
The role of condensin in chromosome resolution - UCL ...
152
The role of condensin in chromosome resolution Adrian Aymard Charbin University College London and Cancer Research UK London Research Institute PhD Supervisor: Dr Frank Uhlmann A thesis submitted for the degree of Doctor of Philosophy University College London September 2012
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of The role of condensin in chromosome resolution - UCL ...

The role of condensin in chromosome resolution
Adrian Aymard Charbin
University College London
and
Cancer Research UK London Research Institute
PhD Supervisor: Dr Frank Uhlmann
A thesis submitted for the degree of
Doctor of Philosophy
University College London September 2012

2
Felix qui potuit rerum cognoscere causas
Happy is he who gets to know the reasons for things
Virgil, Georgics (c. 37BC)

3
Publications arising from this thesis
Condensin aids sister chromatid resolut ion by promoting their
decatenation by topoisomerase II
Adrian Charbin, Céline Bouchoux, and Frank Uhlmann. Genes and Development
(under review)
Faci le synthesis of budding yeast a-factor and i ts use to synchronise
cel ls of α mating type
Nicola O’Reilly, Adrian Charbin, Lidia Lopez-Serra, and Frank Uhlmann. 2012
Yeast 29: 233-40

4
Declaration
I Adrian Charbin confirm that the work presented in this thesis is my own. Where
information has been derived from other sources, I confirm that this has been
indicated in the thesis.

5
Abstract
The condensin complex is a key determinant of mitotic chromosome
architecture. In addition to its role in mitotic chromosome compaction, condensin is
required for resolution of sister chromatid linkages during chromosome segregation
in anaphase. How condensin resolves sister chromatids, and the nature of the
chromosome bridges that are characteristic of cells harboring defective condensin,
have remained topics of debate. Inactivation of topoisomerase II, the main enzyme
that removes topological interlinks that persist between DNA replication products
after their synthesis, leads to similar chromosome bridges. Here, we follow the
catenation status of circular minichromosomes of three sizes during the S.
cerevisiae cell cycle. Catenanes are produced in S-phase and, in part, they are
readily resolved, aided by physical separation of sister chromatids during mitosis.
Complete resolution, however, requires the condensin complex, a dependency that
becomes more pronounced with increasing chromosome size. Condensin and
topoisomerase II directly interact and, using purified proteins, we show that
condensin stimulates DNA decatenation by topoisomerase II in vitro. Therefore, in
parallel to promoting chromosome condensation, condensin facilitates topological
resolution of sister chromatids to secure their successful segregation to daughter
cells during cell division.

6
Acknowledgement
First and foremost, I would like to thank Frank for all his advice, support and
wisdom these past four years. I have been so fortunate to be apart of his research
group, working alongside world-class scientists and learning so much. It has been
a truly great experience.
My deep gratitude goes to everyone in the laboratory who have taught me
so much but also made me laugh. Thank you especially to Chris and Maria for
running the lab so well. Without your help, my project would never have gotten as
far as it did. Thank you to Céline and Sebastian for all your guidance during my
project, I have learnt much from you both and have always enjoyed working with
you. To everyone else in the lab, thank you for all the scientific discussions and
always offering an extra pair of hands during those tricky experiments!
Thanks to all my friends who have been doing PhDs alongside me. Risa and
Vanessa for all our chats in the corridor discussing the perils of graduate work. Ali,
for our morning coffees, scientific discourses and more!
I would also like to say thank you to my family for supporting me throughout
all my studies. It is thanks to your love and encouragement that I have gotten to
where I am today.
Finally I want to thank Susie, who has filled the past four years with so much
happiness, love and laughter. I am so lucky to have you by my side, helping me
follow my dreams.

7
Table of Contents
Abstract ................................ ................................ ..................... 5
Acknowledgement ................................ ................................ ....... 6
Table of Contents ................................ ................................ ....... 7
Table of Figures ................................ ................................ ....... 11
Abbreviations ................................ ................................ ........... 13
Chapter 1. Introduction ................................ ............................. 15
1.1 Historical Perspective ........................................................................................ 15
1.2 The Structural Maintenance of Chromosome Protein Family ...................... 20
1.2.1 Condensin function ....................................................................................... 21 1.2.2 Eukaryotic condensin structure ..................................................................... 22 1.2.3 Prokaryotic condensin structure ................................................................... 25 1.2.4 Condensin’s in vitro biochemical activities .................................................. 25
1.3 Chromosome Condensation .............................................................................. 27
1.3.1 Condensin’s biochemical activities drive condensation ............................... 28 1.3.2 Condensin as a catalyst driving condensation? ............................................ 29 1.3.3 Condensin could behave as a DNA linker to drive condensation ................ 29 1.3.4 Does condensin act through a co-operative mechanism? ............................. 32
1.4 Chromosome Resolution ................................................................................... 33
1.4.1 Proteinaceous linkages .................................................................................. 33 1.4.2 Topological linkages ..................................................................................... 34
1.5 Topoisomerases .................................................................................................. 35
1.5.1 Types of topoisomerase ................................................................................ 35 1.5.2 Topoisomerase II mode of action ................................................................. 37 1.5.3 The physiological importance of topoisomerase II ...................................... 40
1.6 Chromosome resolution, topoisomerase II and condensin - an unresolved
relationship .................................................................................................................. 41
1.6.1 Lessons from the rDNA locus ...................................................................... 45 1.6.2 Condensin is required for complete removal of proteinaceous links ........... 48 1.6.3 Condensin directly partners with topoisomerase II to promote decatenation 49

8
1.6.4 Condensin-driven reconfiguration of mitotic chromosome topology promotes decatenation .............................................................................................. 51
1.7 An introduction to protein inactivations in S. cerevisiae ............................... 52
Chapter 2. Materials & Methods ................................ ................. 56
2.1 Yeast growth and manipulation ....................................................................... 56
2.1.1 Yeast strains .................................................................................................. 56 2.1.2 Yeast strain creation, mating and tetrad dissection ...................................... 57 2.1.3 Yeast media, cultures and synchronizations ................................................. 58 2.1.4 Anchor away strains and nuclear depletion .................................................. 58 2.1.5 Yeast transformations ................................................................................... 59
2.2 General molecular biology techniques ............................................................. 59
2.3 Protein analysis techniques ............................................................................... 59
2.3.1 Protein extract preparation ............................................................................ 59 2.3.2 Chromatin pellets .......................................................................................... 60 2.3.3 SDS-PAGE electrophoresis and western blotting ........................................ 61
2.4 Mini-chromosome purification, electrophoresis and catenane
quantification .............................................................................................................. 62
2.4.1 Catenation assay minichromosomes ............................................................. 63 2.4.2 Zymolyation of yeast cells in agarose plugs and Pulse Field Gel Electrophoresis (PFGE) ............................................................................................ 63
2.5 Cell biology and microscopy ............................................................................. 64
2.5.1 Cell cycle analysis using flow cytometry ..................................................... 64 2.5.2 Bi-nucleate cell counting .............................................................................. 64 2.5.3 In situ immunofluorescence .......................................................................... 65
2.6 Protein purifications .......................................................................................... 65
2.6.1 Purification of S. cerevisiae topoisomerase II .............................................. 65 2.6.2 Purification of S. cerevisiae condensin ......................................................... 66 2.6.3 Interaction analysis between condensin and topoisomerase II ..................... 66
2.7 In vitro assay techniques ................................................................................... 67
2.7.1 kDNA decatenation assays ........................................................................... 67

9
Chapter 3. Results: Catenane Behaviour and Resolution ............. 68
3.1 Development of the catenation assay ............................................................... 68
3.1.1 Optimisation of the catenation assay ............................................................ 71 3.1.2 Identifying the minichromosome’s different topological isoforms .............. 74 3.1.3 Catenation is quantifiable at each time point ................................................ 77
3.2 Spontaneous and assisted minichromosome decatenation: ........................... 81
3.2.1 Temperature’s effect on catenation and isoform distribution ....................... 88 3.2.2 The anchor away system does not effect catenation ..................................... 92
3.3 Catenation of smaller minichromosomes is observable in wild type strains 94
Chapter 4. Results: Condensin’s Influence on Catenation ............ 97
4.1 Condensin promotes completion of minichromosome decatenation ............. 97
4.2 The effect of minichromosome size ................................................................ 105
4.2.1 Visualizing a ring chromosome’s (RCIII) different isoforms .................... 108
4.3 Condensin’s decatenation role is more pronounced on RCIII .................... 111
Chapter 5. Results: Investigating Interactions ............................ 118
5.1 Do condensin and topoisomerase II directly interact? ................................. 118
5.2 Purification of topoisomerase II and condensin ............................................ 121
5.3 Condensin and topoisomerase II directly interact ........................................ 123
5.4 Condensin stimulates in vitro DNA decatenation by topo II ....................... 123
Chapter 6. Results: a-factor Synthesis and Characterization ....... 128
6.1 Synthesis of a-factor ......................................................................................... 128
6.2 Use of a-factor to synchronise cells of α mating type .................................... 128
6.3 Little shmoo formation during a-factor-induced G1 arrest ......................... 131

10
Chapter 7. Discussion ................................ .............................. 133
7.1 Catenation can be observed in minichromosomes ........................................ 133
7.2 Cohesin protects catenation ............................................................................ 133
7.3 Anaphase bridges result from persistent sister chromatid catenanes ........ 134
7.4 Condensin promotes decatenation by topoisomerase II ............................... 135
7.5 Decatenation occurs in steps ........................................................................... 136
7.6 Condensin directly interacts with topoisomerase II ..................................... 137
7.7 Future perspectives .......................................................................................... 137
Reference List ................................ ................................ ......... 141

11
Table of Figures
Figure 1.1 Walther Flemming's observations of chromosomes during mitosis ...... 17 Figure 1.2 Mitotic cycle of rye (Secale cereale) chromosomes .............................. 18 Figure 1.3 Schematic representation of the condensin complex ............................ 24 Figure 1.4 Condensin subunit comparisons among different organisms ............... 24 Figure 1.5 Possible mechanisms of condensation by condensin ........................... 31 Figure 1.6 Topoisomerase II’s mode of action ....................................................... 39 Figure 1.7 Schematic of colliding replication forks, and resultant catenation ......... 44 Figure 1.8 Anaphase bridges in condensin mutants .............................................. 47 Figure 1.9 The anchor away technique of nuclear depletion .................................. 54 Figure 3.1 Map of minichromosome pS14-8 .......................................................... 70 Figure 3.2 Early electrophoresis conditions showed poor isoform resolution ........ 72 Figure 3.3 Background labelling of genomic DNA .................................................. 72 Figure 3.4 Specificity comparison of different AmpR probes .................................. 73 Figure 3.5 Wild type cells display a distinct isoform patterns ................................. 76 Figure 3.6 Enzyme digests allow for isoform identification ..................................... 76 Figure 3.7 Comparison of background deduction techniques ................................ 79 Figure 3.8 Catenane quantification of wild type cell cycle ...................................... 80 Figure 3.9 Topoisomerase II inactivation results in maximum catenane formation 82 Figure 3.10 Nocodazole arrests results in catenane persistence ........................... 84 Figure 3.11 Spindle presence during arrest promotes decatenation ...................... 84 Figure 3.12 Inactivation of cohesin results in fewer observable catenanes ........... 86 Figure 3.13 Catenane quantification for different mitotic inactivations ................... 87 Figure 3.14 Effect of temperature on catenane behaviour ..................................... 89 Figure 3.15 Effect of temperature on different isoforms ......................................... 91 Figure 3.16 Anchor away and rapamycin do not affect catenation ........................ 93 Figure 3.17 Map of minichromosome pRS316 ....................................................... 95 Figure 3.18 Catenation detection and isoform identification for pRS316 ............... 96 Figure 4.1 Condensin ts mutants all display a persistent catenane phenotype ..... 98 Figure 4.2 Condensin is required for complete decatenation ............................... 100 Figure 4.3 brn1-aa triplicates and quantification .................................................. 101

12
Figure 4.4 Condensin contributes to decatenation independently of chromosome
movement ............................................................................................................. 103 Figure 4.5 Decatenation is active prior to anaphase ............................................ 104 Figure 4.6 smc2-8 releases poorly from G1 arrest ............................................... 106 Figure 4.7 Condensin depletion has a smaller effect on pRS316 catenation ....... 107 Figure 4.8 Map of ring chromosome RCIII ........................................................... 110 Figure 4.9 Resolution of RCIII by Pulse Field Gel Electrophoresis (PFGE) ......... 110 Figure 4.10 RCIII wild type timecourse and isoform identification ........................ 113 Figure 4.11 RCIII Quantification profiles .............................................................. 114 Figure 4.12 Nocodazole arrest shows persistent RCIII catenanes ...................... 115 Figure 4.13 Condensin depletion phenotype closes matches nocodazole arrest 115 Figure 4.14 Nuclear depletion of Brn1 .................................................................. 116 Figure 5.1 Co-immunoprecipitation of condensin and topoisomerase II .............. 120 Figure 5.2 Topoisomerase II purification steps .................................................... 122 Figure 5.3 Condensin interacts directly with purified topoisomerase II ................ 125 Figure 5.4 Condensin promotes decatenation of kinetoplasts ............................. 126 Figure 5.5 Condensin stimulates different topoisomerase II enzymes ................. 127 Figure 6.1 Use of a-factor to synchronise cell cycle progression of α cells ......... 130 Figure 6.2 Comparison of pheromone-induced shmoo formation of a and α cells
............................................................................................................................. 132 Figure 7.1 Schematic displaying possible excision and re-ligation of endogenous
chromosome DNA to form de novo ring chromosomes in vivo ............................ 139

13
Abbreviations ARS autonomous replicating origin
ATP adenosine triphosphate
APC anaphase promoting complex
BSA bovine serum albumin
bp base pairs
Cdk Cyclin-dependent kinase
ChIP chromatin immunoprecipitation
CPC Chromosome Passenger Complex
Da Dalton (kDa, kilodalton)
DAPI 4’-6’-diamidino-2-phenylindole
DMSO dimethyl sulfoxide
DNA deoxyribonucleic acid
DNAse deoxyribonuclease
DTT dithiothreitol
ECL enhanced chemiluminescence
EM electron microscopy
EDTA ethylenediamine tetra-acetic acid
FACS fluorescence activated cell sorting
FRAP fluorescence recovery after photobleaching
FEAR Cdc14 (Fourteen) Early Anaphase Release
FRET fluorescence resonance energy transfer
G1 growth phase 1
GAL1 galactose inducible promoter 1
GFP green fluorescent protein
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
HRP horseradish peroxidase
Ig immunoglobin
M phase metaphase
MAT mating type
MEN Mitotic Exit Network
min minute
noc nocodazole

14
OD optical density
ORF open reading frame
PBS phosphate buffered saline
PEG polyethylene glycol
PFGE pulse field gel electrophoresis
PMSF phenylmethane sulfonyl fluoride
rap rapamycin
rDNA ribosomal deoxyribonucleic acid
RNAi RNA interference
rpm revolutions per minute
S phase synthesis phase
SDS sodium dodecyl sulphate
SDS-PAGE sodium dodecyl sulphate-polyacrylamide electrophoresis
SMC structural maintenance of chromosomes
TCA trichloroacetic acid
topo I topoisomerase I
topo II topoisomerase II
ts temperature sensitive
Tris 2-amino-2-hydroxymethyl-1,3-propanediol
WT wild type

Chapter 1 Introduction
15
Chapter 1. Introduction
1.1 Historical Perspective
The cell is the fundamental unit of all life we know, from single-celled bacteria
through to the billions that you consist of, reading this thesis. Yet to describe cells
as mere biological building blocks would be to overlook their immense
sophistication. If there were any word in the English language whose simplicity
most belies the complexity it represents, cell would be it. Since it was first
discovered 350 years ago the cell has remained an object of great intrigue and
even today, thousands of researchers internationally continue to make new
discoveries about how cells work. One of the cell’s most important, beautiful and
poorly understood roles is that of cell division and it is this topic that this thesis
addresses.
In 1665, the English philosopher and polymath Robert Hooke first discovered
cells when examining thin slices of cork under a coarse, compound microscope.
Seeing a multitude of tiny pores, he noted that they looked like the walled
compartments a monk would inhabit in a monastery and so called these pores
‘cells’. While Robert Hooke’s simple microscope was able to see little more than
cell walls, the compound microscope would be continuously improved upon during
the 17th century. During this time, the accumulating observations of cells from
naturalists, philosophers and the curious would eventually lead, in 1824, to Henri
Dutrochet formulating one of the fundamental tenets of modern cell theory, by
declaring that “the cell is the fundamental element of organization” (Nezelof, 2003).
In the almost two hundred years that have passed since then, cell theory has
become one of the foundations of biology. The theory states that all living things
are made of cells, that cells are the basic building units of life and that old cells
dividing into two creates new cells. While this latter point has been studied in great
detail over the years, the mechanisms underlying cell division remain incompletely
understood.
The first recorded observation of the internal organisation of cells during
division came from the second half of the 19th century, when the German biologist
Walther Flemming was using aniline dyes from a nearby chemical factory to stain

Chapter 1 Introduction
16
cells before examining them under the microscope. He observed a substance that
strongly absorbed basophilic dyes, which formed apart of threadlike structures
inside the cell nucleus; he would names these chromatin and chromosomes
respectively. Through studying cell division, Flemming observed and carefully
recorded (Figure 1.1) the distribution of chromosomes into the daughter cells, a
process he called mitosis after the Greek word for thread. Following this, Flemming
concluded for the first time that all cell nuclei came from another predecessor
nucleus (Paweletz, 2001). Meanwhile, at roughly the same time, the Augustinian
friar and scientist Gregor Mendel was studying heredity in pea plants, observing
how his plants inherited certain traits with particular patterns. Unbeknownst to him,
his observations would one day gain him fame as the founder of genetics. However,
Flemming was unaware of Mendel’s work, and the significance of Flemming’s
microscopy observations and their ability to describe a mechanism for inheritance
would remain unrealised until the rediscovery of Mendel’s studies almost two
decades later in the early 1900’s.
In the 20th century, it was shown that Flemming’s chromatin was comprised
of DNA and associated proteins, and that the DNA itself was the hereditary vehicle
behind the process of inheritance Mendel had observed (Morgan, 1915). In turn,
chromosomes would be fully described as organised chromatin structures, with
each chromosome containing a single piece of DNA. However, it was quickly
noticed that the presence of chromosomes was not a permanent feature in the cell.
Indeed, during interphase when the cell is not undergoing cell division, individual
chromosomes are not visible. Upon entry into mitosis, the distinct chromosome
‘threads’ could be seen forming through ‘chromosome condensation’ (Figure 1.2) a
process driving the cytological manifestation of the chromosomes themselves.
Thus, it became apparent that the appearance of distinct chromosomes was
dependent on internal factors that were only active during specific times. This was
supported by demonstrations where interphase cells were merged with mitotic cells
causing condensation of the interphase DNA (Rao and Johnson, 1970).

Chapter 1 Introduction
17
Figure 1.1 Walther Flemming's observations of chromosomes during
mitosis
Hand drawn illustrations of mitotic chromosomes, from Flemming’s 1879 paper “Beitrage zur Kenntniss der Zelle und Ihrer Lebenserscheinungen”.

Chapter 1 Introduction
18
Figure 1.2 Mitot ic cycle of rye (Secale cereale) chromosomes
Scanning electron microscopy (SEM) images of chromosomes undergoing condensation. In interphase (A), DNA is in an uncondensed state, such that individual chromosomes are not identifiable. When condensation begins (B), then individual chromosomes become visible. Condensation is greatest in late metaphase (E), when separation of chromatids becomes visible. After successful segregation of the sister chromatids (F), the chromosomes begin to de-condense. Images from (Zoller et al., 2004).

Chapter 1 Introduction
19
The compaction of chromatin that occurs when cells enter mitosis is
probably the most iconic process of dividing cells and despite its seemingly
superficial nature (Figure 1.2), chromosomes condensation is a ubiquitous process
in eukaryotic cells. Such is the widespread prominence of condensation in the cell
cycle that observers began to suspect that process goes beyond the task of just
making chromosomes smaller. Indeed, it is currently hypothesised that
chromosome condensation is required to resolve several important structural
problems associated with proper DNA segregation in mitosis (Baxter and Aragon,
2012). Firstly and perhaps most obviously, chromosomes are longer than the
length of the cell in which they reside. A typical human cell has a diameter of 10
micrometres, while the approximate length of DNA in its largest chromosome is 2
centimetres. Indeed the total length of cellular DNA is up to a hundred thousand
times the length of the cell itself. Therefore if chromosomes are not properly
condensed, they are unlikely to segregate properly, potentially becoming entrapped
during cytokinesis (Koshland and Strunnikov, 1996a, Hirano, 2000). Secondly,
interphase chromosomes mix, particularly in transcriptionally active areas and this
needs to be separated during mitosis. Finally, after DNA replication is complete the
two newly synthesised DNA strands, or sister chromatids, are extensively linked by
both DNA intertwines, or catenanes (Sundin and Varshavsky, 1980), as well as
proteinaceous links (Michaelis et al., 1997). These topological and proteinaceous
links must be removed if the sister chromatids are to be successfully pulled apart to
opposite poles of the cell by the mitotic spindle. The removal of all topological and
proteinaceous links between sister chromatids is called chromosome resolution, a
process describing the spatial individualization of the chromatids from each other. It
has been proposed that chromosome condensation, an ordered process structuring
DNA into individual rod-shaped chromosomes during mitosis, could drive the
resolution of any catenation between chromatids as well as shortening their length,
thus allowing their proper segregation during anaphase (Hirano, 1999).
Given its importance in chromatid resolution and segregation during mitosis,
chromosome condensation can be unequivocally classified as an essential
housekeeping function, indispensible for cell proliferation. However, the driver of
condensation had not been discovered until recently. About 15 years ago parallel
studies in several laboratories found that a protein complex called condensin was
emerging as the primary, molecular driver of mitotic chromosome condensation

Chapter 1 Introduction
20
(Kimura and Hirano, 1997a, Sutani et al., 1999, Strunnikov et al., 1995a). It is
condensin, with particular regard to its role in chromosome resolution, which will be
the focus of this thesis.
1.2 The Structural Maintenance of Chromosome Protein
Family
Condensin is a member of a family of proteins called the structural
maintenance of chromosome (SMC) proteins. SMC proteins have come to be
recognised as one of the most fundamental classes of proteins that regulate the
structural and functional organisation of chromosomes from bacteria to humans
(Ivanov and Nasmyth, 2005, Hirano, 2006). Given that SMC proteins arguably pre-
date histones, it is not surprising that SMC complexes like condensin have adopted
numerous roles throughout the course of evolution and have increasingly been
viewed as global organisers of the genome (Hirano, 2006).
The best known of the SMC proteins is cohesin, a protein complex containing
SMC1 and SMC3, which binds together sister chromatids from when they are
created in S-phase, until they are destroyed at the onset of anaphase through
cleavage by the protease seperase (Uhlmann, 2003). Sister chromatid cohesion
forms the basis for the pairwise alignment of chromosomes upon the mitotic spindle,
making possible the bi-orientated segregation of chromatids at anaphase (Tanaka
et al., 2000, Toyoda et al., 2002). In addition to cohesin and condensin, a third
SMC complex has been identified consisting of a SMC5/SMC6 heterodimer. This
complex is least well characterised of the three, but it has been implicated in having
a DNA damage prevention role as well as aiding DNA repair (Roy and D'Amours,
2011) In addition, it appears to be required for proper chromosome organisation
during meiosis (Farmer et al., 2011).
Overall, the SMC family has been so interesting to chromosome biologists
owing to several factors. First and foremost, the ubiquitous presence of these
proteins, highly conserved across all kingdoms of life, suggest a pivotal and
essential role for SMCs in the proliferation of cells. Genetic and cell-biology studies
have shown that SMC proteins are involved in chromosome segregation, structure,
regulation and recombinational repair, during both mitosis and meiosis. The next

Chapter 1 Introduction
21
intriguing feature of SMC proteins is their unique protein architecture. While
originally supposed to be some form of chromatin motor, many lines of evidence
have come to show that SMC proteins represent a completely novel form of protein
machine, with a distinct tripartite ring structure, that function as a dynamic linker of
the genome (Hirano, 2006).
1.2.1 Condensin function
While condensin has not been studied in as much depth as cohesin,
multiple studies have shown that the condensin complex plays a crucial role in the
compaction of DNA to form structurally stable mitotic chromosomes and their
segregation in vivo (Strunnikov et al., 1995b, Hagstrom et al., 2002, Saka et al.,
1994a, Hirano, 2005b, Ono et al., 2003, Steffensen et al., 2001, Hudson et al.,
2003). The condensin complex is highly conserved among eukaryotes, and
prokaryotes have a condensin–like complex as well. Condensin mutants in all
eukaryotic model organisms, such as S. pombe, C. elegans or D. melanogaster, all
display similar phenotypes of uncondensed nuclei and the presence of anaphase
bridges (Bhat et al., 1996, Hirota et al., 2004, Saka et al., 1994a, Strunnikov et al.,
1995b). In addition, the inactivation of prokaryotic SMCs in E. coli, B. subtilis and C.
crescentus all show temperature-sensitive colony formation and an increase in the
number of anucleate cells at the permissive temperature, suggesting a deficiency in
chromosome segregation (Niki et al., 1991, Weitao et al., 2000, Moriya et al., 1998,
Jensen and Shapiro, 1999).
Condensin has also been reported to have a range of roles in cellular
functions during interphase, such as in the control of gene expression and
heterochromatin formation (Bhalla et al., 2002) or in X. laevis CENP-A assembly
(Bernad et al., 2011). Condensin plays a role in DNA replication checkpoint
signalling in fission yeast (Aono et al., 2002) and additionally condensin mutants
have defects in DNA damage repair (Akai et al., 2011).

Chapter 1 Introduction
22
1.2.2 Eukaryotic condensin structure
Condensin is a 650 kilodalton pentameric complex consisting of a
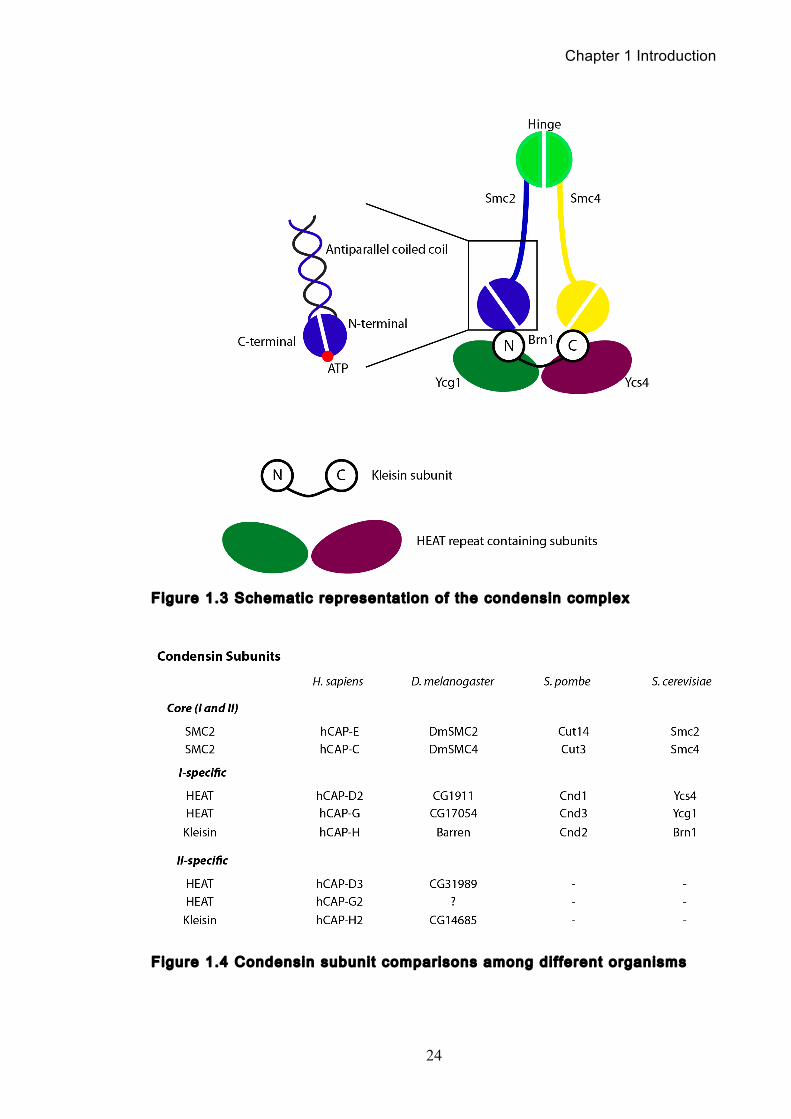
heterodimer of SMC proteins as well as three additional non-SMC subunits (Figure
1.3). The first of the condensin subunits to be identified were proteins of the SMC
family (Strunnikov et al., 1995b), including Cut3 and Cut14 proteins in S. pombe,
their orthologs in X. laevis XCAP-E and XCAP-C and the Smc2 protein in S.
cerevisiae (Hirano and Mitchison, 1994, Saka et al., 1994a, Strunnikov et al.,
1995b). The SMC subunits form the core of condensin’s enzymatic abilities, being
both the DNA-binding subunits (Sutani and Yanagida, 1997a, Cheeseman et al.,
2002) and the DNA-dependent ATPases (Kimura and Hirano, 2000). They have a
unique domain organisation with two canonical nucleotide-binding motifs, known as
the Walker A and B motifs, which are respectively located at the N-terminal and C-
terminal domains. Between the two motifs are two long coiled-coil motifs that are
connected by a non-helical sequence. Biochemical and electron microscopy
studies have shown that the SMC monomer folds back on itself through antiparallel
coiled-coil interactions, thereby creating an ATP-binding ‘head’ domain at one end
and a ‘hinge’ domain at the other. The two SMC monomers can then associate with
each other at the hinge domain to form a V-shaped molecule (Melby et al., 1998,
Haering et al., 2002, Hirano and Hirano, 2002). Additionally, entire condensin and
cohesin complexes from both H. sapiens and X. laevis have been visualised using
electron microscopy. While cohesin displays an open and distinct ring conformation,
condensin’s ring structure appears much more closed with the arms emanating
from the hinge at a smaller angle (Anderson et al., 2002) It has been hypothesised
that these differences in structure could contribute to the different roles of
condensin and cohesin in vivo (Hirano, 2005a).
Meanwhile, the non-SMC subunits are thought to have a regulatory role on
the function of the condensin complex. In vitro studies using X. laevis egg extracts
have shown that the non-SMC subunits of condensin modulate its ATPase activity
(Kimura and Hirano, 2000), though an understanding of how they alter condensin
activity and binding to chromosomes remains to be resolved. Of these three
auxiliary subunits, the CAP-D2 and CAP-G contain HEAT (Huntingtin, elongation
factor 3, the A subunit of protein phosphatase 2A, TOR lipid kinase) repeats
(Neuwald and Hirano, 2000). A number of chromosomal proteins contain these

Chapter 1 Introduction
23
repeats, which are thought to facilitate protein-protein interactions. The two HEAT
repeat containing subunits are recruited by the third subunit, CAP-H (Brn1 in S.
cerevisiae, Barren in D. melanogaster), which belongs to the kleisin family of
proteins (Schleiffer et al., 2003). Kleisins are believed to act as bridging protein
between the two SMC heads as supported by studies where the kleisin subunit was
found to directly bind the SMC head domains of recombinant human condensin
(Onn et al., 2007). For a schematic representation of the known condensin
complexes in different model organisms, please see Figure 1.4.
Given that cohesin exists only as one complex across all organisms it is
found in, the same was assumed for condensin for many years. However, a
genome database search using the human CAP-D2 sequence revealed a distantly
related protein, CAP-D3 (Ono et al., 2003). Immunoprecipitation of CAP-D3
revealed that it also associated with the core condensin subunits (SMC2 and
SMC4) along with two other subunits, CAP-G2 and CAP-H2, the former containing
HEAT repeats and the latter being a kleisin subunit. This new condensin complex
was termed condensin II (Ono et al., 2003, Yeong et al., 2003) and while only
higher eukaryotes have both condensin I and II complexes, the ratio of the two can
vary significantly between different species. Phylogenetic analysis has offered
some insight into how the two complexes might contribute to chromosome
organisation. The model organisms S. pombe, S. cerevisiae, A. nidulans and N.
crassa possess only condensin I and mitotic chromosome condensation is less
dramatic a process in these species when compared to metazoans. This has led to
the speculation that condensin II may allow organisms with larger chromosomes an
additional level of organization (Hirano, 2005b). It has recently been shown using
specific conditional knockouts of the two condensins that depleting either
condensin results in differing mutant phenotypes. Super-resolution microscopy
reveals that condensin I-depleted mitotic chromosomes are wider and shorter, with
a diffuse chromosome scaffold, while condensin II-depleted chromosomes retain a
more defined scaffold, with chromosomes more stretched and seemingly lacking in
axial rigidity (Green et al., 2012).
Chapter 1 Introduction
24
Figure 1.3 Schematic representat ion of the condensin complex
Figure 1.4 Condensin subunit comparisons among dif ferent organisms

Chapter 1 Introduction
25
1.2.3 Prokaryotic condensin structure
SMC proteins are widely conserved across the three domains of life. In
prokaryotes, the best-studied homolog is the SMC-related protein MukB that
functions as the core subunit of a bacterial condensin complex. It is found in a
subclass of γ-proteobacteria that includes E. coli, and despite its limited sequence
homology with other SMC proteins, MukB shares the five-domain structure
common to all SMC family members consisting of a hinge domain, long coiled-coil
arms, and head domains that form the ATP binding pockets (Melby et al., 1998).
MukB forms the complete condensin complex along two other proteins, MukE,
which binds the head domain of MukB via binding to MukF, the kleisin subunit that
is remarkably conserved in bacteria (Yamazoe et al., 1999, Schleiffer et al., 2003).
In B. subtilis the SMC protein also forms a condensin complex with two other
subunits, ScpA and ScpB (Mascarenhas et al., 2002), and the SMC has an intrinsic
DNA binding ability, distinguishable from cohesin by its ability to bind DNA in an
ATP-independent manner (Hirano and Hirano, 1998a).
1.2.4 Condensin’s in vitro biochemical act iv i t ies
Condensin’s ability to reshape chromosomes is both conspicuous and
essential in the cell cycle. While there exist several hypotheses for the mechanistic
basis of condensin’s in vivo actions, which are discussed in greater length in
chapter 1.3.1, several biochemical studies have revealed specific in vitro activities
that presumably underpin condensin’s in vivo functions.
Condensin is able to bind DNA independently of ATP hydrolysis (Kimura
and Hirano, 1997b, Strick et al., 2004), which contrasts with cohesin where it is
required for DNA binding (Onn et al., 2007). While purified condensin complexes
only display low ATPase activity, this is stimulated by the addition of DNA,
increasing ATP turnover rates by up to five-fold (Kimura and Hirano, 1997a, Kimura
and Hirano, 2000, Yoshimura et al., 2002). Interestingly, this stimulation is
dependent on the presence of the non-SMC subunits, both in vitro and in vivo, and
is not seen in their absence (Kimura and Hirano, 2000, Stray and Lindsley, 2003).
While all condensins possess ATPase activities, it remains unclear to what extent
this activity underpins condensin’s cellular functions.

Chapter 1 Introduction
26
In X. laevis, condensin isolated from mitotic cell extracts has the ability to
promote the positive supercoiling of plasmid DNA in the presence of topoisomerase
I (see chapter 1.5 for an introduction to topoisomerases) (Kimura and Hirano,
1997b). A similar activity was detected in a condensin fraction (specifically
condensin II) purified from C. elegans embryos (Hagstrom et al., 2002).
Visualisation by electron spectroscopic imaging suggests this supercoiling is driven
by the wrapping of DNA around the condensin complex in two gyres (Bazett-Jones
et al., 2002). Interestingly, condensin’s supercoiling activity is not constant and
phosphorylation appears to play a major role in its control. Several of condensin’s
non-SMC subunits are targets of mitotic kinases, notably Aurora B (Lavoie et al.,
2004, Takemoto et al., 2006, Lipp et al., 2007) and cyclin-dependent kinase 1
(Cdk1) (Kimura et al., 2001, Kimura et al., 1998, Sutani et al., 1999). Additionally, a
recent study in S. cerevisiae has shown that the Polo kinase Cdc5 directly
phosphorylates all three regulatory subunits of the condensin complex in vivo and
that consequently results in a hyperactivation of condensin’s supercoiling activity
(St-Pierre et al., 2009). Similarly, the MukB subunit of the E. coli SMC complex can
support the formation of supercoils in circular plasmids in the presence of
topoisomerase I. However, this reaction is ATP-independent and produces
negative supercoiling, which is the opposite sign to that generated by eukaryotic
condensin (Petrushenko et al., 2006).
A different change in DNA topology is observed when condensin
holocomplexes immunopurified from X. laevis egg extracts or isolated S. cerevisiae
Smc2/Smc4 dimers are incubated with nicked circular DNA in the presence of ATP
and topoisomerase II. Here, the condensin complex converts the nicked circular
DNA, via topoisomerase II-catalysed strand passage, into a positive three-noded
knot, also known as a trefoil (Stray and Lindsley, 2003, Kimura et al., 1999). This
indicates that condensin cannot only introduce positive supercoils into DNA, but
has the ability to organise two or more supercoils into an ordered, solenoidal form.
Intriguingly, this knotting activity may not require ATP hydrolysis, as evidenced by a
mutant Smc2/Smc4 dimer, defective in ATP hydrolysis, which displays similar if not
identical knotting activity (Stray and Lindsley, 2003). The E. coli MukB dimer, like
its eukaryotic counterpart, can also promote the formation of right-handed DNA
knots in the presence of topoisomerase II (Petrushenko et al., 2006).

Chapter 1 Introduction
27
An additional ATPase-independent activity is the promotion of single-
stranded DNA annealing by the S. pombe Smc2/Smc4 heterodimer (Sakai et al.,
2003, Sutani and Yanagida, 1997b). It has been speculated that this activity could
be required to remove ‘leftover’ interphase products from mitotic chromosomes,
such as RNA-DNA hybrids, in order to allow their correct segregation (Yanagida,
2000). The B. subtilis SMC dimer was shown to promote DNA reannealing in a
similar but ATP-stimulated manner (Hirano and Hirano, 1998b).
1.3 Chromosome Condensation
Chromosomes are composed of roughly equal masses of DNA, histones and
non-histone proteins. Depending on the cell cycle stage, chromosomes adopt
different conformations with varying levels of compaction (see Figure 1.2). Mitotic
chromosomes are the most distinct, reaching their most condensed forms just prior
to anaphase onset. However, even in interphase when chromosomes are most
‘uncondensed’, their length is already 1,000 fold shorter than their linear
counterparts would be. While the hierarchical packaging required to reach the level
of compaction seen in mitotic chromosomes remains a hotly debated area in
chromosome biology (Grigoryev and Woodcock, 2012), it is clear that condensation
of interphase chromatin into mitotic chromosomes is tightly linked with cell cycle
progression and requires condensin.
At the simplest level of packaging, DNA is organised by the histones. 147
base pairs of DNA wrap around each nucleosome, which are comprised of histones,
and each nucleosome is linked to the next by approximately 60 base pairs of linker
DNA (Laemmli et al., 1992). Unsurprisingly, histones were long-standing
candidates for the molecular engine underlying chromosome condensation.
However, when the histone fraction was extracted from mitotic chromosomes in
vitro, the remaining insoluble non-histone fraction, the so-called ‘chromosome
scaffold proteins’, appeared to maintain a structure similar to that of the intact
mitotic chromosomes (Adolphs et al., 1977). While histones do contribute to
condensation in protozoa and higher eukaryotes through phosphorylation of the
core histone tail (Roth and Allis, 1992, Patterton et al., 1998), they are not
responsible mechanically.

Chapter 1 Introduction
28
When high-salt extractions were performed on DNA to reveal what proteins
were tightly associated with it, two of the most abundant components of eukaryotic
chromatin were identified, topoisomerase II and condensin (Lewis and Laemmli,
1982, Earnshaw et al., 1985, Gasser et al., 1986). Soon after, topoisomerase II
dysfunction in S. pombe cells was shown to disrupt both chromosome
condensation and progression through anaphase (Uemura et al., 1987b, Holm et
al., 1985), implicating topoisomerase II as an engine of condensation. However,
topoisomerase II was also known to participate in other functions in chromatin,
such as replication and transcription, and topoisomerase II’s role in chromosome
condensation was shown not to be universal (Hirano and Mitchison, 1994, Lavoie
et al., 2002). Attention then focused onto condensin, a complex that when
inactivated or depleted from eukaryotic cells results in reduced chromosome
compaction (Saka et al., 1994b, Hirano and Mitchison, 1994, Strunnikov et al.,
1995b, Koshland and Strunnikov, 1996b, Hudson et al., 2003, Steffensen et al.,
2001). An overview of the mechanisms by which condensin could drive
condensation can be seen in Figure 1.5.
1.3.1 Condensin’s biochemical act iv i t ies drive condensation
In S. cerevisiae, condensin binding is consistent throughout the cell cycle,
implying that binding itself does not drive condensation, yet its activity is somehow
maximised upon entry into mitosis (Guacci et al., 1994). As discussed in chapter
1.2.4, condensin can drive the supercoiling and knotting of DNA and these
activities are modulated by cell cycle kinases. Indeed, condensin mutants that have
a reduced ability to be phosphorylated in vivo are defective in anaphase-specific
chromosome condensation (St-Pierre et al., 2009). Therefore, it would be
reasonable to propose that the reconfiguration of chromosome topology caused by
condensin’s biochemical activities underpins chromosome condensation. Recent
genome-wide mapping in S. cerevisiae has shown that condensin’s individual
binding sites have an average spacing of 420 kilobases of DNA between them
(D'Ambrosio et al., 2008b). Consequently, between the binding sites of each
condensin complex could be hundreds of nucleosomes that would introduce large
numbers of negative turns to the DNA (Lee et al., 2007). This suggests that taken

Chapter 1 Introduction
29
on its own, condensin’s biochemical activities would not be sufficient to obtain the
required level of chromosome compaction in mitosis. Further experiments are
required to test whether the density of condensin binding is greater on vertebrate
chromosomes, which could account for their stronger compaction during mitosis.
However, while condensin’s biochemical activities may provide an essential
contribution, it is more likely that condensin accomplishes condensation through a
catalytic, linkage or co-operative mechanism.
1.3.2 Condensin as a catalyst driv ing condensation?
If condensin’s biochemical activities alone are not sufficient to explain
condensation then perhaps their action acts as mid-point, catalysing further
topological changes by other chromosomal proteins such as the topoisomerases
(see chapter 1.5). A recent study reported an increase in positive supercoiling of
circular yeast mini-chromosomes preceding their segregation, and this is
dependent on the presence of both the mitotic spindle and condensin function
(Baxter et al., 2011). Most interestingly, they found that positively supercoiled mini-
chromosome dimers isolated from topoisomerase II-deficient cells arrested in
mitosis are more efficiently decatenated in vivo by recombinant topoisomerase II
than negatively supercoiled dimers. This therefore suggests that condensin-driven
changes in topology can render that DNA into a more favourable substrate for
topoisomerase II. However, it is not immediately clear how condensin’s promotion
of decatenation by topoisomerase II could drive chromosome condensation and so
far, there are no further reports of catalytic behaviour in condensin. Nevertheless,
this particular behaviour could have substantial importance during chromosome
resolution, a topic discussed in chapter 1.4.
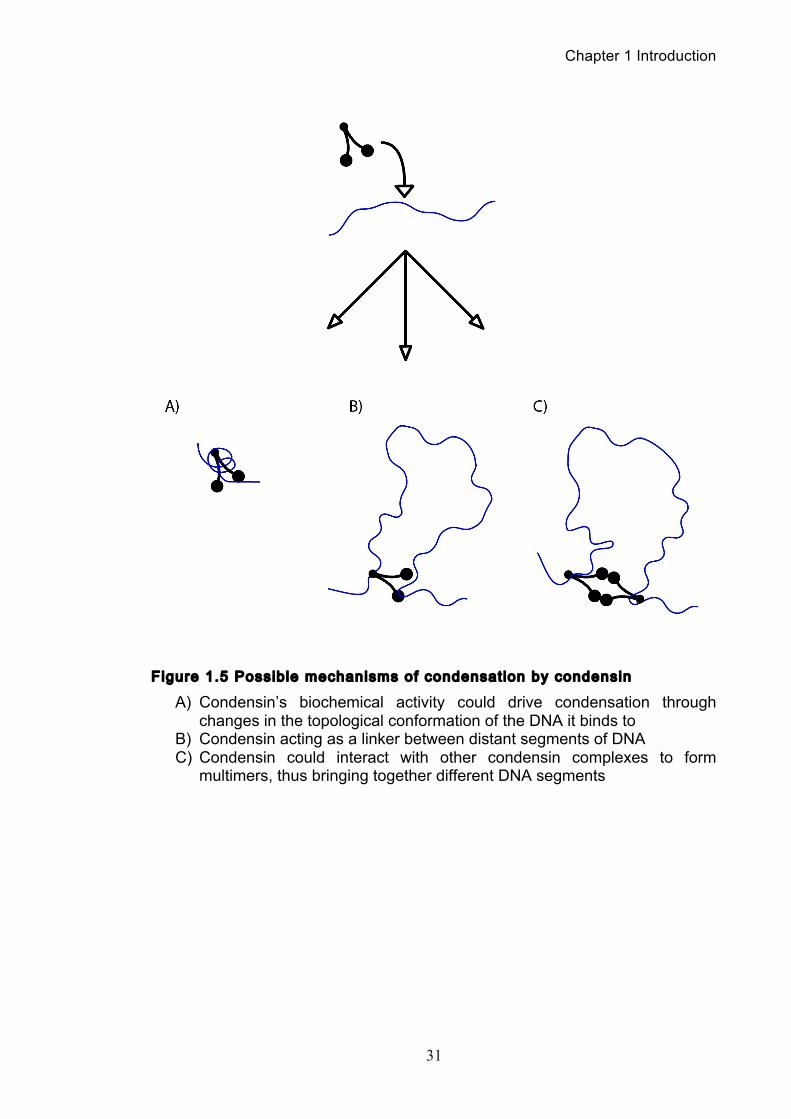
1.3.3 Condensin could behave as a DNA l inker to drive condensation
Another alternative is that condensin could compact DNA by acting as a
molecular linker that brings different segments from within a single DNA strand.
This idea is supported by an interesting experiment where a single DNA strand
tethered to a paramagnetic bead was nano-manipulated allowing measurement of

Chapter 1 Introduction
30
its end-to-end extension and hence level of compaction (Strick et al., 2004). It was
seen that condensin could rapidly physically compact DNA in an ATP-hydrolysis
dependent manner. The compaction reaction was highly dynamic and reversible,
with a kinetics that did not correspond with condensin-driven positive supercoiling.
Rather, the behaviour is better explained through the contraction of the linear DNA
by bringing two segments physically together and looping out the intervening DNA.
Of course the proposal that one condensin complex can link two non-adjacent
segments of DNA leads to the obvious question, is there more than one DNA
binding site on the condensin complex?
Studies of eukaryotic condensin have so far only found evidence for one
binding site. Atomic force microscopy of S. pombe condensin appears to show that
DNA associates with the complex at the hinge domain of the SMC proteins
(Yoshimura et al., 2002). Furthermore, In vitro assays have shown that the
presence of DNA blocks the proteolytic cleavage of the Smc2 hinge domain (Onn
et al., 2007) and furthermore, isolated Smc2/Smc4 hinge domains cause a shift in
DNA electrophoretic mobility suggesting a direct interaction (Griese et al., 2010). In
prokaryotes on the other hand, examination of their SMC proteins identified a
positively charged patch in the structure of the dimerized head domains and well as
in the hinge domain that could interact with DNA (Shin et al., 2009). The
introduction of mutations that negate these positive patches reduce the shift seen
in the electrophoretic mobility of plasmid DNA suggesting that for prokaryotic SMCs,
the possibility remains that they can bind two different segments of DNA at their
head and hinge domains.
Chapter 1 Introduction
31
Figure 1.5 Possible mechanisms of condensation by condensin
A) Condensin’s biochemical activity could drive condensation through changes in the topological conformation of the DNA it binds to
B) Condensin acting as a linker between distant segments of DNA C) Condensin could interact with other condensin complexes to form
multimers, thus bringing together different DNA segments

Chapter 1 Introduction
32
Is this head/hinge double binding of DNA by the condensin complex the only
was it could link DNA? Despite condensin sharing a similar tripartite ring structure
as its fellow SMC complex cohesin, it was assumed that it did not share cohesin’s
ability to capture DNA strands within its ring structure (Ivanov and Nasmyth, 2005,
Anderson et al., 2002). However, using the same techniques used for cohesin, it
has now been shown that condensin rings can also encircle chromosomal DNA
(Cuylen et al., 2011). While cohesin rings may be able to freely slide along
entrapped DNA, condensin binding has been mapped to distinct sites (D'Ambrosio
et al., 2008b). If condensin entraps two DNA strands, this could be explained by the
constant association of condensin with one chromosome segment, while local
chromatin rearrangements are performed through the sliding of the entrapped
second strand. Another possibility is that condensin complexes entrap one DNA
strand, yet bring together separate DNA segments through association with other
complexes.
1.3.4 Does condensin act through a co-operative mechanism?
Irrespective of how condensin complexes bind DNA, it has long been
suspected that condensin function depends on interaction between the complexes.
While it had been assumed that the engagement of SMC head domains always
occurred in a heterotypic fashion (for example, Smc2-Smc4, but not Smc2-Smc2),
the result that two Smc1 head domains had homodimerised in a protein crystal
came as a surprise (Haering et al., 2004). While possibly an artefact of crystal
preparation, it led credence to the idea that condensin complexes may function in a
multimeric manner, with complexes associating to form co-operative condensation
machinery. This proposal was further supported by single molecule studies where
halving the protein concentration can completely eliminate condensation activity
(Strick et al., 2004). Microscopy observations of condensin see its distribution along
the inner axes of mitotic chromosomes, consistent with the notion that condensin
forms part of a chromosome ‘scaffold’ (Maeshima and Laemmli, 2003, Cabello et
al., 2001). However, how condensin complexes could multimerise remains unclear.
Certainly if condensin ‘scaffolds’ were to form, they could not be static structures as
shown by fluorescence recovery after photobleaching (FRAP) experiments, where

Chapter 1 Introduction
33
high turnover of condensin I was observed on mitotic chromosomes (Gerlich et al.,
2006b, Oliveira et al., 2007). While evidence for multimerisation of eukaryotic
condensins remains scarce, electron and atomic force microscopy has observed
prokaryotic SMC complexes forming linear or rosette-like aggregates
(Mascarenhas et al., 2005, Matoba et al., 2005).
In summary, of the possible mechanisms of condensin action discussed, it is
most likely that reality combines aspects from all of them. Current research into
condensin is revealing tantalisingly more about its mode of action, especially with
regards to chromosome resolution. While playing a distinct role from condensation,
it is becoming increasingly clear that condensin’s contribution to both processes
are inexorably linked.
1.4 Chromosome Resolution
Chromosome resolution is a critical step in mitosis if the sister chromatids are
to be successfully segregated and thus correct chromosome segregation is a pre-
requisite for preserving genome integrity. Cohesin and condensin are both required
to ensure faithful chromosome segregation and dysfunction of either can result in
mis-segregations and aneuploidies. While most aneuploidy human embryos are not
viable, aneuploidies that occur later in human life are often associated with the
development of malignant cancer. Chromosome instability, a term referring to a
high rate of loss or gain of whole or parts of chromosomes, is a characteristic of
most human cancers and is associated with their poor prognosis and drug
resistance (McGranahan et al., 2012, Mannini et al., 2012). In summary,
chromosome resolution describes a process of individualization, where all
topological and proteinaceous links between the newly replicated sister chromatids
are removed to produce distinct and spatially isolated entities.
1.4.1 Proteinaceous l inkages
There are two types of link between the sister chromatids that need to be
removed, the first being proteinaceous. These links are predominated by the SMC

Chapter 1 Introduction
34
complex cohesin, which establishes sister chromatid cohesin with a timing that is
tightly coupled with DNA replication so as to not allow replication products to drift
apart (Michaelis et al., 1997, Uhlmann and Nasmyth, 1998).
While cohesin binds to chromosomes before entry into S phase, it is only
during DNA replication that cohesion is established. This is accomplished by a
replication fork-associated acetyltransferase (Eco1 in S. cerevisiae, Eso1 in S.
pombe) that acetylates the Smc3 subunit of cohesin. This protein is essential for
the activation of cohesin and in cells lacking Eco1, cohesin bind the chromosomes,
but the physical linkages between sister chromatids are never established (Ivanov
et al., 2002). Cohesion establishment between sister chromatids is of utmost
importance in mitosis, essential for the correct organisation and bi-orientation of the
chromatids within the mitotic spindle in preparation for segregation during
anaphase. Sister chromatid cohesion is lost upon anaphase onset through
irreversible cleavage of the cohesin subunit Scc1 by the protease separase
(Uhlmann et al., 1999). In vertebrates, the resolution of cohesion has an additional
precursor step where a substantial portion of cohesin is removed from
chromosomes as they condense in prophase in a step mediated by mitotic kinases
(Losada et al., 2000, Losada et al., 2002, Sumara et al., 2000, Sumara et al., 2002).
As soon as cohesin is dissociated from the chromosomes, the class I histone
deacetylase Hos1 deacetylates the cohesin subunit Smc3 (Borges et al., 2010). As
non-acetylated Smc3 is required as a substrate for cohesion establishment in the
following G1 phase, the cycle is completed and ready for the next round of DNA
replication.
1.4.2 Topological l inkages
Topological links describe the physical intertwining of two newly synthesised
DNA strands produced during DNA replication. This occurs during DNA replication
as a consequence of the convergence of replisomes. Initially, the topological
challenge resulting from DNA replication was most obvious in prokaryotes, such E.
coli, which have circular genomes. The unwinding of the parental DNA for
replication in a closed, circular system would inevitably generate new strands of
DNA that were linked, or catenated, together. To overcome this topological

Chapter 1 Introduction
35
constraint of DNA replication, it was first proposed in the 1960s that a nick in one of
the parental DNA strands would eliminate any entanglements by allowing free
rotation of the DNA along its axis (Cairns, 1963). While eukaryotic chromosomes
are linear, it was found that these larger genomes behaved exactly like circular
DNA owing to the existence of domains with fixed ends that prevent any free
rotation of the DNA (Worcel and Burgi, 1972). From early on, predictions were
made that in order for any DNA to be segregated post-replication, the DNA would
need to be cut in several places (Tessman et al., 1957). These initial predictions
were made with endonucleases in mind, but as it would turn out, endonucleases
would not be responsible for these cleavages, but rather a new class of DNA
enzymes called topoisomerases. Specifically, topoisomerase I would serve as a
DNA swivel (Champoux and Dulbecco, 1972) while topoisomerase II would allow
the physical passage of one DNA strand through another via a transient double-
strand break (Brown et al., 1979, Gellert et al., 1976).
1.5 Topoisomerases
Topoisomerases are a very important class of cellular enzymes, required
for the survival of all organisms through their ability to alter DNA topology by
generating transient breaks in the double helix. There are two major classes of
topoisomerases, type I and type II, which are distinguishable by the number of DNA
strands that they cleave and the mechanism by which they alter the topological
properties of the genetic material (Wang et al., 2002, Champoux, 2001, Deweese
and Osheroff, 2009).
1.5.1 Types of topoisomerase
In eukaryotes, type I topoisomerases are monomeric enzymes that require
no high-energy cofactors and can be organised into two subclasses: type IA and
type IB. Type I topoisomerases alter topology by creating transient single-stranded
breaks in the DNA, followed by passage of the opposite intact strand through the
break (type IA) or by controlled rotation of the helix around the break (type IB). As a
consequence of this process, type I topoisomerases can moderate DNA

Chapter 1 Introduction
36
supercoiling, but are not able to remove knots or tangles from duplex DNA
(Leppard and Champoux, 2005). Eukaryotic type II topoisomerases function as
homodimers and require divalent metal ions and ATP for complete catalytic activity.
Topoisomerase II can interconvert different topological forms of DNA (also known
as isoforms) via a ‘double-stranded DNA passage reaction’, which can be broken
down into a series of distinct steps (Wang, 1998). Owing to this DNA passage
mechanism type II topoisomerases cannot only alter DNA supercoiling like the type
I enzymes, but also can remove DNA knots and catenation. It should be noted that
after action upon by topoisomerase II, the chemical structure of the ligated DNA is
identical to that of the original substrate. Hence, only the topological properties of
the double helix are changed by the actions of this enzyme.
Lower eukaryotes and invertebrates, such as S. cerevisiae, encode only a
single topoisomerase II (Goto and Wang, 1984), while vertebrate species encode
two closely related isoforms of the enzyme, topoisomerase IIα and topoisomerase
IIβ. While the isoforms are encoded by separate genes and differ in their protomer
molecular masses (170 and 180 kDa, respectively), they share similar
enzymological characteristics and high sequence homology. However,
topoisomerase IIα and IIβ do have differing patterns of expression and their own
distinct cellular roles. Type IIα is most similar to the type II topoisomerases see in
lower eukaryotes and is essential for proliferating cells to survive, with protein
levels increasing significantly during periods of cell growth. Furthermore, the
enzyme is regulated through the cell cycle with peak concentrations in G2 and M
phase (Heck and Earnshaw, 1986, Heck et al., 1988, Kimura et al., 1994).
Topoisomerase IIα is associated with replication forks and remains tightly bound to
chromosomes throughout mitosis. Thus, it is believed to be the isoform that
functions in the cell’s growth-related processes, such as DNA replication and
chromosome segregation (Wang et al., 2002, Christensen et al., 2002).
Topoisomerase IIβ on the other hand is dispensable on the cellular level and
cannot compensate for the loss of topoisomerase IIα in mammalian cells, implying
that these two isoforms do not play redundant roles in replicative processes
(Sakaguchi and Kikuchi, 2004). Instead, topoisomerase IIβ appears to be required
for proper neural development, most likely through involvement in the transcription
of developmentally or hormonally regulated genes (Ju et al., 2006, Yang et al.,
2000).

Chapter 1 Introduction
37
Prokaryotes encode two distinct type II topoisomerases, gyrase and
topoisomerase IV. Gyrase is unique in that it is the only known topoisomerase that
can actively negatively supercoil DNA and it plays an important role in regulating
the superhelical density of bacterial DNA (Levine et al., 1998). Meanwhile,
topoisomerase IV has a similar role to eukaryotic topoisomerase II, required for
decatenation of sister chromatids after DNA replication and general removal of
knots from the genome (Deweese and Osheroff, 2009).
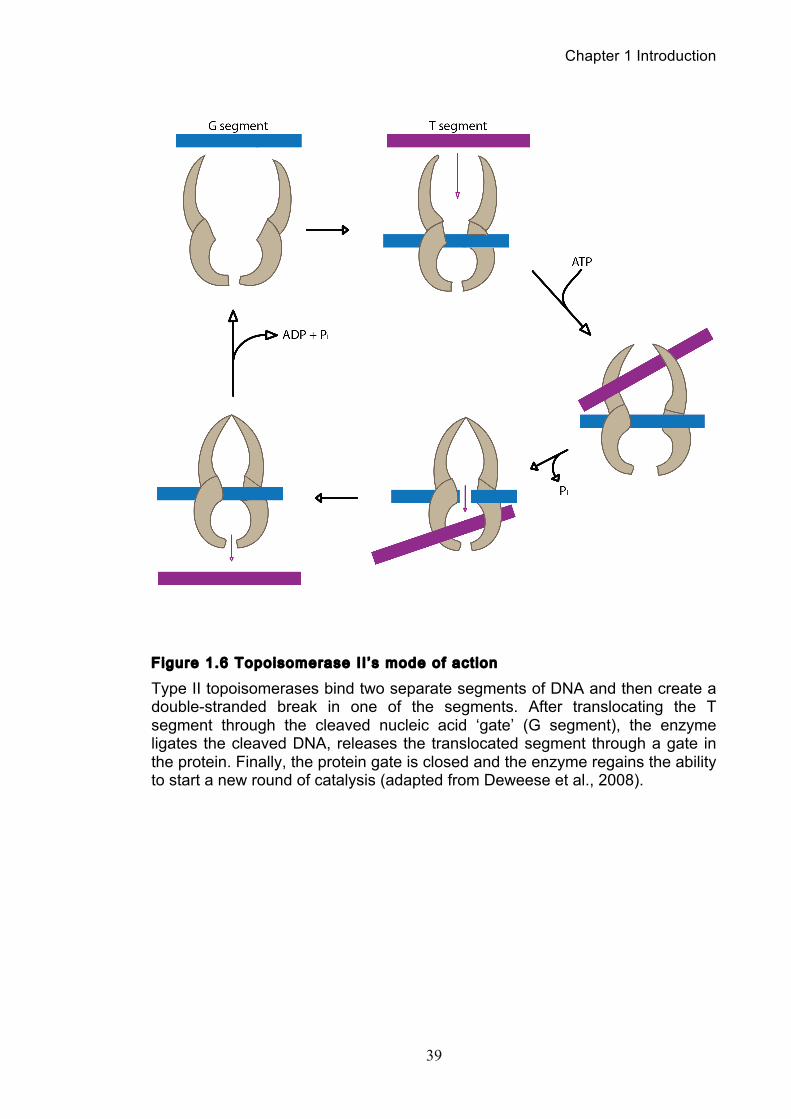
1.5.2 Topoisomerase II mode of act ion
The ability of topoisomerase II to cleave and ligate DNA is central to all of its
catalytic functions. To perform a strand passage reaction, topoisomerase II
interacts with two DNA strands introducing a double strand break in one DNA
strand, termed the gate or G segment, and then passing a second strand termed
the T segment through the break (Figure 1.6). In the presence of Mg2+, the enzyme
can cleave the DNA by nucleophilic attack, forming a phosphotyrosine linkage
between each single strand and a tyrosine in each subunit. The covalent enzyme-
DNA linkage plays two critical roles in the topoisomerase II reaction mechanism.
Firstly, it conserves the bond energy of the sugar-phosphate DNA backbone.
Secondly, the covalent bond does not allow the cleaved DNA chain to dissociate
from the enzyme, thus maintaining the integrity of the genetic material during the
cleavage event (Wang, 1998). This is crucially important as double-stranded
breaks in DNA can have mutagenic and even lethal consequences for cells.
The next step is ATP binding, which causes the enzyme to form a closed
clamp. The closed clamp can also capture another strand (the T strand) that will
then pass through the break made in the G strand. After passing through the break
in the G strand, the T strand exits the enzyme through its carboxy terminus. ATP
hydrolysis occurs at two steps in the reaction cycle, the first ATP hydrolysed likely
assists in the passage of the T segment. The second hydrolysis step allows the
clamp to reopen, releasing the G segment, although the enzyme may initiate
another catalytic cycle without dissociating from the G strand (Harkins and Lindsley,
1998).

Chapter 1 Introduction
38
While it acts globally across all DNA, topoisomerase II appears to cleave at
preferred sites. However, the consensus sequence for cleavage is weak, and many
sites of action do not conform to it (Capranico and Binaschi, 1998). Currently, the
mechanism by which topoisomerase II selects DNA sites to act upon is not
apparent, and it is currently not possible to predict de novo whether a given DNA
sequence will support scission (Deweese et al., 2008). Most likely, the specificity of
topoisomerase II-mediated cleavage is determined by a combination of the local
flexibility, structure or malleability of the DNA that accompanies the sequence, as
opposed to the direct recognition of the bases that comprise that sequence (Velez-
Cruz et al., 2005).
Chapter 1 Introduction
39
Figure 1.6 Topoisomerase II ’s mode of act ion
Type II topoisomerases bind two separate segments of DNA and then create a double-stranded break in one of the segments. After translocating the T segment through the cleaved nucleic acid ‘gate’ (G segment), the enzyme ligates the cleaved DNA, releases the translocated segment through a gate in the protein. Finally, the protein gate is closed and the enzyme regains the ability to start a new round of catalysis (adapted from Deweese et al., 2008).

Chapter 1 Introduction
40
1.5.3 The physiological importance of topoisomerase II
The correlation between faithful segregation of sister chromatids in mitosis
and cancer cell development was touched upon earlier in this introduction. SMC
proteins are required for proper chromosome segregation and their dysfunction can
lead to the mis-segregations and aneuploidies associated with malignant tumours.
However, the unique and critical role topoisomerase II has in managing DNA
topology makes it perhaps the most important enzyme to understand with regards
to chromosome segregation. Indeed, topoisomerase II has become a prime target
for manipulation by anti-cancer drugs (Fortune and Osheroff, 2000, McClendon and
Osheroff, 2007).
Unlike most other protein-targeted drugs, which kill cells by robbing them of
an essential enzyme activity, topoisomerase-targeted drugs exploit the potentially
lethal nature of topoisomerases. During the strand passage reaction, the
topoisomerase II-DNA cleavage complexes are normally short-lived and readily
reversible, with the DNA cleavage/ligation equilibrium of the enzyme greatly
favouring ligation (Bender et al., 2008, Mueller-Planitz and Herschlag, 2008).
Therefore, topoisomerase-targeted drugs ‘poison’ topoisomerase II by increasing
the steady-state levels of the DNA cleavage complexes (Fortune and Osheroff,
2000, McClendon and Osheroff, 2007). This action converts topoisomerases into
potent physiological toxins, resulting in the generation of DNA double strand breaks.
Subsequently, the resultant mass of DNA damage can overwhelm the cell’s repair
pathways resulting in apoptosis (Roos and Kaina, 2012).
These drugs have proven to be powerful tools for oncologists, but growing
evidence now suggests that topoisomerase II-mediated DNA cleavage can trigger
chromosomal translocations that lead to the development of specific types of
leukaemia. It has been shown that up to 3% of patients who take topoisomerase II-
targeted drugs eventually develop acute myeloid leukaemia (Felix et al., 1995, Felix,
1998, Bender et al., 2008). Therefore, given topoisomerase II’s role in both healthy
cell division and the development of malignancies, gaining a better understanding
of this enzyme’s function in chromosome resolution and its relationship to other
chromosomal proteins like condensin, is of great importance.

Chapter 1 Introduction
41
1.6 Chromosome resolution, topoisomerase II and
condensin - an unresolved relationship
From bacteria to humans, DNA is globally underwound, or negatively
supercoiled, by approximately 6% (Bauer et al., 1980). This state is important as
duplex DNA is merely the storage form of the genetic information and in order to
replicate or express this information, the two strands of DNA must be separated. As
global negative supercoiling of the genome imparts increased single-stranded
character to the double helix, it therefore also facilitates strand separation (Espeli
and Marians, 2004). While negative supercoiling may promote many nucleic acid
processes, DNA overwinding, or positive supercoiling, inhibits them. During
replication the linear movement of DNA enzymes, such as the helicases and
polymerases of the replisome, compresses the turns of the double helix into a
shorter region ahead of the replication fork causing it to become increasingly
overwound. The positive supercoiling that results makes it progressively more
difficult to open the two strands of the double helix and eventually this can block
essential nucleic acid processes (Travers and Muskhelishvili, 2007). Normally, type
I topoisomerases are able to relieve this supercoiling but in the latter stages of
replication when replisomes approach each other, the two replication forks impinge
on each other and there is no longer room for a type I topoisomerase to relax the
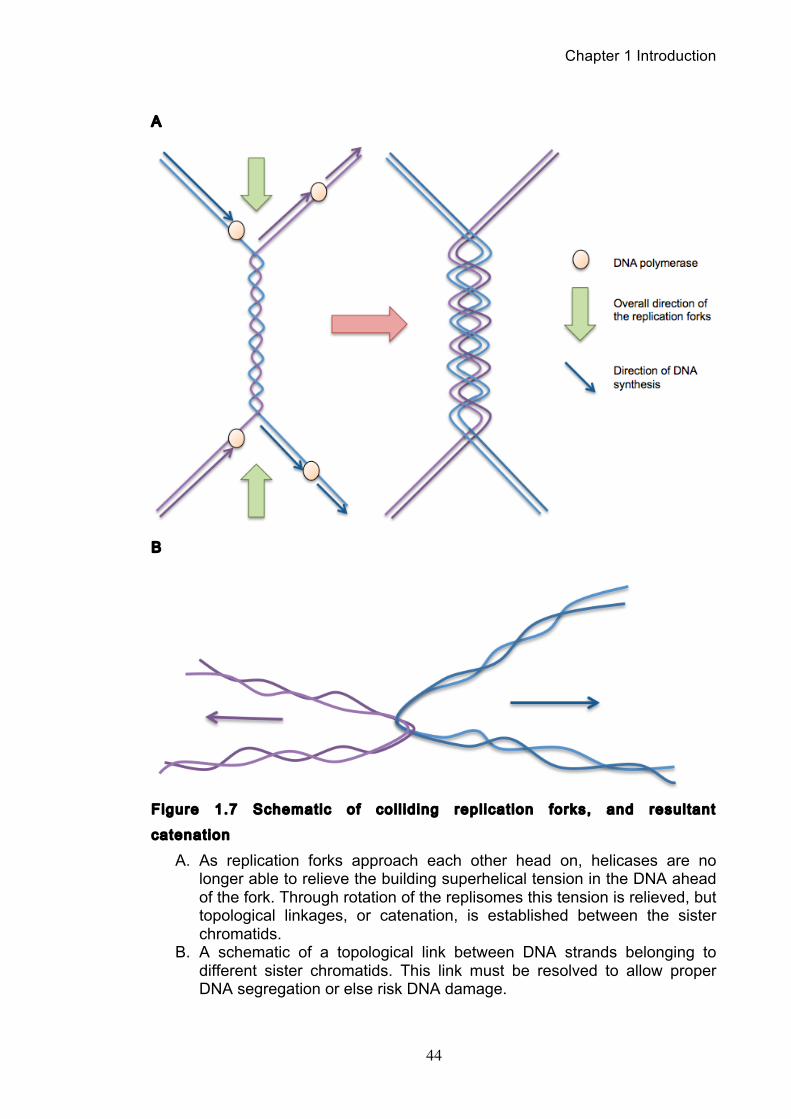
positive supercoiling (Murray and Szostak, 1985, Sundin and Varshavsky, 1980,
Sundin and Varshavsky, 1981). By allowing the replisomes to rotate along the axis
of the DNA strand, the torsion building up ahead of the fork is relieved. A
consequence of this is that pre-catenated DNA molecules are created behind the
fork, that upon replication completion, result in fully catenated DNA daughter
molecules (Figure 1.7) (Zechiedrich and Cozzarelli, 1995, Lucas et al., 2001).
These topological links between the sister chromatids must be removed should
they be successfully segregated during anaphase and this is performed by
topoisomerase II, the only enzyme capable of passing one DNA strand through
another via a transient double strand break (Wang, 2002). Therefore,
topoisomerase I and II mediated topological transitions at the replication forks
ensure proper fork progression and stability and prevent activation of the DNA
damage checkpoint (Bermejo et al., 2007). Topoisomerase II is therefore essential
in mitosis and mutational analysis from various model organisms have shown it to

Chapter 1 Introduction
42
be the main decatenation driver, with mutants having distinct phenotype
characteristics including poorly condensed chromosomes as well as non or mis-
segregation of the sister chromatids in anaphase (Downes et al., 1991, Holm et al.,
1985, Uemura et al., 1987a).
While the importance of topoisomerase II in decatenation was indisputable,
several nagging issues persisted that questioned whether this enzyme could
actually represent the whole story. The most pertinent question was how the cell
could be sure its whole genome had been fully disentangled by anaphase onset?
While topoisomerase II is an exceptional enzyme in its capabilities, its main
shortcoming is that its actions are bi-directional. Not only can it remove topological
linkages, but it can introduce them as well. In most circumstances, when two
entangled DNA strands are decatenated by topoisomerase II, the liberated strands
are then likely to move apart spatially. Not only are the two strands released from
opposing sides of the topoisomerase enzyme, but their further separation is also
energetically favourable through Brownian motion. Therefore, the likelihood that
two strands come close enough together again to be recatenated is low. However,
in a situation where the two strands are physically constrained, such as if a cohesin
complex entraps both, then the probability of recatenation is greatly increased.
Various arguments have been made to overcome this issue, the first being that
since topoisomerases are ATPases, they are capable of catalysing an end point
that is beyond a thermodynamically stable equilibrium (Vologodskii et al., 2001). In
addition, it has been shown that topoisomerase II can detect specific DNA topology
as suggested by their ability to interact with DNA crossovers (Dong and Berger,
2007, Zechiedrich and Cozzarelli, 1995). However, these arguments are not fully
satisfactory in explaining how the complete decatenation of the sister chromatids is
ensured.
The idea that condensin may contribute to resolution of sister chromatids in
addition to its role in chromosome condensation was reinforced with the
characterization of the complex in S. cerevisiae (Freeman et al., 2000). When
compared to vertebrates, the chromosomes of budding yeast do not attain the
same high levels of condensation in mitosis. In fact in S. cerevisiae, condensin-
driven condensation causes only a 1.5-fold compaction of the chromosome arms
and for successful chromosome segregation, only the longest chromosomes
require this shortening (Guacci et al., 1994). Yet in the absence of condensin, all

Chapter 1 Introduction
43
the chromosomes fail to segregate (Bhat et al., 1996). This situation makes the
sister chromatid sorting function of condensin even more apparent. Recently,
studies of S. cerevisiae rDNA have been key in developing our understanding of
condensin’s resolution function.
Chapter 1 Introduction
44
A
B
Figure 1.7 Schematic of col l id ing repl icat ion forks, and resultant
catenation
A. As replication forks approach each other head on, helicases are no longer able to relieve the building superhelical tension in the DNA ahead of the fork. Through rotation of the replisomes this tension is relieved, but topological linkages, or catenation, is established between the sister chromatids.
B. A schematic of a topological link between DNA strands belonging to different sister chromatids. This link must be resolved to allow proper DNA segregation or else risk DNA damage.

Chapter 1 Introduction
45
1.6.1 Lessons from the rDNA locus
The budding yeast rDNA locus consists of between one and two hundred
9.1kb DNA repeats located on the longest arm of chromosome XII. At this site,
ribosome biogenesis occurs as the nucleolus assembles on top of the locus. It has
been previously noted that sister chromatids remain connected at the rDNA until
mid-anaphase independently of the cohesin complex and that to remove this
connection, both topoisomerase II and condensin activities are required (D'Amours
et al., 2004, Sullivan et al., 2004). The phosphatase Cdc14, one of the most
important down regulators of mitotic cyclins during mitotic exit in S. cerevisiae,
appears to activate resolution of the rDNA locus, though a mechanistic explanation
of this remains undetermined. The rDNA locus was interesting to study, as not only
was it easy to visualise using immunofluorescence microscopy, but the condensin-
mediated compaction of the locus could be functionally separated from condensin-
mediated resolution. This is because compaction, but not resolution, depends on
Aurora B kinase. Upon inactivation of the kinase, uncondensed rDNA sister
chromatids succeeded in full rDNA disjunction (D'Amours et al., 2004, Sullivan et
al., 2004). However, despite these uncondensed sister rDNA loci being fully
resolved, they fail to be efficiently segregated in the dividing cell (Sullivan et al.,
2004). Overall, this suggests that the most important function of condensin in rDNA
resolution is not chromosome compaction, but resolution.
Examination of this situation is made even more interesting by considering
the role rDNA plays within the cell. As the site of ribosome biogenesis, the rDNA
locus is area of intense and continual transcription by RNA pol I and RNA pol III. It
is well known that specific DNA structures are formed during the process of
transcription and these are recognised and relaxed by topoisomerases (Mondal
and Parvin, 2001, Osborne and Guarente, 1988). Besides this, in S. cerevisiae
transcription continues non-stop through the cell cycle unlike in higher eukaryotes
where it pauses during anaphase (Elliott and McLaughlin, 1979, Sullivan et al.,
2004). Given that rDNA transcription generates a large number of DNA structural
forms, it is conceivable that this locus would impede its own topological resolution
without the concerted action of condensin and topoisomerase II. Correspondingly, it
has been shown that growing S. cerevisiae cells in the presence of a transcription-
inhibiting drug enhances rDNA segregation (Tomson et al., 2006). Given that the

Chapter 1 Introduction
46
requirement for condensin in rDNA partitioning and its massive enrichment at this
loci (Freeman et al., 2000), it was therefore feasible that condensin’s specific role
was to aid resolution in topologically complex areas of high DNA transcription.
However, a study then demonstrated that cell cycle down-regulation of rDNA
transcription inversely correlated with condensin’s binding efficiency to the rDNA
repeats (Wang et al., 2006). Taken together, these studies painted a conflicting
picture of what was happening. On the one hand, it seemed that high levels of
transcription were conflicting with resolution, perhaps by preventing stable binding
of condensin to the DNA. On the other, while transcription continues unabated at
the rDNA loci, wild type cells can correctly segregate chromosomes during
anaphase. This specific situation was finally clarified in a study where it was shown
that the sister-rDNA segregation defect seen in condensin mutants could be
overcome by ectopic expression of a foreign topoisomerase II (D'Ambrosio et al.,
2008a). This result implied that it was indeed catenation preventing sister-rDNA
segregation but that the endogenous S. cerevisiae topoisomerase II was ineffective
in decatenating the locus without condensin.
Apart from S. cerevisiae, depletion of condensin in a range other cells
resulted in phenotypes that closely resembled those seen in topoisomerase II
inactivations (Strunnikov et al., 1995b, Bhat et al., 1996, Hagstrom et al., 2002,
Hudson et al., 2003). These include poorly condensed and mis-segregated mitotic
chromosomes, but the most striking feature are anaphase bridges (Figure 1.8).
These result from the mitotic spindle pulling incompletely-resolved sister
chromatids apart to opposing poles of the cell and the unresolved DNA is seen
stretched as a bridge between the two spindle poles (Lavoie et al., 2002, Chan et
al., 2007). The exact cause of these bridges has not been determined and their
presence in condensin mutants has been attributed to many different causes, from
the abnormal compaction of DNA in early mitosis (Hirano, 2005a) to the premature
loss of compaction in early anaphase (Gerlich et al., 2006a, Vagnarelli et al., 2006).
Conversely, the similarity between condensin and topoisomerase II inactivation
phenotypes has led to the proposal that condensin may promote DNA decatenation
through interaction with topoisomerase II (Bhat et al., 1996, Coelho et al., 2003,
Sullivan et al., 2004). Despite the diverse range of proposals for condensin’s role in
chromosome segregation, the most likely mechanisms can be summarised in three
different scenarios (Cuylen et al., 2011), discussed in the following three chapters.

Chapter 1 Introduction
47
Figure 1.8 Anaphase bridges in condensin mutants
Immunofluorescence microscopy images of anaphase bridges in DAPI-stained DNA presenting in (top) S. cerevisiae condensin mutants (Lavoie et al., 2002), and in (bottom) H. sapiens condensin mutants (Chan et al., 2007).

Chapter 1 Introduction
48
1.6.2 Condensin is required for complete removal of proteinaceous
l inks
As introduced before, the main proteinaceous linker connecting the sister
chromatids prior to anaphase is cohesin (see chapter 1.4.1). Therefore it is
possible that depletion of condensin results in the incomplete removal of cohesin
and this has been demonstrated in metazoan cells. In nocodazole-arrested HeLa
cells depleted of condensin I (but not condensin II) still retain small amounts of
cohesin on the chromosome arms (Hirota et al., 2004), impairing the resolution of
chromosome arms normally observed under those arrest conditions, suggesting
that condensin is required for complete removal. In S. cerevisiae condensin
mutants, there is also an apparent failure to remove all cohesin from chromosome
arms during mitosis (Renshaw et al., 2010) and even in meiosis, condensin
mutants display telomeric segregation defects that can be reduced by over
expression of seperase (Yu and Koshland, 2005). The kleisin subunit of cohesin is
the cleavage target of separase and it has been shown that kleisin phosphorylation
by PLK1 renders it a better substrate (Alexandru et al., 2001). Correspondingly, a
reduction was observed in condensin mutants in the localization of PLK1 and the
phosphorylation of cohesin’s kleisin subunit during the first meiotic division (Yu and
Koshland, 2005). However it is not clear if condensin could recruit PLK directly to
cohesin, as while both SMC complexes are loaded onto the DNA by a shared
loader complex (Scc2/Scc4) (Ocampo-Hafalla and Uhlmann, 2011), their final
localization on the chromosome arms appear separate (D'Ambrosio et al., 2008b).
Instead of a direct role, condensin could destabilise cohesin binding through the
manipulation of mitotic chromosome structure. In support of this, the anaphase
movement dynamics of fluorescently labelled S. cerevisiae chromosomes was
tracked. Upon anaphase onset, segregation of the sister chromatids begins at the
centromeres before moving along the chromosome arms with delays that increase
towards the telomere. By promoting additional degradation of the kleisin at
anaphase onset, and thus increasing cohesin removal, removes the observed
delays. This implies that an uncleaved population of cohesin underlies the
sequential stretching of DNA along the chromosomes arm. When this residual
cohesin is cleaved by seperase, the chromosome arms spring back. However, in
condensin mutants this elastic action is not seen. This leads to the proposal, which

Chapter 1 Introduction
49
has been supported by mathematical modelling, that condensin-dependent
manipulation of DNA topology promotes removal of these residual uncleaved
cohesins (Renshaw et al., 2010). Countering this, are studies done in human cells
where depletion of Wapl results in much higher levels of cohesin being bound to
the chromosomes. Despite the much higher levels of cohesin present, no
segregation problems are observed (Shintomi and Hirano, 2009). As an interesting
aside, there have also been reports that topoisomerase II functions in the cohesin
cycle, hinting at the possible of a broad relationship between the enzyme and the
SMC family (Tapia-Alveal et al., 2010).
1.6.3 Condensin direct ly partners with topoisomerase II to promote
decatenation
DNA replication results in the topological linkage of the sister chromatids
and this catenation is removed by topoisomerase II prior to anaphase onset.
However, topoisomerase II is a bi-directional enzyme and as such the probability of
these enzymes disentangling entire chromosomes on their own is very unlikely. As
such, condensin has been proposed to promote of complete decatenation via a
direct interaction with topoisomerase II. In prokaryotes, this proposal has recently
received significant credit on the back of two studies demonstrating that the E. coli
SMC protein MukB directly binds to and stimulates the activity of topoisomerase IV
(Li et al., 2010, Hayama and Marians, 2010). In vitro assays demonstrated that the
incubation of DNA with MukB prior to the addition of topoisomerase IV promoted
the relaxation, and to a lesser extent decatenation, abilities of the enzyme. The site
of interaction between the proteins was mapped via mutations to the MukB hinge
domain and the C-terminal domain of the topoisomerase IV ParC subunit. This
stimulatory effect could be a consequence of MukB binding simply boosting
enzyme activity. Alternatively, MukB could preferentially bind sites of DNA
catenation and then recruit topoisomerase IV to them.
Given the interaction observed in prokaryotes, it would not be unreasonable
to predict a similar relationship in eukaryotes. Unfortunately, establishing where
condensin directly interacts with topoisomerase II has been difficult and produced
conflicting reports. The idea was first floated by the findings that mutations in the S.
pombe genes encoding the Smc4 subunit and topoisomerase II are synthetic lethal

Chapter 1 Introduction
50
(Saka et al., 1994b). This was then followed by reports that the D. melanogaster
kleisin subunit Barren co-localised with topoisomerase II and activated the
enzyme’s activity in in vitro assays (Bhat et al., 1996). However, while later studies
confirmed that D. melanogaster cell extracts depleted of Smc4 lose their
decatenation abilities, no direct interaction between condensin and topoisomerase
II was observed (Coelho et al., 2003) and no direct interactions were observed in S.
cerevisiae either (Bhalla et al., 2002). To further muddy the waters, extracts from
mitotic X. laevis cells depleted of condensin showed no reduction in decatenation
ability (Cuvier and Hirano, 2003). As such, there remains no overall consensus in
eukaryotes on whether condensin direct binds topoisomerase II or not, with
variation observed both between and within model organisms.
However, condensin could still directly promote decatenation through the
recruitment of topoisomerase II to catenation sites on sister chromatids. This idea
could explain the reduction in topoisomerase II staining on mitotic chromosomes
spreads seen in S. cerevisiae condensin mutants (Bhalla et al., 2002). In vitro
experiments show that condensins can stimulate plasmid knotting and
preferentially bind structured DNA substrates, which hint at a possible affinity for
DNA crossover sites (Kimura and Hirano, 1997b, Sakai et al., 2003). The
enrichment of condensin in the topologically complex DNA landscape of rDNA loci
in S. cerevisiae also lends credence to this line of thought (Freeman et al., 2000,
Wang et al., 2005, D'Ambrosio et al., 2008b). However, the prospect of condensin
acting as a recruitment agent for topoisomerase II remains unconvincing. Firstly,
under some conditions the requirement for topoisomerase II function is no longer
needed during rDNA segregation in anaphase, while condensin remains essential
(D'Amours et al., 2004). While CHIP on CHIP mapping reveal a loose co-
localization along the chromosomes (D'Ambrosio et al., 2008b), this is conflicted by
immunofluorescence studies of mitotic chromosomes (Maeshima and Laemmli,
2003). Additionally, overexpression of topo II rescues co-localization in condensin
mutants, but not function (Bhalla et al., 2002). Finally, the depletion of condensin
subunits in metazoan cells does not significantly affect chromosomal
topoisomerase II levels (Hudson et al., 2003, Hirota et al., 2004).

Chapter 1 Introduction
51
1.6.4 Condensin-driven reconfiguration of mitot ic chromosome
topology promotes decatenation
If condensin does not directly interact with topoisomerase II, it could still
direct its behaviour. While condensin does not possess topoisomerase’s ability to
cut and translocate DNA strands, it does have its own biochemical activities that
alter DNA topology as introduced in chapter 1.3.1. Condensin’s reconfiguration of
chromosome topology during chromosome condensation could promote
decatenation and proper resolution on both local and global levels.
On a local level, it has been shown that positively supercoiled mini-
chromosome dimers are more efficiently decatenated by topoisomerase II than
negatively supercoiled dimers (Baxter et al., 2011). Given that the positive
supercoiling of these mini-chromosomes is dependent on the presence of
condensin (as well as the mitotic spindle), it suggests that condensin’s supercoiling
activity could generate DNA substrates, which are more amenable to decatenation,
thereby pushing topoisomerase II’s reaction equilibrium towards complete
resolution. On a global level, the compacting activity of condensin within (but not
between) mitotic chromosomes means that catenated sister chromatid DNAs are
being pulled away from each other in a tug-of-war manner. Thus, it is not only
energetically favourable for topoisomerase II to separate the DNAs, but upon
decatenation, the sister chromatid DNAs are immediately physically isolated
making any recatenation impossible.
Of course, condensin-driven condensation could also promote the types of
resolution already discussed. For example, the stiffening of the chromatid fibre
caused chromosome condensation could allow the transmission of mechanical
forces generated at the centromeres by the pulling of the spindle out to the
chromosome arms, thereby tearing apart any residual cohesin linkages that had
not been cleaved by separase (Renshaw et al., 2010).
Condensin is required in three major mitotic events: chromosome
condensation, chromosome resolution of proteinaceous linkages and chromosome
resolution of topological linkages. It has become increasingly apparent that all three
processes are intimately linked, with condensin being the common denominator.
However, the relative contribution of condensin, particularly in regards to

Chapter 1 Introduction
52
chromosome resolution, as well the mechanism of its action remain unclear. This
thesis presents work that aims to address these shortcomings in our knowledge of
condensin.
1.7 An introduction to protein inactivations in S. cerevisiae
In the lab, we possess a range of temperature sensitive (ts) mutants suitable
for use in functional studies and to date, the majority of what is known about yeast
condensin has been learnt from the use of these ts alleles. In a ts mutant, a specific
protein is altered such that at the permissive temperature, usually 25°C, the protein
behaves as if wild type. However, upon temperature shift to the non-permissive
temperature, usually 37°C, then the mutant protein will cease to function and be
inactivated. At the start of our project, we used ts mutants extensively to inactivate
proteins of interest and examine the consequence on catenation. Given the sizable
collection of ts mutants already in possession by the laboratory, we did not need to
create any further ts mutants in order to examine all the proteins we were
interested in.
However, the use of ts mutants does present some problems. Firstly, some
ts mutants have a leaky response to temperature shift, resulting in only a partial
inactivation of the target protein. Secondly, not all the ts mutants are fully
characterised, such that while we know the protein is inactivated at the non-
permissive temperature, the exact mechanism of its inactivation is unclear. For
example, if we shift a condensin ts allele, such as smc2-8, to a non-permissive
temperature we are unsure of the exact effect on the protein. Does the shift cause
the whole condensin pentameric complex to disassemble or does the complex
remain intact but its functional activities are stopped? Does the protein remain
bound to DNA and if so, could it still have some persistent effect on DNA topology,
perhaps through interaction with another chromosomal protein? This lack of
knowledge on the exact nature of the inactivation means that when we started
contemplating the mechanisms behind our results, we were unable to have much
confidence in the models we proposed.
Separate to this uncertainty about the inactivation, the temperature shift
required to inactivate the target protein, usually a change from 25°C to 37°C, will of

Chapter 1 Introduction
53
course itself have an effect on the cells (Morano et al., 2012). At the higher
temperature, the cell’s stress response will be activated and when considered in
the light of our catenation assay, we needed to be sure that the sometimes-subtle
changes observed in our data were a result of a specific target protein inactivation
and not the cell’s innate stress response. While we performed the necessary
control experiments (see chapter 3.2.2 of the results) at the start of our project,
these limitations were deemed acceptable because the only other alternative for
inactivating target proteins was the Degron method (Dohmen et al., 1994), but this
too uses a temperature shift to initiate degradation.
However, shortly after starting this PhD project, a paper was published
detailing a novel technique for the specific depletion of a protein of interest from the
nucleus of S. cerevisiae called anchor away (Haruki et al., 2008b). The concept
was simple: to sequester, in a ligand-dependent manner, a gene product of interest
from its functional compartment to a different one, where it is anchored to a suitable
protein receptor (the anchor) and cannot exert its function. Thus, a target protein
could be specifically depleted from the nucleus by being transported out of the
nucleus and tethered to a receptor protein located in the cytoplasm. This was
achieved through the use of the human 12kDa, FK506 binding protein (FKBP12)
and the 11kDa, FKBP12-rapamycin-binding (FRB) domain of human mTOR, which
were fused to the anchor and target proteins, respectively. Rapamycin binds to the
FKBP12 domain where it forms an interaction surface for the FRB domain to
establish a tight tertiary complex of nanomolar dissociation constant (Chen et al.,
1995). By taking advantage of the massive flow of ribosomal proteins through the
nucleus during maturation, when rapamycin is added and there is formation of a
tertiary complex composed of the anchor, rapamycin, and the target, then this
results in the rapid depletion of the target from the nucleus (Figure 1.9).
The new anchor away technique was exciting as it provided a new means
by which to inactivate a particular protein, in this case through the specific depletion
of the target protein from the nucleus. Given the problems arising from use of ts
alleles, using anchor away offered many advantages, the most important being that
any temperature shifts could be avoided and all experiments performed at 25°C.
Regarding the efficiency of the depletion, by probing Western blot samples of
chromatin pellets, we observed an approximately 80% depletion of the target
protein thirty minutes after rapamycin addition.
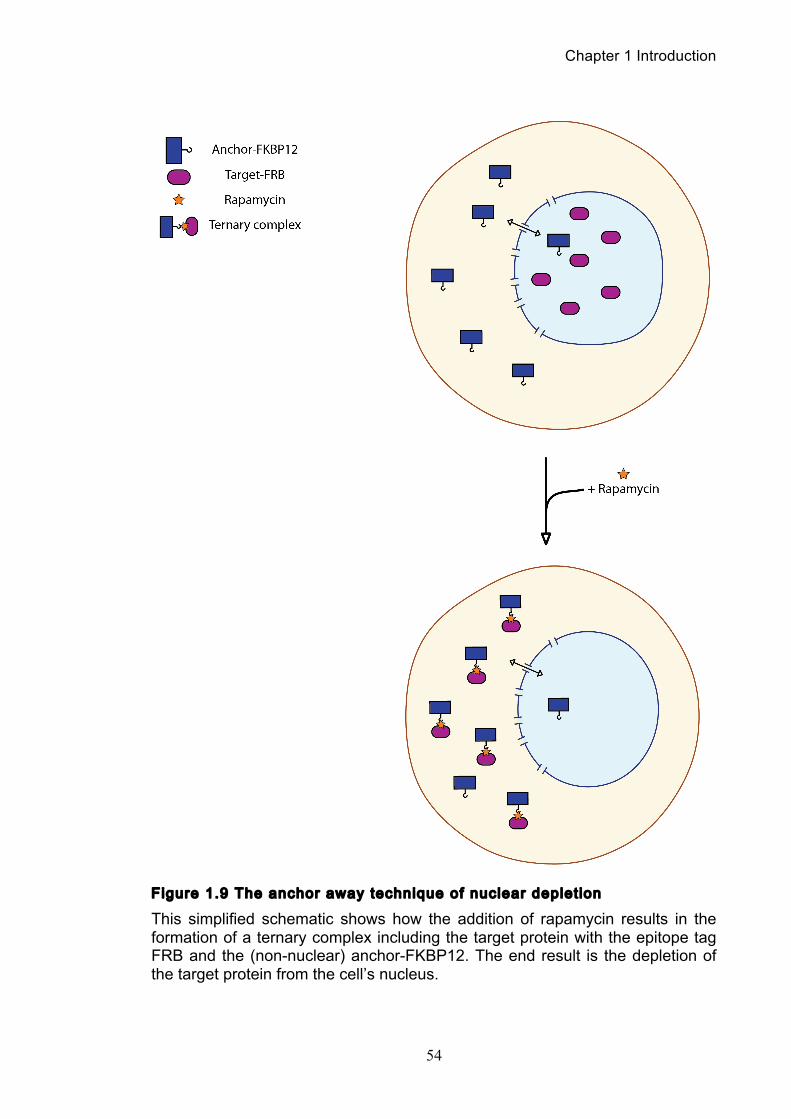
Chapter 1 Introduction
54
Figure 1.9 The anchor away technique of nuclear deplet ion
This simplified schematic shows how the addition of rapamycin results in the formation of a ternary complex including the target protein with the epitope tag FRB and the (non-nuclear) anchor-FKBP12. The end result is the depletion of the target protein from the cell’s nucleus.

Chapter 1 Introduction
55
Therefore, when presenting our results we have often done the inactivation
of the protein using both ts alleles and the anchor away technique and this is
because of several reasons. Firstly, by the time we had obtained the anchor away
strain in the lab we had already done many of experiments using the ts alleles.
Later on in the PhD, it would often be faster to do experiments using the ts strains
as it could take time to correctly tag or cross strains for use with anchor away.
Furthermore, and perhaps most importantly, by repeating key experiments using
both approaches we were able to really ensure that the resulting phenotypes
observed were indeed a consequence of inactivating that specific protein and not a
side effect of temperature, strain sickness or incomplete inactivation of the target
protein. As such, it acted as a control and confirmation of our observations and
thus both approaches were used throughout.
Use of the anchor away strain also resulted in a small side project. In
research labs using S. cerevisiae as their model organism, the ability to
synchronise cell populations using the mating pheromone α-factor has proven
invaluable, especially for cell cycle studies. Given its common use, the α-factor
response pathway has also become an important model to study the molecular
mechanism of G-protein coupled receptor signalling (Vallier et al., 2002). However,
only cells of the a mating type will respond to this pheromone. As it happened, the
anchor away cell lines were of the α mating type, and as such do not respond to α-
factor but rather to a-factor, a farnesylated and C-terminally methylated 12 amino
acid peptide. Because of its more difficult chemical synthesis, a-factor is not readily
available and consequently the a-factor response in S. cerevisiae is poorly
characterised.
Given our extensive use of anchor away strains throughout this project, and
resultant high consumption of a-factor, we collaborated with the peptide synthesis
laboratory in our institute. While they developed a new and improved strategy for a-
factor production based on solid-phase peptide synthesis, we characterised the
successful use of the resultant a-factor in synchronization of cell populations
(O'Reilly et al., 2012). Results from this characterization of a-factor and
comparisons with α-factor can be seen in chapter 6 of the results.

Chapter 2 Materials and Methods
56
Chapter 2. Materials & Methods
2.1 Yeast growth and manipulation
2.1.1 Yeast strains
The strains used in this study were of the W303 background, or were
backcrossed against this background, with the exception of the brn1-9 (Y3939) and
smc4-1 (Y3954) strains that were of S288c background. Strain details are listed
below:
Strain
Number
Genotype
K699 MATa , ade2-1, trp1-1, can1-100, KAN, his3-11, ura3-52, GAL,
psi+, (w303 wild type)
Y216 MATα, SCC1- HA6::HIS3
Y390 MATa ATa, BRN1-HA6::HIS3
Y2665 MATa , w303 wild type containing pS14-8(LEU2)
Y3277 MATa , TOP2-PK3::LEU2
Y3278 MATa , BRN1-HA6::HIS3, TOP2-PK3::LEU2
Y3939 MATa , brn1-9, pS14-8(LEU2) (S288c background)
Y3940 MATa , NET1-GFP::TRP1, ycg1-10, pS14-8(LEU2)
Y3954 MATa , smc4-1, pS14-8(LEU2) (S288c background)
Y3976 Topo II purification strain. See Worland & Wang 1989
Y3993 Condensin purification strain. See St-Pierre et al. 2009
Y4007 MATa , top2-4, pS14-8(LEU2)
Y4029 MATa , scc1-73, pS14-8(LEU2)
Y4031 MATa , SCC1-HA6::HIS3, TOP2-PK3::LEU2
Y4113 MATα, tor1-1, fpr1::NAT, RPL13A-2xFKB12::TRP1, smc2-
FRB::HIS3, MET3pr-HA-CDC20::URA3, pS14-8(LEU2)
Y4129 MATa , MET3pr-HA-CDC20::URA3, pS14-8(LEU2)
Y4201 MATα, tor1-1, fpr1::NAT, RPL13A-2xFKB12::TRP1, scc1-
FRB::HIS3, pS14-8(LEU2)
Y4059 MATα, tor1-1, fpr1::NAT, RPL13A-2xFKB12::TRP1, brn1-
Chapter 2 Materials and Methods
57
FRB::HIS3, pS14-8(LEU2)
Y4199 MATa , pRS316(URA3), (S288c background)
Y4235 MATα, tor1-1, fpr1::NAT, RPL13A-2xFKB12::TRP1, scc1-
FRB::HIS3, smc2-FRB::kanR, pS14-8(LEU2)
Y4236 MATa , smc2-8, pRS316(URA3)
Y4333 MATα, tor1-1, fpr1::NAT, RPL13A-2xFKB12::TRP1, brn1-
FRB::HIS3, pRS316(URA3)
Y4259 MATa , leu2Δ::kanR, RCIII-SUP11-LEU2-3ARS
Y4264 MATa , leu2Δ::kanR, brn1-9::TRP1, RCIII-SUP11-LEU2-3ARS
Y4327 MATα, tor1-1, fpr1::NAT, RPL13A-2xFKB12::TRP1,
leu2Δ::kanR, brn1-FRB::HIS3, RCIII-SUP11-LEU2-3ARS
Y4332 MATα, tor1-1, fpr1::NAT, RPL13A-2xFKB12::TRP1, pS14-
8(LEU2)
Y4333 MATα, tor1-1, fpr1::NAT, RPL13A-2xFKB12::TRP1, brn1-
FRB::HIS3, pRS316(URA3)
Y4334 MATa , tor1-1, fpr1::NAT, RPL13A-2xFKB12::TRP1, top2-
FRB::HIS3, pS14-8(LEU2)
2.1.2 Yeast strain creation, mating and tetrad dissection
Strains were constructed by transformation with the appropriate DNA
integration fragment designed for gene knockout or tagging. Affinity epitope tags
were fused at the gene endogenous loci for Western blot detection, or a 3xGFP
and mRFP cassette for detection by fluorescent microscopy, using polymerase
chain reaction products (Bähler et al., 1998, Knop et al., 1999). In order to cross to
strains, mating was induced through incubation of opposite mating type yeast
strains on YPD plates at 25°C for at least 6 hours. Diploids were then selected by
re-plating the crossed strains onto appropriate selective media and grown again on
YPD overnight. Diploid cells were sporulated on sporulation media (100mM
CH3COONa, 20 nM NaCl, 25 mM KCl, 1.5 mM MgSO4 and 1.5% w/v agar) until
tetrads appear. To break their asci, spores were treated with Zymolase T-20 (MP
Biomedicals) for 10 minutes at 30C. From each ascus, four spores are released
which were then dissected using a Singer-MSM micromanipulator. Finally, spores

Chapter 2 Materials and Methods
58
are incubated at 25°C until colonies formed and from these colonies can then be
re-streaked onto the appropriate selective media to determine the genotypes of the
progeny generated.
2.1.3 Yeast media, cultures and synchronizations
Cells were grown in YP supplemented with 2% w/v glucose (YPD) or 2%
w/v raffinose/galactose (YP-Raff/Gal). Yeast cells of a mating type were arrested in
G1 with the mating pheromone α-factor. To arrest cells in G1, early log phase
cultures (OD600 = 0.1) was treated with 0.5 µg/ml α-factor (provided by peptide
services, Cancer Research UK). Cells of α mating type were synchronised using
0.04 µg/ml a-factor (provided by peptide services, Cancer Research UK), as
described (O'Reilly et al., 2012). One hour after the initial addition of α-factor or a-
factor, the same amount is added to the culture. Within two hours complete
synchronization of the culture in G1 is achieved. Cell cycle arrest was determined
both cytologically by the appearance of a characteristic ‘shmoo’ and by FACS
analysis of DNA content. G1 arrested cells were collected on a membrane filter
(Schleicher & Schuell, ME28, 1.2mm) using a filtration apparatus (Millipore). Cells
were washed with at least five times the initial culture volume of YPD before being
release into fresh YPD media.
For arresting cells at metaphase, 5 µg/ml nocodazole was added to the
culture from a 2 mg/ml stock solution in DMSO. To metaphase arrest strains using
the repression of MET-Cdc20, cells were grown in YNB supplemented with 2%
glucose. To induce arrest, 2 mM methionine was added to the culture.
2.1.4 Anchor away strains and nuclear deplet ion
Anchor-away strains were created by FRB-tagging of the target gene in the
anchor away strain background as described (Haruki et al., 2008b). To deplete
nuclei of cohesin or condensin, rapamycin was added to the respective anchor
away strains at the time of their release from synchronization in G1.

Chapter 2 Materials and Methods
59
2.1.5 Yeast transformations
Transformation of cells with plasmids, minichromosomes or PCR products
was performed using standard lithium acetate transformation. In brief, 50 ml of mid-
log phase culture was pelleted at 3,000 rpm for 5 minutes. The cell pellet is washed
with 1 ml distilled water, then washed with 1 ml 1X TEL (10 mM Tris/HC pH 7.5, 0.1
mM EDTA, 100mM Lithium Acetate) before being re-suspended in a final volume of
100 µl 1X TEL. 1 mg of either linearised DNA vector or PCR product was mixed
with 2 ml of a 10 mg/ml single stranded carrier DNA from salmon sperm and 600 µl
TELP (TEL plus 40% PEG 3350 or 4000). 50 µl of the cell suspension in TEL was
then added to this mix and vortexed for 10 seconds. The mixture is incubated at
25°C for 4 hours and afterwards heat shocked at 42°C for 15 minutes. The cells are
pelleted at 6,000 rpm for 2 minutes, washed with and re-suspended in 1M sorbitol
before being plated onto YNB agar plates lacking the auxotrophic amino acid used
for selection. Transformants were checked for correct integration via either western
blot analysis, Southern blot analysis or in the case of anchor away strains, by death
on rapamycin containing media.
2.2 General molecular biology techniques
All standard molecular biology techniques such as PCR, restriction enzyme
endonuclease digestion or bacterial plasmid purification, were carried out as
described in (Sambrook and Gething, 1989) or as per the supplied manufacturers
protocol.
2.3 Protein analysis techniques
2.3.1 Protein extract preparation
Cell extracts were prepared using the TCA method. 5-10 ml of mid-log
phase cell culture is collected and cells pelleted by centrifugation, 3,000 rpm, 5
minutes at 4°C. Cells are resuspended in 1 ml of 20% trichloroacetic acid (TCA)
and kept on ice until the end of the time course. Cells are spun down for 1 minute
at 13,000 rpm, then washed with 1 ml of 1M Tris-Base before being resuspended in

Chapter 2 Materials and Methods
60
100 µl 2X SDS-PAGE loading buffer containing DTT. Samples are boiled for 2
minutes at 95°C, then 100 µl of 0.5mm glass beads (BioSpec Products, Inc) were
added and the cells broken using a FastPrep FP120 cell breaker (Bio101). To
separate the cell lysate from the glass beads, a few small holes are made in the
bottom of the cell breaker tube using a 27G needle and the tubes are placed inside
15 ml Falcon tubes before being spun at 3,000 rpm, 5 minutes at 4°C. The
collected lysate is then boiled at 95°C for 5 minutes and cleared by centrifugation at
13,000 rpm for 5 minutes before being loaded onto an acrylamide gel.
2.3.2 Chromatin pel lets
Collect 50 ml of mid-log phase culture and centrifuge at 3,000 rpm for 5
minutes at 4°C to pellet cells. If performing timecourse, resuspend cells in 50 mM
HEPES/KOH pH 7.5, 100 mM KCl, 1.5 mM MgCl2, 1 M sorbitol and keep on ice.
When time course is over and samples can be processed, resuspend cells in 3 ml
of 100 mM PIPES/KOH pH9.4, 10 mM DTT, 0.1% Na-Azide) and incubate for 10
minutes at room temperature. Then spin the cells for 2 minutes at 2,000 rpm and
aspirate supernatant. Cells are resuspended in 2 ml of 50 mM KPi pH 7.4, 0.6 M
sorbitol, 10 mM DTT and to each sample 4 µl of 20 mg/ml Zymolyase 100T (MP
Biomedicals) is added for spheroplasting of the cells. After spheroplasting, all work
should be done at 4°C. Cells are spun down for a minute at 4,000 rpm and then
washed with 1 ml of 50 mM HEPES/KOH pH 7.5, 100mM KCl, 2.5 mM MgCl2, 0.4
M sorbitol and then spun again for a minute at 4,000 rpm. Cells are resuspended in
an equal volume of buffer EB (50 mM HEPES/KOH pH 7.5, 100 mM KCl, 2.5 mM
MgCl2, 1 mM DTT, 20 µg/ml leupeptin, 2 mM benzamidine, 2 µg/ml aprotinin, 0.2
mg/ml bacitracin, 2 µg/ml pepstatin A and 1 mM PMSF). To this 10% Triton X-100
is added to a final concentration of 0.25% and then incubated for 3 minutes on ice
with occasional vortexing, which produces the whole cell extract. Prepare 100 µl
EBX-S (EB + 0.25% Triton X-100 + 30% sucrose) in separate Eppendorf tubes and
lay 100 µl of the whole cell extract onto the EBX-S. Spin for 10 minutes at 12,000
rpm, which will produce a white chromatin pellet, a clear sucrose layer and above
this, a yellow supernatant fraction.

Chapter 2 Materials and Methods
61
2.3.3 SDS-PAGE electrophoresis and western blott ing
Protein samples were resolved on acrylamide/bis-acrylamide (37.5:5:1,
amresco) 375 mM Tris-HCl pH 8.8 and 0.1% SDS. Small proteins of less than 30
kDa were typically resolved on 10-12% gels and larger proteins over 100 kDa on
8% gels. A stacking gel was used on top of the separating gel and consisted of 125
mM Tris-HCL pH 6.8, 5% acrylamide/bis-acrylamide and 0.1% SDS.
For electrophoresis a current of 50 mA was applied in an electrophoresis
tank (CBS Scientific) using SDS-PAGE running buffer (25 mM Tris, 250 mM glycine
and 0.1% SDS). To follow electrophoresis progression, and to allow the later size
comparisons of proteins on the gel, a broad range pre-stained protein marker (New
England Biolabs) was used.
After completion of electrophoresis, proteins were transferred onto pre-
equilibrated nitrocellulose membranes (GE Lifesciences) using a wet-transfer tank
(Biorad) and transfer buffer (3.03 g/l Tris base, 14.1 g/l glycine, 0.05% SDS and
20% w/v methanol). To check that the transfer was effective, the membrane is
stained with Ponceau S solution (Sigma). Then the membrane is blocked for an
hour with a 5% milk solution (Marvel) in PBST (170 mM NaCl, 3 mM KCl, 10 mM
Na2HPO4, 2 mM KH2PO4, 0.01% Tween 20) at room temperature. Following this,
membranes were incubated with primary antibodies diluted in milk solution for one
hour at room temperature, or overnight in the cold room. Primary antibodies used
and their final concentrations were a-Pk clone SV5-Pk1 (1:5000, Serotec), a-HA
clone 3F10 (1:5000, Roche), a-HA clone 16B12 (1:5000, Covance) and a-tubulin
clone YOL1/34 (1:1000, Serotec). After primary antibody incubation, membranes
were then washed in an excess of PBST for 30 minutes. Horseradish peroxidase
(HRP) coupled secondary antibodies (anti-mouse or anti-rabbit, 1:5000,
Amersham) were then incubated with the membrane in PBST containing 5% milk
for a further hour. Again membranes were then washed three times with an excess
of PBST for 30 minutes before developing with ECL (Amersham) according the
manufacturer’s instructions.

Chapter 2 Materials and Methods
62
2.4 Mini-chromosome purif ication, electrophoresis and
catenane quantif ication
1 gram of cells were collected at each time point, resuspended in ice cold 1
M sorbitol, 0.1 M EDTA pH 7.5, 0.02% sodium azide, and kept on ice until the end
of the timecourse. Cell pellets were then resuspended in 0.5 ml of the same buffer,
20 µl of 2.5 mg/ml Zymolyase 100T (MP Biomedicals) was added and the
suspension incubated for 1 hour at 37°C with mild agitation. Cells were collected by
centrifugation for 10 minutes at 13,000 rpm, supernatants removed by aspiration
and the pellets resuspended in 0.5 ml of 50 mM Tris/HCl pH 7.4, 20 mM EDTA.
SDS was added to a final concentration of 1% and samples were incubated at
65°C for 15 minutes. Then, 0.2 ml of 5 M potassium acetate was added and the
samples placed on ice for 1 hour. The precipitate was removed by centrifugation for
5 minutes at 14,800 rpm and the supernatants collected. The centrifugation step
was repeated until the supernatant fractions were clear of any debris. 2 volumes of
100% ethanol were now added, and after 5 minutes the DNA was collected by
centrifugation for 1 minute at 13,000 rpm. The supernatants were aspirated and the
pellets allowed to air dry. Once dry, pellets were carefully resuspended in 0.3 ml of
TE, pH 7.4 and 50 mg/ml RNase A added and incubated for 30 minutes at 37°C,
before addition of 100 mg/ml proteinase K for further 30 minutes. Now, 12.6 µl of 5
M sodium chloride was added, and the DNA precipitated by addition of 0.63 ml
100% ethanol. Samples were again centrifuged, supernatants aspirated and the
pellets air-dried. After resuspension in TE, the DNA concentration was measured
(NanoDrop, Thermo Scientific) and adjusted prior to gel electrophoresis.
Samples were resolved on 0.5% agarose/TAE gels at 1.0 V/cm for 48 hours
(pRS316 and pS14-8) at room temperature, or at 0.8 V/cm for 24 hours, followed
by 2.2 V/cm for a further 24 hours (RCIII). Southern transfer was carried out using
capillary blotting onto a positively charged nylon membrane (Hybond-N+, GE
Healthcare) as per (Southern, 1975). The membrane was probed with random-
prime 32P-labeled probes against the AmpR gene (pS14-8 and pRS316) or against
LEU2 (RCIII). The blots were exposed to PhosphorImager screens (GE
Healthcare) that were scanned using a Storm 860 Molecular Imager. Band
intensities were analyzed and quantified using linear background subtraction and
automatic band detection in ImageQuant.

Chapter 2 Materials and Methods
63
Enzymes used to verify the nature of the observed bands were XhoI and PmlI
(New England Biolabs), calf thymus topo I (Invitrogen), human recombinant topo I
and human topo IIa (both Topogen). When statistical analyses have been
performed, these have been done via paired t-tests using Prism software.
2.4.1 Catenation assay minichromosomes
The centromeric plasmid pRS316 (Sikorski and Hieter, 1989) and pS14-8
(containing a genomic region surrounding RAD5 in the centromeric plasmid YCp70
(Aguilera and Klein, 1990), a gift from A.-M. Farcas and K. Nasmyth) were
introduced into yeast using standard lithium acetate transformation. The ring
chromosome III (RCIII) contains 61 kb surrounding centromere III, including
ARS307, 308 and 309 as well as the wild type LEU2 gene (Dershowitz and Newlon,
1993). To identify RCIII by Southern blotting, the leu2-3.112 gene on the authentic
chromosome III in the host strain was replaced with a kanR marker (Wach et al.,
1994).
2.4.2 Zymolyation of yeast cel ls in agarose plugs and Pulse Field Gel
Electrophoresis (PFGE)
Collect approximately 108 cells and fix in 70% ethanol overnight at -20°C.
The following day, prepare the agarose plugs by mixing 1 ml of SEZ buffer (1M
sorbitol, 50 mM EDTA pH 8) with 0.02 g of LOM (low melting point) agarose and
heat to 65°C to melt the agarose. Cells are centrifuged for 1 minute at 13,000 rpm
and the 70% ethanol is aspirated off. Cells are then washed once in 1 ml of
resuspension buffer (1M Tris pH 7.5, 1.2M sorbitol, 0.5M EDTA) before cells are
resuspended in 50 µl SEMZ buffer (1M sorbitol, 50 mM EDTA pH 8, 28mM
mercaptoethanol, 3 mg/ml Zymolyase 100T (MP Biomedicals)). Cells in the SEMZ
buffer are then placed on a shaking heat block set at 37°C for 1 hour. After this
time, the cells are mixed with the melted LOM agarose (with 3 mg/ml Zymolyase
100T added) and pipetted into plug moulds for cooling and setting. Once plugs are
set, they are then incubated for 1 hour in a 37°C shaking heat block in EST buffer
(10 mM Tris pH 8, 100 mM EDTA pH 8, 1% sarcosyl). After an hour, the EST buffer

Chapter 2 Materials and Methods
64
is removed and replaced with 50 mM Tris pH 7.8 and 10mg/ml RNAse A and left at
37°C overnight. The following day the buffer is removed and replaced with ESP
buffer (1% SDS, 1 mg/ml proteinase K, 0.5M EDTA pH 8) and incubated at 50°C
for 2 hours. Finally the buffer is changed to 1X TE and plugs can be stored at 4°C.
Prior to loading, plugs should be washed 3 times for 20 minutes each with 0.5X
TBE or 1X TAE buffer depending on the electrophoresis protocol being used. For
the last 20 minutes prior to loading, the plugs should be kept on ice.
Plugs are then loaded into 0.5% agarose gels and resolved with via PFGE.
In our protocol, we used 1X TAE as the running buffer. PFGE was performed on a
BioRad CHEF DR-III system at 14°C using the following program: Step 1: 24 hours
at 2 V/cm, 96° angle, 1200 seconds switch time; Step 2: 24 hours at 2 V/cm, 100°
angle, 1500 seconds switch time; Step 3: 24 hours at 2 V/cm, 106° angle, 1800
switch time.
2.5 Cell biology and microscopy
2.5.1 Cell cycle analysis using f low cytometry
To determine cell cycle progression by DNA content, 1 ml of culture was
pelleted and fixed in 70% ethanol for at least 2 hours at 4°C. Then cells are
resuspended in 50 mM NaCitrate containing 0.1 mg/ml RNAse A and put on a
shaking heat block at 37°C for an hour. After the hour has passed, proteinase K is
added to a final concentration of 0.1 mg/ml and the mixture left on the shaking heat
block for a further hour. Cells are then pelleted and then resuspended in 50 mM
NaCitrate containing 50 µg/ml propidium iodide. Cells are sonicated (Sanyo,
Soniprep 150) to eliminate clumping and then are analysed on a FACScan (Becton
Dickinson), with final data and graph generation prepared with CellQuest software.
2.5.2 Bi-nucleate cel l counting
The fraction of binucleate cells, an indication for progression through mitosis,
was scored using propidium iodide-stained cells. Scoring was repeated three times
over and an average taken.

Chapter 2 Materials and Methods
65
2.5.3 In situ immunofluorescence
2 ml culture aliquots are collected with an OD600 of at least 0.2. The cells
are spun down and resuspended in 1 ml of ice cold formaldehyde buffer (100 mM
KPO4 pH 6.4, 0.5 mM MgCl2, 3.7% formaldehyde). Cells are fixed overnight in this
buffer at 4°C or can be process immediately by fixing for 2 hours at 30°C. Once
fixed, cells are washed once in 100 mM KPO4 pH 6.4, 0.5 mM MgCl2, then washed
once with 1 ml of spheroplasting buffer (100 mM KPO4 pH 6.4, 0.5 mM MgCl2,
1.2M sorbitol). After washing, cells are resuspended in 200 µl of spheroplasting
buffer plus 2 µl of β-mercaptoethanol and 2 µl of 20 mg/ml Zymolyase 100T (MP
Biomedicals) and incubated for 30 minutes at 30°C. Then cells are spun down for 2
minutes at 4,000 rpm and the supernatant is aspirated. Cells are washed with 0.5
ml of spheroplasting buffer before final resuspension in 0.2 ml of the same buffer. 5
µl of cells are then pipetted onto a polylysine coated 15 multi-well slide (MP
Biomedicals). Slides are washed for 3 minutes in methanol and fixed for 10
seconds in acetone before being blocked with blocking buffer (0.5% Bovine Serum
Albumin). Slides were then incubated with the relevant primary and secondary
antibodies in a humid chamber in the dark for an hour each. In between incubation
with each antibody, the slide wells were washed three times with blocking buffer
and four times before addition of a mounting anti-fade media with 0.1 µg/ml DAPI.
The antibody we used was α-tubulin clone YOL1/34 (Serotec).
2.6 Protein purif ications
2.6.1 Purif icat ion of S. cerevisiae topoisomerase II
Overexpression and purification of S. cerevisiae topoisomerase II followed a
published procedure (Worland and Wang, 1989). 6 hours after induction of topo II
expression by galactose addition, 0.5 grams of cells were collected, washed in
water, then resuspended in 200 µl lysis buffer (50 mM HEPES/KOH, pH 8.0, 150
mM KCl, 1 mM EDTA, 10% glycerol, 5 mM b-mercaptoethanol, 1 mM PMSF, and
an additional protease inhibitor cocktail (CompleteTM, Roche). Cells were broken
with acid washed glass beads to obtain greater than 90% cell lysis and the lysate
collected. A 1 ml phospho-cellulose column (Whatman P11, prepared according to

Chapter 2 Materials and Methods
66
the manufacturer’s instructions) was equilibrated in lysis buffer. The cell lysate was
loaded in batch, then the resin was packed into a column and washed by gravity
flow using lysis buffer containing 200 mM and subsequently 400 mM KCl. Topo II
was eluted in lysis buffer containing 800 mM KCl. This fraction was diluted back to
200 mM KCl and bound to 300 ml Q-Sepharose beads (GE Healthcare). These
were then packed into a column and extensively washed with the loading buffer. A
step gradient with 1M KCl was used for elution. Topo II-containing fractions were
adjusted to 300 mM KCl and 10% glycerol for storage.
2.6.2 Purif icat ion of S. cerevisiae condensin
The 5-subunit condensin complex was overexpressed in S. cerevisiae as
previously described (St-Pierre et al., 2009). Cell extract containing 40 mg protein
in 6 ml of lysis buffer (20 mM HEPES/KOH pH 7.5, 150 mM KCl, 1 mM MgCl2, 5
mM β-mercaptoethanol, 10% glycerol, and protease inhibitors) was cleared by
ultracentrifugation. The supernatant was loaded onto 200 ml of an α-HA affinity
matrix (Roche). After binding, the resin was washed extensively in same buffer.
Condensin was eluted by incubation with 500 µl 1.6 mg/ml HA peptide in lysis
buffer over night at 4°C.
2.6.3 Interaction analysis between condensin and topoisomerase II
To carry out the interaction analysis of condensin with topo II, 25 units/ml
benzonase (Sigma) was added to the cell extract containing overexpressed
condensin. After ultracentrifugation, 400 µl aliquots of the extract were precleared
with 20 µl of IgG-coupled Dynabeads (Invitrogen) for 30 minutes at 4°C. The beads
were removed and 4 mg α-HA antibody (clone 16B12) was added to the
supernatant for 30 minutes at 4°C. Then, 20 µl of protein A-coupled Dynabeads
(Invitrogen) were added for a further 30 minutes. Finally, condensin-bound or
control beads were incubated in the presence of 400 ng purified S. cerevisiae topo
II for 30 minutes at 4°C. After washing, bound protein was elution in SDS-PAGE
loading buffer.

Chapter 2 Materials and Methods
67
2.7 In vitro assay techniques
2.7.1 kDNA decatenation assays
To assess their decatenation activity, 10 ng or 100 ng of purified yeast topo
II, or 0.1 units of E. coli topo IV and human topo II (Topogen), were incubated with
100 ng of kinetoplast DNA (Topogen) in a 20 ml reaction containing 50 mM
Tris/HCl pH 7.8, 150 mM potassium acetate, 6 mM magnesium acetate, 5 mM b-
mercaptoethanol, 0.1 mg/ml BSA and 2 mM ATP. To assay the effect of condensin,
reactions were incubated with the indicated amounts of condensin for 30 minutes at
37°C in the absence of topoisomerase. Then topoisomerase was added and the
reaction incubated at 37°C for further 10 minutes. Reactions were stopped by
addition of an equal volume of stop buffer (2% SDS, 80 mM EDTA, 600 mM NaCl)
and the samples resolved on a 1% agarose/TAE gel. Gels were stained with
ethidium bromide and band intensities were quantified using ImageQuant software.

Chapter 3 Results
68
Chapter 3. Results: Catenane Behaviour and
Resolution
3.1 Development of the catenation assay
As discussed in the introduction, there are two aspects to chromosome
resolution, the cleavage of proteinaceous links and the resolution of topological
links between sister chromatids. It is only once both of these are complete that the
mitotic spindle can successfully pull apart the sister chromatids to the opposite
poles. Therefore in order to study the formation and resolution of sister chromatid
catenanes during the cell cycle, we employed circular yeast minichromosomes of
various sizes in a catenation assay that we tailored to our needs. The assay was
based on a similar protocol originally used to study whether the cohesin ring
physically entraps DNA, holding it together prior to seperase-cleavage and
anaphase onset (Ivanov and Nasmyth, 2007). The assay is a powerful tool for
tracking topological links through the cell cycle, because the minichromosomes,
which unlike linear DNA, maintain any topological links once purified. Moreover, it
has long been observed that larger genomes behave almost exactly like circular
DNA because of the existence of domains whose fixed ends act by preventing any
free rotation (Worcel and Burgi, 1972). However, in order to most closely model the
endogenous chromosomes, we chose to use minichromosomes for our assay that
retained many features of natural yeast chromosomes, including centromeres,
replication origins and actively expressed yeast chromosome arm sequences. Thus,
the minichromosomes would replicate and segregate with the same timing as the
native chromosomes, mimicking their behaviour.
The first substrate we chose to use in our assay was pS14-8, a 21.2 kb
centromeric mini-chromosome that contained a section of chromosome XII arm
sequence around the RAD5 gene (Figure 3.1). We chose to first use such a large
substrate based on prior reports that smaller plasmids are too quickly resolved of
their topological linkages by topoisomerase II to be observed in a normal
timecourse (Baxter et al., 2011), thus maximising our chances of observing
catenanes. For full details on the final protocol that we used, please see chapter
2.4 of the materials and methods. However, unless otherwise detailed, our

Chapter 3 Results
69
timecourses all adhered to the same general following framework: our yeast strain
of interest was transformed with our assay minichromosome and cultures grown
overnight. After dilution to the same optical density (OD) the following morning,
cultures were arrested in G1 through addition of the relevant mating pheromone.
Once arrested, cells were washed and released into pheromone-free media that
would either be shifted to 37°C or contain rapamycin (depending on the method of
protein inactivation). Cells would progress through the cell cycle before re-arrest
back in G1. From the moment cells are released into pheromone-free media,
aliquots for DNA purification are taken every 20 minutes as well as aliquots for
FACS analysis. After the timecourse is complete, the DNA is purified from the cells
and resolved on an ethidium bromide free gel at low voltage. To visualise the
minichromosome we performed a Southern blot and probed against a
minichromosome-specific gene. The end result is a blot showing several bands,
each representing a different topological isoform of the minichromosome in
question.

Chapter 3 Results
70
Figure 3.1 Map of minichromosome pS14-8

Chapter 3 Results
71
3.1.1 Optimisation of the catenation assay
Optimising the assay was a difficult process owing to the size of the plasmid.
Size is an issue because as the plasmid size increases, its electrophoretic mobility
decreases and this applies to all of its topological forms. This means that when
running the DNA on the agarose gel, it can be difficult to cleanly segregate different
isoforms into distinct bands, as they can all run with a similar speed. Initial blots
were difficult to interpret owing to how close bands ran together, making it difficult
to differentiate between them. In Figure 3.2, the first three lanes contain the purified
DNA from a wild type strain containing pS14-8 (Y2665). In comparison to DNA
purified from a wild type strain without pS14-8, we could clearly see some bands
specific to the minichromosome, yet with poor clarity. By careful troubleshooting of
the running conditions, such as the percentage of agarose used and the voltage
applied to the gel, a set of conditions was reached that allowed for improved
segregation of the minichromosome’s topological isoforms. We found that by using
a very low percentage agarose gel (0.5%) and running the samples very slowly
over a long period of time (1.0V/cm for 48hrs), blots were produced that were
generated greater spread of isoforms allowing for easier identification (Figure 3.3).
However looking at the blot, while the bands do segregate more clearly,
there persisted another problem. Despite the specificity of the probe for the AmpR
gene, it became apparent that the first probe we had produced was also weakly
labelling the genomic DNA. This can be seen as a large smeary band in the control
lane where DNA was prepared from wild type cells (K699) that contained no
plasmid or AmpR gene. Unspecific labelling of the genomic DNA was further
confirmed by staining gels after running with ethidium bromide to observe the
genomic band. Indeed, the distance travelled by this band and the distance
travelled by the smeary band on our Southern strongly suggested improper
labelling of the genomic DNA by our probe. Therefore in order to remove this
background labelling, we began testing a set of three new probes (labelled A, B
and C) that we designed against the AmpR gene that varied in length (Figure 3.4).
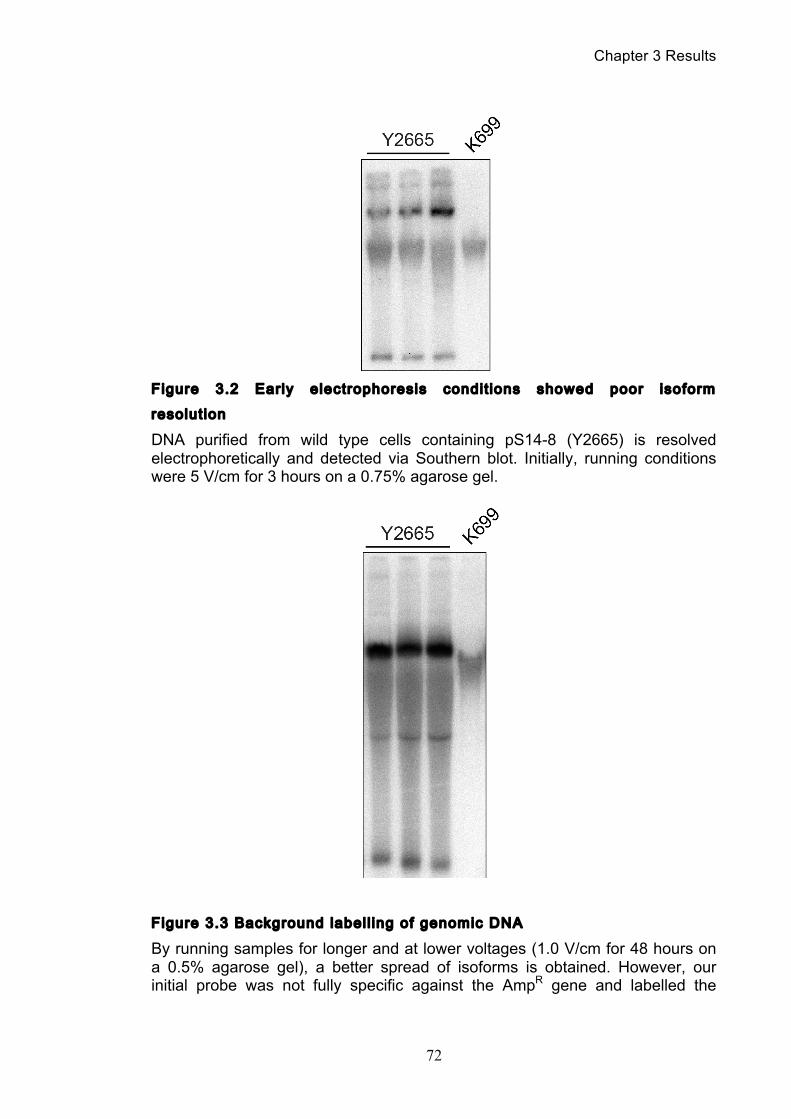
Chapter 3 Results
72
Figure 3.2 Early electrophoresis condit ions showed poor isoform
resolut ion
DNA purified from wild type cells containing pS14-8 (Y2665) is resolved electrophoretically and detected via Southern blot. Initially, running conditions were 5 V/cm for 3 hours on a 0.75% agarose gel.
Figure 3.3 Background labell ing of genomic DNA
By running samples for longer and at lower voltages (1.0 V/cm for 48 hours on a 0.5% agarose gel), a better spread of isoforms is obtained. However, our initial probe was not fully specific against the AmpR gene and labelled the
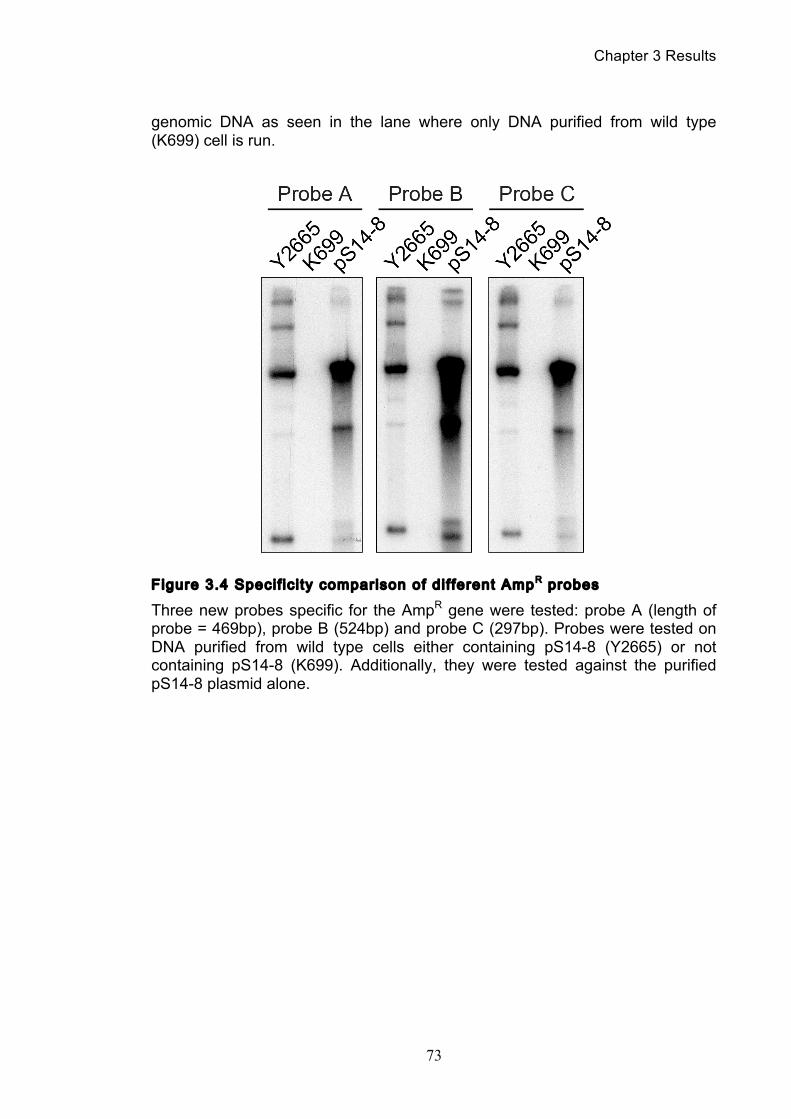
Chapter 3 Results
73
genomic DNA as seen in the lane where only DNA purified from wild type (K699) cell is run.
Figure 3.4 Specif ic i ty comparison of dif ferent AmpR probes
Three new probes specific for the AmpR gene were tested: probe A (length of probe = 469bp), probe B (524bp) and probe C (297bp). Probes were tested on DNA purified from wild type cells either containing pS14-8 (Y2665) or not containing pS14-8 (K699). Additionally, they were tested against the purified pS14-8 plasmid alone.

Chapter 3 Results
74
We used these three new probes to label either purified DNA samples from
Y2665, K699 or purified pS14-8 (i.e. no genomic DNA present) to compare the
intensity of their labelling as well as whether they had any non-specific labelling of
the genomic. Examination of the blots showed that the new set of probes all
seemed to be entirely specific for the AmpR gene with little to no affinity for the
genomic DNA, especially as when compared with our original probe as used in
Figure 3.2 and 3.3. However, they did not all label the AmpR gene with the same
intensity, and we determined that probe B labelled the most clearly and strongly. As
such, for all future experiments where we wished to target AmpR, we used probe B.
3.1.2 Identi fy ing the minichromosome’s dif ferent topological isoforms
Having optimised the conditions for resolving the DNA on the agarose gel so
that we could easily observe all topological isoforms, as well as having produced a
highly specific probe; we could start to use the assay to study the behaviour of
catenanes during mitosis. Having transformed pS14-8 into a wild type strain
(Y2665), we synchronised the culture in G1 using the relevant mating pheromone,
before washing the culture and releasing the cells from their arrest. Cells were
allowed to progress through one cell cycle before being rearrested in G1.
Throughout this timecourse, aliquots of cells were taken every twenty minutes and
were processed to purify the DNA. This DNA was then resolved on an agarose gel
at low voltage and a Southern blot performed, which was then probed against
AmpR using probe B (Figure 3.5).
Examining the blot, we can see a characteristic pattern of bands that
represent the different topological isoforms of the minichromosome pS14-8. While
two abundant bands are consistent throughout the cell cycle, it is clear that three
new bands become apparent at the time of S-phase that then disappear again in
mitosis. In order to identify which isoform each band represented, we performed a
series of enzyme digests upon our purified DNA (Figure 3.6). The first enzyme
used was Xho1, a restriction enzyme that only cuts pS14-8 once, hence linearizing
it. Indeed from the gel, one can see that upon incubation with Xho1, all isoforms
collapsed into a single linear (L) band. Linear DNA is present in all our purified DNA

Chapter 3 Results
75
preps, most likely as an unavoidable artifact of DNA shearing during the DNA
purification process. The next digest performed was with topoisomerase I (topo I),
an enzyme that is capable of relaxing supercoiling in DNA through the transient
‘nicking’ of a single strand of DNA, allowing the DNA helix to rotate and release
tension (see chapter 1.5.1). By treatment with this enzyme, bands representing
supercoiled isoforms of pS14-8 disappeared, with their intensity shifting to two
bands likely representing their relaxed forms, relaxed monomers (RM) and relaxed
catenanes (RC). The final enzyme treatment was with topoisomerase II (topo II),
which differs from topo I in that it can create transient double-strand breaks,
allowing the passage of one DNA strand through another, before re-ligating the
DNA (see chapter 1.5.2). This means that topo II cannot only relax supercoiling
(like topo I), but also can resolve topological links between different DNAs.
Consequently, we saw that treatment with topo II resulted in any bands
representing catenated pS14-8 disappear, leaving one intense band representing
the relaxed monomer form of pS14-8. However, a smear is seen underneath this
band and this likely represents partially supercoiled monomers (PSM). This is a
result of topo II’s bi-directionality, meaning that it can both introduce and remove
supercoiling and catenation. In the lane where the DNA is treated with topo II, the
smear represents monomers of pS14-8 where an equilibrium distribution of partially
supercoiled monomers has been established. The reason that all the monomers
are not partially supercoiled and we have a strong relaxed monomer band, is
because the relaxed monomer band contains plasmids that have likely been
‘nicked’, i.e. had a single strand of DNA broken, either as a result of shearing
during pipetting or as consequence of the enzyme treatment. If nicked, plasmids
are unable to sustain any supercoiling because any tension introduced is
immediately lost. Topo II can also introduce topological links, but the reason we
see no catenanes is because the equilibrium is shifted very much towards
decatenation. For topological links to be introduced between different plasmids,
they need to be physically very close together for the reaction to occur. At their low
concentration and with no proteins holding them together, it is therefore very
unlikely for links to be introduced. For those plasmids that are catenated, once
decatenated by topoisomerase II, the now individualised plasmids will separate
spatially from each other rapidly owing to Brownian motion.
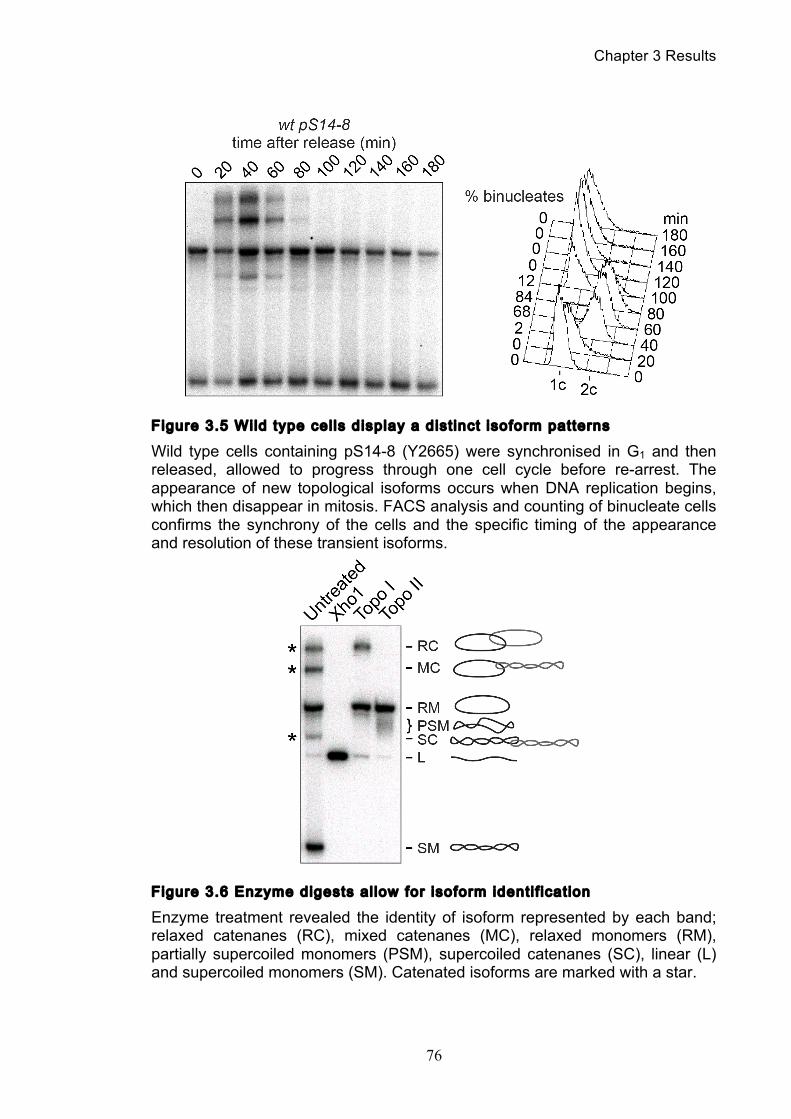
Chapter 3 Results
76
Figure 3.5 Wild type cel ls display a dist inct isoform patterns
Wild type cells containing pS14-8 (Y2665) were synchronised in G1 and then released, allowed to progress through one cell cycle before re-arrest. The appearance of new topological isoforms occurs when DNA replication begins, which then disappear in mitosis. FACS analysis and counting of binucleate cells confirms the synchrony of the cells and the specific timing of the appearance and resolution of these transient isoforms.
Figure 3.6 Enzyme digests al low for isoform identi f icat ion
Enzyme treatment revealed the identity of isoform represented by each band; relaxed catenanes (RC), mixed catenanes (MC), relaxed monomers (RM), partially supercoiled monomers (PSM), supercoiled catenanes (SC), linear (L) and supercoiled monomers (SM). Catenated isoforms are marked with a star.

Chapter 3 Results
77
Having performed these enzyme treatments, we could confidently identify
which topological isoform each band on the Southern blot represents. There are
three bands detectable that represent catenated species and what is immediately
striking when examining the wild type timecourse is how these catenated forms
appear and then disappear with a specific pattern over the course of the cell cycle.
Comparing the blot with the FACS profile, one can see that catenanes appear in S-
phase when DNA replication occurs, matching the prediction made by (Sundin and
Varshavsky, 1980) 30 years ago that catenanes are replication dependent.
Furthermore, one can see that these catenanes are then resolved later in the cell
cycle, approximately around the time of anaphase onset. We will investigate further
the exact timing of this resolution in later experiments.
3.1.3 Catenation is quanti f iable at each t ime point
To be able to observe catenanes using pS14-8, and to measure the specific
timing of their appearance and resolution, was very exciting. However before
investigating the behaviour of these catenanes any further, we required a way to
quantify the relative amount of catenanes present at each time point taken during
the time course. This would allow us to then quantify the effect certain mutants or
nuclear depletions had on catenation creation, resolution and behaviour. We
accomplished this through the use of ImageQuant imaging software, the final
protocol for which is described in the materials and methods (chapter 2.4).
There were many ways in which to calculate the intensities of different
peaks, including factors such as band detection and boundary determination. As far
as possible, all band detections and subsequent calculations was performed by the
program to minimise external influence on the data. However, correctly determining
how to apply the subtraction of background was an area we had to optimise. Figure
3.7 shows a comparison of two methods of background deduction: the rolling ball
and linear background subtraction methods. The rolling ball method is a computer
determined background subtraction that ‘rolls’ an imaginary ball of set diameter
along the underside of the lane profile to determine the background. However, one
can see that this technique is not suited to the types of profiles generated by our
data, with steep gradients seen at the start and end of the gel runs. This results in

Chapter 3 Results
78
abnormally large quantification values for the first and especially the last band. An
improvement on this is the linear background subtraction along the visible
background seen in the lane profile. This deducts the background evenly and fairly
from each band’s quantification. Indeed in Figure 3.7 we can see the differences
between the two methods, which despite both quantifying the same wild-type
timecourse, it is clear that the linear background subtraction method generates
quantification data that best represents what can be seen on the Southern blot.
Once we had optimised the quantification protocol, we were able to start
putting numbers to the observations noted in the catenation assays. Returning to
our wild type timecourse (Figure 3.5), we quantified the amount of catenanes
present to produce a graph showing the amount of catenanes present as a
percentage of the total minichromosome in each lane (Figure 3.8). Therefore
quantification reveals that during DNA replication, the percentage of catenanes
peaks at around 55% before being resolved in mitosis.
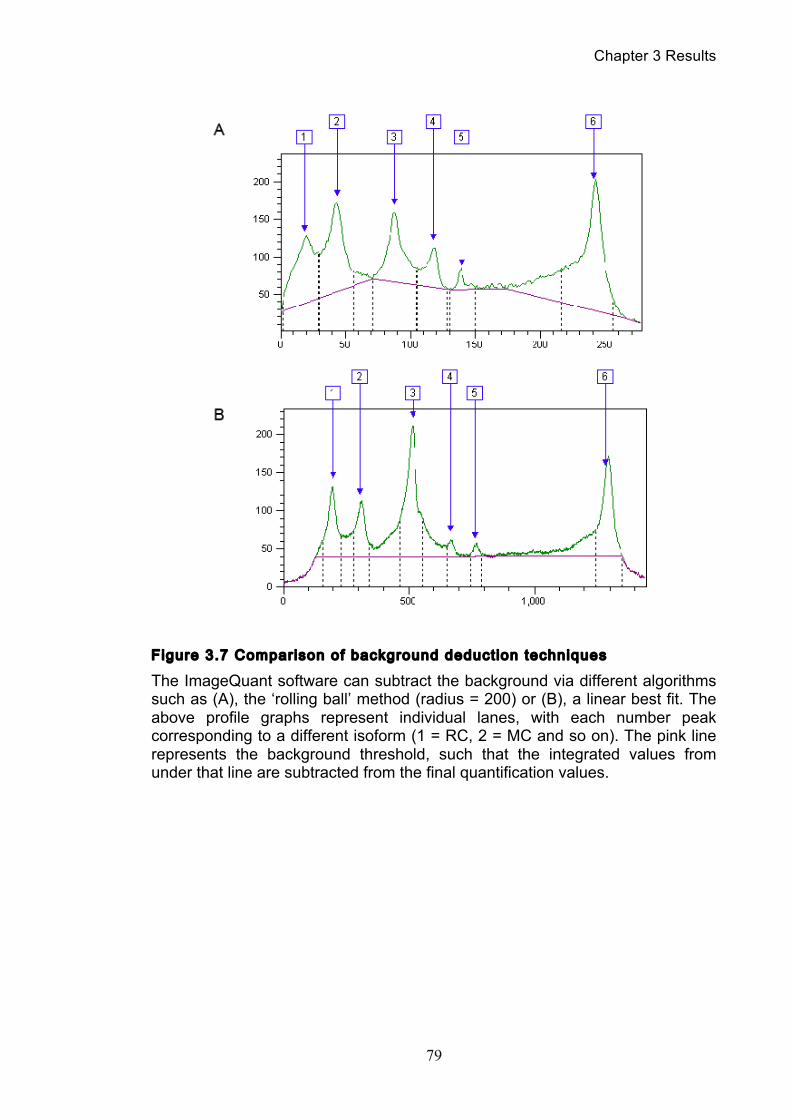
Chapter 3 Results
79
Figure 3.7 Comparison of background deduction techniques
The ImageQuant software can subtract the background via different algorithms such as (A), the ‘rolling ball’ method (radius = 200) or (B), a linear best fit. The above profile graphs represent individual lanes, with each number peak corresponding to a different isoform (1 = RC, 2 = MC and so on). The pink line represents the background threshold, such that the integrated values from under that line are subtracted from the final quantification values.
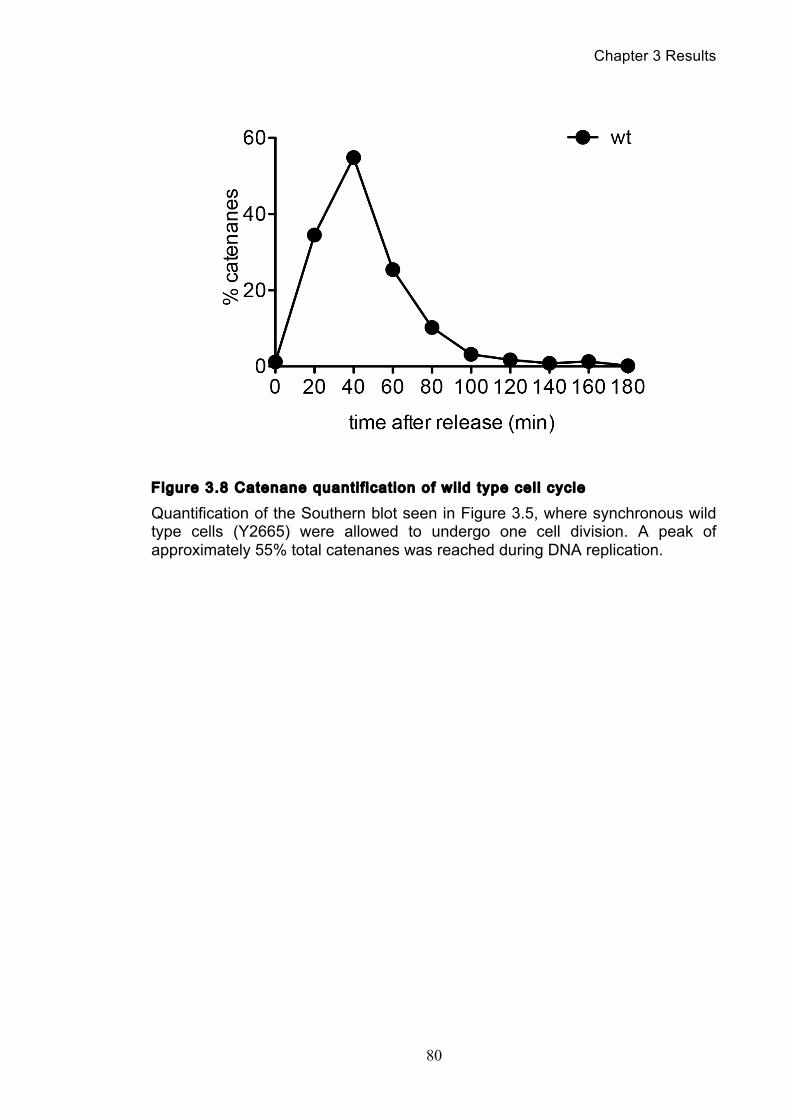
Chapter 3 Results
80
Figure 3.8 Catenane quanti f icat ion of wi ld type cel l cycle
Quantification of the Southern blot seen in Figure 3.5, where synchronous wild type cells (Y2665) were allowed to undergo one cell division. A peak of approximately 55% total catenanes was reached during DNA replication.

Chapter 3 Results
81
3.2 Spontaneous and assisted minichromosome
decatenation:
With the assay established, we were keen to start examining the effects on
catenation by depleting particular components of the mitotic assembly. For most of
our experiments involving the inactivation of a target protein, this has been
achieved through use of both temperature sensitive (ts) alleles and nuclear
depletion via the anchor away technique (see chapter 1.7). For the relevant
controls for both techniques, please see the next chapter 3.2.2.
The pS14-8 mini-chromosome harbours the efficient ARS1 early replication
origin and is expected to replicate and equally segregate during every cell cycle. If
catenation does indeed arise as a consequence of DNA replication, we would
expect to at least transiently observe all of the mini-chromosome to be present in
catenated forms. However, as seen in our wild type time course (Figure 3.5), the
peak was never higher than 60%. This could be because replication does not
always lead to catenation between replication products. Alternatively, it could be
that all replication products are catenated, but that some sister chromatids are
resolved quickly after replication, even before cells enter mitosis. To distinguish
between these two possibilities, we repeated the time course analysis but
inactivated the major endogenous decatenating activity of topo II using both the
temperature sensitive top2-4 mutation (Y4007) (Holm et al., 1985) and nuclear
depletion via anchor away (Y4334) (Figure 3.9). With both types of depletion, we
observed that upon entry into S-phase, virtually all of the minichromosomes
accumulated as catenated species following DNA replication. The fact that almost
no individual plasmids reappear supports the fact that without topoisomerase II,
decatenation does not occur. Additionally, it was reported that without
topoisomerase II, DNA replication does not progress to completion (Baxter and
Diffley, 2008) an idea supported by the observation of plasmid fragmentation during
anaphase as represented by the smear underneath the linear band. When
comparing the ts mutant phenotype with the anchor away, the former appears to
have a stronger phenotype. We believe that given the vast abundance of
topoisomerase II present in the nucleus, anchor away may not be fully able to
completely deplete the nucleus of the enzyme, or do so in a timely enough manner,
while all the ts mutant proteins will be inactivated upon temperature shift.
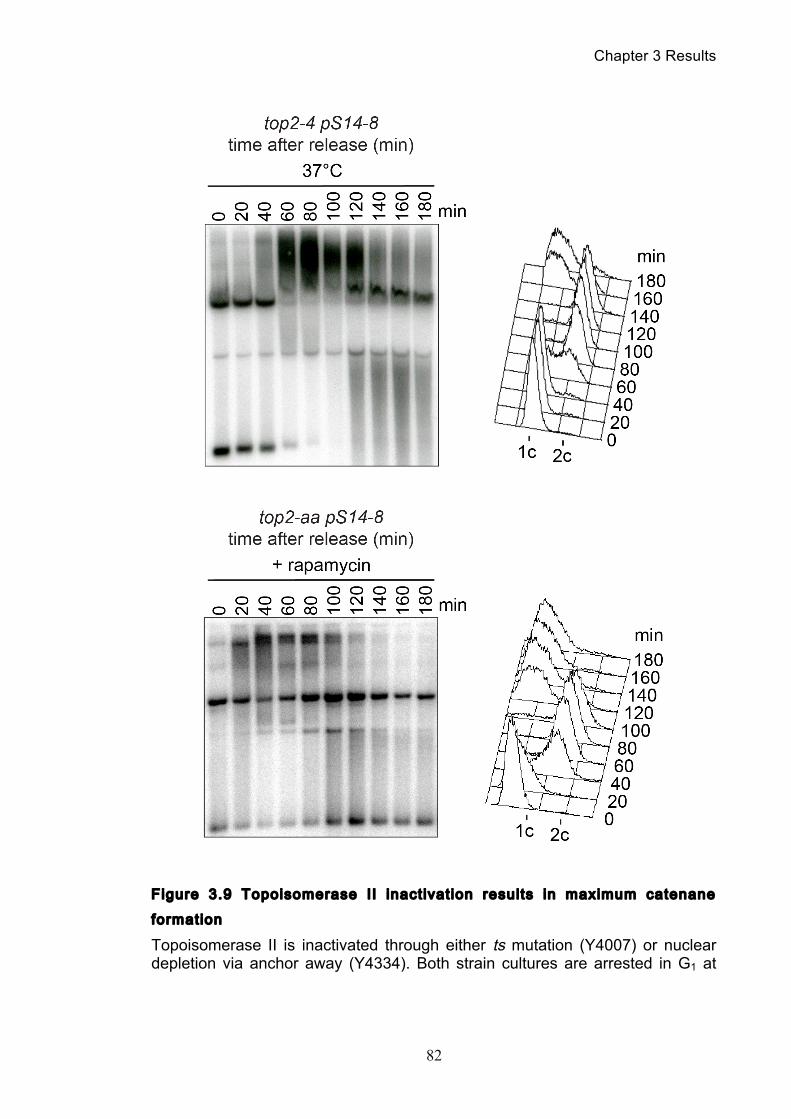
Chapter 3 Results
82
Figure 3.9 Topoisomerase II inactivat ion results in maximum catenane
formation
Topoisomerase II is inactivated through either ts mutation (Y4007) or nuclear depletion via anchor away (Y4334). Both strain cultures are arrested in G1 at

Chapter 3 Results
83
25°C before being washed and released into either media at 37°C or into media containing rapamycin.
Therefore, inactivation of topoisomerase II results in almost complete
conversion of the plasmid monomers during replication to catenated forms,
suggesting that S-phase results in all pS14-8 minichromosomes undergoing
replication and becoming at least transiently catenated.
To estimate which portion of sister minichromosomes are resolved before
progression through mitosis, we quantitatively assessed the catenation status of
pS14-8 in wild type cells (Y2665) arrested in mitosis by nocodazole treatment
(Figure 3.10). After a peak of catenanes during S-phase, over half of sister
minichromosomes appeared to be resolved in the mitotic arrest, with approximately
40% of the minichromosomes persisting as catenanes. Thus substantial
minichromosome decatenation by topo II takes place independently of progression
through mitosis. We then compared a mitotic arrest by depletion of the Cdc20 co-
activator of the anaphase promoting complex (Y4129) (Figure 3.11). In this strain
the endogenous Cdc20 promoter has been replaced such that the addition of 2 mM
methionine will stop Cdc20 expression, resulting in a metaphase arrest. In contrast
to nocodazole treatment, a mitotic spindle is formed in these cells that exerts
tension aimed at separating sister minichromosomes (Tanaka et al., 1999). As a
result, after similar catenane levels during S-phase, only about 20% catenated
molecules persisted in the arrest. This suggests that sister chromatid movement
away from each other, due to tension from the mitotic spindle, promotes their
decatenation.
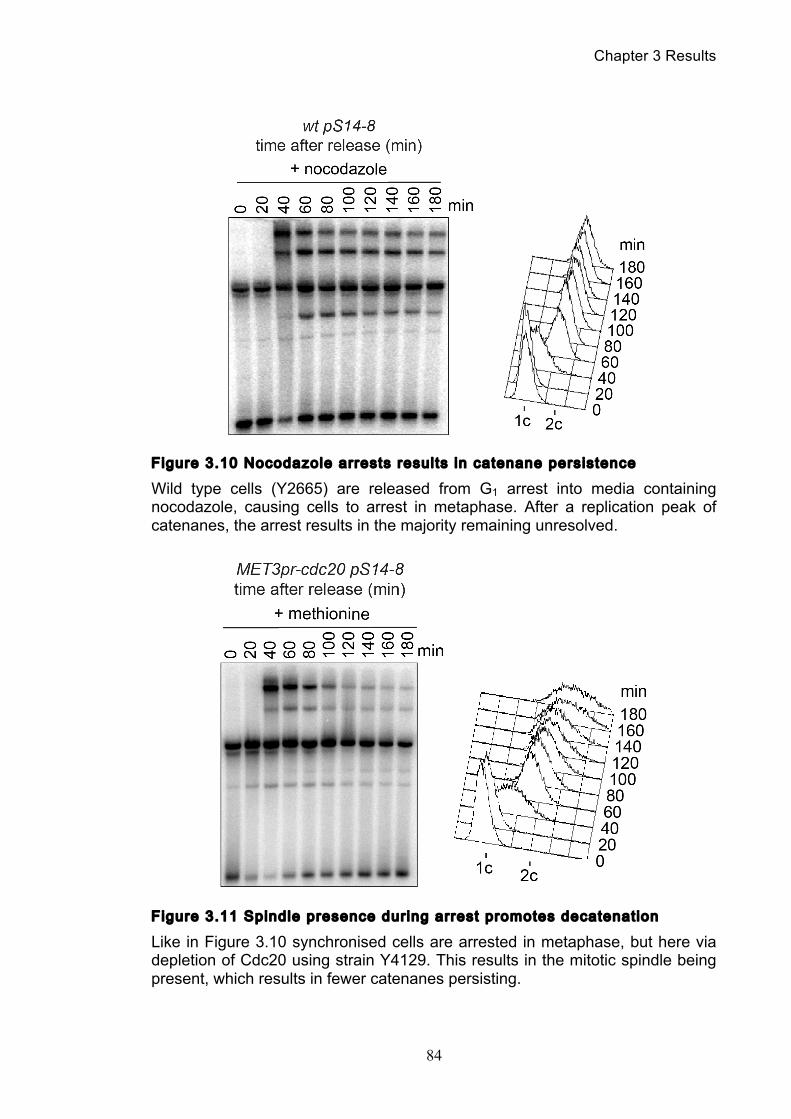
Chapter 3 Results
84
Figure 3.10 Nocodazole arrests results in catenane persistence
Wild type cells (Y2665) are released from G1 arrest into media containing nocodazole, causing cells to arrest in metaphase. After a replication peak of catenanes, the arrest results in the majority remaining unresolved.
Figure 3.11 Spindle presence during arrest promotes decatenation
Like in Figure 3.10 synchronised cells are arrested in metaphase, but here via depletion of Cdc20 using strain Y4129. This results in the mitotic spindle being present, which results in fewer catenanes persisting.

Chapter 3 Results
85
Up until mitosis, movement of sister chromatids away from each other is
restricted by the cohesin complex that maintains sister chromatid cohesion. We
therefore addressed whether removal of cohesin changes catenane maintenance
following DNA replication. After either inactivation via ts allele or nuclear depletion
via anchor away, the abundance of catenanes during the cell cycle was markedly
reduced (Figure 3.12). Even during S-phase, less than 20% of the pS14-8
minichromosomes were detected as catenated species. This suggests that sister
chromatid proximity, afforded by the cohesin complex, protects catenation. In the
absence of cohesin, sister chromatids move apart and are readily decatenated by
topo II. The conclusion that cohesin protects sister chromatid catenation was
recently reached independently (Farcas et al., 2011). Quantification of the
catenanes for all the above time courses can be seen and compared in Figure 3.13.
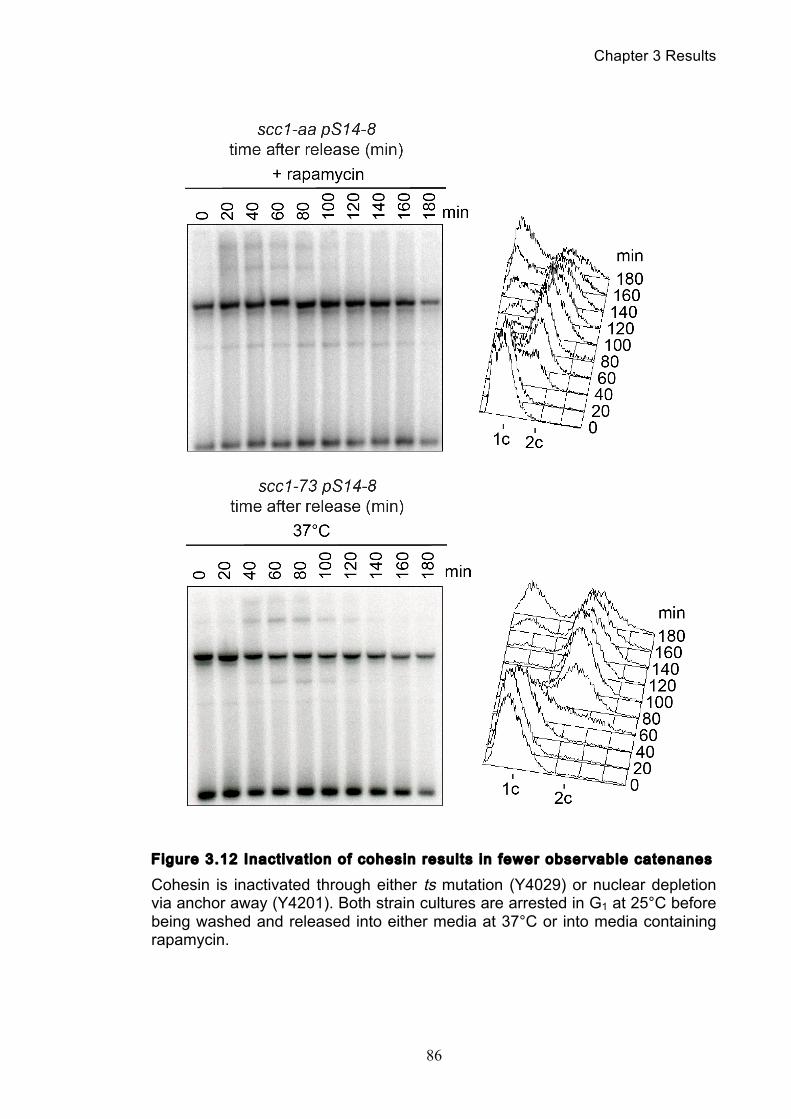
Chapter 3 Results
86
Figure 3.12 Inactivation of cohesin results in fewer observable catenanes
Cohesin is inactivated through either ts mutation (Y4029) or nuclear depletion via anchor away (Y4201). Both strain cultures are arrested in G1 at 25°C before being washed and released into either media at 37°C or into media containing rapamycin.
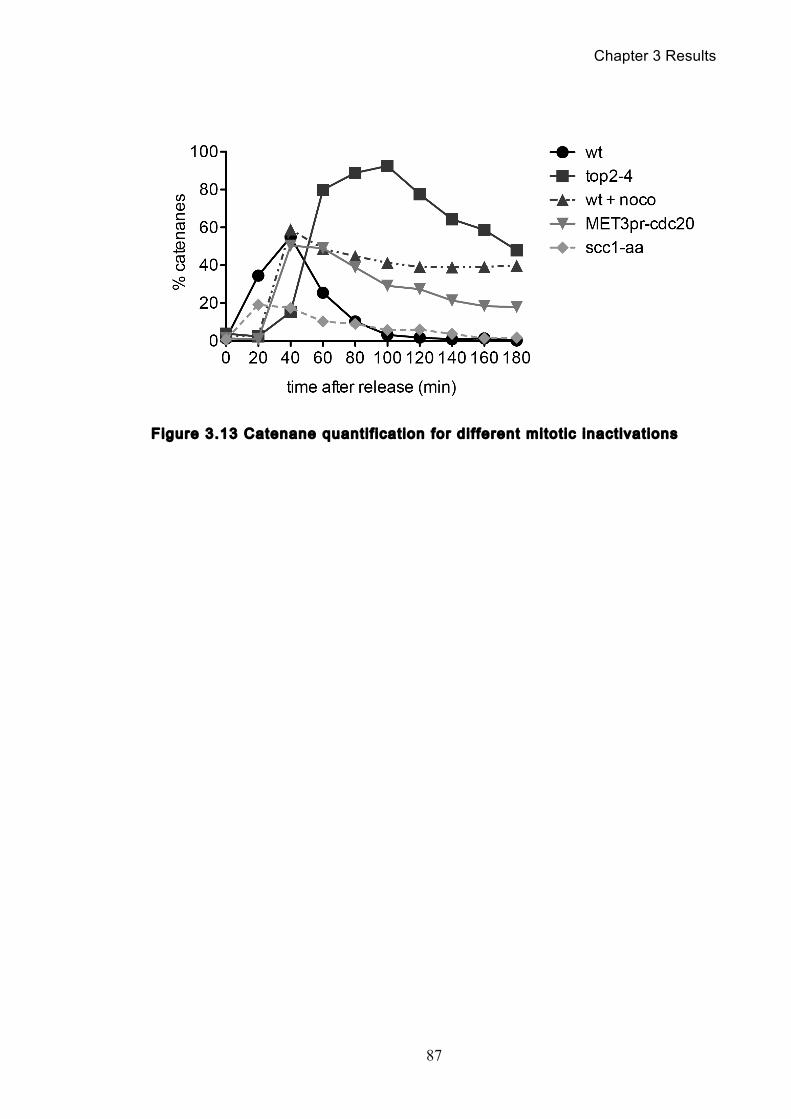
Chapter 3 Results
87
Figure 3.13 Catenane quanti f icat ion for dif ferent mitot ic inactivations

Chapter 3 Results
88
3.2.1 Temperature’s effect on catenation and isoform distr ibution
In the previous chapter, we have been using two different approaches to
inactivate target proteins: protein inactivation via ts alleles and nuclear depletion via
anchor away. Both of these procedures will inactivate the target protein but do so
by differing means, one requires a dramatic temperature shift, the other the
addition of the compound rapamycin. Given that both techniques alter the cellular
environment, we wanted to check that these depletion methods themselves did not
affect our catenation assay via the appropriate control experiments.
Initially, we examined the effect of temperature on our catenation assay. To
examine this, we released synchronous wild type cells (Y2665) from a G1 arrest
and allowed them to proceed through one cell cycle before re-arrest. This time
course was done at both 25°C and 37°C and the Southern blots were quantified
and compared (Figure 3.14). We can see that the pattern and distribution of bands
and their relative intensities is altered between the two different temperatures.
Moreover, a very slight amount of persistent catenation was detected in the 37°C
samples suggesting that the insult of the higher temperature could have some
slight effect on catenation, perhaps via impairment of topo II activity.
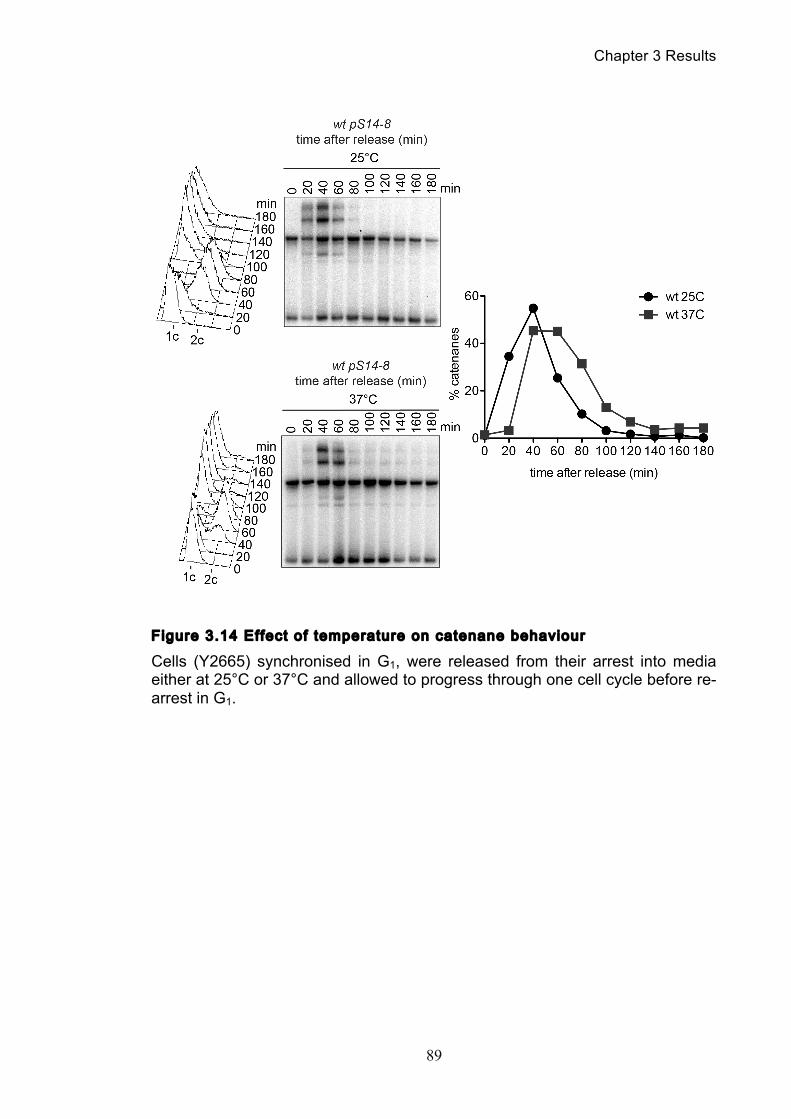
Chapter 3 Results
89
Figure 3.14 Effect of temperature on catenane behaviour
Cells (Y2665) synchronised in G1, were released from their arrest into media either at 25°C or 37°C and allowed to progress through one cell cycle before re-arrest in G1.

Chapter 3 Results
90
Following on from these observations, we also wanted to examine in more
detail the effect of temperature on the different topological forms. In the previous
results chapter 3.2, we found that a nocodazole arrest results in a catenation
equilibrium being established where all the different topological forms are present
and can clearly be seen. We repeated this experiment but this time performing the
experiment at 37°C instead of 25°C then proceeding to quantify and compare the
experiments (Figure 3.15). From this comparison, we made two interesting
observations; the first was that the overall amount of persistent catenation at either
temperature was very similar. The second was that the amount of supercoiled
species, both monomers and catenanes, was markedly reduced at higher
temperatures throughout the entire time course when compared to the levels seen
at 25°C. This is an unsurprising result, considering that increasing the temperature
will increase the kinetic energy of small molecules such as DNA. At higher
temperatures, collisions, interactions and reactions between molecules will occur
with greater energy. It is likely that this could translate to a higher occurrence of
DNA strand-nicking and as discussed earlier, plasmids that contain nicked DNA are
unable to sustain any supercoiling. In conclusion, these temperature comparisons
have shown that while increased temperature does affect the various ratios of
topoisomers present, the overall relative amount of catenation remains very similar.
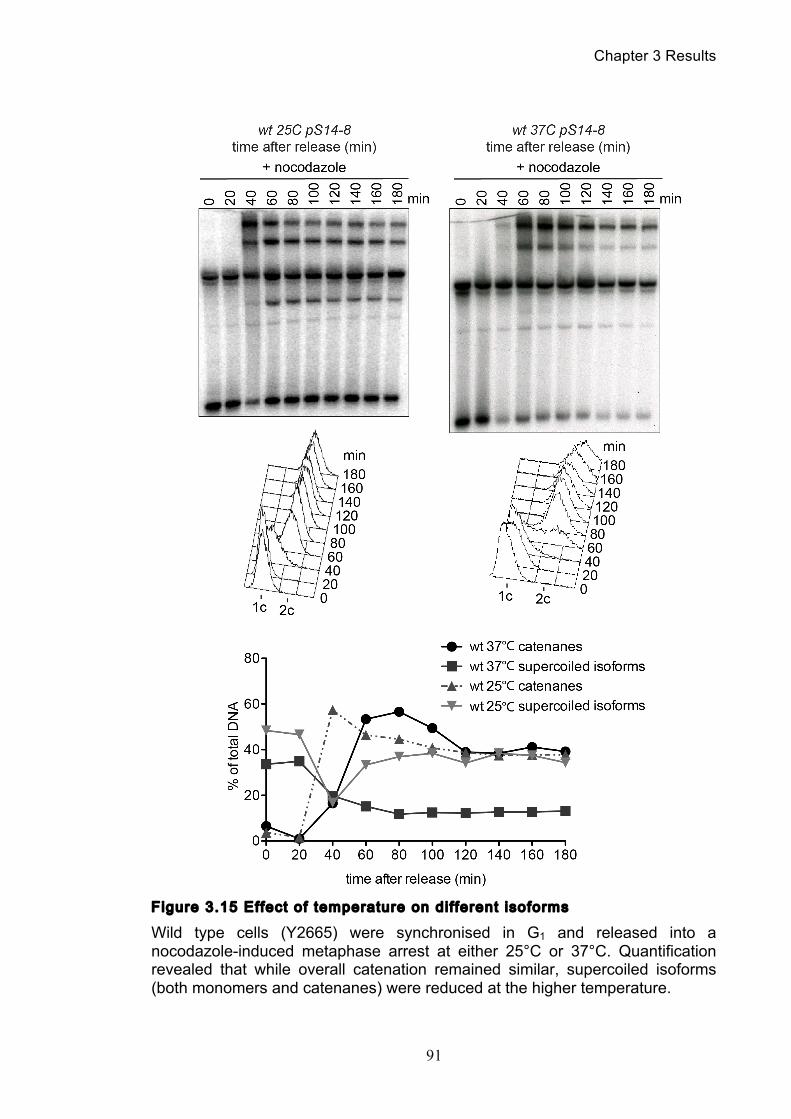
Chapter 3 Results
91
Figure 3.15 Effect of temperature on dif ferent isoforms
Wild type cells (Y2665) were synchronised in G1 and released into a nocodazole-induced metaphase arrest at either 25°C or 37°C. Quantification revealed that while overall catenation remained similar, supercoiled isoforms (both monomers and catenanes) were reduced at the higher temperature.

Chapter 3 Results
92
3.2.2 The anchor away system does not effect catenation
When the new anchor away technique was published (Haruki et al., 2008a),
our lab quickly obtained the strain and modified it for our own ends by tagging our
proteins of interest with the FRB tag (see chapter 1.7 for an introduction to the
anchor away technique). Having tagged our target proteins, we wanted to check
that the parental strain itself, as well as the presence of rapamycin, did not exert
any influence on catenane behaviour. The anchor away strain has certain
mutations inherent to its design. Because rapamycin is toxic to wild-type S.
cerevisiae, the anchor away strains are made rapamycin-resistant through mutation
of TOR1 (tor1-1). Additionally FPR1 (Δfpr1) is deleted, as it is the yeast homolog of
the human FKBP12 gene that encodes the most abundant FK506 and rapamycin
binding protein in S. cerevisiae. This is necessary to reduce competition between
Fpr1p and the anchor-FKBP12 construct for binding to the FRB domain (Heitman
et al., 1991). Therefore given the modifications of the strain, plus the exposure to
the usually toxic rapamycin, we needed to ensure that neither had any effect on the
creation and resolution of catenanes. To test this, we performed a time course
using an anchor away ‘parental’ strain (Y4332) where no target protein has been
FRB-tagged. These cells were synchronised and released into media containing
rapamycin and allowed to progress through one cell cycle (Figure 3.16). As can be
seen, neither the strain nor the presence of rapamycin appears to have any
influence on catenation and the pattern of topological forms looks the same as with
our wild-type strain (Figure 3.5).
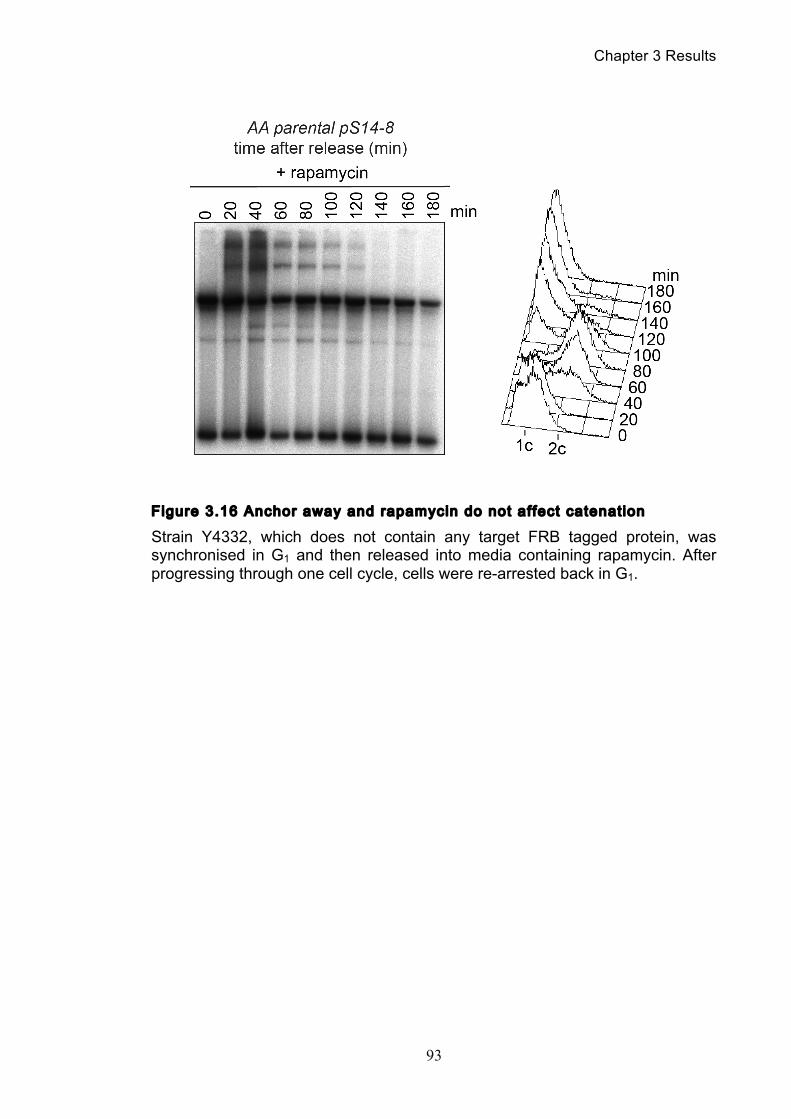
Chapter 3 Results
93
Figure 3.16 Anchor away and rapamycin do not affect catenation
Strain Y4332, which does not contain any target FRB tagged protein, was synchronised in G1 and then released into media containing rapamycin. After progressing through one cell cycle, cells were re-arrested back in G1.

Chapter 3 Results
94
3.3 Catenation of smaller minichromosomes is observable
in wild type strains
Having established the assay using the minichromosome pS14-8, and
having seen how clearly we were able to observe catenanes, we wanted to check
that our results were not unique to this one particular plasmid. Previous catenation
studies involving smaller minichromosomes, specifically pRS316 (4.88kb) (Figure
3.17), have reported that under wild type conditions, catenanes were resolved too
quickly during mitosis to be detected and could only be observed through
inactivation of endogenous topo II (Baxter et al., 2011). However, given the clarity
of our results with pS14-8, we decided to test a smaller minichromosome in the
assay to see if this was indeed the case. We also used pRS316 and apart from
changing the electrophoresis running conditions, used the same protocol for
purification and resolution of the DNA as with pS14-8. We performed a time course
using WT cells (Y4199), quantified the data and were able once again to observe
the appearance and then resolution of catenanes during the cell cycle (Figure 3.18).
To confirm the detection of catenanes, enzyme digests were performed on the
DNA samples to confirm what topological forms each band represented. Digests
with topo I and II revealed that the top two bands with the lowest electrophoretic
mobility indeed represented catenated forms. While the top band contains relaxed
catenanes, the second catenated band most likely contains a mix of both
supercoiled and mixed catenanes. While with pS14-8 these two forms ran
separately, it seems most likely that with the smaller pRS316, the supercoiled and
mixed catenanes have a similar electrophoretic mobility. The linear form of pRS316
was not detectable in our samples, suggesting that these smaller DNA molecules
are less susceptible to shearing during purification than their larger counterparts.
Comparing the results with pS14-8, it was reassuring to obtain similar results with
the much smaller minichromosome and additionally confirm that our catenation
assay was not particular to any one plasmid but rather representative of them all.
However, we can see from the quantification that the smaller pRS316 had a peak
catenation of under 30%, around half of the peak seen for pS14-8, suggesting that
most of the plasmid molecules were decatenated soon after synthesis. So while it
is very much possible to observe the catenanes of smaller plasmids under wild type
conditions, their smaller size seems to result in faster decatenation.
Chapter 3 Results
95
Figure 3.17 Map of minichromosome pRS316
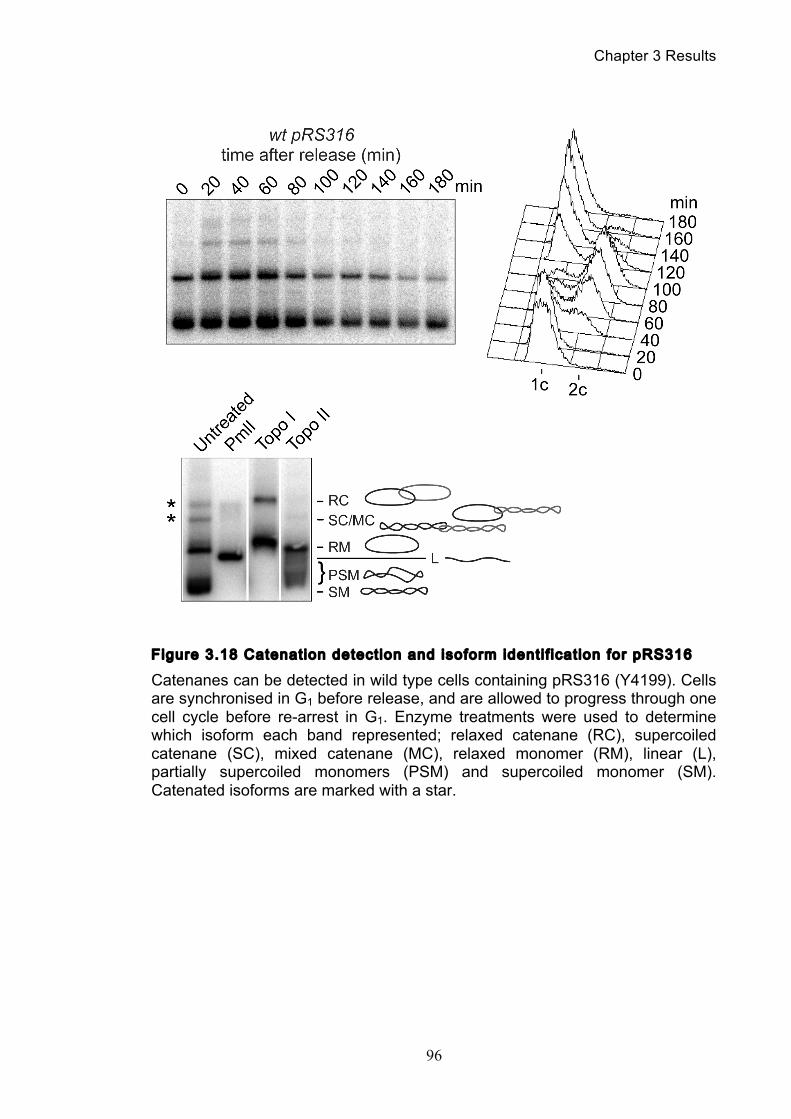
Chapter 3 Results
96
Figure 3.18 Catenation detection and isoform identi f icat ion for pRS316
Catenanes can be detected in wild type cells containing pRS316 (Y4199). Cells are synchronised in G1 before release, and are allowed to progress through one cell cycle before re-arrest in G1. Enzyme treatments were used to determine which isoform each band represented; relaxed catenane (RC), supercoiled catenane (SC), mixed catenane (MC), relaxed monomer (RM), linear (L), partially supercoiled monomers (PSM) and supercoiled monomer (SM). Catenated isoforms are marked with a star.

Chapter 4 Results
97
Chapter 4. Results: Condensin’s Inf luence on
Catenation
4.1 Condensin promotes completion of minichromosome
decatenation
Having established our catenation assay and studied some of the major
mitotic components, it was time to begin studying condensin directly. The first and
simplest approach to take was to inactivate condensin using one of the ts mutants
in our laboratory’s possession. Therefore, we performed timecourses with three
condensin ts alleles: smc4-1 (Y3954), brn1-9 (Y3939) and ycg1-10 (Y3940), each
containing the 21 kb minichromosome, pS14-8. In all cases, cells were
synchronised in G1 at the permissive temperature before being released into media
at the non-permissive temperature (Figure 4.1). Looking at the results for all three
mutants and comparing them with wild type cells shifted to 37°C, one can see a
subtle but constant phenotype of partial persistent catenation once mitosis is
complete. Quantification of the blots reveals that after peaking at approximately
50% catenanes 60 minutes after release from G1, the cells still retain 10-20%
catenanes total 180 minutes after release. When compared to wild type cells, that
have less than 5% catenanes upon re-arrest in G1, these results clearly show that
with condensin inactivated, there is a failure to completely resolve catenation. The
FACS profiles for all three condensin mutants also show a characteristic double
peak or ‘shoulder’ as the cells are re-arrested in G1 at the non-permissive
temperature. This is a characteristic condensin phenotype (D'Ambrosio et al.,
2008a) attributed to chromosome mis-segregation.
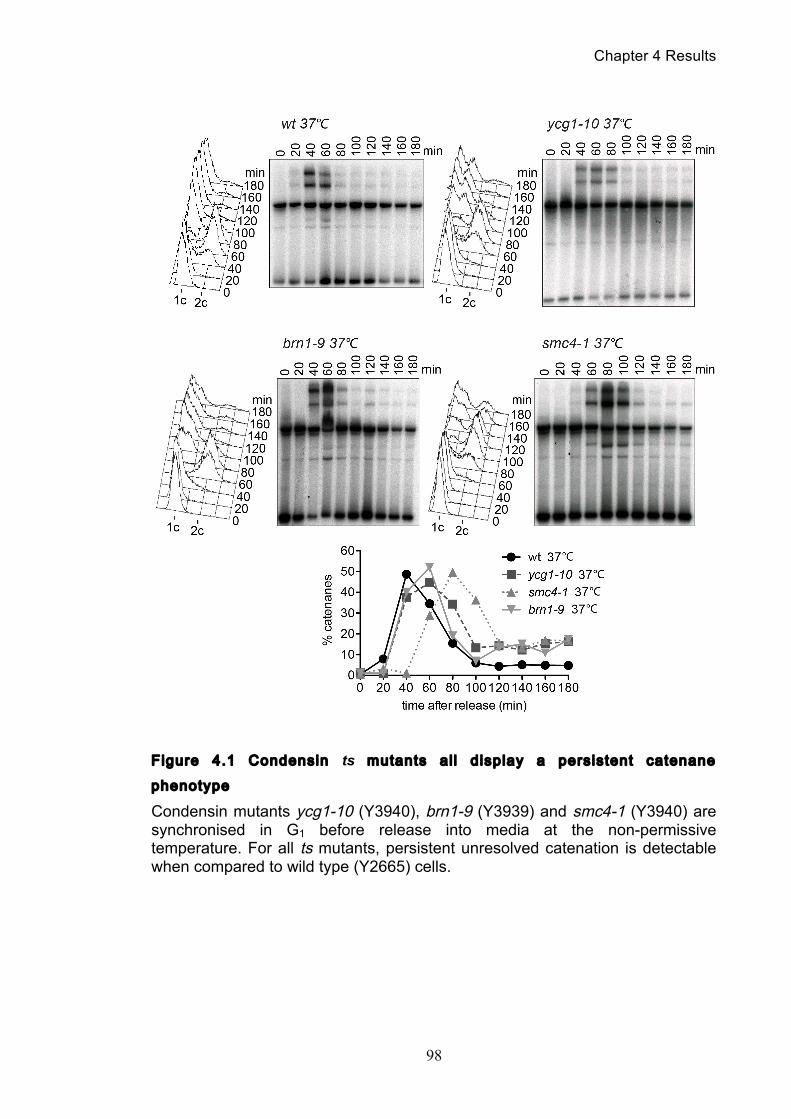
Chapter 4 Results
98
Figure 4.1 Condensin ts mutants al l display a persistent catenane
phenotype
Condensin mutants ycg1-10 (Y3940), brn1-9 (Y3939) and smc4-1 (Y3940) are synchronised in G1 before release into media at the non-permissive temperature. For all ts mutants, persistent unresolved catenation is detectable when compared to wild type (Y2665) cells.

Chapter 4 Results
99
Having obtained these results with the ts mutants, we were then very keen
to see if similar results were obtained via nuclear depletion of condensin using the
anchor away technique. Therefore, we analyzed catenane abundance in
synchronised cells following nuclear depletion of condensin using an anchor away
allele of its Brn1 subunit (brn1-aa (Y4059) Figure 4.2). Catenanes accumulated
during S-phase to comparable levels, irrespective of whether rapamycin was added
to deplete condensin. During G2 and mitosis catenanes started to be resolved
under both conditions but, in contrast to wild type cells that saw complete
decatenation, decatenation progressed more slowly in condensin-depleted cells
and approximately 15% catenanes persisted unresolved until the end of the
experiment. This was not because mitosis was delayed in the absence of
condensin. FACS analysis of the DNA content, as well as the budding index,
demonstrated that both cultures progressed through the cell cycle with similar
kinetics and all cells had returned to G1 by the end of the time course. Anaphase
bridges and chromosome missegregation were abundant after rapamycin addition,
as seen on the FACS profile and in anaphase cells stained with the DNA dye 4',6-
diamidino-2-phenylindole (DAPI; Figure 4.2), verifying that the depletion of
condensin was not just having some minichromosome-specific effect. Persistent
minichromosome catenation after condensin depletion was reproducibly observed
at similar levels in several biological repeats of this experiment (Figure 4.3).
Therefore, these results suggest that condensin promotes resolution of sister
chromatid catenanes and demonstrates that in the absence of condensin a fraction
of catenated minichromosomes persist until the time of cell separation by
cytokinesis.
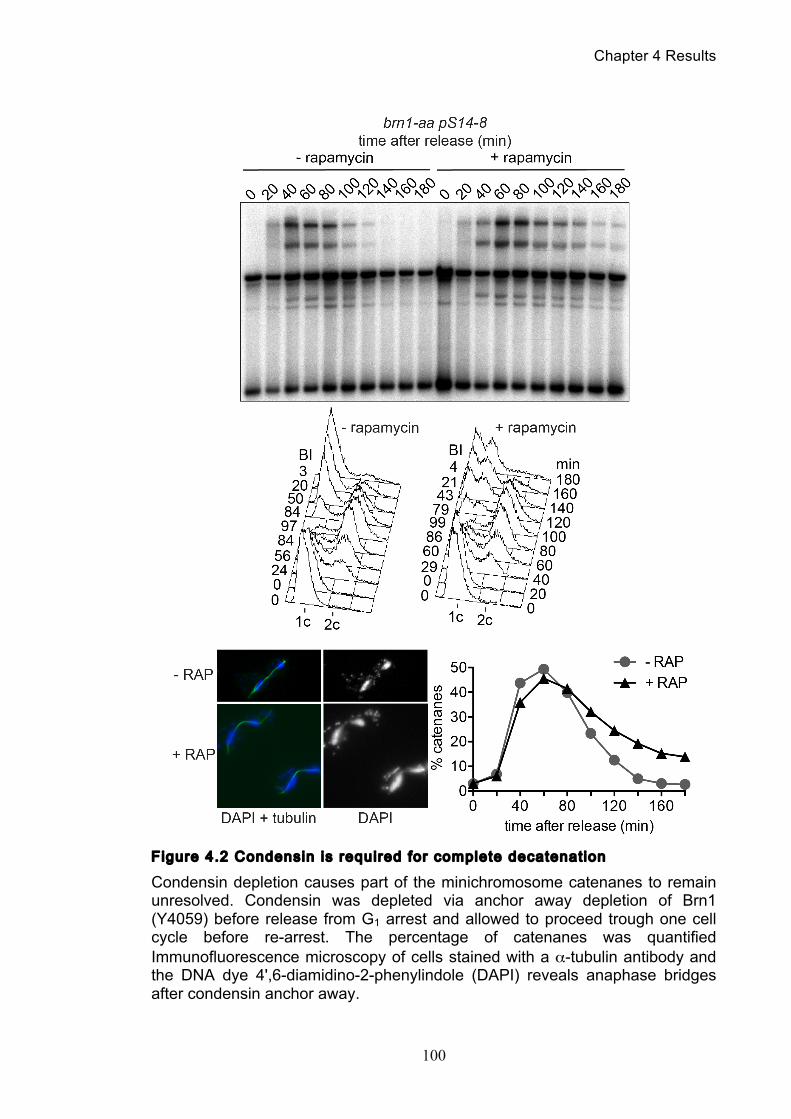
Chapter 4 Results
100
Figure 4.2 Condensin is required for complete decatenation
Condensin depletion causes part of the minichromosome catenanes to remain unresolved. Condensin was depleted via anchor away depletion of Brn1 (Y4059) before release from G1 arrest and allowed to proceed trough one cell cycle before re-arrest. The percentage of catenanes was quantified Immunofluorescence microscopy of cells stained with a α-tubulin antibody and the DNA dye 4',6-diamidino-2-phenylindole (DAPI) reveals anaphase bridges after condensin anchor away.
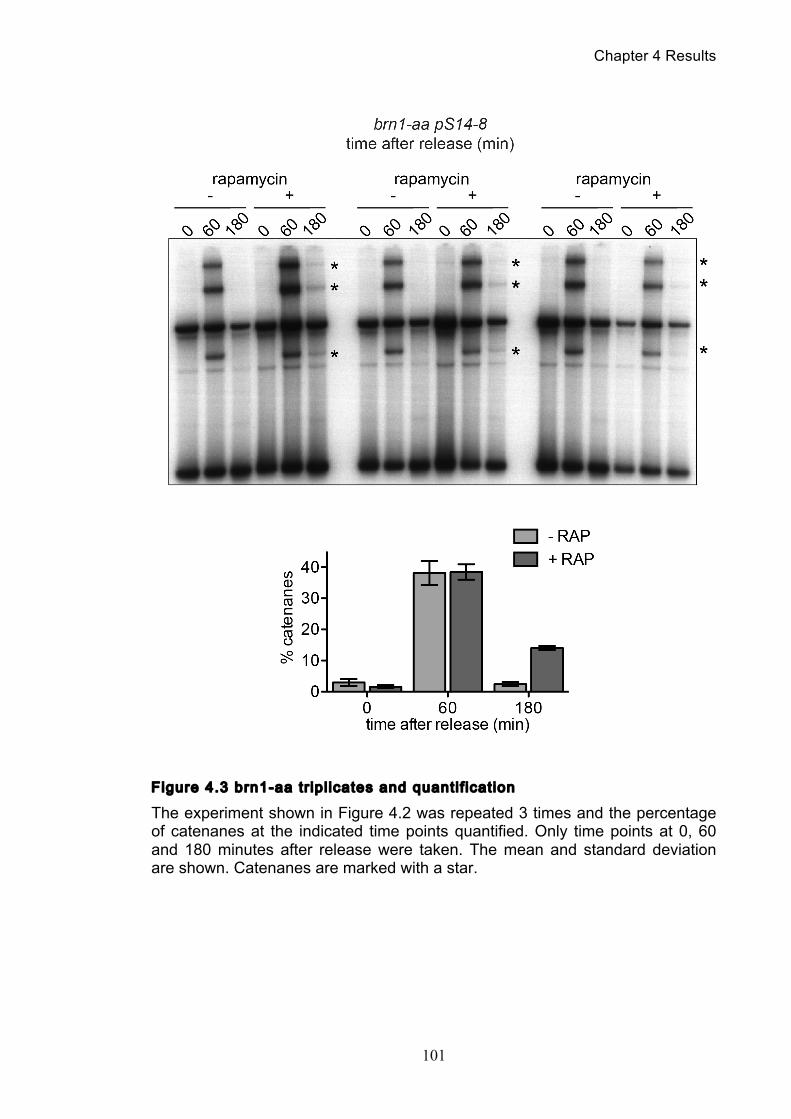
Chapter 4 Results
101
Figure 4.3 brn1-aa tr ipl icates and quanti f icat ion
The experiment shown in Figure 4.2 was repeated 3 times and the percentage of catenanes at the indicated time points quantified. Only time points at 0, 60 and 180 minutes after release were taken. The mean and standard deviation are shown. Catenanes are marked with a star.

Chapter 4 Results
102
If our circular minichromosome is a faithful model for the behavior of authentic
chromosomes, then the condensin requirement for complete decatenation could
explain anaphase bridges and chromosome missegregation in the absence of
condensin. One caveat to this conclusion is that if condensin was required to
resolve different, catenation-independent, linkages on authentic chromosomes,
then such linkages might indirectly hamper minichromosome decatenation. We
therefore addressed whether condensin contributes to chromosome decatenation
independently of promoting chromosome movement. To do this, we arrested cells
in mitosis using nocodazole, a situation where the mitotic spindle is disrupted and
therefore directed chromosome movement no longer contributes to sister chromatid
resolution. Cohesin was depleted from the nucleus using the scc1-aa allele, as
above, allowing spontaneous catenane resolution (Figure 4.4). Condensin
depletion in the same cells, via the brn1-aa allele, led to an increased population of
persisting catenanes. This suggests that condensin supports chromosome
decatenation independently of, or in addition to, promoting efficient chromosome
movement during anaphase. However, paired t-test analysis comparing the two
strains produces a P value of 0.0497, which only just implies a significant difference
at a confidence level of 95%. Following a similar line of thinking, we wanted to
confirm that condensin also supports decatenation independently of mitosis
progression. In Figure 4.5, smc2-aa MET3pr-cdc20 cells were arrested with
addition of methionine either in the presence of rapamycin or not. One can see that
when condensin is depleted, more catenanes persist, thus suggesting that the
mechanism by which condensin promotes decatenation is active prior to the
metaphase to anaphase transition. The effect of condensin here is more significant,
with a paired t-test analysis producing a P value of 0.0010, suggesting that there is
indeed a significant difference resulting from condensin depletion.
Taking these results together, we suggest that persistent catenanes in the
absence of condensin are a likely source of chromosome segregation failures.
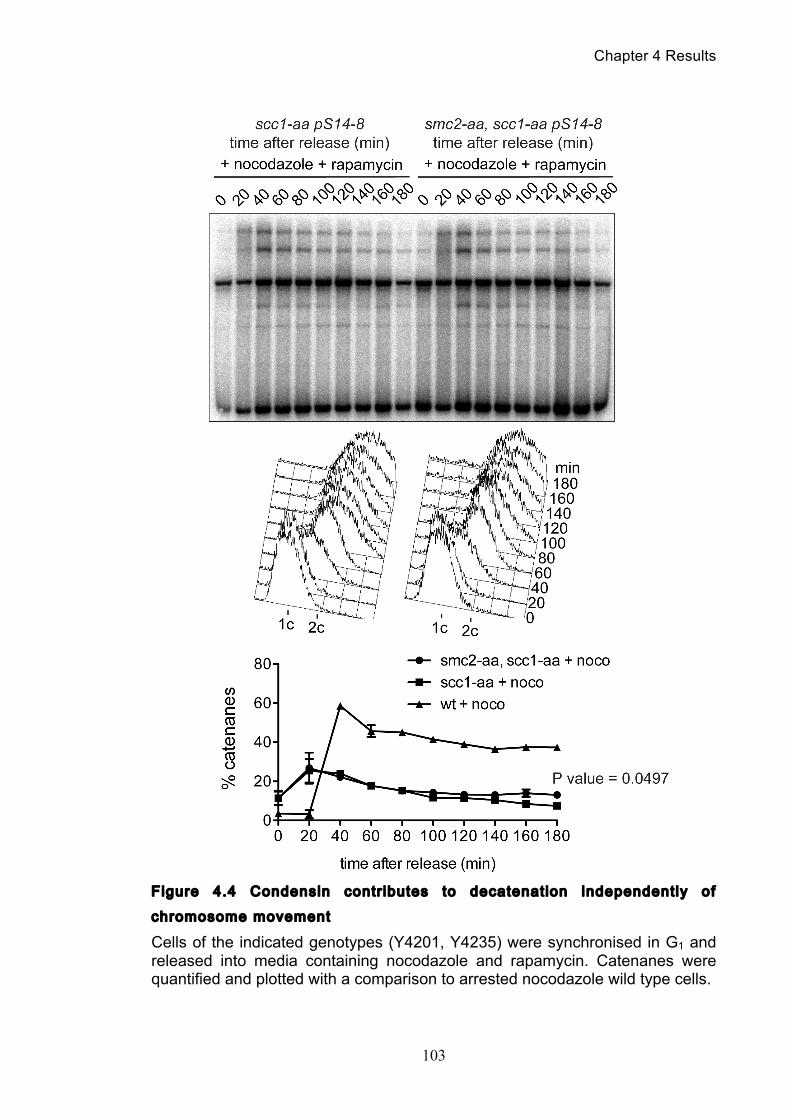
Chapter 4 Results
103
Figure 4.4 Condensin contr ibutes to decatenation independently of
chromosome movement
Cells of the indicated genotypes (Y4201, Y4235) were synchronised in G1 and released into media containing nocodazole and rapamycin. Catenanes were quantified and plotted with a comparison to arrested nocodazole wild type cells.
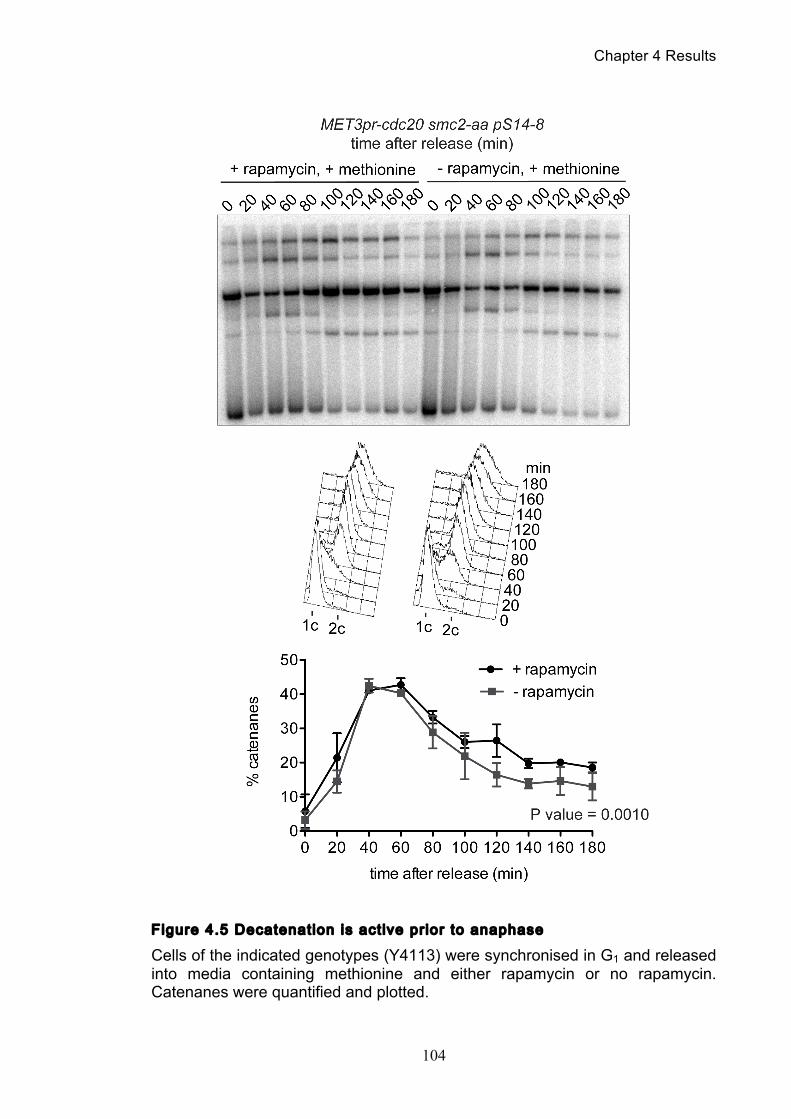
Chapter 4 Results
104
Figure 4.5 Decatenation is active prior to anaphase
Cells of the indicated genotypes (Y4113) were synchronised in G1 and released into media containing methionine and either rapamycin or no rapamycin. Catenanes were quantified and plotted.

Chapter 4 Results
105
4.2 The effect of minichromosome size
This clear observation of persistent catenation resulting from the inactivation
of condensin was stimulus to see if we observed a similar phenotype when
employing the smaller pRS316 plasmid. We began by investigating the effect of the
ts mutant smc2-8, in order to make a direct comparison to results from a previous
study (Baxter et al., 2011) where catenanes had not been seen upon shift to the
non-permissive temperature (Figure 4.6). In our timecourse we do clearly see
catenanes appear and it seems that a portion of these do persist. However, this
particular mutant had difficulty in release synchronously from the G1 arrest such
that we could not be certain that the catenanes observed 120 to 180 minutes after
release were not a result of late comers entering S-phase. The FACS profile is
particularly unclear for this strain (Y4236) making assessment of cell synchrony
difficult. Fortunately, we also had the anchor away method at our disposal (Figure
4.7). Nuclear depletion of condensin revealed a small delay in catenane resolution,
but the effect was less pronounced compared to the larger minichromosome.
However the FACS profile does show the distinct ‘shoulder’ upon re-arrest in G1
that is commonly seen in condensin mutants. By the time cells passed through
mitosis in the absence of condensin, less than 10% of catenanes persisted. This
suggests that condensin makes a contribution to plasmid decatenation, albeit small.
We note that if condensin-dependent positive supercoiling was required for
pRS316 decatenation, which has been observed in topo II depleted cells (Baxter et
al., 2011), we should have observed a greater resolution defect.
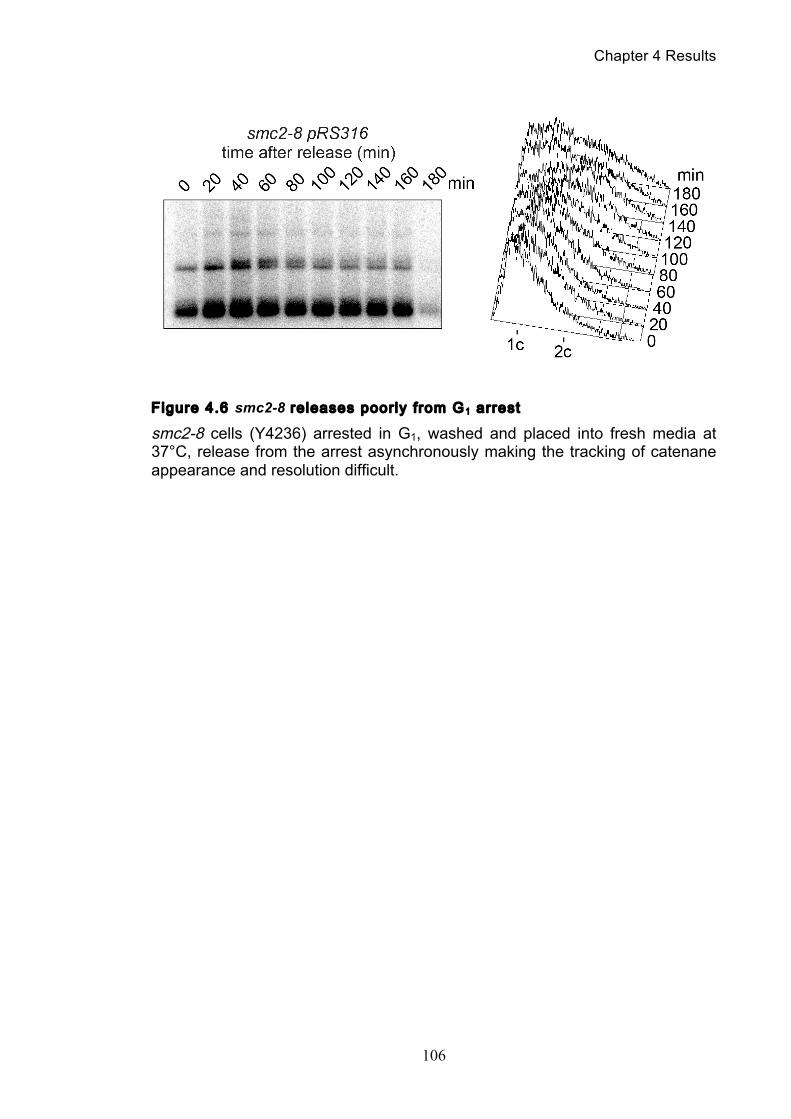
Chapter 4 Results
106
Figure 4.6 smc2-8 releases poorly from G1 arrest
smc2-8 cells (Y4236) arrested in G1, washed and placed into fresh media at 37°C, release from the arrest asynchronously making the tracking of catenane appearance and resolution difficult.
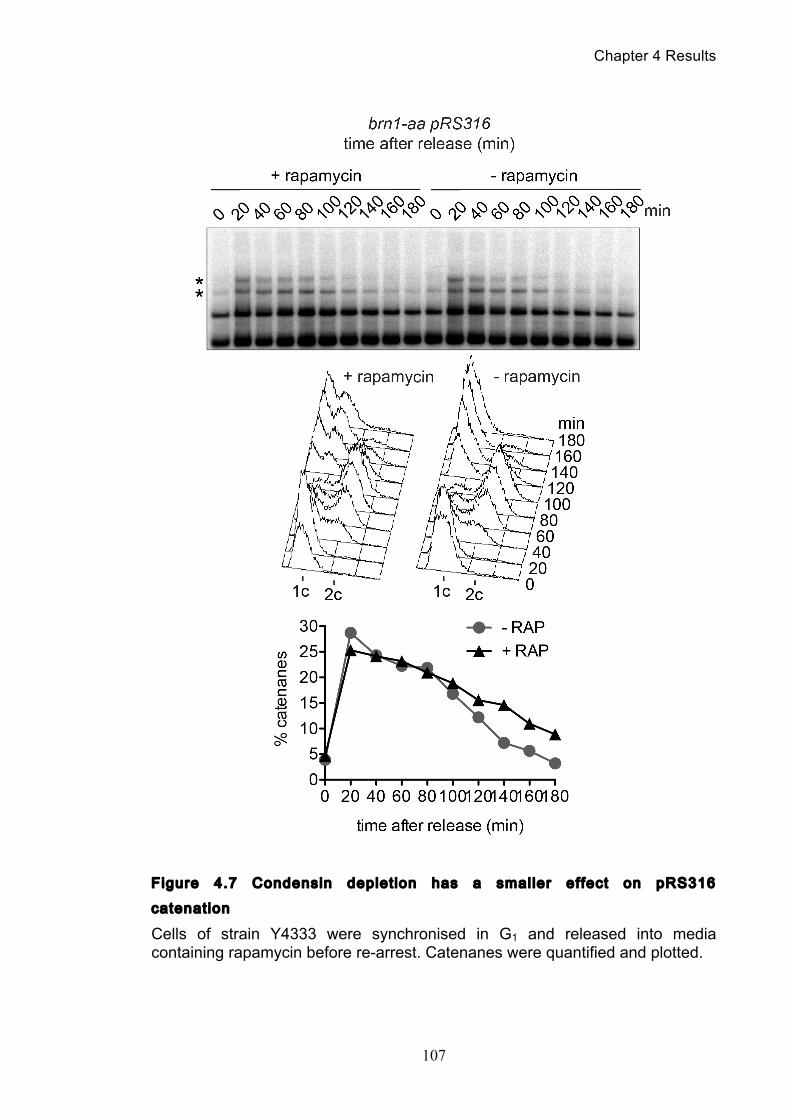
Chapter 4 Results
107
Figure 4.7 Condensin deplet ion has a smaller effect on pRS316
catenation
Cells of strain Y4333 were synchronised in G1 and released into media containing rapamycin before re-arrest. Catenanes were quantified and plotted.

Chapter 4 Results
108
Compared to authentic chromosomes, pRS316 and pS14-8 are small in size
and both contain only one replication origin. Given that catenation results from the
meeting head-on of two replication forks during DNA replication, one can deduce
that the number of topological links between catenated pRS316 and pS14-8
plasmids are few. In addition once a link is removed, if the plasmids are not held
together by further links (or as we demonstrated, cohesin), then they will quickly
physically separate and are unlikely to be recatenated by topoisomerase II. Given
this, we hypothesised that condensin’s contribution to decatenation would be
greater in more complex topological landscapes than small minichromosomes. To
address this prediction, we adapted our catenation assay to use a much larger
substrate. In order to better represent the native chromosomes that this assay is
modelling, we studied a 61 kb ring chromosome (RCIII, Figure 4.8), consisting of a
61kb section of S. cerevisiae chromosome 3, including the centromere, which has
been excised then ligated to form a circular chromosome (Dershowitz and Newlon,
1993). In addition, this ring chromosome contained three origins of replication that
would result in an increased number of topological linkages being formed between
replicated DNAs during S-phase.
However, given the much larger size of this substrate, we found it much
more difficult to separate its topological isoforms using the same purification and
electrophoresis conditions as for pS14-8. Furthermore, owing to its much larger
size, the ring chromosome was more susceptible to shearing during DNA
purification. This resulted in a large fraction being linearised and creating greater
background signal that made band quantification difficult.
4.2.1 Visualiz ing a r ing chromosome’s (RCIII) dif ferent isoforms
In order to address the difficulties in resolving this much larger ring
chromosome, time was spent developing a new protocol to process the cell
samples. In this new protocol, cells were suspended in agarose blocks in which
they would then be lysed and processed to extract their DNA without any pipetting
involved at all, following a specific protocol as described in (Jain et al., 2010). It
was hoped that this gentler approach to DNA extraction would reduce or eliminate
linearization of RCIII. In addition, the DNA from the agarose plugs would then be

Chapter 4 Results
109
resolved using Pulse Field Gel Electrophoresis (PFGE), a technique commonly
used for DNAs of a large size. However, despite extensive troubleshooting the best
results achieved (Figure 4.9) did not offer the resolution we hoped for. While it
appeared that we had been successful in minimising the amount of linear forms
generated (as none can be observed), the clarity of the different bands remained
poor and offered little advantage over our previous DNA purification protocol. As
such, we discontinued using PFGE and agarose gel plugs for DNA extraction and
instead redesigned our previous DNA purification protocol to minimise the number
of steps involving pipetting. In addition, we optimised the electrophoresis running
conditions to give us the best resolution of isoforms possible, as described in
chapter 2.4.
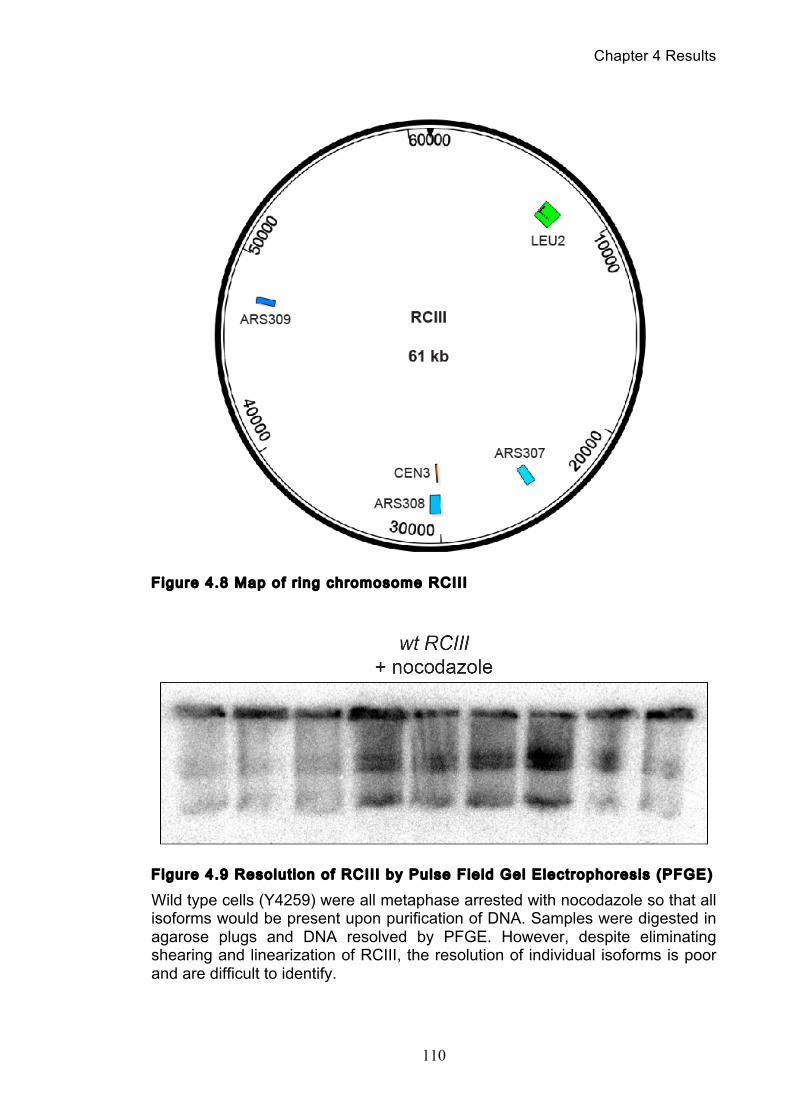
Chapter 4 Results
110
Figure 4.8 Map of r ing chromosome RCIII
Figure 4.9 Resolut ion of RCIII by Pulse Field Gel Electrophoresis (PFGE)
Wild type cells (Y4259) were all metaphase arrested with nocodazole so that all isoforms would be present upon purification of DNA. Samples were digested in agarose plugs and DNA resolved by PFGE. However, despite eliminating shearing and linearization of RCIII, the resolution of individual isoforms is poor and are difficult to identify.

Chapter 4 Results
111
4.3 Condensin’s decatenation role is more pronounced on
RCIII
Having tried PFGE and returned to our original (albeit optimised) purification
and electrophoresis protocol, we were able to find electrophoretic conditions that
revealed wild type catenane formation during DNA replication (Y4259) (Figure
4.10). We were able to observe a discrete band appearing below the relaxed
monomer (RM) band during DNA replication, that we identified as supercoiled
catenanes (SC) via enzyme treatment. Incubation with topo I relaxed these species
into a smear of relaxed and mixed catenanes (MC), probably of varied supercoiling
status, migrating above relaxed monomers. Their catenated nature was confirmed
by topo II treatment, which resolved these species into monomers. The general gel
background in the region of mixed and relaxed catenanes, however, prevented
their reliable analysis. In Figure 4.11 we see a quantification profile generated by
the ImageQuant software. Given that many of the catenated forms run as a smear
overlapping with bands representing monomers, the software is unable to correctly
quantify each topological form. Despite this setback, we proceeded to perform
several timecourse experiments using RCIII.
Focusing on the diagnostic supercoiled catenanes (SC), we observed their
appearance in S-phase, followed by resolution at the time of mitosis. As with the
smaller minichromosomes, only a fraction of the ring chromosome appeared
catenated following S-phase. This suggests that a sizeable portion even of this
large ring chromosome is decatenated rapidly following its replication. When we
blocked mitotic progression using nocodazole, a similar level of catenanes was
detectable during S-phase, but these were no longer resolved and persisted for an
extended period in the arrest (Figure 4.12). The same was observed after
condensin inactivation using the brn1-9 mutation (Figure 4.13). In this case cells
progressed through mitosis, yet catenanes following DNA replication persisted
without sign of resolution for the remainder of the time course. A similar result was
obtained following nuclear depletion of condensin (Figure 4.14). This suggests that
two pools of catenanes are formed in S-phase. One pool is readily resolved
following DNA synthesis, while a second pool of catenanes persists that is helped
by condensin for their decatenation. The condensin requirement for resolution of
this latter pool becomes more pronounced as chromosome size increases. In the

Chapter 4 Results
112
case of the 61 kb ring chromosome, resolution of persistent catenanes showed a
strict condensin requirement.
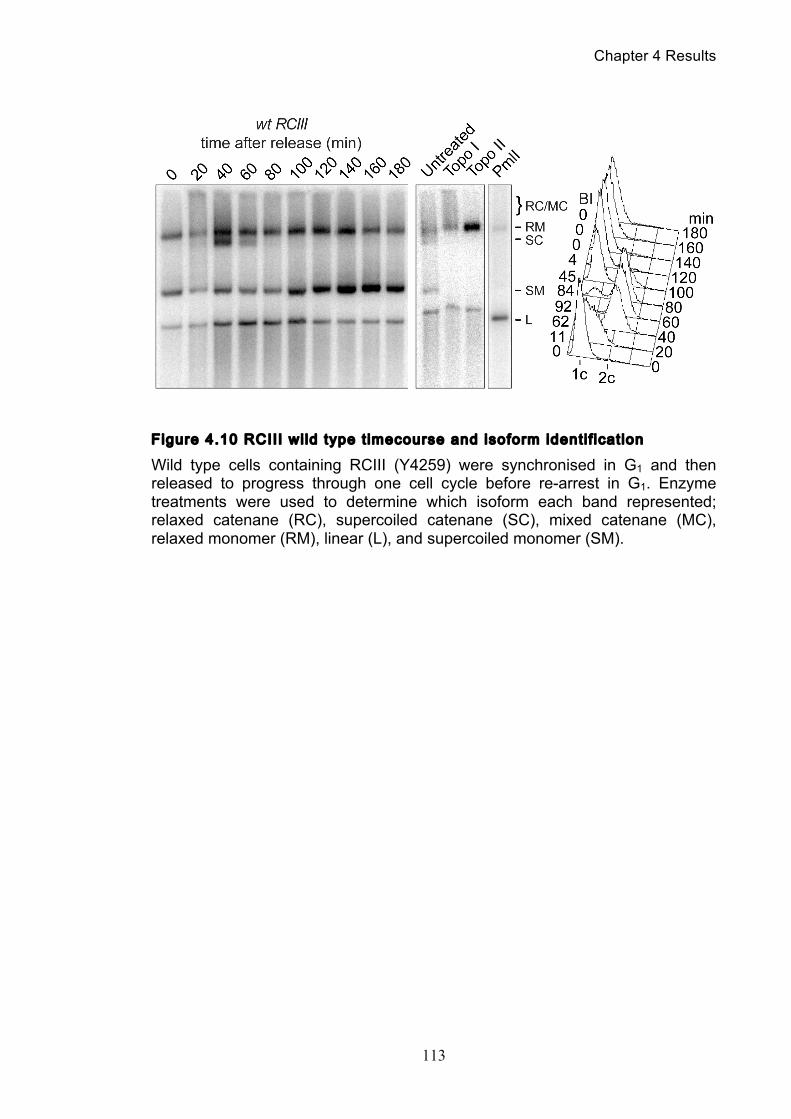
Chapter 4 Results
113
Figure 4.10 RCIII wi ld type t imecourse and isoform identi f icat ion
Wild type cells containing RCIII (Y4259) were synchronised in G1 and then released to progress through one cell cycle before re-arrest in G1. Enzyme treatments were used to determine which isoform each band represented; relaxed catenane (RC), supercoiled catenane (SC), mixed catenane (MC), relaxed monomer (RM), linear (L), and supercoiled monomer (SM).
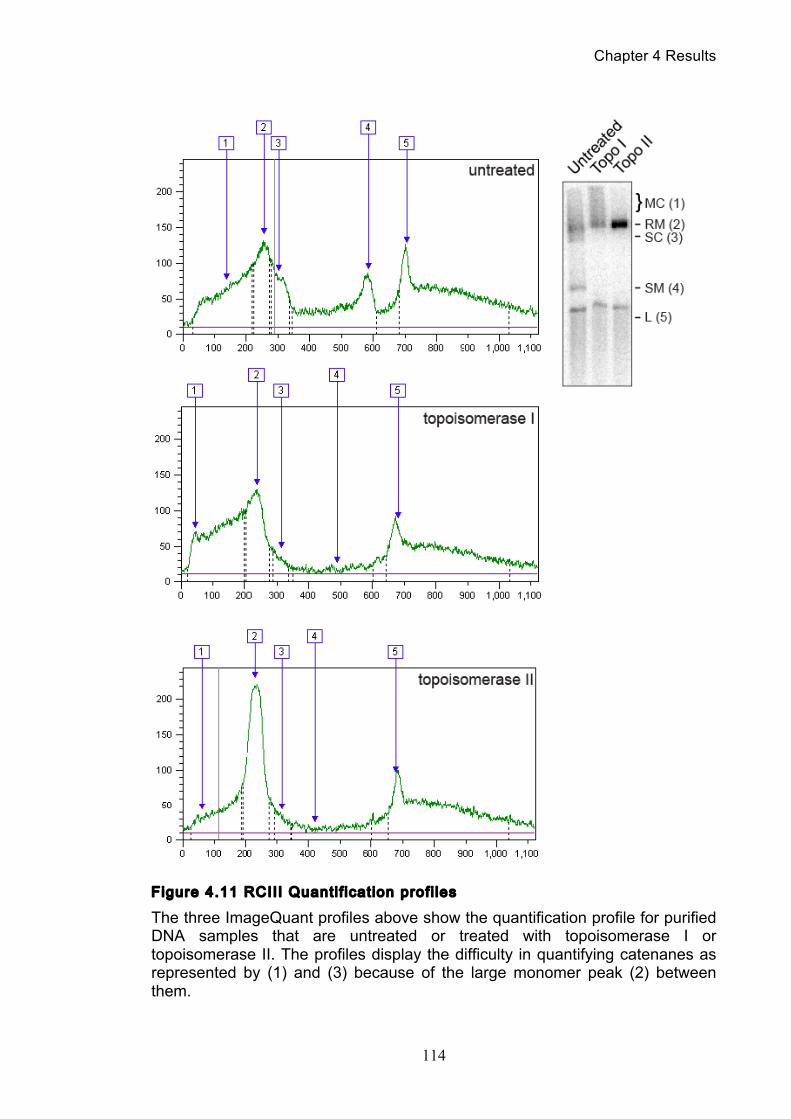
Chapter 4 Results
114
Figure 4.11 RCIII Quanti f icat ion profi les
The three ImageQuant profiles above show the quantification profile for purified DNA samples that are untreated or treated with topoisomerase I or topoisomerase II. The profiles display the difficulty in quantifying catenanes as represented by (1) and (3) because of the large monomer peak (2) between them.
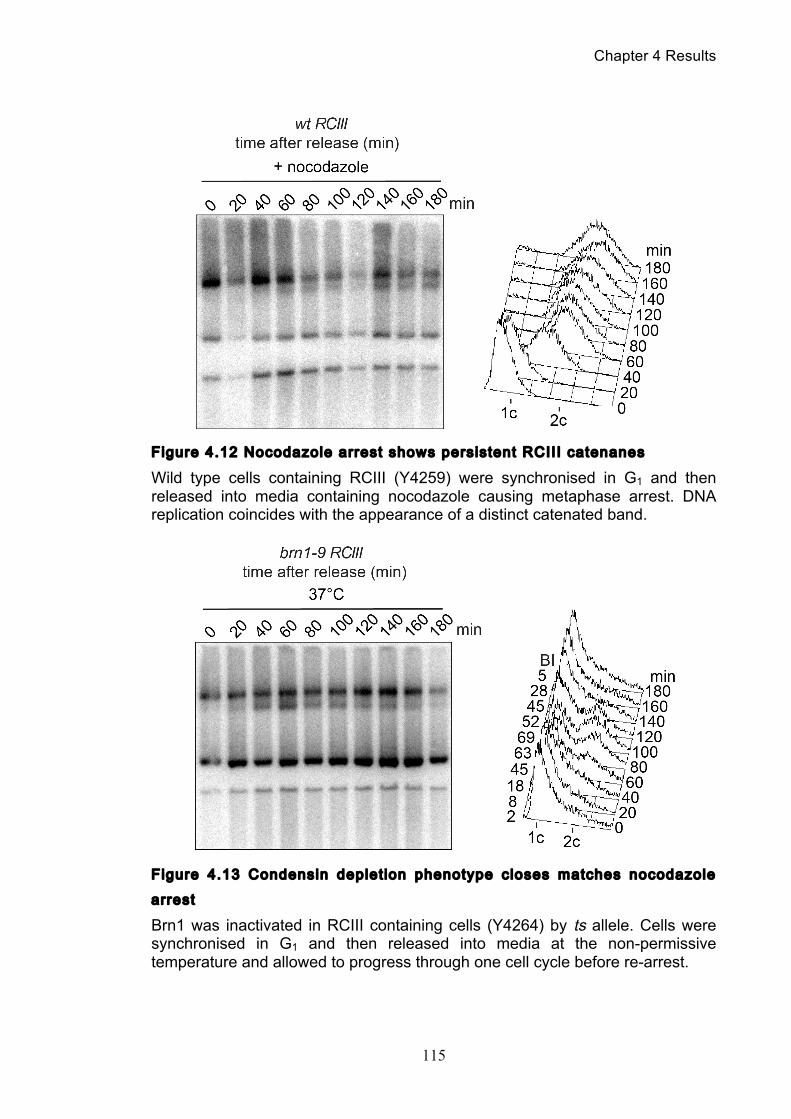
Chapter 4 Results
115
Figure 4.12 Nocodazole arrest shows persistent RCIII catenanes
Wild type cells containing RCIII (Y4259) were synchronised in G1 and then released into media containing nocodazole causing metaphase arrest. DNA replication coincides with the appearance of a distinct catenated band.
Figure 4.13 Condensin deplet ion phenotype closes matches nocodazole
arrest
Brn1 was inactivated in RCIII containing cells (Y4264) by ts allele. Cells were synchronised in G1 and then released into media at the non-permissive temperature and allowed to progress through one cell cycle before re-arrest.
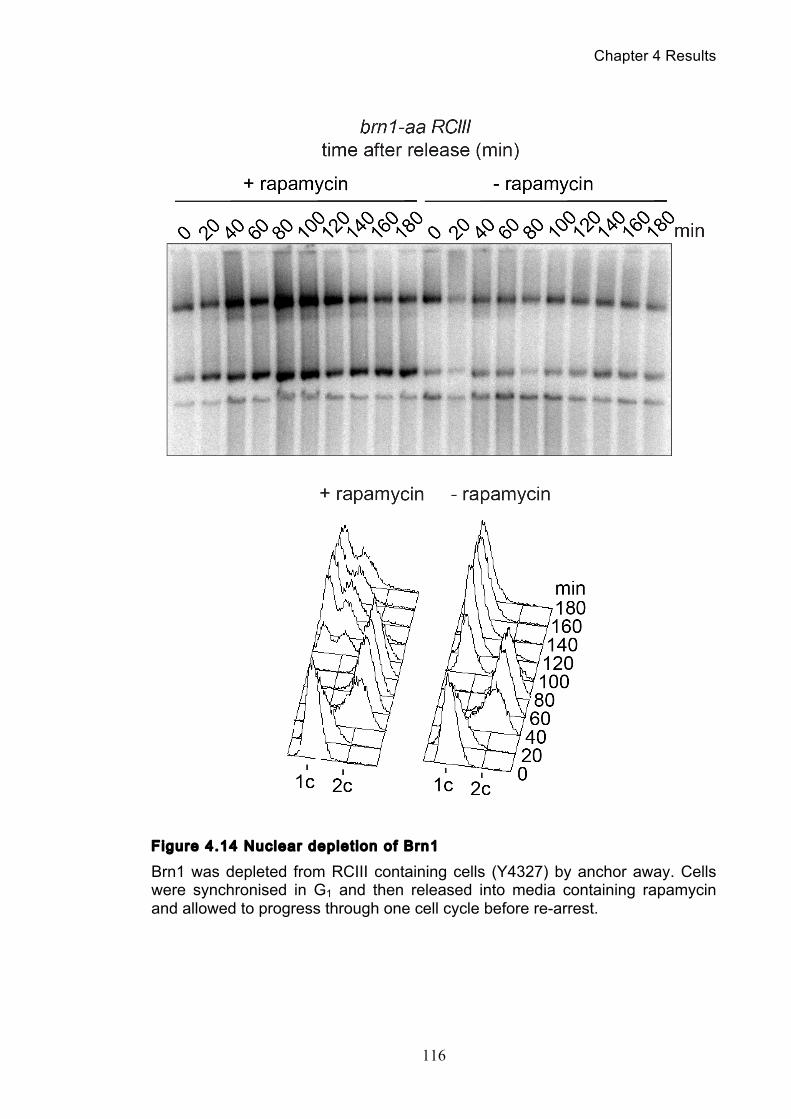
Chapter 4 Results
116
Figure 4.14 Nuclear deplet ion of Brn1
Brn1 was depleted from RCIII containing cells (Y4327) by anchor away. Cells were synchronised in G1 and then released into media containing rapamycin and allowed to progress through one cell cycle before re-arrest.

Chapter 4 Results
117
At this point, we had shown that condensin is required for proper and complete
removal of topological links between sister chromatids and consequently, depletion
of condensin results in chromosome segregation defects. Therefore the next crucial
issue to address was why this occurs. As discussed in the introduction, condensin
itself is incapable of resolving topological linkages and this must be accomplished
by the enzyme topoisomerase II. The question thus remains, how does the removal
of condensin affect topoisomerase II, resulting in unresolved catenation? In the
next results chapter, we will share the results of our investigations into the
mechanics of the relationship between condensin and topoisomerase II.

Chapter 5. Results
118
Chapter 5. Results: Investigating Interactions
5.1 Do condensin and topoisomerase II directly interact?
In order to elucidate the mechanism by which condensin promotes sister
chromatid decatenation, we asked whether we could detect a direct protein
interaction between condensin and topo II. We fused the endogenous TOP2 gene,
encoding topo II, to a Pk epitope tag to facilitate immunoprecipitation and detection
(Craven et al., 1998). The condensin subunit Brn1 was fused to an HA epitope.
Brn1-HA was efficiently recovered in topo II-Pk immunoprecipitates, but not in a
control immunoprecipitate from a strain lacking the topo II Pk tag (Figure 5.1A).
This suggests that condensin interacts with topo II in yeast cell extracts.
However, doing the co-precipitation experiments by just using cell lysate
could be generating false positives. As has been shown previously in the lab
(D'Ambrosio et al., 2008b), the distribution of condensin and topoisomerase II is
very similar along the DNA. Therefore, positive results for co-precipitation could be
generated not by genuine interaction between the proteins, but instead by
condensin and topoisomerase II both binding the same fragments of DNA in close
proximity, effectively co-precipitating them via their association with the same
stretch of DNA.
To overcome this potential false positive, we used a technique that more
gently lysed the cells and allowed us to pellet the chromatin without fragmenting
the DNA. In this way we collected the supernatant fraction, which would contain no
DNA, and using this fraction only, tested for the interaction of condensin and
topoisomerase II (Figure 5.1B). In this experiment, we now performed a Brn1-HA
immunoprecipitation. However, in the HA pull-down, neither Brn1-HA nor Top2-Pk
was recovered. It seems that protein stability was compromised in this type of
extract.
To test this possibility, we returned to our original method of lysing the cells
such that DNA was present in the whole cell extract and could mediate any
possible interaction between the two proteins. However, we required an additional
control to ensure that the original interaction observed was specific, and not just a
consequence of the abundance of the two proteins. For this, we included the
additional control of tagging cohesin (specifically Scc1) with the same HA epitope

Chapter 5. Results
119
tag that Brn1 was tagged with. By performing a topo II-Pk immunoprecipitation, we
could see if the enzyme pulled down both proteins with it, or specifically just one
(Figure 5.1C). Examining the results, we could again clearly detect condensin in
the pull-down but we saw cohesin being pulled down as well. However, the single-
tagged Scc1-HA6 control strain also exhibited a faint band in the pull down,
indicating that there was some unspecific binding of cohesin by the beads. Despite
further repeats, we were unable to get rid of the background cohesin binding by the
beads, leaving us unable to make a clear decision on the nature of interaction
between condensin and topoisomerase II. At this point, it seemed clear that we
would be unable to obtain much more information from these sets of co-
immunoprecipitation experiments, so we considered how else biochemically we
could investigate their interaction.
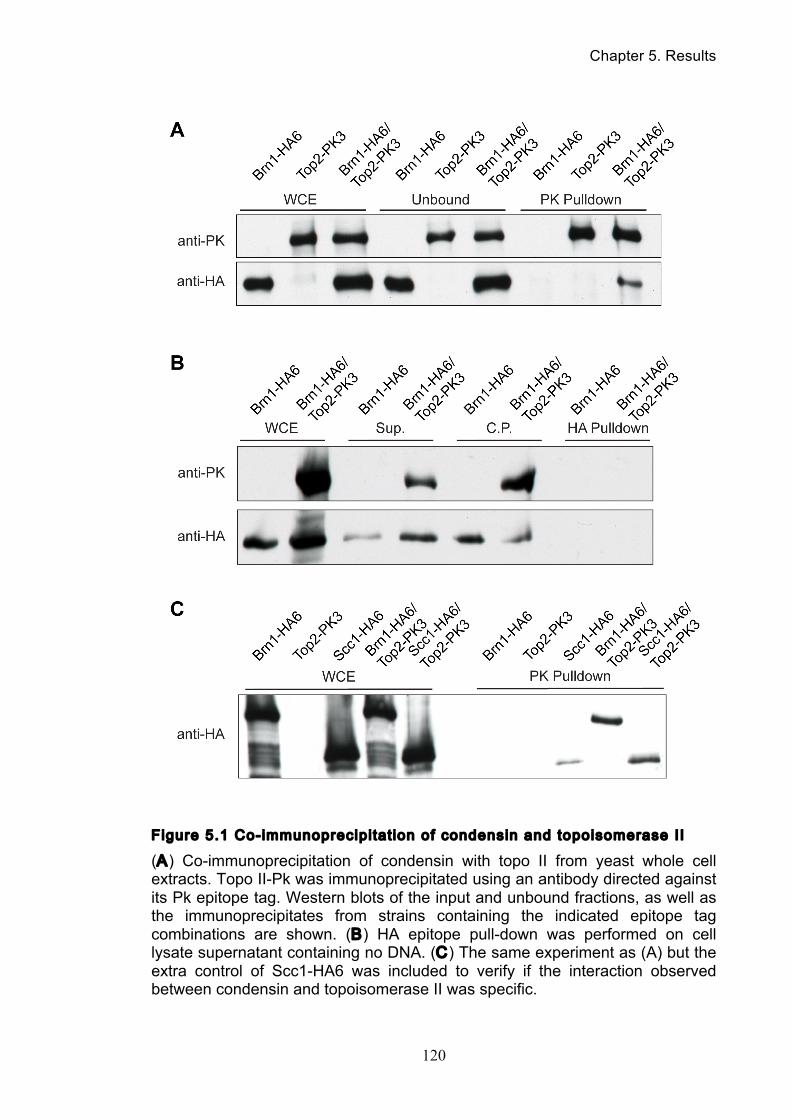
Chapter 5. Results
120
Figure 5.1 Co-immunoprecipitat ion of condensin and topoisomerase II
(A) Co-immunoprecipitation of condensin with topo II from yeast whole cell extracts. Topo II-Pk was immunoprecipitated using an antibody directed against its Pk epitope tag. Western blots of the input and unbound fractions, as well as the immunoprecipitates from strains containing the indicated epitope tag combinations are shown. (B) HA epitope pull-down was performed on cell lysate supernatant containing no DNA. (C) The same experiment as (A) but the extra control of Scc1-HA6 was included to verify if the interaction observed between condensin and topoisomerase II was specific.

Chapter 5. Results
121
5.2 Purif ication of topoisomerase II and condensin
To really be able to determine whether and how condensin and topo II
interact, we decided to investigate whether biochemically purified condensin and
topo II interact. To do this, we first purified topo II from yeast cells overexpressing
the enzyme, following a published protocol (Worland and Wang, 1989). In Figure
5.2 we can see the steps of this effective purification technique, which yielded
enzyme preparations in which topo II was the only protein detectable by
Coomassie blue staining and was functionally active in trial decatenation and
relaxation assays.
For the purification of condensin, the protein was affinity purified from yeast
strain overexpressing the five condensin subunits (St-Pierre et al., 2009). Initially
we attempted to purify condensin using the two-step purification protocol detailed in
their paper, but had faced problems with low yields of condensin that consisted of
subunits that were not stoichiometrically correct. In collaboration with Céline
Bouchoux in the lab, we were instead able to purify condensin in a one step
procedure using an anti-HA affinity matrix that specifically bound condensin.
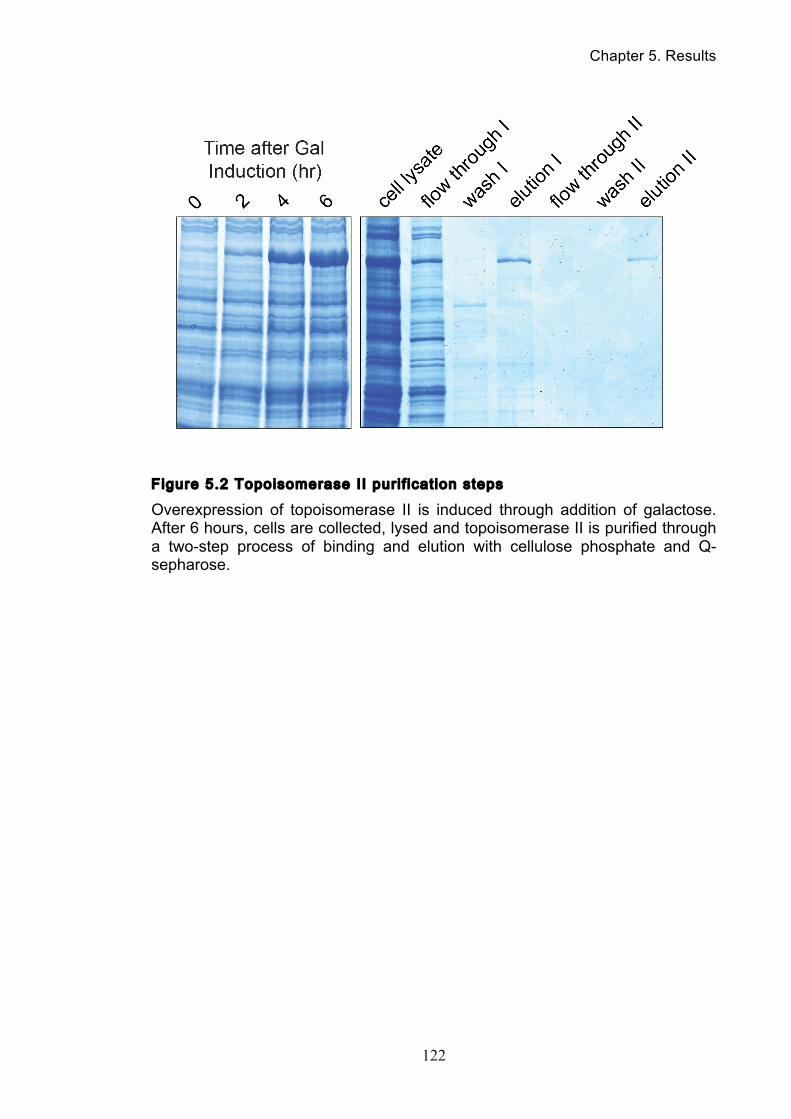
Chapter 5. Results
122
Figure 5.2 Topoisomerase II purif icat ion steps
Overexpression of topoisomerase II is induced through addition of galactose. After 6 hours, cells are collected, lysed and topoisomerase II is purified through a two-step process of binding and elution with cellulose phosphate and Q-sepharose.

Chapter 5. Results
123
5.3 Condensin and topoisomerase II directly interact
Having purified both protein complexes, we were now able to determine the
nature of their interaction. To do this, condensin was bound via an HA epitope-
tagged Brn1 subunit to α-HA antibody-coupled affinity beads, then purified topo II
was added (Figure 5.3). Before performing the pull-down, both preparations
underwent nuclease treatment. As a control, we added topo II to the α-HA affinity
beads in the absence of condensin. After washes, bound proteins were eluted and
analyzed. Topo II was efficiently recovered from condensin-bound, but not control,
beads. This suggests that budding yeast topo II has a direct and physical
interaction with condensin.
5.4 Condensin stimulates in vitro DNA decatenation by
topo II
Given the physical interaction between condensin and topo II, we wondered
whether condensin has a direct impact on the ability of topo II to resolve catenated
DNA. As an assay to measure the decatenation activity of topo II, we used
kinetoplast DNA (kDNA) as the substrate (Marini et al., 1980). kDNA is a
mitochondria-derived network of intercatenated DNA minicircles of approximately
2.5 kb in size. Prior to decatenation, kDNA will remain in the wells during
electrophoresis, owing to its high aggregate molecular weight. In the presence of
topo II, decatenated minicircles are released that enter the gel. Incubation with our
purified yeast topo II preparation led to efficient kDNA decatenation (Figure 5.4).
DNA decatenation by topo II is an ATP-dependent reaction, and omission of ATP
from the incubation prevented minicircle release. This serves as a control to ensure
that we are observing topo II-associated decatenation activity and that gel entry is
not due to a contaminating nuclease activity.
To study the effect of condensin on decatenation, we reduced the topo II
concentration in the reaction such that, in the absence of condensin, approximately
10% of the kDNA substrate was decatenated during the incubation. Addition of
increasing concentrations of condensin led to a marked, dose-dependent,
stimulation of kDNA decatenation. In the presence of 20 nM condensin,

Chapter 5. Results
124
decatenation was stimulated by almost 3-fold compared to the reaction lacking
condensin. Incubation of kDNA with condensin in the absence of topo II did not
result in minicircle release. This suggests that condensin stimulates kDNA
decatenation by topo II.
The reaction mix in which we observed the greatest stimulation of
decatenation by condensin contained approximately equimolar amounts of kDNA,
topo II and condensin. Considering that condensin and topo II interact, condensin
might therefore directly stimulate the catalytic activity of topo II by binding to it.
Alternatively, condensin might introduce a conformational change to the DNA
substrate that facilitates its decatenation by topo II. To differentiate between these
possibilities, we investigated whether the ability of budding yeast condensin to
enhance kDNA decatenation is restricted to topo II from the same species, or
whether it extends to type 2 topoisomerases from other organisms. We used
commercial preparations of human topo IIα and E. coli topo IV in this comparison
(Figure 5.5). Topoisomerase concentrations were again titrated such that in the
absence of condensin approximately 10% of the kDNA substrate was resolved.
Addition of condensin in each case led to a reproducible stimulation of the kDNA
decatenation reaction. Human topo IIα shows 49% sequence identity and can
functionally complement the essential in vivo function of yeast topo II (Jensen et al.,
1996). A protein interaction between the two proteins could therefore be conserved
between the species. This is less likely the case for E. coli topo IV, even though it’s
parE subunit shows 24% identity and 43% similarity to the N-terminal half of
budding yeast topo II. Condensin might thus promote decatenation both by
establishing a DNA substrate geometry that facilitates decatenation as well as
recruit or activate topo II to assist the decatenation reaction.

Chapter 5. Results
125
Figure 5.3 Condensin interacts direct ly with purif ied topoisomerase II
Purified topo II binds to condensin that is retained on an antibody affinity matrix. Purification and interaction analysis was performed as described in Materials and Methods, the input and eluates were analyzed by SDS-PAGE and proteins visualised by Coomassie blue staining.
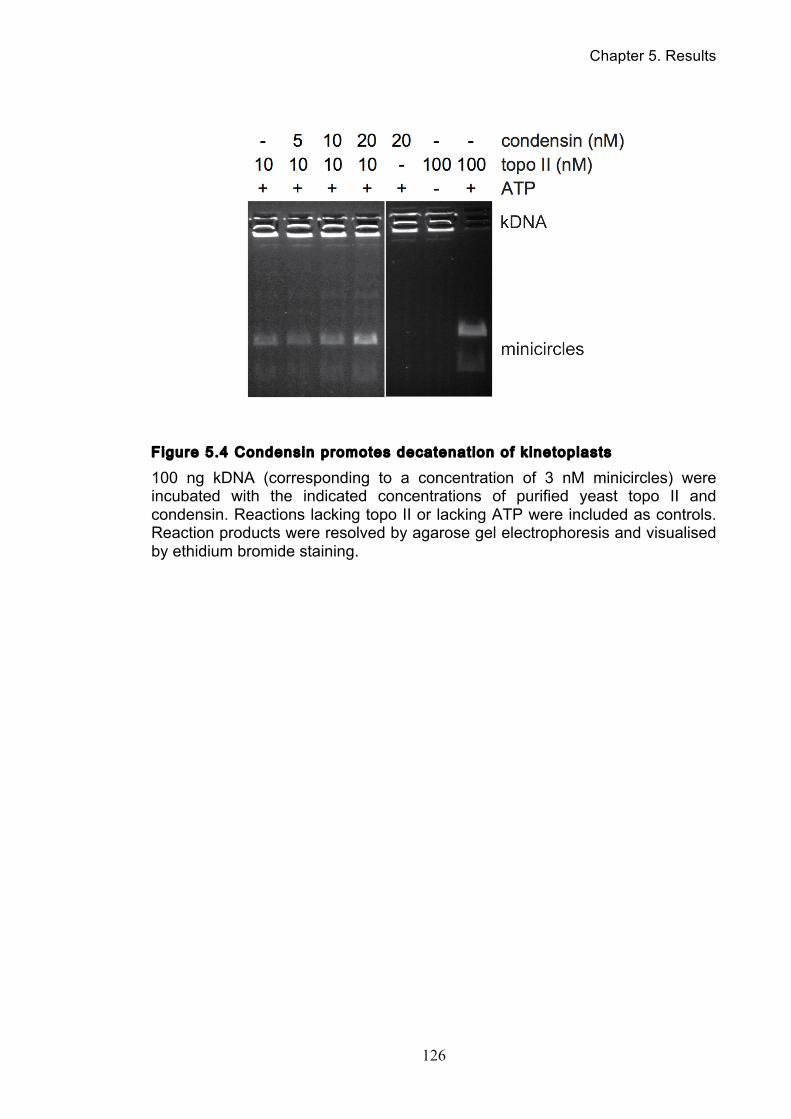
Chapter 5. Results
126
Figure 5.4 Condensin promotes decatenation of kinetoplasts
100 ng kDNA (corresponding to a concentration of 3 nM minicircles) were incubated with the indicated concentrations of purified yeast topo II and condensin. Reactions lacking topo II or lacking ATP were included as controls. Reaction products were resolved by agarose gel electrophoresis and visualised by ethidium bromide staining.
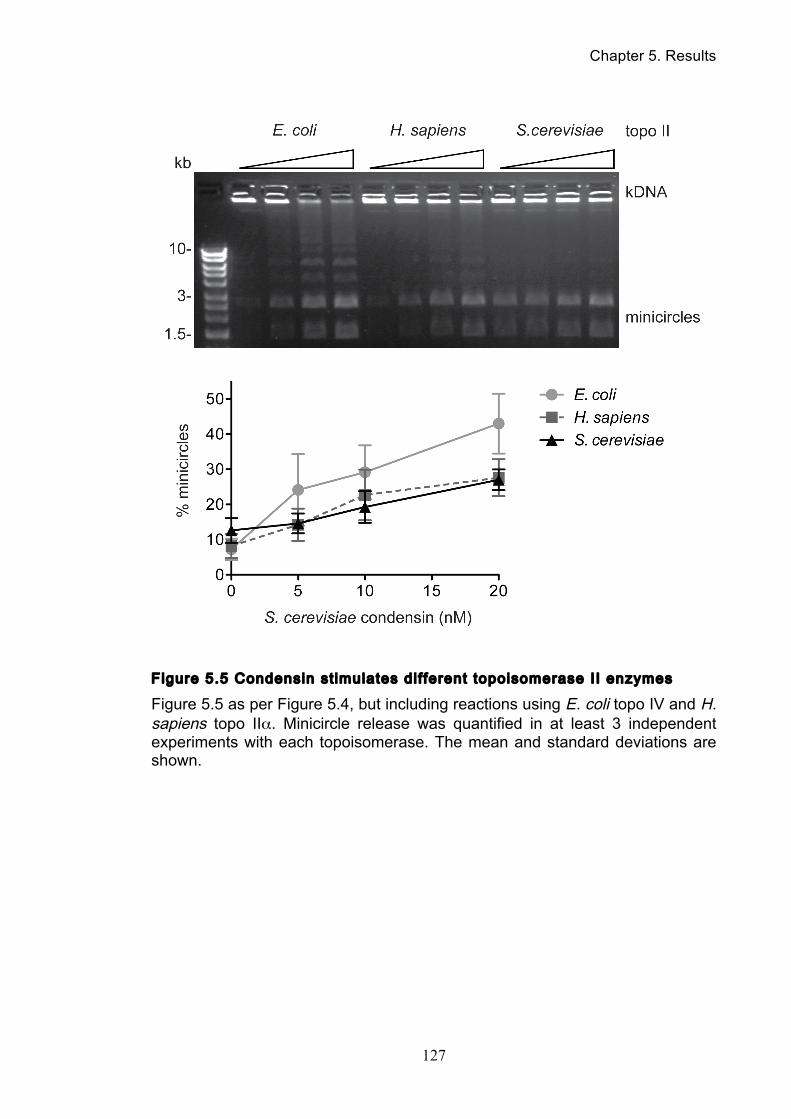
Chapter 5. Results
127
Figure 5.5 Condensin st imulates dif ferent topoisomerase II enzymes
Figure 5.5 as per Figure 5.4, but including reactions using E. coli topo IV and H. sapiens topo IIα. Minicircle release was quantified in at least 3 independent experiments with each topoisomerase. The mean and standard deviations are shown.

Chapter 6. Results
128
Chapter 6. Results: a-factor Synthesis and
Characterization
6.1 Synthesis of a-factor
As was introduced in chapter 1.7, the anchor away strains we used were
mostly mating type α , which meant that to synchronise the cells in G1 the mating
pheromone a-factor had to be used. However, while α-factor is readily produced by
peptide synthesis laboratories in many research institutes as well as being
commercially available, a-factor is not. This is because α-factor lacks any peptide
modifications while a-factor has both farnesylation and carboxyl methylation, both
of which are important for the biological potency of a-factor (Anderegg et al., 1988)
(Marcus et al., 1991). This makes α-factor readily accessible by automated solid-
phase peptide synthesis and much easier to manufacture. While over the years
many a-factor synthesis strategies have been published, they typically involve
liquid-phase reactions that are a hallmark of expert organic chemistry laboratories
and thus out of reach for many users who do not have access to such expertise
(Xue et al., 1989) (Sherrill et al., 1995) (Mullen et al., 2011).
In collaboration with the Peptide Synthesis laboratory at our institute, a
novel strategy for the synthesis of a-factor was developed. The process is based
on solid-phase peptide synthesis, followed by two simple steps in solution using
widely available reagents and apparatus, thus making a-factor accessible to
laboratories with standard peptide synthesis facilities (O'Reilly et al., 2012). Our
contribution was to demonstrate the successful use of our synthetic a-factor to
synchronise cell cycle progression of cells of the α mating type.
6.2 Use of a-factor to synchronise cells of α mating type
Initially we tried to synchronise haploid cells of the α mating type, using a-
factor concentrations and conditions similar to what is used for α-factor
synchronization of a mating type cells. Adding a-factor at a concentration of 0.5
µg/ml to asynchronously growing cells in early exponential phase led to efficient
cell cycle arrest in G1. However, upon filtration and washout of the a-factor, these

Chapter 6. Results
129
cells resumed cell cycle progression only later and very inefficiently. We therefore
reduced the a-factor concentration through a series of optimisation experiments.
Eventually we identified that addition of a total concentration of 0.04 µg/ml a-factor,
in two halves at the beginning and after 1 h of arrest, efficiently arrested cells in G1.
Washout of the a-factor and resuspension in fresh pheromone-free medium now
resulted in synchronous release from the arrest (Figure 6.1). We followed cell cycle
progression by FACS analysis of DNA content, as well as by scoring the budding
index and the fraction of binucleate cells. 60 minutes after release, when most cells
showed a new bud, we added a-factor back to the culture to re-arrest cells after
completion of mitosis in the following G1. This analysis showed that arrest of α cells
with a-factor and release by filtration gives rise to a cell population that transverses
the cell cycle with very good synchrony.
The a-factor concentration we used in this protocol is 10-fold lower than the
concentration of 2-factor that we routinely use to synchronise a mating type cells. It
is 100-fold lower than recommended α-factor concentrations in some
synchronization protocols (Breeden, 1997). When we tried to use α-factor at a low
concentration to arrest a mating type cells, we found that 0.04 µg/ml α-factor was
not sufficient to impose a stable cell cycle arrest. We do not know the reason for
the greater specific biological activity of a-factor as compared to α-factor. Its
hydrophobic nature and farnesylation might enhance its affinity for lipid membranes
and thereby increase its local concentration close to the target cell a-factor receptor.
In addition, the absence of a known a-factor protease, equivalent to the Bar1 α-
factor protease, may mean that a-factor is more stable in the culture medium or in
the vicinity of the receptor.
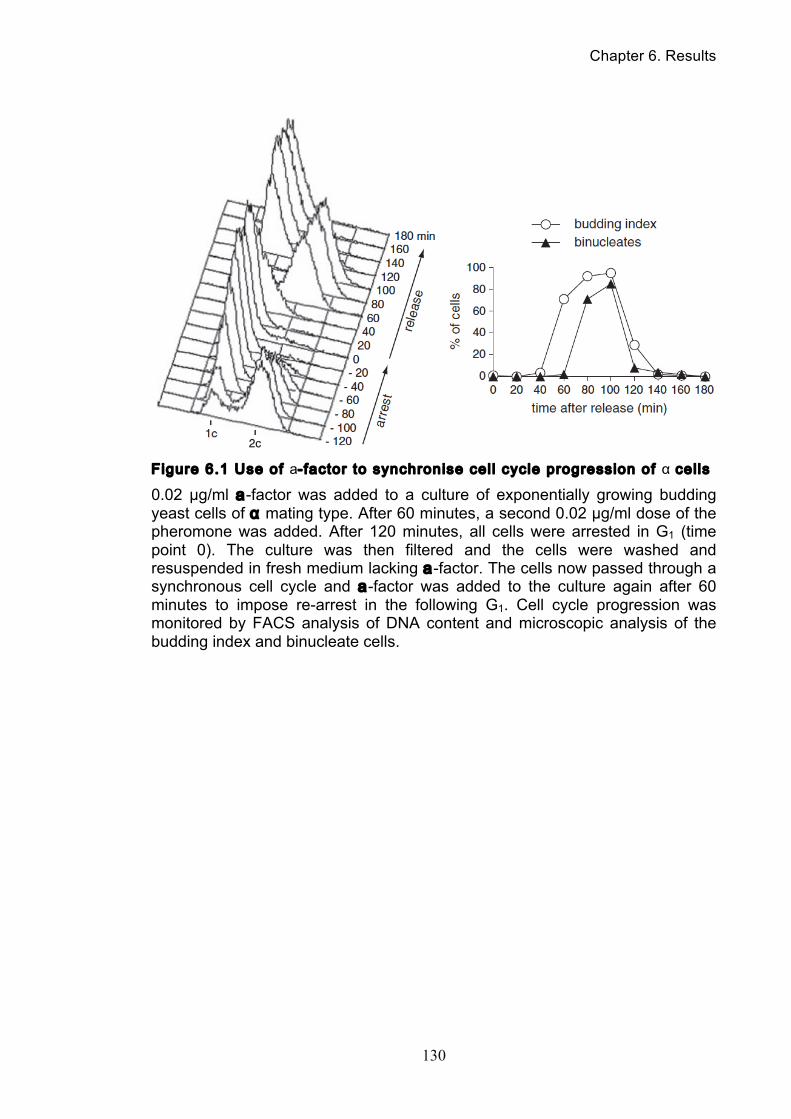
Chapter 6. Results
130
Figure 6.1 Use of a-factor to synchronise cel l cycle progression of α cel ls
0.02 µg/ml a-factor was added to a culture of exponentially growing budding yeast cells of α mating type. After 60 minutes, a second 0.02 µg/ml dose of the pheromone was added. After 120 minutes, all cells were arrested in G1 (time point 0). The culture was then filtered and the cells were washed and resuspended in fresh medium lacking a-factor. The cells now passed through a synchronous cell cycle and a-factor was added to the culture again after 60 minutes to impose re-arrest in the following G1. Cell cycle progression was monitored by FACS analysis of DNA content and microscopic analysis of the budding index and binucleate cells.

Chapter 6. Results
131
6.3 Litt le shmoo formation during a-factor-induced G1
arrest
When we microscopically followed the a-factor response during cell
synchronization, we noticed that even though cells arrested unbudded, they
showed hardly any signs of shmoo formation. This was unlike α-factor-treated a
mating type cells, where G1 arrest is typically accompanied by noticeable shmoo
formation (Figure 6.2). In our cultures, α cells treated with synthetic a-factor lost
their oval shape and took on a more rounded, sometimes lemon-shaped,
appearance. The cell volume increased over time in the arrest, but directed shmoo
formation was barely observed. This was even the case at higher pheromone
concentrations, similar to those that elicit efficient shmoo formation of α-factor-
treated a mating type cells (Figure 6.2). While it has been previously noticed that α
cell shmoos differ from the typical a cell projections (Wilkinson and Pringle, 1974)
(Betz et al., 1977), our observations suggest that as yet poorly understood
differences exist in the way the opposite mating pheromones act on yeast cells, or
in the way that cells of the opposite mating types react to pheromone exposure.

Chapter 6. Results
132
Figure 6.2 Comparison of pheromone-induced shmoo formation of a and α
cel ls
Haploid cells of either a or α mating type were grown in early exponential phase. Mating pheromones α- and a-factor, respectively, were added at the indicated concentrations. Cells were photographed before and 2.5 hours after pheromone addition, using a Zeiss Axioplan 2 microscope equipped with differential interference contrast optics.

Chapter 7. Discussion
133
Chapter 7. Discussion
7.1 Catenation can be observed in minichromosomes
In our experiments, we have shown the utility of our catenation assay in
observing and tracking catenanes for a range of minichromosome sizes. Despite
previous reports that replication-dependent catenation of smaller minichromosomes
was too transient to be detectable in wild type cells (Baxter et al., 2011), we could
observe catenation with all our minichromosome substrates. By careful optimisation
of our DNA purification process and electrophoresis conditions, we established
parameters that enabled us to clearly track catenane behaviour through the cell
cycle and our purified minichromosomes show distinct patterns of replication-
dependent catenation that resolve with a specific timing.
The significance of our assay results was further supported by the dual
approaches we took towards protein inactivation. By being able to inactivate
proteins by both ts allele disruption and anchor away-mediated nuclear depletion,
we were able to verify that the resultant phenotypes were indeed a specific result of
the target protein’s inactivation.
7.2 Cohesin protects catenation
When cohesin was inactivated through either use of a ts Scc1 allele or
nuclear depletion via anchor away, we saw a significant reduction in the abundance
of catenanes. During S-phase, when we normally see the greatest proportion of
catenanes, less than 20% of minichromosomes were detected as catenated. Thus
it appears that cohesin plays a substantial role in the protection of catenation.
There are differing mechanisms by which cohesin could exert this influence; an
‘indirect’ mechanism whereby cohesin might retard topo II-driven decatenation
hence maintaining topological linkages, or ‘directly’ through the entrapment of sister
DNAs within the cohesin tripartite ring. In the former mechanism, cohesin could
retard decatenation by topo II through some sort of interaction. In our co-
immunoprecipitation studies in chapter 5.1, we used cohesin as a control and saw
an unexpected interaction between Scc1 and Top2, which suggests that some

Chapter 7. Discussion
134
direct interaction between the two proteins is at least feasible. Another possibility is
that the cohesin complex blocks topo II’s access to sites of intertwining, inhibiting
decatenation. As the cohesin tripartite ring physically entraps the sister DNAs, it is
possible that the sites of entrapment coincide with where the sister DNAs are
physically closest, or catenated. The presence of cohesin could therefore block
access until the time it is cleaved by separase.
However, if we take into account the effect of other mitotic components on
catenation, it seems that cohesin may play a more indirect role. If we compare
catenane levels between cells arrested in metaphase either by nocodazole or by
inactivation of Cdc20, we can see that there is a marked decrease in catenanes
when the spindle is present. In both situations cohesin is still present and
uncleaved, yet the presence of the mitotic spindle seems to promote decatenation,
implying that the cohesin complex isn’t blocking topo II. Thus, cohesin could be
playing a more direct role in maintaining catenation. We hypothesize that while
topo II is able to access and act upon the sister DNAs, prior to cohesin cleavage,
those sister DNAs will remain physically intimate owing to their entrapment by the
cohesin ring. Therefore, the reintroduction of topological linkages owing to the bi-
directionality of topo II is possible. As long as cohesin remains uncleaved, an
equilibrium of decatenation and recatenation will be established between sister
DNAs. The influence of other factors, such as the tension exerted by the mitotic
spindle or the cleavage of cohesin allowing sister DNAs to move away from each
other, will alter the balance of that equilibrium. A recent study has come to a similar
conclusion on cohesin’s catenane-protection properties (Farcas et al., 2011).
7.3 Anaphase bridges result from persistent sister
chromatid catenanes
Chromosome segregation failure and anaphase bridges are a hallmark
phenotype of cells with compromised condensin function, yet prior to our study, the
reason underlying this failure has remained undetermined. We now provide
evidence that, at least in the case of circular minichromosomes in budding yeast,
condensin is required to promote complete resolution of catenanes that are
retained between sister chromatids following their replication. Persistent sister

Chapter 7. Discussion
135
chromatid catenanes have previously been proposed as a source for anaphase
bridges in a condensin mutant (Bhat et al., 1996), though it is to our knowledge the
first time that these catenanes have been directly observed. An important
prediction from a model where sister chromatid catenation prevents chromosome
segregation is that ectopic decatenation should resolve the anaphase bridges in
the absence of condensin. This has been achieved in case of the budding yeast
rDNA locus where anaphase bridges in a condensin mutant were resolved by
ectopic expression of a viral topo II enzyme. Though again the catenations status
of the locus could not be observed at the time (D'Ambrosio et al., 2008a). Taken
together with the direct visualization of persistent catenanes between circular
chromosomes that we now report, we can suggest the chromosome bridges that
arise when condensin function is compromised are indeed due to persistent DNA
catenation.
7.4 Condensin promotes decatenation by topoisomerase
II
An important area for future research is to investigate whether catenation
between linear chromosomes is regulated in the same way as we have observed
here in the case of circular substrates. Catenation, in theory, should not exist
between linear pieces of DNA, as any intertwining could easily slide off the DNA
ends. This is a reason why catenation of linear chromosomes cannot be studied
after DNA isolation by gel electrophoresis. In vivo, however, chromosome
movement is constrained such that catenation between linear sister chromatids is
maintained and has the ability to restrain chromosome segregation during
anaphase (Holm et al., 1985, Uemura et al., 1987b). Whether, and how far,
topological links between sister chromatids might be able to translocate along DNA
strands is not known. Helical tension along individual sister strands is constrained
to regions of approximately 100 kb in budding yeast (Joshi et al., 2010). It seems
likely that sister chromatid catenation is similarly constrained. Topologically isolated
domains along chromosomes are probably the consequence of protein-mediated
DNA interactions, as has been demonstrated at least in the case of mitotic
chromosomes (Kawamura et al., 2010). How topological interlinks between linear

Chapter 7. Discussion
136
chromosomes arise, where and for how long they are maintained, and how they
are eventually resolved are important question. To address these will likely require
the topological isolation of sections of linear chromosomes by excision in a circular
form to facilitate their analysis.
7.5 Decatenation occurs in steps
While studying the sister chromatid catenation status of circular
chromosomes during the cell cycle, we found that most catenanes are rapidly
resolved by topo II soon after DNA replication. When topo II was active, at most
half of the chromosomes were seen catenated in our synchronised cell population
at any one time. Only 10-20% catenanes persisted until mitosis, or in the absence
of cohesin when proteinaceous sister links were removed. This fraction is similar to
the fraction of catenanes that persisted throughout mitosis and into G1 of the next
cell cycle in cells depleted of condensin. This raises the question whether there is a
distinction between catenanes that are readily resolved and those that are
decatenated later in the cell cycle and require condensin for their resolution? We
can see two possible scenarios. In the first, all replication products are left
catenated in the same way, possibly by a small number of interlinks per replication
termination site. Topo II will act on these based on chance encounters sister DNA
strands. Based on its mode of DNA binding, topo II favors decatenation over
catenation at such crossing points (Vologodskii et al., 2001, Dong and Berger,
2007). Resolution is limited in this scenario by Brownian movement of sister
chromatids, constrained by cohesin-dependent cohesion. Condensin’s role would
be to accelerate resolution by stimulating topo II activity and promoting a substrate
geometry conducive to decatenation. In an alternative scenario, topological
interlinks between sister chromatids might exist in different forms. Depending on
the reactions involved in replication termination and constraints imposed by the
chromosome environment the catenanes produced might differ in the number of
interlinks or additional topological features, e.g. knots. Simple catenanes could be
good substrates for rapid decatenation, while condensin would aid the resolution of
more complex topologies. In S. cerevisiae, the rDNA shows the greatest resolution
problems in condensin mutants (D'Ambrosio et al., 2008a). This area of DNA is

Chapter 7. Discussion
137
highly transcribed, even during mitosis, possibly generating topologically complex
catenanes that benefit more from condensin activity. Condensin binding to DNA
introduces positive-handed writhe, which in the presence of topo II allows knot
formation (Kimura et al., 1999). Equally, by stabilizing transient DNA conformations
that rarely form spontaneously, condensin might facilitate resolution of complex
structures. In support of this possibility, the fraction of catenanes that require
condensin for resolution appear to be refractory to decatenation in its absence and
persist for extended time periods. Such structures might also pose the greatest
danger to chromosome segregation.
7.6 Condensin directly interacts with topoisomerase II
How does condensin promote sister chromatid decatenation? As discussed
above condensin might act at the level of the DNA substrate to facilitate catenane
resolution, maybe in particular those of a complex topological nature. Our
observation that budding yeast condensin facilitates the in vitro decatenation of
kDNA circles not only by budding yeast topo II but also by topoisomerases of
human and bacterial origin is consistent with such an activity. In addition to
affecting DNA conformation, we also observed a direct protein interaction between
budding yeast condensin and topo II. Such an interaction could directly stimulate
the catalytic activity of topo II as has been recently observed in the bacterial system
(Li et al., 2010, Hayama and Marians, 2010). Yet another way how condensin’s
interaction with topo II could facilitate sister chromatid decatenation is by
recruitment of topo II to its sites of action. Condensin and topo II show significant
overlap in their chromosomal association pattern (D'Ambrosio et al., 2008b),
consistent with a role in topo II recruitment.
7.7 Future perspectives
Our catenation assay has proved to be a useful and flexible tool in our
investigation, having produced an array of interesting results. Coupled with our in
vitro biochemical experiments using purified proteins, we have been able to make
some novel insights into condensin’s and topoisomerase II’s role in chromosome

Chapter 7. Discussion
138
resolution. However, looking to the future we believe that the next logical step
would be to move on from using minichromosomes as models and to develop
techniques to study catenation on the native chromosomes themselves. We believe
that we have reached the limit of what can be accurately observed using
minichromosomes. In our experiments, as minichromosome size increased so did
linearization of the substrate, most likely owing to an increased vulnerability to
pipette-induced shearing. We found that by minimizing the steps in our purification
protocol that involved pipette use, as well as implementing other purification
optimizations, we could minimise the amount linearised. However, it is likely that
our assay has pushed the limits of what is observable with large sized
minichromosomes. RCIII, being over 60 kb in size, already presented many
difficulties in resolution and was very susceptible to damage. While we found that
pulse field gel electrophoresis did not yield satisfactory resolution for our assay, it is
likely that any larger substrates would have to be resolved via this technique.
However, we feel that the next logical step for future investigations would be the
formation of a technique that could manipulate the native chromosomes
themselves, a direction that some labs have already tentatively begun moving in
(Farcas et al., 2011). It is feasible that native chromosomes could be engineered
such that sections could be cut out and ligated in vivo to create circular DNAs that
are easily assayable by adapting our current protocols, perhaps through adaption
of the Cre/lox recombinase system commonly used in mammalian and plant cells
(Figure 7.1). With the availability of condensin binding maps, replication origins and
knowledge of areas of potential catenation, interesting sites could be marked for
excision and study finally allowing for the direct observation of endogenous
chromosomes.
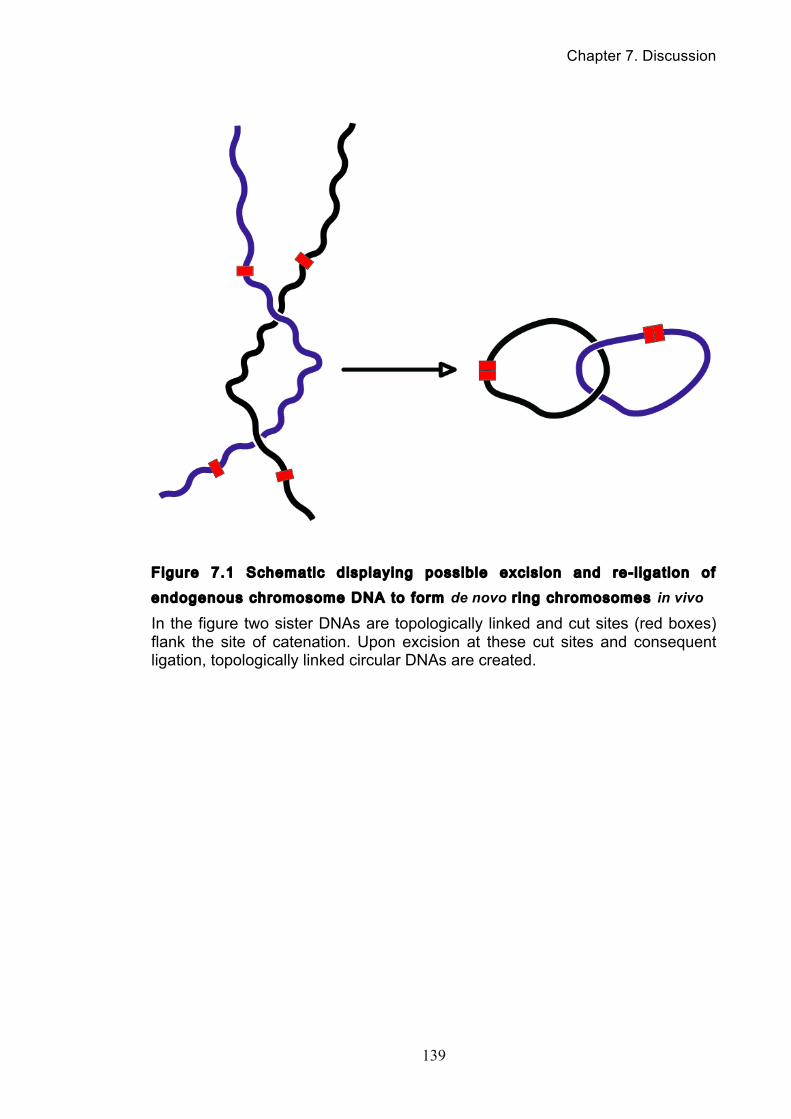
Chapter 7. Discussion
139
Figure 7.1 Schematic displaying possible excision and re-l igation of
endogenous chromosome DNA to form de novo r ing chromosomes in vivo
In the figure two sister DNAs are topologically linked and cut sites (red boxes) flank the site of catenation. Upon excision at these cut sites and consequent ligation, topologically linked circular DNAs are created.

Chapter 7. Discussion
140
We are still very naïve about how cells deal with the immense topological
challenge of replicating millimeter-long chromosomes in micrometer-sized nuclei
and ensuring the smooth resolution of DNA interlinks during the segregation of
sister chromatids during mitosis. Cell division underpins the propagation of all life
on earth and unraveling this fundamental process is both fascinating and essential.
Chromosome resolution and segregation lies at the heart of human growth,
reproduction and regeneration and the danger of persisting anaphase bridges to
genome stability makes this an important area of further study.

Reference List
141
Reference List
ADOLPHS, K. W., CHENG, S. M., PAULSON, J. R. & LAEMMLI, U. K. 1977. Isolation of a protein scaffold from mitotic HeLa cell chromosomes. Proc Natl Acad Sci U S A, 74, 4937-41.
AGUILERA, A. & KLEIN, H. L. 1990. HPR1, a novel yeast gene that prevents intrachromosomal excision recombination, shows carboxy-terminal homology to the Saccharomyces cerevisiae TOP1 gene. Mol. Cell. Biol., 10, 1439-1451.
AKAI, Y., KUROKAWA, Y., NAKAZAWA, N., TONAMI-MURAKAMI, Y., SUZUKI, Y., YOSHIMURA, S. H., IWASAKI, H., SHIROIWA, Y., NAKAMURA, T., SHIBATA, E. & YANAGIDA, M. 2011. Opposing role of condensin hinge against replication protein A in mitosis and interphase through promoting DNA annealing. Open Biol, 1, 110023.
ALEXANDRU, G., UHLMANN, F., POUPART, M.-A., MECHTLER, K. & NASMYTH, K. 2001. Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc5 kinase regulates sister chromatid separation in yeast. Cell, 105, 459-472.
ANDEREGG, R. J., BETZ, R., CARR, S. A., CRABB, J. W. & DUNTZE, W. 1988. Structure of Saccharomyces cerevisiae mating hormone a-factor: Identification of S-farnesyl cysteine as a structural component. J. Biol. Chem., 263, 18236-18240.
ANDERSON, D. E., LOSADA, A., ERICKSON, H. P. & HIRANO, T. 2002. Condensin and cohesin display different arm conformations with characteristic hinge angles. J Cell Biol, 156, 419-24.
AONO, N., SUTANI, T., TOMONAGA, T., MOCHIDA, S. & YANAGIDA, M. 2002. Cnd2 has dual roles in mitotic condensation and interphase. Nature, 417, 197-202.
BÄHLER, J., WU, J. Q., LONGTINE, M. S., SHAH, N. G., MCKENZIE III, A., STEEVER, A. B., WACH, A., PHILIPPSEN, P. & PRINGLE, J. R. 1998. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast, 14, 943-951.
BAUER, W. R., CRICK, F. H. & WHITE, J. H. 1980. Supercoiled DNA. Sci Am, 243, 100-13.
BAXTER, J. & ARAGON, L. 2012. A model for chromosome condensation based on the interplay between condensin and topoisomerase II. Trends Genet, 28, 110-7.
BAXTER, J. & DIFFLEY, J. F. 2008. Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol Cell, 30, 790-802.
BAXTER, J., SEN, N., MARTÍNEZ, V. L., DE CARANDINI, M. E., SCHVARTZMAN, J. B., DIFFLEY, J. F. X. & ARAGÓN, L. 2011. Positive supercoiling of mitotic DNA drives decatenation by topoisomerase II in eukaryotes. Science, 331, 1328-1332.
BAZETT-JONES, D. P., KIMURA, K. & HIRANO, T. 2002. Efficient supercoiling of DNA by a single condensin complex as revealed by electron spectroscopic imaging. Mol. Cell, 9, 1183-1190.
BENDER, R. P., JABLONKSY, M. J., SHADID, M., ROMAINE, I., DUNLAP, N., ANKLIN, C., GRAVES, D. E. & OSHEROFF, N. 2008. Substituents on etoposide that interact with human topoisomerase IIalpha in the binary enzyme-drug complex: contributions to etoposide binding and activity. Biochemistry, 47, 4501-9.
BERMEJO, R., DOKSANI, Y., CAPRA, T., KATOU, Y. M., TANAKA, H., SHIRAHIGE, K. & FOIANI, M. 2007. Top1- and Top2-mediated topological transitions at

Reference List
142
replication forks ensure fork progression and stability and prevent DNA damage checkpoint activation. Genes Dev, 21, 1921-36.
BERNAD, R., SANCHEZ, P., RIVERA, T., RODRIGUEZ-CORSINO, M., BOYARCHUK, E., VASSIAS, I., RAY-GALLET, D., ARNAOUTOV, A., DASSO, M., ALMOUZNI, G. & LOSADA, A. 2011. Xenopus HJURP and condensin II are required for CENP-A assembly. J Cell Biol, 192, 569-82.
BETZ, R., MACKAY, V. L. & DUNTZE, W. 1977. a-factor from Saccharomyces cerevisiae: partial characterization of a mating hormone produced by cell of mating type a. J. Bacteriol., 132, 462-472.
BHALLA, N., BIGGINS, S. & MURRAY, A. W. 2002. Mutation of YCS4, a budding yeast condensin subunit, affects mitotic and nonmitotic chromosome behavior. Mol. Biol. Cell, 13, 632-645.
BHAT, M. A., PHILP, A. V., GLOVER, D. M. & BELLEN, H. J. 1996. Chromatid segregation at anaphase requires the barren product, a novel chromosome-associated protein that interacts with topoisomerase II. Cell, 87, 1103-1114.
BORGES, V., LEHANE, C., LOPEZ-SERRA, L., FLYNN, H., SKEHEL, M., ROLEF BEN-SHAHAR, T. & UHLMANN, F. 2010. Hos1 deacetylates Smc3 to close the cohesin acetylation cycle. Mol. Cell, 39, 677-688.
BREEDEN, L. L. 1997. Alpha-factor synchronization of budding yeast. Methods Enzymol., 283, 332-341.
BROWN, P. O., PEEBLES, C. L. & COZZARELLI, N. R. 1979. A topoisomerase from Escherichia coli related to DNA gyrase. Proc Natl Acad Sci U S A, 76, 6110-4.
CABELLO, O. A., ELISEEVA, E., HE, W. G., YOUSSOUFIAN, H., PLON, S. E., BRINKLEY, B. R. & BELMONT, J. W. 2001. Cell cycle-dependent expression and nucleolar localization of hCAP-H. Mol Biol Cell, 12, 3527-37.
CAIRNS, J. 1963. The bacterial chromosome and its manner of replication as seen by autoradiography. J Mol Biol, 6, 208-13.
CAPRANICO, G. & BINASCHI, M. 1998. DNA sequence selectivity of topoisomerases and topoisomerase poisons. Biochim Biophys Acta, 1400, 185-94.
CHAMPOUX, J. J. 2001. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem, 70, 369-413.
CHAMPOUX, J. J. & DULBECCO, R. 1972. An activity from mammalian cells that untwists superhelical DNA--a possible swivel for DNA replication (polyoma-ethidium bromide-mouse-embryo cells-dye binding assay). Proc Natl Acad Sci U S A, 69, 143-6.
CHAN, K. L., NORTH, P. S. & HICKSON, I. D. 2007. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J, 26, 3397-409.
CHEESEMAN, I. M., ANDERSON, S., JWA, M., GREEN, E. M., KANG, J., YATES ILL, J. R., CHAN, C. S., G., D. D. & BARNES, G. 2002. Phospho-regulation of kinetochore-microtubule attachments by the Aurora kinase Ipl1p. Cell, 111, 163-172.
CHEN, J., ZHENG, X. F., BROWN, E. J. & SCHREIBER, S. L. 1995. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci U S A, 92, 4947-51.
CHRISTENSEN, M. O., LARSEN, M. K., BARTHELMES, H. U., HOCK, R., ANDERSEN, C. L., KJELDSEN, E., KNUDSEN, B. R., WESTERGAARD, O., BOEGE, F. & MIELKE, C. 2002. Dynamics of human DNA topoisomerases IIalpha and IIbeta in living cells. J Cell Biol, 157, 31-44.
COELHO, P. A., QUEIROZ-MACHADO, J. & SUNKEL, C. E. 2003. Condensin-dependent localisation of topoisomerase II to an axial chromosomal structure is

Reference List
143
required for sister chromatid resolution during mitosis. J. Cell Sci., 116, 4763-4776.
CRAVEN, R. A., GRIFFITH, D. J. F., SHELDRICK, K. S., RANDALL, R. E., HAGAN, I. M. & CARR, A. M. 1998. Vectors for the expression of tagged proteins in Schizosaccharomyces pombe. Gene, 221, 59-68.
CUVIER, O. & HIRANO, T. 2003. A role of topoisomerase II in linking DNA replication to chromosome condensation. J. Cell. Biol., 160, 645-655.
CUYLEN, S., METZ, J. & HAERING, C. H. 2011. Condensin structures chromosomal DNA through topological links. Nat Struct Mol Biol, 18, 894-901.
D'AMBROSIO, C., KELLY, G., SHIRAHIGE, K. & UHLMANN, F. 2008a. Condensin-dependent rDNA decatenation introduces a temporal pattern to chromosome segregation. Curr. Biol., 18, 1084-1089.
D'AMBROSIO, C., SCHMIDT, C. K., KATOU, Y., KELLY, G., ITOH, T., SHIRAHIGE, K. & UHLMANN, F. 2008b. Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes Dev., 22, 2215-2227.
D'AMOURS, D., STEGMEIER, F. & AMON, A. 2004. Cdc14 and condensin control the dissolution of cohesin-independent linkages at repeated DNA. Cell, 117, 455-469.
DERSHOWITZ, A. & NEWLON, C. S. 1993. The effect on chromosome stability of deleting replication origins. Mol. Cell. Biol., 13, 391-398.
DEWEESE, J. E., OSHEROFF, M. A. & OSHEROFF, N. 2008. DNA Topology and Topoisomerases: Teaching a "Knotty" Subject. Biochem Mol Biol Educ, 37, 2-10.
DEWEESE, J. E. & OSHEROFF, N. 2009. The DNA cleavage reaction of topoisomerase II: wolf in sheep's clothing. Nucleic Acids Res, 37, 738-48.
DOHMEN, R. J., WU, P. & VARSHAVSKY, A. 1994. Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science, 263, 1273-1276.
DONG, K. C. & BERGER, J. M. 2007. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature, 450, 1201-1206.
DOWNES, C. S., MULLINGER, A. M. & JOHNSON, R. T. 1991. Inhibitors of DNA topoisomerase II prevent chromatid separation in mammalian cells but do not prevent exit from mitosis. Proc. Natl. Acad. Sci. USA, 88, 8895-8899.
EARNSHAW, W. C., HALLIGAN, B., COOKE, C. A., HECK, M. M. S. & LIU, L. F. 1985. Topoisomerase II is a structural component of mitotic chromosome scaffolds. J. Cell Biol., 100, 1706-1715.
ELLIOTT, S. G. & MCLAUGHLIN, C. S. 1979. Regulation of RNA synthesis in yeast III. Mol. Gen. Genet., 169, 237-243.
ESPELI, O. & MARIANS, K. J. 2004. Untangling intracellular DNA topology. Mol Microbiol, 52, 925-31.
FARCAS, A.-M., ULUOCAK, P., HELMHART, W. & NASMYTH, K. 2011. Cohesin's concatenation of sister DNAs maintains their intertwining. Mol. Cell, 44, 97-107.
FARMER, S., SAN-SEGUNDO, P. A. & ARAGON, L. 2011. The Smc5-Smc6 complex is required to remove chromosome junctions in meiosis. PLoS One, 6, e20948.
FELIX, C. A. 1998. Secondary leukemias induced by topoisomerase-targeted drugs. Biochim Biophys Acta, 1400, 233-55.
FELIX, C. A., HOSLER, M. R., WINICK, N. J., MASTERSON, M., WILSON, A. E. & LANGE, B. J. 1995. ALL-1 gene rearrangements in DNA topoisomerase II inhibitor-related leukemia in children. Blood, 85, 3250-6.
FORTUNE, J. M. & OSHEROFF, N. 2000. Topoisomerase II as a target for anticancer drugs: when enzymes stop being nice. Prog Nucleic Acid Res Mol Biol, 64, 221-53.

Reference List
144
FREEMAN, L., ARAGON-ALCAIDE, L. & STRUNNIKOV, A. 2000. The condensin complex governs chromosome condensation and mitotic transmission of rDNA. J. Cell Biol., 149, 811-824.
GASSER, S. M., LAROCHE, T., FALQUET, J., BOY DE LA TOUR, E. & LAEMMLI, U. K. 1986. Metaphase chromosome structure. Involvement of topoisomerase II. J Mol Biol, 188, 613-29.
GELLERT, M., MIZUUCHI, K., O'DEA, M. H. & NASH, H. A. 1976. DNA gyrase: an enzyme that introduces superhelical turns into DNA. Proc Natl Acad Sci U S A, 73, 3872-6.
GERLICH, D., HIROTA, T., KOCH, B., PETERS, J.-M. & ELLENBERG, J. 2006a. Condensin I stabilizes chromosomes mechanically through a dynamic interaction in live cells. Curr. Biol., 16, 333-344.
GERLICH, D., KOCH, B., DUPEUX, F., PETERS, J.-M. & ELLENBERG, J. 2006b. Live-cell imaging reveals a stable cohesin-chromatin interaction after but not before DNA replication. Curr. Biol., 16, 1571-1578.
GOTO, T. & WANG, J. C. 1984. Yeast DNA topoisomerase II is encoded by a single-copy, essential gene. Cell, 36, 1073-80.
GREEN, L. C., KALITSIS, P., CHANG, T. M., CIPETIC, M., KIM, J. H., MARSHALL, O., TURNBULL, L., WHITCHURCH, C. B., VAGNARELLI, P., SAMEJIMA, K., EARNSHAW, W. C., CHOO, K. H. & HUDSON, D. F. 2012. Contrasting roles of condensin I and condensin II in mitotic chromosome formation. J Cell Sci, 125, 1591-604.
GRIESE, J. J., WITTE, G. & HOPFNER, K. P. 2010. Structure and DNA binding activity of the mouse condensin hinge domain highlight common and diverse features of SMC proteins. Nucleic Acids Res, 38, 3454-65.
GRIGORYEV, S. A. & WOODCOCK, C. L. 2012. Chromatin organization - the 30 nm fiber. Exp Cell Res, 318, 1448-55.
GUACCI, V., HOGAN, E. & KOSHLAND, D. 1994. Chromosome condensation and sister chromatid pairing in budding yeast. J. Cell Biol., 125, 517-530.
HAERING, C. H., LÖWE, J., HOCHWAGEN, A. & NASMYTH, K. 2002. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol. Cell, 9, 773-788.
HAERING, C. H., SCHOFFNEGGER, D., NISHINO, T., HELMHART, W., NASMYTH, K. & LÖWE, J. 2004. Structure and stability of cohesin's Smc1-kleisin interaction. Mol. Cell, 15, 951-964.
HAGSTROM, K. A., HOLMES, V. F., COZZARELLI, N. R. & MEYER, B. J. 2002. C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes Dev, 16, 729-42.
HARKINS, T. T. & LINDSLEY, J. E. 1998. Pre-steady-state analysis of ATP hydrolysis by Saccharomyces cerevisiae DNA topoisomerase II. 1. A DNA-dependent burst in ATP hydrolysis. Biochemistry, 37, 7292-8.
HARUKI, H., NISHIKAWA, J. & LAEMMLI, U. K. 2008a. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell, 31, 925-32.
HARUKI, H., NISHIKAWA, J. & LAEMMLI, U. K. 2008b. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell, 31, 925-932.
HAYAMA, R. & MARIANS, K. J. 2010. Physical and functional interation between the condensin MukB and the decatenase topoisomerase IV in Escherichia coli. Proc. Natl. Acad. Sci. USA, 107, 18826-18831.
HECK, M. M. & EARNSHAW, W. C. 1986. Topoisomerase II: A specific marker for cell proliferation. J Cell Biol, 103, 2569-81.

Reference List
145
HECK, M. M., HITTELMAN, W. N. & EARNSHAW, W. C. 1988. Differential expression of DNA topoisomerases I and II during the eukaryotic cell cycle. Proc Natl Acad Sci U S A, 85, 1086-90.
HEITMAN, J., MOVVA, N. R. & HALL, M. N. 1991. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science, 253, 905-9.
HIRANO, M. & HIRANO, T. 1998a. ATP-dependent aggregation of single-stranded DNA by a bacterial SMC homodimer. EMBO J, 17, 7139-48.
HIRANO, M. & HIRANO, T. 1998b. ATP-dependent aggregtion of single-stranded DNA by a bacterial SMC homodimer. EMBO J., 17, 7139-7148.
HIRANO, M. & HIRANO, T. 2002. Hinge-mediated dimerization of SMC protein is essential for its dynamic interaction with DNA. EMBO J, 21, 5733-44.
HIRANO, T. 1999. SMC-mediated chromosome mechanics: a conserved scheme from bacteria to vertebrates? Genes Dev, 13, 11-9.
HIRANO, T. 2000. Chromosome Cohesion, Condensation, and Separation. Annu. Rev. Biochem., 69, 115-144.
HIRANO, T. 2005a. Condensins: organizing and segregating the genome. Curr Biol, 15, R265-75.
HIRANO, T. 2005b. SMC proteins and chromosome mechanics: from bacteria to humans. Philos Trans R Soc Lond B Biol Sci, 360, 507-14.
HIRANO, T. 2006. At the heart of the chromosome: SMC proteins in action. Nat Rev Mol Cell Biol, 7, 311-22.
HIRANO, T. & MITCHISON, T. J. 1994. A heterodimeric coiled-coil protein required for mitotic chromosome condensation in vitro. Cell, 79, 449-458.
HIROTA, T., GERLICH, D., KOCH, B., ELLENBERG, J. & PETERS, J.-M. 2004. Distinct functions of condensin I and II in mitotic chromosome assembly. J. Cell Sci., 117, 6435-6445.
HOLM, C., GOTO, T., WANG, J. C. & BOTSTEIN, D. 1985. DNA topoisomerase II is required at the time of mitosis in yeast. Cell, 41, 553-563.
HUDSON, D. F., VAGNARELLI, P., GASSMANN, R. & EARNSHAW, W. C. 2003. Condensin is required for nonhistone protein assembly and structural integrity of vertebrate mitotic chromosomes. Dev. Cell, 5, 323-336.
IVANOV, D. & NASMYTH, K. 2005. A topological interaction between cohesin rings and a circular minichromosome. Cell, 122, 849-860.
IVANOV, D. & NASMYTH, K. 2007. A physical assay for sister chromatid cohesion in vitro. Mol. Cell, 27, 300-310.
IVANOV, D., SCHLEIFFER, A., EISENHABER, F., MECHTLER, K., HAERING, C. H. & NASMYTH, K. 2002. Eco1 is a novel acetlytransferase that can acetylate proteins involved in cohesion. Curr. Biol., 12, 323-328.
JAIN, D., HEBDEN, A. K., NAKAMURA, T. M., MILLER, K. M. & COOPER, J. P. 2010. HAATI survivors replace canonical telomeres with blocks of generic heterochromatin. Nature, 467, 223-7.
JENSEN, R. B. & SHAPIRO, L. 1999. The Caulobacter crescentus smc gene is required for cell cycle progression and chromosome segregation. Proc Natl Acad Sci U S A, 96, 10661-6.
JENSEN, S., REDWOOD, C. S., JENKINS, J. R., ANDERSEN, A. H. & HICKSON, I. D. 1996. Human DNA topoisomerases IIa and IIb can functionally substitute for yeast TOP2 in chromosome segregation and recombination. Mol. Gen. Genet., 252, 79-86.
JOSHI, R. S., PINA, B. & ROCA, J. 2010. Positional dependence of transcriptional inhibition by DNA torsional stress in yeast chromosomes. EMBO J., 29, 740-748.

Reference List
146
JU, B. G., LUNYAK, V. V., PERISSI, V., GARCIA-BASSETS, I., ROSE, D. W., GLASS, C. K. & ROSENFELD, M. G. 2006. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science, 312, 1798-802.
KAWAMURA, R., POPE, L. H., CHRISTENSEN, M. O., SUN, M., TEREKHOVA, K., BOEGE, F., MIELKE, C., ANDERSEN, A. H. & MARKO, J. F. 2010. Mitotic chromosomes are constrained by topoisomerase II-sensitive DNA entanglements. J. Cell Biol., 188, 653-663.
KIMURA, K., CUVIER, O. & HIRANO, T. 2001. Chromosome condensation by a human condensin complex in Xenopus egg extracts. J. Biol. Chem., 276, 5417-5420.
KIMURA, K., HIRANO, M., KOBAYASHI, R. & HIRANO, T. 1998. Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science, 282, 487-90.
KIMURA, K. & HIRANO, T. 1997a. ATP-Dependent positive supercoiling of DNA by 13S condensin: a biochemical implication for chromosome condensation. Cell, 90, 625-634.
KIMURA, K. & HIRANO, T. 1997b. ATP-dependent positive supercoiling of DNA by 13S condensin: a biochemical implication for chromosome condensation. Cell, 90, 625-34.
KIMURA, K. & HIRANO, T. 2000. Dual roles of the 11S regulatory subcomplex in condensin functions. Proc. Natl. Acad. Sci. USA, 97, 11972-11977.
KIMURA, K., NOZAKI, N., SAIJO, M., KIKUCHI, A., UI, M. & ENOMOTO, T. 1994. Identification of the nature of modification that causes the shift of DNA topoisomerase II beta to apparent higher molecular weight forms in the M phase. J Biol Chem, 269, 24523-6.
KIMURA, K., RYBENKOV, V. V., CRISONA, N. J., HIRANO, T. & COZZARELLI, N. R. 1999. 13S condensin actively reconfigures DNA by introducing global positive writhe: implications for chromosome condensation. Cell, 98, 239-248.
KNOP, M., SIEGERS, K., PEREIRA, G., ZACHARIAE, W., WINSOR, B., NASMYTH, K. & SCHIEBEL, E. 1999. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast, 15, 963-972.
KOSHLAND, D. & STRUNNIKOV, A. 1996a. Mitotic chromosome condensation. Annu Rev Cell Dev Biol, 12, 305-33.
KOSHLAND, D. & STRUNNIKOV, A. 1996b. Mitotic chromosome condensation. Annu. Rev. Cell Dev. Biol., 12, 305-333.
LAEMMLI, U. K., KAS, E., POLJAK, L. & ADACHI, Y. 1992. Scaffold-associated regions: cis-acting determinants of chromatin structural loops and functional domains. Curr Opin Genet Dev, 2, 275-85.
LAVOIE, B. D., HOGAN, E. & KOSHLAND, D. 2002. In vivo dissection of the chromosome condensation machinery: reversibility of condensation distinguishes contributions of condensin and cohesin. J. Cell Biol., 156, 805-815.
LAVOIE, B. D., HOGAN, E. & KOSHLAND, D. 2004. In vivo requirements for rDNA chromosome condensation reveal two cell-cycle-regulated pathways for mitotic chromosome folding. Genes Dev., 18, 76-87.
LEE, W., TILLO, D., BRAY, N., MORSE, R. H., DAVIS, R. W., HUGHES, T. R. & NISLOW, C. 2007. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet, 39, 1235-44.
LEPPARD, J. B. & CHAMPOUX, J. J. 2005. Human DNA topoisomerase I: relaxation, roles, and damage control. Chromosoma, 114, 75-85.
LEVINE, C., HIASA, H. & MARIANS, K. J. 1998. DNA gyrase and topoisomerase IV: biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim Biophys Acta, 1400, 29-43.

Reference List
147
LEWIS, C. D. & LAEMMLI, U. K. 1982. Higher order metaphase chromosome structure: evidence for metalloprotein interactions. Cell, 29, 171-81.
LI, Y., STEWART, N. K., BERGER, A. J., VOS, S., SCHOEFFLER, A. J., BERGER, J. M., CHAIT, B. T. & OAKLEY, M. G. 2010. Escherichia coli condensin MukB stimulates topoisomerase IV activity by a direct physical interaction. Proc. Natl. Acad. Sci. USA, 107, 18832-18837.
LIPP, J. J., HIROTA, T., POSER, I. & PETERS, J. M. 2007. Aurora B controls the association of condensin I but not condensin II with mitotic chromosomes. J Cell Sci, 120, 1245-55.
LOSADA, A., HIRANO, M. & HIRANO, T. 2002. Cohesin release is required for sister chromatid resolution but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev., 16, 3004-3016.
LOSADA, A., YOKOCHI, T., KOBAYASHI, R. & HIRANO, T. 2000. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J. Cell Biol., 150, 405-416.
LUCAS, I., GERME, T., CHEVRIER-MILLER, M. & HYRIEN, O. 2001. Topoisomerase II can unlink replicating DNA by precatenane removal. EMBO J, 20, 6509-19.
MAESHIMA, K. & LAEMMLI, U. K. 2003. A two-step scaffolding model for mitotic chromosome assembly. Dev. Cell, 4, 467-480.
MANNINI, B., CASCELLA, R., ZAMPAGNI, M., VAN WAARDE-VERHAGEN, M., MEEHAN, S., ROODVELDT, C., CAMPIONI, S., BONINSEGNA, M., PENCO, A., RELINI, A., KAMPINGA, H. H., DOBSON, C. M., WILSON, M. R., CECCHI, C. & CHITI, F. 2012. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc Natl Acad Sci U S A, 109, 12479-84.
MARCUS, S., CALDWELL, G. A., MILLER, D., XUE, C.-B., NAIDER, F. & BECKER, J. M. 1991. Significance of C-terminal cystein modifications to the biological activity of the Saccharomyces cerevisiae a-factor. Mol. Cell. Biol., 11, 3603-3612.
MARINI, J. C., MILLER, K. G. & ENGLUND, P. T. 1980. Decatenation of kinetoplast DNA by topoisomerases. J. Biol. Chem., 255, 4976-4979.
MASCARENHAS, J., SOPPA, J., STRUNNIKOV, A. V. & GRAUMANN, P. L. 2002. Cell cycle-dependent localization of two novel prokaryotic chromosome segregation and condensation proteins in Bacillus subtilis that interact with SMC protein. EMBO J, 21, 3108-18.
MASCARENHAS, J., VOLKOV, A. V., RINN, C., SCHIENER, J., GUCKENBERGER, R. & GRAUMANN, P. L. 2005. Dynamic assembly, localization and proteolysis of the Bacillus subtilis SMC complex. BMC Cell Biol, 6, 28.
MATOBA, K., YAMAZOE, M., MAYANAGI, K., MORIKAWA, K. & HIRAGA, S. 2005. Comparison of MukB homodimer versus MukBEF complex molecular architectures by electron microscopy reveals a higher-order multimerization. Biochem Biophys Res Commun, 333, 694-702.
MCCLENDON, A. K. & OSHEROFF, N. 2007. DNA topoisomerase II, genotoxicity, and cancer. Mutat Res, 623, 83-97.
MCGRANAHAN, N., BURRELL, R. A., ENDESFELDER, D., NOVELLI, M. R. & SWANTON, C. 2012. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep, 13, 528-38.
MELBY, T. E., CIAMPAGLIO, C. N., BRISCOE, G. & ERICKSON, H. P. 1998. The symmetrical structure of structural maintenance of chromosomes (SMC) and MukB proteins: long, antiparallel coiled coils, folded at a flexible hinge. J. Cell Biol., 142, 1595-1604.
MICHAELIS, C., CIOSK, R. & NASMYTH, K. 1997. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell, 91, 35-45.

Reference List
148
MONDAL, N. & PARVIN, J. D. 2001. DNA topoisomerase IIalpha is required for RNA polymerase II transcription on chromatin templates. Nature, 413, 435-8.
MORANO, K. A., GRANT, C. M. & MOYE-ROWLEY, W. S. 2012. The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics, 190, 1157-95.
MORGAN, T. H. 1915. Localization of the Hereditary Material in the Germ Cells. Proc Natl Acad Sci U S A, 1, 420-9.
MORIYA, S., TSUJIKAWA, E., HASSAN, A. K., ASAI, K., KODAMA, T. & OGASAWARA, N. 1998. A Bacillus subtilis gene-encoding protein homologous to eukaryotic SMC motor protein is necessary for chromosome partition. Mol. Microbiol., 29, 179-187.
MUELLER-PLANITZ, F. & HERSCHLAG, D. 2008. Coupling between ATP binding and DNA cleavage by DNA topoisomerase II: A unifying kinetic and structural mechanism. J Biol Chem, 283, 17463-76.
MULLEN, D. G., KYRO, K., HAUSER, M., GUSTAVSSON, M., VEGLIA, G., BECKER, J. M., NAIDER, F. & DISTEFANO, M. D. 2011. Synthesis of a-factor peptide from Saccharomyces cerevisiae and photoactive analogues via Fmoc solid phase methodology. Bioorg. Med. Chem., 19, 490-497.
MURRAY, A. W. & SZOSTAK, J. W. 1985. Chromosome segregation in mitosis and meiosis. Ann. Rev. Cell Biol., 1, 289-315.
NEUWALD, A. F. & HIRANO, T. 2000. HEAT repeats associated with condensins, cohesins, and other complexes involved in chromosome-related functions. Genome Res., 10, 1445-1452.
NEZELOF, C. 2003. Henri Dutrochet (1776-1847): an unheralded discoverer of the cell. Ann Diagn Pathol, 7, 264-72.
NIKI, H., JAFFE, A., IMAMURA, R., OGURA, T. & HIRAGA, S. 1991. The new gene mukB codes for a 177 kd protein with coiled-coil domains involved in chromosome partitioning of E. coli. EMBO J, 10, 183-93.
O'REILLY, N., CHARBIN, A., LOPEZ-SERRA, L. & UHLMANN, F. 2012. Facile synthesis of budding yeast a-factor and its use to synchronise cells of a mating type. Yeast, in press.
OCAMPO-HAFALLA, M. T. & UHLMANN, F. 2011. Cohesin loading and sliding. J Cell Sci, 124, 685-91.
OLIVEIRA, R. A., HEIDMANN, S. & SUNKEL, C. E. 2007. Condensin I binds chromatin early in prophase and displays a highly dynamic association with Drosophila mitotic chromosomes. Chromosoma, 116, 259-74.
ONN, I., AONO, N., HIRANO, M. & HIRANO, T. 2007. Reconstitution and subunit geometry of human condensin complexes. EMBO J., 26, 1024-1034.
ONO, T., LOSADA, A., HIRANO, M., MYERS, M. P., NEUWALD, A. F. & HIRANO, T. 2003. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell, 115, 109-121.
OSBORNE, B. I. & GUARENTE, L. 1988. Transcription by RNA polymerase II induces changes of DNA topology in yeast. Genes Dev, 2, 766-72.
PATTERTON, H. G., LANDEL, C. C., LANDSMAN, D., PETERSON, C. L. & SIMPSON, R. T. 1998. The biochemical and phenotypic characterization of Hho1p, the putative linker histone H1 of Saccharomyces cerevisiae. J Biol Chem, 273, 7268-76.
PAWELETZ, N. 2001. Walther Flemming: pioneer of mitosis research. Nat Rev Mol Cell Biol, 2, 72-5.
PETRUSHENKO, Z. M., LAI, C. H., RAI, R. & RYBENKOV, V. V. 2006. DNA reshaping by MukB. Right-handed knotting, left-handed supercoiling. J Biol Chem, 281, 4606-15.

Reference List
149
RAO, P. N. & JOHNSON, R. T. 1970. Mammalian cell fusion: studies on the regulation of DNA synthesis and mitosis. Nature, 225, 159-64.
RENSHAW, M. J., WARD, J. J., KANEMAKI, M., NATSUME, K., NEDELEC, F. J. & TANAKA, T. U. 2010. Condensins promote chromosome recoiling during early anaphase to complete sister chromatid separation. Dev Cell, 19, 232-44.
ROOS, W. P. & KAINA, B. 2012. DNA damage-induced apoptosis: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett.
ROTH, S. Y. & ALLIS, C. D. 1992. Chromatin condensation: does histone H1 dephosphorylation play a role? Trends Biochem Sci, 17, 93-8.
ROY, M. A. & D'AMOURS, D. 2011. DNA-binding properties of Smc6, a core component of the Smc5-6 DNA repair complex. Biochem Biophys Res Commun, 416, 80-5.
SAKA, Y., SUTANI, T., YAMASHITA, Y., SAITOH, S., TAKEUCHI, M., NAKASEKO, Y. & YANAGIDA, M. 1994a. Fission yeast cut3 and cut14, members of a ubiquitous protein family, are required for chromosome condensation and segregation in mitosis. EMBO J, 13, 4938-52.
SAKA, Y., SUTANI, T., YAMASHITA, Y., SAITOH, S., TAKEUCHI, M., NAKASEKO, Y. & YANAGIDA, M. 1994b. Fission yeast cut3 and cut14, members of a ubiquitous protein family, are required for chromosome condensation and segregation in mitosis. EMBO J., 13, 4938-4952.
SAKAGUCHI, A. & KIKUCHI, A. 2004. Functional compatibility between isoform alpha and beta of type II DNA topoisomerase. J Cell Sci, 117, 1047-54.
SAKAI, A., HIZUME, K., SUTANI, T., TAKEYASU, K. & YANAGIDA, M. 2003. Condensin but not cohesin SMC heterodimer induces DNA reannealing through protein-protein assembly. EMBO J., 22, 2764-2775.
SAMBROOK, J. & GETHING, M. J. 1989. Protein structure. Chaperones, paperones. Nature, 342, 224-5.
SCHLEIFFER, A., KAITNA, S., MAURER-STROH, S., GLOTZER, M., NASMYTH, K. & EISENHABER, F. 2003. Kleisins: A superfamily of bacterial and eukaryotic SMC protein partners. Mol. Cell, 11, 571-575.
SHERRILL, C., KHOURI, O., ZEMAN, S. & ROISE, D. 1995. Synthesis and biological activity of fluorescent yeast pheromones. Biochemistry, 34, 3553-3560.
SHIN, H. C., LIM, J. H., WOO, J. S. & OH, B. H. 2009. Focal localization of MukBEF condensin on the chromosome requires the flexible linker region of MukF. FEBS J, 276, 5101-10.
SHINTOMI, K. & HIRANO, T. 2009. Releasing cohesin from chromosome arms in early mitosis: opposing actions of Wapl-Pds5 and Sgo1. Genes Dev, 23, 2224-36.
SIKORSKI, R. S. & HIETER, P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics, 122, 19-27.
SOUTHERN, E. M. 1975. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol, 98, 503-17.
ST-PIERRE, J., DOUZIECH, M., BAZILE, F., PASCARIU, M., BONNEIL, E., SAUVÉ, V., RATSIMA, H. & D'AMOURS, D. 2009. Polo kinase regulates mitotic chromosome condensation by hyperactivation of condensin DNA supercoiling activity. Mol. Cell, 34, 416-426.
STEFFENSEN, S., COELHO, P. A., COBBE, N., VASS, S., COSTA, M., HASSAN, B., PROKOPENKO, S. N., BELLEN, H., HECK, M. M. S. & SUNKEL, C. E. 2001. A role for Drosophila SMC4 in the resolution of sister chromatids in mitosis. Curr. Biol., 11, 295-307.
STRAY, J. E. & LINDSLEY, J. E. 2003. Biochemical analysis of the yeast condensin Smc2/4 complex. An ATPase that promotes knotting of circular DNA. J. Biol. Chem., 278, 26238-26248.

Reference List
150
STRICK, T. R., KAWAGUCHI, T. & HIRANO, T. 2004. Real-time detection of single-molecule DNA compaction by condensin I. Curr Biol, 14, 874-80.
STRUNNIKOV, A., HOGAN, E. & KOSHLAND, D. 1995a. SMC2, a Saccharomyces cerevisiae gene essential for chromosome segregation and condensation, defines a subgroup within the SMC family. Genes Dev., 9, 587-599.
STRUNNIKOV, A. V., HOGAN, E. & KOSHLAND, D. 1995b. SMC2, a Saccharomyces cerevisiae gene essential for chromosome segregation and condensation, defines a subgroup within the SMC family. Genes Dev, 9, 587-99.
SULLIVAN, M., HIGUCHI, T., KATIS, V. L. & UHLMANN, F. 2004. Cdc14 phosphatase induces rDNA condensation and resolves cohesin-independent cohesion during budding yeast anaphase. Cell, 117, 471-482.
SUMARA, I., VORLAUFER, E., GIEFFERS, C., PETERS, B. H. & PETERS, J.-M. 2000. Characterization of vertebrate cohesin complexes and their regulation in prophase. J. Cell Biol., 151, 749-761.
SUMARA, I., VORLAUFER, E., STUKENBERG, P. T., KELM, O., REDEMANN, N., NIGG, E. A. & PETERS, J.-M. 2002. The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol. Cell, 9, 515-525.
SUNDIN, O. & VARSHAVSKY, A. 1980. Terminal stages of SV40 DNA replication proceed via multiply intertwined catenated dimers. Cell, 21, 103-114.
SUNDIN, O. & VARSHAVSKY, A. 1981. Arrest of segregation leads to accumulation of highly intertwined catenated dimers: dissection of the final stages of SV40 DNA replication. Cell, 25, 659-69.
SUTANI, T. & YANAGIDA, M. 1997a. DNA renaturation activity of the SMC complex implicated in chromosome condensation. Nature, 388, 798-801.
SUTANI, T. & YANAGIDA, M. 1997b. DNA renaturation activity of the SMC complex implicated in chromosome condensation. Nature, 388, 798-801.
SUTANI, T., YUASA, T., TOMONAGA, T., DOHMAE, N., TAKIO, K. & YANAGIDA, M. 1999. Fission yeast condensin complex: essential roles of non-SMC subunits for condensation and Cdc2 phosphorylation of Cut3/SMC4. Genes Dev., 13, 2271-2283.
TAKEMOTO, A., KIMURA, K., YANAGISAWA, J., YOKOYAMA, S. & HANAOKA, F. 2006. Negative regulation of condensin I by CK2-mediated phosphorylation. EMBO J., 25, 5339-5348.
TANAKA, T., COSMA, M. P., WIRTH, K. & NASMYTH, K. 1999. Identification of cohesin association sites at centromeres and along chromosome arms. Cell, 98, 847-858.
TANAKA, T., FUCHS, J., LOIDL, J. & NASMYTH, K. 2000. Cohesin ensures bipolar attachment of microtubules to sister centromeres and resists their precocious separation. Nat Cell Biol, 2, 492-9.
TAPIA-ALVEAL, C., OUTWIN, E. A., TREMPOLEC, N., DZIADKOWIEC, D., MURRAY, J. M. & O'CONNELL, M. J. 2010. SMC complexes and topoisomerase II work together so that sister chromatids can work apart. Cell Cycle, 9, 2065-70.
TESSMAN, I., TESSMAN, E. S. & STENT, G. S. 1957. The relative radiosensitivity of bacteriophages S13 and T2. Virology, 4, 209-15.
TOMSON, B. N., D'AMOURS, D., ADAMSON, B. S., ARAGON, L. & AMON, A. 2006. Ribosomal DNA transcription-dependent processes interfere with chromosome segregation. Mol. Cell. Biol., 26, 6239-6247.
TOYODA, Y., FURUYA, K., GOSHIMA, G., NAGAO, K., TAKAHASHI, K. & YANAGIDA, M. 2002. Requirement of chromatid cohesion proteins Rad21/Scc1 and Mis4/Scc2 for normal spindle-kinetochore interaction in fission yeast. Curr. Biol., 12, 347-358.

Reference List
151
TRAVERS, A. & MUSKHELISHVILI, G. 2007. A common topology for bacterial and eukaryotic transcription initiation? EMBO Rep, 8, 147-51.
UEMURA, T., OHKURA, H., ADACHI, Y., MORINO, K., SHIOZAKI, K. & YANAGIDA, M. 1987a. DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. Cell, 50, 917-925.
UEMURA, T., OHKURA, H., ADACHI, Y., MORINO, K., SHIOZAKI, K. & YANAGIDA, M. 1987b. DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. Cell, 50, 917-25.
UHLMANN, F. 2003. Chromosome cohesion and separation: from men and molecules. Curr Biol, 13, R104-14.
UHLMANN, F., LOTTSPEICH, F. & NASMYTH, K. 1999. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature, 400, 37-42.
UHLMANN, F. & NASMYTH, K. 1998. Cohesion between sister chromatids must be established during DNA replication. Curr. Biol., 8, 1095-1101.
VAGNARELLI, P., HUDSON, D. F., RIBEIRO, S. A., TRINKLE-MULCAHY, L., SPENCE, J. M., LAI, F., FARR, C. J., LAMOND, A. I. & EARNSHAW, W. C. 2006. Condensin and Repo-Man-PP1 co-operate in the regulation of chromosome architecture during mitosis. Nat Cell Biol, 8, 1133-42.
VALLIER, L. G., SEGALL, J. E. & SNYDER, M. 2002. The alpha-factor receptor C-terminus is important for mating projection formation and orientation in Saccharomyces cerevisiae. Cell Motil Cytoskeleton, 53, 251-66.
VELEZ-CRUZ, R., RIGGINS, J. N., DANIELS, J. S., CAI, H., GUENGERICH, F. P., MARNETT, L. J. & OSHEROFF, N. 2005. Exocyclic DNA lesions stimulate DNA cleavage mediated by human topoisomerase II alpha in vitro and in cultured cells. Biochemistry, 44, 3972-81.
VOLOGODSKII, A. V., ZHANG, W., RYBENKOV, V. V., PODTELEZHNIKOV, A. A., SUBRAMANIAN, D., GRIFFITH, J. D. & COZZARELLI, N. R. 2001. Mechanism of topology simplification by type II DNA topoisomerases. Proc. Natl. Acad. Sci. USA, 98, 3045-3049.
WACH, A., BRACHAT, A., PÖHLMANN, R. & PHILIPPSEN, P. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast, 10, 1793-1808.
WANG, B.-D., BUTYLIN, P. & STRUNNIKOV, A. 2006. Condensin function in mitotic nucleolar segregation is regulated by rDNA transcription. Cell Cycle, 5, 2260-2267.
WANG, B.-D., EYRE, D., BASRAI, M., LICHTEN, M. & STRUNNIKOV, A. 2005. Condensin binding at distinct and specific chromosomal sites in the Saccharomyces cerevisiae genome. Mol. Cell. Biol., 25, 7216-7225.
WANG, J. C. 1998. Moving one DNA double helix through another by a type II DNA topoisomerase: the story of a simple molecular machine. Q Rev Biophys, 31, 107-44.
WANG, J. C. 2002. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol, 3, 430-40.
WANG, S.-W., READ, R. L. & NORBURY, C. J. 2002. Fission yeast Pds5 is required for accurate chromosome segregation and for survival after DNA damage or metaphase arrest. J. Cell Sci., 115, 587-598.
WEITAO, T., DASGUPTA, S. & NORDSTROM, K. 2000. Role of the mukB gene in chromosome and plasmid partition in Escherichia coli. Mol Microbiol, 38, 392-400.
WILKINSON, L. E. & PRINGLE, J. R. 1974. Transient G1 arrest of S. cerevisiae cells of mating type a by a factor produced by cells of mating type a. Exp. Cell Res., 89, 175-187.

Reference List
152
WORCEL, A. & BURGI, E. 1972. On the structure of the folded chromosome of Escherichia coli. J Mol Biol, 71, 127-47.
WORLAND, S. T. & WANG, J. C. 1989. Inducible overexpression, purification, and active site mapping of DNA topoisomerase II from the yeast Saccharomyces cerevisiae. J. Biol. Chem., 264, 4412-4416.
XUE, C.-B., CALDWELL, G. A., BECKER, J. M. & NAIDER, F. 1989. Total synthesis of the lipopeptide a-mating factor of Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun., 162, 253-257.
YAMAZOE, M., ONOGI, T., SUNAKO, Y., NIKI, H., YAMANAKA, K., ICHIMURA, T. & HIRAGA, S. 1999. Complex formation of MukB, MukE and MukF proteins involved in chromosome partitioning in Escherichia coli. EMBO J, 18, 5873-84.
YANAGIDA, M. 2000. Cell cycle mechanism of sister chromatid separation; roles of Cut1/separin and Cut2/securin. Genes Cells, 5, 1-8.
YANG, H., JIANG, W., GENTRY, M. & HALLBERG, R. L. 2000. Loss of a protein phosphatase 2A regulatory subunit (Cdc55p) elicits improper regulation of Swe1p degradation. Mol. Cell. Biol., 20, 8143-8156.
YEONG, F. M., HOMBAUER, H., WENDT, K. S., HIROTA, T., MUDRAK, I., MECHTLER, K., LOREGGER, T., MARCHLER-BAUER, A., TANAKA, K., PETERS, J.-M. & OGRIS, E. 2003. Identification of a subunit of a novel Kleisin-beta/SMC complex as a potential substrate of protein phosphatase 2A. Curr. Biol., 13, 2058-2064.
YOSHIMURA, S. H., HIZUME, K., MURAKAMI, A., SUTANI, T., TAKEYASU, K. & YANAGIDA, M. 2002. Condensin architecture and interaction with DNA: regulatory non-SMC subunits bind to the head of SMC heterodimer. Curr. Biol., 12, 508-513.
YU, H.-G. & KOSHLAND, D. 2005. Chromosome morphogenesis: condensin-dependent cohesin removal during meiosis. Cell, 123, 397-407.
ZECHIEDRICH, E. L. & COZZARELLI, N. R. 1995. Roles of topoisomerase IV and DNA gyrase in DNA unlinking during replication in Escherichia coli. Genes Dev., 9, 2859-2869.
ZOLLER, J. F., HERRMANN, R. G. & WANNER, G. 2004. Chromosome condensation in mitosis and meiosis of rye (Secale cereale L.). Cytogenet Genome Res, 105, 134-44.




















