
post-anesthetic recovery room registered nurses' experiences

Original Paper
Pharmacology 2005;74:65–78DOI: 10.1159/000083705
Local Anesthetic Procaine Protects RatPheochromocytoma PC12 Cells againstß-Amyloid-Induced Neurotoxicity
Laurent Lecanua,b Yao Wenguoa,b Jing Xua,b Janet Greesonc
Vassilios Papadopoulosa,b
aDepartment of Biochemistry and Molecular Biology and bSamaritan Research Laboratories,Georgetown University Medical Center, Washington, D.C., and cSamaritan Pharmaceuticals,Las Vegas, Nev., USA
Received: August 27, 2004Accepted after revision: December 6, 2004Published online: January 31, 2005
Dr. L. LecanuDepartment of Biochemistry and Molecular BiologyGeorgetown University Medical CenterWashington, DC 20057 (USA)Tel. +1 202 687 4625, Fax +1 202 687 7186, E-Mail [email protected]
ABCFax + 41 61 306 12 34E-Mail [email protected]
© 2005 S. Karger AG, Basel0031–7012/05/0742–0065$22.00/0
Accessible online at:www.karger.com/pha
Key WordsAlzheimer’s disease W Muscarinic receptor W Û1 receptor W
HMG-CoA reductase W Procaine W Glutamate
AbstractAlzheimer’s disease (AD) is the most common dementiaoccurring in elderly. We report herein the neuroprotec-tive properties of procaine and other anesthetic agentsagainst ß-amyloid-induced neurotoxicity. Procaine dis-played strong neuroprotective properties against theamyloid peptide Aß1–42 and preserved Aß1–42-inducedATP depletion on rat pheochromocytoma PC12 cells.Procaine also inhibited the neurotoxic effect that gluta-mate displayed on PC12 cells, suggesting that the reduc-tion of glutamate-induced neurotoxicity may be themechanism by which these compounds exert their ‘an-tiamyloid’ effects. In search of a mechanism of action weobserved that procaine is a ligand for the Û1 receptor, aprotein which ligands have been shown to protect mito-chondrial function and to exert antidepressant proper-ties. Procaine binds also to muscarinic receptors but thetrue meaning of this feature needs to be clarified. In con-clusion, these data suggest that procaine exerts neuro-
protective properties and may serve either as a treat-ment for AD or as a starting point for the development ofnovel therapies for AD.
Copyright © 2005 S. Karger AG, Basel
Introduction
Alzheimer disease (AD) neuropathology is histological-ly characterized by an increase of brain ß-amyloid (Aß)peptide levels accompanied by the formation of senileplaques [1] and the appearance of neurofibrillary tangles,due to a hyperphosphorylation of the Ù protein [2]. Aß isproduced by proteolytic cleavage of the ß-amyloid precur-sor protein (ß-APP) by the membrane enzymes ß- and Á-secretase. Aß exists either as the most commonly found40 amino acid length Aß1–40 form or the 42 amino acidAß1–42 form, reported to be more neurotoxic thanAß1–40. Although our understanding of Aß-mediated neu-rotoxicity has dramatically increased during the last de-cade, no Aß1–42 targeting therapeutic strategy has beenshown yet to successfully slow down the progression of thedisease.

66 Pharmacology 2005;74:65–78 Lecanu/Wenguo/Xu/Greeson/Papadopoulos
Procaine-containing formulations have been tested fordecades as a possible treatment of cognitive disorders ingeriatric subjects including depression [3]. Several possi-ble mechanisms mediating the effects of procaine havebeen studied, such as cerebral blood vessel dilation andmonoamine oxidase inhibition but none were really con-vincing. We recently demonstrated that procaine chlorhy-drate itself down-regulated the stressor-induced increaseof adrenal corticosteroid synthesis in vitro and in vivo inrats [4]. Such a stressor-induced unbalance of the hypo-thalamo-pituitary-adrenal axis has been described inmany diseases including AD [5, 6]. High concentrationsof cortisol have been reported to contribute to the neuro-degeneration that occurs in AD either by a direct effect onthe neuronal cells or by sensitizing them to Aß1–42 andglutamate neurotoxicity [7]. In addition, excessive serumconcentrations of cortisol have been associated with im-paired memory processes and suggested to trigger non-cognitive conditions like depression and mood variability[8]. The mechanism by which procaine reduced cortico-sterone blood level involves a reduction of the cholesterolsynthesis consecutive to the reduction of the rate-limitingenzyme HMG-CoA reductase mRNA expression [4], pre-senting therefore procaine as an interesting new approachto treat AD pathology. In addition, procaine and othercompounds members of the local anesthetic family ofdrugs have been shown to regulate calcium homeostasis[9]. Dysregulation of calcium homeostasis has been alsoproposed to be responsible, at least in part, for the onset ofAD [10]. Indeed, Aß has been described to alter many cal-cium-controlling systems eliciting an important rise of theintracellular calcium concentration in various cell types.There is also evidence about the alteration of the gluta-matergic transmission [11] and an overactivation of thevoltage-dependent calcium channels type [12], possiblythrough the direct activation of the G protein-coupledextracellular Ca2+-sensing receptor [13]. Local anestheticswere shown to exhibit neuroprotective properties in vivo,during cerebral ischemia in gerbils [14], and in vitro, dur-ing a hypoxic episode in hippocampal neurons [9, 15].Concomitantly, procaine and lidocaine have been showedto inhibit NMDA receptor activity [16], suppress theanoxia-induced increase of the intracellular calcium con-centration in gerbil hippocampus [9] and prevent the isch-emia-triggered increase of extracellular glutamate concen-tration in gerbil brain [14].
These considerations led us to assess the neuroprotec-tive properties of procaine against Aß toxicity and toattempt to identify the mechanisms underlying the neuro-protective action of procaine.
Methods and Materials
MaterialsAß1–42 peptide was purchased from American Peptide Co. (Sun-
nyvale, Calif., USA). Procaine, tetracaine, lidocaine, procainamide,the antioxidant tert-butyl-phenylnitrone (PBN), the N-methyl-D-aspartate (NMDA) receptor antagonist (+)-MK801, ryanodine andtetrodotoxin (TTX) were purchased from Sigma (St. Louis, Mo.,USA). Cell culture supplies were purchased form Gibco (GrandIsland, N.Y., USA) and cell culture plasticware was from Corning(Corning, N.Y., USA) and Packard BioSciences Co. (Meriden,Conn., USA). RNA STAT-60 was from Tel-Test, Inc. (Friendswood,Tex., USA). TaqMan® Reverse Transcription Reagents, randomhexamers, and SYBR® Green PCR Master Mix were from AppliedBiosystems (Foster City, Calif., USA).
Cell Culture and TreatmentsPC12 cells (rat pheochromocytoma) (ATCC, Manassas, Va.,
USA) were cultured in RPMI 1640 without glutamine medium con-taining 10% of fetal bovine serum and 5% of horse serum at 37°Cand 5% CO2. PC12 cells were incubated for 24 h in 96-well plates(5 ! 104 cells per well) with increasing concentrations (1, 10 and100 Ìmol/l) of procaine, procainamide, lidocaine or tetracaine.Aß1–42 was incubated overnight at 4 °C and then added to the cells at0.1, 1 or 10 Ìmol/l final concentrations for a 24-hour time period.The different treatments have been added to the complete mediumused to culture cells. The rationale for having chosen a 24-hour pre-incubation period with the local anesthetics was that a pre-incuba-tion was necessary to observe a reduction of the HMG-CoA reduc-tase activity. Therefore, to assess whether an eventual metaboliceffect was involved in the neuroprotective activity of procaine andother local anesthetics, we chose to pre-incubate these compoundsrather than adding them just before Aß1–42.
To study the role played by the NMDA receptor in the Aß1–42-induced neurotoxicity, increasing concentrations of the NMDAreceptor antagonist (+)-MK801 were added to the cell media imme-diately before Aß1–42. Cell viability was assessed 4 h later as describedbelow. To assess the effect of procaine on the glutamate-inducedexcitotoxicity, PC12 cells were pre-treated with 0.3, 1, 3, 10 and30 Ìmol/l procaine for 24 h and then submitted to glutamate expo-sure for another 24-hour time period. Cell viability was subsequentlyassessed. To assess the role of sodium channels in Aß1–42-inducedneurotoxicity, PC12 cells were incubated for 4 h with 3, 30 or300 Ìmol/l of the sodium-channel blocker TTX followed by additionof Aß1–42. The involvement of the oxidative stress in the toxicity ofAß1–42 was assessed by incubating the PC12 in presence of 10, 100 or500 Ìmol/l PBN for 24 h. Aß1–42 was then added to the incubationmedia. Cell viability was assessed by 24 h later.
Cell Viability DeterminationThe cellular toxicity of Aß was assessed using the 3-(4,5-dimethyl-
thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Trevi-gen, Gaithersburg, Md., USA) as previously described [17]. Briefly,10 Ìl of the MTT solution was added to the cells cultured in 100 Ìl ofmedium. After an incubation period of 4 h using the same conditionsas described above, 100 Ìl of detergent were added and cells incu-bated overnight at 37°C. The blue color formation was quantified at600 and 690 nm using the Victor spectrophotometer (EGG-Wallac,Gaithersburg, Md., USA) and DO600–DO690 was quantified. To com-pare the protective effect of the compounds tested, the decrease of

Procaine in Neuroprotection against Aß Pharmacology 2005;74:65–78 67
MTT signal observed with Aß1–42 was considered to be the 100%inhibition of the NADPH diaphorase activity and the effect of thecompounds tested is shown as an increase or decrease of this percent-age.
ATP MeasurementATP concentrations were measured using the ATPLite-MTM
assay (Packard BioSciences Co.), as previously described [17]. Inbrief, cells were cultured on black 96-well ViewPlateTM and the ATPconcentrations measured on a TopCount NXTTM counter (PackardBioSciences Co.) according to the manufacturer’s recommendations.The effect of Aß1–42 was expressed in arbitrary units. To compare thepotential protective effect of the compounds tested on ATP recovery,the decrease of ATP concentration induced by Aß1–42 was consideredto be the 100% reduction and the effects of the compounds tested areshown as changes of this percentage.
Free Radical ProductionOxidative stress was assessed by measuring the free radical pro-
duction using the fluorescent probe di-hydroxy di-chlorofluoresceindiacetate (2,7-DCF) (Molecular Probes, Eugene, Oreg., USA), as pre-viously described [17]. For these experiments, cells were cultured inpolylysine-coated microplates. Cells were washed once with RPMI1640 and medium was then replaced by 100 Ìl RPMI 1640. Cellswere incubated 45 min at room temperature in the dark with 100 Ìlof 2,7-DCF 50 Ìmol/l and the fluorescence (excitation Ï = 485 nm,emission Ï = 535 nm) was measured using the Victor multilabelcounter (EGG-Wallac).
Radioligand Binding StudiesÛ1 and Û2 Receptors. Radioligand binding studies were per-
formed using human recombinant Û1 receptor expressed in Jurkatcells. Increasing concentrations of procaine ranging from 3 ! 10–10
to 1 ! 10–5 mol/l were incubated for 120 min at 22°C in presence ofthe specific Û1-receptor ligand [3H]-(+)-pentazocine at 8 nmol/l todetermine procaine IC50 and Hill value nH. The Û2 receptor bindingstudy was performed using Û2 receptors from rat cerebral cortex.Procaine was incubated in presence of the Û2 receptor agonist [3H]-1,3-di(2-tolyl)guanidine 5 nmol/l with pentazocine 300 nmol/l toblock the Û1 receptors.
·-Bungarotoxin-Insensitive Nicotinic Receptors. Procaine was in-cubated for 75 min at 4°C on rat cerebral cortex ·-bungarotoxin(·-BGTX)-insensitive nicotinic receptor in presence of 1.5 nmol/l[3H]-cytisine.
Muscarinic (Non-Selective) Receptor. Procaine was incubated for120 min at 22°C on rat cerebral cortex non-selective receptor in pres-ence of 0.05 nmol/l [3H]-quinuclidinyl benzilate (QNB).
Central Imidazoline I2 Receptor. Procaine was incubated for30 min at 22°C on rat cerebral cortex ss receptor in presence of2 nmol/l [3H]-idazoxan.
Real-Time Quantitative RT-PCR (Q-PCR)PC12 cells cultured in 6-well plates for 18 h were treated with
increasing concentrations of procaine for the indicated time period.After treatment, cells were exposed to Aß1–42 1 Ìmol/l for 24 h. At theend of the incubation, total cell RNA was extracted using RNASTAT-60 (Tel-Test Inc, Friendswood, Tex., USA) according to themanufacturer’s instructions. HMG-CoA reductase mRNA was quan-tified by Q-PCR using the ABI Prism 7700 sequence detection sys-tem (Perkin-Elmer/Applied Biosystems, Foster City, Calif., USA).
RT reaction was performed using TaqMan® Reverse TranscriptionReagents with 1 Ìg total RNA and random hexamers as primers foreach reaction, as we previously described [4]. For quantifying ratHMG-CoA reductase mRNA with Q-PCR, the primers were de-signed according to GenBank Accession No. BC 019782 using PE/AB Primer Express software, which is specifically designed for theselection of primers and probes. The forward primer was 5)-GACTGT GGT TTG TGA AGC TGT CAT-3) (24 nucleotides) andreverse primer was 5)-AAT ACT TCT CTC ACC ACC TTG GCT-3)(24 nucleotides), respectively. The primers were synthesized by Bio-Synthesis Inc. (Lewisville, Tex., USA). Reactions were performed ina reaction mixture consisting of a 20-Ìl solution containing 10 ÌlSYBR® Green PCR Master Mix and 1 Ìl primers mix (5 Ìmol/l each)with 2 Ìl cDNA. The cycling conditions were: 15 s at 95°C and 1 minat 60°C for 40 cycles following an initial step of 2 min at 50°C and10 min at 95 °C. AmpliTaq Gold polymerase was activated at 95°Cfor 10 min. The 18S RNA was amplified at the same time and used asan internal control. To exclude the contamination of unspecific PCRproducts such as primer dimers, a melting curve analysis was appliedto all final PCR products after the cycling protocol. Also, PCR reac-tions without the RT reaction were performed for each sample inorder to exclude genomic DNA contamination. The PCR productswere collected and run on a 3% (w/v) agarose/TAE gel to confirm theproduct size. The threshold cycle (Ct) values for 18S RNA and sam-ples were calculated using the PE/AB computer software. Ct wasdetermined at the most exponential phase of the reaction. Relativetranscript levels were calculated as x = 2¢¢Ct, in which ¢¢Ct =¢E – ¢C, and ¢E = Ct experiment – Ct 18S, ¢C = Ct control – Ct 18s.
Statistical AnalysisData are expressed as mean B SD. Data obtained were assessed
between experimental groups by a one-way ANOVA and Dunnett’stest was used for comparison. A difference was considered significantwhen p ! 0.05.
Results
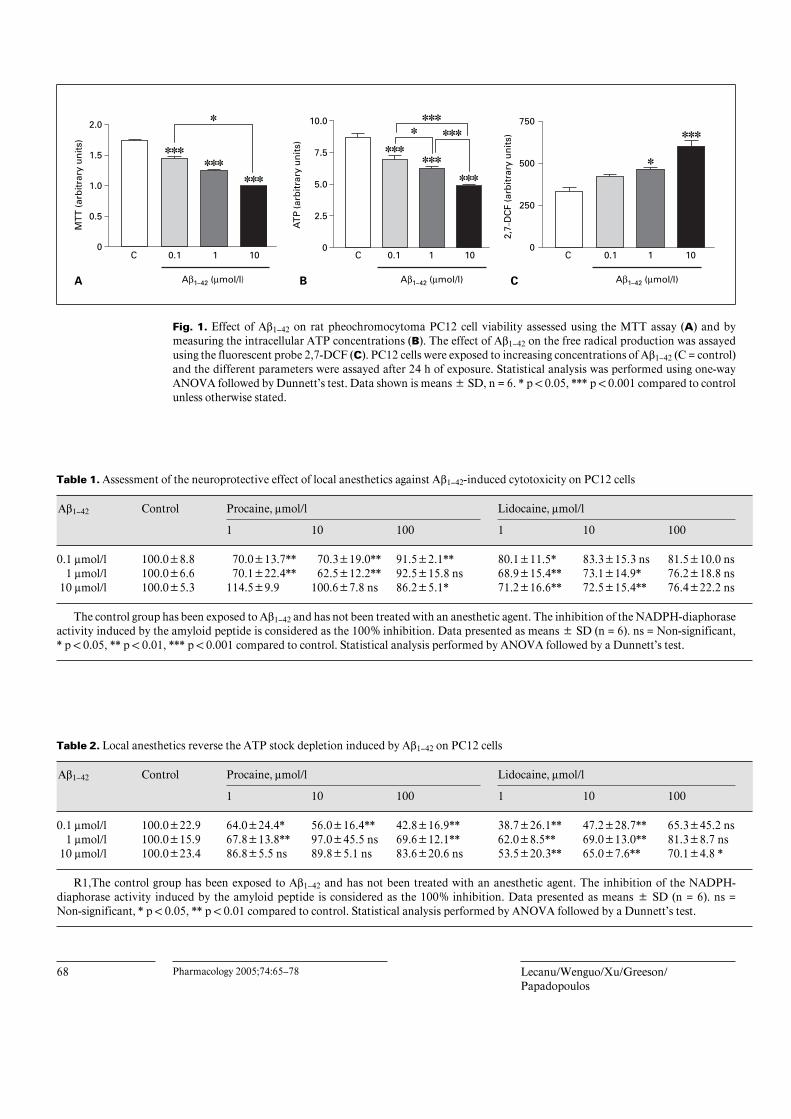
Aß1–42 Neurotoxicity Assessed by MTT Assay, ATPMeasurement and Free Radical Production in PC12Cells (fig. 1)Aß1–42 induces a dose-dependent decrease of PC12 cell
viability (p ! 0.001) (fig. 1A) and of the intracellular ATPconcentrations (p ! 0.001) (fig 1B). A dose-dependentrelationship is also observed on the free radical produc-tion as Aß1–42 at 1 and 10 Ìmol/l concentrations induced asignificant increase of the oxidative stress (p ! 0.05 andp ! 0.001 respectively) (fig. 1C).
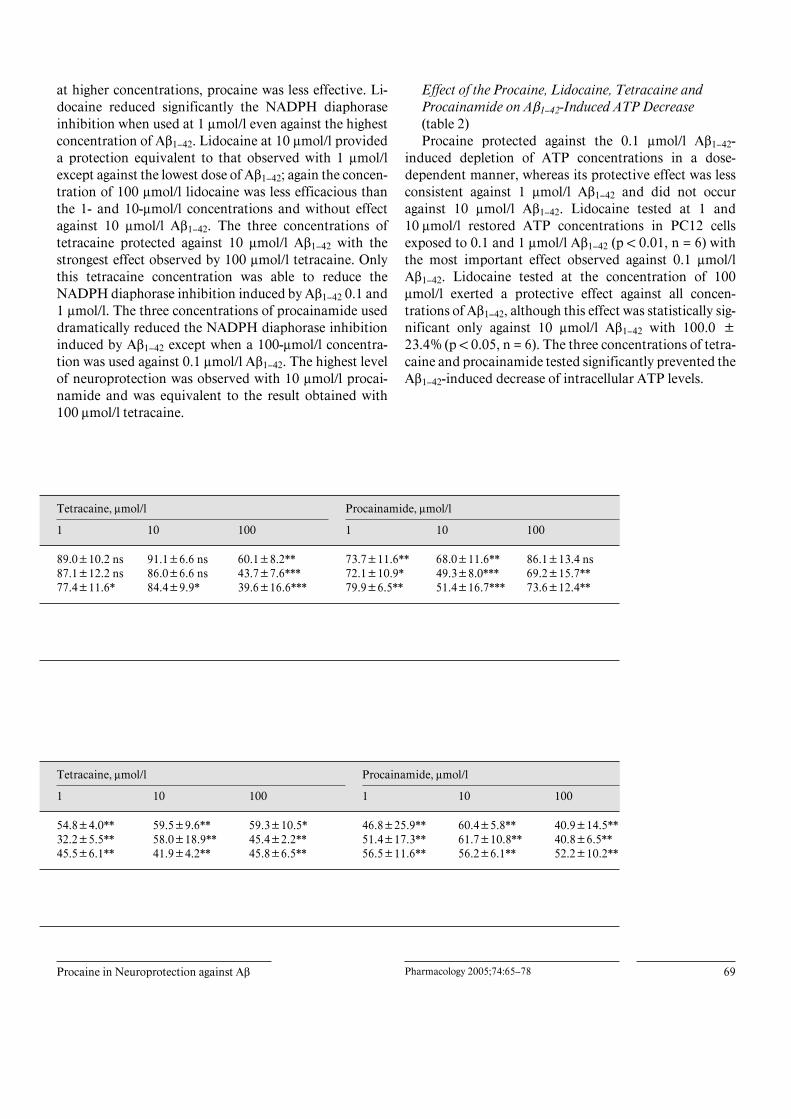
Effect of Local Anesthetics on Aß1–42-InducedNeurotoxicity (table 1)Procaine displays an important protective effect
against 0.1 and 1 Ìmol/l Aß1–42-induced toxicity assessedusing the MTT assay. Treatment with 1 and 10 Ìmol/lprocaine resulted in a reduction of the NADPH diapho-rase inhibition induced by Aß1–42 at least 30% (p ! 0.01);
68 Pharmacology 2005;74:65–78 Lecanu/Wenguo/Xu/Greeson/Papadopoulos
Fig. 1. Effect of Aß1–42 on rat pheochromocytoma PC12 cell viability assessed using the MTT assay (A) and bymeasuring the intracellular ATP concentrations (B). The effect of Aß1–42 on the free radical production was assayedusing the fluorescent probe 2,7-DCF (C). PC12 cells were exposed to increasing concentrations of Aß1–42 (C = control)and the different parameters were assayed after 24 h of exposure. Statistical analysis was performed using one-wayANOVA followed by Dunnett’s test. Data shown is means B SD, n = 6. * p ! 0.05, *** p ! 0.001 compared to controlunless otherwise stated.
0
0.5
1.0
1.5
2.0
✽✽✽
✽✽✽
✽✽✽
✽
MT
T (a
rbit
rary
un
its)
0
2.5
5.0
7.5
10.0
✽✽✽✽✽✽
✽✽✽
✽✽✽
✽✽✽
✽
AT
P (a
rbit
rary
un
its)
C 0.1 1 100
250
500
750✽✽✽
2,7–
DC
F (a
rbit
rary
un
its)
✽
Aβ1–42 (µmol/l)
C 0.1 1 10
Aβ1–42 (µmol/l)
C 0.1 1 10
Aβ1–42 (µmol/l)A B C
Table 1. Assessment of the neuroprotective effect of local anesthetics against Aß1–42-induced cytotoxicity on PC12 cells
Aß1–42 Control Procaine, Ìmol/l
1 10 100
Lidocaine, Ìmol/l
1 10 100
0.1 Ìmol/l 100.0B8.8 70.0B13.7** 70.3B19.0** 91.5B2.1** 80.1B11.5* 83.3B15.3 ns 81.5B10.0 ns1 Ìmol/l 100.0B6.6 70.1B22.4** 62.5B12.2** 92.5B15.8 ns 68.9B15.4** 73.1B14.9* 76.2B18.8 ns
10 Ìmol/l 100.0B5.3 114.5B9.9 100.6B7.8 ns 86.2B5.1* 71.2B16.6** 72.5B15.4** 76.4B22.2 ns
The control group has been exposed to Aß1–42 and has not been treated with an anesthetic agent. The inhibition of the NADPH-diaphoraseactivity induced by the amyloid peptide is considered as the 100% inhibition. Data presented as means B SD (n = 6). ns = Non-significant,* p ! 0.05, ** p ! 0.01, *** p ! 0.001 compared to control. Statistical analysis performed by ANOVA followed by a Dunnett’s test.
Table 2. Local anesthetics reverse the ATP stock depletion induced by Aß1–42 on PC12 cells
Aß1–42 Control Procaine, Ìmol/l
1 10 100
Lidocaine, Ìmol/l
1 10 100
0.1 Ìmol/l 100.0B22.9 64.0B24.4* 56.0B16.4** 42.8B16.9** 38.7B26.1** 47.2B28.7** 65.3B45.2 ns1 Ìmol/l 100.0B15.9 67.8B13.8** 97.0B45.5 ns 69.6B12.1** 62.0B8.5** 69.0B13.0** 81.3B8.7 ns
10 Ìmol/l 100.0B23.4 86.8B5.5 ns 89.8B5.1 ns 83.6B20.6 ns 53.5B20.3** 65.0B7.6** 70.1B4.8 *
R1,The control group has been exposed to Aß1–42 and has not been treated with an anesthetic agent. The inhibition of the NADPH-diaphorase activity induced by the amyloid peptide is considered as the 100% inhibition. Data presented as means B SD (n = 6). ns =Non-significant, * p ! 0.05, ** p ! 0.01 compared to control. Statistical analysis performed by ANOVA followed by a Dunnett’s test.
Procaine in Neuroprotection against Aß Pharmacology 2005;74:65–78 69
at higher concentrations, procaine was less effective. Li-docaine reduced significantly the NADPH diaphoraseinhibition when used at 1 Ìmol/l even against the highestconcentration of Aß1–42. Lidocaine at 10 Ìmol/l provideda protection equivalent to that observed with 1 Ìmol/lexcept against the lowest dose of Aß1–42; again the concen-tration of 100 Ìmol/l lidocaine was less efficacious thanthe 1- and 10-Ìmol/l concentrations and without effectagainst 10 Ìmol/l Aß1–42. The three concentrations oftetracaine protected against 10 Ìmol/l Aß1–42 with thestrongest effect observed by 100 Ìmol/l tetracaine. Onlythis tetracaine concentration was able to reduce theNADPH diaphorase inhibition induced by Aß1–42 0.1 and1 Ìmol/l. The three concentrations of procainamide useddramatically reduced the NADPH diaphorase inhibitioninduced by Aß1–42 except when a 100-Ìmol/l concentra-tion was used against 0.1 Ìmol/l Aß1–42. The highest levelof neuroprotection was observed with 10 Ìmol/l procai-namide and was equivalent to the result obtained with100 Ìmol/l tetracaine.
Effect of the Procaine, Lidocaine, Tetracaine andProcainamide on Aß1–42-Induced ATP Decrease(table 2)Procaine protected against the 0.1 Ìmol/l Aß1–42-
induced depletion of ATP concentrations in a dose-dependent manner, whereas its protective effect was lessconsistent against 1 Ìmol/l Aß1–42 and did not occuragainst 10 Ìmol/l Aß1–42. Lidocaine tested at 1 and10 Ìmol/l restored ATP concentrations in PC12 cellsexposed to 0.1 and 1 Ìmol/l Aß1–42 (p ! 0.01, n = 6) withthe most important effect observed against 0.1 Ìmol/lAß1–42. Lidocaine tested at the concentration of 100Ìmol/l exerted a protective effect against all concen-trations of Aß1–42, although this effect was statistically sig-nificant only against 10 Ìmol/l Aß1–42 with 100.0 B23.4% (p ! 0.05, n = 6). The three concentrations of tetra-caine and procainamide tested significantly prevented theAß1–42-induced decrease of intracellular ATP levels.
Tetracaine, Ìmol/l
1 10 100
Procainamide, Ìmol/l
1 10 100
89.0B10.2 ns 91.1B6.6 ns 60.1B8.2** 73.7B11.6** 68.0B11.6** 86.1B13.4 ns87.1B12.2 ns 86.0B6.6 ns 43.7B7.6*** 72.1B10.9* 49.3B8.0*** 69.2B15.7**77.4B11.6* 84.4B9.9* 39.6B16.6*** 79.9B6.5** 51.4B16.7*** 73.6B12.4**
Tetracaine, Ìmol/l
1 10 100
Procainamide, Ìmol/l
1 10 100
54.8B4.0** 59.5B9.6** 59.3B10.5* 46.8B25.9** 60.4B5.8** 40.9B14.5**32.2B5.5** 58.0B18.9** 45.4B2.2** 51.4B17.3** 61.7B10.8** 40.8B6.5**45.5B6.1** 41.9B4.2** 45.8B6.5** 56.5B11.6** 56.2B6.1** 52.2B10.2**
70 Pharmacology 2005;74:65–78 Lecanu/Wenguo/Xu/Greeson/Papadopoulos
0 0.3 1 10 301.1
1.2
1.3
1.4
MT
T a
bso
rban
ce(D
O60
0–D
O69
0) ✽✽✽✽✽✽✽✽✽
✽✽✽
Procaine (µmol/l)
Glutamate 100 (µmol/l)MK801 (µmol/l)
Control Control0 1 5 10 25 1001.0
1.2
1.4
1.6
1.8
2.0
2.2✽✽✽
✽✽✽
✽✽✽✽
✽
MT
T a
bso
rban
ce(D
O60
0–D
O69
0)
0
50
100
✽✽✽
Aβ1–42
1 (µmol/l)
NA
DP
H d
iap
ho
rase
act
ivit
y(%
inh
ibit
ion
)
Aβ 0.1 µmol/lAβ 1 µmol/lAβ 10 µmol/l
ControlProcaine 1 µmol/lProcaine 1 µmol/l 1 MK801 100 µmol/l
A
C
B
Fig. 2. Protective effect of the non-competitive NMDA antagonist(+)-MK801 against Aß1–42 neurotoxicity (A) and neuroprotectiveeffect of procaine against glutamate-induced cell death on PC12 cells(B). A PC12 cells were pre-incubated for 24 h with increasing concen-trations of (+)-MK801 before being exposed for 24 h to increasingconcentrations of Aß1–42. B PC12 cells were pre-incubated withincreasing concentrations of procaine for 24 h before being exposedto 100 Ìmol/l glutamate for 24 h. Cell viability was assessed using theMTT assay. Control cells treated with vehicle only. C Interestingly,
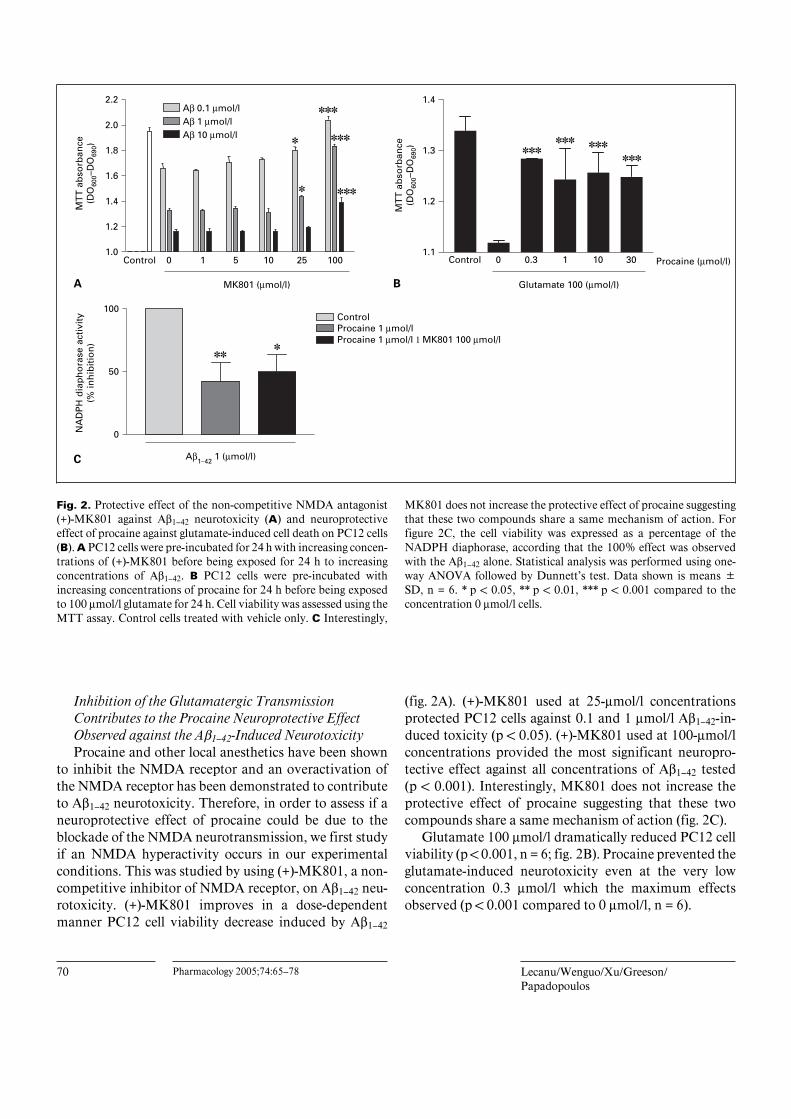
MK801 does not increase the protective effect of procaine suggestingthat these two compounds share a same mechanism of action. Forfigure 2C, the cell viability was expressed as a percentage of theNADPH diaphorase, according that the 100% effect was observedwith the Aß1–42 alone. Statistical analysis was performed using one-way ANOVA followed by Dunnett’s test. Data shown is means BSD, n = 6. * p ! 0.05, ** p ! 0.01, *** p ! 0.001 compared to theconcentration 0 Ìmol/l cells.
Inhibition of the Glutamatergic TransmissionContributes to the Procaine Neuroprotective EffectObserved against the Aß1–42-Induced NeurotoxicityProcaine and other local anesthetics have been shown
to inhibit the NMDA receptor and an overactivation ofthe NMDA receptor has been demonstrated to contributeto Aß1–42 neurotoxicity. Therefore, in order to assess if aneuroprotective effect of procaine could be due to theblockade of the NMDA neurotransmission, we first studyif an NMDA hyperactivity occurs in our experimentalconditions. This was studied by using (+)-MK801, a non-competitive inhibitor of NMDA receptor, on Aß1–42 neu-rotoxicity. (+)-MK801 improves in a dose-dependentmanner PC12 cell viability decrease induced by Aß1–42
(fig. 2A). (+)-MK801 used at 25-Ìmol/l concentrationsprotected PC12 cells against 0.1 and 1 Ìmol/l Aß1–42-in-duced toxicity (p ! 0.05). (+)-MK801 used at 100-Ìmol/lconcentrations provided the most significant neuropro-tective effect against all concentrations of Aß1–42 tested(p ! 0.001). Interestingly, MK801 does not increase theprotective effect of procaine suggesting that these twocompounds share a same mechanism of action (fig. 2C).
Glutamate 100 Ìmol/l dramatically reduced PC12 cellviability (p ! 0.001, n = 6; fig. 2B). Procaine prevented theglutamate-induced neurotoxicity even at the very lowconcentration 0.3 Ìmol/l which the maximum effectsobserved (p ! 0.001 compared to 0 Ìmol/l, n = 6).
Procaine in Neuroprotection against Aß Pharmacology 2005;74:65–78 71
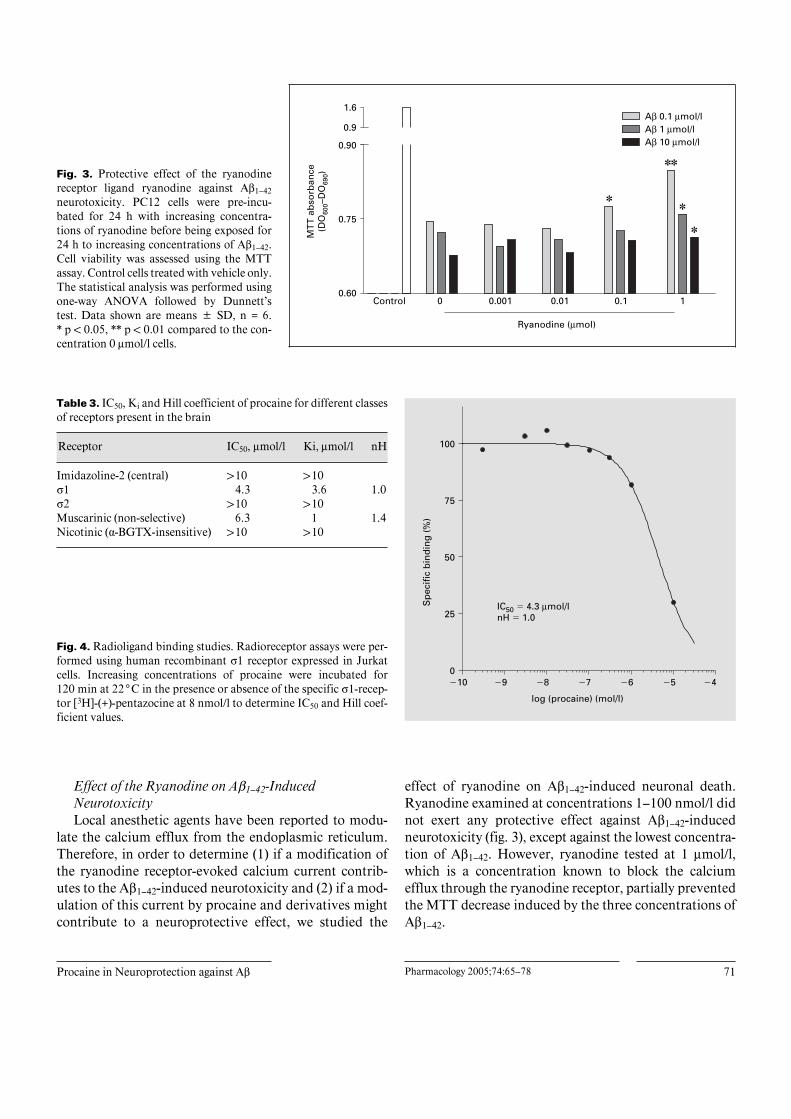
Fig. 3. Protective effect of the ryanodinereceptor ligand ryanodine against Aß1–42neurotoxicity. PC12 cells were pre-incu-bated for 24 h with increasing concentra-tions of ryanodine before being exposed for24 h to increasing concentrations of Aß1–42.Cell viability was assessed using the MTTassay. Control cells treated with vehicle only.The statistical analysis was performed usingone-way ANOVA followed by Dunnett’stest. Data shown are means B SD, n = 6.* p ! 0.05, ** p ! 0.01 compared to the con-centration 0 Ìmol/l cells.
0.60
0.75
0.90
✽✽
✽
0.9
1.6
✽
✽MT
T a
bso
rban
ce(D
O60
0–D
O69
0)Ryanodine (µmol)
Control 0 0.001 0.01 0.1 1
Aβ 0.1 µmol/lAβ 1 µmol/lAβ 10 µmol/l
Table 3. IC50, Ki and Hill coefficient of procaine for different classesof receptors present in the brain
Receptor IC50, Ìmol/l Ki, Ìmol/l nH
Imidazoline-2 (central) 110 110Û1 4.3 3.6 1.0Û2 110 110Muscarinic (non-selective) 6.3 1 1.4Nicotinic (·-BGTX-insensitive) 110 110
Fig. 4. Radioligand binding studies. Radioreceptor assays were per-formed using human recombinant Û1 receptor expressed in Jurkatcells. Increasing concentrations of procaine were incubated for120 min at 22°C in the presence or absence of the specific Û1-recep-tor [3H]-(+)-pentazocine at 8 nmol/l to determine IC50 and Hill coef-ficient values.
IC50 � 4.3 µmol/lnH � 1.0
log (procaine) (mol/l)
Sp
ecif
ic b
ind
ing
(%
)
�10 �9 �8 �7 �6 �5 �40
25
50
75
100
Effect of the Ryanodine on Aß1–42-InducedNeurotoxicityLocal anesthetic agents have been reported to modu-
late the calcium efflux from the endoplasmic reticulum.Therefore, in order to determine (1) if a modification ofthe ryanodine receptor-evoked calcium current contrib-utes to the Aß1–42-induced neurotoxicity and (2) if a mod-ulation of this current by procaine and derivatives mightcontribute to a neuroprotective effect, we studied the
effect of ryanodine on Aß1–42-induced neuronal death.Ryanodine examined at concentrations 1–100 nmol/l didnot exert any protective effect against Aß1–42-inducedneurotoxicity (fig. 3), except against the lowest concentra-tion of Aß1–42. However, ryanodine tested at 1 Ìmol/l,which is a concentration known to block the calciumefflux through the ryanodine receptor, partially preventedthe MTT decrease induced by the three concentrations ofAß1–42.
72 Pharmacology 2005;74:65–78 Lecanu/Wenguo/Xu/Greeson/Papadopoulos
Control 0 1 10 100250
550
850
Procaine (µmol/l)
Control 0 1 10 100
Tetracaine (µmol)
Control 0 1 10 100
Lidocaine (µmol/l)
Control 0 1 10 100
Procainamide (µmol/l)
2,7-
DC
F (a
rbit
rary
un
its)
250
550
850
2,7-
DC
F (a
rbit
rary
un
its)
250
550
850
2,7-
DC
F (a
rbit
rary
un
its)
250
550
850
2,7-
DC
F (a
rbit
rary
un
its)
Aβ 0.1 µmol/lAβ 1 µmol/lAβ 10 µmol/l
Aβ 0.1 µmol/lAβ 1 µmol/lAβ 10 µmol/l
Aβ 0.1 µmol/lAβ 1 µmol/lAβ 10 µmol/l
Aβ 0.1 µmol/lAβ 1 µmol/lAβ 10 µmol/l
A B
C D
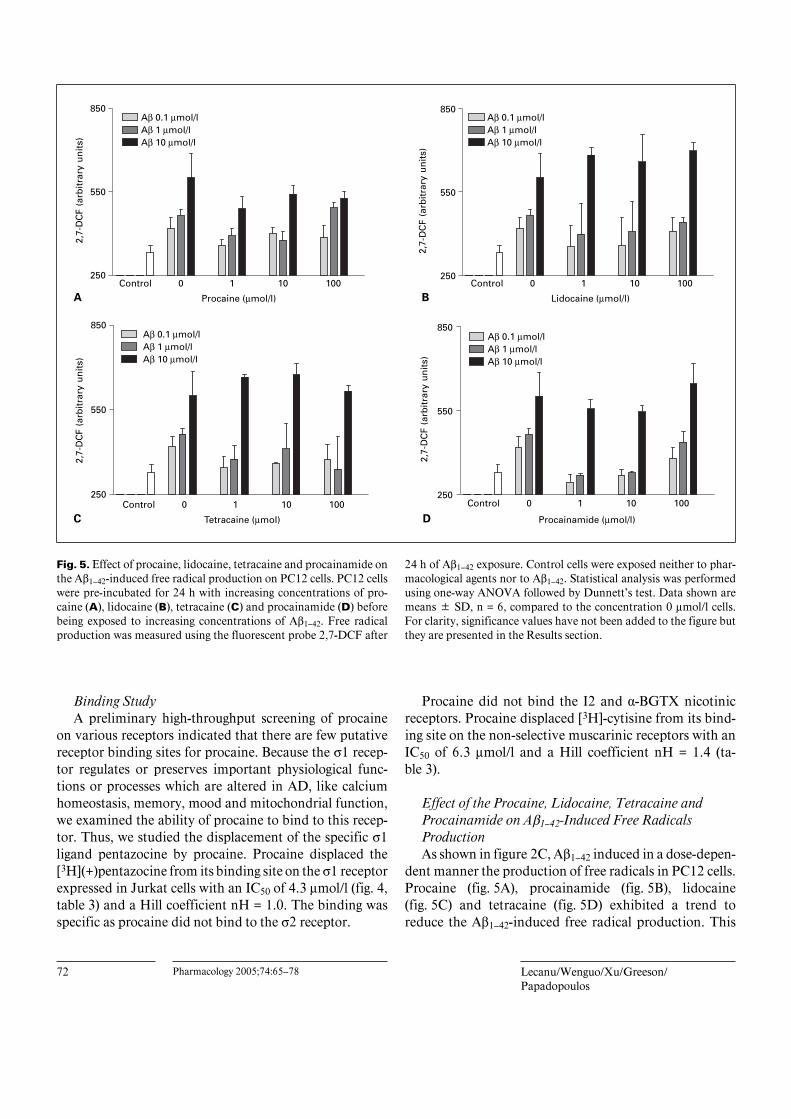
Fig. 5. Effect of procaine, lidocaine, tetracaine and procainamide onthe Aß1–42-induced free radical production on PC12 cells. PC12 cellswere pre-incubated for 24 h with increasing concentrations of pro-caine (A), lidocaine (B), tetracaine (C) and procainamide (D) beforebeing exposed to increasing concentrations of Aß1–42. Free radicalproduction was measured using the fluorescent probe 2,7-DCF after
24 h of Aß1–42 exposure. Control cells were exposed neither to phar-macological agents nor to Aß1–42. Statistical analysis was performedusing one-way ANOVA followed by Dunnett’s test. Data shown aremeans B SD, n = 6, compared to the concentration 0 Ìmol/l cells.For clarity, significance values have not been added to the figure butthey are presented in the Results section.
Binding StudyA preliminary high-throughput screening of procaine
on various receptors indicated that there are few putativereceptor binding sites for procaine. Because the Û1 recep-tor regulates or preserves important physiological func-tions or processes which are altered in AD, like calciumhomeostasis, memory, mood and mitochondrial function,we examined the ability of procaine to bind to this recep-tor. Thus, we studied the displacement of the specific Û1ligand pentazocine by procaine. Procaine displaced the[3H](+)pentazocine from its binding site on the Û1 receptorexpressed in Jurkat cells with an IC50 of 4.3 Ìmol/l (fig. 4,table 3) and a Hill coefficient nH = 1.0. The binding wasspecific as procaine did not bind to the Û2 receptor.
Procaine did not bind the I2 and ·-BGTX nicotinicreceptors. Procaine displaced [3H]-cytisine from its bind-ing site on the non-selective muscarinic receptors with anIC50 of 6.3 Ìmol/l and a Hill coefficient nH = 1.4 (ta-ble 3).
Effect of the Procaine, Lidocaine, Tetracaine andProcainamide on Aß1–42-Induced Free RadicalsProductionAs shown in figure 2C, Aß1–42 induced in a dose-depen-
dent manner the production of free radicals in PC12 cells.Procaine (fig. 5A), procainamide (fig. 5B), lidocaine(fig. 5C) and tetracaine (fig. 5D) exhibited a trend toreduce the Aß1–42-induced free radical production. This
Procaine in Neuroprotection against Aß Pharmacology 2005;74:65–78 73
A� 0.1 �mol/lA� 1 �mol/lA� 10 �mol/l
Aβ 0.1 µmol/lAβ 1 µmol/lAβ 10 µmol/l
450
650
850
2,7-
DC
F (a
rbit
rary
un
its)
0
0.5
1.0
1.5
MT
T (a
rbit
rary
un
its)
PBN (�mol/l)
Control 0 10 100 500
PBN (µmol/l)
Control 0 10 100 500
A B
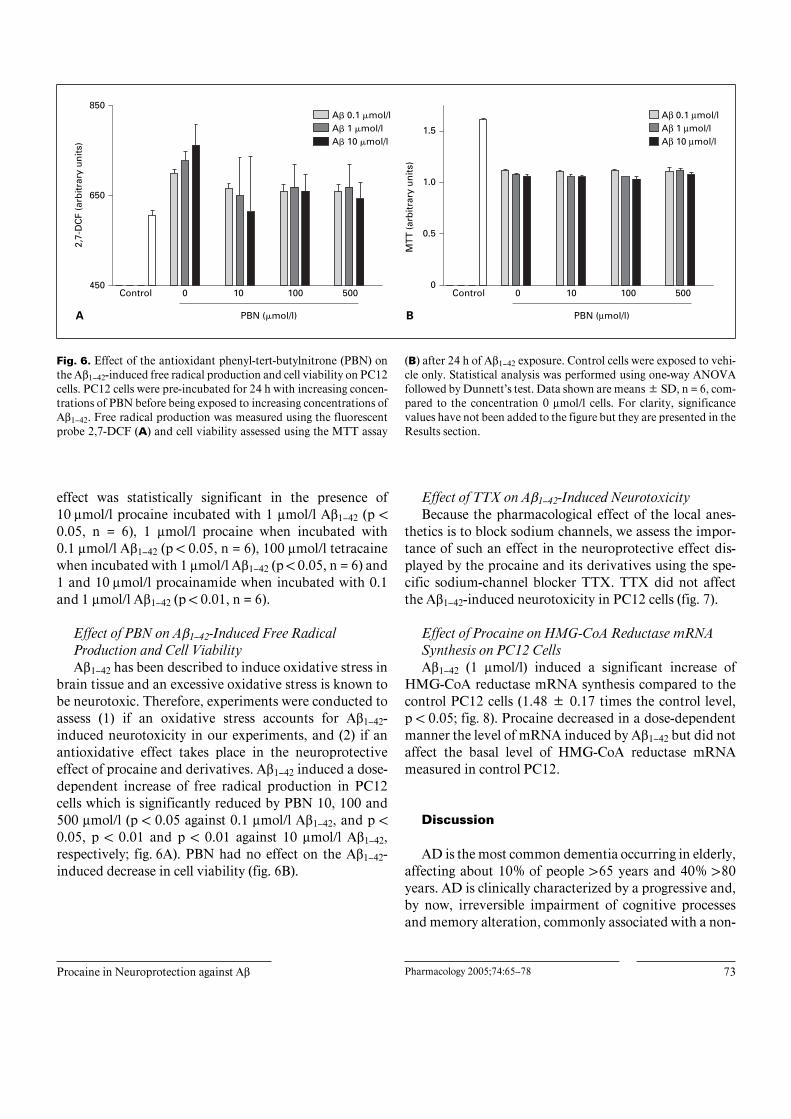
Fig. 6. Effect of the antioxidant phenyl-tert-butylnitrone (PBN) onthe Aß1–42-induced free radical production and cell viability on PC12cells. PC12 cells were pre-incubated for 24 h with increasing concen-trations of PBN before being exposed to increasing concentrations ofAß1–42. Free radical production was measured using the fluorescentprobe 2,7-DCF (A) and cell viability assessed using the MTT assay
(B) after 24 h of Aß1–42 exposure. Control cells were exposed to vehi-cle only. Statistical analysis was performed using one-way ANOVAfollowed by Dunnett’s test. Data shown are means B SD, n = 6, com-pared to the concentration 0 Ìmol/l cells. For clarity, significancevalues have not been added to the figure but they are presented in theResults section.
effect was statistically significant in the presence of10 Ìmol/l procaine incubated with 1 Ìmol/l Aß1–42 (p !0.05, n = 6), 1 Ìmol/l procaine when incubated with0.1 Ìmol/l Aß1–42 (p ! 0.05, n = 6), 100 Ìmol/l tetracainewhen incubated with 1 Ìmol/l Aß1–42 (p ! 0.05, n = 6) and1 and 10 Ìmol/l procainamide when incubated with 0.1and 1 Ìmol/l Aß1–42 (p ! 0.01, n = 6).
Effect of PBN on Aß1–42-Induced Free RadicalProduction and Cell ViabilityAß1–42 has been described to induce oxidative stress in
brain tissue and an excessive oxidative stress is known tobe neurotoxic. Therefore, experiments were conducted toassess (1) if an oxidative stress accounts for Aß1–42-induced neurotoxicity in our experiments, and (2) if anantioxidative effect takes place in the neuroprotectiveeffect of procaine and derivatives. Aß1–42 induced a dose-dependent increase of free radical production in PC12cells which is significantly reduced by PBN 10, 100 and500 Ìmol/l (p ! 0.05 against 0.1 Ìmol/l Aß1–42, and p !0.05, p ! 0.01 and p ! 0.01 against 10 Ìmol/l Aß1–42,respectively; fig. 6A). PBN had no effect on the Aß1–42-induced decrease in cell viability (fig. 6B).
Effect of TTX on Aß1–42-Induced NeurotoxicityBecause the pharmacological effect of the local anes-
thetics is to block sodium channels, we assess the impor-tance of such an effect in the neuroprotective effect dis-played by the procaine and its derivatives using the spe-cific sodium-channel blocker TTX. TTX did not affectthe Aß1–42-induced neurotoxicity in PC12 cells (fig. 7).
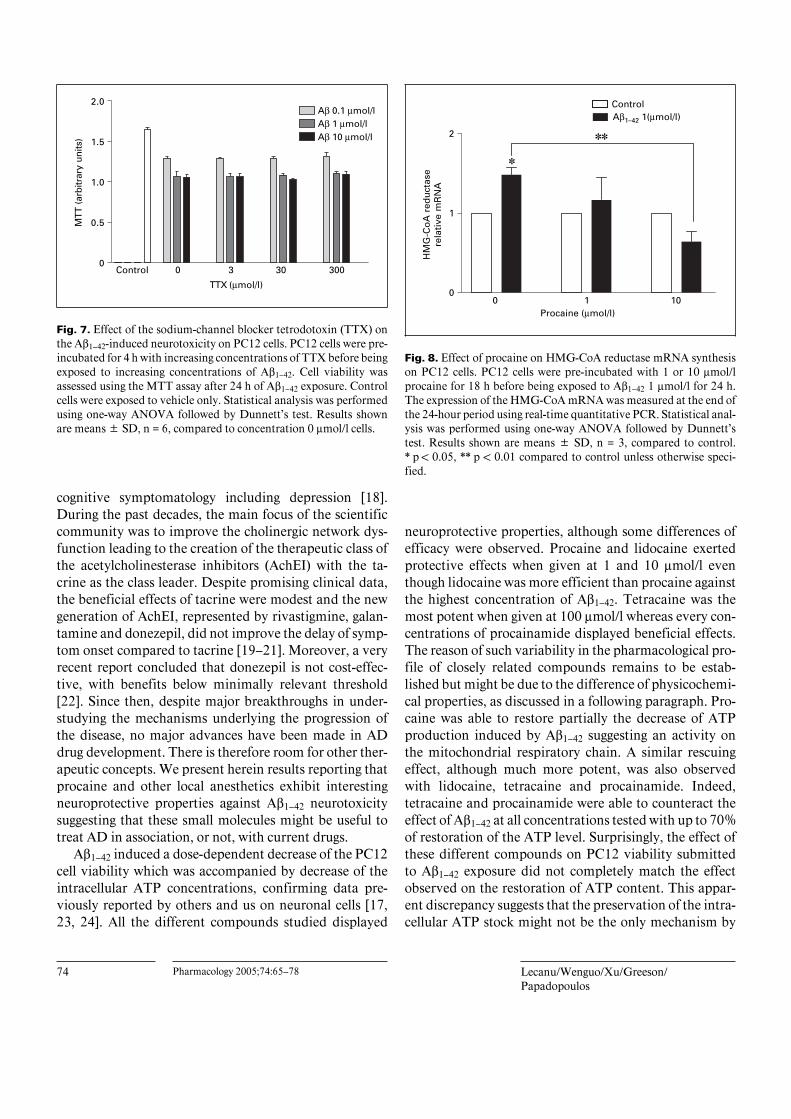
Effect of Procaine on HMG-CoA Reductase mRNASynthesis on PC12 CellsAß1–42 (1 Ìmol/l) induced a significant increase of
HMG-CoA reductase mRNA synthesis compared to thecontrol PC12 cells (1.48 B 0.17 times the control level,p ! 0.05; fig. 8). Procaine decreased in a dose-dependentmanner the level of mRNA induced by Aß1–42 but did notaffect the basal level of HMG-CoA reductase mRNAmeasured in control PC12.
Discussion
AD is the most common dementia occurring in elderly,affecting about 10% of people 165 years and 40% 180years. AD is clinically characterized by a progressive and,by now, irreversible impairment of cognitive processesand memory alteration, commonly associated with a non-
74 Pharmacology 2005;74:65–78 Lecanu/Wenguo/Xu/Greeson/Papadopoulos
Fig. 7. Effect of the sodium-channel blocker tetrodotoxin (TTX) onthe Aß1–42-induced neurotoxicity on PC12 cells. PC12 cells were pre-incubated for 4 h with increasing concentrations of TTX before beingexposed to increasing concentrations of Aß1–42. Cell viability wasassessed using the MTT assay after 24 h of Aß1–42 exposure. Controlcells were exposed to vehicle only. Statistical analysis was performedusing one-way ANOVA followed by Dunnett’s test. Results shownare means B SD, n = 6, compared to concentration 0 Ìmol/l cells.
Control 0 3 30 3000
0.5
1.0
1.5
2.0
TTX (µmol/l)
MTT
(ar
bitr
ary
units
)
Aβ 0.1 µmol/lAβ 1 µmol/lAβ 10 µmol/l
Fig. 8. Effect of procaine on HMG-CoA reductase mRNA synthesison PC12 cells. PC12 cells were pre-incubated with 1 or 10 Ìmol/lprocaine for 18 h before being exposed to Aß1–42 1 Ìmol/l for 24 h.The expression of the HMG-CoA mRNA was measured at the end ofthe 24-hour period using real-time quantitative PCR. Statistical anal-ysis was performed using one-way ANOVA followed by Dunnett’stest. Results shown are means B SD, n = 3, compared to control.* p ! 0.05, ** p ! 0.01 compared to control unless otherwise speci-fied.
0 1 100
1
2
✽
✽✽
Procaine (µmol/l)
HM
G-C
oA
red
uct
ase
rela
tive
mR
NA
ControlAβ1–42 1(µmol/l)
cognitive symptomatology including depression [18].During the past decades, the main focus of the scientificcommunity was to improve the cholinergic network dys-function leading to the creation of the therapeutic class ofthe acetylcholinesterase inhibitors (AchEI) with the ta-crine as the class leader. Despite promising clinical data,the beneficial effects of tacrine were modest and the newgeneration of AchEI, represented by rivastigmine, galan-tamine and donezepil, did not improve the delay of symp-tom onset compared to tacrine [19–21]. Moreover, a veryrecent report concluded that donezepil is not cost-effec-tive, with benefits below minimally relevant threshold[22]. Since then, despite major breakthroughs in under-studying the mechanisms underlying the progression ofthe disease, no major advances have been made in ADdrug development. There is therefore room for other ther-apeutic concepts. We present herein results reporting thatprocaine and other local anesthetics exhibit interestingneuroprotective properties against Aß1–42 neurotoxicitysuggesting that these small molecules might be useful totreat AD in association, or not, with current drugs.
Aß1–42 induced a dose-dependent decrease of the PC12cell viability which was accompanied by decrease of theintracellular ATP concentrations, confirming data pre-viously reported by others and us on neuronal cells [17,23, 24]. All the different compounds studied displayed
neuroprotective properties, although some differences ofefficacy were observed. Procaine and lidocaine exertedprotective effects when given at 1 and 10 Ìmol/l eventhough lidocaine was more efficient than procaine againstthe highest concentration of Aß1–42. Tetracaine was themost potent when given at 100 Ìmol/l whereas every con-centrations of procainamide displayed beneficial effects.The reason of such variability in the pharmacological pro-file of closely related compounds remains to be estab-lished but might be due to the difference of physicochemi-cal properties, as discussed in a following paragraph. Pro-caine was able to restore partially the decrease of ATPproduction induced by Aß1–42 suggesting an activity onthe mitochondrial respiratory chain. A similar rescuingeffect, although much more potent, was also observedwith lidocaine, tetracaine and procainamide. Indeed,tetracaine and procainamide were able to counteract theeffect of Aß1–42 at all concentrations tested with up to 70%of restoration of the ATP level. Surprisingly, the effect ofthese different compounds on PC12 viability submittedto Aß1–42 exposure did not completely match the effectobserved on the restoration of ATP content. This appar-ent discrepancy suggests that the preservation of the intra-cellular ATP stock might not be the only mechanism by

Procaine in Neuroprotection against Aß Pharmacology 2005;74:65–78 75
which local anesthetics exert their neuroprotective prop-erties.
The disruption of calcium homeostasis is one of thehypotheses that have emerged in an attempt to explain thepathophysiology of AD [10]. Previous reports have clearlyindicated that Aß1–42 increases intraneuronal calciumconcentrations leading to an excitotoxicity-type neuronaldeath [25] and the intracellular calcium raise seems tohave multiple extra- and intracellular origins. The gluta-matergic network is targeted by the ß-amyloid peptidessince Aß1–40 [26] and Aß25–35 [27] have been described toselectively augment NMDA receptor-mediated synaptictransmission in rat hippocampus. Interestingly, theNMDA receptor antagonist MK801 protected cholinergicnucleus basalis neurons and striatal neurons from amy-loid peptides neurotoxicity in vivo [11, 28] and in vitro onneuroblastoma cells whereas AP-5, which binds specifi-cally the glutamate site, did not [29]. These results ledthese authors to conclude that amyloid peptides might actmore by stabilizing the opening state of the NMDA-asso-ciated calcium channel after inserting the plasma mem-brane rather than by directly binding the glutamate site.MK801 reduced in a dose-dependent manner the neuro-toxicity induced by Aß1–42, suggesting therefore the in-volvement of an overstimulation of the NMDA receptorsin the neurotoxicity we report herein. Moreover, procainereduced the glutamate-induced excitotoxicity on thePC12 cells, indicating that the inhibition of the NMDA-induced calcium inward current might account for theprotective effect provided by the local anesthetics in ourexperiments. Our data are further reinforced by recentfindings reporting that local anesthetic agents inhibitNMDA receptor channel in mouse CA1 pyramidal neu-rons [16]. Interestingly, an overactivation of the rat hip-pocampus NMDA receptors by Aß1–42 has been describedto affect the long-term depression and, in turn, the long-term potentiation [30], the two main forms of synapticplasticity in the brain. This deleterious pathway has beenproposed to contribute to the memory processes ham-pered in AD and therefore raise the interest of procaine asdrug candidates for AD therapy. However, procaine0.3 Ìmol/l which succeeded in protecting against gluta-mate failed to protect neuronal cells against Aß1–42, sug-gesting therefore that the antiexcitotoxic effect of pro-caine might not be the only mechanism involved in theneuroprotective properties procaine displayed against theamyloid peptide.
The regulation of calcium signaling also involves intra-cellular ryanodine receptors (RyR) whose role is to triggercalcium release from intracellular storages to amplify var-
ious transduction pathways. Local anesthetics, and in par-ticular procaine, have been described to reduce the caf-feine-evoked calcium release from RyR type 2 in gerbilhippocampal neurons [31], but also from sarcoplasmicreticulum [32]. Because RyR plays a critical role in theregulation of calcium release from the endoplasmic retic-ulum in the brain, impairment of which is believed to con-tribute to the development of AD, the effect of ryanodineon the Aß1–42-induced neurotoxicity was tested on PC12cells. Ryanodine displayed neuroprotective propertieseven against the highest concentration of Aß1–42 tested.The profile of ryanodine activity in our experiments isvery interesting because it shows a trend to exacerbateAß1–42 toxicity at low concentrations whereas at 1 Ìmol/lconcentration it was neuroprotective. This biphasic phar-macological behavior has already been described and isdue to the ability of ryanodine to activate RyR and ampli-fy the calcium release at concentrations up to 0.01 Ìmol/l,whereas an inhibitory effect has been documented athigher concentrations [33]. Therefore it is likely that anactivation of the RyR by Aß1–42 contributes to the neuro-toxic effect observed in our experiments and that a block-ade of the RyR by procaine and its derivatives mayaccount for their neuroprotective properties. Interesting-ly, alterations in the RyR calcium release channel havebeen demonstrated to correlate with AD pathology [34].These authors described an augmentation of the RyR2binding in the cortex of AD patients displaying an earlystage of the disease whereas the opposite was reported atthe late stage probably due to the progressive disappear-ance of the receptor. This early binding increase might beresponsible for a disruption of the cellular calcium ho-meostasis leading to the cell death. Recent studies havealso shown that RyR-controlled calcium release is in-volved in both amyloid precursor protein (APP) process-ing and in presenilin 1 (PS1) function. Further in vitrostudies have shown that calcium released from RyR-sen-sitive ER intracellular pools induced a fourfold increasein the production of Aß1–42 [35]. In addition, PS1 muta-tion has been shown to increase the level of RyR and cal-cium release by PC12 cells and cortical neurons [36] andto sensitize PC12 cells to Aß1–42 toxicity by a mechanismblocked by dantrolene, an inhibitor of RyR-controlledcalcium release [37]. Because RyR is also involved inlong-term memory formation, spatial learning and cogni-tive processes [38], the modulation of this family of recep-tors is a very attractive therapeutic target and raises there-fore the interest for procaine and the procaine-derivativesas drug-candidates to treat AD.

76 Pharmacology 2005;74:65–78 Lecanu/Wenguo/Xu/Greeson/Papadopoulos
Since local anesthetic agents, like procaine, and class-Iantiarrhythmic drugs, like procainamide, are sodium-channel blockers, a blockade of the sodium channels inPC12 cannot be excluded for mediating the neuroprotec-tive effects of these compounds against Aß1–42-inducedtoxicity. However, as no involvement of such channelshas been described, so far, in AD pathology development,and as the TTX, a specific sodium-channel antagonist,failed in rescuing the PC12 cells exposed to Aß1–42, it isunlikely that such an effect takes place in the protectiveproperties displayed by the compounds assessed herein.
An intriguing observation we made is the ability ofprocaine to bind to the Û1 receptor with an IC50 of4.3 Ìmol/l and a Hill coefficient of 1.0, although the ques-tion whether procaine acts as an agonist or an antagonistremains. However, the binding seems to be specific of Û1as procaine does not bind the Û2 receptor. Several Û1-receptor agonists have been described to reverse in a dose-dependent manner the scopolamine-induced amnesia inrats [39]. One of them, the SA4503, enhanced the Achrelease in the hippocampus of rat brain slices [40] and invivo [41], suggesting that the antiamnesic effect could bedue in part to the activation of the cholinergic pathway. Inaddition, the effect of procaine binding on the Û1 receptoron the Ach release seems to be much more pronounced inthe hippocampus when compared to tacrine [41]. In addi-tion, igmesine, a Û1-receptor agonist, was recently dem-onstrated to exert an antidepressant activity in mice intra-cerebroventrically injected with the amyloid fragmentAß25-35 [42]. This antidepressant effect has also beenobserved with another Û1-receptor agonist, PRE-084, inmice submitted to the forced swimming test [43], there-fore reinforcing the interest of procaine as a potential ADtreatment as depression is a non-cognitive condition thatis very often experienced by AD patients. Interestingly,the antidepressant activity displayed by Û1-receptor ago-nists involves a modulation of the intracellular calciummobilization [42] in part mediated by the regulation ofRyR, reinforcing therefore the assumption we make re-garding a down-regulation of RyR activity as part of theneuroprotective effect displayed by procaine.
The involvement of an oxidative stress has been pro-posed as one of the fundamental pathogenic mechanismsoccurring in AD [44, 45], although its real importanceremains unclear. Thus, we still do not know if the oxida-tive stress reported in AD is due to an increase of freeradical production or to an increased sensitivity to anaging-related oxidative process. However, because in bothcases it might be of interest to reduce the formation of thefree radicals, we studied the implication of a possible
antioxidant property in the neuroprotective activity of thecompounds tested herein. The three local anestheticagents and procainamide reduced significantly free radi-cal production induced by Aß1–42, but only procaine, at allconcentrations examined, was able to reduce the oxida-tive stress induced by Aß1–42 10 Ìmol/l. Surprisingly, onlyprocaine given at 100 Ìmol/l was protective against thehighest concentration of Aß1–42, whereas all the doses oftetracaine and procainamide were active despite a lack ofantioxidant properties. This observation was further con-firmed by the total absence of a rescuing effect by thepotent antioxidant PBN on PC12 cells despite a dramaticdecrease of the Aß1–42-induced free radical productionand by previous data reporting that free radicals and lipidperoxidation do not mediate the acutely induced neuro-nal cell death by Aß1–42 [46]. These data suggest thereforethat the neuroprotective effect of procaine and other localanesthetics is not mediated by their antioxidant proper-ties.
Hormonal unbalance, and in particular hypercortisol-ism, is commonly described in patients suffering from AD[5, 6] and increasing evidence support a role for this raiseof serum cortisol concentrations in the worsening of thedisease [7, 8]. Procaine was recently demonstrated todown-regulate the stress-induced cortisol increase in vivoin rats and in vitro in dbcAMP-stimulated adrenal corti-cal cells [4]. These data indicated that the decrease of thecortisol production by the adrenal cortical cells was due tothe decreased expression of the cholesterol synthesis rate-limiting enzyme HMG-CoA reductase mRNA and corre-lated with the restoration of cell viability. The effect ofprocaine on HMG-CoA reductase mRNA levels in PC12cells ‘stressed’ by Aß1–42 exposure reported herein isequivalent to that previously reported for adrenal cells‘stressed’ by cAMP [4]. The ability of Aß1–42 to modulatethe HMG-CoA reductase activity through an increase ofthe expression of its mRNA was complementary to recentfindings on the physiological function of the ß-amyloidpeptide in the control of neuronal cholesterol levels andtransport [47]. However, in our model it is very unlikelythat any reduction of corticosteroid synthesis accounts forthe protective effect of procaine against Aß1–42 as PC12pheochromocytoma cells do not produce steroids. It ismore likely that the dose-dependent reduction of HMG-CoA reductase mRNA expression by procaine results,first, in a decrease of the cholesterol production with, as adirect consequence, a modification of the membranefluidity and an alteration of Aß1–42 trafficking through thecell membrane. These modifications might therefore ren-der the cell less sensitive to Aß1–42-induced neurotoxicity.

Procaine in Neuroprotection against Aß Pharmacology 2005;74:65–78 77
In addition, the reduction of cholesterol synthesis hasbeen shown to reduce APP cleavage and ß-amyloid pep-tide production by reducing Á-secretase activity [48], fur-ther strengthening the interest of procaine as a treatmentfor AD pathology.
Procaine displayed the properties to bind the non-selective muscarinic receptors. Although the IC50 seemsmodest (6.3 Ìmol/l), this result does not take into consid-eration the possibility of a much higher affinity of pro-caine towards specific subtypes of muscarinic receptors.Moreover, procaine has been recently reported to bind invivo with a high affinity the M2 receptor located in theparalimbic area of the monkey brain [49]. This affinity forthe muscarinic M2 receptor has been presented by theseauthors to have a role in the emotions and mood modula-tion which, therefore, could have an interest to treat thenon-cognitive conditions which occur during AD.
AD is an evolving neurodegeneration involving manypathological pathways and for which there is not yet anavailable cure. The difficulty to find a treatment able toblock with efficacy the evolution of the disease might findan answer in that the drugs actually used aim only at onetherapeutic target. Because the onset and the progression
of the disease are probably due to the synergistic action ofhighly intricate networks (i.e. calcium homeostasis dis-ruption, amyloid hypothesis, oxidative stress, alterationof cholesterol and steroid metabolism), the use of a multi-targeting drugs might be an interesting approach to treatAD pathology. The neuroprotective effect displayed byprocaine against Aß1–42 in our experiments is probably theresult of the synergistic effect on different mechanisms ofaction including a regulation of the calcium homeostasisthrough a modulation of RyR, of calcium channels andglutamate receptors, a modulation of the Û1-receptoractivity and a down-regulation of the stress-induced in-crease of cholesterol biosynthesis. Procaine might repre-sentative of what could be the lead compound of a newtherapeutic class aiming at multiple targets at the sametime and. However, this very interesting pharmacologicalprofile is hampered by a very high metabolic rate and,therefore, a better alternative might be the use of procaineas a substructure to screen natural compounds databasesto isolate stable natural analogs. Moreover, procainestructure might be used to start structure/activity relation-ship studies in order to improve the stability/efficiencyleading to development of more attractive analogs.
References
1 Nikaido T, Austin J, Trueb L, Hutchison J,Rinehart R, Stuckenbrok H, Miles B: Isolationand preliminary characterization of Alzheimerplaques from presenile and senile dementia.Trans Am Neurol Assoc 1970;95:47–50.
2 Kosik KS, Joachim CL, Selkoe DJ: Microtu-bule-associated protein Ù is a major antigeniccomponent of paired helical filaments in Alz-heimer disease. Proc Natl Acad Sci USA 1986;83:4044–4048.
3 Ostfeld A, Smith CM, Stotsky BA: The sys-temic use of procaine in the treatment of theelderly: A review. J Am Geriatr Soc 1977;25:1–19.
4 Xu J, Lecanu L, Han Z, Yao Z, Greeson J,Papadopoulos V: Inhibition of adrenal corticalsteroid formation by procaine is mediated byreduction of the cAMP-induced 3-hydroxy-3-methylglutaryl-coenzyme A reductase messen-ger ribonucleic acid levels. J Pharmacol ExpTher 2003;307:1148–1157.
5 Swaab DF, Raadsheer FC, Endert E, HofmanMA, Kamphorst W, Ravid R: Increased corti-sol levels in aging and Alzheimer’s disease inpostmortem cerebrospinal fluid. J Neuroendo-crinol 1994;6:681–687.
6 O’Brien JT, Ames D, Schweitzer I, MastwykM, Colman P: Enhanced adrenal sensitivity toadrenocorticotrophic hormone is evidence ofHPA axis hyperactivity in Alzheimer’s disease.Psychol Med 1996;26:7–14.
7 Abraham I, Harkany T, Horvath KM, Veene-ma AH, Penke B, Nyakas C, Luiten PG:Chronic corticosterone administration dose-dependently modulates Aß1–42- and NMDA-induced neurodegeneration in rat magnocellu-lar nucleus basalis. J Neuroendocrinol 2000;12:486–494.
8 Tafet GE, Toister-Achituv M, Shinitzky M:Enhancement of serotonin uptake by cortisol:A possible link between stress and depression.Cogn Affect Behav Neurosci 2001;1:96–104.
9 Liu K, Adachi N, Yanase H, Kataoka K, AraiT: Lidocaine suppresses the anoxic depolariza-tion and reduces the increase in the intracellu-lar Ca2+ concentration in gerbil hippocampalneurons. Anesthesiology 1997;87:1470–1478.
10 Kachaturian ZS: Hypothesis on the regulationof cytosol calcium concentration and the agingbrain. Neurobiol Aging 1987;8:345–346.
11 Harkany T, Mulder J, Sasvari M, Abraham I,Konya C, Zarandi M, Penke B, Luiten PG,Nyakas C: N-methyl-D-aspartate receptor an-tagonist MK801 and radical scavengers protectcholinergic nucleus basalis neurons against ß-amyloid neurotoxicity. Neurobiol Dis 1999;6:109–121.
12 MacManus A, Ramsden M, Murray M, Hen-derson Z, Pearson HA, Campbell VA: En-hancement of 45Ca2+ influx and voltage-depen-dent Ca2+ channel activity by ß-amyloid-(1–40)in rat cortical synaptosomes and cultured corti-cal neurons. Modulation by the proinflamma-tory cytokine interleukin-1ß. J Biol Chem2000;275:4713–4718.
13 Ye C, Ho-Pao CL, Kanazirska M, Quinn S,Rogers K, Seidman CE, Seidman JG, BrownEM, Vassilev PM: Amyloid-ß proteins activateCa2+-permeable channels through calcium-sensing receptors. J Neurosci Res 1997;47:547–554.
14 Fujitani T, Adachi N, Miyazaki H, Liu K,Nakamura Y, Kataoka K, Arai T: Lidocaineprotects hippocampal neurons against ischemicdamage by preventing increase of extracellularexcitatory amino acids: A microdialysis studyin Mongolian gerbils. Neurosci Lett 1994;179:91–94.
15 Raley-Susman KM, Kass IS, Cottrell JE, New-man RB, Chambers G, Wang J: Sodium influxblockade and hypoxic damage to CA1 pyrami-dal neurons in rat hippocampal slices. J Neuro-physiol 2001;86:2715–2726.
16 Nishizawa N, Shirasaki T, Nakao S, MatsudaH, Shingu K: The inhibition of the N-methyl-D-aspartate receptor channel by local anes-thetics in mouse CA1 pyramidal neurons.Anesth Analg 2002;94:325–330.

78 Pharmacology 2005;74:65–78 Lecanu/Wenguo/Xu/Greeson/Papadopoulos
17 Lecanu L, Yao W, Teper GL, Yao ZX, GreesonJ, Papadopoulos V: Identification of naturallyoccurring spiroatenils preventing ß-amyloid-induced neurotoxicity. Steroids 2004;69:1–16.
18 Robert P: Alzheimer disease; in Becker R, Gia-cobini E (eds): From Molecular Biology toTherapy. Boston, Birkhäuser, 1996, pp 487–493.
19 Grossberg G, Irwin P, Satlin A, Mesenbrink P,Spiegel R: Rivastigmine in Alzheimer disease:Efficacy over two years. Am J Geriatr Psychia-try 2004;12:420–431.
20 Tariot P, Winblad B: Alzheimer’s disease; inIqbal K, Sisodia SS, Winblad B (eds): Advancesin Etiology, Pathogenesis and Therapeutics.Chichester, Wiley, 2001, pp 707–723.
21 Waldemar, et al: Alzheimer’s disease; in IqbalK, Sisodia SS, Winblad B: Advances in Etiolo-gy, Pathogenesis and Therapeutics. Chichester,Wiley, 2001, pp 725–738.
22 AD2000 Collaborative Group: Long-termtreatment in 565 patients with Alzheimer’s dis-ease (AD2000): Randomized double-blindtrial. Lancet 2004;363:2105–2115.
23 Casley CS, Land JM, Sharpe MA, Clark JB,Duchen MR, Canevari L: ß-Amyloid fragment25–35 causes mitochondrial dysfunction in pri-mary cortical neurons. Neurobiol Dis 2002;10:258–267.
24 Kuhla B, Loske C, Garcia De Arriba S, Schin-zel R, Huber J, Munich G: Differential aspectsof ‘advanced glycation end products’ and ß-amyloid peptide on glucose utilization andATP levels in the neuronal cell line SH-SY5Y.J Neural Transm 2004;111:427–439.
25 Olney JW, Wozniak DF, Farber NB: Excito-toxic neurodegeneration in Alzheimer disease.New hypothesis and new therapeutic strategies.Arch Neurol 1997;54:1234–1240.
26 Wu J, Anwyl R, Rowan MJ: ß-Amyloid selec-tively augments NMDA receptor-mediatedsynaptic transmission in rat hippocampus.Neuroreport 1995;6:2409–2413.
27 Mogensen HS, Beatty DM, Morris SJ, Jorgen-sen OS: Amyloid ß-peptide(25–35) changes[Ca2+] in hippocampal neurons. Neuroreport1998;9:1553–1558.
28 Parks JK, Smith TS, Trimmer PA, Bennett JPJr, Parker WD Jr: Neurotoxic Aß peptidesincrease oxidative stress in vivo throughNMDA-receptor and nitric-oxide-synthasemechanisms, and inhibit complex IV activityand induce a mitochondrial permeability tran-sition in vitro. J Neurochem 2001;76:1050–1056.
29 Le WD, Colom LV, Xie WJ, Smith RG, Alex-ianu M, Appel SH: Cell death induced by ß-amyloid 1–40 in MES 23.5 hybrid clone: Therole of nitric oxide and NMDA-gated channelactivation leading to apoptosis. Brain Res1995;686:49–60.
30 Kim JH, Anwyl R, Suh YH, Djamgoz MB,Rowan MJ: Use-dependent effects of amy-loidogenic fragments of ß-amyloid precursorprotein on synaptic plasticity in rat hippocam-pus in vivo. J Neurosci 2001;21:1327–1333.
31 Chen J, Xu W, Jiang H: The effects of localanesthetics on intracellular Ca2+ release fromryanodine-sensitive Ca2+ stores in gerbil hippo-campal neurons. Chin Med J (Engl) 2002;115:1542–1544.
32 Shoshan-Barmatz V, Zchut S: The interactionof local anesthetics with the ryanodine receptorof the sarcoplasmic reticulum. J Membr Biol1993;133:171–181.
33 Lai FA, Misra M, Xu L, Smith HA, MeissnerG: The ryanodine receptor-Ca2+ release chan-nel complex of skeletal muscle sarcoplasmicreticulum. Evidence for a cooperatively cou-pled, negatively charged homotetramer. J BiolChem 1989;264:16776–16685.
34 Kelliher M, Fastbom J, Cowburn RF, BonkaleW, Ohm TG, Ravid R, Sorrentino V, O’NeillC: Alterations in the ryanodine receptor cal-cium release channel correlate with Alzhei-mer’s disease neurofibrillary and ß-amyloidpathologies. Neuroscience 1999;92:499–513.
35 Querfurth HW, Jiang J, Geiger JD, Selkoe DJ:Caffeine stimulates amyloid ß-peptide releasefrom ß-amyloid precursor protein-transfectedHEK293 cells. J Neurochem 1997;69:1580–1591.
36 Chan SL, Mayne M, Holden CP, Geiger JD,Mattson MP: Presenilin-1 mutations increaselevels of ryanodine receptors and calcium re-lease in PC12 cells and cortical neurons. J BiolChem 2000;275:18195–18200.
37 Guo Q, Sopher BL, Furukawa K, Pham DG,Robinson N, Martin GM, Mattson MP: Alz-heimer’s presenilin mutation sensitizes neuralcells to apoptosis induced by trophic factorwithdrawal and amyloid ß-peptide: Involve-ment of calcium and oxyradicals. J Neurosci1997;17:4212–4222.
38 Salinska EJ, Bourne RC, Rose SP: Long-termmemory formation in the chick requires mobi-lization of ryanodine-sensitive intracellular cal-cium stores. Neurobiol Learn Mem 2001;75:293–302.
39 Maurice T, Phan VL, Privat A: The anti-amnesic effects of Û1-receptor agonists con-firmed by in vivo antisense strategy in themouse. Brain Res 2001;898:113–121.
40 Horan B, Gifford AN, Matsuno K, Mita S,Ashby CR: Effect of SA4503 on the electricallyevoked release of 3H-acetylcholine from striataland hippocampal rat brain slices. Synapse2002;46:1–3.
41 Kobayashi T, Matsuno K, Nakata K, Mita S:Enhancement of acetylcholine release bySA4503, a novel Û1-receptor agonist, in the ratbrain. J Pharmacol Exp Ther 1996;279:106–113.
42 Urani A, Romieu P, Roman FJ, Maurice T:Enhanced antidepressant effect of Û1-receptoragonists in ß25–35-amyloid peptide-treatedmice. Behav Brain Res 2002;134:239–247.
43 Urani A, Romieu P, Portales-Casamar E, Ro-man FJ, Maurice T: The antidepressant-likeeffect induced by the Û1-receptor agonist igme-sine involves modulation of intracellular cal-cium mobilization. Psychopharmacology 2001;163:26–35.
44 Volicer L, Crino PB: Involvement of free radi-cals in dementia of the Alzheimer type: Ahypothesis. Neurobiol Aging 1990;11:567–571.
45 Pappolla MA, Omar RA, Kim KS, RobakisNK: Immunohistochemical evidence of oxida-tive (corrected) stress in Alzheimer’s disease.Am J Pathol 1996;149:1770.
46 Yao ZX, Drieu K, Szweda LI, Papadopoulos V:Free radicals and lipid peroxidation do notmediate ß-amyloid-induced neuronal celldeath. Brain Res 1999;847:203–210.
47 Yao ZX, Papadopoulos V: Function of ß-amy-loid in cholesterol transport: A lead to neuro-toxicity. FASEB J 2002;16:1677–1679.
48 Buxbaum JD, Cullen EI, Friedhoff LT: Phar-macological concentrations of the HMG-CoAreductase inhibitor lovastatin decrease the for-mation of the Alzheimer ß-amyloid peptide invitro and in patients. Front Biosci 2002;7:50–59.
49 Benson BE, Carson RE, Kiesewetter DO,Herscovitch P, Eckelman WC, Post RM, Ket-ter TA: A potential cholinergic mechanism ofprocaine’s limbic activation. Neuropsycho-pharmacology 2004;29:1239–1250.
Copyright © 2022 FDOKUMEN




















