weight. Am J Primatol 1992; 27: 189–204.
18 Burbacher T, Shen D, Grant K, Sheppard L, Damian D, Ellis S, Liberato N: Reproductive and offspring developmental effects follow-ing maternal inhalation exposure to metha-nol in nonhuman primates. Res Rep Health Eff Inst 1999;i–ii:1–117; discussion 119–133.
19 Brazelton TB (ed): Brazelton Neonatal Be-havioral Assessment Scale (National Spas-tics Monographs). London, William Heine-man and Sons, 1973.
20 Clarren SK, Bowden DM: Fetal alcohol syn-drome: a new primate model for binge drink-ing and its relevance to human ethanol tera-togenesis. J Pediatr 1982; 101: 819–824.
21 Sackett GP, Fahrenbruch CE, Ruppenthal GC (eds): Development of Basic Physiologi-cal Parameters and Sleep-Wakefulness Pat-terns in Normal and At-Risk Neonatal Pig-tail Macaques (Macaca nemestrina) . New York, Plenum Press, 1979.
22 Dobson V, McDonald MA, Kohl P, Stern N, Samek M, Preston K: Visual acuity screening of infants and young children with the acuity card procedure. J Am Optom Assoc 1986; 57:
284–289. 23 Teller DY: The forced-choice preferential
looking procedure: a psychophysical tech-nique for use with human infants. Infant Be-hav Dev 1979; 2: 135–153.
24 Teller DY: The development of visual acuity in human and monkey infants. Trends Neu-rosci 1981; 4: 21–24.
25 Mishkin M, Appenzeller T: The anatomy of memory. Sci Am 1987; 256: 80–89.
26 Diamond A, Goldman-Rakic P: Comparison of human infants and rhesus monkeys on Piaget’s AB task: evidence for dependence on dorsolateral prefrontal cortex. Exp Brain Res 1989; 74: 24–40.
27 Fagan JF, Singer LT (eds): Infant Recognition Memory as a Measure of Intelligence. New York, Ablex, 1983.
28 Gunderson VM, Sackett GP: Development of pattern recognition in infant pigtailed ma-caques (Macaca nemestrina) . Dev Psychol 1984; 20: 418–426.
29 Rose SA, Feldman JF: Prediction of IQ and specific cognitive abilities at 11 years from infancy measures. Dev Psychol 1995; 31: 685–696.
30 Sackett GP, Stephenson E, Ruppenthal GC: Digital data acquisition systems for observ-ing behavior in laboratory and field settings. Behav Res Methods Instrum 1973; 5: 344–348.
31 Barta PE, Pearlson GD, Powers RE, Richards SS, Tune LE: Auditory hallucinations and smaller superior temporal gyral volume in schizophrenia. Am J Psychiatry 1990; 147:
1457–1462. 32 Aylward EH, Anderson NB, Bylsma FW,
Wagster MV, Barta PE, Sherr M, Feeney J, Davis A, Rosenblatt A, Pearlson GD, Ross CA: Frontal lobe volume in patients with Huntington’s disease. Neurology 1998; 50:
252–258. 33 Aylward EH, Li Q, Honeycutt NA, Warren
AC, Pulsifer MB, Barta PE, Chan MD, Smith PD, Jerram M, Pearlson GD: MRI volumes of the hippocampus and amygdala in adults with Down’s syndrome with and without de-mentia. Am J Psychiatry 1999; 156: 564–568.
34 Webb PG, Sailasuta N, Kohler SJ, Raidy T, Moats RA, Hurd RE: Automated single-vox-el proton MRS: technical development and multisite verification. Magn Reson Med 1994; 31: 365–373.
35 Provencher SW: Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med 1993;30:672–679.
36 Stewart WB, Ment LR, Schwartz M: Chronic postnatal hypoxia increases the numbers of cortical neurons. Brain Res 1997; 760: 17–21.
37 Ment LR, Schwartz M, Makuch RW, Stewart WB: Association of chronic sublethal hy-poxia with ventriculomegaly in the develop-ing rat brain. Brain Res Dev Brain Res 1998;
111: 197–203. 38 Ashwal S, Cole DJ, Osborne S, Osborne TN,
Pearce WJ: A new model of neonatal stroke: reversible middle cerebral artery occlusion in the rat pup. Pediatr Neurol 1995; 12: 191–196.
39 Sola A, Rogido M, Lee BH, Genetta T, Wen TC: Erythropoietin after focal cerebral isch-emia activates the Janus kinase-signal trans-ducer and activator of transcription signal-ing pathway and improves brain injury in postnatal day 7 rats. Pediatr Res 2005; 57:
481–487.
40 Rice JE 3rd, Vannucci RC, Brierley JB: The influence of immaturity on hypoxic-isch-emic brain damage in the rat. Ann Neurol 1981; 9: 131–141.
41 Ment LR, Stewart WB, Fronc R, Seashore C, Mahooti S, Scaramuzzino D, Madri JA: Vas-cular endothelial growth factor mediates re-active angiogenesis in the postnatal develop-ing brain. Brain Res Dev Brain Res 1997; 100:
52–61. 42 Ment LR, Stewart WB, Duncan CC, Pitt BR,
Cole JS: Beagle puppy model of perinatal ce-rebral infarction. Regional cerebral prosta-glandin changes during acute hypoxemia. J Neurosurg 1986; 65: 851–855.
43 Adcock LM, Yamashita Y, Goddard-Fine-gold J, Smith CV: Cerebral hypoxia-ischemia increases microsomal iron in newborn pig-lets. Metab Brain Dis 1996; 11: 359–367.
44 Stave U: Age-dependent changes of metabo-lism. II. Influences of hypoxia on tissue en-zyme patterns of newborn and adult rabbits. Biol Neonat 1965; 8: 114–130.
45 Vannucci RC: Experimental models of peri-natal hypoxic-ischemic brain damage.APMIS Suppl 1993; 40: 89–95.
46 Myers RE: Four patterns of perinatal brain damage and their conditions of occurrence in primates. Adv Neurol 1975; 10: 223–234.
47 Sackett GP, Ruppenthal GC, Davis AE: Sur-vival, growth, health, and reproduction fol-lowing nursery rearing compared with mother rearing in pigtailed monkeys (Ma-caca nemestrina) . Am J Primatol 2002; 56:
165–184. 48 Behrman RE: Asphyxia in the primate foe-
tus. Acta Endocrinol Suppl (Copenh) 1972;
166: 258–259. 49 Myers RE: Cerebral ischemia in the develop-
ing primate fetus. Biomed Biochim Acta 1989; 48:S137–142.
50 Malik GK, Pandey M, Kumar R, Chawla S, Rathi B, Gupta RK: MR imaging and in vivo proton spectroscopy of the brain in neonates with hypoxic ischemic encephalopathy. Eur J Radiol 2002; 43: 6–13.
51 Robertson NJ, Lewis RH, Cowan FM, Allsop JM, Counsell SJ, Edwards AD, Cox IJ: Early increases in brain myo-inositol measured by proton magnetic resonance spectroscopy in term infants with neonatal encephalopathy. Pediatr Res 2001; 50: 692–700.
52 Kato T, Nishina M, Matsushita K, Hori E, Mito T, Takashima S: Neuronal maturation and N-acetyl- L -aspartic acid development in human fetal and child brains. Brain Dev 1997; 19: 131–133.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:321–330 DOI: 10.1159/000105473
Erythropoietin Enhances Long-Term Neuroprotection and Neurogenesis in Neonatal Stroke
Fernando F. Gonzalez
a, b Patrick McQuillen
a Dezhi Mu
b, d Yunsil Chang
b, e
Michael Wendland
c Zinaida Vexler
b Donna M. Ferriero
a, b
Departments of a Pediatrics, b
Neurology, and c Radiology, University of California, San Francisco, Calif. , USA;
d Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu , China;
e Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul ,
South Korea
Introduction
Stroke occurs in approximately 1 in 4,000 live full-term births [1] . This leads to adverse changes in central nervous system development and, consequently, in-creased mortality and long-term neurological morbidity. Despite these lifelong effects, no adequate postinjury strategy for treatment is currently available.
We have previously described a nonhemorrhagic stroke model in the neonatal rat using transient middle cerebral artery occlusion (MCAO) [2, 3] . This ischemia-reperfusion injury results in damage to the ipsilateral striatum and parietotemporal cortex. Erythropoietin (EPO) has been shown to play an important role in neu-ronal survival in both mature and immature rodent mod-els of brain injury [4–11] . Recent studies also demonstrate a beneficial effect of exogenous EPO administration in reducing infarct volume [12] , and improving short and long-term functional performance [12–14] . EPO may mediate protection via downregulation of proapoptotic genes [15, 16] , modulation of the inflammatory response [17–19] , reduction of glutamate toxicity [5, 20–22] , vaso-active and proangiogenic effects [13, 23, 24] , or direct stimulation of neuronal production from precursors [25, 26] .
Key Words
Ischemia � Neonatal brain injury � Neonatal stroke � Cell fate � Stem cell � Neurogenesis
Abstract
Neonatal stroke leads to mortality and severe morbidity, but there is no effective treatment currently available. Erythro-poietin (EPO) has been shown to promote cytoprotection and neurogenesis and decrease subventricular zone mor-phologic changes following brain injury. The long-term cel-lular response to EPO has not been defined, and local changes in cell fate decision may play a role in functional improve-ment. We performed middle cerebral artery occlusion in P10 rats. EPO treatment (5 U/g IP) significantly preserved hemi-spheric brain volume 6 weeks after injury. Furthermore, EPO increased the percentage of newly generated neurons while decreasing newly generated astrocytes following brain in-jury, without demonstrating long-term differences in the subventricular zone. These results suggest that EPO may neuroprotect and direct cell fate toward neurogenesis and away from gliogenesis in neonatal stroke.
Copyright © 2007 S. Karger AG, Basel
Received: September 6, 2006 Accepted after revision: October 7, 2006
Fernando F. Gonzalez, MD Departments of Neurology and Pediatrics, University of California, San Francisco 521 Parnassus Avenue, C215 San Francisco, CA 94143-0663 (USA) Tel. +1 415 502 5822, Fax +1 415 502 5821, E-Mail [email protected]
© 2007 S. Karger AG, Basel
Accessible online at:www.karger.com/dne

Gonzalez /McQuillen /Mu /Chang /Wendland /Vexler /Ferriero
Dev Neurosci 2007;29:321–330322
Focal ischemic injury has been found to temporarily stimulate precursor cell proliferation in the adult rodent forebrain subventricular zone (SVZ), which is a germina-tive area for new neurons that migrate to the olfactory bulb throughout life [27–30] . EPO is known to promote neurogenesis in vitro and in vivo [25] , and has also been shown to enhance neurogenesis in the SVZ following stroke in the adult rat [13] . We have also seen increased SVZ hypertrophy at an early time point in neonatal ro-dents, 2 weeks following MCAO, but response was de-creased in EPO-treated rats [12] , possibly from enhanced early migration of cells or decreased SVZ response sec-ondary to preservation of local tissue.
Given the changes in infarct and SVZ volumes ob-served with exogenous EPO, a question arises regarding this preserved tissue, and what types of cells populate the injured area. Neurogenesis has been demonstrated fol-lowing EPO treatment with an increase in newly gener-ated cells from precursors [13, 25, 26] , and possibly also an effect on cell fate in vitro [13, 25] . However, a variety of cell types besides neurons express EPO and EPO recep-tor (EPO-R) following brain injury [5] , including astro-cytes, endothelial cells, and microglia, and a glial response may play a role in volume preservation after injury. We set out to determine if EPO alters cell fate decision 6 weeks after transient MCAO by quantifying the proportion of newly generated neurons and astrocytes in the injured ar-eas, and we used unbiased stereology to calculate pre-served brain volume in vehicle and EPO-treated brains.
Materials and Methods
The University of California, San Francisco Institutional An-imal Care and Use Committee approved all animal research, and every effort was made to minimize animal suffering and reduce the number of animals used.
Cerebral Focal Ischemia – Reperfusion All surgical procedures were performed in 10-day-old Sprague-
Dawley rats; this age was chosen to approximate the development of the term human newborn [31] . Female rats with an 8- to 9-day old litter ( � 10 pups per litter) were bought from Simonson Labs (Gilroy, Calif., USA). Mothers were housed in a temperature and light-controlled facility and given food and water until pups were 10 days old. Transient focal cerebral ischemia was produced using the MCAO method with some modifications [2, 3] . Briefly, each pup was weighed and anesthetized with 3% isoflurane in a mix-ture of 70% N 2 O and 30% O 2 . Following induction of anesthesia, 1.5% isoflurane was maintained and rectal temperature was mon-itored and maintained at 36–37 ° C with a combination of heating blanket and overhead light. With the animal supine, the right common carotid artery, external carotid artery (ECA), and inter-
nal carotid artery were exposed with a midline cervical incision. The pterygopalatine, occipital, superior thyroid, maxillary, and lingual arteries were coagulated. After ligation of the ECA, a 5-0 nylon monofilament suture with blunted tip was inserted into the ECA lumen and gently advanced through the internal carotid ar-tery up to the middle cerebral artery until slight resistance was felt. Sham controls did not have the suture advanced. After place-ment of the suture, the wound was closed.
MR Imaging and EPO Treatment Each animal was examined by diffusion-weighted spin echo
planar imaging (SE EPI) at 25–30 min after MCAO. The entire brain was imaged with serial 2-mm thick coronal sections as pre-viously described [2, 12] using the following pulse sequence set-tings: TR/TE = 5,000/60 ms, 4 averages, field of view = 35 mm, data matrix 128 ! 128, diffusion gradient duration = 20 ms, sep-aration = 29.7 ms, amplitude = 70 mT/m, b-factor = 1,045 s/mm 2 . Reperfusion was achieved after 45 min of occlusion by removing the suture under isoflurane anesthesia. In a previous study, we found that an occlusion time of 45 min produced a moderate lev-el of injury involving the ipsilateral striatum and parietotemporal cortex, and reperfusion was confirmed with contrast study [12] . Animals that exhibited ischemic injury in atypical regions, such as brainstem, or that showed lack of cortical involvement were excluded from the study. Immediately upon reperfusion, a single dose of either vehicle [0.1% bovine serum albumin (Sigma, St. Louis, Mo., USA) in saline] or recombinant human EPO (a gift from Johnson and Johnson) at a dose of 5 units per gram of body weight was injected intraperitoneally. Following surgery, animals recovered from anesthesia and were returned to the dam until weaning. Most pups that received MCAO at P10 showed poor suckling during the first 2–3 days following surgery and were ga-vage fed, with daily weights measured for the first week to ensure adequate weight gain. All animals were injected with BrdU (5-bromo-2 � -deoxy-uridine; Roche, Indianapolis, Ind., USA) at a dose of 50 mg/kg/dose twice per day on post-MCAO days 7 through 9. This dosing regimen was chosen to limit labeling of proliferating glial cells just after brain injury, and for the maximal period of neurogenesis following adult brain injury [32] .
Histology For histopathologic examination, animals were anesthetized
with sodium pentobarbital (100 mg/kg; Nembutal, Abbott Labs, Abbott Park, Ill., USA) and sacrificed 6 weeks following surgery by transcardiac perfusion with ice-cold 4% paraformaldehyde in 0.1 M phosphate buffered saline (PBS, pH 7.4). Brains were care-fully removed and postfixed overnight, equilibrated in 30% su-crose in 0.1 M PBS and left at 4 ° C for a maximum of 72 h. Free-floating serial 40- � m coronal sections were collected throughout the brain in each animal using a freezing microtome. Sections were stored at 4 ° C in 0.1 M phosphate buffer with 0.1% sodium azide for a maximum of 2 weeks or stored in cryoprotectant at –20 ° C until staining.
Volumetric Analysis of Bilateral Brain Hemispheres and SVZ For volumetric analysis, sections were mounted and air-dried,
stained with cresyl violet, dehydrated in graded ethanol solutions, cleared in Citrisolv (Fisher Scientific, Pittsburgh, Pa., USA) and cover-slipped in Permount (Fisher Scientific, Pittsburgh, Pa., USA). Using systematic random sampling, a series representing

Erythropoietin in Neonatal Stroke Dev Neurosci 2007;29:321–330 323
every 12th section was selected, stained, and analyzed. Sections encompassed the whole striatum from the genu of the corpus cal-losum rostrally to the rostral hippocampus caudally. Volumetric quantifications were performed using a Nikon Eclipse E600 pho-tomicroscope equipped with a high-resolution CCD camera, a motorized XYZ axis computer-controlled stage, and Neurolucida and Neuroexplorer morphometry software package (Micro-BrightField, Inc., Colchester, Vt., USA). The cross-sectional areas of the region of interest (ROI) in each section was traced on the computer screen at low power using a 2!/4! lens and the volume of the ROI was calculated according to the Cavalieri principle [33] . For the ROI, the right and left hemisphere and SVZ were traced. Morphological criteria were used consistently in all ani-mals to determine the boundaries of the SVZ. Dorsolateral stria-tal extension of the SVZ was outlined, which resembles the dark band and thin long triangle in the sections corresponding to Plate 10–20 of the Paxinos Rat Brain Atlas [34] . Briefly, the superior-medial boundary of the SVZ was defined by the corpus callosum, the lateral boundary by the striatum, and the inferior boundary by the lateral ventricular margin. By using this sampling strategy, approximately 11 histological sections per brain for hemispheric measurement and 6 sections per brain for SVZ measurements were analyzed. Quantification was conducted by an examiner blinded to treatment group. Damage due to stroke was deter-mined quantitatively by calculating the percent preserved volume in the ipsilateral, or lesioned, hemisphere vs. the contralateral, control hemisphere. Size alteration of SVZ was investigated by calculating the percent SVZ volume in lesioned vs. control hemi-sphere in each animal.
Immunophenotyping and Confocal Analysis For quantititative analysis of cell types, the entire striatum was
sectioned coronally and a systematic random sampling of every 24th section (3–4 sections per animal) was double-immuno-stained for BrdU/NeuN, or BrdU/GFAP. Briefly, after overnight incubation in either mouse anti-NeuN (1: 800, Chemicon, Tem-ecula, Calif., USA) or rabbit anti-GFAP (1: 400, Chemicon, Tem-ecula, Calif., USA), sections were fixed in 4% PFA, denatured by incubation in 2 M hydrochloric acid at 37 ° C for 30 min, washed in 0.1 M Borate buffer and blocked in 25% goat serum (in TBS with 0.1% Triton-X) for 1 h before BrdU staining. Sections were incu-bated overnight with rat anti-BrdU (1: 200, Abcam, Cambridge, Mass., USA) and NeuN or GFAP in blocking solution. Sections were rinsed and incubated for 1 h with goat anti-rat Alexa 594 (1: 800, Molecular Probes, Eugene, Oreg., USA) and Alexa 488 sec-ondary antibodies in blocking solution. Sections were then mounted on Superfrost slides (Fisher Scientific, Pittsburgh, Pa., USA) and coverslipped using Vectashield (Vector Laboratories, Burlingame, Calif., USA).
Double-immunostained sections were imaged using a confo-cal scanning laser microscope (Zeiss LSM 510, Jena, Germany) at 20 ! objective (Plan-Apo lens, NA 0.75, camera resolution 1,024 ! 1,024 pixels, field dimensions 412 ! 412 ! 25 � m), and ana-lyzed using the LSM software (Zeiss LSM Image Browser Version 3.5.0.376). On each section, a tile scan of the entire hemisphere was imaged, the ROI (striatum) was outlined, and the 20 ! field was randomly placed within the ROI for imaging. Following im-aging of the full thickness z stack (2- � m steps) of the 20 ! field, the field was manually moved a fixed distance of approximately 400 � m in the horizontal and then vertical axis, resulting in 4–6
counting images per striatum per section. BrdU+ cells were quan-tified using Metamorph Offline (6.0, Universal Imaging Corpo-ration, Downington, Pa., USA) and double-labeled cells were counted manually. Cells were considered double-labeled if cola-beling with relevant morphology was seen throughout the extent of the nucleus for BrdU and NeuN, or if the cytoplasmic GFAP markings surrounded the nuclear BrdU marker, when viewed in x–y cross-section, as well as in x–z and y–z cross-sections pro-duced by reconstructions from z stacks taken with the 20 ! objec-tive. Cell density was calculated as average number of cells per 20 ! field, and cell percentage was calculated as total number of double-labeled cells per total number of BrdU+ cells.
Data Analysis Results are expressed as mean 8 SD. For volumetric analysis,
one-way analysis of variance (ANOVA) was performed with Stu-dent-Newman-Keuls post-hoc test for multiple comparisons. For cell counts, unpaired t test with Fisher’s PLSD was used. Pearson correlation coefficients compared statistical relationships. p val-ues below 0.05 were considered significant. All statistical analyses were performed using StatView (version 5.0.1, SAS software, Cary, N.C., USA).
Results
Twenty animals underwent transient MCAO and 8 underwent sham surgery; there was no mortality from the procedure. According to the inclusion criterion by MRI imaging, 17 of 20 MCAO animals were included and randomly allocated to MCAO with vehicle (n = 9) or MCAO with EPO treatment (n = 8). Two pups from the MCAO vehicle group and one from the MCAO EPO group died during the week following the procedure. The remaining survivors were divided as follows: sham sur-gery with vehicle (VS, n = 4), sham surgery with EPO (ES, n = 4), MCAO with vehicle (VO, n = 7), and MCAO with EPO (EO, n = 7). Male and female rats were evenly dis-tributed between the 4 groups, with no statistical signifi-cance between males and females in any of the analyses (data not shown).
EPO Preserves Hemispheric Brain Volume following MCAO Animals that received transient MCAO had signifi-
cant tissue loss in the ipsilateral striatum and parietotem-poral cortex at 6 weeks after injury ( fig. 1 a). In animals that underwent sham surgery, there was no difference in hemispheric volume between VS and ES treatment. In the VO group, there was a significant decrease in percent pre-served volume, but this tissue loss was significantly re-duced in the EO group, demonstrating a marked protec-tive effect ( fig. 1 b, c).

Gonzalez /McQuillen /Mu /Chang /Wendland /Vexler /Ferriero
Dev Neurosci 2007;29:321–330324
No Difference in SVZ Volume 6 Weeks after MCAO On cresyl violet staining, the SVZ is a darkened, nar-
row band between the corpus callosum and subcallosal striatum adjacent to the ventricular margin rostrally, which widens dorsolaterally beneath the corpus callosum through caudal sections ( fig. 2 a). Previously, we have shown an increase in SVZ size ipsilaterally 2 weeks after MCAO, with marked expansion rostrally and laterally, which decreased with EPO treatment [12] . In this study, there was no statistically significant difference in SVZ volume between the 4 groups at 6 weeks after injury ( fig. 2 b). Animals with severe injury did show an increase in SVZ size, with widened morphology ( fig. 2 c), which correlated with size of injury ( fig. 2 d); however, there was no difference between EPO and vehicle treatment.
EPO Did Not Increase Density of Newly Generated Cells in the Damaged Striatum On days 7 through 9 following MCAO, animals were
injected with BrdU, a thymidine analog incorporated during S phase, to label cells newly generated during this time period after injury. Given previous studies suggest-
ing an increase in production of cells from precursors in the SVZ following MCAO and EPO [12, 13, 25] , we looked at the production of all BrdU+ cells in the injured areas at 6 weeks. We did not find an increase in the density of newly generated cells following MCAO, in either the VO or the EO group ( fig. 3 ), suggesting that a single dose of EPO may have a larger effect on cell fate than number on days 7 through 9 after injury.
EPO Increases Neurogenesis in Damaged Striatum Sections were double-immunostained with antibody
to BrdU and NeuN, a neuronal nuclei monoclonal anti-body that is a marker for mature neurons, to identify newly generated cells that survived to become neurons in and near the site of injury ( fig. 4 ). EPO treatment led to a significant increase in both the density ( fig. 4 c) and pro-portion ( fig. 4 d) of newly generated neurons in the in-jured striatum, whereas the numbers in the contralateral striatum were not different from controls (data not shown). In addition, in the sham groups, EPO treatment significantly increased the density of newly generated cells that became neurons ( fig. 4 c), suggesting that EPO
VS0
c
0.2
0.4
0.6
0.8
1.2
Ipsi
/con
tra
hem
isp
her
e vo
lum
e1.0
ES
Group
VO
#
EO
, #*
Fig. 1. Evidence that exogenous EPO has a neuroprotective effect at 6 weeks after in-jury. a Cresyl violet-stained coronal sec-tion shows MCAO causes persistent injury and volume loss in the ipsilateral striatum ( * ) and cortex ( * * ) at 6 weeks. b Volume loss is improved following EPO treatment. Hole in contralateral hemisphere repre-sents left hemisphere identifier. c No dif-ference in ratio of ipsilateral, lesioned hemisphere vs. contralateral, control hemisphere volume between the ES and VS sham groups. There is a significant de-crease in ipsi/contra hemispheric brain volume in MCAO vs. sham animals ( # p ! 0.05 vs. VS, ES), with a significant protec-tive effect against hemispheric volume loss in EO vs. VO rats ( * p ! 0.004 vs. VO).

Erythropoietin in Neonatal Stroke Dev Neurosci 2007;29:321–330 325
plays a role in cell fate decision, even in the absence of injury, but that MCAO treatment results in a larger in-crease of new neurons following EPO treatment.
EPO Leads to a Decrease in GFAP-Positive Astrocytes in the Damaged Striatum A large number of BrdU+ cells in the injured striatum
did not express NeuN, perhaps indicative of gliosis or ac-cumulation of other cell types in the ischemic brain tis-sue. Therefore, GFAP, an anti-glial fibrillary acidic pro-tein polyclonal antibody that labels astrocytes, was used to examine the role of gliosis following MCAO ( fig. 5 ). In the MCAO groups, there was a significantly reduced pro-portion ( fig. 5 c) of newly generated cells that become as-trocytes in the striatum following EPO treatment. Like-wise, in the sham groups, EPO treatment reduced the percentage of newly generated astrocytes, suggesting a role for EPO in determining cell fate and favoring a neu-rogenic outcome.
VS0
a b
c d
0.5
1.0
1.5
2.0
2.5
00 0.1 0.2
Ipsi SVZ volume
0.3 0.4 0.5 0.6
20
40
60
80
100
120
140
160
180
200
Ipsi
/con
tra
SVZ
vol
ume
Ipsi
hem
isp
her
e vo
lum
e
ES
Group
VO EO
Fig. 2. Neither MCAO nor exogenous EPO has an overall significant effect on SVZ volume 6 weeks after injury. a Cresyl vio-let-stained coronal section showing the right SVZ (marked by arrows) in a sham animal, bordered by the corpus callosum superiomedially, the striatum laterally, and the lateral ventricle medially. b There was no difference in SVZ volumes at 6 weeks after injury between the 4 groups. c SVZ (arrows) in a severely injured ani-mal, with widened morphology rostrally and triangular shape caudally. d SVZ size correlates with severity of hemispheric in-jury (r 2 = 0.19, p ! 0.04).
VS0
5
10
15
20
25
30
35
40
45
Brd
U+
cel
ls/h
pf
ES
Group
VO EO
Fig. 3. EPO has no effect on density of newly generated cells in either the sham or MCAO groups. hpf = 20 ! field (412 ! 412 ! 25 � m).

Gonzalez /McQuillen /Mu /Chang /Wendland /Vexler /Ferriero
Dev Neurosci 2007;29:321–330326
Discussion
The results of this study demonstrate that a single dose of exogenous EPO immediately following transient MCAO preserves brain tissue and decreases infarct vol-ume 6 weeks after injury. EPO treatment leads to an in-crease in the percentage of newly born neurons in the injured areas, while decreasing the percentage of astro-cytes, without changing the overall density of newly born (BrdU+) cells. This suggests a dual role for EPO, promot-
ing not just neuroprotection, but also neurogenesis pos-sibly via the alteration of cell fate decisions, as opposed to increased production of all cell types.
EPO has previously been shown to preserve brain vol-ume following neonatal hypoxia-ischemia (HI) [6, 8, 14] , and to decrease infarct volume following transient MCAO in P7 rats [15] . We chose a single dose of EPO immedi-ately following injury, as opposed to during or prior to injury, to more closely approximate a clinical scenario where EPO may be used for treatment. This model, simi-
VS0
2
4
6
8
10
12
14
a
c d
b
(Neu
N +
Brd
U)/
hp
f
ES
#
Group
VO EO
* *
VS0
0.1
0.2
0.3
0.4
0.5
0.6
(Neu
N +
Brd
U)/
Brd
U
ES
Group
VO EO
Merge
BrdU
NeuN
Fig. 4. Evidence for exogenous EPO as a regulator of neurogenesis. Coronal 40- � m section of striatum immu-nostained with antibody to BrdU (red) and NeuN (green) to label newly generated cells that survived to become mature neurons by 6 weeks of age. a 20 ! confocal projection image. b Selection of individual 2- � m z steps of NeuN+ cells (top), BrdU+ cells (middle), and merge images (bottom). Colabeled cell marked by arrow. c EPO increased the density of newly generated neurons in sham ( # p ! 0.05) and MCAO animals ( * p ! 0.001 vs. VO). d EPO increased the proportion of newly generated cells that become neurons in MCAO animals ( * p ! 0.03 vs. VO).

Erythropoietin in Neonatal Stroke Dev Neurosci 2007;29:321–330 327
lar to other postinjury dosing regimens [14, 15] , has pre-viously been demonstrated to confer short-term histolog-ical and functional benefit [12] . In our model, similar to our findings at 2 weeks, there still persists preservation of brain tissue at 6 weeks. While EPO had no effect on volumes in sham animals, the significant difference in injured animals suggests neuroprotection. The mecha-nism by which this occurs has not been entirely elucidat-ed, but it may involve a decrease in intracellular calcium [21] , inhibition of nitric oxide-induced death [22] , or modulation of the inflammatory [17–19] or angiogenic responses [13, 23, 24] . In transient MCAO, Sola et al. [15] found a significant decrease in TUNEL-positive cells with EPO, suggesting an antiapoptotic effect. EPO in-duces an increase in phosphorylated Janus kinase 2 and signal transducer and activator of transcription-5 (STAT5) expression, resulting in upregulation of anti-apoptotic genes such as Bcl-xL [15, 16, 25, 35] and NF- � B [25] . EPO does not appear to protect against early injury
in the first 6 h [8] , rather at later time points, suggesting a delay is required for the responsible mechanism. This may be related to upregulation of EPO-R, synthesis of protein, or activation of a cascade that results in neuro-protection.
While these results suggest a role for EPO in preserva-tion of tissue, this may not be sufficient for long-term improvement, and neurogenesis may be a necessary re-sponse. In rodents, neurogenesis is thought to primarily occur during embryogenesis, with the exceptions being the olfactory bulb and dentate gyrus [36, 37] . The SVZ is a source of neurons that migrate to the olfactory bulb [38, 39] , and this neurogenesis peaks during the first 2 post-natal weeks [40] . Previously, the SVZ was found to be-come less cellular 3 weeks after HI [41] , but other studies have found an increase in ipsilateral SVZ size, propor-tional to the size of hemispheric damage [27, 28, 42] , with an increase in progenitor cells [43] and neuroblast migra-tion toward the site of injury [27, 28] ; however, these in-
ba
Merge
BrdU
GFAP
VS0
c
0.2
0.1
0.3
0.4
0.5
0.7
(GFA
P +
Brd
U)/
Brd
U
0.6
ES
Group
VO
#
�
EO
*
Fig. 5. EPO decreases astrogliosis follow-ing MCAO. a 20 ! confocal projection im-age. b Individual z steps of individually la-beled GFAP+ cells (top) and BrdU+ cells (middle), with merged images (bottom). Colabeled cells are marked by arrows. c MCAO increases proportion of newly generated astrocytes, while EPO decreases astrogliosis in both MCAO and sham ani-mals ( # p ! 0.04 vs. VS, VO, EO; * p ! 0.03 vs. VO, VS, ES; ^ p ! 0.03 vs. VS, ES, EO).

Gonzalez /McQuillen /Mu /Chang /Wendland /Vexler /Ferriero
Dev Neurosci 2007;29:321–330328
creases in SVZ size and cellularity following injury do not persist [27, 28] . Increases in BrdU+ density in the ipsilat-eral SVZ at 2 weeks following HI [44] , quinolinic acid-induced seizure [45] , and MCAO [27, 30, 46, 47] decrease to or near control levels 2–4 weeks later.
Local injury may initiate pathways that increase pre-cursor proliferation, but also enhance migration and de-termine cell fate. Parent et al. [47] found a transient in-crease in SVZ neurogenesis 2 weeks after MCAO, without changes in astrocytic markers, and migration and differ-entiation of region appropriate neurons in the peri-in-farct striatum. Collin et al. [45] also found an increase in SVZ cell proliferation and newly generated neuroblasts in the ipsilateral striatum following quinolinic acid-induced injury, with a gradient from medial to lateral, suggesting neurogenesis through recruitment of neuroblasts into the striatum. Arvidsson et al. [46] noticed an increase in BrdU+/NeuN+ cells in the lesioned striatum after stroke, as well as massive gliosis, but 80% of the newly generated neurons disappeared between 2 and 6 weeks. This sug-gests that following brain injury, new neurons migrate into the damaged areas, but local trophic factors/signals that play a role in migration and differentiation may be inadequate for survival.
EPO does show promise as a trophic factor that may support cell differentiation, survival, and possible incor-poration into neural networks. EPO increases the density of newly born cells and oligodendrocyte precursors in the striatum, corpus callosum, and SVZ [48] . In our study, there was no overall difference in SVZ volume between the groups at 6 weeks after injury, regardless of MCAO or EPO treatment. Hypertrophy was still seen in the ani-mals with the most severe injury, suggesting a relation-ship between injury size and precursor proliferation. Consistent with this, MCAO caused an increase in ipsi-lateral SVZ size and change in morphology at 2 weeks after injury, which was reduced with EPO treatment [12] . At 6 weeks, the overall lack of difference may represent a return of SVZ size to normal following migration [30] or a decrease in precursor cell proliferation and number af-ter the infarct has evolved. In addition, while newly gen-erated neurons migrate from the SVZ following injury, new astrocytes may proliferate locally as well, so the exact role of the SVZ in producing different cell types following EPO is not clear. To differentiate between these hypoth-eses will require careful quantification of cell type and number in the SVZ and surrounding infarct areas at se-quential time points following injury.
The most striking finding of our study is a shift from astrocytic to neuronal cell fate. In this model, despite a
lack of effect on SVZ size, there was an increased density and percentage of newly generated neurons (co-labeled with BrdU and NeuN) in the injured striatum. We did not, however, find an increased density of newly born (BrdU+) cells. Normally after brain injury, extensive gli-osis occurs near the site of injury [27, 28, 44] , which per-sists in contrast to newly generated neurons. Plane et al. [27] found increased BrdU+ and GFAP+ cells in the in-jured hemisphere at 1 week following HI, concentrated in the ipsilateral white matter and periventricular stria-tum. At 2 weeks, many newly generated neurons and as-trocytes were located near the lesioned striatum, and at 3 weeks widespread gliosis remained while new neurons decreased. Ong et al. [28] saw an increase in newly gen-erated neuroblasts and astrocytes 2 weeks following HI, but only astrocytes persisted at 4 weeks. Shingo et al. [25] found that hypoxia, via elevated EPO expression and blocked by anti-EPO antibody, induced a two- to three-fold increase in numbers of neurons/sphere in vitro. This increase in neuronal production was mimicked by EPO in a dose-dependent manner, but did not increase total cell number or astroglial cells vs. controls, and decreased the production of secondary stem cells. In vivo, intraven-tricular EPO increased newly generated neurons that migrated to the olfactory bulb, with rapid phosphoryla-tion of STAT5 and upregulation of NF- � B. This resulted in an increase in neurogenesis and a corresponding de-crease in secondary stem cells, suggesting a role for NF- � B in directing multipotent precursors to the neuronal lineage [25] . In addition, TUNEL labeling did not show differences between EPO and non-EPO treated, signify-ing a role for EPO in neurogenesis as opposed to cell sur-vival.
What mechanism underlies this change in cell fate commitment? EPO is increased by hypoxia and brain in-jury, and EPO and EPO-R have been detected in multiple cell lines in the brain, suggesting EPO could work in au-tocrine-paracrine fashion and result in neuroprotection and neurogenesis [5, 25] . EPO and EPO-R are weakly ex-pressed in the adult brain [49] , but are upregulated in the ischemic penumbra following permanent focal ischemia in adult mice [5] . EPO and EPO-R play important roles in embryonic development, including radial glia develop-ment, starting at E11 [50] . Upregulation of astrocytes and their precursors, such as radial glia, may be the source of newly generated neurons in the postnatal brain [51] . Fa-gel et al. [42] found an increased number of cells of the astroglial lineage proliferating in the neurogenic zones during recovery from perinatal hypoxia. These cells, via glial-derived factors or neuroblast production from ra-

Erythropoietin in Neonatal Stroke Dev Neurosci 2007;29:321–330 329
dial glia, may influence production and migration of cells to injured areas.
In this model of neonatal brain injury, a single dose of EPO immediately after injury preserved brain volume and increased neurogenesis while decreasing the glial re-sponse in the injured area. An important question to an-swer involves the long-term functional outcomes in this model. We have previously seen improved behavioral performance at 2 weeks after injury. Others have shown long-term improvements with EPO in visuospatial learn-ing and memory [14] . Given the evolution of injury over time [8] , it remains to be seen what dose and treatment regimen results in the best long-term outcomes. Taken together with previous studies, our observations suggest that EPO may be useful both early, for its direct neuro-
protective effect, and late, to influence progenitor cell fate and maximize neurogenesis. The findings of this study will need to be correlated with long-term behavioral per-formance, and the optimal dosing regimens defined to maximize the neurogenesis that occurs a week or later after injury.
Acknowledgements
This work was supported by a grant from Johnson & Johnson Pharmaceutical Research and Development, L.L.C., Raritan, N.J.; March of Dimes Birth Defect Foundation 6-FY 2006-465, andNS 35902 to D.M.F. The authors would like to acknowledge the technical assistance of Joel Faustino, BS.
References
1 Lynch JK, Nelson KB: Epidemiology of peri-natal stroke. Curr Opin Pediatr 2001; 13:
499–505. 2 Derugin N, Wendland M, Muramatsu K,
Roberts TP, Gregory G, Ferriero DM, Vexler ZS: Evolution of brain injury after transient middle cerebral artery occlusion in neonatal rats. Stroke 2000; 31: 1752–1761.
3 Mu D, Jiang X, Sheldon RA, Fox CK, Ham-rick SE, Vexler ZS, Ferriero DM: Regulation of hypoxia-inducible factor 1alpha and in-duction of vascular endothelial growth fac-tor in a rat neonatal stroke model. Neurobiol Dis 2003; 14: 524–534.
4 Sadamoto Y, Igase K, Sakanaka M, Sato K, Otsuka H, Sakaki S, Masuda S, Sasaki R: Erythropoietin prevents place navigation disability and cortical infarction in rats with permanent occlusion of the middle cerebral artery. Biochem Biophys Res Commun 1998;
253: 26–32. 5 Bernaudin M, Marti HH, Roussel S, Divoux
D, Nouvelot A, MacKenzie ET, Petit E: A po-tential role for erythropoietin in focal per-manent cerebral ischemia in mice. J Cereb Blood Flow Metab 1999; 19: 643–651.
6 Aydin A, Genc K, Akhisaroglu M, Yoruko-glu K, Gokmen N, Gonullu E: Erythropoie-tin exerts neuroprotective effect in neonatal rat model of hypoxic-ischemic brain injury. Brain Dev 2003; 25: 494–498.
7 Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, Itri LM, Cerami A: Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 2000; 97:
10526–10531. 8 Matsushita H, Johnston MV, Lange MS, Wil-
son MA: Protective effect of erythropoietin in neonatal hypoxic ischemia in mice. Neu-roreport 2003; 14: 1757–1761.
9 Kumral A, Ozer E, Yilmaz O, Akhisaroglu M, Gokmen N, Duman N, Ulukus C, Genc S, Ozkan H: Neuroprotective effect of erythro-poietin on hypoxic-ischemic brain injury in neonatal rats. Biol Neonate 2003; 83: 224–228.
10 Sun Y, Zhou C, Polk P, Nanda A, Zhang JH: Mechanisms of erythropoietin-induced brain protection in neonatal hypoxia-isch-emia rat model. J Cereb Blood Flow Metab 2004; 24: 259–270.
11 Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, Keenan S, Gleiter C, Pasquali C, Capobianco A, Mennini T, Heumann R, Cerami A, Ehrenreich H, Ghezzi P: Erythropoietin prevents neuronal apoptosis after cerebral ischemia and meta-bolic stress. Proc Natl Acad Sci USA 2001; 98:
4044–4049. 12 Chang YS, Mu D, Wendland M, Sheldon RA,
Vexler ZS, McQuillen PS, Ferriero DM: Erythropoietin improves functional and histological outcome in neonatal stroke. Pe-diatr Res 2005; 58: 106–111.
13 Wang L, Zhang Z, Wang Y, Zhang R, Chopp M: Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke 2004; 35: 1732–1737.
14 Kumral A, Uysal N, Tugyan K, Sonmez A, Yilmaz O, Gokmen N, Kiray M, Genc S, Du-man N, Koroglu TF, Ozkan H, Genc K: Erythropoietin improves long-term spatial memory deficits and brain injury following neonatal hypoxia-ischemia in rats. Behav Brain Res 2004; 153: 77–86.
15 Sola A, Rogido M, Lee BH, Genetta T, Wen TC: Erythropoietin after focal cerebral isch-emia activates the Janus kinase-signal trans-ducer and activator of transcription signal-ing pathway and improves brain injury in postnatal day 7 rats. Pediatr Res 2005; 57:
481–487. 16 Digicaylioglu M, Lipton SA: Erythropoietin-
mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature 2001; 412: 641–647.
17 Villa P, Bigini P, Mennini T, Agnello D, Lar-agione T, Cagnotto A, Viviani B, Marinovich M, Cerami A, Coleman TR, Brines M, Ghezzi P: Erythropoietin selectively attenuates cy-tokine production and inflammation in ce-rebral ischemia by targeting neuronal apop-tosis. J Exp Med 2003; 198: 971–975.
18 Agnello D, Bigini P, Villa P, Mennini T, Ce-rami A, Brines ML, Ghezzi P: Erythropoie-tin exerts an anti-inflammatory effect on the CNS in a model of experimental autoim-mune encephalomyelitis. Brain Res 2002;
952: 128–134. 19 Arvin B, Neville LF, Barone FC, Feuerstein
GZ: The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev 1996;
20: 445–452. 20 Dzietko M, Felderhoff-Mueser U, Sifringer
M, Krutz B, Bittigau P, Thor F, Heumann R, Buhrer C, Ikonomidou C, Hansen HH: Erythropoietin protects the developing brain against N-methyl- D -aspartate recep-tor antagonist neurotoxicity. Neurobiol Dis 2004; 15: 177–187.
21 Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R: Erythropoietin receptor is ex-pressed in rat hippocampal and cerebral cor-tical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 1997; 76: 105–116.

Gonzalez /McQuillen /Mu /Chang /Wendland /Vexler /Ferriero
Dev Neurosci 2007;29:321–330330
22 Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R: In vivo evi-dence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA 1998; 95: 4635–4640.
23 Yamaji R, Okada T, Moriya M, Naito M, Tsu-ruo T, Miyatake K, Nakano Y: Brain capillary endothelial cells express two forms of eryth-ropoietin receptor mRNA. Eur J Biochem 1996; 239: 494–500.
24 Chong ZZ, Kang JQ, Maiese K: Angiogenesis and plasticity: role of erythropoietin in vas-cular systems. J Hematother Stem Cell Res 2002; 11: 863–871.
25 Shingo T, Sorokan ST, Shimazaki T, Weiss S: Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci 2001; 21: 9733–9743.
26 Lu D, Mahmood A, Qu C, Goussev A, Schal-lert T, Chopp M: Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J Neu-rotrauma 2005; 22: 1011–1017.
27 Plane JM, Liu R, Wang TW, Silverstein FS, Parent JM: Neonatal hypoxic-ischemic in-jury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis 2004; 16: 585–595.
28 Ong J, Plane JM, Parent JM, Silverstein FS: Hypoxic-ischemic injury stimulates subven-tricular zone proliferation and neurogenesis in the neonatal rat. Pediatr Res 2005; 58: 600–606.
29 Jin K, Minami M, Lan JQ, Mao XO, Batteur S, Simon RP, Greenberg DA: Neurogenesis in dentate subgranular zone and rostral sub-ventricular zone after focal cerebral isch-emia in the rat. Proc Natl Acad Sci USA 2001;
98: 4710–4715. 30 Zhang RL, Zhang ZG, Zhang L, Chopp M:
Proliferation and differentiation of progeni-tor cells in the cortex and the subventricular zone in the adult rat after focal cerebral isch-emia. Neuroscience 2001; 105: 33–41.
31 Hagberg H, Bona E, Gilland E, Puka-Sund-vall M: Hypoxia-ischaemia model in the 7-day-old rat: possibilities and shortcomings. Acta Paediatr Suppl 1997; 422: 85–88.
32 Parent JM: Injury-induced neurogenesis in the adult mammalian brain. Neuroscientist 2003; 9: 261–272.
33 Regeur L, Pakkenberg B: Optimizing sam-pling designs for volume measurements of components of human brain using a stereo-logical method. J Microsc 1989; 155: 113–121.
34 Paxinos G: The Rat Brain in Stereotaxic Coordinates. San Diego, Academic Press, 1986.
35 Wen TC, Sadamoto Y, Tanaka J, Zhu PX, Na-kata K, Ma YJ, Hata R, Sakanaka M: Eryth-ropoietin protects neurons against chemical hypoxia and cerebral ischemic injury by up-regulating Bcl-xL expression. J Neurosci Res 2002; 67: 795–803.
36 Luskin MB, Parnavelas JG, Barfield JA: Neu-rons, astrocytes, and oligodendrocytes of the rat cerebral cortex originate from separate progenitor cells: an ultrastructural analysis of clonally related cells. J Neurosci 1993; 13:
1730–1750. 37 Lois C, Alvarez-Buylla A: Proliferating sub-
ventricular zone cells in the adult mamma-lian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci USA 1993; 90:
2074–2077. 38 Levison SW, Chuang C, Abramson BJ, Gold-
man JE: The migrational patterns and devel-opmental fates of glial precursors in the rat subventricular zone are temporally regulat-ed. Development 1993; 119: 611–622.
39 Levison SW, Goldman JE: Both oligoden-drocytes and astrocytes develop from pro-genitors in the subventricular zone of post-natal rat forebrain. Neuron 1993; 10: 201–212.
40 Bayer SA: 3H-thymidine-radiographic stud-ies of neurogenesis in the rat olfactory bulb. Exp Brain Res 1983; 50: 329–340.
41 Levison SW, Rothstein RP, Romanko MJ, Snyder MJ, Meyers RL, Vannucci SJ: Hypox-ia/ischemia depletes the rat perinatal sub-ventricular zone of oligodendrocyte progen-itors and neural stem cells. Dev Neurosci 2001; 23: 234–247.
42 Fagel DM, Ganat Y, Silbereis J, Ebbitt T, Stewart W, Zhang H, Ment LR, Vaccarino FM: Cortical neurogenesis enhanced by chronic perinatal hypoxia. Exp Neurol 2006;
199: 77–91.
43 Felling RJ, Snyder MJ, Romanko MJ, Roth-stein RP, Ziegler AN, Yang Z, Givogri MI, Bongarzone ER, Levison SW: Neural stem/progenitor cells participate in the regenera-tive response to perinatal hypoxia/ischemia. J Neurosci 2006; 26: 4359–4369.
44 Zaidi AU, Bessert DA, Ong JE, Xu H, Barks JD, Silverstein FS, Skoff RP: New oligoden-drocytes are generated after neonatal hypox-ic-ischemic brain injury in rodents. Glia 2004; 46: 380–390.
45 Collin T, Arvidsson A, Kokaia Z, Lindvall O: Quantitative analysis of the generation of different striatal neuronal subtypes in the adult brain following excitotoxic injury. Exp Neurol 2005; 195: 71–80.
46 Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O: Neuronal replacement from en-dogenous precursors in the adult brain after stroke. Nat Med 2002; 8: 963–970.
47 Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM: Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol 2002; 52: 802–813.
48 Zhang PB, Liu Y, Li J, Kang QY, Tian YF, Chen XL, Zhao JJ, Shi QD, Song TS, Qian YH: Ependymal/subventricular zone cells migrate to the peri-infarct region and differ-entiate into neurons and astrocytes after fo-cal cerebral ischemia in adult rats. Di Yi Jun Yi Da Xue Xue Bao 2005; 25: 1201–1206.
49 Siren AL, Knerlich F, Poser W, Gleiter CH, Bruck W, Ehrenreich H: Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol (Berl) 2001; 101: 271–276.
50 Knabe W, Knerlich F, Washausen S, Ki-etzmann T, Siren AL, Brunnett G, Kuhn HJ, Ehrenreich H: Expression patterns of eryth-ropoietin and its receptor in the developing midbrain. Anat Embryol (Berl) 2004; 207:
503–512. 51 Morshead CM, Garcia AD, Sofroniew MV,
van Der Kooy D: The ablation of glial fibril-lary acidic protein-positive cells from the adult central nervous system results in the loss of forebrain neural stem cells but not ret-inal stem cells. Eur J Neurosci 2003; 18: 76–84.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:331–340 DOI: 10.1159/000105474
Perinatal Hypoxic/Ischemic Brain Injury Induces Persistent Production of Striatal Neurons from Subventricular Zone Progenitors
Zhengang Yanga Steven W. Levisonb a Institutes of Brain Science and State Key Laboratory of Medical Neurobiology, Fudan University, Shanghai, PR China, and b Department of Neurology and Neurosciences, UMDNJ – New Jersey Medical School, Newark, N.J. , USA
Introduction
Ischemia-induced production of new striatal neurons has been described in both young and adult rodent mod-els. It has been considered transient, and some studies have suggested that it is of limited value as the vast num-bers of newly born neurons appear to die [Arvidsson et al., 2002; Parent et al., 2002; Jin et al., 2003; Plane et al., 2004; Ong et al., 2005; Felling et al., 2006; Yang and Levison, 2006]. However, a recent study showed that there is long-lasting ischemia-induced neuronogenesis (the process of generating new neurons from progenitors, versus neuro-genesis – the production of new neurons and glial cells [Caviness et al., 1995]) in the striatum of adult rats [Thored et al., 2006]. Studies have also suggested that there are multipotential progenitors scattered throughout the ner-vous system in addition to the stem/progenitor cells resid-ing in the subventricular zone (SVZ) [Palmer et al., 1995]. Moreover, the numbers of parenchymal progenitors are greater in the newborn than the adult brain [Laywell et al., 2000]. Because most studies to date have used parenchy-mally administered tracers for dividing cells, it has been impossible to establish whether the neurons that inhabit the ischemic penumbra were derived from local progeni-tors versus cells that migrated from more distant germinal zones. If we intend on devising pharmacological therapies to enhance brain repair after injury, it is essential to know which precursors to stimulate.
Key Words
Hypoxia � Ischemia � Neuronogenesis � Striatum � Stroke � Stem cells
Abstract
Ischemia-induced production of new striatal neurons in young and adult rodents has been studied. However, it is unclear whether neonatal hypoxic/ischemic (H/I) brain in-jury-induced neuronogenesis in the striatum is transient or sustained, nor has it been established whether these new neurons arise from progenitors within the striatum or from precursors residing in the adjacent subventricular zone. Here, we report that from 2 weeks to 5 months after H/I there are more doublecortin-positive (Dcx+) cells and Dcx+/NeuN+ cells in the damaged striatum compared to the con-tralateral striatum. After the S-phase marker 5-bromo-2 � -de-oxyuridine (BrdU) was injected at both short and long inter-vals (2 days and 2 months) after H/I to label newly born cells, more BrdU+/Dcx+ and BrdU+/NeuN+ cells were observed in the ipsilateral striatum compared to the contralateral stria-tum. Retroviral fate-mapping studies demonstrated that these newly born striatal neurons are generated from pre-cursors within the subventricular zone. Altogether, these ob-servations indicate the neonatal brain initiates a prolonged regenerative response from the precursors of the subven-tricular zone (SVZ) that results in persistent production of new striatal neurons. Copyright © 2007 S. Karger AG, Basel
Received: December 2, 2006 Accepted after revision: January 31, 2007
Steven W. Levison, PhD, Laboratory for Regenerative NeurobiologyDepartment of Neurology and Neuroscience and NJMS-UH Cancer Center UMDNJ – New Jersey Medical School 205 South Orange Avenue, H-1226, Newark, NJ 07103 (USA) Tel. +1 973 972 5162, Fax +1 973 972 2668, E-Mail [email protected]
© 2007 S. Karger AG, Basel
Accessible online at:www.karger.com/dne

Yang/Levison
Dev Neurosci 2007;29:331–340332
In this study, we assessed neuronogenesis in the stria-tum during acute recovery (2 weeks and 5 weeks) and de-layed recovery (2 months, 3 months and 5 months) from brain injury using the widely used Vannucci rat model of perinatal hypoxia/ischemia (H/I) [Vannucci et al., 2005]. The paradigm employed produces unilateral brain dam-age as a consequence of an acute reduction of blood flow and oxygenation to mimic the disruption in the delivery of nutrients and oxygen to the brain that is the primary cause of neurological injury during the perinatal period.
Materials and Methods
Animals Experimental animals were housed and cared for by the De-
partment of Comparative Medicine at the Veteran’s Administra-tion Animal care facility under the care of UMDNJ New Jersey Medical School Veterinarians. This facility is accredited by the Association for Assessment and Accreditation of Laboratory An-imal Care. All experimental animal protocols were approved by the UMDNJ IACUC Committee and animal experimentation was in accordance with the Society for Neuroscience’s policy on the appropriate use of animals for neuroscience research. The au-thors also certify that all experiments on animals were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication 80-23, rev. 1996), and the authors further attest that all efforts were made to minimize the number of animals used and their suffering.
Perinatal H/I Timed pregnant Wistar rats (Charles River, Wilmington, Del.,
USA) were maintained in the Laboratory Animal Care accredited facility for at least 3 days. After normal delivery, the litter size was adjusted to 12 pups per litter. Cerebral H/I was produced in 6-day-old rats (day of birth being P0), by a permanent unilateral com-mon carotid artery ligation followed by systemic hypoxia [Van-nucci et al., 2005]. Briefly, pups were lightly anesthetized with isoflurane (4% induction, 2% maintenance). Once fully anesthe-tized, a midline neck incision was made and the right common carotid artery was isolated by blunt dissection and then ligated using 3-0 silk. The incision was then sutured, and animals were returned to the dam for 1.5 h. The pups were prewarmed for 20 min in jars submerged in a 37 ° C water bath. They were then exposed to 80 min of humidified 8% O 2 /92% N 2 . Control animals were separated from the dam for the same amount of time as ex-perimental animals, but were otherwise not manipulated. Ap-proximately 60% of the animals sustained infarcts that involved both the ipsilateral striatum and neocortex and these animals were used in this study.
BrdU Injections Intraperitoneal injections of 5-bromo-2 � -deoxyuridine (BrdU;
50 mg/kg, Sigma, St. Louis, Mo., USA) were given twice daily for 3 days after 2 days (acute group) or 2 months (delayed group) re-covery from H/I ( fig. 1 A, F). For the acute group, animals were perfused 2 weeks (n = 4) or 5 weeks (n = 3) after H/I (days 9 or 30
after the last BrdU injection). For the delayed group, animals were killed 1 week (n = 4) or 5 weeks (n = 3) after the last BrdU injec-tion. At 2 days of recovery from H/I, another 3 rats each received a single injection of BrdU. Rats were perfused 2 h later. This short interval is sufficient for BrdU to incorporate into cells in S-phase but too brief for migration to occur [Nowakowski et al., 1989; Gould et al., 1999]. Another 2 rats were perfused at 5 months of recovery from H/I.
Retrovirus Injections Replication-incompetent retroviruses encoding the marker
gene human placental alkaline phosphatase (AP) were harvested from the psi2 DAP cell line (ATCC CRL-1949), concentrated, ti-tered, and tested for helper virus as described previously [Levison and Goldman, 1993]. The titer was 1 ! 10 5 colony-forming units/ml. P5 rat pups were anesthetized and 2 � l of retrovirus with 8 � g/ml of polybrene was injected stereotaxically into lateral ven-tricles at A: 1.2, L: 8 1.3, D: 2.5 mm. Rats were killed 1.5 days (n = 3), 2 weeks (n = 5), or 5 weeks (n = 5) after DAP virus injec-tions. Sections at 240- � m intervals were collected and processed for either AP histochemistry or AP immunofluorescence in com-bination with neuronal markers.
Immunohistochemistry Animals were anesthetized with a mixture of ketamine (75
mg/kg) and xylazine (5 mg/kg) prior to intracardiac perfusion with 4% paraformaldehyde. Brains were postfixed with 4% para-formaldehyde overnight and then cryoprotected for at least 24 h in 30% sucrose in PBS. The brain samples were frozen in embed-ding medium (OCT, Sakura Finetek, Torrance, Calif., USA) on a dry ice/ethanol slush.
Immunofluorescence staining was performed on 40- � m free-floating sections. Sections for BrdU staining were pretreated with 2 N HCl for 1 h at room temperature to denature DNA. Sections were blocked for 1 h in TBS with 10% donkey serum and 1–5% BSA. Primary antibodies were incubated for 24 h at 4 ° C. The fol-lowing antibodies were used: anti-doublecortin (Dcx; goat poly-clonal, Dcx-COOH terminus, 1: 100, Santa Cruz); anti-NeuN (mouse monoclonal, 1: 100, Chemicon); anti-BrdU (rat monoclo-nal, 1: 30, Accurate); anti-AP (rabbit polyclonal, 1: 30, Accurate). Secondary antibodies against the appropriate species were incu-bated for 2 h at RT (all from Jackson, 1: 200). All secondary anti-body combinations were carefully examined to ensure that there was no cross-talk between fluorescent dyes or cross-reactivity be-tween secondary antibodies, especially for anti-rat and anti-mouse secondary antibodies. DAPI (Sigma, 1 � g/ml) was used for 10 min (acute animals) or 15 min (chronic animals) to counter-stain nuclei.
Microscopy Fluorescently immunolabeled sections were analyzed on a
Zeiss LSM410 confocal laser scanning microscope using the fol-lowing filter sets with the indicated wavelengths (in nm) for the excitation laser line and emission filters: Cy2, 488/(510/540); rho-damine, 568/(590–610). Confocal Z sectioning was performed for double labeling. Images were acquired and three-dimensionally reconstructed using the Zeiss LSM software, cropped, adjusted and optimized in Photoshop 9.0. Images of enzyme histochemis-try labeled sections and some fluorescently immunolabeled sec-tions were acquired using an Olympus BX 41 microscope.

Striatal Neuronogenesis after H/I Dev Neurosci 2007;29:331–340 333
Cell Quantification Since the posterior part of the striatum was severely damaged
in this animal model, rostral striatal regions were analyzed (coro-nal sections were collected from the anterior tip of the corpus cal-losum). BrdU+/NeuN+ cells in the striatum were counted using
a Zeiss LSM410 confocal laser scanning microscope. Briefly, 6 sections at 240- � m intervals were quantified per brain. For each section, 6 fields in the dorsolateral striatum of both contralateral and ipsilateral hemispheres were analyzed. Confocal Z sectioning was performed at 0.5- � m intervals using Plan-Apochromat 63 !
Fig. 1. Newly born striatal neurons are generated within the SVZ. A , B Replication-incompetent retroviruses encoding the marker gene AP were bilaterally injected into the ventricles one day before inducing H/I to label dividing cells in the SVZ. AP histochemistry ( A , AP+ cells are blue) and immunofluorescence ( B , AP+ cells are red: the dorsolateral SVZ has slightly higher background staining, and therefore, appears as the slightly brighter red area) of the SVZ 1.5 days after injection. C1–C3 Single and double immunolabeling of the same microscopic field for AP/Dcx in the SVZ 1.5 days after injection of DAP retrovirus. D1 AP histochemistry at 2 weeks in the ipsilateral striatum. Neuron-like cells are identified by arrows.
D2 AP+ neuron-like cells in the ipsilateral striatum (arrows) and the neocortex (arrowhead). E1–E3 Immunofluorescence at 2 weeks for AP+ and Dcx+ cells in the striatum. F , G Five weeks after H/I, AP histochemistry ( F1–F3 ) and immunofluorescence ( G1–G2 ) re-veals cells with more mature neuronal morphologies in the ipsi-lateral striatum. H , I AP and NeuN double immunofluorescence in the ipsilateral striatum at 5 weeks recovery from H/I. Insets in H reveal AP+ spine-like appendages. J An AP+ but NeuN– cell in the ipsilateral striatum. K Image showing an AP/NeuN double+ cell in the contralateral striatum at 5 weeks recovery from H/I. Scale bars: 100 � m ( A , D ); 20 � m ( B , C ); 10 � m ( E–K ).

Yang/Levison
Dev Neurosci 2007;29:331–340334
oil-immersion objective (NA = 1.40) in each field (200 ! 200 � m; fig. 1 D). This method enabled us to accurately count the BrdU+/NeuN+ cells in the damaged striatum, including cells heavily la-beled with BrdU and strongly expressing NeuN, cells lightly la-beled with BrdU but strongly expressing NeuN, and cells heavily labeled with BrdU but lightly expressing NeuN. The total volume analyzed of each striatum was 6 fields ! 6 sections ! 40 � m sec-tion thickness ! 200 � m ! 200 � m = 0.0576 mm 3 .
Cell counts for AP+ neuron-like cells and AP+/NeuN+ cells were obtained in 6 sections of each brain at 240- � m intervals in the contralateral and ipsilateral striatum using a 60 ! objective on an Olympus BX41 microscope. The total number of cells was calculated by multiplying the number of cells counted per stria-tum and the sampling frequency.
Statistical Analysis Results from the cell counting were analyzed for statistical sig-
nificance using Student’s paired t test. All data are presented as means 8 SEM. Comparisons were interpreted as significant when associated with p ! 0.05.
Results
More Dcx+ and Dcx+/NeuN+ Cells Were Observed in the Ipsilateral Striatum Compared to the Contralateral Striatum of Young and Adult Rats after Perinatal H/I To investigate whether there is increased striatal neu-
ronogenesis after perinatal rat H/I, we evaluated immu-nostaining of Dcx-expressing cells in the striatum. Dcx is a microtubule-associated protein expressed by neuronal-ly committed precursors and immature neurons and has been used in numerous studies as a marker of adult neu-ronogenesis. Within the undamaged striatum of 3-week-old rats (2 weeks of recovery), Dcx+ cells could be found, but they were sparse. The processes of these cells were observed in both the striatal matrix and patch ( fig. 2 A, C). At 2 and 5 weeks of recovery from perinatal H/I, a readily visible increase in Dcx immunostaining was ob-served in the ipsilateral hemisphere compared to the con-tralateral hemisphere. In the ipsilateral striatum, large numbers of Dcx+ cells with morphologies of migrating neuroblasts were interspersed between the ipsilateral SVZ and striatum ( fig. 2 B, D1, D2, E, F2 and F3). In con-trast, Dcx+ cells were rare in the striatum of the contra-lateral striatum ( fig. 2 A, C, E). There was also a visible increase in the number of Dcx+/NeuN+ cells in the ipsi-lateral striatum compared to the contralateral striatum suggesting Dcx+ cells were maturing after H/I ( fig. 2 C, D, F).
To determine whether neonatal H/I not only initiates but sustains a regenerative response, we analyzed ani-
mals after 2 months, 3 months, and 5 months recovery from H/I. Although the brains were still damaged after long survival intervals after H/I, surprisingly, large num-bers of Dcx+ cells with an immature neuronal morphol-ogy were still present in the ipsilateral striatum compared to the contralateral striatum ( fig. 2 G–I). More Dcx+/NeuN+ cells were also observed in the ipsilateral stria-tum at 2, 3, and 5 months of recovery from H/I ( fig. 2 G–I). Dcx+ cells also migrated into the damaged neocortex ( fig. 2 F1), as we have recently described [Yang and Levi-son, 2006; Yang et al., 2007]).
BrdU-Labeled Neurons Are Continuously Generated in the Striatum of Young and Adult Rats after Perinatal H/I To determine whether these new striatal neurons are
continuously produced after perinatal H/I, the S-phase marker BrdU was injected into the peritoneal cavity dur-ing recovery to label newly born cells. Two hours after a single BrdU injection at 2 days recovery from H/I, BrdU+/Dcx+ cells were found in the SVZ. In contrast, few if any BrdU+/Dcx+ cells were detected in the striatum indicat-ing that Dcx+ cells are readily proliferating in the SVZ, but not in the striatum. BrdU was then injected into the animals for 3 days beginning at 2 days recovery from H/I, and BrdU/Dcx double immunofluorescence was per-formed at 2 weeks recovery ( fig. 3 A). More BrdU+/Dcx+ cells were observed in the ipsilateral striatum compared to the contralateral striatum ( fig. 3 B, C). BrdU was also injected into young adult animals for 3 days beginning at 2 months recovery from H/I, and BrdU/Dcx double im-munofluorescence was performed at 1 week after the last
Fig. 2. Dcx+ and Dcx/NeuN double+ cells are more abundant in the ipsilateral striatum of young and adult rats after perinatal H/I. A , B Dcx/NeuN double immunofluorescence in the contralat-eral ( A ) and ipsilateral ( B ) periventricular white matter and stria-tum at 2 weeks after H/I. LV = Lateral ventricle. C Confocal image of the boxed area of A reveals Dcx+ cells in the contralateral stri-atum. D1–D2 Confocal images of the boxed areas of B reveal large numbers of Dcx+ and Dcx/NeuN double+ cells in the ipsilateral striatum. E Dcx/NeuN double immunofluorescence in the con-tralateral and ispilateral hemisphere at 5 weeks after H/I. F Im-ages of the boxed areas in E showing large numbers of Dcx+ cells migrating into the ispilateral neocortex ( F1 ) and striatum ( F2 , F3 ). G–I Dcx/NeuN double immunofluorescence in the ipsilat-eral striatum at 2 months ( G ), 3 months ( H ) and 5 months ( I ) after H/I. Insets in G–I show Dcx/NeuN double+ cells. Scale bars: 100 � m in B for A and B ; 20 � m in D2 for C , D1 and D2 ; 500 � m in E ; 50 � m in F1 and F2 ; 10 � m in F3 ; 50 � m in G , H and I ; 10 � m in insets in G , H and I .

Striatal Neuronogenesis after H/I Dev Neurosci 2007;29:331–340 335

Yang/Levison
Dev Neurosci 2007;29:331–340336

Striatal Neuronogenesis after H/I Dev Neurosci 2007;29:331–340 337
BrdU injection ( fig. 3 F). This experiment revealed that the ipsilateral striatum contained a larger number of BrdU+/Dcx+ cells than the contralateral striatum ( fig. 3 G, H). Compared to the young animals (2 weeks recovery from H/I), the H/I-induced increase in BrdU+/Dcx+ cells was even more pronounced in the longer recovery group (2 months recovery from H/I). This may in part be due to the extremely low numbers of BrdU+/Dcx+ cells in the adult contralateral striatum compared to the juvenile striatum (compare fig. 3 G with fig. 2 B). These results in-dicate that there is protracted production of new neuro-blasts in the striatum during recovery from perinatal H/I.
To assess whether these newly born neuroblasts (BrdU+/Dcx+ cells) in the striatum of young and adult brain mature, BrdU/NeuN double immunofluorescence was performed at longer intervals after BrdU injections. At 5 weeks recovery from H/I (acutely analyzed animals, 30 days after the last BrdU injection), large numbers of BrdU+/NeuN+ cells were found in the ipsilateral stria-tum compared to the contralateral striatum ( fig. 3 E1–E6). Similarly in the adult rat brain, 5 weeks after the last BrdU injection at 2 months of recovery from H/I, many BrdU+/NeuN+ cells were found in the damaged striatum ( fig. 3 I–L). Quantifying the numbers of double-positive cells revealed a 7-fold and a 27-fold increase in the num-ber of newly born neurons (BrdU+/NeuN+ cells) in the
ipsilateral striatum compared to the contralateral stria-tum in both the acute and delayed recovery animals ( fig. 4 A; p ! 0.05, Student’s paired t test). These results suggest that some of the neuroblasts that are continuous-ly produced after perinatal H/I in the young and adult striatum differentiate into mature neurons.
The SVZ Is the Source of the Newly Born Striatal Neurons after H/I To establish whether these newly born neurons in the
striatum are descendants of the SVZ, we injected replica-tion-incompetent retroviruses encoding the marker gene AP bilaterally into the ventricles one day before inducing H/I to label dividing cells in the SVZ. When retrovirally infected animals were examined at 1.5 days survival, AP+ cells were mainly observed in the SVZ. Few if any AP+ cells were located outside of the SVZ ( fig. 1 A–C). Most of these AP+ cells expressed Dcx in the SVZ at this time point suggesting they are neuroblasts ( fig. 1 C). By 2 weeks of recovery from H/I, AP+ cells were found in the stria-tum and most of the AP+ cells with neuronal morpholo-gies were still expressing Dcx ( fig. 1 D, E).
By 5 weeks after H/I, cells with the typical morphol-ogy of mature neurons that were retrovirally labeled could be found in the striatum ( fig. 1 F–K). Cells with elaborately branched dendrites that were decorated with spine-like appendages could be observed ( fig. 1 F1, F3 and H). AP/NeuN double immunofluorescence revealed that a subset of these cells expressed NeuN ( fig. 1 H, I, K). Quantifying the numbers of AP-expressing neuron-like cells and AP+/NeuN+ immunofluorescently labeled cells in the ipsilateral striatum versus the contralateral stria-tum showed that there were significantly more neurons in the ipsilateral striatum ( fig. 4 B; p ! 0.005, Student’s paired t test). Newly generated astrocytes and oligoden-drocytes were also present within the retrovirally labeled cell population and AP-labeled myelinated axons were not uncommon within the pencil fibers ( fig. 1 F2).
Discussion
Neuronogenesis Is Sustained and May Be More Robust in the Immature than in the Adult Brain after Ischemic Injury The data that we provide here show that robust pro-
duction of new striatal neurons occurs after perinatal H/I. These new neurons are descendants of the SVZ and they migrate towards regions left cell sparse by the injury. Surprisingly, this neuronogenesis is sustained for months.
Fig. 3. Persistent production of neurons in the striatum of young and adult rats after perinatal H/I. A BrdU was injected on days 3–5 of recovery from H/I (P9–11), and animals were sacrificed 2 weeks (P20) and 5 weeks (P41) after H/I. B , C BrdU/Dcx double immunofluorescence was performed at 2 weeks after H/I. Repre-sentative examples of BrdU/Dcx double+ cells in the ipsilateral striatum ( C1–C4 ) and contralateral striatum ( B ). D Image of a representative section that was intensively scanned using the con-focal microscope to quantify numbers of BrdU/NeuN double im-munofluorescently labeled cells at 5 weeks post H/I. E1–E6 Con-focal images of BrdU/NeuN double+ cells in the ipsilateral stria-tum at 5 weeks recovery from H/I. E1–E5 Each set of 3 panels depicts single labels for BrdU and NeuN, with the larger panel the merged image. F Animals were injected with BrdU for 3 days be-ginning at 2 months after H/I and were sacrificed 1 and 5 weeks after the last BrdU injection. G , H BrdU/Dcx immunofluores-cence in the striatum showing at 2 months of recovery BrdU/Dcx double+ cells in the ipsilateral striatum ( H ) and the contralateral striatum ( G ) 1 week after last BrdU injection. I Nine consecutive 0.5- � m Z series, confocal images. J Z-stack reconstruction of pre-vious panels showing BrdU/NeuN double+ cells. K , L BrdU/NeuN double+ cells in the ipsilateral striatum 5 weeks after last BrdU injection at 2 months after H/I. Scale bars: 10 � m in all image ex-cept D , 100 � m.

Yang/Levison
Dev Neurosci 2007;29:331–340338
Our results are consistent with results from studies of adult animals after ischemia [Arvidsson et al., 2002; Par-ent et al., 2002; Thored et al., 2006]. As with the neuronal replacement that has been documented in the adult stria-tum [Thored et al., 2006], the neuronogenetic response of neonatal brain to ischemic injury is long-lasting. In-deed, the data presented here suggest that this regenera-tive response may be more robust in the immature than in the adult brain. In our study, we injected BrdU 6 times for 3 days beginning at 2 days of recovery from H/I. At 5 weeks after H/I, we found 23 BrdU+/NeuN+ cells in 0.0576 mm 3 , or approximately 400 BrdU+/NeuN+ cells per mm 3 in the damaged striatum. Arvidsson et al. [2002] reported 136 BrdU+/NeuN+ cells per mm 3 in the stria-tum when they administered BrdU 6 times for 3 days dur-ing days 4–6 after focal stroke. BrdU was available for an equivalent period of time, but we observed a greater num-ber of BrdU+/NeuN+ cells; therefore, the neuronogenesis may be more robust in the striatum of neonates after in-jury. This interpretation of our data is entirely consistent with the widely held view that the young nervous system possesses a greater capacity to recover from injury than the adult nervous system, although factors other than cell replacement may account for any functional recovery (Kennard principle) [Kennard, 1936].
Our data are complementary to and yet different from 2 other reports that used a similar animal model of peri-natal H/I. In those studies, an expansion of the SVZ was observed, and a similar increase in striatal neuronogen-esis was reported, but the newly generated neurons did
not appear to mature into NeuN+ cells [Plane et al., 2004; Ong et al., 2005]. On that basis, they concluded that most of the newly born neurons die from lack of trophic sup-port. It is not entirely clear why BrdU+/NeuN+ neurons were not found in the striatum after H/I in those studies. One possibility is that there are species differences in the neurogenetic response since perinatal mice and Sprague Dawley rats were used in those studies whereas perinatal Wistar rats were used in our study [Sheldon et al., 1998; Ray and Gage, 2006]. Another possibility is that in those studies animals were examined only 2 or 3 weeks after BrdU injection, which may be too short for BrdU-labeled neuroblasts to differentiate and express NeuN protein. Indeed, in our study, the majority of the NeuN+ cells re-tained their expression of Dcx, indicating that these cells are slow to mature. In a companion paper, we analyzed the extent to which newly generated neocortical inter-neurons are retained after perinatal H/I. In that study, we found that approximately 85% of the neurons produced by the SVZ after H/I do not survive [Yang et al., 2007]. Thus, while we agree with the overall conclusions of these authors that the extent of repair is limited by a lack of requisite trophic factors, it is not entirely absent as they have suggested.
The SVZ Appears to Be the Predominant Source of the New Striatal Neurons after Injury In the normal adult brain, not all neuroblasts from the
SVZ join the rostral migratory stream, some instead mi-grate into the striatum; however, this number is low
0
Nu
mb
er
of
Brd
U+
/Ne
uN
+ce
lls
5
10
15
20
25
30
Young
*
Adult
*
A
0
Nu
mb
er
of
cell
s
10
20
30
40
60
70
AP+
**
AP+/NeuN+
**
B
50
Fig. 4. Quantitative analysis of neurono-genesis after perinatal H/I. A BrdU was ad-ministered for 3 consecutive days either acutely or at a 2 months’ delay and the number of BrdU+/NeuN+ cells in the stri-atum (0.0576 mm 3 ) was quantified 4–5 weeks later. Cell counts were obtained from 3 animals at each age. B Number of AP+ neuronal cells and AP+/NeuN+ cells in the striatum at 5 weeks after perinatal H/I (n = 5). ) = Contralateral; $ = ipsilat-eral. * p ! 0.05; * * p ! 0.005, Student’s paired t test.

Striatal Neuronogenesis after H/I Dev Neurosci 2007;29:331–340 339
[Nacher et al., 2001]. At this time, we do not know how these neuroblasts migrate into the damaged striatum; however, we and others have documented a local expan-sion of the SVZ subsequent to injury, and in sagittal sec-tions of retrovirally injected animals there is a strong stream of labeled cells migrating rostrally to the olfactory bulb in the ipsilateral hemisphere that is not evidently different from the rostral migratory stream in the contra-lateral hemisphere [Plane et al., 2004; Ong et al., 2005; Fagel et al., 2006; Felling et al., 2006; Yang and Levison, 2006]. These observations suggest that the neuronogen-esis reported here is not entirely due to diverted rostral migration. Several reports have shown the presence of radial glia-like cells extending from the SVZ into the damaged striatum after neonatal injury; thus, these cells may provide passage from the SVZ to their destination [Ganat et al., 2002; Ong et al., 2005].
While present, the radial fibers do not typically extend far beyond the boundaries of the SVZ, thus other mecha-nisms must come into play. � -Chemokine stromal cell-derived factor-1 (SDF-1; CXCL-12) has been shown to regulate normal interneuron migration to the neocortex [Stumm et al., 2003] and an immunohistochemical anal-ysis of SDF-1 after perinatal H/I found that SDF-1 was induced significantly within the striatum [Miller et al., 2005]. Analogously, in an adult model of ischemic brain damage, SDF-1 was increased in the striatum and the SDF-1 receptor antagonist, AMD3100 abrogated the colo-nization of the striatum by Dcx+ cells [Thored et al., 2006]. As SDF-1 attracts CXC-motif receptor 4 (CXCR-4)-expressing adult neural stem cells [Imitola et al., 2004], these data lead to the conclusion that SDF-1 is likely an important regulator of the migration of these new neu-rons to the damaged striatum.
It has been suggested that newly-generated neurons may be derived from precursors residing outside the SVZ [Palmer et al., 1995]. However, our analysis of BrdU in-corporation at short survival intervals indicate that few Dcx+ cells outside of the SVZ are proliferating. We ob-served large numbers of BrdU+/Dcx+ cells within the SVZ and large numbers of Dcx+ cells with migratory pro-files distributed between the SVZ and the damaged stria-tum. This conclusion is further supported by the retrovi-rus fate mapping studies, which showed early and spe-cific labeling of SVZ cells, with virally labeled cells subsequently appearing within the striatum and possess-ing both morphological and antigenic features of mature neurons. Our data are also supported by studies of neu-ronogenesis after adult ischemia, which demonstrated that newly born striatal neurons are generated from
GFAP-expressing SVZ cells [Yamashita et al., 2006]. Nonetheless, we cannot exclude the possibility that some of the newly-generated neurons are derived from local precursors or from glial progenitors that de-differentiate and become re-specified as neurons.
The Regenerative Capacity of the Neonatal SVZ Our group has studied the effects of perinatal H/I on
the SVZ at both early and late recovery time points after mild, moderate and severe insults. These studies have re-vealed that during the first 2 days of recovery from H/I both restricted neural progenitors [Romanko et al., 2004] and restricted oligodendrocyte progenitors within the SVZ are vulnerable to insult [Levison et al., 2001; Ness et al., 2001]. By contrast, the cells that inhabit the most me-dial aspect of the SVZ (which is where the putative neural stem cells reside) appear to tolerate the insult with few adverse effects [Romanko et al., 2004]. Using immuno-histochemical markers for stem cells and progenitors, we found that cells in the most medial region of the SVZ, which express nestin, but do not express PSA-NCAM, survive severe perinatal H/I insults, whereas PSA-NCAM+ cells and oligodendrocyte progenitors are more vulnerable [Skoff et al., 2001; Rothstein and Levison, 2002; Romanko et al., 2004]. Our most recent studies in-dicate that as early as 3 days after an insult, and sustained for the first week of recovery from perinatal H/I, there is an expansion in the numbers of tri-potential neural stem-like cells [Felling et al., 2006; Yang and Levison, 2006]. We also established that the precursors within the SVZ after H/I have a greater potential to differentiate into both neurons and oligodendrocytes in vitro [Yang and Levi-son, 2006]. Here, we demonstrate that this potential is indeed realized in vivo, where during the weeks and months after injury, the expanded pool of the SVZ pre-cursors sustains a regenerative process aimed at replacing deleted neurons and glial cells. These observations sup-port the view that the neural stem/progenitors of the SVZ are a viable target for therapeutic strategies to enhance recovery after neonatal brain injury.
Acknowledgements
This work was supported by a grant from the NIH (MH 59950 awarded to S.W.L.). We thank P. Gupta for assistance in titering the retroviruses and J. Menonna for assistance with the confocal imaging. We thank Matt Covey for comments on the manuscript and also thank P. Dowling for his support and encouragement.

Yang/Levison
Dev Neurosci 2007;29:331–340340
References
Arvidsson A, Collin T, Kirik D, Kokaia Z, Lind-vall O (2002): Neuronal replacement from endogenous precursors in the adult brain af-ter stroke. Nat Med 8: 963–970.
Caviness VS Jr, Takahashi T, Nowakowski RS (1995): Numbers, time and neocortical neu-ronogenesis: a general developmental and evolutionary model. Trends Neurosci 18:
379–383. Fagel DM, Ganat Y, Silbereis J, Ebbitt T, Stewart
W, Zhang H, Ment LR, Vaccarino FM (2006): Cortical neurogenesis enhanced by chronic perinatal hypoxia. Exp Neurol 199: 77–91.
Felling RJ, Snyder MJ, Romanko MJ, Rothstein RP, Ziegler AN, Yang Z, Givogri MI, Bongar-zone ER, Levison SW (2006): Neural stem/progenitor cells participate in the regenera-tive response to perinatal hypoxia/ischemia. J Neurosci 26: 4359–4369.
Ganat Y, Soni S, Chacon M, Schwartz ML, Vac-carino FM (2002): Chronic hypoxia up-reg-ulates fibroblast growth factor ligands in the perinatal brain and induces fibroblast growth factor-responsive radial glial cells in the sub-ependymal zone. Neuroscience 112:
977–991. Gould E, Reeves AJ, Graziano MS, Gross CG
(1999): Neurogenesis in the neocortex of adult primates. Science 286: 548–552.
Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD, Frenkel D, Li J, Sidman RL, Walsh CA, Snyder EY, Khoury SJ (2004): Di-rected migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci USA 101: 18117–18122.
Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S, Greenberg DA (2003): Directed migration of neuronal precursors into the ischemic cere-bral cortex and striatum. Mol Cell Neurosci 24: 171–189.
Kennard MA (1936): Age and other factors in motor recovery from precentral lesions in monkeys. Am J Physiol 115: 138–146.
Laywell ED, Rakic P, Kukekov VG, Holland EC, Steindler DA (2000): Identification of a mul-tipotent astrocytic stem cell in the immature and adult mouse brain. Proc Natl Acad Sci USA 97: 13883–13888.
Levison SW, Goldman JE (1993): Both oligoden-drocytes and astrocytes develop from pro-genitors in the subventricular zone of post-natal rat forebrain. Neuron 10: 201–212.
Levison SW, Rothstein RP, Romanko MJ, Snyder MJ, Meyers RL, Vannucci SJ (2001): Hypox-ia/ischemia depletes the rat perinatal sub-ventricular zone of oligodendrocyte progen-itors and neural stem cells. Dev Neurosci 23:
234–247. Miller JT, Bartley JH, Wimborne HJ, Walker AL,
Hess DC, Hill WD, Carroll JE (2005): The neuroblast and angioblast chemotaxic factor SDF-1 (CXCL12) expression is briefly upreg-ulated by reactive astrocytes in brain follow-ing neonatal hypoxic-ischemic injury. BMC Neurosci 6: 63.
Nacher J, Crespo C, McEwen BS (2001): Double-cortin expression in the adult rat telencepha-lon. Eur J Neurosci 14: 629–644.
Ness JK, Romanko MJ, Rothstein RP, Wood TL, Levison SW (2001): Perinatal hypoxia-isch-emia induces apoptotic and excitotoxic death of periventricular white matter oligodendro-cyte progenitors. Dev Neurosci 23: 203–208.
Nowakowski RS, Lewin SB, Miller MW (1989): Bromodeoxyuridine immunohistochemis-try determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. J Neuro-cytol 18: 311–318.
Ong J, Plane JM, Parent JM, Silverstein FS (2005): Hypoxic-ischemic injury stimulates subven-tricular zone proliferation and neurogenesis in the neonatal rat. Pediatr Res 58: 600–606.
Palmer TD, Ray J, Gage FH (1995): FGF-2-re-sponsive neuronal progenitors reside in pro-liferative and quiescent regions of the adult rodent brain. Mol Cell Neurosci 6: 474–486.
Parent JM, Vexler ZS, Gong C, Derugin N, Fer-riero DM (2002): Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol 52: 802–813.
Plane JM, Liu R, Wang TW, Silverstein FS, Par-ent JM (2004): Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis 16: 585–595.
Ray J, Gage FH (2006): Differential properties of adult rat and mouse brain-derived neural stem/progenitor cells. Mol Cell Neurosci 31:
560–573.
Romanko MJ, Rothstein RP, Levison SW (2004): Neural stem cells in the subventricular zone are resilient to hypoxia/ischemia whereas progenitors are vulnerable. J Cereb Blood Flow Metab 24: 814–825.
Rothstein RP, Levison SW (2002): Damage to the choroid plexus, ependyma and subependy-ma as a consequence of perinatal hypoxia/ischemia. Dev Neurosci 24: 426–436.
Sheldon RA, Sedik C, Ferriero DM (1998): Strain-related brain injury in neonatal mice subjected to hypoxia-ischemia. Brain Res 810: 114–122.
Skoff RP, Bessert DA, Barks JD, Song D, Cerghet M, Silverstein FS (2001): Hypoxic-ischemic injury results in acute disruption of myelin gene expression and death of oligodendro-glial precursors in neonatal mice. Int J Dev Neurosci 19: 197–208.
Stumm RK, Zhou C, Ara T, Lazarini F, Dubois-Dalcq M, Nagasawa T, Hollt V, Schulz S (2003): CXCR4 regulates interneuron mi-gration in the developing neocortex. J Neu-rosci 23: 5123–5130.
Thored P, Arvidsson A, Cacci E, Ahlenius H, Kallur T, Darsalia V, Ekdahl CT, Kokaia Z, Lindvall O (2006): Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells 24: 739–747.
Vannucci RC, Brucklacher RM, Vannucci SJ (2005): Glycolysis and perinatal hypoxic-ischemic brain damage. Dev Neurosci 27:
185–190. Yamashita T, Ninomiya M, Hernandez Acosta P,
Garcia-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K (2006): Subventricular zone-derived neuro-blasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J Neurosci 26: 6627–6636.
Yang Z, Covey M, Bitel C, Ni L, Jonakait G, Le-vison SW (2007): Sustained neocortical neu-rogenesis after neonatal hypoxic/ischemic injury. Ann Neurol, in press.
Yang Z, Levison SW (2006): Hypoxia/ischemia expands the regenerative capacity of progen-itors in the perinatal subventricular zone. Neuroscience 139: 555–564.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:341–354 DOI: 10.1159/000105475
Uteroplacental Inflammation Results in Blood Brain Barrier Breakdown, Increased Activated Caspase 3 and Lipid Peroxidation in the Late Gestation Ovine Fetal Cerebellum
Lisa C. Hutton Margie Castillo-Melendez David W. Walker
Fetal and Neonatal Research Group, Department of Physiology, Monash University – Clayton Campus, Melbourne , Australia
Introduction
Children born preterm or with low birth weights have a high risk of cerebellar damage [1–3] . The alterations seen in the cerebella of preterm infants have been shown to persist into adolescence and are associated with cogni-tive deficits [4] . Furthermore, a consistent neurological abnormality present in autistic individuals is Purkinje cell loss, which is thought to arise not from cell death af-ter birth, but as a developmental abnormality during ges-tation [reviewed in 5 ]. Postmortem studies of still-born human fetal brains reveal strong 4-hydroxynonenal (4HNE) immunoreactivity – a marker of lipid peroxida-tion – in cerebellar Purkinje cells [6] .
In the human fetus, the critical period of cerebral neu-ronal development is up to 28 weeks of gestation, where-as the period of rapid cerebellar growth is from approxi-mately 28 weeks gestation to term [2] . The rapid develop-ment of the cerebellum in late gestation may make it vulnerable to infectious and hypoxic challenges in utero, as well as disturbances in the trajectory of development caused by premature birth [2] . The brain stem and cere-bellum share the same arterial blood supply, and yet blood supply to the brainstem is higher than for other brain regions [7] and usually preserved or increased dur-ing hypoxic fetal stress [7–9] . In contrast, blood flow and oxygen transport to the cerebellum is reduced in response
Key Words
Uteroplacental inflammation � Blood brain barrier breakdown � Perinatal brain damage � Caspase 3 � Lipid peroxidation � Lipopolysaccharide
Abstract
Maternal infection is associated with perinatal brain dam-age, but effects on the cerebellum are not known in detail. In this study, we examined the effects of placental inflamma-tion induced by administering lipopolysaccharide into the uterine artery of pregnant sheep at 134–136 days gestation. The fetal brain was collected 72 h later and compared to brains collected from age-matched untreated fetuses. Pla-cental lipopolysaccharide treatment had substantial effects on the fetal cerebellum, including increasing the number of cells undergoing apoptosis, widespread lipid peroxidation, and extravasation of plasma albumin, suggesting compro-mise of the cerebellar blood-brain barrier. These effects may account for some of the learning and motor deficits that emerge in neonates from pregnancies compromised by in-fection. Copyright © 2007 S. Karger AG, Basel
Received: September 5, 2006 Accepted after revision: December 8, 2006
Lisa Hutton Department of Physiology, Monash University – Clayton Campus Wellington RoadClayton, Victoria 3800 (Australia) Tel. +61 3 9905 2356, Fax +61 3 9905 2547, E-Mail [email protected]
© 2007 S. Karger AG, Basel0378–5866/07/0295–0341$23.50/0
Accessible online at:www.karger.com/dne

Hutton/Castillo-Melendez/Walker
Dev Neurosci 2007;29:341–354342
to hypoxia [7] . The extensive synaptic connections of Purkinje cells result in high metabolic demand and a high level of calcium uptake [10] . The high metabolic rate of the cerebellum may increase the vulnerability of Purkin-je cells to oxidative and metabolic stress. Purkinje cell damage has been linked to many adult diseases such as epilepsy, Huntington’s disease, Alzheimer’s disease and mitochondrial disorders [reviewed in 11 ]. In animal models of experimental intrauterine infection and in-flammation, cerebellar injury is often overlooked.
In the current study, a range of immunohistochemical markers are used in order to investigate the cerebellar re-sponse to uteroplacental inflammation. Lectin is pre-dominantly used as a marker of inflammatory cells, such as microglia and macrophages. Whilst playing an impor-tant role in the phagocytosis of cellular debris, upon re-cruitment or activation, these cells are also associated with the production of neurotoxic substances such as proinflammatory cytokines, quinolinic acid and nitric oxide [12–15] . Astrocytes have been shown to respond to inflammatory stimuli, with marked astrogliosis (hyper-trophy and hyperplasia of astrocytes). The integrity of the blood brain barrier (BBB) may be investigated by the de-tection of large plasma proteins (such as albumin) in the brain, which is indicative of BBB breakdown, allowing passage of these proteins into the brain. Lipid peroxida-tion (indicated by 4HNE immunohistochemical stain-ing) is an important pathological process as a result of inflammatory insults.
Whilst the developing brain contains a large number of cells in the process of proliferation and apoptosis, al-terations in the balance between the two processes may be associated with pertubations of normal brain develop-ment. The utilization of markers such as activated cas-pase 3 and Ki67 provide an indication of whether this balance is altered, in addition to information regarding the recovery of the brain by an increase in proliferating cells to replace those which may succumb to cell death. An understanding of the types of cells which are particu-larly susceptible to apoptosis is useful in the investigation of the pathology associated with inflammatory induced brain damage.
Placental inflammation has been proposed to be the link between maternal infection and fetal brain damage, given the ability of the placenta to produce proinflamma-tory cytokines and other potentially neurotoxic substanc-es that can be detrimental to the fetal brain [16, 17] . Many previous studies investigating the effects of inflammatory stimuli on the developing brain have administered lipo-polysaccharide (LPS) directly to the fetus or neonate [18–
21] , thus mimicking a situation that is likely to be the end-point of an infection cascade that begins as a maternal infection, and progresses to profound intra-amniotic and placental infection. In the current study, we administered LPS into the uterine artery (UTA) of pregnant sheep. As evidence suggests that LPS does not cross the ovine pla-centa [22] , we sought evidence for the hypothesis that fetal cerebellar damage can arise as a consequence of uteropla-cental inflammation, rather than as a result of the direct effect of the endotoxin on the fetal brain.
Methods
Animals and Surgical Procedures Twenty Merino/Border-Leicester cross, time-mated pregnant
ewes with singleton fetuses were housed in individual cages in a controlled environment at 22 ° C with a 12 h light/dark cycle, and at least one other sheep present at all times. Sheep were fed Lu-cerne chaff between 09: 00 and 11: 00 h and with access to water at all times. All experimental procedures had received prior approv-al from the Standing Committee on Ethics and Animal Experi-mentation of Monash University. All experiments and animal care were conducted according to the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. At 124–126 days of gestation (term is approximately 147 days) each ewe was anesthetized by inhalation of 2% halothane (Fluothane; Me-rial, Australia) in oxygen for surgical implantation of polyvinyl catheters into a maternal carotid artery and jugular vein. A mid-line laparotomy was performed, a fetal hind limb exteriorized and a catheter inserted into the pedal artery and advanced until the tip was judged to have entered the dorsal aorta. The uterus and uterine membranes were then closed. Suitable tributaries of the uterine vein and UTA were catheterized with the tip of the cath-eter advanced until identified to be in a main vein or artery of the uterine horn, with the tip of the catheter located at the level of the cervix. A bolus i.v. injection of ampicillin (1 g; Aspen Pharmacare Australia Pty, Ltd.) was given to the ewe during the surgery. Cath-eters were flushed daily with heparinized saline (50 U/ml; Hepa-rin, Pharmacia, Australia) to maintain catheter patency.
Experimental Protocol At 134–136 days of gestation, 30 (n = 12) or 60 � g (8 singleton
pregnancies) LPS (derived from Eschericia coli , Serotype 0127:B8, Sigma, St. Louis, Mo., USA) was administered in 3 ml sterile saline into the UTA. Maternal and fetal arterial blood samples were tak-en at intervals before and after the LPS treatments for measure-ment of blood gases and acid-base status. All fetuses and ewes were killed by an i.v. overdose of pentobarbitone sodium (Lethabarb; Virbac Pty Ltd, Australia) 72 h after the administration of LPS into the UTA. Fetal brains were collected from fetuses that survived until 72 h after LPS administration (30 � g, n = 7; 60 � g, n = 6), for subsequent analysis of brain histology. Brains were perfused trans-cardially with approximately 1 l of normal saline containing 50 U/ml heparin (Pharmacia, Australia) at 90 ml/min and 40 mm Hg, followed by 1 l 4% paraformaldehyde (PFA; Probing & Structure, Australia) in 0.1 M phosphate buffer (pH 7.4). The brains were re-moved and postfixed in 4% PFA for 24 h. Brains were also col-

Uteroplacental Inflammation and the Fetal Cerebellum
Dev Neurosci 2007;29:341–354 343
lected from age-matched untreated fetuses (n = 6) with collection and processing identical to that for the LPS-treated fetuses. The cerebellum was then bisected into hemispheres and embedded in paraffin wax. Sagittal sections (10 � m) were cut for the histologi-cal and immunohistochemical procedures outlined below.
Immunohistochemistry Sections were dewaxed and rehydrated through graded etha-
nols before commencing the procedures outlined below. All washes were carried out in phosphate-buffered saline (PBS, 0.1 M , pH 7.4). For each immunohistochemical protocol, sections con-taining a particular brain region from all treatment groups were included in a single run to eliminate variations between runs.
Microglia and Macrophages Lectin immunohistochemistry was used to detect microglia
and macrophages. Briefly, antigen retrieval was performed using microwave irradiation (3 bursts of 5 minutes’ duration) in 0.01 M citric acid buffer (pH 6). Sections were then allowed to cool in buffer for 20 min, prior to blocking of endogenous peroxidases by incubation in 0.3% H 2 O 2 in 50% methanol for 15 min. Sections were thoroughly washed and nonspecific binding was blocked by incubation in 5% normal goat serum (NGS) and 2% bovine serum albumin (BSA) in 0.1% Triton-X100 in PBS for 60 min. Peroxi-dase-labeled isolectin B4 derived from Bandeiraea simplicifolia ( Griffonia simplicifolia ; Sigma Aldrich, USA) diluted 1: 200 in PBS was added and the sections were incubated overnight at 4 ° C. Lec-tin was then washed from sections prior to the addition of diami-nobenzidine (DAB; Pierce, USA).
Astrocytes Glial fibrillary acidic protein (GFAP) immunohistochemistry
was performed by incubating sections in 0.3% H 2 O 2 in 50% meth-anol for 20 min, prior to blocking with 5% normal rabbit serum in PBS for 45 min. Mouse monoclonal anti-GFAP primary antibody (Sigma, USA) was added (diluted 1: 400 in PBS, with 0.3% Triton-X100) and left overnight at 4 ° C. Sections were washed thoroughly prior to the addition of biotinylated rabbit anti-mouse secondary antibody (1: 400, DAKO, USA) in PBS for 1 h. Sections were washed and incubated in streptavidin horseradish peroxidase (Strep HRP; 1: 200; GE Healthcare, Australia) followed by staining with DAB.
Lipid Peroxidation 4HNE is a by-product of lipid peroxidation. 4HNE immuno-
histochemistry was performed using antigen retrieval (3 ! 4 min microwave irradiation in citric acid buffer) followed by blocking of endogenous peroxidases using 0.3% H 2 O 2 in 50% methanol for 10 min. Sections were incubated in 5% NGS and 5% BSA blocking solution in 0.3% Triton-X100 for 90 min followed by overnight incubation in rabbit anti-4HNE primary antibody (1: 1250; Alex-is Biochemicals, Switzerland) in PBS with 0.3% Triton-X100, 5% NGS and 2% BSA. Biotinylated goat anti-rabbit secondary anti-body (Vector, USA) diluted 1: 200 in PBS was added for 1 h. Sec-tions were then washed and incubated in Strep HRP (1: 200) fol-lowed by staining with DAB.
Albumin Albumin immunohistochemistry was done to determine the
extent of plasma protein extravasation into the brain, as a marker of compromise of the BBB. Briefly, sections were blocked for en-
dogenous peroxidases using 0.3% H 2 O 2 in 50% methanol. Serum-free protein block (DAKO) was used to prevent background stain-ing. Sections were then incubated overnight in rabbit anti-sheep albumin primary antibody (Accurate Chemical and Scientific Corp., USA) diluted 1: 1,000 in PBS with 0.5% fish gelatin and 0.2% Triton-X100. Biotinylated goat anti-rabbit secondary anti-body (1: 200) was used followed by incubation in Strep HRP. Sec-tions were washed and stained with DAB.
Apoptosis Activated caspase 3 immunohistochemistry was used as a
marker of cells in the apoptotic pathway. Antigen retrieval was conducted using microwave irradiation (2 ! 5 min, in citric acid buffer), followed by cooling for 15 min in the buffer, and subse-quent blocking of endogenous peroxidase with 0.3% H 2 O 2 in 50% methanol for 20 min. Sections were washed and blocked using 5% NGS with 1% BSA in PBS with 0.3% Triton-X100 for 60 min. Sec-tions were incubated overnight in rabbit anti-activated caspase 3 primary antibody (1: 1,000; R&D Systems, USA) followed by in-cubation for 1 h in biotinylated goat anti-rabbit secondary anti-body. Sections were then incubated in Strep HRP and stained with DAB. In order to determine the type of cell expressing caspase 3 protein, each of the following primary antibodies was used in con-junction with activated caspase 3 primary antibody: MAP1b(1: 100; Neomarkers, USA) and MAP2a,b (1: 100; Neomarkers) were used to detect neurons at various stages of development and differentiation, GFAP (1: 400) for astrocytes, and CNPase (1: 200; Sigma, USA) for oligodendrocytes. Antigen retrieval was con-ducted using microwave irradiation in citric acid buffer (3 !
4 min) followed by blocking of endogenous peroxidases in 0.3% H 2 O 2 with 50% methanol. Autofluoresence was blocked using so-dium borohydride (10 mg/ml) in PBS and nonspecific staining was blocked with serum-free protein block (DAKO). Sections were then incubated in a mixture containing activated caspase 3 primary antibody (1: 1,000) with one of the cell type-specific pri-mary antibodies overnight. Sections were then incubated in a mixture of rabbit anti-mouse Alexa Fluor A488 (1: 800; Molecu-lar Probes, The Netherlands) and goat anti-rabbit Alexa Fluor 594 (1: 1,000, Molecular Probes) for the detection of cell type markers and activated caspase 3, respectively.
Proliferation Antigen retrieval was performed using microwave irradiation
(3 ! 5 min) followed by a further 30 min in hot citric acid buffer. Sections were immersed in 0.3% H 2 O 2 in 50% methanol for 20 min followed by blocking with 5% NGS and 0.5% BSA in PBS with 0.3% Triton-X100. Sections were incubated overnight in rabbit monoclonal anti-Ki67 (1: 2, Neomarkers) primary antibody in PBS. Sections were then washed and incubated in biotinylated goat anti-rabbit secondary antibody (1: 200) in PBS for 1 h fol-lowed by washes and the addition of Strep HRP and subsequent visualization with DAB.
Histological Analysis Lectin-positive cells were analyzed to distinguish between
macrophages, and resting vs. activated microglia ( fig. 1 ), using morphological criteria as described previously using this isolectin [23] . To our knowledge, no reliable and specific markers that dif-ferentiate between resting microglia, activated microglia and macrophages in paraffin-embedded ovine tissue exist. Resting or

Hutton/Castillo-Melendez/Walker
Dev Neurosci 2007;29:341–354344
ramified microglia were determined based upon the presence of fine cytoplasmic processes, and small (often elongated) cell bod-ies. Activated or amoeboid microglia were determined based on the presence of short or long thick processes, usually with a large cell body of irregular shape. In contrast to the previous study [23], we also identified macrophages, which were determined by a lack of any cellular processes, consistent cell body size and regular cell body shape. Cellular inclusions were often seen in this cell type.
The number of lectin-positive cells, resting or activated mi-croglia, and macrophages were counted in 3 fields of view within a given brain region, on 2 slides per animal (i.e. a total of 6 fields of view per brain region per animal). The number of cells counted
in this field of view was averaged and expressed as the number of cells per mm 2 for each animal. The percentage of Purkinje cells which were in contact with lectin-positive cells was also calculated on 5 fields of view on two slides (a total of 10 fields of view) per animal. These data were then averaged and expressed as the per-centage of Purkinje cells with spatially-associated lectin-positive cells. The number of cells immunopositive for GFAP, 4HNE or activated caspase 3 was counted in 3 fields of view within a given brain region, on 2 slides per animal (a total of 6 fields of view per brain region per animal). In addition, the percentage of Purkinje cells that were immunopositive for 4HNE or activated caspase 3 was determined from 5 fields of view, on 2 slides (a total of 10 fields of view per animal). The number of albumin-positive cells per sagittal section (within 2 mm of the cerebellar midline) was count-ed. The number of cells that were immunoreactive for Ki67 was counted in the region 1 mm from the tip of 10 folia on two slides in different regions of the cerebellum, as shown in figure 2 .
Statistical Evaluation All data are presented as mean 8 SEM. The Mann-Whitney
U test was used to determine significant differences between groups for cell number, or percentage of area occupied by spe-cific staining, in each brain region for the untreated (control), 30 � g LPS and 60 � g LPS treatments, using the Bonferroni-Holm adjustment for multiple comparisons.
Results
The 30 � g LPS treatment produced mild and transient reductions in maternal pH (7.479–7.434) and PO 2 (112.8–77.8 mm Hg) for 1 h following LPS administration, and O 2 saturation was reduced by approximately 8% (data not shown). Two hours after LPS treatment, these parameters
Ramified (resting)microglia
Amoeboid (activated)microglia Macrophages
Absence of processesConsistent cell body size
Regular shape
Thick processesLarge, irregular cell body
Margination of chromatin masses
Thin, ramified processesSmall cell body
Fig. 1. Microglia and macrophages in cerebellar white matter of fetal sheep after treatment with 30 � g LPS into the uteroplacental circulation. Note the ramified processes of resting microglia, the ‘amoeboid’ shape of acti-vated microglia, and the distinctive regular, rounded shape of macrophages. Scale bar = 10 � m.
1 mm
Molecular layer
Purkinje layer
Granular layer
Cerebellar white matter
Fig. 2. Diagram showing the method of quantification of Ki67 immunoreactivity in the fetal cerebellum. The number of Ki67-positive cells was counted 1 mm from the tip of a folium, in each of the regions depicted.
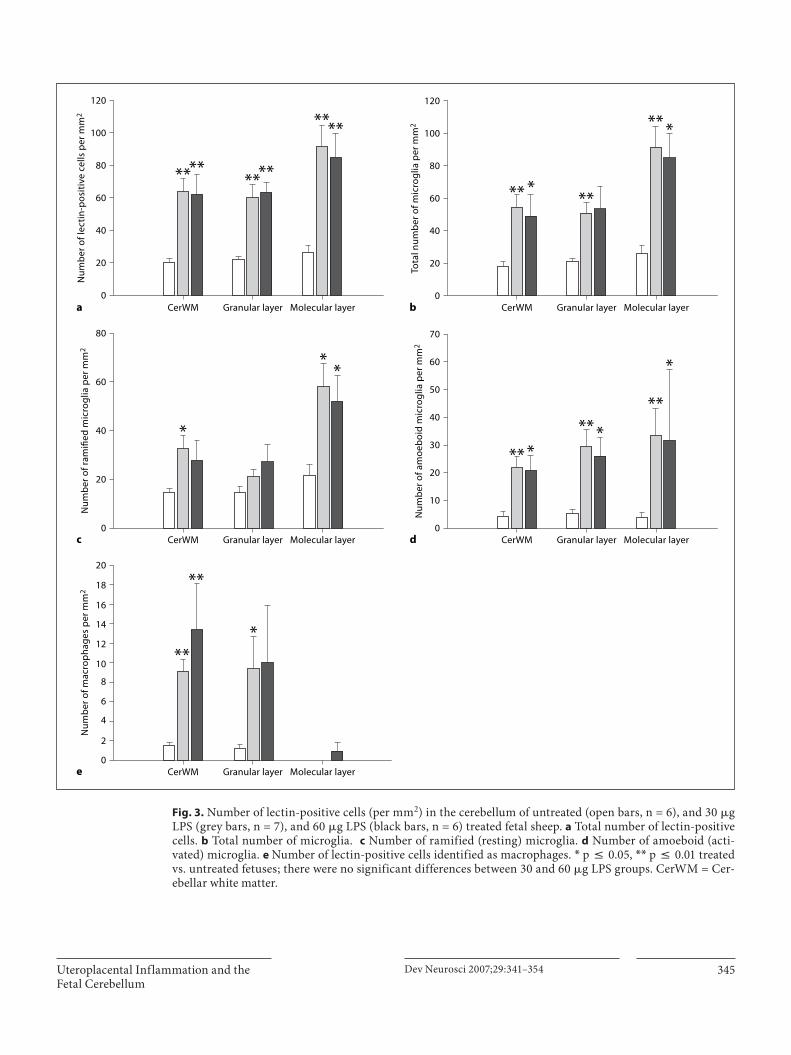
Uteroplacental Inflammation and the Fetal Cerebellum
Dev Neurosci 2007;29:341–354 345
CerWM0
20
a b
c d
e
40
60
80
100
********
****
120
Num
ber
of l
ecti
n-p
osit
ive
cells
per
mm
2
Granular layer Molecular layer CerWM0
20
40
60
80
100
** ***
** *
120
Tota
l num
ber
of m
icro
glia
per
mm
2
Granular layer Molecular layer
CerWM0
10
20
30
40
50
60
** *** *
**
*
70
Num
ber
of a
moe
boi
d m
icro
glia
per
mm
2
Granular layer Molecular layerCerWM0
20
40
60
*
**
80
Num
ber
of r
amifi
ed m
icro
glia
per
mm
2
Granular layer Molecular layer
CerWM0
**
**
*
20
18
16
14
12
10
8
6
4
2
Num
ber
of m
acro
ph
ages
per
mm
2
Granular layer Molecular layer
Fig. 3. Number of lectin-positive cells (per mm 2 ) in the cerebellum of untreated (open bars, n = 6), and 30 � g LPS (grey bars, n = 7), and 60 � g LPS (black bars, n = 6) treated fetal sheep. a Total number of lectin-positive cells. b Total number of microglia. c Number of ramified (resting) microglia. d Number of amoeboid (acti-vated) microglia. e Number of lectin-positive cells identified as macrophages. * p ̂ 0.05, * * p ̂ 0.01 treated vs. untreated fetuses; there were no significant differences between 30 and 60 � g LPS groups. CerWM = Cer-ebellar white matter.

Hutton/Castillo-Melendez/Walker
Dev Neurosci 2007;29:341–354346
were not significantly different from pre-LPS treatment values, or compared to the saline treatment at 2 h. The 60 � g LPS treatment did not result in any significant differ-ences in maternal pH, PO 2 or O 2 saturation at these time points. Fetal pH was maintained in the normal range im-mediately after treatment, although it fell significantly at 8–12 h after both the 30 and 60 � g LPS treatments; there-after, pH was within normal physiological range. Fetal PO 2 and O 2 saturation fell progressively, more rapidly and to a greater extent after the 30 � g compared to the60 � g LPS dose.
The total number of lectin-positive cells, irrespective of their classification by morphology as either microglia or macrophages, was significantly increased in all regions of the cerebellum examined following the administration of LPS into the uteroplacental circulation ( fig. 3 a). In the cerebellar white matter and granular layers, this increase was largely attributed to increases in the number of acti-vated microglia and macrophages, whilst in the molecu-lar layer the increase was attributed primarily to an in-crease in the number of resting and activated microglia, with minimal detection of macrophages ( fig. 3 b–e). The percentage of Purkinje cells in close proximity (contact) with microglia was significantly increased ( fig. 4 a–c) fol-lowing the administration of either 30 � g (85.3 8 3.4%, p = 0.009) or 60 � g LPS (83.8 8 2.7%, p = 0.008), com-pared to cerebella from untreated control fetuses (35.9 8 7.3%). Macrophages were rarely observed in close prox-imity to Purkinje cells.
There was a dose-dependent increase in the number of GFAP-positive cell bodies in the cerebellar white matter, although this reached statistical significance only follow-ing administration of the 60 � g LPS dose ( fig. 5 j). In the granular layer, there was a significant increase in the number of astrocytes following administration of 30 � g LPS. The GFAP-positive cell bodies seen in the granular layer following the administration of 30 � g LPS were more intensely stained for GFAP than in the untreated or 60 � g LPS-treated groups ( fig. 5 h compared with g and i). The number of GFAP-positive cells in the molecular layer could not be determined as they were difficult to differentiate accurately even at high magnifications due to the dense distribution of Bergmann fibers. GFAP immunoreactivity associated with Bergmann glia was usually more intense and the fibers more densely distrib-uted following the administration of either dose of LPS ( fig. 5 a–c).
The number of cells immunopositive for 4HNE was significantly increased in all regions of the cerebellum examined, with an approximately 10-fold increase in the cerebellar white matter ( fig. 6 a). The percentage of Pur-kinje cells immunopositive for 4HNE ( fig. 6 b) was sig-nificantly increased following the administration of ei-ther 30 � g LPS (18.0 8 6.4%, p = 0.014) or 60 � g LPS (16.2 8 4.3%, p = 0.018) compared to cerebella from untreated control fetuses (4.9 8 0.7%).
Albumin-immunopositive cells were identified in a number of untreated and LPS treated fetal brains ( table 1
a b
c 0
** **100
80
60
40
20Purk
inje
cel
ls w
ith
sp
atia
lly
asso
ciat
ed m
icro
glia
(%)
Fig. 4. Microglia in close association with Purkinje cells. Micrographs of sections from the cerebellum of a con-trol ( a ) and 60 � g LPS ( b ) treated fetuses showing microglia (arrowheads) in contact with Purkinje cells (PC). Scale bar = 10 � m. c Percentage of Purkinje cells spatially associated with microglia. * * p ̂ 0.01 treated vs. untreated fetuses; there were no significant differences between 30 and 60 � g LPS groups.

Uteroplacental Inflammation and the Fetal Cerebellum
Dev Neurosci 2007;29:341–354 347
a b c
d e f
g h i
j0
CerWM Granular layer
*
*
250
200
#150
100
50
Num
ber
of G
FAP-
pos
itiv
e ce
ll b
odie
s p
er m
m2
Fig. 5. GFAP immunoreactivity in fetal cerebellum. Micrographs of sections from the cerebellum of a control ( a , d , g ), 30 � g LPS ( b , e , h ) and 60 � g LPS ( c , f , i ) treated fetuses. Increased GFAP staining in Bergmann glia is seen following administration of LPS ( a , compared with b and c ). Micrographs of sections from the cer-ebellum showing GFAP staining (cell bodies indicated by arrow-heads) in the cerebellar white matter ( d–f ), and the granular lay-er ( g–i ). Scale bar = 50 � m. j Number of GFAP-positive cell bod-ies (per mm 2 ) in cerebellar white matter and the granular layer of untreated (open bars, n = 6), 30 � g LPS (grey bars, n = 7), and 60 � g LPS (black bars, n = 6) treated fetal sheep cerebellum. * p ̂ 0.05 treated vs. untreated fetuses; # p ̂ 0.05 30 � g vs. 60 � g LPS groups.

Hutton/Castillo-Melendez/Walker
Dev Neurosci 2007;29:341–354348
and fig. 7 ). When the number of albumin-positive cells was determined, a significantly greater number were found in the cerebellar white matter and molecular layer follow-ing the administration of both 30 � g LPS, and 60 � g LPS, compared to untreated controls. The albumin immunore-activity was predominantly intracellular, suggesting cel-lular sequestration of albumin by cells, which based on morphology, appeared to be neurons ( fig. 7 b, c). Other cells, likely to be glia, also showed albumin immunostain-ing, but this occurred in a minority of fetuses (30 � g LPS, n = 2; 60 � g LPS, n = 1). The number of Purkinje cells that were immunopositive for albumin was significantly in-creased following administration of 30 � g LPS ( fig. 7 h).
In the cerebellar white matter, the number of cells immunopositive for activated caspase 3 was significant-ly increased following the administration of 30 and 60 � g LPS compared to controls, the greatest effect occur-ring after the 30 � g LPS treatment ( fig. 8 b, g). In the molecular layer, the number of cells immunopositive for activated caspase 3 was significantly increased in the 30 � g LPS-treated group compared to untreated controls. The percentage of Purkinje cells immunopositive for ac-tivated caspase 3 was significantly increased following
the administration of 30 � g LPS ( fig. 8 i). In the granular layer, the high density of activated caspase 3-positive cells (seen in both controls and LPS-treated groups) made it difficult to accurately count the number of ac-tivated caspase 3-positive cells even at high magnifica-tions. Thus, the area occupied by activated caspase 3 staining was calculated in this region. The area occu-pied by immunopositive staining in the granular layer in both LPS-treated groups was not significantly differ-ent to that seen in controls ( fig. 8 h).
Table 1. Proportion (in %) and number of animals showing intra-cellular albumin staining within the cerebellum
Untreated LPS, �g
30 60
Cerebellar white matter 66.8 (4/6) 100.0 (7/7) 100.0 (6/6)Cerebellar granular layer 66.8 (4/6) 100.0 (7/7) 100.0 (6/6)Cerebellar molecular layer 33.4 (2/6) 100.0 (7/7) 83.3 (5/6)Purkinje cells 33.4 (2/6) 83.3 (6/7) 100.0 (6/6)
a b0
CerWM Granular layer Molecular layer
**
**
**
**
****
800
600
400
200
Num
ber
of 4
HN
E-p
osit
ive
cells
per
mm
2
0Purkinje cells
*
*
30
25
20
10
5
Purk
inje
cel
ls p
osit
ive
for 4
HN
E (%
)
15
Fig. 6. Number of 4-HNE-positive cell bodies (per mm 2 ) in, cerebellar white matter, granular layer, and molecu-lar layer ( a ), and percentage of Purkinje cells immunopositive for 4-HNE ( b ) in untreated (open bars, n = 6), 30 � g LPS (grey bars, n = 7), and 60 � g LPS (black bars, n = 6) treated fetal sheep cerebellum. * * p ̂ 0.01, * p ̂ 0.05, treated vs. untreated fetuses; there were no significant differences between 30 � g and 60 � g LPS groups.

Uteroplacental Inflammation and the Fetal Cerebellum
Dev Neurosci 2007;29:341–354 349
g h0
CerWM Granular layer Molecular layer
70
60
50
40
30
20
10
Num
ber
of a
lbum
in-p
osit
ive
cells
per
mm
2
0Purkinje cells
*
***
**
*
350
250
150
50
300
200
100
Num
ber
of a
lbum
in-p
osit
ive
cells
per
sag
itta
l sec
tion
Fig. 7. Albumin immunoreactivity in fetal cerebellum. Micro-graphs of sections from the cerebellum of a control ( a , d ), 30 � g LPS ( b , e ) and 60 � g LPS ( c , f ) treated fetuses showing albumin immunoreactivity in cerebellar white matter ( a–c ), and in Pur-kinje cells and granular layer (GL; d–f ). Scale bar = 50 � m. Num-ber of albumin-immunopositive cells (per mm 2 ) in cerebellar
white matter, granular layer, and molecular layer ( g ) and number of albumin-positive Purkinje cells per sagittal section ( h ) in un-treated (open bars, n = 6), 30 � g LPS (grey bars, n = 7), and 60 � g LPS (black bars, n = 6) treated fetal sheep cerebellum. * p ̂ 0.05, * * p ̂ 0.01, treated vs. untreated fetuses; there were no signifi-cant differences between 30 � g and 60 � g LPS groups.

Hutton/Castillo-Melendez/Walker
Dev Neurosci 2007;29:341–354350
dominant caspase 3-positive cell in the molecular layer and were also seen in cerebellar white matter ( fig. 9 ). This was the case for both 30 and 60 � g LPS treatment groups. Few CNPase-positive cells were positive for activated caspase 3.
When double-label immunohistochemistry was used to investigate the types of cells that expressed the acti-vated caspase 3 protein, GFAP-positive cells were most commonly positive for activated caspase 3 in the cerebel-lar white matter. MAP2a,b-positive cells were the pre-
g h0
CerWM Molecular layer
**
*
2,000 **#
1,500
500
1,000
Num
ber
of a
ctiv
ated
casp
ase
3-p
osit
ive
cells
per
mm
2
Tota
l are
a oc
cup
ied
by
acti
vate
dca
spas
e 3-
pos
itiv
e st
ain
ing
(%)
0Granular layer
*100
60
40
20
80
i
Purk
inje
cel
ls p
osit
ive
for
acti
vate
d c
asp
ase
3 (%
)
0Purkinje cells
100
60
40
20
80
Fig. 8. Activated caspase 3 immunoreactivity in fetal cerebellum. Micrographs of sections from the cerebellum of a control ( a , d ), 30 � g LPS ( b , e ) and 60 � g LPS ( c , f ) treated fetuses showing ac-tivated caspase 3 immunoreactivity in cerebellar white matter ( a–c ), and in granular layer, molecular layer (ML) and Purkinje cells ( d–f ). Scale bar = 50 � m. Number of activated caspase 3-im-munopositive cells (per mm 2 ) in cerebellar white matter and mo-
lecular layer ( g ), area occupied by activated caspase 3-positive staining in the granular layer ( h ) and the percentage of Purkinje cells positive for activated caspase 3 ( i ) in untreated (open bars,n = 6), 30 � g LPS (grey bars, n = 7), and 60 � g LPS (black bars, n = 6) treated fetal sheep cerebellum. * p ̂ 0.05, * * p ̂ 0.01, un-treated vs. treated fetuses; # p ̂ 0.05, 30 � g vs. 60 � g LPS groups.

Uteroplacental Inflammation and the Fetal Cerebellum
Dev Neurosci 2007;29:341–354 351
In both untreated controls and LPS-treated groups, the number of Ki67-immunopositive cells was greatest in cells surrounding the Purkinje cells, although Purkinje cells themselves were not seen to be proliferating. The to-tal number of Ki67-positive cells within 1 mm from the tip of the folia was not significantly altered by the utero-placental LPS treatments (control, 29.52 8 4.75 cells 1 mm from folia tip; 30 � g LPS, 39.91 8 7.26 cells 1 mm from folia tip; 60 � g LPS, 41.03 8 4.23 cells 1 mm from folia tip).
There were no significant differences in the number of cells undergoing proliferation, indicated by Ki67 immu-noreactivity (data not shown), between untreated and LPS treated groups. Molecular layer (control, 12.05 8 4.15 cells 1 mm from folia tip; 30 � g LPS, 16.54 8 7.13 cells 1 mm from folia tip; 60 � g LPS, 20.16 8 9.47 cells 1 mm from folia tip) and the granular layer (control, 10.01 8 4.74 cells 1 mm from folia tip; 30 � g LPS, 13.27 8 7.35 cells 1 mm from folia tip; 60 � g LPS, 17.88 8 12.30 cells 1 mm from folia tip) of the cerebellum tended to show a dose-dependent increase in the number of Ki67-positive cells; however, this did not reach statistical significance.
Fig. 9. Double-label immunohistochemistry for caspase 3 (green label) and specific cell markers (red label) identifying astrocytes and Bergmann glia (GFAP; a , d , g ) and mature neurons (MAP2a, b; b, e, h ) in cerebellar white matter and mature neurons (MAP2a,b; c, f, i ) in the molecular layer of untreated ( a , b , c ), 30 � g LPS ( d , e , f ) and 60 � g LPS ( g , h , i ) treated fetuses. White arrowheads indicate cells positive for the cell marker and for activated caspase 3. Scale bar = 50 � m.

Hutton/Castillo-Melendez/Walker
Dev Neurosci 2007;29:341–354352
Discussion
It is evident that this model of LPS administration – where endotoxin is given into the uteroplacental circula-tion and none can be detected in the fetal circulation [Hutton and Walker, unpubl. data] – results in a number of alterations in the fetal cerebellum suggestive of an in-flammatory reaction. Specifically, there was widespread microglial activation, macrophage infiltration, increased lipid peroxidation, albumin uptake by Purkinje and other cells, and increased expression of a late-stage apoptotic protein – cleaved, or activated caspase 3. It is not clear how these reactive changes are brought about – LPS could not be detected in the fetal circulation, and only modest changes occurred in the levels of cytokines such as TNF � , IL-1 � and IL-6 [Hutton and Walker, unpubl. data]. Tran-sient fetal hypoxemia, acidemia, and maternal (and there-fore, fetal) hyperthermia [22] have been recorded in re-sponse to LPS administration, and while it cannot be ruled out that any one or a combination of these could ac-count for the changes, the fetal cerebellum has not hith-erto been regarded as being particularly sensitive to these factors. Further work will be necessary to identify the pro-cesses that connect the inflammatory reactions in the uterus and placenta to the cells in the fetal brain.
The current study shows that uteroplacental inflam-mation resulted in a significant increase in the total num-ber of lectin-positive cells in the fetal cerebellum, com-prising increased numbers of ‘activated’ and ‘resting’ mi-croglia, as well as macrophages. In the granular layer, 30 � g LPS resulted in an increase in the total number of mi-croglia, and in the number of activated (amoeboid) mi-croglia, but without a clear reduction in the number of resting (ramified) microglia, suggesting that some mi-croglia must have migrated into this region from else-where. The absence of an increase in the number of pro-liferating cells in the fetal cerebellum suggests that mi-totic division of microglia does not contribute to the increased numbers of this cell type. To confirm this ob-servation, double-label immunohistochemistry with B. simplicifolia isolectin and Ki67 is required. Alternatively, some resident microglia might react to the B. simplicifolia isolectin only after the LPS treatments, but we are un-aware of any other results to suggest this is a likely expla-nation. In addition, it should be noted that in the absence of a reliable marker to differentiate macrophages from mi-croglia in the ovine brain, it is possible a number of lectin-positive cells, which demonstrate morphology associated with macrophages may in fact be microglia. Previous studies have differentiated resting and activated microglia
and not taken into account the contribution from macro-phages. Thus in the current study, we included these as a third group of lectin-positive cells. It should be noted that it remains possible that a small proportion of those cells which are classified as macrophages may be microglia lacking processes, although we believe this method of classification is more informative than grouping macro-phages into the ‘amoeboid microglia’ classification.
Cells that enter the apoptotic pathway display a range of ‘eat me’ signals which attract microglia and macro-phages, resulting in engulfment and phagocytosis [24–26] . This is of interest as it has recently been demonstrat-ed that microglia are responsible for the programmed cell death of Purkinje cells during normal development by a mechanism that involves superoxide production [27] . In the untreated, control fetuses, approximately 36% of Pur-kinje cells were immunopositive for activated caspase 3 and 5% were positive for 4HNE, a by-product of lipid per-oxidation typically caused by superoxide ion. Thus, the proportion of Purkinje cells closely associated with mi-croglia may be seen as an indicator of the microglial-in-duced apoptosis of Purkinje cells.
The number of Purkinje cells closely associated with microglia was significantly greater after the uteroplacen-tal LPS treatments, and the proportion of Purkinje cells that were immunopositive for activated caspase 3 and 4HNE was also increased. Many Purkinje cells were also immunopositive for albumin. It has been shown that Pur-kinje cells are particularly prone to sequestering albumin [28] , which may be a protective response and an attempt to abrogate or forestall apoptosis [29] . Given that Pur-kinje cells form a population of cells that, after a certain time in development are not replaced, a deficit in these cells may result in significant life-long functional deficits. The absence of Ki67-positive Purkinje cells in this study suggests that this is likely to be true for the fetal sheep cerebellum in late gestation.
The cerebellar white matter and granular layer also showed significant infiltration of macrophages following administration of LPS. This demonstrates that while a mi-croglial response occurred widely in the cerebellum, the infiltration of macrophages was more region- and dose-specific. The absence of macrophages in the molecular layer following the 30- � g dose is interesting because this region of the cerebellum showed increased albumin im-munoreactivity, possibly due to alterations in permeabil-ity of the BBB, yet significant macrophage invasion was only observed to occur after the higher LPS dose.
In addition to a well-characterized phagocytic role [30, 31] , the accumulation of activated microglia and

Uteroplacental Inflammation and the Fetal Cerebellum
Dev Neurosci 2007;29:341–354 353
macrophages in the brain has a number of other effects, attributable to the secretion of substances such as nitric oxide, reactive oxygen species, proinflammatory cyto-kines, and the NMDA receptor agonist quinolinic acid, which follows their activation [12–15] . The increase in 4HNE immunoreactivity may be related to the increased neurotoxic and pro-oxidative status of the tissue result-ing from the presence of larger numbers of activated mi-croglia and macrophages. Further studies should deter-mine the types of cell that show the increase in 4HNE immunoreactivity to elucidate the cell type susceptible to this form of insult.
There was a clear astrocytic response in the granular layer following 30 � g LPS, and in the white matter fol-lowing the administration of 60 � g LPS, with a corre-sponding increase in the staining of GFAP in Bergmann glia. Bergmann cells have soma which are spatially asso-ciated with Purkinje cells and extend radial fibers that ensheathe synapses on Purkinje cell dendrites. Bergmann fibers have a role in neuronal migration and phagocytosis during development [32–35] . It is possible that damage to Purkinje cells and cells of the molecular layer, as shown by the increased expression of activated caspase 3 protein, resulted in a reactive response in astrocytes and Berg-mann fibers. Increased neuronal death produced by kai-nic acid or methylazoxymethanol significantly increased the GFAP immunoreactivity of Bergmann glia in the cer-ebellum of 5-day-old rats [32, 36] .
Accumulation of albumin by cerebellar cells has been described previously [28] . In the present study, the cells that accumulated albumin were mainly neurons, such as Purkinje cells and cells in the granular layer; the major-ity of albumin-positive cells in white matter also had a neuron-like morphology. The presence of albumin in the parenchyma of the cerebellum may indicate breakdown of the BBB. It has also been shown that the compromise of the BBB following infection with the rabies virus in adult mice results in greater disruption of the BBB in the cerebellum, compared to the cerebral cortex [37] .
The presence of a large number of cells expressing the late-stage apoptotic protein – activated caspase 3 – in the fetal cerebellum, particularly in GFAP-positive cells, is consistent with our previous findings for the sheep fetus [38] and is consistent with the extensive growth and re-modeling of the cerebellum that occurs at this stage of gestation in sheep [39] . There was a significant increase in the number of cells expressing caspase 3 in white mat-ter following both the low and high dose of LPS, again primarily associated with GFAP-immunopositive cells. MAP2a,b-positive cells, observed in cerebellar white
matter of both untreated and treated fetal sheep brains, were only colocalized with activated caspase 3 following the LPS treatments. In the molecular layer, an increase in the number of activated caspase 3-positive cells was seen following the 30 � g, but not 60 � g LPS treatment. In con-trast to these changes, the expression of activated caspase 3 in the granular layer was largely unaffected by the utero-placental LPS, although it is possible that the method of analysis (spatial quantification) may not have been sensi-tive enough to detect alteration in the degree or number of activated caspase 3-positive cells.
There was no increase in the number of cells express-ing the proliferation marker Ki67 in the cerebellum af-ter the LPS treatments. This is in contrast to prosence-phalic areas such as cortical and periventricular WM where this uteroplacental treatment significantly in-creased the number of Ki67-positive cells [Hutton and Walker, unpubl. data]. Thus, while supratentorial re-gions of the fetal brain appear to have a proliferative re-sponse that may replace the cells lost by increased apop-tosis, this is not the case for the cerebellum where all regions except the granular layer showed an increase in the expression of activated caspase 3 protein. It is pos-sible that proliferative activity occurs later than the posttreatment time examined in this study (72 h), or that there is significant migration of cells from sur-rounding areas to repopulate the regions where apop-totic cell death had increased. Alternatively, regions of the developing cerebellum that lose cells to inflamma-tory insult may remain depleted. The consequence of such cell deficit remains unknown.
An inverse effect between the dose of LPS and a num-ber of markers seen in the fetal cerebellum was often ob-served in this study. In addition, it was found that the lower dose of LPS resulted in a higher rate of fetal mortal-ity. These data suggest that a multimodal mechanism of action may be responsible for the translation of uteropla-cental inflammation to damage in the fetal cerebellum. The data suggest that pathways activated in response to each dose of LPS may be different and thus they result in different degrees of brain damage and threat to fetal well-being. These mechanisms required further investigation.
Acknowledgments
We thank Alex Satragno for help with the surgical preparation of the sheep, and Dr. Edwin Yan for help with conducting the ex-periments. This project was supported by a grant from the Na-tional Health and Medical Research Council of Australia to D.W.W.

Hutton/Castillo-Melendez/Walker
Dev Neurosci 2007;29:341–354354
1 Bodensteiner JB, Johnsen SD: Cerebellar in-jury in the extremely premature infant: new-ly recognized but relatively common out-come. J Child Neurol 2005; 20: 139–142.
2 Limperopoulos C, Soul JS, Gauvreau K, Huppi PS, Warfield SK, Bassan H, Robertson RL, Volpe JJ, du Plessis AJ: Late gestation cer-ebellar growth is rapid and impeded by pre-mature birth. Pediatrics 2005; 115: 688–695.
3 Limperopoulos C, Soul JS, Haidar H, Huppi PS, Bassan H, Warfield SK, Robertson RL, Moore M, Akins P, Volpe JJ, du Plessis AJ: Impaired trophic interactions between the cerebellum and the cerebrum among pre-term infants. Pediatrics 2005; 116: 844–850.
4 Allin M, Matsumoto H, Santhouse AM, Nosarti C, AlAsady MH, Stewart AL, Rifkin L, Murray RM: Cognitive and motor func-tion and the size of the cerebellum in adoles-cents born very pre-term. Brain 2001; 124:
60–66. 5 Kern JK: Purkinje cell vulnerability and au-
tism: a possible etiological connection. Brain Dev 2003; 25: 377–382.
6 Itakura A, Kurauchi O, Takashima S, Uchida K, Ito M, Mizutani S: Immunological detec-tion of 4-hydroxynonenal protein adducts in developing pontine and Purkinje neurons and in karyorrhexis in pontosubicular neu-ronal necrosis. Early Hum Dev 2002; 67: 19–28.
7 Gleason CA, Hamm C, Jones MD Jr: Effect of acute hypoxemia on brain blood flow and oxygen metabolism in immature fetal sheep. Am J Physiol 1990; 258:H1064–H1069.
8 Jensen A, Klonne HJ, Detmer A, Carter AM: Catecholamine and serotonin concentra-tions in fetal guinea-pig brain: Relation to regional cerebral blood flow and oxygen de-livery in the growth-restricted fetus. Reprod Fertil Dev 1996; 8: 355–364.
9 Ashwal S, Dale PS, Longo LD: Regional cere-bral blood flow: studies in the fetal lamb dur-ing hypoxia, hypercapnia, acidosis, and hy-potension. Pediatr Res 1984; 18: 1309–1316.
10 Welsh JP, Yuen G, Placantonakis DG, Vu TQ, Haiss F, O’Hearn E, Molliver ME, Aicher SA: Why do Purkinje cells die so easily after global brain ischemia? Aldolase C, EAAT4, and the cerebellar contribution to posthy-poxic myoclonus. Adv Neurol 2002; 89: 331–359.
11 Sarna JR, Hawkes R: Patterned Purkinje cell death in the cerebellum. Prog Neurobiol 2003; 70: 473–507.
12 Guillemin GJ, Kerr SJ, Brew BJ: Involvement of quinolinic acid in AIDS dementia com-plex. Neurotox Res 2005; 7: 103–123.
13 Banati RB, Gehrmann J, Schubert P, Kreutz-berg GW: Cytotoxicity of microglia. Glia 1993; 7: 111–118.
14 Raivich G, Banati R: Brain microglia and blood-derived macrophages: molecular pro-files and functional roles in multiple sclero-sis and animal models of autoimmune demy-elinating disease. Brain Res Brain Res Rev 2004; 46: 261–281.
15 Hendriks JJ, Teunissen CE, de Vries HE,Dijkstra CD: Macrophages and neurodegen-eration. Brain Res Brain Res Rev 2005; 48:
185–195. 16 Manuelpillai U, Ligam P, Smythe G, Wallace
EM, Hirst J, Walker DW: Identification of kynurenine pathway enzyme mRNAs and metabolites in human placenta: up-regula-tion by inflammatory stimuli and with clin-ical infection. Am J Obstet Gynecol 2005;
192: 280–288. 17 Urakubo A, Jarskog LF, Lieberman JA, Gil-
more JH: Prenatal exposure to maternal in-fection alters cytokine expression in the pla-centa, amniotic f luid, and fetal brain. Schizophr Res 2001; 47: 27–36.
18 Yan E, Castillo-Melendez M, Nicholls T, Hirst J, Walker D: Cerebrovascular respons-es in the fetal sheep brain to low-dose endo-toxin. Pediatr Res 2004; 55: 855–863.
19 Garnier Y, Berger R, Alm S, von Duering MU, Coumans AB, Michetti F, Bruschettini M, Lituania M, Hasaart TH, Gazzolo D: Sys-temic endotoxin administration results in increased S100B protein blood levels and periventricular brain white matter injury in the preterm fetal sheep. Eur J Obstet Gynecol Reprod Biol 2006; 124: 15–22.
20 Duncan JR, Cock ML, Scheerlinck JP, West-cott KT, McLean C, Harding R, Rees SM: White matter injury after repeated endotox-in exposure in the preterm ovine fetus. Pedi-atr Res 2002; 52: 941–949.
21 Stolp HB, Dziegielewska KM, Ek CJ, Potter AM, Saunders NR: Long-term changes in blood-brain barrier permeability and white matter following prolonged systemic in-flammation in early development in the rat. Eur J Neurosci 2005; 22: 2805–2816.
22 McClure L, O’Connor AE, Hayward S, Jen-kin G, Walker DW, Phillips DJ: Effects of age and pregnancy on the circulatory activin re-sponse of sheep to acute inflammatory chal-lenge by lipopolysaccharide. J Endocrinol 2005; 185: 139–149.
23 Hewicker-Trautwein M, Schultheis G: Lec-tin labelling of amoeboid and ramified mi-croglial cells in the telencephalon of ovine fetuses with the B4 isolectin from Griffonia simplicifolia. J Comp Pathol 1994; 111: 21–31.
24 Ravichandran KS: ‘Recruitment signals’ from apoptotic cells: invitation to a quiet meal. Cell 2003; 113: 817–820.
25 Elward K, Gasque P: ‘Eat me’ and ‘don’t eat me’ signals govern the innate immune re-sponse and tissue repair in the CNS: empha-sis on the critical role of the complement sys-tem. Mol Immunol 2003; 40: 85–94.
26 Lauber K, Blumenthal SG, Waibel M, Wes-selborg S: Clearance of apoptotic cells: get-ting rid of the corpses. Mol Cell 2004; 14:
277–287. 27 Marin-Teva JL, Dusart I, Colin C, Gervais A,
van Rooijen N, Mallat M: Microglia promote the death of developing Purkinje cells. Neu-ron 2004; 41: 535–547.
28 Fishman PS, Farrand DA, Kristt DA: Inter-nalization of plasma proteins by cerebellar Purkinje cells. J Neurol Sci 1990; 100: 43–49.
29 Tabernero A, Granda B, Medina A, Sanchez-Abarca LI, Lavado E, Medina JM: Albumin promotes neuronal survival by increasing the synthesis and release of glutamate. J Neu-rochem 2002; 81: 881–891.
30 Ashwell K: Microglia and cell death in the developing mouse cerebellum. Brain Res Dev Brain Res 1990; 55: 219–230.
31 Ferrer I, Bernet E, Soriano E, del Rio T, Fon-seca M: Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience 1990; 39: 451–458.
32 Lafarga M, Andres MA, Calle E, Berciano MT: Reactive gliosis of immature Bergmann glia and microglial cell activation in re-sponse to cell death of granule cell precur-sors induced by methylazoxymethanol treat-ment in developing rat cerebellum. Anat Embryol (Berl) 1998; 198: 111–122.
33 Yamada K, Watanabe M: Cytodifferentia-tion of Bergmann glia and its relationship with Purkinje cells. Anat Sci Int 2002; 77: 94–108.
34 Muller T, Kettenmann H: Physiology of Bergmann glial cells. Int Rev Neurobiol 1995; 38: 341–359.
35 Hatten ME, Mason CA, Liem RK, Edmond-son JC, Bovolenta P, Shelanski ML: Neuron-astroglial interactions in vitro and their im-plications for repair of CNS injury. Cent Nerv Syst Trauma 1984; 1: 15–27.
36 Milenkovic I, Nedeljkovic N, Filipovic R, Pe-kovic S, Culic M, Rakic L, Stojiljkovic M: Pattern of glial fibrillary acidic protein ex-pression following kainate-induced cerebel-lar lesion in rats. Neurochem Res 2005; 30:
207–213. 37 Phares TW, Kean RB, Mikheeva T, Hooper
DC: Regional differences in blood-brain barrier permeability changes and inflamma-tion in the apathogenic clearance of virus from the central nervous system. J Immunol 2006; 176: 7666–7675.
38 Castillo-Melendez M, Chow JA, Walker DW: Lipid peroxidation, caspase-3 immunoreac-tivity, and pyknosis in late-gestation fetal sheep brain after umbilical cord occlusion. Pediatr Res 2004; 55: 864–871.
39 Rees S, Harding R: The effects of intrauterine growth retardation on the development of the Purkinje cell dendritic tree in the cere-bellar cortex of fetal sheep: a note on the on-togeny of the Purkinje cell. Int J Dev Neuro-sci 1988; 6: 461–469.
References

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:355–362 DOI: 10.1159/000105476
Identification of POSH2, a Novel Homologue of the c-Jun N-Terminal Kinase Scaffold Protein POSH
Michael Wilhelm
a Nickolay V. Kukekov
b Zhiheng Xu
b Lloyd A. Greene
b
Departments of a Pediatrics and b
Pathology, Columbia University Health Sciences, New York, N.Y. , USA
Introduction
The evolutionarily conserved mitogen-activated pro-tein kinase (MAPK) cascades allow cells to respond to a variety of extracellular stimuli. MAPKs control such di-verse signals as cellular proliferation, cell migration, dif-ferentiation, and survival/apoptosis [1] . In mammals, three MAPK pathways exist: the extracellular signal-re-lated kinases, c-Jun N-terminal kinases (JNKs) and the p38 MAPKs [1, 2] . The p38 MAPKs and JNKs are collec-tively called the stress-activated protein kinases to sig-nify their role in responding to cellular stressors such as hypoxia, DNA damage and oxidative insults [1, 3] .
Three genes encode ten different isoforms of JNKs, which are required for neuronal apoptosis following a va-riety of stimuli. Jnk1 and jnk2 play a key role in region-specific apoptosis during brain development [4] . The neuron-specific isoform jnk3 is required for apoptosis fol-lowing excitotoxicity and trophic factor deprivation in vi-tro [5] . Furthermore, inhibition of the JNK pathway pro-tects against acoustic trauma and cerebral hypoxia-isch-emia (HI) in adult animals and juvenile animals [6, 7] .
Scaffold proteins organize kinase cascades into func-tional modules, promoting interactions between the in-dividual kinases and enhancing signaling. In yeast, the scaffold protein Ste5 is required for efficient signaling
Key Words
Apoptosis � c-Jun N-terminal kinase � Scaffold proteins
Abstract
The c-Jun N-terminal kinase (JNK) pathway plays an impor-tant role in neuronal apoptosis both during normal CNS de-velopment and following stroke in adult animals. As with other MAP kinase pathways, scaffold proteins regulate JNK signaling. The scaffold protein POSH (Plenty of SH3s) en-hances JNK activation and apoptosis. We identified a POSH homologue, POSH2, which was cloned from rat brain and is present in cortical neurons in vitro. POSH2 mRNA is ex-pressed in a variety of tissues including brain, and this dis-tribution partially overlaps with that of POSH. POSH2 over-expression promotes JNK activation in HEK293 cells and promotes apoptosis in neuronal PC12 cells, which is blocked by a dominant-negative c-Jun. Finally POSH2 contains a functional RING domain and enhances the stability of coex-pressed mixed-lineage kinases. These results indicate that POSH2 may regulate JNK activation and consequent apop-tosis under conditions of increased expression.
Copyright © 2007 S. Karger AG, Basel
Received: September 7, 2006 Accepted after revision: January 8, 2007
Dr. Michael Wilhelm Department of Pediatrics, Pediatric Critical CareColumbia University Health Sciences, 3959 Broadway, BHN 10–24 New York, NY 10032 (USA) Tel. +1 212 342 4087, Fax +1 212 342 2293, E-Mail [email protected]
© 2007 S. Karger AG, Basel0378–5866/07/0295–0355$23.50/0
Accessible online at:www.karger.com/dne

Wilhelm /Kukekov /Xu /Greene
Dev Neurosci 2007;29:355–362356
through the MAPK pheromone response pathway [8] . In mammals, the JNK-interacting proteins (JIPs) form a family of homologous scaffold proteins for the JNK path-way [2] . The JIPs interact with MKK4/7 and the JNKs to enhance JNK signaling [2] and are required for JNK ac-tivation and apoptosis following excitotoxicity and oxy-gen-glucose deprivation in hippocampal neurons [9] .
Another JNK scaffold, POSH (Plenty of SH3s) also po-tentiates JNK signaling through an interaction with the mixed lineage kinases (MLKs) and the JIPs [10, 11] . POSH also promotes the stability of JNK pathway proteins through activation of JNKs, resulting in a feed-forward loop that amplifies apoptotic signaling [12] . POSH is re-quired for JNK activation and apoptosis following tro-phic factor deprivation of neuronal PC12 cells and sym-pathetic neurons in vitro [10] . More recently, POSH has also been implicated in neuronal apoptosis following HI in adult animals [13] .
Here, we report the identification of a novel POSH ho-mologue, POSH2, which can also act as a scaffold for the JNK pathway. POSH2 promotes JNK activation and apoptosis of neuronal PC12 cells when overexpressed. This apoptosis appears to be dependent on c-Jun activa-tion. Finally, POSH2 promotes stabilization of JNK path-way proteins when overexpressed, providing one possible mechanism of JNK activation. Given the role of the JNKs in HI in adult animals and during normal CNS develop-ment, the function of POSH2 and the other JNK scaffolds may have important implications for therapy of neona-tal HI.
Materials and Methods
Materials Adult rat brain RNA was obtained from Clontech (Mountain
View, Calif., USA). Cell culture media RPMI 1640 and Dulbecco’s Modified Eagle’s Medium were obtained from Mediatech (Hern-don, Va., USA). Lactacystin was from Calbiochem (San Diego, Calif., USA). Lipofectamine 2000 was obtained from Invitrogen (Carlsbad, Calif., USA). Recombinant human NGF was kindly supplied by Genentech (South San Francisco, Calif., USA). Hoechst dye 33342 was obtained from Sigma (St Louis, Mo., USA). JNK/phospho-JNK antibodies were from Cell Signaling (Den-vers, Mass., USA). Anti-Flag and anti-HA were obtained from Santa Cruz (Paso Robles, Calif., USA).
Generation of Plasmids and Primers The sequence of all newly generated constructs was confirmed
by sequencing at the DNA facility of the Columbia University Health Sciences campus using appropriate primers. Internal primers were designed based on generated sequences when neces-sary. The primers used to amplify POSH2 from rat brain mRNA
were: 5 � -ATGGATGATTTGACGTTACTTGATCTCCTG and3 � -TTTGCTGGAAATAAGGTCTGTGAGACTGA. The 5 � Flag tag was added using the primer AACCATGGACTACAAGGA-CGATGATGACAAAGATGATTTGACGTTACTT. The RING mutant of POSH2 was generated using the Stratagene (La Jolla, Calif., USA) QuikChange Site-Directed Mutagenesis kit and the following primers: 5 � -GCCAAAGTTCTCCCCAGCCAGCAC-ACCTTCAGCAAACCAA GTCTACAGCGGATTTTC and 3 � -GAAAATCCGCTGTAGACTTGGTTTGCTGAAGGTGTGCT-GGCTGGGG AGAACTTTGGC. POSH and dominant-negative c-Jun constructs have been previously described [10] , as have MLK2, MLK3 and DLK constructs [14] .
Multiple Tissue Northern Blots Rat Multiple Tissue Northern (MTN TM ) blots were obtained
from Clontech (Mountain View, Calif., USA) and were probed following the manufacturer’s instruction. For probing MTN blots, the POSH2 probe (bp 1,812–2,205) was generated using the following primers 5 � -TTCCATCATCATGGAAGGCAAAG and 3 � -TTTGCTGGGAA ATAAGGTCTG using GFP-POSH2 as the template. Primers for the POSH probe were 5 � -TTCCATCAT-CATGGAAGGCAAAG and 3 � -TTCGCTGGGAAATAAGGTC-TG, and GFP-POSH was used as a template. PCRs were per-formed in the presence of [ 32 P]CTP.
Cell Culture and Transfections HEK293 cells were maintained in Dulbecco’s Modified Eagle’s
Medium 1 ! supplemented with 10% fetal bovine serum. Culture of PC12 cells, both naïve and differentiated (‘primed’) was per-formed as previously described as was NGF deprivation of PC12 cells [10] . Primary cortical neurons were cultured as previously described [15] . HEK293 cells were transfected using the calcium chloride method as previously described [10] . PC12 cells were transfected 2 days after priming using Lipofectamine 2000 ac-cording to the manufacturer’s instructions. The PC12 cells gener-ally transfected with and efficiency between 10–20%.
Determination of Cell Death Strip counting to determine cell survival was performed as
previously described [10] . Nuclear morphology was examined us-ing Hoechst 33342 staining (1: 1,000 dilution for staining live cells).
Statistical Analysis All comparisons between groups were performed using un-
paired t tests, with significance set at the p ! 0.05 level. Where appropriate, one-directional p values were used.
Results
A Homologue of POSH, POSH2, Is Expressed in Neurons Using the sequence for rat POSH, we performed an
NCBI Blast search for possible POSH homologues. We identified a POSH-related sequence with three SH3 do-mains (one less than POSH) that was predicted from the

JNK Scaffold Protein POSH2 Dev Neurosci 2007;29:355–362 357
rat genome sequence. Primers were designed to amplify this predicted sequence as in ‘Materials and Methods’. We successfully amplified a 2.2-kb cDNA from adult rat brain total mRNA, added a 5 � Flag-tag and cloned this cDNA into the pCMS-EGFP vector (pCMS-EGFP.POSH2). The sequence of this cDNA corresponds to a
revised, predicted rat sequence (accession: BC100155.1). The predicted protein encoded by this sequence has an estimated molecular weight of � 82 kDa and three SH3 domains and a RING domain. Figure 1 shows an align-ment of POSH2 protein with POSH, using ClustalW [16] , with the known domains indicated. In addition to clon-
POSH 1 MDESALLDLLECPVCLERLDASAKVLPCQHTFCKRCLLGIVGSRNELRCPECRTLVGSGVDELPSNILLVRLLDGIKQRPWK 82 POSH2 1 MDDLTLLDLLECPVCFEKLDVTAKVLPCQHTFCKPCLQRIFKAHKELRCPECRTLVFCSIEALPANLLLVRLLDGVRSG--- 79 ----------------RING DOMAIN-------------- POSH 83 PGPGGGGSTTCTNVLRAQGSTVVNCGSKDLQSPQCGQQPRVQAWSPPVRGIPQLPCAKALYNYEGKEPGDLKFSKGDIIILR 164 POSH2 80 HNSWRGGSFRRPRILTLQDNRKAKSSPRSLQASPFQLGPTVRIH------MDGVPRAKALYNYRGKNPGDLKFNKGDVILLQ 155 ----------------------SH3--- POSH 165 RQVDENWYHGEVNGVHGFFPTNFVQIIKPLPQPPPQCKALYDFEVKDKE--ADKDCLPFAKDDVLTVIRRVDENWAEGMLAD 244 POSH2 156 RQLDENWYQGEINGVSGFFPASSVEVIKQLPQPPPLCRALYNFDLRDKDKSENQDCLTFLKDDVITVISRVDENWAEGKLGD 237 --------------------------- --------------------------------SH3--------------- POSH 245 KIGIFPISYVEFNSAAKQLIEWDK----PPVPGVDTAECPSATAAQSSSASKHS--DTKKNTRKRHSFTSLTMANKSSQASQ 320 POSH2 238 KVGIFPILFVEPNLSARHLLEKSKGHQLSRTKHLSLMSSPSRGKATNTSTLRKSPGSRRKGSGQFAMTTALNTLNRMVHSPE 319 ----------- ------------------------------- POSH 321 NRHSMEISPPVLISSSNPTAAARISELSGLSCSAPSQVHISTTGLIVTPPPSSPVTTGPSFTFPTDVPYQAALGTMNPPLPL 402 POSH2 320 GHQMVEISTPVLISSTSPSMLTQHGDRADFPASSAGQVSTS------HPAPASPGHSTAMVSVPSSQQHLSTN--------- 386 ---RAC BINDING DOMAIN-------------------- POSH 403 QPPLLATTVARLYTVRRYAAAVAAAAAAVAAGVGPRPAVGSTEQIAHLRPQTRPSVYVAIYPYTPRKEDELELRKGEMFLVF 484 POSH2 387 -------------------------------------------------------MFVALHTYSAQGPEELDLKKGEGIRVL 413 --------------------------- POSH 485 ERCQDGWYKGTSMHTSKIGVFPGNYVAPVTRAVTNASQAKVPMSTAGQASRGVTMVSPSTAGGPAQKPQGNGVAGNP-SVVP 565 POSH2 414 GKNQDGWLRGVSLVTGRTGIFPSDYVIPVFSSTARKTSSFP-------DSRHPTVCTTWALSTSSVSSQGSFSEGDPRQSGP 488 -SH3------------------------- POSH 566 TAVVSAAHIQTSPQAKVLLHMTGQMTVNQARNAVRTVAAHNQERPTAAVTPIQVQNAACIGPASVGLPHHSLASQPLPPMVG 647 POSH2 489 FRSVFVPTAVNPPRSTSGPGTSGQGSLRKVRSSMR--KNGSLQRPVQSGIPTFMVGSLRCSPAMVIRPQKFQFYQPQ----- 563 POSH 648 PAAHIAAVNINRTSVPLACAAGASSLASPNMTTAALETEPSGRTVTILPGLPTSPESAASACGNSSAVKPDKDSKKEKKGLL 729 POSH2 564 --------GMTPSPTPIMVEIGSKSISTG---------------EPALTCINRGGKTRTHSAGNSIIMEGKE---------- 612 POSH 730 KLLSGASTKRKPRVSPPASPTLDVELGSGEVPLQGAVGPELPLGGVHGRVGSCPTDGDGP-VAAGTAALAQDAFHRKTSSLD 810 POSH2 613 -----TPIKSEPPPKPPAS-----------APPSILVKPENSKNGIEKQVKTVRFQNYSPPPTKHSASGPTSGKHEQPATLK 678 POSH 811 SAVPIAPPPRQACSSLGPVMNEARPVVCERHRVVVSYPPQSEAELELKEGDIVFVHKKREDGWFKGTLQRNGKTGLFPGSFV 892 POSH2 679 GSQPEAVSSEGEMTILFAHRSGCHSGQQTDLRRKSAFSKTTPPVSTASVSQTLFPSK------------------------- 735 --------------------------------------SH3 #4----------- POSH 893 ENI 895 POSH2 ---
-
Fig. 1. Protein domains of POSH and POSH2. ClustalW was used to align the protein sequence of POSH and POSH2. Identical amino acids are highlighted in black, while shades of grey indicate homologous amino acids. The known domains of each protein are indicated underneath the sequences.

Wilhelm /Kukekov /Xu /Greene
Dev Neurosci 2007;29:355–362358
ing POSH2 from rat brain RNA, we were able to use PCR to amplify a fragment from rat cortical neurons in culture using the following primers: 5 � -GAGTGAGAACCAG-GATTGCCTGACCTTCCTC and 3 � -GGCTG TA ATTC-TGAAATCTCACGGTTTTGAC. This produced a PCR product of the predicted 1.3-kb size and sequencing con-firmed identity with POSH2 (data not shown). POSH2 is also expressed in HEK293 cells and in naïve PC12 cells ( fig. 2 b). Thus, we have identified a structural homologue of POSH, which lacks the fourth SH3 domain and is ex-pressed in brain, including in cortical neurons.
To further determine the tissue expression of POSH2, we probed rat (adult) MTN blots with a radiolabeled probe specific for POSH2 ( fig. 2 a). For comparison, we reprobed the same blots with a radiolabeled probe for POSH. Note that some tissues express both mRNAs at detectable levels, while some preferentially express one gene or the other. Also note the similar distribution of POSH, and similar size of the mRNA to that previously reported in mouse [17] .
POSH2 POSH
9.5 kb
7.5 kb
4.4 kb
2.4 kb
1.35 kb
0.24 kb
2.0 kb1.2 kb0.8 kb
HEK
293
Cor
tica
l
Nai
ve P
C12
1 765432 1312
1110
9816
1514
1 765432a
b
1312
1110
9816
1514
Fig. 2. Expression of POSH and POSH2. a Rat MTN blots were probed as in ‘Materials and Methods’. Note that some tissues ex-press both mRNAs at detectable levels, while some preferentially express one or the other. 1: Large intestine, 2: prostate, 3: stomach,
4: thyroid, 5: spinal cord, 6: eye, 7: bladder, 8: adrenal, 9: heart, 10: brain, 11: spleen, 12: lung, 13: liver, 14: smooth muscle, 15: kidney, 16: testis. b PCR was performed as in the text, and the products resolved on a 1% agarose gel.
�-P-JNK
�-JNK
GFP
POSH
POSH
2
Fig. 3. POSH2 activates JNK. The indicated cDNA in the pCMS-EGFP vector was expressed in HEK293 cells. Lysates were sub-jected to Western immunoblot with a phosphor-specific JNK an-tibody. Membranes were stripped and reprobed with an antibody to total JNK.

JNK Scaffold Protein POSH2 Dev Neurosci 2007;29:355–362 359
Overexpression of POSH2 Promotes JNK Activation Given the structural similarity of POSH2 to POSH, we
examined whether POSH2 promotes JNK activation when overexpressed. We transfected HEK293 cells with either pCMS-EGFP.POSH2 or empty pCMS-EGFP (ef-ficiency � 90%). As shown in figure 3 , POSH2 promotes phosphorylation of JNK under these conditions. Further-more, JNK activation by POSH2 is at least as robust as that induced by POSH ( fig. 3 ).
Overexpression of POSH2 Promotes Apoptosis in Neuronal PC12 Cells through JNK Activation We next examined whether POSH2 promotes apopto-
sis of neuronal cells. We transfected neuronal PC12 cells with either pCMS-EGFP.POSH2 or pCMS-EGFP. As shown in figure 4 a, there was a significant reduction in survival of cells expressing POSH2, similar to levels of cells expressing POSH. Though the individual experi-ment has a p value that does not quite reach significance, using pooled data from two, consecutive experiments (each performed in triplicate) the p value was less than 0.04. This reduced survival was accompanied by an in-
crease in the number of GFP+ cells exhibiting apoptotic nuclei ( fig. 4 b).
To determine whether JNK activation played a role in this apoptosis, we cotransfected dominant-negative c-Jun (d/n-c-Jun) or empty vector with either pCMS-EGFP.POSH2 or empty pCMS-EGFP. Again, as shown in figure 3 c, cells coexpressing pCMS-EGFP.POSH2 with empty CMV vector exhibited reduced survival when compared to control cells. However, when d/n-c-Jun was coex-pressed with POSH2, survival was restored to basal levels ( fig. 4 c).
POSH2 Contains a Functional RING Domain The RING domain of POSH controls its own protea-
somal degradation [10] . To determine the role of the RING domain of POSH2, Flag-tagged POSH2 was ex-pressed in HEK 293 cells with or without lactacystin (20 � M ) in the medium for 16 h. As with POSH [10] , POSH2 levels increase in the presence of this proteasome in-hibitor ( fig. 5 a). We then used site-directed mutagenesis to inactivate the E3-ligase function of the RING do-main [10] (mRing2, fig. 5 b). When equal amounts of
10
a b c
20
40
60
80
100
120
2 3
Day
Surv
ival
(%)
POSHPOSH2
GFP
*
*
**
0
100
200
300
400
500
Ap
opto
sis
(%)
*
**
GFPPOSHPOSH2
0
20
40
60
80
100
140
120
1 2 3
Day
Surv
ival
(%)
GFP/CMV
POSH2/CMV
POSH2/dn-cJun
Fig. 4. Overexpression of POSH2 promotes apoptosis dependent on c-Jun activation. a 48 h after exposure to NGF, neuronally dif-ferentiated PC12 cells were transfected with the indicated cDNAs in the pCMS-EGFP vector. GFP+ cells were counted daily, start-ing 24 h after transfection (day 1). Data are normalized to 100% survival in cells expressing pCMS-EGFP vector. POSH- and POSH2-expressing cells exhibited reduced survival. Shown is one experiment, performed in triplicate ( * p = 0.051, * * p = 0.03). b Live cells, treated as in a , were stained with Hoechst 33342 24 h after transfection to determine nuclear morphology. The percent-
age of GFP+ cells with condensed, fragmented nuclei was deter-mined 48 h later. More than 100 cells were counted in each well. Shown are the data from three consecutive experiments ( * p ! 0.01, * * p ! 0.02). c PC12 cells were cotransfected with the indi-cated constructs 48 h after priming. To ensure coexpression in GFP+ cells, CMV or d/n-c-Jun were expressed in fourfold excess relative to the EGFP-containing vector. Cells were counted as in a , and survival normalized to pCMS-EGFP/d/n-c-Jun-expressing cells. Shown is one experiment performed in triplicate ( * p ! 0.02).

Wilhelm /Kukekov /Xu /Greene
Dev Neurosci 2007;29:355–362360
vectors encoding POSH2 and mRing2 were expressed in HEK 293 cells, levels of mRing2 protein were signif-icantly greater than for wild-type POSH2, and compa-rable to levels of POSH2 in the presence of lactacystin ( fig. 5 a).
POSH2 Controls the Stability of MLK Family Proteins POSH participates in a feed-forward stabilization loop
with proteins in the JNK pathway [12] . We therefore ex-amined whether POSH2 promotes reciprocal stabiliza-tion of MLK family proteins when they are coexpressed in HEK293 cells. As shown in figure 6 a, coexpression of MLK2, MLK3 and DLK with POSH2 promotes increased
levels of POSH2. This occurs despite a reduction in GFP expression as shown in the lower panel. This is due to dramatically enhanced apoptosis in these cells, even by 20 h after transfection (data not shown). Conversely, POSH2 promotes enhanced stability of the MLKs. Figure 6 b shows that POSH2 enhances the stability of MLK2 and DLK when coexpressed. Similar results were ob-tained for MLK3 (data not shown). Thus, like POSH, un-der conditions of enhanced POSH2 expression, the MLKs would appear to become stabilized and they in turn fur-ther enhance POSH2’s expression. Thus overexpressed POSH2 appears to promote a feed-forward stabilization loop promoting JNK activation and apoptosis.
�-Flag
�-GFP
Lactacystin(μM)
0
a
20 0
POSH2 –++mRing +– –
cDNA:
b
POSH2 79 ccctgccagcacaccttctgcaaaccatgtc 109mRing 70 cccagccagcacaccttcagcaaaccaagtc 109
Protein:POSH2 27 P C Q H T F C K P C 36mRing 27 P S Q H T F S K P S 36
Fig. 5. POSH2 controls its own stability through the RING do-main. a POSH2 or the RING mutant (mRing) were expressed in HEK293 cells. The proteasome inhibitor lactacystin was added 4 h after transfection as indicated. Cells were collected 20 h after
transfection and immunoblots performed. Membranes were re-probed for EGFP to ensure equal loading of transfected proteins. b The point mutations generated for mRing are shown. The cDNA is shown above the corresponding amino acid sequence.
Flag-POSH2
EGFP
pCMS-EGFP.POSH2
pcDNA3.1
a b
+
++ +++
++
DLK
MLK2MLK3
Flag-POSH2 ++
pCMS-EGFP.MLK2 + +
HA-DLKHA-MLK2
++pCMS-EGFP.DLK
EGFP
Fig. 6. POSH2 and the MLKs stabilize each other. a Flag-tagged POSH2 in the pCMS-EGFP vector was coexpressed with the in-dicated cDNAs in the pcDNA3.1 vector. Lysates were collected 20 h later and subjected to immunoblot as indicated. Membranes were reprobed with GFP to determine equal quantity of the POSH construct in each sample. Note the robust increase in POSH2 sig-nal despite a marked reduction in GFP. b Cotransfections were
performed as indicated and immunoblot performed as in a . In this case, MLK2 and DLK were expressed in the pCMS-EGFP vec-tor and POSH2 in the pcDNA vector. Again, note the increase in MLK2 and DLK signals despite nearly complete absence of the GFP signal. As noted in the text, coexpression of POSH2 and the MLKs induces marked apoptosis, which accounts for the reduced GFP signal.

JNK Scaffold Protein POSH2 Dev Neurosci 2007;29:355–362 361
Discussion
JNKs play important roles in normal CNS develop-ment [4] , as well as the response to HI in the juvenile and adult brain [7, 18] , though their role in neonatal brain in-jury remains less clear. While p38 MAPK [19] and extra-cellular signal-related kinases [20–22] have been impli-cated in neuronal injury following neonatal HI, previous work suggested that phospho-JNK is decreased following neonatal HI [19] . However, a recent paper suggests that phospho-JNK increases in the injured regions [23] , simi-lar to our own unpublished data, and deletion of jnk3 protects neonatal mice from HI [23] . Much less is known about JNK scaffolds in HI, particularly in the developing brain. Different JNK scaffolds may regulate JNK activa-tion under different conditions or in specific cell types. Therefore, understanding how these scaffolds regulate JNK signaling may have important implications for ther-apy of HI in the developing brain.
Here, we report the identification of a POSH homo-logue that promotes JNK signaling and apoptosis in neu-ronal cells when overexpressed. Under these conditions, POSH2 induces robust JNK activation and neuronal apoptosis to levels similar to that seen with POSH. Fur-thermore, coexpression of a dominant-negative isoform of c-Jun completely inhibits POSH2-induced apoptosis. Thus, under conditions of enhanced expression, POSH2 appears to act similarly to POSH.
Using ClustalW [16] , alignment of predicted POSH2 sequences from multiple species shows that POSH2 is highly conserved between rat, mouse, monkey and hu-
man. Using the ClustalW utility, these sequences have similarity scores of approximately 90% with each other. The predicted POSH2 sequences in Xenopus laevis and Tribolium castaneum are significantly more dissimilar (scores of � 25), but do support a common evolutionary ancestor gene. Interestingly, Xenopus has a gene encoding a POSH homologue with four SH3 domains, and a pos-sible POSH2 homologue with only two, as predicted in the NCBI library. Given the similar functions between POSH and POSH2 when overexpressed, the differences in their functions are not yet clear. Several possibilities exist including roles in different cell types, following dif-ferent death stimuli or during different stages of develop-ment. Work is currently underway to investigate each of these possibilities.
Our work supports a model in which, following en-hanced expression, POSH2 promotes stabilization of MLKs and subsequent JNK activation and apoptosis. Furthermore, it seems likely that POSH and POSH2 could interact when coexpressed in a given cell, and this could provide a further level of regulation of this path-way. The differential expression of these scaffold proteins under normal and pathologic conditions could have dra-matic effects on JNK activity and neuronal apoptosis. Work is underway in our lab to clarify the interaction be-tween these proteins and how this affects JNK signaling. Further efforts will be needed to determine the roles that JNK scaffolds, individually or in combination, play in apoptosis in the brain during normal development and following neonatal and pediatric HI.
References
1 Chang L, Karin M: Mammalian MAP kinase signalling cascades. Nature 2001; 410: 37–40.
2 Yasuda J, Whitmarsh AJ, Cavanagh J, Shar-ma M, Davis RJ: The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol 1999; 19: 7245–7254.
3 Irving EA, Bamford M: Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab 2002; 22: 631–647.
4 Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA: The Jnk1 and Jnk2 protein kinases are required for regional spe-cific apoptosis during early brain develop-ment. Neuron 1999; 22: 667–676.
5 Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA: Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997; 389: 865–870.
6 Wang J, Van De Water TR, Bonny C, de Rib-aupierre F, Puel JL, Zine A: A peptide inhib-itor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J Neurosci 2003; 23: 8596–8607.
7 Borsello T, Clarke PG, Hirt L, Vercelli A, Re-pici M, Schorderet DF, Bogousslavsky J, Bon-ny C: A peptide inhibitor of c-Jun N-termi-nal kinase protects against excitotoxicity and cerebral ischemia. Nat Med 2003; 9:
1180–1186. 8 Choi KY, Satterberg B, Lyons DM, Elion EA:
Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S. cerevisiae . Cell 1994; 78: 499–512.
9 Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP, Appel M, Rossini AA, Jones SN, Flavell RA, Rakic P, Davis RJ: Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev 2001; 15: 2421–2432.
10 Xu Z, Kukekov NV, Greene LA: POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J 2003; 22: 252–261.
11 Kukekov NV, Xu Z, Greene LA: Direct inter-action of the molecular scaffolds POSH and JIP is required for apoptotic activation of JNKs. J Biol Chem 2006; 281: 15517–15524.
12 Xu Z, Kukekov NV, Greene LA: Regulation of apoptotic c-Jun N-terminal kinase signal-ing by a stabilization-based feed-forward loop. Mol Cell Biol 2005; 25: 9949–9959.

Wilhelm /Kukekov /Xu /Greene
Dev Neurosci 2007;29:355–362362
13 Zhang QG, Wang RM, Yin XH, Pan J, Xu TL, Zhang GY: Knock-down of POSH expres-sion is neuroprotective through down-regu-lating activation of the MLK3-MKK4-JNK pathway following cerebral ischaemia in the rat hippocampal CA1 subfield. J Neurochem 2005; 95: 784–795.
14 Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA: The MLK family mediates c-Jun N-terminal kinase activation in neuro-nal apoptosis. Mol Cell Biol 2001; 21: 4713–4724.
15 Park DS, Morris EJ, Padmanabhan J, Shelan-ski ML, Geller HM, Greene LA: Cyclin-de-pendent kinases participate in death of neu-rons evoked by DNA-damaging agents. J Cell Biol 1998; 143: 457–467.
16 Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD: Mul-tiple sequence alignment with the Clustal se-ries of programs. Nucleic Acids Res 2003;31:
3497–3500. 17 Tapon N, Nagata K, Lamarche N, Hall A: A
new rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-kappaB signalling pathways. EMBO J 1998;
17: 1395–1404. 18 Kuan CY, Whitmarsh AJ, Yang DD, Liao G,
Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, Davis RJ, Rakic P: A critical role of neural-specific JNK3 for isch-emic apoptosis. Proc Nat Acad Sci USA 2003;
100: 15184–15189. 19 Hee Han B, Choi J, Holtzman DM: Evidence
that p38 mitogen-activated protein kinase contributes to neonatal hypoxic-ischemic brain injury. Dev Neurosci 2002; 24: 405–410.
20 Han BH, Holtzman DM: BDNF protects the neonatal brain from hypoxic-ischemic inju-ry in vivo via the ERK pathway. J Neurosci 2000; 20: 5775–5781.
21 Jones NM, Bergeron M: Hypoxia-induced ischemic tolerance in neonatal rat brain in-volves enhanced ERK1/2 signaling. J Neuro-chem 2004; 89: 157–167.
22 Cho D-G, Mulloy MR, Chang PA, Johnson MD, Aharon AS, Robison TA, Buckles TL, Byrne DW, Drinkwater DC Jr: Blockade of the extracellular signal-regulated kinase pathway by U0126 attenuates neuronal dam-age following circulatory arrest. J Thorac Cardiovasc Surg 2004; 127: 1033–1040.
23 Pirianov G, Brywe KG, Mallard C, Edwards D, Flavell RA, Hagberg H, Mehmet H: Dele-tion of the c-Jun N-terminal kinase 3 gene protects neonatal mice against cerebral hy-poxic-ischaemic injury. J Cereb Blood Flow Metab 2006, E-pub ahead of print.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:363–372 DOI: 10.1159/000105477
Pomegranate Polyphenols and Resveratrol Protect the Neonatal Brain against Hypoxic-Ischemic Injury
Tim West
a, b Madeliene Atzeva
a, b David M. Holtzman
a–c
a Department of Neurology, b
Hope Center for Neurological Disorders and c Molecular Biology and Pharmacology,
Washington University School of Medicine, St. Louis, Mo. , USA
Introduction
Neonatal hypoxia-ischemia (H-I) is a major cause of morbidity and mortality in human newborns with motor and cognitive sequelae frequently seen in survivors [Rob-ertson et al., 1989; Shankaran et al., 1991; Volpe, 1995; Back, 2001]. Recent advances in neonatal care have im-proved the survival of severely prematurely born babies. These very low birth weight babies have a greatly in-creased risk of perinatal brain injury, including hypoxic-ischemic brain injury [Back and Rivkees, 2004; Gieron-Korthals and Colon, 2005]. Neonatal H-I has been associ-ated with increases in reactive oxygen species, and recent studies suggest that new avenues of treatments of neona-tal H-I should include selective responses to the genera-tion of oxidative damage [Singh et al., 1999; Hamrick and Ferriero, 2003; Gulcan et al., 2005]. Current clinical treat-ments that appear successful in initial studies include hy-pothermia, which is thought to slow down the neuronal cell death processes and attenuate the generation of reac-tive oxygen species [Kil et al., 1996; Thoresen et al., 1997; Hashimoto et al., 2003; Gluckman et al., 2005; Shankaran et al., 2005].
Our lab and others have utilized an animal model of neonatal H-I in mice to determine mechanisms of injury as well as address potential treatments [Gibson et al., 2001; Han et al., 2001; West et al., 2006]. In a recent study using this model, we found that continuous pre- and postinjury ingestion of pomegranate juice by the dam
Key Words
Pomegranate juice � Polyphenols � Resveratrol � Hypoxic-ischemic injury � Caspase 3 activation
Abstract
A previous study from our lab has shown that the polyphe-nol-rich pomegranate juice can protect the neonatal mouse brain against hypoxic-ischemic (H-I) injury when given to mothers in their drinking water. To test the hypothesis that this protection is due to the polyphenols in the juice, we studied the effects of the pomegranate polyphenol extract in the same neonatal H-I model. To further explore the role of a specific polyphenol in neonatal H-I we investigated the effects of resveratrol. The neuroprotective effects of resve-ratrol have been demonstrated in adult models of stroke, but had not previously been examined in neonates. We show that pomegranate polyphenols and resveratrol reduce caspase-3 activation following neonatal H-I. Resveratrol re-duced caspase-3 activation when given before the injury but not when given 3 h after the injury. In addition to preventing caspase-3 activation, resveratrol also reduced calpain activa-tion. Finally, we show that resveratrol can protect against tis-sue loss measured at 7 days after the injury. These and other recent findings suggest that polyphenols should be further investigated as a potential treatment to decrease brain in-jury due to neonatal H-I.
Copyright © 2007 S. Karger AG, Basel
Received: September 22, 2006 Accepted after revision: November 22, 2006
David M. Holtzman Washington University, Department of Neurology Box 8111 660 S Euclid Saint Louis, MO 63110 (USA) Tel. +1 314 747 0286, Fax +1 314 362 2826, E-Mail [email protected]
© 2007 S. Karger AG, Basel
Accessible online at:www.karger.com/dne

West /Atzeva /Holtzman
Dev Neurosci 2007;29:363–372364
leads to significant protection of the neonatal brain [Lo-ren et al., 2005]. The mechanism of neuroprotection af-forded by pomegranate juice is unknown but it could po-tentially involve the antioxidant or other properties of polyphenols.
One of the most well studied polyphenols is resveratrol which is found in grapes, several types of nuts and kojo-hkon (Japanese Knotweed), an oriental medicine used to treat diseases of the blood vessels [Sato et al., 1997; Faus-tino et al., 2003; Tokusoglu et al., 2005]. Resveratrol is an antioxidant but its in vivo effects reach far beyond that of other antioxidants. Most famously, resveratrol is thought to be the compound responsible for the low incidence of heart disease in the French population due to the high intake of red wine in France [Kopp, 1998]. Besides car-diovascular effects, resveratrol may have prophylactic ef-fects against other human diseases, such as cancer [Bi-anchini and Vainio, 2003] and dementia [Leibovici et al., 1999; Truelsen et al., 2002]. In rats, resveratrol has been studied extensively in adult models of stroke and has been found to protect the brain when the rodents have been pre-treated for 3 weeks or longer with resveratrol con-taining drinking water [Virgili and Contestabile, 2000] or when resveratrol is administered by interperitoneal in-jection [Huang et al., 2001; Gupta et al., 2002; Sinha et al., 2002]. However, there have to our knowledge been no studies looking at the neuroprotective effects of resvera-trol in neonatal brain injury.
In this study, we found that pomegranate polyphenol extract (PPE) as well as resveratrol can protect the neo-natal rodent brain against H-I brain injury. When ad-ministered before injury, resveratrol can protect against both caspase-3 activation at 24 h after the injury and tis-sue loss at 7 days after injury. These studies suggest that polyphenols should be considered for further evaluation as potential treatments to lessen the effects of neonatal H-I brain injury.
Materials and Methods
Animals and Surgical Procedures All rats and mice were kept under 12/12 light/dark cycles with
ad libitum access to food and water. For studies of polyphenol ex-tract of pomegranates and resveratrol in mice, male and female C57BL/6-J mice were interbred. For pups used in H-I experi-ments, the date of delivery was noted and at postnatal day 7 (P7) the pups underwent hypoxia-ischemia as described [Gibson et al., 2001; Han et al., 2001; West et al., 2006]. Briefly, mice were anaes-thetized by inhalation of 5% halothane (balance room air) for induction and 1.5% for maintenance. An incision was made on the left side of the neck and the carotid artery isolated, exposed
and permanently ligated. The pups were then put at 37 ° C to wake up and finally returned to the dam for a 2 h recovery period. Hy-poxia was induced by putting pups in temperature-controlled chambers (37 ° C) through which humidified 8% oxygen flowed for 45 min. A similar protocol was used to induce H-I injury in P7 Sprague-Dawley rats; however, for the surgery on rats 2% halo-thane was required for maintenance of anesthesia and the rat pups were subjected to 8% oxygen for 2.5 h.
PPE Ingestion The pomegranate polyphenol-enriched extract (PPE) was ob-
tained from Pom Wonderful. PPE was produced from the skin and the aril of Punica granatum L. Wonderful variety and con-tains 0.9 mg polyphenols per mg powder. 96 mg of PPE was di-luted in 100 ml of sugar water containing the same sugar compo-sition as used in the previous experiments with pomegranate juice (a 1: 160 dilution of 12.4% sucrose, 1.1% fructose and 1.1% glucose) [Loren et al., 2005]. This corresponds to an adult mouse ingesting 4.8 mg polyphenols per day (each mouse drinks about 5 ml of wa-ter per day). This dose of polyphenols was estimated to be similar to the amount of polyphenols ingested by mice drinking pome-granate juice in our previous study [Loren et al., 2005]. PPE di-luted in sugar water or straight sugar water alone was given to the pregnant females as the sole source of drinking water throughout pregnancy and following the delivery, for the duration of the life of the pups. Drinking water was prepared fresh twice a week and kept in UV opaque bottles to prevent breakdown of the polyphe-nols. Since littermate controls were not possible for this experi-ment, we used 4 litters for each group to minimize the effect of litter to litter variability.
Resveratrol and Vehicle Injections Before each experiment, resveratrol (Cayman Chemicals) was
freshly dissolved in 100% dimethyl sulfoxide (DMSO, Sigma) at a final concentration of 70 mg/ml. At this concentration, a 3.5-gram mouse would receive a 1- � l injection to give a final concen-tration of 20 mg/kg (high dose). For 200- � g/kg (medium-dose) and 2- � g/kg (low-dose) injections the 70 mg/ml stock was diluted 1: 100 and 1: 10,000, respectively. Drug or vehicle was injected into the intraperitoneal space using a 26-gauge needle and a 5- � l Hamilton syringe (Hamilton). For initial studies in mice, 3 dif-ferent time points of injection were chosen: 24 h before the start of the hypoxic episode, 10 min before hypoxia or 3 h after comple-tion of the hypoxic episode. At each time point, half the litter would be injected with one concentration of resveratrol and the other half injected with vehicle (DMSO) alone. All rats were in-jected with 20 mg/kg at 10 min before injury, or 3 h after injury using a freshly prepared 70 mg/ml stock as for mice.
Tissue Lysis and DEVD Cleavage Activity At 24 h following the end of the hypoxic episode, mice and rats
were sacrificed by lethal injection of 200 mg/kg pentobarbital. Following transcardial perfusion with heparinized saline, the brains were extracted and the left and right hippocampi dissected on ice and frozen using dry ice. For each individual experiment, left and right hippocampi from all animals were lysed on the same day in the same lysis buffer (Cell Signaling) containing protease inhibitor cocktail (Roche). Lysates were cleared by spinning at 20,000 g for 15 min at 4 ° C. Protein concentration in each lysate was determined using a BCA kit (Pierce) and DEVD cleavage ac-

Polyphenols Protect against Neonatal Hypoxic-Ischemic Injury
Dev Neurosci 2007;29:363–372 365
tivity determined as described [West et al., 2006]. Briefly, lysates were incubated with DEVD-AMC (Calbiochem) in assay buffer (10 m M HEPES, pH 7.4, 42 m M KCl, 5 m M MgCl 2 , 3 m M DTT, and 10% sucrose) and fluorescence was measured every 5 min using a fluorescence plate reader (BioTek). DEVD cleavage activity was normalized to total protein concentration for each lysate and is reported as pmol AMC generated per mg total protein per min-ute.
Western Blotting Remaining tissue lysates from the hippocampi that had been
used for DEVD cleavage assays were pooled together into groups containing lysates from 4 individual mice. Based on protein con-centration, each pool contains the same amount of protein lysate from the left or right hippocampus of 4 different mice. This allows for comparison of lysates from several different mice on the same gel. For Western blotting, 20 � g of total protein per lane was sep-arated on a 4–12% Bis-Tris NuPage gel (Invitrogen). Proteins were transferred onto Immobilon P membranes (Millipore) and blocked in 2% ECL blocking reagent (Amersham). Membranes were then incubated with primary antibody overnight. Antibod-ies used were: Spectrin (1: 1k, Chemicon), tubulin (1: 4k, Sigma). Membranes were then incubated with HRP-labeled secondary antibody, developed using SuperSignal (Pierce), and visualized on an ImageStation (Kodak).
Histology and Tissue Loss Calculation For histological assessment of the extent of H-I injury, mice
were sacrificed at P14 and their brains extracted and sectioned into 50- � m sections. Slices 300- � m apart were mounted, stained with cresyl violet and digitized on an Expression 1680 transpar-ency scanner (Epson). Percentage volume loss in the hippocam-pus, cortex and striatum was calculated by comparing the area of remaining tissue in the injured and noninjured hemispheres as described [West et al., 2006]. For hippocampal tissue loss, 6 con-secutive sections were measured, for cortical tissue loss 4 con-secutive sections were measured and for striatal tissue loss 2 con-secutive sections were measured.
Statistics All data are presented as mean 8 SEM and comparisons be-
tween drug and vehicle groups were done using a t test if the data were parametric, or a Mann-Whitney U test for nonparametric data. Statistical significance was set at p ! 0.05. Statistics were performed using GraphPad Prism (GraphPad Software, Inc.).
Results
Ingestion of PPE by the Dam Lowers Caspase-3Activation in Hippocampus of Pups following Neonatal H-I Pregnant female mice were given either vehicle (sugar
water) or PPE in vehicle as the sole supply of drinking water during pregnancy and following delivery. Neonatal H-I was performed on the pups at postnatal day 7 (P7) and the pups were sacrificed 24 h following the injury.
Caspase-3 activity (measured as rate of DEVD cleavage) was measured in the left and right hippocampus. In this model of neonatal H-I in our prior studies, the majority of tissue loss and cell death is in the hippocampus with � 40% tissue loss 7 days after H-I, � 20% tissue loss in the striatum, and � 5% in the cortex [Han et al., 2001; West et al., 2006]. In previous studies using this model, cas-pase-3 activation in the hippocampus has been shown to correlate well with injury in other brain regions [Han et al., 2001; West et al., 2006]. Caspase-3 activation is a reli-able and readily quantifiable measure of the extent of apoptotic neuronal cell death in response to neonatalH-I injury [Han et al., 2000; Gibson et al., 2001]. Thus, we assessed caspase-3 activation in the hippocampus af-ter H-I. Pups of dams that have been administered PPE had significantly less caspase-3 activity in the hippocam-pus than did pups of dams subjected to sugar water alone ( fig. 1 ). This suggests that the polyphenols in the pome-granate juice were responsible for the neuroprotection observed in the previous study.
Resveratrol Protects against Caspase-3 and Calpain Activation in Mice in a Dose- and Time-Dependent Manner To further explore the role of polyphenols in protect-
ing the neonatal brain following neonatal H-I, we inves-
**
0
DEV
Das
e ac
tivi
ty(p
mol
/min
/mg
pro
tein
)
25
50
75
100
125
150
PPE L VEH L VEH RPPE R
Fig. 1. PPE protects against caspase-3 activation following neona-tal H-I. Female mice had their drinking water replaced with either sugar water (vehicle, VEH) or sugar water containing the PPE during pregnancy and following delivery. Pups from these dams were subjected to neonatal H-I at P7 and caspase-3 activity was measured in the left (L) and right (R) hippocampus at 24 h follow-ing the injury by DEVD cleavage assay. Pups of dams drinking PPE (n = 25) had significantly less caspase-3 activation in the left hippocampus than pups of dams drinking sugar water (VEH, n = 21). * * p = 0.0024 comparing PPE vs. vehicle-treated mice.

West /Atzeva /Holtzman
Dev Neurosci 2007;29:363–372366
tigated the effect of the specific polyphenol resveratrol. Previous studies using resveratrol in adult rat stroke models have focused on high concentrations and the ef-fect of treatment before the injury. Since the effect of res-veratrol has not been tested in neonatal H-I model, we wanted to investigate the dose and time dependence of protection by resveratrol in this injury paradigm in mice. We tested 3 different concentrations of resveratrol: 20 mg/kg, 200 � g/kg and 2 � g/kg. These concentrations were administered at 3 different time points: 24 h before
the start of hypoxia, 10 min before the start of hypoxia, and 3 h after hypoxia. As an indicator of the level of in-jury we measured caspase-3 activity at 24 h after injury. When administered 24 h before the hypoxic insult, res-veratrol decreased DEVD cleavage activity in a dose-de-pendent manner, with 20 mg/kg and 200 � g/kg being significantly protective against caspase-3 activation and 2 � g/kg having no significant effect ( fig. 2 a). If adminis-tered 10 min before injury, resveratrol is protective in the same concentration-dependent manner as if adminis-
RES high VEH high RES med VEH med RES low VEH low
0
a
b
c
DEV
Das
e ac
tivi
ty(p
mol
/min
/mg
pro
tein
)
100
200
300
**
12 10 8
*
8 8 9
RES high VEH high RES med VEH med RES low VEH low
0
DEV
Das
e ac
tivi
ty(p
mol
/min
/mg
pro
tein
)
100
200
300
**
12 13 12
*
10 14 13
RES high VEH high RES med VEH med RES low VEH low
0
DEV
Das
e ac
tivi
ty(p
mol
/min
/mg
pro
tein
)
100
200
300
12 11 13 11 8 9
Fig. 2. Resveratrol protects the neonatal brain against H-I-induced caspase-3 acti-vation in a time- and dose-dependent manner. At different time points before or after injury, littermate mouse pups were injected interperitoneally with vehicle (DMSO) or resveratrol at high (20 mg/kg), medium (200 � g/kg) or low (2 � g/kg) dos-es. Caspase-3 activation was measured as DEVD cleavage activity in the injured hip-pocampus at 24 h following neonatal H-I injury given at P7. The number of animals in each treatment group is indicated inside each bar in the graph: 55 pups were given either resveratrol or vehicle at 24 h before injury, 74 pups were given resveratrol or vehicle 10 min before injury, and 64 mice were treated with resveratrol or vehicle 3 h after injury. There is no difference in DEVD cleavage activity in the noninjured hippocampus, data not shown for clarifi-cation. a If injected at 24 h before hypoxia, resveratrol protects against caspase-3 acti-vation in the hippocampus in a dose-de-pendent manner, when compared to litter-mate mice injected with vehicle. The high and the medium dose of resveratrol signif-icantly reduce the amount of caspase-3 ac-tivation, while the low dose of resveratrol does not provide significant protection. b If injected 10 min before hypoxia resve-ratrol reduces caspase-3 activation at the high and medium doses, but not at the low dose. c When injected at 3 h after injury, resveratrol does not reduce caspase-3 ac-tivity at any of the doses utilized. * p ! 0.05 and * * p ! 0.01 resveratrol vs. vehicle-in-jected littermates.

Polyphenols Protect against Neonatal Hypoxic-Ischemic Injury
Dev Neurosci 2007;29:363–372 367
tered 24 h prior to injury ( fig. 2 b). However, when admin-istered 3 h after injury, resveratrol has no effect on cas-pase-3 ( fig. 2 c). The level of protection is the same at 20 mg/kg and 200 � g/kg, showing that resveratrol could po-tentially protect at physiologically relevant levels. Inter-estingly, resveratrol is protective even when given at 24 h before the injury. This could either be due to slow me-tabolism of resveratrol [Yu et al., 2002] in the mouse pup or that resveratrol can mimic the effects of precondition-ing, as shown in a rat brain slice model of ischemia [Ra-val et al., 2006]. To see if there was a difference in protec-tion afforded by resveratrol administered at different time points, we compared the level of caspase-3 activa-tion between time points by one-way ANOVA. While cas-pase-3 activation in mice receiving preinjury administra-tion is significantly different from postadministration, there is no statistical difference in the results obtained when resveratrol was administered either 24 h or 10 min prior to H-I.
To further investigate the cell death pathways inhib-ited by resveratrol, we investigated calpain activation in the injured and noninjured hippocampus following H-I. Spectrin is cleaved by both calpain and caspase-3, giving rise to specific cleavage products, p145 and p150 for cal-
pain and p120 for caspase-3. We have previously shown that calpain cleavage of spectrin is caspase-3 independent [Han et al., 2002; West et al., 2006]. Thus, calpain cleav-age of spectrin is a measure of a nonapoptotic and likely necrotic type of cell death. In brain lysates assessed 24 h after H-I, the calpain cleavage products of spectrin were markedly decreased in the hippocampus of mice that had received resveratrol 10 min before the onset of hypoxia ( figure 3 ). This shows that in addition to decreasing apop-totic cell death, resveratrol also decreases the amount of necrotic cell death following H-I.
Resveratrol Protects the Neonatal Brain against Tissue Loss following H-I To correlate the decrease in molecular markers of
apoptosis and necrosis in mice injected with resveratrol with longer term protection of the brain, we measured the percentage tissue loss at P14. For this study, littermate mice received either 20 mg/kg resveratrol or vehicle at 10 min before the start of the hypoxic period at P7. At 7 days after injury (P14), the mice were sacrificed and their brains extracted and processed for histological analysis. Percentage volume tissue loss was calculated by compar-ing the ipsi- and contralateral areas of the brain in con-
RES
L R L R L R L R L R L R
p150
p145
p120
b
Tubulin
Rela
tive
inte
nsi
ty
*** ***
RES L
a
RES R VEH L
p150 p145
VEH R RES L RES R VEH L VEH R
VEH RES VEH RES VEH
Fig. 3. Resveratrol reduces caspase-3 and calpain cleavage of spectrin following neo-natal H-I. Spectrin cleavage products were investigated at 24 h after injury in mice in-jected with 20 mg/kg resveratrol or vehicle at 10 min before exposure to hypoxia and after carotid ligation. Pooled lysates from 4 different animals were run in each lane. Injured (left, L) hippocampus or nonin-jured (right, R) were run side by side on SDS PAGE and probed with antibodies against spectrin. Spectrin is cleaved by cal-pain to give rise to a p150 and a p145 band and by caspase-3 to give rise to a p120 band. These bands are strongly present in lysates from vehicle-injected mice but to a much lesser extent in lysates from resvera-trol-injected mice. Relative intensity of the p150 and p145 bands was calculated by comparison to the intensity of the tubulin loading control. The intensities of the p150 and p145 bands were significantly reduced in the mice that had received resveratrol injection, suggesting that calpain activa-tion is prevented by resveratrol. * * * p ! 0.001 resveratrol vs. vehicle by t test.

West /Atzeva /Holtzman
Dev Neurosci 2007;29:363–372368
secutive coronal sections 300- � m apart. We have previ-ously shown that this is an accurate way to measure the amount of tissue lost following neonatal H-I injury [Cheng et al., 1997; West et al., 2006]. Administration of 20 mg/kg of resveratrol resulted in significant protection against tissue loss in the hippocampus and striatum ( fig. 4 ). There is also a decrease in the cortical tissue loss; however, this is not statistically significant due to the low amount of injury in the cortex.
Resveratrol Reduces Caspase-3 Activation in the Hippocampus of Neonatal Rats following H-I The patterns and mechanisms of brain injury in the
neonatal rat and mouse are similar but not the same. To further investigate the neuroprotective effects of resvera-trol, we tested the ability of resveratrol to inhibit caspase-3 activation in a rat model of neonatal H-I. Although neo-natal H-I injury in our protocol is performed at P7 in both rats and mice, it is likely that the brain of the two rodents are at slightly different developmental stages at this age [Hagberg et al., 1997]. An example of differences between species is that minocycline has been found to be neuro-protective in the rat but not in the mouse neonatal H-I
model [Tsuji et al., 2004]. Also, the time course of cas-pase-3 activation in rats and mice following neonatal H-I is different [Cheng et al., 1998; Han et al., 2001]. Finally, there is a difference in the fatality rate during exposure to hypoxia between rats and mice. In mice the fatality rate
0
Perc
ent
tiss
ue lo
ss
10
Hippocampus Cortex Striatum
20
30
40
50
60
**
***
n.s.
Fig. 4. Resveratrol decreases the amount of tissue loss at P14. P7 mice were injected with resveratrol ( $ ; n= 24) or vehicle ( ) ; n = 26) following carotid ligation and 10 min before hypoxia and tis-sue loss was assessed at 7 days after injury. This is a good measure of overall brain injury following neonatal H-I. Percentage tissue loss was determined by comparing the area of the brain region in the left and the right hemisphere through several serial coronal brain sections as described in the ‘Materials and Methods’ sec-tion. When compared to mice injected with vehicle, mice receiv-ing resveratrol were significantly protected against tissue loss in the hippocampus and striatum. Resveratrol also appears to pro-tect the cortex, but this protection is not statistically significant (p = 0.1). * * p ! 0.01 and * * * p ! 0.001 resveratrol vs. vehicle in-jected littermates.
*
0a
DEV
Das
e ac
tivi
ty(p
mol
/min
/mg
pro
tein
)
10
20
30
40
50
60
70
80
90
100
110
RES L VEH L VEH RRES R
0
DEV
Das
e ac
tivi
ty(p
mol
/min
/mg
pro
tein
)
25
50
75
100
125
150
b RES L VEH L VEH RRES R
Fig. 5. Resveratrol protects the neonatal rat brain against caspase-3 activation following H-I injury. Rats underwent neonatal H-I at P7 and were injected with 20 mg/kg resveratrol or vehicle follow-ing carotid ligation and 10 min before hypoxia ( a ) or 3 h after hypoxia ( b ). DEVD cleavage activity was measured at 24 h after injury in hippocampal lysates. As seen in the mice, rats that re-ceived resveratrol before injury are protected against caspase-3 activation following neonatal H-I. Due to a higher incidence of no detectable caspase-3 activation following neonatal H-I in rats, a larger number of rats were enrolled in this study to determine whether there was a statistical difference between resveratrol-treated (n = 18) and vehicle-treated (n = 16) rats. * p ! 0.05 resve-ratrol vs. vehicle injected littermates. When administered 3 h af-ter hypoxia, there was no significant difference in tissue loss be-tween rats that received resveratrol (n = 19) vs. vehicle (n = 18).

Polyphenols Protect against Neonatal Hypoxic-Ischemic Injury
Dev Neurosci 2007;29:363–372 369
is below 10%, probably because the hypoxic episode is relatively short. However, in our protocol that results in a similar amount of brain injury, rats are hypoxic for 150 min and this is associated with a higher fatality rate. In this study, 4 of 22 (18%) rats injected with resveratrol died during hypoxia while 6 of 22 (27%) of rats injected with vehicle died during hypoxia.
To test if resveratrol specifically protects the neonatal rat brain, we measured caspase-3 activation at 24 h fol-lowing neonatal H-I in rats. We found that administra-tion of 20 mg/kg resveratrol to rats 10 min before the start of the hypoxic period leads to a significant decrease in caspase-3 activation in rat pups as it did in mice ( fig. 5 a). We also tested if 20 mg/kg of resveratrol is protective when administered 3 h after the injury since the time course of caspase-3 activation is different between rats and mice and since there has been reports of neuropro-tective agents working in the rat when administered after the injury [Wei et al., 2004; Shin et al., 2006]. However, at this time point, resveratrol is not protective against caspase-3 activation ( fig. 5 b). Thus, it appears that resve-ratrol protection of the rat brain follows similar time de-pendence as in the mouse brain.
Discussion
Dietary supplementation with foods rich in polyphe-nols – pomegranates, blueberries, green tea, and apple juice – has been shown to provide neuroprotection in an-imal models of focal brain ischemia, of periventricular white matter injury, and of Alzheimer’s disease [Levites et al., 2001; Sweeney et al., 2002; Dajas et al., 2003; Etus et al., 2003; Ortiz and Shea, 2004; Loren et al., 2005; Hart-man et al., 2006]. Polyphenols have been found to possess antioxidant properties as well as to have effects on gene expression [Kostrzewa and Segura-Aguilar, 2003]. Spe-cifically, one polyphenol, resveratrol, has been shown to increase activity of members of the sirtuin gene class, blunting p53 action and blocking apoptosis [Latruffe et al., 2002; Hall, 2003; Howitz et al., 2003]. Recent studies indicate that among foods that contain polyphenols, juice extracted from the pomegranate has the highest concen-tration of measurable polyphenols [Gil et al., 2000; Kela-wala and Ananthanarayan, 2004]. The pharmacologic actions of pomegranate juice include antiatherosclerotic, antibacterial, and antiproliferative properties [Anesini and Perez, 1993; Kim et al., 2002]. We recently found that when the polyphenol-rich pomegranate juice is con-sumed by the dam polyphenols from the juice are present
in the pup and protected the pup against H-I brain injury [Loren et al., 2005]. Other studies have shown that the polyphenols caffeic acid phenylethyl ester and amentofla-vone are also protective against neonatal H-I brain injury [Wei et al., 2004; Shin et al., 2006]. To test the hypothesis that it is the polyphenols of pomegranate juice that are responsible for neuroprotection, we tested the effect of PPE in the neonatal H-I mouse model. Supplementation of PPE to the drinking water of pregnant and nursing dams resulted in significantly decreased H-I-induced caspase-3 activation. This suggests that it is the polyphe-nols of the pomegranate juice that are responsible for the neuroprotection.
To further investigate the role of polyphenols in neo-natal H-I, we focused on the specific polyphenol resvera-trol. This naturally occurring compound has been found to be neuroprotective in adult ischemia in rats when ad-ministered before the injury, but to our knowledge resve-ratrol has never been tested in neonatal H-I [Virgili and Contestabile, 2000; Huang et al., 2001; Gupta et al., 2002; Sinha et al., 2002]. By examining a variety of different concentrations at several different time points, we found that IP injection of resveratrol leads do decreased cas-pase-3 activation in the P7 mouse in a concentration- and time-dependent manner. At doses of 200 � g/kg or great-er, resveratrol leads to decreased caspase-3 activation but only when resveratrol is injected prior to the injury. In addition to decreasing the caspase-3 activation, resvera-trol also decreases the calpain activation following neo-natal H-I, suggesting that it works as a generally neuro-protective agent and not just on the apoptotic pathway.
In addition to finding that resveratrol is protective in the neonatal mouse, we also demonstrated that resvera-trol protects the neonatal rat against H-I-induced cas-pase 3 activation. Although the injury paradigm is sim-ilar in rats and mice, there are several neuroprotective agents that have been found to work only in one species. Since resveratrol has been found to protect against stroke in neonatal rats and mice as well as in adults, it could potentially be considered for further investiga-tions in humans. Interestingly, we did not find resvera-trol to be protective in the rat when given after the in-jury. Since the apoptotic cell death in the rat starts much later in the rat than in the mouse, and several drugs have been shown to be protective in the rat when given after the insult, we thought that resveratrol might follow the same pattern. The fact that resveratrol does not protect when given after the injury suggests that it is acting through proximal mechanisms in the cell death path-way initiated by H-I. One pathway that may be involved

West /Atzeva /Holtzman
Dev Neurosci 2007;29:363–372370
References
Aggarwal BB, Shishodia S (2004): Suppression of the nuclear factor-kappaB activation path-way by spice-derived phytochemicals: rea-soning for seasoning. Ann N Y Acad Sci 1030: 434–441.
Anesini C, Perez C (1993): Screening of plants used in Argentine folk medicine for antimi-crobial activity. J Ethnopharmacol 39: 119–128.
Araki T, Sasaki Y, Milbrandt J (2004): Increased nuclear NAD biosynthesis and SIRT1 activa-tion prevent axonal degeneration. Science 305: 1010–1013.
Back SA (2001): Recent advances in human peri-natal white matter injury. Prog Brain Res 132: 131–147.
Back SA, Rivkees SA (2004): Emerging concepts in periventricular white matter injury. Semin Perinatol 28: 405–414.
Bianchini F, Vainio H (2003): Wine and resvera-trol: mechanisms of cancer prevention? Eur J Cancer Prev 12: 417–425.
Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mosto-slavsky R, Cohen HY, Hu LS, Cheng HL, Je-drychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME (2004): Stress-depen-dent regulation of FOXO transcription fac-tors by the SIRT1 deacetylase. Science 303:
2011–2015.
Cheng Y, Deshmukh M, D’Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, Johnson EM, Holtzman DM (1998): Caspase inhibitor affords neuroprotection with de-layed administration in a rat model of neo-natal hypoxic-ischemic brain injury. J Clin Invest 101: 1992–1999.
Cheng Y, Gidday JM, Yan Q, Shah AR, Holtzman DM (1997): Marked age-dependent neuro-protection by brain-derived neurotrophic factor against neonatal hypoxic-ischemic brain injury. Ann Neurol 41: 521–529.
Dajas F, Rivera F, Blasina F, Arredondo F, Ech-everry C, Lafon L, Morquio A, Heizen H (2003): Cell culture protection and in vivo neuroprotective capacity of f lavonoids. Neu-rotox Res 5: 425–432.
Etus V, Altug T, Belce A, Ceylan S (2003): Green tea polyphenol (–)-epigallocatechin gallate prevents oxidative damage on periventricu-lar white matter of infantile rats with hydro-cephalus. Tohoku J Exp Med 200: 203–209.
Faustino RS, Sobrattee S, Edel AL, Pierce GN (2003): Comparative analysis of the phenolic content of selected Chilean, Canadian and American Merlot red wines. Mol Cell Bio-chem 249: 11–19.
Gibson ME, Han BH, Choi J, Knudson CM, Korsmeyer SJ, Parsadanian M, Holtzman DM (2001): BAX contributes to apoptotic-like death following neonatal hypoxia- isch-emia: evidence for distinct apoptosis path-ways. Mol Med 7: 644–655.
Gieron-Korthals M, Colon J (2005): Hypoxic-ischemic encephalopathy in infants: new challenges. Fetal Pediatr Pathol 24: 105–120.
Gil MI, Tomas-Barberan FA, Hess-Pierce B, Hol-croft DM, Kader AA (2000): Antioxidant ac-tivity of pomegranate juice and its relation-ship with phenolic composition and pro-cessing. J Agric Food Chem 48: 4581–4589.
Gluckman PD, Wyatt JS, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, Polin RA, Robertson CM, Thoresen M, Whitelaw A, Gunn AJ (2005): Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet 365: 663–670.
Gulcan H, Ozturk IC, Arslan S (2005): Altera-tions in antioxidant enzyme activities in ce-rebrospinal f luid related with severity of hy-poxic ischemic encephalopathy in newborns. Biol Neonate 88: 87–91.
Gupta YK, Briyal S, Chaudhary G (2002): Pro-tective effect of trans-resveratrol against kai-nic acid-induced seizures and oxidative stress in rats. Pharmacol Biochem Behav 71:
245–249.
in the effects of polyphenols is via activation of the sir-tuins such as SIRT1.
Polyphenols such as resveratrol may have beneficial effects on health via their antioxidant properties, sup-pression of inflammatory pathways, or other pathways such as activation of the sirtuin pathway [Aggarwal and Shishodia, 2004]. Included in the sirtuin family is SIRT1, a human protein deacetylase that promotes cell survival by mechanisms such as negatively regulating the p53 tu-mor suppressor [Luo et al., 2001; Vaziri et al., 2001; Lang-ley et al., 2002], deacetylating the transcription factor FOXO3 [Brunet et al., 2004; Motta et al., 2004], repress-ing PPAR � signaling [Picard et al., 2004], and modula-tion of NF- � � -dependent transcription [Yeung et al., 2004]. Modulation of these pathways may provide a means to protect the developing brain against neonatal H-I-induced brain damage. Recent studies show that polyphenols, including resveratrol, increase cell survival via activation of SIRT1 [Howitz et al., 2003]. Parker et al. [2005] found that increased sir2 gene dosage or treatment with resveratrol in Caenorhabditis elegans blocked neu-ronal dysfunction and cell death induced by polygluta-
mine expansion. Suggesting that resveratrol may act through a similar pathway in mammals, resveratrol pro-tected mammalian neuronal cell lines from mutant hun-gtingtin-induced cell death, and this effect was inhibited by sirtuin inhibitors [Parker et al., 2005]. There is also evidence that resveratrol can block axonal degeneration via SIRT1 in the mammalian peripheral nervous system [Araki et al., 2004]. While increasing evidence suggests that resveratrol and other polyphenols are neuroprotec-tive, whether their protective actions in the CNS in vivo are via SIRT1 has not been directly assessed. Determin-ing the mechanism of protection of resveratrol, pome-granate polyphenols, and other polyphenols may lead to novel insights into both pathogenesis and treatment of neonatal H-I brain injury.
Acknowledgements
This research was supported by NIH NS35902 and by funds from the Stewart and Linda Resnick Revocable Trust.

Polyphenols Protect against Neonatal Hypoxic-Ischemic Injury
Dev Neurosci 2007;29:363–372 371
Hagberg H, Bona E, Gilland E, Puka-Sundvall M (1997): Hypoxia-ischaemia model in the 7-day-old rat: possibilities and shortcomings. Acta Paediatr Suppl 422: 85–88.
Hall SS (2003): Longevity research. In vino vi-talis? Compounds activate life-extending genes. Science 301: 1165.
Hamrick SE, Ferriero DM (2003): The injury re-sponse in the term newborn brain: can we neuroprotect? Curr Opin Neurol 16: 147–154.
Han BH, D’Costa A, Back SA, Parsadanian M, Patel S, Shah AR, Gidday JM, Srinivasan A, Deshmukh M, Holtzman DM (2000): BDNF blocks caspase-3 activation in neonatal hy-poxia-ischemia. Neurobiol Dis 7: 38–53.
Han BH, DeMattos RB, Dugan LL, Kim-Han JS, Brendza RP, Fryer JD, Kierson M, Cirrito J, Quick K, Harmony JA, Aronow BJ, Holtzman DM (2001): Clusterin contributes to caspase-3-independent brain injury following neo-natal hypoxia-ischemia. Nat Med 7: 338–343.
Han BH, Xu D, Choi J, Han Y, Xanthoudakis S, Roy S, Tam J, Vaillancourt J, Colucci J, Siman R, Giroux A, Robertson GS, Zamboni R, Nicholson DW, Holtzman DM (2002): Selec-tive, reversible caspase-3 inhibitor is neuro-protective and reveals distinct pathways of cell death after neonatal hypoxic-ischemic brain injury. J Biol Chem 277: 30128–30136.
Hartman RE, Shah A, Fagan AM, Schwetye KE, Parsadanian M, Schulman RN, Finn MB, Holtzman DM (2006): Pomegranate juice decreases amyloid load and improves behav-ior in a mouse model of Alzheimer’s disease. Neurobiol Dis 24: 506–515.
Hashimoto T, Yonetani M, Nakamura H (2003): Selective brain hypothermia protects against hypoxic-ischemic injury in newborn rats by reducing hydroxyl radical production. Kobe J Med Sci 49: 83–91.
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA (2003): Small molecule activators of sir-tuins extend Saccharomyces cerevisiae life-span. Nature 425: 191–196.
Huang SS, Tsai MC, Chih CL, Hung LM, Tsai SK (2001): Resveratrol reduction of infarct size in Long-Evans rats subjected to focal cere-bral ischemia. Life Sci 69: 1057–1065.
Kelawala NS, Ananthanarayan L (2004): Anti-oxidant activity of selected foodstuffs. Int J Food Sci Nutr 55: 511–516.
Kil HY, Zhang J, Piantadosi CA (1996): Brain temperature alters hydroxyl radical produc-tion during cerebral ischemia/reperfusion in rats. J Cereb Blood Flow Metab 16: 100–106.
Kim ND, Mehta R, Yu W, Neeman I, Livney T, Amichay A, Poirier D, Nicholls P, Kirby A, Jiang W, Mansel R, Ramachandran C, Rabi T, Kaplan B, Lansky E (2002): Chemopreven-tive and adjuvant therapeutic potential of pomegranate (Punica granatum) for human breast cancer. Breast Cancer Res Treat 71:
203–217.
Kopp P (1998): Resveratrol, a phytoestrogen found in red wine. A possible explanation for the conundrum of the ‘French paradox’? Eur J Endocrinol 138: 619–620.
Kostrzewa RM, Segura-Aguilar J (2003): Novel mechanisms and approaches in the study of neurodegeneration and neuroprotection. A review. Neurotox Res 5: 375–383.
Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T (2002): Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular se-nescence. EMBO J 21: 2383–2396.
Latruffe N, Delmas D, Jannin B, Cherkaoui Mal-ki M, Passilly-Degrace P, Berlot JP (2002): Molecular analysis on the chemopreventive properties of resveratrol, a plant polyphenol microcomponent. Int J Mol Med 10: 755–760.
Leibovici D, Ritchie K, Ledesert B, Touchon J (1999): The effects of wine and tobacco con-sumption on cognitive performance in the elderly: a longitudinal study of relative risk. Int J Epidemiol 28: 77–81.
Levites Y, Weinreb O, Maor G, Youdim MB, Mandel S (2001): Green tea polyphenol (–)-epigallocatechin-3-gallate prevents N-meth-yl-4-phenyl-1,2,3,6-tetrahydropyridine-in-duced dopaminergic neurodegeneration. J Neurochem 78: 1073–1082.
Loren DJ, Seeram NP, Schulman RN, Holtzman DM (2005): Maternal dietary supplementa-tion with pomegranate juice is neuroprotec-tive in an animal model of neonatal hypoxic-ischemic brain injury. Pediatr Res 57:
858–864. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh
A, Guarente L, Gu W (2001): Negative con-trol of p53 by Sir2alpha promotes cell sur-vival under stress. Cell 107: 137–148.
Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L (2004): Mammalian SIRT1 re-presses forkhead transcription factors. Cell 116: 551–563.
Ortiz D, Shea TB (2004): Apple juice prevents oxidative stress induced by amyloid-beta in culture. J Alzheimers Dis 6: 27–30.
Parker JA, Arango M, Abderrahmane S, Lam-bert E, Tourette C, Catoire H, Neri C (2005): Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet 37: 349–350.
Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L (2004): Sirt1 promotes fat mobilization in white adipo-cytes by repressing PPAR-gamma. Nature 429: 771–776.
Raval AP, Dave KR, Perez-Pinzon MA (2006): Resveratrol mimics ischemic precondition-ing in the brain. J Cereb Blood Flow Metab 26: 1141–1147.
Robertson CM, Finer NN, Grace MG (1989): School performance of survivors of neonatal encephalopathy associated with birth as-phyxia at term. J Pediatr 114: 753–760.
Sato M, Suzuki Y, Okuda T, Yokotsuka K (1997): Contents of resveratrol, piceid, and their iso-mers in commercially available wines made from grapes cultivated in Japan. Biosci Bio-technol Biochem 61: 1800–1805.
Shankaran S, Laptook AR, Ehrenkranz RA, Ty-son JE, McDonald SA, Donovan EF, Fanaroff AA, Poole WK, Wright LL, Higgins RD, Fin-er NN, Carlo WA, Duara S, Oh W, Cotten CM, Stevenson DK, Stoll BJ, Lemons JA, Guillet R, Jobe AH (2005): Whole-body hy-pothermia for neonates with hypoxic-isch-emic encephalopathy. N Engl J Med 353:
1574–1584. Shankaran S, Woldt E, Koepke T, Bedard MP,
Nandyal R (1991): Acute neonatal morbidity and long-term central nervous system se-quelae of perinatal asphyxia in term infants. Early Hum Dev 25: 135–148.
Shin DH, Bae YC, Kim-Han JS, Lee JH, Choi IY, Son KH, Kang SS, Kim WK, Han BH (2006): Polyphenol amentoflavone affords neuro-protection against neonatal hypoxic-isch-emic brain damage via multiple mecha-nisms. J Neurochem 96: 561–572.
Singh SK, Dua T, Tandon A, Kumari S, Ray G, Batra S (1999): Status of lipid peroxidation and antioxidant enzymes in hypoxic isch-emic encephalopathy. Indian Pediatr 36:
561–566. Sinha K, Chaudhary G, Gupta YK (2002): Pro-
tective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life Sci 71: 655–665.
Sweeney MI, Kalt W, MacKinnon SL, Ashby J, Gottschall-Pass KT (2002): Feeding rats di-ets enriched in lowbush blueberries for six weeks decreases ischemia-induced brain damage. Nutr Neurosci 5: 427–431.
Thoresen M, Satas S, Puka-Sundvall M, Whitelaw A, Hallstrom A, Loberg EM, Ungerstedt U, Steen PA, Hagberg H (1997): Post-hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. Neuroreport 8:
3359–3362. Tokusoglu O, Unal MK, Yemis F (2005): Deter-
mination of the phytoalexin resveratrol (3,5,4 � -trihydroxystilbene) in peanuts and pistachios by high-performance liquid chro-matographic diode array (HPLC-DAD) and gas chromatography-mass spectrometry (GC-MS). J Agric Food Chem 53: 5003–5009.
Truelsen T, Thudium D, Gronbaek M (2002): Amount and type of alcohol and risk of de-mentia: the Copenhagen City Heart Study. Neurology 59: 1313–1319.
Tsuji M, Wilson MA, Lange MS, Johnston MV (2004): Minocycline worsens hypoxic-isch-emic brain injury in a neonatal mouse mod-el. Exp Neurol 189: 58–65.

West /Atzeva /Holtzman
Dev Neurosci 2007;29:363–372372
Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA (2001): hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107: 149–159.
Virgili M, Contestabile A (2000): Partial neuro-protection of in vivo excitotoxic brain dam-age by chronic administration of the red wine antioxidant agent, trans-resveratrol in rats. Neurosci Lett 281: 123–126.
Volpe J (1995): Neurology of the Newborn. Phil-adelphia, WB Saunders.
Wei X, Zhao L, Ma Z, Holtzman DM, Yan C, Dodel RC, Hampel H, Oertel W, Farlow MR, Du Y (2004): Caffeic acid phenethyl ester prevents neonatal hypoxic-ischaemic brain injury. Brain 127: 2629–2635.
West T, Atzeva M, Holtzman DM (2006): Cas-pase-3 deficiency during development in-creases vulnerability to hypoxic-ischemic injury through caspase-3-independent path-ways. Neurobiol Dis 22: 523–537.
Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW (2004): Mod-ulation of NF-kappaB-dependent transcrip-tion and cell survival by the SIRT1 deacety-lase. EMBO J 23: 2369–2380.
Yu C, Shin YG, Chow A, Li Y, Kosmeder JW, Lee YS, Hirschelman WH, Pezzuto JM, Mehta RG, van Breemen RB (2002): Human, rat, and mouse metabolism of resveratrol. Pharm Res 19: 1907–1914.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:373–384 DOI: 10.1159/000105478
Mast Cell Stabilization LimitsHypoxic-Ischemic Brain Damagein the Immature Rat
Yuxuan Jin
a Ann-Judith Silverman
a Susan J. Vannucci
b
a Department of Pathology and Cell Biology, b
Department of Pediatrics, Columbia University Medical Center, New York, N.Y. , USA
with the MCs stabilizer, disodium cromoglycate (cromolyn), prior to and/or following hypoxia-ischemia. The cromolyn treatment inhibited MC migration into the CNS (p ! 0.05) and limited brain damage more than 50% (p ! 0.01) vs. saline controls. These data support the hypothesis that MCs are key contributors to the extent of brain damage due to hypoxia-ischemia in the immature animal.
Copyright © 2007 S. Karger AG, Basel
Introduction
Perinatal hypoxic-ischemic (HI) brain damage is a ma-jor cause of acute mortality and chronic neurological mor-bidity in infants and children. Statistics show that 20–50% of asphyxiated newborns with HI encephalopathy die within the newborn period [Vannucci, 2000]. Up to 25% of the survivors exhibit permanent neurological handi-caps, including cerebral palsy, epilepsy and mental retar-dation [Volpe, 1992; Vannucci, 2000]. Most of the current understanding of the mechanisms underlying HI brain damage and subsequent strategies for potential therapy in the neonate derive from extensive experimental studies in adult stroke models. However, in recent years it has be-come evident that immature brains differ from those of adults in many aspects [Vannucci and Hagberg, 2004]. Immature brains can withstand longer periods of HI
Key Words
Astroglia � Blood-brain barrier � Cromolyn � Endothelia � Microglia
Abstract
Perinatal hypoxic-ischemic (HI) brain damage is a major cause of mortality and neurological morbidity in infants and children. Using an established model of unilateral hypoxia-ischemia in neonatal rats, the present study focused on mast cells (MCs), important regulators of inflammatory processes, as potential contributors to HI damage. MCs are present in the pia of the neonatal rat, entering the central nervous sys-tem (CNS) during cerebral development along penetrating blood vessels. Following hypoxia-ischemia, MC numbers in-creased dramatically in the ipsilateral (ischemic) hemisphere (p ! 0.01). In animals exposed to hypoxia only, the numbers of MCs were elevated in both hemispheres to an extent equal to that observed in the contralateral hemisphere of HI animals (p ! 0.05 vs. control). Within damaged areas (ipsilat-eral only), MCs were observed in regions of activated mi-croglia and astroglia that characterize the ischemic hemi-sphere. Using a triple-label paradigm, MCs were observed along elongating blood vessels, some of which express the GLUT1 isoform of the glucose transporter protein, indicative of blood-brain barrier vessels. To determine whether MC ac-tivation has a role in HI brain damage, rat pups were treated
Received: September 6, 2006 Accepted after revision: January 3, 2007
Yuxuan Jin Department of Pathology and Cell Biology, Columbia University Medical Center 650 W168th Street, BB1524New York, NY 10032 (USA) Tel. +1 212 305 3453, Fax +1 212 305 3970, E-Mail [email protected]
© 2007 S. Karger AG, Basel
Accessible online at:www.karger.com/dne

Jin /Silverman /Vannucci
Dev Neurosci 2007;29:373–384374
than adults. Neonates also exhibit periods of increased sensitivity to injury, depending on the developmental state of the brain and site of injury [McQuillen et al., 2003; Van-nucci and Hagberg, 2004]. Therefore, a thorough under-standing of the pathophysiology of perinatal HI brain damage is essential to design effective intervention.
Inflammation plays an important role in the patho-genesis of cerebral ischemic injury [Dirnagl et al., 1999]. The cascade of inflammatory reactions in adult and neo-natal brains includes inflammatory cell infiltration, ede-ma, and the synthesis of specific cytokines/chemokines and endothelial adhesion molecules. Extensive animal studies have shown that in neonatal brain HI initiates both acute and prolonged responses, which progress over days to weeks and result in secondary development of in-jury. These inflammatory responses include glial activa-tion and elevated expression of proinflammatory media-tors [Silverstein et al., 1997; Bona et al., 1999].
Mast cells (MCs) are multifunctional immune cells derived from the hematopoetic stem cell. In normal ro-dent brain, MCs are located in the dura mater, leptomen-inges, choroid plexus, diencephalon and along blood ves-sels on the brain side of the blood-brain barrier (BBB) [Dimitriadou et al., 1990]. MCs are found in the pia mater beginning in late fetal life in the rat and enter the central nervous system (CNS) parenchyma (primarily the thala-mus) at approximately postnatal days (P) 7–8 [Lam-bracht-Hall et al., 1990] in parallel with the growth of the vascular tree [Lambracht-Hall et al., 1990; Michaloudi et al., 2003]. The close proximity of MCs to the other mem-bers of the neurovascular unit (neurons, astrocytes and endothelia) places them in an ideal location to respond rapidly to physiological and pathological changes in brain and to influence the neural cells around them.
Although recent studies suggest MCs are important players in adult ischemia brain damage models [Hu et al., 2004; Strbian et al., 2006], little is known about the role of MCs in the pathogenesis of HI brain damage in new-borns. MCs are linked to damage following ischemia in the adult heart [Singh and Saini, 2003] and other organs. Cardiac MCs accumulated in massive numbers in the in-farct region after acute myocardial infarction [Engels et al., 1995] and can rapidly degranulate, releasing hista-mine, tumor necrosis factor (TNF)- � and thromboxane B 2 shortly after myocardial ischemia [Frangogiannis et al., 1998]. Similarly, oxygen-glucose deprivation induces MCs to degranulate [Hu et al., 2005].
We hypothesized that MCs residing in the pia and neural tissue of the neonatal rat brain respond to HI and contribute to the initial damage. To test this hypothesis,
we subjected neonatal rats to unilateral cerebral HI [Rice et al., 1981; Vannucci et al., 1996] and examined brains for changes in the number and distribution of brain MCs relative to neuronal damage, glial activation, and barrier properties of blood vessels on which they reside. We dem-onstrate that MC stabilization in conjunction with tran-sient cerebral HI reduces both MC migration into the brain and their degranulation, resulting in a reduction in brain damage.
Material and Methods
Animals Timed pregnant Wistar rats (Charles River, Mass., USA) were
purchased and allowed to deliver in individual cages. On day of birth, 2–3 litters were randomized and each dam was given 10 pups. Dams had free access to rat chow and water. Animal hous-ing and procedures described below were approved by Institu-tional Animal Care and Use Committee of Columbia University, and follow the guidelines set by the NIH.
Animal Model of Unilateral Cerebral Hypoxia-Ischemia Three groups of P7 pups were used: (1) unilateral hypoxia-
ischemia (HI); (2) hypoxia treatment only (H) and (3) untreated controls (C). Unilateral HI was induced according to our standard methodology [Rice et al., 1981; Vannucci et al., 1996]. Pups were anesthetized with isoflurane (4% for induction of anesthesia; 1% for maintenance of anesthesia)/balance oxygen and the right common carotid artery permanently ligated. The animals were returned to their dams for 2 h to allow for recovery and suckling. Prior to hypoxic exposure, all animals were placed in open glass containers submerged in a 37 ° C water bath to maintain a constant thermal environment. The HI and H pups were exposed to the hypoxic gas mixture (8% O 2 /balance N 2 ) for 90 min, following which they were allowed to recover in the open jars in the water bath. Control animals were placed in an identical chamber into which room air was delivered at the same flow rate. Pups were returned to their dams 15 min after the end of the hypoxicperiod.
Cromolyn Treatment In these experiments, 75 min of HI was used to produce mod-
erate and consistent damage (see results). To investigate the effect of MC stabilization, cromolyn (CR; Sigma, Mo., USA) was admin-istered subcutaneously (SC) 30 min prior to HI, immediately af-ter, and at 1 and 24 h after HI. Saline-injected HI animals served as controls. Initially, two doses of CR were tested: 5 and 50 mg/kg. There was no difference in animal survival, but 50 mg/kg was more effective in reducing MC number and brain injury severity. Therefore, 50 mg/kg was used in experiments reported here. In a subsequent set of experiments, CR was administered SC immedi-ately after HI and again following 1 and 24 h of reperfusion.
Tissue Preparation At 48 h of reperfusion, pups were anesthetized with isoflurane
and decapitated. The brains were then rapidly removed, sub-merged in 4% paraformaldehyde overnight and fixation contin-

CNS Mast Cells and PerinatalHypoxia-Ischemia
Dev Neurosci 2007;29:373–384 375
ued by microwave (80 s). Following cryoprotection, frozen sec-tions (75 � m) were cut on a sliding microtome and collected in serial sets of 5 sections.
MC Quantification One section per set (1/5) was stained with acidic toluidine blue
(TB; Electron Microscopy Sciences, Hatfield, Pa., USA; pH = 2.0) to visualize MCs. Quantitative data were obtained by counting MCs from immediately posterior to the crossing of the anterior commissure through the entire extent of the hippocampus [cor-responding to Bregma 1.80–8.80 mm, Paxinos and Watson, 1986]. Two observers performed these counts and inter-rater reliability was determined by the Pearson correlation. Cell counts were made using a modified optical dissect method, an unbiased ste-reological approach particularly appropriate for thick sections [Coggeshall and Lekan, 1996; Asarian et al . , 2002]. Only cells cut through the plane of the nucleus were included. Raw counts were multiplied by 5; the Abercrombie factor was applied to raw counts to correct for double counting using the formula N = 2n (T/T + D), where N is the corrected cell number, n is the uncorrected cell number, T is the section thickness, and D is the mean diameter of cell nucleus. Equal numbers of male and female animals were used; no sex difference in MC numbers was found for any treat-ment group and data were subsequently merged. Data were seg-regated by hemisphere and tabulated by brain region: hippocam-pus, thalamus, cortex and fornix. For controls and hypoxia-only brains, only one hemisphere was counted (there was no signifi-cant difference between left and right hemisphere in these ani-mals) and the results compared statistically with the ipsilateral or contralateral hemispheres of HI brains.
Assessment of Neuronal Cell Death Dying neurons were identified using Fluoro-Jade B (Chemi-
con, Calif., USA), a dye specific for dead or dying neurons, accord-ing to the protocol developed by Schmued and Hopkins [2000]. Briefly, sections mounted on slides were immersed in a solution of basic alcohol, followed by incubation in 70% alcohol and a dis-tilled water wash. Sections were then incubated for 20 min in a 0.06% solution of potassium permanganate to reduce background staining followed by incubation for 20 min in a 0.0004% solution of Fluoro-Jade B. Slides were then rinsed in distilled water, drained of excess water and air dried. The sections were cleared with His-toclear (National Diagnostic, Ga., USA), coverslipped with D.P.X., nonfluorescent mounting media (Fluka/Sigma, Mo., USA), and examined by fluorescence microscopy (Olympus, Japan) with a QImaging CCD camera (QImaging, Canada) using blue light ex-citation. The morphology of the Fluoro-Jade B-positive cells indi-cates that most, if not all, of the stained material is neuronal in origin.
Quantification of Brain Damage Fluoro-Jade B staining was performed on sections from the
same set as for the TB staining described above. Sections were collected serially from anterior to posterior as for TB above and 1/10 were processed and scored (9–10 sections per brain). Analy-ses of regions containing dying neurons were performed by as-sessing the percentage of Fluoro-Jade B-positive cells in sections. A scale of fluorescence intensity from 0 and 4 ( fig. 1 ) was estab-lished to correspond with the percentage of area damaged per
brain region. A score of 0 indicates no, or very few, dying neurons; a score of 1, less than 20%; 2, 20–50%; 3, 50–80%; 4, more than 80% of the brain region containing dying neurons ( fig. 1 ). Dam-age was scored separately in each brain region (e.g. cortex) and segregated by hemisphere. Fluoro-Jade B stains damaged and dy-ing neurons and is a more sensitive indicator of neuronal damage than standard hematoxylin and eosin histology. Staining inten-sity increases with the extent of damage allowing for relative quantification of fluorescent signal.
Immunochemistry Brain sections were incubated in one or more of the following
primary antibodies at 4 ° C for 24–48 h: rabbit anti-glial fibrillary acidic protein (GFAP) specific for the intermediate filaments of astrocytes (1: 1,000, gift from R. Liem, Columbia University, N.Y., USA); mouse anti-rat OX42, a marker for activated microglia/monocytes (1: 500); mouse anti-rat RECA-1 for blood vessel endo-thelium (1: 100, both from Serotec, UK), and rabbit anti-GLUT-1 for glucose transporter-1 (1: 1,000) [Vannucci et al., 1996]. Over-night incubation (4 ° C) was followed with appropriate fluorescent secondary antibodies (Jackson Labs, Pa., and Invitrogen, Calif., USA) and fluorophore-conjugated egg white avidin (1: 600, Sig-ma), which binds to heparin, an MC-specific glycosaminoglycan [Lindahl and Kjellen, 1991]. Sections were examined using fluo-rescence microscopy (Olympus) fitted with a QImaging CCD camera linked to the Openlab program (Improvision, UK), or by laser scanning confocal microscopy LSM 510 (Carl Zeiss, Ger-many) with associated LSM 5 image browser (Carl Zeiss).
Statistical Analysis Each experimental group consisted of at least 7 animals. All
values were expressed as the mean 8 SEM. One-way ANOVA was used to test for overall significance followed by the Tukey test as a Y post-hoc test for significance (p ! 0.05).
Results
Alteration of Number and Distribution of Brain MCs after Hypoxia and HI The distribution and numbers of MCs were quantified
in TB-stained sections. As previously described, intra-cranial MCs were found in the pia associated with the hippocampus, choroid plexus ( fig. 2 ) and the choroid fis-sure (data not shown). Within the CNS proper, small numbers of MCs were present in the thalamus, hippo-campus, fornix, medial habenula and cerebral cortex of P9 control animals. The number of MCs increased sig-nificantly in the brain in response to hypoxia and HI at 48 h of reperfusion, with the largest accumulation occur-ring in HI brains in the hemisphere ipsilateral to the liga-tion (p ! 0.01; fig. 3 ). In addition, the MC populations in the contralateral hemisphere of HI, and both hemispheres of H brains, were significantly greater than in controls(p ! 0.05). There was no difference in this parameter be-

Jin /Silverman /Vannucci
Dev Neurosci 2007;29:373–384376
Fig. 1. Assessment of brain damage using Fluoro-Jade B to detect dying neurons following HI. Shown are representative micrographs taken at the level of the hippocampus and thalamus. These micrographs illus-trate the psychophysical scale (0–4) used for quantifi-cation of neuronal death (damage score). The assigned number (lower right corner) represents degree of brain damage (i.e. percent of brain region damaged). The damage score was recorded for each section through each brain region studied. Fig. 2. Distribution of mast cells in the ipsilateral hemi-sphere following HI. Mast cells at 48 h after HI were found in the pia (arrow), particularly associated with the hippocampus. Mast cells were also present within the thalamus and hippocampus, particularly along penetrating blood vessels (arrowhead). Areas of dam-age are marked with an asterisk.
1
2

CNS Mast Cells and PerinatalHypoxia-Ischemia
Dev Neurosci 2007;29:373–384 377
tween hypoxia alone and the contralateral hemispheres of HI pups. Post-hoc analysis showed that significant in-creases in MCs occurred in the hippocampus, thalamus, cingulate cortex and fornix in the H and HI brains (p ! 0.05; fig. 3 ). In HI brains, MCs were also found occasion-ally in the hypothalamus, dorsal to the third ventricle (data not shown).
In control brains, MCs were filled with TB-positive, metachromatic granules ( fig. 4 a). In comparison, signs of MC degranulation (activation) were observed in HI and H brains ( fig. 4 b–d). Two patterns of TB staining signaled degranulation. (1) MCs in H brains were characterized by regions of cytoplasm that contained swollen, hydrated granules. Such areas appeared pink/purple comparedto the intense blue-purple of intact granules ( fig. 4 b).(2) MCs in HI brains exhibited ‘explosive’ exocytosis ( fig. 4 c) in which a portion of the proteoglycan core of the secretory granule remained intact (called granule rem-nant), reacted with the dye, and was found outside of the MCs ( fig. 4 c, d).
Regions of Tissue Damage and Brain MC Localization The dye Fluoro-Jade B was used to detect dying neurons
( fig. 5 a–c). Forty-eight hours following HI, numerous dy-ing neurons were detected in the cortex, hippocampus and thalamus in the hemisphere ipsilateral to the ligation. Dy-ing neurons were minimal or absent in the contralateral hemisphere, H alone, and control animals ( fig. 5 a–c).
The astrocytic marker, GFAP ( fig. 5 d–f), and the mi-croglial/monocyte marker, OX42 (recognizes CD11b) ( fig. 5 g–i), were used to map regions of glial activation rel-ative to regions of neuronal death. In the ipsilateral hemi-sphere both activated astroglia and microglia filled the
site of damage, as delineated by Fluoro-Jade B-positive neurons. OX42- and GFAP-positive cells were present in the pia, thalamus, hippocampus, cortex and striatum. MC number was greatest in these damaged areas (see below). In both the contralateral hemispheres and in H brains, cells immunoreactive for either marker were few in num-ber and were found predominately in the pia. OX42-posi-tive cells were absent in the regions surveyed in control brains ( fig. 5 g). MCs were not absent from either control or H brains, however their phenotype as determined by TB was different from that seen in regions of HI damage. At higher magnification, microglia ( fig. 6 a) and astroglia ( fig. 6 b) made close contact with MCs within both the pia and the brain tissue of the ipsilateral hemisphere.
MCs and Brain Blood Vessels We next asked if MCs were in position to alter the per-
meability or other characteristics of both barrier and nonbarrier blood vessels. Using triple labeling for RECA-1 (endothelial marker), GLUT1 (glucose transporter 1, marker of BBB vessels) and avidin, MCs were found to be associated with both barrier (RECA-1+/GLUT1+) and nonbarrier (RECA-1+/GLUT1–) vessels ( fig. 7 a, b, re-spectively). The association of MCs with both barrier and nonbarrier vessels was also observed in H and control animals (data not shown).
MC Stabilization and HI Brain Damage Given the anatomical localization and degranulation
of MCs described above, and the fact that MC stabiliza-tion has a positive effect following other ischemic situa-tions [e.g. Strbian et al., 2006], we tested whether the ad-ministration of MC stabilizer CR would alter the outcome
0
500
1,000
1,500
2,000
2,500
Total number Hippocampus Thalamus Cortex Fornix
Bra
inM
Cs
Control (n = 9)H (n = 7)HI contralateral hemisphere (n = 22)HI ipsilateral hemisphere (n = 22)
* *
**
* ***
* *
**
* *
**
* ***
Fig. 3. Changes in brain mast cell number 48 h after H or HI. Mast cell numbers increased significantly in the brain following HI or H compared to controls in all brain regions studied; the largest numbers were found in the ipsilateral HI hemisphere. The bar graph repre-sents mean numbers of brain mast cells (per brain re-gion) 8 SEM ( * * p ! 0.01, * p ! 0.05).
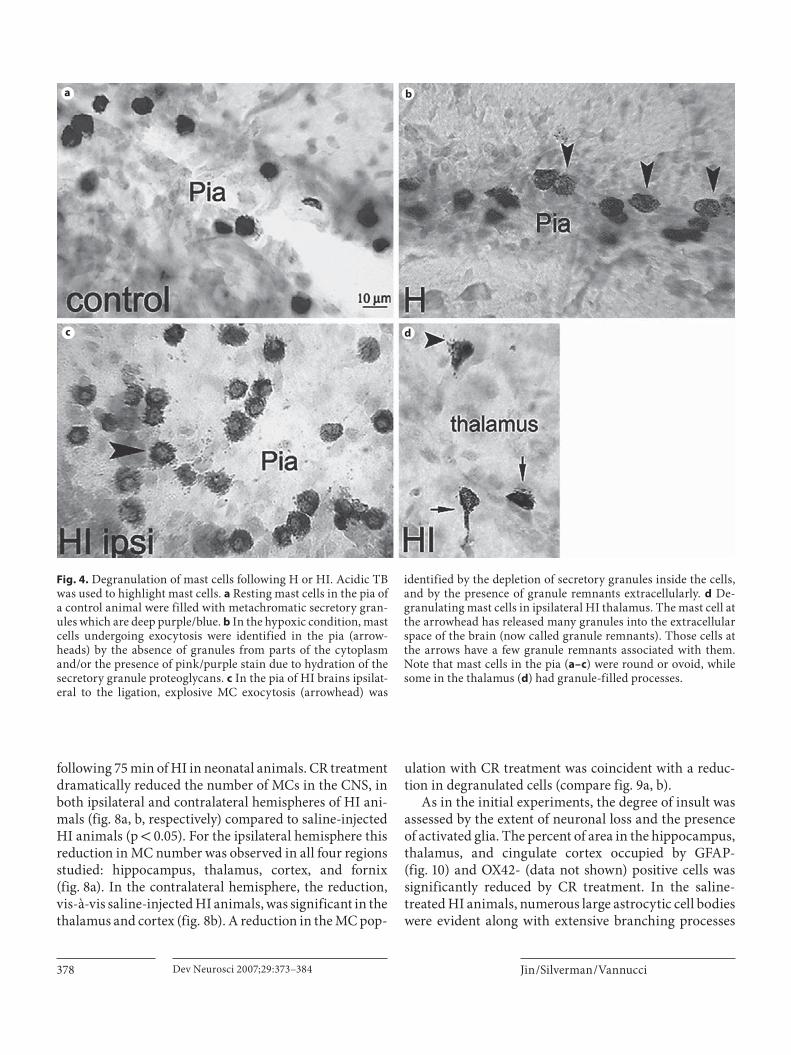
Jin /Silverman /Vannucci
Dev Neurosci 2007;29:373–384378
following 75 min of HI in neonatal animals. CR treatment dramatically reduced the number of MCs in the CNS, in both ipsilateral and contralateral hemispheres of HI ani-mals ( fig. 8 a, b, respectively) compared to saline-injected HI animals (p ! 0.05). For the ipsilateral hemisphere this reduction in MC number was observed in all four regions studied: hippocampus, thalamus, cortex, and fornix ( fig. 8 a). In the contralateral hemisphere, the reduction, vis-à-vis saline-injected HI animals, was significant in the thalamus and cortex ( fig. 8 b). A reduction in the MC pop-
ulation with CR treatment was coincident with a reduc-tion in degranulated cells (compare fig. 9 a, b).
As in the initial experiments, the degree of insult was assessed by the extent of neuronal loss and the presence of activated glia. The percent of area in the hippocampus, thalamus, and cingulate cortex occupied by GFAP- ( fig. 10 ) and OX42- (data not shown) positive cells was significantly reduced by CR treatment. In the saline-treated HI animals, numerous large astrocytic cell bodies were evident along with extensive branching processes
a b
c d
Fig. 4. Degranulation of mast cells following H or HI. Acidic TB was used to highlight mast cells. a Resting mast cells in the pia of a control animal were filled with metachromatic secretory gran-ules which are deep purple/blue. b In the hypoxic condition, mast cells undergoing exocytosis were identified in the pia (arrow-heads) by the absence of granules from parts of the cytoplasm and/or the presence of pink/purple stain due to hydration of the secretory granule proteoglycans. c In the pia of HI brains ipsilat-eral to the ligation, explosive MC exocytosis (arrowhead) was
identified by the depletion of secretory granules inside the cells, and by the presence of granule remnants extracellularly. d De-granulating mast cells in ipsilateral HI thalamus. The mast cell at the arrowhead has released many granules into the extracellular space of the brain (now called granule remnants). Those cells at the arrows have a few granule remnants associated with them. Note that mast cells in the pia ( a–c ) were round or ovoid, while some in the thalamus ( d ) had granule-filled processes.

CNS Mast Cells and PerinatalHypoxia-Ischemia
Dev Neurosci 2007;29:373–384 379
Fig. 5. Neuronal death and glial activation following HI. These micrographs illustrate sections cut through the thalamus and hip-pocampus ( a–c ) or thalamus only ( d–i ). a–c Numerous dying neurons (Fluoro-Jade B+, green) were found in HI hippocampus and thalamus but were minimal/absent in H and control animals. Large numbers of activated astrocytes (GFAP+, red; d–f ) and ac-
tivated microglia (OX42+, red; g–i ) were observed in the ipsilat-eral hemisphere of HI brains, but few in H or control animals. Mast cells (avidin+, green) were present along blood vessels (BV). Fields containing mast cells overlapped those of activated astro-cytes and microglia in HI brains. The scale bar in a is for a–c , while that in g is for d–i .

Jin /Silverman /Vannucci
Dev Neurosci 2007;29:373–384380
( fig. 10 a). This specific population was much reduced or absent after CR treatment ( fig. 10 b). With CR treatment, GFAP staining was confined to the processes and endfeet surrounding blood vessels. Most importantly, there was also a dramatic reduction in Fluoro-Jade B staining in the ipsilateral hemisphere, indicating that neuronal cell death was also prevented or reduced during this acute period ( fig. 11 a). The protection afforded by CR was found across all brain regions studied (p ! 0.01).
To be clinically relevant, CR administration after HI must be neuroprotective as well. To test this, we admin-
istered CR (50 mg/kg, SC) immediately following 75 min of HI. Fluoro-Jade B staining of the CNS 48 h later dem-onstrated that damage was significantly reduced by more than 50% in the ipsilateral hemisphere of brains from an-imals receiving the drug. The protection was also found across all brain regions studied (p ! 0.05; fig. 11 b).
Discussion
Recently, studies of ischemic brain damage have fo-cused less on the neuronal response alone and more on the roles of all cell types that comprise the neurovascular unit, i.e. neurons, glia, and endothelial cells. This study is the first demonstration that an additional cell type, the brain MC, also plays a role in cerebral ischemia in the im-mature brain. Following unilateral cerebral HI in the P7 rat, we report rapid increases in the intracerebral MC population and increases in MC activation (degranula-tion). The origin of the ‘new’ MC is not known but they could be derived from those in the pia [Kitamura et al., 1979] or by the entry of circulating precursors in the blood [Rodewald et al., 1996]. Activated MCs were ob-served in both the pia, where the population is extensive, and in the brain parenchyma. Activated MCs were con-sistently associated with regions of neuronal loss and re-active gliosis. To test our hypothesis that MCs contribute to the damage following HI, neonatal rats were treated with the MC stabilizer CR. In those experiments which utilized both pre- as well as post-HI injection protocols, MC migration into brain was reduced in CR-treated ani-mals and there were fewer degranulated cells in both in-tracerebral populations. In support of our hypothesis, brain damage, as measured by neuronal loss and gliotic
a b
Fig. 6. Identification of brain mast cells and activated microglia and astrocytes in the HI thalamus. a At higher magnifica-tion, mast cells (avidin+, green) appeared to contact (arrowheads) or be in close proximity to activated microglia/mono-cytes (OX42+, red). b In the dorsal thala-mus near the pia, processes (arrow) of ac-tivated astrocytes (GFAP+, red) wrapped around thalamic mast cells.
a
b
Fig. 7. Localization of mast cells and thalamic microvessels. Triple staining for RECA-1 (endothelial marker, blue), GLUT1 (marker of BBB vessels, red) and avidin (mast cells, green) was used to determine the maturity of blood vessels with mast cells. In theHI thalamus, mast cells were found along side of barrier ves-sels (RECA-1+/GLUT1+; a ) and nonbarrier vessels (RECA-1+/GLUT1–; b ).

CNS Mast Cells and PerinatalHypoxia-Ischemia
Dev Neurosci 2007;29:373–384 381
rak, 1992]. The results presented here support the hy-pothesis that in response to HI, MC migration and de-granulation are causative to the initiation of the proin-flammatory cascade that results in cell death. Following hypoxia alone, which does not result in gliosis or neuro-nal death, the brain MC population increases and MCs do degranulate, although not to the same ‘explosive’ state as seen in HI. Future studies are required to address the differential effects of hypoxia alone.
Inflammatory responses occur quickly after neonatal HI [Silverstein et al., 1997; Bona et al., 1999]. MCs are known to play a central role in inflammation [reviewed in Galli et al., 2005], and the increased MC numbers in the immature brain put them in a key position to func-tion here as well. MCs store and produce upon demand a broad array of proinflammatory cytokines (e.g. TNF- � , IL1–6), vasoactive mediators (histamine, serotonin and leukotrienes) and unique serine proteases through which they can alter the brain’s microenvironment and have the potential to recruit other elements of the immune system [Silver et al., 1996]. Molecular evidence of MC activation was found in neonatal mouse brain, where the expression of MC-associated genes (i.e. glycoprotein 49A, 49B, lym-
responses, was limited. Most importantly, CR treatment following the HI insult was equally effective in dramati-cally limiting neuronal loss, making this drug more at-tractive as a potential therapeutic agent.
Interestingly, hypoxia alone elevated MC number in the brain although to a lesser extent than HI, and was not accompanied by brain damage. We hypothesize that for each stimulus condition (H vs. HI) the state of activation/degranulation and, potentially, the nature of mast cells’ secretions are determined by the precise stimulus. MCs are known to be very plastic and heterogeneous in pro-duction of mediators. In addition, MC secretion can be selective rather than an all or none phenomenon [Dvo-
0
500
1,000
1,500
2,000
Total
number
Hippo-
campus
Thalamus Cortex FornixBra
inM
Cs
inip
sila
tera
l he
mis
ph
ere HI with saline (n = 10)
HI with CR 50 mg/kg (n = 11)
**
** **
*
*
a
Bra
inM
Cs
inco
ntr
ala
tera
l he
mis
ph
ere
**
* *
HI with saline (n = 10)HI with CR 50 mg/kg (n = 11)
0
500
1,000
1,500
2,000
Total
number
Hippo-
campus
Thalamus Cortex Fornix
b
a
b
Fig. 9. CR treatment reduced mast cell degranulation in HI brains. Acidic TB was used to determine the effectiveness of CR treat-ment on mast cell degranulation. a Brains of saline-treated ani-mals had MCs with obvious signs of degranulation including ex-truded granules (arrows). b MCs were fully granulated in HI an-imals given CR. There is little evidence of extracellular granule remnants in drug-treated animals.
Fig. 8. CR treatment reduced MC numbers following HI. The bar graph illustrates the size of the brain mast cell population in the ipsilateral ( a ) and contralateral ( b ) hemispheres of HI animals treated with saline or CR at 48 h after HI. There is a significant reduction in MC number with drug treatment on both the ipsi-lateral and contralateral sides when compared to equivalent hemi-spheres of the saline-treated HI pups. This decrease was found in the ipsilateral side in the thalamus, hippocampus, cortex and for-nix; contralaterally, the reduction was significant in the thalamus and cortex. The bar represents mean brain MC number 8 SEM ( * * p ! 0.01, * p ! 0.05).

Jin /Silverman /Vannucci
Dev Neurosci 2007;29:373–384382
phocyte cytosolic protein 2 and paired-Ig-like receptor A3) was upregulated in the ipsilateral hemisphere follow-ing HI [Hedtjarn et al., 2004].
Histamine has long been associated with increased vascular permeability and edema. It has been estimated that in the adult rat brain 80% of the thalamic histamine is attributable to MCs [Goldschmidt et al., 1985]. In-creases in MCs and/or histamine have been identified in other brain injury models. For example, histamine levels, postulated to be derived from MCs, increase in
the basal ganglia after focal brain ischemia [Subrama-nian et al., 1981]. Following fluid percussion injury to the cortex, MCs infiltrate the damaged region [Lozada et al., 2005; Strbian et al., 2006]. The enzymatic activi-ties of MC-specific serine proteases such as tryptase and chymase can also enhance vascular permeability via en-dothelial basal lamina degradation during the early phase of ischemia [Saarinen et al., 1994; Imamura et al., 1996]. Destruction or diminution of the basal lamina creates a permissive environment for plasma constitu-
0
1
2
3
4
Whole brain Hippocampus Thalamus Cortex
Bra
ind
am
ag
esc
ore
** ** ****
HI with saline (n = 10)HI with CR 50 mg/kg (n = 11)
a
a b
Fig. 10. Reduction of glial activation in HI animals after CR treatment. a Astrocytes (GFAP+) in the ipsilateral thalamus of saline-treated animals show the activated phenotype of large cell bodies (arrow) and thick pro-cesses that ramify throughout the region. b Astrocytes with an activated morphology were few or absent in the ipsilateral thalamus of CR-treated, HI animals. Glial processes pictured here are the normally occurring glial endfeet confined to and investing the walls of large blood vessels (BV).
0
1
2
3
4
Whole brain Hippocampus Thalamus Cortex
***
**
HI with saline (n = 4)CR (50 mg/kg) after HI (n = 5)
b
Bra
ind
am
ag
esc
ore
Fig. 11. CR treatment limited brain damage following HI. There was a significant reduction in the brain dam-age score (measured with Fluoro-Jade B) in the ipsilateral hemisphere with CR given prior to ( a ) or after ( b ) HI compared to ipsilateral hemisphere of HI pups treated with saline only. The bar graph represents mean brain damage score 8 SEM ( * p ! 0.05, * * p ! 0.01).

CNS Mast Cells and PerinatalHypoxia-Ischemia
Dev Neurosci 2007;29:373–384 383
ents as well as inflammatory cells and blood-borne in-flammatory mediators or cytokines to enter the CNS. Edema of the ipsilateral hemisphere is a standard feature of this animal model as a hallmark of ultimate damage. Although we did not specifically measure histamine re-lease in this study, an MC-mediated contribution to the development of edema would be quite consistent with these observations.
TNF- � is a powerful proinflammatory cytokine that initiates the postischemic inflammatory cascade and increases brain damage in adult stroke models [Barone et al., 1997]. MCs are the only cells that contain pre-formed stores of TNF- � in CNS. MCs, therefore, are in a position to release TNF- � immediately upon stimula-tion [Gordon and Galli, 1991] as has been shown for pu-rified brain MCs stimulated by substance P [Cocchiara et al., 1999]. Szaflarski et al. [1995] reported a transient stimulation of TNF- � mRNA expression in ischemic cortex and hippocampus shortly after initiation of HI in neonatal rats.
Our central hypothesis is that MCs play a causal role in neonatal brain damage following HI. This hypothesis is strongly supported by the outcome of the experiments using CR, an inhibitor of MC degranulation. CR treat-ment alleviated all signs of damage following HI com-
pared to saline-treated pups. CR effectively reduced MC migration into brain and inhibited their degranulation. This would suggest that MCs release mediators that ei-ther alter the extracellular environment and make it con-ducive for cellular movement and/or alter the properties of the MC plasma membrane so it can interact with ele-ments of the extracellular matrix.
Conclusion
The results of these studies point to a new cellular mechanism associated with perinatal brain damage fol-lowing HI. We hypothesize that MCs contribute directly to the inflammatory response to neonatal HI and may be the initiators of this response. If MCs do play a central role as additional members of the neurovascular unit, they would provide a novel target for clinical intervention in perinatal HI brain injury.
Acknowledgements
This project was supported by NIH RO1 MH 54088 (A.J.S.) and HD 30704 (S.J.V.).
References
Asarian L, Yousefzadeh E, Silverman AJ, Silver R (2002): Stimuli from conspecifics influ-ence brain mast cell population in male rats. Horm Behav 42: 1–12.
Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ (1997): Tumor necrosis factor-alpha. A me-diator of focal ischemic brain injury. Stroke 28: 1233–1244.
Bona E, Andersson AL, Blomgren K, Gilland E, Puka-Sundvall M, Gustafson K, Hagberg H (1999): Chemokine and inflammatory cell response to hypoxia-ischemia in immature rats. Pediatr Res 45: 500–509.
Cocchiara R, Albeggiani G, Lampiasi N, Boni-ovanni A, Azzolina A, Geraci D (1999): His-tamine and tumor necrosis factor alpha pro-duction from purified rat brain mast cells mediated by substance P. Neuroreport 10:
575–583. Coggeshall RE, Lekan HA (1996): Methods for
determining numbers of cells and synapses: a case for more uniform standards of review. J Comp Neurol 364: 6–15.
Dimitriadou V, Lambracht-Hall M, Reichler J, Theoharides TC (1990): Histochemical and ultrastructural characteristics of rat brain perivascular mast cells stimulated with com-pound 48/80 and carbachol. Neuroscience 9:
209–224. Dirnagl U, Iadecola C, Moskowitz MA (1999):
Pathobiology of ischaemic stroke: an inte-grated view. TINS 9: 391–397.
Dvorak AM (1992): Basophils and mast cells: piecemeal degranulation in situ and ex vivo: a possible mechanism for cytokine-induced function in disease. Immunol Ser 57: 169–271.
Engels W, Reiters PH, Daemen MJ, Smits JF, van der Vusse GJ (1995): Transmural changes in mast cell density in rat heart after infarct in-duction in vivo. J Pathol 177: 423–429.
Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH,Spengler RN, Smith CW, Entman ML (1998): Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circula-tion 98: 699–710.
Galli SJ, Kalesnikoff J, Grimbaldeston MA, Pili-ponsky AM, Williams CM, Tsai M (2005): Mast cells as ‘tunable’ effector and immuno-regulatory cells: recent advances. Annu Rev Immunol 23: 749–786.
Goldschmidt RC, Hough LB, Glick SD (1985): Rat brain mast cells: contribution to brain histamine levels. J Neurochem 44: 1943–1947.
Gordon JR, Galli SJ (1991): Release of both pre-formed and newly synthesized tumor necro-sis factor alpha (TNF-alpha)/cachectin by mouse mast cells stimulated via the Fc epsi-lon RI. A mechanism for the sustained action of mast cell-derived TNF-alpha during IgE-dependent biological responses. J Exp Med 174: 103–107.
Hedtjarn M, Mallard C, Hagberg H (2004): In-flammatory gene profiling in the developing mouse brain after hypoxia-ischemia. J Cereb Blood Flow Metab 24: 1333–1351.
Hu W, Shen Y, Fu Q, Dai H, Tu H, Wei E, Luo J, Chen Z (2005): Effect of oxygen-glucose de-privation on degranulation and histamine release of mast cells. Cell Tissue Res 322:
437–441.

Jin /Silverman /Vannucci
Dev Neurosci 2007;29:373–384384
Hu W, Xu L, Pan J, Zheng X, Chen Z (2004): Ef-fect of cerebral ischemia on brain mast cells in rats. Brain Res 1019: 275–280.
Imamura T, Dubin A, Moore W, Tanaka R, Tra-vis J (1996): Induction of vascular perme-ability enhancement by human tryptase: de-pendence on activation of prekallikrein and direct release of bradykinin from kinino-gens. Lab Invest 74: 861–870.
Kitamura Y, Hatanaka K, Murakami M, Shibata H (1979): Presence of mast cell precursors in peripheral blood of mice demonstrated by parabiosis. Blood 53: 1085–1088.
Lambracht-Hall M, Dimitriadou V, Theoharides TC (1990): Migration of mast cells in the de-veloping rat brain. Brain Res Dev Brain Res 56: 151–159.
Lindahl U, Kjellen L (1991): Heparin or heparan sulfate – what is the difference? Thromb Haemost 66: 44–48.
Lozada A, Maegele M, Stark H, Neugebauer EM, Panula P (2005): Traumatic brain injury re-sults in mast cell increase and changes in reg-ulation of central histamine receptors. Neu-ropathol Appl Neurobiol 31: 150–162.
McQuillen PS, Sheldon RA, Shatz CJ, Ferriero DM (2003): Selective vulnerability of sub-plate neurons after early neonatal hypoxia-ischemia. J Neurosci 23: 3308–3315.
Michaloudi H, Grivas I, Batzios C, Chiotelli M, Papadopoulos GC (2003): Parallel develop-ment of blood vessels and mast cells in the lateral geniculate nuclei. Brain Res Develop Brain Res 140: 269–274.
Paxinos G, Watson C (1986): The Rat Brain in Stereotaxic Coordinates, ed 2. Orlando, Aca-demic Press.
Rice JE 3rd, Vannucci RC, Brierley JB (1981): The influence of immaturity on hypoxic-isch-emic brain damage in the rat. Ann Neurol 9:
131–141. Rodewald HR, Dessing M, Dvorak AM, Galli SJ
(1996): Identification of a committed pre-cursor for the mast cell lineage. Science 271:
818–822. Saarinen J, Kalkkinen N, Welgus HG, Kovanen
PT (1994): Activation of human interstitial procollagenase through direct cleavage of the Leu83-Thr84 bond by mast cell chymase. J Biol Chem 269: 18134–18140.
Schmued LC, Hopkins K (2000): Fluoro-Jade B, a high affinity fluorescent marker for the lo-calization of neuronal degeneration. Brain Res 874: 123–130.
Silver R, Silverman AJ, Vitkovic L, Lederhendler II (1996): Mast cells in the brain: evidence and functional significance. TINS 19: 25–31.
Silverstein FS, Barks JD, Hagan P, Liu XH, Ivacko J, Szaflarski J (1997): Cytokines and perina-tal brain injury. Neurochem Int 30: 375–383.
Singh M, Saini HK (2003): Resident cardiac mast cells and ischemia-reperfusion injury. J Car-diovasc Pharmacol Ther 8: 135–148.
Strbian D, Karjalainen-Lindsberg ML, Tatlisu-mak T, Lindsberg PJ (2006): Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. J Cereb Blood Flow Metab 26: 605–612.
Subramanian N, Theodore D, Abraham J (1981): Experimental cerebral infarction in pri-mates: regional changes in brain histamine content. J Neural Transm 50: 225–232.
Szaflarski J, Burtrum D, Silverstein FS (1995): Cerebral hypoxia-ischemia stimulates cyto-kine gene expression in perinatal rats. Stroke 26: 1093–1100.
Vannucci RC (2000): Hypoxic-ischemic enceph-alopathy. Am J Perinatol 17: 113–120.
Vannucci SJ, Hagberg H (2004): Hypoxia-isch-emia in the immature brain. J Exp Biol 207:
3149–3154. Vannucci SJ, Seaman LB, Vannucci RC (1996):
Effects of hypoxia-ischemia on GLUT1 and GLUT3 glucose transporters in immature rat brain. J Cereb Blood Flow Metab 16: 77–81.
Volpe JJ (1992): Brain injury in the premature infant – current concepts of pathogenesis and prevention. Biol Neonate 62: 231–242.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:385–392 DOI: 10.1159/000105479
Gender-Dependent Pathways ofHypoxia-Ischemia-Induced Cell Death and Neuroprotection in the Immature P3 Rat
Cora H.A. Nijboer
a, b Annemieke Kavelaars
b Frank van Bel
a Cobi J. Heijnen
b
Floris Groenendaal
a
a Department of Neonatology and b
Laboratory of Psychoneuroimmunology, Wilhelmina Children’s Hospital, University Medical Center Utrecht, Utrecht , The Netherlands
Introduction
Perinatal hypoxia-ischemia (HI) can lead to serious and permanent brain injury in the human neonate [1] . The pattern of injury appears to be dependent on the ges-tational age at the time of the insult: human neonates born prematurely suffer mainly from injuries to the peri-ventricular white matter, whereas in full-term neonates more often the gray matter of the cerebral cortex or basal ganglia is affected [2–4] . These differences could also be demonstrated in animal models of brain HI [5] . Further-more, gender also seems to play a key role; female preterm neonates show a better survival and less handicaps com-pared to males, which suggests gender-dependent differ-ences in brain injury in the human neonate [6] . Gender differences in pathways leading to cerebral injury after HI have been examined recently in the neonatal animal. We and others have demonstrated that apoptotic mecha-nisms involved in neuronal injury differ between male and female neonatal rodents and between neuronal cells from female and male animals in culture [7–10] . Caspase-independent apoptosis, associated with mitochondrial to nuclear translocation of apoptosis-inducing factor (AIF) is mainly observed in males. Caspase-dependent apopto-sis, initiated by translocation of cytochrome c from the mitochondria to the cytosol with consequent caspase 3
Key Words
Apoptosis � Cytochrome c � Heat shock protein 70 � Hypoxia-ischemia � Immature brain � Neonate � Braininjury � Neuroprotection � Rat brain
Abstract
Previously, we demonstrated neuroprotection with 2-imino-biotin (2-IB) after cerebral hypoxia-ischemia (HI) in female, but not in male P7 rats. Given the different patterns of brain injury in more immature rats, we examined whether these gender differences could also be observed in P3 rats. HI was induced by unilateral carotid ligation and FiO 2 reduction, fol-lowed by 2-IB administration. HSP70 protein expression and cytochrome c release from the mitochondria, markers of short-term outcome, were induced by HI to the same extent in male and female animals. However, reduction in HSP70 production and cytochrome c release by 2-IB was seen in fe-male rats only. Long-term cerebral injury after HI, assessed with histology, was similar in male and female P3 rats, but long-term neuroprotection by 2-IB was observed in female rats only. In conclusion, 2-IB provides neuroprotection after cerebral HI in female, but not in male immature P3 rats.
Copyright © 2007 S. Karger AG, Basel
Received: September 22, 2006 Accepted after revision: December 22, 2006
Dr. Floris Groenendaal Department of Neonatology, Wilhelmina Children’s Hospital University Medical Center Utrecht, Room KE.04.123.1, PO Box 85090 NL–3508 AB Utrecht (The Netherlands) Tel. +31 30 250 4545, Fax +31 30 250 5320, E-Mail [email protected]
© 2007 S. Karger AG, Basel0378–5866/07/0295–0385$23.50/0
Accessible online at:www.karger.com/dne

Nijboer /Kavelaars /van Bel /Heijnen /Groenendaal
Dev Neurosci 2007;29:385–392386
activation was seen in both male and female animals in our in vivo study in P7 rats, although neurons from fe-male animals have been described to use this pathway with higher proclivity in vitro and in vivo [7–9] .
Previously, we have demonstrated neuroprotection af-ter HI in the P12 rat, and in neonatal piglets after treat-ment with 2-iminobiotin (2-IB) [11, 12] . 2-IB was de-scribed to be an inhibitor of neuronal and inducible nitric oxide (NO) synthase (nNOS and iNOS), enzymes cata-lyzing the production of NO. However, in vitro and in vivo data from our recent study strongly indicate that at the dose used in vivo, the protective effect of 2-IB is not mediated via NOS inhibition [10] .
Moreover, gender differences in the effects of 2-IB af-ter HI have been reported by us: neuroprotection was ob-tained only in female but not in male P7 rats [10] . Gender differences have also been demonstrated for other neuro-protective strategies such as brain cooling [13] .
It has been reported recently that pathways leading to cell death after HI are dependent on the developmental stage of the brain in the mouse: AIF translocation, cyto-chrome c release, and caspase 3 activation were far more pronounced in immature as opposed to juvenile or adult brains [14] . It is unknown if the previously observed gen-der differences in the neuroprotective properties of 2-IB at P7 exist also at earlier developmental stages. In the present study, we therefore examined whether gender dif-ferences in neuroprotection by 2-IB were present in the P3 rat pup, which can be regarded as a model of the very preterm born human neonate [15] . The so-called Van-nucci-Rice model of HI and reperfusion is an established newborn rodent model to mimic human birth asphyxia which has been used widely during the last decade. Long-term neuroprotection can be assessed using this model [16] . Knowledge of potential gender differences is rele-vant for planning of neuroprotective strategies and trials in human full-term as well as preterm neonates with peri-natal HI.
Materials and Methods
Animals Experiments were performed in accordance with internation-
al guidelines and approved by the University Medical Center Utrecht experimental animal committee. Timed-pregnant Wi-star rats (Charles River, Sulzfeld, Germany) delivered their pups at the Central Laboratory Animal Institute (Utrecht, the Nether-lands); the day of birth was considered day 0. Animals were kept on a 12: 12 h light:dark cycle and were weaned at 3 weeks of age.
To induce HI, we used a modification of the original Van-nucci and Rice model [17] . At P3, pups were anesthetized (5–10
min) with isoflurane (5.0% induction, 1.5% maintenance) in O 2 :N 2 O (1: 1). Blood flow in the right common carotid artery was permanently interrupted by thermocauterization, xylocaine spray (100 mg/ml; AstraZeneca, Zoetermeer, the Netherlands) was applied and wounds were closed. Animals were kept warm on a heated water mattress (37 ° C) during surgery. After at least 1 and maximal 3 h of recovery, pups were exposed to 8% O 2 in N 2 for 60 min. The gas mixture was humidified and preheated. During hypoxia, the animals were kept on a heated water mat-tress in a child incubator with a temperature of 37 ° C. After hy-poxia, the animals were returned to their dams and were kept at room temperature. Sham-treated controls underwent anesthesia and incision without further surgical procedures and without hypoxia.
2-IB (Sigma-Aldrich, St. Louis, Mo., USA) was dissolved at 1 mg/ml in 0.01 M HCl and pH was adjusted to 4.2 with 0.1 M NaOH. Pups received 2-IB (10 mg/kg) or vehicle (10 ml/kg) s.c. at 0, 12 and 24 h after hypoxia. The dose and treatment schedule are identical to the dose used in our earlier studies using P7 and P12 rat pups [10, 12] .
Histology Six weeks after HI, rats received an overdose pentobarbital
(300 mg/kg) and were perfused with 4% paraformaldehydein PBS. Brains were paraffin-embedded and coronal sections(8 � m) were cut at approximately –3.30 mm from the bregma and stained with hematoxylin-eosin. Sections were scored in a blind-ed way in 6 regions of the parietal cortex and in 5 regions of the hippocampus [10,12] . Scores ranged from 0 to 3: 3 = normal, ! 10% of neurons damaged; 2 = 10–50% of neurons damaged;1 = 50–90% of neurons damaged; 0 = almost all neurons dam-aged (90–100%), gliosis and/or cystic infarction; the maximal total score being 33. All measurements were performed by one investigator (C.N.).
Deparaffinized sections were incubated with mouse anti-my-elin basic protein (MBP) antibody (Sternberger Monoclonals In-corporated, Lutherville, Md., USA), followed by incubation with biotinylated horse anti-mouse antibody (Vector Laboratories, Burlingame, Calif., USA). Visualization was performed using Vectastain ABC kit (Vector Laboratories) and diaminobenzami-dine. MBP staining was quantified using image processing tools in ImageJ software (Rasband WS, ImageJ, U.S. National Institutes of Health, Bethesda, Md.; http://rsb.info.nih.gov/ij/, 1997–2006).
Western Blot Analysis Pups were decapitated at 24 or 48 h after HI, cerebellum was
removed and left and right hemispheres were frozen in liquid ni-trogen. Hemispheres were pulverized using a liquid nitrogen-cooled mortar and pestle, divided in fractions and stored at –80 ° C.
Pulverized hemisphere fractions were processed by homoge-nization in buffer containing 70 m M sucrose, 210 m M mannitol, 5 m M HEPES, 1 m M EDTA and protease inhibitors using a Potter homogenizer (Heidolph, Schwabach, Germany). Homogenates were incubated on ice for 30 min, followed by 10 min centrifuga-tion at 800 g at 4 ° C, leading to a nuclear pellet (P1). Supernatants (S1) were collected and centrifuged at 10.000 g for 10 min at 4 ° C to obtain mitochondrion-free supernatant (S2) and a mitochon-drial pellet (P2). Protein concentration was determined using a protein assay (BioRad, Hercules, Calif., USA) with BSA as stan-

Neuroprotection and Gender in Asphyxiated P3 Rats
Dev Neurosci 2007;29:385–392 387
dard. Proteins from fraction S2 were separated by SDS-PAGE and transferred to nitrocellulose membranes (Hybond-C, Amersham Biosciences, Roosendaal, the Netherlands). Equal protein loading was verified by Ponceau-S staining. Membranes were stained with mouse anti-HSP70 (Stressgen Biotech, Victoria, Canada), mouse anti-cytochrome c (Pharmingen, San Diego, Calif., USA), mouse anti-nitrotyrosine (Biomol, Plymouth, Pa., USA) or rabbit anti-iNOS (Santa Cruz Biotechnology, Santa Cruz, Calif., USA) fol-lowed by incubation with goat � -mouse-HRP (Jackson Immu-noresearch, UK) or donkey � -rabbit-HRP (Amersham Bioscien-ces). Specific bands were visualized by chemiluminescence detection (ECL, Amersham Biosciences) with X-ray film expo-sure. Films were scanned with a GS-700 Imaging Densitometer and analyzed with Quantity One Software (both BioRad).
Statistical Analysis Power analysis was performed to enable adequate analysis of
gender and treatment effects. Western Blot and MBP data are pre-sented as mean and SEM. Paired t tests were used to analyze dif-ferences between contra- and ipsilateral hemispheres. Two-way ANOVA with Bonferroni posttests was used to analyze treatment
and gender effects. Histological scores are presented as individu-al data with median and were analyzed using Kruskal-Wallis tests. Mann-Whitney U tests were used for further analysis of 2-IB treatment and gender effects.
Results
Short-Term Outcome Heat-Shock Protein 70 (HSP70) and Cytochrome c HSP70, a cell stress marker, is known to be upregu-
lated in the ipsilateral hemisphere after HI and was used as an early indicator of brain damage. Results of the HSP70 analysis are presented in figure 1 a. HSP70 was el-evated to a similar extent after HI in the ipsilateral hemi-spheres of male and female animals at 48 h after HI. Ad-ministration of 2-IB significantly prevented the increase of HSP70 in female (p ! 0.001 vs. vehicle-treated females),
0
a b
c
Sham HI-veh HI-2-IB
Male Female
Male
Veh 2-IB Veh 2-IBFemale
HI-veh HI-2-IB
i c i c i c i c i
c i c i c i c i
1
2
3
4
HSP
70 (A
U)
5
n.s.
n.s.
***6
7
0c
Sham HI-veh HI-2-IB
Male Female
Male
Veh 2-IB Veh 2-IB
Female
HI-veh HI-2-IB
i c i c i c i c i
c i c i c i c i
1
Cyt
osol
ic c
ytoc
hro
me
c (A
U)
2
n.s.
n.s.
**3
4
Fig. 1. The effect of 2-IB treatment on HSP70 expression ( a ) and cytosolic cytochrome c ( b ) level at 48 and 24 h after HI in P3 rats. After HI, mitochondria-free cytosolic preparations (S2) of contra- ( ) ) and ipsilateral ( $ ) hemispheres were processed for Western blot analysis. Data are presented for all groups as mean 8 SEM in AU. Post-HI treatment with 2-IB only reduced HSP70 ( a ) and cytosolic cytochrome c ( b ) significantly in females ( * * * p ! 0.001; * * p ! 0.01 vs. vehicle-treated controls). c = Contralateral; i = ipsilateral; veh = vehicle. Animal numbers: sham-operated (n = 6), males treated with vehicle (n = 5–6) or 2-IB (n = 7–9), females treated with vehicle (n = 6–9) or 2-IB (n = 6–7).

Nijboer /Kavelaars /van Bel /Heijnen /Groenendaal
Dev Neurosci 2007;29:385–392388
but not in male P3 rats. A very low level of HSP70 was detected in contralateral hemispheres of HI rat pups of both genders and in both hemispheres of sham-operated rat pups.
Translocation of cytochrome c from the mitochondria to the cytosol is known to be one of the key events during
(caspase-dependent) apoptotic cell death. HI induced ip-silateral cytochrome c release to the cytosol at 24 h after HI at similar levels in both genders. 2-IB significantly prevented mitochondrial cytochrome c translocation in female rats only (p ! 0.01 vs. vehicle-treated females; fig. 1 b). Cytosolic cytochrome c levels in contralateral
0
a
Sham
Male Female
HI-veh HI-2-IB HI-veh HI-2-IB
Tota
l his
tolo
gic
al s
core
5
20
25
30
35
n.s.
n.s.
**Veh
Male Female
2-IB
b
0.5
c
0.6
0.7
MBP
ipsi
late
ral/
con
tral
ater
al
0.8
0.9
1.0
1.1n.s.
n.s.
**
Sham
Male Female
HI-veh HI-2-IB HI-veh HI-2-IB
Fig. 2. Long-term effect of 2-IB treatment on histological scores and white matter damage in P3 rats. a Brain sections were stained with hematoxylin-eosin. Brain damage was scored at 6 weeks af-ter HI in 6 areas of the parietal cortex and 5 regions of the hippo-campus using a 4-point scale (0–3). The total histological score is presented for all individual P3 animals, including the median for each group (horizontal bars). The maximum total histological score for normal animals is 33. Post-HI treatment with 2-IB had long-term neuroprotective effects on female rat pups only ( * * p ! 0.01 vs. vehicle-treated animals). b Examples of neuronal damage in parietal cortex. c MBP immunohistochemistry was performed on brain sections at 6 weeks after HI. MBP staining in ipsilateral and contralateral hemispheres was measured and the ratio is shown. Post-HI treatment with 2-IB reduced MBP loss in female rats only ( * * p ! 0.01 vs. vehicle-treated animals). Animal num-bers: sham-operated (n = 11), males treated with vehicle (n = 10) or 2-IB (n = 8), females treated with vehicle (n = 8) or 2-IB(n = 7).

Neuroprotection and Gender in Asphyxiated P3 Rats
Dev Neurosci 2007;29:385–392 389
hemispheres of all HI rat pups were comparable to levels in sham-operated littermates.
Long-Term Outcome: Cerebral Damage Eighteen male and 15 female P3 rats were exposed to
HI, and 6 weeks later cerebral damage was analyzed his-tologically. The data in figure 2 a show that HI induced a similar reduction in ipsilateral histological scores in both male and female P3 rats (p ! 0.001 vs. sham-operated animals). Importantly, long-term neuroprotective effects in ipsilateral histological scores after treatment with 2-IB were observed in female rats only (p ! 0.01). The histo-logical score of 2-IB-treated P3 females did not differ from sham-operated animals. The conditions used to in-duce HI in our P3 model resulted in relatively mild brain damage in parietal cortex and hippocampus, which is il-lustrated in figure 2 b.
As the P3 rat is a model for the preterm-born human neonate, we also determined white matter damage by an-alyzing loss of MBP staining at 6 weeks after the insult. White matter damage after HI was similar in both gen-ders; 8.3 8 1.2% in males and 10.3 8 1.9% in females in vehicle-treated animals ( fig. 2 c). 2-IB treatment did not prevent loss of MBP staining in male rats (9.8 8 2.5%), whereas treatment did completely prevent loss of MBP staining in female rats (p ! 0.01).
iNOS and Nitrotyrosine Levels Although we showed before that it is unlikely that in
vivo 2-IB has its protective effects via inhibition of NOS activity [10] , we did analyze expression of iNOS after HI in male and female animals of different postnatal age. Figure 3 a shows that iNOS levels are high in P3 rats and decline with age in P7 and P12 rat pups (p ! 0.001, P3 vs.
0
a b
HI-veh
P3 P7
P3 P7
P12
P3 P7 P12
HI-2-IB HI-veh HI-2-IB HI-veh HI-2-IB
c i
c i c i c i c i c i c i
c i c i c i c i c i
2.5
5.0
7.5
10.0
12.5
15.0
iNO
S (A
U)
17.5
20.0
22.5
25.0
27.5 ***
*
Veh 2-IB Veh 2-IB
c i c i c i c i
45 kDa
36 kDa
32 kDa
28 kDa
Veh 2-IB Veh 2-IB
Veh 2-IBBSA NT-BSA
Fig. 3. The effect of 2-IB treatment on iNOS ( a ) and NT ( b ) ex-pression at 24 h after HI at different developmental ages. a After HI, contra- ( ) ) and ipsilateral ( $ ) hemispheres were processed for Western blot analysis. No effect of HI or 2-IB treatment was observed on iNOS levels in P3, P7 and P12 female rat pups. The levels of iNOS expression were high in P3 pups and declined with age (p ! 0.001, P3 vs. P7; p ! 0.05, P7 vs. P12). No difference in expression between females and males was observed (data not
shown). There were 5 animals/group. Data are presented for all groups as mean 8 SEM in AU. b Neither HI nor post-HI treat-ment with 2-IB affected NT levels in P3 or P7 female animals. A marked difference in NT intensity and pattern is observed be-tween P3 and P7 animals. No difference in expression between females and males was observed (data not shown). Inset shows specificity of the NT antibody. Representative example from 5 animals/group.

Nijboer /Kavelaars /van Bel /Heijnen /Groenendaal
Dev Neurosci 2007;29:385–392390
P7; p ! 0.05, P7 vs. P12). In line with earlier observations in the P12 rat [18] , we did not observe an HI-induced change in iNOS level in the ipsilateral hemisphere in P3 or P7 pups at 24 h after HI, nor was there an effect of 2-IB treatment ( fig. 3 a). No gender differences on the levels of iNOS expression were observed at the different gestation-al ages (data not shown).
In addition, cerebral nitrotyrosine (NT) protein levels were determined at 24 h after HI. We did not observe any effect of HI on nitration of tyrosine residues in brain pro-teins nor any effect of 2-IB treatment or gender as was de-scribed earlier [10] ( fig. 3 b). Interestingly, however, we did observe differences in the intensity and pattern of NT-pos-itive proteins between different gestational ages; NT stain-ing was more intense at P7 compared to P3 and band pat-terns were different (e.g. � 45 kDa and 37 kDa; fig. 3 b).
Discussion
In the present study, we observed gender differences in short-term and long-term neuroprotection after 2-IB treat-ment between the immature male and female P3 rat: fe-male animals could be protected by 2-IB, whereas male animals could not. These differences are strikingly similar to our recent observations in the P7 rat [10] and our stud-ies in the P12 rat [van den Tweel et al., unpubl. data].
In human neonates, patterns of brain injury after HI are very much dependent on the developmental stage at the time of the insult [2–4] . Whereas in preterm human neo-nates the white matter appears to be particularly vulner-able, leading to periventricular leukomalacia, in full-term neonates the basal ganglia, hippocampus and cortex ap-pear to be most affected by HI. Recently, patterns of brain injury in P3 rats demonstrating white matter injury and columnar injury to the cerebral cortex were different from those in P7 rats, demonstrating laminar cortical necrosis and hippocampal injury [15, 19, 20] . Although in our pres-ent model slightly different conditions of HI were used, mild cerebral cortical injury was comparable to that ob-served by Sizonenko et al. [15] who also used P3 rats. Sub-tle white matter injury as determined by loss of MBP stain-ing was also observed in our P3 rats in the internal and external capsule as well as in the cingulum. Interestingly, 2-IB treatment completely prevented the loss of MBP stain-ing in females. This observation suggests that 2-IB has gen-der-dependent protective effects via the same pathway in both neurons as well as oligodendroglial cells.
Not only patterns of brain injury, but also pathways leading to HI-induced cell death may be dependent on the
developmental stage [14] . For example, it has been de-scribed that cerebral HI-induced increases in transloca-tion of AIF and cytochrome c release, as well as caspase 3 activation were far more pronounced in immature ani-mals (P5 and P9 mice) than in juvenile animals (P21 and P60 mice) [14] . Sex differences in AIF translocation (male) and caspase 3 activation (female) were examined by this group in P9 mice only [9] .
When comparing P3 and P7 male and female animals, we found striking similarities in gender effects on neuro-protection by 2-IB after HI. 2-IB reduced cytochrome c release in female, but not in male P3 and P7 rats. Path-ways leading to cell death after HI differ between male and female animals at P7. AIF translocation from the mi-tochondria to the nucleus after HI was observed in males, but not in females [9, 10] . This suggests that the PARP-1-AIF pathway associated with caspase-independent apop-tosis is more activated in males than in females. The ob-servation that AIF plays a more prominent role in males is supported by a recent study showing that PARP-1 dele-tion protects neonatal male, but not female mice from HI-induced cerebral injury [8, 21] . Not only in intact an-imals but also in cell culture were differences in apop-totic pathways between male and female cell lines report-ed recently. Neurons from female animals predominant-ly use the cytochrome c caspase 3-dependent pathway of apoptosis after exposure to a number of cell death-induc-ing agents. In contrast, neurons from male animals show a proclivity for the use of the caspase-independent path-way of apoptosis that is characterized by PARP-1 activa-tion and AIF translocation from mitochondria to the nu-cleus [7] . Other differences between males and females in brain damage after HI have been reported. In animal stroke models, adult females are relatively protected against cerebral ischemia compared to males. Female re-productive hormones are responsible for the protective effects [22–24] . It is unlikely that female sex hormones have played a role in the P3 rat, since endogenous estro-gen production does not start earlier than at P21 [25] . In addition, maternally acquired estrogens are bound to � -fetoprotein in the circulation and are unlikely to enter the P3 brain [26] .
Previously, gender differences in other neuroprotec-tive strategies have been described. Bona et al. [13] men-tioned better effects of post-HI hypothermia treatment in female than in male P7 rats, which emphasizes the need for gender-specific analysis in trials aiming at neuropro-tection.
The precise mechanism of action of 2-IB is currently under our investigation. Previously, it has been suggested

Neuroprotection and Gender in Asphyxiated P3 Rats
Dev Neurosci 2007;29:385–392 391
to be an inhibitor of nNOS and iNOS. Our recent work indicated that NOS inhibition is unlikely to occur in vivo after administration of 2-IB in the dose used in this study [10] . In accordance with those earlier data, we show here that there was no effect of 2-IB treatment on iNOS ex-pression after HI in P3 rats, which express high levels of iNOS. Furthermore, there was no effect of 2-IB treatment on NT levels after HI. Expression of both iNOS and NT did not differ between female or male rat pups. Together, these data further strengthen the notion that the gender-dependent protective effects of 2-IB do not involve NOS inhibition.
As described before, 2-IB can prevent the release of cytochrome c from the mitochondria in females but not males. Moreover, the translocation of AIF from the mito-chondria to the nucleus that occurs selectively in males is not affected by 2-IB [10] . From these two observations, we must conclude that 2-IB works upstream of cyto-chrome c release induced by mitochondrial outer mem-brane permeabilization. However, it is not likely that 2-IB has a general effect on mitochondrial protein release, since AIF translocation is not affected by 2-IB treatment, as we described in our previous study [10] . Interestingly, cytochrome c release was observed in both genders, whereas 2-IB only prevents cytochrome c in female ani-mals . These data suggest that the mechanisms leading to cytochrome c release differ between genders. One possi-bility is that cytochrome c release in males is secondary to AIF translocation and mitochondrial damage [27, 28] . In addition, it has been suggested that AIF and cyto-chrome c are released from the mitochondria via differ-ent pores, e.g. the Bax/Bak pore or the VDAC pore, and it is possible that 2-IB only affects one of these pores.
Furthermore, it can be speculated that more upstream events that lead to mitochondrial outer membrane per-meablization are gender dimorphic. There are a number
of potential targets for 2-IB that are upstream of the HI-induced release of cytochrome c and caspase-dependent apoptosis, but independent of the AIF-PARP pathway. Recently, Renolleau et al. [29] described that only females were protected from neonatal HI brain damage by a broad-spectrum caspase inhibitor. The gender-depen-dent working mechanism of 2-IB might also involve cas-pase inhibition, e.g. inhibition of death receptor-associ-ated initiator caspases like 8 and 10 or the less investi-gated caspase 2. Caspase 2 is thought to be a nuclear initiator caspase, activated by genotoxic stress or DNA damage, which is likely to occur during HI. Caspase 2 has been described to be responsible for cytochrome c release and permeabilization of the mitochondria, possibly via effects on the formation of the Bax pore [30, 31] .
Finally, we cannot exclude that more upstream exter-nal proapoptotic signals like Fas ligand and Fas death re-ceptors [32] or effects on cerebral leukocyte infiltration are responsible for the gender-dependent effect of 2-IB.
We conclude that the influence of gender on neuro-protection was strikingly similar in both P3 and P7 rats. Short-term and long-term neuroprotection with 2-IB could be obtained in female P3 rats, but not in male P3 rats. These gender differences were also observed in old-er rats, and may be relevant for neuroprotective strategies in preterm human neonates.
Acknowledgements
This study was funded by the Wilhelmina Children’s Hospital Research Fund. The premixed gas (8% oxygen/92% nitrogen) used in this study was kindly provided by HoekLoos Medical B.V., Eindhoven, the Netherlands. The authors would like to thank A. Faber, S. Bakker, E. van Zutphen and W. Woudstra for their excel-lent technical assistance regarding histological preparations.
References
1 Ferriero DM: Neonatal brain injury. N Engl J Med 2004; 351: 1985–1995.
2 Okumura A, Hayakawa F, Kato T, Kuno K, Watanabe K: MRI findings in patients with spastic cerebral palsy. I. Correlation with gestational age at birth. Dev Med Child Neu-rol 1997; 39: 363–368.
3 Sie LT, van der Knaap MS, Oosting J, de Vries LS, Lafeber HN, Valk J: MR patterns of hy-poxic-ischemic brain damage after prenatal, perinatal or postnatal asphyxia. Neuropedi-atrics 2000; 31: 128–136.
4 Volpe JJ: Neurology of the Newborn, ed 4, revised. Philadelphia, Saunders, 2001.
5 Myers RE: Four patterns of perinatal brain damage and their conditions of occurrence in primates. Adv Neurol 1975; 10: 223–234.
6 Marlow N, Wolke D, Bracewell MA, Samara M: Neurologic and developmental disability at six years of age after extremely preterm birth. N Engl J Med 2005; 352: 9–19.
7 Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, Graham SH, Clark RS: Innate gender-based proclivity in response to cyto-toxicity and programmed cell death pathway. J Biol Chem 2004; 279: 38563–38570.
8 Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M, Poitras MF, Dawson TM, Dawson VL, Northington F, Johnston MV: PARP-1 gene disruption in mice preferentially protects males from peri-natal brain injury. J Neurochem 2004; 90:
1068–1075.

Nijboer /Kavelaars /van Bel /Heijnen /Groenendaal
Dev Neurosci 2007;29:385–392392
9 Zhu C, Xu F, Wang X, Shibata M, Uchiyama Y, Blomgren K, Hagberg H: Different apop-totic mechanisms are activated in male and female brains after neonatal hypoxia-isch-aemia. J Neurochem 2006; 96: 1016–1027.
10 Nijboer CH, Groenendaal F, Kavelaars A, Hagberg HH, Van Bel F, Heijnen CJ: Gender-specific neuroprotection by 2-iminobiotin after hypoxia-ischemia in the neonatal rat via a nitric oxide independent pathway. J Cereb Blood Flow Metab 2007; 27: 282–292.
11 Peeters-Scholte C, Koster JG, Veldhuis W, van den Tweel E, Zhu C, Kops N, Blomgren K, Bär D, van Buul-Offers SC, Hagberg H, Nicolay K, Van Bel F, Groenendaal F: Neuro-protection by selective nitric oxide synthase inhibition at 24 h after perinatal hypoxia-ischemia. Stroke 2002; 33: 2304–2310.
12 van den Tweel ERW, Van Bel F, Kavelaars A, Peeters-Scholte CMPCD, Haumann J, Nij-boer CHA, Heijnen CJ, Groenendaal F: Long-term neuroprotection with 2-iminio-biotin, an inhibitor of neuronal and induc-ible nitric oxide synthase, after cerebral hy-poxia-ischemia in neonatal rats. J Cereb Blood Flow Metab 2005; 25: 67–74.
13 Bona E, Hagberg H, Loberg EM, Bagenholm R, Thoresen M: Protective effects of moder-ate hypothermia after neonatal hypoxia-ischemia: short- and long-term outcome. Pe-diatr Res 1998; 43: 738–745.
14 Zhu C, Wang X, Xu F, Bahr BA, Shibata M, Uchiyama Y, Hagberg H, Blomgren K: The influence of age on apoptotic and other mechanisms of cell death after cerebral hy-poxia-ischemia. Cell Death Differ 2005; 12:
162–176. 15 Sizonenko SV, Kiss JZ, Inder T, Gluckman
PD, Williams CE: Distinctive neuropatho-logic alterations in the deep layers of the pa-rietal cortex after moderate ischemic-hy-poxic injury in the P3 immature rat brain. Pediatr Res 2005; 57: 865–872.
16 Vannucci RC, Connor JR, Mauger DT, Pal-mer C, Smith MB, Towfighi J, Vannucci SJ: Rat model of perinatal hypoxic-ischemic brain damage. J Neurosci Res 1999; 55: 158–163.
17 Rice JE, Vannucci RC, Brierley JB: The influ-ence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 1981; 9:
131–141. 18 van den Tweel ER, Nijboer C, Kavelaars A,
Heijnen CJ, Groenendaal F, van Bel F: Ex-pression of nitric oxide synthase isoforms and nitrotyrosine formation after hypoxia-ischemia in the neonatal rat brain. J Neuro-immunol 2005; 167: 64–71.
19 Towfighi J, Mauger D, Vannucci RC, Van-nucci SJ: Influence of age on the cerebral le-sions in an immature rat model of cerebral hypoxia-ischemia: a light microscopic study. Brain Res Dev Brain Res 1997; 100: 149–160.
20 Sizonenko SV, Sirimanne E, Mayall Y, Gluck-man PD, Inder T, Williams C: Selective cor-tical alteration after hypoxic-ischemic inju-ry in the very immature rat brain. Pediatr Res 2003; 54: 263–269.
21 McCullough LD, Zeng Z, Blizzard KK, Deb-choudhury I, Hurn PD: Ischemic nitric oxide and poly (ADP-ribose) polymerase-1 in cere-bral ischemia: male toxicity, female protec-tion. J Cereb Blood Flow Metab 2005; 25:
502–512. 22 Alkayed NJ, Harukuni I, Kimes AS, London
ED, Traystman RJ, Hurn PD: Gender-linked brain injury in experimental stroke. Stroke 1998; 29: 159–165.
23 Toung TJ, Traystman RJ, Hurn PD: Estro-gen-mediated neuroprotection after experi-mental stroke in male rats. Stroke 1998; 29:
1666–1670. 24 McCullough LD, Hurn PD: Estrogen and
ischemic neuroprotection: an integrated view. Trends Endocrinol Metab 2003; 14:
228–235.
25 MacLusky NJ, Chaptal C, McEwen BS: The development of estrogen receptor systems in the rat brain and pituitary: postnatal devel-opment. Brain Res 1979; 178: 143–160.
26 Bakker J, De Mees C, Douhard Q, Balthazart J, Gabant P, Szpirer J, Szpirer C: Alpha-feto-protein protects the developing female mouse brain from masculinization and de-feminization by estrogens. Nat Neurosci 2006; 9: 220–226.
27 Wang H, Yu SW, Koh DW, Lew J, Coombs C, Bowers W, Federoff HJ, Poirier GG, Dawson TM, Dawson VL: Apoptois-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci 2004; 24: 10963–10973.
28 Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Daw-son TM, Dawson VL: Mediation of poly (ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002; 297: 259–263.
29 Renolleau S, Fau S, Goyenvalle C, Joly LM, Chauvier D, Jacotot E, Mariani J, Charriaut-Marlangue C: Specific caspase inhibitor Q-VD-OPh prevents neonatal stroke in P7 rat: a role for gender. J Neurochem 2006; 100:
1062–1071. 30 Guo Y, Srinivasula SM Druilhe A, Fer-
nandes-Alnemri T, Alnemri ES: Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem 2002; 277: 13430–13437.
31 Robertson JD, Enoksson M, Suomela M, Zhi-votovsky B, Orrenius S: Caspase-2 acts up-stream of mitochondria to promote cyto-chrome c release during etoposide-induced apoptosis. J Biol Chem 2002; 277: 29803–29809.
32 Graham EM, Sheldon RA, Flock DL, Ferrie-ro DM, Martin LJ, O’Riordan DP, Northing-ton FJ: Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol Dis 2004;
17: 89–98.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:393–402 DOI: 10.1159/000105480
Delayed Peripheral Administration of a GPE Analogue Induces Astrogliosis and Angiogenesis and Reduces Inflammation and Brain Injury following Hypoxia-Ischemia in the Neonatal Rat
Pernilla Svedin
b Jian Guan
a Sam Mathai
a Rong Zhang
a Xiaoyang Wang
b
Malin Gustavsson
a Henrik Hagberg
c Carina Mallard
b
a The Liggins Institute, University of Auckland, Auckland , New Zealand; b
Perinatal Center, Department of Neuroscience and Physiology, and c
Department of Clinical Science, Sahlgrenska Academy, Göteborg University, Göteborg , Sweden
astrocytosis was also determined in the hippocampus. Ani-mals treated with multiple doses of G-2mPE demonstrated reduced overall brain injury 7 days after HI, particularly in the hippocampus and thalamus compared to vehicle-treated rats. The expression of IL-6 was decreased in G-2mPE-treated animals compared to vehicle-treated pups, and both the capillary length and astrogliosis were increased in the drug-treated animals. There was no effect on caspase 3 activity. This study indicates that peripheral administration of G-2mPE, starting 2 h after a hypoxic-ischemic insult, reduces the degree of brain injury in the immature rat brain. The nor-malization of IL-6 levels and the promotion of both neovas-cularization and reactive astrocytosis may be potential mechanisms that underlie its protective effects.
Copyright © 2007 S. Karger AG, Basel
Introduction
Perinatal hypoxia-ischemia (HI) remains a significant risk factor of acute mortality and chronic neurologic morbidity in infants and children [Johnston et al., 2001]. As more information about perinatal brain injury mech-anisms has evolved during the last decades, it has become clear that although the development of tissue damage
Key Words
Hypoxia-ischemia � Glycine 2-methyl proline glutamate � Neonatal brain
Abstract
Glycine 2-methyl proline glutamate (G-2mPE) is a proline-modified analogue to the naturally existing N-terminal tri-peptide glycine-proline-glutamate that is a cleaved product from insulin-like growth factor-1. G-2mPE is designed to be more enzymatically resistant than glycine-proline-gluta-mate and to increase its bioavailability. The current study has investigated the protective effects of G-2mPE following hy-poxic-ischemic brain injury in the neonatal brain. On postna-tal day 7, Wistar rats were exposed to hypoxia-ischemia (HI). HI was induced by unilateral ligation of the left carotid artery followed by hypoxia (7.7% O 2 , 36 ° C) for 60 min. The drug treatment started 2 h after the insult, and the pups were giv-en either 1.2 mg/kg (bolus), 1.2 mg/ml once a day for 7 days, or vehicle. The degree of brain damage was determined his-tochemically by thionin/acid fuchsin staining. G-2mPE’s anti-inflammatory properties were investigated by IL-1 � , IL-6, and IL-18 ELISA, and effects on apoptosis by caspase 3 activity. Vascularization was determined immunohistochemically by the total length of isolectin-positive blood vessels. Effect on
Received: September 12, 2006 Accepted after revision: January 5, 2007
Dr. Jian Guan The Liggins Institute, University of Auckland Private Bag 92019 Auckland (New Zealand) Tel. +64 9373 7599, ext. 86134, Fax +64 9373 7497, E-Mail [email protected]
© 2007 S. Karger AG, Basel0378–5866/07/0295–0393$23.50/0
Accessible online at:www.karger.com/dne

Svedin /Guan /Mathai /Zhang /Wang /Gustavsson /Hagberg /Mallard
Dev Neurosci 2007;29:393–402394
starts during the insult, injurious processes proceed for several days afterwards [Ferriero, 2004]. Recent random-ized controlled trials have shown that postasphyxial cool-ing of either the head or whole body reduce mortality and attenuate neurological deficits [Gluckman et al., 2005; Shankaran et al., 2005]. These are key studies as they sup-port the notion of a therapeutic window, with the possi-bility to treat the perinatal brain after the insult. There-fore, a major challenge today is to find neuroprotective drugs that can be administered after the primary insult and which provide lasting effects.
Insulin-like growth factor-1 (IGF-1) is a naturally ex-isting hormone with antiapoptotic properties. Central administration of IGF-1 has been shown to be neuropro-tective after cerebral ischemia in both the mature and the immature brain [Johnston et al., 1996; Guan et al., 2003; Brywe et al., 2005]. In plasma and brain tissue, endoge-nous IGF-1 is believed to be degraded into des-N-(1-3)-IGF-1 and a tripeptide glycine-proline-glutamate (GPE) [Sara et al., 1989; Yamamoto and Murphy, 1995]. With its small molecular weight, GPE crosses the blood-brain barrier and has been shown to have neuroprotective properties in the adult and juvenile brain after HI follow-ing peripheral administration [Sizonenko et al., 2001; Guan et al., 2004]. The exact neuroprotective mecha-nisms of the GPE peptide is not known, but it is thought that it may act through the NMDA receptor which in turn reduces the excitotoxicity [Sara et al., 1989, 1993]. One limitation of GPE in clinical applications is the short half-life when administered systemically (approximately 2 min), although once in the brain, the half-life is in-creased to 30 min [Batchelor et al., 2003; Baker et al., 2005]. To overcome the problem with the short half-life of GPE in plasma, intravenous infusions of the drug have been used to achieve maximum neuroprotective effects [Guan et al., 2004]. Recently, a proline-modified ana-logue, G 2-methyl PE (G-2mPE) [Harris and Brimble, 2006] was designed to increase the enzymatic resistance to proteases in plasma and thereby increase the half-life [Guan et al., 2004; Baker et al., 2005]. The role of G-2mPE as a neuroprotectant in the immature brain is not known.
The purpose of this study was to evaluate the neuro-protective effects of G-2mPE following delayed adminis-tration after HI brain injury in 7-day-old rats. Secondly, we wanted to investigate the role of G-2mPE in delayed injury mechanisms including inflammation (IL-1 � , IL-6, and IL-18), apoptosis (caspase 3 activity) and astrocytic activation and angiogenesis.
Material and Methods
Induction of HI in Postnatal Day 7 Rats Pregnant Wistar rats were purchased from Charles River Lab-
oratories (Sulzfeld, Germany), and housed at Experimental Bio-medicine, Göteborg University, Sweden. Rats were housed with a 12-hour light-dark cycle, free access to a standard laboratory chow diet (B&K, Solna, Sweden) and deionized drinking water. The animal experiments were approved by the local Animal Eth-ical Committee of Göteborg.
At postnatal day 7, pups were exposed to HI [Rice et al., 1981; Hedtjarn et al., 2002] with some modifications. Briefly, the pups were anesthetized with isoflurane (Forene � , 3.5% for induction, 1.5% for maintenance) in a mixture of nitrous oxide and oxygen (1: 1), and the duration of anesthesia was ! 5 min. The left com-mon carotid artery was permanently ligated between double liga-tures of prolene sutures (6–0). After the surgical procedure, the wounds were infiltrated with a local anesthetic, and the pups were allowed to recover for 1 h after which the pups were placed in a chamber perfused with a humidified gas mixture (7.7% oxygen in nitrogen) for 60 min. The temperature in the incubator, and the temperature of the water used to humidify the gas mixture, was kept at 36 ° C. After hypoxic exposure, the pups were returned to their dam.
G-2mPE Treatment for the Neuroprotection Study Pups were randomly divided into 3 groups for administration
of G-2mPE. One group was treated with 1.2 mg/kg G-2mPE as a single dose at 2 h after the HI insult. The second group was given 1.2 mg/kg G-2mPE starting 2 h after the insult, followed by ad-ministration of 1.2 mg/kg once a day for 7 days. Pups in the con-trol group were given a saline injection 2 h after HI. All injections were given s.c. in a volume of 200 � l. All pups that received injec-tions were handled and weighed every day. Naïve pups were not handled before sacrifice.
Tissue Preparation and Histochemical Procedures Pups were sacrificed on postnatal day 14. The animals were
anesthetized with tiopenthal (Pentothal � Sodium) and perfusion fixed with 4% paraformaldehyde (Histofix, Histolab, Göteborg, Sweden). The brains were then dehydrated and embedded in par-affin and coronal sections at the level of striatum and the hippo-campus were cut (6 � m) for further evaluation. Three coronal sections (including striatum, hippocampus, thalamus and cere-bral cortex) from each brain were mounted on glass slides and stained with thionine/acid fuchsin for morphological assessment of the regional brain injury as previously described [Guan et al., 1993, 1996].
GFAP-positive cells were immunohistochemically stained on paraffin tissues [Guan et al., 2004]. Coronal sections (6 � m) con-taining the level of the hippocampus were cut and mounted on chrome-alum-coated slides. The sections were deparaffinized and incubated in 0.1 M PBS. All sections were pretreated with 1% H 2 O 2 in 50% methanol and then 1.5% normal horse serum. The sections were incubated with primary antibody (monoclonal mouse anti-GFAP, 1: 500, at 4 ° C for 2 days; Sigma-Aldrich, Saint Louis, Mo., USA). The sections were then incubated with biotinyl-ated secondary antibody (horse anti-mouse biotinylated, 1: 200, overnight at 4 ° C; Sigma-Aldrich), then ExtrAvidin (1: 200, 3 h at RT, Sigma-Aldrich) followed by DAB reaction.

G-2mPE Prevent Brain Injury following HI Injury in Neonatal Rat
Dev Neurosci 2007;29:393–402 395
For specific visualization of cerebral capillaries, isolectin B4 from Griffonia simplicifolia seeds (Sigma-Aldrich) was used as the marker [Ismail et al., 2003]. The sections were pretreated with 1% H 2 O 2 in 50% methanol after being deparaffinized, then incubat-ed overnight at 4 ° C with the isolectin B-4 (1: 4) in Tris-buffered saline before being developed in DAB.
Fluorometric Assay of Caspase 3-Like Activity At 8 and 24 h after the hypoxic-ischemic insult, pups (n = 9/
time point/group) were killed with an overdose of tiopenthal (Pentothal � Sodium, i.p.) and perfused with cold saline. The brains were rapidly removed and divided into ipsilateral ischemic hemisphere and contralateral nonischemic hemisphere and fur-ther dissected into cortex/hippocampus and striatum/thalamus. The hemispheric tissue was frozen immediately in isopenthane and stored at –80 ° C. The frozen brain tissue was homogenized in cold homogenizing buffer (50 m M Tris, pH 7.3, 5 m M EDTA, 1% proteinase inhibitor cocktail; Sigma-Aldrich). Total protein con-centration in the homogenates was measured by the Soft Max PRO 3.0. The caspase 3 activity was measured as described by Wang et al. [2001] with some modifications. Homogenate was mixed with extraction buffer (50 m M Tris-HCl, pH 7.3, 100 m M NaCl, 5 m M EDTA, 1 m M EGTA, 1 m M PMSF, 1% proteinase in-hibitor cocktail; Sigma-Aldrich) and 0.2% CHAPS on a microtiter plate (Microfluor, Dynatech). After 15 min of incubation in RT, 100 � l of assay buffer (m M Tris-HCl, pH 7.3, 100 m M NaCl, 5 m M EDTA, 1 m M EGTA, 1 m M PMSF, and 10 m M DTT) containing 25 � M peptide substrate (AC-DEVD-AMC; Enzyme Systems Products, Livermore, Calif., USA) was added into each well. Cleavage of the substrate was measured at 37 ° C using a Spectra-max Gemini microplate flourometer with an excitation wave-length of 380 nm and an emission wavelength of 460 nm, 2-min interval for 2 h. The V max was calculated from the entire linear part of the curve. Standard curves with AMC in the appropriate buffer were used to express the data in pmole of AMC formed per minute and milligram of protein. All samples were done in dupli-cate.
Enzyme-Linked Immunosorbent Assay IL-1 � and IL-6 activity was measured 8 h after HI (n = 8/
group) and active IL-18 was measured 24 h after HI (n = 8/group). The brain tissue was prepared as described for caspase 3-like ac-tivity measurements, except the cortex/hippocampus and stria-tum/thalamus samples were pooled. The homogenates were cen-trifuged at 10,000 g at 4 ° C for 10 min, and the supernatants were saved and used for the ELISA analysis. Cytokines were measured by rat-specific immunoassay kits (IL-1 � by Quantikine from R&D systems and IL-6 and IL-18 from Biosource). All standards were diluted in the homogenization buffer, and the samples were done in duplicates. The assays were performed as recommended by the manufacturers.
Neuropathological Analysis Histopathological analysis was performed on coded slides,
with the observer unaware of the experimental grouping. Dead neurons were identified as those with acidophilic (red) cytoplasm and contracted nuclei [Brown and Brierley, 1972; Auer et al., 1985]. Brain tissue with selective neuronal death, cellular reaction and/or pan-necrosis were considered to be damaged [Guan et al., 2000]. The severity of brain damage in the lateral cortex was as-
sessed using three levels, the dentate gyrus (DG) and the CA1–2, 3 and 4 subregions of the hippocampus using two levels and the striatum and thalamus using one level as follows: 0 = no damage; 1 = ! 5% tissue damaged; 2 = ! 50% tissue damaged; 3 = 1 50% tis-sue damaged and 4 = 1 95% damaged [Guan et al., 2000].
GFAP Measurement The average density of GFAP-positive staining in ipsilateral
hippocampus of HI-injured groups and the right side hippocam-pus of normal control groups was measured using an image ana-lyzer (SigmaScan Pro 5.0; SPSS, Chicago, Ill., USA) and light mi-croscopy ( ! 4; Nikon 800, Tokyo, Japan).
Capillary Vessel Length Measurement The total length of cerebral capillaries (tube images only) was
determined using a high throughput image analysis assay devel-oped in the High Content Screening Laboratory, Department of Pharmacology, The University of Auckland (http://www.health.auckland.ac.nz/pharmacology/discovery-1/) using the Meta-morph v.6.2.6 Image analysis software (Molecular Devices). Images were acquired at ! 100 magnification and stored as JPEGs. The AddInvertFlattenBgdAngiogenesis assay automati-cally opened each image, converted the image to 16-bit, inverted each 16-bit brightfield image, performed a Flatten Background transformation to even the intensity of immunostaining across the image (to correct for background unevenness), and then per-formed the Metamorph Angiogenesis Tube Formation applica-tion on each image. Total Tubule Length (in pixels) was then au-tomatically logged into an Excel spreadsheet. To avoid the back-ground staining in the damaged brain regions, the isolectin B-4-positive capillaries were digitally photographed in the ipsi-lateral thalamus and the contralateral cerebral cortex, hippocam-pus and thalamus.
Statistics ANOVA (Fisher’s PLSD) was used for statistical comparisons
between study groups. Values were considered significant at p ! 0.05, and data presented as mean 8 SEM.
Results
Multiple Doses of G-2mPE Reduce Neuropathology The hypoxic-ischemic insult resulted in severe brain
damage in the ipsilateral hemisphere, particularly in the cerebral cortex and the subregions of the hippocampus ( fig. 1 a–d). Loss of cortical tissue resulted due to infarc-tion and atrophy was found in the ipsilateral hemispheres. There was no histological damage in the contralateral hemispheres. The neuropathological scoring was per-formed in the striatum, the hippocampus including the CA1/2, CA3, CA4 subregions, DG, the thalamus and the cerebral cortex. No significant difference in neuropathol-ogy score was observed in animals that received a single bolus injection of G-2mPE 2 h after the hypoxic-ischemic insult (2.67 8 0.29, n = 26) compared to vehicle-treated

Svedin /Guan /Mathai /Zhang /Wang /Gustavsson /Hagberg /Mallard
Dev Neurosci 2007;29:393–402396
animals (2.87 8 0.28, n = 25; fig. 1 e, f). In the group that received repeated doses of G-2mPE (n = 26), there was an attenuation of the neuropathological score compared to vehicle-treated animals (n = 25) in several brain regions, including the CA1/2 (2.76 8 0.22 vs. 3.32 8 0.11, p =
0.0388), CA3 (3.03 8 0.25 vs. 3.64 8 0.09, p = 0.0268) and thalamus (1.60 8 0.14 vs. 2.00 8 0.07, p = 0.0096; fig. 1 e) and the total neuropathological score (2.37 8 0.25 vs. 2.87 8 0.28, p = 0.0240; fig. 1 f). Since there was no neuroprotective effect in the group that received the
0
20
Total
*
4
8
12
16
0
1
2
3
4
Tis
sue
da
ma
ge
sco
re
Striatum CA 1/2 CA 3 CA 4 DG Cortex
**
Brain regions
Vehicle Single Multiple
e f
Fig. 1. Photomicrographs demonstrating the injury at the level of the hippocampus in vehicle-treated ( a , c ) and G-2mPE-treated ( b , d ) animals. Tissue damage in the striatum, thalamus, hippocampus (CA1/2, CA3, CA4 and DG), cerebral cor-tex ( e ), and the total score ( f ) after treat-ment (s.c.) with either vehicle (n = 25), one single bolus dose of G-2mPE (n = 26), or repeated doses of G-2mPE (n = 26). The neuropathological score in the CA1/2 and CA3 regions of the hippocampus and the total score was reduced after repeated in-jections of G-2mPE. Results are expressed as mean 8 SEM; * p ! 0.05 compared to vehicle.

G-2mPE Prevent Brain Injury following HI Injury in Neonatal Rat
Dev Neurosci 2007;29:393–402 397
single bolus dose of G-2mPE, no further experiments were carried out using this treatment regimen.
Caspase 3-Like Activity following G-2mPE Treatment The caspase 3 activity was detected by the degradation
of the substrate Ac-DVED-AMC at 8 and 24 h after the HI insult in tissue from the striatum/thalamus ( fig. 2 a, c) and from the hippocampus/cortex ( fig. 2 b, d). Caspase 3 activity was increased in the ipsilateral hemisphere in striatum/thalamus in both the G-2mPE and vehicle-treated animals at 8 h after HI (vehicle ipsilateral: 24.56 8 3.99 vs. vehicle contralateral: 6.22 8 0.52 p ! 0.0001; G-2mPE ipsilateral: 25.12 8 2.16 vs. G-2mPE contralat-eral: 6.10 8 0.33, p ! 0.0001) and in hippocampus/cortex (vehicle ipsilateral: 28.36 8 4.50 vs. vehicle contralateral: 7.38 8 0.44, p ! 0.001; G-2mPE ipsilateral: 27.81 8 2.14 vs. G-2mPE contralateral: 7.93 8 0.30, p ! 0.0001; fig. 2 a, b) and at 24 h after HI in striatum/thalamus (vehicle ip-silateral: 39.59 8 8.94 vs. vehicle contralateral: 7.25 8 1.38 p ! 0.01; G-2mPE ipsilateral: 25.66 8 3.88 vs. G-2mPE contralateral: 6.51 8 0.62, p ! 0.001) and in hip-pocampus/cortex (vehicle ipsilateral: 78.65 8 5.35 vs. ve-hicle contralateral: 7.47 8 1.58, p ! 0.0001; G-2mPE ip-silateral: 72.68 8 8.29 vs. G-2mPE contralateral: 6.47 8 0.95, p ! 0.0001; fig. 2 c, d). There was no difference in caspase 3 activity in the ipsilateral hemisphere between treatment groups at either 8 h in the hippocampus/cortex (p = 0.7846) or striatum/thalamus (p = 0.9043) or at 24 h in the hippocampus/cortex (p = 0.5535) or striatum/thal-amus (p = 0.1721).
Contralateral Ipsilateral Contralateral Ipsilateral0
a b
c d
5101520253035
pm
ol A
MC
/min
× m
gp
rote
in
05
101520253035
pm
ol A
MC
/min
× m
gp
rote
inp
mol
AM
C/m
in ×
mg
pro
tein
Contralateral Ipsilateral0
102030405060708090
pm
ol A
MC
/min
× m
gp
rote
in
Contralateral Ipsilateral0
102030405060708090 Fig. 2. Caspase 3 activity in the striatum/
thalamus ( a , c ), the hippocampus/cortex ( b , d ) at 8 h ( a , b ) and 24 h ( c , d ) after HI. Caspase 3 activity was significantly upreg-ulated in the ipsilateral hemispheres com-pared to the contralateral hemispheres. There were no differences between G-2mPE-treated ( ) ; n = 9) and vehicle-treat-ed ( $ ; n = 9) animals. Results are expressed as mean 8 SEM.
*
Contralateral Ipsilateral0
a
b
c
102030405060708090
IL-1
� p
rote
in (p
g/m
gto
tal p
rote
in)
Contralateral Ipsilateral0
102030405060708090
IL-6
pro
tein
(pg
/mg
tota
l pro
tein
)
Contralateral Ipsilateral0
10
20
30
IL-1
8 p
rote
in (p
g/m
gto
tal p
rote
in)
Fig. 3. IL-1 � ( a ), IL-6 ( b ) activity at 8 h and IL-18 activity ( c ) at24 h after HI. The activity of IL-6 was significantly decreased in the ipsilateral hemisphere in G-2mPE-treated animals ( ) ) com-pared to animals that received vehicle ( $ ; p = 0.0205). Results are expressed as mean 8 SEM; * p ! 0.05 compared to vehicle; n = 8 animals/group.

Svedin /Guan /Mathai /Zhang /Wang /Gustavsson /Hagberg /Mallard
Dev Neurosci 2007;29:393–402398
G-2mPE Treatment Reduced IL-6 Activity following HI The expression of IL-1 � and IL-6 was measured 8 h
after the HI insult. There was a 6.1-fold increase in IL-1 � activity in the ipsilateral hemisphere (42.5 8 10.6 pg/mg) compared to the contralateral hemisphere (7.0 8 1.1 pg/mg) in vehicle-treated animals (n = 8) and a 5.3-fold in-crease in IL-1 � activity in the ipsilateral hemispheres (41.5 8 13.8 pg/mg) compared to the contralateral hemi-sphere (7.9 8 1.6 pg/mg) in the G-2mPE-treated animals (n = 8; fig. 3 a). There was no difference in IL-1 � activity in the ipsilateral hemisphere in vehicle-treated compared to G-2mPE-treated animals (p = 0.9539; fig. 3 a). IL-6 ac-tivity was reduced by 32% in the ipsilateral hemisphere in G-2mPE-treated (51.0 8 4.7 pg/mg) compared to vehi-cle-treated animals (74.8 8 7.8 pg/mg, p = 0.0205; fig. 3 b). The activity of IL-18 was increased 1.9-fold in the ipsilat-eral hemispheres (vehicle: 21.7 8 3.3 pg/mg; G-2mPE: 21.8 8 3.8 pg/mg) compared to the contralateral hemi-spheres (vehicle: 11.7 8 1.3 pg/mg; G-2mPE: 11.3 8 0.9
pg/mg) in both the G-2mPE-treated and vehicle-treated animals at 24 h after HI ( fig. 3 c). There were no signifi-cant differences in IL-18 activity in the ipsilateral hemi-spheres in the vehicle-treated compared to G-2mPE-treated animals (p = 0.9781; fig. 3 c).
G-2mPE Treatment Promoted Vessel Length following HI Isolectin B-4-positive staining was apparent in cere-
bral capillaries in control animals ( fig. 4 a). In injured ar-eas, isolectin-positive staining was also seen in activated microglia; however, at 1 week after HI, there were rela-tively few microglia cells. The total length of cerebral cap-illaries was significantly reduced in the ipsilateral thala-mus compared to the contralateral thalamus following HI in vehicle-treated animals (3,624 8 234 pixels vs. 4,711 8 343.4 pixels, p ! 0.001, n = 13; fig. 4 b). In the contralateral cortex (4,647 8 245 pixels, fig. 4 e) and hip-pocampus ( fig. 4 f), hypoxia increased vessel length com-pared to normal controls, (3,486 8 164 pixels, p ! 0.05,
Contralate
ral
Ipsil
atera
l0
2,500
5,000
7,500
**
Tota
l len
gth
of
cap
illar
ies
(pix
els)
Normal
Vehicle
G-2m
PE0
2,500
5,000
7,500
#
Tota
l len
gth
of
cap
illar
ies
(pix
els)
Normal
Vehicle
G-2m
PE0
2,500
5,000
7,500
#
Tota
l len
gth
of
cap
illar
ies
(pix
els)
Normal
Vehicle
G-2m
PE0
2,500
5,000
7,500
*#
Tota
l len
gth
of
cap
illar
ies
(pix
els)
Normal
Vehicle
G-2m
PE0
2,500
5,000
7,500
a
c
e f
b
d
*
Tota
l len
gth
of
cap
illar
ies
(pix
els)
Fig. 4. a Photomicrograph shows the isolectin B-4-positive vessels in the cere-bral cortex of a normal control rat. b Ef-fects of HI injury on vessel length in the thalamus. Effects of G-2mPE (n = 27) on vessel length in the ipsilateral ( c ) and the contralateral ( d ) thalamus compared to normal (n = 10) and vehicle controls (n = 21) and in the contralateral cortex ( e ) and contralateral hippocampus ( f ). # p ! 0.05, one-way ANOVA, * p ! 0.05, * * p ! 0.001, paired t test.

G-2mPE Prevent Brain Injury following HI Injury in Neonatal Rat
Dev Neurosci 2007;29:393–402 399
n = 10). The vessel length was further increased in the contralateral cortex by G-2mPE compared to vehicle-treated animals (5,541 8 335 vs. 4,647 8 245 pixels, p ! 0.05; fig. 4 e). G-2mPE treatment increased the total vessel length in both the ipsilateral thalamus (4,430 8 255 vs. 3,624 8 234 pixels, p ! 0.05, n = 15; fig. 4 c) and the con-tralateral thalamus (6,505 8 554 vs. 4,711 8 343 pixels, p ! 0.05; fig. 4 d) compared with vehicle-treated ani-mals.
Increased Astrogliosis following G-2mPE Treatment Morphologically, the GFAP-positive cells in the ipsi-
lateral hemisphere demonstrated hypertrophic processes with much denser immunostaining ( fig. 5 b) compared to the contralateral hemisphere ( fig. 5 a). GFAP-positive cells were also closely associated with cerebral vascula-ture. The average density of GFAP-positive staining in the hippocampus was significantly increased after HI in-jury in the vehicle-treated group (14.2 8 1.1, n = 21, p ! 0.001) compared to normal naïve controls (2.3 8 0.3, n = 10; fig. 5 c). The treatment with G-2mPE significantly in-creased the GFAP density (22.6 8 1.8, n = 17, p ! 0.001) in the hippocampus compared to the vehicle-treated rats ( fig. 5 c).
Discussion
This study shows that treatment with G-2mPE, an an-alogue derived from IGF-1, attenuates neuronal injury moderately, even when the drug is administered periph-
erally and treatment is started 2 h after the hypoxic-isch-emic insult. G-2mPE treatment also reduced IL-6 activ-ity and promoted astrogliosis and possible angiogenesis.
Neuroprotection has previously been observed follow-ing treatment with the tripeptide GPE in both the adult and juvenile brain following HI [Guan et al., 1999; Si-zonenko et al., 2001]. In this study, we show neuroprotec-tion after treatment with G-2mPE, a GPE analogue. The treatment effect was rather modest, which is possibly as-sociated with the severe brain injury and acute irrevers-ible damage of neurons during and immediately after the insult before treatment was initiated. Similar to previous studies using GPE, the most pronounced protection was observed in the thalamus and hippocampal regions such as CA1/2 and CA3. While GPE has a very short half-life in plasma [Batchelor et al., 2003; Guan et al., 2004; Baker et al., 2005], which limits its clinical use, the tripeptide G-2mPE was designed to increase the half-life in plasma [Harris and Brimble, 2006] to allow for a more stable drug administration. Although we do not know how pe-ripherally administered G-2mPE accesses or affects the brain, our results indicate similar properties as the GPE peptide based on the regional distribution of the protec-tive effects.
Neonatal HI results in prominent caspase 3 activation, which is believed to be an important apoptotic cell death mechanism, particularly in the immature brain [Wang et al., 2001; Zhu et al., 2005]. Similarly, we find an increase in caspase 3 activity in the ischemic ipsilateral hemi-sphere; however, there was no treatment effect of G-2mPE, suggesting that the G-2mPE neuroprotective ef-
Normalc Vehicle G-2mPE0
5
10
15
20
25
###
***
Ave
rag
e d
ensi
ty o
f GFA
P-p
osit
ive
stai
nin
g
Fig. 5. Photomicrographs of GFAP-positive cells, in the contralateral ( a ) and the ipsilateral ( b ) hippocampus, 1 week following HI. c The density of GFAP-positive cells in the hippocampus was significantly increased after HI injury in the vehicle-treated group (n = 21) compared to normal controls (n = 10), and treatment with G-2mPE (n = 17) further increased GFAP density. ***p ! 0.001; ###p ! 0.001.

Svedin /Guan /Mathai /Zhang /Wang /Gustavsson /Hagberg /Mallard
Dev Neurosci 2007;29:393–402400
fects are not via inhibition of caspase 3. This may seem surprising as IGF-1 administration reduced brain injury following neonatal HI, at least partly via caspase 3-medi-ated pathways [Brywe et al., 2005] and GPE reduced cas-pase 3-positive cells in adult rats after HI injury [Guan et al., 2004]. These results indicate that IGF-1 and GPE may act via different mechanisms compared to G-2mPE. However, the lack of effect on caspase 3 activity may also relate to that caspase 3 was measured in the whole hemi-sphere, while neuroprotection was mainly observed in the hippocampus. Furthermore, we cannot rule out the possibility that the majority of cell death in the present study was caused by more immediate death as the brain damage was very severe. In general, severe brain damage is more likely to be associated with rapid cell death where-as the more delayed and/or programmed cell death may be the result of a mild injury [Beilharz et al., 1995]. Given that the single treatment of G-2mPE did not statistically prevent tissue damage, the chronic treatments may have targeted the delayed brain damage to a certain degree. Delayed or programmed neuronal/glial death can be me-diated through necrotic, apoptotic and more recently suggested necroptotic pathways [Degterev et al., 2005]. The downstream cascades of these cell death pathways, however, are largely overlapping [Yuan et al., 2003]. Fur-thermore, recent studies also suggest that caspase-inde-pendent mechanisms, including apoptosis-inducing fac-tor, may significantly contribute to neuronal death after neonatal HI [Zhu et al., 2003].
HI triggers microglia cells to become activated and to produce cytokines, such as IL-1 � , IL-6, and IL-18. Sev-eral clinical studies show that IL-6 is upregulated, both in plasma and CSF, in children with hypoxic-ischemic encephalopathy [Silveira and Procianoy, 2003; Bartha et al., 2004], and plasma levels of IL-6 have been positively correlated with the severity of stroke [Smith et al., 2004]. Furthermore, the expression of IL-6 is enhanced in the brain following neonatal HI in rats [Hagberg et al., 1996]. At the same time, inhibition of IL-6 appears to be detri-mental and increases brain injury following stroke in mice [Yamashita et al., 2005]. In the present study, G-2mPE treatment normalized IL-6 activity, indicating anti-inflammatory properties of the peptide, which may be important for the neuroprotective effects observed. Similar to our previous study, we found an upregulation of both IL-1 � and IL-18 activity after HI in the neonatal brain [Hedtjarn et al., 2002]. Adult studies indicate that IL-1 � plays an important role in the development of in-jury after cerebral ischemia [Allan et al., 2005]. In con-trast, in the neonatal rat, IL-1 � deficiency does not confer
protection following HI [Hedtjarn et al., 2005], which is in support of our results in the present study where G-2mPE-mediated protection did not affect IL-1 � levels. Hedtjärn et al. [2002, 2005], showed that IL-18 is particu-larly activated at 3–14 days after HI and that IL-18-defi-cient mice are moderately protected from neonatal HI. Studies in adult brain have shown that IL-18 does not contribute to acute brain injury after focal cerebral isch-emia, but it might be more important at a later stage [Wheeler et al., 2003]. In the present study, G-2mPE did not seem to affect the early activation of IL-18 at 24 h af-ter HI; however, we cannot rule out the possibility that IL-18 may be affected at later time points. Taken together, the cytokine measurements indicate that the immuno-modulatory effects of G-2mPE may be specific.
HI resulted in severe injury in the ipsilateral hemi-sphere, but with mild injury in the thalamus, where the isolectin B-4-positive microglia was absent. The length of blood vessels measured in 2-D profile was reduced in the ipsilateral thalamus compared to the contralateral thala-mus, suggesting the loss of cerebral blood vessel could be part of the pathology of HI injury. However, hypoxic in-sult only caused an upregulation in lectin-positive blood vessels as an increase in capillary length (2-D) in both contralateral cortex and hippocampus, where histologi-cal damage was absent compared to the normal control rats. G-2mPE treatment increased the total length of cap-illaries in selective brain regions and the effects were not associated with histological injury. We are not able to de-termine in the present study whether the treatment ef-fects in capillary length are due to a higher degree of re-vascularization or preservation of blood vessels following HI. However, the increased capillary length may be a con-tributing factor of the neuroprotection as in the thalamus the neuroprotection and increased capillary length were coherent. In addition, it has been demonstrated that an increase in the density of blood vessels is associated with improved blood flow and contributes to the neuroprotec-tive effects of hypoxic preconditioning in neonatal rats [Gustavsson et al., 2006].
Angiogenesis is a major pathway for injury-associated neovascularization by endothelial sprouting and divid-ing existing vessel lumens for newly formed vessels [Frontczak-Baniewicz and Walski, 2002]. The formation of new cerebral vessels is fundamental for providing nu-tritional and trophic support to adjacent tissues, main-taining BBB integrity and tissue repair after injury. As-trocytes have an important role in vascularization [Sal-hia et al., 2000; Acker et al., 2001; Frontczak-Baniewicz and Walski, 2002]. Ischemic brain injury can cause neu-

G-2mPE Prevent Brain Injury following HI Injury in Neonatal Rat
Dev Neurosci 2007;29:393–402 401
roendothelial damage and provoke astrocytosis [Brault et al., 2003]. The injury-induced increase in GFAP-reactive astrocytosis has been found to be associated with neoan-giogenesis [Salhia et al., 2000]. Our data demonstrate that the GFAP-positive staining was clearly associated with vascular morphology. A role for GFAP-reactive astro-cytes in angiogenesis has been well documented by in-ducing key vascular regulators, including VEGF and an-giopoietin 1 (Ang-1) and its receptors [Acker et al., 2001]. Treatment with G-2mPE significantly increased GFAP density and revascularization, probably through promot-ing astrocytic angiogenesis. IGF-1, the parent peptide of GPE, has been reported to have a critical role in vascular remodeling by increasing vessel growth in the perilesion-al area after injury in adult mice brain [Lopez-Lopez et al., 2004]. The role of G-2mPE on angiogenesis needs to be further investigated.
In conclusion, this study demonstrates that peripheral delayed post-HI treatment with the IGF-1 tripeptide ana-logue G-2mPE attenuates neuronal injury and inflam-mation after HI in the immature brain and promotes as-trogliosis and probably blood vessel growth. Even though the neuroprotective effect was modest, these results are encouraging and indicate that G-2mPE may have the po-tential as a future candidate for treatment of newborn children suffering from birth asphyxia.
Acknowledgments
This work was supported by the Swedish Medical Research Council (K2004-33X-14185-03A), Sven Jerring Foundation, Wil-helm and Martina Lundgren Science Foundation, Frimurare Barnhus Foundation and the Åhlén Foundation. Neuren Phar-maceuticals, Ltd., provided the compounds.
References
Acker T, Beck H, Plate KH (2001): Cell type spe-cific expression of vascular endothelial growth factor and angiopoietin-1 and -2 sug-gests an important role of astrocytes in cer-ebellar vascularization. Mech Dev 108: 45–57.
Allan SM, Tyrrell PJ, Rothwell NJ (2005): Inter-leukin-1 and neuronal injury. Nat Rev Im-munol 5: 629–640.
Auer RN, Kalimo H, Olsson Y, Siesjo BK (1985): The temporal evolution of hypoglycemic brain damage. I. Light- and electron-micro-scopic findings in the rat cerebral cortex. Acta Neuropathol (Berl) 67: 13–24.
Baker AM, Batchelor DC, Thomas GB, Wen JY, Rafiee M, Lin H, Guan J (2005): Central pen-etration and stability of N-terminal tripep-tide of insulin-like growth factor-I, glycine-proline-glutamate in adult rat. Neuropeptides 39: 81–87.
Bartha AI, Foster-Barber A, Miller SP, Vigneron DB, Glidden DV, Barkovich AJ, Ferriero DM (2004): Neonatal encephalopathy: associa-tion of cytokines with MR spectroscopy and outcome. Pediatr Res 56: 960–966.
Batchelor DC, Lin H, Wen JY, Keven C, Van Zijl PL, Breier BH, Gluckman PD, Thomas GB (2003): Pharmacokinetics of glycine-pro-line-glutamate, the N-terminal tripeptide of insulin-like growth factor-1, in rats. Anal Biochem 323: 156–163.
Beilharz EJ, Williams CE, Dragunow M, Siri-manne ES, Gluckman PD (1995): Mecha-nisms of delayed cell death following hypox-ic-ischemic injury in the immature rat: evidence for apoptosis during selective neu-ronal loss. Brain Res Mol Brain Res 29: 1–14.
Brault S, Martinez-Bermudez AK, Marrache AM, Gobeil F Jr, Hou X, Beauchamp M, Quiniou C, Almazan G, Lachance C, Rob-erts J 2nd, Varma DR, Chemtob S (2003): Se-lective neuromicrovascular endothelial cell death by 8-Iso-prostaglandin F2alpha: pos-sible role in ischemic brain injury. Stroke 34:
776–782. Brown AW, Brierley JB (1972): Anoxic-ischaemic
cell change in rat brain light microscopic and fine-structural observations. J Neurol Sci 16:
59–84. Brywe KG, Mallard C, Gustavsson M, Hedtjarn
M, Leverin AL, Wang X, Blomgren K, Is-gaard J, Hagberg H (2005): IGF-I neuropro-tection in the immature brain after hypoxia-ischemia, involvement of Akt and GSK3beta? Eur J Neurosci 21: 1489–1502.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Mos-kowitz MA, Yuan J (2005): Chemical inhibi-tor of nonapoptotic cell death with therapeu-tic potential for ischemic brain injury. Nat Chem Biol 1: 112–119.
Ferriero DM (2004): Neonatal brain injury. N Engl J Med 351: 1985–1995.
Frontczak-Baniewicz M, Walski M (2002): Non-sprouting angiogenesis in neurohypophysis after traumatic injury of the cerebral cortex. Electron-microscopic studies. Neuro Endo-crinol Lett 23: 396–404.
Gluckman PD, Wyatt JS, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, Polin RA, Robertson CM, Thoresen M, Whitelaw A, Gunn AJ (2005): Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet 365: 663–670.
Guan J, Bennet L, Gluckman PD, Gunn AJ (2003): Insulin-like growth factor-1 and post-ischemic brain injury. Prog Neurobiol 70: 443–462.
Guan J, Gunn AJ, Sirimanne ES, Tuffin J, Gun-ning MI, Clark R, Gluckman PD (2000): The window of opportunity for neuronal rescue with insulin-like growth factor-1 after hy-poxia-ischemia in rats is critically modulat-ed by cerebral temperature during recovery. J Cereb Blood Flow Metab 20: 513–519.
Guan J, Thomas GB, Lin H, Mathai S, Bachelor DC, George S, Gluckman PD (2004): Neuro-protective effects of the N-terminal tripep-tide of insulin-like growth factor-1, glycine-proline-glutamate (GPE) following intrave-nous infusion in hypoxic-ischemic adult rats. Neuropharmacology 47: 892–903.
Guan J, Waldvogel HJ, Faull RL, Gluckman PD, Williams CE (1999): The effects of the N-ter-minal tripeptide of insulin-like growth fac-tor-1, glycine-proline-glutamate in different regions following hypoxic-ischemic brain injury in adult rats. Neuroscience 89: 649–659.
Guan J, Williams C, Gunning M, Mallard C, Gluckman P (1993): The effects of IGF-1 treatment after hypoxic-ischemic brain in-jury in adult rats. J Cereb Blood Flow Metab 13: 609–616.
Guan J, Williams CE, Skinner SJ, Mallard EC, Gluckman PD (1996): The effects of insulin-like growth factor (IGF)-1, IGF-2, and des-IGF-1 on neuronal loss after hypoxic-isch-emic brain injury in adult rats: evidence for a role for IGF binding proteins. Endocrinol-ogy 137: 893–898.

Svedin /Guan /Mathai /Zhang /Wang /Gustavsson /Hagberg /Mallard
Dev Neurosci 2007;29:393–402402
Gustavsson M, Mallard C, Vannucci SJ, Wilson MA, Johnston MV, Hagberg H (2006): Vas-cular response to hypoxic preconditioning in the immature brain. J Cereb Blood Flow Metab, E-pub ahead of print.
Hagberg H, Gilland E, Bona E, Hanson LA, Ha-hin-Zoric M, Blennow M, Holst M, McRae A, Soder O (1996): Enhanced expression of in-terleukin (IL)-1 and IL-6 messenger RNA and bioactive protein after hypoxia-isch-emia in neonatal rats. Pediatr Res 40: 603–609.
Harris PW, Brimble MA (2006): Synthesis of macrocyclic analogues of the neuroprotec-tive agent glycyl-L-prolyl-L-glutamic acid (GPE). Org Biomol Chem 4: 2696–2709.
Hedtjarn M, Leverin AL, Eriksson K, Blomgren K, Mallard C, Hagberg H (2002): Interleu-kin-18 involvement in hypoxic-ischemic brain injury. J Neurosci 22: 5910–5919.
Hedtjarn M, Mallard C, Iwakura Y, Hagberg H (2005): Combined deficiency of IL-1beta18, but not IL-1alphabeta, reduces susceptibility to hypoxia-ischemia in the immature brain. Dev Neurosci 27: 143–148.
Ismail JA, Poppa V, Kemper LE, Scatena M, Giachelli CM, Coffin JD, Murry CE (2003): Immunohistologic labeling of murine endo-thelium. Cardiovasc Pathol 12: 82–90.
Johnston BM, Mallard EC, Williams CE, Gluck-man PD (1996): Insulin-like growth factor-1 is a potent neuronal rescue agent after hy-poxic-ischemic injury in fetal lambs. J Clin Invest 97: 300–308.
Johnston MV, Trescher WH, Ishida A, Nakajima W (2001): Neurobiology of hypoxic-isch-emic injury in the developing brain. Pediatr Res 49: 735–741.
Lopez-Lopez C, LeRoith D, Torres-Aleman I (2004): Insulin-like growth factor I is re-quired for vessel remodeling in the adult brain. Proc Natl Acad Sci USA 101: 9833–9838.
Rice JE 3rd, Vannucci RC, Brierley JB (1981): The influence of immaturity on hypoxic-isch-emic brain damage in the rat. Ann Neurol 9:
131–141. Salhia B, Angelov L, Roncari L, Wu X, Shannon
P, Guha A (2000): Expression of vascular en-dothelial growth factor by reactive astro-cytes and associated neoangiogenesis. Brain Res 883: 87–97.
Sara VR, Carlsson-Skwirut C, Bergman T, Jorn-vall H, Roberts PJ, Crawford M, Hakansson LN, Civalero I, Nordberg A (1989): Identifi-cation of Gly-Pro-Glu (GPE), the aminoter-minal tripeptide of insulin-like growth fac-tor 1 which is truncated in brain, as a novel neuroactive peptide. Biochem Biophys Res Commun 165: 766–771.
Sara VR, Carlsson-Skwirut C, Drakenberg K, Giacobini MB, Hakansson L, Mirmiran M, Nordberg A, Olson L, Reinecke M, Stahlbom PA, et al. (1993): The biological role of trun-cated insulin-like growth factor-1 and the tripeptide GPE in the central nervous sys-tem. Ann N Y Acad Sci 692: 183–191.
Shankaran S, Laptook AR, Ehrenkranz RA, Ty-son JE, McDonald SA, Donovan EF, Fanaroff AA, Poole WK, Wright LL, Higgins RD, Fin-er NN, Carlo WA, Duara S, Oh W, Cotten CM, Stevenson DK, Stoll BJ, Lemons JA, Guillet R, Jobe AH (2005): Whole-body hy-pothermia for neonates with hypoxic-isch-emic encephalopathy. N Engl J Med 353:
1574–1584. Silveira RC, Procianoy RS (2003): Interleukin-6
and tumor necrosis factor-alpha levels in plasma and cerebrospinal f luid of term new-born infants with hypoxic-ischemic enceph-alopathy. J Pediatr 143: 625–629.
Sizonenko SV, Sirimanne ES, Williams CE, Gluckman PD (2001): Neuroprotective ef-fects of the N-terminal tripeptide of IGF-1, glycine-proline-glutamate, in the immature rat brain after hypoxic-ischemic injury. Brain Res 922: 42–50.
Smith CJ, Emsley HC, Gavin CM, Georgiou RF, Vail A, Barberan EM, del Zoppo GJ, Hallen-beck JM, Rothwell NJ, Hopkins SJ, Tyrrell PJ (2004): Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol 4: 2.
Wang X, Karlsson JO, Zhu C, Bahr BA, Hagberg H, Blomgren K (2001): Caspase-3 activation after neonatal rat cerebral hypoxia-isch-emia. Biol Neonate 79: 172–179.
Wheeler RD, Boutin H, Touzani O, Luheshi GN, Takeda K, Rothwell NJ (2003): No role for interleukin-18 in acute murine stroke-in-duced brain injury. J Cereb Blood Flow Metab 23: 531–535.
Yamamoto H, Murphy LJ (1995): Enzymatic conversion of IGF-I to des(1–3)IGF-I in rat serum and tissues: a further potential site of growth hormone regulation of IGF-I action. J Endocrinol 146: 141–148.
Yamashita T, Sawamoto K, Suzuki S, Suzuki N, Adachi K, Kawase T, Mihara M, Ohsugi Y, Abe K, Okano H (2005): Blockade of inter-leukin-6 signaling aggravates ischemic cere-bral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J Neurochem 94: 459–468.
Yuan J, Lipinski M, Degterev A (2003): Diversity in the mechanisms of neuronal cell death. Neuron 40: 401–413.
Zhu C, Qiu L, Wang X, Hallin U, Cande C, Kroe-mer G, Hagberg H, Blomgren K (2003): In-volvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J Neurochem 86: 306–317.
Zhu C, Wang X, Xu F, Bahr BA, Shibata M, Uchi-yama Y, Hagberg H, Blomgren K (2005): The influence of age on apoptotic and other mechanisms of cell death after cerebral hy-poxia-ischemia. Cell Death Differ 12: 162–176.

Fax +41 61 306 12 34E-Mail [email protected]
Dev Neurosci 2007;29:403–411 DOI: 10.1159/000105481
Antioxidant Status Alters Levels of Fas-Associated Death Domain-Like IL-1B-Converting Enzyme Inhibitory Protein following NeonatalHypoxia-Ischemia
Kurlen S.E. Payton
a R. Ann Sheldon
b Devin W. Mack
a Chainglian Zhu
c
Klas Blomgren
c Donna M. Ferriero
b Frances J. Northington
a
a Department of Pediatrics, Eudowood Neonatal Pulmonary Division, Neonatal Research Laboratory,
Johns Hopkins University School of Medicine, Baltimore, Md. , b Departments of Neurology and Pediatrics,
Neonatal Brain Disorders Laboratory, University of California San Francisco, San Francisco, Calif. , USA; c
Institute of Neuroscience and Physiology, Göteborg University, Göteborg , Sweden
expressing mice and significant differences between WT and SOD-overexpressing mice (ANOVA, p ! 0.01). There is no difference in FADD expression among the 3 groups 24 h after HI. At 24 h following HI, the ratio of FLIP to Fas DR ex-pression supports a significant negative correlation with in-jury score (r 2 = 0.99, slope = –4.01), and expression of both the active fragment of caspase 8 and caspase 8 activity is increased in SOD overexpressors compared to GPx overex-pressors at 24 h after HI (ANOVA, p ! 0.05). The overall de-gree of injury previously seen in these 3 strains correlates well with changes in expression of Fas DR signaling proteins favoring neuroprotection in the GPx-overexpressing mice, i.e. increased FLIP expression and decreased caspase 8 ac-tivity compared to SODtg mice. The mechanism by which antioxidant status alters FLIP levels following neonatal HI may be related to the ability to detoxify H 2 O 2 produced fol-lowing neonatal HI. Copyright © 2007 S. Karger AG, Basel
Key Words
Hypoxia-ischemia � Neonatal brain injury � Fas-associated death domain-like IL-1 � -converting enzyme inhibitory protein � Fas death receptor signaling � Superoxide dismutase � Glutathione peroxidase
Abstract
Activation of Fas death receptor (Fas DR) signaling cascade is seen after neonatal hypoxia-ischemia (HI). Cell survival is favored when signaling through the death-inducing signal-ing complex and cleavage of caspase 8 to its active form is blocked by FLIP, a dominant negative of caspase 8. H 2 O 2 quickly downregulates expression of FLIP. Neonatal mice overexpressing glutathione peroxidase (GPx) have less in-jury and less H 2 O 2 accumulation compared with neonatal mice overexpressing superoxide dismutase (SOD) or wild-type (WT) littermates. Expression of both FLIP L and FLIP S is increased in GPx-oxerexpressing mice relative to WT mice at 24 h and relative to SOD-overexpressing mice at 2 and 24 h following neonatal HI (ANOVA, p ! 0.05). There is an in-crease in Fas DR expression at 24 h in both WT and GPx-over-
Received: June 28, 2006 Accepted after revision: September 15, 2006
Frances J. Northington, MD Eudowood Neonatal Pulmonary Division Department of Pediatrics, CMSC 6-104, 600 N. Wolfe St. Baltimore, MD 21287 (USA) Tel. +1 410 955 4576, Fax +1 410 614 8388, E-Mail [email protected]
© 2007 S. Karger AG, Basel
Accessible online at:www.karger.com/dne

Payton /Sheldon /Mack /Zhu /Blomgren /Ferriero /Northington
Dev Neurosci 2007;29:403–411404
Introduction
Perinatal-neonatal brain injury as a result of hypoxia-ischemia (HI), stroke, or chorioamnionitis leads to cere-bral palsy and other neurodevelopmental abnormalities [Hagberg et al., 2002; Ferriero, 2004; Folkerth, 2005; Rees and Inder, 2005; Kirton and Deveber, 2006]. Hypoxia-ischemia, which accompanies most types of neonatal brain injury, triggers biochemical cascades that ultimate-ly lead to neuronal necrosis and apoptosis [McLean and Ferriero, 2004; Northington et al., 2005]. HI causes early accumulation of reactive oxygen species which cause oxi-dative damage of cellular lipids, proteins, and nucleic ac-ids [Peeters-Scholte et al., 2002; Ogihara et al., 2003; Cal-vert and Zhang, 2005; Marzocchi et al., 2005; Blomgren and Hagberg, 2006]. The antioxidant capacity of imma-ture neurons is easily overwhelmed by hypoxia-induced oxygen free radicals [Lievre et al., 2001] and oxidative stress-regulated release of pro-apoptotic factors from mi-tochondria plays a much more important role in injury in the immature brain [Blomgren and Hagberg, 2006]. In vitro studies reveal that hydrogen peroxide (H 2 O 2 ) is more toxic to immature murine neurons [Mischel et al., 1997], and the immature brain accumulates more H 2 O 2 after HI than the mature brain [Lafemina et al., 2006]. Studies have also shown that neonatal mice overexpressing hSOD are more vulnerable to HI injury than wild-type (WT) mice [Ditelberg et al., 1996] and neonatal mice overex-pressing GPx1 are protected from HI injury compared to WT mice [Sheldon et al., 2004]. In addition, in vitro stud-ies show that neurons from glutathione peroxidase trans-genic (GPxtg) mice are uniquely resistant to H 2 O 2 toxic-ity [McLean et al., 2005]. It has been hypothesized that the ability to effectively scavenge H 2 O 2 leads to differences in vulnerability to HI but how this translates into increased neuroprotection is poorly understood.
Reactive oxygen species also participate in Fas death receptor (DR)-mediated cell death [Banki et al., 1999; Gouaze et al., 2002; Sato et al., 2004], and Fas DR signal-ing plays a role in neurodegeneration following HI injury in the neonatal brain [Northington et al., 2001; Graham et al., 2004]. Fas DR propagates its signal via an intra-cellular death domain (DD) which recruits other DD-containing proteins to form a multimolecular complex, the death-inducing signal complex (DISC) [Peter and Krammer, 2003]. Formation of the DISC results in auto-cleavage of pro-caspase 8 to its active form [Peter and Krammer, 2003]. Fas-associated death domain-likeIL-1 � -converting enzyme inhibitory protein (FLIP), a DD-containing protein, may be recruited to the DISC.
Because FLIP is a pro-caspase homologue, it inhibits for-mation of active caspase 8, and caspase 8-mediated apop-tosis [Yang et al., 2005]. FLIP expression is strongly in-hibited in the presence of H 2 O 2 [Nitobe et al., 2003]; thus, it may be an upstream step in Fas DR signaling that can be regulated by antioxidant status. Therefore, we tested the hypothesis that neonatal mice overexpressing GPx (GPxtg) known to be neuroprotected from HI, have in-creased levels of FLIP relative to mice overexpressing su-peroxide dismutase (SODtg) and WT mice, both of which are more injured following HI.
Material and Methods
These animal studies were performed with approval by the Institutional Animal Care and Use Committee at the University of California San Francisco and carried out with standards of care and housing in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, U.S. Depart-ment of Health and Human Services 85-23, 1985.
Animal Models hSOD1 Transgenic Pups As previously described, WT CD1 mice were bred with hetero-
zygote transgenic mice with the human cytosolic SOD1 gene to yield transgenic and WT mice for SOD1 [Ditelberg et al., 1996]. The heterozygotes expressed threefold higher SOD activity. Pups were identified at P6 by sampling tail blood using qualitative non-denaturing gel electrophoresis followed by nitroblue tetrazolium staining for the human SOD protein as previously described [Ep-stein et al., 1987; Huang et al., 2002].
hGPx1 Transgenic Pups As previously described, GPxtg mice were obtained by breed-
ing WT CD1 mice with heterozygote human GPx1 mice [Sheldon et al., 2004]. Genotype was identified via tail clippings on P6.
Hypoxic-Ischemic Injury P7 mice were exposed to HI as previously described using the
Vannucci model [Rice et al., 1981; Ditelberg et al., 1996]. The mice were anesthetized with 2.5% halothane and 30% nitrous oxide. The right common carotid artery was exposed and ligated by elec-trical coagulation. After a recovery period with the dam, the mice were placed in separate but interconnected chambers (two per chamber) partially submerged in a 37 ° C water bath, through which flowed a humidified gas mixture containing 8% oxygen/balance nitrogen for 45 min. Genetically identical littermates were exposed to an identical procedure except that the carotid artery was not ligated and they were not exposed to hypoxia to serve as sham controls. Temperature was monitored by rectal probe (Physitemp) during the procedure to maintain a tempera-ture of 36–37 ° C.
Sample Preparation and Immunoblotting Cortical samples were obtained at 2 and 24 h following the end
of hypoxia or sham hypoxia. Mice were killed by rapid decapita-

Antioxidant Status Alters FLIP Levels following Neonatal Hypoxia-Ischemia
Dev Neurosci 2007;29:403–411 405
tion, brains were removed and ipsilateral cortex was separated from the remainder of the brain and immediately frozen in iso-pentane (–30 ° C). In order to have enough protein for subcellular fractionation, samples from genetically identical mice were pooled. Because of the small size of ipsilateral cortex samples, samples from 6–8 mice were pooled. Although originally the samples were separated by gender, sample size requirement was such that male and female samples had to be combined. Mem-brane-enriched and soluble protein fractions were prepared as previously described [Northington et al., 1996, 2001]. Twenty mi-crograms of membrane-enriched or soluble protein samples were electroeluted onto nitrocellulose membranes. Each blot contained paired sham and HI samples for each genotype so that the effect of hypoxia-ischemia on protein expression could be determined. Blots were performed to assess expression of both the long and short splice variants of FLIP (FLIP L , 55 kDa; FLIP S , 28 kDa), Fas DR, the p20 active fragment of caspase 8 and FADD. Blots were performed on samples obtained at 24 h after HI for each protein and additionally for FLIP L and FLIP S at 2 h after HI. Blots were repeated 3–6 times each. The membranes were stained with Pon-ceau S after transfer to evaluate the efficiency of sample loading and protein transfer. Blots were blocked with 2.5% nonfat dry milk with 0.1% Tween 20 in 50 m M Tris buffered saline, pH 7.4, and incubated overnight with primary antibody at 4 ° C. Blots were washed, exposed to secondary antibody, and developed with enhanced chemiluminescence. To quantitate protein loading, membranes were stripped (Pierce, Rockford, Ill., USA) and incu-bated again with anti- � actin. Blots were repeated 3–6 times each using all samples at least twice to confirm results.
Antibodies Anti-FLIP G-11 antibody (Santa Cruz Biotechnology, Inc.,
Santa Cruz, Calif., USA) is a mouse monoclonal antibody raised against amino acids 1–202 of FLIP of human origin. It recognizes both FLIP L and FLIP S 55 and 28 kDa, respectively. Anti-Fas C-20 antibody (Santa Cruz) is a rabbit polyclonal antibody raised against a peptide mapping near the carboxy terminus of human FAS. It recognizes a single band at 48 kDa. Jurkat cells known to express high levels of the Fas DR were used as a positive control. Anti-FADD, clone 1F7, antibody (Upstate, Lake Placid, N.Y., USA) is a mouse antibody raised against a full-length fusion protein corresponding to human FADD. It recognizes FADD at 28 kDa. Anti-caspase 8 antibody (Chemicon International, Inc., Temecu-la, Calif., USA) is a rabbit polyclonal antibody raised against a synthetic peptide of caspase 8. It recognizes pro-caspase 8, the intermediate cleavage products and the p18–20 active fragment of caspase 8. Anti- � actin antibody (Sigma, St. Louis Mo., USA) is a mouse monoclonal antibody and recognizes a single band. Anti-bodies were used in the following amounts for immunoblotting: anti-Fas 0.1 � g/ml, anti-FLIP 2 � g/ml, anti-FADD 0.5 � g/ml, anti-caspase 8 1 � g/ml and anti- � actin 0.13 � g/ml.
Caspase 8 Activity Assay Twenty-five microliters of cytosolic samples from pooled sam-
ples of SODtg and GPxtg, sham and 24 h post-HI mice were mixed with 75 � l of extraction buffer (50 m M Tris-HCl (pH 7.3), 100 m M NaCl, 5 m M EDTA, 1 m M EGTA, 3 m M NaN 3 , 1 m M PMSF, 1% protease inhibitor cocktail (Sigma) and 0.2% CHAPS on microtiter plates (Microfluor, Dynatech, Va., USA). After incubation for 15 min at room temperature, 100 � l of assay buffer (including 50 m M
Ac-IETD-AFC; Enzyme System Products, Livermore, Calif., USA) and 4 m M DTT, but without protease inhibitors or CHAPS were added. Cleavage of Ac-IETD-AFC was measured with an excita-tion wavelength of 400 nm and an emission wavelength of 505 nm, and expressed as pmol AFC released per mg protein and minute.
Analysis and Statistics To quantify protein immunoreactivity, films were scanned us-
ing Epson Scan, saved as TIFF files and optical densitometry (OD) performed with IP Lab Gel H software. The OD of � -actin in the corresponding lanes was used to correct the OD of the pro-tein immunoreactivity for differences in protein loading. To de-termine the effect of HI on caspase 8 activation, corrected OD for the p20 caspase 8 fragment from the HI sample was divided by corrected OD of the corresponding sham sample and this ratio compared across strains. To determine the effect of HI on expres-sion of FLIP, Fas DR, and FADD, corrected OD of the respective sham sample was subtracted from corrected OD of the HI sample of the same genotype and time point and the result divided by the corrected OD of the paired sham sample. This resulted in a ratio that if 1 0 meant there was an increase above sham levels in re-sponse to HI, or if ! 0 indicated that there was a decrease below corresponding sham levels in response to HI. This ratio is referred to subsequently as HI effect for Fas DR, FLIP and FADD proteins. HI effect for FLIP is calculated using the expression of both FLIP S and FLIP L . When expression of each of the splice variants was analyzed separately, results were the same as for total FLIP. HI/sham ratio or HI effect for each protein, at each time point, was compared across the three genotypes with ANOVA. When a sig-nificant difference was found, post-hoc testing with Student-Newman Keuls test was performed to identify specific differenc-es between genotypes. If data did not pass the test for being nor-mally distributed or if there were not enough samples to test for normal distribution, Kruskal-Wallis, nonparametric ANOVA, was utilized and post-hoc testing was performed with Dunn’s multiple comparisons test. Significance was set at p ! 0.05.
Results
Early and Sustained Increase in FLIP Expression in GPxtg Mice following Neonatal HI There is a significant difference between the mean HI
effect of WT, SODtg, and GPxtg at 2 and 24 h following HI. Mean HI effect is –0.11, –0.42, and 10.35 in WT, SOD-tg, and GPxtg (p = 0.001, n = 4) compared to their sham controls at 2 h after HI and 0.79, 0.14 and 2.84 in WT, SODtg, and GPxtg at 24 h after HI (p = 0.031, n = 6). Post-hoc testing reveals between-group differences in GPxtg and SODtg mice at 2 h and between GPxtg and WT and SODtg mice at 24 h after neonatal HI (p ! 0.05; fig. 1 a). Western blot of soluble protein samples from naïve WT, WT, SODtg, and GPxtg shams and WT, SODtg and GPxtg HI samples for FLIP L and FLIP S are shown ( fig. 1 b). The significant increase in FLIP expression is due to less FLIP expression in GPxtg shams and to a robust induc-

Payton /Sheldon /Mack /Zhu /Blomgren /Ferriero /Northington
Dev Neurosci 2007;29:403–411406
tion of FLIP expression following HI in GPxtg mice. The data showed that similar trends in expression of both FLIP splice variants occurred following HI injury. There-fore, data were combined from both FLIP L and FLIP S and presented as total FLIP expression in figure 1 a.
Differences in Fas DR Expression following HI; FLIP/Fas DR Ratios Are Consistent with Significant Neuroprotection in GPxtg Mice There is significant variation in HI effect between the
WT, SODtg and GPxtg mice in Fas DR expression at24 h following neonatal HI (p = 0.0009, n = 6). The HI effect means for Fas DR expression are 2.2, –0.17 and 1.07 in WT, SODtg, and GPxtg compared to their sham con-trols. Post-hoc testing reveals that Fas DR levels are lower in SODtg mice relative to WT (p ! 0.01; fig. 2 a). When Fas DR expression is combined with total FLIP expres-sion as a reflection of the balance between pro-death sig-naling and inhibition of death signaling, a ratio of pro- and anti-death signaling is created. The greater the ratio, the stronger the inhibitory component of the Fas signal-ing pathway is following neonatal HI. FLIP/Fas DR ex-pression ratios are 0.37 for WT mice, –0.82 for SODtg mice and 2.65 for GPxtg mice ( fig. 2 b). To determine if
the FLIP/Fas DR ratio is predictive of injury in the pres-ent model, the FLIP/Fas DR ratio versus previously pub-lished injury scores for p7 mice of the same genotypes was correlated [Ditelberg et al., 1996; Sheldon et al., 2004] ( fig. 2 c). The line of best fit shows a strong negative cor-relation between FLIP/Fas DR ratio and increasing injury score, r 2 = 0.99 and slope = –4.01.
Caspase 8 Activity and Cleavage of Pro-Caspase 8to Its Active Form Is Suppressed in GPxtg Mice Compared to SODtg Mice following Neonatal HI To determine if altered FLIP expression and FLIP/Fas
DR ratio have a functional significance, immunoblotting and caspase 8 activity assays were performed on pooled cytosolic samples from GPxtg and SODtg mice at 24 h after HI and compared to sham controls. There is sig-nificant variation in the HI to sham ratio between the WT, SODtg and GPxtg mice in expression of the p20 ac-tive fragment of caspase 8 at 24 h following neonatal HI (p = 0.0383, n = 5). The ratio of HI to sham expression of caspase 8 p20 is 1.13, 1.35 and 0.32 in WT, SODtg, and GPxtg. Post-hoc testing reveals that caspase 8 p20 is low-er in GPxtg mice compared to SODtg mice (p ! 0.05; fig. 3 a). Additionally, caspase 8 activity assay showed that
–5a
0
5
10
15
20
25
FLIP
HI e
ffec
t at
2 a
nd
24
h a
fter
HI
WT SOD* *
*
GPx
FLIPL
GPx HI
SOD HI
WT H
I
GPx Sham
SOD Sham
WT Sham
FLIPS
�-Actin
b
Fig. 1. a FLIP expression at 2 and 24 h after HI. There is a signifi-cant difference between the mean HI effect of WT, SODtg, and GPxtg at 2 and 24 h following HI. Mean HI effect is –0.11, –0.42, and 10.35 in WT, SODtg, and GPxtg, respectively (p = 0.001), at2 h after HI and 0.79, 0.14, and 2.84 in WT, SODtg, and GPxtg at 24 h after HI (p = 0.031). There are within-group differences be-tween GPxtg and SODtg mice at 2 h and between GPxtg and WT
and SODtg mice at 24 h after neonatal HI ( * p ! 0.05 vs. GPxtg). b Immunoblotting for FLIP S and FLIP L at 2 h after HI. In GPxtg, both FLIP splice variants (FLIP L – 55 kDa and FLIP S – 30 kDa) increase in expression after HI compared to sham control. In SODtg, there is decreased expression of both splice variants after HI compared to sham controls. � -Actin is used as a loading con-trol.

Antioxidant Status Alters FLIP Levels following Neonatal Hypoxia-Ischemia
Dev Neurosci 2007;29:403–411 407
–1
Fas
DR
HI e
ffec
t
a
0
1
2
3
4
5
WT
*
GPxSOD
–2FL
IP/F
as D
R ra
tio
–1
0
1
2
3
4
5
WT
b
GPxSOD 0
5
10
15
20
25
–1 0
c
1 2 3 4
SODtg
WT
GPxtg
FLIP/Fas DR ratio
Inju
ry s
core
Fig. 2. a Fas DR expression 24 h after HI. There is significant variation in the HI effect between WT, SODtg, and GPxtg mice (p = 0.0009). At 24 h, Fas DR expression is 2.2, –0.17 and 1.07 in WT, SODtg, and GPxtg, respectively, compared to their sham controls. There is greater Fas DR expression in WT mice relative to SODtg mice ( * p ! 0.01 vs. WT). b Ratio of FLIP/Fas DR at24 h after HI. Using protein expression data, FLIP to Fas DR ex-pression ratio was determined to assess the levels of FLIP and Fas DR in relation to one another. This ratio is 0.37 for WT, –0.82 for
SODtg, and 2.65 for GPxtg mice, indicating an abundance of FLIP protein in relation to Fas DR protein in GPxtg mice, more Fas DR protein than FLIP protein in SODtg mice and an intermediate ratio in WT mice. c Correlation between FLIP/Fas DR expression ratio and injury score. FLIP/Fas DR ratio was plotted versus in-jury score previously determined for WT, SOD, and GPxtg mice following neonatal HI. The line of best fit shows a strong inverse relationship between FLIP/FAS DR expression ratio and injury score, r 2 = 0.99, slope = –4.01.
0
0.5
1.0
1.5
2.0
2.5
WTa SOD GPx
*
Cas
pas
e 8
p20
exp
ress
ion
HI/
sham
rati
o
0b
5
10
15
20
25
Sham 24 h after HI
SODtg
GPxtg
Cas
pas
e 8
acti
vity
(pm
ole
AFC
rele
ased
per
min
/mg
pro
tein
–1)
Fig. 3. a Cleavage of pro-caspase 8 to its active form is suppressed in GPxtg mice compared to SODtg mice at 24 h after HI. Thereis significant variation in the HI/sham ratio between the WT, SODtg and GPxtg mice in expression of the p20 active fragment of caspase 8 at 24 h following neonatal HI (p = 0.0383, n = 5). The ratio of HI/sham expression of caspase 8 p20 is 1.13, 1.35, and 0.32 in WT, SODtg, and GPxtg, respectively. Post-hoc testing reveals that expression of caspase 8 p20 is lower in GPxtg mice compared
to SODtg mice (* p ! 0.05 vs. SODtg). b Caspase 8 activity is in-creased in SODtg at 24 h after HI. Caspase 8 activity detected as cleavage of Ac-IETD-AFC showed that there was no increase in caspase 8 activity in GPxtg mice at 24 h after HI compared to sham controls (11.1 vs. 12.0 pmole AFC/min/mg protein –1 ). Con-versely, in SODtg mice exposed to HI, caspase 8 activity was twice that found in SODtg sham controls (11.2 vs. 22.5 pmole AFC/min/mg protein –1 ).

Payton /Sheldon /Mack /Zhu /Blomgren /Ferriero /Northington
Dev Neurosci 2007;29:403–411408
there was no increase in caspase 8 cleavage activity in GPxtg mice at 24 h after HI compared to sham controls but in SODtg mice exposed to HI, caspase 8 cleavage ac-tivity doubled that found in the sham controls ( fig. 3 b).
FADD Expression at 24 h after HI Does Not Vary Based on Genetically Determined Antioxidant Status There is no difference in FADD HI effect among the 3
groups (p = 0.373, n = 3). The FADD HI effect is 0.024, –0.101, and 1.06 in WT SODtg, and GPxtg, respectively, compared to their sham controls ( fig. 4 ).
Discussion
This study is the first to investigate the Fas DR signal-ing pathway after HI in animals with altered antioxidant status. The most important finding of the present study is that antioxidant status alters the levels of three major proteins of the Fas DR signaling pathway following neo-natal HI, favoring pro-survival signaling in GPxtg mice and favoring pro-death signaling in WT and SODtg mice. These differences occur primarily because there is a marked increase in FLIP expression compared to sham controls in GPxtg mice compared to WT and SODtg mice at 2 h after HI, and this increase is sustained through
24 h after HI. In addition, Fas DR protein increases sig-nificantly in WT mice and the effect of the changes in Fas DR expression when combined with FLIP expression produces a rank order (GPxtg 1 1 WT 1 SODtg) exactly the inverse of the susceptibility to injury of the 3 strains, following neonatal HI. The suppression of caspase 8 ac-tivity in GPxtg mice demonstrates that enhanced expres-sion of FLIP in these mice results in a functional outcome favoring neuroprotection. FADD expression is not sig-nificantly altered by antioxidant status. These findings are the first to translate effects of reactive oxygen species on Fas DR signaling, previously shown only in vitro , to an in vivo model of neonatal brain injury and suggest an important mechanism of H 2 O 2 -mediated injury in the neonatal brain.
Although Fas DR expression and its signaling inter-mediates are known to be upregulated following neonatal HI [Northington et al., 2001; Graham et al., 2004], the exact mechanisms of Fas DR-mediated neuronal injury especially in relation to antioxidant status following brain injury is unknown. FLIP is a pro-caspase 8 homologue and impairs processing to active caspase 8. Active cas-pase 8 is the primary upstream executioner of Fas DR-mediated cell death [Peter and Krammer, 2003]. FLIP ex-ists as a long splice variant (FLIP L , 55 kDa) and a short splice variant (FLIP S , 28 kDa), and these splice variants block apoptosis via different mechanisms. FLIP S com-petes with caspase 8 for binding to FADD within the DISC, while FLIP L disrupts FADD self-association pre-venting higher-order oligomerization of DISC necessary for effective caspase 8 autocleavage [Yang et al., 2005].
The large ratio of HI to sham levels of FLIP at both 2 and 24 h following HI in mice overexpressing GPx dem-onstrates that this protein is highly inducible in response to stress. This finding is in good agreement with in vitro studies showing that there is a high turnover rate for FLIP. Metabolic inhibitors selectively downregulate FLIP expression in neuroblastoma and EL-4 thymoma cells within 2–4 h of exposure to cycloheximide or actinomy-cin D [Fulda et al., 2000; Kataoka et al., 2002]. The de-crease in FLIP levels in GPxtg mice at 24 compared to2 h may be explained by a high turnover rate for FLIP in an environment in which GPx activity falls dramatically between 2 and 24 h following neonatal HI, even in GPxtg animals [Fullerton et al., 1998]. Even though GPx activ-ity is markedly enhanced in GPxtg mice, the decrease in GPx activity between 2 and 24 h after HI is functionally manifest in loss of a protein, FLIP, that is short-lived and markedly sensitive to levels of H 2 O 2 .
–1
0
1
2
FAD
D H
I eff
ect
at 2
4 h 3
4
5
WT GPxSOD
Fig. 4. FADD HI effect at 24 h after HI. FADD expression at 24 h after HI is 0.024, –0.101, and 1.06 in WT, SODtg, and GPxtg mice, respectively, compared to their sham controls. No effect of geno-type on FADD expression following HI is found (p = 0.37).

Antioxidant Status Alters FLIP Levels following Neonatal Hypoxia-Ischemia
Dev Neurosci 2007;29:403–411 409
Reactive oxygen species have been repeatedly shown to be essential for Fas-mediated cell death in the immune system. Production of oxygen free radicals is linked to cross-talk between extrinsic and intrinsic apoptosis cas-cades via caspase 8-mediated activation of the BH 3 only pro-apoptotic bcl 2 family protein, Bid, and subsequent mitochondrial free radical leak, mitochondrial mem-brane permeability transition, and apoptosome forma-tion and to an early less well described upstream mecha-nism directly related to ligation of the Fas DR [Banki et al., 1999; Gouaze et al., 2002; Sato et al., 2004]. The im-portance of reactive oxygen species in Fas DR signaling has been verified in studies showing enhanced antioxi-dant status protects against Fas DR-mediated cell death. In addition to their free radical scavenging activity, vita-min C and GPx directly prevent caspase 8 activation [Per-ez-Cruz et al., 2003], and cellular glutathione content correlates directly with susceptibility to Fas DR-induced cell death [Banki et al., 1996; Chiba et al., 1996; Watson et al., 1997]. A positive relationship between FLIP expres-sion and antioxidant status is not found in all experimen-tal systems. For example, FLIP expression is not enhanced in human breast cancer cell lines protected from Fas DR injury by overexpression of GPx1 [Gouaze et al., 2002].
Hydrogen peroxide causes a decrease in FLIP expres-sion in neonatal rat ventricular myocytes [Nitobe et al., 2003; Yaniv et al., 2005] and sensitizes these normally resistant cells to Fas ligand-mediated apoptosis. Protec-tion against Fas-mediated cardiac myocyte cell death en-hanced by H 2 O 2 and other sources of oxygen free radicals is reversed with either N-acetylcysteine or a combination of SOD and catalase [Nitobe et al., 2003]. Based on these data, the hypothesis that the neuroprotection seen in GPxtg mice following neonatal HI may result from en-hanced FLIP expression because of their increased ability to scavenge H 2 O 2 compared with WT and SODtg mice was tested. The results support this hypothesis and sug-gest that the combined effect of antioxidant status on the ratio of FLIP/Fas DR expression is a predictor of injury susceptibility in these neonatal mouse strains. This con-clusion is strengthened by the caspase 8 cleavage activity data showing minimal change from sham at 24 h after HI in GPxtg mice compared to the increase seen after HI in SODtg mice. Further evidence that enhanced levels of FLIP block DR signaling is the decrease in expression of the active p20 fragment of caspase 8 at 24 h after HI in GPxtg mice compared to SODtg and WT mice.
Free radicals may also exert control over intracellular levels of FLIP via other mechanisms, such as nitric oxide inhibition of proteasome degradation, as has been shown
in bronchial epithelial cells where NO-liberating agents protected against FLIP downregulation and caspase 8 ac-tivation via S-nitrosylation of FLIP, making it resistant to ubiquitination and subsequent proteasomal degradation [Chanvorachote et al., 2005]. The role of proteasomal degradation of FLIP in the setting of altered antioxidant status was not investigated in the present study.
Levels of FLIP and Fas DR expression may also par-ticipate in determining regional vulnerability to injury following neonatal HI. The neonatal hippocampus sus-tains more severe injury than the thalamus and cortex [Ferriero et al., 1996; Sheldon et al., 2001; Graham et al., 2004]. The hippocampus has higher basal levels of Fas expression than either the thalamus or cortex, and it has minimal baseline expression of FLIP relative to cortical and thalamic samples. After HI injury, there is some up-regulation of Fas and FLIP in all regions [Graham et al., 2004]. Though expression of the Fas DR signaling inter-mediates was measured differently than in the present study, previous data suggest that a relative lack of FLIP expression in relation to Fas DR expression, especially at baseline, may cause regions to respond differently to HI. A regional investigation was not performed in this first study of the effects of altered antioxidant status on Fas DR signaling; however, this is likely to be an important area of study in fully understanding the selective vulner-ability of the neonatal brain to HI. It is unknown if there is a gender effect of antioxidant status on FLIP expres-sion because although gender was checked, the amount of tissue required for subcellular fractionation required pooling of brain tissue from both male and female mice.
Though FLIP levels alone provide the expected inverse correlation with injury score following neonatal HI, in-clusion of relative changes in Fas DR expression adds strength to this relationship, ( fig. 2 c). This relationship between the pro-death signaling caused by upregulation of Fas DR and pro-survival signaling of increased FLIP expression was shown directly in vitro in murine thy-moma cells [Kataoka et al., 2002] and can be inferred from our previous findings in C57/B6 neonatal mice [Graham et al., 2004]. Fas DR upregulation and recruit-ment to the cellular membrane appears to be under con-trol of p53 [Muller et al., 1998; Chandrasekaran and Richburg, 2005]. A recent study in neonatal mice has confirmed that p53 expression is robust and peaks early, 3–8 h following neonatal HI [Xu et al., 2006]. Thus, mechanisms are intact in the neonatal brain for the tran-scriptional and translocational upregulation of Fas DR expression, and in combination with rapidly acting mech-

Payton /Sheldon /Mack /Zhu /Blomgren /Ferriero /Northington
Dev Neurosci 2007;29:403–411410
anisms for regulation of FLIP expression [Fulda et al., 2000] provide for a highly responsive signaling mecha-nism which can either promote or inhibit cell death fol-lowing neonatal HI.
FADD expression is not altered by HI and there are no differences in FADD expression between the WT, SODtg and GPxtg mice in the present study. In the same group of experiments in which FLIP expression was rapidly downregulated by metabolic inhibitors, neither FADD nor pro-caspase 8 expression was affected by cyclohexi-mide or actinomycin D [Fulda et al., 2000]. Similarly, in cardiac myocytes FADD levels are stable, despite a fall in FLIP expression and enhanced cell death secondary to exposure to oxygen free radicals and Fas ligand [Nitobe et al., 2003]. Both structural units of the FADD, the DD and the death effector domain (DED) are resistant to pro-teolysis and overexpression of FADD-DED causes cell death via a ROS-based mechanism [Lee et al., 2000], making it unlikely that FADD protein would itself, be sensitive to H 2 O 2 toxicity. Based on the results of these in vitro studies and the current study, it is doubtful that ox-ygen free radicals alter Fas DR signaling through the DISC via changes in levels of FADD .
In summary, FLIP levels appear to be regulated by the oxidant status after HI injury. Specifically, FLIP expres-sion is upregulated more efficiently after HI injury in mice that are more capable of scavenging H 2 O 2 . SODtg mice accumulate H 2 O 2 after injury and this is associated with decreased expression of FLIP and increased caspase 8 cleavage and activity and may explain their increased vulnerability to neonatal HI. Conversely, GPxtg mice are protected from HI injury and exhibit greater levels of FLIP at 2 and 24 h after HI injury and less activation of caspase 8 to its active form. Further understanding of ad-ditional modes of antioxidant protection outside those of direct free radical scavenging such as the complex inter-action between antioxidant status, FLIP, and other Fas DR signaling proteins could lead to novel therapies for neonatal HI.
Acknowledgements
This study was supported by NS 45059 (F.J.N.), and NS 33997 (D.M.F.). The authors would like to thank Debbie Flock for expert technical assistance and Estelle Gauda for her insights in review of these data.
References
Banki K, Hutter E, Colombo E, Gonchoroff NJ, Perl A (1996): Glutathione levels and sensi-tivity to apoptosis are regulated by changes in transaldolase expression. J Biol Chem 271:
32994–33001. Banki K, Hutter E, Gonchoroff NJ, Perl A (1999):
Elevation of mitochondrial transmembrane potential and reactive oxygen intermediate levels are early events and occur indepen-dently from activation of caspases in Fas sig-naling. J Immunol 162: 1466–1479.
Blomgren K, Hagberg H (2006): Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic Biol Med 40:
388–397. Calvert JW, Zhang JH (2005): Pathophysiology
of an hypoxic-ischemic insult during the perinatal period. Neurol Res 27: 246–260.
Chandrasekaran Y, Richburg JH (2005): The p53 protein influences the sensitivity of testicu-lar germ cells to mono-(2-ethylhexyl) phthalate-induced apoptosis by increasing the membrane levels of Fas and DR5 and de-creasing the intracellular amount of c-FLIP. Biol Reprod 72: 206–213.
Chanvorachote P, Nimmannit U, Wang L, Steh-lik C, Lu B, Azad N, Rojanasakul Y (2005): Nitric oxide negatively regulates Fas CD95-induced apoptosis through inhibition of ubiquitin-proteasome-mediated degrada-tion of FLICE inhibitory protein. J Biol Chem 280: 42044–42050.
Chiba T, Takahashi S, Sato N, Ishii S, Kikuchi K (1996): Fas-mediated apoptosis is modulated by intracellular glutathione in human T cells. Eur J Immunol 26: 1164–1169.
Ditelberg JS, Sheldon RA, Epstein CJ, Ferriero DM (1996): Brain injury after perinatal hy-poxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr Res 39: 204–208.
Epstein CJ, Avraham KB, Lovett M, Smith S, El-roy-Stein O, Rotman G, Bry C, Groner Y (1987): Transgenic mice with increased Cu/Zn-superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc Natl Acad Sci U S A 84: 8044–8048.
Ferriero DM (2004): Neonatal brain injury. N Engl J Med 351: 1985–1995.
Ferriero DM, Holtzman DM, Black SM, Sheldon RA (1996): Neonatal mice lacking neuronal nitric oxide synthase are less vulmerable to hypoxic-ischemic injury. Neurobiol Dis 3:
64–71.
Folkerth RD (2005): Neuropathologic substrate of cerebral palsy. J Child Neurol 20: 940–949.
Fulda S, Meyer E, Debatin KM (2000): Metabol-ic inhibitors sensitize for CD95 (APO-1/Fas)-induced apoptosis by down-regulating Fas-associated death domain-like interleu-kin 1-converting enzyme inhibitory protein expression. Cancer Res 60: 3947–3956.
Fullerton HJ, Ditelberg JS, Chen SF, Sarco DP, Chan PH, Epstein CJ, Ferriero DM (1998): Copper/zinc superoxide dismutase trans-genic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Ann Neu-rol 44: 357–364.
Gouaze V, Andrieu-Abadie N, Cuvillier O, Mal-agarie-Cazenave S, Frisach MF, Mirault ME, Levade T (2002): Glutathione peroxidase-1 protects from CD95-induced apoptosis. J Biol Chem 277: 42867–42874.
Graham EM, Sheldon RA, Flock DL, Ferriero DM, Martin LJ, O’Riordan DP, Northington FJ (2004): Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol Dis 17: 89–98.

Antioxidant Status Alters FLIP Levels following Neonatal Hypoxia-Ischemia
Dev Neurosci 2007;29:403–411 411
Hagberg H, Peebles D, Mallard C (2002): Models of white matter injury: comparison of infec-tious, hypoxic-ischemic, and excitotoxic in-sults. Ment Retard Dev Disabil Res Rev 8:
30–38. Huang TT, Raineri I, Eggerding F, Epstein CJ
(2002): Transgenic and mutant mice for oxy-gen free radical studies. Methods Enzymol 349: 191–213.
Kataoka T, Ito M, Budd RC, Tschopp J, Nagai K (2002): Expression level of c-FLIP versus Fas determines susceptibility to Fas ligand-in-duced cell death in murine thymoma EL-4 cells. Exp Cell Res 273: 256–264.
Kirton A, Deveber G (2006): Cerebral palsy sec-ondary to perinatal ischemic stroke. Clin Perinatol 33: 367–386.
Lafemina MJ, Sheldon RA, Ferriero DM (2006): Acute hypoxia-ischemia results in hydrogen peroxide accumulation in neonatal but not adult mouse brain. Pediatr Res 59: 680–683.
Lee SW, Ko YG, Bang S, Kim KS, Kim S (2000): Death effector domain of a mammalian apoptosis mediator, FADD, induces bacterial cell death. Mol Microbiol 35: 1540–1549.
Lievre V, Becuwe P, Bianchi A, Bossenmeyer-Pourie C, Koziel V, Franck P, Nicolas MB, Dauca M, Vert P, Daval JL (2001): Intracel-lular generation of free radicals and modifi-cations of detoxifying enzymes in cultured neurons from the developing rat forebrain in response to transient hypoxia. Neuroscience 105: 287–297.
Marzocchi B, Perrone S, Paffetti P, Magi B, Bini L, Tani C, Longini M, Buonocore G (2005): Nonprotein-bound iron and plasma protein oxidative stress at birth. Pediatr Res 58:
1295–1299. McLean C, Ferriero D (2004): Mechanisms of
hypoxic-ischemic injury in the term infant. Semin Perinatol 28: 425–432.
McLean CW, Mirochnitchenko O, Claus CP, No-ble-Haeusslein LJ, Ferriero DM (2005): Overexpression of glutathione peroxidase protects immature murine neurons from ox-idative stress. Dev Neurosci 27: 169–175.
Mischel RE, Kim YS, Sheldon RA, Ferriero DM (1997): Hydrogen peroxide is selectively tox-ic to immature murine neurons in vitro. Neurosci Lett 231: 17–20.
Muller M, Wilder S, Bannasch D, Israeli D, Lehl-bach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH (1998): p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 188: 2033–2045.
Nitobe J, Yamaguchi S, Okuyama M, Nozaki N, Sata M, Miyamoto T, Takeishi Y, Kubota I, Tomoike H (2003): Reactive oxygen species regulate FLICE inhibitory protein (FLIP) and susceptibility to Fas-mediated apoptosis in cardiac myocytes. Cardiovasc Res 57: 119–128.
Northington FJ, Ferriero DM, Flock DL, Martin LJ (2001): Delayed neurodegeneration in neonatal rat thalamus after hypoxia-isch-emia is apoptosis. J Neurosci 21: 1931–1938.
Northington FJ, Graham EM, Martin LJ (2005): Apoptosis in perinatal hypoxic-ischemic brain injury: How important is it and should it be inhibited? Brain Res Brain Res Rev 50:
244–257. Northington FJ, Koehler RC, Traystman RJ,
Martin LJ (1996): Nitric oxide synthase 1 and nitric oxide synthase 3 protein expression is regionally and temporally regulated in fetal brain. Dev Brain Res 95: 1–14.
Ogihara T, Hirano K, Ogihara H, Misaki K, Hi-roi M, Morinobu T, Kim HS, Ogawa S, Ban R, Hasegawa M, Tamai H (2003): Non-pro-tein-bound transition metals and hydroxyl radical generation in cerebrospinal f luid of newborn infants with hypoxic ischemic en-cephalopathy. Pediatr Res 53: 594–599.
Peeters-Scholte C, Koster J, Veldhuis W, van den Tweel E, Zhu C, Kops N, Blomgren K, Bar D, van Buul-Offers S, Hagberg H, Nicolay K, van Bel F, Groenendaal F (2002): Neuropro-tection by selective nitric oxide synthase in-hibition at 24 h after perinatal hypoxia-isch-emia. Stroke 33: 2304–2310.
Perez-Cruz I, Carcamo JM, Golde DW (2003): Vitamin C inhibits FAS-induced apoptosis in monocytes and U937 cells. Blood 102:
336–343.
Peter ME, Krammer PH (2003): The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 10: 26–35.
Rees S, Inder T (2005): Fetal and neonatal ori-gins of altered brain development. Early Hum Dev 81: 753–761.
Rice JE, Vannucci RC, Brierley JB (1981): The in-fluence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9: 131–141.
Sato T, Machida T, Takahashi S, Iyama S, Sato Y, Kuribayashi K, Takada K, Oku T, Kawano Y, Okamoto T, Takimoto R, Matsunaga T, Takayama T, Takahashi M, Kato J, Niitsu Y (2004): Fas-mediated apoptosome formation is dependent on reactive oxygen species de-rived from mitochondrial permeability tran-sition in Jurkat cells. J Immunol 173: 285–296.
Sheldon RA, Hall JJ, Noble LJ, Ferriero DM (2001): Delayed cell death in neonatal mouse hippocampus from hypoxia-ischemia is nei-ther apoptotic nor necrotic. Neurosci Lett 304: 165–168.
Sheldon RA, Jiang X, Francisco C, Christen S, Vexler ZS, Tauber MG, Ferriero DM (2004): Manipulation of antioxidant pathways in neonatal murine brain. Pediatr Res 56: 656–662.
Watson RW, Rotstein OD, Jimenez M, Parodo J, Marshall JC (1997): Augmented intracellular glutathione inhibits Fas-triggered apoptosis of activated human neutrophils. Blood 89:
4175–4181. Xu FL, Zhu CL, Wang XY, Qiu L, Ji L, Cheng XY,
Luan B (2006): Expression of p53 in neonatal mice following hypoxia-ischemia and effects of its inhibitor on neonatal brain injury (in Chinese). Zhongguo Dang Dai Er Ke Za Zhi 8: 137–140.
Yang JK, Wang L, Zheng L, Wan F, Ahmed M, Lenardo MJ, Wu H (2005): Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol Cell 20: 939–949.
Yaniv G, Shilkrut M, Larisch S, Binah O (2005): Hydrogen peroxide predisposes neonatal rat ventricular myocytes to Fas-mediated apop-tosis. Biochem Biophys Res Commun 336:
740–746.

Fax +41 61 306 12 34E-Mail [email protected]
Author Index Vol. 29, No. 4–5, 2007
Holtzman, D.M. 363 Hutton, L.C. 341 Jin, Y. 373 Juul, S.E. 311 Kavelaars, A. 385 Kukekov, N.V. 355 Kuratani, J. 311 Levison, S.W. 279, 302, 331 Leviton, A. 280 Loladze, V. 302 Mack, D.W. 403 MacKendrick, W. 289 Mallard, C. 302, 393 Mathai, S. 393 McPherson, R.J. 311 McQuillen, P. 321 Meyer, J. 289 Mu, D. 321 Nijboer, C.H.A. 385 Northington, F.J. 403 Payton, K.S.E. 403
Altieri, S. 302 Atzeva, M. 363 Aylward, E. 311 Blomgren, K. 403 Bregman, J. 289 Brywe, K.G. 302 Burbacher, T.M. 311 Castillo-Melendez, M. 341 Chang, Y. 321 Dammann, O. 280 Derrick, M. 289 Drobyshevsky, A. 289 Ferriero, D.M. 321, 403 Gangoli, N. 302 Gonzalez, F.F. 321 Greene, L.A. 355 Groenendaal, F. 385 Guan, J. 393 Gustavsson, M. 393 Hagberg, H. 302, 393 Heijnen, C.J. 385
© 2007 S. Karger AG, Basel
Accessible online at:www.karger.com/dne
Prasad, P.V. 289 Richards, T. 311 Sheldon, R.A. 403 Silverman, A.-J. 373 Storey, P. 289 Svedin, P. 393 Tan, S. 279, 289 van Bel, F. 385 Vannucci, S.J. 373 Vexler, Z. 321 Walker, D.W. 341 Wang, X. 393 Wendland, M. 321 West, T. 363 Wilhelm, M. 355 Wood, T.L. 302 Xu, Z. 355 Yang, Z. 331 Zhang, R. 393 Zhu, C. 403
Subject Index Vol. 29, No. 4–5, 2007
Apoptosis 302, 355, 385 Astroglia 373 Blood brain barrier breakdown 341 Blood-brain barrier 373 Brain damage 280 – development 289 – injury 385 Caspase 3 341 – – activation 363 Causal inference 280 Causation 280 Cell fate 321 c-Jun N-terminal kinase 355 Cromolyn 373 Cytochrome c 385 Endothelia 373 Erythropoietin 311 Excitotoxicity 302 Fas death receptor signaling 403 Fas-associated death domain-like IL-1 � -
converting enzyme inhibitory protein 403
Fetus 280 Glutathione peroxidase 403 Glycine 2-methyl proline glutamate 393 Heat shock protein 70 385 Hypoxia 331 – -ischemia 289, 311, 385, 393, 403 Hypoxic-ischemic injury 363 Immature brain 280, 385 Insulin-like growth factor 302 Intraventricular hemorrhage 289 Ischemia 321, 331 Lipid peroxidation 341 Lipopolysaccharide 341 Microglia 373 Myelin 302 Neonatal brain 393 – – injury 280, 321, 403 – stroke 321 Neonate 385 Neurodevelopment 311 Neurogenesis 321 Neuronogenesis 331
Neuroprotection 385 Perinatal brain 280 – – damage 341 Periventricular leukomalacia 302 Polyphenols 363 Pomegranate juice 363 Premature infant 289 Rat brain 385 Resveratrol 363 Scaffold proteins 355 Stem cell(s) 321, 331 Striatum 331 Stroke 331 Superoxide dismutase 403 Trophic factors 302 Uteroplacental inflammation 341 White matter damage 302




















