
Tissue plasminogen activator and urokinase plasminogen activator in human epileptogenic pathologies
17
TISSUE PLASMINOGEN ACTIVATOR AND UROKINASE PLASMINOGEN ACTIVATOR IN HUMAN EPILEPTOGENIC PATHOLOGIES A. M. IYER, a1 E. ZUROLO, a1 K. BOER, a J. C. BAAYEN, b F. GIANGASPERO, c,d A. ARCELLA, d G. C. DI GENNARO, d V. ESPOSITO, d,e W. G. M. SPLIET, f P. C. VAN RIJEN, g D. TROOST, a J. A. GORTER h,i AND E. ARONICA a,i * a Department of (Neuro) Pathology, Academic Medical Center, Univer- sity of Amsterdam, The Netherlands b Department of Neurosurgery, VU University Medical Center, Amster- dam, The Netherlands c Department of Experimental Pathology, University Sapienza, Rome, Italy d IRCCS Neuromed, Pozzilli, Isernia, Italy e Department of Neuroscience, University Sapienza, Rome, Italy f Department of Pathology, Rudolf Magnus Institute for Neuroscience, University Medical Center Utrecht, The Netherlands g Department of Neurosurgery, Rudolf Magnus Institute for Neuro- science, University Medical Center Utrecht, The Netherlands h Swammerdam Institute for Life Sciences, Center for Neuroscience, University of Amsterdam, The Netherlands i Epilepsy Institute in The Netherlands Foundation (Stichting Epilepsie Instellingen Nederland, SEIN), Heemstede, The Netherlands Abstract—A growing body of evidence demonstrates the in- volvement of plasminogen activators (PAs) in a number of physiologic and pathologic events in the CNS. Induction of both tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA) has been observed in different experimental models of epilepsy and tPA has been implicated in the mecha- nisms underlying seizure activity. We investigated the expres- sion and the cellular distribution of tPA and uPA in several epileptogenic pathologies, including hippocampal sclerosis (HS; n6), and developmental glioneuronal lesions, such as focal cortical dysplasia (FCD, n6), cortical tubers in patients with the tuberous sclerosis complex (TSC; n6) and in ganglio- gliomas (GG; n6), using immuno-cytochemical, western blot and real-time quantitative PCR analysis. TPA and uPA immuno- staining showed increased expression within the epileptogenic lesions compared to control specimens in both glial and neu- ronal cells (hippocampal neurons in HS and dysplastic neurons in FCD, TSC and GG specimens). Confocal laser scanning mi- croscopy confirmed expression of both proteins in astrocytes and microglia, as well as in microvascular endothelium. Immu- noblot demonstrated also up-regulation of the uPA receptor (uPAR; P<0.05). Increased expression of tPA, uPA, uPAR and tissue PA inhibitor type mRNA levels was also detected by PCR analysis in different epileptogenic pathologies (P<0.05). Our data support the role of PA system components in different human focal epileptogenic pathologies, which may critically influence neuronal activity, inflammatory response, as well as contributing to the complex remodeling of the neuronal net- works occurring in epileptogenic lesions. © 2010 IBRO. Pub- lished by Elsevier Ltd. All rights reserved. Key words: hippocampal sclerosis, focal cortical dysplasia, tuberous sclerosis, ganglioglioma, epilepsy. The plasminogen (fibrinolytic) system comprises the inactive proenzyme, plasminogen, that can be converted to the active enzyme plasmin by two serine proteases, the tissue-type plasminogen activator (tPA) and the urokinase-type plasmin- ogen activator (uPA) (Collen, 1999). TPA and uPA elicit various cellular responses, involving the activation of distinct signaling pathways. While several of these pathways have been described (Maupas-Schwalm et al., 2004; Benarroch, 2007), their interactions and the link to specific biological responses are only partly understood. Attention has been recently focused on the role of uPA receptor (uPAR) indicat- ing that it may act as signaling receptor, also independently of uPA-mediated proteolysis (for review see Smith and Mar- shall, 2010). In the CNS, increasing evidence suggests a crucial role of the plasminogen system in a broad range of physiological and pathological processes ranging from neu- ronal development, cell migration and invasion, cell growth and apoptosis, immune responses, inflammation, angiogen- esis and regulation of synaptic remodeling and neuronal plas- ticity (Seeds et al., 1999; Tsirka, 2002; Powell et al., 2003; Alfano et al., 2005; Benarroch, 2007). TPA is widely expressed by many types of neurons in the human CNS, including the neocortical and hippocam- pal pyramidal neurons (Teesalu et al., 2004). Activation of the plasminogen system, involving neurons, reactive glial cells and vascular endothelium, as source of plasminogen activators, has been reported in different neurological dis- orders such as stroke and other forms of acute brain injury, as well as in patients with inflammatory disorders (Gveric et al., 2001; Teesalu et al., 2002; Benarroch, 2007). A complex deregulation of the plasminogen system may also be in- volved in neurodegenerative disorders, such as Alzheimer’s disease (Tucker et al., 2002; Fabbro and Seeds, 2009). Several experimental findings identified a role for tPA in the mechanisms underlying seizure activity (Tsirka et al., 1995; Pawlak and Strickland, 2002; Benarroch, 2007). Inter- estingly, induction of plasminogen activators (PAs) has been 1 The first two authors contributed equally to the present work. *Correspondence to: E. Aronica, Department of (Neuro) Pathology, Ac- ademic Medical Center, Meibergdreef 9, 1105 AZ Amsterdam, The Neth- erlands. Tel: 31-20-5662943; fax: 31-20-5669522. E-mail address: [email protected] (E. Aronica). Abbreviations: DG, dentate gyrus; FCD, focal cortical dysplasia; GFAP, glial fibrillary acidic protein; GG, ganglioglioma; HS, hippocam- pal sclerosis; IR, immunoreactivity; MCD, malformations of cortical development; NMDA, N-methyl-D-asparte; PAI1, plasminogen activa- tor inhibitor type-1; PAs, plasminogen activators; SE, status epilepti- cus; TBP, TATA-binding protein; TLE, temporal lobe epilepsy; tPA, tissue-type plasminogen activator; TSC, tuberous sclerosis complex; uPA, urokinase-type plasminogen activator. Neuroscience 167 (2010) 929 –945 0306-4522/10 $ - see front matter © 2010 IBRO. Published by Elsevier Ltd. All rights reserved. doi:10.1016/j.neuroscience.2010.02.047 929
Transcript of Tissue plasminogen activator and urokinase plasminogen activator in human epileptogenic pathologies

TA
AFGPEa
sb
dc
Id
e
f
Ug
sh
Ui
I
Avptamnse(fwgaslrica
1
*aeEAGpdtctu
Neuroscience 167 (2010) 929–945
0d
ISSUE PLASMINOGEN ACTIVATOR AND UROKINASE PLASMINOGEN
CTIVATOR IN HUMAN EPILEPTOGENIC PATHOLOGIESn(tadhicwl
Kt
Tpepovsb2rriuscpraetA
tptcaoaadvd
t1
. M. IYER,a1 E. ZUROLO,a1 K. BOER,a J. C. BAAYEN,b
. GIANGASPERO,c,d A. ARCELLA,d
. C. DI GENNARO,d V. ESPOSITO,d,e W. G. M. SPLIET,f
. C. VAN RIJEN,g D. TROOST,a J. A. GORTERh,i AND
. ARONICAa,i*
Department of (Neuro) Pathology, Academic Medical Center, Univer-ity of Amsterdam, The Netherlands
Department of Neurosurgery, VU University Medical Center, Amster-am, The Netherlands
Department of Experimental Pathology, University Sapienza, Rome,taly
IRCCS Neuromed, Pozzilli, Isernia, Italy
Department of Neuroscience, University Sapienza, Rome, Italy
Department of Pathology, Rudolf Magnus Institute for Neuroscience,niversity Medical Center Utrecht, The Netherlands
Department of Neurosurgery, Rudolf Magnus Institute for Neuro-cience, University Medical Center Utrecht, The Netherlands
Swammerdam Institute for Life Sciences, Center for Neuroscience,niversity of Amsterdam, The Netherlands
Epilepsy Institute in The Netherlands Foundation (Stichting Epilepsienstellingen Nederland, SEIN), Heemstede, The Netherlands
bstract—A growing body of evidence demonstrates the in-olvement of plasminogen activators (PAs) in a number ofhysiologic and pathologic events in the CNS. Induction of bothissue plasminogen activator (tPA) and urokinase plasminogenctivator (uPA) has been observed in different experimentalodels of epilepsy and tPA has been implicated in the mecha-isms underlying seizure activity. We investigated the expres-ion and the cellular distribution of tPA and uPA in severalpileptogenic pathologies, including hippocampal sclerosisHS; n�6), and developmental glioneuronal lesions, such asocal cortical dysplasia (FCD, n�6), cortical tubers in patientsith the tuberous sclerosis complex (TSC; n�6) and in ganglio-liomas (GG; n�6), using immuno-cytochemical, western blotnd real-time quantitative PCR analysis. TPA and uPA immuno-taining showed increased expression within the epileptogenicesions compared to control specimens in both glial and neu-onal cells (hippocampal neurons in HS and dysplastic neuronsn FCD, TSC and GG specimens). Confocal laser scanning mi-roscopy confirmed expression of both proteins in astrocytesnd microglia, as well as in microvascular endothelium. Immu-
The first two authors contributed equally to the present work.Correspondence to: E. Aronica, Department of (Neuro) Pathology, Ac-demic Medical Center, Meibergdreef 9, 1105 AZ Amsterdam, The Neth-rlands. Tel: �31-20-5662943; fax: �31-20-5669522.-mail address: [email protected] (E. Aronica).bbreviations: DG, dentate gyrus; FCD, focal cortical dysplasia;FAP, glial fibrillary acidic protein; GG, ganglioglioma; HS, hippocam-al sclerosis; IR, immunoreactivity; MCD, malformations of corticalevelopment; NMDA, N-methyl-D-asparte; PAI1, plasminogen activa-or inhibitor type-1; PAs, plasminogen activators; SE, status epilepti-us; TBP, TATA-binding protein; TLE, temporal lobe epilepsy; tPA,
eissue-type plasminogen activator; TSC, tuberous sclerosis complex;PA, urokinase-type plasminogen activator.
306-4522/10 $ - see front matter © 2010 IBRO. Published by Elsevier Ltd. All rightoi:10.1016/j.neuroscience.2010.02.047
929
oblot demonstrated also up-regulation of the uPA receptoruPAR; P<0.05). Increased expression of tPA, uPA, uPAR andissue PA inhibitor type mRNA levels was also detected by PCRnalysis in different epileptogenic pathologies (P<0.05). Ourata support the role of PA system components in differentuman focal epileptogenic pathologies, which may critically
nfluence neuronal activity, inflammatory response, as well asontributing to the complex remodeling of the neuronal net-orks occurring in epileptogenic lesions. © 2010 IBRO. Pub-
ished by Elsevier Ltd. All rights reserved.
ey words: hippocampal sclerosis, focal cortical dysplasia,uberous sclerosis, ganglioglioma, epilepsy.
he plasminogen (fibrinolytic) system comprises the inactiveroenzyme, plasminogen, that can be converted to the activenzyme plasmin by two serine proteases, the tissue-typelasminogen activator (tPA) and the urokinase-type plasmin-gen activator (uPA) (Collen, 1999). TPA and uPA elicitarious cellular responses, involving the activation of distinctignaling pathways. While several of these pathways haveeen described (Maupas-Schwalm et al., 2004; Benarroch,007), their interactions and the link to specific biologicalesponses are only partly understood. Attention has beenecently focused on the role of uPA receptor (uPAR) indicat-ng that it may act as signaling receptor, also independently ofPA-mediated proteolysis (for review see Smith and Mar-hall, 2010). In the CNS, increasing evidence suggests arucial role of the plasminogen system in a broad range ofhysiological and pathological processes ranging from neu-onal development, cell migration and invasion, cell growthnd apoptosis, immune responses, inflammation, angiogen-sis and regulation of synaptic remodeling and neuronal plas-
icity (Seeds et al., 1999; Tsirka, 2002; Powell et al., 2003;lfano et al., 2005; Benarroch, 2007).
TPA is widely expressed by many types of neurons inhe human CNS, including the neocortical and hippocam-al pyramidal neurons (Teesalu et al., 2004). Activation ofhe plasminogen system, involving neurons, reactive glialells and vascular endothelium, as source of plasminogenctivators, has been reported in different neurological dis-rders such as stroke and other forms of acute brain injury,s well as in patients with inflammatory disorders (Gveric etl., 2001; Teesalu et al., 2002; Benarroch, 2007). A complexeregulation of the plasminogen system may also be in-olved in neurodegenerative disorders, such as Alzheimer’sisease (Tucker et al., 2002; Fabbro and Seeds, 2009).
Several experimental findings identified a role for tPA inhe mechanisms underlying seizure activity (Tsirka et al.,995; Pawlak and Strickland, 2002; Benarroch, 2007). Inter-
stingly, induction of plasminogen activators (PAs) has beens reserved.

osgvuavw2seuettoioCti
ush
S
Tott(Pscirdbpptscn
tfccrtTDpdp
3BWFaweoepmphw
H
Twltcfm�MN
G1I(m1DCc
(
T
P
HNF
C
GC
uberous
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945930
bserved in different experimental models of epilepsy (Luka-iuk et al., 2003; Gorter et al., 2006; Lahtinen et al., 2006) andene expression profile analysis of gangliogliomas (GG) re-ealed that both tPA and uPA represent one of the mostp-regulated genes in these epileptogenic lesions (Aronica etl., 2008). In rat hippocampus, tPA and uPA were both acti-ated at one day after induction of status epilepticus (SE) andere still elevated during epileptogenesis (Gorter et al., 2006,007; Lahtinen et al., 2006). Expression of tPA mRNA wastill increased in the chronic phase in the CA3 region (Gortert al., 2007). Recently, the cellular distribution of uPA andPAR has been characterized in rat hippocampus duringpileptogenesis (Lahtinen et al., 2006). However, whetherhe up-regulation of plasminogen activator proteins persists inhe chronic phase in epileptic human brain is unclear. More-ver, a detailed analysis of tPA and uPA cellular expression
n human epileptogenic pathologies is still lacking. Becausef the functional redundancy among PAs (Carmeliet andollen, 1995), characterization of the expression in human
issue is important for the correct interpretation of the exper-mental observations.
In the present study we have examined the tPA andPA tissue distribution, evaluated their degree of expres-ion and defined their cellular origin in common causes ofuman focal chronic refractory epilepsy.
EXPERIMENTAL PROCEDURES
ubjects
he human cases included in this study were obtained from the filesf the departments of neuropathology of the Academic Medical Cen-
er (AMC, University of Amsterdam), the VU University Medical Cen-er (VUMC) in Amsterdam, the University Medical Center in UtrechtUMC) and the Neuromed Neurosurgery Center for Epilepsy,ozzilli-Isernia, Italy. We examined a total of 29 surgical epilepsypecimens, six hippocampal sclerosis (HS), five hippocampal surgi-al specimens of patients without HS (non-HS; with a focal lesion notnvolving the hippocampus proper; no appreciable neuronal loss andeactive gliosis are observed in the hippocampus), six focal corticalysplasia (FCD) type IIB, six cortical tubers from patients with Tu-erous Sclerosis Complex (TSC), six ganglioglioma (GG). In sixatients (two FCD, two GG and two TSC) a significant amount oferilesional tissue (normal-appearing cortex/white matter adjacent to
he lesion) was resected as well. This material was carefully in-pected by microscopy, using both histological and immunocyto-hemical stainings (HE, luxol-PAS, GFAP, vimentin, neurofilament,
able 1. Summary of clinical details of cases studied according to pa
athology type (pm or s) Number of cases Mean age at s
S (pm) 6 26.3 (12–42)on-HS (s) 5 29.5 (18–41)CD IIB (s) 6 27.3 (14–48)
ortical tubers (TSC; s) 6 17.8 (5–35)
anglioglioma (GG; s) 6 32 (16–49)ontrol neocortex (pm) 6 31.6 (18–35)
HS, Hippocampal Sclerosis; FCD, Focal Cortical Dysplasia; TSC, T
euronal nuclear protein, NeuN and phosphorylated ribosomal pro- o
ein S6 and CD34) to differentiate the lesion (tumor or malformation)rom the perilesional control cortex, defined as cortical region adja-ent the lesion, but histologically normal (i.e. not containing tumorells, dysplastic neurons and without appreciable neuronal loss andeactive gliosis). Informed consent was obtained for the use of brainissue and for access to medical records for research purposes.issue was obtained and used in a manner compliant with theeclaration of Helsinki. The clinical characteristics derived from theatient’s medical records are summarized in Table 1. Patients un-erwent therapeutic surgical resection for refractory epilepsy and hadredominantly medically intractable complex partial seizures.
The HS specimens include four cases of classical HS (grade; Wyler et al., 1992; Mesial Temporal Sclerosis (MTS) type 1a;lumcke et al., 2007) and two cases of severe HS (grade IV;yler et al., 1992; MTS type 1b; Blumcke et al., 2007). For the
CD we followed the classification system proposed by Palmini etl. for grading the degree of FCD (Palmini et al., 2004). All patientsith cortical tubers fulfilled the diagnostic criteria for TSC (Gomezt al., 1999). For the GG we used the revised WHO classificationf tumors of the CNS (Louis et al., 2007). The patients undergoingpilepsy surgery predominantly had medically intractable complexartial seizures. In addition, normal-appearing control cortex/whiteatter was obtained at autopsy from six young adult controlatients (male/female: 3/3; mean age 31; range 18–35), without aistory of seizures or other neurological diseases. All autopsiesere performed within 12 h after death.
uman tissue preparation and immunocytochemistry
he tissue was fixed in 10% buffered formalin (autopsy tissue, for 2eeks; surgical specimens, for 24 hours). To detect differences in
abeling related to technical variables such as tissue fixation, we alsoested the antibodies in specimens of selected regions (temporalortex/ hippocampus) collected at autopsy and immediately fixed inormalin for 24 h (same fixation time used for the surgical speci-ens). Formalin fixed, paraffin-embedded tissue was sectioned at 6m and mounted on organosilane-coated slides (SIGMA, St. Louis,O, USA). Specimens were processed for haematoxylin/eosin andissl, as well as for immunocytochemical reactions.
Glial fibrillary acidic protein (GFAP; polyclonal rabbit, DAKO,lostrup, Denmark; 1:4000), vimentin (mouse clone V9, DAKO;:400), neuronal nuclear protein (NeuN; mouse clone MAB377,gG1; Chemicon, Temecula, CA, USA; 1:1000), neurofilamentNF, SMI311; Sternberger Monoclonals, Lutherville, MD; 1:1000),icrotubule-associated protein (MAP2; mouse clone HM2; Sigma
:100), (HLA)-DP, DQ, DR (mouse clone CR3/43; DAKO, Glostrup,enmark, 1:400), CD68 (mouse clone PG-M1, DAKO; 1:200) andD31 (mouse JC/70A; 1:100), were used in the routine immuno-ytochemical analysis of epilepsy specimens.
For the detection of tPA we used a mouse anti-human tPAAmerican Diagnostica Inc., Greenwich, CT, USA; 1:50; previ-
nge/y) Localization Mean duration of epilepsy (range/y)
Temporal 17 (11–32)Temporal 15.2 (6–22)Temporal (4) 19.2 (5–25)Frontal (2)Frontal (3) 13.5 (2.8–34)Temporal (2)Parietal (1)Temporal 16.1 (12–26)Temporal —
Sclerosis; pm, post-mortem; s, surgical specimens.
thology
urgery (ra
usly used and characterized; Teesalu et al., 2004), a rabbit

aaMcuGSutspsoosttmtdcDc
pFmwme
E
Apss(sitfhn(tg
tnnrfiuctae
W
Fh(tNaP(
(awsCmpHIftBcAlh(mp
Ra
FRgucm(wddbb
fcggrhgmm0ia(latosdustan
S
S(dA
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945 931
nti-human tPA (Santa Cruz Bio., CA, USA; 1:50) and a rabbitnti-human tPA (1:100; kindly provided by Dr. Lijnen, Center forolecular and Vascular Biology, Leuven, Belgium and previously
haracterized (Lijnen et al., 1988)). For the detection of uPA wesed a mouse anti-human uPA (American Diagnostica Inc.,reenwich, CT, USA; 1:50; previously used and characterized;chmitt et al., 1991; Wagner et al., 1996) and a rabbit anti-humanPA (1:500; provided by Dr. H.R. Lijnen and previously charac-
erized; Lijnen et al., 1988). To evaluate the specificity of thetaining the following control experiments were performed onaraffin-embedded human temporal cortex and hippocampalpecimens: (1) omission of the primary antibody; (2) substitutionf the primary antibody with a rabbit pre-immune or non-immune IgGr a monoclonal mouse IgG of irrelevant specificity; and (3) pread-orption of tPA and uPA polyclonal antibodies using an excess ofPA- and uPA antigen, or using the specific immunogen peptide forhe anti-human tPA (from Santa Cruz Bio). These control experi-
ents resulted in the absence of staining. Immunocytochemis-ry was carried out on paraffin-embedded tissue as previouslyescribed (Aronica et al., 2001b). Single-label immunocyto-hemistry was performed with Powervision (Immunologic,uiven, The Netherlands). 3,3-Diaminobenzidine was used ashromogen. Sections were counterstained with Haematoxylin.
For double-labeling studies, sections, after incubation withrimary antibodies, were incubated for 2 h at RT with Alexaluor® 568 and Alexa Fluor® 488 (anti-rabbit IgG or anti-ouse IgG; 1:200; Molecular probes, Eugene, USA). Sectionsere then analyzed by means of a laser scanning confocalicroscope (Bio-Rad, Hercules, CA, USA; MRC1024)quipped with an argon-ion laser.
valuation of immunostaining
ll labeled tissue sections were evaluated with respect to theresence or absence of various histopathological parameters andpecific immunoreactivity (IR) for the different markers. The inten-ity of tPA and uPA staining was evaluated using a scale of 0–30: no; 1: weak; 2: moderate; 3: strong staining). All areas of thepecimen were examined and the score represents the predom-nant cell staining intensity found in each case for the different cellypes (neurons, astrocytes, microglial cells and balloon cells). Therequency of tPA and uPA positive cells ((1) rare; (2) sparse; (3)igh) was also evaluated to give information about the relativeumber of positive cells within the specimen. As proposed beforeVandeputte et al., 2002; Ravizza et al., 2006), the product ofhese two values (intensity and frequency scores) was taken toive the overall score (total score).
To analyze the percentage of GFAP or NeuN positive cellshat express tPA or uPA within the epileptogenic lesions (HS,�5; FCD, n�5) in each specimens two representative adjacenton-overlapping fields of the areas of interest (CA1, CA3 and hilaregion for HS; dysplastic cortex for FCD) were captured (magni-cation 40�; total area of each field: 165.250 �m2) and digitizedsing a laser scanning confocal microscope. The total number ofells stained with tPA (or uPA) and GFAP (or NeuN), as well ashe number of double labelled cells were counted and percent-ges were calculated (expressed as mean�SEM) of cells co-xpressing tPA (or uPA) and GFAP (or NeuN).
estern blot analysis
or immunoblot analysis, freshly frozen histologically normal autopsyippocampus and cortex (n�5), HS (n�6), GG (n�6) and FCDn�5) specimens; samples were homogenized in lysis buffer con-aining 10 mM Tris (pH 8.0), 150 mM NaCl, 10% glycerol, 1% NP-40,a-orthevanadate (10.4 mg/ml), 5 mM EDTA (pH 8.0), 5 mM NaFnd protease inhibitor cocktail (Boehringer Mannheim, Germany).rotein content was determined using the bicinchoninic acid method
Smith et al., 1985). For electrophoresis, equal amounts of proteins M
30 �g/lane) were separated by sodium dodecylsulfate-polyacryl-mide gel electrophoretic (SDS-PAGE) analysis. Separated proteinsere transferred to nitrocellulose paper for 1 h and 30 min, using aemi-dry electroblotting system (BioRad, Transblot SD, Hercules,A, USA). Blots were incubated over night in TTBS (20 mM Tris, 150M NaCl, 0.1% Tween, pH 7.5)/5% non fat dry milk, containing therimary antibody (tPA, rabbit anti-human, 1:1000, provided by Dr..R. Lijnen; uPA mouse anti-human, 1:500, American Diagnostica
nc; uPA receptor (uPAR), 1:5000, American Diagnostica Inc., usingor uPAR non-reducing conditions). After several washes in TTBS,he membranes were incubated in TTBS/5% non fat dry milk/1%SA, containing the goat anti-rabbit or goat anti-mouse antibodiesoupled to horse radish peroxidase (1:2500; Dako, Denmark) for 1 h.fter washes in TTBS, immunoreactivity was visualized using Lumi–
ight PLUS western blotting substrate (Roche Diagnostics, Mann-eim, Germany) and digitized using a Luminescent Image AnalyzerLAS-3000, Fuji Film, Japan). Expression of �-actin (monoclonalouse, Sigma, St. Louis, MO, USA 1:50.000) was used as referencerotein.
NA isolation and real-time quantitative PCRnalysis (RT-PCR)
or RNA isolation, frozen material was homogenized in Trizol LSeagent (Invitrogen, Carlsbad, CA, USA). After addition of 200 �glycogen and 200 �l chloroform, the aqueous phase was isolatedsing Phase Lock tubes (Eppendorf, Hamburg, Germany). Theoncentration and purity of RNA were determined spectrophoto-etrically at 260/280 nm using a nanodrop spectrophotometer
Ocean Optics, Dunedin, FL, USA). Five micrograms of total RNAere reverse-transcribed into cDNA using oligo dT primers asescribed previously (Aronica et al., 2004). Specific primers wereesigned for the genes of interest using the Universal ProbeLi-rary of Roche (https://www.roche-applied-science.com) on theasis of the reported cDNA sequences.
The following primers were used: TATA-binding protein (TBP;orward: caggagccaagagtgaagaac; reverse: aggaaataactctggct-ataactact), tPA (forward: cgggtggaatattgctggt; reverse: cccgtt-aaacaccttgg), uPA (forward: ttgctcaccacaacgacatt; reverse:gcaggcagatggtctgtat), uPAR (forward: acaccaccaaatgcaacga;everse: ccccttgcagctgtaacac) and the plasminogen activator in-ibitor type-1 (PAI1; forward: tccagcagctgaattcctg; reverse: gctg-agacatctgcatcct). For 1 �l sample a Light Cycler SYBR green Iaster mix (Roche Applied Sciences, Indianapolis, IN, USA) wasade containing in a total volume of 5 �l: 0.2 �l forward primer;.2 �l reverse primer and 2.5 �l master mix. A total of 6 �l was run
n duplicates in a 384 -well plate (Roche Applied Sciences, Indi-napolis, IN, USA) in the LighCycler® 480 Real-Time PCR SystemRoche Applied Sciences, Indianapolis, IN, USA) under the fol-owing conditions: A 5 minute denaturing step at 95 °C followed by
total of 45 amplification cycles consisting of 15 seconds dena-uring at 95 °C, 5 seconds of annealing at 60 °C and 10 secondsf extension at 72 °C. Fluorescent product was measured by aingle acquisition mode at 72 °C after each cycle. Quantification ofata was performed described previously (Aronica et al., 2008),sing the computer program LinRegPCR in which linear regres-ion on the Log (fluorescence) per cycle number data are appliedo determine the amplification efficiency per sample (Ramakers etl., 2003). The starting concentration of each specific product wasormalised to the amount of the reference gene TBP.
tatistical analysis
tatistical analyses were performed with SPSS for WindowsSPSS 11.5, SPSS Inc., Chicago, IL, USA) using two-tailed Stu-ent’s t-test; to assess differences between more than two groupsNOVA and a non-parametric Kruskal–Wallis test followed by
ann–Whitney U-test. P�0.05 was considered significant.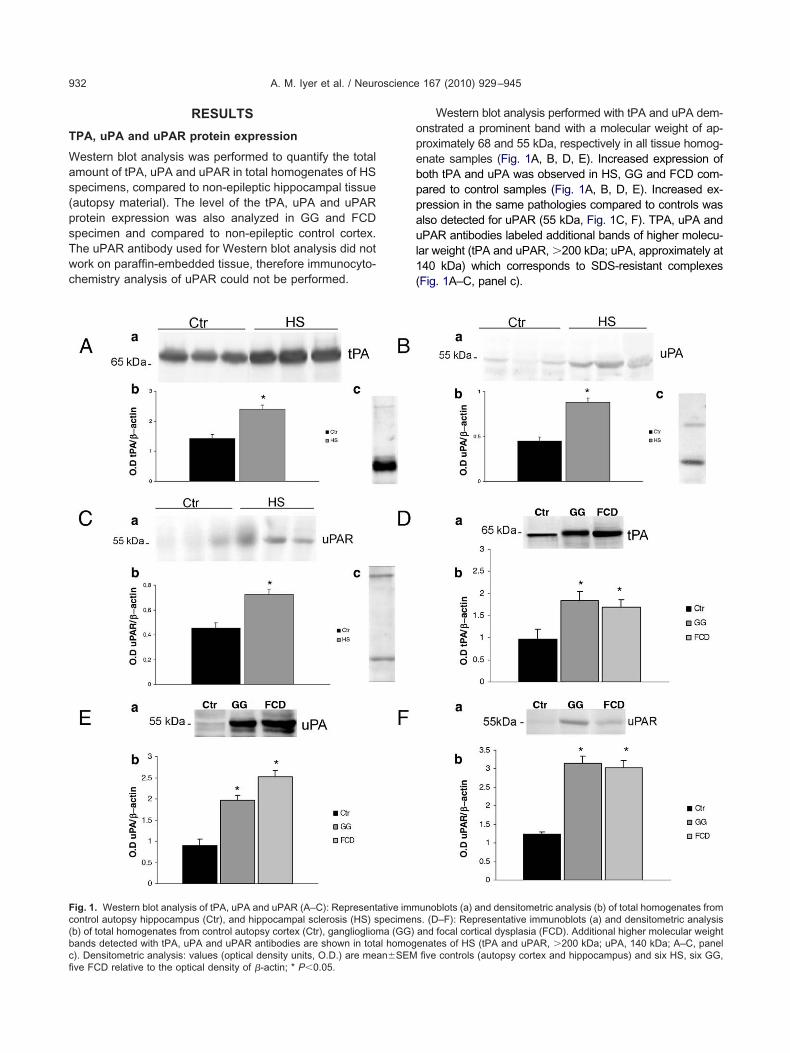
T
Was(psTwc
opebppaul1(
Fc(bcfi
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945932
RESULTS
PA, uPA and uPAR protein expression
estern blot analysis was performed to quantify the totalmount of tPA, uPA and uPAR in total homogenates of HSpecimens, compared to non-epileptic hippocampal tissueautopsy material). The level of the tPA, uPA and uPARrotein expression was also analyzed in GG and FCDpecimen and compared to non-epileptic control cortex.he uPAR antibody used for Western blot analysis did notork on paraffin-embedded tissue, therefore immunocyto-hemistry analysis of uPAR could not be performed.
ig. 1. Western blot analysis of tPA, uPA and uPAR (A–C): Representontrol autopsy hippocampus (Ctr), and hippocampal sclerosis (HS) sb) of total homogenates from control autopsy cortex (Ctr), gangliogliomands detected with tPA, uPA and uPAR antibodies are shown in tota
). Densitometric analysis: values (optical density units, O.D.) are mean�SEMve FCD relative to the optical density of �-actin; * P�0.05.Western blot analysis performed with tPA and uPA dem-nstrated a prominent band with a molecular weight of ap-roximately 68 and 55 kDa, respectively in all tissue homog-nate samples (Fig. 1A, B, D, E). Increased expression ofoth tPA and uPA was observed in HS, GG and FCD com-ared to control samples (Fig. 1A, B, D, E). Increased ex-ression in the same pathologies compared to controls waslso detected for uPAR (55 kDa, Fig. 1C, F). TPA, uPA andPAR antibodies labeled additional bands of higher molecu-
ar weight (tPA and uPAR, �200 kDa; uPA, approximately at40 kDa) which corresponds to SDS-resistant complexesFig. 1A–C, panel c).
unoblots (a) and densitometric analysis (b) of total homogenates froms. (D–F): Representative immunoblots (a) and densitometric analysisand focal cortical dysplasia (FCD). Additional higher molecular weightenates of HS (tPA and uPAR, �200 kDa; uPA, 140 kDa; A–C, panel
ative immpecimena (GG)
l homog
five controls (autopsy cortex and hippocampus) and six HS, six GG,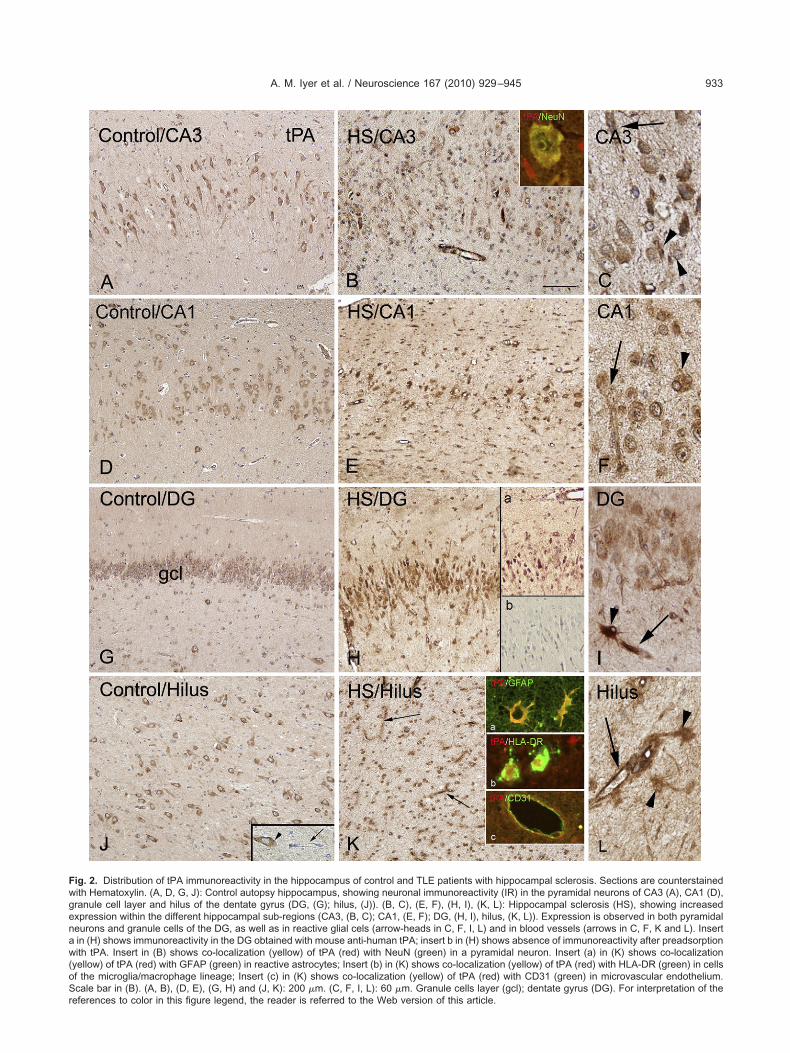
Fwgenaw(oSr
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945 933
ig. 2. Distribution of tPA immunoreactivity in the hippocampus of control and TLE patients with hippocampal sclerosis. Sections are counterstainedith Hematoxylin. (A, D, G, J): Control autopsy hippocampus, showing neuronal immunoreactivity (IR) in the pyramidal neurons of CA3 (A), CA1 (D),ranule cell layer and hilus of the dentate gyrus (DG, (G); hilus, (J)). (B, C), (E, F), (H, I), (K, L): Hippocampal sclerosis (HS), showing increasedxpression within the different hippocampal sub-regions (CA3, (B, C); CA1, (E, F); DG, (H, I), hilus, (K, L)). Expression is observed in both pyramidaleurons and granule cells of the DG, as well as in reactive glial cels (arrow-heads in C, F, I, L) and in blood vessels (arrows in C, F, K and L). Insertin (H) shows immunoreactivity in the DG obtained with mouse anti-human tPA; insert b in (H) shows absence of immunoreactivity after preadsorptionith tPA. Insert in (B) shows co-localization (yellow) of tPA (red) with NeuN (green) in a pyramidal neuron. Insert (a) in (K) shows co-localization
yellow) of tPA (red) with GFAP (green) in reactive astrocytes; Insert (b) in (K) shows co-localization (yellow) of tPA (red) with HLA-DR (green) in cellsf the microglia/macrophage lineage; Insert (c) in (K) shows co-localization (yellow) of tPA (red) with CD31 (green) in microvascular endothelium.cale bar in (B). (A, B), (D, E), (G, H) and (J, K): 200 �m. (C, F, I, L): 60 �m. Granule cells layer (gcl); dentate gyrus (DG). For interpretation of the
eferences to color in this figure legend, the reader is referred to the Web version of this article.
Ts
TltathhPmbtst
tpHi
uppgsp
ssdE(c3th
ne
F((n
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945934
PA IR in control hippocampus and in hippocampalclerosis
o evaluate the changes in expression and the cellularocalization of tPA in the hippocampus of patients withemporal lobe epilepsy (TLE), immunocytochemicalnalysis was performed in surgical specimens of pa-
ients with HS and in surgical and autopsy specimens ofistologically normal hippocampus. Three different anti-uman tPA antibodies were used (see Experimentalrocedures), with similar results; Fig. 2 shows the im-unostaining performed with anti-tPA polyclonal anti-ody ((Lijnen et al., 1988); a representative micropho-ograph of the dentate gyrus (DG) in HS specimenstained with the monoclonal antibody against humanPA is shown in panel H, insert a).
In agreement with previous observations in humanissue (Teesalu et al., 2004) in histologically normal hip-ocampus (autopsy and non-sclerotic hippocampus, non-S), tPA IR was mainly observed in neuronal cells, includ-
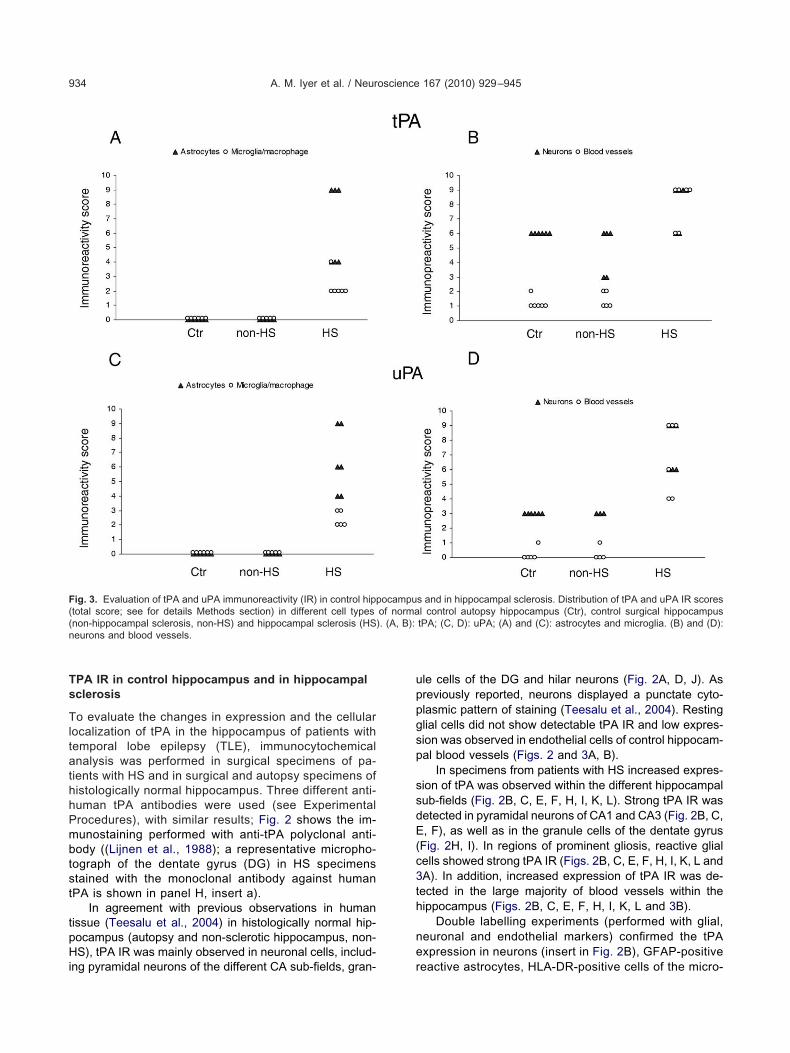
ig. 3. Evaluation of tPA and uPA immunoreactivity (IR) in control hipptotal score; see for details Methods section) in different cell typesnon-hippocampal sclerosis, non-HS) and hippocampal sclerosis (HSeurons and blood vessels.
ng pyramidal neurons of the different CA sub-fields, gran- r
le cells of the DG and hilar neurons (Fig. 2A, D, J). Asreviously reported, neurons displayed a punctate cyto-lasmic pattern of staining (Teesalu et al., 2004). Restinglial cells did not show detectable tPA IR and low expres-ion was observed in endothelial cells of control hippocam-al blood vessels (Figs. 2 and 3A, B).
In specimens from patients with HS increased expres-ion of tPA was observed within the different hippocampalub-fields (Fig. 2B, C, E, F, H, I, K, L). Strong tPA IR wasetected in pyramidal neurons of CA1 and CA3 (Fig. 2B, C,, F), as well as in the granule cells of the dentate gyrus
Fig. 2H, I). In regions of prominent gliosis, reactive glialells showed strong tPA IR (Figs. 2B, C, E, F, H, I, K, L andA). In addition, increased expression of tPA IR was de-ected in the large majority of blood vessels within theippocampus (Figs. 2B, C, E, F, H, I, K, L and 3B).
Double labelling experiments (performed with glial,euronal and endothelial markers) confirmed the tPAxpression in neurons (insert in Fig. 2B), GFAP-positive
s and in hippocampal sclerosis. Distribution of tPA and uPA IR scoresl control autopsy hippocampus (Ctr), control surgical hippocampustPA; (C, D): uPA; (A) and (C): astrocytes and microglia. (B) and (D):
ocampuof norma). (A, B):
eactive astrocytes, HLA-DR-positive cells of the micro-

gi
er9tC
Tl
Tcdicsib
tit(da
Tpbvgdeleco
cpi
Us
TlTgaTEsccmH
h
dtsaGat
ssdE(cima
npHe
er7uC
Ul
Tccstosj
ctgIss
TprbaGlafi
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945 935
lia/macrophage lineage and in endothelial cells (insertsn Fig. 2K).
The percentage of glial cells positive for tPA and co-xpressing GFAP was quantified in CA1, CA3 and hilaregion in five HS specimens (84�5, CA1; 86�6, CA3;7�3, hilus); the percentage of neuronal cells positive forPA and co-expressing NeuN was quantified in CA1 andA3 (98�3, CA1; 97�4, CA3).
PA IR in control cortex and focal developmentalesions
o evaluate the occurrence of changes in expression andellular localization of tPA in focal malformations of corticalevelopment (MCD), we studied tPA expression patterns
n surgical specimens of patients with FCD (type IIB),ortical tubers (TSC), GG and in surgical and autopsypecimens of histologically normal cortex. Fig. 4 shows themmunostaining performed with anti-tPA polyclonal anti-ody (Lijnen et al., 1988).
Low tPA staining was observed within the normal cor-ex (autopsy material; Fig. 4A, B). The staining was local-zed in pyramidal neurons; resting glial cells did not showPA IR and low expression was observed in blood vesselsand Fig. 5A, B). Histologically normal perilesional cortexisplayed a pattern of IR similar to that observed in controlutopsy cortex.
In FCD and TSC cases (Fig. 4C–E, FCD; Fig. 4F,SC), intense tPA IR was observed throughout the dys-lastic cortex. Staining was detected in dysplastic neurons,alloon and giant cells, reactive glial cells and in bloodessels (Figs. 4C–F and 5A, B). Both the neuronal andlial components, as well as the blood vessels of GGisplayed tPA IR (Figs. 4G and 5A, B). Double labellingxperiments (performed with glial, neuronal and endothe-
ial markers in FCD, TSC and GG) confirmed the tPAxpression in neurons, GFAP-positive cells, in endothelialells (inserts in Fig. 4), as well as in HLA-DR-positive cellsf the microglia/macrophage lineage (not shown).
The percentage of glial cells positive for tPA ando-expressing GFAP (89�5) and neuronal cells co-ex-ressing NeuN (96�4) was quantified in five FCD spec-
mens.
PA IR in control hippocampus and in hippocampalclerosis
o evaluate the changes in expression and the cellularocalization of uPA in the hippocampus of patients withLE, immunocytochemical analysis was performed in sur-ical specimens of patients with HS and in surgical andutopsy specimens of histologically normal hippocampus.hree different anti-human uPA antibodies were used (seexperimental Procedures), with similar results; Fig. 6hows the immunostaining performed with anti-uPA poly-lonal antibody (Lijnen et al., 1988); a representative mi-rophotograph of the DG in HS specimens stained with theonoclonal antibody against human uPA is shown in panel, insert (a).
In agreement with previous observations in control rat
ippocampus (Lahtinen et al., 2006), low uPA IR was tetected in histologically normal human hippocampus (au-opsy and non-sclerotic hippocampus, non-HS). The inten-ity of staining was light in CA1-3 pyramidal cells, as wells in granule cells of the DG and hilar neurons (Fig. 6A, D,, J). Resting glial cells did not show detectable uPA IRnd low expression was observed occasionally in endo-helial cells (Figs. 6 and 3C, D).
In specimens from patients with HS increased expres-ion of uPA was observed within the different hippocampalub-fields (Fig. 6B, C, E, F, H, I, K, L). Strong uPA IR wasetected in pyramidal neurons of CA1 and CA3 (Fig. 6B, C,, F), as well as in the granule cells of the dentate gyrus
Fig. 6H, I). In regions of prominent gliosis, reactive glialells showed strong uPA IR (Figs. 6L and 3C). In addition,
ncreased expression of uPA IR was detected in the largeajority of blood vessels within the hippocampus (Figs. 6Knd 3D).
Double labelling experiments (performed with glial,euronal and endothelial markers) confirmed the uPA ex-ression in neurons, GFAP-positive reactive astrocytes,LA-DR-positive cells of the microglia/macrophage lin-age and in endothelial cells (inserts in Fig. 6K).
The percentage of glial cells positive for uPA and co-xpressing GFAP was quantified in CA1, CA3 and hilaregion in five HS specimens (74�2, CA1; 73�5, CA3;9�4, hilus); the percentage of neuronal cells positive forPA and co-expressing NeuN was quantified in CA1 andA3 (97�3, CA1; 98�2, CA3).
PA IR in control cortex and focal developmentalesions
o evaluate the occurrence of changes in expression andellular localization of uPA in focal malformations of corti-al development, we studied uPA expression patterns inurgical specimens of patients with FCD (type IIB), corticalubers (TSC), GG and in surgical and autopsy specimensf histologically normal cortex. Fig. 7 shows the immuno-taining performed with anti-uPA polyclonal antibody (Li-nen et al., 1988).
Very low uPA staining was observed within the normalortex (autopsy material; Fig. 7A, B). Occasionally scat-ered lightly labeled neurons were detected, but restinglial cells and blood vessels did not display detectable uPAR (Figs. 7A, B and 5C, D). Histologically normal perile-ional cortex displayed a pattern of IR similar to that ob-erved in control autopsy cortex.
In FCD and TSC cases (Fig. 7C–E, FCD; Fig. 7F,SC), intense uPA IR was observed throughout the dys-lastic cortex. Staining was detected in dysplastic neu-ons, balloon and giant cells, reactive glial cells and inlood vessels (Figs. 7C–F and 5C, D). Both the neuronalnd glial components, as well as the blood vessels ofG displayed uPA IR (Figs. 7G and 5C, D). Double
abelling experiments (performed with glial, neuronalnd endothelial markers in FCD, TSC and GG) con-rmed the uPA expression in neurons and GFAP-posi-
ive cells, in endothelial cells, as well as in HLA-DR-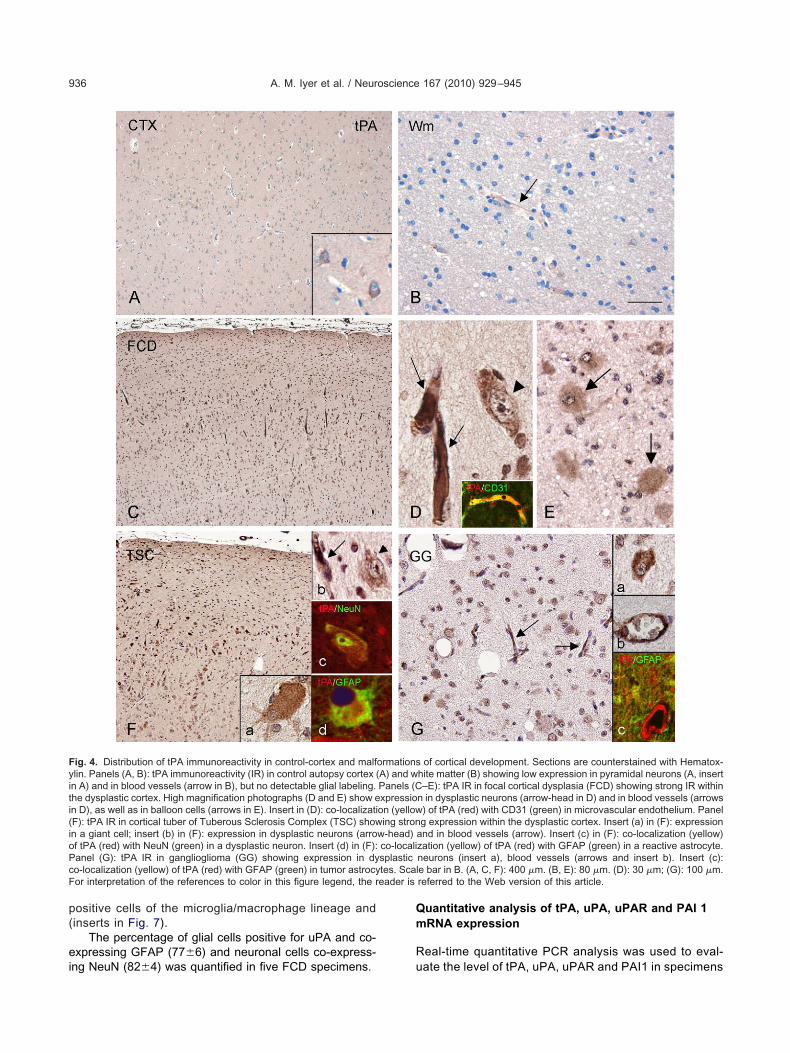
p(
ei
Qm
R
Fyiti(ioPcF eader is
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945936
ositive cells of the microglia/macrophage lineage andinserts in Fig. 7).
The percentage of glial cells positive for uPA and co-xpressing GFAP (77�6) and neuronal cells co-express-
ig. 4. Distribution of tPA immunoreactivity in control-cortex and malflin. Panels (A, B): tPA immunoreactivity (IR) in control autopsy cortex (n A) and in blood vessels (arrow in B), but no detectable glial labeling.he dysplastic cortex. High magnification photographs (D and E) show en D), as well as in balloon cells (arrows in E). Insert in (D): co-localizatiF): tPA IR in cortical tuber of Tuberous Sclerosis Complex (TSC) shown a giant cell; insert (b) in (F): expression in dysplastic neurons (arrof tPA (red) with NeuN (green) in a dysplastic neuron. Insert (d) in (F):anel (G): tPA IR in ganglioglioma (GG) showing expression in dyo-localization (yellow) of tPA (red) with GFAP (green) in tumor astrocyor interpretation of the references to color in this figure legend, the r
ng NeuN (82�4) was quantified in five FCD specimens. u
uantitative analysis of tPA, uPA, uPAR and PAI 1RNA expression
eal-time quantitative PCR analysis was used to eval-
of cortical development. Sections are counterstained with Hematox-hite matter (B) showing low expression in pyramidal neurons (A, insert–E): tPA IR in focal cortical dysplasia (FCD) showing strong IR within
n in dysplastic neurons (arrow-head in D) and in blood vessels (arrows) of tPA (red) with CD31 (green) in microvascular endothelium. Panelg expression within the dysplastic cortex. Insert (a) in (F): expression
and in blood vessels (arrow). Insert (c) in (F): co-localization (yellow)zation (yellow) of tPA (red) with GFAP (green) in a reactive astrocyte.neurons (insert a), blood vessels (arrows and insert b). Insert (c):e bar in B. (A, C, F): 400 �m. (B, E): 80 �m. (D): 30 �m; (G): 100 �m.referred to the Web version of this article.
ormationsA) and wPanels (Cxpressioon (yellowing stron
w-head)co-localisplastictes. Scal
ate the level of tPA, uPA, uPAR and PAI1 in specimens
omwt8
DcotGwsh
hie
tmepto1sab
H
Hcsigt
FMS , D): uPAv
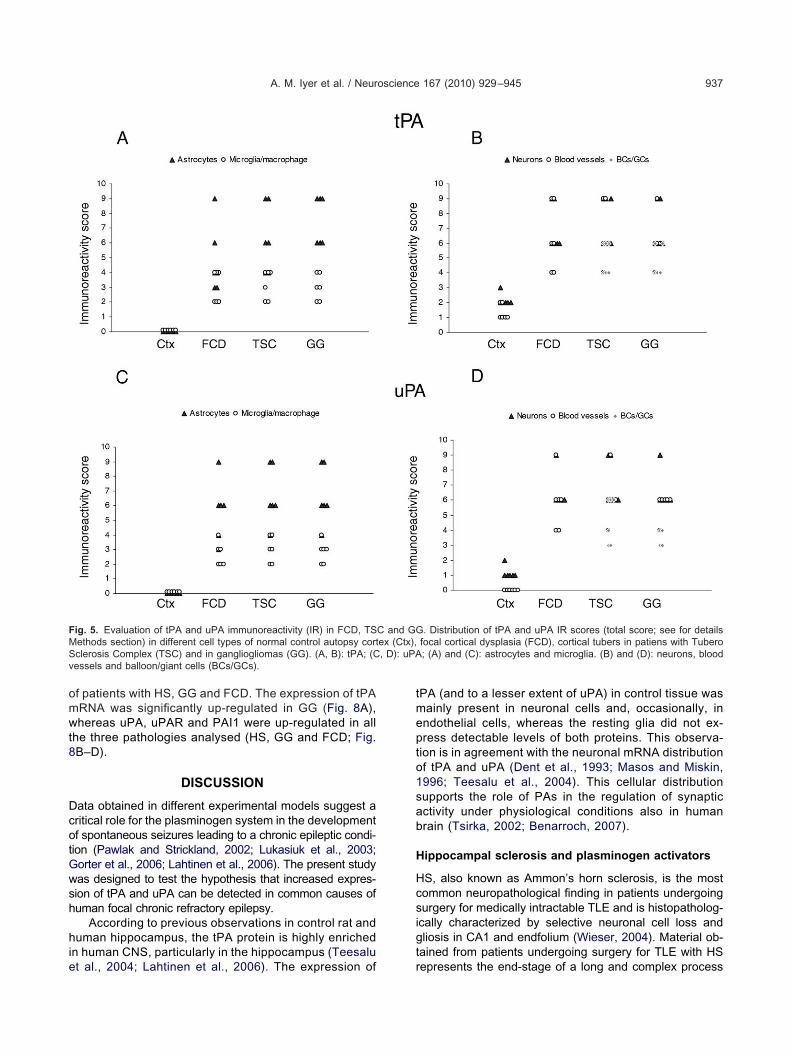
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945 937
f patients with HS, GG and FCD. The expression of tPARNA was significantly up-regulated in GG (Fig. 8A),hereas uPA, uPAR and PAI1 were up-regulated in all
he three pathologies analysed (HS, GG and FCD; Fig.B–D).
DISCUSSION
ata obtained in different experimental models suggest aritical role for the plasminogen system in the developmentf spontaneous seizures leading to a chronic epileptic condi-
ion (Pawlak and Strickland, 2002; Lukasiuk et al., 2003;orter et al., 2006; Lahtinen et al., 2006). The present studyas designed to test the hypothesis that increased expres-ion of tPA and uPA can be detected in common causes ofuman focal chronic refractory epilepsy.
According to previous observations in control rat anduman hippocampus, the tPA protein is highly enriched
n human CNS, particularly in the hippocampus (Teesalu
ig. 5. Evaluation of tPA and uPA immunoreactivity (IR) in FCD, TSethods section) in different cell types of normal control autopsy cortclerosis Complex (TSC) and in gangliogliomas (GG). (A, B): tPA; (Cessels and balloon/giant cells (BCs/GCs).
t al., 2004; Lahtinen et al., 2006). The expression of r
PA (and to a lesser extent of uPA) in control tissue wasainly present in neuronal cells and, occasionally, inndothelial cells, whereas the resting glia did not ex-ress detectable levels of both proteins. This observa-
ion is in agreement with the neuronal mRNA distributionf tPA and uPA (Dent et al., 1993; Masos and Miskin,996; Teesalu et al., 2004). This cellular distributionupports the role of PAs in the regulation of synapticctivity under physiological conditions also in humanrain (Tsirka, 2002; Benarroch, 2007).
ippocampal sclerosis and plasminogen activators
S, also known as Ammon’s horn sclerosis, is the mostommon neuropathological finding in patients undergoingurgery for medically intractable TLE and is histopatholog-
cally characterized by selective neuronal cell loss andliosis in CA1 and endfolium (Wieser, 2004). Material ob-ained from patients undergoing surgery for TLE with HS
. Distribution of tPA and uPA IR scores (total score; see for detailsfocal cortical dysplasia (FCD), cortical tubers in patiens with Tubero; (A) and (C): astrocytes and microglia. (B) and (D): neurons, blood
C and GGex (Ctx),
epresents the end-stage of a long and complex process
Fw(i(ciIo(d
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945938
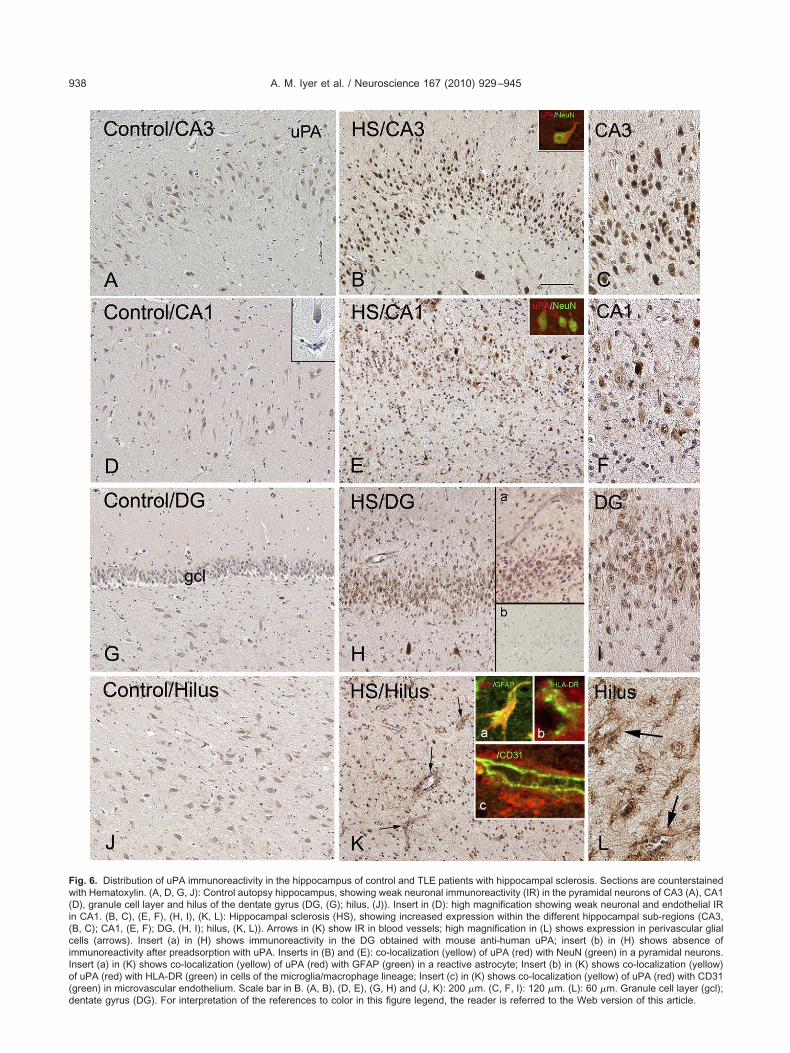
ig. 6. Distribution of uPA immunoreactivity in the hippocampus of control and TLE patients with hippocampal sclerosis. Sections are counterstainedith Hematoxylin. (A, D, G, J): Control autopsy hippocampus, showing weak neuronal immunoreactivity (IR) in the pyramidal neurons of CA3 (A), CA1
D), granule cell layer and hilus of the dentate gyrus (DG, (G); hilus, (J)). Insert in (D): high magnification showing weak neuronal and endothelial IRn CA1. (B, C), (E, F), (H, I), (K, L): Hippocampal sclerosis (HS), showing increased expression within the different hippocampal sub-regions (CA3,B, C); CA1, (E, F); DG, (H, I); hilus, (K, L)). Arrows in (K) show IR in blood vessels; high magnification in (L) shows expression in perivascular glialells (arrows). Insert (a) in (H) shows immunoreactivity in the DG obtained with mouse anti-human uPA; insert (b) in (H) shows absence ofmmunoreactivity after preadsorption with uPA. Inserts in (B) and (E): co-localization (yellow) of uPA (red) with NeuN (green) in a pyramidal neurons.nsert (a) in (K) shows co-localization (yellow) of uPA (red) with GFAP (green) in a reactive astrocyte; Insert (b) in (K) shows co-localization (yellow)f uPA (red) with HLA-DR (green) in cells of the microglia/macrophage lineage; Insert (c) in (K) shows co-localization (yellow) of uPA (red) with CD31green) in microvascular endothelium. Scale bar in B. (A, B), (D, E), (G, H) and (J, K): 200 �m. (C, F, I): 120 �m. (L): 60 �m. Granule cell layer (gcl);
entate gyrus (DG). For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.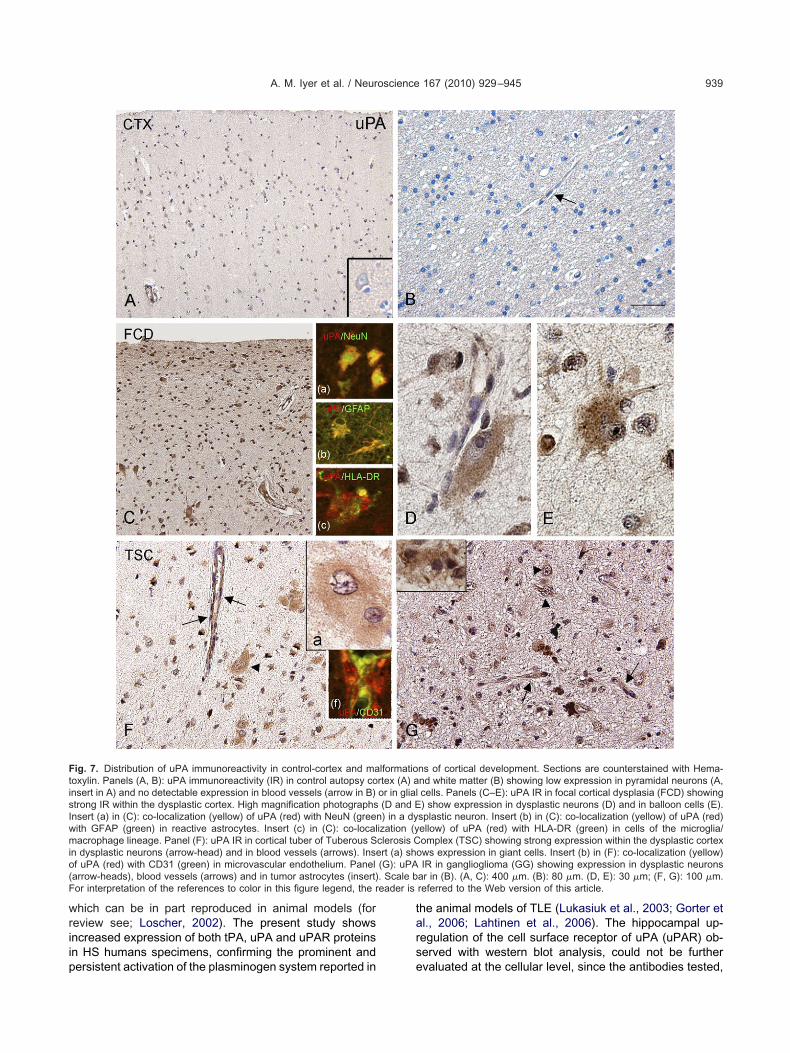
wriip
tars
FtisIwmio(F eader is
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945 939
hich can be in part reproduced in animal models (foreview see; Loscher, 2002). The present study showsncreased expression of both tPA, uPA and uPAR proteinsn HS humans specimens, confirming the prominent and
ig. 7. Distribution of uPA immunoreactivity in control-cortex and maoxylin. Panels (A, B): uPA immunoreactivity (IR) in control autopsy consert in A) and no detectable expression in blood vessels (arrow in B)trong IR within the dysplastic cortex. High magnification photographsnsert (a) in (C): co-localization (yellow) of uPA (red) with NeuN (greenith GFAP (green) in reactive astrocytes. Insert (c) in (C): co-localacrophage lineage. Panel (F): uPA IR in cortical tuber of Tuberous S
n dysplastic neurons (arrow-head) and in blood vessels (arrows). Insef uPA (red) with CD31 (green) in microvascular endothelium. Panelarrow-heads), blood vessels (arrows) and in tumor astrocytes (insert)or interpretation of the references to color in this figure legend, the r
ersistent activation of the plasminogen system reported in e
he animal models of TLE (Lukasiuk et al., 2003; Gorter etl., 2006; Lahtinen et al., 2006). The hippocampal up-egulation of the cell surface receptor of uPA (uPAR) ob-erved with western blot analysis, could not be further
ns of cortical development. Sections are counterstained with Hema-nd white matter (B) showing low expression in pyramidal neurons (A,cells. Panels (C–E): uPA IR in focal cortical dysplasia (FCD) showing) show expression in dysplastic neurons (D) and in balloon cells (E).
splastic neuron. Insert (b) in (C): co-localization (yellow) of uPA (red)ellow) of uPA (red) with HLA-DR (green) in cells of the microglia/omplex (TSC) showing strong expression within the dysplastic cortex
ows expression in giant cells. Insert (b) in (F): co-localization (yellow)IR in ganglioglioma (GG) showing expression in dysplastic neurons
ar in (B). (A, C): 400 �m. (B): 80 �m. (D, E): 30 �m; (F, G): 100 �m.referred to the Web version of this article.
lformatiortex (A) aor in glial(D and E) in a dy
ization (yclerosis Crt (a) sh(G): uPA. Scale b
valuated at the cellular level, since the antibodies tested,
webashpa
rltrnttscta
scoi2isr
uPachcd2dcsts(
rmblet(e2ephw
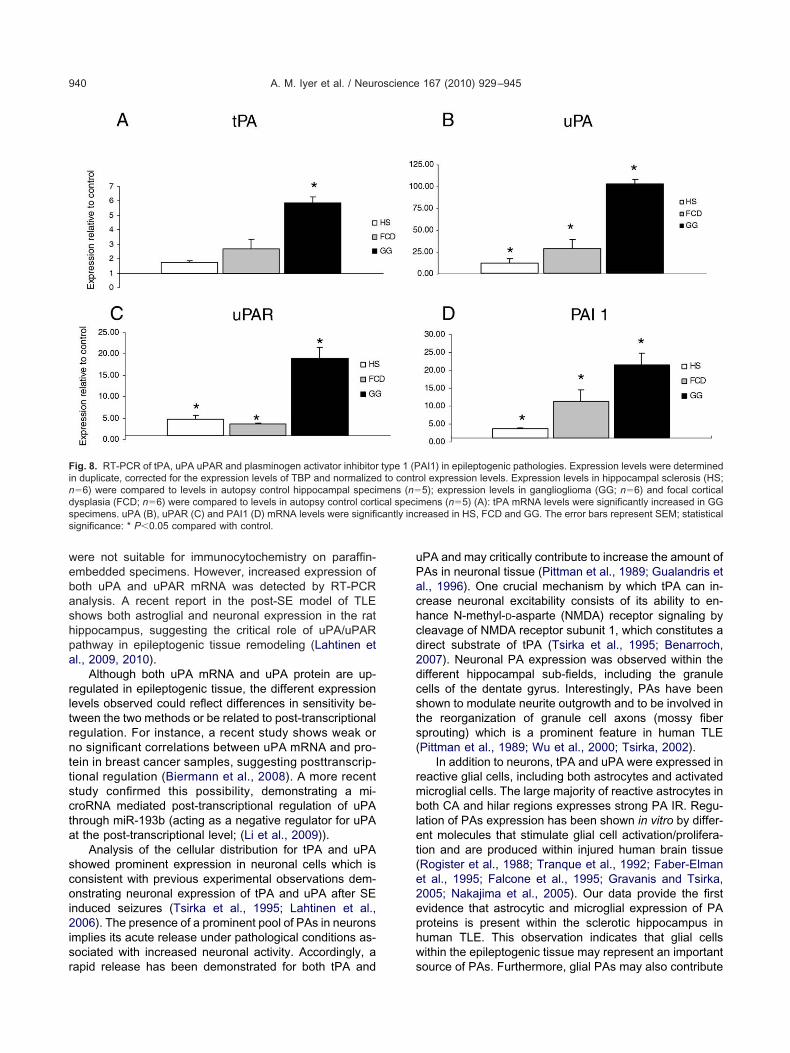
Finds cantly incs
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945940
ere not suitable for immunocytochemistry on paraffin-mbedded specimens. However, increased expression ofoth uPA and uPAR mRNA was detected by RT-PCRnalysis. A recent report in the post-SE model of TLEhows both astroglial and neuronal expression in the ratippocampus, suggesting the critical role of uPA/uPARathway in epileptogenic tissue remodeling (Lahtinen etl., 2009, 2010).
Although both uPA mRNA and uPA protein are up-egulated in epileptogenic tissue, the different expressionevels observed could reflect differences in sensitivity be-ween the two methods or be related to post-transcriptionalegulation. For instance, a recent study shows weak oro significant correlations between uPA mRNA and pro-ein in breast cancer samples, suggesting posttranscrip-ional regulation (Biermann et al., 2008). A more recenttudy confirmed this possibility, demonstrating a mi-roRNA mediated post-transcriptional regulation of uPAhrough miR-193b (acting as a negative regulator for uPAt the post-transcriptional level; (Li et al., 2009)).
Analysis of the cellular distribution for tPA and uPAhowed prominent expression in neuronal cells which isonsistent with previous experimental observations dem-nstrating neuronal expression of tPA and uPA after SE
nduced seizures (Tsirka et al., 1995; Lahtinen et al.,006). The presence of a prominent pool of PAs in neurons
mplies its acute release under pathological conditions as-ociated with increased neuronal activity. Accordingly, a
ig. 8. RT-PCR of tPA, uPA uPAR and plasminogen activator inhibitorn duplicate, corrected for the expression levels of TBP and normalize�6) were compared to levels in autopsy control hippocampal speciysplasia (FCD; n�6) were compared to levels in autopsy control cortpecimens. uPA (B), uPAR (C) and PAI1 (D) mRNA levels were signifiignificance: * P�0.05 compared with control.
apid release has been demonstrated for both tPA and s
PA and may critically contribute to increase the amount ofAs in neuronal tissue (Pittman et al., 1989; Gualandris etl., 1996). One crucial mechanism by which tPA can in-rease neuronal excitability consists of its ability to en-ance N-methyl-D-asparte (NMDA) receptor signaling byleavage of NMDA receptor subunit 1, which constitutes airect substrate of tPA (Tsirka et al., 1995; Benarroch,007). Neuronal PA expression was observed within theifferent hippocampal sub-fields, including the granuleells of the dentate gyrus. Interestingly, PAs have beenhown to modulate neurite outgrowth and to be involved inhe reorganization of granule cell axons (mossy fiberprouting) which is a prominent feature in human TLEPittman et al., 1989; Wu et al., 2000; Tsirka, 2002).
In addition to neurons, tPA and uPA were expressed ineactive glial cells, including both astrocytes and activatedicroglial cells. The large majority of reactive astrocytes inoth CA and hilar regions expresses strong PA IR. Regu-
ation of PAs expression has been shown in vitro by differ-nt molecules that stimulate glial cell activation/prolifera-ion and are produced within injured human brain tissueRogister et al., 1988; Tranque et al., 1992; Faber-Elmant al., 1995; Falcone et al., 1995; Gravanis and Tsirka,005; Nakajima et al., 2005). Our data provide the firstvidence that astrocytic and microglial expression of PAroteins is present within the sclerotic hippocampus inuman TLE. This observation indicates that glial cellsithin the epileptogenic tissue may represent an important
AI1) in epileptogenic pathologies. Expression levels were determinedol expression levels. Expression levels in hippocampal sclerosis (HS;5); expression levels in ganglioglioma (GG; n�6) and focal cortical
mens (n�5) (A): tPA mRNA levels were significantly increased in GGreased in HS, FCD and GG. The error bars represent SEM; statistical
type 1 (Pd to contrmens (n�ical speci
ource of PAs. Furthermore, glial PAs may also contribute

tTtettPtaoaii
wcdpmvaupas1i2ahbc(2hdctme
D
Ftriltctpteaadtmb
aRimoiGidhcisppicbagttiacmFodamn(tba2iciseg
lpiAdcaca(aGim
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945 941
he activation of glial cells (Gravanis and Tsirka, 2005).PA has been proposed as key regulator of microglia at
he site of injury, promoting additional tissue damage (Siaot al., 2003; Gravanis and Tsirka, 2005). In contrast, as-rocytes have been suggested to be involved in the neu-ralization of tPA toxicity, through the uptake of the tPA/AI1 complex via an endocytic tPA receptor, the lipopro-
ein receptor-related protein (LRP) (Fernandez-Monreal etl., 2004). Increased expression of PAI1 mRNA has beenbserved in both experimental and human TLE (Gorter etl., 2006 and present results). Whether the tPA expression
n astrocytes indicates sequestration in the form of annactive complex with PAI1 in vivo remains to be explored.
Expression of PAs was also increased in blood vesselsithin the sclerotic hippocampus, indicating that the vas-ular endothelium represents an additional source of en-ogenous PAs within the human epileptogenic tissue. Ex-osure of endothelial cells to angiogenic and/or pro-inflam-atory mediators differentially regulates the PA expression in
itro (Gerritsen et al., 1993; Niedbala, 1993; Mandriota etl., 1995; Larsson et al., 2008). Recently, an increase inPA and uPAR expressing blood vessels has been re-orted in rat hippocampus after SE induction (Lahtinen etl., 2006, 2009, 2010). The plasminogen system has beenhown to play a critical role in angiogenesis (Pepper et al.,996; Brodsky et al., 2001; Rakic et al., 2003), as well as
n the regulation of the BBB permeability (Yepes et al.,003). Interestingly, alterations of the BBB permeabilitynd angiogenesis have been recently observed in bothuman and experimental TLE with a positive correlationetween the increased vascular permeability and the oc-urrence of spontaneous seizures in chronic epileptic ratsRigau et al., 2007; van Vliet et al., 2007; Ravizza et al.,008). In addition, release of PAs by brain endothelial cellsas been suggested to critically regulate monocyte diape-esis through the BBB (Reijerkerk et al., 2008). Thus PAsould indirectly play a pro-epileptogenic role and contributeo the persistent inflammatory reactions observed in hu-an and experimental TLE (Fabene et al., 2008; Ravizzat al., 2008; Vezzani et al., 2008).
evelopmental lesions and plasminogen activators
ocal developmental lesions, including FCD, TSC corticalubers and glioneuronal tumors (i.e. gangliogliomas) rep-esent another frequent finding in patients with medicallyntractable TLE (Blumcke et al., 2002; Thom, 2004). Theseesions share a number of clinical and pathological fea-ures and have been recently included among the MCDharacterized by active proliferation and abnormal cellypes (Barkovich et al., 2005). Our large-scale gene ex-ression profile analysis recently demonstrated that bothPA and uPA are strongly up-regulated within GG (Aronicat al., 2008). Increased expression was also observed fornnexin II, a membrane protein which has been identifieds binding partner of tPA, possibly involved in the tPA-ependent microglial activation (Siao and Tsirka, 2002). Inhe present study we confirmed the activation of the plas-inogen system in GG, showing increased expression of
oth tPA and uPA proteins and mRNAs. Increased tPA pnd uPA protein expression was also detected in FCD.T-PCR analysis showed up-regulation of uPA and uPAR
n both GG and FCD specimens. Similarly to HS speci-ens, PA IR was observed in both neuronal and glial cellsf MCD cases. The large majority of dysmorphic neurons
n FCD and TSC specimens, as well as neuronal cells ofG specimens displayed strong uPA and tPA expression,
ndicating that neurons are a major source of PAs also inevelopmental lesions. The presence of tPA within theseighly epileptogenic lesions may exacerbate neuronal ex-itability, increasing NMDA receptor signaling. Interest-
ngly, both dysplastic neurons and neuronal cells in GGpecimens contain high levels of NMDA receptor subunitroteins (Ying et al., 1999; Aronica et al., 2001a,c). Therominent neuronal expression of PAs in MCD is also
nteresting considering the increasing evidence that indi-ates a crucial role for the plasminogen system duringrain development (Zhang et al., 2005; Royer-Zemmour etl., 2008). Results obtained using mice lacking the tPAene indicate a role for tPA in facilitating neuronal migra-ion (Seeds et al., 1999), whereas selective alteration ofhe distribution of cortical interneurons have been reportedn mice lacking the uPAR (Powell et al., 2003; Eagleson etl., 2005). Thus, the induction of PA proteins, producinghanges in extracellular matrix and activation of matrixetalloproteinases (Irigoyen et al., 1999; Dityatev andellin, 2008) could contribute to the abnormal morphologyf dysplastic cells and their abnormal positioning within theysplastic cortex. Moreover, increasing evidence indicatescritical role for uPAR signaling, independently of uPA-ediated proteolysis (Smith and Marshall, 2010). Recentlyew uPA ligands have been detected, such as the SRPX2Sushi-Repeat Protein, X-linked 2). Interestingly, a muta-ion in SRPX2 is associated with Rolandic epilepsy andilateral perisylvian polymicrogyria and leads to a gain offfinity of SRPX2 with uPAR (Royer-Zemmour et al.,008). In addition, considerable evidence indicates that
ntegrins, a major family of extracellular matrix (ECM) re-eptors (such as �1 and �3 integrins) represent the mostmportant transmembrane receptors associated with uPARignalling (reviewed in (Smith and Marshall, 2010)). Sev-ral genes associated with cell adhesion, including inte-rins, are up-regulated in cortical tubers (Boer et al., 2009).
Expression of tPA and uPA was also observed in bal-oon cells in FCD and giant cells in TSC specimens. Theresence of PAs in balloon/giant cells may reflect the
mmature phenotype of these cells (Cepeda et al., 2003;lonso-Nanclares et al., 2005; Cepeda et al., 2005; Kal-eron et al., 1990). Furthermore, analysis of tPA and uPAellular distribution in MCD demonstrated both astroglialnd microglial cells IR, supporting the contribution of theseells to the amount of PAs that is present in these lesions,s well as the potential role of PAs in glial activationGravanis and Tsirka, 2005). The large majority of reactivestrocytes in FCD and TSC, as well as tumor astrocytes inG, expressed tPA and uPA. Expression of PAs was also
ncreased in blood vessels within the different develop-ental lesions studied. Interestingly, alterations of the BBB
ermeability and prominent inflammatory reactions have
a2
tht2i(vmGeGcdcretc2ipp
IpsrePtoater(tbihrJtct2maP
AtFmA
A
A
A
A
A
A
A
A
A
B
B
B
B
B
B
B
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945942
lso been observed in focal MCD (Aronica et al., 2005,007; Boer et al., 2006, 2008; Ravizza et al., 2006).
Concerning the expression patterns of PAs in glialumors, increased levels of uPA have been observed inigh-grade gliomas, whereas the data on the expression ofPA are still controversial (for review see; Levicar et al.,003). In particular attention has been focused on their role
n tumor invasion and angiogenesis in high grade tumorsYamamoto et al., 1994; Levicar et al., 2003). Our obser-ations indicate that induction of the plasminogen systemay also occur in low-grade glioneuronal tumors, such asG. Although a rapid induction of PAs can be triggered inxperimental epilepsy models (Lukasiuk et al., 2003;orter et al., 2006; Lahtinen et al., 2006), seizures aloneannot account for the increased PA expression in theseevelopmental lesions since the analysis of the histologi-ally normal perilesional cortex showed a pattern of neu-onal IR similar to that observed in control cortex. How-ver, recent evidence suggests that molecular and func-ional alterations in the perilesional region, may alsoontribute to the epileptogenicity of focal MCD (Wong,008). Thus, evaluation of a larger number of samples,
ncluding also areas of electrocortico-graphically definederilesional epileptogenic regions, is required to exclude aossible occurrence of perilesional tPA and uPA changes.
CONCLUSION
n summary, our findings demonstrate that activation of thelasminogen system, as exemplified by increased expres-ion of tPA and uPA, indeed occurs in human focal chronicefractory epilepsy, confirming the studies performed inxperimental models of TLE. The chronic expression ofAs together with the up-regulation of uPAR might con-
ribute to the mechanisms underlying the epileptogenicityf focal lesions, through direct modulation of neuronalctivity or, indirectly, through regulation of the inflamma-
ory response and/or epiletogenic tissue remodeling. Sev-ral mechanisms are possibly involved, including a directegulation of neuronal excitability via NMDA receptorsTsirka et al., 1995; Benarroch, 2007). Additionally, bothPA and uPA may contribute to the disruption of blood–rain barrier integrity and amplification of inflammatory
nfiltrates (Lo et al., 2002; Del Rosso et al., 2008), whichave been recently shown to play a critical role in chronicefractory epilepsy (Vezzani and Granata, 2005; Oby andanigro, 2006). Finally we have to consider the key role ofhe PA system in the complex tissue remodeling that oc-urs during epileptogenesis, such as axonal reorganiza-ion, angiogenesis and astrogliosis (Dityatev and Fellin,008). Further research, particularly using different animalodels, including models of MCD, might help to developnti-epileptic treatments that target specific components ofA/uPAR-mediated signaling.
cknowledgments—We are grateful to J.T. van Heteren for herechnical help. This work has been supported by National Epilepsyund, NEF 09-05 (EA); EU FP7 project NeuroGlia, Grant Agree-ent N° 202167 and Stichting Michelle M06.011, M07.016(EA,
MY).REFERENCES
lfano D, Franco P, Vocca I, Gambi N, Pisa V, Mancini A, Caputi M,Carriero MV, Iaccarino I, Stoppelli MP (2005) The urokinase plas-minogen activator and its receptor: role in cell growth and apopto-sis. [see comment]. Thromb Haemost 93:205–211.
lonso-Nanclares L, Garbelli R, Sola RG, Pastor J, Tassi L, SpreaficoR, DeFelipe J (2005) Microanatomy of the dysplastic neocortexfrom epileptic patients. Brain 128:158–173.
ronica E, Boer K, Becker A, Redeker S, Spliet WG, van Rijen PC,Wittink F, Breit T, Wadman WJ, Lopes da Silva FH, Troost D,Gorter JA (2008) Gene expression profile analysis of epilepsy-associated gangliogliomas. Neuroscience 151:272–292.
ronica E, Boer K, van Vliet EA, Baayen JC, Redeker S, Spliet WGM,Lopes da Silva FH, Wadman WJ, Troost D, Gorter JA (2007)Complement activation in experimental and human temporal lobeepilepsy. Neurobiol Dis 26:497–511.
ronica E, Catania MV, Geurts J, Yankaya B, Troost B (2001a) Im-munohistochemical localization of Group I and II metabotropicglutamate receptors in control and ALS human spinal cord: up-regulation in reactive astrocytes. Neuroscience 105:509–520.
ronica E, Gorter JA, Ramkema M, Redeker S, Ozbas-Gerceker F,van Vliet EA, Scheffer GL, Scheper RJ, van der Valk P, Baayen JC,Troost D (2004) Expression and cellular distribution of multidrugresistance-related proteins in the hippocampus of patients withmesial temporal lobe epilepsy. Epilepsia 45:441–451.
ronica E, Gorter JA, Redeker S, Ramkema M, Spliet WG, van RijenPC, Leenstra S, Troost D (2005) Distribution, characterization andclinical significance of microglia in glioneuronal tumours from pa-tients with chronic intractable epilepsy. Neuropathol Appl Neurobiol31:280–291.
ronica E, Leenstra S, Jansen GH, van Veelen CW, Yankaya B,Troost D (2001b) Expression of brain-derived neurotrophic factorand tyrosine kinase B receptor proteins in glioneuronal tumors frompatients with intractable epilepsy: colocalization with N-methyl-D-aspartic acid receptor. Acta Neuropathol 101:383–392.
ronica EM, Leenstra S, Jansen G, van Veelen CW, Yankaya B,Troost D (2001c) Expression of BDNF and tyrosine kinase B (TrkB)receptor proteins in glioneuronal tumors from patients with intrac-table epilepsy: colocalization with NMDA receptor. Acta Neuro-pathol 101:383–392.
arkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB(2005) A developmental and genetic classification for malforma-tions of cortical development. Neurology 65:1873–1887.
enarroch EE (2007) Tissue plasminogen activator: beyond thrombol-ysis. Neurology 69:799–802.
iermann JC, Holzscheiter L, Kotzsch M, Luther T, Kiechle-Bahat M,Sweep FC, Span PN, Schmitt M, Magdolen V (2008) QuantitativeRT-PCR assays for the determination of urokinase-type plasmin-ogen activator and plasminogen activator inhibitor type 1 mRNA inprimary tumor tissue of breast cancer patients: comparison toantigen quantification by ELISA. Int J Mol Med 21:251–259.
lumcke I, Pauli E, Clusmann H, Schramm J, Becker A, Elger C,Merschhemke M, Meencke HJ, Lehmann T, von Deimling A,Scheiwe C, Zentner J, Volk B, Romstock J, Stefan H, HildebrandtM (2007) A new clinico-pathological classification system for me-sial temporal sclerosis. Acta Neuropathol 113:235–244.
lumcke I, Thom M, Wiestler OD (2002) Ammon’s horn sclerosis: amaldevelopmental disorder associated with temporal lobe epi-lepsy. Brain Pathol 12:199–211.
oer K, Crino PB, Gorter JA, Nellist M, Jansen FE, Spliet WG, vanRijen PC, Wittink F, Breit T, Troost D, Wadman WJ, Aronica E(2009) Gene expression analysis of tuberous sclerosis complexcortical tubers reveals increased expression of adhesion and in-flammatory factors. Brain Pathol 2009, Oct 8; [Epub ahead of print];doi:10.1111/j.1750-3639.2009.00341.
oer K, Jansen K, Nellist M, Redeker M, van den Ouweland AMW,
Spliet WGM, van Nieuwenhuizen O, Troost D, Crino PB, Aronica E
B
B
C
C
C
C
D
D
D
E
F
F
F
F
F
G
G
G
G
G
G
G
I
K
L
L
L
L
L
L
L
L
L
L
L
M
M
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945 943
(2008) Inflammatory processes in cortical tubers andsubependymal giant cell tumors of tuberous sclerosis complex.Epilepsy Res 78:7–21.
oer K, Spliet WG, van Rijen PC, Redeker S, Troost D, Aronica E(2006) Evidence of activated microglia in focal cortical dysplasia.J Neuroimmunol 173:188–195.
rodsky S, Chen J, Lee A, Akassoglou K, Norman J, Goligorsky MS(2001) Plasmin-dependent and �independent effects of plasmin-ogen activators and inhibitor-1 on ex vivo angiogenesis. Am JPhysiol Heart Circ Physiol 281:H1784–H1792.
armeliet P, Collen D (1995) Gene targeting and gene transfer studiesof the biological role of the plasminogen/plasmin system. ThrombHaemost 74:429–436.
epeda C, Andre VM, Vinters HV, Levine MS, Mathern GW (2005) Arecytomegalic neurons and balloon cells generators of epileptic ac-tivity in pediatric cortical dysplasia? Epilepsia 46:82–88.
epeda C, Hurst RS, Flores-Hernandez J, Hernandez-Echeagaray E,Klapstein GJ, Boylan MK, Calvert CR, Jocoy EL, Nguyen OK,Andre VM, Vinters HV, Ariano MA, Levine MS, Mathern GW (2003)Morphological and electrophysiological characterization of abnor-mal cell types in pediatric cortical dysplasia. J Neurosci Res 72:472–486.
ollen D (1999) The plasminogen (fibrinolytic) system. Thromb Hae-most 82:259–270.
el Rosso M, Fibbi G, Pucci M, Margheri F, Serrati S (2008) Theplasminogen activation system in inflammation. Front Biosci13:4667–4686.
ent MA, Sumi Y, Morris RJ, Seeley PJ (1993) Urokinase-type plas-minogen activator expression by neurons and oligodendrocytesduring process outgrowth in developing rat brain. Eur J Neurosci5:633–647.
ityatev A, Fellin T (2008) Extracellular matrix in plasticity and epilep-togenesis. Neuron Glia Biol 4:235–247.
agleson KL, Bonnin A, Levitt P (2005) Region- and age-specificdeficits in gamma-aminobutyric acidergic neuron development inthe telencephalon of the uPAR(-/-) mouse. J Comp Neurol 489:449–466.
abbro S, Seeds NW (2009) Plasminogen activator activity is inhibitedwhile neuroserpin is up-regulated in the Alzheimer disease brain.J Neurochem 109:303–315.
abene PF, Navarro MG, Martinello M, Rossi B, Merigo F, Ottoboni L,Bach S, Angiari S, Benati D, Chakir A, Zanetti L, Schio F, OsculatiA, Marzola P, Nicolato E, Homeister JW, Xia L, Lowe JB, McEverRP, Osculati F, Sbarbati A, Butcher EC, Constantin G (2008) A rolefor leukocyte-endothelial adhesion mechanisms in epilepsy. [seecomment]. Nat Med 14:1377–1383.
aber-Elman A, Miskin R, Schwartz M (1995) Components of theplasminogen activator system in astrocytes are modulated by tu-mor necrosis factor-alpha and interleukin-1beta through similarsignal transduction pathways. J Neurochem 65:1524–1535.
alcone DJ, McCaffrey TA, Mathew J, McAdam K, Borth W (1995)THP-1 macrophage membrane-bound plasmin activity is up-regu-lated by transforming growth factor-beta 1 via increased expres-sion of urokinase and the urokinase receptor. J Cell Physiol164:334–343.
ernandez-Monreal M, Lopez-Atalaya JP, Benchenane K, Leveille F,Cacquevel M, Plawinski L, MacKenzie ET, Bu G, Buisson A, VivienD (2004) Is tissue-type plasminogen activator a neuromodulator?Mol Cell Neurosci 25:594–601.
erritsen ME, Niedbala MJ, Szczepanski A, Carley WW (1993) Cyto-kine activation of human macro- and microvessel-derived endo-thelial cells. Blood Cells 19:325–339; discussion 340–322.
omez M, Sampson J, Whittemore V (1999) The tuberous sclerosiscomplex. Oxford: Oxford University Press.
orter JA, Van Vliet E, Aronica E, Rauwerda H, Breit T, Lopes da SilvaFH, Wadman WJ (2006) Potential new antiepileptogenic targetsindicated by microarray analysis in a rat model for temporal lobe
epilepsy. J Neurosci 26:11083–11110.orter JA, Van Vliet E, Rauwerda H, Breit T, van Schaik L, Vreugden-hil E, Redeker S, Aronica E, Lopes da Silva FH, Wadman WJ(2007) Dynamic changes of proteases and protease inhibitorsrevealed by microarray analysis in CA3 and temporal lobe duringepileptogenesis in the rat. Epilepsia 40:1–12.
ravanis I, Tsirka SE (2005) Tissue plasminogen activator and glialfunction. Glia 49:177–183.
ualandris A, Jones TE, Strickland S, Tsirka SE (1996) Membranedepolarization induces calcium-dependent secretion of tissue plas-minogen activator. J Neurosci 16:2220–2225.
veric D, Hanemaaijer R, Newcombe J, van Lent NA, Sier CF, CuznerML (2001) Plasminogen activators in multiple sclerosis lesions:implications for the inflammatory response and axonal damage.Brain 124:1978–1988.
rigoyen JP, Munoz-Canoves P, Montero L, Koziczak M, Nagamine Y(1999) The plasminogen activator system: biology and regulation.Cell Mol Life Sci 56:104–132.
alderon N, Ahonen K, Fedoroff S (1990) Developmental transition inplasticity properties of differentiating astrocytes: age-related bio-chemical profile of plasminogen activators in astroglial cultures.Glia 3:413–426.
ahtinen L, Lukasiuk K, Pitkanen A (2006) Increased expression andactivity of urokinase-type plasminogen activator during epilepto-genesis. Eur J Neurosci 24:1935–1945.
ahtinen L, Huusko N, Myohanen H, Lehtivarjo AK, Pellinen R, Tu-runen MP, Yla-Herttuala S, Pirinen E, Pitkanen A (2009) Expres-sion of urokinase-type plasminogen activator receptor is increasedduring epileptogenesis in the rat hippocampus. Neuroscience163:316–328.
ahtinen L, Ndode-Ekane XE, Barinka F, Akamine Y, Esmaeili MH,Rantala J, Pitkänen A (2010) Urokinase-type plasminogen activa-tor regulates neurodegeneration and neurogenesis but not vascu-lar changes in the mouse hippocampus after status epilepticus.Neurobiol Dis 37:692–703.
arsson P, Ulfhammer E, Karlsson L, Bokarewa M, Wahlander K, JernS (2008) Effects of IL-1beta and IL-6 on tissue-type plasminogenactivator expression in vascular endothelial cells. Thromb Res123:342–351.
evicar N, Nuttall RK, Lah TT (2003) Proteases in brain tumour pro-gression. [erratum appears in Acta Neurochir (Wien). 2003 Nov;145(11):1023 Note: Nutall R K [corrected to Nuttall R K]]. ActaNeurochirurgica 145:825–838.
i XF, Yan PJ, Shao ZM (2009) Downregulation of Mir-193b contrib-utes to enhance urokinase-type plasminogen activator (uPA) ex-pression and tumor progression and invasion in human breastcancer. Oncogene 28:3937–3948.
ijnen HR, Nelles L, Van Hoef B, Demarsin E, Collen D (1988) Char-acterization of a chimeric plasminogen activator consisting ofamino acids 1 to 274 of tissue-type plasminogen activator andamino acids 138 to 411 of single-chain urokinase-type plasmino-gen activator. J Biol Chem 263:19083–19091.
o EH, Wang X, Cuzner ML (2002) Extracellular proteolysis in braininjury and inflammation: role for plasminogen activators and matrixmetalloproteinases. J Neurosci Res 69:1–9.
oscher W (2002) Animal models of drug-resistant epilepsy. NovartisFound Symp 243:149–159; discussion 159–166.
ouis DN, Ohgaki H, Wiestler OD, Cavanee WK (2007) WHO classi-fication of tumours of the central nervous system. Lyon: IARC.
ukasiuk K, Kontula L, Pitkanen A (2003) cDNA profiling of epilepto-genesis in the rat brain. Eur J Neurosci 17:271–279.
andriota SJ, Seghezzi G, Vassalli JD, Ferrara N, Wasi S, Mazzieri R,Mignatti P, Pepper MS (1995) Vascular endothelial growth factorincreases urokinase receptor expression in vascular endothelialcells. J Biol Chem 270:9709–9716.
asos T, Miskin R (1996) Localization of urokinase-type plasminogenactivator mRNA in the adult mouse brain. Brain Res Mol Brain Res
35:139–148.
M
N
N
O
P
P
P
P
P
R
R
R
R
R
R
R
R
S
S
S
S
S
S
T
T
T
T
T
T
T
v
V
V
V
W
W
W
W
W
Y
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945944
aupas-Schwalm F, Auge N, Robinet C, Cambus JP, Parsons SJ,Salvayre R, Negre-Salvayre A (2004) The sphingomyelin/cer-amide pathway is involved in ERK1/2 phosphorylation, cell prolif-eration, and uPAR overexpression induced by tissue-type plasmin-ogen activator. FASEB J 18:1398–1400.
akajima K, Tohyama Y, Kurihara T, Kohsaka S (2005) Enhancementof urokinase-type plasminogen activator (uPA) secretion, but notthat of substrate plasminogen (PGn), by rat microglia stimulatedwith neuronal conditioned medium. Neurosci Lett 378:13–17.
iedbala MJ (1993) Cytokine regulation of endothelial cell extracellularproteolysis. Agents Actions Suppl 42:179–193.
by E, Janigro D (2006) The blood-brain barrier and epilepsy. Epilep-sia 47:1761–1774.
almini A, Najm I, Avanzini G, Babb T, Guerrini R, Foldvary-SchaeferN, Jackson G, Luders HO, Prayson R, Spreafico R, Vinters HV(2004) Terminology and classification of the cortical dysplasias.Neurology 62:S2–S8.
awlak R, Strickland S (2002) Tissue plasminogen activator and sei-zures: a clot-buster’s secret life. [comment]. J Clin Invest 109:1529–1531.
epper MS, Montesano R, Mandriota SJ, Orci L, Vassalli JD (1996)Angiogenesis: a paradigm for balanced extracellular proteolysisduring cell migration and morphogenesis. Enzyme Protein 49:138–162.
ittman RN, Ivins JK, Buettner HM (1989) Neuronal plasminogenactivators: cell surface binding sites and involvement in neuriteoutgrowth. J Neurosci 9:4269–4286.
owell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, LevittP (2003) Genetic disruption of cortical interneuron developmentcauses region- and GABA cell type-specific deficits, epilepsy, andbehavioral dysfunction. J Neurosci 23:622–631.
akic JM, Maillard C, Jost M, Bajou K, Masson V, Devy L, Lambert V,Foidart JM, Noel A (2003) Role of plasminogen activator-plasminsystem in tumor angiogenesis. Cell Mol Life Sci 60:463–473.
amakers C, Ruijter JM, Deprez RH, Moorman AF (2003) Assump-tion-free analysis of quantitative real-time polymerase chain reac-tion (PCR) data. Neurosci Lett 339:62–66.
avizza T, Boer K, Redeker S, Spliet WG, van Rijen PC, Troost D,Vezzani A, Aronica E (2006) The IL-1beta system in epilepsy-associated malformations of cortical development. Neurobiol Dis24:128–143.
avizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A (2008)Innate and adaptive immune mechanisms during epileptogenesisand spontaneous seizures: evidence from experimental modelsand human temporal lobe epilepsy. Neurobiol Dis 29:142–160.
eijerkerk A, Kooij G, van der Pol SM, Leyen T, van Het Hof B,Couraud PO, Vivien D, Dijkstra CD, de Vries HE (2008) Tissue-type plasminogen activator is a regulator of monocyte diapedesisthrough the brain endothelial barrier. J Immunol 181:3567–3574.
igau V, Morin M, Rousset MC, de Bock F, Lebrun A, Coubes P, PicotMC, Baldy-Moulinier M, Bockaert J, Crespel A, Lerner-Natoli M(2007) Angiogenesis is associated with blood-brain barrier perme-ability in temporal lobe epilepsy. Brain 130:1942–1956.
ogister B, Leprince P, Pettmann B, Labourdette G, SensenbrennerM, Moonen G (1988) Brain basic fibroblast growth factor stimulatesthe release of plasminogen activators by newborn rat culturedastroglial cells. Neurosci Lett 91:321–326.
oyer-Zemmour B, Ponsole-Lenfant M, Gara H, Roll P, Leveque C,Massacrier A, Ferracci G, Cillario J, Robaglia-Schlupp A, Vincen-telli R, Cau P, Szepetowski P (2008) Epileptic and developmentaldisorders of the speech cortex: ligand/receptor interaction of wild-type and mutant SRPX2 with the plasminogen activator receptoruPAR. Hum Mol Genet 17:3617–3630.
chmitt M, Chucholowski N, Busch E, Hellmann D, Wagner B,Goretzki L, Janicke F, Gunzler WA, Graeff H (1991) Fluorescentprobes as tools to assess the receptor for the urokinase-typeplasminogen activator on tumor cells. Semin Thromb Hemost
17:291–302.eeds NW, Basham ME, Haffke SP (1999) Neuronal migration isretarded in mice lacking the tissue plasminogen activator gene.Proc Natl Acad Sci U S A 96:14118–14123.
iao CJ, Fernandez SR, Tsirka SE (2003) Cell type-specific roles fortissue plasminogen activator released by neurons or microglia afterexcitotoxic injury. J Neurosci 23:3234–3242.
iao CJ, Tsirka SE (2002) Tissue plasminogen activator mediatesmicroglial activation via its finger domain through annexin II. J Neu-rosci 22:3352–3358.
mith HW, Marshall CJ (2010) Regulation of cell signalling by uPAR.Nat Rev Mol Cell Biol 11:23–36.
mith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, ProvenzanoMD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurementof protein using bicinchoninic acid. Anal Biochem 150:76–85.
eesalu T, Kulla A, Asser T, Koskiniemi M, Vaheri A (2002) Tissueplasminogen activator as a key effector in neurobiology and neu-ropathology. Biochem Soc Trans 30:183–189.
eesalu T, Kulla A, Simisker A, Siren V, Lawrence DA, Asser T, VaheriA (2004) Tissue plasminogen activator and neuroserpin are widelyexpressed in the human central nervous system. Thromb Haemost92:358–368.
hom M (2004) Recent advances in the neuropathology of focallesions in epilepsy. Expert Rev Neurother 4:973–984.
ranque P, Robbins R, Naftolin F, Andrade-Gordon P (1992) Regula-tion of plasminogen activators and type-1 plasminogen activatorinhibitor by cyclic AMP and phorbol ester in rat astrocytes. Glia6:163–171.
sirka SE (2002) Tissue plasminogen activator as a modulator ofneuronal survival and function. Biochem Soc Trans 30:222–225.
sirka SE, Gualandris A, Amaral DG, Strickland S (1995) Excitotoxin-induced neuronal degeneration and seizure are mediated by tissueplasminogen activator. Nature 377:340–344.
ucker HM, Kihiko-Ehmann M, Estus S (2002) Urokinase-type plas-minogen activator inhibits amyloid-beta neurotoxicity and fibrillo-genesis via plasminogen. J Neurosci Res 70:249–255.
an Vliet EA, da Costa Araújo S, Redeker S, van Schaik R, Aronica E,Gorter JA (2007) Long-lasting increased permeability of the blood-brain barrier may contribute to seizure progression in temporal lobeepilepsy. Brain 130:521–534.
andeputte DA, Troost D, Leenstra S, Ijlst-Keizers H, Ramkema M,Bosch DA, Baas F, Das NK, Aronica E (2002) Expression anddistribution of id helix-loop-helix proteins in human astrocytic tu-mors. Glia 38:329–338.
ezzani A, Granata T (2005) Brain inflammation in epilepsy: experi-mental and clinical evidence. Epilepsia 46:1724–1743.
ezzani A, Ravizza T, Balosso S, Aronica E (2008) Glia as a source ofcytokines: implications for neuronal excitability and survival. Epi-lepsia 49(Suppl 2):24–32.
agner SN, Atkinson MJ, Wagner C, Hofler H, Schmitt M, Wilhelm O(1996) Sites of urokinase-type plasminogen activator expressionand distribution of its receptor in the normal human kidney. Histo-chem Cell Biol 105:53–60.
ieser HG (2004) ILAE Commission Report. Mesial temporal lobeepilepsy with hippocampal sclerosis. Epilepsia 45:695–714.
ong M (2008) Mechanisms of epileptogenesis in tuberous sclerosiscomplex and related malformations of cortical development withabnormal glioneuronal proliferation. Epilepsia 49:8–21.
u YP, Siao CJ, Lu W, Sung TC, Frohman MA, Milev P, Bugge TH, DegenJL, Levine JM, Margolis RU, Tsirka SE (2000) The tissue plasminogenactivator (tPA)/ plasmin extracellular proteolytic system regulates seizure-induced hippocampal mossy fiber outgrowth through a proteoglycansubstrate. J Cell Biol 148:1295–1304.
yler AR, Dohan C, Schweitzer JB, Berry AD III (1992) A gradingsystem for mesial temporal pathology (hippocampal sclerosis) fromanterior temporal lobectomy. J Epilepsy 5:220–225.
amamoto M, Sawaya R, Mohanam S, Bindal AK, Bruner JM, Oka K,
Rao VH, Tomonaga M, Nicolson GL, Rao JS (1994) Expression
Y
Y
Z
A. M. Iyer et al. / Neuroscience 167 (2010) 929–945 945
and localization of urokinase-type plasminogen activator in humanastrocytomas in vivo. Cancer Res 54:3656–3661.
epes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Law-rence DA (2003) Tissue-type plasminogen activator induces open-ing of the blood-brain barrier via the LDL receptor-related protein.
J Clin Invest 112:1533–1540.ing Z, Babb TL, Mikuni N, Najm I, Drazba J, Bingaman W (1999) Selectivecoexpression of NMDAR2A/B and NMDAR1 subunit proteins in dysplas-tic neurons of human epileptic cortex. Exp Neurol 159:409–418.
hang Y, Pothakos K, Tsirka SA (2005) Extracellular proteases: bio-logical and behavioral roles in the mammalian central nervous
system. Curr Top Dev Biol 66:161–188.(Accepted 19 February 2010)(Available online 26 February 2010)








![Accuracy of distinguishing between dysembryoplastic neuroepithelial tumors and other epileptogenic brain neoplasms with [11C]methionine PET](https://static.fdokumen.com/doc/165x107/63360da5cd4bf2402c0b568c/accuracy-of-distinguishing-between-dysembryoplastic-neuroepithelial-tumors-and-other.jpg)











