
Multiple_antibiotic_resistance.pdf - UCL Discovery
347
i mu him i 2807400862 MULTIPLE ANTIBIOTIC RESISTANCE IN METHICILLIN-AMINOGLYCOSIDE RESISTANT STAPHYLOCOCCUS AUREUS Thesis siubmitted by PETER ALAN CHRISTOPHER MAPLE for the Degree of Doctor of Philosophy in the Faculty of Medicine of the University of London. MEDICAL LIBRARY. ROYAL FREE HOSPITAL HAMPSTEAD Department of Medical Microbiology, Royal Free Hospital School of Medicine, London NW3.
-
Upload
khangminh22 -
Category
Documents
-
view
6 -
download
0
Transcript of Multiple_antibiotic_resistance.pdf - UCL Discovery

i m u h i m i2807400862
MULTIPLE ANTIBIOTIC RESISTANCE
IN METHICILLIN-AMINOGLYCOSIDE RESISTANT
STAPHYLOCOCCUS AUREUS
Thesis siubmitted by
PETER ALAN CHRISTOPHER MAPLE
for the Degree of Doctor of Philosophy
in the Faculty of Medicine of the
University of London.
MEDICAL LIBRARY.
ROYAL FREE HOSPITAL
HAMPSTEAD
Department of Medical Microbiology,
Royal Free Hospital School of M e d i c i n e ,
London N W 3 .

ProQuest Number: U552871
All rights reserved
INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a com p le te manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uestProQuest U552871
Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author.
All rights reserved.This work is protected against unauthorized copying under Title 17, United States C ode
Microform Edition © ProQuest LLC.
ProQuest LLC.789 East Eisenhower Parkway
P.O. Box 1346 Ann Arbor, Ml 48106- 1346

ABSTRACT
Antibiotic susceptibility profiles of 100 strains
of methicillin and gentamicin resistant
Staphylococcus aureus (MGRSA) from 32 centres in 23
countries were determined. This is the first
survey to document the international problem of
multiple antibiotic resistance in MGRSA. Many
differing susceptibility profiles were found, some
strains being sensitive to a range of currently
available antistaphylococcal agents; others were
resistant to many of these agents.
More than 50% of MGRSA studied were non-typable
with the "International Set" of phages. Those
strains which typed, mostly reacted with phage 85
alone, or with other Group III phages as well.
Typing with supplementary phages revealed many of
the non-typable strains to possess Group Ill-
related patterns.
The variety of phage-typing and antibiotic
resistance patterns seen suggests that the
worldwide occurrence of MGRSA is probably not due
to widespread dissemination of a single clone. The
aminoglycoside modifying enzyme APH (2")/AAC ( 6 f) was
found in 44% of strains, while 56% produced APH
(2")/AAC ( 6 T) and APH ( 3 ’)-IV. Gene probing
experiments showed the same gene for APH (2")/AAC
(6') in MGRSA worldwide.
2

Options for treating MGRSA infections are
limited, currently few agents can reliably be used
in place of vancomycin. Fosfomycin and
pristinamycin appear to be promising. For treating
MGRSA carriage, azelaic acid and nitrofurazone may
be useful alternatives to mupirocin. Few, if any,
agents currently under development appear to be
promising alternatives to vancomycin.
Widespread resistance was found to the
fluoroquinolones (eg. ciprofloxacin) in MGRSA.
Experimental studies showed fluoroquinolone
resistance to readily occur. Analysis of resistant
clinical isolates showed the high incidence of
ciprofloxacin resistance in the MGRSA studied
resulted from independent evolution and not cross-
infection. Strains were least able to develop
resistance to ofloxacin, and newer fluoroquinolones
(eg. sparfloxacin) have improved activity against
MGRSA.
Multiple antibiotic resistance in MGRSA is a
major problem which could become significantly
worse should vancomycin resistance develop.
3

Publications arising from this thesis:
1. MAPLE, P.A.C., BRUMFITT, W., HAMILTON-MILLER, J.M.T.(1989). Comparative in vitro activity of vancomycin, teicoplanin, ramoplanin (formerly A16686), paldimycin, DuP 721 and DuP 105 against methicillin and gentamicin resistant Staphylococcus aureus collected worldwide. Journal of Antimicrobial Chemotherapy 23: 517-525.
2. MAPLE, P.A.C., HAMILTON-MILLER, J . M . T . , BRUMFITT, W. (1989). World-wide antibiotic resistance in methicillin- resistant Staphylococcus aureus. Lancet i: 537-540.
3. MAPLE, C.A.P., HAMILTON-MILLER, J.M.T., BRUMFITT, W. (1989). Ciprofloxacin resistance in methicillin- resistant Staphylococcus aureus. European Journal of Clinical Microbiology and Infectious Diseases 8:622-624.
4. MAPLE, P.A.C., HAMILTON-MILLER, J.M.T., BRUMFITT, W. (1991). Differing activity of quinolones against ciprofloxacin- sensitive and ciprofloxacin- resistant,methicillin- resistant Staphylococcus_______a u r e u s .Antimicrobial Agents and Chemotherapy 35: 345-350.
ABSTRACTS
1. BRUMFITT, W., MAPLE, P.A.C., HAMILTON-MILLER, J.M.T.(1989). Antibiotic Sensitivity Patterns ofMethicillin-Resistant Staphylococcus aureus and their use for Biotyping. Abstract no. 778/PP40, 4th European Congress of Clinical Microbiology, Nice.
2. MAPLE, P.A.C., HAMILTON-MILLER, J.M.T., BRUMFITT, W.(1990). Alternative topical agents to mupirocin for the eradication of staphylococcal carriage. Oralpresentation at the 160th Meeting of thePathological Society of Great Britain and Ireland.
4

CONTENTSfage
TITLE 1
ABSTRACT 2
PUBLICATIONS 4
CONTENTS 5
ACKNOWLEDGEMENTS 17
INTRODUCTION
1.0 Taxonomy and nomenclature of Staphylococcus aureus
1.1 Historical Introduction 18
2.0 The occurrence and pathogenicity of S. aureus
2.1 Distribution of S. aureus in nature 21
2.2 The human Staphylococcus aureus
i. Occurrence 22
ii. Factors influencing the incidence of 23
nasal carriage
2.3 Pathogenicity of Staphylococcus aureus 24
3.0 Staphylococcal infections and their treatment
3.1 The spectrum of staphylococcal infections 26
3.2 Treatment of staphylococcal infections in 27
the pre-antibiotic era
3.3 The antibiotic era 1940-1960
i. The sulphonamides 30
ii. The introduction of penicillin 31
iii. The use of streptomycin, tetracycline, 33
erythromycin and chloramphenicol against S .aureus
iv. The introduction of methicillin 34
v. The efficacy of methicillin and its 35
derivatives

4.0 Staphylococcus aureus resistant to methicillin
4.1 Occurrence of MRSA - 1960-1975 37
4.2 Treatment of MRSA - 1960-1975 39
i. Therapeutic options and the need 39
for combination therapy
ii. Treatment with topical antibiotics 40
iii. Treatment of MRSA infections with 42
penicillinase-resistant penicillins and/or
cephalosporins
4.3 The detection and nature of methicillin 44
resistance
i. Detection 44
ii. Nature of methicillin resistance 46
5.0 Use of gentamicin and the development of resistance
i. Use of gentamicin 49
ii. Development of resistance to gentamicin 51
6.0 Increasing hospital problems due to MGRSA
i. Problems due to MGRSA in Great Britain 53
ii. Problems due to MGRSA in Australia 56
iii. Problems due to MGRSA in the USA 57
iv. Problems due to MGRSA in Europe 60
v. Problems due to MGRSA in other countries 62
OBJECTIVES 64
MATERIALS AND METHODS
1.0 Strains 65
1.1 Strain origins and phage types 65
2 .0 Antibiotics
2.1 Antibiotics used in susceptibility and 72
studies of antibiotic properties

3.0 Media and Reagents
3.1 Media used in susceptibility and other 79
studies of antibiotic properties
3.2 Media used in biotyping studies
i . Egg-yolk agar 79
i i . Ly sed blood DST agar 79
i i i . Milk agar 79
i v . Sheep blood agar 80
v . Tween 80 agar 80
vi . Staph-■typing agar 80
3.3 Antibiotic containing discs for determining
sensitivity profiles and identifying
aminoglycoside-modifying enzymes
i. Antibiotic discs used for antibiotic 81
sensitivity profiles
ii. Antibiotic discs used for identifying 81
aminoglcoside-modifying enzymes
3.4 Materials and reagents used for detection 82
of specific haemolysins
4.0 Buffers used in plasmid isolation studies
i. Buffers used in Takahashi and Nagano method 83
ii. Buffers used in PHLS (Johnson) method 83
iii. Loading buffer 84
5.0 Identification of S. aureus 85
6.0 Detection of methicillin resistance 86
7.0 Determination of susceptibility to antimicrobial
agents
i. Determination of MIC by agar dilution 86
ii. Determination of MIC by broth dilution 87
7

iii. Selection of antibiotic break-points 88
7.1 Determination of bactericidal activity
i. Determination of minimum bactericidal 90
concentration (MBC)
ii. Determination of rate of killing by 90
time-kill curves
7.2 Determination of mutation rates to resistance 91
7.3 Microbiological assay of fluoroquinolones 92
8.0 Development of a system for typing MGRSA
8.1 Phage-typing MGRSA 93
8.2 Identification of aminoglycoside-modifying enzymes
i. Van de Klundert’s method 94
ii. Gene probing aminoglycoside resistance genes 95
8.3 Determination of antibiotic sensitivity profiles 96
8.4 Identification of physical properties of
use in biotyping
i. Identification of haemolysins by titration 96
ii. Haemolysis on sheep blood agar 98
iii. Egg-yolk reaction 99
iv. Tween 80 hydrolysis 99
v. Pigmentation on milk agar 99
8.5 Plasmid isolation by gel electrophoresis
i. Takahashi and Nagano method 101
ii. PHLS (Johnson) method 103
iii. Agarose gel electrophoresis 104
iv. Plasmid-sizing studies 104
APPENDICES
A,B,C Further information on methods used 308
8

RESULTS AND DISCUSSION
1.0 Antibiotic resistance in MGRSA - how 105
serious is the problem?
1.1 Resistance to aminoglycosides in MGRSA 118
1.2 Resistance to tetracyclines in MGRSA 122
1.3 Resistance to macrolides, lincosamides 124
and streptogramins
1.4 Resistance to trimethoprim in MGRSA 127
1.5 Resistance to chloramphenicol in MGRSA 128
1.6 Resistance to rifampicin in MGRSA 129
1.7 Resistance to ciprofloxacin in MGRSA 130
1.8 Resistance to fosfomycin in MGRSA 131
1.9 Resistance to fusidic acid in MGRSA 133
1.10 Resistance to novobiocin in MGRSA 135
1.11 Resistance to bacitracin in MGRSA 136
1 .12 Multiple antibiotic resistance in MGRSA 138
1 .13 Do MGRSA pose a therapeutic problem? 141
2.0 Therapeutic options for the treatment of
MGRSA infections or colonisation
2.1 Antibiotics available for treatment of 143
systemic infections
i. Antistaphylococcal activity of fosfomycin 144
ii. Antistaphylococcal activity of fusidic acid 149
iii. Antistaphylococcal activity of nitrofurantoin 151
iv. Antistaphylococcal activity of novobiocin 152
v. Antistaphylococcal activity of pristinamycin 153
v i . Antistaphylococcal activity of rifampicin 155
vii. Antistaphylococcal activity of teicoplanin 156
and vancomycin9

2.2 New antibiotics active against MGRSA 168
i. Antistaphylococcal activity of daptomycin 168
ii. Antistaphylococcal activity of DuP721 & DuP105 171
iii. Antistaphylococcal activity of paldimycin 172
iv. Antistaphylococcal activity of ramoplanin 173
v. Antistaphylococcal activity of new 14-, 174
15-, and 16- membered macrolides
vi. Antistaphylococcal activity of RP 59500- 175
an injectable streptogramin
vii. The lack of new agents available for 177
use in the treatment of infections due to MGRSA
2.3 Topical antibiotics for treatment of 179
MRSA carriage
i. Antistaphylococcal activity of azelaic 181
acid, nitrofurazone and silver sulphadiazine
compared to mupirocin
3.0 Do fluoroquinolones have a useful therapeutic
role against MGRSA?
3.1 Activity of the present clinically used
fluoroquinolones against MGRSA
i. Inhibitory and bactericidal activity of 190
ciprofloxacin, enoxacin, norfloxacin, ofloxacin
and pefloxacin against MGRSA
ii. Time-kill curves for ciprofloxacin, 193
ofloxacin and pefloxacin against MGRSA
iii. Spontaneous plate mutation rates to 200
resistance for ciprofloxacin, enoxacin,
norfloxacin, ofloxacin and pefloxacin compared
10

to nalidixic acid.
3.2 Fluoroquinolone resistance in MGRSA- development
of resistance in v i t r o , and properties of resistant
strains derived in vitro and from clinical sources
i. Development of fluoroquinolone resistance 202
in vitro during time-kill experiments
ii. Quinolone susceptibility patterns of clinical 210
isolates of ciprofloxacin-resistant MGRSA
iii. Are the reports of high incidences of 216
fluoroquinolone resistance in hospitals due
to the propensity of MGRSA to develop
fluoroquinolone resistance, or are they due to
the epidemic spread of resistant strains?
3.3 New fluoroquinolones with improved 225
activity against MGRSA
GENERAL DISCUSSION AND CONCLUSIONS
4.1 "Barber’s Law - the spread of resistant 229
staphylococci can be controlled either by not giving
antibiotics or by preventing the transmission of
the resistant organisms between persons"
4.2 Antibiotic options for the treatment of 239
MRSA infections and/or colonisation
i. Options available for treatment of 239
systemic infections
ii. Options for treatment of MGRSA carriage 243
4.3 Role of fluoroquinolones against MGRSA 244
11

4.4 The future problem of antibioticresistance in MGRSA
249
REFERENCES 252
APPENDICES 308
A. Influence of antibiotic carry-over in 308
fluoroquinolone MBC and time-kill experiments
B . Development of a biotyping system for MGRSA
i. Use of API STAPH profiles 309
ii. Use of antibiotic susceptibility profiles 310
iii. Determination of haemolysin profiles by 310
microtitre method
iv. Sheep blood haemolysis 312
v. Egg-yolk reaction 312
vi. Tween 80 reaction 313
vii. Pigmentation on Milk agar 313
viii. Plasmid-typing 314
C. Detection and Isoelectric focusing of 317
type SI enzyme
D. Suppliers of strains of MGRSA 319
SUMMARY OF CONCLUSIONS AND SUGGESTIONS FOR 320
FUTURE WORK
INSERTS Papers published
12

List of Tables
Number
I
II
III
IV
V
VIA
VIB
VII
Heading Page
Origins and phage-types of strains 66in study to determine extent ofantibiotic resistance in MGRSA
Antibiotics used in susceptibility and 73 other studies of antibiotic properties
Minimum inhibitory concentrations (MICs) 89 used to classify strains as sensitive, moderately resistant or resistant
Extent of antibiotic resistance 106to currently available antibiotics
Degree of resistance (%) to 23 non 117beta-lactam antibiotics
Inhibitory activity of fosfomycin, 145fusidic acid, nitrofurantoin, novobiocin, pristinamycin, rifampicin and teicoplanin compared to vancomycin against 100 strains of MGRSA from 26 centres in 19 countries
Influence of inoculum size on the 146 activity of fosfomycin, fusidic acid, novobiocin, pristinamycin, rifampicin, teicoplanin and vancomycin against 40 strains of MGRSA
Mutation rates to resistance to 147fosfomycin, fusidic acid, nitrofurantoin, novobiocin, pristinamycin, teicoplanin or vancomycin for 3 sensitive strains of MGRSA
13

VIII MICs and MBCs of teicoplanin and 159vancomycin at different times against 20 strains of MGRSA
X
XI
XII
XIII
XIV
Inhibitory and bactericidal activity 170determined against 80 strains of MGRSA of five new antibiotics possessingnovel chemical structures
Activity of 14-, 15-, and 16- membered 176macrolides, pristinamycin and RP 59500 against an international collectionof MGRSA comprising 13 erythromycin- sensitive strains, 30 inducibly resistant strains and 35 constitutivelyresistant strains
Inhibitory and bactericidal activity 182of azelaic acid, nitrofurazone , and silver sulphadiazine compared to that of mupirocin against 80 strains of MGRSA
Inhibitory and bactericidal activity of 191 clinically used fluoroquinolones compared to nalidixic acid against 160 strains of MGRSA
Pharmacokinetic properties of the 192fluoroquinolones
Fluoroquinolone susceptibilities ofparent and overgrowing strains following exposure to concentrationso f :
A: ciprofloxacinB: enoxacinC: ofloxacin
203204
D: pefloxacin205206
14

XVI
XVII
XVIII
XIX
XX
XXI
List
Number
1
Patterns of quinolone resistance in 212an international collection ofciprofloxacin-resistant MGRSA of clinical origin
Susceptibilities to acrosoxacin, 215fluoroquinolones and nalidixic acid of mutants of strain RFH 10 isolated from time-kill experiments
Phage-types of ciprofloxacin-resistant 219 and sensitive MGRSA from Texas andciprofloxacin-resistant MGRSA from 2 centres in Israel
Antibiotic susceptibility types and 221and biotypes of ciprofloxacin-resistant and sensitive MGRSA from Texas andciprofloxacin-resistant MGRSA from 2 centres in Israel
Susceptibilities of 160 ciprofloxacin- 227sensitive MGRSA and 40 ciprofloxacin-resistant MGRSA to current and futurequinolone agents
Supplementary phage-types of MGRSA 231used in antibiotic resistance studies Plasmid contents and sizes of a 315selection of MGRSA of Worldwide Origins
of figures
Heading Page
Problems due to MRSA in North-East 55Thames Regional Health Authority (RHA) compared to those in Yorkshire RHA from 1986-1990
15

2 Degree of multiresistance in MGRSA strains 139
A. killing curves for fosfomycin at 25 mg/1 161B. killing curves for fusidic acid 162
at 10 mg/1C. killing curves for nitrofurantoin 163
at 32 mg/1D. killing curves for novobiocin at 1.0 mg/1 164E. killing curves for pristinamycin 165
at 4.0 mg/1F. killing curves for rifampicin at 5.0 mg/1 166G. killing curves for strain SA 1 exposed 167
to various concentrations ofteicoplanin and vancomycin
A. killing curves for azelaic acid at 2.5 g/1 186B. killing curves for nitrofurazone at 60 mg/1 186C. killing curves for silver sulphadiazine 187
at 250 mg/1D. killing curves for mupirocin at 4.0 mg/1 187E. killing curves for ramoplanin at 188
1.0 mg/1 and 2.0 mg/1
A. killing curves of ciprofloxacin at 194various concentrations
B. killing curves of enoxacin at 195various concentrations
C. killing curves of ofloxacin at 196various concentrations
D. killing curves of pefloxacin at 197various concentrations
Plate
Plate 1 Growth of a number of different 100strains of MGRSA on Milk agar,Tween 80 agar, Egg-yolk glucose agar and Sheep blood agar
16

ACKNOWLEDGEMENTS
I am very grateful to my supervisors, Professor
W. Brumfitt and Professor J.M.T. Hamilton-Miller for
their support and advice. We are most grateful
to all those microbiologists worldwide who
supplied us with strains of MGRSA.
Three periods of working were spent away from
the Royal Free Hospital. Firstly, I am most
grateful to Dr S.G.B Amyes for allowing me to
spend 3 months at his laboratory in the Old
Medical School, University of Edinburgh. During
this stay I learnt various molecular biological
techniques and isolation and characterisation of
dihydrofolate reductases by isoelectric focussing.
Short periods of time were spent working in the
laboratories of Dr Alan Johnson (Antibiotic
Reference Laboratory) and Dr R. R. Marples
(Staphylococcal Reference Laboratory) at the Central
Public Health Laboratories, Colindale, London. I am
indebted to Dr Johnson for showing me his method
of staphylococcal plasmid isolation, and to Dr
Marples for his considerable help with phage-
typing .
Finally, I would like to thank my family for
their encouragement, perseverence and support during
my studies.
17

1. Taxonomy and Nomenclature of Staphylococcusaureus.
1.1 Historical Introduction,
Cocci were first classified on the basis of
their microscopically observed cellular arrangements
by Billroth in 1874 as part of his treatise on
Coccobacteria septica. Billroth believed that all the
round and rod-shaped bacterial forms were stages in
the development of a plant ("Coccobacteria septica” )
and he used the term "coccos" (seed) to describe the
smallest observed forms of this "plant” . He
differentiated these forms in terms of size or
arrangement into "micrococcos", "monococcos" ,
"diplococcos", "streptococcos", "gliacoccos" etc.
(Bulloch, 1960) .
Sir Alexander Ogston (Ogston, 1882) used the
descriptive term "staphylococcus" in his paper
entitled "Micrococcus Poisoning" to describe cluster
forming cocci observed in certain human pyogenic
abscesses. Ogston proposed this name, which is
derived from the Greek noun s taphy le ("a bunch of
grapes"); to differentiate these organisms from the
chain-forming cocci (streptococci) described by
Billroth. Rosenbach is credited as the first worker
to characterise Staphylococcus. In his original paper
published in 1884 , he initially proposed the names
Staphylococcus pyogenes aureus and Staphylococcus
18

pyogenes albus for orange and white staphylococci
which were indistinguishable from each other except
in colour. Hoewever, as trinomial names are invalid
in bacterial nomenclature, and as Rosenbach used the
binomial Staphylococcus aureus in a later part of
the same paper (Baird-Parker, 1972), Staphylococcus
aureus Rosenbach is accepted as the validly
published name for the nomenclatural type species of
the genus Staphylococcus (Editorial Board, 1958).
In 1906, Winslow and Rogers (Winslow & Rogers, 1906)
classified the staphylococci in the Subfamily
Paracoccaceae (parasites thriving well under anaerobic
conditions). The family was composed of four genera:
Diplococcus, Streptococcus, Aurococcus (staphylococci
producing orange pigment including S . aureus
Rosenbach) and Albococcus (staphylococci producing
porcelain white growth). In the other Subfamily,
Metacoccaceae (facultative parasites or saprophytes
thriving best under aerobic conditions) were the
genera Micrococcus, Sarcina and Rhodococcus. However,
in response to criticism from a number of
colleagues this scheme was modified in 1920 (Winslow
et a l ., 1920) by dispensing with the separate genera
Aurococcus and Albococcus in favour of the genus
Staphylococcus .
During the second quarter of the 20th Century
it was realised that micrococci and staphylococci
were closely related, so much so, that in the 1948
edition of Bergey * s Manual o_f Determinative
19

Bacteriology (Hucker, 1948) the staphylococci were
reclassified under the genus Micrococcus. Conversely,
although Shaw, Stitt and Cowan (1951) believed that
staphylococci and micrococci should belong to the
same genus they preferred the use of the generic
title of Staphylococcus to that of Micrococcus (Shaw
et a l ., 1951). By 1957, there had been yet another
change of view, in that staphylococci were found to
be substantially different from micrococci because of
the ability of the former to anaerobically ferment
glucose. Evans reintroduced the genus Staphylococcus
in the 7th edition of Bergey * s_______ Manual____of
Determinative Bacteriology (Breed et a l . , 1957), and
this distinction has remained until the present day.
Currently (Schleifer, 1986), the family
Micrococcaceae is composed of the genera Micrococcus,
Stomato coccus, Planococcus, and Staphylococcus.
20

2. The Occurrence and Pathogenicity ofStaphylococcus aureus
2.1 Distribution of S. aureus in na t u r e .
Following the reported isolation by Foggie in
1947 of staphylococci from the mouth, nose, vagina
and skin of ewes, Rountree et a l . (1956) were
prompted to screen various domestic and laboratory
animals for S . a ureus. These workers found nasal
carriage in a selection of dogs, guinea pigs, horses
and monkeys, but not in cows, rabbits and sheep, and
they concluded that the staphylococci appeared human
in character because of their similar phage types
to human isolates. Marandon & Oeding (1966, 1967)
established that there were biochemical and antigenic
differences between human and animal strains. Hajek
and Marsalek (1971) proposed that animal strains be
designated into six biotypes for inclusion into the
Baird-Parker classification of staphylococci (Baird-
Parker, 1963). These biotypes included strains from
sources as follows: A human, B swine and poultry, C
cattle and sheep, D hares, E dogs, and F pigeons.
Biotypes E and F were subsequently transferred to
S. intermedius (Hajek, 1976), however, apart from this,
the scheme remains largely unchanged (Parker, 1983).
21

2.2 The Human Staphylococcus aureus
i . Occurrence:
In man, S. aureus has a predeliction for the
anterior nares, although it can be isolated from a
number of other body sites (Williams, 1946).
Frequently, these organisms are commensals in the
healthy host - and their isolation under these
circumstances is referred to (by microbiologists) as
"staphylococcal carriage11. The carriage of S. aureus
in healthy people was first shown by Hallman
(1937). Subsequently, the incidence of nasal carriage
in healthy populations has been studied by a number
of workers (Williams, 1963). Usually, between 35 and
50 per cent of normal adults can be expected to
be nasal carriers of S . aur e u s . Rates of nasal
carriage have been found to be different in various
age groups. Casewell and Hill (1986) have estimated
the following prevalences with age: newborn (less than
5%), babies 2 weeks old (60-70%), 6-24 months (15-
25%), 5-6 year-olds (35-50%), normal adults (30-50%), and
the elderly (20-25%).
Aly et a l . (1977) found in vitro evidence that
S. aureus has a greater affinity for nasal
epithelial cells derived from carriers than from
non-carriers. A genetic predisposition to carriage
has been suggested by the findings of Noble et a l .
(1967) who provided evidence of a familial link.
22

i i . Factors influencing the incidence of nasal
ca rriage.
A number of studies (Williams, 1963, Noble et a l . ,
1964) have shown that exposure to antibiotics and
the hospital environment significantly increases the
rate of nasal carriage of S. a ureus. Casewell & Hill
(1986) estimate prevalences of nasal carriage of 20-
70% for nurses, 40-70% for patients two weeks in
hospital and 80-100% for babies two weeks in
hospital. Noble et a l . (1964) found that patients who
were not nasal carriers of staphylococci on
admission to hospital acquired nasal staphylococci
more often than those admitted carrying
staphylococci, regardless of antibiotic therapy. In
both groups higher incidences of acquisition were
found following antibiotic treatment. These findings
suggested that disruption of normal host flora
predisposes to staphylococcal colonisation. Carriers
of S . aureus have, following admission to hospital,
commonly become colonised with strains different to
those carried prior to admission and it has been
suggested that some strains of hospital staphylococci
have enhanced colonising ability (Williams, 1963).
Tuazon et a l . ( 1975) have reported an increased
rate of carriage compared to control populations in
regularly injecting drug abusers, and also in insulin
injecting diabetics. Patients undergoing long-term
haemodialysis have also been reported to have
23

increased rates of carriage (Kirmani et a l . , 1978).
2.3 Pathogenicity of Staphylococcus aureus.
The production of a staphylococcal infection is
determined by a complex interaction of host and
bacterial factors (Verhoef & Verbrugh, 1981). In
general, overt infection does not occur in the
healthy host with most strains. Predisposing factors
are required, for example skin trauma which can be
due to skin disorders/disease (eg. eczema), or can be
as a result of surgery, accident, or self induced
injuries such as intravenous drug abuse. Other
predisposing factors to infection are neutropenia, the
presence or insertion of foreign bodies, the use of
steroids, or virus induced damage to respiratory
mucosa. Certain genetic (e.g. chronic granulomatous
disease) or pathologically induced (e.g. J o b ’s syndrome)
defects in the h o s t ’s intracellular killing of
bacteria also predispose to infection (Quie et a l . ,
1974) .
Jeljaszewicz (1983) and many microbiologists are of
the opinion that S. aureus are ubiquitous micro
organisms of rather low virulence and pathogenicity
which possess the capability to cause severe even
lethal disease in the compromised host. This has
not always been so, during the 1950s a particularly
virulent form of S. aureus known as type 80/81
24

spread epidemically amongst hospitals (Rountree &
Freeman, 1955, Hennessey & Miles, 1958; Williams, 1959).
Strain 80/81 caused numerous infections in patients
with none of the above predisposing factors.
25

3. Staphylococcal Infections and their Treatment.
3.1 The Spectrum of Staphylococcal Infections.
S . aureus most often produces acute inflammatory
lesions, often mild and localized at the point of
entry of the bacteria. Such lesions may progress
(especially in compromised patients) to produce
generalized infection. This picture can be
modified by the production at the site of
localized lesions of certain toxins such as
epidermolytic toxin or the toxic shock syndrome
toxins which produce either extensive lesions or
acute toxaemia even in healthy patients. The
introduction of S. aureus into the blood stream
may also produce generalized infection such as
osteomyelitis, perinephric abscesses, septicaemia and
endocarditis. Such introduction can be as a result
of wound sepsis, pneumonia or osteomyelitis, or it
can be due to direct inoculation as in the case
of intravenous drug administration.
Most commonly, S . aureus is associated with the
production of pustular lesions, the most
characteristic of which is the skin boil. These
lesions are usually self limiting, and if this is
not so, the most effective treatment is drainage
of the pus. Following surgery, wound infection with
S . aureus can result in prolonged healing times, or
at the worst, extensive wound breakdown.
26

Staphylococcal pneumonia is a major problem inpatients requiring mechanical ventilation, and it is
a common cause of pneumonia in lungs debilitated
by bacterial or viral infection e.g. post
influenzal pneumonia.
S. aureus is a major cause of hospital
(nosocomial) sepsis. In a survey of the prevalence
of nosocomial infection in 47 hospitals in 14
countries by the World Health Organisation (Mayon-
White et a l ., 1988) S. aureus was found to be the
most common cause of surgical wound infection (14%)
and skin infection (18%). The opportunities
presented by the hospital environment for the
dissemination of S. aureus (e.g. via the hands of
staff) are numerous and epidemics of infection due
to particular S. aureus strains have occurred. For
example, in a study of the epidemiology and
control of staphylococcal infection in a maternity
hospital, Gillespie et a l . (1958) found there to be
several sources and modes of spread of
staphylococci which made the task of preventing
cross-infection all the more difficult.
3.2 Treatment of Staphylococcal Infections in the
Pre-Antibiotic Era.
Medical bacteriology was first practised in the
last quarter of the 19th Century following the
27

observations of Pasteur and Koch pertaining to the
germ theory of disease. Before this time the
concept of "contagion" had existed, and physicians
had adopted measures such as venesection and
treatment with various herbs and potions (see
Culpeper’s Herbal) to treat the diseased. The
following abridged case report (Paul, 1833) published
in "The Lancet” of 1833 illustrates what nowadays
might be considered as a typical case of post
operative staphylococcal (or streptococcal) sepsis.
"Case of Congenital Fungus Haematodes in whichAmputation of the Thigh was performed in the tenth week of the C h i l d ’s life".
THE LANCET 1833
A male child, seven weeks old, was brought tome by his parents with an enormous swelling ofthe right leg, which had all the characteristic marks of fungus haematodes.... The whole leg being involved in the morbid action of the disease, nothing but amputation above the knee even required the consideration of a moment....On the operation being fin ished...the child sucked almost immediately... and improved daily. The ligatures were all away by the tenth day after the operation,and the greater portion of the stump had united. A few days later the stump became very tense and hot. The next day a blush of erysipelatous redappeared over it and the edges of the woundappeared livid, there was scanty purulent discharge. Following this the erysipelatous inflammation extended to the scrotum and abdomen, and then upthe back and across the abdomen. The childdeveloped a high fever and died.
The surgeon who performed the operation was of
the opinion that the operation was a complete
success at the tenth day and begged the mother
to remain only another week in the hospital.
Following the c h i l d ’s illness, the surgeon learnt

that the c h i l d ’s mother had often taken him to
sit with, and be cradled by a nurse suffering
from aggravated erysipelas. In the surgeons own
words ”To my mind the evidence is here quite
conclusive, that the disease was communicated to
the child by the sick nurse”
Despite the great discoveries made during the
Golden Age of bacteriology from 1875-1925 and the
advances made in medicine with regard to surgical
technique and the need for antisepsis at
operations, by 1935 there were still no treatments
of proven efficacy for acute, invasive
staphylococcal (or streptococcal) infection (Topley and
Wilson, 1936). With this background, it is easy to
appreciate the significance of the introduction of
the first antimicrobial agents - the sulphonamides.
29

3.3 The Antibiotic Era 1940 - 1960
i . The Sulphonamides.
In 1935, a startling chemotherapeutic discovery
was announced by Domagk in Germany. He reported
that injection of the hydrochloride of the dye -
"4- sulphamido-2: 4-diaminoazobenzol" (Prontosil
rubrum) protected mice against streptococcal
infection. In 1936, Colebrook and Kenny (Colebrook &
Kenny, 1936) reported that injection of this dye
had a remarkable curative effect both in mice
infected with haemolytic streptococcus and in human
puerperal infections due to haemolytic
streptococcus. The active principle of Prontosil
rubrum was soon discovered to be p-amino-benzene
sulphonamide - "sulphanilamide", and from this
initial structure many hundreds of analogues were
prepared. Following the introduction into clinical
use in 1937 of sulphanilamide and its subsequent
analogues, the number of deaths in the U.K. due
to puerperal pyrexia in 1935 were halved by 1940
(Barber, 1960) .
Unfortunately, the sulphonamides were to prove
less successful against staphylococcal infections.
Not only was clinical experience in treating
severe staphylococcal sepsis disappointing, bacterial
resistance also readily occurred (Spink, 1954).
30

ii. The Introduction of Penicillin.
In 1929, Fleming reported the production of a
bacteriolytic substance (which he called penicillin)
from a mould contaminating a culture plate of
staphylococcus (Fleming, 1929). Further development of
penicillin was hindered due to problems of
producing sufficient quantities of pure substance.
In 1941, Abraham et a l . reported the results of
the first therapeutic trial with penicillin. They
found penicillin to be of low toxicity, and also
effective in treating staphylococcal and
streptococcal infections (Abraham et al. , 1941). It
was still difficult to produce penicillin in large
quantities, and because of the War, information
concerning the development of penicillin was
restricted (Clarke et a l . , 1949).
From 1942 onwards, increasing supplies of
penicillin became available, and usage of this drug
has had a most profound effect on mortality due
to staphylococcal infections. For instance, a
reduction of mortality from 80% in the pre
penicillin era to 28% was seen for patients with
staphylococcaemia following the introduction of
penicillin into clinical use at the University of
Minnesota (Spink & Hall, 1945). Penicillin has had a
major and lasting effect on the treatment of
previously fatal diseases such as haemolytic
streptococcal infection, pneumococcal pneumonia,
31

bacterial endocarditis, gas gangrene and a variety
of other infections. However, against staphylococci
the efficacy of penicillin was short-lived, because
of the development of bacterial resistance.
Therapeutic failure due to development of
staphylococcal penicillin resistance during
penicillin treatment was first reported in 1942
(Rammelkamp and Maxon, 1942). According to the
authors the penicillin resistance was not due to
production of a "penicillinase". It had previously
been shown (Abraham et a l . , 1941) that staphylococci
could acquire penicillin resistance in vitro by
serial exposure to increasing concentrations of
penicillin. This "acquired" resistance was believed
to be responsible for the early therapeutic
failures found with penicillin. In 1944, Kirby
reported the isolation of staphylococci naturally
resistant to penicillin, in which resistance was
due to the production of a "penicillinase".
Subsequently, Bondi and Dietz (1945) showed that
penicillin-resistant clinical isolates of S. aureus
did not acquire resistance during therapy, but were
naturally resistant due to the production of
penicillinase.
In 1947, Barber announced that the incidence of
S . aureus resistant to penicillin was increasing at
an alarming rate (Barber, 1947). In a survey
conducted from April-N ovember, 1946 an incidence of
32

12.5% staphylococcal penicillin resistance was
found, which had increased in a second survey
conducted from February to June, 1947 to 38%.
Increasing staphylococcal penicillin resistance was
was also reported from hospitals in the USA
(Finland & Haight, 1953), Australia (Rountree & Thomson,
1949), France (Chabbert & Terrial, 1952), Scandinavia
(Laurell & Wallmark, 1953) and India (Gupta &
Chakravati, 1954). By the late 1940s, staphylococcal
resistance to penicillin had increased to such an
extent that the use of penicillin for treating
infections due to S. aureus virtually had to be
abandoned (Barber & Rozwadowska-Dowzenko, 1948).
iii . The use o_f Streptomycin, Tetracycline,
Erythromycin and Chloramphenicol against S. aureus.
Streptomycin was introduced in 1944, and because
of its activity against Mycobacterium tuberculosis
its use was reserved for the treatment of
tuberculosis. When streptomycin was used for the
treatment of staphylococcal infections resistant
strains rapidly emerged. For example, no
streptomycin-resistant S. aureus were isolated at
the Royal Prince Alfred Hospital in Sydney,
Australia in May 1949, whereas over 20% of S .
aureus isolated in January, 1950 were streptomycin-
resistant. Furthermore, all the streptomycin-resistant
strains were also penicillin-resistant (Rountree ejt
33

a l . , 1951).
Similarly, following the introduction of
chloramphenicol and tetracycline increasing levels
of resistance were found in hospitals in France,
Australia and the USA (Barber, 1960). Once the
toxic effect of chloramphenicol was realised, its
use was restricted, and following this,
staphylococcal resistance to chloramphenicol declined
(Kirby & Ahern, 1953). These experiences with new
antibiotics led to the realisation that there was
a correlation between the extent of antibiotic
usage and development of staphylococcal resistance.
This led to calls that new antibiotics with good
antistaphylococcal activity (e.g erythromycin) be
held in reserve (Barber & Burston, 1955; Beavan &
Burry, 1956). Unfortunately, even when erythromycin
usage was restricted, the ability of S. aureus to
develop resistance to this compound was such that
resistant strains readily appeared (Lepper et a l . ,
1954).
i v . The Introduction of Me thicillin.
By the mid- 1950s, the problems of antibiotic
resistance in S . aureus were so serious that there
was great concern regarding the future effective
use of antibiotics for treating staphylococcal
infections (Barber & Burston, 1955; Finland, 1955).
Furthermore, as the 1950s progressed, there were
34

increasing reports of epidemics of infection
involving particularly virulent ("type 80") strains
(Williams, 1959). A considerable number of the
staphylococci from outbreaks during this period
were multiply resistant to four or more
antibiotics (Leading Article, 1965; Bulger & Sherris,
1968; Ridley et a l ., 1970). In view of the above
problems, the introduction of methicillin in 1960
was most welcome, especially on account of its
resistance to "penicillinase". In association with
methicillin, the implementation of stricter cross
infection control measures and antibiotic control
policies resulted in a major decrease in the
number of multiple resistant staphylococci isolated
during the 1960s (Bulger & Sherris, 1968; Ridley e_t
a l ., 1970).
v . The Ef f icacy of Methicillin and its
Derivatives.
In the British Medical Journal of September 3,
1960, a series of papers was published detailing
the in v i t r o , in vivo and clinical properties of
a new antibiotic, BRL 1241. BRL1241 was
subsequently named methicillin and commercially
called "Celbenin". Methicillin was resistant to
inactivation by staphylococcal penicillinase. In
terms of antibacterial activity, methicillin was
active against staphylococci and streptococci, but
35

not Gram-negative organisms (Knox, 1960). In clinical
use, methicillin was found to be non-toxic, and was
effective in the treatment of infections due to
penicillin-resistant staphylococci (Douthwaite &
Trafford, 1960; Stewart et_____a l ., 1960) including
staphylococcaemia (Allen et a l ., 1962).
A disadvantage of methicillin was its
instability in the presence of acid, thus it had
to be injected. The introduction of the acid-
stable isoxazolyl penicillins (cloxacillin , oxacillin,
dicloxacillin and flucloxacillin) overcame this
problem (Leading Article, 1964). The isoxazolyl
penicillins possess contrasting properties, for
example flucloxacillin is more active than
methicillin, whereas methicillin is more stable to
staphylococcal penicillinase than flucloxacillin
(Frimodt-Moller et a l . , 1986). Nevertheless, these
contrasting properties are largely self-cancelling,
and such is the efficacy of these compounds that
even more than 25 years since their introduction
they are still recommended for the initial
treatment of severe staphylococcal infections (Garrod
et a l ., 1981; Eykyn, 1988).
36

4.0 Staphylococcus aureus Resistant to Methicillin(MRSA).
4.1 Occurrence of MRSA - 1960-1975.
When methicillin was introduced into clinical
use in 1960, no strains of S. aureus were found
to be resistant to this compound (Thompson et a l .,
1960). However, it was not long until resistant
strains were reported. In 1961 Jevons (Jevons, 1961)
found three strains from a total of 5,440
screened to be methicillin-resistant. These strains
all originated from the same hospital, but
methicillin had only been used to treat one
patient at this institution. There was no evidence
to suggest that the resistant strains had arisen
as a consequence of use of methicillin. Rolinson
(1961) showed that the methicillin resistance in
the strains reported by Jevons was heterogeneous,
that is most cells in a culture appeared
sensitive, however a small proportion were highly
resistant. Furthermore, the highly resistant
organisms did not destroy methicillin by virtue of
production of a methicillin degrading enzyme.
By 1963, Jevons (Jevons et al . , 1963) had
screened 27,479 cultures sent to the Staphylococcus
Reference Laboratory, and this revealed a gradually
increasing incidence of methicillin-resistant S .
aureus (MRSA). In the period Jan-March 1961 the
37

incidence of methicillin resistance was 0.055%, and
from July-September 1962 it was 0.81%. According
to Parker & Hewitt (1970) two independent surveys of
the occurrence of MRSA indicated that there was
a moderate increase until 1963 followed by a
stationary period until 1967. Following 1967 there
was a further increase in MRSA. In 1967 the
incidence of MRSA was 1.0%, however in 1969 it
was 4.1%. These figures refer mainly to isolates
from centres in the U.K.
During the late 1960s, the problems posed by
MRSA were more serious in some countries e.g.
Switzerland, Denmark, and France than others e.g. USA.
For instance, Kayser (1975) reported that from 1966-
1971 around 20% of staphylococcal disease in
hospital inpatients was due to MRSA. In Denmark
between 1966 and 1970 from 10-15% of
staphylococcal strains were methicillin-resistant
(Rosendal et a l . , 1976) . This contrasts with the
USA where the first outbreak of MRSA infection
did not occur until 1967, before which only a few
cases of clinical illness due to MRSA had been
reported (Barrett et a l . , 1968). During the first
half of the 1970s, there was a noticeable decline
in the incidence of MRSA in many countries
(Kayser, 1975; Rosendal et a l . , 1976; Ayliffe et a l . ,
1979).
38

4.2 Treatment of MRSA 1960-1975.
i . Therapeutic options and the need for combination
therapy.
The introduction of methicillin combined with
more rigorous hospital infection and antibiotic
control measures resulted in a reduction of the
multiple antibiotic resistant hospital staphylococci
which had caused so many problems during the late
1950s (Parker et a l . , 1974). Although the frequency
of isolation of MRSA increased during the 1960s
the clinical problems posed by these organisms in
terms of the frequency, severity and options for
the treatment of infections were not comparable to
those presented by the multiple antibiotic
resistant staphylococci of the late 1950s.
MRSA isolated during the 1960s and early 1970s
often displayed a variety of reactions with Group
III phages or they were non- typable (Parker &
Hewitt, 1970; Kayser, 1975). They were nearly always
resistant to streptomycin and tetracycline, and
commonly resistant to erythromycin (Ridley et a l .,
1970; Parker & Hewitt, 1970). Resistance to other
antibiotics usually reflected antibiotic usage
adopted at particular institutions. For instance,
Ridley et al. , (1970) showed that use of
chloramphenicol in just one ward of St Thomas's
39

Hospital resulted in more than half the hospital's
chloramphenicol-resistant S. aureus isolates. From
1966 onwards, this hospital restricted the
prescription of erythromycin-like drugs (e.g
spiramycin, oleandomycin and lincomycin), novobiocin
and sodium fusidate primarily for use in MRSA
infections. Furthermore, use of either erythromycin,
erythromycin-like drugs, novobiocin, rifamycins or
sodium fusidate as monotherapy had often resulted
in the development of bacterial resistance, and
combination therapy using these agents was strongly
recommended (Jensen, 1968; Garrod, 1968; Jensen & Lassen,
1969).
Vancomycin, was known to be effective in the
treatment of severe staphylococcal infections, but
was regarded as too toxic compared to the above
agents, hence during the 1960s it was often kept
as a reserve drug (Garrod, 1968; Kucers, 1972).
i i . Treatment with topical antibiotics.
The topical antibiotics available for use
against MRSA during the first half of the 1960s
were primarily neomycin, bacitracin and Fucidin
(fusidic acid). To prevent the emergence of
resistance, these agents were often used in
combination, for example the cream "Naseptin”
consisted of neomycin sulphate and chlorhexidene
40

hydrochloride. Following the introduction of
neomycin and bacitracin into clinical use during
the early 1950s there were few reports of
resistance for several years. It was believed that
by using the agents in combination with another
antibiotic (e.g. neomycin + bacitracin) or antiseptic
(e.g "Naseptin” ) that even if resistance developed
to one agent the other would prevent growth of
the resistant organism. However, during the early
1960s there were increasing reports of neomycin-
resistant S. a ureus, and by 1965 neomycin-resistant
strains were widespread (Leading Article, 1965).
Lowbury et a l . (1964) suggested that the spread
of neomycin resistance was probably due to the
widespread dissemination of a resistant strain, and
not the development of resistance during individual
treatment episodes . Once in the hospital
environment use of neomycin favoured the spread of
this strain (Mitchell, 1964). In 1965, Rountree &
Beard (1965) reported the spread of S. aureus which
was initially resistant to neomycin, but then
became resistant to bacitracin as well. This has
been attributed to the treatment of patients
harbouring neomycin-resistant strains with neomycin-
bacitracin ointment. Hence, the spread of neomycin-
resistant strains limited the use of such
ointments.
Fusidic acid is an irritant of mucous
membranes, and is consequently inappropriate for
41

topical use against S. aureus nasal carriage.
Staphylococci resistant to fusidic acid have been
isolated from patients receiving topical Fucidin, or
oral fusidic acid therapy (Pattison & Mansell, 1973;
Lowbury et a l ., 1962). Ayliffe et a l . t (1977) have
correlated the amount of fusidic acid used with
the appearance of resistant strains in a burns
unit, hence on restricting topical fusidic acid
usage there was a decline in the number of
fusidic acid resistant strains. In some centres
during the 1960s there was a reluctance to use
fusidic acid topically because of its use for
treating infections due to MRSA. When gentamicin
ointment became available in the late 1960s, and
was found to be more effective than "Naseptin”
(Williams et a l ., 1967) there was considerable usage
of gentamicin (Editorial, 1977) with consequences
which are described later.
iii . Treatment______of_____ MRSA______infections_____ with
penicillinase-resistant__________penicillins__________and/or
cephalosporins.
In 1968 Benner and Kayser reported their
experiences for 26 patients with significant
infections due to MRSA who had been given
intensive therapy with penicillinase-resistant
penicillins and/or cephalosporins. They found that
although 18 of the 26 patients were cured or
42

improved by this therapy, all those patients
infected with highly resistant organisms (oxacillin
MICs of 62.5 mg/1 or greater) died of infection.
Benner and Kayser had believed that treatment of
infections with penicillinase-resistant penicillins
or cephalosporins would be effective because only
a few cells (usually less than 1%) in a population
of MRSA possessed high-level resistance to
methicillin or oxacillin. Following Benner and
Ka y s e r ’s paper, and also other studies by French
workers most microbiologists were left in little
doubt that methicillin is inappropriate for
treating MRSA infections (Leading Article, 1968). In
1970, Acar et a l . (1970) concluded that primary
treatment either with a cephalosporin (cephalothin,
cephaloridine, or cephalexin) or with a cephalosporin
combined with an aminoglycoside (kanamycin or
gentamicin) failed to eradicate staphylococcaemia
after three days treatment.
Despite the evidence that treating
staphylococcal infections with penicillinase-resistant
penicillins or cephalosporins gave less than
optimal results, some workers still saw fit to re
evaluate the efficacy of these agents in
eradicating MRSA. For example, two trials from the
MRC Industrial Injuries and Burns Unit (Lowbury e_t
a l ., 1977; Kidson et a l ., 1979) concluded that oral
flucloxacillin for treatment of burns eliminated or
reduced MRSA in significantly greater numbers
43

compared to untreated controls. Furthermore,
cefamandole either alone or in combination (with
tobramycin) has been found to be effective in MRSA
infections (Coppens et a l . , 1983; Frongillo et a l . ,
1986). When assessing such findings it is
important to consider that "cure" or "improvement" is
a complex interaction depending upon the severity
of infection, the nature of the infecting organism
and the condition of the host. Many workers still
retain the opinion that penicillinase-resistant
penicillins or cephalosporins are not appropriate
for the treatment of infections due to MRSA
(Hackbarth & Chambers, 1989a).
4.3 The Detection and Nature of Methicillin-
Resistance.
i . Detection
In 1964, Barber reported that the growth of
MRSA on nutrient agar containing methicillin (even
at subinhibitory concentrations) was much less
luxuriant than that seen on nutrient agar plates
containing no antibiotic. Further work showed that
growth in the presence of methicillin was enhanced
by an excess of electrolytes (5% NaCl or 7.5%
( N H ^ ^ S O ^ ) or decrease in agar concentration. In
the same year Sutherland and Rolinson (1964) confirmed
44

Rolinson's earlier investigations showing that
cultures of MRSA consisted of mixed populations in
which the majority of cells were of normal
sensitivity to methicillin with a minority showing
methicillin resistance. Furthermore, the resistance
of MRSA was "intrinsic" and was not due to an
increased ability to inactivate the drug. These
workers also showed the importance of using a
large inoculum to detect methicillin resistance.
Subsequently, it has been shown that temperature,
pH, visible light, growth in anaerobic conditions,
exposure to chelating agents and pre-exposure to
beta-lactam antibiotics all influence the expression
of methicillin resistance (Mathews & Stewart, 1984).
Parker and Hewitt investigated the influence of
temperature on the detection of methicillin
resistance (Parker & Hewitt, 1970) and showed that
initial surveys (e.g. Jevons, 1961) to detect the
prevalence of MRSA under-estimated the true numbers
of these organisms. This was because in earlier
surveys susceptibilities were read after 18 hours
incubation at 37°C, and under these conditions only
a few cells in a culture appear resistant
(Rolinson, 1961, Sutherland & Rolinson, 1964). Annear
(1968) showed that methicillin resistance was much
greater at 30°C, because a greater proportion of
cells expressed methicillin resistance at 30°C
(Dyke, 1969; Parker & Hewitt, 1970).
A great many reports assessing methodologies for
45

detecting methicillin resistance have appeared in
the literature over the years (Hansen & Freedy, 1984;
Jolly & Goldberg, 1989; Mouton et a l ., 1989). In the
USA attempts have been made to introduce standard
tests for determining methicillin resistance
(McDougal & Thornsberry, 1984). However, in the U.K
no such guidelines exist and the techniques used
to conduct and interprate methicillin sensitivity
testing can vary considerably between laboratories.
i i . Nature of Methicillin Resistance
Throughout the 1960s there was considerable
confusion regarding the nature of methicillin
resistance and the genetic factors governing it.
Although many workers (e.g. Rolinson, 1961; Barber,
1964) reported that methicillin resistance was not
due to a "methicillinase” , others suggested that
enzymatic inactivation might be responsible (Stewart
& Holt, 1963; Eriksen & Erichsen, 1963). Dyke (1969) in
a comprehensive survey of the penicillinases of
MRSA found none with an increased efficiency of
hydrolysis of methicillin. Staphylococcal beta-
lactamase has been shown to hydrolyse methicillin
slowly at MIC concentrations of the drug ie.
0.0019% the rate of benzylpenicillin (Hamilton-
Miller & Ramsay, 1967). Lacey & Stokes (1977) have
suggested that staphylococcal beta-lactamase
significantly inactivates flucloxacillin, however, few
46

other workers seem to share this opinion.
During the 1970s it was shown that penicillin
(and other beta-lactam antibiotics) kill bacteria by
inhibiting penicillin-sensitive enzymes involved in
the final stages of peptidoglycan synthesis (Spratt,
1978). In 1980, Brown and Reynolds suggested that
methicillin resistance was due to a modification
of cellular penicillin binding proteins (PBPs), or
the presence of a new PBP with a reduced
affinity for methicillin. Following this observation
a great deal of work has been performed on the
PBPs of S. aureus (Hackbarth & Chambers, 1989b).
Currently, it is believed that methicillin
resistance is due to the production of a modified
protein- P B P 2 f- which is both thermosensitive and
acid-sensitive. Hence, at 30°C P B P 2 T is expressed
in greater amounts than at 37°C, similarly at pH
5.2, PBP 2 f is not expressed (Lyon & Skurray, 1987).
This theory applies to "true methicillin-resistant"
strains. Tomasz et a l . (1989) have suggested that
"borderline" resistance to methicillin may be due
to a number of different types of mechanism such
as hyperproduction of beta-lactamase. Beta- lactamase
hyperproducers can be distinguished from "true" MRSA
because methicillin resistance due to beta-lactamase
hyper-production is lost in the presence of
clavulanate. The explanation that methicillin-
resistance is due to the production of PBP 2* may
be too simplistic. For example, using this theory
47

Madiraju et a l . (1987) could not explain the
influence of NaCl on methicillin resistance.
Neither could Murakami and Tomasz (1989) account for
strains with high homogeneous expression of
methicillin resistance containing the same amounts
of PBP 2' as isolates in which 99.99% of the
cells had MICs of 2.0 or 4.0 mg/1.
Because PBP 2 f is considered to have a reduced
affinity for all beta-lactams, methicillin resistance
results in cross-resistance to all beta-lactam
agents. However, differences in affinities or modes
of action may result in some beta-lactams (e.g.
certain cephalosporins, imipenem) possessing greater
activities against MRSA than others (Chambers &
Sachdeva, 1990). Furthermore, the development of a
beta-lactam with increased affinity for PBP 2 T is
a real possibility e.g. BRL 44154 (Blake et a l . ,
1990).
48

5.0 Use of Gentamicin and the Development ofResistance
i . Use of Gentamicin.
The antimicrobial activity of gentamicin was
first reported in 1963 by a number of workers
who showed it to be a bactericidal antibiotic
possessing broad-spectrum activity (Finland, 1974).
Gentamicin was first given to four patients in
March 1962, and although it proved succesful it
also produced vestibular toxicity (Jackson, 1969).
Subsequent decreases in dosage, and the introduction
of laboratory monitoring of blood levels have
greatly reduced this risk. During the 1960s, the
primary systemic use of gentamicin was in the
treatment of urinary tract infections or
septicaemia due to multiresistant Gram-negative
organisms (Neu, 1974). Comparatively little data was
compiled of gentamicin's activity against
staphylococci, and it was not regarded as amongst
the systemic agents of first choice for the
treatment of staphylococcal infections. In 1966,
Barber and Waterworth reported gentamicin to have
good activity against neomycin-sensitive and
neomycin-resistant hospital staphylococci, and
suggested that use of gentamicin as a spray for
clearing staphylococcal carriage should be
considered.
49

In a trial by White (1964) of the use of
gentamicin as a nasal ointment it was found that
topical application was effective in reducing
carrier rate and dissemination of staphylococci.
Gentamicin was regarded as an excellent agent for
the treatment of skin infections due to its
broad-spectrum activity (including Ps. aeruginosa), and
lack of non-irritant and skin sensitizing effects.
Because of these properties use of gentamicin
ointment steadily increased. For example, at the
Belfast City Hospital gentamicin usage increased
from 15,804 x 15 gram units in 1974-75 to 23,020 x
15 gram units in 1975-1976 (Wyatt et a l . , 1977).
In 1968, at an international symposium on
gentamicin it was reported that gentamicin was
active against multi-resistant strains of MRSA
(Hoeprich, 1969). Furthermore, Waitz and Weinstein
(1969) found that only 4 of 1352 isolates of S .
aureus were not inhibited by 10 mg/l gentamicin.
Despite its activity against multiple-resistant
staphylococci, clinical trials assessing gentamicin
for treating staphylococcal infections produced
contrasting results. Richards et a l . (1971) found
gentamicin to be satisfactory in the treatment of
non life-threatening infections, however reservations
were expressed over gentamicin's efficacy in
serious infections. Klastersky et a l . (1975) reported
gentamicin to be less efficacious than
cephaloridine in the treatment of staphylococcal
50

infections and suggested that gentamicin should not
be used as a first-line drug for such infections.
Because of in vitro observations of synergy
between cephalosporins and aminoglycosides a number
of trials have been performed using such
combinations. Against MRSA cefamandole and
tobramycin were found to be nearly as effective
as vancomycin (Klastersky & Van der Auwera, 1986).
i i . Development of Resistance to Ge ntamicin.
Prior to 1975, staphylococcal resistance to
gentamicin was virtually unheard of (Lacey &
Mitchell, 1969). However, because of the increasing
topical usage of this compound it was predicted
that as with neomycin, resistance would also occur
to gentamicin (Lacey, 1975). Soon after this
prophecy single strains of S. aureus possessing
plasmid-mediated gentamicin resistance were reported
in the U.K and France (Porthouse et a l . , 1976;
Soussy et a l . , 1975). In February 1976, Speller ejt
a l . (1976) reported an outbreak of
colonization/infection at the Westminster Hospital
due to gentamicin-r esis tant S. a u r e u s , and by the
end of the year MRSA resistant to gentamicin had
been reported in the U.K and France (Shanson .et
a l . , 1976; Soussy et a l . , 1976). Whereas the early
1970s had seen the decline of the MRSA, these new

reports of gentamicin resistant MRSA - (MGRSA) were
to herald a new era where once again S. aureus
possessing resistance to many antibiotics was to
become a major problem in hospital medicine.
52

6.0 Increasing Hospital Problems due to M G R S A .
Casewell has suggested that the perception of
the problems caused by MRSA depends upon o n e ’s
historical and geographical viewpoint (Casewell,
1986). Epidemiological data shows that the problems
caused by MRSA are greater in some hospitals than
others. Where outbreaks occur, the institution of
control measures can result in considerable
fluctuations in the number of MRSA isolated over
a few months. Hence, the incidence of MRSA can be
influenced by many factors including the frequency
and time of screening, the inclusion of multiple
isolates from the same patient, and the nature
(burns, surgical etc) of the wards or hospitals
under study.
i . Problems due to MGRSA in Great B r i t a i n .
For the period 1976-1980 few serious problems
due to MGRSA were reported from hospitals in the
United Kingdom, however, in Eire 55 patients in
Dublin hospitals were reported to have had a
MGRSA bacteraemia between 1976 and 1979 (Leading
Article, 1981). Cafferkey et a l . (1985a) showed that
the number of new patients from whom MRSA was
isolated peaked at c. 120 early in 1980 and
then gradually declined to c. 40 in 1984 following
the introduction of effective control measures. The
53

Dublin MRSA was characteristically gentamicin
resistant (Cafferkey et a l . , 1983), whereas in the
U.K the presence of gentamicin resistance has been
more variable (Kerr et a l ., 1990).
In a questionnaire survey conducted for the
period 1982-1983 it was found that although MRSA
were widely distributed in the U.K., they occurred
infrequently in many hospitals and only caused
serious problems in a small number of hospitals,
mainly located in the Thames Regional Health
Authority (Cooke et a l . , 1986). A two region survey
of the occurrence of MRSA in hospitals in the
North East Thames (NETRHA) and Yorkshire Regional
Health Authorities over the last quarter of 1985
showed 190 new cases of colonisation and 136 new
cases of infection in NETRHA compared to 10 and
21 respectively in Yorkshire (Communicable Disease
Report, 1986). The high frequency of MRSA in
hospitals in the South East of England was
attributed to the spread of a single strain
termed "epidemic MRSA" or EMRSA (Marples et a l . ,
1986) .
In 1986, a national surveillance scheme to
detect the prevalence of MRSA was initiated, and
Figure 1 compares the number of new cases of
colonisation or infection in NETRHA and Yorkshire
regions at 6 monthly intervals between 1986 and
1989. From Figure 1 it can be seen that there
is a continuing problem due to MRSA in the South
54
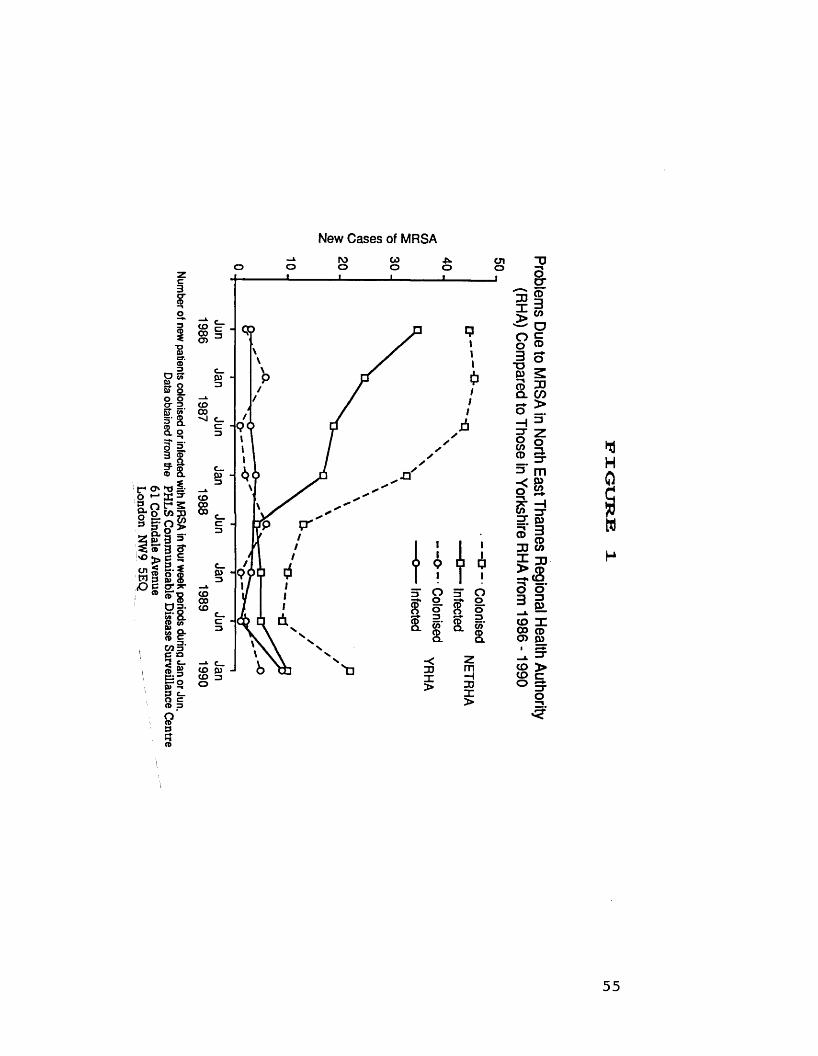
Number of new
patients colonised
or infected with
MRSA in
four week periods
during Jan
or Jun.Data
obtained from
the PHLS
Comm
unicable Disease
Surveillance Centre
61 Colindale
Avenue London
NW9
5EQ —
-
New Cases of MRSAIV)o 09o o oio
CO00CT)
CO00'nJc
3CO8
CO00CO
mCOCOo
33X>oo3TDP)—nCDQ.
133g;CD3CO
OcCD
30 CD >O _
ZTOCOCD
2S 'g;i"CD33X>
oZTmo>CO
pj3CDCO33CD
- CS-o o3 g5 x22 ® o> p>
rr>cCOCOo
55

East, so much so that a combined working party of
the Hospital Infection Society and British Society
for Antimicrobial Chemotherapy issued guidelines in
(Working Party, 1986 and 1990) for the control and
eradication of these organisms.
i i . Problems due to MGRSA in Australia.
Gentamicin resistance rapidly appeared in
Australian strains of MRSA. For example at the
Royal Melbourne Hospital 6% of MRSA were
gentamicin-resistant at the end of 1978, however
one year later more than 70% of isolates were
gentamicin-resistant (McDonald et______ a l . , 1981).
Similarly, at St Vincent's Hospital, Darlinghurst,
New South Wales 32% of MRSA were gentamicin-
resistant in 1978 increasing to 96% in 1981 (King
e t a l . t 1981). From 1979 onwards, teaching hospitals
in Eastern Australia encountered serious problems
due to MRSA (Medical Journal of Australia, 1982).
For example, six university teaching hospitals in
the Melbourne metropolitan area reported MRSA to
comprise 20% to 40% of S. aureus isolates in
1979, however in rural hospitals MRSA seldom posed
a problem (Pavillard e t a l . t 1982). During 1981,
some university teaching hospitals in Melbourne
reported 50% of all S. aureus isolates to be
MRSA, and over a 12 month period (Jan. 1979- Jan
1980) in one representative 700-bed hospital MRSA
56

was isolated from 545 patients (Pavillard et a l .,
1982). The seriousness of this problem attracted
a great deal of publicity. Headlines in the
"popular press" such as ’Killer bug in NSW
hospitals - epidemic warning* and ’Shock killer
infection’s 12 v ictims’ generated increased public
awareness of the problem to the extent of causing
widespread alarm (Blum, 1982).
MRSA continues to be a major problem in
teaching hospitals in Eastern Australia. In 1986-
1987 MRSA comprised c. 25% of S. aureus isolates
from Queensland and Victoria (Turnidge et a l . ,
1989). Genetic analysis has shown that one strain
- "eastern Australian MRSA" has been largely
responsible for the continuing epidemic in Eastern
Australia. It has also been shown that this
strain is closely related to the epidemic MRSA
isolated in South-East England (Townsend et a l . ,
1987) .
iii. Problems due to MGRSA in the U S A .
MRSA resistant to gentamicin first started to
cause problems in hospitals in the USA in 1975
(Crossley et a l ., 1979) . Unlike the situation in
Australia and Great Britain where aminoglycoside-
resistant strains were characterised by resistance
to gentamicin and tobramycin, in the USA there
were outbreaks due to strains possessing a number
57

of different aminoglycoside resistance patterns. For
this reason the Americans have often used the
term MARSA - methicillin-aminoglycoside resistant S .
au r e u s . In the first outbreak of MRSA resistant
to aminoglycosides, 75% of the MARSA were resistant
to tobramycin, amikacin and kanamycin yet
susceptible to gentamicin, sissomicin and netilmicin
(Crossley et a l . , 1979). Similarly, Craven et al
(1981) reported an extensive outbreak over a 16
month period (Sept. 1978- Jan. 1980) in a surgical
building due to tobramycin-resistant MRSA, 30% of
which were sensitive to gentamicin.
In other areas a similar pattern of events
occurred to that experienced in Australia and
Great Britain. Schaefler et _al_. (1981) first
detected MGRSA from hospitals in New York City in
1978, and by the spring of 1980 more than 80% of
MRSA received from hospitals in the New York city
area were gentamicin-resistant. Similarly, Dunkle et
a l . (1981), Graham et a l . (1980), Linnemann et a l .
(1982) and Saroglou et a l . (1980) have all reported
hospital outbreaks due to the emergence of MGRSA.
Haley et al (1982) reviewed the epidemiology of
MRSA infections in United States hospitals up
until 1981, and concluded that the occurrence of
MRSA had reached epidemic proportions. However,
major problems were only experienced by a small
number of large teaching hospitals. Unlike the
situation in Australia and Great Britain there is

no evidence to suggest that the problems in USA
hospitals are due to the spread of a single
epidemic strain.
Since 1981, problems due to MRSA have increased.
In a survey by Wakefield et a l . (1987) of 136
hospital laboratories throughout the United States,
S. aureus isolates reported as resistant to
methicillin ranged from 0% to 52% (percentage of
S. aureus isolates tested) with a mean value of
10%. Unlike the findings of Haley et a l . (1982)
major problems were not restricted to a small
number of large teaching hospitals. 18% of
hospitals in W a k e f i e l d fs survey reported MRSA
isolation rates of at least 20%, although there
was geographic clustering with distinct areas of
very high (mainly in Eastern areas of USA) and
very low (mainly in Western areas of USA) rates
of isolation of MRSA. Furthermore, in certain areas
of the USA, MRSA has spread into the community
(Saravolatz et a l .t 1982; McGowan Jr., 1988) where it
is a particular problem amongst intravenous drug
abusers (Markowitz et a l . t 1983; Craven et a l . ,
1986). In a recent personal communication from
Professor R. A. Weinstein of the Michael Reese
Hospital in Chicago, during 1988 of approximately
26,000 inpatient admissions, 80-90% of patients were
infected with MRSA and over half of these
isolates were derived from the community.
59

iv . Problems due to MGRSA in Europe.
Because there has been no central agency for
the collection of data on MRSA throughout Europe,
it is difficult to assess the problems caused by
MGRSA from 1976 to present. However, reports from
individual countries do reveal problems due to
MRSA of differing extents. In France, MGRSA were
first reported by Soussy et a l . in 1976 and since
this time strains resistant to all aminoglycosides,
including gentamicin, netilmicin and amikacin have
become endemic in many hospitals (Acar & B uu-Hoi,
1988). At the Hopital Saint- Joseph in Paris
during 1988 approximately 80% of MRSA were
resistant to aminoglycosides (Acar & Buu-Hoi, 1988).
The incidence of antibiotic resistance in
Austria, Switzerland and West Germany has been
monitored by the Paul Ehrlich Society (Kresken &
Wiedemann, 1986). The overall rate of isolation of
MRSA was less than 10 %, and no increase was
found between the years 1976- 1984. Only in one
hospital (in Vienna) were problems with MRSA
encontered (Wiedemann & Kresken, 1984) and our
laboratory testing has shown strains from this
hospital to be gentamicin-resistant. In 1988,
Borowski et a l . reported an overall isolation rate
of 17% MRSA for 1283 strains of S. aureus from
15 Polish hospitals, 87% of these MRSA were
gentamicin-resistant.
60

In Italy, the isolation of MRSA has increased
from 6% in 1981 to 26% in 1986 according to
data presented by Schito and Varaldo (1988).
Unfortunately, these authors did not specify the
levels of gentamicin resistance in the different
species studied, however for all strains of
Staphylococcus monitored, gentamicin resistance
increased from 1% in 1981 to 21% in 1986 (Schito
& Varaldo, 1988). According to Varaldo et a l .
(1984), 49% of methicillin-resistant staphylococci
were gentamicin-resistant in 1983, although again
the authors fail to distinguish between levels in
MRSA and other species of methicillin-resistant
staphylococci.
The first report of MGRSA in a Portuguese
hospital was made by Melo Cristino et a l . in
1985. MGRSA first started causing problems in
Spanish hospitals in 1978, for example an outbreak
occurred in a newborn nursery resulting in an
overall hospital rate of MRSA isolation of 23% in
1978 (Trallero et a l ., 1988). In Greece, the first
serious problems due to MRSA appeared in 1978
(Giamarellou et a l ., 1981) and since then MGRSA has
become an important nosocomial pathogen (Kosmidis,
1988).
The experiences in Southern Europe contrast with
those in Scandinavia. For example, in the early
1970s MRSA was a major problem in Danish
hospitals, however, during the late 1970s and
61

continuing into the 1980s problems due to these
organisms have diminished, so as to be virtually
non-existent (Jepsen, 1986; Ipsen & Gahrn- Hansen, 1988).
v . Problems due to MGRSA in other countries.
MGRSA pose considerable hospital problems in
Southeast Asia. For instance, in Hong Kong 25-30%
of all hospital isolates of S. aureus are MRSA,
and these are commonly resistant to gentamicin
(French et a l . , 1988). In Singapore, isolation rates
of MRSA have steadily increased from 12% of all
S. aureus isolates in 1982 to 27% in 1985, these
MRSA are also typically gentamicin-resistant (Grubb
et a l ., 1986).
In Israel, MGRSA has emerged as an important
and common pathogen responsible for serious endemic
and epidemic nosocomial infections. For example,
between 1984 and 1985, in one hospital an
isolation rate of 29% was found (Finkelstein ejt
al. , 1989). In South Africa, MGRSA is also causing
serious hospital problems (Pochee et a l . t 1988;
Coovadia et a l . , 1989). MRSA are also causing
problems in countries in northern Africa eg. Egypt
(Khalifa et a l . , 1989), Ethiopia (Gedebou et a l . ,
1987) and Nigeria (Montefiore et a l . , 1989). High
rates of isolation of MRSA have been reported,
however, it is not stated whether these strains
are gentamicin-resistant or not.

MGRSA is also causing problems in hospitals in
South America. In a Chilean hospital from 1984-
1988 isolations of MRSA have oscillated between 10
and 23%, and these strains are usually gentamicin-
resistant (Monteil et a l ., 1988). In Brazil, MGRSA
is an established problem, for example in hospitals
in Porto Alegre and Rio de Janeiro isolation
rates of 35% - 50% have been found (Marques et_
al. , 1989).
63

Ob jectives
1 . To assess the problem of multiple antibiotic
resistance in methicillin and gentamicin resistant
Staphylococcus aureus worldwide.
2. To consider current and foreseeable antibiotic
therapeutic options in the light of resistance
found at present, and that which might occur in
the future.
As a result of finding widespread ciprofloxacin
resistance early in the above studies, a third
objective was added.
3. To determine possible reasons for the high
incidence of fluoroquinolone resistance found in
MGRSA.
64

MATERIALS AND METHODS
MATERIALS1. Strains
1.1 Strain Origins and Phage T y p e s .
Epidemiologically distinct strains of methicillin
and gentamicin resistant Staphylococcus aureus were
requested from centres worldwide. The origins and
phage types of these strains are shown in Table I.
MGRSA resistant to ciprofloxacin were
specifically obtained from two centres in Israel,
strains IS 1, IS 2, IS 3 were supplied by Professor
D. Merzbach (Rambam Medical Center, Haifa) and strains
IS A - IS 8 were supplied by Dr M. Dan (Edith
Wolfson Medical Center, Holon). Ciprofloxacin-
resistant and ciprofloxacin-sensitive MGRSA (TEX 1 -
TEX 24) from one of the first reported outbreaks
of ciprofloxacin resistance in MRSA (Isaacs et a l . ,
1988) were supplied by Dr R. L. Cohen of the
Veterans Administration Medical Center, Dallas, Texas.
The phage types of these strains are shown in
Table XVII.
Throughout our antibiotic susceptibility studies,
S. aureus NCTC 6571 (strain SOX) was used as a
control. For plate antimicrobial assays of the
fluoroquinolones, Escherichia coli NCTC 10418 was
the indicator strain.

Paulista BZ12
53/85BZ15
85BZ16
NTBZ17
NTBZ18
84
DO Dd > >W W cp cp> tr1 CO COINI CT3 H H1—1 M Wr-1 C5 > H
3 t -1 >M>
EC CO hh C5cp O rt P PP P EC ■-» H-
1 w H- DO Dd H- Dd O 3 <cn < O •-J rt •-J w < P P <rt P rt P P P p H' M O i-l H*
, (33 P 00 I-* w H- P cr ET w PPu W o 3 cn r+ o o P H- PP H- p P p P p p 3 rt PP P- rt P cp t—1 I-1 p l-t O P Ph-» P P P P cn rt P rt rt
(=L H- - P ET COP < CO P **
• M I—1P H-
p pH* H-p
DO Dd Dd N N N^ M H
55 55 5;t-3 H H
OO9r+a .
td to to It- t-1 t-_p co 1—1
S3vO
CnS 3>
vO\ S 3 00 vO O \ \ O ' vO \ Cn -P" \ S 3 O ' M
C/3 C/3 C/3 C/3 CO
H O ' CO S H
O'
- >S 3w
■p-
CnCO
Cn4S" JCn
ooCn
4SS 3M \ \ Cn ■P- P' "J \Cn Cn CO \\ ooCn CO -P> >-o ooCn i—1
sz: 55 oo 55 5-5 H H Cn H H
< < i—* ooCn
55 55 h-3 H
OOPPr t
Oppr t►1CD
CO O r t O ►-« cu p fl> H-
P
•XIP*
03 P I—‘00a>
coCD H r t v-< TP
C ®? oHa
oo
ofjH*30H*Pcn
ol-hPPr tH -C7*H*OrtH*O
PP*P[30CD
r t■<PPW
O("h
pGOCDa *
COr tea--<
r tO
PuPr tP•13H -PP
H
66
Table

Contd.
aW Oo ^ a c/330
W H 3 H - P -a o03 a 3 h-1 1-4
3 3
30 a a a 3d 3d aa a a a a a aW-H h -l I—pH H H H-pH »—pH H-rH►—<-H t—l-H K- H K-l-H H-LH *-±H J-MH H in 4S U3 N3 Hi— O
so ooH3 Cn
so so a so so H H H H H
30O
'O03
DO I—1 t -1 O H*C/3 <S
a t -1 3 H - H * *-»a < a 03 CD O I—1 •“! O X3 I—1
O O
CO COa ai—1 i—* '-J CT'
oocn
MSOcna>s :to
sCDa* td h- os cnO *d 1-4 03 3 CDhoi s;
< to o
cn oCD &3rt>-4CD
o
soH
oaMatd
C/133rtH-30QO
cn cn a a
F - CO
a oo H Cn
oo33rt•-4
cnCD3rt•-4CD
a n a O ' I a 3 3 H-
3
aa
(13 3 (—> 0Q 3
C/33 1-3 rt
a^ 3ai-3a
xi—*oo
o•-4H -JOH -33
OHi33rtH*aH*oaH*o
aa3103
soaC/3>
a
a3C/3
oa
c33a
3a3a
ao
<=u3a3a3H-33
67
Table I
contd
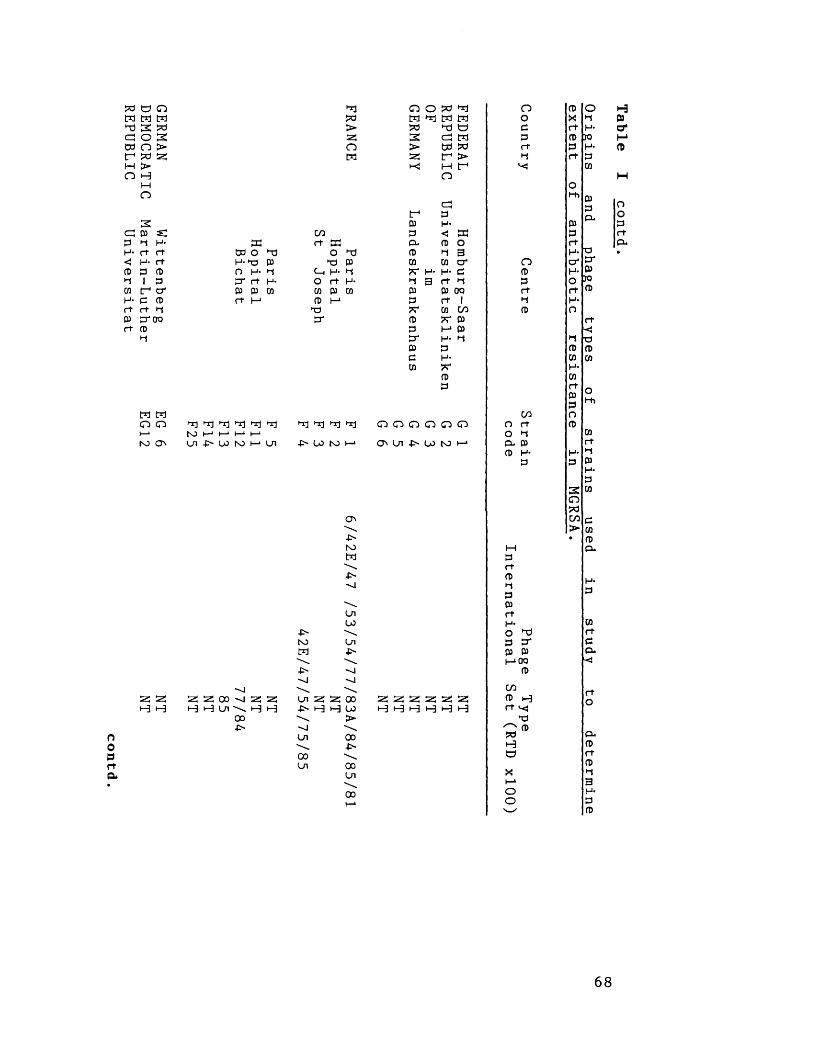
GERMAN Wittenberg
EG 6
DEMOCRATIC Martin-Luther
EG12REPUBLIC
Universitat
tiw>scnt*a
scW O T ) H-T3 Pn h - i-iU rt H-03 03 CO r t |->
It ) Tj i-rj hrj hr) rtcjN3 h-* I—1 H-* l—■U l -p" LO K3 I—1 U l
s; s; S! 25 OO S5 2; i-3i-3 00■C-OO
PrtP*
cort SC
O T) T ) 03 C—i H' T
O rt H- CO 03 CO 03 I - 1 T)P*
T1 T1 Tl Tl-C> (jO N 3 I— 1
-P~N3M■P-•JLn-c>-Ul00Ul
S3 S3 i-3 H
-P'N3M•P'
UiU3
Ul■P*
ooU3>oo-o-
ooUi00
O O W T l n 03 oo X •-J
W TJ W c r t H-2 a m p 03 JO> w p r t P H-S3 t - > i-j r t Pi-3 M f W
OO
cCt-h P
3f P CL03 H- 03P < SC Pa. o) o r tCD P 3 H- O(» co c r O O ' s r? r H* H- c CD H- PP 3 r t P P O JO03 03 00 r t r t CDP rr 1 P H-
co co CD OCD 3C P I r tP T * P ■<s r H- P ►t ■o03 P CD pe h - CO coCO 77* H*
03 COp r t
PP
of
CO oO G~3 G"3 C~3 O r t p
O P COP W to H P - P r t
CD H- pP P P
H-P
2 COOTPCO P► CO• p
25 S3 !23 ss 53 25 H H i-3 i-3 i-3 H
Prt03PP03rtH-OP
TJP*
03 p I—*00 03
C/30) H rt v<*T)SP ®HOXi—*Oo
COrtPa-
68
Table I
contd

oo3rtPu
a C_ l—l a cn oC3 > H o a os: a > a M 3> > tr- cn M 3H a o rtH M t*
aoacna H3 3 cnC/J H- 3 «o a3 3 a H- ® 3 a a h -H- i-h 'O < O 3 o o 3 >rt 3 o a 3 a 3 3 3 3 OO rtrt H 3 I-J 3 1=) 3 OO a a O
1 o < 3 < H- H* 3 3<=! a cn a H- H- H* rt rt a 3 33 3 3 ^ 3 rt 3 3 3 3 3 rtH- < H- o 3 t-»3 o h-»3 •-t<J 3 < 3 M 33 H- 3 'C OO -►1 rt 3. 33 3 H-H- H-rt rt
a a a C_ 1—1 1—1 1—1l—lM a a a > > C/0s: s: s: T) H i-3 H H t-3 a a a 1-3i-3 O rt
H-1 I—* O tl-J Ui ■p* tO l-i to t—* 00 -o O'* w to I—* W i—* a 3
Uizoo z
OOUi
OOo
00o-P-tOM-P~-OUl■p"
1Ui00
CTv■P-
00 Ul -P> -O-
a a ooH H Ul
-o00-C>
Ul Ul Ul -P* -P> 4>»-o -o --J "J -ooo oo ooW W W > > >00 00 00 Ul Ul Ul
tov£> 00 \ •P' Ulto
3rt(Dl-t30)rtH-O3 TJ3*3 3 1—1OQ 3C/33 H rt v- X3 ^ 3aHOXi—*oo
ol-JH-3 JO3r+Ot-h
i3P*3O wrtH-O
3cnaC/3>
rt■333
Ot-h
333a*
3rt3o--<4
rtO
Pu3rt3•33H-33
69
Table I
contd
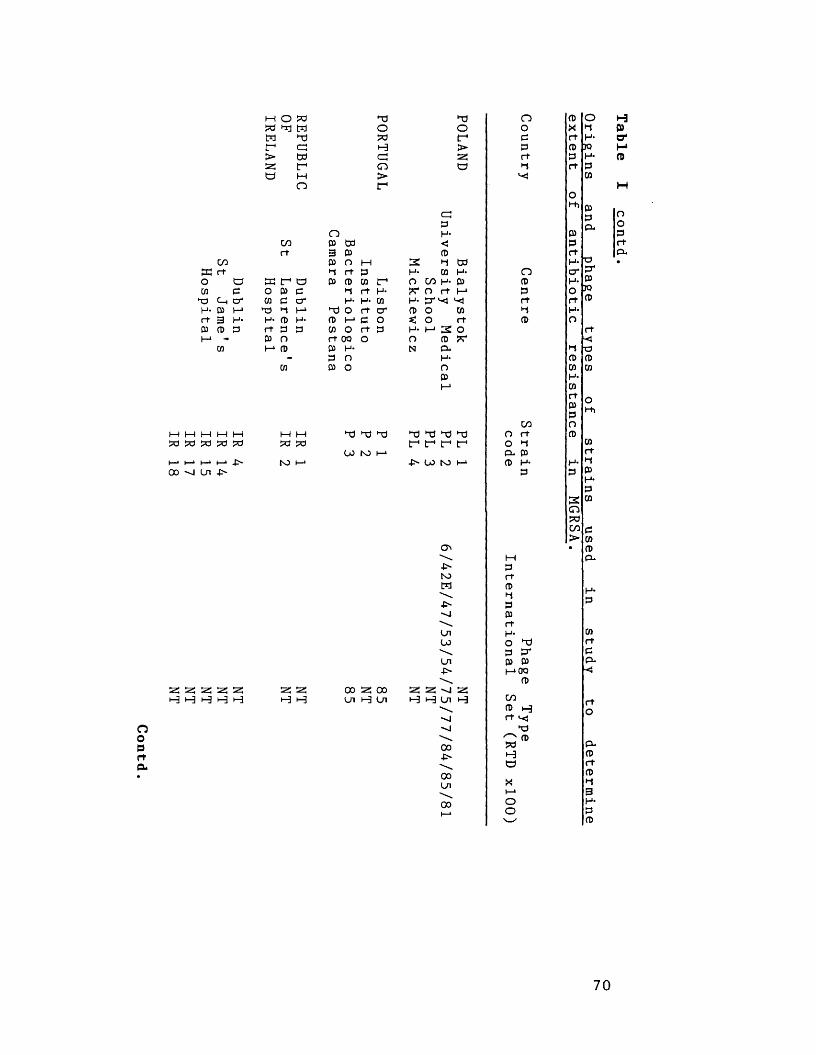
Contd
m o adad trj tdw ddr*>astd
cdtdMn
ooffi r t O Ui dd H- rt
td £c_ cr* pa m 3 H*
oor t
K f td
03 (D £
OCOTdH-rt03 O M P
ddOadt-3cdo>r 1
o03 w3 0303 O MId r t £03 P W
d rtH- H-
t ) O r t c r n>co ort 00 03 H'£ O03 O
r *H*(0
M £ O r t £ O
00 -Si Ln
as as as « « H H H H H ss a* M H
oo s; oo Ui H U i
ddOt ->astd
2H-
<=:£H-<CD•d03o c/a h- 03
?r O rt H
o coO rt M 2 O
cd ar p *H*O 03
M M M M M M M X) dd dd dd dJ dd ddad ad ad ad ad ad ao f tr* f tr*U3 toi—1 i— 1 I—* i— » .p- N3 i—1 u> to i—*
-P-toM
-P-
UlU3Ul-P'
as as -o as H H U i H
oo■P-
ooUl
00
oo££rt•d
OCD£r t•d(D
C/3 O rt O £ P- 03 CD H- £
£rtCD•d£03rtH-O£ dd
£*03 03 MOO
CD
COCD t-3 r t v<
ddC naoHtd
xi—*oo
o•dH-JOH-£CO
ohh
03 £ rtH.r£
2cnadco>
p *03
poCD
rt<tdCDUi
Ot-h
£UiCDCu
Uirt£P*
rtO
P-CDrtP•d3H-£P
70
Table I
contd
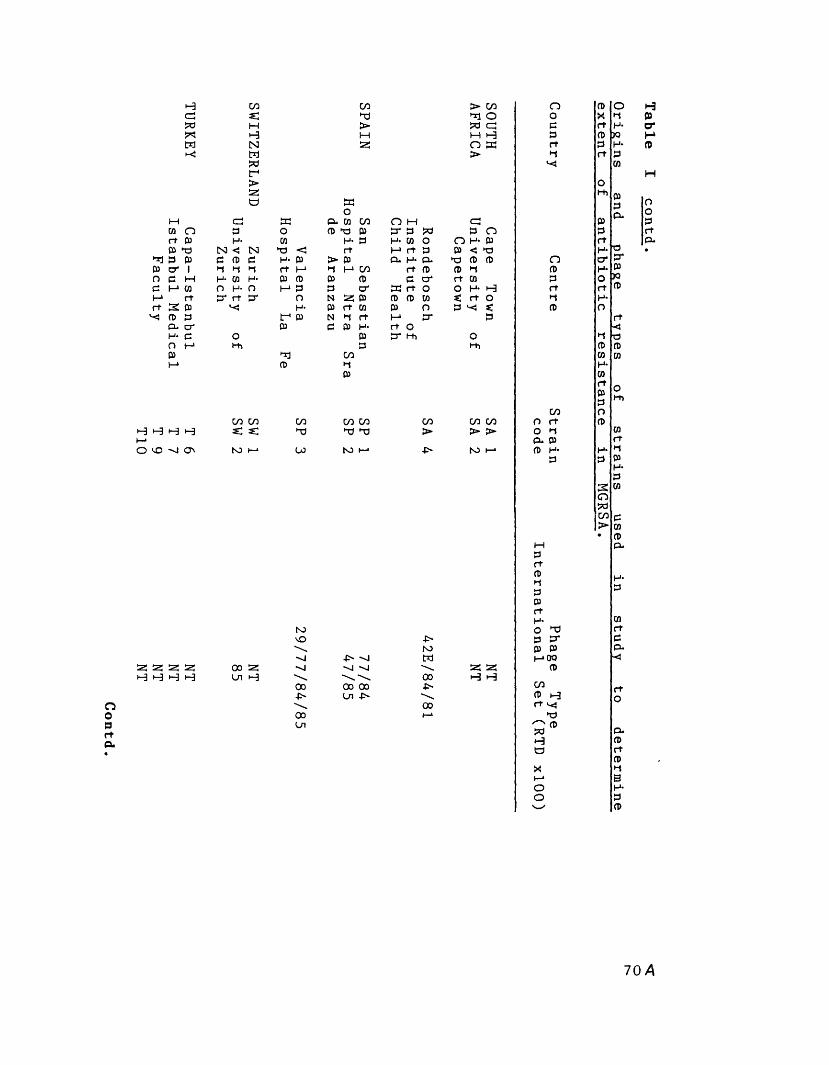
TURKEY Capa-Istanbul
Istanbul Medical
Faculty
H *-3 t-3 Ht— *O VO Ni O'
as as as as t-3 »-3 H H
COs:Mt-3NMWr1>aso
CO COc s:NO I—*
asHOoprtCL
COTd>
S3O
COTdto
NOvO"vl-vl00-O'ooLn
COM05
CO CO Td TJ
NO
CO>
■P- 'v j >v4
oo ooLn -O'
P>NOW00p~00
> COoW CJ M H O S3>
cd S3 CL Ui CO O M Cpp O P Td P cr P W p OH- 05 H- P H* co O O H- P
N < N Td < rt (-» rt P P < TdP (0 P H* P > P CL H* CL Td p PM M M rt I - 1 M (-* CO rt P p MH- 05 H- P fl> P p P O' rt 05O H- O (-* P P c r S3 rt o O H* t-3P* rt P* O N a: p P P Ui t , rt O
H- P rt Ui P o P 'O «:f P N M rt M c r pP P P H* rt O
O P cr rh Ot-t> P t-t>
CO CO > >NO
as as t-3 H
nopprtM
O(0PrtM(0
co O rt O *1 P* p (t> H-
P
TdP*
05 05 I-* 00 (1)COP i-3 rt ^ Tdc ®Htd
Xt—1oo
OMH-50H-P05
OMi
rop*05ooP
rt<■pPUi
OMi
P05PCL
05rtpCL
rtO
CLprt0)M3H-Pn>
704
Table I
contd

Abbreviations: NT
= not
typable with
International Set
oHrtcr
M O P to 03 CD rt P H* P-rtCrt0)
cn
cncn
pa
i-3h03e3os cnrt 03 O H H* 03 O rt
pacn
to
cnH
o 3Hi p
P- H-
cn n o o 03 cr e h o i rt Hcr mcn fB
P w O H rt P < O H P P O H I—1 03 H* H- P rt (33 h-
cn cn o o
-O'HiLn-O-h I to Ln vOh ih j
oo00>
25cn cn (BO P m p td o h- ort p
s:wo
W 0) P rt
P P 00 O t—‘ W t—1 P
CO p PPL
pa pa pa pa pa 3 3 S 3 3
h-1 O Ln •£'■ 00 NO I— 1
Ln Ln NO NO
vO vO ^ ^ Ln Ln Ln NO NO \ w^ ^ -O' -0- -O'
NO NO NOoo oo w M M o o ^ w
-O' -O- -O'Hi Hi HiKO 'JO Ln Ln^ "'Ui Ln Ln
-O' -O' -O' -O''J Hi \ \^ — -J
V 1 ^ Ln Ln Ln ■O' O '- . ^
Hi HiLn Ln00 00 00 00 > >
h! h I -vj "J H| Hioo oo
>3WpaHO>
cnpH-<CB OH
O CO P H- P rt rt H P H
H- P O H*P P P
3 rt P H- P*H-OPH*
cn cn cn cni—> i—1 00 NO
On00oo oo oo oo >■ c»00 00 On on
o cn cn O p o Hh s: O X H P> M C rt H- a*H H P P JO MM W rt P H- Pcn cn H rt PHI W
MO
cn Hh mrt P
P-OO
P PM P rt3 M H| rt P-P H- O H- o •P. N p n cr CrH- P P p H- Po cr oo p O JOP P w rt rt pH rt rt H H-
cr o P ot, rt
n p -<p cn H TdP o P Prt CO OT COH to H*P H* cn
rt P H1
rtPP
of
cn ncn O rt pcn O H 03
Cu p rthi P H- H- H
P p PH*P
3 goocnp► 03• P
M a*PrtPHP
in
PrtH- wO TJ rtp cr ep p a.1—‘OO
p
25 cn f~ri-3 P h ort v<-
X)^ ppa P-i-3 Pcn rt
PX Hi—* 3o H-o P'—✓ P
71

For the haemolysin identification studies S .
aureus NCTC 7121, S. aureus NCTC 10345 and S .
aureus NCTC 5664 were used as control organisms.
For short term storage (up to 6 months) strains
were kept on nutrient agar slopes (Southern Group
Laboratories, Lewisham & Southwark), for longer term
storage strains were kept in liquid nitrogen.
2. Antibiotics
2.1 Antibiotics used in susceptibility and other
studies of antibiotic properties.
The antibiotics used in susceptibility, and other
studies of antibiotic properties are shown in
Table II. In Table II the sources of antibiotics,
and the solvents used to dissolve them are given.
Antibiotic stock solutions were formulated according
to batch potency, and all solutions were used
within five hours of preparation.
3. Media and Reagents
3.1 Media used in susceptibility and other studies
of antibiotic properties.
When required strains were subcultured onto 7%
horse blood agar (7% horse blood in Columbia Blood
Agar Base, Oxoid, Basingstoke). For susceptibility
72

CIPROFLOXACIN ciprofloxacin
hydrochloride, lab.
waterstandard-
Bayer AG,
Wuppertal, FRG.
oaaow>3TJaM25Moot-*
w>oMHa>O
>INIHHaao3>-<o
>INIMt->MO
>O
>3MX>OMa
>oaocnoX>o
►prrH*O'H*0rtan
M a p O P N a P O P a c p cn pP p p a o a a n a n x a 3 a ocn *-i a p *: p a a p p a p a p ar t a p *: CL o n a 3 a a a a a oa p p P p a a p a P a cnCD i •» a a a o a p. - o a oa a n a o p o 00 > a P X
00 p a o 1 P 3 a p 3 P 00 Pa < h * a o a x >» a l o<* a O p t-3 a P o p a d a
W a cn a a o a O n 3 a 03 a P Oa P a p a • p o a p* p e p - ap 3 H- P V - . p . p. = a a a3 a p a o <• p . a a 00P p a p SC H* O a a i a a p acn cn p • P P a p cnT3 p p o a pa p p a o a a o 03 aH- P H- a a • p CP P -a a o o a o p . a cnCD o o o a p cn a p . a o a
a a 3P
• » aP
O Po a C 1 a
insto
e p a p a p .
a p 3 O P - cn cn a cl pP o a a p P - a I a a aa s: o o p a 00 3 P o P .cn a p a . a . 3 3 x a. a i• P p o - I a p P P a- i-i p *
ppa a
cn03
a o 03 a *
1 r t cn cn o a cna p > e a o eo p. . i-j a p ap a p . a
px
0)pr taO'aOr tH-O■Pao■pa>•-«r tH-pcn
cndcnocnpr tH*P *H*aH*r t■<
PPP.
Or tacna
i-3Po'H*n
H
ooprta
cnr+a05po
05r tCDa
3 P cnP a s: o oa P p 52! • p aa a a P O 03 <p Ln p p O a CD pp o a a a 3 P< Po o aa o
cnr tPaacncn
ohti
73

tdr*wt>oox>o
oprtCL
MWk )H33WOsXo
dd O S3 W P•d O P M •dO 3 3 H-CL td td rtP O co P*o P P* t- •drt P H- H- oCO CL *d I-1 3
P• L<J Otrdd H-et o Co 3CL• to O- 00 O td1 • dds: •p to r--i—14> rt* . O CL i—*X \ • Pp O' - cro •
4> Wo 1 P CDp CO rt*d txl H- PCL O P Pp O 00 CLP P* CO P
P rt •dO CLo X* 1
H- Prt -
Ms:oX>o
C-hPtd0)P
td fD p* p
tdpdd'■sito
33 oCD O3 3O td P O >d P CO P
CL
tdcdd
OLn
« nCD O 3 3 o tdc o >d c Cfl P
CL
rt C/0 00H* td • td • tdO o > c > pP *: • dd • ddl-> CL
P -o•d o to O (->
o 1 p i—* P Oo p 1 P tn• td p P 1
p < td <t- H- p p P tdrt p ■• » pCL H-• td C/0 dd CO- td K o s; dd
o H- p H- oO P rt rt rt P05 N N rtP P Pf*r *d CL •dp P M CL* P
PPP
p
td>ddHOsr<oMS3
td o i- 1 o H- 3
X) O
t-4 P H- P I—1 CL M
t”4 Co H<CD i-1 O -O • O'1- o00 M NOP -CLH- M 03 05 P CT 05 . PO CO I—1 rt H- P W P ~ CL
pcp <-iC/0 CL> I
nr-i-tS5td>3►-<o
K w O (D rt H CO P H- rt p p
CL CLp p w 1 3 C CL^
co I oCO H *(D c px td• C—I.
O P* p*1 P CL •d
otr- n rt pr c l i— 1 • O - i-i
H- O CL •d P 03 - *:I—* I-* p p ^ cr
PrtH-crH*ortH*n
o►iH*0QH*P
Prt O t. < *. C/3O
O P* 25 • P P P P P H^ • P P O rt rt rt rt 03 «P o P O !-» P P P P P PO 1-* S3 S OI-1 33 3 •d ►d •d •d CL 0 ft
P > HP P prt rt crH- H* Hcr cr pH- H-O ort rt HH- H- Hn o
wtd O*d OO p P■p w rtP p CL•d CL •rtH-PCO H -
COPCOnpTdrtH -crH*i-1H-rt
X
74

> 53 3 3 3 3 oo > cr; M H M HM tr" XJ O 53 H sz:a h i—i n O EC H
a X) > n P >M o 3 k ! O 3X! o t < o P MM 1—1 O f f On 53 M 1—1 f IP
53 32! P sgM
s: p XJ P P « 3 IP 3 CP CO 00P p P n P P P P P P o o pP i—* X P I-4 P O cr p P CL d prt P K CL I—1 m o co o O P CD rtP* CL «• 1 P cr P • o cr d CD PM P rt P 3 P 3 CD 3O X CO td CD /->. o 3 P H*P P d P O a. p n
n X p IP P P P 3 H-X o n rt p < p tP P IP P
O P p* o O-* - P P P ;t PP p p p • co - w c r o r• o • 3 rt - !-> P P H* CD CO
P P P O P P O • dt- CL P •-3 c r P P M P prt XI P O • c r n p IPX3P. P* P ** • cr p rt cr• P P P •p co o H- CL p>• O X ao O rt P> CD p « rt
* 3 ■. Q3 rt tp - <• PO CL P P P P <•e P n H-* c_, cl O P cr p cCP M p K3 p p ^ a* CO P X PP c • P X p p • c r a * PP. rt 00 p cl P X >» • X c rM> CP P p I P CL p •O XI O 3 • 3 1 CP CD CLM • P 00 3 P X rt 00 CDCL 1 P p CL tP P P p r t- co P p P P P
CO - p C_i. CL rt CL PCOrt CD P O P P P 3 CLe P w CD CP M O X H- PX X X d w p X CL CL XM 1—1 O cl CO P CL | CL CLP P O o p IP • CD 1
P ?r 3 P O X• I30 CT O <*r P •
P P p CL3 P o
n p•
•ncccoMOMO>O
t -1 COp o o a*
PC
XJ 3 P*PX l-h 3 d 0} CO O P a) c ld 03 r t r t p a> o <•03I-1 P
03O' X) •XO CO CL f tC 03 r> pr t p . CD 03 - 1-1
CLa ipp3px
x ioCOXIo3•-<o
O CL . H- M CO• OX) CL. h- > C - 3
303 t-hp . oX 03 P M i CL O• 3
CO O XJ P P P P - P• PP
c r
CDr tPPp .PXcl
► P > Hp P P Pr t r t r t o*H- P P po* o r cr PH* P p0 O or t r t r t HH* P P M0 O n
CO
■p OX Oo d Pp CD rtp CD CLX ex. •rtPPCD
pp
OXP00P0
noprtP.
o s: 3P *. *: s:CO0
C H53 • p r t p P p p p CO <P O r t cr r t r t r t r t r t 0 PO h- P P P P P P P CL Pcn 3 X p X X X X X r t
CDcCDOa>■or tPc rPPPrtH4
75

Michigan, USA
OoprtP.
>ftdMsXn
t Td 03 c i-* h 0) n> 3 PN ^ O 03 O X - P -
X3 x O X P
TdO*PphI
a►oC—i.op *p
no
*:prf03H
O►dt - 1OX>o
S3 O O H i C I-1 P O 03 X l_i (aO O < X - P
2X I—1 P . P P O ' X •p03 P
r fPPPPHPIKOPnP -cor f
c3
O s*.P o O f-* K 3
8<otdMOo
zpr fPH
s:otd►dfOX>o
23HHtdO►dcdtd>INOs :td
ssXHtdo►d<=:to>s :Ho
tz:tdHMfSXO(-1
O S3 •P O O i-* K 3
X _° X * a3 m{33 P 3 rtX ^ d,XP M
H iO X3Pr fP*
s :tdo3Xo
Cd p CO P td^o n d td H- P O PTd o P* o P c O p. P p P H pt_l. p. p H rf H 3 rf H rf P oo X H Hi O P 'CT h X H- *: 3p* P Td X P P o H* X X Xp 3 o 3 Hi P 3 p o
fio X P X c COC-u H- X H-P 1—1X -* H rf P O >• P
t-' p td n P rf P O H*rf O O X O H CO p rf P s :P < p* p • o rt rf td H- p P• o 3 - X O p. O CO P>• CT* p d H- 3 P Oi rf X
X - X S3 H IT- d O c Tdo o P O p O P X p*H O td cr H N e p. < *CT CO pp X P • *: o H*'ct w H* P* c rfs: P P* X P CO o » P P CO PI-* - Z P n P * «: X rf pp p rf p* p. CO 1 P pX X x P - 3 03 c X td- P - P Td H- ^ f-h X • 'd
CT* p s; o “ 1 X H- P 1?: • s: p p s; CO O H Pp P H z p. ° CO M CT* cpp CO «: p p c H. td X z Tdrf rf 1 Kj P oo • 1 s: X.
P X 1 H* 3 s: p oP Ch 3 o - 03 p rf p*CO P p P d H H* p
e P H *1 td o cn z Xp ►1 P O >• H* Xp P p td «:
H*O H- tr*
p 1 X * p td P rfX
cpCO>• O
P“ 3H-O r* =
p.•CO P
Xrf
> P.
>prtXo 'X0rtXo
o•1X0QXP
C/3O
< Z C XP P P <r f r f P PP P P- PH •d rt
P > h3P P Prf X O'X X XCT* CT* pX Xo orf rf HX X Ho o
p
CT On Oo p PCT p rfP p P*H p. •rtH-PP X
p
COepopor fXCT*XXXr f
X
76

SPARFLOXACIN compound
RP 64206,
lab. standard-
0.1MRhone
Poulenc Sante,
Vitry Sur
Seine. NaOH
CO CO <=3 H t- t- Tl < td M> toaM>NM23M
Td COo H-s: (-»p * <p (Dhi
H
co co3 CH- I-1r t tdt r CL
(0Qo P.
H-2 ! COp N
03 H-t r Pa> CDs:
3to H-o O3 HHi Oo PH H-CL CO
CDCLto
COCOPX
ONO
rtcl
S*Td H O
o o t1
GO
toTd
LnvOLnOO
toO«MH>2*-<o
toM>2TdMoM2!
to>29Tdf>25
TdtoMCOHM25>2•-<n
Tdto
tr1ox>o
< (0 to t-3 H CD H 2 2 0 tOTd <3 P TdH - rt td O O H H - H - P O cr h H- rt Prt P tv tv C H> I - 1 H 3 O H - rt P H iH P Ln X H - Td P P H Td P p H P I - 1X CL vO O rt Td 3 P P O P rt X CL O
P Ln <. p O Td >* I - 1 C H- P XH O 3 H - H* P p H P
CO CL O U X n M CL T) P CO CL Oe 1 p o f H - rt O 3 e H -H Td H- p p P td e x T P
to g P P Td I—1 O > I - 1 o tot r CD P - p X S3 1 P H- t r
CO o rt • rt Td . - 1— ‘ P P CO O 3CD p t r h-» H - O O' o p p PH - CD p P rt t. f O' H- p rtP p cr CL P 00 Td P t rCD CD • p Td O' CO O P P- Td CO H p - p S3 >. Td p
o CO Id - rt P CL o PP CO rt p H- Td rt p c P
H H* e P V I—* rt c P H H I—1 cP CD i—1 P p H V. « p p I-*P P Hi CL 2 cr P p p H iO n O P H - • to < H* o o OCD P H I—1 P H- P p p• P CL P w W Td rt cr . p
CO rt 1 P rt P O H • CO rtP CD - 0) P *: X P pp )-3 p H CL P P >»rt O 1—1 CL O p rt rtCD CO *-< rt p t r T P P P f—1- P O P H 1 C P - p
t-> 1—1 CL H CL crrt X | O P .- c-t p H
o p CO CLM N rt P 1P O T H -cr - P P• *• P
3 3 3P P S3 P *3
K rt rt P rt PP t r cr rt t r rtrt p P P p PP p P H p HH o o O
>pf tH*O*H*Or tH*O
o•1H-09H-P
COop I-* GO < ft) ft)p . p
f t
P > h3P p Prt rt CH* H- Mcr cr PH* H-o Ort rt HH- H- HO O
P
Td OT Oo C P■p P rtp P CL►T CL •rtH-PP H-■ P
COPCOoCDTdrtH-crH-I - 1H-rtX
77

VANCOMYCIN vancomycin
hydrochloride, lab.
standard- water
Eli Lilly
& Co.,
Indianapolis, In.,
USA.
cn < o toP 03 r+ to or03 =3 2 to o rf Po op r t • to3_
I©ctoor03>-»to03cro>-»03r tO*1to03CO
>-3 H H H C/3 C/3Od O o to to H toto CO W •-3 M W to3 C5 to to O to toW to > > o to >H to 2 O to H 2g O >-< •-< to O© JxJ O n > 2 Oto > M to 25 toto o 25 h M n 52!M M 25 2 : i-i3 25 to 25
rtrt > O to rt W rt 2 w O CD < to O
cr co I-1 O rt P P o or rt to or Oto cr e to cr P rt H P. p <-< rt o 33 O to •t P fi to 3 P •1 p to03 rt to P P- P P C to to p o: rt rt o to 3 P O to 3 O rt por X to ^ rj ^ to P O po 03 to O P. O to 3 C/3 to p*to to o l_i |_i. 1 to rt P o»i 03 to ^ P to © P o ►t pto cr 3 to P O to n to to to3 O tot P S n o P p to
n co to o 3 C/3 pp rt o o N to to P o Ln
: l—1 rt ° • to p or to to p w to LO03 o w - c p ^ p p p e P U>o rj v<* rf p. to p ^ to P C/3rt to 1—1 to to •1 p to - to P <•03
° S P O to o rt P or Prt CD ^ D. P rt O H- - C/3 P rtj rt to03 •. 03 H- - p. or rt rt rt ►t P P
P • to - to p P V crM ^ p to - o P P •
toM P
03 p 2 cr to OP 1-1S- to O' C/3 to to • O to P CD
to ^ o • P P. to P o • rtP * to P P P CO to P
to ° OS to CO p u v. P rt 03 (P PP H* rt co rt <• p - P P*(_i. CO pi v. p to to P •-J P03 * P p O P to p. 2 1 •IO (IL. to P- or cr rt p to P-rt CO 03 co p » • p •-» CO c/1 1to CO > p to P. CD toO >*■ c • eu to ^ 1 O OQP 1 I p • C 3
P r» Prt to
0Q • .
> p > h3P p P Pr+ rt rt o*to H- to too* o ' cr PH- H- toO O ort rt rt Hto H* H- Ho O O
m
to O»-j Oo p Pto w rtP p P.*1 p* •rftoPCO H*
P
O►1to00toP
H* 3 C/3+ P P O
< to *. K «: rt P toP «: P P P p or CO <rt p r f rt rt rt p p pP n s P P P P p a p
p 2 1-1 03r+
•-« •1 •-« oto
rt
WecooCDtor ttocrtoI-1tor t
78

testing, strains were grown up in peptone water
broth (Southern Group Laboratories).
IsoSensitest Agar (Oxoid, Basingstoke) was used
for all determinations of antibiotic susceptibility
(except for methicillin and paldimycin) by agar
incorporation. Nutrient Agar (Oxoid) was used for
methicillin susceptibility determinations and in
the case of paldimycin, Antibiotic Medium No. 2
(Oxoid) was used because of the improved stability
found for paldimycin at pH 6.5. IsoSensitest Broth
(Oxoid) was used for determination of MICs by
broth dilution studies, and also in kill curve
studies. IsoSensitest Agar was also used in time-
kill and population structure experiments for
viable counting, and in plate antibiotic assays.
3.2 Media used in biotyping studies.
i . Egg Yolk Agar
Egg Yolk Emulsion (Oxoid) was added to molten
(50°C) nutrient agar (Oxoid) to a final concentration
of 10% v/v. The medium was cleared by addition of
1% NaCl, and sterile 40% D-glucose solution was
then added to a final concentration of 1% w/v.
i i . Lysed Blood DST Agar
Lysed horse blood (Tissue Culture Services,
Botolph Claydon, Bucks) was added to DST Agar
(Oxoid) held at 50°C to a final concentration of 7%.
79

i i i . Milk Agar
"Nestles Ideal Evaporated Milk" was added to
molten nutrient agar to produce a final
concentration of 20% w/v.
i v . Sheep Blood Agar
Sheep blood (Oxoid) was added to IsoSensitest
agar held at 48°C to a final concentration of 5%
v/v. This mixture was then layered (c. 10 ml) onto
plates prepoured with 10 ml IsoSensitest agar.
v . Tween 80 Agar
Tween 80 (polyoxyethylene-20-sorbitan mono-oleate)
was added to nutrient agar to a final
concentration of 1% v/v.
v i . Staph-Typing A g a r .
This consisted of 20 g/1 Oxoid Nutrient Broth
no. 2, 5.0 g/1 NaCl and 7.5 g/1 Oxoid Agar no. 1.
After autoclaving (121°C for 15 minutes) the medium
was supplemented with C a C ^ solution to a final
concentration of 1.2 g/1.
80

3.3 Antibiotic-containing discs for determining
sensitivity profiles and identifying aminoglycoside-
modifying enzymes.
i Antibiotic discs used for Antibiotic Sensitivity
Profiles.
The nine antibiotics (disc strengths in
parentheses) used in our antibiotic sensitivity
typing scheme were as follows: amikacin (10 ug) ,
chloramphenicol (10 ug), ciprofloxacin (5 u g ) ,
clindamycin (2 u g ) , neomycin (10 ug), netilmicin (10 ug),
rifampicin (5 ug), tetracycline (10 ug) and trimethoprim
(1.25 u g ) . Netilmicin discs were supplied by Oxoid
(Basingstoke), all the others were supplied by Mast
Laboratories, Merseyside.
ii . Antibiotic discs used for identifying
Aminoglycoside-modifying enzymes.
The antibiotic discs (strengths in parentheses)
used for identifying aminoglycoside-modifying enzymes
were: amikacin (10 u g ) , gentamicin (10 u g ) , kanamycin (30
ug), neomycin (30 ug), netilmicin (30 ug) and sissomicin
(10 ug) . These were supplied in the form of
specially prepared antibiotic disc "rings” by Mast
Laboratories, Merseyside.
81

3.4 Materials and Reagents used for detection ofspecific haemolysins.
The micro-titration assay of Jordens et a l . ,
(1989) was used to detect specific haemolysins.
Human blood (from volunteer), rabbit blood (from
animal house) and sheep blood (in Alsever solution
from Oxoid, Basingstoke) were used. Human blood was
collected in Lithium Heparin LH/10 "Monovette" tubes
(Sarstedt UK Ltd., Leicester) and EDTA KE/2.7
"Monovette" tubes (Sarstedt). Rabbit blood (5 ml) was
collected in 5 ml Alsever solution (glucose 2.05%,
NaCl 0.42% and sodium citrate 0.8% in sterile
distilled water). Rabbit and human blood were used
within 6 days of collection. For use in assays
blood was washed thrice in phosphate buffered
saline (NaCl 8.0 g/1, K2HP04 1.21 g/1 and 0.34 g/1
KH2P0^) supplemented with 1 mM MgSO^.
Heparin ("Monoparin", CP Pharmaceuticals Ltd.,
Wrexham) and bovine blood (fraction 1) fibrinogen (BDH
Chemicals Ltd., Poole) were also used in these
experiments. Assays were performed in "v" well
microtiter plates (Sterilin, Feltham, Middlesex).
82

4.0. Buffers used in plasmid isolation studies.
i . Buffers used in Takahashi and Nagano m e t h o d .
Buffer A. 40 mM TRIZMA base and 20 mM disodium EDTA
in sterile distilled water, adjusted to pH 8.0
with glacial acetic acid.
Buffer B. 3.0 M sodium acetate in sterile
distilled water, adjusted to pH 5.5 with glacial
acetic acid.
Buffer C. 10 mM TRIZMA base and 2.0 mM disodium
EDTA in sterile distilled water, adjusted to pH
8.0 with glacial acetic acid.
Buffer D. 1.0 M sodium acetate, 10 mM TRIZMA base
and 2.0 mM disodium EDTA in sterile distilled
water, adjusted to pH 8.0 with glacial acetic
acid .
i i . Buffers used in PHLS (Johnson) m e t h o d .
Suspending Buffer. 2.5 M NaCl and 50 mM disodium
EDTA, p H 7 .5.
Lysis Buffer. 50 mM Tris- acetate, 50 mM disodium
EDTA, pH 8.0 Brij-58 added to 1% and sodium
deoxycholate added to 0.4%.
83

Tris-Borate Buffer x 20 (for electrophoresis). 216 g
TRIZMA base and 9.3 g disodium EDTA was added to
1.0 litre sterile distilled water, and pH was
adjusted to 9.8 with boric acid.
Buffers were autoclaved at 115°C for 10 mins.
TRIZMA-base, sodium acetate, disodium EDTA and NaCl
(molecular biology grade reagents) and sodium
deoxycholate and Brij-58 were obtained from the
Sigma Chemical Company (Sigma Chemicals), Poole,
Dorset. Glacial acetic acid (AnalaR grade) was
obtained from British Drug Houses (BDH), Poole.
iii. Loading b uffer.
Loading buffer consisted of 0.1% bromocresol
purple and 50% glycerol in water.
84

METHODS
5.0 Identification of S. aureus.
Cultures were identified as S . aureus on the
basis of Gram-stain, catalase production, coagulase
production and "API STAPH" biochemical profile.
Gram-staining and catalase production were performed
by standard laboratory techniques (Cowan & Steel,
1974).
Coagulase production was detected by the tube
coagulase method of Gillespie (Cruickshank et a l . ,
1975). 0.1 ml of 18-24 hour old nutrient broth
culture of bacteria was added to 1 ml of a 1 in
10 dilution of rabbit plasma (Difco Laboratories,
Michigan, USA) in plastic bijoux. The bijoux were
incubated at 37°C and checked at 1 , 3 and 6 hours
for the presence of coagulum (ie. conversion of
plasma to a soft or stiff gel). If negative,
bijoux were left overnight at room temperature and
examined next morning. Weakly positive (S. aureus
NCTC 6571) and negative (S. epidermidis) strains were
run as controls.
API STAPH biochemical test strips were obtained
from API (Montalieu Vercieu, France) and processed
according to the manufacturer’s instructions.
85

6.0 Detection of Methicillin Resistance.
Methicillin resistance was detected by streaking
isolates and appropriate controls onto nutrient
agar (Oxoid). Methicillin (25 ug) strips (Mast
Laboratories, Merseyside) were layed perpendicularly
to the inocula. Following incubation for 40 hours
at 30°C, methicillin-resistant strains grew to
within 3 mm or less of the edge of the strip.
7.0 Determination of Susceptibility to Antimicrobial
A g e n t s .
i . Determination of minimum inhibitory concentration
(MIC) by agar dilution.
Except where stated doubling dilutions of
agent were incorporated into bottles containing
molten IsoSensitest agar (Oxoid, UK) held at 50°C.
A 1:10 to 1:100 ratio of agent to agar was used
to ensure uniform dissolution after mixing. Plates
were immediately poured and were usually used
within 6 hours of pouring. Bacterial isolates were
subcultured from blood agar plates into 3 ml
volumes of peptone water broth (Southern Group
Laboratories, Lewisham & Southwark) which were
incubated for 18-22 hours at 37°C unless otherwise
stated. These broth cultures contained approximately
5.0 x 10® cfu/ml, and using a Multipoint Inoculator86

(Denley Instruments, Billingshurst) which deposited
1.0 ul of broth onto the surface of dried
plates, an initial inoculum of c. 5.0 x 10^ cfu
was delivered. Using dilutions of broth (i.e. 1/100)
the inoculum effect of antibiotics could be
studied .
Inoculated plates were incubated for 24 hours
at 37°C prior to reading. The MIC of an agent
was the lowest concentration at which visible
growth was inhibited. S. aureus NCTC 6571 was used
as a control in all tests.
i i . Determination of minimum inhibitory concentration
(MIC) by broth dilution.
Broth cultures were prepared as stated above.
Doubling dilutions of antibiotic were made in 0.9
ml IsoSensitest broth (Oxoid) in bijoux tubes. 0.1
ml of a 1/100 dilution of broth culture was
added to each tube. Antibiotic-free and organism-
free controls were included in each test. After
24 hours incubation at 37°C the tube containing
the lowest concentration of antibiotic to inhibit
visible growth was taken as the MIC. S. aureus
NCTC 6571 (strain SOX) was used as a control.
87

iii. Selection of Antibiotic break-points.
The MICs of 23 antimicrobial agents against 100
strains of MGRSA were determined in our survey of
antimicrobial resistance (Table IV). Strains were
classified as sensitive (S), moderately resistant ( M ) ,
or resistant (R) to each agent according to the
MICs (see Table III). This classification was made
with different criteria depending on the agent
tested. For fosfomycin breakpoints suggested by an
International Study Group were adopted (Andrews et
al.t 1983).
A number of compounds could be grouped in
relation to the distribution of recorded MICs.
MICs for mupirocin, nitrofurantoin, pristinamycin,
teicoplanin and vancomycin were narrow with a
single peak, and strains were all classified as
sensitive. Separate distributions (bimodal) of MICs
were observed for streptomycin, neomycin, bacitracin,
tetracycline, minocycline, erythromycin, clindamycin,
chloramphenicol and novobiocin. In these instances
strains were classified as sensitive or resistant.
For other antibiotics (trimethoprim, rifampicin,
fusidic acid and ciprofloxacin) three separate
distributions (trimodal) of MICs were found, and
strains were classified as sensitive, moderately
resistant and resistant.
For the aminoglycosides, gentamicin, tobramycin,
netilmicin and amikacin there was no obvious
88
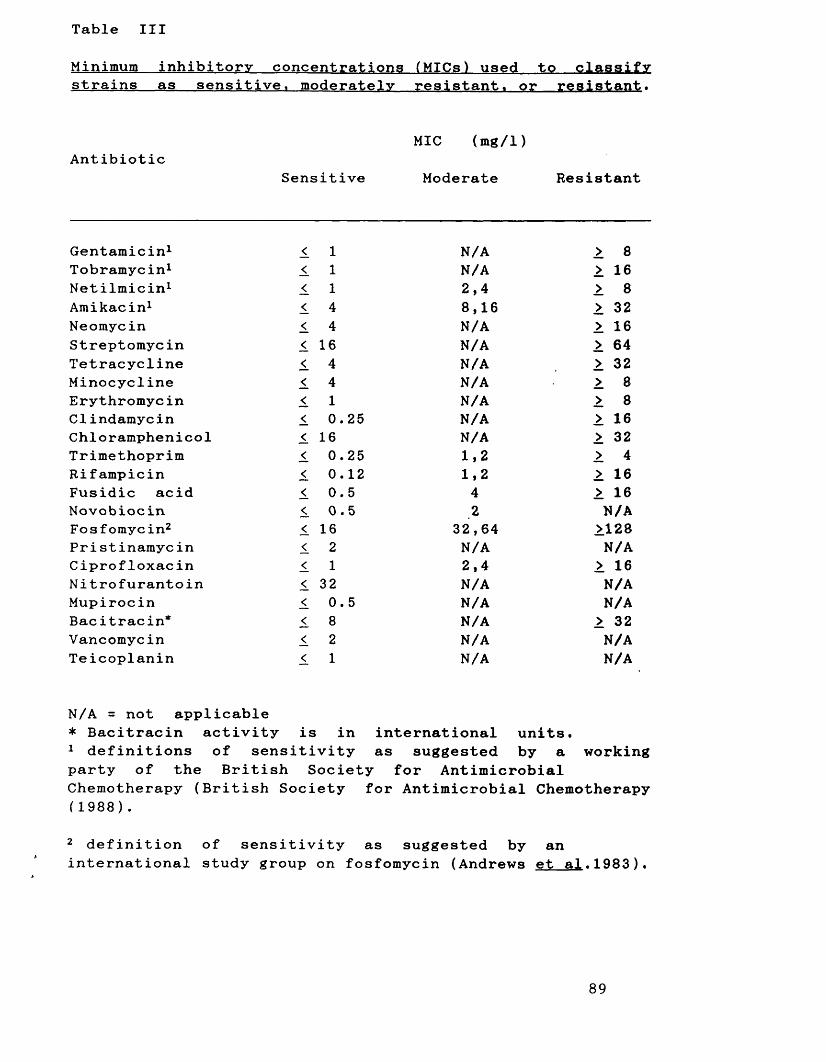
Table III
Minimum inhibitory concentrations (MICs) used to classifystrains as sensitive, moderatelv resistant, or resistant.
AntibioticMIC (mg/1)
Sensitive Moderate Resistant
Gentamicin1 < 1 N/A > 8Tobramycin1 <. 1 N/A > 16Netilmicin1 1 1 2,4 > 8Amikacin1 < 4 8,16 > 32Neomycin 1 4 N/A > 16Streptomycin < 16 N/A > 64Tetracycline < 4 N/A > 32Minocycline 1 4 N/A >. 8Erythromycin < 1 N/A > 8Clindamycin < 0.25 N/A > 16Chloramphenicol 1 16 N/A > 32Trimethoprim 1 0.25 1,2 > 4Rifampicin 1 0.12 1,2 > 16Fusidic acid < 0.5 4 > 16Novobiocin < 0.5 2 N/AFosfomyc in2 < 16 32,64 >128Pristinamycin < 2 N/A N/ACiprofloxacin < 1 2,4 > 16Nitrofurantoin < 32 N/A N/AMupirocin 1 0.5 N/A N/ABac itracin* 1 8 N/A > 32Vancomycin < 2 N/A N/ATeicoplanin < 1 N/A N/A
N/A = not applicable* Bacitracin activity is in international units.1 definitions of sensitivity as suggested by a working party of the British Society for Antimicrobial Chemotherapy (British Society for Antimicrobial Chemotherapy (1988).
2 definition of sensitivity as suggested by an international study group on fosfomycin (Andrews et a l .1983).
89

pattern of distribution, and breakpoints suggested
by the British Society for Antimicrobial
Chemotherapy (1988) were used.
7.1 Determination of bactericidal activity.
i . Determination______ of.______ minimum______ bactericidal
concentration ( M BC).
Minimum bactericidal concentrations (MBCs) were
measured by replicating (with velvet pads) from 24
hour-old MIC plates (initially inoculated with 5.0
x 105 cfu) onto fresh IsoSensitest agar (Elek &
Hilson, 1954). Replica-plates were incubated for 24
hours prior to reading. The transfer efficiency of
this method is believed to be 1% (Elek & Hilson,
1954). Because the MBC is defined as 99.9%
killing, with this method the MBC was the lowest
concentration of agent at which 5 or fewer
colonies grew.
i i . Determination of rate of killing by time-kill
c u r v e s .
Timed killing curves were performed with
selected strains. The strains were grown for 24
hours at 37°C in 10 ml of IsoSensitest broth, and
0.5 ml aliquots were used to inoculate 500 ml
Erlenmeyer flasks containing 100 ml IsoSensitest
90

broth. Antibiotics were then added to the flasks
to achieve the desired concentrations, and 0.5 ml
samples were removed for viable counting. The
flasks were then incubated at 37°C in a rotary
incubator (100 rpm) and sampled at desired time
intervals. Viable counts were performed by
spreading 0.1 ml of sample (or 1 in 10 dilutions
of sample) on IsoSensitest agar plates. After 24
hours incubation at 37°C the numbers of colonies
on plates were counted.
Antibiotic-free control flasks and strain S .
aureus NCTC 6571 were used as controls in these
experiments.
7.2 Determination of mutation rates to r e s i s t a n c e .
Selected strains were shaken at 100 rpm for
18-22 hours in 10 ml IsoSensitest broth at 37°C.
Cultures were centrifuged (3,600 rpm for 12
minutes) and resuspended in phosphate buffered
saline (NaCl 8.0 g/1, K 2HP04 , 1.21 g/1, K H 2P04 , 0.34
g/1). 0.1 ml amounts were spread on IsoSensitest
agar plates containing various concentrations of
agent. The plates were then incubated for a total
of 48 hours at 37°C and read after 24 hours and
48 hours. Colonies which grew were picked off,
reidentified as MRSA and resistance confirmed by
MIC determination. Mutation rates to resistance
were calculated by averaging the results obtained
91

in two separate experiments.
7.3 Microbiological assay of fluoroquinolones
To detect whether fluoroquinolone activity was
lost in time-kill broths as a result of
incubation conditions, or other non-microbiological
factors, fluoroquinolone levels were measured in
IsoSensitest broth after 0, 24 and 48 hours
incubation at 37°C.
Assay plates were prepared by pouring 150 ml
molten IsoSensitest agar into "Nunc" (24 cm x 24 cm
x 1.8 cm) Bio-Assay dishes (A/S Nunc, Denmark) on a
level surface. After cooling and drying, plates
were seeded with approximately 5.0 ml of 1/100
dilution of E. coli NCTC 10418 previously grown for
c. 20 hours in IsoSensitest broth at 37°C. The
agar surface was evenly covered with culture, and
the plates were then dried. 36 x 6 mm wells (6 x 6)
were cut in the agar using a no. 4 size borer.
These holes were filled with either test or
standard solutions according to a random number
pattern. Following incubation for 24 hours at 37°C
zone sizes were measured, and fluoroquinolone
concentrations in the test samples were determined
by interpolation from the standard curve (ie. a
graph of log^Q fluoroquinolone concentration versus
zone diameter).
92

8.0 Development of a system for typing MGRSA.
8.1 Phage-typing of M G R S A .
Phage-typing of the MGRSA was performed by the
author whilst visiting the Staphylococcal Reference
Laboratory (SRL), P H L S , Colindale and also by staff
of the SRL at intermittent periods.
Isolated colonies (not more than 24 hours old)
from blood agar plates were inoculated into 5 ml
volumes of nutrient broth. After incubation at
37°C for 4-6 hours these broths (which should just
be starting to show visible growth) were used to
flood dried plates of "Staph Typing" agar. Excess
culture was pipetted from the plates, which were
left to dry and then inoculated with 0.01 ml of
phage suspension using a multiple-loop applicator.
Plates were then incubated at 30°C overnight.
Next morning the plates were examined for lysis
with a xlO hand lens and the following reactions
were recorded irrespective of the size of plaques
produced.
1-19 plaques +/- weak reaction
20-50 plaques + weak reaction
50 plaques, confluent lysis ++ strong reaction
Phage typing was performed with the
International Set of phages at routine test93

dilution (RTD) and lOOx R T D . Slight variations in
patterns of lysis may be obtained with cultures
of the same organism tested on separate occasions.
Usually strains must differ by at least two strong
reactions to be regarded as epidemiologically
distinct. As an aid to discrimination, we have
also typed strains with complimentary phages (88A,
90, 83C and 932) and also supplementary ("Nuan") phages
(Richardson et a l . , 1988).
8 . 2 Identification______ of_ aminoglycoside-modifying
e nzymes.
i . Van de Klundert's me t h o d .
Dried IsoSensitest agar plates (agar depth 4.0
mm) were seeded with 1.0 ml of a 1/1000 dilution
of 18-hour old culture of organism grown in
IsoSensitest broth at 37°C. Plates were dried and
specially prepared "rings" (Mast Laboratories)
consisting of 6 aminoglycoside-containing paper
discs were placed on their surfaces. Following
incubation at 37°C for 18-22 hours zone diameters
(if any) produced by the discs were measured.
The stepwise determination scheme proposed by
Van de Klundert et a l . (1984) was used to infer
the presence of particular aminoglycoside-modifying
enzymes. Strains produced APH (2")/AAC ( 6 f) + APH
( 3 ’)-IV if inhibition zone diameters of less than
94

18 mm, 19 mm and 20 mm were found to gentamicin,kanamycin and neomycin respectively. If the strains
were sensitive to neomycin (ie. zone diameter of 20
mm or greater) the production of APH (2M )/AAC ( 6 f)
was inferred. Strains sensitive to gentamicin and
sissomicin (ie. zone diameters equal or greater than
18 mm and 21 mm respectively), but resistant to
tobramycin (zone diameter less than 11 mm) produced
the enzyme ANT ( 4 ?,4n ).
i i . Gene probing aminoglycoside resistance g e n e s .
By arrangement with Dr G. Miller of Schering-
Plough Research (Schering-Plough Corporation,
Bloomfield, New Jersey) "dot-blots" prepared by
ourselves were probed. Dot-blots were prepared by
spotting approximately 10 ul of 48 hour-old
cultures (grown in 5 ml Nutrient Broth no. 2 at
37°C and shaken at 100 rpm) onto GeneScreen paper.
The papers were left to dry at room temperature
and then placed for 5 minutes (culture side up) on
Whatman 3MM filter paper saturated with 0.5 M
NaOH. The dot-blot papers were transferred onto
filter paper soaked with 1.0 M Tris pH 7.0, and 5
minutes later they were placed for 3 minutes in
a container with 500 ml of 1.0 M Tris pH 7.0. The
dot-blot papers were then removed and left to dry
at room temperature. Gene probing was then
performed at Schering Research in New Jersey.
95

8. 3 Determination of Antibiotic Susceptibility
P r o f i l e s .
Lysed blood DST agar was seeded with
approximately 10^ cfu of organism (ie. 1 ml of a
1/1000 dilution of 18-22 hour-old culture grown in
peptone water broth at 37°C), and antibiotic-
containing discs (Media & Reagents, 3.3i) were placed
5 on one plate, and 4 on another. After
incubation for 24 hours at 37°C the plates were
read according to the following criteria. Strains
were classified as "sensitive" if the zone diameter
produced by an antibiotic was more than 66% that
produced against S. aureus NCTC 6571. Three digit
code numbers were assembled using this system, and
sensitivity was scored as follows -
netilmicin (1), amikacin (2), neomycin (4) - 1st digit
tetracycline(1) , clindamycin(2), chloramphenicol(4) - 2nd
digit
trimethoprim (1), rifampicin (2), ciprofloxacin (4) - 3rd
digit
8.4 Identification of Physiological Properties of
use in B i o t y p i n g .
i . Identification of Haemolysins by titration.
A micro-titration assay developed by Jordens
96

et a l . (1989) was used to detect and quantifyhaemolysin production in our strains.
Cultures of organisms grown (shaking at 100 rpm)
in 10 ml IsoSensitest broth for 18-22 hours at
37°C were centrifuged down (3,600 rpm for 20
minutes) and the broth supernatant collected. This
was then serially diluted (to 1 in 256) in 100 ul
volumes of phosphate buffered saline (containing 1
mM MgSO^) in a microtitre tray. Dilutions of each
culture supernatant were prepared in triplicate.
Heparin (20 ul of 1000 units/ml) was added to one
set of dilutions and fibrinogen ( 20 ul of 10
rag/ml) was added to another. The plates were then
incubated for 15 minutes at 37°C. The haemolysin
assays were then performed by adding 100 ul of
washed (2% in MgSO^ PBS) human, rabbit or sheep
erythrocytes to the plates. These were then
incubated for 80 minutes at 37°C.
The assays were read using a Titertek (Flow
laboratories) microtitre plate viewer. The highest
dilution showing definite (ie. approximately 50%)
lysis compared to a control using broth instead
of broth supernatant was recorded as the
haemolytic titre of the organism. This was a
subjective assessment based upon appearance of
diffuse haemoglobin in the well (ie. a diffuse red
colouration) and size of the "button" of red cells
at the bottom of the well. Because heparin
inhibits the action of gamma haemolysin and
97

fibrinogen inhibits delta toxin (Jordens et a l .
1989), the titre of individual toxins was
calculated as follows:
alpha-titre = titre of supernate (for rabbit
erythrocytes) not inhibited by fibrinogen.
beta-titre = increase in titre for sheep
erythrocytes after 1 hour at 4°C.
gamma-titre = titre of supernate (for human
erythrocytes) inhibited by heparin.
delta-titre = titre of supernate (for human
erythrocytes) inhibited by fibrinogen.
Three organisms producing known haemolysins i.e
S. aureus strains NCTC 7121 (alpha haemolysin), NCTC
10345 (delta haemolysin) and NCTC 5664 (beta and
gamma haemolysins) were used as controls in these
as s a y s .
i i . Haemolysis on Sheep Blood A g a r .
Sheep blood agar (5%) was inoculated with 18-22
hour-old cultures grown at 37°C using a Denley
Multipoint Inoculator. After 24 hours incubation at
37°C zones of haemolysis (if produced) were
measured. We differentiated strains on the basis
98

of zone size produced, those producing zones of
clearing of greater than 10 mm in diameter were
regarded as positive. A typical sheep blood
haemolysis plate is shown in plate 1.
iii. Egg Yolk React i o n .
We tested our strains for the egg-yolk reaction
by inoculating (with a Denley Multipoint Inoculator)
egg yolk glucose agar with 18-22 hour-old cultures
grown in IsoSensitest broth at 37°C. The plates
were incubated at 37°C for 48 hours and read.
Production of a zone of opacity was taken as a
positive reaction. An example of an egg yolk
glucose agar plate is shown in plate 1.
i v . Tween 80 hydrolysis.
Lipase production was also detected by
inoculating (as in the egg-yolk reaction) strains
onto Tween 80 medium and incubating plates for 48
hours at 37°C. Tween 80 hydrolysis was detected
as a zone of opacity (due to formation of small
crystals) around colonies. A Tween 80 plate is
shown in plate 1.
v . Pigmentation on Milk A g a r .
Lacey's Milk Agar (Lacey et a l . . 1970) was used
99
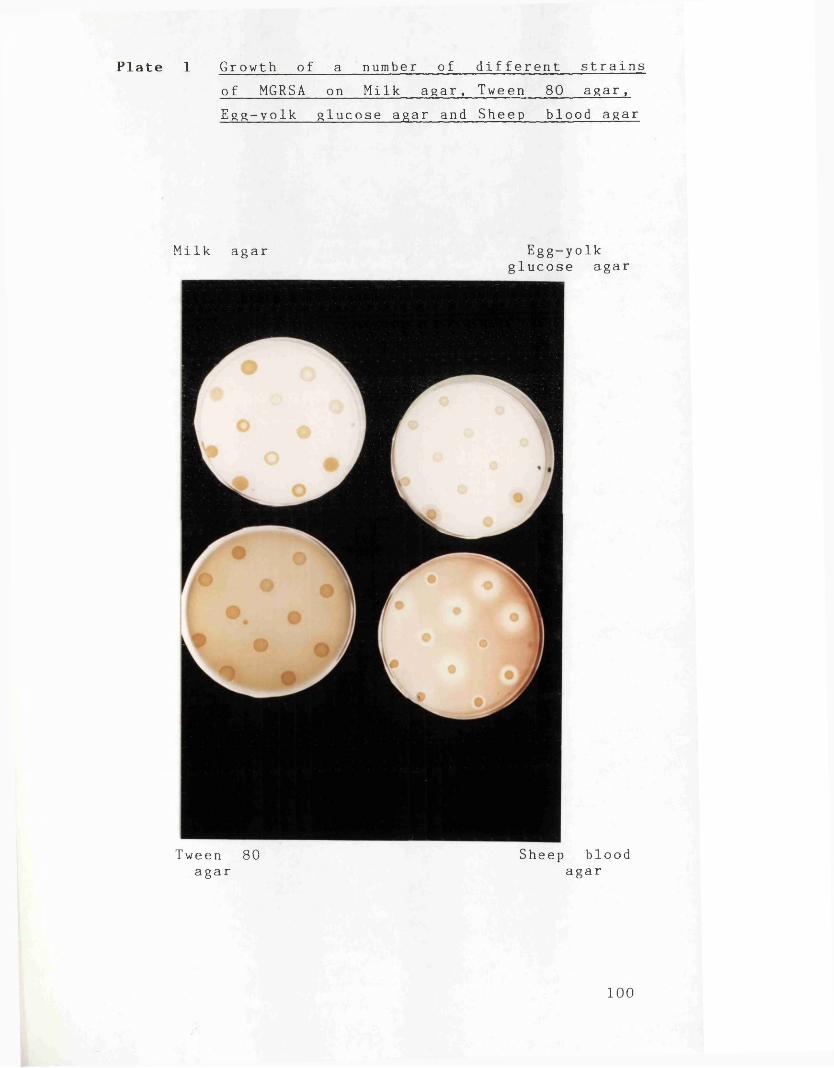
Plate 1 Growth of a number of different strains of MGRSA on Milk agar, Tween 80 agar, Egg-yolk glucose agar and Sheep blood agar
Milk agar Egg-yolkglucose agar
Tween 80 Sheep bloodagar agar
100

for pigmentation studies. Milk agar plates were
inoculated using a Denley Multipoint Inoculator
with organisms grown for 18-22 hours at 37°C in
IsoSensitest broth. The plates were incubated for
12-16 hours at 37°C and then left at room
temperature for a further 12-16 hours before
reading. An example of a milk agar plate is
shown in plate 1.
8.5 Plasmid isolation by Gel Electrophoresis,
i . Takahashi and Nagano M e t h o d .
This method is a rapid procedure for isolation
of plasmid DNA and its application to
epidemiological analysis (Takahashi & Nagano, 1984). It
involves alkaline lysis and extraction of denatured
chromosomal (linear) DNA and RNA followed by salt
precipitation of chromosomal and other protein
material.
Cells were grown for 12 hours (shaking at 100
rpm) at 37°C in Nutrient broth no. 2 (Oxoid) and
harvested by centrifugation (3,600 rpm for 15
m inutes). Cell pellets were resuspended in 200 ul
buffer A and transferred to "Eppendorf"
polypropylene centrifuge tubes. These were
centrifuged (low-speed) for 4 minutes in a MSE
Micro Centaur centrifuge, buffer A was decanted and
the cell pellet was thoroughly resuspended in 200
101

ul of lysostaphin solution. This contained 12
units/ml lysostaphin (Sigma Chemicals) in 100 mM
NaCl, 40 mM Tris-NaOH, and 50 mM disodium EDTA at
pH 6.9. The tubes were incubated at 37°C for 10
minutes, and then, 400 ul lysing solution (4% sodium
dodecyl sulphate with NaOH added to a final
concentration of 0.2N) was added. The tubes were
gently rotated so as to mix their contents, and
then they were left to stand at room temperature
for 5 minutes. 300 ul of cold (4°C) buffer B was
added and the tube contents mixed by gentle
rotation, then after standing for 5 minutes on ice
the tubes were centrifuged at low-speed (rtp for
5 minutes). The tubes were then stood on ice for
a further 10 minutes, and then centrifuged at low
speed for 5 mins at 4°C. The supernatant was
decanted to new tubes taking care not to carry
over any white flocculate. An equal volume of
chloroform was added to the supernatant, and
following emulsification (by gently inverting 10
times) the tubes were centrifuged at low speed for
5 minutes at 4°C. 500 ul of the upper aqueous
layer was carefully transferred to a new tube to
which 1 ml of cold (-20°C) ethanol was added.
Following mixing and standing on ice for 5 mins
the precipitate was collected by centrifugation at
low speed for 5 mins at 4°C. After drying to
remove traces of ethanol, the pellet was
redissolved in buffer C. For gel electrophoresis
102

25 ul of sample was mixed with 5.0 ul loading
buffer.
i i . PHLS "Johnson" me t h o d .
This method differed from the Takahashi and
Nagano method primarily in that it uses RNase
(bovine pancreas RNase, Sigma Chemicals) and Protease
(Type XIV, Sigma Chemicals) to remove RNA and
protein impurities.
Strains were grown on blood agar overnight, and
thick suspensions were made in Eppendorf tubes
containing 0.25 ml lysis buffer (2.5 M NaCl, 50 mM
EDTA, pH 7.5). Lysostaphin was added to a final
concentration of 10 units/ml. The tubes were
placed in a 37°C water bath for 20 minutes and
then 0.4 ml lysis buffer (see "Materials 4.0ii) was
added. Following mixing the tubes were centrifuged
at high speed (Eppendorf microfuge) for 50 minutes.
The supernatant was carefully decanted into a new
tube to which RNase (final conc. 100 ug/ml) was
added. After incubation at 37°C for 30 minutes
Protease (final conc. 100 ug/ml) was added, and the
tubes were incubated for a further 30 minutes. An
equal volume of cold (-20°C) isopropanol was added
to the tubes which were left overnight at -20°C.
Plasmid DNA was pelleted by centrifuging (high
speed) for 15 minutes, excess isopropanol was
drained off and the tubes left to dry for 30103

minutes. 10 ul loading buffer was added to the
pellet, time was left for it to dissolve and then
agarose gel electrophoresis was performed on these
mixtures.
iii. Agarose gel electrophoresis.
Electrophoresis was performed using a BIO-RAD
DNA sub cell. Tris-acetate pH 7.8 (buffer A) was
used in the Takahashi and Nagano method and Tris-
borate buffer was used for the PHLS method. 0.9%
Agarose (Sigma Chemicals, molecular biology grade)
gels were used. Electrophoresis was usually carried
out overnight. Gels were stained with ethidium
bromide (0.5 ug/ml) for 30 minutes, rinsed in water
and the DNA visualised for photography (with orange
filter) by a shortwave UV transilluminator.
i v . Plasmid sizing studies.
Three plasmids of known size were run in
parallel with the strains to be tested. The
plasmids used (Sykes & Matthew, 1976) were - plasmid R6K
(26 Md), plasmid RP 4 (36 Md) and plasmid R1 (61 Md).
The size of plasmids isolated from test strains
was estimated by interpolation from a graph of
log^Q size (Md) against distance migrated.
104

RESULTS AND DISCUSSION
1. Antibiotic Resistance in MGRSA — how serious is the problem?
As the 1970s progressed, concern over the
problem of multiple antibiotic resistance in S .
aureus declined to such an extent that this
decade has been referred to as the "Decade of
Complacency" (Shanson, 1981). With the emergence of
MGRSA, the 1980s have witnessed increased activity
and debate pertaining to the problems posed by
multi-resistant S . aur e u s . In particular,
strategies for eliminating and controlling MRSA
have received renewed interest from
microbiologists, clinicians, hospital administrators
and pharmaceutical companies alike (Working Party,
1986 and 1990). Because of the differing
geographical incidence of MGRSA, opinions vary as
to their importance, for example, Lacey (1987)
believes that little action is needed to control
these organisms. Because there has been no
international survey to examine the problem of
antibiotic resistance in MGRSA, we have collected
MGRSA from hospitals worldwide and determined
their susceptibility to a wide range of
antibiotics.
In Table IV the extent of resistance to 21
antibiotics is shown for 100 isolates of MGRSA
105

w s ssP p H- n ►oH- H' rt H H O co nO H'H- 3
O Tl H- h O Hd H- CO 4 CO H i O rt O Hi H- 3I-1 P H -O to o X 3 H- 03 ^ P O O H- H*P P
as o c< CO O H- CH P- H- H*
CP H H- H Hi H- 03 3
OH-Hd P H
H-
o n m s H co s:pH H* H H' fD rt CD(—1 P rt H OO P rt o H 0) 3H P P O 0) P X p p H ej O rt O3 3 O O ^ O H-*P *-< 3 l—1 O 3 P
H* l—1 P H- OCD P H-
P n ^P H* O P P H
> PS H O 3 P O P H> rt p p JP H- H <tp l_i P p0 3 3 3 H- H*P O O O
H* H- H* P P P
WCOW2:c/3C/lWWC/3?dWcOfd^?0?3WS^W|P
GOCOGOCSCOCOCOCOCOCPCPCOCPCPCPCPCPCPCPCPCP
C O W M 2 W W W C O W P 1 P W P P P P W 2 W P P
COCOCOcOGOtOCOCOC/OGOGOGOCOCPCPCPGOSJdCPCPCP
W M W W C O W C / 3 W W p O C / l ? d p i P P ? d W W W P ? a
C/^COCOCOGOCOCOCOpdCOCPCOC/^CPSPCPCPSSCPCP
<PwI—1CTi
wI—*'■vl
n
CPHjS3
CPHIcpro
CPHjCP00CPHjCP-P"
CPHjCPon
>prtH-o*H*ortH*o
wrt>-»pH-p
C/3rtppH-pW
HiHo3
oo
opprtHpw
w i-3X P>rt a 4P H*P Prt
C O W W S c n C n W w C O W W C d C P C P f p C P W S W C O C d
CPHdCP
CPHjCP
COCOCOCOCOCOCOCOCOCOCPCPCPCOCPCPCOSCPCPCP
106

w 2 s:3 3 H- O *3 H- H
3o nH 3
H- 3 O *d H- CD h 01 hhO rt o l-h H- 3 I—1 3 ^ 0 3 0 X 3 H* 3 ^ 3 O O l_u H-3 3
^ *3 3 H- 3 Hi H' 3 3- 3 H->3 O H-
O H-
3 3 O H- <=u
H n 3 3* H- M 3 O3 3 rt 3 3* 3 O T3 >3 3 “
n M SI—1 •-* M- 3
3 rt o 3 “ O 3 O O 3 Mp.O 3 H- 3 3
H CO 3 3 rt 3 rt 3 O 3 3 3 3 *d ^ o rt o< O H* 0 3 3
H- O 3 H- 3 3
> s; H O 3 3 0 3 H- rt o' 3*T H- 3 rt3 H» 3 3 0 3 3 3H* H*3 0 0 0 H- H* H*
3 3 3
C/lCOCOCOC/l2SJtiWSCOCOUdW31«CO?0333
C/lCOMC/lCOCOC/lpdCOCOWWWCOWWWfdS^^
cococococococococowcoco^djsdfsa^cosdtsajtfpd
COCOCOCOCOCOCOCOC030COCO?3j50>0j50CO3d?!d30S^
c/ic/iwc/ic/iwc/ic/ico^wc/ifd?3»?dw?d?djfljd
C / l C O C / l C O C / l W W ^ W S C O C O ? d 3 3 3 W 3 3 3 3
COCOC/lWC/lWW?3C/lS:WC/l?dW>d?dC/lWW^3?d
cococococococococoGococoj3>c)£ci:?a£a:?dS=dt3:3
► CO m
P rt Xrt 3 rtH- 3 3O* H- 3H- 3 rtO 3rtH* OO Hi Hi
3O3
33
to H*3
1—1 rtP3 o 3I—» 3 3
3 Ort 33
M 3S3 3 rtto O
H-3 O
1—1 33
■P' M 33 33 3h-1 rt
M 3 H*>3 3 <t—» P.-P> CO
rt 33 3 <3 3 31—1 H- 3* H-
P3 P (—1I—* 3Ul to o*
1—*3
O1—1 3*3 3 >I—* rt 3"J 3 rt
3 H*3 a*
H*M oP3 H* rtI—1 3 H*00 O
3CO
X )CO 3TJ H*t—1 3•
H3C7*
H<
cococo3:cococoaico3:co3d!?03ds=03a?333?d?dj3dCO3jto
COLOC033COWCOCOpdCOC03d33!7d>C?3C03df5d3d33CO*TJ(jO
107
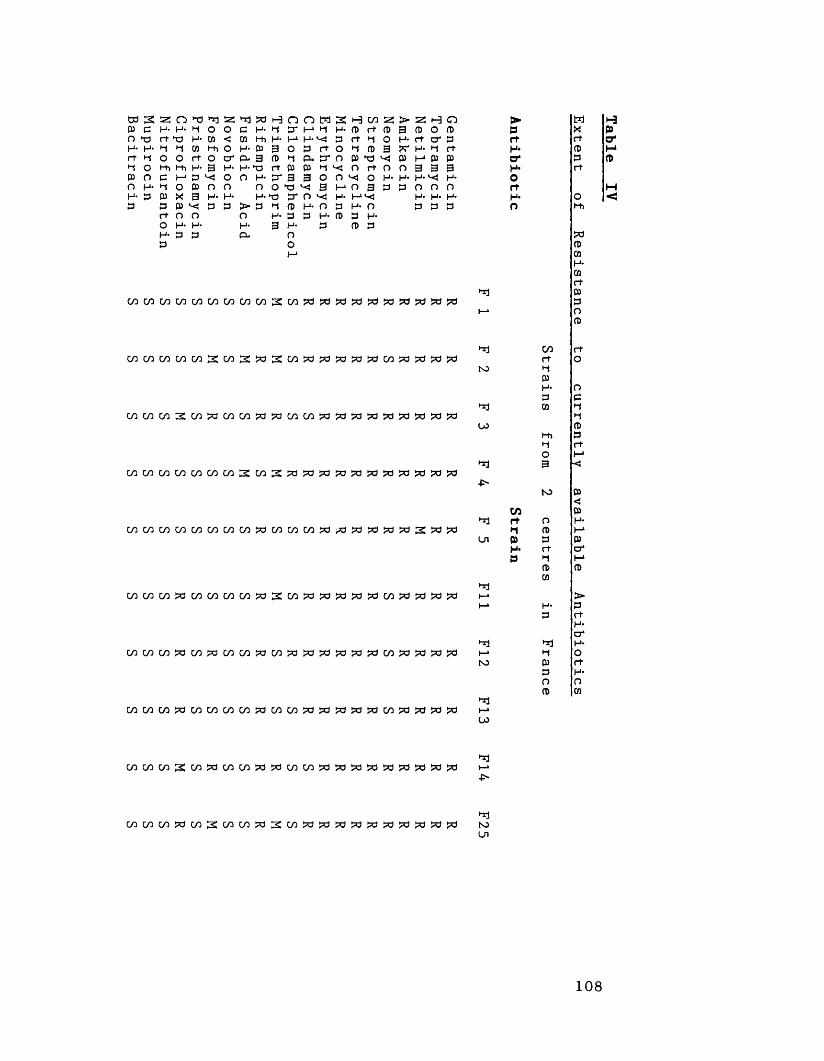
tusz:05 C H- n >oH* H'H O O H'3
n •D *rl H- O O ►O H- W
(0 Hi O rt O l-h H- 3 M P *-«Jo (a n X 3 H- P ^ Po oH* H-P P
C H-03 Hi H- P CL. 3 H-X3 O H-n
H-> PoH*a*
n oO' i-* (_i h- O P
P* P 3
o
M 2 H W S ! H H- P rf p
h- p rt H ort O H P 3c n Pti^ H ^ o rt oO O o H-3 I-* o 3 p^ H- l-“<jO P H- OH- P P H- P P P
> 2! H O3 P O P H1 ft O* P ?T H- H rt P M P P 0 3 3 3H- H* *"< H- P O O O
H- H* H- P P P
03 03 03 03 03 03 03 W 0 3 S 0 3 ? O P P P P « P P P P
cncAicococ/32:co3:>02:cn?d^3>d?d^cn?d>c3pd?a
M W C / ) M W M M W P W M C / 3 W ^ a P P P P 2 P P
0 3 0 3 W P C / D 0 3 0 3 C / 3 P S 0 3 ? d ? O P P P 0 3 P P P P
oococo?dco?a(y3co>acojd>d>a^?o^cy3?d?d>d^
C/3C/3C/3?dC/3WW03?dC/3W?d?d?a?d?ClC/3?dW?d^
•n
>prtH*o'H*OrtH*O
M i-3X Prt O'P MP Prt
*n C/0 rtrt O
to HPH* oP pCO n
Hco P
Hi PH rtO I-13 ■<*
■P-to P
<w P
*1 rt o H**-» p 1—1
Cn P p PH* rt O'P H 1-*
P PW
I—* >1—1 H- P
P rtH-O'
Hj trj H-1—> H Oto P rt
P H*O nP cn*1i—*
OJ
hji—*■P>
W W W W M 2 W M » 3 : w ? d ? d ? d ? d ? d » ? d ^ ? c l ? d HjtoCn
108

(U C H-o tsH- H' rt H H O 00 O O H'H- 3
O tjH' *1 O *d H* (D H M H i O rt O Hi H- 3 I—* £3 *-<J O 03 O X 3 H- 3 ^ 3 O O H* H- 3 3
Hj tsd 3 H- 03 H i H- 3 3* 3 H-X3 O H<
O H* 3 3
O H»CX
o o3 I-1 I-1 H- O 3 Q-
3 3
o
M 2 H W 2 4 H- (D ft (D^ 3 rt H Ort o H 3 30*0 3 ‘dv- H ^ O rt nO O O H*3 M O 3 3^ H- H 1O 3 H- OH- 3 3 H-3 3 3
> as h o3 3 0 3 H- rt o* 3 X- H* H rt 3 I—* 3 3 0 3 3 3 H* H* ^ H* 3 0 0 0
H- H- H- 3 3 3
cococo3cocococococo>dco>ci:*3S3>d!?a3:£dj3!*j
cncnw^cncncncncncnzocnxjzoxi&zo^pcipdpa
GocococoGocococoGoco>dCO>ci:?oj3a>dp3>d:?dpa:?o
Gococo2:c/ocyococococA)OdCo?ot3dpci?ciodOdpdpd?>d
cococ/02:cococncoooc/)0dcy00clj5d?d?cl0cl0>d0d?a0d
cococowcossacocococo^acowtsaiadi^JfjapasdWfa
cyococotyocotyocoooco3:>o?=dociodj3dpdoo2:oooci>d
cococococococoscocostfSJdwcocowcoiPdssJpd:^
> CO Mp rt Xrt H rtH* 3 3o* H* 3H* 3 rto 3rtH* Oo H i H i
HO3
33
i—* H-3rt
o o 33 3
I—* 3 Ort 3H3
o rtO
NJ H*3
OC
o 3S H3 H
OJ 3 3rt 3
rtI—1
o O3
•IN H3 3
w 3 <rt 3 3
o H*3 <• H>
Ul H* 3p > cr
3 i-»3 3
o rtH
ON H- >3 3
rtH-
< 3 cr3 H-oo 3* o
rtH-
CO O< *: 3
H*1—> rtUl N3
HM
CO 3«; 3I—1 0-
H3o'
M<
W W W M W C O W 2 3 W 3 0 l 3 3 3 3 M 3 : 3 3 3cos:ho
109

a s a;0) 3 H- n 'TdH* H' r t H H OCO O O H- H* 3 3
o "d *r| H' *1 O d H- CO ^ CD Hi O rt O Hi H- 3 1—1 3 *-< O P Ox a h -03 ^ 3o nH* H- 3 3
T| |Pd 3 H- CD Hi H* 3 P- 3
D H-nH-
> 3OH*P-
o n cr i-1l_» h* O 3
P* 3 3
o
W 2 H C D 3 H H* CD rt (13^ 3 rt 4 Ort o H CD 3crn oti'-o n rf n O O ^ O H-3 M O a 3^ H- (-‘VJO 3 H- OH* CD 3 H*3 CD 3
> H O 3 CD O CD H- rt cr 3** H- *1 rtP 1-1 03 030 3 3 3 H* H-'-eJ H* 3 0 0 0
H* H- H* 3 3 3
cowcyocoai2:c^2:co?dco?djd?apd?a?a?apdjd>d
WCDWU3CDWWC/)C/3SWJd?dCDaiMfO?d>Cl33
cococococococococo2:co:?a:?dcococo!?citd:?cii=dw
C D C D C D C D C D C D C D C D C D S C D 3 3 3 3 3 3 S W W W
cococococococococosco!*3!?dcococosd?d!*3!?af5d
cococococococococo3co!!d:*3cococo:?d?dSd!?d:*3
c jco
C=JCOI—*S3
C3COI—*w
5d3S
*32to
300
5*33On
►3r tH-o1H*OrtH*O
COrtn0)H*3
COr tH3H*3CO
HiO3
OCD3rtHCDCD
cdCO>
w HX 03rt O'<D I-13 CDrt
o3H*-{CD3r tI-*
c o c o c o c o c o c o c o c ocococosdtdcococotJd SS’ci^jsdcoO
C O C O C O ? d C O C O C O C O C O C D C O W ? d C O C O W ? d S W ? C l W
COo
110

W 2 a p p h*OH* H'
t OnH'P
o a H- *-j O •o H- 01 ^ M Mi O rt O Hi H- 3 I—* P <4o t» o X 3 H- P ^ P O O H- H- P P
Hr)P H- W Hi H- P CL g H-H3 O H-
O H-
> P O H- CL
n o p* h*I—1 H- O P
& p 3
o
H W 2 ! P rt P ft 1 O H{ P P ►P O rt v<j o O 3
h* n P H- p p
3
oH-P
> z; h o3 P O P
rt cr p1-4 rt
M P p3 3 3H- H*n o nH- H* H*P P P
c/iC/sc/scncoacoGAayPac/spaas’a a a a S ’o a a
co co co co co po co co cra^papaopcioopcicocopppat^ppc)
oococococooooooooocococo^dcooopapdSZcizci^a
ooc/3c/3cocowcoooza?pcoc/3?d?d?d?pfp!7dfpi5c(>d
MWWC/lWW(/)W?OWWW?ctW?OW?dtti?OJd?a
coGoc/QGOGnzac/ooopaazacn^dpci^^d^aaazci
{► C/3 WP rt Xrt *-j rtH* P Po' H* PH* P rt0 COrtH* On l-h l-h
•-4O3 a
Pcn
t—1 H-ww rt
N o Pi—» p P
p Ort Pt-4W p
N rtNJ OH-
POw P
N W H■P* H »-4
P PN PH* rt
W I-1 l-1N <i—*N) P
P PGO P. <Jrt P
W •-i H*CS3 P h-» Mi—* H* PUl p cr
o i-1p ppw rt
N *-4 >I—* P po rt
H-H- crP H*
w oN rtI—* O H-"J a O
H- COI-*P
W
Hpo*
H<3
N
00
oaCO
W W W M W M W W W W W t / l | ^ W a i W ^ ? d ? 0 ? d ? d oa-P'
111
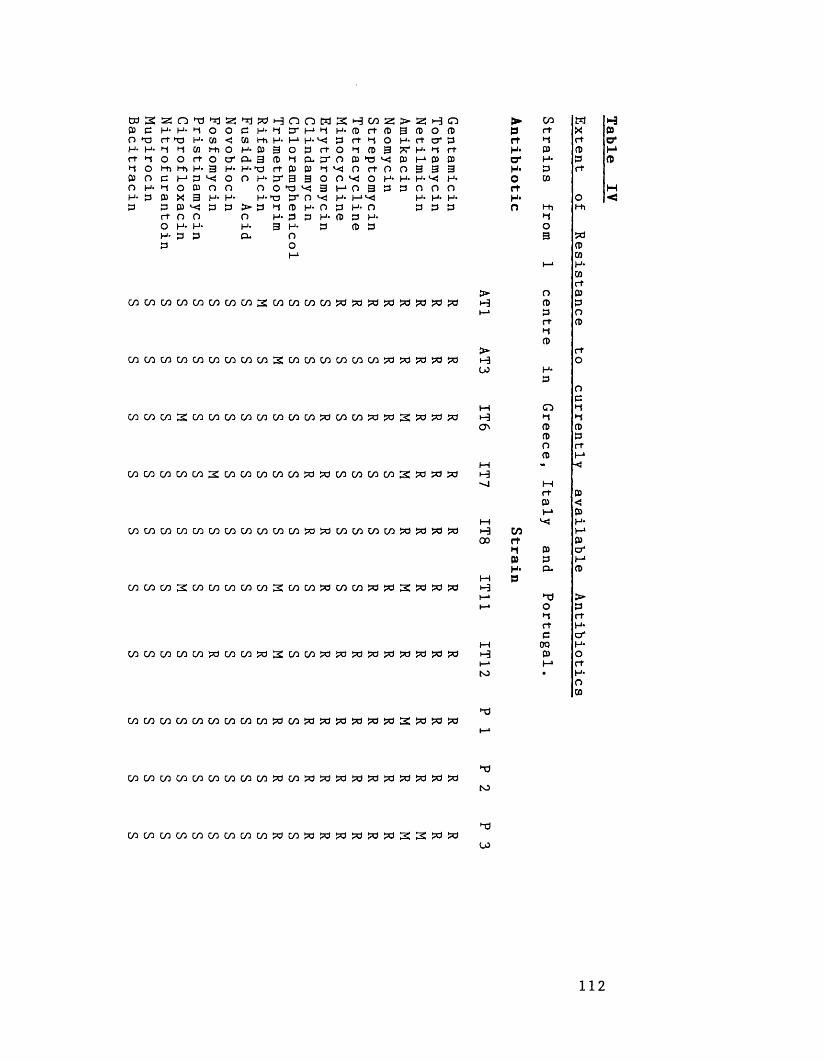
W S 2 DJ C H- O hd H- H
3 O O H 3
O >d rrj H* 4 O hO H* CO ^ CO l-h O rt O l-h H- 3 M 3 >■<( O 03 O X 3 H- 03 ^ 3 O O H- H- 3 3
►d PH 3 H- CO l-h H* 03 o- 3 H*hd O H*
O H-
> 3 O H- 3*
H O 3 3* H- H-> 3 O CD i-f rt 3 3* 3 O hd *3 3 "I 3 H- 3 3 H*
Oo
o wH* 3 H-'-eJ3 rt
S H W 2 S H- CD rt CD 3 rt •-$ O O h (D 3 O< O rt O O ^ O H- I-1 O 3 3 H- t—“<53 h- o3 3 H-
CD 3
> as 1-3 o 3 CD O CD H- ft O' 3 S' M- h rt 3 I-* 3 3 0 3 3 3 H. H. H. 3 0 0 0 H- H- H*
3 3 3
GOGACOCOCOCOGOOO^COcncoco^dXiSdjdSdSdSdjd
WCOCOWWCOCOWC/3SCOCOCOCOCOW3S33S3S3
C 0 C 0 W 3 : c / l C 0 M I / l M W W C 0 3 C 0 C 0 f d 3 S 3 3 3
C O C O C O C O W 2C O C O C O W W 33C O C O C O W 2333
C O C O C O C O C O W C O C O C O W W 3 3 W W C O C O W 3 3 S 3
w c o c o 2 c o c o c o c o c o 2 c o c o s o c o c o » 3 S W 3 3
C/)01COC^C/lCOCyDC/103t3COSdSdi^3S3ScJSc3S»S3?d
>H
>■u>
HO'
MH•o
HI— *to
•-d
► CO H Hp ft X 3rt •d rt O'H* 3 3 1—*O' H* 3 3H* 3 rtO 3rt HH- O <0 l-h l-h
o33rtrt3
ort33O3
rt 33 <}M 3
l—l H*>-a CO Moo rt 3*1 3 a*
3 3 I-1H- a. 3
M PTJOrtrt3003
cococococoGncococosacosdSdtdsdfdtdSdSdSdfd "dt o
C O C O C O C O C / l W C O W M 3 C 0 3 3 3 3 3 3 2 : 2 3 3 ►d00
112
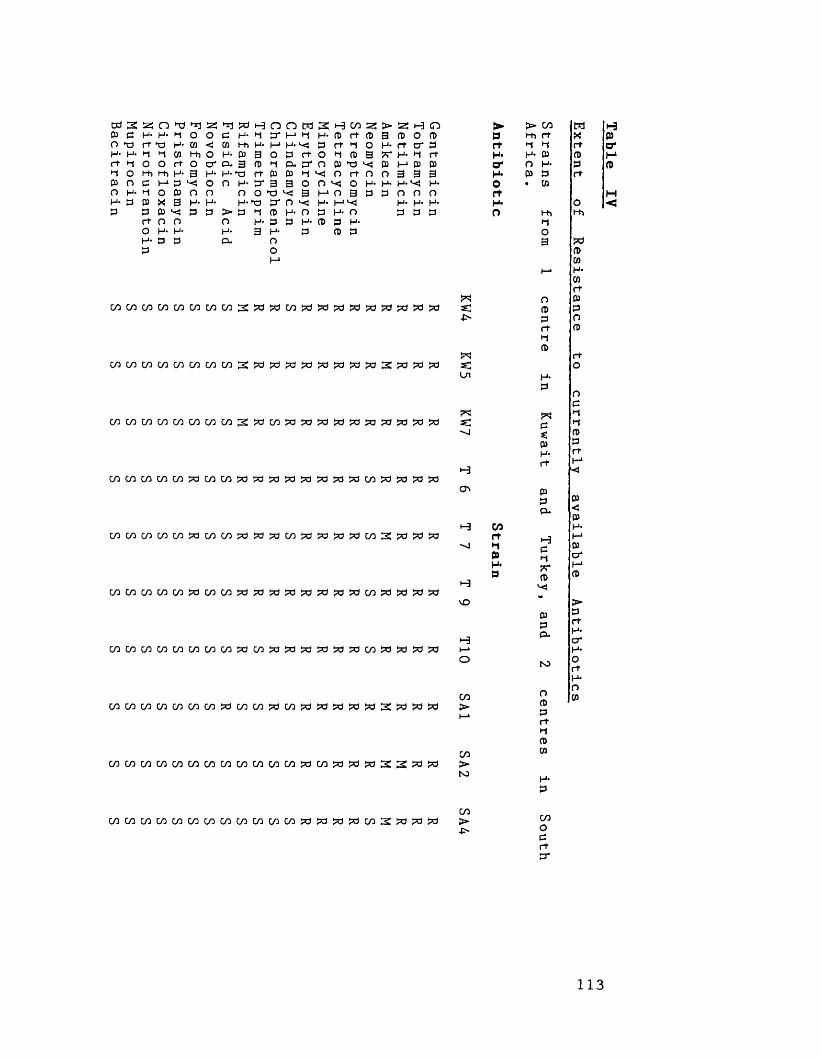
05 3 O hd H- H* r t •-! t O
H- t O *0 H‘ CD t 3 l"h O r t O l-h H* 3 I—1 3 *-<O 0) o X 3 H- P ><! 3o nH- H- 3 3
<i 05 i -h h - I—1 H* 3 r t h O H- r t o ' 30 H ’ P 3 0 3 r t 0 hl ( t 9 R ‘ H ' h r t O' & 3 3 h a n C55 hr) ^ 05 I— 1 (55 05
H - 3 r t p P h X O r t O O O H- M O 3 3
O H* 3 * 3 3 O O O T ) ^ 3H-h3 3 O X
> 3 O H- 3*
t 3 H - O H- 3 3 H- 3 H* 3 O O
H- O 3 H- 0) 3
F'n0 5 0 5 0 5 0 5 0 5 0 5 0 5 0 5 2 3 ^ 0 5 3 3 3 3 3 3 3 3 3 Si-P-
OOCOCOCy5GOC/500COS?33333>Cl33>dpC)3d2:?d?=d>d s;Ul
0 5 0 5 0 5 0 5 0 5 0 5 0 5 0 5 2 3 0 5 3 3 3 3 3 3 3 3 3 3 S]"J
C / 5 0 5 C / 5 0 5 W ? d 0 5 W 3 3 3 3 W 3 3 3 0 5 3 3 3 3
cococoty5co^coco?c)?d3dco^W53d>aco2:?ci?033
coc/5c/5c/5c/5?>dc/5cn?d?df3d?=a>dpd?d3doo3cl?d>dj5c)
cr»
vo
cocococococococo?dco>a!?aso?'d*3:?aco!?a:3dWS5a i—*O
cocncncncncncncnzawcnpocn&zapapo&^s&i&pa >
coC/50505050505C/5050505W053W33322:3I3 >t o
> > CO M Hp l-h rt X 3rt •t t rt o*H* H* 3 3 1—*O* O H- 3 3H* 3 3 rtO • 3rt HH* O <O l-h l-h
COrtti09H*P
O(153r trt(l>
PICs:3H-r t
333*
H3Ct?r3
333 *
to
O33r tr-t33
COC/5C/5 0 5 0 5 C / 5 C / 5 C 0 0 5 0 5 C 0 C / 5 0 5 3 3 3 3 C / 5 2 3 3 3 >-P*
COOcr t3 *
113

a 3 a3 3 H- O X)H* H'
M OoH'3
n |-d w 'ss,H- M O O ►a h* 3 <j M 3 l-h O o 3 o cr Hi H- 3 H- I—1 3 *-< O 0 3 0 0X 3 3 O O H- H- 3 3
W a3 H- 3 Mi H- 3 3- 3 H-X3 O H-
O H*
> 3 O H- O-
n o w3* I-1 M M H- 0 3 3
3- 3 3oH 3
3*OO3
' oH*3
H- 3 3 3 3 3 M O O 3 3 3 O 3 t l '< j ■ < 0 3 0 O ^ O H- M O 3 3
3 H- O 3 3 H-
3 3
t> « H O 3 3 0 3 H - 3 0*3 ?r H- M 3 3 3* 3 3 0 3 3 3|_l. H .V«- H .
3 0 0 0 H* H- H* 3 3 3
W M M W W W W W C / 3 2 W M W W W > d W » ? 3 1 t ! ! d
M W M M W W M M M ^ M W W M ! X I 3 W 2 » » 3
W W W M M W W M W S W W W W J d J d M S W ^ W
> CO Wp 3 X3 •-J 3H* 3 3O* H> 3H- 3 30 33H- OO 3 3
>-»O3 a
33
i—* H*33
o 3> 3 300 3 Ot—• 3 3
M3
3> OCO H-NO 3
O3
> M> 3 •-{C/3 3 300 3 3
•-» 33 MI-* ■<
> H*CO 3ON >• 3
<CO 33 3 H-
> •I a MCO 3 3 3I—1 H* 3 cro P i-1
333
33 a* >« 3I—* 3
a H-o cr3 H*
a 00 oa 3NO H*
a Oo 33
a 00a •
i-33a*I-*3
M<
<j0
c_,a
w w w w w w m m w 3 ! w ^ ? o w w w ! ! f l ^ w ^ » Ch"dNO
114

3 3 H - H - » ~ 1 0 0 0 H - '-1 3 'l-1 '-»H-3 O *3 r+ T3 H* 3 < W fh H- H H' ^ 3 rtH - H ' 3 |- < 3 l - t i 0 H - 3 3 0 3 r t 0 ' - < f t i o o r t o t f a g o ^ a D ' O P i 3 O l-ht-hH*a H-H*T3 rt 3 3 t-jv- o3 r > 3 i - ‘ 3 v< 0 0 H - 3 ' 3 3 0 0 i<i O H- •-» O 3 O O O O >3 <5 3 MOH- 3 3 X 3 H- H- H-t) t r n H- H3 3 3 l< * 3 3 > 3 ' - t 3 H * O p H -
rt O O O H- 3 3 H* 3 3O H- H- H- 3 H- 3 (DH- 3 3 O* O3 O
W W W W M M M W W W 3 W 3 3 3
W W W C / 3 W C / 3 C / 3 M M W M M 3 3 3
M W W 0 1 W W W W W 2 M 3 3 3 3
c o c o c o c o c o c o c o c o c o c o s d s d s d s d s d
W C / ] W W C / 3 M W W W W ? 3 ? C l ? d 3 3
W C / 3 W S W W W W M W ( y 3 3 3 3 3
W W W W W W W W M C / 3 C O C O W 3 3
W W W C / 3 W W C / 3 W W ? 3 ? d W | P d ? d 3
W W W W W W W W W W W W M 3 W
CO 25 > 25 t-3 O f> CO W t-3rt CD 3 3 O 3 3 rt X 3O H- rt cr 3 rt ►t rt O'CD 3 sc H- •-J rt H* 3 3 H►3 3 I-13 3 o' H- 3 3rt o o 3 3 3 H* 3 rtO H- H- H- H- O 33 3 3 O o O rt H
H* H* H- H* O •<o 3 3 3 O rt> rhH- ►13 O
3 S333
i—* H*3rt
td o 3RR
S3 S3
RR
tr" 3 3t—* 3 O
rt 3•-»3
to rtRRR
S3 S3 S3 tr1 OCO H-
3 O3w td 3RRSMSR tr* 3 *-{■P* I—1 3OQ 3H* rt3 I—*"d 3 ■<RRRRSR tr <•
I—1TJ 3O <CO I-* 3
TJ rt 3 H-RRRRRR
f ►1 3 I-1CO 3 3. 3
H* >• crP rM 3
n 3RRRRSR tr 3CO rt >
3rt
O H--d 3 crRRRRSR tr* •-J H--P* 3 O
3 rt3 H*
OM 3RRRMSR
oON 3
3P-
MRRRRRR
o S3(-* 3N3 3
3H-3
S3 •SRSRRR
CONO
115

from 32 centres in 23 countries. Table V
summarises these findings in terms of the
percentage of isolates resistant to individual
antibiotics. Although the incidence of antibiotic
resistance in the MGRSA studied has been
quantified, it is intended that our figures
present a qualitative assessment of the extent
of antibiotic resistance in MGRSA. In
epidemiological parlance "incidence" refers to the
number of new cases (or strains) in a specific
population over a defined time period, and
"prevalence" refers to the total number of
current cases (or strains) of an infection in a
defined population at one point in time. Our
data satisfy neither of these requirements. We
wish to show the nature of particular isolates
that are present, or have been present, in
hospitals in different countries and regions, and
hence obtain an indication of the problem of
antibiotic resistance worldwide. Because of the
ability of MGRSA to cross-infect, and the
increasing international transfer of patients
between hospitals, introduction and spread of
multiple-resistant MGRSA from "foreign" hospitals is
already becoming a problem (Carroll et a l ., 1989).
116

O H 3* ft I-1 H*o g►i033>i=i3'P i-J
n m sI-1 *1 H- H- ^ 3 3 rt O 0-3 0 03 *-J '-O 3 0 0 ^ 3 I—1 O ^ H- H- O 3 3 H- P
3
H C/3 2 cd rt n> rt ^ O ^ CD 3 P 3 "<S O r+ O ^ O H- 0 3 3 l-1H- O 3 H*CD 3
> 2 H O 3 CD O CD H- rt cr 3 fT H- O rt 03 1-1 03 03 0 3 3 3 H- H-l< H* 3 0 0 0
H* H- H- 3 3 3
J ^ U i - D ' v O - v J C C v O O i v O v O O v OH O O s j H W W O H O O U i O O O
>3rtH-crH-OrtH-O
•-$CDCOH*COrtP3OCD
aCDJO1-1CDCD
Ol-h
2S3cn>t-h
O3
u>N3OCD3rt
CDCO
toU»
0)cr
< •-3 00 ss •n >3 n S3 >p P 3 H- 3 P o 3 o H- H- 33 H- H- rt 3 O < CO CD3 l-h rtO O co O H- H* o H- t-h •1 P H*O O rt O •-$ rt cr CL O O 3 cr3 >3 H* t-h O 3 H- H- 3 l-h 3 H*
I- 1 3 3 O P o O I—1 H- oO P P ►1 H- O o o O O rtH- 3 3 P 3 H- H- H- X H- H-3 H- 5 3 3 3 P 3 p 3 O
3 O rt o oH- o H- H-3 H- CL 3
O O O O O I—1 I—*H N3 S3 to Ln I—* to 00
o9
CDCOH-COrtP3OCD
clH-cortrjH-cr3rtCDCL
oMCLs:H*CLP
toOJoo33rtr<H*PCO
30 3I
crprtP1_tPOrtP3
OO
117

1.1. Resistance to Aminoglycosides in MGRSA.
From the hospitals surveyed, we requested
epidemiologically distinct strains of MRSA which
were resistant to gentamicin. In Europe and
Australia the term "MGRSA" is commonly used,
whereas in the USA the term MARSA is preferred.
This is because in the USA, strains of MRSA
sensitive to gentamicin, yet resistant to other
aminoglycosides (e.g. tobramycin) have been reported
on a number of occasions (Crossley et a l . , 1979;
Rimland, 1987). Consequently, in terms of
aminoglycoside resistance patterns, our collection
is biased to those displayed by MGRSA. Two
strains (thought to be gentamicin-resistant by
their senders) which appeared in our tests to be
gentamicin-sensitive but resistant to tobramycin
and other aminoglycosides have been included for
interest.
Strains exhibiting low-level and high-level
gentamicin resistance have been found. High-level
gentamicin resistance was found in many strains.
MGRSA from Australia and England possessed low-
level (MIC less than 32 mg/1) resistance to
gentamicin. Townsend et a l . (1987) have shown that
strains of MGRSA from hospitals in London and
Eastern Australia are very similar, and these
"epidemic" strains characteristically show low-
gentamicin resistance (Cookson £t a l ., 1986;
118

Townsend et a l ., 1984).
Using the method of Van de Klundert et a l .
(1984) the identity of aminoglycoside-modifying
enzymes responsible for aminoglycoside resistance
has been determined. 43% of gentamicin-resistant
strains produced the bifunctional enzyme APH
( 2 ’’)/AAC ( 6 !), whereas 55% of strains produced
A P H ( 2 f,)/AAC ( 6 ’) and the phosphor ylase APH (31)-
IV. These enzymes are unique to staphylococci
(Miller et______ al. , 1980). APH (2’’)/AAC ( 6 f) has 6-
N-acetyltransferase activity combined with an
ability to phosphorylate gentamicin-kanamycin type
antibiotics (Dowding, 1977).
The 3 ’- phosphotransferase - APH ( 3 ’)-IV
determines high levels of resistance to neomycin
B and kanamycins A, B and C, additionally this
enzyme phosphorylates amikacin although this may
not always result in amikacin resistance
(Courvalin & Davies, 1977). A P H (2")/AAC( 6 ’) and
APH(2")/AAC(6 1 ) + APH(3)-IV were ubiquitous in
occurrence, and no marked geographical trends in
their distribution were apparent. The production
of the adenylating enzyme ANT ( 4 T 4 ff) is
responsible for the susceptibility to gentamicin
and resistance to tobramycin seen in the
Australian and Russian MRSA.
The genetic determinants for the production of
APH ( 2 ,?)/AAC ( 6 f) are located on a transposon- Tn
4001 (Lyon et. a_l. , 1987)- which may be
119

chromosomally inserted or plasmid-located, and
there is considerable evidence that this
transposon is disseminated worldwide (Skurray e_t
al. , 1988; Storrs e_t . , 1988). No specific
survey of the aminoglycoside-modifying enzymes
present in MGRSA worldwide has been published,
however Dornbusch et a l . (1990) have found APH
(2 f?)/AAC (6f) to be the most prevalent enzyme in
aminoglycoside resistant staphylococci, whereas APH
( 2 ,f)/AAC ( 6 ’) + APH (3’)-IV was rarely found.
The aminoglycoside-modifying enzyme profiles
found can be used to interpret the incidences
of resistance to the aminoglycosides shown in
Table IV. Tobramycin and kanamycin modification
is mediated primarily by acetylation- AAC (6’),
whereas gentamicin resistance is produced by
phosphorylation by APH ( 2 ff). As both these
enzymes were found in 98 of the strains studied
there is good agreement with the 98% incidence
of gentamicin/kanamycin (data not shown)/tobramycin
resistance. Both amikacin and netilmicin possess
an amido bond at position 6 of the
aminocyclitol ring which is susceptible to
acetylation by AAC (6'). Similar levels of
resistance to gentamicin, amikacin and netilmicin
have been found, however the degree of amikacin
and netilmicin resistance depends upon the
acetylating activity possessed by APH (2f1)/AAC( 6 f )
of individual strains. The presence of APH ( 3 1)-
120

IV mediates high level resistance to neomycinalthough more attention has been paid to the
phosphorylation of amikacin by this enzyme. There
is good correlation between neomycin resistance
and the presence of APH (3’)-IV, however there is
less correlation between amikacin-resistance and
the presence of this enzyme. Devaud et a l .
(1977) have shown the production of APH ( 3 T)-IV
to have little influence on amikacin resistance.
MRSA are characteristically resistant to
streptomycin, and have been so since their first
isolation in the 1960s. MRSA isolated during the
1960s and early 1970s very often had high level
streptomycin resistance. This resistance was due
to chromosomal mutation, and streptomycin MICs of
greater than 10,000 mg/1 were found (Lacey &
Grinstead, 1973). In strains of epidemic MRSA
isolated from Australia and the U.K since the
mid-1970s low-level streptomycin resistance (ie.
MICs of less than 100 mg/1) has been found
(Cookson e t a l . , 1986; Townsend e t a l . , 1984;
Townsend et a l . t 1987). Many of the MGRSA
from Australia and the U.K which we studied
possessed low-level streptomycin resistance. Low-
level streptomycin resistance was also found in
MGRSA from centres in Brazil, Italy, Japan,
Portugal, South Africa, Switzerland and Turkey.
Low-level streptomycin resistance is due to
121

antibiotic modification, and can be plasmid- or
chromosomally-mediated (Lyon & Skurray, 1987).
1 .2 Resistance to Tetracyclines in M G R S A .
The tetracyclines are a family of closely
related antibiotics which act by attaching to
the bacterial 30S ribosomal subunit preventing
protein transcription resulting in inhibition of
protein synthesis. Tetracycline was first
introduced in 1948, and since this time a number
of derivatives have been developed eg. doxycycline
and minocycline.
A c c o r d i n g to Lacey (1975), MRSA
characteristically showed uniform levels (MIC c.
100 mg/1) of plasmid-mediated tetracycline
resistance. More recently, tetracycline resistance
in MRSA has been reported to be chr omosomally
mediated (Townsend et a l ., 1987). We found 83% of
our MGRSA to be tetracycline-resistant
(MICs of 64-256 mg/1). Tetracycline resistance in
our strains was geographically widespread. Most
of the tetracycline-sensitive strains found were
from countries in Southern Europe (Italy and
Greece) and from centres in the USA.
Minocycline may be active against
tetracycline-resistant S. aureus (Minuth et a l . ,
1974). Two types of tetracycline resistance in
S . aureus have been described (Asheshov, 1975).
122

Firstly, there is plasmid-mediated, inducible
resistance to tetracycline where strains appear
t e t r a c y c l i n e - r e s i s t a n t , m i n o c y c l i n e - sensitive.
Alternatively, there is chromosomally-mediated
constitutive resistance in which strains are
resistant to both tetracycline and minocycline.
A range of minocycline MICs (0.25- 64 mg/1) were
found for the MGRSA we studied. 27 strains were
susceptible to 0.25-4.0 mg/1 minocycline, 10 were
susceptible to 8.0 mg/1, and 61 strains were
susceptible to 16-32 mg/1 minocycline.
Bismuth et a l . (1990) have studied the gene
heterogeneity for tetracycline resistance in MRSA
of French origin. An excellent correlation was
found between tetracycline and minocycline MICs
of the strains studied, and their genotypes
determined by DNA-DNA hybridization. According to
these criteria 52% of our tetracycline/minocycline
resistant strains contained tet(K ) and te t ( M ) , and
48% contained tet(M ) . Both of these genotypes
were geographically widespread. Unlike Bismuth e_t
a l . (1990) who found no minocycline-sensitive,
tetracycline-resistant MRSA, 10% of our strains
were of this phenotype.
123

1 . 3 Resistance to Macrolides, Lincosamides andStreptogramins.
The macrolides, lincosamides and streptogramins
are a structurally unrelated group of antibiotics
which act on the 50S ribosomal site to arrest
protein synthesis. The macrolides have large
oxygen-containing ring structures (a macrocyclic
lactone ring) to which sugars are attached, and
the most widely used representative is
erythromycin. The lincosamides are pyrrolidine-
carbohydrate structures of which lincomycin and
(the more active) clindamycin are members. The
streptogramins (or "synergimycins") contain two
components A and B. Component A is a
"depsipeptideM and component B is a macrocyclic
lactone moiety similar to erythromycin. When
combined in certain proportions, components A and
B act synergistically. The antibiotic
pristinamycin is a streptogramin.
Staphylococcal resistance to the macrolide,
lincosamide and streptogramin (MLS) antibiotics can
be due to alteration (methylation) of the
ribosomal target site resulting in decreased
antibiotic binding, or it can be due to
enzymatic inactivation of the antibiotic (Arthur
e t . » 1987). Methylation of ribosomal RNA
results in resistance to the macrolides,
lincosamides and streptogramin B antibiotics and
124

is referred to as MLSg resistance. MLSg
resistance can be constitutive or inducible. In
the latter case resistance is seen to
erythromycin (a highly active inducer), but not
to other MLS antibiotics (poor inducers).
However, in the presence of erythromycin induction
of resistance takes place to the other MLS
antibiotics (Weisblum, 1984).
Organisms with induced MLSg resistance are
resistant to macrolides, lincosamides and the B-
(erythromycin-like) component of the streptogramins.
Constitutively-resistant organisms show the same
resistance pattern as "induced" organisms. Bacteria
exhibiting MLSg resistance are still susceptible
to the A-component of the streptogramin
antibiotics and the synergistic activity between
A and B components appears to be retained.
91% of our MGRSA were erythromycin-resistant.
Nearly all the erythromycin-resistant strains grew
well in the presence of 125 mg/1 erythromycin,
however strains from Brazil showed reduced growth
in the presence of erythromycin at concentrations
of 8.0 mg/1 or greater. Constitutive resistance
to clindamycin was found in 47 strains, and all
these strains grew well in the presence of 32
mg/1 clindamycin. On disc testing for inducible
resistance 44 strains which had appeared
clindamycin-sensitive by MIC were shown to have
inducible clindamycin resistance. Only 9% of
125

strains were completely sensitive to the
mac rolides.
Inducible or constitutive resistance was found
in strains from many different geographical
regions. Initially, "epidemic" MRSA in the U.K and
Australia were typically constitutively resistant
to the MLS antibiotics (Marples et a l . , 1986) ,
however strains possessing inducible resistance
have since occurred (Cookson et a l . , 1986; Kerr e_t
al. , 1990; Townsend et a l . , 1984). We have also
observed this phenomenon. Our Australian strains,
AS 1 , AS 2 and AS 3 were isolated in 1990 and
showed inducible MLS resistance, whereas strains
AS 6 and AS 10 were isolated in 1984 and were
constitutively resistant. MGRSA from the USA were
constitutively resistant to clindamycin, and this
is a common property of many strains from the
USA (Schaefler et a l . . 1984; Hackbarth & Chambers,
1989a). Nevertheless, clindamycin-sensitive MRSA do
occur in the USA (Smith et a l ., 1988).
Constituitive and inducible resistance to the
MLSg antibiotics may be chromosomally or plasmid
mediated, and a transposon (Tn 551) has been
identified which mediates such resistance (Lyon &
Skurray, 1987).
Resistance to pristinamycin has not been
found. Staphylococcal resistance due to enzymatic
inactivation of pristinamycin has been reported
from centres in France (Le Goffic et a l . , 1977),126

but so far, is a rare phenomenon (Duval, 1985).
1 . 4 . Resistance to Trimethoprim in M G R S A .
Trimethoprim resistance in S. aureus may be
chromosomally or plasmid-mediated. The plasmid-
mediated resistance is due to the production of
a novel dihydrofolate reductase (type SI)
possessing a much reduced affinity for
trimethoprim (Young et a l . , 1987). Plasmid-mediated
trimethoprim resistance was first found in
Australian MRSA and was located on plasmid pSKl
which also carried aminoglycoside resistance (Tn
4001) and resistance to quaternary ammonium
compounds (Tennent et a l . , 1985). Trimethoprim MICs
against strains with plasmid-mediated resistance
are characteristically greater than 512 mg/1.
Prior to the appearance of plasmid-mediated
resistance during the early 1980s, trimethoprim-
resistance in MRSA was characteristically low-
level with MICs ranging from 10-250 mg/1.
Chromosomally-mediated overproduction of bacterial
dihydrofolate reductase is responsible for low-
level trimethoprim resistance, and this type of
resistance has been found worldwide (Amyes & Tait,
1989) .
Only low-level resistance was found in the
strains we studied, and our results in terms of
the distribution of MIC values resembles those
127

obtained by Burdeska and Then (1989) who also
investigated trimethoprim susceptibility patterns
in an international collection of MRSA. Like
these workers we found trimethoprim MICs of 64-
128 mg/1 against the majority of trimethoprim-
resistant strains. For smaller numbers of
strains, MICs ranging from 1.0-32 mg/1 were
found. Burdeska and Then also stress that large
differences occur in the incidence of
trimethoprim resistance between centres and we
have also found this trend. No strains resistant
to greater than 512 mg/1 trimethoprim have been
found. Trimethoprim MICs of 512 mg/1 were found
for seven MGRSA (three from Ireland, two from
Australia and two from France). Performance of
isoelectric focusing by Amyes, Tait and Maple (see
Appendix C) using an in-house developed technique
for the selective detection of type SI enzyme
failed to show this enzyme in the aforementioned
s trains.
1.5 Resistance to Chloramphenicol in M G R S A .
Chloramphenicol is a protein-synthesis inhibitor
which acts by binding to the 50S ribosomal
subunit to prevent transpeptidation. Only one
mechanism of chloramphenicol resistance is
believed to occur in S. aureus (and many other
bacterial species as well). This involves the
128

production of chloramphenicol acetyltransferase
which catalyses an acetyl coenzyme A dependent
acetylation of the antibiotic (Shaw, 1984). In S .
aureus chloramphenicol resistance is invariably
associated with a group of small plasmids of
2.9- 5.1 kilobases in size (Tennent et a l . , 1986;
Lyon & Skurray, 1987). Resistance to chloramphenicol
is known to rapidly appear following substantial
usage of the drug and be lost with decreased
usage (Kirby & Ahern, 1953).
41% of the MGRSA strains we studied were
chloramphenicol-resistant. Chloramphenicol resistance
was found in strains from Africa, Asia, Australia
and Europe. Although no chloramphenicol-resistant
MGRSA were found from the USA, resistant strains
have been reported (Locksley e_t a_l. , 1982;
Schaefler et a l ., 1984).
1.6 Resistance to Rifampicin in M G R S A .
Rifampicin binds specifically to bacterial RNA
polymerase, thus inhibiting DNA transcription.
Bacterial resistance to rifampicin is produced by
chromosomal mutations resulting in decreased
antibiotic binding. Mutation to rifampicin
resistance usually results in high-level
resistance (MICs greater than 512 mg/1 for S .
aureus). However, according to Wehrli (1983)
mutation to rifampicin resistance is not an all-129

or-nothing phenomenon. The susceptibility ofrifampicin-resistant mutants can vary , depending
upon the sensitivity of the bacterial RNA
polymerase to the antibiotic, and also on the
interaction of other factors eg. cellular
permeability.
We found four rifampicin-resistant MGRSA (1
from Greece, 3 from Kuwait) to be susceptible to
1.0 mg/1 rifampicin, and 24 MGRSA resistant to
more than 8 mg/1 rifampicin. In terms of
geographic distribution, most of our rifampicin-
resistant MGRSA came from Brazil, France and
Turkey. The development of rifampicin resistance
is governed by usage of the antibiotic itself.
It is often advised that rifampicin be used in
combination with another antibiotic because large
populations of staphylococci contain resistant
variants which can overgrow following rifampicin
monotherapy.
1. 7 . Resistance to Ciprofloxacin in M G R S A .
The first fluoroquinolone to enter widespread
clinical use was ciprofloxacin during the second
half of the 1980s. Bacterial resistance to the
fluoroquinolones was not expected because of
their unique mechanism of action. In our first
survey (Maple et a l . , 1989c) of the prevalence of
ciprofloxacin resistance in MGRSA we found 17%
130

of strains to be ciprofloxacin-resistant. We have
investigated fluoroquinolone resistance in MGRSA
in much greater detail and our findings are
given in section 3.
1 . 8 . Resistance to Fosfomycin in M G R S A .
Fosfomycin has a totally different structure
from those of other clinically used antibiotics.
It is a comparatively simple molecule - cis- 1,2-
epoxypropylphosphonic acid. Fosfomycin inhibits
cell wall synthesis by irreversibly binding to
phosphoenolpyruvate transferase which is essential
for the synthesis of UDP-N-acetylmuramic acid
(Kahan ejt a_l. , 1974) . For binding to
phosphoenolpyruvate transferase to occur, fosfomycin
must traverse the cell membrane which requires
active transport of the antibiotic. The alpha-
glycerophosphate system, which is constitutive in
fosfomycin-susceptible bacteria has been shown to
transport fosfomycin into the cell. An
additional, inducible pathway- the hexosephosphate
system can also transport fosfomycin when it is
induced e.g by glucose-6-phosphate . Phosphate or
glucose inhibit fosfomycin uptake by the glucose-
6-phosphate pathway, and their presence in varying
amounts in different susceptibility testing media
has produced differing MIC values (Goto, 1977).
Bacterial resistance to fosfomycin can result
131

from prevention of fosfomycin uptake by thesepathways as a result of mutation. Fosfomycin
resistance can be of two types (Courtieu et a l .,
1977): certain mutants are sensitive to fosfomycin
in the presence of glucose-6-phosphate (i.e the
hexosephosphate pathway is still operative) whereas
others are totally resistant (i.e both pathways
are inoperative). We have followed international
guidelines for the determination of fosfomycin
susceptibility (Andrews et a l . , 1983) which specify
the incorporation of glucose-6-phosphate into the
susceptibility testing medium, hence only totally
resistant strains are shown in our results.
Another problem concerning the detection of
resistance is that bacterial populations contain
highly-resistant variants which are selected out
when using inocula of greater than 10~* cfu
(Woodruff et a l . , 1977). For the strains against
which fosfomycin MICs of 32 mg/1 or 64 mg/1
were recorded at an inoculum of c. 10^ cfu, on
repeat testing with an inoculum of 10^ cfu
fosfomycin MICs of less than 32 mg/1 were
found. This inoculum effect was not seen with
strains classified as fully resistant ie. MICs
of greater than 64 mg/1. For the 21 strains of
MGRSA which were classified as moderately or
fully fosfomycin-resistant, only those 14 which
were fully resistant can be regarded as genuine
clinically resistant mutants. These strains came
132

from Brazil (5), Italy (1), Federal Republic of
Germany (1), France (3), Spain (1) and Turkey (3).
S. aureus mutates to fosfomycin resistance at
a high frequency. Courtieu et a l . (1977) have
recommended that fosfomycin should be used in
combination with other appropriate antibiotics.
Plasmid-mediated fosfomycin resistance may also
exist, but this has been difficult to prove
because of problems encountered with recipient
strains mutating to resistance (Baquero et a l . ,
1977). In countries where fosfomycin has been
clinically used, there have been widely differing
reports of the incidence of fosfomycin
resistance. For example, Rodriguez et a l . (1986)
reported no fosfomycin resistance in MRSA from
several Spanish hospitals whereas Igari et a l .
(1988) reported over 80% fosfomycin resistance in
MRSA from Japanese hospitals.
1.9 Resistance to Fusidic Acid in MGRSA.
Fusidic acid belongs to a group of several
structurally related, naturally occuring antibiotics
- the ’fusidanes’. Against bacteria, fusidic acid
acts as a protein-synthesis inhibitor by
interfering with the functioning of ribosomally
associated elongation factor G - "translocase"
(Cundliffe, 1972). Two mechanisms of fusidic acid
resistance in S. aureus have been described133

(Chopra, 1976). Resistance may be due to
chromosomal mutation leading to structural
modification of factor G, or plasmid-mediated
exclusion of the antibiotic from the cell.
According to Shanson (1990) the majority of
clinically isolated resistant strains possess the
plasmid-mediated resistance. Reducing fusidic acid
usage can limit the development and spread of
chromosomal fusidic acid resistance, however, this
doesn't apply to plasmid-mediated resistance.
Plasmid-mediated fusidic acid resistance can be
disseminated on plasmids carrying linked
resistance determinants eg. B- lactamase, heavy-
metal, aminoglycoside resistance (Lyon & Skurray,
1987). Consequently, fusidic acid resistance
(plasmid-mediated) can remain stable and even
increase despite reduced usage of the drug.
This may account for the reports of 80% - 100%
fusidic acid resistance in MRSA from Dublin
(Coleman et a l ., 1985; Morgan & Harte-Barry, 1989).
An overall incidence of fusidic acid
resistance of 15% was found, and this was mainly
due to clusters of resistant strains from
centres in Austria, France, Ireland, Switzerland, USA
and the U.K. Phage typing and biotyping of
these strains suggests that strains in the
clusters are unrelated. This suggests that
resistance has spread to strains, and is not a
result of the spread of a single strain.
134

Shanson (1990) has found a low rate ofresistance emergence for acute courses of fusidic
acid monotherapy, which does not correlate with
the higher rates of resistance implicated by in
vitro testing, and he suggests that the plasmid-
mediated form of resistance is the clinically
significant form.
1 . 10 Resistance to Novobiocin in M G R S A .
Novobiocin is a dihydroxy-glycosylated coumarin
derivative discovered in 1955, and used during
the late 1950s and early 1960s for the
treatment of multi-resistant staphylococcal
infections. Although highly active against
staphylococci usage of novobiocin was greatly
curtailed following the introduction of the
penicillinase-resistant penicillins because of the
development of resistance (Kirby et a l . , 1956).
Treatment with novobiocin also gave rise to an
unacceptable incidence of hepatotoxicity and
sensitivity reactions (Bridges et a l ., 1957).
The mechanism of action of novobiocin was not
fully understood until the discovery of DNA
gyrase in 1976. It was then reported that
novobiocin inhibits DNA gyrase to prevent
supercoiling of bacterial DNA (Gellert et a l .,
1976). It is now known that novobiocin acts on
the beta subunits of DNA gyrase whereas the
135

quinolones act on the alpha subunits (Wolfson &
H o o p e r , 1985) .
Only one strain (strain IR 1) showed reduced
novobiocin susceptibility (MIC 2.0 mg/1) in our
survey. Kirby et a l . (1956) have also reported
low-level resistance (less than 5.0 mg/1) occuring
in patients treated with novobiocin.
Strain IR 1 was completely susceptible to the
fluoroquinolones (ciprofloxacin MIC 0.5 mg/1),
which is as expected because of the different
sites of action of these agents.
There have been no reports of plasmid-mediated
novobiocin resistance, however Schaefler (1982) has
reported the phage-mediated transfer of
novobiocin-resistance at high rates between S .
aureus strains. The clinical impact of transfer
of novobiocin resistance in this manner is not
known. The lack of use of novobiocin since the
1960s may account for the low incidence of
resistance we have found. Similarly, Turnidge et
a l . (1989) only reported 1.2% novobiocin resistance
in 4537 isolates of S. aureus screened in a
national survey of resistance in Australian
hospitals.
1.11 Resistance to Bacitracin in MGRSA.
Bacitracin is a peptide antibiotic, first
discovered in 1945 in culture filtrates of a
136

strain of Bacillus licheniformis isolated from a
tibial wound sustained by a young patient-
Margaret Tracy. Commercial bacitracin is a
heterogeneous mixture of related peptides of
which 60-80% is made up of Bacitracin A.
Bacitracin is too toxic for systemic use,
although it has found application in a number
of topical preparations often in combination with
other antibiotics such as neomycin or rifampicin.
According to Jawetz (1961) bacitracin resistance
is infrequently encountered in susceptible
species, and resistance does not emerge rapidly
during clinical infections. Nevertheless, bacitracin
resistance has been reported in S . a u r e u s , for
example, in neomycin-resistant S. aureus which
caused hospital epidemics during the early and
mid-1960s (Leading Article, 1965). We found
bacitracin resistance in only one strain of
MGRSA.
Currently, bacitracin resistance is only found
in sporadic strains of MRSA in the U.K (Kerr ejt
a l ., 1990). Data relating to the overall
prevalence of bacitracin resistance in other
countries is extremely limited, although when
MGRSA are tested for such resistance they are
usually sensitive (Graham et a l ., 1980).
137

Resistance_________to_________Mupirocin,____ Nitrofurantoin,
Pristinamycin, Teicoplanin and Vancomycin in MGRSA
No resistance was found to mupirocin,
nitrofurantoin, pristinamycin, teicoplanin and
vancomycin in the collection of MGRSA studied.
These results will be discussed in the following
sections together with our further studies of
the antimicrobial properties of these compounds.
1.12. Multiple Antibiotic Resistance in M G R S A .
From Table IV the degree of multiple
resistance in individual isolates to 21
antibiotics has been calculated. Our results are
depicted in Figure 2. The most highly multiple
resistant strains (resistant to 13 or 14
antibiotics) originated from Brazil (3 strains),
France (6 strains), Kuwait (1 strain), Turkey (2
strains), and 1 strain each from Austria,
Switzerland and the USA. The least resistant
strains (resistant to 8 or fewer antibiotics)
originated from Hong Kong (3 strains), Italy (3
strains), U.K (5 strains), USA (4 strains) and there
were also single strains from Brazil, Chile, East
Germany, Russia and South Africa.
Few national surveys of multiple antibiotic
resistance in S. aureus have been reported. The
138
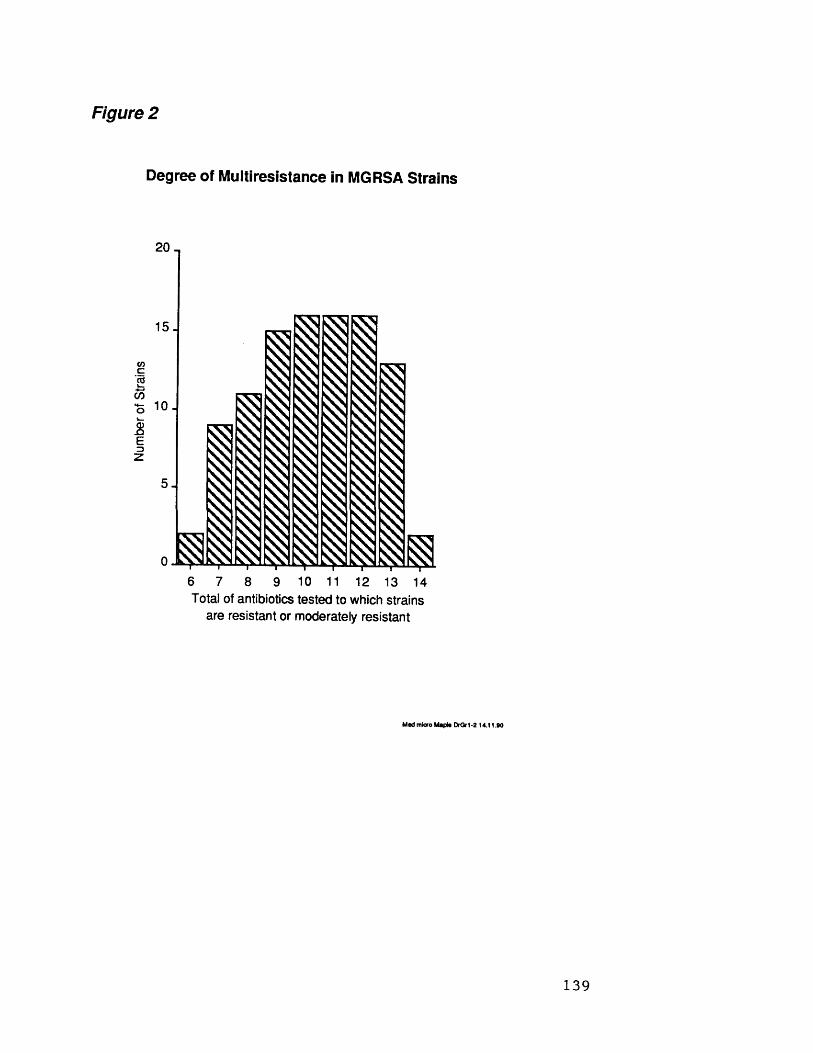
Num
ber
of St
rain
s
Figure 2
Degree off Multiresistance in MGRSA Strains
20 -
15 J
imi l l II1
IPI II IIII iil IIIIII
10 J
6 7 8 9 10 11 12 13 14Total of antibiotics tested to which strains
are resistant or moderately resistant
Med micro Maple DrOrl -2 14.11.90
139

sources and nature of isolates examined in such
surveys can greatly influence the prevalences of
resistances seen. For instance, isolates from out
patients possess much less resistance than
isolates from in-patients (Kayser, 1975).
Furthermore, MRSA tend to possess more resistance
than MSSA. For example, in a national survey of
S. aureus antimicrobial resistance in Australian
teaching hospitals, Turnidge et a l . (1989) found
that gentamicin and novobiocin resistance were
predominantly restricted to MRSA. The types of
hospitals surveyed in studies of the prevalence
of antimicrobial resistance may also significantly
influence the incidence of antimicrobial
resistance found. Haley et a l . (1982) in a survey
of the emergence of methicillin resistance in
the USA reported that such resistance was
virtually restricted to large tertiary referral
and teaching centres, and that the overall
increases in methicillin resistance seen from
1974 to 1981 were due to substantial increases
in only four hospitals.
Within hospitals, the incidence of
antimicrobial resistance can vary widely between
different specialities; in general, high levels of
antibiotic resistance are seen in staphylococci
from dermatological wards, burns and plastic
surgery units where topical antibiotics are
commonly used and the nature of treatments
140

adopted predisposes to cross-infection. The
adoption of antibiotic policy and use of
effective infection control facilities have
considerably reduced the appearance of multi-
resistant strains (Shanson, 1981). With regard to
all these variables, in our survey of multiple
antibiotic resistance in MGRSA we decided that
in order to reduce bias from any one particular
centre it was necessary to look at small
numbers of isolates from a large number of
centres. Using the data thus obtained we could
then consider the overall problem of multiple
antibiotic resistance in MGRSA.
1.13 Do MGRSA pose a therapeutic problem?
Multiple antibiotic resistance in MGRSA is a
variable phenomenon, and so attitudes to the
treatment of infections due to these organisms
may vary from centre to centre. We have found
numerous patterns of resistance to the antibiotics
studied. Although the degree of multiple
antibiotic resistance observed ranged from 6 to
14 antibiotics, the choice of effective recognised
chemotherapy for systemic infections in many
instances was restricted to rifampicin and
fusidic acid, or to vancomycin. For the highly
multi-resistant strains vancomycin was often the
only available recognised therapeutic option.
141

Little resistance was found to fosfomycin and
novobiocin, and no resistance was found to
nitrofurantoin, pristinamycin and teicoplanin.
Currently, knowledge of the in vitro and in vivo
activity of these agents against MGRSA is
limited. Disturbing levels of ciprofloxacin
resistance were also found, and a re-evaluation
of the use of fluoroquinolones against MGRSA is
necessary. Because of these problems, vancomycin
is still regarded as the antibiotic of choice
for treating infections due to MRSA (Milatovic,
1986) .
142

2. Therapeutic Options for the Treatment of MGRSA Infections or Colonization.
2.0 Current Therapeutic Options for the Treatment
of Infections or Carriage Due to M G R S A .
2.1 Antibiotics Available for Treatment of Systemic
Infections
Clindamycin (Smith et a l . t 1988) and trimethoprim
(Elwell et al. , 1986) have been used successfully
to treat infections due to MRSA. Unfortunately,
staphylococcal resistance greatly limits the use of
these agents. For instance, Markowitz et a l . (1983)
was reluctant to use clindamycin to treat MRSA
infections because of the rapid development of
resistance. Trimethoprim (frequently combined with
sulphame thoxazole in the USA) is as effective as
vancomycin in the treatment of serious MRSA
infections, but widespread trimethoprim resistance
prevents more extensive use of this treatment
regimen (Hackbarth & Chambers, 1989a). Less resistance
was found to chloramphenicol. Rapid development of
resistance following chloramphenicol use combined
with fears over its toxicity, and lack of efficacy
against MRSA (Cafferkey et a_l. , 1985) make
chloramphenicol an unattractive therapeutic option.
Fosfomycin, fusidic acid, nitrofurantoin, novobiocin,
pristinamycin, rifampicin and teicoplanin appear to
143

be antistaphylococcal agents of potential value in
the treatment of MRSA infections. Little resistance
was found to these agents, and relative to
vancomycin they are less toxic. The inhibitory
activities of fosfomycin, fusidic acid,
nitrofurantoin, novobiocin, pristinamycin, rifampicin
and teicoplanin compared to vancomycin against 100
strains of MGRSA of diverse origins are shown in
Table Via. In Table VIb the influence of inoculum
size on inhibitory activity is shown, and in Table
VII the spontaneous rates of mutation to
resistance to low and high concentrations of
antibiotic are given. In figure 3, the bactericidal
activity of the antibiotics with time is
portrayed.
i . Antistaphylococcal Activity of Fosfomycin.
The activity of fosfomycin against MGRSA is
substantially decreased on increasing inoculum size
(Table VIb). Goto (1977) has found likewise.
Fosfomycin was bactericidal at concentrations two
to four times MIC in replica-plating studies.
Graninger et a l . (1984) and Lau et a l . (1986) have
also reported fosfomycin to be bactericidal. In
time-kill studies (figure 3a), fosfomycin initially
reduced the inoculum (10^ cfu) 10 to 100-fold,
however regrowth by resistant mutants then took
p l a c e .
144

< H S3 S3 Z 31 3 )p CD H - 3 0 H - 3 o3 H - H i H - < r+ CO COO o P CO o 3 H* H iO o 3 c+ c r O a o5 >d t3 H - H - H i H* 3
* < M H* 3 o c o * <O P O p o 3 oH - 3 H - 3 H - P H*3 H - 3 3 3 p 3
3 O r+ oH» O H-3 H* a
3
o o o o 00 o CO• • • • • • • •o 25 oo
CO
12 06 o 12 o 3p31 i 1
V
1 1 100
1V
1Von
ana>
CO CO CO CO CO 00 h-kO o o o o o o CO
s:i—io o o o o CO o 00 O• • • • • • • • u»00 on o CO t-i o h-* o oon o
ont—1 05 CO
asV i—‘ CO M
1—1 o CO o o o o Q• • • • • • • • <0-o CO o on 23 o -o o ©
> O H as t-H Han H i P H * 3 Pa> H* C+ sr c r3 O 3 H - t—1c+ K O O c r pQ d H> H *
S3 t-< 3 C+CO P 3 O <> 3 P 3 M
H * 3 ^ >3 c+H i O3 H * >o O 3 O3 0 • c+9 H *d as <JCO P 0 H *05 3 < C+
P o <a c ras o H*H* p o o3 3 c+ o H iH- O H*B 3 33 CD * 3jB CO < O
P S3 CO3 3 H ih-i H* O H - o3 3 O CO 3tr B c+W H- '< H* o3 O' i—‘ o 3 H *O H - CO H * p 3
O C+ 3 3 •c o '<1—1 3 o O *1c << O > H* 3B c n 3 CO
3 P • H»O ct H* a
H* O . 3 3 S3 H *O 3 H* CO oV. O • P c+ H i
<D CO P3 • 3 >O c+ M d o
H> 3 o H* H *3 P o o ac+ H* •H - 3O CO3 c+
3 PP 3
B H* &on 3s (0M
145
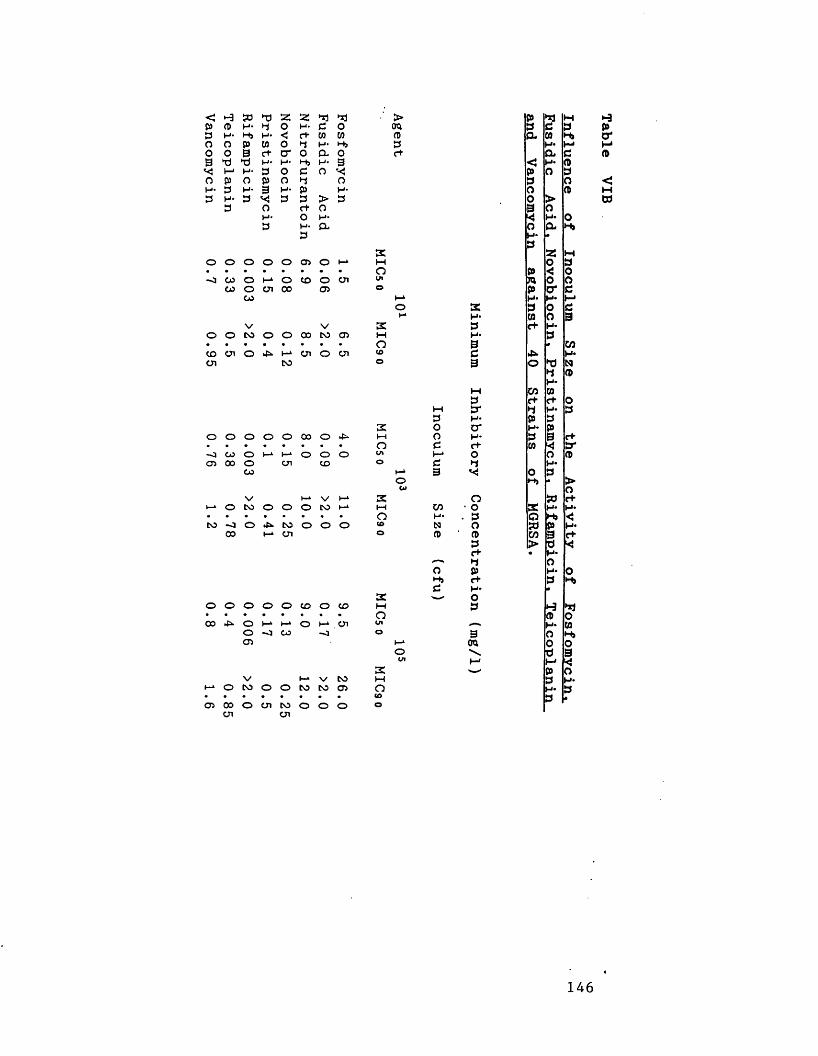
< -3 50 ►d 55 55 d j d j ■ > P d j MA A H- d o H* c o era 3 d 30 H* M> H* < e+ CO CO A a cn M>o o P CO o d H- H> 3 H* Mo o 9 c+ cr O a o c+ a C
>d t j H ' H- H- 3 c H* AVj M H* 3 o c o << p O 3o A O p o d 0 3 aH- 3 H- 3 H* p H* O A3 H- 3 V* 3 3 > 3 o >
3 o c+ o 9 oH* O H- *< H* 03 H- a o a H»
3 H - •3
2 25 wO O o o o 05 o i-* t—( O 3• • • • • • • • o P < 0-J CO o I-* o CO o cn in m o o
CO o cn 00 05 o P o* cCO h-k H* H* M
O 2 3 o dI-* H* CO 0 9
V V s 3 d* H*o o to o o 00 to 05 M H - 3• • • • • • • • o 9 cnCD cn o I-* cn o cn CD d M*U1 to O 9 O ►d N
d AH*
M CO CO3 c+ c t 0
M 3 * d H* 33 H* p 3
3C O c r H* PO o o o o oo o t—( O H* 3 9 r+• • • • • • • • O e C+ CO < 3 --O CO o t—1 I—1 o o O cn H-* o o A05 00 o cn CD o c d H -
CO h-L 9 v{ o 3O * >u» O
\ / V—‘ V I-* 2 o 50 c+1—‘ o to o o o to i-± M cn • o 2 H* M** • • • • • • • o H- . 3 Q H j <5to -0 o to o o o CD N o 5d P
00 H* cn O A ■ A cn 9 C+3 > d *r*- • H*
-—* d OO P H* O
c+ 3 H>d H* •
sc w> Oo o o o O CD o CD i—i 3 •-3 d j• • • • • • • • o A O00 o t-* ►-* o cn cn H* COo -o CO -o o 9 O H»
05 b-* oa 0 oo d 9cn r - ' Ms p o
V I-* V to t—1 3 M*t—* o to o o to to 05 o H* 3• • • • • • • • CD 3 A05 00 o cn to o o O o
cn cn
•-3PO'MA<MW
146

Table VIIMutation Rates to Resistance to Fosfomycin, FusidicAcid , Nitrofurantoin. Novobiocin , Pristinamycin,Teicoplanin or Vancomycin for 3 Sensitive Strains of MGRSA.
Antibiotic
Fosfomycin + G6P*
C o n c n . (mg/1)
100500
Mutation rates after 48h incubn
RLG
6.0 x 10-7 0
AUS3 RM1
1.5 x 10~6 5.3 x 10"70 0
Fosfomycin 100- G6P 500
1.1 x 10"6 1.5 x 10“ 7
1.8 x 10“60
1.21 . 0
x 10“ 6 x 10"6
Fusidic acid 2 10
6.8 x 10“ 7 4.0 x 10~8 1.5 x 10"71.9 x 10"7 1.3 x 10"8 4.0 x 10~8
Nitrofurantoin 60 300
Novobiocin
Pristinamycin
Rifampicin
Teicoplanin
Vancomycin
4204
202
10
5 25
525
00
7.6 x 10-9 0
00
7.6 x 10"82.3 x 10~8
00
00
00
2.5 x 10~8 0
00
7.7 x5.1 x
00
00
10” 810“ 8
00
00
00
3.4 x 10"72.2 x 10"7
00
00
* G6P = glucose-6-phosphate
0 = no mutant colonies grew from an inoculum of approximately 5.0 x 109 cfu.
147

High spontaneous rates of mutation to resistancewere determined (Table VII) for fosfomycin in the
absence of glucose-6-phosphate. Phenotypically, the
mutant colonies varied in size between pin-point
and small, and 48 hours incubation was required to
enable accurate counting of the number of mutants
produced. In the presence of glucose-6-phosphate (25
mg/1) fewer mutant colonies were found, and these
were mostly of pin-point size. These observations
can be explained as follows. For staphylococci in
the absence of glucose-6-phosphate (G6P) prevention
of fosfomycin uptake by the alpha-glycerophosphate
system is only required, whereas in the presence
of G6P prevention of uptake by the hexosephosphate
system is necessary as well. Inhibition of one
pathway will allow some growth, however inhibition
of both will enable growth to proceed only very
slowly. According to Woodruff et al (1977) such
fosfomycin-resistant mutants are of reduced
virulence, and in vivo do not appear to present
the number of problems that would be expected on
the basis of in vitro data.
Fosfomycin has been used extensively in Japan
and Spain, however it is not available for
clinical use in countries such as Australia, U.K
and the U.S.A. Published reports of the clinical
efficacy of fosfomycin in the treatment of
staphylococcal infections are sparse. However, Lau
e t a l . (1986) have used fosfomycin successfully to148

treat three patients with MRSA septicaemia.
Fosfomycin appears to be a promising
antibiotic, but more information on its clinical
efficacy is required together with guidelines
suggesting the most appropriate antibiotics with
which to combine it.
i i . Antistaphylococcal Activity of Fusidic A c i d .
Using an inoculum of 10^ cfu, 90% of the 100
strains of MGRSA studied (Table Via) were inhibited
by 0.7 mg/1 of fusidic acid. Reports of similar
good activity against MRSA have been made by
Guenther and Wenzel (1984) and Verbist (1990). No
substantial increase in MIC was found on
increasing the inoculum size from 10^ to 10^ cfu
(Table VIb) , however there was a two-fold increase
in MIC on increasing the inoculum from 10^ to
10^ cfu. Hilson (1962) reported fusidic acid to
produce a striking inoculum effect with large
inocula (10^ cfu) which he believed was due to an
alkaline reaction in the nutrient medium resulting
in fusidic acid degradation.
Various claims have been made concerning the
bactericidal activity displayed by fusidic acid.
Initially, fusidic acid was shown to be
bactericidal (Chabbert 1953), however the cellophane
transfer method he used to obtain these results
149

was unreliable because of a high degree of
antibiotic carry-over (Barber & Waterworth, 1962).
Chokkavelu et a l . (1984) have also reported fusidic
acid to be bactericidal against MRSA, however many
workers consider fusidic acid to be bacteriostatic
(Hilson, 1962; Verbist, 1990).
In our replica-plating experiments, fusidic acid
was not bactericidal, although at 8.0 mg/1 a
reduction in bacterial numbers was apparent. In
the time-kill studies (figure 3b) fusidic acid at
10 mg/1 reduced the initial inoculum 10 to 100 -
fold (see figure 3b) after 8 hours, but by 24
hours regrowth by resistant mutants occurred.
Godtfredsen et al. (1962) performed time-kill
experiments with an inoculum of 10^ cfu/ml and
found a similar initial rate of killing to ours
with no subsequent overgrowth, however with larger
inocula overgrowth by resistant mutants took place.
Mutation to fusidic acid resistance readily
occurs in vitro (Table VII), but according to
Shanson (1990) such variants are not as significant
a cause of clinical resistance as would be
expected. Fusidic acid has been used with success
to treat MRSA infections although to prevent the
development of resistance its use in appropriate
combination is recommended (Jensen, 1968).
150

iii. Antistaphylococcal Activity of Nitrofurantoin.
In the 1940s, the nitrofurans were first
recognised as potentially useful antimicrobial
agents, and since this time several thousand
compounds belonging to this class have been
synthesized (Chamberlain, 1976). Despite the large
number of nitrofuran compounds which have been
evaluated as antimicrobial agents, only three -
furazolidone, nitrofurantoin and nitrofurazone have
seen widespread clinical use. Nitrofurazone is a
topical agent (see section 2.3i), furazolidone is
only used for the treatment of gastrointestinal
infections, and nitrofurantoin only achieves
therapeutically useful levels in urine and bile.
Nitrofurantoin has good antistaphylococcal
activity (Table Via). Like us, Chamberlain (1976) has
found S. aureus to be inhibited by 6.0- 12 mg/1
nitrofurantoin. No inoculum effect was detected for
nitrofurantoin. Nitrofurantoin was bactericidal in
both replica-plating (MBCq q , 20 mg/1) and time-kill
studies (figure 3c). Herlich and Schweiger (1976) have
reported nitrofurantoin to exert a bacteriostatic
effect by blocking the initiation of translation,
and also to be bactericidal by inducing non
r e p a y a b l e DNA lesions. In our time-kill studies
(figure 3c) one strain (SA 1) overgrew in the
presence of 32 mg/1 nitrofurantoin. This strain
had developed reduced susceptibility to
151

nitrofurantoin (MIC = 160 mg/1). Overgrowth bystrain SA1 did not occur in broth containing 64
mg/1 nitrofurantoin.
An attractive property of nitrofurantoin is the
lack of development of bacterial resistance in the
clinical situation despite 40 years of use. For
example, Schneierson (1960) in surveys conducted
during the 1950s found little resistance to
nitrofurantoin in S. au r e u s . No nitrofurantoin
resistance was found in the MGRSA we studied.
Peacock et a l . (1980) reported nitrofurantoin to be
active against more than 90% of MGRSA tested,
however, on the basis of disc susceptibility
testing 7.5% of MGRSA were resistant. McOsker e_t
al (1990) have suggested that the lack of bacterial
resistance is because nitrofurantoin binds non-
specifically to almost every ribosomal protein thus
having multiple sites of action.
i v . Antistaphylococcal Activity of Novobiocin.
Novobiocin was uniformly active against the
MGRSA studied (Table Via). Walsh et a l . (1985)
reported a novobiocin MIC q q of 0.25 mg/1 against
MRSA, we obtained a MIC^ q of 0.23 mg/1 against
our strains. No inoculum effect was seen for
novobiocin (Table V I b ) . In replica-plating studies
novobiocin was mainly bacteriostatic, although at
higher concentrations (eg. 8.0 mg/1) some bactericidal
152

activity was observed. In time-kill studies (figure
3d), novobiocin initially slowly killed bacteria,
however this was then masked by overgrowth of
resistant mutants. Johnston et a l . (1987) have also
reported overgrowth by resistant mutants in time-
kill studies.
A spontaneous frequency of mutation to
novobiocin resistance of 2.5 x 10“ ® - 7.6 x 10“9 was
detected for low concentrations of novobiocin (4 mg/1),
but no resistant mutants grew at higher novobiocin
concentrations (20 mg/1). Previous experience with
novobiocin monotherapy has shown that staphylococcal
resistance to novobiocin can readily develop (Kirby
et a l . . 1956; Ward et a l .. 1981). Use of novobiocin
in appropriate combinations is recommended. Arathorn
et a l . (1990) have reported that short courses of
oral novobiocin-rifampicin are effective in
eradicating MRSA carriage.
v . Antistaphylococcal activity of Pristinamycin.
Pristinaraycin belongs to a group of antibiotics
formerly known as the "Virginiamycins", but nowadays
more often referred to as the "Streptogramins" or
"Synergistins” .
Few reports of the properties of pristinamycin
are available in English literature. This is
perhaps because pristinamycin has only been used
in France and a number of other continental
153

European countries. Pristinamycin had good activity
against MGRSA (Table V i a ) , and both erythromycin-
sensitive and erythromycin-resistant strains were
susceptible. Our findings agree with those of
Maskell et a l . (1988) who reported pristinamycin
M I C q q S of 0.4 mg/1 against erythromycin-sensitive
and erythromycin-resistant MRSA. No inoculum effect
was found for pristinamycin (Table V I b ) , and this
agrees with the findings of Barber and Waterworth
(1964) .
Pristinamycin was bactericidal against some
strains in both replica-plating and kill-curve
studies. This was investigated further to determine
whether there was a correlation with macrolide and
lincosamide sensitivity status. In replica-plating
studies pristinamycin was bactericidal against
erythromycin-sensitive and inducibly (MLSg) resistant
strains. Pristinamycin was not bactericidal against
strains possessing constitutive MLSg resistance.
Similar trends were found in our time-kill studies
(figure 3e) with pristinamycin at a concentration
of 4.0 mg/1.
Pristinamycin resistance was not found in the
MGRSA we surveyed, and no resistant mutants were
obtained in our spontaneous mutation rate
experiments (Table VII). Clinically, resistance to
pristinamycin does not appear to be a problem as
there have only been a few, sporadic reports of
resistant clinical isolates (Petit et a l . , 1983;
154

Duval, 1985). Pristinamycin is a safe, well-tolerated
antibiotic useful in the treatment of
staphylococcal infections (Bastin et a l ., 1982).
v i . Antistaphylococcal activity of Rifamp icin.
Like us (Table V i a ) , numerous workers have found
rifampicin to be extremely active against MRSA.
Rifampicin is bactericidal at near MIC
concentrations, however in time-kill studies (figure
3f) an initial reduction of inoculum was followed
by overgrowth of resistant mutants even at
concentrations substantially greater than MIC.
A spontaneous frequency of mutation to
resistance of c. 10“® was found in our experiments
(Table VII). Moorman and Mandell (1981) have
detected similar mutation rates to ours.
Unfortunately, development of staphylococcal
resistance to rifampicin in the clinical
situation (Binda et a l ., 1971) has necessitated that
rifampicin be used in appropriate combination
(Kapusnik et a l . t 1984; Eng et a l . , 1985). Despite
its good antistaphylococcal activity, rifampicin was
initially regarded as primarily an anti-tuberculous
agent (Garrod et a l . , 1981), and even today its
potential efficacy in treating staphylococcal
infections is not fully recognized (eg. Chambers,
1988). Other workers (e.g. Acar et a l ., 1983; Clumeck
et a l . , 1984) have shown rifampicin to be a most155

useful component of successful combined chemotherapy
against MRSA.
v i i . Antistaphylococcal activity of Teicoplanin and
Vancom ycin.
Vancomycin is currently the antibiotic of choice
for treating systemic infections caused by
methicillin-resistant staphylococci (Watanakunakorn,
1982; Kucers, 1984; Cafferkey et a l . , 1985b). When
vancomycin was first introduced into clinical use
a significant number of side effects were reported
(e.g. thrombophlebitis, flushing and rashes were
common). Additionally, vancomycin was also shown
to be nephrotoxic and ototoxic, and there had been
a number of reports of histamine-release reactions
-"red- man syndrome" (Farber, 1984). Initially,
vancomycin preparations were of poor purity, the
term "Mississippi Mud" was used to describe some of
the earliest. Nowadays with advances in production
and purification methods vancomycin is available as
a white powder. Slow intravenous vancomycin
infusion over a period of one hour along with
monitoring of antibiotic levels has resulted in a
major reduction of antibiotic-associated side
effects.
Teicoplanin, like vancomycin, is a glycopeptide
antibiotic, however teicoplanin can be injected (once
daily). There is some debate as to the clinical
156

efficacy of teicoplanin (Calain & Waldvogel, 1990).
Some workers (eg. Glupczynski et a l . t 1986) have
reported an unacceptable number of treatment
failures in serious infections which have been
mostly attributed to inadequate serum levels.
We found MIC q q S for teicoplanin and vancomycin
of 0.9 and 1.7 mg/1 respectively (Table Via). Many
other workers have reported similar results for S .
aureus (Watanakunakorn, 1981; Greenwood, 1988).
Vancomycin was slightly less active than
teicoplanin, and for both antibiotics a negligible
inoculum effect was found (Table VIb).
In our replica-plating studies teicoplanin and
vancomycin had variable bactericidal activity (Maple
et a l . , 1989a). The degree of bactericidal activity
exhibited by these antibiotics depended upon the
time at which replica-plating was performed (see
Table VIII). For example, teicoplanin and vancomycin
had MBC q q S of 19.0 and 13 mg/1 respectively at
24 hours incubation, however on replica-plating
after 26 hours incubation MBCq q S of 4.0 mg/1 and
2.5 mg/1 respectively were found.
Time-kill studies (figure 3g) also showed a
variable bactericidal effect depending upon
glycopeptide concentration, and the time of exposure
to glycopeptide. Vancomycin produced a greater rate
of killing than teicoplanin, and for both
antibiotics the time required to achieve a 99.9%
kill depended upon the concentration used (figure 3g).
157

Variable MBCs of vancomycin against MGRSA have
also been reported by Foldes et a l . (1983) and
Traub et a l . (1984). Lagast et a l . (1986) have also
shown vancomycin to be more rapidly bactericidal
than teicoplanin. On the basis of our 22 hour
readings for teicoplanin and vancomycin (Table VIII)
some of our strains could be described as
"tolerant" (a ratio of MBC to MIC of greater than
32). The finding of "tolerance” has been implicated
as a cause of sub-optimal clinical response with
glycopeptide therapy (Sorell et a l . , 1982). Kaye
(1980) has questioned the clinical significance of
"tolerance". Some of our strains could be
described as "tolerant" at 22 hours but not after
26 hours exposure to vancomycin. Like others
(Pelletier, 1984), we feel that the laboratory
aspects of "tolerance" need further clarification.
Vancomycin is the mainstay of chemotherapy of
serious infections due to MRSA, and also of
empirical therapy in the absence of susceptibility
data. It is of major importance to microbiologists
and clinicians alike that vancomycin resistance
does not develop in clinical isolates of S .
a ureus. In vitro vancomycin resistance is very
difficult to produce (Griffith, 1984; Watanakunakorn,
1988). A number of workers (Watanakunakorn, 1990)
have found it easier to produce in vitro
resistance to teicoplanin. Furthermore, of great
concern is a recent report by Kaatz et a l .(1990a)
158
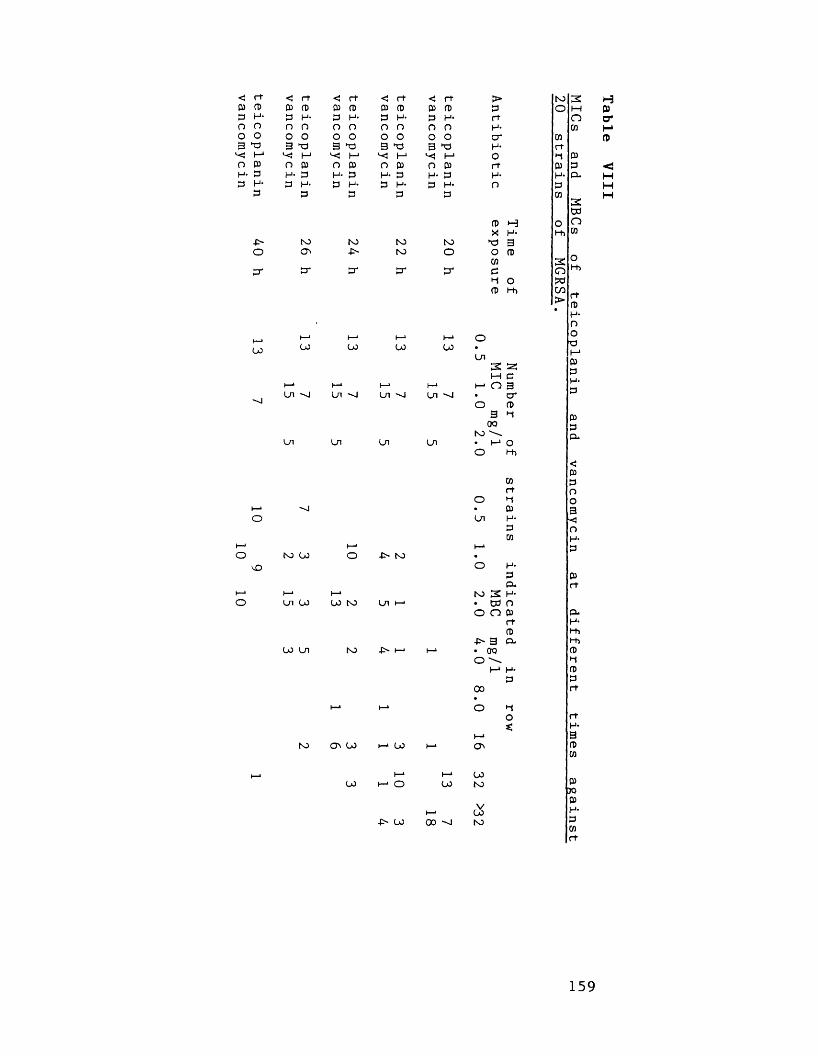
< rt < rt <! rt < rt < rt > NO3 t-3p P P P P P P P P P P O H Pp H- P H- P H- p H- p H- rt O cr0 0 n n O 0 0 O 0 n H- CO0 0 0 0 O 0 0 O 0 0 o' W p3 p 3 p 3 p 3 P 3 P H- rt
M m I-1 "<S 1—1 O P0 p O p O p O P n p rt P P <H- p H- p H- p H* P H* p H- H- CL HP H- P H- P H- P H* P H- n P H
P p P P P CO H3CO
P H O 0X H- t-h CO
-P~ NO NO NO NO P 3O O' •P' NO O O P
M 3 0CT cr cr cr cr o
fu
re
OPC/3>
l-h
r tP
LO
vO
LO
Ln "O
Ln
NO LO
Ln LO
LO Ln
LO
Ln
Ln
LO NO
CT\ LO
LO
LO
Ln -O
Ln
Ln
JN- i—J
LO
LO
Ln ■'-J
Ln
-P> LO
LO
00 "0
OLn 3 2; m e 1— 0 3• cr o P
3 ^ 00no
• I - * o O 1-h
mr t
o *-iP
Ln H-P wH-pCLH*oNO 3!W
> O Pr tP
• 3 CL 00
> \I-1 H-P
00
L0NO
L0NO
H-nop t—1 p p H* P
PpoPH-PWr t
159

of the clinical development of teicoplanin
resistance in S. a ureus. Sporadic resistance to
teicoplanin and vancomycin has been reported in
coagulase-negative staphylococci (Johnson et a l . ,
1990). Also of great concern to microbiologists
are recent reports of outbreaks of infection with
enterococci possessing transferable vancomycin
resistance (Johnson et a l .t 1990).
S. aureus has a remarkable capacity for
developing resistance to a wide range of
antimicrobial compounds. The emergence of vancomycin
resistance in these organisms would seem to be
inevitable.
160
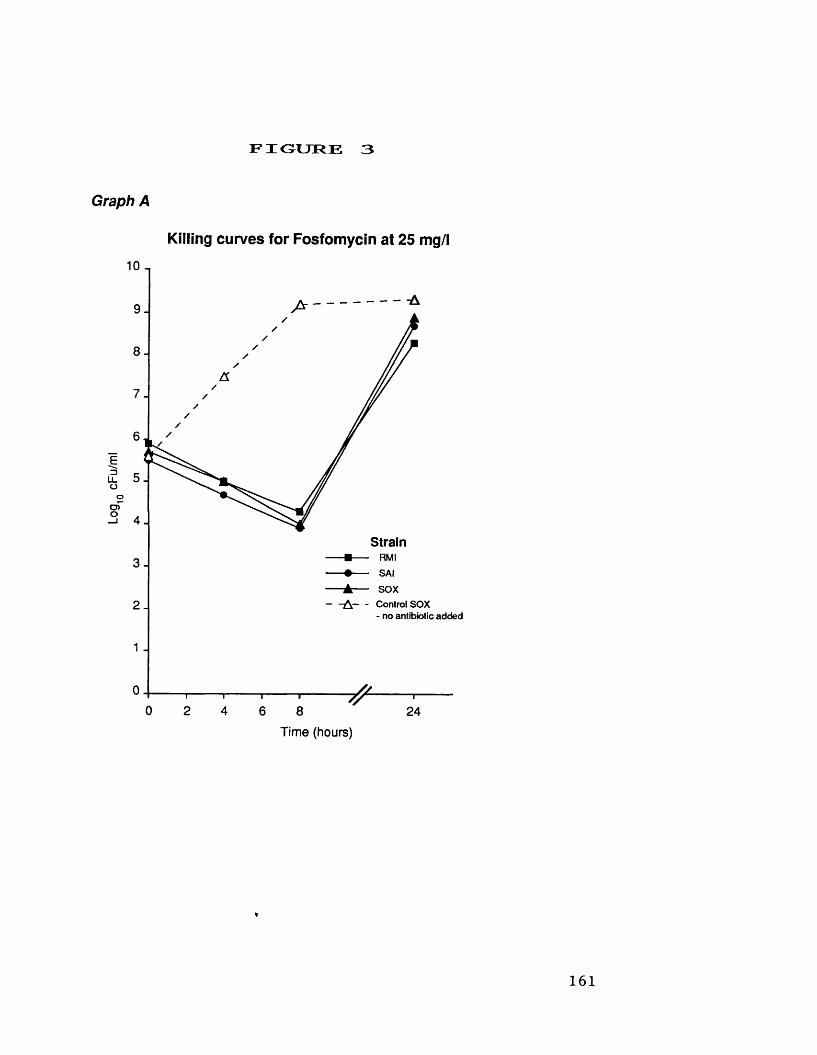
Log
cFu/ml
F I G U R E 3
Graph A
Killing curves for Fosfomycin at 25 mg/l
______________-A
Strain3-
A C o n t r o l S O X- n o a n t i b i o t i c a d d e d
1 .
T --------1---------- 7 ---------1 -6 8 24
Time (hours)
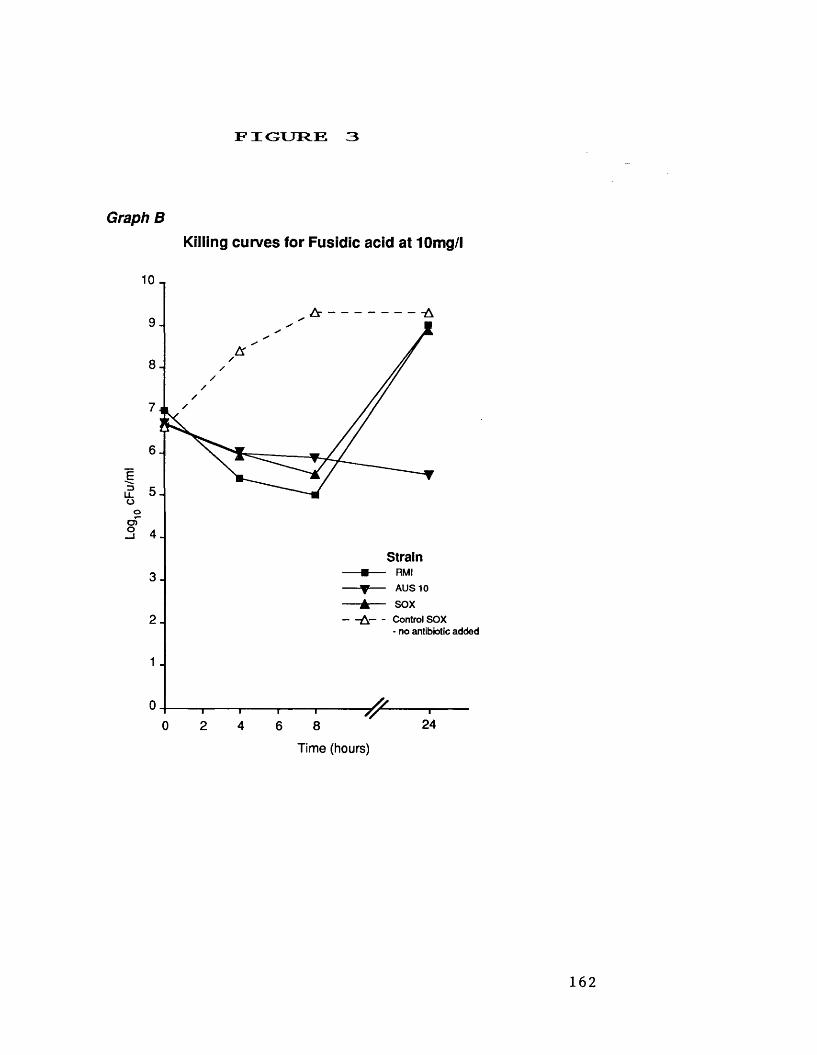
Log1
0 cF
u/m
lF I G U R E 3
Graph BKilling curves for Fusidic acid at 10mg/l
4.
2 .
Strain-■-- RMI
A U S 1 0-A— sox- A C o n t r o l S O X
- n o a n t i b i o t i c a d d e d
1 .
~ l --------------1-------------------1--------------T "
2 4 6 8 24
Time (hours)

Log1
0 cF
u/m
l
F I G U R E 3
Graph CKilling curves for Nitrofurantoin at 32 mg/l
1 0-,
5-
StrainR M IS A IS O X
^ C o n t r o l S O X- n o a n t i b i o t i c a d d e d
,_ _ K illing c u r v e o f 6 4 m g / l n i t r o f u r a n t i o na g a i n s t s t r a i n S A I
24Time (hours)
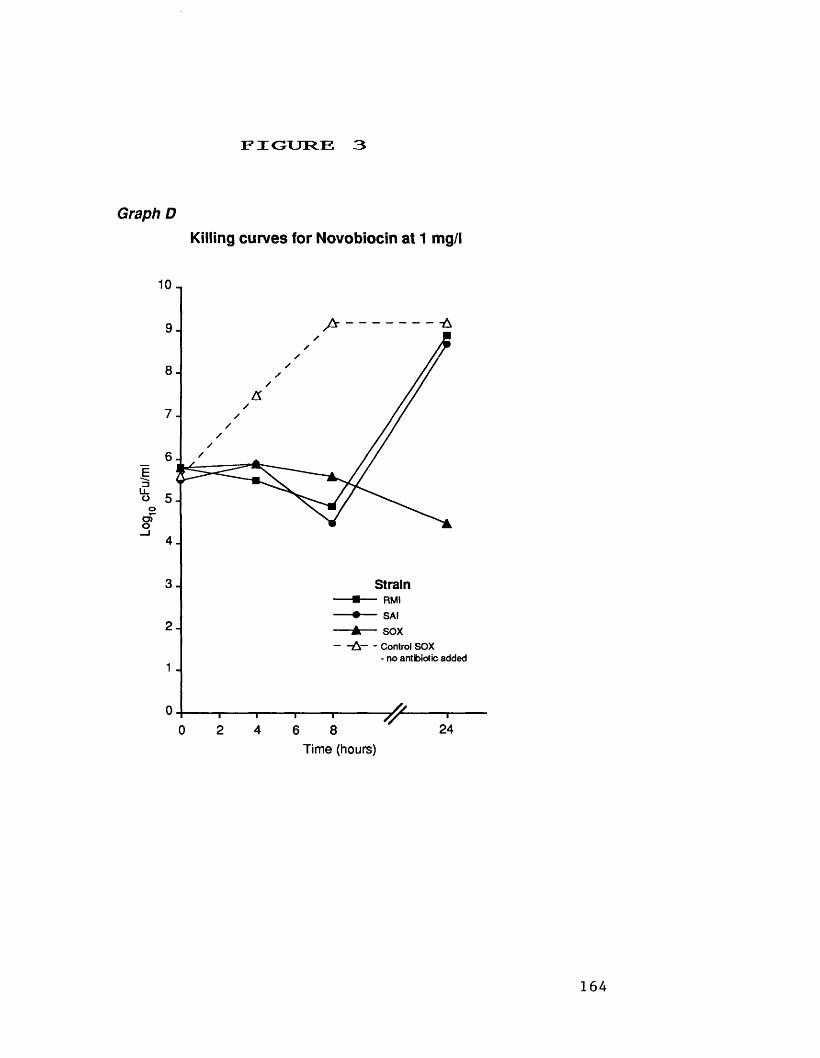
Log1
0 cF
u/m
lF I O U T R E 3
Graph DKilling curves for Novobiocin at 1 mg/l
10
2 -
1 .
Strain- ■ - - R M I- • - - S A I- A — S O X
A C o n t r o l S O X- n o a n t b i o t i c a d d e d
Time (hours)
— r~24
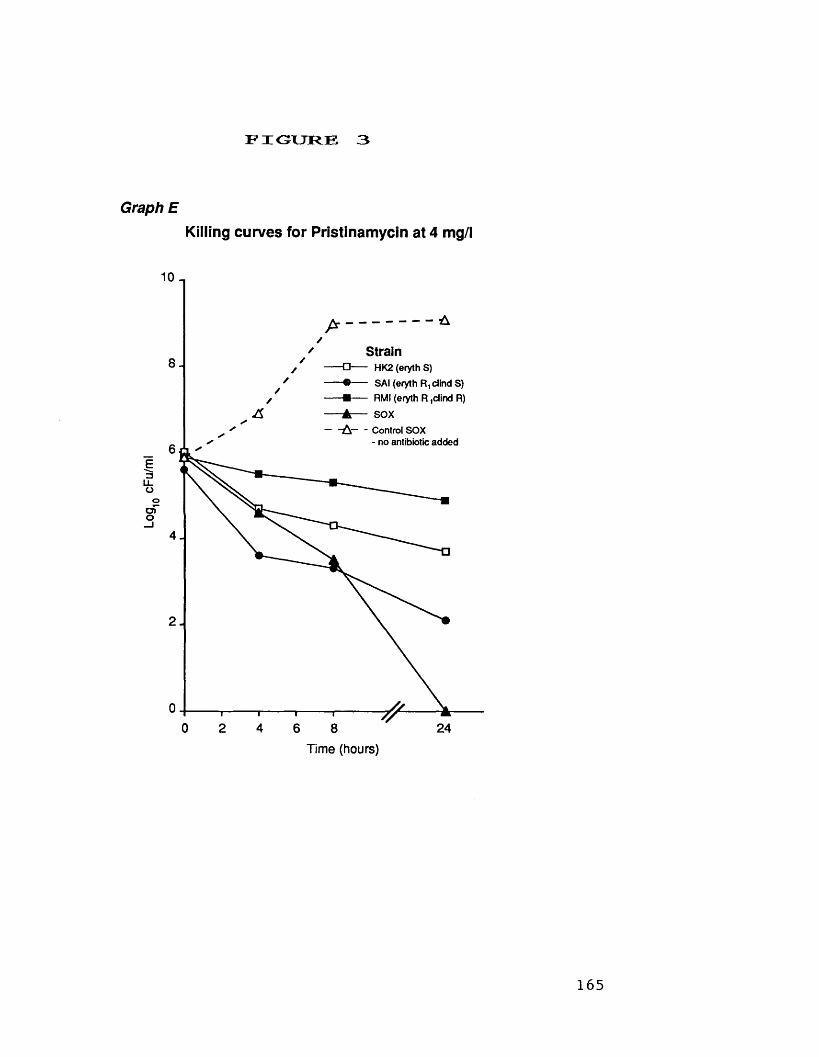
Log.
cFu/
ml
F I G U R E 3
Graph EKilling curves for Pristinamycin at 4 mg/l
10
Strain □ - - - H K 2 ( e r y t h S ) ® - - - S A I ( e r y t h R , d i n d S ) ■ - - R M I ( e r y t h R , c l i n d R ) ▲ - - S O X— - A C o n t r o l S O X
- n o a n t i b i o t i c a d d e d
4.
24Time (hours)
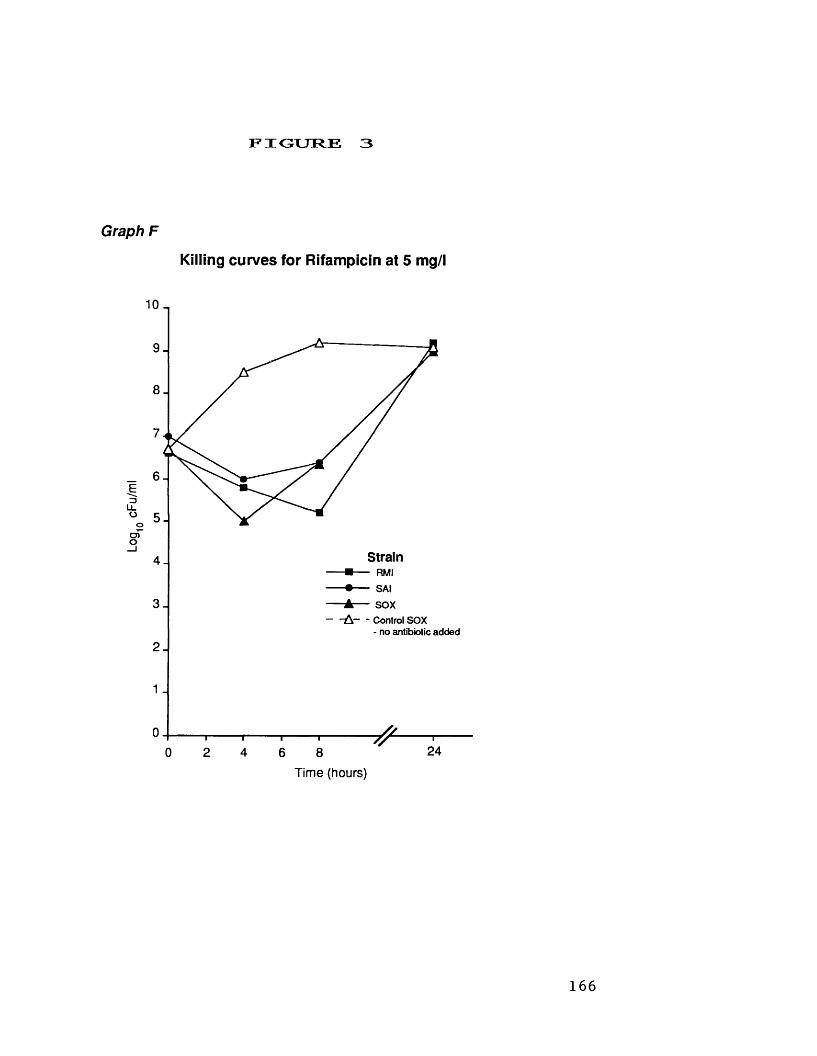
Log
cFu/
ml
FIGURE 3
Graph F
Killing curves for Rifampicin at 5 mg/l
Strain ■ - - R M I • - - S A I ▲ - - S O X A C o n t r o l S O X
- n o a n t i b i o t i c a d d e d
24Time (hours)
166
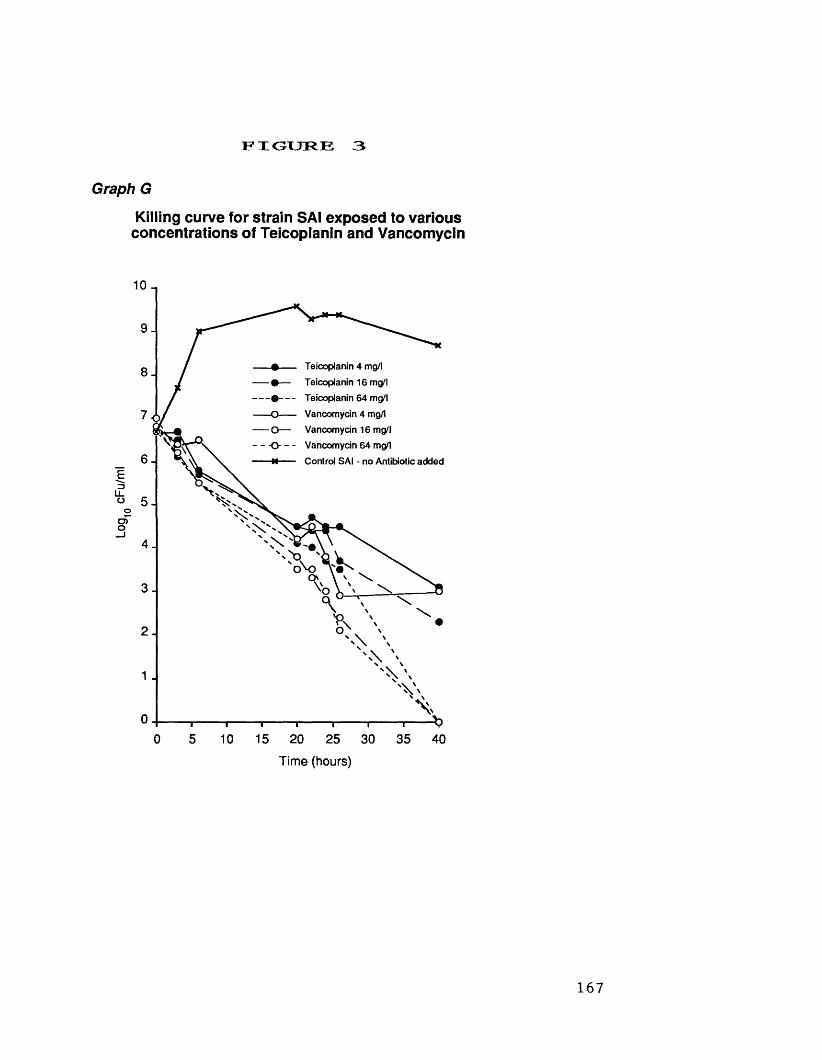
Log1
0 cF
u/m
l
F I G U R E 3
Graph G
Killing curve for strain SAI exposed to various concentrations of Teicoplanin and Vancomycin
- # - - T e i c o p l a n i n 4 m g / l- • — T e i c o p l a n i n 1 6 m g / l- • T e i c o p l a n i n 6 4 m g / l- O V a n c o m y c i n 4 m g / l- O — V a n c o m y c i n 1 6 m g / lO - - V a n c o m y c i n 6 4 m g / l- n C o n t r o l S A I - n o A n t i b i o t i c a d d e d
Time (hours)
167

2.2 New Antibiotics active against MGRSA.
During recent years a number of new antibiotic
moieties have been discovered which have shown
good activity against MRSA. Most notable of these
are the daptomycins (daptomycin), lipoglycodepsipeptides
(ramoplanin), oxazolidinones (DuP 105 and DuP 721) and
paulomycins (paldimycin). The activity of these
antibiotics against MGRSA is shown in Table IX.
There have also been attempts to modify the
structure of existing antibiotics to produce
compounds possessing greater antistaphylococcal
activity, or more amenable pharmacokinetics for the
treatment of staphylococcal infections. Examples of
these are the chemical synthesis of a water
soluble pristinamycin (RP 59500), and the development
of new 14, 15 or 16-membered macrolides.
i . Antistaphylococcal Activity of Daptomycin.
The antibacterial activity of daptomycin (an
acidic lipopeptide compound) is primarily due to an
interaction with cellular lipoteichoic acid (Canepari
e t a l ., 1990). Daptomycin is active against both
aerobic and anaerobic Gram-positive bacteria, however
it has little activity against Gram-negative
organisms. The in vitro activity of daptomycin is
enhanced by supplementing susceptibility testing
media with calcium chloride (Eliopoulos et a l . ,
168

1986). Andrew et a l . (1987) have reported that the
enhancement of activity is specifically produced by
calcium ions. Hence, daptomycin MICs vary with the
calcium ion content of the susceptibility testing
medium used, and these can vary greatly. For
example, IsoSensitest agar contains only 1.55 mg
Ca++ /1 compared with 26.4 Ca++ in Mueller Hinton
agar, furthermore these amounts may show
considerable batch to batch variation.
In our studies (Table IX) daptomycin was
uniformly active against MGRSA of diverse origins.
We obtained a MIC^ q of 1.4 mg/l and MIC^ q of
2.6 mg/l in IsoSensitest agar supplemented with
10 mg/l Ca + + (as recommended by Andrew et a l . ,
1987) compared to MICs of greater than 8 mg/l in
non-supplemented media. Our results are similar to
those obtained by Andrew et al (1987) against MRSA.
In replica-plating studies (Table I X ) , daptomycin was
bactericidal at MIC or near MIC concentrations.
Van der Auwera (1989) has reported daptomycin to
be rapidly bactericidal against MRSA.
Preliminary human pharmacokinetic data indicate
that daptomycin achieves plasma levels of 8.5 mg/l
one hour following intravenous dosing of lmg/kg
(Black et a l . , 1986). In an animal model of
methicillin-resistant S. aureus pneumonia, treatment
with daptomycin was of similar efficacy to
treatment with vancomycin (Kephart & Esposito, 1988).
Initial clinical trials with daptomycin were
169

» © © © o > £ Wp P C C p 3 O 33 © © © r+ S3 3*o a r+ M- C/3 H4© H* O cr > O'i—4 3 t-* -o 3 H* H4P O CO*< 0 C+3 o cn I—4 o c+ O OH- H' H- H- H>3 3 3 O <
H»H* P< 3
O o A acn I—4 DO h-4 t—4 ►1
CO P 3 O'1 1 I 1 1 3 A pTO *: o
DO DO 00 £» 4s. A c+Ap ►13c+ OH* H43: O' a
O o cn h-4 J-4 M H* p• • • • • o O MCTl to CO OS 4s. Ul <7+
<1 c H4O P
£ CO OM c+o H 4
3 <O H4
3 CO t+sc TO CO <
h—4 o -o CO CO M \ A• • • • • o l—t COo -o 4 o OS to C0 a
cn o H- H* A3 3 reO n Ao 3
/\ C 3A t—* 3 H4
CO V V • 3 3 O 3t -4 CO . P 3 < A• 1 I—1 I- 4 3 A ao DO CO 1 TO M
V 00 00 (D f - 4os 4s. o pcn O
3* pA H*O 3 3H» H* CO
V V K c O c+A A W P
H-* I - 4 I—4 O M• CO CO CO CO U1 00o 00 00 o o
OS co to ^ 00 00 4^
3tz totd \o mto
170
Table IX

prematurely suspended in June 1988 because of
unexplained treatment failures in patients with
endocarditis or bacteraemias caused by Gram-positive
cocci. It is believed that these failures were
due to a profound diminution of bactericidal
activity in the presence of albumin (Garrison ejt
a l . , 1990). Currently, clinical trials are underway
in which a more aggressive dosing strategy has
been adopted, however problems with toxicity at
these increased doses have been encountered.
i i . Antistaphylococcal Activity of DuP 721 and DuP 10 5 .
DuP 721 and DuP 105 are members of a new
series of synthetic antimicrobial agents - the
oxazolidinones. These agents are principally active
against Gram-positive cocci (Slee et a l ., 1987).
DuP 721(p-acetylphenyloxooxazolidinylmethylacetamide)
has consistently been shown to be more active
than DuP 105 which is a methy1-sulfinyl derivative
(Slee et a l . , 1987; Neu et a l . , 1988). Like Daly
et a l . (1988) we found DuP 721 and DuP 105 to be
bacteriostatic (Table IX).
Both DuP 721 and DuP 105 showed uniform
activity against MGRSA of diverse geographical
origins which possessed resistance to many classes
of antimicrobial agents. The absence of cross
resistance may be because the functional
oxazolidinone group of the oxazolidinones is only
171

rarely found in pharmaceuticals (Brumfitt & Hamilton-
Miller, 1988). Little human pharmacokinetic data is
available for DuP 721 and DuP 105, and there have
been no reports of clinical trials conducted with
these agents.
iii. Antistaphylococcal Activity of Paldimycin.
Paldimycin is a paulomycin-related antibiotic
composed of two closely related substances (U 67963
and U 67964) derived from paulomycins A and B
(Argoudelis et a l . , 1987a). Paldimycin, like the
corresponding paulomycins, is sensitive to heat and
acidic or alkaline environments (Argoudelis et a l .,
1987b); furthermore, the activity of paldimycin is
also medium dependent (Rolston et a l . , 1987).
Paldimycin showed greatest activity in Antibiotic
Medium No. 2 (pH 6.8), in which we determined a
MIC^ q of 0.27 mg/l and MIC^ q of 0.75 mg/l
following incubation at 37°C for 18 h (Table IX).
In replica-plating studies, paldimycin was
bactericidal against most strains tested at near
MIC concentrations. Using nutrient broth (pH 6.8)
Chandrasekar and Sluchak (1989) have also reported
similar paldimycin activity against MRSA. Very
little pharmacokinetic or clinical data is
available on paldimycin, and it seems that the
commercial development of this agent for
clinical use will not be progressed.
172

i v . Antistaphylococcal Activity of Ramoplanin.
Ramoplanin (formerly called A 16686 and MDL 62198)
is a complex of three closely-related polypeptides
produced by Actinoplanes sp. ATCC 33076 (Cavalleri
et a l . , 1984). The manufacturers of ramoplanin
recommend that it be called a
"lipoglycodepsipeptide" . Ramoplanin specifically
inhibits bacterial cell wall synthesis (Cavalleri e_t
a l . , 1984), and its activity is primarily restricted
to Gram-positive bacteria (Pallanza et a l . t 1984).
We have found ramoplanin to be highly, and
uniformly active against MGRSA (Table IX). In
replica-plating studies ramoplanin was bactericidal
at near MIC concentrations (Table IX), and time-
kill studies (figure IVe) showed it to produce
99.9% killing in 4 hours or less. A number of
workers (Neu & Neu, 1986; Jones & Barry, 1989) have
reported similar findings.
Cross-resistance of ramoplanin with currently
used antibiotics has not been found. Little data
is available regarding the pharmacokinetics and
clinical efficacy of ramoplanin, although it has
been suggested by O'Hare et a l . (1990) that
ramoplanin will probably be used as a topical
ag e n t .
173

v • Ant istaphylococcal Activity of new 14, 15 and16-membered macro lides.
Recently, a number of new macrolides have been
developed which can be differentiated by the
number of carbon atoms constituting the macrocyclic
lactone ring. Erythromycin is a 14-membered
macrolide, and although alterations have been made
to its formulation it is believed that new
compounds with improved activity (eg. against H .
influenzae) and pharmacological properties (eg. longer
serum half-life) can be developed to replace it.
A number of new 14-membered macrolides such as
clarithromycin, dirithromycin and roxithromycin have
been found to possess improved pharmacokinetics
relative to erythromycin. None of these compounds
are markedly more active than erythromycin against
S . a ureus, and none are active against erythromycin
resistant strains. Similarly, azithromycin (a 15-
membered macrolide) showed little activity against
MGRSA (Table X).
The 16-raembered macrolides such as miocamycin,
rokitamycin and spiramycin are active against MRSA
possessing inducible MLSg-type resistance. Our
findings on the activity of these compounds
against MGRSA are shown in Table X. MGRSA are
most susceptible to rokitamycin followed by
miocamycin and then spiramycin. Table X shows the
importance of determining the erythromycin
174

sensitivity status of the test panel of strains.If the majority of strains used are constitutively
resistant then neither agent would appear to have
useful activity against MRSA. Hardy et a l . (1988)
reported miocamycin, rokitamycin and spiramycin to
have little activity against MRSA, because most of
the strains tested were constitutively resistant.
Spiramycin has been succesfully used to treat
staphylococcal infections (Macfarlane et a l . , 1968).
Spiramycin is not a new antibiotic, however it is
only available in certain countries (eg. France). We
have found (Table V) constitutive MLSg resistance in
47% of MGRSA from worldwide sources, and in view
of this there is only limited scope for use of
16-membered macrolides in the treatment of MGRSA
infections.
v i . Ant istaphylococcal Activity of RP 59500 - an
injectable streptogramin.
Streptogramins (eg. pristinamycin) can only be
administered by the oral route, which limits their
use in severe infections. We have previously
observed that pristinamycin has good activity
against MGRSA (Table Via) and that staphylococcal
resistance does not appear to be a problem (Table
V). To utilise these attributes of pristinamycin,
Rhone Poulenc Sante are in the process of
developing a water soluble "pristinamycin"- RP 59500.
175

to ►a CO to 3E > M > o CD 53 >►d 3 >d o H* N 3 z o d »d oH' H- *- o H- *< H 3 pi-cn co d H- o pi rt t—t CO pi cn H*CD c+ p c+ p s' 3* w c+ s' CD <Ol H- B p B 3 d M H* d cn H*o 3 B *< 0 0 o pi- o o pi-o p o *< o B B H 3 3 o <3 H* o H- *< *< M t+ '<Vj 3 H- 3 o 0 o H* oo 3 H- H' < H* p 0H- 3 3 CD 3 R Hj3 M p
CO 3 I-4o o 4- o o o o 3 CD CO 4»-• • • • • • • w d 3 c+ »h-4 I—4 CO I—4 CD CO CO o CD CO00 00 cn CO cn CO CO H* H-*o 3 co CD H- pi- p cnc+ 3 CO H* 3II d CO e+ <!p H* p A pI-4 H* c+ 3 H* 3►—4 3 H* Pi 3 o*3 0) < CO pi-o o -0 o I—4 o o w CD c+ CD• • • • • • • o co d d I-4to to o 4*. ~o 4- 4* (£> pi- p 3 osCO CO Oi CD © d H- p 1p 3 c+H- CO H* 33 • 0 (DCO 3 B3 • CO p o'3 CD H* o I-4 CDV V M CO 3 do o 4*. o I-4 o H- a CD• • • • • CO CO cn co 3 H* o aH-4 I—4 00 I—4 o CO to o r+ o 3 o00 cn CD 3 p H- 0-> 1—13 cr 3 M 3II r+ I-1 o a> pCD H- o oCO o' c+ dCO CO -—- I-4 H* o3 c+ a: < o I-*V V w d t-4 • 3 H*o o -o o H4 o p CO- a• • • • • CO CO CO H- w d CDto to to £» 00 CO CO o 3 >_- CD o CO4- to CO CO Hj •H*CO dc* 3p o H*3 53 COc+ CO C+3 > H-3 CD 3o o V V V V V M CO o CO p• • o H« o pi- o B4>> CO CO CO CO CO CO cn CO 3 d o <00 CD to to to to CO o c+ co p B o3 p pi- H* d H*3 H* 3 d 3II r+ c+ CO H*3 COCO c+ H* pcn CO H- p 3 33 r+ < 3 r ao o V V V V V M 3 CD a• • o p00 CD CO CO CO CO CO CO H- CO h-4CO to to to CO CO o 3 cn CO
176
Table

RP 59500 has been produced from semi-synthetic
modifications of the two major components of
pristinamycin, and the selection and combination of
two promising derivatives RP 57669 and RP 54476 to
yield RP 59500 methanesulfonate (Barriere et a l . ,
1990).
RP 59500 showed comparable activity to
pristinamycin against MGRSA possessing inducible and
constitutive MLS resistance (Table X). Like us,
other workers have reported RP 59500 to have good
activity against MRSA (Mitsuhashi et a l .,1990).
v i i . The lack of new agents available for use in
the treatment of infections due to M G R S A .
Although daptomycin, the oxazolidinones, paldimycin,
ramoplanin and the new macrolides showed good
activity against MGRSA, none of these agents
currently appears to offer the clinician a future
alternative choice to vancomycin. Daptomycin has
shown disappointing efficacy in clinical trials, and
there is concern over toxicity associated with
higher doses. The development of the oxazolidinones
and paldimycin does not appear to have been
progressed by their respective manufacturers.
Ramoplanin is a promising compound, but will most
likely be only available for topical use. The new
14- and 15-raembered macrolides, although having
improved pharmacological properties are no more
177

active against MGRSA than erythromycin. The 16-
membered macrolides (josamycin, miocamycin and
rokitamycin were more active against MGRSA, however
approximately 50% of the strains were still
resistant. Finally, RP 59500 may be an alternative
agent to vancomycin in the future, but its
efficacy still has to be determined by clinical
trials, at least in countries where it is still
not available.
178

2.3 Topical Antibiotics for the Treatment of MRSAC a r r i a g e .
The control of outbreaks of MRSA
infection/colonisation is expensive both in terms
of the logistical requirements necessary for
preventing cross-infection (Mehtar et a l , 1989), and
the increased costs in terms of prolonged length
of stay and more expensive antibiotic treatments
associated with such outbreaks (Cheng & French, 1988).
Guidelines (Working Party, 1986; Working Party, 1990)
issued by a combined working party of the
Hospital Infection Society and British Society for
Antimicrobial Chemotherapy recommend that patients
whether infected or colonised with MRSA should be
immediately isolated, and that every attempt should
be made to eradicate carriage by use of topical
antibiotics and/or antiseptics. Furthermore, staff
may be carriers of MRSA, and it is imperative in
order to control the dissemination of infection
and prevent their prolonged removal from duty, that
effective eradication can be achieved.
There have been conflicting reports of the
efficacy of antiseptics in eradicating MRSA
carriage. Bartzokas et a l . (1984) and Brady et a l .
(1990) have reported triclosan to be effective in
clearing MRSA carriage, whereas Pearraan et a l .
(1985) and Cookson and Phillips (1989) have found
various antiseptic regimens ineffective in clearing
179

MRSA. The use of topical antibiotic preparations
such as bacitracin, tetracycline and neomycin,
together or in combination with antiseptics is of
limited efficacy in clearing carriage (Casewell &
Hill, 1986; Chow & Yu, 1989). Topical therapy with
fusidic acid (White et a l . , 1989) or systemic
therapy with rifampicin (Yu et a l ., 1986) has been
found to be effective in clearing staphylococcal
carriage, however the emergence of resistance to
these antibiotics following such use is a cause
for concern. Recently, the new topical agent
mupirocin (pseudomonic acid) has been introduced into
clinical use, and reports of its efficacy in
clearing MRSA carriage have been most encouraging
(Casewell & Hill, 1987). Unfortunately, following the
clinical use of mupirocin the emergence of
bacterial resistance has been reported (Rahman e_t
a l . , 1987; Baird & Coia, 1987). In this section, the
antistaphylococcal activity of mupirocin is compared
to that of potential alternative topical agents.
180

i . Antistaphylococcal Activity of Azelaic Acid,Nitrofurazone and Silver Sulphadiazine compared toMupirocin.
The in vitro activity in terms of M I C ^ q , M I C q q ,
M B C50 , MBC90 of azelaic acid, nitrofurazone and
silver sulphadiazine compared to mupirocin against
80 strains of MGRSA is shown in Table XI.
Azelaic acid is a saturated dicarboxylic acid.
Topical azelaic acid has a beneficial effect in
the treatment of acne, where it has been shown
to produce on average, a 224-fold reduction in
micrococcaceae and 30-fold reduction in
Propionobacterium spp. (Bladon et a l . , 1986). Azelaic
acid is commonly used as a 15-20% cream with no
deleterious effect, furthermore, oral administration
of up to 20 grams a day is tolerated (Breathnach
et a l ., 1984). There have been few reports of the
antibacterial activity of azelaic acid, and we have
found none which show its activity against MGRSA.
We have found azelaic acid to be uniformly
active against MGRSA from many different
geographical sources (Table XI), and it is
bactericidal both in replica-plating studies and
time-kill studies (figure 4a). The efficacy of
azelaic acid in clearing MRSA carriage would seem
well worth investigating.
Nitrofurazone ("Furacin") is a nitrofuran
derivative possessing broad-spectrum activity which
181

2 >C H- H* NPI I—1rt fD HH- < H I—1 1—1H p O 03 tdO h Hi H* Mn P O oH- H H03 M 03 M
p N 03 Oi—1O O•p 3 H-cr fD CO03P-H-03NH*PfD
2o 00 M• 00 i—*On OI—* On vO O OnOn O
tO00
to
to to O VO On
vO
00to On O 00 O O O O
3OQ
OvOO
i—> 200 td(—» o OO to o Ono O O
2tdn300
2tdOvOO
03 03 Mrt P PH CL cr03 H*H- C7*P 03 H*cn H- rt
I-* O< Ho fD -<
t-h ►1
P2 03 PO P CL
MC/3P>• p*
p cr pCL oH* rtP PN H
H* H*P ofD H-CLP
OO3Td
1—*
P PH OP rtCL H-<
H-to rt■<
rt OP*Prt
H i
PN
O PH i 1-*P
H*3CP
O
H* PH OO H-O CLH*P P
H-P rt30 HP OH* H iP eW Hrt p
NO
00 PO P
H03o*I-1PM
182

has been used in various clinical situations since
its introduction in 1944. Currently, nitrofurazone
is recommended primarily for topical use in
wounds, second and third degree burns, and as
prophylaxis in skin grafting. Nitrofurazone solution
has been used as a bladder, eye and mouth
irrigant, and nitrofurazone cream has been used to
treat bacterial vaginitis and cervicitis. We found
nitrofurazone to be uniformly active against all
strains tested. Replica-plating studies (Table XI)
and time-kill studies (figure 4b) showed
nitrofurazone to be bactericidal at near MIC
concentrations. A considerable number of clinical
reports have shown nitrofurazone to be effective
against infections caused by Gram-positive and
Gram-negative bacteria, and despite over 30 years
of use clinical resistance is still not a problem
(Hooper & Covarrubias, 1983). Should mupirocin be
ineffective in clearing MRSA wound carriage,
nitrofurazone would appear to be an appropriate
alternative, however we are not aware of the
results of any clinical trials using nitrofurazone
in this indication.
Silver sulphadiazine ("Flamazine") was uniformly
active against the MGRSA studied (Table XI) despite
many of the strains being sulphonamide-resistant.
Silver sulphadiazine was less active than
nitrofurazone, however like nitrofurazone , replica-
plating and time-kill studies (Figure 4c) showed
183

silver sulphadiazine to be bactericidal at near
MIC concentrations. No difference in the rate of
killing was observed between sulphonamide-sensitive
and sulphonamide-resistant strains. Silver
sulphadiazine is a broad-spectrum agent which has
been shown to be beneficial in the treatment and
prevention of burn infections (Carr et a l . t 1973).
In the U.K, "Flamazine" is recommended (Lowbury e£
a l ., 1982) as first-line treatment for the
prevention of burn wound infection, however,
following its prophylactic use a preponderant flora
of sulphonamide-resistant Gram-negative bacilli can
emerge. There is little reported data regarding
the antibacterial activity of silver sulphadiazine
despite the widespread clinical use of this agent.
Rode et al (1989) have reported that a combination
of 1% silver sulphadiazine and 0.2% chlorhexidene
as topical therapy in a burns unit failed to
control MRSA wound colonisation/infection which was
subsequently brought under control on replacement
by mupirocin.
Mupirocin (Table XI) is highly active against
MGRSA, although concentrations substantially greater
than MIC are required for a bactericidal effect.
The consistent success of mupirocin in clearing
staphylococcal carriage has led to its
recommendation (Working Party, 1986; Working Party
1990) for the treatment of MRSA colonisation. No
mupirocin resistance was found in the MGRSA we
184

tested. High-level (greater than 1000 mg/1)
resistance to mupirocin in MRSA is increasingly
been found, and has been shown to be plasmid-
mediated (Rahman et a l ., 1990).
185
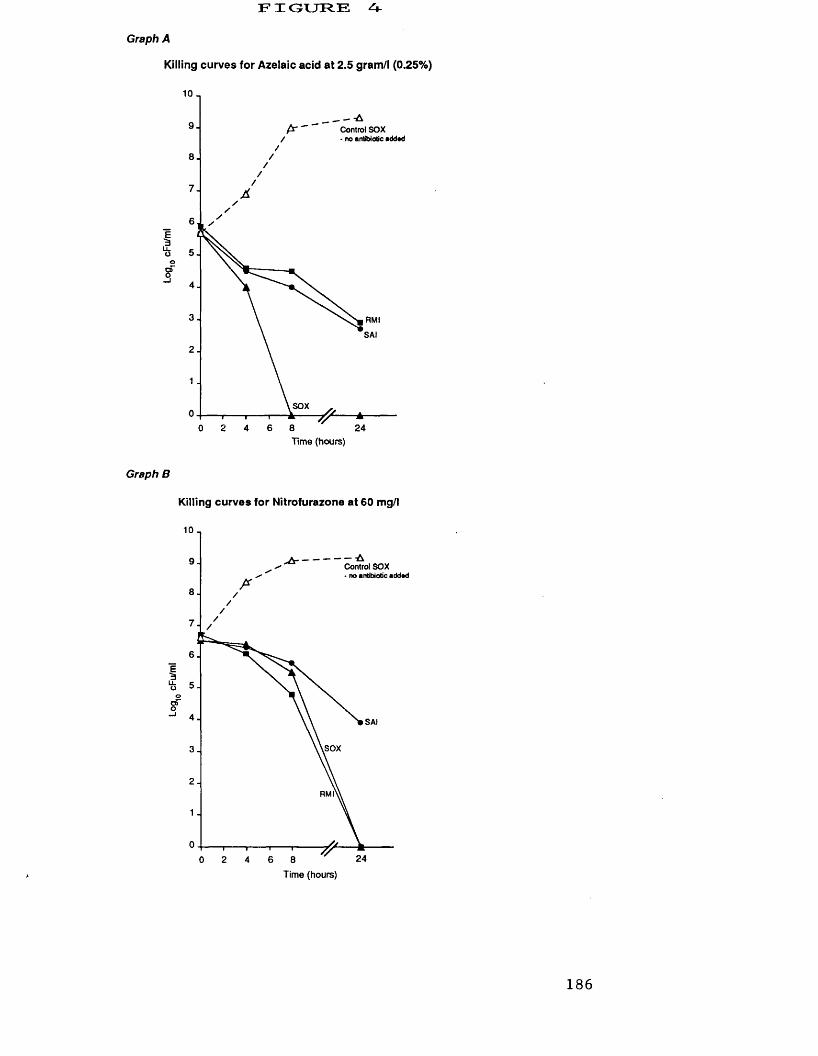
FIGURE A-Graph A
Killing curves for Azelaic acid at 2.5 gram/l (0.25%)
10 _
Control SOX - no antibiotic addad
Eao
RMISAI
SOX
24T i m e ( h o u r s )
Graph B
Killing curves for Nitrofurazone at 60 mg/l
10
— -A Control SOX - no antibiotic addad
9
8
7
6
5
4 SAI
iSOX3
2RM
1
0T i m e ( h o u r s )
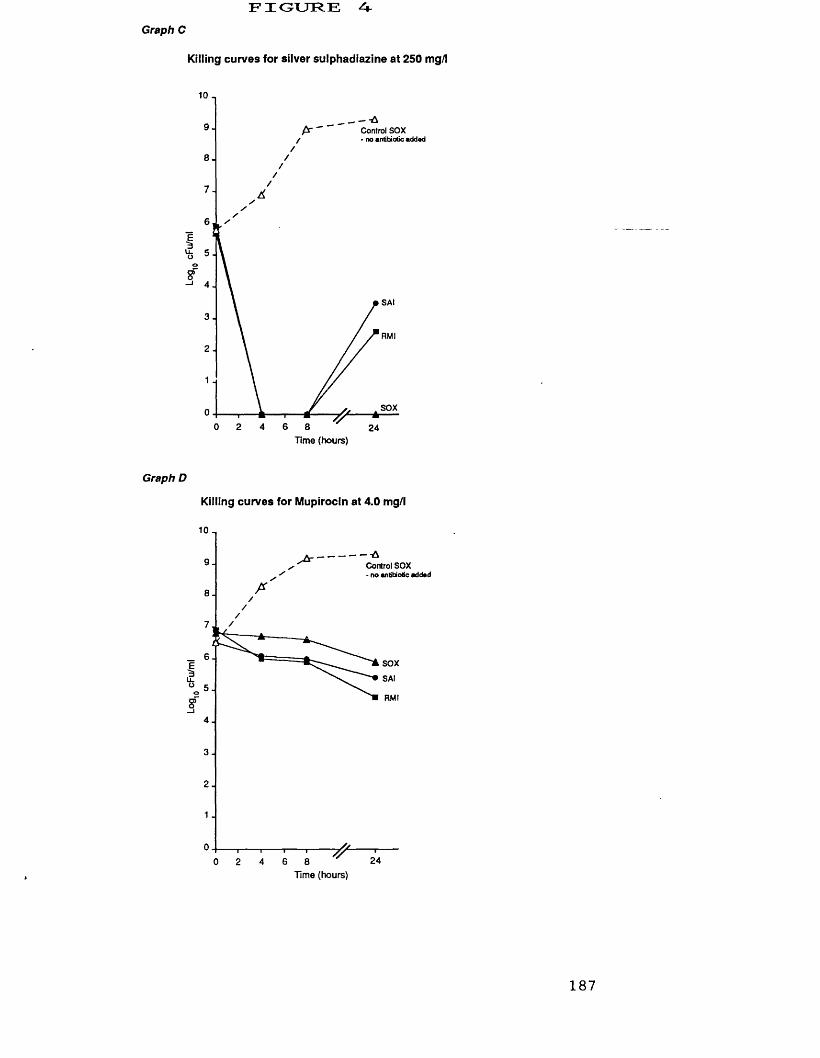
FIGURE AGraph C
Killing curves for silver sulphadiazine at 250 mg/l
10
9 Control SOX ■ no antibiotic addad
8
7
6
5
4SAI
3RMI
2
1
SOX02 4
T i m e ( h o u r s )
Graph D
Killing curves for Mupirocin at 4.0 mg/l
10
-■A Control SOX - no antibiotic addad
9
8
7
6SOX
SAI5o
RMI
4
3
2
1
02 4
T i m e ( h o u r s )
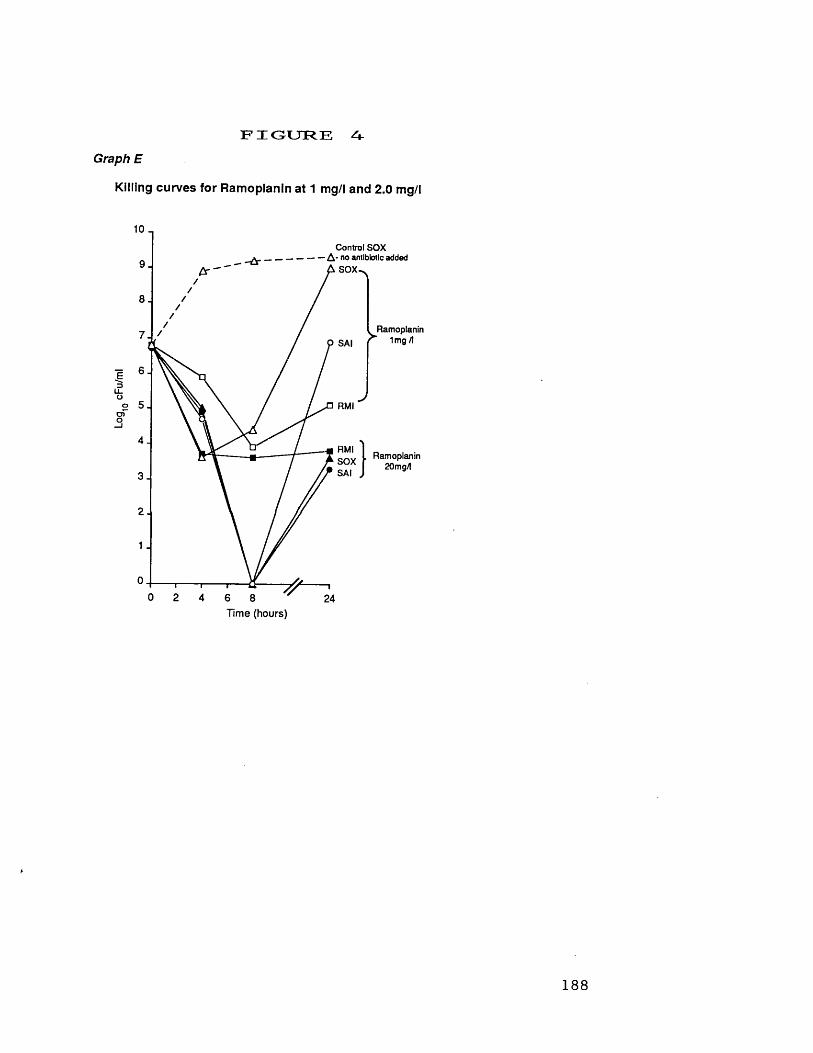
Log
cFu
/ml
FIGURE ^Graph E
Killing curves for Ramoplanin at 1 mg/l and 2.0 mg/l
10 _Control SOX
A - no antibiotic added A S O X ^
Ramoplanin 1mg /ISAI
RM I
RM ISO XSAI
Ramoplanin20mg/l
T im e (hours)
188

3. Do fluoroquinolones have a useful therapeutic role against MGRSA?
Ciprofloxacin was the first fluoroquinolone to
be developed for general systemic use, and became
clinically available during the mid-1980s.
Ciprofloxacin possessed uniform inhibitory activity
against MRSA resistant to multiple antibiotics eg.
aminoglycosides and macrolides. Additionally,
ciprofloxacin was rapidly bactericidal against these
organisms (Smith & Eng, 1985).
The novel mode of action of the
fluoroquinolones, whose primary site of action is
subunit A of DNA gyrase means that cross
resistance with other antibiotics was not found.
Furth ermore,it was believed that bacterial
resistance to these agents would not be a
clinical problem (Smith, 1984). In general,
fluoroquinolones are non-toxic, can be given orally
or by injection, and achieve good tissue levels.
In view of the problems of resistance and
toxicity encountered with other available
antistaphylococcal agents there was initial
enthusiasm for using fluoroquinolones against MRSA.
This early optimism has been tempered by recent
reports of widespread fluoroquinolone resistance in
MRSA. In this section the antistaphylococcal
properties of selected fluoroquinolones are
described, followed by an assessment of the problem
189

of development of resistance, and finally, promising
new fluoroquinolones are discussed.
3.1 Activity of the present clinically used
fluoroquinolones against M G R S A .
i . Inhibitory and bactericidal activity of
ciprofloxacin, enoxacin, norfloxacin, ofloxacin and
pefloxacin against M G R S A .
Currently, five fluoroquinolones, ciprofloxacin,
enoxacin, norfloxacin, ofloxacin and pefloxacin are
available for general clinical use. The inhibitory
and bactericidal activity of these agents compared
to nalidixic acid is shown in Table XII.
Ciprofloxacin, ofloxacin and pefloxacin are similarly
active against MGRSA, and enoxacin and norfloxacin
are less active. The non-fluorinated quinolone,
nalidixic acid is inactive against staphylococci.
By replica-plating, the fluoroquinolones were shown
to be bactericidal at near MIC concentrations (Table
XII). Our findings agree with those reported by
many other workers (eg. Smith, 1986; Fass & Helsel,
1987) .
The relative activity of fluoroquinolones in
vivo depends upon their pharmacokinetic properties.
These are summarised in Table XIII. Norfloxacin has
the lowest ratio of in vitro activity relative to
achievable serum levels (i.e therapeutic ratio), and
190
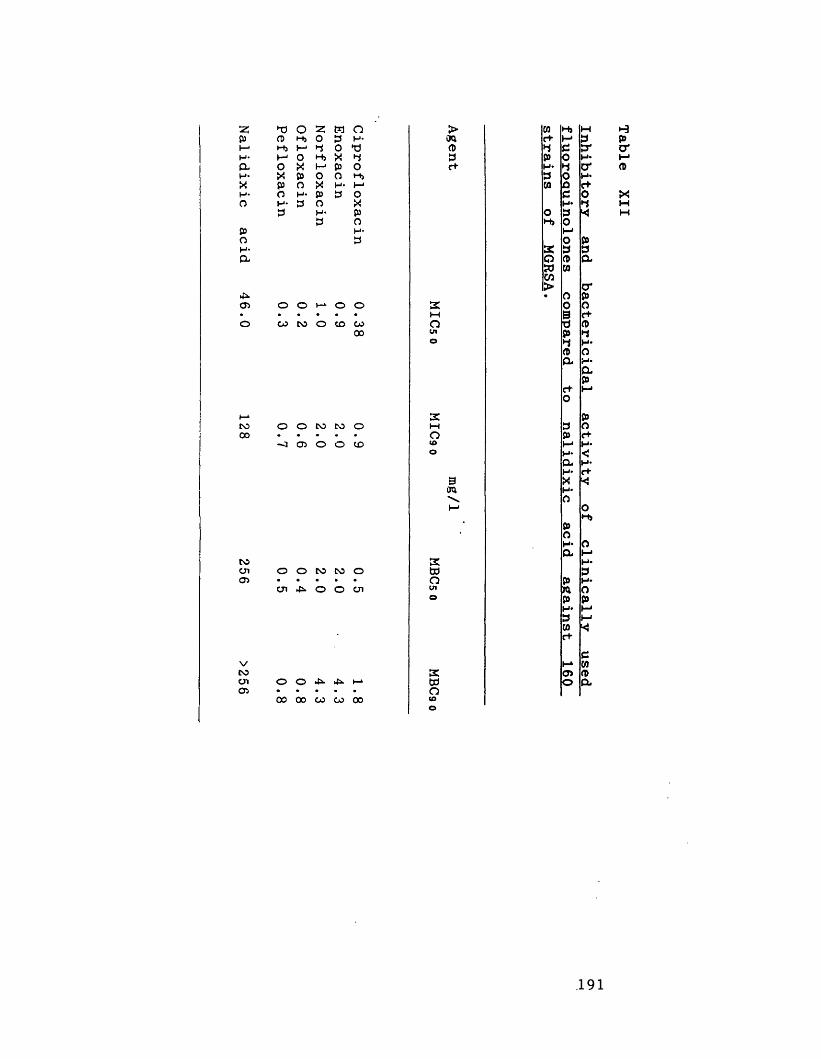
2: »T3 o z M Op a> H j 0 3 H*i—1 H j I - 1 3 0H* 1—1 0 H j X 3a o X h-* P 0H* X p o o H jX p o X H -H - o H* p 3 Oo H - 3 o X
3 H ' p3 o
p H*o 3H -a
>TOa>3c+
4OS 0 0 1-^00
CO CO O CO CO00
CO00
O O CO CO o -~3 O O O CO
o
BTO
COcnOS
O O CO CO o cn 4*. o O cn
vCOcnOs
O O 4*. 4s» i-*00 00 CO CO 00
£tdolO
CO Hj Me+ h-« 33 3 trP o H*H* 3 cr3 o H*CO o C+
c oH*
o 3 *Hj 0I-*o p£ 3 3O O a» to
CO> cr• O P
0 os c+d CDp 33 H*p Oa H*
ap
C+“ H*o
p3 OP c+
H*H* <a H*H* r+XH*Ci O
HjPOH* oa M
H*3
p H*TO 0p pH* M3 t—1CO *c+
ch-» WOS fl>o a
.191
Table XII

These values
can be
increased with
multiple dosing
to different
extents.
* P I r t X O td O I-* "I n<j 03 X I-1 (0
03 XCL Ow cn
c r fl> n> ° " 2
r tO
>crcrm03<H-03r tH-OPcn
i—i< p.p
03P&
o o pr>
r+ p
•• S0)aSx O M X 03 X f
03 pj
2 ° H-° c O cn 3CL° o* rtcn03 cncn 03
Hcr03
cncX03Mcno►iH-tdr t
I03 ^3H-pjgxH- IPr tH* td O 03
X
O vO X \0 03 O n ^H- .P
O
Pxcrpi-*MiI
-H*H-Mi
CL P OP >• OP Po as or t > pP pcn i r t
Mp P
r t o r tcr r t H-P Or t P
P<
-*
r t P r tH-3»—*P 03 Xcr
< i-* 1P pl—1<• r te H-p X 3cn O P
Mi M tn c r td P c r r t r t O op p m H- M < H* X 3 3 op p H . o O P o P P cnC3 P 3 cl r t H- 1 X X PP I—1 H- H- P I - 1 00I-* p p H- P S~~\ p
✓—\ P oo p c r p* 3MS S'S r t 1 H- ^ N_ c r 00 /—N3-9 ^ H- X a-g \ 3
o / s H- w h-» 00p r t v-x
X */—"So 00 I—* p as
N3 1 On • o oto On NO M* 1 00 I—1 p o X00 1 P> -F' P* 1 1 1 X
00 On X 00 ►—» X I fO h-» ■ o X
c r 00ocM o
ON cn NO HO ON O X
as 1 1 • o X> "J NO 00 H o
On O oo < X1 1 X
00 On ot-1 00 On ON i—» I—* NO On XX o i—* O 1 1 O >1 1 1 I—* NO O o
00 4> 00 . • X 1—1O On O On NO O 53
P32: -F> On O X> ON 1 • o as
oo On M ooo ■ < XOn P >1 1 o
■P- ON NO P XI►—1 ON O . i—* 1 O 5300 NO i—* 00 P • oo o
o 1—* NO • XX o
On NOoo 1 • O o
as I NO o X> 00 H f
NO On < oNO 1 XOn X 1—* OO >i—* i—• ■ • P n
-O' 00 oo 1—» On o MO On 1 1 o as1 1 1—* On X
NO NO • • oO On P 00
pOn o X
as as 1 1 • o X> > ON oo H X
1 < r*I—* oP X
as u> NO l—* 00 P >> i—* O nO M* • o o
i 1 O • 00 o MOn 00 1 On 1 X asNO o i—> p o
O •o I—*
HPcr
x
XP*P•d3Poo7?H-P03r tH*OX
O■op►1r tH*P03
OMi
r tcrp
xiMCO►tOnpH-POI-*OPPcn
192

is not used for treating systemic infections.Ciprofloxacin, ofloxacin and pefloxacin have higher
serum concentrations and greater in vitro activity,
compared to norfloxacin. In Table XIII the peak
serum concentrations of these agents are shown.
Multiple oral dosing with ofloxacin or pefloxacin
can produce higher peak serum levels (c. 8.0 mg/l).
For many Gram-negative bacteria these blood levels
greatly exceed MIC, however for Gram-positive
bacteria the therapeutic ratio is much less.
i i . Time-kill curves for ciprofloxacin, enoxacin,
ofloxacin and pefloxacin against M G R S A .
The killing kinetics against MGRSA of
ciprofloxacin, enoxacin, ofloxacin and pefloxacin have
been studied using various concentrations of agent.
Results shown in figures V ai, bi, ci and di were
with the fluoroquinolones at MIC (determined by
broth dilution studies) concentrations. Here killing
was observed for up to 8 hours following
inoculation, however, by 24 hours all the strains
had overgrown producing turbid broths. In figure
V aii, bii, cii and dii, fluoroquinolones at 4 x MIC
concentrations were used. At these concentrations
ciprofloxacin and j ofloxacin were usually
bactericidal, while enoxacin and pefloxacin showed
an initial bactericidal effect followed by
overgrowth by all strains after 24 hours.
193
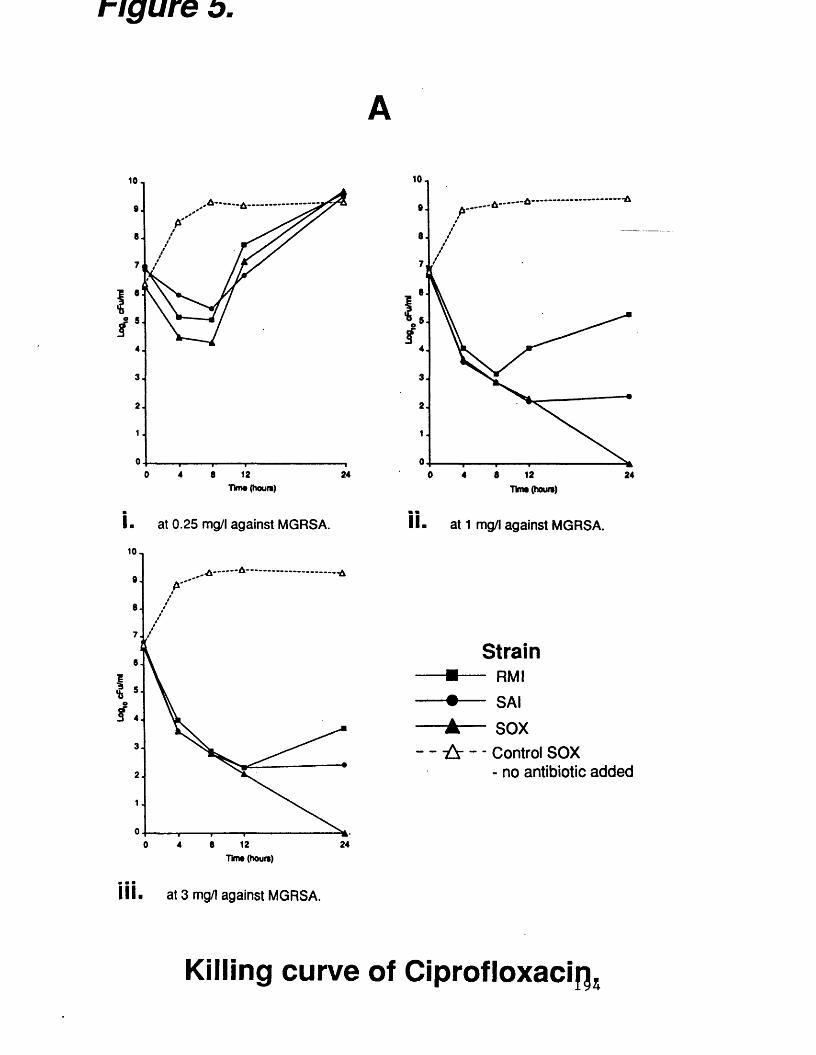
Figure o
10
9.
0 8 12 244Tim* (hour*)
I ■ at 0.25 mg/l against MGRSA.
10,
8 12 240 4Tim* (hours)
• ■ ■
I I I . at 3 mg/l against MGRSA.
A1ft
ef
3.
0 8 124 24Tim. (hours)
■ •
I I - at 1 mg/l against MGRSA.
Strain- RMI
- SAI
' SOX- Control SOX
- no antibiotic added
Killing curve of Ciprofloxacin
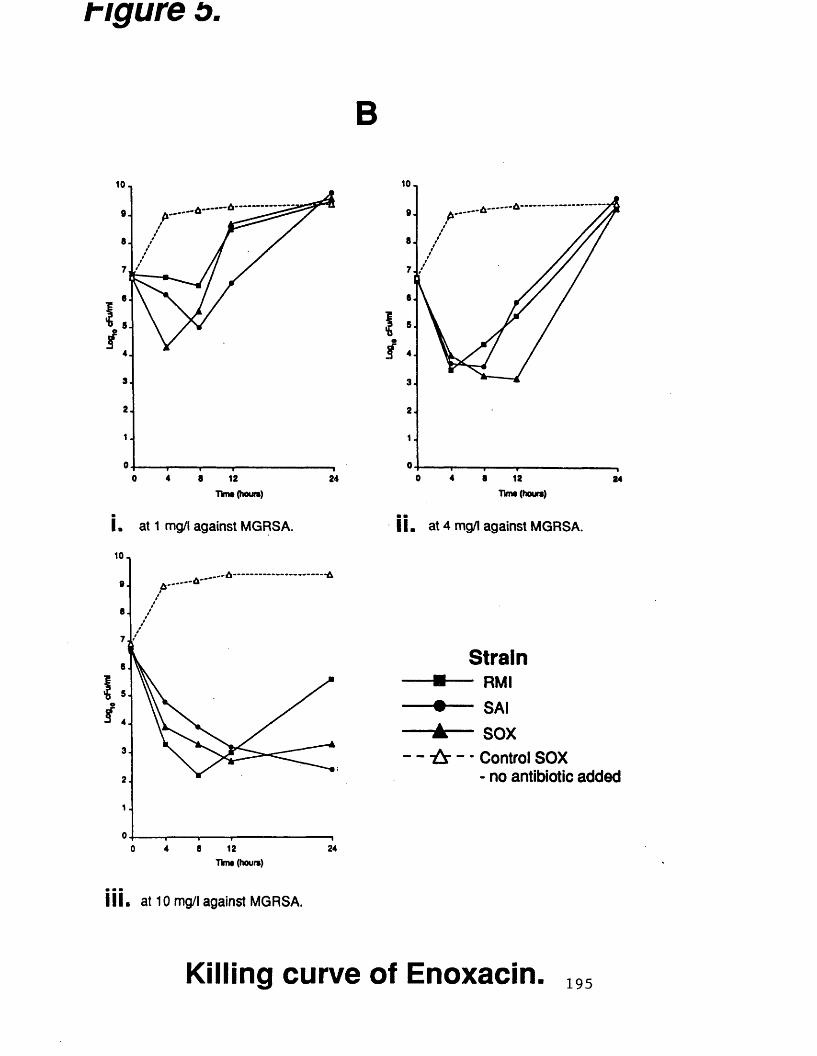
t-igure o
B10-.
40 8 12 24Tim* (hours)
I . at 1 mg/l against MGRSA.
10-i
24o 4 8 12Tim* (hour*)
I I I . at 10 mg/l against MGRSA.
10
e
f
o 4 8 12 24Tim* (hours)
■ mI I . at 4 mg/l against MGRSA.
StrainRMI
SAI
SOX
Control SOX - no antibiotic added
Killing curve of Enoxacin. 195

r i g u r e o
10 _
12 24eo 4Tim* (hours)
m
I . at 0.5 mg/l against MGRSA.
10,
o
f
o 4 6 12 24Tim* (hours)
i l l . at 8 mg/l against MGRSA.
10,
o 4 8 24
I I . at 2 mg/l against MGRSA.
Strain■m— rm i
+ ------ SAI
A — soxControl SOX - no antibiotic added
Killing curve of Ofloxacin. 196
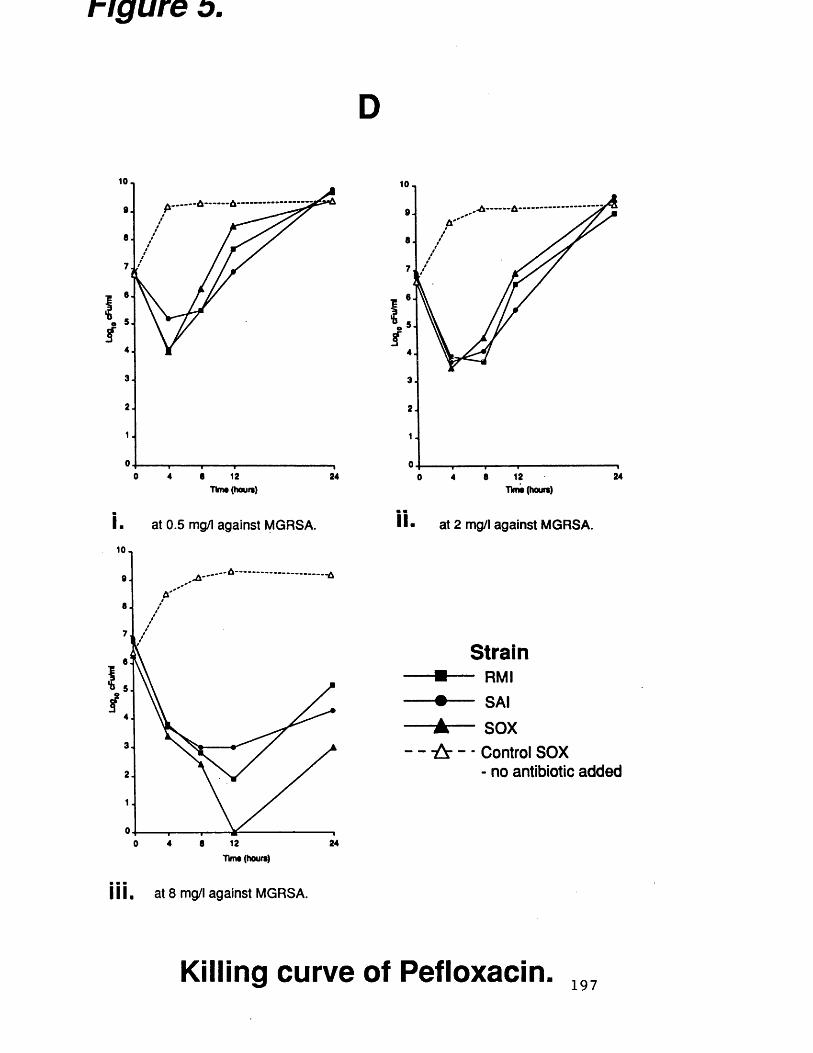
figure o
D10 -
of
o s 12 244
I.
Tims (houn)
at 0.5 mg/l against MGRSA.
10.
3.
12 244 80Tim* (hours)
j j j . at 8 mg/l against MGRSA.
10-19 .
241280 4
II.
Tims (hours)
at 2 mg/l against MGRSA.
StrainRMI
• ------ SAI
± — SOX
Control SOX - no antibiotic added
Killing curve of Pefloxacin. 197

Overgrowth by strain RMI sometimes occurred with
ciprofloxacin and ofloxacin.
The highest fluoroquinolone concentrations used
in the time-kill studies were ciprofloxacin (3.0
mg/l), enoxacin (10 mg/l), ofloxacin (8.0 mg/l) and
pefloxacin (8.0 mg/l). For ciprofloxacin, ofloxacin
and pefloxacin these concentrations were similar to
the respective peak serum levels following oral
dosing. The corresponding figure for enoxacin is
c. 4.0 mg/l, but at this concentration (i.e 4 x MIC)
overgrowth by all strains had been found in the
previous experiments. In order to establish whether
overgrowth could be prevented by a higher
concentration, an arbitrary concentration of 10 mg/l
enoxacin was used in the final time-kill
experiments. With these higher concentrations of
ciprofloxacin, enoxacin, ofloxacin and pefloxacin a
99.9% reduction in the initial inoculum was
usually maintained for 24 hours incubation (figure
V aiii, biii, ciii and diii) and broth turbidity
was not apparent even after 48 hours incubation.
On repeating certain time-kill experiments we
found considerable variation in the viable counts
obtained after 24 hours incubation. We have used
median data values (compiled from three separate
experiments) for 24-hour data points in figure V.
The production of broth turbidity was also a
variable phenomenon. Sometimes broths appeared
turbid at 24 hours although on viable counting
198

only 10^ - 1C)6 cfu/ml were present. Normally, broth
turbidity does not become apparent until a viable
count of c. 10® cfu/ml is achieved. Smith et a l .
(1986) who also observed this phenomenon, attributed
it to cell swelling and the production of multi-
cellular forms. When we have quoted the finding
of broth turbidity, viable counting has always been
used to confirm that the turbidity was due to a
viable count of 10® - 10^ cfu/ml.
To assess whether overgrowth was due to
fluoroquinolone instability in IsoSensitest broth
following prolonged incubation at 37°C (in the
dark) fluoroquinolone levels were assayed in
uninoculated broth before and after incubation. No
significant difference (t-test, p = 0.9) in
fluoroquinolone activity was found in pre-incubation
broth compared to post-incubation broth. The
development of resistance during the time-kill
studies is discussed in the next section (3.2i).
There have been differing reports of the
activity of ciprofloxacin in time-kill studies. For
example, with ciprofloxacin at 4 x MIC, Smith and Eng
(1985) reported initial killing with no overgrowth,
whereas Foster e t a l . (1986) found overgrowth
following initial killing. Antibiotic carry-over
resulting in inhibition of growth on viable
counting plates could significantly influence
experimental findings. We investigated the
significance of antibiotic carry-over in our
199

experiments. If 0.1 ml of 4 x MIC, or higher
concentrations, of fluoroquinolones was dropped onto
a plate seeded with S. aureus NCTC 6571 (ie. strain
SOX), after 24 hours incubation inhibition and
clearing of growth could be seen where the drop
fell. However, if 0.1 ml aliquots of the same
fluoroquinolone concentrations were thoroughly spread
on plates, no significant difference (t-test, p = 0.9)
in viable counts on these treated plates compared
to non-treated plates was found.
i i i . Spontaneous plate mutation rates to resistance
for ciprofloxacin, enoxacin, norfloxacin, ofloxacin
and pefloxacin compared to nalidixic a c i d .
For strains RM1 , SA1 and SOX spontaneous
mutation rates were determined by plating out c.
lO^ cfu onto agar containing ciprofloxacin at 5
mg/1 and 25 mg/1, enoxacin at 10 mg/1 and 50
mg/1, norfloxacin at 10 mg/l and 50 mg/1, ofloxacin
at 5 mg/l and 25 mg/l and pefloxacin at 5 and
25 mg/l. Resistant mutants were only obtained at
frequencies of 3.0 x 10“ 9 to 5.0 x 10“ 9 on plates
containing 10 mg/l norfloxacin. In no case were
resistant mutants found to the other
fluoroquinolones. For the older non-fluorinated
quinolone, nalidixic acid at 256 mg/l, frequencies
of mutation to resistance of 10“ - 10“ ® were
observed. Numerous in vitro studies (Wolfson &
200

Hooper, 1985) have reported much lower mutation
rates to fluoroquinolone resistance compared to
those found for the older quinolones (eg. nalidixic
acid ) .
According to Smith (1990) frequencies of mutation
to resistance of 10“ ^ - 10“® occur for S. aureus in
the presence of 5 x MIC concentrations of enoxacin
and norfloxacin, and 10~9 _ 10“ 10 in the presence
of 5 x MIC concentrations of ciprofloxacin and
ofloxacin. At 10 x MIC concentrations of
ciprofloxacin, enoxacin, norfloxacin and ofloxacin,
resistant mutants could only be obtained with
norfloxacin (frequency 10” ^). We have made similar
findings. Kayser and Novak (1987) and French et al
(1988) showed that high-level fluoroquinolone
resistance can easily be produced by passaging
strains in the presence of two-fold increasing
drug concentrations.
201

3 . 2 Fluoroquinolone____resistance____ in_____MGRSA -
development of resistance in vitro, and properties
of resistant strains derived in vitro and from
clinical sources.
i . Development of fluoroquinolone resistance in
vitro during time-kill experiments.
As an extension of our findings of regrowth
in the time-kill experiments (figure V), four
strains (RFH 10, AUS 10, SA 1 and SOX) were grown for
48 hours in the presence of the same
concentrations of ciprofloxacin, enoxacin, ofloxacin
and pefloxacin as previously used. Strains RFH 10,
AUS 10 and SA 1 were chosen because they possessed
a range of susceptibilities to ciprofloxacin.
Strain RFH 10 was moderately susceptible/resistant
to ciprofloxacin (MIC 2.0 mg/l) and strains AUS 10
and SA 1 were susceptible to ciprofloxacin (MIC 0.5
mg/l and 0.25 mg/l respectively). Strain SOX
(ciprofloxacin MIC, 0.25 mg/l) was used as a
control in these experiments.
Viable counts were performed on the time-kill
broths at 24 and 48 hours incubation, and the
susceptibilities to fluoroquinolones of any
organisms isolated were determined. Our results
are shown in Table XIV (a, b, c and d).
For ciprofloxacin at 0.25 rag/1, there was
initial killing followed by regrowth, and by 24
202
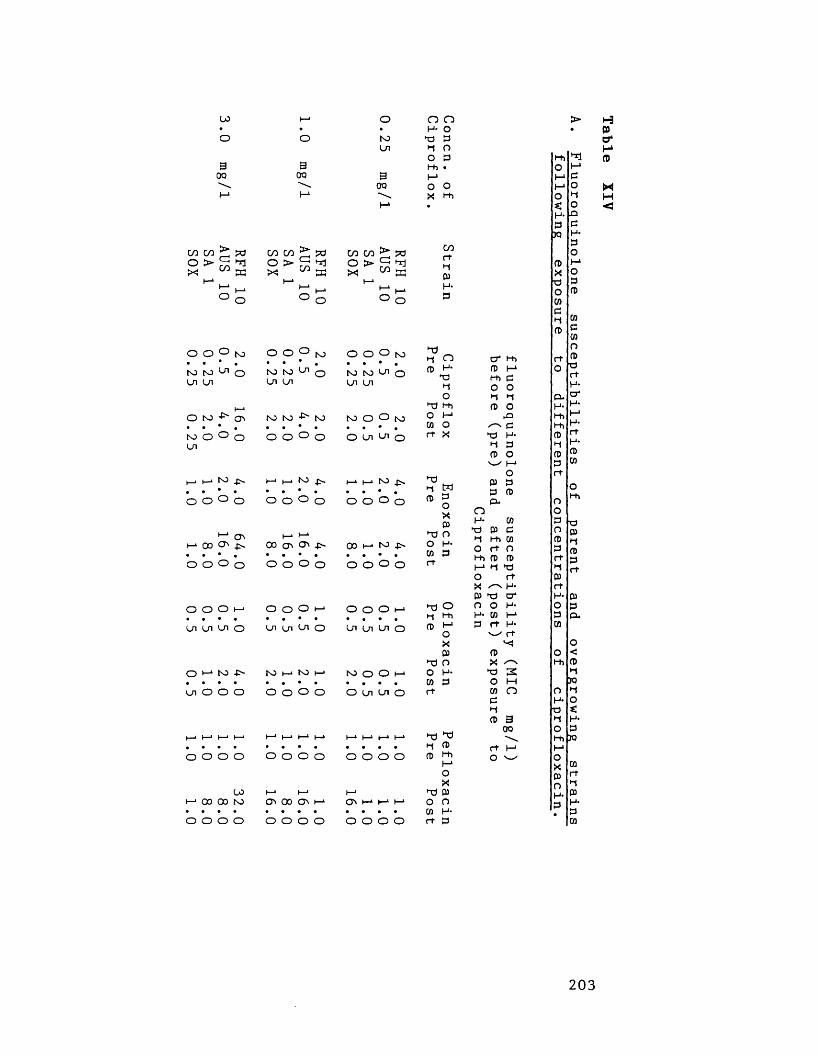
L0 I—*• •o o3 300 00M H*
C/3 CO > pa CO CO > pao > Cd HI o > cd HIX C/J EC X CO W
I—1 1—1I—1 i—* I—1 i—>o O o O
o o o to o o o to• . • . • • •CO to Ln O to to Ln oLn Ln Ln Ln
t—1O to -O- CTi to to -P' to• • • . • . • •to O O O o O o oLn
(—* t—1 to -P' t—1 H-* to ■p-. . • • • « • .O o O o o o o o
i—* d I—1 t—1i—100d ■P- 00 d d -P~• • • , • • • •o o o O o o o o
o o o h-> o o o 1—*• « • • • * • •Ln Ln Ln o Ln Ln Ln o
O i—1 to -P- to i—1 to f—*• • • * • • • •Ln O O o O O O o
i—* i—1 h—> 1—1 t—* i—1 i—* (—». • • • • • • •O o o o o o o o
00 I—1 t—1i—10000 to d 00 d 1—>• • • • • • • •o o o o o o o o
o O O• H- oto H) £Ln H O
O £hh •3 M00 O O\ X Hi1-* m
O O O jsj • * * •M K) ^ O Ln Ln
to O O to • • • •O Ln U 1 o
I—‘I—> ho • • • •O O O o
00 h-> M • • • *O O O O
O O o I-* • « • •in ui Ui o
to O o h-» • . • •O U i U i O
o o o o
d I—1 I—1 I—1 • • • •o o o o
COHHCDH*£
Hih nCD H- HI H O
H H i O H-* 73 O rt X
H3H WCD £
OXCD
HJ OO73 £H
HJ OH HiCD h-1
oXCD
HJ OO H-73 £H
HI HJH CDCD Hi
1—*OX
HJ CDO n73 H-H £
O' H> CD H* Hi £ O O H H CD O £3 /-s P HI H- H £ CD O ^ M
OCD £ £ CD £*n
h - m t3 01 c H Hi 73 O H O Hi CD CD I—1 H ’"£
HOXcd hi o o
CO
H-crH*I—4
H H- v_x H XCDX ^ H 3 O M CO OcHCD 3
00H I—1 O
CDXX)oCO£HCD
HII—*£OHOn£H*£OI-1o£CD
CO£COnCD■£HH-dH-I-1H-HH*CDCO
OHi
■£CDHCD£H
(D££-
Hi JO
HCDO'I-4CD
MH<
203

1—* ■C> t—> M Oo • • P Oo o O P X O3 CD POO 3 3 o •00 00 H-l-> \ \ P oM I-* Hi
CO CO > pa CO CO > pa CO CO > wOX > ci!
co S3o >X
oCO t a
OX > C2CO S3
1—1i—*o
10
1—1 10 i—io1—1 i—io
10
O O O f o • • * .N) K) O UI UI
O O o • • • •to NJ Ui O ui UI
I—i l—' to . • • •o o o o
o o o I—• • • • •UI Ui Ui o
o o O 00 • • • •UI Ui Ui o
ooto
o o o o
o O ° to . . * .to IO U1 O On Ui
to to
o o ° o
(—1 H-* NO • . • «o o ° o
O' '— ■ vj'I—‘I—*N0^ 00 Oi O' -N• • • . - *O O O o O o O o
O o o t—* • • • •On On On o
to to to • * • •o o o o
i—* oo I—* oo O ' to O M O
o o o o
o o ° to • • • .to On Ui oOn
to to ■P' to • • • .o o ° o
I—> I— 1 to _p«. . • • .o o o 0
h—* t-*oo O' O' •C' • • • •O O o o
o o O t—1 • • • •On on On o
to to to h • * • •O O O o
o o o o o o o o o o o o
I—1 00 I—1 O' to O' H
o o o o
C/0rtH05H*P
TJH o cd h- nj
•-to•"d Hi O I-* CO O rt X
>-0H M CD 3 O
X CD*-d oO H- (0 P rt
p TJH CDCD Hi
I-1OX
"d CDO oCO H-rt P
cr hi n) i-1 t-h po oH H CD O
►£1 c
►p H- H P CD Ow |-i
OCD P P CD CO
MPO CD P X Hi CO P rt O O CD CD H* H P rt
H-P cr
P O O H*rj Hi CO I-1CD t—1 rt H*
O rtX XCD CD*P o X ^
O H- P 3CO P O Mrt CO O
rt i—1 O o-/
td
*1I-1PotloLapH-POH*OP(D
COPCOoCDPrtH*c rH-t->H*rtH-CD(0
O O
CD P O X CDO pQ H*
CO rt H CD H* P CO
1-3P>a*MCD
2 0 4

00 to O O O• • • Hi Oo o Ln M P
o n X P3 3 3 P •00 OO 00 nH- o
I—1 M I- 1 3 Hi
o h3Q)a*
COCO > Sd CO CO>f \ >0 COCO> r 1Sd
O > >0 C/3 K
o > C-iCOHjEC
o > C-HCO**1W
1—*I—*o i—1o
I—* 10 K-*o
I—1
o
10
O O O fo O o °M M '-H OLn Ln
O O ° to . . * .to to L/l O Ln Ln
O O ^ O
i—1 i—i •—1 • . • •o o ° o
O O o h-j• I • •Ln Ln Ln o
O O O ‘• • • •Ln Ln Ln o
t o. . • .to to ^ oLn ui
O O CT>to toLn Ln
Ln
LOH H tO |o • • * •O O o o
O o ° H-• • • • •Ln Ln ^ O
O O O • • * •Ln Ln Ln o
O O ° to • • * •to to ^ OLn Ln
to to to • • * •o o ° o
I—I to. . • .O O o o
I—1 l-‘CD O '• • • •O o O o
O o O h • • * •Ln ui Ln O
to to to l—* • • • •O O O O
cortH03H*P
T)H OCD H-
npHO
Tl HiO (—1CO Ort X
HdH WCD P
OX03
TJ OO H-CO Prt
t—11—* I—1I—1 I—11—* t—1t—1 I—*t—1 I—11—1 "d TJ• • • • . . • . • • • • H CDO o o o o o o o o o o o CD Hi|—io
XI—* t—* I—* “d 03
t—* I—1I—* h-> h-«I—11—* 00 o 0-1 O' I—1 o o• • • • • • • • • • • • CO H-o o o o o o o o o o o o rt P
cr t-h CD M Hi p O O H H CD O ►a✓—N CP H* H P CD O
Ot-t> coH 0) C O Hi CO X rt O 03 CD CD O H P H- rtP H-na cr
•n O O H-H Hi CO !-*(D I-1 rt H-
O rtX03 03
"d O X ^o H- P 2CO P O Hrt co n
rt l - 1 O ^
Hi ^ CDo MM PM o HO ^ H< O <H* jQP PJO H*
PO
CD I-1X OP PO CD
coPCOoCD■artH-crH-t-*H-rtH*CDCO
2 0 5

oo to o hd n• • • p oo o Ln hh P I-1 O
3 O P3 3 X •OO OO oo Po oI-* l-» I-1 H- hh P
co in > w co co > w CO CO >/■H saoX > co •n
EC o >XGCO
Hr]EC
O > Gco hrjECi—•10 10
t—>(—> o 1—» oH-* 10 i—*
O
o o ° m o o °to to OLn Ln
O O ° o • • * .to to OLn Ln
toO O
O'! to ^
o O O t— • • • •Ln Ln Ln o
O O O O
LOto
O O O O
toto to Ln Ln
Ln
to toO O
to
0 0 ° 0 O O o o
o o o (—> • • • •Ln ui Ln o
O O O to to to 00« t * • • # • •Ln Ln Ln o O O O O
(— * i— 1 i— * LO O ' O ' C M OO O O O
O O ° to • • • .to to oLn Ln
to to -£~ 00 . . * .o o ° o
| ‘I »• • • •o o o 0
h—‘ H-11 CD O' O'• • • •O O o o
O O o (-» • • • •to to Ln oLn Ln
to to to t—> • • • •o o o o
o o o o o o o o
o o o o • • • •o o o o
00rtHJPH-p
■nHJ Op H-
HdPO
hd Hho 1-*cn Ort X
hdHJ MP P
OXPhd OO H*03 Prt
Hd OHJ HhP H-*
OXPhd oo H*CO Prt
X) hdHJ pP tt>i—1oXhd Po ow H-rt P
O l-h P t-1 t-h C O O h« *-J fD O
kQ /-N C Hd H- HJ P 03 O w |-io03 P P 03 P*
h303 03H i 01 ci—* t-h cn O rt O X CD 03 P h| Hd O rt H- H- P P D*
O H- C0 H-» rt H-
rt03X HP S O M 03 OcHI03 300rt h-» O ^
t-t> hd Po I-1t—1 et—1o xo ^ Hs: O <H- idP P30 H-
POP I—1X op po p
COc(0oCD ■p rt H- cr H- t—* H* rt H- CD CO
X pQ P
H0)o*
2 0 6

hours incubation
were found. The
the overgrowing
and SA 1) was
NCTC 6571) a
susceptibility
ciprofloxacin,
turbid broths,
only 2.6 x 10^
incubation, and
hours incubation,
isolated at the end of
2-16 fold decrease
susceptibility. MICs
unchanged. At 3.0
RFH 10 overgrew
strain possessed
fluoroquinolone
only 102 - 105
10, SA 1 and
fluoroquinolone
were unchanged. Strain
48 hours incubation,
ciprofloxacin (data
produced mutants
susceptibilities.
Similar
have been
XIV b
108 - 109 cfu/ml
susceptibility of
RFH 10, AUS 10
SOX (S. aureus
fluoroquinolone
.0 mg/l
and SOX produced
a viable count of
found after 24 hours
not appear until 36
AUS 10, SA 1 and SOX
the experiment showed a
fluoroquinolone
RFH 10 were
only strain
broth. This
decrease in
counts of
strains AUS
incubation. The
these organisms
isolated after
of 5.0 mg/l
strain RFH 10
fluoroquinolone
slight variations
pefloxacin (Table
mutants with
2 0 7
turbid broths of
fluoroquinolone
strains of MGRSA (ie.
unchanged. For strain
8-16 fold decrease in
occurred (Table X l V a ) . With 1
strains RFH 10, SA 1
For strain AUS 10
cfu/ml was
turbidity did
Strains
in
for strain
mg/l ciprofloxacin
to produce a turbid
a 2-32 fold
susceptibility. Viable
cfu/ml were found for
SOX after 48 hours
susceptibilities of
SOX was not
In the presence
not shown) only
with decreased
trends, although with
found for enoxacin and
and d). Overgrowth by

decreased fluoroquinolone susceptibilities occurred
at 0.5 and 2.0 mg/l pefloxacin, and 1.0 and 4.0
mg/l enoxacin. In the presence of 10 mg/l
enoxacin and 8.0 mg/l pefloxacin, only strain RFH
10 could produce mutants and overgrow. A different
trend was observed for ofloxacin, strains AUS 10, SA 1
and SOX only overgrew in broth containing 0.5
mg/l. At 2,0 mg/l ofloxacin only strain RFH 10
overgrew, and at 8.0 mg/l even strain RFH 10 could
not produce mutants.
These findings show that fluoroquinolone
resistance can be easily selected in the presence
of therapeutic concentrations of ciprofloxacin,
enoxacin, ofloxacin and pefloxacin. However, the
propensity of strains to become resistant may be
different for individual fluoroquinolones. Thus,
selection of resistance was least with ofloxacin,
which may be due to the fact that ofloxacin
possesses an extra mechanism of killing (Lewin &
Smith, 1988) against staphylococci not found for
ciprofloxacin. This extra mechanism of killing of
ofloxacin may also account for the observation
that this antibiotic was the most active of the
agents studied against the fluoroquinolone-resistant
mutants.
Irrespective of the particular fluoroquinolone
or concentration used, resistant mutants were found
to possess similar reductions in susceptibility,
suggesting that only a one-step mutation could
2 0 8

take place. Furthermore, decreases in
fluoroquinolone susceptibility were proportional to
the initial susceptibilities of strains. For
example, with ciprofloxacin strain SOX could only
mutate to a ciprofloxacin susceptibility of 2.0
mg/l (a 8-fold decrease in susceptibility) and
strain RFH 10 could only mutate to a
ciprofloxacin susceptibility of 16.0 mg/l (again a
8-fold decrease in susceptibility). Generally,
mutation to resistance in the presence of one
fluoroquinolone resulted in cross-resistance to the
others. The degree of reduction in susceptibility
differred between the fluoroquinolones. The least
reduction in susceptibility was found for ofloxacin
followed by ciprofloxacin. The greatest reduction
in susceptibility was found for pefloxacin.
Fluoroquinolone resistance appears to develop in
a stepwise manner. For example, following exposure
to 1.0 mg/l ciprofloxacin, strains initially
sensitive to ciprofloxacin (MIC 0.25 - 0.5 mg/l) may
develop borderline resistance (i.e ciprofloxacin MICs
2.0-4.0 mg/l). In the clinical situation such
strains exist (eg. RFH 10), and these can then
mutate to clinical resistance (ie. ciprofloxacin MIC
= 16 mg/l) following further exposure to
fluoroquinolones. Laboratories may well fail to
detect those strains possessing subclinical
resistance using current disc testing techniques.
As a consequence of failing to detect borderline
2 0 9

resistance a permanent reservoir of organisms with
the potential to rapidly develop clinical
resistance may build up following widespread
fluoroquinolone usage.
Ofloxacin appears to be the most appropriate
fluoroquinolone to use against MGRSA, at least with
a view to minimising the risk of emergence of
resistance. Furthermore, depending upon the initial
fluoroquinolone MICs of strains which mutate to
resistance different levels of fluoroquinolone
resistance might be expected in a diverse
collection of resistant strains. In order to
determine whether the phenomena discovered in our
time-kill experiments are relevant to the clinical
situation, we have investigated the properties of
an internationally diverse collection of
fluoroquinolone-resistant MGRSA.
i i . Quinolone susceptibility patterns of clinical
isolates of ciprofloxacin-resistant M G R S A .
Our in vitro studies show that fluoroquinolone
resistance can emerge in the presence of
therapeutic concentrations of fluoroquinolones.
Irrespective of the fluoroquinolones used, the same
patterns of resistance are obtained, and the level
of this resistance depends upon the initial
susceptibility of the strains. Thus, in v i v o , it
might be expected that clinical isolates of
2 1 0

fluoroquinolone-resistant MGRSA would possess similar
susceptibility patterns. To investigate this
possibility, we have collected MGRSA from 7 centres
worldwide (two centres in France and Israel,and one
centre in Germany, Italy and the USA) and have
determined their susceptibility to a range of
fluoroquinolone agents. Our findings are shown in
Table XV.
From the MICs in Table XV different levels
and patterns of fluoroquinolone resistance are
apparent. Even strains from the same centres
showed different patterns and levels of resistance
eg. strain IS1 vs. strain IS2, strain TEX1 vs
strain TEX2. Other workers (Kojima e t a l ., 1990;
Yamamoto, 1990) have also reported different
fluoroquinolone susceptibility patterns in clinical
isolates. Kojima et al (1990) have proposed that
different mechanisms of resistance may occur (i.e
mutation of DNA gyrase, or reduced permeability due
to an alteration in the cytoplasmic membrane). In
v i t r o , French et a l . (1988) and Limb et a l . (1987)
have shown that passage in subinhibitory
concentrations of fluoroquinolones can result in a
variety of levels and patterns of fluoroquinolone
resistance. Thus, it would appear from our results
with the clinical isolates of ciprofloxacin-
resistant MGRSA that a number of different
mechanisms of fluoroquinolone resistance exist, or
that fluoroquinolone resistance has evolved along a
211

- 3 •-3 i-3 •-3 h 9 i-3 •-3 •-3 ►-3 *-3 »-3 t-3 k—1 M M M *H M w MW W M W W W W W W M W M W CO CO CO CO CO CO CO COX X X X X X X X X X X X X
00 - 3 05 05 £>- 00 CO I-4to >—* t—4 i—1 k-4 k-4 k—4 k -4 —) 05 oo to i - 4o -0 05 O l 4 * 03 k-4 o
G C G a g g g G G G G G G M 1—1 HH l- l M w M MCO CO CO CO CO CO CO CO CO CO CO CO CO CO CO CO CO CO W CO CO> > > > > > > > > > > > > 3 3 3 4 3 3 3 3p j p j p j j B p f j p p<T>(T><T><T>(I>00<D (—' I—> 1—1 I—■ t—• t—* t—' I—•
V V V V V V V V V V V V Vcn cn cn cn cn cn cn cn cn cn cn cn cn cn cn cn cn cn cn cn cn S5k-4 i—4 k-4 5—4 i—4 j- 4 t—4 5—4 i - 4 i—4 t-4 »—4 i—4 5—4 5—4 t-4 i—4 I - 4 k-4 i - 4 k-4 •to to to CO CO CO CO CO CO to to CO CO CO CO CO CO CO CO CO CO >
O' V V V V V V V V V V V V V V V V V1—4 I—4 t—4 I—4 I—4 k-4 I—4 k-4 k-4 k-4 I—4 k-4 k-4 k-4 k-4 k-4 k—4 k—4 >CO CO CO CO to CO CO 00 CO to 00 00 CO CO CO CO CO CO CO CO CO o00 00 00 00 00 00 00 00 00 00 00 00 00 00 00 00 00 00 3
VCO CO CO CO 05 5—4 CO 4» co CO 4*. 4» i-4 05 I- 4 05 05 V-4 I- 4 k-4 I - 4 OCO CO to CO 4*- CO CO CO to 05 4 . CO 4 - 4». 05 05 CO 05 H *00 00 00
k—4 k—4 k—4 03 k—4 V k—4 k-4 t—4 k—4CO CO CO CO CO CO CO 05 COoo oo oo 00 oo 00 00 oo
o c n u u c n o c n o ) 4- to to 4^ »£*•
V/ V V V V V V V V VI—4 k—4 I—4 k—4 k—4 k-4 t—4 00 I-4 05 co k—4 CO 05 k-4 I—4 k—4 05 05 k-4 05CO to to CO CO CO CO to to CO 05 CO 4 CO CO CO 4. 4* CO 4>-00 00 00 00 00 00 00 00 00 00 00 00
H M M H H O I H N H H M N C O 05 05 05 O) 05 ^ 05 O) 05 DO
DODC*a O ) 0 ) 0 5 0 1 U h * 0 ) 0505 k—4
oo 00
M C O U U H P U M 05 CO tO tO 05 05 tO 05
V V V VCOk-4 k-4 k-4 COCOk-4 COt o t o t o t o t o t o t o t oOO 00 00 00
tz: coO c+ • p H- 3
o oH> O
c3O t+
3 3H- V*TOH*3
w
5SHJ
oX
►dX
H*33*H*crH*c+o3
oo3oa3c+p<+H*o3sTO
ooMMAo«+H*o 13oH>
oH**do*-boXp0H*1
3ACOH*ac*p3c+a:QwCO>oH>
H*3H*op
o3H*TOH*3
2 1 2
TABLE XV.
Patterns of
quinolone resistance
in an
international
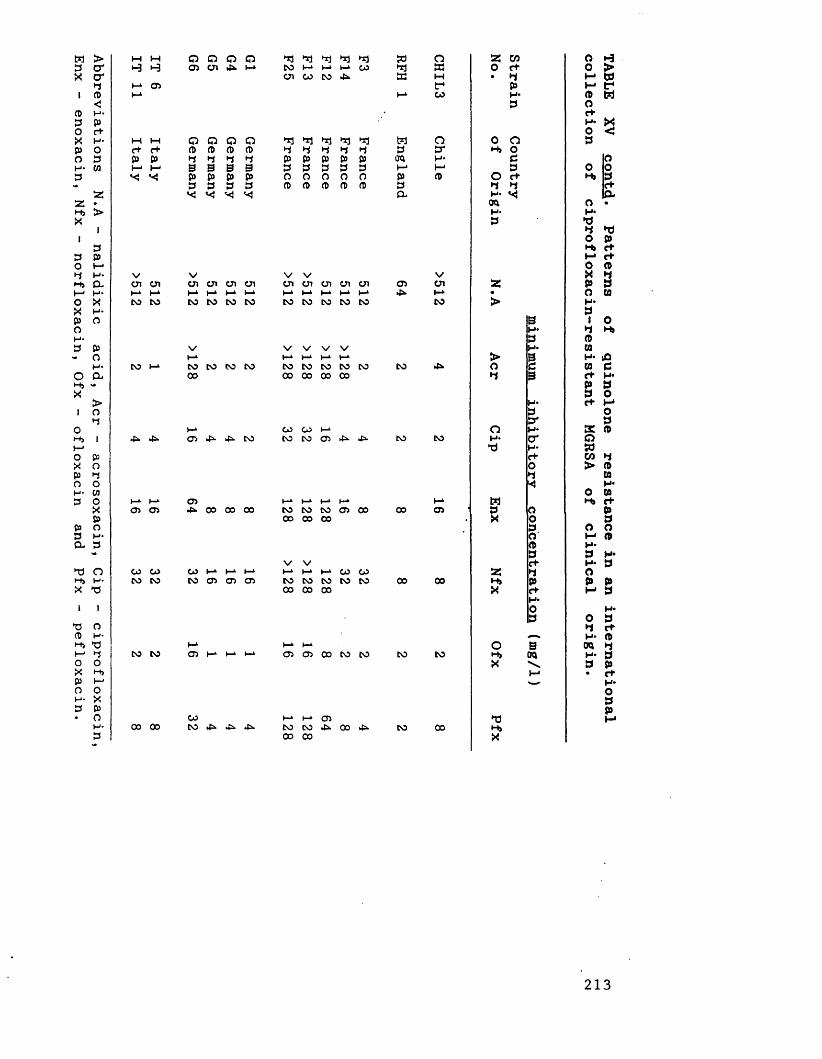
Abbreviations N.A
- nalidixic
acid, Acr
- acrosoxacin,
Cip -
ciprofloxacin Enx
- enoxacin,
Nfx -
norfloxacin, Ofx
- ofloxacin
and Pfx
- pefloxacin.
w M o o Q o ►d *3 ►S3 50 o a w o>-3 »-3 OS U i 4». CO 1—‘ l- l I—1 CO a o c+ 0 >
U i CO CO 4k a w • d M at-1 OS f p H* a
H* CO H* CD Mp ac+H* Xo <
i—i M o Q Q a *1 *3 *3 *3 w o 0 o pc+ C+ a A A CD d d *1 d p a HJ oP P d d d d p p p p p TO H* c at—1 I - 1 B 0 B B p p p p p I—1 p o o
P P P p o o o o o p A o «+ H i p3 p P p CD CD CD CD a p d d av* v j a H- a
TO o •H- H*p *d
d ►do pH , <+
c+o A
V V V V V X dU I U I U I u i U I U i U I U I U I U i U i os U I a p pt—1 1—‘ h-* t—1 I—4 t—‘ h-* »-» I-* l-k ►-* 4k. I-* • o COto CO CO CO CO CO CO CO CO CO CO CO > H*p
B 1 0H* d H»p A
V V V V V H* wH-1 h-* I—1 I—1 > 0 h - id
CO (-» CO CO CO CO CO CO CO CO CO CO 4k. o p ca c00 00 00 00 00 d 0 c+ H*p pp o
H* c+ t-*p oa p
h-* co CO (-* a H* a A4k. 4k. OS 4^ 4k. CO CO CO OS 4k. 4 - CO CO a Q
•d H - w<+ w do > Ad 09
H*o w
h-L I—1 OS 1—‘ H-1 H-* h-k I-* M H i c+cn OS 4k. 00 00 00 CO CO CO OS 00 00 OS p a p
oo 00 00 X o pp o oo M AA H *p p
V V c+ H* pCO CO CO l-k h-1 I—‘ »-* H* »-* CO CO a d oCO CO CO os OS OS CO CO CO CO CO 00 00 H i p p p
00 00 00 X c+ I-* pH*o H*p o p
d CtH* A
I—1 t—1 I—1 o 5 TO dCO CO OS h-* I—1 OS OS 00 CO CO CO CO H i TO H* p
X p pM • rc
H*opp
CO 1—‘ 1-1 OS ►d M00 00 CO £>• 4k. 4k. CO CO 41. 00 4>. CO 00 H i
00 00 X
213

number of separate evolutionary pathways, or a
combination of both. We have been unable to
determine the fluoroquinolone resistance mechanisms
in the strains studied because of the practical
difficulties encountered in investigating them in S .
aureus (Ubukata et a l . 1989).
Against the ciprofloxacin-resistant MGRSA,
ciprofloxacin and ofloxacin were found to be the
most active (median MIC = 16 mg/l) of the
fluoroquinolones tested, followed by pefloxacin
(median MIC = 32 mg/l) and enoxacin and norfloxacin
(median MICs = 64 mg/l). The older, non-fluorinated
quinolone - acrosoxacin was also tested against the
ciprofloxacin-resistant strains, and contrary to our
expectations some strains were found to be
sensitive (eg. RFH 1, F3, IT6). The phenomenon of
"reversed incomplete cross-resistance" between the
fluoroquinolones and older non-fluorinated quinolones
has been seldom reported. Van Caekenberghe and
Pattyn (1987) have reported "reversed incomplete
cross-resistance" with acrosoxacin for a strain of
Flavobacterium multivorans. Fluoroquinolone-resistant
mutants derived in vitro showed little acrosoxacin
resistance. Our results for strain RFH 10 are
shown in Table XVI.
There have been few reports which have
monitored the clinical emergence of fluoroquinolone
resistance during treatment. Milne and Faiers (1988)
reported the rapid development of ciprofloxacin
2 1 4
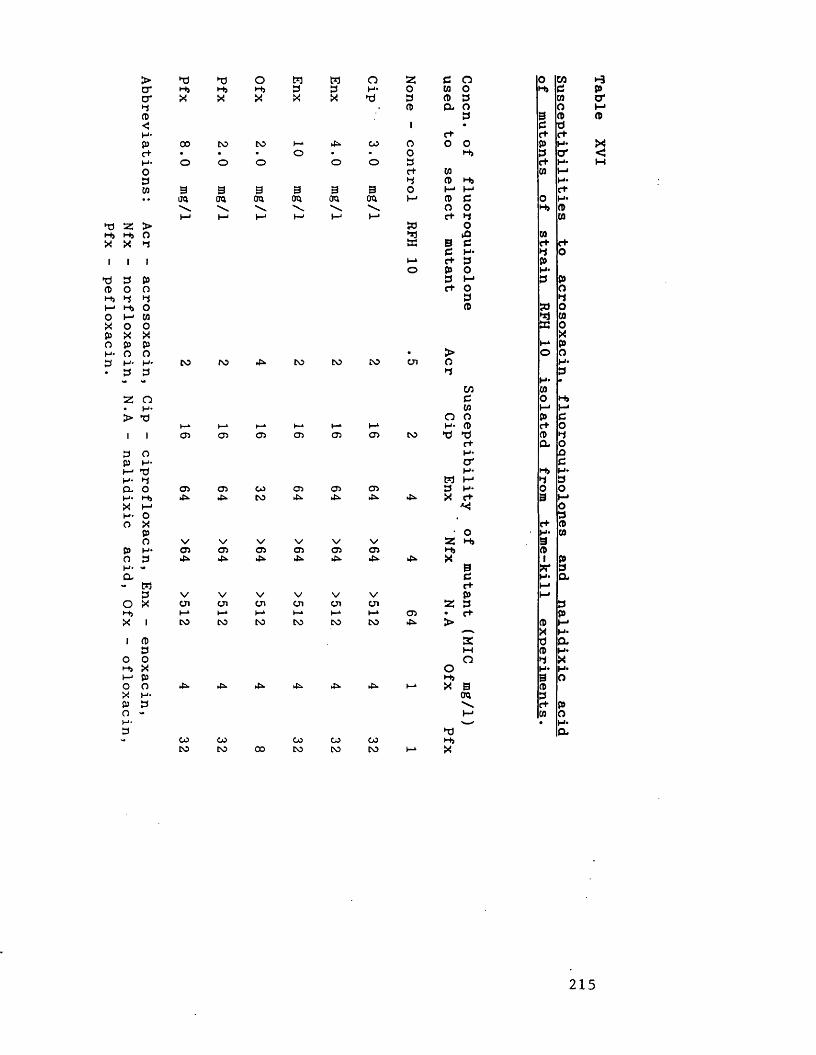
Abbreviations: Acr
- acrosoxacin,
Cip - ciprofloxacin,
Enx -
enoxacin,Nfx
- norfloxacin,
N.A - nalidixic
acid, Ofx
- ofloxacin,
Pfx - pefloxacin.
p p o H W O s: 3 o 0 U iHj Hj Hj 3 3 H 4 0 to o Hj 3X X X X X P 3 CD 3 tocd a o O3 9 CDi • 3 Pc+ t+ c+00 to CO I-4 4. OO o O 0 P H-. • • o • • o Hj 3 tro o o O o 3 c+ H4rt" to to j—•3 CD Hj H*
9 9 9 9 9 9 O M h-< ctTO TO TO TO TO TO t—' CD 3 0 H*\ \ \ \ N N O O Hj CDt—1 K—1 t—' l—1 I—1 c+ 3 COW OP 43 toffi 9 3 c+ C+3 H4 3 Oi—4 c+ 3 Po P O H43 I-* 3 Pc+ o o3 3CD » OCO53 0XK4 p• > O oto ro 4 to CO CO 071 a H43 3H4 *Ui CO3 o Hjto M MO o P 3h-4 i—4 b-L I—4 I—4 H 4 a> c+ O
05 05 05 05 05 05 CO P P ct> 3c+ a OH 4 Qcr 3H4 Hj H*t*3 I-* 3 3
05 05 OO 05 05 05 3 H4 O O4* 4. CO 4*. 4. 4*. 4. X C+ 9 MO3c+ CDO H4 toN/ o V \/ V V ss Hj 905 05 05 05 05 05 Hj cd4* 4* 4*. 4*. 4- 45. 4». X i P9 X* 33 H4 at+ MV V V V V V P Mon CJ\ 071 Ol on 071 3 3i—1 I—4 I—4 i—4 I-4 05 • t+ pto to CO CO CO CO 4. > CD H-'—. X H-K d aM CD H4O 3 Xo H4 H4H> 9 o4 4*. 4* 4. 4*. 4=. I—4 X 9 CDTO 3S e+ pCO o>w* • H*p aOO OO OO oo 00 HjCO CO 00 CO to CO l—4 X
•-3Pa*
X<
2 1 5

resistance in MRSA following use of ciprofloxacin
to treat MRSA septicaemia. Initially, this isolate
had a ciprofloxacin MIC of 0.5 mg/l which
increased to 4.0 mg/l, and finally 16 mg/l, before
the patient died. The serum ciprofloxacin level 2
hours after a 100 mg intravenous dose was only
0.7 mg/l. This dosage appeared inadequate, and it
may be appropriate to monitor ciprofloxacin levels
in blood to ensure that satisfactory concentrations
are maintained when dealing with staphylococcal
infections.
i i i . Are the reports of high incidences of
fluoroquinolone resistance in hospitals due to the
propensity of MGRSA to develop fluoroquinolone
resistance or are they due to epidemic spread of
resistant strains?
According to Shalit et a l . (1989) during a
widespread epidemic of ciprofloxacin-resistant MRSA
in a general hospital in no instance was a
quinolone-resistant organism isolated from a patient
following fluoroquinolone treatment. Raviglione et
a l . (1990) found exactly the opposite: ciprofloxacin-
resistant MRSA being isolated only from patients
in whom there had been prior use of
ciprofloxacin. Daum et a l . (1990) and Smith et a l .
(1990) found initial cases of colonization or
2 1 6

infection with ciprofloxacin-resistant MRSA followed
use of ciprofloxacin. Subsequent cases were not
related to ciprofloxacin therapy rather hospital
transmission of existing strains. We have made our
own assesment of whether the problem of
ciprofloxacin resistance is primarily one of cross
infection or is due to development of resistance
in separate strains.
We have examined ciprofloxacin-sensitive and
ciprofloxacin-resistant MGRSA from one of the first
reported outbreaks of ciprofloxacin-resistant MRSA
(Isaacs et a l . , 1988) which occurred in a Texas
hospital. Ciprofloxacin-resistant MGRSA from two
centres in Israel which have been suggested to be
cross-infecting strains have also been studied.
The results of phage-typing are shown in Table
XVII. Many of the strains from Texas gave a
strong reaction with phage 75 at 100 x R T D ,
although at RTD this reaction was weak or non
existent. All the ciprofloxacin-sensitive MGRSA from
Texas showed the same phage-type, however a number
of different patterns were observed for the
ciprofloxacin-resistant strains. Strains TEX 2, TEX 10
and TEX 16 reacted with a variety of phages from
lytic groups I, II and III of the International
Set at RTD, although slight differences in patterns
were observed between these strains. Four strains
from Israel (IS 1, IS 2, IS 3, IS 4) were non-typable
with the International Set even at RTD x 100.
2 1 7

However, 3 of these 4 did react with supplementary
or Nuan phages (Richardson et a l . , 1988) . The
remaining 4 Israeli strains showed different typing
patterns.
In order to type non phage-typable strains, and
possibly differentiate strains of the same phage
type, plasmid analysis, antibiotic susceptibility
typing and biotyping was performed on the strains.
The results of antibiotic susceptibility typing, and
biotyping are shown in Table XVIII. The non phage-
typable Texas strains (ie. strains 13 and 20) were
different from each other in terms of
ciprofloxacin MIC, plasmid content, antibiotic
susceptibility profile and biotype. A 36 md
plasmid was found in all Texas strains, however in
strain 13, a 2.5 md plasmid was also present.
Strain 13 was resistant to tetracycline and also
rifampicin. The ciprofloxacin-resistant, phage-type 75
strains (i.e 1,3,7,11,17 and 5 and 14) could be
split into 3 groups on the basis of antibiotic
susceptibility profiles and biotyping. Strains
1,7,11 and 17 had similar ciprofloxacin MICs (16 or
32 mg/l) and produced sheep blood haemolysis but
not egg yolk lipase. Strain 3 had a similar
biotype to strains 1,7,11 and 17, however a
ciprofloxacin MIC of only 4.0 mg/l was found.
Strains 5 and 14 differed from 1,7,11 and 17 because
they did not produce sheep blood haemolysis.
Strains 2, 10 and 16 were widely susceptible to
2 1 8

ciprofloxacin sensitive
strains
ooprtCL
•—3 <—3 1—3 W H H X X X00 O' -X
H H H H H H H H H H H H H ooM M M M M M M M W M M M M r tX! X X X X X X X X X X X X P
PH-P
20 17 16 15 14 13 11 10 X On 00 to h-»
Ui Ln Ln as x t-3 Ln
tovOLntoLnto>xVO00o
VOLnLn£-XLn00OO>00Ln
as as asH H H H *-3
as as as •-3 H H
as as•-3 H
Ln-f>- X x. Ln X Ln
to vOLn toLn to >00 o53 X X. X X X H Ln vO Ln Ln Ln LnOvtoW-O-xLn-P"XLn00
x to Ln vO00 Ln 00 to > \ \ Ln 00 to I—* J>vO — I Q\ vO
0000ovD00to
vOas Si as 25 00 •—3 <—3 •—3 1-3 to
25 525 SS H H H
Ovi—1X Ov\to2;siZ C^Ln H H H to Ln
Ovas Si as H H H
x x 00 on OvOLnOv-P'tom£-xLn
0000>0000ovO00to
ovOVOV
avtoooovtoV0
asH
prtP►1pprtH*OPP
00prt
PCHa
oo
PP* asP p0Q PP PCO
p hd Hp P* Pp- P O*
JO HP P
oH*P rt M•1 ■< <O p Hl-h P HI-* oiOX
P oO hhH*
C/0 O p*c Pp Pp rt W
p h-> •-J Pp* p P PP 3 cn W
00 P H*p P rtco rt H* H*
P P <*1
M
P
W 2hj Op wp C/0h-1>
oH*P►tOHih-*OXp0H-P1PPwH*WrtPPr t
2 1 9

ciprofloxacin sensitive
strains.
i—i i—i i—ii—i i—i i—ii—ii—iW W W W W CO cn CO00 Nl a\ Ui CO tO
H H H H i-3 H H COM M M t*J M M W rtX X X X X X ►1
Pto to to to 1—1 1—1CD H--P' CO to I—100 to * P* * * # * *
oo o o c n - o s : U c n 4>> -v l H > \
00£-
—I —I ~-J ''JCn Cn Cn Cn Ui Cn Cn
00COOvOCOto
CD CD CD 25 CO CO COH M to W
CDw 2 : z to H H
« « 25 a; s 25 25 H H H *-E H H H
Q\ O' ON
MPr tp
p05rtH-OP05
COPr t
!5dH
OO
»—*I—1I—1 P-O O' 00 O' Ch O' P* 2S\ \ \ Z co to 2 ; to 25 « 2 ! « 2* 25 P c0 0 O' H O O H O H H t-3 H H H H 00 pCO to to p p0 co 0 co
p TJ 1-3p P* Pp. P O*
K> MP P0
H-P rt M•t ■< <O P Hhh P HI-* 05OX OP O OO l-h PH- rtP P.
CO O p.p Pp Pp rt 05
p t—1 •-» Pp* p P PP B CO CO00 P H-p p rtW rt H- H-
P P <•-t P
Hco 3•-» OP pap C/5
>•
to
oH-p•tol-hI-1OX050 H* P1 ►1 0) CO H* CO r t P P r t
220
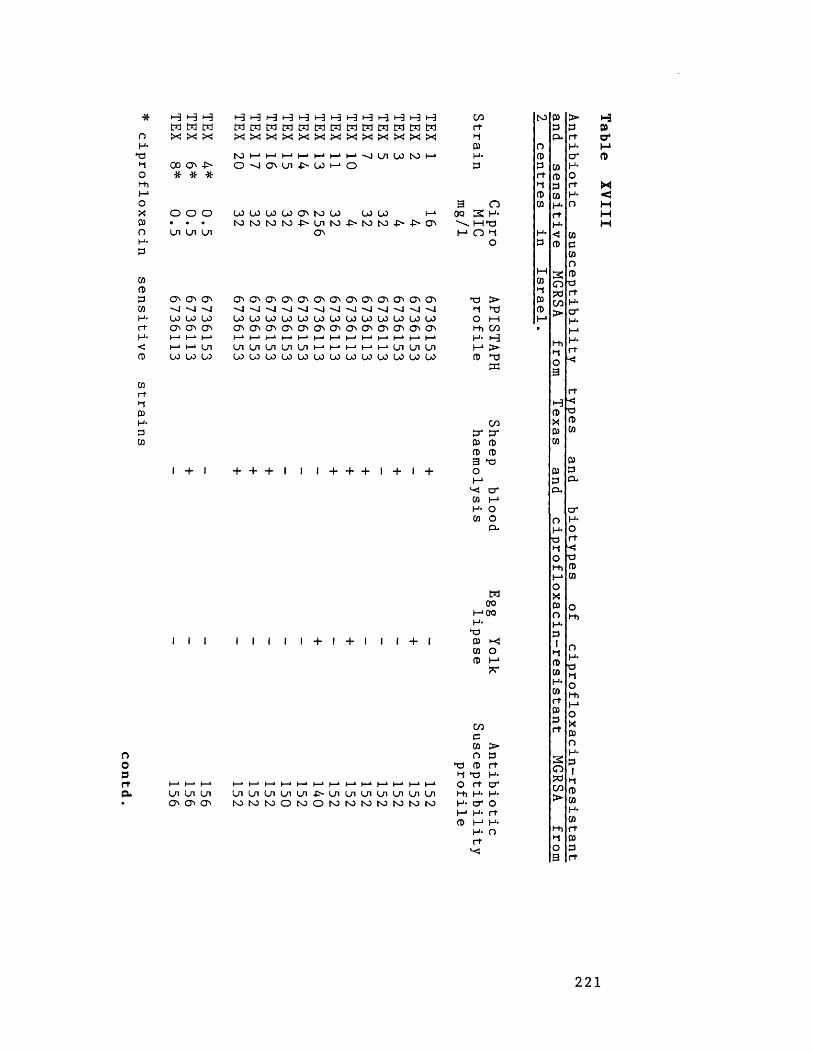
ciprofloxacin sensitive
strains
oopr t
H H H H t-3 H H •-3 H H H H H 1-3 H H cn toCd cd Cd Cd M tn Cd Cd Cd Cd Cd Cd Cd Cd Cd Cd rtX X X X X X X X X X X X X X X X p
(33 oto i—* i—* I—* I—1 t—1 1—»1—1X Ui 0 0 to 1—* H* CD
oo ON -p> o X ON On -O 00 h—1o p P* * * rt
•dCD
3 n CDo o o oo 00 0O 00 ON to oo oo 00 I—* 00 S H-• • • to to to to -P~ On to -P~ to to -P> ON M tPLn Ln L n ON M n ■d H*
o P
M03
ON O n ON ON ON ON ON ON O n ON ON ON ON ON ON ON np > 03X X X X X X x x x X x x X x X X •d TJ CDOO oo 00 oo 00 00 00 00 00 00 00 00 00 00 00 03 O M I-1ON ON ON ON ON ON ON O n O n ON ON ON ON ON ON ON l-h cn •I—* 1—* t—1 h—1 I—1 ‘ 1—* t—1 I—* t—»h-‘ I—* I—* t—* (—* H - >-3I—1 I—1 On On On On On On 1—* I—* 1—* I—* h-» On on On I-1>LO oo 00 00 oo oo 00 00 oo 00 oo 00 00 00 00 00 CD TJ
EC
I + I
I i I
Ln Cn Ln O'* O n O'*
+ + + I I I + + + I + I +
l l l l l + l + l l l + l
cnET ET03 CDCD CD3 ipOI-1X crCD H-1H- OCD O
D-
Cd OO I—* OO H- npP Xw oCO t-»
LnLnLnLnLnf'LnLnLnLnLnLnLnt O M t O O t O O t O r O f O W t O M M
cndCO >o P
tp CD rt■d hp H*O rt crl-h H* H-H- cr oh-1H- rtCD I-1H*
H- ortX
01dCOO(0■ortH*crH-I—1H-rtX
03Peu
crH-or+X■aCDCD
Ol-h
OH-■a•dol-hI-10 X 03 O H- P1•d0303H*CD
rt03pft
i-3Pcr
x<HM
22 1

ciprofloxacin sensitive
strains
* 1—1h-1M M M M M l-l i-3 H H t-3 H H H c/o N3CO CO CO CO CO CO CO CO M W M M W W M rt
X! X! X X X X X ■1oo X O' Ln -P" L0 NO i—1 3 O
NO NO NO NO i—1I—* vO H* 3-P~ LO NO 1—1oo NO * 3 3
* * * * ■* rtt3I—1 I—1 3 n CO
O' N3 O' O' h-> h-» NO t—1 O O O O 000 oo 3 H--P- 00 -O' -O- O' O' oo O' • • • • • • • HtJLn Ln Ln Ln Ln Ln Ln I-1O h» H*o 3
M03t
O' O' O' O' O' O' O' O' O' O' O' O' O' O' O' to > 3X X X X X X -0 X X X X X X X X to 3LO LO LO LO LO LO LO L0 L0 LO L0 L0 L0 L0 L0 O M (-*O' O' O' O' O' O' O' O' O' O' O' O' O' O' O' hh co •f—1i—1i—1t—1I—1 (—* I—*I—1 (—*i—»I—*i—*i—*I—1t—* H» 1-3Ln Ln Ln Ln Ln Ln Ln Ln Ln Ln Ln Ln Ln Ln Ln I-1>03 LO L0 LO L0 L0 L0 LO L0 LO L0 L0 L0 L0 L0 3 to
i i i i i i i i i i i i i i
+ + + + + + i + i i i i i i i
COO' cr3 33 33 too1-*
X o '3 h-»H * O3 O
3 *
M OO I-100 H-to03 X CO O 3 I—1
O M t o O O M M O M M
o o oho NJ W
L n L n i n L n L n U i U i O ' O ' O ' O ' O ' O ' O '
CO33 >O 3
tO 3 r tt to H-o r t O 'hh H- H*H* O ' Oh-1H- r t3 h-1H -
H -r t
X
O
CO3COoCDTOr tH-O'H-I-*H -r tHi
033
OHi
oH-OhjOh hH-*OX030 H- 31 ►1 CD (0 H- CO r t 3 3 r t
i-3CDo'
X<MMHOO3rta.
222

a variety of phages, and had different biotypes.
The ciprofloxacin- sensitive MGRSA from Texas
showed much less variation in typing properties
compared to the ciprofloxacin-resistant strains.
They were negative for egg-yolk lipase, had a 36
md plasmid, and possessed the same antibiotic
sensitivities. Strains 6 and 18 differed from the
other ciprofloxacin sensitive strains because they
produced haemolysis on sheep blood agar.
All the strains from Israel had identical
biotypes except for the non phage-typable strain
IS 2 which was the only Israeli strain to produce
a negative egg yolk lipase reaction. Despite
identical biotypes and plasmid-types (a cryptic
plasmid was found in all strains) strains 6 and 7
differed from the others in terms of antibiotic
sensitivity profiles. Combining our phage typing,
biotyping and antibiotic sensitivity profile data
of the Israeli strains, no two strains were found
to be identical.
The results of these studies suggest that the
problem of ciprofloxacin resistance in the
hospitals we have studied is due more to the
development of resistance in separate strains
rather than cross-infection due to a single strain.
Daum et a l . (1990) have shown that ciprofloxacin-
resistant MGRSA causing outbreaks of hospital
infection in the USA can be of a number of
different phage types. Hadorn et a l . (1990) have
223

used ribotyping (probing of DNA coding for
ribosomal RNA) to confirm that their ciprofloxacin-
resistant Israeli strains are all the same. The
results for our Israeli strains suggest that
independent evolutionary events have occurred. Some
workers question the discriminatory power of
ribotyping (Judith Richardson, PHLS Colindale, personal
communication), and our typing methods may
differentiate strains of the same ribotype.
In our typing studies we have found that
methods such as biotyping, phage typing with the
International Set (even at RTD x 100), determination
of antibiotic susceptibility profiles or plasmid-
typing, used on their own sometimes fail to
provide sufficient strain differentiation. However,
when a variety of methods are combined the
prospects of strain differentiation are much
improved. Thus, we agree with Cookson et a l .
(1986) in that a combination of strategies is
required to distinguish between strains so as to
confirm known or suspected routes of acquisition.
Finally, exposure to fluoroquinolones and/or
mutation to fluoroquinolone resistance has been
reported to alter the phenotypes of strains.
Crumplin (1990) has suggested that mutation to
fluoroquinolone resistance can result in pleiotropic
effects. This is because DNA gyrase has a
significant role in sustaining normal bacterial
function, any alteration in DNA gyrase activity
224

would disrupt "normal" function. We have found
similar physiological profiles (in terms of APISTAPH
profile) for ciprofloxacin- sensitive and
ciprofloxacin-resistant MGRSA. Smith (1990) has
reported loss of coagulase activity in S. aureus
following mutation to resistance in v i t r o , however
both our in vitro derived mutants, and clinically
resistant strains produced coagulase. Should loss
of coagulase production occur in the clinical
situation, S. aureus may be misidentif ied as S .
epidermidis. We have also found that the phage-
types of laboratory derived fluoroquinolone-resistant
mutants were no different from those of parent
strains .
3.3 New fluoroquinolones with improved activity
against M G R S A .
Since the introduction of ciprofloxacin, and
other related fluoroquinolones in the mid-1980s,
considerable effort has been expended by many
pharmaceutical companies in developing new
fluoroquinolones. Initially, many of these new
compounds, such as fleroxacin (Leigh et a l ., 1988),
possessed only improved pharmacokinetics compared to
ciprofloxacin.
Recently, newer compounds have been developed
with much improved antistaphylococcal activity eg.
tosufloxacin (Barry & Jones, 1989) and sparfloxacin
2 2 5

(Kojima et a l . , 1989). The activities of these
agents against ciprofloxacin-sensitive and
ciprofloxacin-resistant MGRSA are shown in Table
XIX. Compared to ciprofloxacin, sparfloxacin and
tosufloxacin showed approximately a ten-fold
improvement in activity against ciprofloxacin-
sensitive MGRSA. Against ciprofloxacin-resistant
MGRSA, sparfloxacin and tosufloxacin were active
against strains with low-level (MIC 2-8 mg/1)
resistance, but not against those showing higher
levels of resistance. Our results for sparfloxacin,
obtained against isolates of ciprofloxacin-resistant
MGRSA are similar to those reported by Chaudhry
et a l . (1990). Tosufloxacin only attains peak serum
levels of c. 0.25 mg/1 (personal communication,
Abbott Laboratories, Illinois). Hence, despite its
excellent in vitro activity tosufloxacin has a
similar therapeutic ratio to ciprofloxacin. According
to Montay et a l . (1990), sparfloxacin attains peak
serum levels of 1.0-2.0 mg/1 depending on the
dose given. Thus, sparfloxacin may be more active
in vivo than ciprofloxacin or ofloxacin. Another
new fluoroquinolone with excellent in vitro (MIC^q
0.004 mg/1) activity against MGRSA is WIN 57273
(Kaatz & Seo. 1990b), however, we have been unable to
obtain supplies of this compound for laboratory
testing .
In the future there may be new fluoroquinolones
available with greatly enhanced antistaphylococcal
226
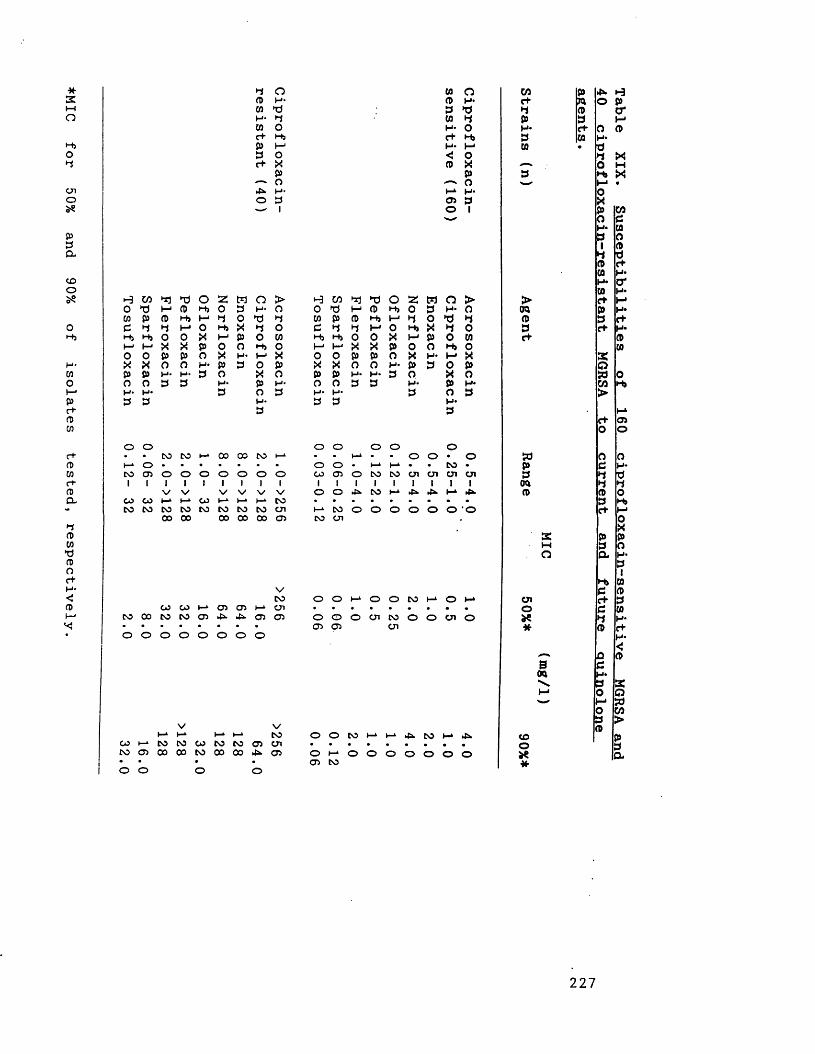
♦MIC for
50% and
90% of
isolates tested,
respectively.
4*O
OH*
3oP -OXp0H'31
w o(!) P *3 >d
H U) 33 O Z M O >0 d P ft H> 0 3 p . oCO p f t H» I - 1 3 O *d 33 3 3 P 0 H j X 3 oH> H i O 0 X p P o wP p X X p 0 o H j oo O p p o X P* t—1 XX X o o P * P 3 o pp P H* H- 3 o X ao o 3 3 P * p p -P» P * 3 o 33 3 p.
H W 3jo 1j Hw p ft 3 3 3 Hj H) O I—1 h-1 X O O P X X o P P H- O O PH- H>3 3
*0 0 2 CD h, o H i H 4 M O H>O X P x p oP O X O H- p H* 3 O 3 N- 3
0 o P oCO 051 Itd O) H H
r+ h > p- I—1 < O CD X P OP H- 05 3 O I
w o >3 H- Oo >d 3X 4 o P O P O Hi o P- P X 3 O p X o P H* o 3P*3
o o o o oCO CO 1—* 00 00 CO p • • p • • o o • o• • • • • • • o o • p p • • CO •01 01 01 01 01 01 01 co1 051 0
1CO1 CO1 051 cn1 Ol1 Ol11
V1V
1 1V
1V
1V 1V 1o 1o 145. 1CO
1p
14*. 145. 1p 145.
p 1—1 CO I—1 p. p CO • • . • • • • . •CO CO CO CO CO CO cn p CO o o o o o o • O00 00 00 00 00 05 CO Ol
wc+►1PH*3CO
>Od<D3e+
Sdp3Odo>
nft3c+w oH*d3oH>POXp0H*313ftCOH*COc+p3c+
a:Q»wctOoc33ft3c+
2Mop3a
VCOO) U M O) ® M Ol
CO 00 CO CO 05 45 45 05 05
o o o o o o o o
O O P O O C O P C 5 Po o o c n c o o o o i o05 05 ot
Oloae*
3c+33ft
sOd
V Vp p p p CO
co p CO CO CO CO CO 05 OlCO 05 00 00 CO 00 00 45. 05• • • •o o o o
O O C O P P 4 5 C O P 4 5 • • • • • • • • •O P O O O O O O O 05 CO
COo*
2 2 7
Table XIX.
Susceptibilities of
160 ciprofloxacin-sensitive
MGRSA and

activities compared to ciprofloxacin and ofloxacin.
If these new agents can attain satisfactory serum
levels so that their increased activity is
reproduced in vivo (i.e they have an improved
therapeutic ratio) their use may result in reduced
development of fluoroquinolone resistance. This is
because, as we have shown, if fluoroquinolone
concentrations are high enough sensitive strains
cannot mutate to resistance. Additionally, strains
with decreased fluoroquinolone susceptibilities (eg.
RFH 10) may prove susceptible.
228

4.0 GENERAL DISCUSSION AND CONCLUSIONS
4.1 "Ba r b e r ’s Law - the spread of resistant
staphylococci can be controlled either by not
giving antibiotics or by preventing the
transmission of the resistant organisms between
persons". Parker et a l ., 1974.
The antibiotic susceptibility profiles (Table IV)
of 100 strains of MGRSA from 32 centres in 23
countries have been determined. This is the first
survey to document the international problem of
multiple antibiotic resistance in MGRSA. Many
differing susceptibility profiles were found, some
strains being sensitive to a range of currently
available antistaphylococcal agents, others resistant
to many of these agents.
During the late 1960s and continuing into the
1970s multiple antibiotic resistance in
Staphylococcus aureus declined (Parker et a l . , 1974).
Initially, it was thought that a decrease in
tetracycline usage (although not an overall
reduction in use of antibiotics) was responsible
for the decline in staphylococcal multiple
antibiotic resistance. This explanation was rejected
by Ayliffe and his colleagues (Ayliffe et a l . ,
1979) who also found that use of one antibiotic
(in their case- erythromycin) selected an outbreak
strain resistant to tetracycline, erythromycin and
229

novobiocin. While rigid antibiotic policies
significantly reduced multiple antibiotic resistance
in S . au r e u s , Ridley ^t al. (1970) reported
isolations of MRSA to increase. It would appear
that the spread of MRSA is not directly
influenced by antibiotic usage, which sharply
contrasts with the role of penicillin in promoting
the spread of penicillin-resistant staphylococci.
The reasons why methicillin and gentamicin
resistant S. aureus so rapidly appeared worldwide
following the initial discovery of these organisms
during the mid-1970s are not clear. Only the
phage type 80/81 S . aureus of the 1950s has shown
a similar capacity for rapid, worldwide spread
(Shanson, 1981). More than 50% of our MGRSA were
non phage-typable with the "International Set" of
phages even at RTD x 100. 23 of the 42 typable
strains were lysed by phage 85 alone, or phage 85
and other Group III phages as well (Table I). The
remaining typable strains had mixed Group I + III
lysis pat terns.
The results of typing with supplementary
phages are shown in Table XX. Four reserve, non
specific phages (88A, 90, 83C and 932) were used
together with a new set of experimental ("Nuan")
phages (616, 617, 618, 620, 622, 623, 625, 626, 629 and
630) developed by Richardson e_t al. (1988).
According to these workers all of the "Nuan" phages
were lytic group-III related except phage 625
230

Table XX.Supplementary phage-types of MGRSA used in antibiotic resistance studies.
Country Strain SupplementaryCode phage-type
"Nuan"phage-type
AUSTRIA
AUSTRALIA
BELGIUM
BRAZIL
CHILE
ENGLAND
V 8V 15
AS 1 AS 2
AS 3
AS 6 AS 10
BL 1 BL 3
BL 4
BZ 1 BZ 2 BZ 4 BZ 1 2 BZ15 BZ16 BZ17 BZ18
CH 3 CH 4
C 1 UK16 UK1 7
F.R.G.
RFH RFH RFH RFH RFH RFH10 RFH11
G 1 G 2 G 3
NTNT
NT88A/932
90/932
NTNT
90/932 88A/90/83C/932
90/932
NTNTNT
90/932932NT
932NT
90/932NT
932NTNT932932
88A/93288A/932
93288A/90/932
83C
932932932
NTNT
618/630616/617/620/622/626/630
616/617/618/620/622/625/626/630
NTNT
618/620/623616/617/618/620/622/623617/620/622/623/629/630
NTNT
629 618/629
NTNT
617/618/620/622/630618
630 NT
617/620/622/626NT
625/629617/620/622/626617/620/622/626616/617/622/626
617/620/622/626/630616/617/622/630
616/617/620/622/626/630618
617/618/620/622/626618/620618/620
C o n t d .
231

Table XX. contd .
Supplementary phage-types of MGRSA used in antibiotic resistance studies.
Country Strain Supplementary "Nuan"Code phage-type phage-type
F.R.G. G 4 932 617/618/620/622/626/630contd . G 5 NT 618/620
G 6 NT 618/620
FRANCE F 1 NT 623F 2 NT NTF 3 NT 623F 4 NT 623F 5 NT NTF 11 NT NTF 1 2 90/932 NTF 13 NT NTF14 NT NTF25 932 NT
GDR EG 6 NT NTEG 1 2 NT NT
GREECE AT 1 NT 620AT 3 NT NT
HONG KONG HK 1 90/932 617/618/622/629HK 2 90/932 617/618/622/626HK 3 90/932 617/618/622
ITALY IT 6 NT 629IT 7 NT NTIT 8 NT NTIT11 932 618/620/623/625/629IT1 2 88A/90 NT
JAPAN JP 1 NT NTJP 2 NT NT
KUWAIT KW 4 NT NTKW 5 NT 616/617/618/620/622/630KW 7 NT 617/618/620/622/626
POLAND PL 1 88A/932 617/620/622/626PL 2 88A 623/625PL 3 88A/932 616/617/620/622/626/630PL 4 88A/932 617/620/622/626
C o n t d .
232

Table XX. contd.Supplementary phage- types of MGRSA used in antibioticresistance stud ies.
Country Strain Supplementary "Nuan"Code phage-type phage-type
PORTUGAL P 1 NT 616/617/622/623P 2 NT NTP 3 NT NT
REPUBLIC IR 1 NT 617/622OF IR 2 NT 617/622IRELAND IR 4 NT 617/620/622/626
IR14 NT 617/620/622/626IR 15 NT 617/620/622/626IR 17 NT NTIR18 NT 617/620/622
SOUTH SA 1 932 NTAFRICA SA 2 932 618
SA 4 NT 618/629SPAIN SP 1 88A/932 NT
SP 2 NT NTSP 3 90/932 NT
SWITZERLAND SW 1 NT NTSW 2 90/932 617/622/623/626
TURKEY T 6 NT 625T 7 NT 617/620/626T 9 NT NTT10 NT 625
USA US 7 NT NTUS 12 88A/83C/932 616/617/618/620/622/
623/625/626/630US 13 88A/83C/932 616/617/618/620/622/
623/625/626/630RM 1 NT 618/623/629RM 2 NT 623/629RM 3 NT 618/623/629RM 4 NT 617/620/622/623RM 5 NT 617/620/622/623SC10 NT NTSC11 83C 623/629
U.S.S.R RS 2 NT 616/617/620/622/625/626/630
233

which was Group-I related. Many of the previously
non-typable MGRSA could be typed using these
phages, and by inference many of these strains
would appear to be Group Ill-related.
A questionnaire survey conducted in the UK and
Ireland between 1982 and 1983 established a high
incidence of MGRSA in hospitals in South-East
England (Cooke et.______ a_l. , 1986). Subsequent
bacteriological examination of these strains by
Marples et a l . (1986) showed that a single epidemic
isolate, EMRSA-1 was primarily responsible for the
high incidence seen. A very similar strain was
also causing an epidemic of infections in Eastern
Australian hospitals (Townsend et a l . , 1987). The
properties of EMRSA-1 have been studied in depth
by Cookson and Phillips (1988) who stated EMRSA-
1 to have chromosomally-mediated resistance to
tetracycline, minocycline, erythromycin (MIC greater
than 1000 mg/1), clindamycin (MIC 250 mg/1) and
streptomycin (MIC 60 mg/1). EMRSA-1 is usually
sensitive to fusidic acid, neomycin and rifampicin.
Aminoglycoside resistance (due to production of APH
(2M )/AAC(6’) is often encoded on a 15-23 Md plasmid
commonly found in EMRSA-1.
Richardson et al (1988) have shown strains of
EMRSA-1 to have characteristic phage-types. Some
strains react with phage 85 (usually weakly). These
Mtype-85 strains have a Nuan phage profile of
616/617/622/626/630, and may react with phages
234

88A/932. Other EMRSA may react with phages 84 and
85, sometimes with the supplementary phages 88A/932,
and these 84/85 strains show a wider pattern of
Nuan phage lysis ie. 616/617/618/620/622/626/630.
Unfortunately, Richardson and her colleagues (1988)
also observed that similar profiles with the
"Nuan" phages could be obtained in apparently
unrelated strains. The variety of phage-types
(Tables I and XX) and antibiotic resistance
patterns (Table IV) found in the strains we studied
suggests that the spread of MGRSA is probably not
due to the widespread dissemination of a single
clone .
We have found that 44% of MGRSA produce the
bifunctional aminoglycoside-modifying enzyme APH
(2")/AAC ( 6 1), while 56% produce this enzyme along
with APH(3')-IV as well. These mechanisms were
inferred from the results of disc sensitivity test
data to six aminoglycoside antibiotics (Van de
Klundert et a l ., 1984). Attempts were also made to
detect the genes responsible for aminoglycoside
resistance using specific probes currently under
development by Dr G. Miller of Schering-Plough
Research, Bloomfield, New Jersey, USA. These studies
are still under way, however preliminary results
show agreement with the mechanisms inferred using
Van de Klun dert’s method. Ounissi et a l . (1990)
reported virtually a 100% correlation between the
presence of enzymes APH (3f) and APH (2")/AAC (6*) in
235

S. aureus as deduced from susceptibility data toaminoglycoside agents and the detection of these
enzymes by specific probes. For streptomycin the
correlation is not so good, as streptomycin
resistance may be due to a number of enzymes or
ribosomal mutation.
In our strains the presence of APH (2” )/AAC (6’)
positively correlated with sensitivity to neomycin
and resistance to gentamicin (sisomicin), kanamycin
and tobramycin. The presence of APH ( 3 f) and APH
(2")/AAC ( 6 ’) positively correlated with resistance
to gentamicin (sisomicin), kanamycin and neomycin.
The finding that the same enzyme - APH (2")/AAC ( 6 ’)
was responsible for gentamicin, kanamycin and
tobramycin resistance in MGRSA worldwide suggests
that the genes for this resistance have
disseminated worldwide. This might at first seem
unlikely because gentamicin resistance genes have
been found to be chr omosomally located in some
strains (Storrs et a l . , 1988), while in others they
may be carried on completely different plasmids.
For example, gentamicin resistance in Australian
strains is commonly carried on non-conjugative Inc 1
plasmids, whereas in strains from the USA the
resistance determinant is usually found on large
conjugative plasmids (Lyon & Skurray, 1987). These
differences have been explained by the
identification of a transposon (Tn 4 0 0 1 ) carrying
the genetic determinants of gentamicin resistance
236

in the aforementioned Australian and U.S. strains
(Byrne et a l ., 1990).
Our results, together with the genetic and
molecular findings of Byrne et a l . (1990) suggest
that gentamicin resistance in MRSA has been
produced by the worldwide dissemination of a
transposon (Tn 4001) into MRSA, and not the spread
of a resistant clone. The factors responsible for
such widespread dissemination are not clear. Thus,
B a r b e r ’s law does not seem to apply to the spread
of gentamicin, kanamycin and tobramycin resistance
in MRSA.
Parker et a l . (1974) believed that multiple
antibiotic-resistant hospital strains of S. aureus
declined during the 1960s because of reduced
transmission by improved infection control measures,
rather than decreased antibiotic usage. Such
measures may have been effective against the
multi-resistant S . au r e u s , but they were not
effective against MRSA, which increased in numbers
as the 1960s progressed (Parker & Hewitt, 1970). With
this in mind we totally agree with the following
statement made by Parker and his colleagues (Parker
et a l . , 1974).
’’The days are past when the microbiologistcould attribute resistance problems solely to the misuse of antibiotics by clinicians, and then retire to his laboratory. We must accept that a great deal of antibiotic treatment will be given in hospitals, and see that this is done with as little ill effect as possible. The first element in our policy must be to have an efficient infection control organisation. In planning the use of individual agents, it is insufficient to ensure
237

that an effective agent is used for the treatment of all clinical infections, because this ignores the action of the agent on organisms at carrier sites; it is the total exposure of the hospital population to the agent that matters. The use of antibiotics in combination has so often failed to prevent the accumulation of resistant strains,notably in staphylococci with neomycin and bacitracin, and more recently with cotrimoxazole, that it cannot be relied upon in the future. On the other hand, we have several effective and unrelated agents for the treatment of staphylococcal infection, and resistance to these is at present infrequent or irregular in distribution. This situation offers some hope that an intelligent and flexible policy of diversification in the use of antibiotics might be a useful means of slowing up the rate at which resistances accumulate."
In this study, we have shown multiple
antibiotic resistance to be a particular problem
in MGRSA, for example more than 60% of strains
examined were resistant to 10 or more antibiotics.
With this in mind do we still (like Parker e_t
a l . , 1974) have "several effective and unrelated
agents" available to treat infections due to MGRSA?
238

4.2 Antibiotic options for the treatment of MRSAinfections and/or colonisation.
i . Options available for treatment of systemic
infections.
Table V shows that over 50% of MGRSA tested
were resistant to tobramycin, netilmicin, amikacin,
neomycin, streptomycin, tetracycline, minocycline,
erythromycin, clindamycin (including inducible
resistant strains) and trimethoprim. 41% of strains
were resistant to chloramphenicol, 28% were
resistant to rifampicin, 15% were resistant to
fusidic acid, and one strain was resistant to
novobiocin. No resistance was found to
nitrofurantoin, pristinamycin, teicoplanin and
vancomycin. Use of chloramphenicol often results in
rapid emergence of resistance, and this agent is
nowadays considered as sub-optimal therapy for
treatment of staphylococcal infections. Although
little resistance was found to novobiocin, its use
has often been associated with the rapid
development of resistance. Novobiocin has been
shown to be hepatotoxic, and its manufacturers
(Upjohn) seem most unwilling to encourage its use.
Currently, fusidic acid and rifampicin are
regarded as acceptable alternative agents to use
in place of vancomycin. From our in vitro studies
these agents are highly active against MGRSA.
239

However, because of the propensity of MGRSA todevelop resistance following exposure to either of
these agents, their use in appropriate combination
is recommended. 28% of MGRSA were resistant to
rifampicin, and for many of these strains the only
available recognised alternative chemotherapy is
vancomyc i n .
Because of the current lack of resistance to
vancomycin, and its proven efficacy in the
treatment of MRSA infections, vancomycin is the
first choice antibiotic to use against MGRSA.
Clinically, vancomycin use does have its problems,
because of its ototoxicity and nephrotoxicity
levels have to be monitored, and it has to be
slowly infused so as to avoid thrombophlebitis.
However, of greater concern to us is the
increasing reliance placed on vancomycin for
treating many Gram-positive infections. The resulting
greater bacterial exposure to vancomycin in the
hospital environment increases the likelihood of
development of resistance. Already increasing
problems with vancomycin resistance are being
encountered. There have recently been reports not
only of outbreaks of infection with enterococci
possessing plasmid-mediated vancomycin resistance, but
also of the isolation of vancomycin-resistant
coagulase-negative staphylococci (Johnson et a l . 1990) .
If P a r k e r ’s predictions are correct it only seems
to be a matter of time before vancomycin
240

resistance occurs in MRSA. With this in mind wehave attempted to identify agents which might be
used in place of vancomycin.
Fosfomycin, nitrofurantoin and pristinamycin had
good activity against MGRSA. Resistance to
fosfomycin was found in 21% of MGRSA studied,
however no resistance was found to nitrofurantoin
and pris tinamycin. Nitrofurantoin use is limited to
urinary tract infections, although the related
nitrofuran- furazolidone could be used to treat
MGRSA bowel carriage or enterocolitis. Intravenous
sodium nitrofurantoin has also been used in the
past. Fosfomycin and pristinamycin have been used
in some countries (eg. Spain, France), although they
are not available in others (eg. U.K) and
comparatively little is known of their
antistaphylococcal activity. We have shown
pristinamycin to be uniformly active against all
strains and are most encouraged by these findings.
Clinical resistance to fosfomycin (MIC greater than
64 mg/1) was only found in 14 strains of MGRSA.
Development of fosfomycin resistance in vitro was
found to readily occur, however the relative lack
of clinically isolated strains suggests that in
vitro findings may not necessarily be relevant to
the in vivo situation. Fosfomycin has been used
with success in the treatment of MRSA septicaemia
(Lau et a l . , 1986) and it could become a useful
alternative agent to vancomycin.

The activity of antimicrobial agents currently
under development eg. daptomycin, the oxazolidinones
(DuP 105 and DuP 721), paldimycin, ramoplanin, new 14,
15- and 16- membered macrolides and an injectable
streptogramin (RP 59500) have been assessed against
MGRSA. We have found daptomycin, the oxazolidinones,
paldimycin, ramoplanin and RP 59500 to be uniformly
active against MGRSA. However, only daptomycin and
RP 59500 (an injectable pristinamycin) are currently
being progressed as far as clinical trials.
Preliminary clinical trials with daptomycin were
disappointing and re-evaluation of this compound is
currently being undertaken. Rokitamycin was the
most active of the macrolides tested, however it
will only be of limited use because even though
it had good activity against erythromycin-sensitive
and inducible resistant MGRSA it was still
inactive against clindamycin-resistant strains.
From these studies it appears that few agents
are presently under development which could be
used as alternatives to vancomycin for systemic
therapy. Hence, P a r k e r ’s aim of "diversification in
the use of antibiotics" does not appear to be a
current reality.
242

i i . Options for treatment of MGRSA carriage.
Much consideration has been given to infection
control measures directed towards the elimination
of MRSA from the hospital environment (Working
Party, 1986 & 1990). Infection control measures can
range from treatment of colonised individuals with
antiseptics and/or topical antibiotics in an
attempt to rid them of carriage to closure of
whole wards and removal of colonised patients to
specialist isolation facilities.
Until recently, no single agent has consistently
eradicated MRSA from carriage sites, and there has
been considerable debate as to the efficacy of
antiseptics or topical antibiotics in such
indications (Casewell & Hill, 1986; Brumfitt & Hamilton-
Miller, 1989). Now that mupirocin is available (both
in skin and nasal preparations) we have an agent
of consistent and proven efficacy in clearing
nasal and skin carriage (Casewell & Hill, 1987).
Initial screening studies of staphylococci failed
to detect resistance to mupirocin (Casewell & Hill,
1985), and in our own survey of 100 MGRSA no
resistance was found. However, staphylococcal
resistance to mupirocin may become an increasing
problem, and naturally occurring resistant strains
have even been found (Rahman et a l . , 1990). Because
of emerging bacterial resistance, and the potential
toxicity (due to the polyethylene glycol base in
243

skin formulations) of mupirocin when applied to
extensive burns (Rode et a l . , 1989) other alternative
topical agents have been looked for. We have
found azelaic acid, nitrofurazone, ramoplanin, and
silver sulphadiazine to be uniformly active against
the MGRSA tested. We were particularly interested
by azelaic acid, a naturally occurring compound
reported to possess a considerable number of
probiotic effects (Breathnach et al . , 1984).
Antiseptics and topical antibiotics may hinder
wound healing (Leaper & Simpson, 1986), and it would
be interesting to learn of the influence of
azelaic acid on such processes.
4.3 Role of fluoroquinolones against M G R S A .
Ciprofloxacin was the first fluoroquinolone
possessing broad-spectrum activity and
therapeutically useful blood-levels to be developed
and enter clinical use. There was some initial
enthusiasm that ciprofloxacin might be efficacious
against MRSA in view of its novel mode of
action, uniform activity and rapid killing against
these organisms (Smith & Eng, 1985). Other
fluoroquinolones- enoxacin, ofloxacin and pefloxacin-
soon followed ciprofloxacin into clinical use, and
were similarly regarded as potentially useful
antistaphylococcal agents. Early in v i t r o , in vivo
244

and clinical studies were fairly optimistic with
regard to the efficacy of ciprofloxacin in
resolving S. aureus skin structure infections and
osteomyelitis (Neu, 1987). Specifically, ciprofloxacin
was shown to successfully eradicate MRSA
colonisation (Mulligan et a l . , 1987) and it was
thought to be a major advance in the treatment
of MRSA osteomyelitis (Neu, 1987).
Staphylococcal resistance to ciprofloxacin did
not appear to be a problem until 1989. Isaacs e_t
a l . (1988) are usually credited as the first
workers to report an outbreak of infection with
ciprofloxacin-resistant MRSA. However, in French
hospitals pefloxacin-resistant MRSA had posed
serious problems since 1986 (Acar & Buu-Hoi, 1988;
Jean-Pierre et a l . , 1988). In 1989, we showed
ciprofloxacin resistance in MGRSA to be an
international problem (Maple et a l . , 1989c), and
warned of the dangers of inappropriate
ciprofloxacin usage (Maple et a l . , 1989b). Since
this time there have been numerous reports of
ciprofloxacin resistance in MRSA.
Our studies outlined in Section 3 determined
which, if any, fluoroquinolone was most appropriate
for treating MGRSA infections, and sought to
explain the high incidences of ciprofloxacin
resistance reported for MRSA. Ciprofloxacin,
ofloxacin and pefloxacin were all highly active
against MGRSA, however because the peak serum levels
245

of these agents differ (Table XIII), ofloxacin and
pefloxacin appear to have a greater therapeutic
index than ciprofloxacin. The fluoroquinolones were
rapidly bactericidal, and only low mutation rates
to resistance were found.
Fluoroquinolone-resistant strains emerged during
our time-kill experiments, and this resistance could
readily develop following exposure to therapeutic
concentrations of ciprofloxacin, enoxacin and
pefloxacin. Resistance did not readily emerge in
the presence of therapeutic concentrations of
ofloxacin. Our results using strains of MGRSA with
different initial fluoroquinolone susceptibilities
showed that development of fluoroquinolone
resistance occurred in a stepwise manner.
The ability of a strain to become resistant
depended upon its initial fluoroquinolone
susceptibility and the concentration of
fluoroquinolone to which it was exposed. In this
manner strains with initial ciprofloxacin MICs of
0.25 mg/1 and 2.0 mg/1 could increase their
resistance to 2.0 mg/1 and 16.0 mg/1 respectively.
If this was repeated in v i v o , clinical isolates of
ciprofloxacin-resistant MRSA showing different levels
of resistance would be expected. Of more concern
is that any strain can develop fluoroquinolone
resistance in this way, and presumably the extent
of such resistance will depend on the amount of
fluoroquinolone usage. Perhaps these observations
246

might explain Schaefler’s (1989) observations with
ciprofloxacin-resistant MRSA in New York.
Ciprofloxacin-resistant MRSA isolated in 1987 were
characteristically susceptible to 2.0-4.0 mg/1
ciprofloxacin, whereas strains isolated after January
1988 were often more resistant (ciprofloxacin MICs
of 12.5-25 mg/1). Ciprofloxacin-resistant strains
appeared simultaneously in many different hospitals
and phage-typing and antibiotic resistance profiles
suggested they had independently evolved.
The same pattern of fluoroquinolone resistance
was selected in vitro by ciprofloxacin, enoxacin,
ofloxacin and pefloxacin. On the other hand,
different levels and patterns of fluoroquinolone
resistance were found in the clinical isolates of
MGRSA studied. The latter may be a result of
different mechanisms of fluoroquinolone resistance.
Of particular interest was the observation of
reversed incomplete resistance to acrosoxacin in
some of our isolates. Kojima e t a l . (1990) have
suggested that fluoroquinolone resistance can be
due to altered DNA gyrase or permeability/efflux
mechanisms.
There is some controversy at present as to
whether the high incidence of ciprofloxacin-
resistant MRSA is due to independent evolution of
strains or cross-infection by a few resistant
strains. Central to this debate is our current
ability to differentiate MGRSA. We have shown
247

(Table I) that a considerable number of MGRSA are
non-typable by the International Set of phages, and
new typing methods are required to differentiate
such strains. We have typed many of our strains
by using supplementary phages, antibiotic
susceptibility profiles, plasmid-content and a small
number of biological tests (sheep blood haemolysis
and egg yolk lipase production). Using this system
we have differentiated between many ciprofloxacin-
resistant isolates showing that the spread of
ciprofloxacin resistance is due to development of
resistance in individual strains as well as cross
infection by resistant strains.
Due to the ease with which fluoroquinolone
resistance apparently develops in MGRSA, we are
pessimistic over the future therapeutic usefulness
of these agents against MGRSA. If fluoroquinolone
use is considered necessary for treatment then we
would suggest that ofloxacin is the most
appropriate agent to use. In the future new
fluoroquinolones may become available (eg.
sparfloxacin) which have a better therapeutic ratio
than ofloxacin, and such agents may prove more
effective in preventing the development of
resistant strains.
248

4.4 The future problem of antibiotic resistance inM G R S A .
In this work we have shown that multiple
antibiotic resistance in MGRSA presents a
significant therapeutic problem. Of great concern
is our observation that there are very few
currently available alternative agents for the
treatment of MGRSA carriage or infection. The
significance of these findings depends upon the
environment in which MGRSA is being isolated. In
countries where health care budgets are sufficient
for hospitals to devote time and expenditure to
control of MGRSA by implementation of rigorous
infection control measures the problems posed be
even highly multiple resistant strains of MGRSA
can be limited. However, in countries where such
measures cannot be financed although new
generations of increasingly potent antibiotics can
be purchased, the conditions are ripe for the
development and spread of highly multiple resistant
strains. Should vancomycin resistance emerge the
consequences for treatment of MGRSA infections
could be disastrous.
We have also seen that certain types of
antibiotic resistance can rapidly spread worldwide,
as in the case of gentamicin resistance in MRSA.
The rapid spread of this resistance is most
probably due to dissemination of the resistance
genes on a transposable genetic element. As such,
249

spread of resistance cannot be directly controlled
by antibiotic policy or even cross-infection
control measures. The location of resistance genes
on transposable genetic elements poses serious
potential resistance problems (Lyon & Skurray, 1987).
For S. aureus there is already considerable
evidence that such elements can alternate between
chromosomal or plasmid sites, be rapidly
disseminated between strains, and can be tranferred
to or acquired from different species (eg. S .
epidermidi s ) and even different genera (eg.
Enterococcus faecalis) .
It has been suggested (Koch, 1981; Foster, 1983)
that the genes responsible for antibiotic
resistance have evolved over many thousands of
years amongst soil organisms (particularly antibiotic
producers). Thus, by developing totally synthetic
antimicrobial agents with novel mechanisms of
action eg. the fluoroquinolones, it was believed
that bacterial resistance would not be a problem.
Our experience with ciprofloxacin resistance in
MGRSA shows that, as with trimethoprim and the
s ulphonamides, this is not the case.
Since the introduction of penicillin into
clinical use during the 1940s and the subsequent
development of other antibiotics there has been a
continual conflict with hospital staphylococci which
have shown a fecundity for developing resistance
to each new antibiotic. The impact of S. aureus
250

on the hospital environment has waxed and waned
over the years, as have the types of strain
responsible for such problems.
In the late 1940s extensive problems were
caused by Group I type (eg. type 52A) penicillinase
producing S. aureus which had a prediliction for
maternity and neonatal units. During the 1950s
these and type 80/81 strains were a cause of
major concern. Late in the 1950s multiple
antibiotic-resistant Group III strains became a
significant problem. In the 1960s Group I strains
ceased to appear in the hospital environment, and
following the introduction of methicillin and its
congeners most multiple resistant Group III strains
could be effectively dealt with. During the late
1960s and into the 1970s the influence of S .
aureus on hospital practice declined, however in
the late 1970s and continuing into the 1980s
major hospital problems due to MGRSA emerged.
These continue, for example, recently a small
outbreak of infections (one fatality) occurred in
our hospital due to a highly multiple-resistant
MGRSA. This MGRSA was resistant to many
antibiotics including ciprofloxacin, fusidic acid and
mupirocin, and could only be treated with vancomycin.
If vancomycin resistance does appear in MGRSA
our assessment of the current therapeutic options
available suggests that we are ill equipped for
this eventuality.
251

REFERENCES
ABRAHAM, E. P., CHAIN, E., FLETCHER, C. M . , GARDNER, A. D., HEATLEY, N. G., JENNINGS, M. A. (1941). Further observations on Penicillin. Lancet ii: 177-189.
ACAR, J. F., COURVALIN, P., CHABBERT, Y. A. (1970). Methicillin-resistant staphylococcaemia: bacteriological failure of treatment with cephalosporins, pp. 280- 285. Antimicrobial Agents and Chemotherapy (1971).
ACAR, J. F., GOLDSTEIN, F. W., DUVAL, J. (1983). Use of rifampicin for the treatment of serious staphylococcal ond Gram- negative bacillaryinfections. Reviews of Infectious Diseases 5 (S u p p l . 3), 502- 506.
ACAR, J. F., B U U - H O I , A. Y. (1988). Resistance patterns of important Gram- positive pathogens. Journal of Antimicrobial Chemotherapy 21: (S u p p l . C ), 41- 47.
ACAR, J. F., CLUZEL, R., COURVALIN, P., DUVAL, J., FLEURETTE, J., MEGRAUD, F., MEYRAND, M . , SOUSSY, C. J., THABAUT, A. (1990). RP 59500: a collaborative study of a semi-synthetic streptogramin in seven general hospitals in France. Abstracts of the 30th International Conference of Antimicrobial Agents and Chemotherapy (Atlanta), Abstract no. 780.
ALLEN, J. D., EVANS ROBERTS, C., KIRBY, W. M. M. (1962). Staphylococcal septicaemia treated with methicillin. Report of twenty-two cases. New England Journal of Medicine 266: 111-116.
ALY, R., SHINEFIELD, H. I., STRAUSS, W. G., M A I B A C H , H.I. (1977). Bacterial adherence to nasal mucosal cells. Infection and Immunity 17: 546-549.
252

A M Y E S , S. G. B., TAIT, S. (1990). Trimethoprimresistance in staphylococci. Journal of Medical Microbiology 31: 4-7.
ANDREW, J. H., WALE, M. C. J., WALE, L. J., GREENWOOD, D.(1987). The effect of cultural conditions on the activity of LY 146032 against staphylococci and streptococci. Journal of Antimicrobial Chemotherapy 20: 213- 221.
ANDREWS, J. M., B A Q U E R O , F., BELTRAN, J. M., CANTON, E., CROKAERT, F., G O B E R N A D O , M., GOMEZ- L U S , R., LOZA, E., NAVARRO, M., OLAY, T., RODRIGUEZ, A., VICENTE, M. V., WISE, R., YOURASSOWSKY, E. (1983). Internationalcollaborative study on standardization of bacterial sensitivity to fosfomycin. Journal of Antimicrobial Chemotherapy 12: 357- 361.
ANNEAR, D. I. (1968). The effect of temperature on resistance of Staphylococcus aureus to methicillin and some other antibiotics. Medical Journal ofAustralia 1: 444-446.
ARATH00N, E. G., HAMILTON, J. R., HENCH, C. E., STEVENS, D. A. (1990). Efficacy of short courses of oral novobiocin-rifampin in eradicating carrier state of methicillin-resistant Staphylococcus aureus and in vitro killing studies of clinical isolates. Antimicrobial Agents and Chemotherapy 34: 1655-1659.
ARGOUDELIS, A. D., BACZYNSKYJ, L., B U E J E , J. A.,MARSHALL, V. P., MIZSAK, S. A., WILEY, P. F. (1987a). Paulomycin-related antibiotics: paldimycins andantibiotics 273a2» Isolation and characterization. Journal of Antibiotics 40: 408- 418.
253

ARGOUDELIS, A. D., BACZYNSKYJ, L., MIZSAK, S. A., SHILLIDAY, F. B., SPINELLI, P. A., DEZWAAN, J. (1987b). Paldimycins A and B and antibiotics 273a2a and 273aab» Synthesis and characterization. Journal of Antibiotics 40: 419- 436.
ARTHUR, M., BRISSON-NOEL, A., COURVALIN, P. (1987). Origin and evolution of genes specifying resistance to macrolide, lincosamide and streptograminantibiotics: data and hypotheses. Journal ofAntimicrobial Chemotherapy 20: 783- 802.
ASHESHOV, E. H. (1975). The genetics of tetracyclineresistance in Staphylococcus a ureus. Journal ofMedical Microbiology 88: 132- 140.
A YLIFFE, G. A. G., GREEN, W., LIVINGSTON, R., L O W B U R Y , E. J. L. (1977). Antibiotic- resistant Staphylococcus aureus in dermatology and burn wounds. Journal ofClinical Pathology 30: 40-44.
AYLIFFE, G. A. G., LILLY, H. A., LOWBURY, E. J. L. (1979). Decline of the hospital staphylococcus?Incidence of multi-resistant S . aureus in threeBirmingham hospitals. Lancet i: 538-541.
BAIRD, D., C0IA, J. (1987). Mupirocin-resistant Staphylococcus au r e u s . Lancet ii: 387-388.
BAIRD-PARKER, A. C. (1963). A Classification ofMicrococci and Staphylococci based on Physiologicaland Biochemical tests. Journal oj GeneralMicrobiology 30: 409-427.
BAIRD-PARKER A. C. (1972). Classification andIdentification of Staphylococci and their Resistanceto Physical Agents ^In: COHEN, J. 0. (ed). TheStaphylococci. New York, Wiley-Interscience Press, p. 1- 2 0 .
254

BAQUERO, F., LOPEZ-BREA, M., V A L L S , A., CANEDO, T.(1977). Fosfomycin and plasmidic resistance. Chemotherapy 23 (Suppl. 1) t 133-140.
BARBER, M. (1947). Staphylococcal infection due to penicillin-resistant strains. British Medical Journal 2: 863-865.
BARBER, M. (1960). Drug-resistant staphylococcal infection. British Journal of Obstetrics and Gynaecology 67: 727-732.
BARBER, M. (1964). Naturally occurring methicillin-resistant staphylococci. Journal o_f GeneralMicrobiology 35: 183-190.
BARBER, M., R0ZWAD0WSKA-D0WZENK0, M. (1948). Infection by penicillin-resistant staphylococci. Lancet ii: 641-644.
BARBER, M., B U R S T O N , J. (1955). Antibiotic-resistant staphylococcal infection. A study of antibiotic sensitivity in relation to bacteriophage types. Lancet ii: 578-583.
BARBER, M., WATERWORTH, P. M. (1962). Antibacterialactivity in vitro of fucidin. Lancet i: 931- 932.
BARBER, M., WATERWORTH, P. M. (1964). Antibacterialactivity of lincomycin and pristinamycin: acomparison with erythromycin. British MedicalJournal 2: 603- 606.
BARBER, M., WATERWORTH, P. M. (1966). Activity ofgentamicin against pseudomonas and hospital staphylococci. British Medical Journal 1: 203-205.
255

BARRETT, F. F., McGEHEE, R. F., FINLAND, M. (1968). Methicillin-resistant Staphylococcus aureus at Boston City Hospital. Bacteriologic and epidemiologic observations. New England Journal of Medicine 279: 441-448.
BA R R I E R E , J. C., BOUANCHAUD, D. H., HARRIS, N. V., PARIS, J. M., ROLIN, 0., SMITH, C. (1990). The design, synthesis and properties of RP 59500 and related semi-synthetic streptogramin antibiotics. Abstracts of the 30th International Congress on Antimicrobial Agents and Chemotherapy (Atlanta), Abstract No. 768.
BARRY, A. L., JONES, R. N. (1989). In-vitro activities of temafloxacin, tosufloxacin (A-61827) and five other fluoroquinolone agents. Journal of Antimicrobial Chemotherapy 23:527-536.
BARTZOKAS, C. A., PAT0N, J. H., GIBSON, M. F., GRAHAM,R., McLOUGHLIN, G. A., CROTON, R. S. (1984). Control and eradication of methicillin-resistantStaphylococcus aureus on a surgical unit. NewEngland Journal of Medicine 311: 1422-1425.
BASTIN, R., RAPIN, M., KERNBAUM, S. (1982). Controlled study of pristinamycin versus oxacillin in staphylococcal infections. Pathologie et Biologie(Paris) 6: 473- 475.
BEAVEN, D. W., BURRY, A. F. (1956). Staphylococcalpneumonia in the newborn. An epidemic with 8 fatalcases. Lancet ii: 211-215.
BENNER, E. J., KAYSER, F. H. (1968). Growing clinical significance of methicillin-resistant Staphylococcus aureus. Lancet ii: 741-744.
256

B I N D A , G., DOMENICHINI, E., GOTTARDI, A., ORLA N D I , B., ORTELLI, E., PACINI, B., FOWST, G. (1971). Rifampicin, a general review. Arzneimittelforschung 12a: p.1962.
BISMUTH, R., ZILHAO, R., SAKAMOTO, H., GUESDON, J - L . , COURVALIN, P. (1990). Gene heterogeneity for tetracycline resistance in Staphylococcus spp. Antimicrobial Agents and Chemotherapy 34: 1611—1614.
BLACK, H. R., BRIER, G. L., W O L N Y , J. D., N Y H A R T , E. H. (1986). Preliminary pharmacology and pharmacokinetics of LY 146032, a new peptolide antibiotic. Abstracts of the 26th Interscience Conference onAntimicrobial Agents and Chemotherapy, New Orleans, Abstract 894.
BLADON, P. T., BURKE, B. M., CUNLIFFE, W. J., FORSTER, R. A., HOLLAND, K. T., KING, K. (1986). Topical azelaic acid and the treatment of acne: a clinical andlaboratory comparison with oral tetracycline.British Journal of Dermatology 114: 493-499.
BLAKE, S. C., CHAPMAN, P. C., EATON, N, MERRI K I N , D. J. (1990). BRL 44154 - A novel penicillin activeagainst methicillin- resistant staphylococci - in vitro and in vivo properties.Abstracts of the 7th Mediterranean Congress ofChemotherapy, Barcelona, Spain, 20- 25th May, 1990.
BLUM, A. (1982). Culturing an epidemic of staphylococci in the mass media. Medical Journal of Australia 1: 475- 479.
BONDI A., Jr., DIETZ, C. C. (1945). Penicillin resistant staphylococci. Proceeedings of the Society for Experimental Biology and Medicine 60: 55-58.
257

BOROWSKI, J., JAKUBICZ, P., ZAREMBA, M. et al. (1988).Nationwide studies on occurrence of methicillin resistant Staphylococcus aureus and Staphylococcuscoagulase-negative strains in Polish hospitals. Some characteristics of MRSA strains. Abstract P2 / 12. 1st International Conference of the Hospital Infection Soc i e t y , 1988.
BRADY, L. M., THOMSON, M., PALMER, M. A., HARKNESS, J. L. (1990). Succesful control of endemic MRSA in a cardiothoracic surgical unit. Medical Journal of Australia 152: 240-245.
BREATHNACH, A. S., NAZZAR0-P0RR0, M. , PASSI, S. (1984). Azelaic acid. British Journal of Dermatology 111: 115-120.
BREED, R.S., MURRAY, E.G.D., SMITH, N.R. (1957). Bergey's Mannual of Determinative Bacteriology.Seventh Edition, London, E. & S. Livingstone.
BRIDGES, R. A., B E R E N D E S , H., GOOD, R. A. (1957).Serious reactions to novobiocin. Journal___ ofPediatrics 50: 579- 585.
BRITISH SOCIETY FOR ANTIMICROBIAL CHEMOTHERAPY(1988). Break-points in in____ vitro antibioticsensitivity testing. Journal_____of AntimicrobialChemotherapy 21: 701-710.
BROWN, D. F. J., REYNOLDS, P. E. (1980). Intrinsic resistance to B-lactam antibiotics in Staphylococcus au r e u s . FEBS Letters 122: 275-278.
BRUMFITT, W., HA MILTON-MILLER, J. M. T. (1988). In vitro microbiological activities of DuP 105 andDuP 721, novel synthetic oxazolidinones. Journal of Antimicrobial Chemotherapy 21: 711- 720.
258

BRUMFITT, W., MAPLE, P. A. C., HAMILTON-MILLER, J. M. T.(1989). Antibiotic sensitivity patterns of methicillin-resistant Staphylococcus aureus and their use in biotyping. Abstract 778/PP40, p. 325, Abstracts of 4th European Congress of Clinical Micro biology, Nice, France.
BRUMFITT, W., HAMILTON-MILLER, J. M. T. (1989).Methicillin-resistant Staphylococcus a u r e u s . NewEngland Journal of Medicine 320: 1188-1196.
BULGER, R. J., SHERRIS, J. C. (1968). Decreasedincidence of antibiotic resistance amongStaphylococcus au r e u s . A study in a university hospital over a 9-year period. Annals of Internal Medicine 69: 1099-1107.
BULLOCH, W. (1938). The History of Bacteriology.First Edition, London, Oxford University Press.
BURDESKA, A., THEN, R. L. (1990). Clinical importance of trimethoprim resistance in staphylococci isolated in Europe. Journal of Medical Microbiology 31: 11-14.
BYRNE, M. E., GILLESPIE, M. T., SKURRAY, R. A. (1990). Molecular analysis of a gentamicin resistancetransposonlike element on plasmids isolated fromNorth American Staphylococcus aureus strains.Antimicrobial Agents and Chemotherapy 34: 2106-2113.
CAFFERKEY, M. T., HONE, R., FALKI N E R , F. R., KEANE, C. T., POMEROY, H. (1983). Gentamicin and methicillin- resistant Staphylococcus aureus in Dublin hospitals: clinical and laboratory studies. Journal of Medical Microbiology 16: 117-127.
259

CAFFERKEY, M. T., HONE, R., COLEMAN, D . , POMEROY, H.,McGRATH, B., RUDDY, R., KEANE, C. T. (1985a).Methicillin-resistant Staphylococcus aureus in Dublin1971-1984. Lancet ii: 705-708.
CAFFERKEY, M. T., HONE, R., KEANE, C. T. (1985). Antimicrobial chemotherapy of septicaemia due tomethicillin-resistant Staphylococcus_______ aureus.Antimicrobial Agents and Chemotherapy 28: 819- 823.
CALAIN, P., WALDVOGEL, F. (1990). Clinical efficacy ofteicoplanin. European_____Journal_____ of_____ClinicalMicrobiology and Infectious Disease 9: 127- 129.
CANEPARI, P., BOARETTI, M., DEL MAR LLEO, M., SATTA, G.(1990). Lipoteichoic acid as a new target foractivity of antibiotics: mode of action ofdaptomycin (LY 146032). Antimicrobial Agents and Chemotherapy 34: 1220- 1226.
CARR, H. S., WL0DK0WSKI, T. J., ROSENKRANZ, H. S. (1973). Silver sulfadiazine: in vitro antibacterial activity. Antimicrobial Agents and Chemotherapy 4: 585-587.
CARROLL, J. D., POMEROY, H. M., RUSSELL, R. J.,ARBUTHN0TT, J.P., KEANE, C. T., McCORMICK, 0. M. ,COLEMAN, D. C. (1989). A new methicillin- andgentamicin- resistant Staphylococcus____ aureus inDublin: molecular genetic analysis. Journal___ ofMedical Microbiology 28: 15-23.
CASEWELL, M. W., HILL, R. L. R. (1985). In-vitroactivity of mupirocin ("pseudomonic acid") againstclinical isolates of Staphylococcus aureus. Journal of Antimicrobial Chemotherapy 15: 523-531.
2 6 0

CASEWELL, M. W. (1986). Epidemiology and control of the ’modern’ methicillin-resistant Staphylococcusaureus. Journal of Hospital Infection 7: (Suppl. A)t 1- 1 1 .
CASEWELL, M. W., HILL, R. L. R. (1986). The Carrier State: methicillin-resistant Staphylococcus aureus.Journal of Antimicrobial Chemotherapy 18: (Suppl. A), 1-12.
CASEWELL, M. W. , HILL, R. L. R. (1987). Mupirocin (”pseudomonic acid”)- a promising new topicalantimicrobial agent. Journal of AntimicrobialChemotherapy 19: 1-5.
CAVALLERI, B., PAGANI, H., VOLPE, G., SELVA, E., PARENTI,F. (1984). A-16686, a new antibiotic fromActinoplanes. I. Fermentation, isolation andpreliminary physico-chemical characteristics. Journal of Antibiotics 37: 309-317.
CHABBERT, Y., TERRIAL, G. (1952). Evolution actuelledes types de resistance aux antibiotiques chez lesstaphylocoques pathogenes. Annales De L f InstitutPasteur 83: 499-505.
CHAMBERLAIN, R. E. (1976). Chemotherapeutic properties of prominent nitrofurans. Journal of Antimicrobial Chemotherapy 2: 325-336.
CHAMBERS, H. F. (1988). Methicillin-resistantstaphylococci. Clinical Microbiology Reviews 1: 173-186.
CHAMBERS, H. K., SACHDEVA, M. (1990). Binding of B- lactam antibiotics to penicillin-binding proteins in methicillin-resistant Staphylococcus aureus. Journal of Infectious Diseases 161: 1170-1176.
261

CHANDRASEKAR, P. H., SLUCHAK, J. A. (1989). Susceptibility of staphylococci to paldimycin and emergence of resistance in vitro. Journal of Antimicrobial Chemotherapy 24: 821-824.
CHAUDHRY, A. Z., KNAPP, C. C., SIERRA-MADERO, J.,WASHINGTON, J. A. (1990). Antistaphylococcal activities of sparfloxacin (CI-978; AT-4140), ofloxacin andciprofloxacin. Antimicrobial Agents and Chemotherapy 34: 1843-1845.
CHENG, A. F., FRENCH, G. (1988). Methicillin-resistant Staphylococcus aureus bacteraemia in Hong Kong. Journal of Hospital Infection 12: 91-101.
CHOKKAVELU, V., CHANDRASEKAR, P., R0LST0N, K., LeFROCK, J. L., SCHELL, R. F. (1984). Activity of eleven antimicrobial agents against methicillin-,methicillin- and rifampicin- resistant Staphylococcus aureus . Chemotherapy 30: 97-101.
CHOPRA, I. (1976). Mechanisms of resistance to fusidic acid in Staphylococcus aureus. Journal of General Microbiology 96: 229-238.
CHOW, J. W., YU, V. L. (1989). Staphylococcus aureus nasal carriage in haemodialysis patients, its role in infection and approaches to prophylaxis. Archives of Internal Medicine 149: 1258-1262.
CLARKE, H. T., JOHNSON, J. R., ROBINSON, R. (1949). The chemistry of Penicillin. Princeton University Press, New Jersey.
CLUMECK, N., MARCELIS, L., AMRI-LAMRASKI, M. H., GORDTS,B. (1984). Treatment of severe staphylococcal infections with a rifampicin-minocycline association. Journal of Antimicrobial Chemotherapy 13 (Suppl. C), 17- 22.
262

COCITO, C. (1979). Antibiotics of the virginiamycin family, inhibitors which contain synergisticcomponents. Microbiological Reviews 43: 145- 198.
COLEBROOK, L., KENNY, M. (1936). Treatment of human puerperal infections, and experimental infections in mice, with prontosil. Lancet i: 1279-1286.
COLEMAN, D. C., POMEROY, H., ESTRIDGE, J. K., KEANE, C. T., CAFFERKEY, M. T., HONE, R., FOSTER, T. J. (1985). Susceptibility to antimicrobial agents and analysis of plasmids in gentamicin- and methicillin- resistant Staphylococcus aureus from Dublin hospitals. Journal of Medical Microbiology 20: 157- 167.
COMMUNICABLE DISEASE REPORT (1986). Two- region survey of methicillin-resistant Staphylococcus aureus (MRSA). Communicable Disease Report 86/10.
COOKE, E. M., CASEWELL, M. W., EMMERSON, A. M., GASTON,M., de SAXE, M., MAYON-WHITE, R. T., GALBRAITH, N. S.(1986). Methicillin-resistant Staphylococcus aureus in the U.K. and Ireland. A questionaire survey. Journal of Hospital Infection 8: 143-148.
C00KS0N, B., TALSANIA, H., NAIDOO, J., PHILLIPS, I.(1986). Strategies for typing and properties of epidemic methicillin- resistant Staphylococcus aureus. European Journal of Clinical Microbiology 5: 702-709.
C00KS0N, B. D., PHILLIPS, I. (1988). Epidemicmethicillin-resistant Staphylococcus aureus. Journal of Antimicrobial Chemotherapy 21, (Suppl. C), 57-65.
C00KS0N, B. D., PHILLIPS, I. (1989). Letter to the editor. New England Journal of Medicine 321: 1344.
263

COOVADIA, Y. M.f BHANA, R. H., JOHNSON, A. P., HAFFEJEE,I., MARPLES, R. R. (1989). A laboratory- confirmed outbreak of rifampicin- methicillin resistant Staphylococcus aureus (RMRSA) in a newborn nursery. Journal of Hospital Infection 14: 303- 312.
COPPENS, L., HANSON, B., KLASTERSKY, J. (1983). Therapy of staphylococcal infections with cefamandole orvancomycin alone or with a combination ofcefamandole and tobramycin. Antimicrobial Agents and Chemotherapy 23: 36-41.
COURTIEU, A. L., DRUGEON, H., BILLAUDEL, S. (1977).Susceptibility to fosfomycin of hospital strainsisolated in Nantes (France). Frequency of mutation to resistance. Chemotherapy 23 (Suppl. 1), 25- 36.
COURVALIN, P., DAVIES, J. (1977). Plasmid- mediated aminoglycoside phosphotransferase of broad substrate range that phosphorylates amikacin. Antimicrobial Agents and Chemotherapy 11: 619- 624.
COWAN, S. T., STEEL, K. J. (1974). Identification of Medical Bacteria. 2nd edition, Cambridge University Press (Cambridge).
CRAVEN, D. E., REED, C., KOLLISCH, N., DeMARIA, A.,LICHTENBERG, D., SHEN, K., McCABE, W. R. (1981). A large outbreak of infections caused by a strain of Staphylococcus aureus resistant to oxacillin and aminoglycosides. American Journal of Medicine 71: 53-58.
CRAVEN, D. E., RIXINGER, A. I., GOULARTE, T. A., McCABE, W. R. (1986). Methicillin- resistant Staphylococcus aureus bacteraemia linked to intravenous drug abusers using a "shooting gallery". American Journal of Medicine 80: 770-776.
2 6 4

CROSSLEY, K., LOESCH, D., LANDESMAN, B., MEAD, K., CHERN, M. , STRATE, R. (1979). An outbreak of infectionscaused by strains of Staphylococcus aureusresistant to methicillin and aminoglycosides. 1.Clinical studies. Journal of Infectious Diseases139: 273-279.
CRUICKSHANK, R., DUGUID, J. P., MARMION, B. P., SWAIN, R. H. A. (1975). Medical Microbiology (The Practice ofMedical Microbiology). 12th edition, ChurchillLivingstone (Edinburgh).
CRUMPLIN, G. C. (1990). Mechanisms of resistance to the 4-quinolone antibacterial agents. Journal of Antimicrobial Chemotherapy 26 (Suppl. F), 131-144.
CULLMANN, W., STIEGLITZ, M., BAARS, B., OPFERKUCH, W.(1985). Comparative evaluation of recently developed fluoroquinolone compounds - with a note on thefrequency of resistant mutants. Chemotherapy 31:19-28.
CUNDLIFFE, E. (1972). The mode of action of fusidicacid. Biochemical and Biophysical ResearchCommunications 46: 1794-1801.
DALY, J. S., ELI0P0UL0S, G. M., REISZNER, E., MOELLERING Jr., R. C. (1988). Activity and mechanism of actionof DuP 105 and DuP 721, new oxazolidinonecompounds. Journal of Antimicrobial Chemotherapy 21: 721-730.
DAUM, T. E., SCHABERG, D. R., TERPENNING, M. S., SOTTILE, W. S., KAUFFMAN, C. A. (1990). Increasing resistanceof Staphylococcus aureus to ciprofloxacin.Antimicrobial Agents and Chemotherapy 34: 1862-1863.
265

DEVAUD, M., KAYSER, F. H., HUBER, R. (1977). Resistance of bacteria to the newer aminoglycoside antibiotics: an epidemiological and enzymatic study. Journal of Antibiotics XXX: 655- 664.
DORNBUSCH, K., MILLER, G. H., HARE, R. S., SHAW, K. J. and the ESGAR Study Group (1990). Resistance to aminoglycoside antibiotics in Gram- negative bacilliand staphylococci isolated from blood. Report froma European collaborative study. Journal ofAntimicrobial Chemotherapy 26: 131- 144.
DOUTHWAITE, A. H., TRAFFORD, J. A. P. (1960). A new penicillin. British Medical Journal 2: 687-690.
DOWDING, J. E. (1977). Mechanisms of gentamicin resistance in Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 11: 47- 50.
DUGUID, J. P. (1951). The demonstration of bacterial capsules and slime. Journal of Pathology andBacteriology 63: 673-685.
DUNKLE, L. M., NAQVI, S. H., McCALLUM, R., LOFGREN, J. P.(1981). Eradication of epidemic methicillin- gentamicin resistant Staphylococcus aureus in anintensive care nursery. American Journal ofMedicine 70: 455- 458.
DUVAL, J. (1985). Evolution and epidemiology of MLSresistance. Journal of Antimicrobial Chemotherapy 16: (Suppl. A), 137- 149.
DYKE, K. G. H. (1969). Penicillinase production andintrinsic resistance to penicillins in methicillin-resistant cultures of Staphylococcus aureus. Journalof Medical Microbiology 2: 261-278.
266

EDITORIAL (1977). Topical Antibiotics. British Medical Journal 1: 1494.
EDITORIAL BOARD (1958). Conservation of the generic name Staphylococcus Rosenbach, designation of thename Staphylococcus aureus Rosenbach as thenoraenclatural type of the genus StaphylococcusRosenbach, and designation of a neotype culture ofStaphylococcus____ aureus Rosenbach. Opinion 17.International_____ Bulletin_______ of______ BacteriologicalNomenclature and Taxonomy 8: 138-153.
ELEK, S. D., HILSON, G. R. F. (1954). Combined agardiffusion and replica plating techniques in the study of antibacterial substances. Journal ofClinical Pathology 7: 37-44.
ELIOPOULOS, G. M., WILLEY, S., REISZNER, E., SPITZER, P.G., CAPUTO, G., MOELLERING Jr., R. C. (1986). In vitroand in vivo activity of LY 146032, a new cycliclipopeptide antibiotic. Antimicrobial Agents andChemotherapy 30: 532-535.
ELWELL, L. P., WILSON, H. R., KNICK, V. B.,KEITH, B. R.(1986). In vitro and in vivo efficacy of the combination trimethoprim-sulfamethoxazole againstclinical isolates of methicillin-resistantStaphylococcus aureus. Antimicrobial Agents andChemotherapy 29: 1092-1094.
ENG, R. K., SMITH, S. M., TILLEM, M., CHERUBIN, C.(1985). Rifampicin resistance: development ofresistance during the therapy of methicillin-resistant Staphylococcus aureus infection. Archives of Internal Medicine 145: 146-148.
ERIKSEN, K. R., ERICHSEN, I. (1963). Resistance to the newer penicillins. British Medical Journal 1: 746.
2 6 7

EYKYN, S. J. (1988). Staphylococcal sepsis. The changing pattern of disease and therapy. Lancet i: 100-104.
FARBER, B. B. (1984). Vancomycin: renewed interest inan old drug. European Journal____ of___ClinicalMicrobiology 3: 1-3.
FASS, R. J., HELSEL, V. L. (1987). In vitro antistaphylococcal activity of pefloxacin alone andin combination with other antistaphylococcal drugs.Antimicrobial Agents and Chemotherapy 31: 1457-1460.
FINKELSTEIN, R., MARKEL, A., REINHERZ, G., HASHMAN, N., MERZBACH, D. (1989). The emergence of methicillin- resistant Staphylococcus aureus infections in an Israeli hospital. Journal of Hospital Infection 14: 55-61.
FINLAND, M., HAIGHT, T. H. (1953). Antibioticresistance of pathogenic staphylococci. Archives of Internal Medicine 91: 143-158.
FINLAND, M. (1955). Emergence of antibiotic-resistant bacteria. New England Journal of Medicine 253: 909-922.
FINLAND, M. (1974). An Historical Introduction I_n JACKSON, G. G., ESCARZAGA, E., GARR0D, L. P., NEU, H., UEDA, Y. (eds). Gentamicin: review and commentary on selected world literature. New York, Council forInterdisciplinary Communication in Medicine Ltd.,pp. iii- viii .
FLEMING, A. (1929). On the antibacterial action of cultures of a Penicillium, with special referenceto their use in the isolation of B. influenzae. British Journal of Experimental Pathology 10: 226-236.
268

FOLDES, M . , MUNRO, R., SORRELL, T. C., SHANKER, S., TOOHEY, M. (1983). In vitro effects of vancomycin, rifampicin, and fusidic acid, alone and in combination, against methicillin- resistantStaphylococcus aureus. Journal of AntimicrobialChemotherapy 11: 21-26.
FOSTER, J. K., LENTINO, J. R., STRODTMAN, R., DiVINCENZO, C. (1986). Comparison of in vitro activity of quinolone antibiotics and vancomycin against gentamicin- and methicillin-resistant Staphylococcus aureus by time-kill studies. Antimicrobial Agents and Chemotherapy 30: 823-827.
FOSTER, T. J. (1983). Plasmid-determined resistance to antimicrobial drugs and toxic metal ions inbacteria. Microbiological Reviews 47: 361-409.
FRENCH, G. L., LING, J., LING, T., HUI, Y. W. (1988). Susceptibility of Hong Kong isolates ofmethicillin- resistant Staphylococcus aureus toantimicrobial agents. Journal____ of AntimicrobialChemotherapy 21: 581-588.
FRIMODT-MOLLER, N., THAMDRUP ROSDAHL, V., SORENSEN, G., HARTVIG HARTZEN, S., WEIS BENTZON, M. (1986).Relationship between penicillinase production andthe in-vitro activity of methicillin, oxacillin, cloxacillin, dicloxacillin, flucloxacillin, and cephalothin against strains of Staphylococcus aureus of different phage patterns and penicillinase activity. Journal of Antimicrobial Chemotherapy 18: 27-33.
269

FRONGILLO, R. F., DONATI, L., FEDERICO, G., MARTINO, P., MORONI, M., ORTONA, L., PALUMBO, M. , PASTICCI, B. M. , PIZZIGALLO, E., PRIVITERA, G., SERRA, P., SIGNORINI, M. , VENDITTI, M., PAULUZZI, S. (1986). Clinical comparative study on the activity of cefamandole in the treatment of serious staphylococcal infections caused by methicillin-susceptible and methicillin-resistant strains. Antimicrobial Agents andChemotherapy 29: 789-796.
GARGAN, R. A., BRUMFITT, W., HAMILTON-MILLER, J. M. T.(1982). A concise biotyping system differentiating strains of Escherichia coli. Journal of ClinicalPathology 35: 1366-1369.
GARRISON, M. W., VANCE-BRYAN, K., LARSON, T. A., TOSCANO, J. P., ROTSCHAFER, J. C. (1990). Assesment of effects of protein binding on daptomycin and vancomycinkilling of Staphylococcus aureus by using an invitro pharmacodynamic model. Antimicrobial Agentsand Chemotherapy 34: 1925-1931.
GARROD, L. P. (1944). The laboratory control ofPenicillin treatment. British Medical Journal 1:528-530.
GARROD, L. P. (1968). Methicillin-resistantstaphylococci. Lancet ii: 871.
GARROD, L. P., LAMBERT, H. P., 0 fGRADY, F. (1981).Antibiotic and Chemotherapy. 5th edition, Edinburgh, Churchill Livingstone, pp. 73-77.
GEDEBOU, M., KRONVALL, G., HABTE-GABR, E., RINGERTZ, S.(1987). The bacteriology of nosocomial infections at Tikur Anbessa teaching hospital, Addis Ababa.Acta Pathologica, Micr obiologica , et ImmunologicaScandinavica, Section B 95: 331- 336.
270

GELLERT, M. , O ’ DEA, M. H., ITOH, T., TOMIZAWA, J. (1976). Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proceedings of the National Academy of Sciences, USA. 73: 4474- 4478.
GIAMARELLOU, H., PAPAPETROPOULOU, M., DAIKOS, G. K.(1981). ’Methicillin resistant’ Staphylococcus aureus infections during 1978- 1979: clinical andbacteriological observations. Journal_______ofAntimicrobial Chemotherapy 7: 649-655.
GILLESPIE, W. A., ALDER, V. G. (1952). Production of opacity in egg-yolk media by coagulase-positive staphylococci. Journal of Pathology and Bacteriology 64: 187-199.
GILLESPIE, W. A., SIMPSON, K. (1958). Staphylococcal infection in a maternity hospital, epidemiology and control. Lancet ii: 1075-1080.
GLUPCZYNSKI, Y., LAGAST, H., VAN DER AUWERA, P., THYS, J. P., CROKAERT, F., YOURASSOWSKY, E., MEUNIER-CARPENTIER, F., KLASTERSKY, J., KAINS, J. P., LEGRAND, J. C. (1986). Clinical evaluation of teicoplanin for therapy of severe infections caused by Gram-positive bacteria. Antimicrobial Agents and Chemotherapy 29: 52-57.
GODTFREDSEN, W., R0H0LT, K., TYBRING, L. (1962).Fucidin, a new orally active antibiotic. Lancet i: 928-931.
GOTO, S. (1977). Fosfomycin, antibacterial activity in vitro and in vivo. Chemotherapy 23 (Suppl. 1), 63-74.
GRAHAM, D. R., CORREA- VILLASENOR, A., ANDERSON, R. L.,VOLLMAN, J. H., BAINE, W. B. (1980). Epidemic neonatalgentamicin- methicillin- resistant Staphylococcusaureus infection associated with nonspecific topical
271

use of gentamicin. Journal of Pediatrics 97: 972-978.
GRANINGER, W. , LEITHA, T., HAVEL, M., GEORGOPOULOS, A.(1984). In vitro activity of fosfomycin against methicillin-susceptible and methicillin-resistant Staphylococcus aureus. Infection 12: 293- 295.
GREENWOOD, D. (1988). Microbiological properties ofteicoplanin. Journal of Antimicrobial Chemotherapy 21 (Suppl. A), 1- 13.
GRIFFITH, R. S. (1984). Vancomycin use - an historical review. Journal of Antimicrobial Chemotherapy 14(Suppl. D), 1- 5.
GRUBB, W. B., TOWNSEND, D.E., ASHDOWN, N., TJIA, T., McGLASHAN, C., LENG, T. (1986). Genetic analysis ofmethicillin-resistant Staphylococcus aureus fromSingapore Hospitals. European Journal of ClinicalMicrobiology 5: 728-730.
GUENTHNER, S. H., WENZEL, R. P. (1984). In vitroactivities of teichomycin, fusidic acid,flucloxacillin, fosfomycin, and vancomycin againstmethicillin- resistant Staphylococcus______aureus .Antimicrobial Agents and Chemotherapy 26: 268- 269.
GUPTA, S. P., CHAKRAVATI, R. N. (1954). Determination of bacterial sensitivity to common antibiotics.Indian Journal of Medical Research 42: 159-164.
HACKBARTH, C. J., CHAMBERS, H. F. (1989a). Methicillin-resistant staphylococci: detection methods andtreatment of infections. Antimicrobial Agents andChemotherapy 33: 995-999.
272

HACKBARTH, C. J., CHAMBERS, H. F. (1989b). Methicillin- resistant staphylococci: genetics and mechanisms ofresistance. Antimicrobial Agents and Chemotherapy33: 991-994.
HADORN, K., LENZ, W. , KAYSER, F. H., SHALIT, I., KRASEMANN, C. (1990). Use of a ribosomal RNA gene probe for the epidemiological study of methicillin and ciprofloxacin resistant Staphylococcus aureus. European Journal of Clinical Microbiology and Infectious Diseases 9: 649-653.
HAJEK, V., MARSALEK, E. (1971). The Differentiation of Pathogenic Staphylococci and a Suggestion for theirTaxonomic Classification. Zentralblatt furBakteriologie Mikrobiologie und Hygiene, I. Abt. Originale (Series) A 217: 176-182.
HAJEK, V. (1976). Staphylococcus intermedius, a New Species isolated from Animals. International Journal of Systematic Bacteriology 26: 401-408.
HALEY, R. W., HIGHTOWER, A. W., KHABBAZ, R. F., THORNSBERRY, C., MARTONE, W. J., ALLEN, J. R., HUGHES, J. M. (1982). The emergence of methicillin- resistant Staphylococcus aureus infections in United Stateshospitals. Possible role of the house staff-patient transfer circuit. Annals___ of InternalMedicine 97: 297-308.
HALLMAN, F.A. (1937). Pathogenic staphylococci in the anterior nares: their incidence and diffentiation.Proceedings of____ the Society___ for ExperimentalMedicine and Biology 36: 789-794.
HAMILTON-MILLER, J. M. T. (1982). B- lactamases andtheir clinical significance. Journal_____ ofAntimicrobial Chemotherapy 9: (Suppl. B) , 11-19.
273

HAMILTON-MILLER, J. M. T., RAMSAY, J. (1967). Stability of cephaloridine and cephalothin to staphylococcalpenicillinase. Journal of General Microbiology 49: 491-501.
HANSEN, S. L., FREEDY, P. K. (1984). Variation in the abilities of automated, commercial, and referencemethods to detect methicillin-resistant(heteroresistant) Staphylococcus aureus.Journal of Clinical Microbiology 20: 494-499.
HARDY, D. J., SWANSON, R. N., HENSEY, D. M., RAMER, N.R., BOWER, R. R., HANSON, C. W., CHU, D. T. W., FERNANDES, P. B. (1987). Comparative antibacterialactivities of temafloxacin hydrochloride (A-62254) and two reference fluoroquinolones. Antimicrobial Agents and Chemotherapy 31: 1768-1774.
HARDY, D. W., HENSEY, D. M., BEYER, J. M. , VO JTKO, C., McDo n a l d , E. J., FERNANDES, P. V. (1988). Comparative in vitro activities of new 14, 15 and 16- memberedmacrolides. Antimicrobial Agents and Chemotherapy 32: 1710-1719.
HENNESSEY, R. S. F., MILES, R. A. (1958). Staphylococcus aureus type 80 and human infections in Uganda. British Medical Journal 2: 893-895.
HERRLICH, P., SCHWEIGER, M. (1976). Nitrofurans, agroup of synthetic antibiotics, with a new mode of action: discrimination of specific messenger RNAclasses. Proceedings of the National Academy of Sciences, USA 73: 3386-3390.
HILS0N, G. R. F. (1962). In-vitro studies of a new antibiotic (fucidin). Lancet i: 932-933.
274

HOEPRICH, P. D. (1969). Gentamicin versusStaphylococcus aureus . Journal o_f InfectiousDiseases 119: 391-392.
HOOPER, G., COVARRUBIAS, J. (1983). Clinical use and efficacy of furacin: a historical perspective.Journal of International Medical Research 11: 289-293.
HUCKER, G. J. (1948). In BREED, R.S., MURRAY, E.G.D.,HITCHENS, A.P. (eds.), Ber gey1 s______Mannual______ofDeterminative Bacteriology. Sixth Edition, London, Balliere, Tindall & Cox, p 235.
IGARI, J., SHIMOJI, K., UEZU, N., NAKASONE, I, TAIRA, K., KAKUZU, R. (1988). In vitro susceptibilities of clinical isolates of Staphylococcus aureus. Japan Journal of Antibiotics 41: 1205-1211.
IPSEN, T., GAHRN- HANSEN, B. (1988). Occurrence of methicillin- resistant Staphylococcus aureus in a department of orthopedic surgery 1970- 1986.European Journal of Clinical Microbiology and Infectious Diseases 7: 400-403.
ISAACS, R. D., KUNKE, P. J., COHEN, R. L., SMITH, J. W.(1988). Ciprofloxacin resistance in epidemic methicillin-resistant Staphylococcus aureus. Lancet ii: 843.
JACKSON, G. G. (1969). Introduction. Journal of Infectious Diseases 119: 341.
JAWETZ, E. (1961). Polymixin, colistin and bacitracin. Pediatric Clinics of North America 8: 1067-1071.
JEAN-PIERRE, H., BOYER, G., JEAN, A., DARBAS, H. (1988).Evolution de la resistance des Staphylococcusaureus a la pefloxacine. Etude portant sur 782souches isolees en 1985 et 1986. Pathologie et
275

Biologie 36: 956-958.
JEFFERSON, J., McKNIGHT, A. G. (1969). Topical Gentamicin. British Journal of Clinical Practice 23: 133-138.
JELJASZEWICZ ( 1972). Toxins (Hemolysins) In: COHEN J.0. (ed) The Staphylococci. New York, WileyInterscience Press, p 249-280.
JELJASZEWICZ, J. (1983). Infections Caused by Staphylococci. Infection 11 (Suppl. 2), 109-111.
JENSEN, K. (1968). Methicillin-resistant staphylococci. Lancet ii: 1078.
JENSEN, K., LASSEN, H. C. A. (1969). Combined treatment with antibacterial chemotherapeutical agents in staphylococcal infections. Quarterly Journal of Medicine, New Series 38: 91-106.
JEPSEN, 0. B. (1986). The demise of the "old”methicillin-resistant Staphylococcus aureus. Journal of Hospital Infection 7: (Suppl. A), 13-17.
JEV0NS, M. P. (1961). nCelbeninM-resistantstaphylococci. British Medical Journal i: 124-125.
JEV0NS, M. P., COE, A. W., PARKER, M. T. (1963). Methicillin resistance in staphylococci. Lancet i: 904-907.
JOHNSON, A. P., UTTLEY, A. H., WOODFORD, N., GEORGE, R. C. (1990). Resistance to vancomycin and teicoplanin: an emerging clinical problem. Clinical Microbiology Reviews 3: 280-291.
276

JOHNSTON, B. L., KWOK, R. Y. Y., MULLIGAN, M. E. (1987). In vitro activity of novobiocin and rifampin alone and in combination against oxacillin-resistant Staphylococcus aureus. Diagnostic Microbiology and Infectious Diseases 8: 137-147.
JOLLY, J., GOLDBERG, M. (1989). Methicillin resistance in staphylococci: an evaluation of conditions for detection. Medical Laboratory Sciences 46: 2-5.
JONES, R. N., BARRY, A. L. (1989). In vitro evaluation of ramoplanin (A 16686 or MDL 62198). A new depsipeptide complex for potential topical use. Diagnostic Microbiology and Infectious Diseases 12:279-282.
JORDENS, J. Z., DUCKWORTH, G. J., WILLIAMS, R. J. (1989). Production of "virulence factors" by "epidemic" methicillin-resistant Staphylococcus aureus in vitro. Journal of Medical Microbiology 30: 245-252.
KAATZ, G. W., SEO, S. M., DORMAN, N. J., LERNER, S. A. (1990a). Emergence of teicoplanin resistance duringtherapy of Staphylococcus aureus endocarditis.Journal of Infectious Diseases 162: 103- 108.
KAATZ, G. W., SEO, S. M. (1990b). WIN 57273, a new fluoroquinolone with enhanced in vitro activity versus Gram-positive pathogens. Antimicrobial Agents and Chemotherapy 34: 1376-1380.
KAHAN, F. M., KAHAN, J. S., CASSIDY, P. J., KR0PP, H. (1974). The mechanism of action of fosfomycin (Phosphonomycin) . Annals of the New York Academy of Sciences 235: 364- 385.
KAPLAN, M. P., TENENBAUM, M. J. (1982). Staphylococcus aureus: Cellular Biology and Clinical Application. American Journal of Medicine 72: 243-258.
277

KAPUSNIK, J. E., PARENTI, F., SANDE, M. A. (1984). The use of rifarapicin in staphylococcal infections- a review. Journal of Antimicrobial Chemotherapy 13 (Suppl. C), 61-66.
KAYE, D. (1980). The clinical significance oftolerance of Staphylococcus aureus. Annals of Internal Medicine 93: 924-926.
KAYSER, F. H. (1975). Methicillin-resistantstaphylococci 1965-1975. Lancet ii: 650-653.
KAYSER, F., NOVAK, J. (1987). In vitro activity of ciprofloxacin against Gram-positive bacteria.American Journal of Medicine 82 (Suppl. 4A), 33-39.
KEPHART, P. A., ESPOSITO, A. L. (1988). Comparison of the investigational drug, LY 146032, with vancomycin in experimental pneumonia due to methicillin-resistant Staphylococcus aureus . Journal ofAntimicrobial Chemotherapy 21: 33-39.
KERR, S., KERR, G. E., MACKINTOSH, C. A., MARPLES, R. R. (1990). A survey of methicillin- resistant Staphylococcus aureus affecting patients in England and Wales. Journal of Hospital Infection 16: 35-48.
KHALIFA, K. I., HEIBA, A. A., HANCOCK, G. (1989). Nontypable bacteriophage patterns of methicillin- resistant Staphylococcus aureus involved in ahospital outbreak. Journal of Clinical Microbiology 27: 2249-2251.
KIDSON, A., LILLY, H. A., LOWBURY, E. J. L. (1979). Flucloxacillin treatment of methicillin-,,resistant" and sensitive staphylococcal infection. Journal of Antimicrobial Chemotherapy 5: 359-364.
278

KING, K., BRADY, L. M., HARKNESS, J. L. (1981). Gentamicin-resistant staphylococci. Lancet ii: 698-699.
KIRBY, W. M. M. (1944). Extraction of highly potent penicillin-inactivator from penicillin-resistantstaphylococci. Science 99: 452-3.
KIRBY, W. M. M., AHERN, J. J. (1953). Changing pattern of resistance of staphylococci to antibiotics. Antibiotics and Chemotherapy I I I : 831-835.
KIRBY, W. M. M., HUDSON, D. G., NOYES, W. D. (1956). Clinical and laboratory studies of novobiocin, a new antibiotic. Archives of Internal Medicine 98: 1-7.
KIRMANI, N., TUAZON, C. U., MURRAY, H. W. , PARRISH, A.E., SHEAGREN, J. N. (1978). Staphylococcus aureus carriage rate of patients receiving long-term haemodialysis. Archives of Internal Medicine 138: 1657-1659.
KLASTERSKY, J., HENSGENS, C., DANEAU, D. (1975). Therapy of staphylococcal infections: a comparative study of cephaloridine and gentamicin. American Journal ofthe Medical Sciences 269: 201-207.
KLASTERSKY, J., VAN DER AUWERA, P. (1986).Cephalosporins, vancomycin, aminoglycosides and other drugs, especially in combination, for the treatment of methicillin-resistant staphylococcal infections. Journal of Antimicrobial Chemotherapy 17: (Suppl. A) 19-24.
KNOX, R. (1960). A new penicillin (BRL 1241) active against penicillin-resistant staphylococci. British Medical Journal 2: 690-693.
KOCH, A. L. (1981). Evolution of antibiotic resistancegene function. Microbiological Reviews 45: 355-378.
279

KOJIMA, T., INOUE, M. , MITSUHASHI, S. (1989). In vitro activity of AT-4140 against clinical bacterial isolates. Antimicrobial Agents and Chemotherapy 34: 1123-1127.
KOJIMA, T., INOUE, M., MITSUHASHI, S. (1990). In vitro activity of AT-4140 against quinolone- andmethicillin- resistant Staphylococcus______aureus.Antimicrobial Agents and Chemotherapy 34: 1123-1127.
KOSMIDIS, J. (1988). Staphylococcal infections in hospital: the Greek experience. Abstract S4/ 2. 1st International Conference of the Hospital Infection Society, 1988.
KRESKEN, M., WIEDEMANN, B. (1986). Development ofresistance in the past decade in central Europe. Journal of Antimicrobial Chemotherapy 18: (Suppl. C), 235-242.
KUCERS , A. (1972). The Use of Antibiotics, aComprehensive Review with Clinical Emphasis. London, William Heinemann Books Ltd.
KUCERS, A. (1984). Vancomycin. Journal______ofAntimicrobial Chemotherapy 14: 564-567.
LACEY, R. W. (1975). Antibiotic resistance plasmidsof Staphylococcus aureus and their clinicalimportance. Bacteriological Reviews 39: 1-32.
LACEY, R. W. (1984). Antibiotic resistance inStaphylococcus aureus and streptococci. British Medical Bulletin 40: 77-83.
LACEY, R. W. (1987). Multi- resistant Staphylococcus aureus - a suitable case for inactivity? Journal of Hospital Infection 9: 103-105.
280

LACEY, R. W., MITCHELL, A. A. B. (1969). Gentamicin- resistant Staphylococcus aureus. Lancet ii: 1425-1426.
LACEY, R. W., ALDER, V. G., GILLESPIE, W. A. (1970). The survival of Staphylococcus aureus on human skin. An investigation using mixed cultures. British Journal of Experimental Pathology 51: 305-313.
LACEY, R. W., GRINSTEAD, J. (1973). Genetic analysis of methicillin- resistant strains of Staphylococcus aureus; evidence for their evolution from a single clone. Journal of Medical Microbiology 6: 511-526.
LACEY, R. W., STOKES, A. (1977). Susceptibility of the 'penicillinase- resistant' penicillins andcephalosporins to penicillinase of Staphylococcus aureus. Journal of Clinical Pathology 30: 35-39.
LACEY, R. W., STOKES, A. (1979). Studies on recently isolated cultures of methicillin-resistantStaphylococcus_____aureus . Journal_____ of_____GeneralMicrobiology 114: 329-339.
LACEY, R. W., LORD, V. L., H0WS0N, G. L. (1984). In vitro evaluation of miokamycin: bactericidal activityagainst streptococci. Journal of AntimicrobialChemotherapy 13: 5-13.
LAGAST, H., DODIAN, P., KLASTERSKY, J. (1986).Comparison of pharmacokinetics and bactericidal activity of teicoplanin and vancomycin. Journal of Antimicrobial Chemotherapy 18: 513-520.
LAU, W. Y., TEOH-CHAN, C. H., FAN, S. T., LAU, K. F.(1986). In vitro and in vivo study of fosfomycinin methicillin-resistant Staphylococcus____ aureussepticaemia. Journal of Hygiene, Cambridge 96: 419- 423.
281

LAURELL, G., WALLMARK, G. (1953). Studies on Staphylococcus aureus pyogenes in a children's hospital. Acta Pathologica et MicrobiologicaScandinavia 32: 438-447.
LEADING ARTICLE (1964). New Penicillins. BritishMedical Journal 2: 323-324.
LEADING ARTICLE (1965). Staphylococci resistant toneomycin and bacitracin. Lancet ii: 421-422.
LEADING ARTICLE (1968). Methicillin-resistantstaphylococci. Lancet ii: 759.
LEADING ARTICLE (1981). Of gentamicin and staphylococci. Lancet ii: 127-128.
LEAPER, D. J., SIMPSON, R. A. (1986). The effect of antiseptics and topical antimicrobials on wound healing. Journal of Antimicrobial Chemotherapy 17: 135-137.
LECLERCQ, R., NANTAS, L., SOUSSY, C. J., DUVAL, J.(1990). Activity of RP 59500 (RP), a new parenteral semisynthetic streptogramin (Sg) againstStaphylococcus aureus strains according tomechanisms of resistance to macrolide-lincosamide- streptogramin antibiotics (MLS). Abstracts of the 30th International Conference on Antimicrobial Agents and Chemotherapy (Atlanta), Abstract no. 786.
LE GOFFIC, F., CAPMAU, M. L., BONNET, D., CERCEAU, C.,SOUSSY, C., DUBLANCHET, A., DUVAL, J. (1977). Plasmid-mediated pristinamycin resistance PAC IIA: a newenzyme which modifies Pristinamycin IIA. Journal ofAntibiotics XXX: 665-669.
282

LEIGH, D., BINT, A., GOULD, I. (Eds). Fleroxacin, a long acting fluoroquinolone with broad spectrum activity. Journal of Antimicrobial Chemotherapy 22 (Supplement D ), 1988.
LEPPER, M. H., MOULTON, B., DOWLING, H. F., JACKSON, G.C, KOFMAN, S. (1954). Epidemiology of erythromycin- resistant staphylococci in a hospital population -effect on therapeutic activity of erythromycin.Antibiotics Annual 1953-1954, 308-313 (1954).
LEWIN, C., SMITH, J. T. (1988). Bactericidal mechanisms of ofloxacin. Journal of Antimicrobial Chemotherapy22 (Suppl. C ), 1-8.
LIMB, D. I., DABBS, D. J. W., SPENCER, R. C. (1987). In- vitro selection of bacteria resistant to the 4-quinolone agents. Journal_____of_____AntimicrobialChemotherapy 19: 65-71.
LINNEMANN Jr. C. C., MASON, M., MOORE, P., KORFHAGEN, T. R., STANECK, J. L. (1982). Methicillin- resistantStaphylococcus aureus: experience in a generalhospital over four years. American Journal ofEpidemiology 115: 941- 950.
LOCKSLEY, R. M., COHEN, M. L., QUINN, T. C., TOMPKINS, L.S., COYLE, M. B., KIRIHARA, J. M., COUNTS, G. W. (1982). Multiply antibiotic-resistant Staphylococcus aureus: Introduction, transmission, and evolution ofnosocomial infection. Annals of Internal Medicine 97: 317-324.
LOWBURY, E. J. L., CASON, J. S., MacG. JACKSON, D., MILLER, R. W. S. (1962). Fucidin for staphylococcal infection of burns. Lancet ii: 478-480.
283

LOWBURY, E. J. L., BABB, J. R., BROWN, V. I., COLLINS, B. J. (1964). Neomycin-resistant Staphylococcus aureus in a burns unit. Journal of Hygiene, Cambridge 62: 221-228.
LOWBURY, E. J. L., LILLY, H. A., KIDSON, A. (1977)."Methicillin- resistant" Staphylococcus______ aureus:reassesment by controlled trial in burns unit. British Medical Journal 1: 1054-1056.
LOWBURY, E. J. L., AYLIFFE, G. A. J., GEDDES, A. M.,WILLIAMS, J. D. (1982). Control of hospitalinfection, a practical handbook. 2nd edition, Chapman & Hall Ltd. (London).
LYON, B. R., GILLESPIE, M. T., BYRNE, M. E., MAY, J. W., SKURRAY, R. A. (1987). Plasmid- mediated resistance to gentamicin in Staphylococcus aureus: the involvement of a transposon. Journal of Medical Microbiology 23: 101-110.
LYON, B. R., SKURRAY, R. (1987). Antimicrobial resistance of Staphylococcus aureus: genetic basis.Microbiological Reviews 51: 88-134.
MACFARLANE, J. A., MITCHELL, A. A. B., WALSH, J. M. , ROBERTSON, J. J. (1968). Spiramycin in the prevention of postoperative staphylococcal infection. Lancet i: 1-4.
MADIRAJU, M. V. V. S., BRUNNER, D. P., WILKINSON, B. J.(1987). Effects of temperature, NaCl, and methicillin on penicillin-binding proteins, growth, peptidoglycan synthesis, and autolysis in methicillin-resistant Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 31: 1727-1733.
284

MAPLE, P. A. C., HAMILTON-MILLER, J. M. T., BRUMFITT, W. (1989a). Comparative in vitro activity of vancomycin, teicoplanin, ramoplanin (formerly A 16686), paldimycin, DuP 721 and DuP 105 against methicillinand gentamicin resistant Staphylococcus aureus.Journal of Antimicrobial Chemotherapy 23: 517-525.
MAPLE, P. A. C., HAMILTON-MILLER, J. M. T., BRUMFITT, W. (1989b). World-wide antibiotic resistance in methicillin- resistant Staphylococcus aureus. Lancet i: 537-540.
MAPLE, P. A. C., HAMILTON-MILLER, J. M. T., BRUMFITT, W. (1989c). Ciprofloxacin resistance in methicillin- and gentamicin-resistant Staphylococcus aureus. European Journal of Clinical Microbiology and InfectiousDiseases 8: 622-623.
MAPLE, P., BRUMFITT, W., HAMILTON-MILLER, J. M. T.(1990d). A review of the antimicrobial activity ofthe fluoroquinolones. Journal of Chemotherapy 2:280-294.
MARANDON, J. L., OEDING, P. (1966). Investigations onanimal Staphylococcus aureus strains. 1. Biochemical characteristics and phage typing. Acta Pathologica et Microbiologica Scandinavia 67: 149-156.
MARANDON, J. L., OEDING, P. (1967). Investigations onanimal Staphylococcus aureus strains. 2. Antigens.Acta Pathologica et Microbiologica Scandinavia 70: 300-304.
MARKOWITZ, N., P0HL0D, D. J., SARAVOLATZ, L. D., QUINN, E. L. (1983). In vitro susceptibility patterns of methicillin- resistant and - susceptible Staphylococcus aureus strains in a population of parenteral drug abusers from 1972- 1981. Antimicrobial Agents and Chemotherapy 23: 450-457.
285

MARPLES, R. R., RICHARDSON, J. F., de SAXE, M. (1986). Bacteriological characters of strains ofStaphylococcus aureus submitted to a referencelaboratory related to methicillin resistance. Journal of Hygiene, Cambridge 96: 217-223.
MARQUES, A. R., PETRILLO, V., HOEFEL, H. (1989).Methicillin- resistant Staphylococcus aureus in ageneral hospital in Brazil. Journal of HospitalInfection 14: 380-381.
MASKELL, J. P., SEFTON, A. M. , YONG, J., CHI, S. J., WILLIAMS, J. D. (1988). Comparative in vitro activityof erythromycin, vancomycin and pristinamycin.Infection 16: 365-370.
MATHEWS, P. R., STEWART, P. R. (1984). Resistanceheterogeneity in methicillin-resistant Staphylococcus aureus. FEMS Microbiology Letters 22: 161-166.
MAYON-WHITE, R.T., DUCEL, G., KERESELIDZE, T., TIKOMIROV, E. (1988). An international survey of the prevalence of hospital-acquired infection. Journalof Hospital Infection 11 (Suppl. A): 43-48.
MCDONALD, M., HURSE, A., SIM, K. N. (1981). Methicillin- resistant Staphylococcus aureus bacteraemia. Medical Journal of Australia 2: 191-194.
McDOUGAL, L. K., THORNSBERRY, C. (1984). Newrecommendations for disk diffusion antimicrobial susceptibility tests for methicillin-resistant(heteroresistant) staphylococci. Journal of Clinical Microbiology 19: 482-488.
McGOWAN Jr., J. E. (1988). Gram-positive bacteria: spread and antimicrobial resistance in university and community hospitals in the USA. Journal of Antimicrobial Chemotherapy 21: (Suppl. C) , 49-55. 286

McOSKER, C. C., POLLACK, J. R., ANDERSEN, J. A. (1990). Inhibition of bacterial protein synthesis bynitrofurantoin macrocrystals: an explanation for thecontinued efficacy of nitrofurantoin Iii HARRISON, L.H. (ed.), Management of Urinary Tract Infections.Royal Society of Medicine Services International Congress and Symposium Series No. 154.
MEDICAL JOURNAL OF AUSTRALIA (1982). Methicillin- resistant Staphylococcus aureus. Medical Journal ofAustralia 1: 445-480.
MEHTAR, S., DRABU, Y. J., MAYET, F. (1989). Expenses incurred during a 5 week epidemic methicillin- resistant Staphylococcus aureus outbreak. Journal ofHospital Infection 13: 199-200.
MELO CRISTINO, J. A. G., TORRES PEREIRA, A., AFONSO, F.(1985). Infection with methicillin- gentamicin- resistant Staphylococcus aureus strains in apaediatric surgery unit in Lisbon. Journal ofHospital Infection 6: 426-428.
MILATOVIC, D. (1986). Vancomycin for treatment ofinfections with methicillin- resistant Staphylococcus aureus: are there alternatives? European Journal of Clinical Microbiology 5: 689-692.
MILLER, G. H., SABATELLI, F. J., HARE, R. S., WAITZ, J. A. (1980). Survey of aminoglycoside resistance patterns. Developments in Industrial Microbiology 21: 90-104.
MILNE, L. M., FAIERS, M. C. (1988). Letter to the editor. Lancet ii: 843.
287

MINUTH, J. N., HOLMES, T.M., MUSHER, D. M. (1974). Activity of tetracycline, doxycycline, and minocycline against methicillin- susceptible and -resistant staphylococci. Antimicrobial Agents and Chemotherapy 6: 411-414.
MITCHELL, A. A. B. (1964). New epidemic strain of Staphylococcus aureus. Emergence and spread in a General Hospital. Lancet i: 859-862.
MITSUHASHI, S., INOUE, M., INOUE, K. (1990). In vitro evaluation of RP 59500 (a streptogramin family). Abstracts of the 30th International Conference on Antimicrobial Agents and Chemotherapy (Atlanta), Abstract no. 772.
MOLLBY, R., WADSTROM, T. (1973). Purification of staphlyococcal beta-, gamma- and delta-hemolysins In: JELJASZEWICZ, J., HRYNIEWICZ, W. (eds) Staphylococci and Staphylococcal Infections. Warsaw, Polish Medical Publishers, 298-313.
MOLLBY, R. (1983). Isolation and properties of membrane damaging toxins Ijas EASMON C. S. F., ADLAM,C. (eds) Staphylococci and Staphylococcal Infections. The organism in vivo and in vitro. Academic Press (London), 619-669.
MONTAY, G., BRUNO, R., THEBAULT, J. J., VERGINOL, J. C., CHASSARD, D . , EBMEIER, M., GAILLOT, J. (1990). Dose- dependent pharmacokinetic study of sparfloxacin (SPX) in healthy young volunteers. Abstracts of the 30th International Congress on Antimicrobial Agents and Chemotherapy, Atlanta, Abstract no. 1248.
288

MONTEFIORE, D., ROTIMI, V. 0., ADEYEMI-DORO, F. A. B.(1989). The problem of bacterial resistance to antibiotics among strains isolated from hospital patients in Lagos and Ibadan, Nigeria. Journal of Antimicrobial Chemotherapy 23: 641-651.
MONTIEL, F., KALTWASSER, G., VALDIVIESO, C., LAM, M.(1988). In vitro susceptibility to 10 antibiotics of methicillin- resistant (MRSA) Staphylococcus aureus in Chile. Diagnostic Microbiology and Infectious Diseases 10: 145- 148.
MOORMAN, D. R., MANDELL, G. L. (1981). Characteristics of rifampin-resistant variants obtained fromclinical isolates of Staphylococcus____ aureus.Antimicrobial Agents and Chemotherapy 20: 709- 713.
MORGAN, M. G., HARTE- BARRY, M. J. (1989). Methicillin- resistant Staphylococcus aureus: a ten- year survey in a Dublin hospital. Journal of Hospital Infection 14: 357- 362.
M0UT0N, R. P., MULDERS, S. L. T., de KNIFF, J., HERMANS, J. (1989). Comparison of test systems for recognition of methicillin resistance inStaphylococcus aureus. European Journal of Clinical Microbiology and Infectious Diseases 8: 968-973.
MULLIGAN, M. E., RUANE, P. J., JOHNSTON, L., WONG, P., WHEEL0CK, J. P., MacDONALD, K., REINHARDT, J. F., JOHNSON, C. C., STATNER, B., BLOMQUIST, I., McCARTHY, J., O ’BRIEN, W., GARDNER, S., CITRON, D.M.(1987). Ciprofloxacin for eradication ofmethicillin-resistant Staphylococcus________aureuscolonization. American Journal of Medicine 82,(Suppl. 4A), 215-219.
289

MURAKAMI, K., TOMASZ, A. (1989). Involvement of multiple genetic determinants in high-level methicillin resistance in Staphylococcus aureus. Journal of Bacteriology 171: 874-879.
NEU, H. C. (1974). Gentamicin, an overview In: JACKSON, G. G., ESCARZAGA, E., GARROD, L. P., NEU, H., UEDA, Y. (eds). Gentamicin; review and commentary on selected world literature. New York, Council for Interdisciplinary Communication in Medicine, pp 179-191.
NEU, H. C. (1987). Ciprofloxacin: an overview andprospective appraisal. American Journal of Medicine 82, (Suppl. 4A), 395-404.
NEU, H. C., NEU, N. M. (1986). In vitro activity of A 16686, a new glycopeptide. Chemotherapy 32: 453-457.
NEU, H. C., NOVELLI, A., SAHA, G., CHIN, N-X. (1988). In vitro activities of two oxazolidinone antimicrobial agents, DuP 721 and DuP 105. Antimicrobial Agents and Chemotherapy 32: 580-583.
NOBLE, W. C., WILLIAMS, R.E.O., JEVONS, M. P., SHOOTER, R. A. (1964). Some aspects of nasal carriage of staphylococci. Journal of Clinical Pathology 17: 79-83.
NOBLE, W. C., VALKENBURG, H. A., WOLTERS, C. H. L. (1967). Carriage of Staphylococcus aureus in random samples of a normal population. Journal of Hygiene, Cambridge 65: 567-573.
0GST0N, A. (1882). Micrococcus Poisoning. Journal of Anatomy (London) 17: 24-58.
O ’HARE, M. D., GHOSH, G., FELMINGHAM, D., GRUNEBERG, R.N. (1990). In vitro studies with ramoplanin (MDL62198); a novel lipoglycopeptide antimicrobial.Journal of Antimicrobial Chemotherapy 25: 217-220.
290

OUNISSI, H., DERLOT, E., CARLIER, C., COURVALIN, P.(1990). Gene homogeneity for aminoglycoside-modifying enzymes in Gram-positive cocci. Antimicrobial Agents and Chemotherapy 34: 2164-2168.
PALLANZA, R., BERTI, M., SCOTTI, R., RANDISI, E., ARIOLI, V. (1984). A-16686, a new antibiotic fromActinoplanes. II. Biological properties. Journal of Antibiotics 37: 318-324.
PARKER, M. T., HEWITT, J. H. (1970). Methicillin resistance in Staphylococcus aureus. Lancet i: 800-804.
PARKER, M. T., ASHESHOV, E. H., HEWITT, J. H., NAKHLA, L.S., BROCK, B. M. (1974). Endemic staphylococcal infections in hospitals. Annals of the New YorkAcademy of Sciences 236: 466-484.
PARKER, M. T. (1983). Staphylococcus and Micrococcus; the anaerobic gram-positive cocci JLn: WILSON, G. S., MILES, A. A., PARKER, M. T. (ed.) Topley and Wilson’s principles of bacteriology, virology and immunity. London, Edward Arnold, 218-245.
PATTIS0N, J. R., MANSELL, P. E. (1973). Fucidin-resistant staphylococci in current hospitalpractice. Journal of Medical Microbiology 6: 235-244.
PAUL, J. (1833). Case of congenital fungus haematodes in which amputation of the thigh was performed in the tenth week of the child’s life.Lancet i: 439-445.
PAVILLARD, R., HARVEY, K., DOUGLAS, D., HEWST0NE, A.,ANDREW, J., COLLOPY, B., ASCHE, V., CARSON, P., DAVIDSON,A., GILBERT, G., SPICER, J., T0S0LINI, F. (1982).Epidemic of hospital-acquired infection due tomethicillin-resistant Staphylococcus aureus in majorVictorian hospitals. Medical Journal of Australia 1:
291

451-454.
PEACOCK, J. E., MARSIK, F. J., WENZEL, R. P. (1980).Methicillin-resistant Staphylococcus_______ aureus:Introduction and spread within a hospital. Annals of Internal Medicine 93: 526-532.
PEARMAN, J. W., CHRISTIANSEN, K. J., ANNEAR, D. I., GOODWIN, C. S., METCALF, C., DONOVAN, F. P., MACEY, K. L., BASSETTE, L. D., POWELL, I. M., GREEN, J. M., HARPER, W. E., McKELVIE, M. S. (1985). Control of methicillin-resistant Staphylococcus aureus (MRSA) in anAustralian metropolitan teaching hospital complex. Medical Journal of Australia 142: 103-108.
PELLETIER, L. L. (1984). Lack of reproducibility ofmacrodilution MBCs for Staphylococcus aureus.Antimicrobial Agents and Chemotherapy 26: 815-818.
PETIT, J-C, COHEN, F., COUCHOUD, S., DAGUET, G. L.(1983). Staphylococcus aureus oxacilline etgentamicine-resistants: survenue et evolution sur une periode de 6 mois. Biomedicine & Pharmacotherapy 37: 429-433.
PIPER, J., HADFIELD, T., McCLESKEY, F., EVANS, M.,FRIEDSTROM, S., LAUDERDALE, P., WINN, R. (1988). Efficacies of Rapid Agglutination Tests for Identification of Methicillin-Resistant StaphylococcalStrains as Staphylococcus aureus. Journal ofClinical Microbiology 26: 1907-1909.
P0CHEE, E., CREWE-BROWN, H. H. (1988). Methicillinresistant Staphylococcus aureus at Ga-Rankuwahospital. Abstract 09/ 4. 1st____ InternationalConference of the Hospital Infection Society, 1988.
292

POHLOD, D. J., SARAVOLATZ, L. D., SOMERVILLE, M. M.(1987). In vitro susceptibility of Gram-positive cocci to LY 146032, teicoplanin, sodium fusidate, vancomycin and rifampicin. Journal of Antimicrobial Chemotherapy 20: 197-202.
P0RTH0USE, A., BROWN, D. F. J., GRAEME SMITH, R., ROGERS, T. (1976). Gentamicin resistance in Staphylococcus aureus. Lancet i: 20-21.
PUTLAND, R. A., GUINNESS, M. D. (1985). Autobac susceptibility testing of methicillin-resistant Staphylococcus aureus isolated in an Australian hospital. Journal of Clinical Microbiology 22: 822-827.
QUIE, P. G., HILL, H. R., TODD DAVIS, A. (1974). The Defective Phagocytosis of Staphylococci. Annals of the New York Academy of Sciences 236: 233-243.
RAHMAN, M., NOBLE, W. C., C00KS0N, B. (1987). Mupirocin- resistant Staphylococcus aureus. Lancet ii: 387.
RAHMAN, M., CONNOLLY, S., NOBLE, W. C., C00KS0N, B., PHILLIPS, I. (1990). Diversity of staphylococci exhibiting high-level resistance to mupirocin. Journal of Medical Microbiology 33: 97-100.
RAMMELKAMP, C. H., MAX0N, T. (1942). Resistance of Staphylococcus aureus to the action of Penicillin. Proceedings of the Society for Experimental Biology and Medicine 51: 386-389.
RAVIGLIONE, M. C., BOYLE, J. F., MARIUZ, P., PABL0S- MENDEZ, A.,CORTES, H., MERL0, A. (1990). Ciprofloxacin- resistant methicillin-resistant Staphylococcus aureus in an acute-care hospital. Antimicrobial Agents and Chemotherapy 34: 2050-2054.
293

REPORT (1986). Guidelines for the control ofepidemic methicillin-resistant Staphylococcus aureus. Journal of Hospital Infection 7: 193-201.
RICHARDS, F., McCALL, C., COX, C. (1971). Gentamicin treatment of staphylococcal infections. Journal of the American Medical Association 215: 1297-1300.
RICHARDSON, J. F., CHITTASOBHON, N., MARPLES, R. R.(1988). Supplementary phages for the investigationof methicillin-resistant Staphylococcus aureus.Journal of Medical Microbiology 25: 67-74.
RIDLEY, M., BARRIE, D., LYNN, R., STEAD, K. C. (1970).Antibiotic-resistant Staphylococcus_____ aureus andhospital antibiotic policies. Lancet i: 230-233.
RIMLAND, D. (1987). Nosocomial infections withmethicillin and tobramycin resistant Staphylococcusaureus: implications of physiotherapy in hospital-wide dissemination. American Journal of the MedicalSciences 290: 91-97.
RODE, H., HANSL0, D., DE WET, P. M., MILLAR, A. J. W., CYWES, S. (1989). Efficacy of mupirocin inmethicillin-resistant Staphylococcus_____ aureus burnwound infection. Antimicrobial______ Agents____ andChemotherapy 33: 1358-1361.
RODRIGUEZ, A., OLAY, T., VICENTE, M. V. (1986). Invitro activity of fosfomycin alone and combinedagainst methicillin- resistant staphylococci In.: Fosfomycin, Proceedings of the internationalsymposium. Instituto de Farmacologia Espaniola (CEPA), Madrid, 1987.
R0G0LSKY, M. (1979). Nonenteric Toxins ofStaphylococcus aureus. Microbiological Reviews 43: 320-360.
294

ROLINSON, G. N. (1961). "Celbenin"-resistantstaphylococci. British Medical Journal 2: 125-126.
ROLSTON, K. V. I., LeBLANC, B., HO, D. H., BODEY, G. P. (1987). In vitro activity of paldimycin (U- 70138F) against Gram-positive bacteria isolated from patients with cancer. Antimicrobial Agents and Chemotherapy 31: 650-652.
ROSENDAL, K., BULOW, P., BENTZON, M. W., ERIKSEN, K. R. (1976). Staphylococcus aureus strains isolated in Danish hospitals from January 1st, 1966, to December31st, 1974. Acta_____ Pathologica MicrobiologicaScandinavica Section B 84: 359-368.
ROUNTREE, P. M., THOMSON, E. F. (1949). Penicillin- resistant and streptomycin-resistant staphylococci in a hospital. Lancet ii: 501-504.
ROUNTREE, P. M., BARBOUR, R. G. H., THOMSON, E. F. (1951). Incidence of penicillin-resistant and streptomycin-resistant staphylococci in a hospital. Lancet i: 435-436.
ROUNTREE, P. M., FREEMAN, B. M. (1955).Infections caused by a particular phage type of Staphylococcus aureus. Medical Journal of Australia 2: 157-161.
ROUNTREE, P. M., FREEMAN, B. M., JOHNSTON, K. G. (1956). Nasal carriage of Staphylococcus aureus by variousdomestic and laboratory animals. Journal ofPathology and Bacteriology 72: 319-321.
ROUNTREE, P. M., BEARD, M. A. (1965). The spread of neomycin-resistant staphylococci in a hospital. Medical Journal of Australia i: 498-502.
295

SARAVOLATZ, L. D., MARKOWITZ, N., ARKING, L. M., POHLOD,D., FISHER, E. (1982). Methicillin-resistantStaphylococcus aureus. Epidemiologic observationsduring a community acquired outbreak. Annals of Internal Medicine 96: 11-16.
SAROGLOU, G., CROMER, M., BISNO, A. L. (1980).Methicillin- resistant Staphylococcus______aureus:interstate spread of nosocomial infections withemergence of gentamicin- methicillin- resistantstrains. Infection Control 1: 81-89.
SCHAEFLER, S. (1982). Bacteriophage-mediatedacquisition of antibiotic resistance byStaphylococcus aureus type 88. Antimicrobial Agents and Chemotherapy 21: 460-467.
SCHAEFLER, S. (1989). Methicillin-resistant strains ofStaphylococcus aureus resistant to quinolones.Journal of Clinical Microbiology 27: 335-336.
SCHAEFLER, S., JONES, D., PERRY, W., RUVINSKAYA, P. L., BARADET, T., MAYR, E., WILSON, M. E. (1981). Emergence of gentamicin- and methicillin -resistantStaphylococcus aureus strains in New York Cityhospitals. Journal of Clinical Microbiology 13:754-759.
SCHAEFLER, S., JONES, D., PERRY, W., BARADET, T., MAYR,E., RAMPERSAD, C. (1984). Methicillin- resistant Staphylococcus aureus strains in New York Cityhospitals: inter- hospital spread of resistantstrains of type 88. Journal of ClinicalMicrobiology 20: 536-538.
SCHITO, G. C., VARALDO, P. E. (1988). Trends in theepidemiology and antibiotic resistance of clinicalStaphylococcus strains in Italy. Journal__ofAntimicrobial Chemotherapy 21: (Suppl. C), 67- 78.
296

SCHNEIERSON, S. S. (1960). 1959 bacterialsusceptibility to nitrofurantoin as compared to 1953 to 1955. Antibiotics and Chemotherapy 10: 747-752.
SCHLEIFER, K. H. (1986). In BUTLER, J.P. (ed.), Bergey's Mannual of Systematic Bacteriology. Baltimore,Williams & Wilkins, p 1003.
SHALIT, I., BERGER, S. A., GOREA, A., PRIMERNMAN, H.(1989). Widespread quinolone resistance amongmethicillin-resistant Staphylococcus aureus isolates in a general hospital. Antimicrobial Agents andChemotherapy 33: 593-594.
SHANSON, D. C. (1981). Antibiotic- resistantStaphylococcus aureus. Journal of Hospital Infection2: 11-36.
SHANSON, D. C. (1990). Clinical relevance ofresistance to fusidic acid in Staphylococcusaureus. Journal of Antimicrobial Chemotherapy 25 (Suppl. B), 15-21.
SHANSON, D. C. KENSIT, J. G., DUKE, R. (1976). Outbreak of hospital infection with a strain of Staphylococcus aureus resistant to gentamicin andmethicillin. Lancet ii: 1347-1348.
SHAW, C., STITT, J.M., COWAN, S.T. (1951). Staphylococci and their Classification. Journal of General Microbiology 5: 1010-1023.
SHAW, W. V. (1984). Bacterial resistance to chloramphenicol. British Medical Bulletin 40: 36-41.
SLEE, A. M., WU0N0LA, M. A., McRIPLEY, R. J., ZAJAC, I.,ZAWADA, M. J., BARTHOLOMEW, P. T., GREGORY, W. A.,FORBES, M. (1987). Oxazolidinones, a new class of
297

synthetic antibacterial agents: in vitro and invivo activities of DuP 105 and DuP 721. Antimicrobial Agents and Chemotherapy 31: 1791- 1797.
SKURRAY, R. A., ROUCH, D. A., LYON, B. R., GILLESPIE, M. T., TENNENT, J. M., BYRNE, M. E., MESSEROTTI, L. J., MAY, J. W. (1988). Multi- resistant Staphylococcus aureus: genetics and evolution of Australian strains. Journal of Antimicrobial Chemotherapy 21: (Suppl. C) , 19-38.
SMITH, J. T. (1984). Awakening the slumbering potential of the 4-quinolone antibacterials. The Pharmaceutical Journal 233: 299-305.
SMITH, J. T. (1990). Mutation rates to 4-quinolone resistance. Arzneimittel-Forschung/Drug Research 40: 1- 1 1 .
SMITH, S. M. (1986). In vitro comparison of A- 56619, A-56620, amifloxacin, ciprofloxacin, enoxacin,norfloxacin, and ofloxacin against methicillin- resistant Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 29: 325-326.
SMITH, S. M., ENG, R. H. K. (1985). Activity of ciprofloxacin against methicillin-resistantStaphylococcus aureus. Antimicrobial Agents and Chemotherapy 27: 688-691.
SMITH, S. M., ENG, R. H. K., BERMAN, E. (1986). The effect of ciprofloxacin on methicillin-resistant Staphylococcus aureus. Journal of Antimicrobial Chemotherapy 17: 287-295.
SMITH, S. M., MANGIA, A., ENG, R. H. K., RUGGERI, P., CYTRYN, A., TECSON-TUMANG, F. (1988). Clindamycin for colonization and infection by methicillin-resistant Staphylococcus aureus. Infection 16: 95-97.
298

SMITH, S. M., ENG, R. H. K., BAIS, P., FAN-HAVARD, P., TECSON-TUMANG, F. (1990). Epidemiology ofciprofloxacin-resistance among patients withmethicillin-resistant Staphylococcus aureus. Journal of Antimicrobial Chemotherapy 26: 567-572.
SOMPOLINSKY, D., SAMRA, Z., KARAKAWA, W. W., VANN, W. F., SCHNEERSON, R., MALIK, Z. (1985). Encapsulation and Capsular Types in Isolates of Staphylococcus aureus from different Sources and Relationship to Phage Types. Journal of Clinical Microbiology 22: 828-834.
SORRELL, T. C., PACKHAM, D. R., SHANKER, S., FOLDES, M., MUNRO, R. (1982). Vancomycin therapy for methicillin- resistant Staphylococcus aureus. Annals of Internal Medicine 97: 344-350.
SOUSSY, C. J., BOUANCHAUD, D. H., FOUACE, J., DUBLANCHET, A., DUVAL, J. (1975). A gentamycin resistance plasmid in Staphylococcus aureus. Annales de Microbiologie 126 B: 91-94.
SOUSSY, C. J., DUBLANCHET, A., CORMIER, M., BISMUTH, R., MIZON, F., CHARDON, H., DUVAL, J., FABIANI, G. (1976). Nouvelles resistances plasmidiques de Staphylococcus aureus aux aminosides (gentamicine, tobramycine, amikacine). Nouvelle Presse Medicale 5: 2599-602.
SPELLER, D. C. E., RAGHUNATH, D., STEPHENS, M., VIANT, A.C., REEVES, D. S., WILKINSON, P. J., BROUGHALL, J. M. , HOLT, H. A. (1976). Epidemic infection by a gentamicin-resistant Staphylococcus aureus in three hospitals. Lancet i: 464-466.
SPINK, W. W. (1954). Staphylococcal infections and the problem of antibiotic-resistant staphylococci. Archives of Internal Medicine 94: 167-196.
299

SPINK, W. W., HALL, W. H. (1945). Penicillin therapy at the University Hospitals. Annals of Internal Medicine 22: 510-525.
SPRATT, B. G. (1978). Escherichia coli resistance to B-lactam antibiotics through a decrease in the affinity of a target for lethality. Nature 274:713-715.
STEWART, G. T., NIXON, H. H., COLES, H. M. T. (1960). Report on clinical use of BRL 1241 in children with staphylococcal and streptococcal infections. British Medical Journal 2: 703-706.
STEWART, G. T., HOLT, R. J. (1963). Evolution of natural resistance to the newer penicillins. British Medical Journal 1: 308-311.
ST0RRS, M. J., C0URVALIN, P., FOSTER, T. J. (1988). Genetic analysis of gentamicin resistance in methicillin- and gentamicin- resistant strains of Staphylococcus aureus isolated in Dublin hospitals. Antimicrobial Agents and Chemotherapy 32: 1174-1181 .
SUBCOMMITTEE (1965). Subcommittee on Taxonomy of Staphylococci and Micrococci - Minutes of First Meeting. International Bulletin of Bacteriological Nomenclature and Taxonomy 15: 107-108.
SUTHERLAND, R., R0LINS0N, G. N. (1964). Characteristics of methicillin-resistant staphylococci. Journal of Bacteriology 87: 887-899.
SYKES, R. B., MATTHEW, M. (1976). The B-lactamases of Gram-negative bacteria and their role in resistance to B-lactam antibiotics. Journal of Antimicrobial Chemotherapy 2: 115-157.
300

TENNENT, J. M., LYON, B. R., GILLESPIE, M. T., MAY, J. W., SKURRAY, R. A. (1985). Cloning and expression of Staphylococcus aureus plasmid-mediated quaternaryammonium resistance in Escherichia_____ coli.Antimicrobial Agents and Chemotherapy 27: 79-83.
TENNENT, J. M., MAY, J. W., SKURRAY, R. A. (1986). Characterisation of chloramphenicol resistance plasmids of Staphylococcus aureus and S. epidermidis by restriction enzyme mapping techniques. Journal of Medical Microbiology 22: 79-84.
THELESTAM, M. (1983). Modes of membrane damagingaction of staphylococcal toxins I_n: EASMON, C. S.F., ADLAM, C. (eds) Staphylococci and staphylococcal infections. London, Academic Press, 2: 705-744.
THOMPSON R. E. M., HARDING, J. W. , SIMON, R. D. (1960).Sensitivity of Staphylococcus_____pyogenes tobenzyl penicillin and BRL 1241. British MedicalJournal 2: 708-709.
TIMBURY, M. C., WILSON, T. S., HUTCHINSON, J. G. P., GOVAN, A. D. T. (1958). A staphylococcus type-80 epidemic in a maternity hospital illustrating some special features. Lancet ii: 1081-1084
T0MASZ, A., DRUGE0N, H. B., deLENCASTRE, H. M., JABES,D., McDOUGAL, L., BILLE, J. (1989). New mechanism for methicillin resistance in Staphylococcus aureus: clinical isolates that lack the PBP 2a gene and contain normal penicillin-binding proteins with modified penicillin-binding capacity. Antimicrobial Agents and Chemotherapy 33: 1869-1874.
T0PLEY, W. W. C., WILSON, G. S. (1936). The principles of bacteriology and immunology. Second Edition, London, Edward Arnold & Co.
301

TOSCANO, W. A., STORM, D. R. (1982). Bacitracin. Pharmacology and Therapeutics 16: 199-210.
TOWNSEND, D. E., ASHDOWN, N., BRADLEY, J. M., PEARMAN, J. W., GRUBB, W. B. (1984). "Australian” methicillin-resistant Staphylococcus aureus in a Londonhospital? Medical Journal of Australia 141: 339-340.
TOWNSEND, D. E., ASHDOWN, N., BOLTON, S., BRADLEY, J., DUCKWORTH, G., M00RH0USE, E. C., GRUBB, W. B. (1987). The International spread of methicillin-resistant Staphylococcus aureus. Journal of Hospital Infection 9: 60-71.
TRALLERO, E. P., ARENZANA, J. M. G., EGUILUZ, G. C., CANCER, R. C. (1988). Prevalence of methicillin-resistant Staphylococcus aureus in a Spanishhospital. Reviews of Infectious Diseases 10: 627-628.
TRAUB, W., SPOHR, M., BAUER, D. (1984). Gentamicin and methicillin resistant, clinical isolates ofStaphylococcus aureus: comparative in vitro and in vivo efficacy of alternative antimicrobial drugs. Chemotherapy 30: 102-112.
TUAZ0N, C. U., PEREZ, A., KISHABA, T., SHEAGREN, J. N. (1975). Staphylococcus aureus among insulin-injecting diabetic patients: an increased carrier rate.Journal of the American Medical Association 231: 1272.
TURNIDGE, J., LAWSON, P., MUNRO, R., BENN, R. (1989). A national survey of antimicrobial resistance inStaphylococcus aureus in Australian teachinghospitals. Medical Journal of Australia 150: 65-72.
UBUKATA, K., ITOH-YAMASHITA, N., KONNO, M. (1989).Cloning and expression of the nor A gene forfluoroquinolone resistance in Staphylococcus aureus.Antimicrobial Agents and Chemotherapy 33: 1535-1539.
302

VAN DER AUWERA, P. (1989). Ex vivo study of serum bactericidal titers and killing rates of daptomycin (LY 146032) combined or not combined with amikacin compared with those of vancomycin. Antimicrobial Agents and Chemotherapy 33: 1783-1790.
VAN CAEKENBERGHE, D. L., PATTYN, S. R. (1987). Reversed incomplete cross-resistance among the older andnewer quinolone antibiotics. Journal______ ofAntimicrobial Chemotherapy 19: 404.
VAN DE KLUNDERT, J. M., VIEGENTHART, J. S., VAN D00RN,E., BONGAERTS, G. P. A., MOLENDIJK, L., M0UT0N, R. P.(1984). A simple method for the identification ofaminoglycoside- modifying enzymes. Journal____ ofAntimicrobial Chemotherapy 14: 339-348.
VARALDO, P. E., CIPRIANI, P., FOCA, A., GERACI, C.,GIORDANO, A., MADEDDU, M. A., ORSI, A., POMPEI, R.,PRENNA, M., REPETTO, A. et al. (1984). Identification, clinical distribution, and susceptibility tomethicillin and 18 additional antibiotics ofclinical Staphylococcus isolates: nationwideinvestigation in Italy. Journal of ClinicalMicrobiology 19: 838-843.
VERBIST, L. (1990). The antimicrobial activity of fusidic acid. Journal of Antimicrobial Chemotherapy 25 (Suppl. B), 1-5.
VERHOEF, J., VERBRUGH, H. A. (1981). Host Determinants in Staphylococcal Disease. Annual Reviews of Medicine 32: 107-122.
WADSTR0M, T. (1983). Biological effects damaging toxins JEn: EASM0N, C. S. F.,Staphylococci and staphylococcal infections Academic Press, 2: 671-704.
303
of cell ADLAM, C.
London,

WAITZ, J. A., WEINSTEIN, M. J. (1969). Recent microbiological studies with gentamicin. Journal of Infectious Diseases 119: 355-360.
WAKEFIELD, D. S., PFALLER, M., MASSANARI, M. , HAMMONS, G. T. (1987). Variation in methicillin- resistantStaphylococcus aureus occurrence by geographiclocation and hospital characteristics. Infection Control 8: 151-157.
WALSH, T. J., HANSEN, S. L., TATEM, B. A., AUGER, F., STANDIFORD, H. C. (1985). Activity of novobiocin against methicillin-resistant Staphylococcus aureus. Journal of Antimicrobial Chemotherapy 15: 435-440.
WARD, T. T., WINN, R. E., HARTSTEIN, A. I., SEWELL, D. L. (1981). Observations relating to an inter- hospital outbreak of methicillin- resistant Staphylococcus aureus: role of antimicrobial therapy in infectioncontrol. Infection Control 2: 435-439.
WATANAKUNAKORN, C. (1981). The antibacterial actionof vancomycin. Reviews of Infectious Diseases 3 (Suppl.), 210-215.
WATANAKUNAKORN, C. (1982). Treatment of infections due to methicillin-resistant Staphylococcus aureus. Annals of Internal Medicine 97: 370-378.
WATANAKUNAKORN, C. (1988). In vitro induction ofresistance in coagulase-negative staphylococci to vancomycin and teicoplanin. Journal of Antimicrobial Chemotherapy 22: 321-324.
WATANAKUNAKORN, C. (1990). In vitro selection ofresistance of Staphylococcus aureus to teicoplaninand vancomycin. Journal_____ of_____ AntimicrobialChemotherapy 25: 69-72.
304

WEHRLI, W. (1983). Rifampicin: mechanisms of action and resistance. Journal of Infectious Diseases 5: 407-411.
WEISBLUM, B. (1984). Inducible erythromycin resistance in bacteria. British Medical Bulletin 40: 47-53.
WHITE, A. (1964). The use of gentamicin as a nasal ointment. American Journal of the Medical Sciences 248: 52-55.
WHITE, D. G., COLLINS, P. 0., BOSWELL, R. B. (1989).Topical antibiotics in the treatment of superficial skin infections in general practice - a comparison of mupirocin with sodium fusidate. Journal of Infection 18: 221-229.
WIEDEMANN, B., KRESKEN, M. (1984). The incidence anddevelopment of resistance in Staphylococcus aureusfrom three European countries. Journal ofAntimicrobial Chemotherapy 14: (Suppl. D), 27-34.
WILLIAMS, J. D., WALTHO, C. A., AYLIFFE, G. A. J.,LOWBURY, E. J. L. (1967). Trials of fiveantibacterial creams in the control of nasal carriage of Staphylococcus aureus. Lancet ii: 390-392.
WILLIAMS, R. E. 0. (1946). Skin and Nose Carriage of bacteriophage types of Staph, aureus. Journal of Pathology and Bacteriology 58: 259-268.
WILLIAMS, R. E. 0. (1959). Epidemic Staphylococci.Lancet i: 190-195.
WILLIAMS, R. E. 0. (1963). Healthy carriage ofStaphylococcus aureus: its prevalence and importance. Bacteriological Reviews 27: 56-71.
305

WILLIAMS, R. E. 0., HARPER, G. J. (1946). Determination of coagulase and alpha-haemolysin production bystaphylococci. British Journal of ExperimentalPathology 27: 72-81.
WINSLOW, C.-E. A., ROGERS, A. F. (1906). A Statistical Study of Generic Characters in the Coccaceae. Journal of Infectious Diseases 3: 485-546.
WINSLOW, C.-E. A., ROTHBERG, W., PARSONS, E.I. (1920). Notes on the Classification of the White and Orange Staphylococci. Journal of Bacteriology 5: 145-168.
WISE, R. (1990). Macrolide progress. Journal ofAntimicrobial Chemotherapy 26: 5-6.
W0LFS0N, J. S., HOOPER, D. C. (1985). Thefluoroquinolones: structures, mechanisms of action and resistance, and spectra of activity in vitro. Antimicrobial Agents and Chemotherapy 28: 581-586.
WOODRUFF, H. B., MATA, J. M. , HERNANDEZ, S., MOCHALES,S., RODRIGUEZ, A., STAPLEY, E. 0., WALLICK, H., MILLER,A. K., HENDLIN, D. (1977). Fosfomycin: laboratorystudies. Chemotherapy 23 (Suppl. 1) , 1-22.
WORKING PARTY (1986). Guidelines for the control of epidemic methicillin-resistant Staphylococcus aureus. Journal of Hospital Infection 7: 193-201.
WORKING PARTY (1990). Revised guidelines for thecontrol of epidemic methicillin-resistantStaphylococcus aureus. Journal of Hospital Infection 16: 351-357.
WYATT, T. D., FERGUSON, W. P., WILSON, T. S., McCORMICK,E. (1977). Gentamicin resistant Staphylococcus aureusasssociated with the use of topical gentamicin.Journal of Antimicrobial Chemotherapy 3: 213-217.
306

YAMAMOTO, T., TAKUBO, S., FUJITA, K., OGURI, T., YOKOTA, T. (1990). Cloning and restriction analysis of DNA conferring new quinolone antimicrobial agent resistance from Staphylococcus aureus and other coagulase-negative Staphylococcus species. FEMSMicrobiology Letters 68: 335-340.
YOUNG, H. K., SKURRAY, R. A., AMYES, S. G. B. (1987).Plasmid-mediated trimethoprim resistance inStaphylococcus aureus. Characterisation of the firstGram-positive plasmid dihydrofolate reductase (typeSI). Biochemical Journal 243: 309-312.
YU, V. L., GOETZ, A., WAGENER, M., SMITH, P. B., RUES, J.D., HANCHETT, J., ZURAVLEFF, J. J. (1986).Staphylococcus aureus nasal carriage and infectionin patients on haemodialysis. New England Journal of Medicine 315: 91-96.
307

Appendices
Further Information on Methods Used.
A . Influence____ o_f__ antibiotic____ carry-over___ in
fluoroquinolone MBC and time-kill experiments.
Some workers (eg. Kaatz & Seo, 1990b; Hardy et al.,
1987) have reported that antibiotic carry-over
influenced their results in time-kill experiments.
One of the advantages of the replica-plating
method of Elek and Hilson was that it avoided
the broth antibiotic carry-over associated with
viable counting from fluoroquinolone-containing
broth. Broth carry-over could not be avoided in
the time-killing experiments. Some workers have
found that they can only eliminate the influence
of fluoroquinolone carry-over by diluting broth
100-fold in water. However, this approach has
limitations when counting small numbers of
organisms. We observed no fluoroquinolone inhibitory
effects (ie. areas of reduced or no growth on
counting plates) in our viable counting from
fluoroquinolone-containing broths. However, to
determine the influence of fluoroquinolone carry
over on our viable counts the following experiment
was performed.
0.1 ml samples of IsoSensitest broth and
IsoSensitest broth containing the highest
308

concentrations of ciprofloxacin, enoxacin, ofloxacin
and pefloxacin used were spread in triplicate onto
IsoSensitest agar- plates. Immediately after, the
plates were spread with 0.1 ml of a bacterial
suspension containing approximately 500 cfu/ml of
S. aureus NCTC 6571. After 24 hours incubation at
37°C the plates were counted. On comparison of
the viable counts on antibiotic spread plates to
those on non-antibiotic spread plates no difference
(Student’s T-test, p = 0.9) in viable count was
found.
B . Development of a biotyping system for MGRSA.
i . Use of API STAPH profiles.
’’API STAPH” is a biochemical test system
designed to identify staphylococci and micrococci.
While it is designed to facilitate speciation
rather than sub-speciation (the latter being the
aim of biotyping), our previous experience (Gargan
et al. , 1982) with Gram-negative species is that
this type of system can be of some use in
biotyping. We obtained 11 different biotypes (ie.
API codes) for the MGRSA we identified. However
55% of strains fell into two biotypes (6736153 and
6736113). A variety of other profiles (eg. 6776153,
6736115, 6736112, 6734113 etc.) were found for small
numbers of strains.
309

i i. Use of antibiotic susceptibility profiles.
In a previous study (Brumfitt et al. , 1989) we
found that of 23 antibiotics tested, sensitivity to
netilmicin, amikacin, neomycin, tetracycline,
clindamycin, chloramphenicol, trimethoprim, rifampicin
and ciprofloxacin provided most discrimination (ie.
between 15 and 85% of strains were sensitive).
Using this system 59 antibiotypes were obtained,
and the greatest number of strains of any one
type was 4.
iii . Determination of____ haemolysin___ profiles by
microtitre method.
Staphylococci produce a variety of haemolysins
(alpha, beta, gamma and delta), each of which is
capable of lysing erythrocytes of different species
origins to varying degrees (Mollby, 1983). To
identify and quantify production of these
haemolysins we used the micro-titration method
reported by Jordens et al. (1989).
Three known specific haemolysin producing
organisms, S . aureus NCTC 7121, S . aureus NCTC 5664
and S . aureus NCTC 10345 were used as controls in
the assay. S. aureus NCTC 7121 (Wood 46) only
produces alpha haemolysin and in our assay had a
titre of 128 against rabbit erythrocytes in the
presence of fibrinogen. S. aureus NCTC 5664 (Y2,
310

Sweden) produced beta and gamma haemolysins. Broth
supernatant from this strain was active against
human, rabbit and sheep erythrocytes, and this
activity was reduced in the presence of heparin.
Following incubation at 0°C an increase in titre
was found against all erythrocytes, but particularly
with sheep erythrocytes. S. aureus NCTC 10345
produced delta haemolysin and broth supernatants of
this organism had little activity against rabbit
or sheep erythrocytes (titre 0-2), and not much
more against human erythrocytes (titre 2-8). Little
reduction in titre ( 1 or 2 wells ) occurred in the
presence of fibrinogen. On cooling, an increase in
titre was found similar to that seen with S .
aureus NCTC 5664.
Using this method we tested strains from Israel
and Texas. Titres of 4-32 were obtained with
rabbit blood + fibrinogen, however little activity
(maximum titre of 4) was found with human or
sheep erythrocytes, and this was not changed in
the presence of heparin or fibrinogen. Because we
only reliably detected alpha haemolysin in these
strains using this method, we felt that because of
its expense and lack of discrimination it was not
worthwhile to continue. Also from previous
biotyping studies we had found that strains could
be differentiated on the basis of sizes of zones
of haemolysis on sheep blood agar. There was a
correlation between sizes of zones of haemolysis
311

on sheep blood agar and titre of alpha
haemolysin. Strains producing large zones, had
greater titres than those producing small zones.
Elek and Levy (1954) reported that discrepancies
can occur between the haemolysins detected using
culture filtrates compared to those determined by
growth on agar. According to Mollby (1983) agar
plate tests are more reliable than broth methods
for detecting mixtures of haemolysins: for example
beta-haemolysin production may readily be lost by
growth in liquid media.
iv. Sheep blood haemolysis.
Williams and Harper (1946) found 93% of S. aureus
of human origin produced alpha-haemolysin as
detected by antitoxin neutralization on sheep blood
agar. These workers also found other haemolysins
responsible for sheep blood lysis and suggested
that other toxins may be produced. We are of the
same opinion; however, using the method of Jordens
et al. (1989) we have failed to identify them.
v . Egg-yolk reaction.
The observations that most strains of coagulase-
positive staphylococci produced opacity when grown
in media containing egg yolk prompted Gillespie
and Alder (1952) to study this reaction in detail.
312

They found that it was probably due to production
of a lipase, and that isolates of S. aureus from
hospital inpatients were often egg yolk negative
as were most penicillin-resistant staphylococci. On
the contrary, most S. aureus from outpatients were
egg-yolk positive.
Approximately 60% of MGRSA tested were egg-yolk
positive, which contrasts with Gillespie and
Alder’s findings because these strains were
predominantly in-patient isolates resistant to many
antibiotics including penicillin. We found the egg-
yolk reaction most useful in differentiating
strains.
vi. TWEEN 80 reaction.
The results for Tween 80 hydrolysis mirrored
those obtained for the egg-yolk reaction.
vii. Pigmentation on Milk Agar.
The interpretation of colour varies between
workers, for example Lacey and Stokes (1979)
classified strains as orange or yellow whereas
Putland and Guinness (1985) classified strains as
gold, cream or white. Lacey and Stokes (1979) found
most (c. 90%) MRSA produced orange pigment and
Putland and Guinness (1985) observed that most (67%)
of their strains produced gold pigment. Most (c.
313

70%) of our MGRSA produced orange/gold pigment,
however this figure was highly subjective as
considerably different results were obtained when
different people read the plates. We are of the
opinion that this method is too subjective, and
the results we obtained were not used to
differentiate strains in our fluoroquinolone
resistance studies. Interestingly, two of the Irish
MGRSA produced a lime yellow pigment.
viii. Plasmid-typing.
The Takahashi and Nagano (1984) method was
inconsistent in reliably detecting plasmids.
Although small and large plasmids were isolated,
there was considerable variability in results on
repeating experiments. Large plasmids (30 mD or
larger) were sometimes observed, yet on other
occassions they were not. A further problem was
the low plasmid yield (ie. production of only faint
bands) obtained. The PHLS (Johnson) method
consistently detected large and small plasmids.
The plasmid contents and sizes of 45 strains of
MGRSA used in the resistance survey (Table IV) were
studied, and Table XXI shows our findings for 33 of
those strains. It is evident that the MGRSA
314
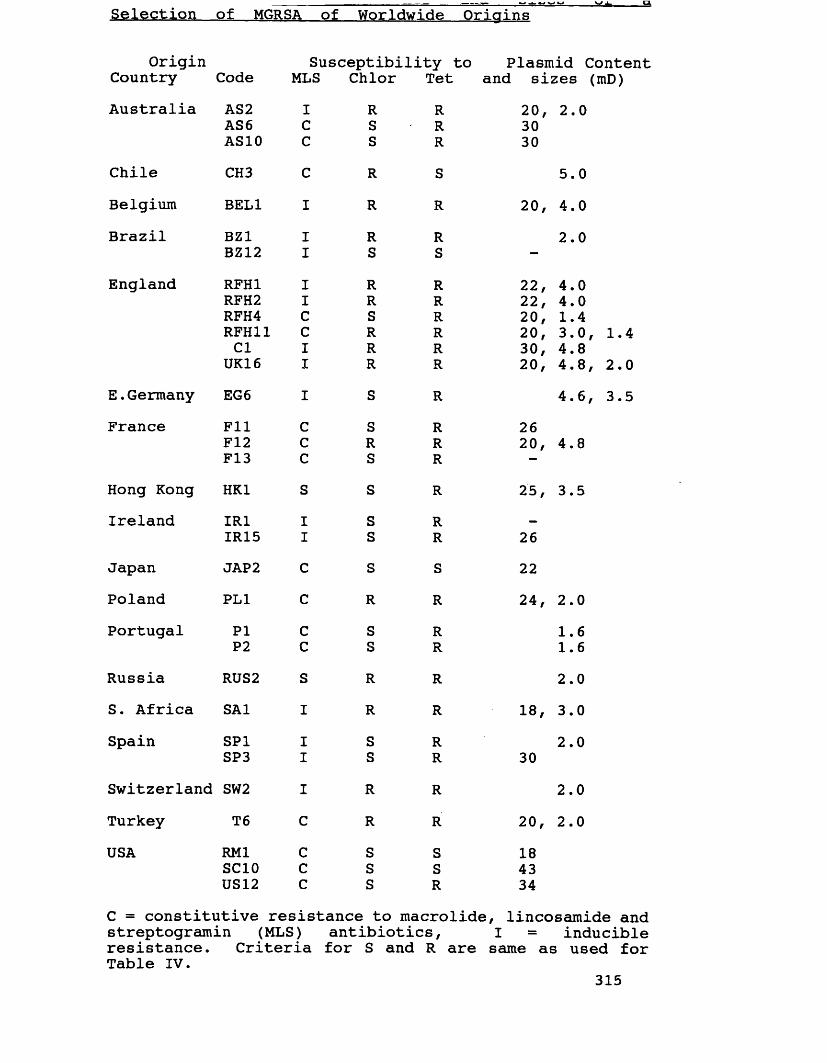
Selection of MGRSA of Worldwide Origins
Origin Susceptibility to Plasmid ContentCountry Code MLS Chlor Tet and sizes (mD)Australia AS2 I R R 20, 2.0AS6 C S R 30
AS10 c S R 30Chile CH3 c R S 5.0Belgium BEL1 I R R 20, 4.0Brazil BZ1 I R R 2.0
BZ12 I S S —
England RFH1 I R R 22, 4.0RFH2 I R R 22, 4.0RFH4 c S R 20, 1.4RFH11 c R R 20, 3.0, 1.4Cl I R R 30, 4.8UK16 I R R 20, 4.8, 2.0
E .Germany EG 6 I S R 4.6, 3.5France Fll c S R 26
F12 c R R 20, 4.8F13 c S R —
Hong Kong HK1 s S R 25, 3.5Ireland IR1 I s R —
IR15 I s R 26Japan JAP 2 c s S 22Poland PL1 c R R 24, 2.0Portugal PI c S R 1.6
P2 c S R 1.6Russia RUS2 s R R to » o
S. Africa SA1 I R R 00 > 3.0Spain SP1 I S R 2.0SP3 I S R 30Switzerland SW2 I R R o•CM
Turkey T6 c R R 20, 2.0USA RM1 c S S 18
SC10 c S S 43US12 c S R 34C = constitutive resistance to macrolide, lincosamide and streptogramin (MLS) antibiotics, I = inducibleresistance. Criteria for S and R are same as used for Table IV.
315

studied contained a variety of plasmids of
different sizes. For the 45 strains studied, only
three strains (BZ12, F13 and IR1) were found to
contain no plasmids, all the other strains were
found to contain from 1-3 plasmids. In Table XXI
the susceptibility to macrolide, lincosamide and
streptogramin (MLS) antibiotics, chloramphenicol and
tetracycline of strains is compared to plasmid
content. Chloramphenicol resistance was found to be
always plasmid-mediated, whilst tetracycline resistance
or resistance to MLS antibiotics could have been
plasmid or chromosomally mediated in different
strains .
Although this work was interesting it was felt
that further plasmid analysis studies were
inappropriate because of the difficulties in
transferring or curing single plasmids from strains
containing multiple plasmids which is requisite for
assigning the location of resistance factors.
Furthermore, following Lyon and Skurray's excellent
review of the genetic basis of antimicrobial
resistance in S. aureus (Lyon & Skurray, 1987) we came
to the opinion that further studies of the genetic
basis of resistance in our strains would be of
limited significance to the project as a whole.
316

C . Detection and Isoelectric focusing of type SI
enzyme.
Detection and isoelectric focusing of type SI
enzyme was carried out at the Bacteriology
Research Laboratory, Old Medical School, Edinburgh
University. The method used was being developed by
S. Tait, and formed a major part of his PhD
thesis. Because of the complexity of the method,
the fact that it is as yet unpublished, and
because the results obtained were of limited
significance to this study, the methodology shall
only be briefly outlined.
Cultures were initially grown in 10 ml
IsoSensitest broth containing 2.0 mg/1 trimethoprim
for 12 hours at 37°C. These starter cultures were
then aseptically added to 2.0 litre flasks
containing 1.0 litre IsoSensitest broth. After
shaking overnight (80 rpm) at 37°C, bacteria were
harvested by centrifugation (6000 rpm for 10
minutes). They were washed in saline and
resuspended in 10 ml phosphate-mercaptoethanol
buffer. The bacteria were lysed with lysostaphin
and then sonicated. The extracts were centrifuged
at 16,000 rpm for 1 hour, and the supernatant
collected. Nucleic acids in these crude
preparations were precipitated by addition of
streptomycin sulphate, they were then centrifuged
(10,000 rpm for 30 minutes at 4°C) and the
I 317

supernatant collected. Ammonium sulphate was added
to 50% saturation (to remove protein contaminants)
and the supernatant decanted. This was then
saturated to 80% with ammonium sulphate and the
precipitate (containing the proteins we do want) was
collected by centrifugation at 4°C for 30 minutes
at 10,000 rpm. The pellet was resuspended and
dialysed in phosphate-mercaptoethanol buffer. It was
then ready for isoelectric focusing. Polyacrylamide
gels were pre-run for 20 minutes (1500 volts,15
watts), and samples (and controls containing purified
type SI) were streaked over the surface at the
anode. When the standards had focussed the gel
was sequentially stained with NADPH solution
followed by dihydrofolate and then mercaptoethanol
solution.
318

D . Suppliers of Strains of MGRSA
We are indebted to the following who supplied
the strains of MGRSA used for this work: J.F.
Acar, 0. Ang, G.F. Ara j , C.A. Bartzokas, E. Bergogne-
Berezin, J. Borowski, M.T. Cafferkey, P.Y. Chau, R.L.
Cohen, J. Cooper, M. Dan, S. Dixson, A.A. Forder, A.
Georgopoulos, H. Giamarellou, M. Gobernado, F.W.
Goldstein, W. Graninger, G.G. Grassi, D. Hanslo, C. Hohne,
J.F. John Jr, F.H. Kayser, C.T. Keane, C.C. Linnemann, D.
Merzbach, R.C. Moellering Jr, E.C. Moorhouse, H.C. Neu,
C.E.O. Pires de Campos, V. Prado, G.W. Smith, K.H. Spitzy,
J.L. Staneck, A, Torres Pereira, E.P. Trallero, W.H.
Traub, K. Ubukata, C. Watanakunakorn, E. Yourassowsky.
319

Summary of Conclusions and Suggestions for futurew o r k .
Although a substantial amount of work has been
published in recent years concerning the genetics
and mechanisms of antibiotic resistance possessed
by MGRSA, few workers have attempted to answer the
question as to whether antibiotic resistance in
MGRSA poses a significant clinical problem. In
endeavoring to answer this we discovered a number
of new facts regarding the emergence of antibiotic
resistance in MGRSA, and specifically the development
of fluoroquinolone resistance.
i . New facts emerging.
From the international survey of antibiotic
resistance in MGRSA we showed that these organisms
possessed differing degrees of multiple antibiotic
resistance. Some strains were highly multiple
antibiotic resistant whilst others were considerably
less so. Most of the highly multiple resistant
strains emerged from Brazil, France and Turkey,
countries which have a reputation for failing to
control antibiotic usage. Strains showing least
multiple resistance originated from Chile, East
Germany, Hong Kong, Italy, Russia, U.K and the USA.
In these countries antibiotic usage is either
320

controlled or newer classes of antibiotic are
simply not available.
In terms of MIC a considerable diversity of
resistance phenotypes was seen, both with reference
to the variety of different antibiotic resistance
patterns, and the levels (low or high) of resistance
to individual antibiotics such as ciprofloxacin,
fusidic acid, gentamicin, minocycline and streptomycin.
Resistance to macrolide, lincosamide and streptogramin
(MLS-type) antibiotics was closely associated with
methicillin and gentamicin resistance. Gentamicin
resistance in strains of worldwide origin was
associated with the same enzyme - APH(2")/A A C (6f) .
Phage typing with the International Set and
additional experimental phages revealed most strains
to belong to Group III or be Group III-related.
A comprehensive assesment of the antebacterial
activity of currently available antistaphylococcal
agents was carried out by studying MIC, MBC and
killing curves. Fosfomycin and pristinamycin appeared
as potentially useful alternatives to vancomycin.
Further evidence was obtained that the phenomenon
of tolerance to vancomycin and teicoplanin was due
to methodological variables. New data were obtained
on the activity against MGRSA of azelaic acid,
nitrofurazone and silver sulphadiazine.
We were among the first workers to find
resistance to fluoroquinolones in MGRSA, and showed

it to be present in strains from a number of
countries. Ciprofloxacin-resistant MGRSA from clinical
sources had different levels and patterns of
quinolone resistance. The phenomenon of "reversed
incomplete cross-resistance" between fluoroquinolones
and older non-fluorinated quinolones was seen in a
number of strains. In broth time-kill experiments
fluoroquinolone resistance readily emerged in the
presence of peak serum levels of ciprofloxacin,
enoxacin and pefloxacin. Resistance did not readily
emerge in the presence of peak serum concentrations
of ofloxacin. No change in phage-type or
susceptibility to agents other than quinolones was
detected following mutation to fluoroquinolone
resistance. Fluoroquinolone resistance developed in
a stepwise manner, and irrespective of the
particular concentration or fluoroquinolone used,
resistant mutants possessed similar reductions in
susceptibility indicating that only an initial one-
step mutation could occur. Typing of ciprofloxacin-
sensitive and resistant MGRSA from one of the
first outbreaks to be reported showed that
resistance had evolved in a number of different
strains.
322

i i . New thoughts on Antibiotic Resistance in MGRSA.
It has previously been stated that a single
clone of MRSA has spread worldwide. Our work
showed MGRSA from several sources to be different
and we suggest that antibiotic resistance has
evolved in separate strains. The aminoglycoside
modifying enzyme A P H (2")/AAC( 6 ?) was present in all
MGRSA which indicates that the gene for this
enzyme has become disseminated worldwide. Genes
coding for resistance to aminoglycosides and MLS
antibiotics have been found in antibiotic producing
Streptomyces spp., and also other Gram-positive
bacteria such as streptococci and coagulase negative
staphylococci. These organisms may have acted as a
reservoir for gene transfer to MGRSA. The
unrestricted use of antibiotics provides the
selective pressure for multiple resistance to appear
in MRSA, and that in the case of aminoglycoside
resistance and resistance to MLS antibiotics this
resistance may readily be acquired through
interstrain, interspecies or intergeneric transfer.
iii. Future work to be d o n e .
The data from the survey of antibiotic
resistance in MGRSA enabled us to qualitatively
assess the problem of multiple antibiotic resistance
323

in MGRSA. What is needed is a survey to
quantitatively determine the international problem of
antibiotic resistance in MRSA and relate this to
patterns of antibiotic usage and clinical origin of
strains. Currently, the European Society for Clinical
Microbiology and Infectious Diseases is attempting
to survey the incidence of MRSA in different
countries, however antibiotic usage patterns are not
being ascertained.
Recently, much work has been published on the
genetic control of resistance in MGRSA and it
appears that a great deal of antibiotic resistance
is transposon-mediated. Certain chromosomal "hot
spots" for the integration of transposons have been
identified and a further understanding of these
would be most useful. Determination of the location
(chromosome or plasmid) of resistances in the
strains we studied would be useful as a means of
further understanding how resistance has evolved and
spread.
The most important aim for the future is to
attain an international concensus regarding the
appropriate use of antibiotics with a view to
limiting the opportunities for emergence of multiple
resistance .

WORLD-WIDE ANTIBIOTIC RESISTANCE IN METfflCIIXIN-RESISTANT
STAPHYLOCOCCUS AUREUS
P. A. C. M a p l e J. M. T. H a m i l t o n - M i l l e r W . B r u m f i t t
Departtnent of Medical Microbiology, Royal Free Hospital School of Medicine, London NW3 2QG
Summary Antibiotic resistance patterns were determined for 106 strains of methicillin-
resistant Staphylococcus aureus (MRSA) from 21 countries. Resistance to gentamicin, tobramycin, netilmicin, amikacin, streptomycin, or erthromycin was recorded in more than 90% of strains. Resistance to the other compounds tested was as follows: tetracycline 86%, minocycline 76%, trimethoprim 69%, clindamycin 66%, neomycin 59%, chloramphenicol 39%, rifampicin 26%, fosfomycin 22%, ciprofloxacin 17%, fusidic acid 12%, bacitracin 2%, and novobiocin 1%. All the stains were sensitive to mupirocin, pristinamycin, ramoplanin, teicoplanin, and vancomycin. There were geographical patterns of resistance: MRSA from the UK and Australia were predominantly resistant to trimethoprim, whereas many strains from centres in Europe and the USA were sensitive. MRSA that were resistant to ciprofloxacin were of French and German origin. 15 strains, 12 of which came from France, Turkey, or Brazil, were resistant either to thirteen or to fourteen agents.
INTRODUCTION
O u t b r e a k s of infection attributable to methicillin- resistant Staphylococcus aureus (MRSA) have been reported world wide, and, since 1976, aminoglycoside-resistant strains have become increasingly prevalent.1,2 Many strains of MRSA are multiresistant, and vancomycin is at present the antibiotic of choice for systemic infection.3 Resistance to vancomycin would be extremely worrying: a report of plasmid mediated resistance in enterococci4 has fuelled this concern.
During the past three years, we have collected strains of MRSA from 28 centres in 21 countries. We have looked at the chemotherapeutic options and we have determined the incidence and degree of multiresistance in different geographical regions.
MATERIALS AND METHODS
Bacterial StrainsW e studied 106 strains of M R S A from the following 21 countries
(if more than one centre, number in parentheses): Australia, Austria, Belgium, Brazil, England (3), Federal Republic of

Germany, France (2), German Democratic Republic, Greece, Hong Kong, Ireland (2), Italy, Japan, Kuwait, Poland, Portugal, South Africa (2), Spain, Switzerland, Turkey, and the U S A (3).
MicrobiologyMinimum inhibitory concentrations (MIC) of the 23 antibiotics
listed in table I were determined by the agar dilution method. Briefly, doubling dilutions of antibiotic were incorporated into ‘IsoSensitest Agar’ (Oxoid, UK). A Denley Multipoint Inoculator (Denley Instruments, UK) was used to inoculate the agar plates with 10s organisms from peptone water broths grown at 37°C for 24 h. The inoculated plates were incubated for 24 h at 37°C before recording MICs. MICs of fosfomydn were done in the presence of glucose-6-phosphate (25 mg/1).
Methicillin resistance was confirmed by incubation of strains on nutrient agar against 25 jig methicillin strips (Mast Laboratories, U K ) for 40 h at 30°C.
RESULTS
The MICs of 23 antimicrobial agents against 106 strains of MRSA are shown in tables II and ill. We classified strains as sensitive, moderately resistant, or resistant to each agent according to the MICs (see table i). Compounds could be
TABLE I—MICS USED TO CLASSIFY STRAINS AS SENSITIVE, MODERATELY RESISTANT, OR RESISTANT
Antibiotic
MIC (mg/1)
Sensitive Moderate Resistant
Gentamicin* <1 N/A 2*8Tobramycin* <1 N/A ^ 16Netilmicin* <1 2,4 2*8Amikacin* <4 8,16 ^32Streptomycin* <16 N/A ^64Neomycin* <4 N/A ^ 16Bacitradnt ^8 N/A ^32Tetracycline <4 N/A 2*32Minocycline <4 N/A ^8Erythromycin <1 N/A 2? 8Chloramphenicol < 16 N/A 2*32Fosfomycint <16 32,64 2*128Clindamycin <0-25 N/A ^16Trimethoprim <0-25 1,2 2*4Rifampirin <012 1 ^16Fusidic acid <0-5 4 2*16Novobiocin <0-5 2 N/APristmamycin <2 N/A N/ACiprofloxacin <1 2,4 2*16Mupirocin <0-5 N/A N/ARamoplanin <1 N/A N/AVancomycin <2 N/A N/ATeicoplanin <1 N/A N/A
MIC = minimum inhibitory concentration.N/A = not applicable. tIU.Definitions of sensitivity as suggested by *a working party of the British Society of Antimicrobial Chemotherapy5 and by tan international study group.6
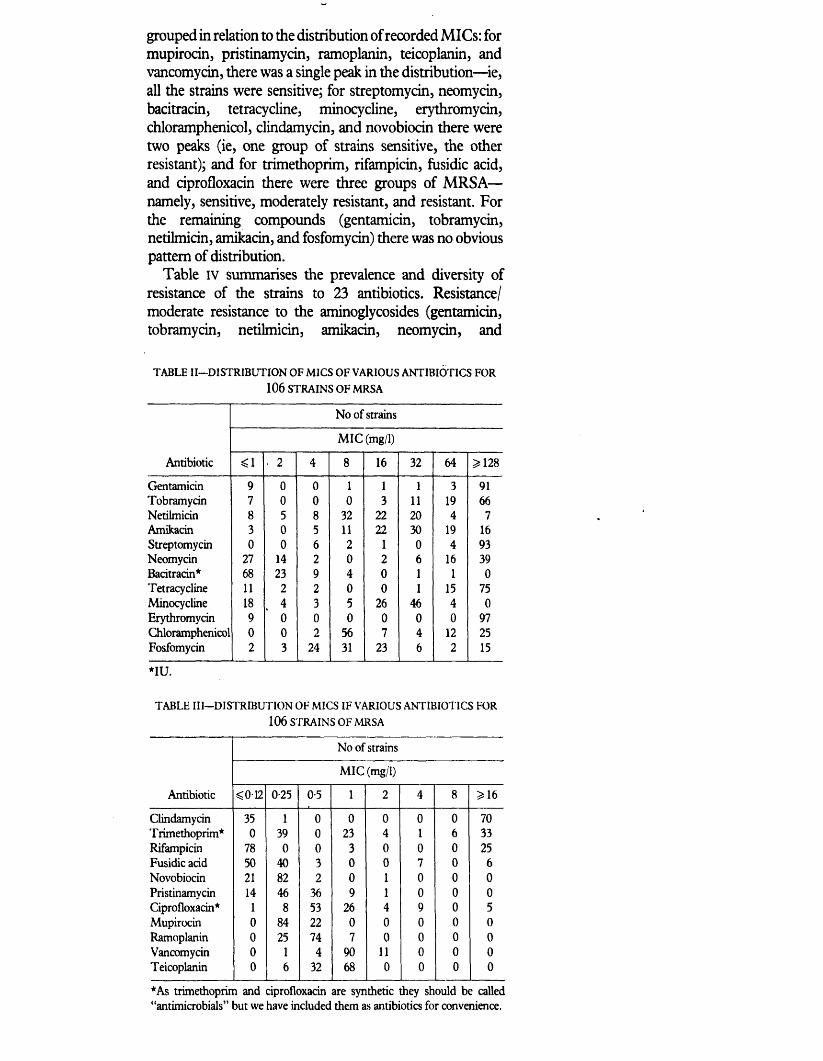
grouped in relation to the distribution of recorded MICs: for mupirocin, pristinamycin, ramoplanin, teicoplanin, and vancomycin, there was a single peak in the distribution—ie, all the strains were sensitive; for streptomycin, neomycin, bacitracin, tetracycline, minocycline, erythromycin, chloramphenicol, clindamycin, and novobiocin there were two peaks (ie, one group of strains sensitive, the other resistant); and for trimethoprim, rifampicin, fusidie acid, and ciprofloxacin there were three groups of MRSA— namely, sensitive, moderately resistant, and resistant. For the remaining compounds (gentamicin, tobramycin, netilmicin, amikacin, and fosfomycin) there was no obvious pattern of distribution.
Table IV summarises the prevalence and diversity of resistance of the strains to 23 antibiotics. Resistance/ moderate resistance to the aminoglycosides (gentamicin, tobramycin, netilmicin, amikacin, neomycin, and
TABLE II—DISTRIBUTION OF MICS OF VARIOUS ANTIBIOTICS FOR 106 STRAINS OF MRSA
No of strains
MIC (mg/1)
Antibiotic <1 2 4 8 16 32 64 3*128
Gentamicin 9 0 0 1 1 1 3 91Tobramycin 7 0 0 0 3 11 19 66Netilmicin 8 5 8 32 22 20 4 7Amikacin 3 0 5 11 22 30 19 16Streptomycin 0 0 6 2 1 0 4 93Neomycin 27 14 2 0 2 6 16 39Bacitracin* 68 23 9 4 0 1 1 0Tetracycline 11 2 2 0 0 1 15 75Minocycline 18 4 3 5 26 46 4 0Erythromycin 9 0 0 0 0 0 0 97Chloramphenicol 0 0 2 56 7 4 12 25Fosfomycin 2 3 24 31 23 6 2 15
*IU.
TABLE III—DISTRIBUTION OF MICS IF VARIOUS ANTIBIOTICS FOR 106 STRAINS OF MRSA
No of strains
MIC (mg/1)
Antibiotic <012 0-25 05 1 2 4 8 3*16
Clindamycin 35 1 0 0 0 0 0 70Trimethoprim* 0 39 0 23 4 1 6 33Rifampicin 78 0 0 3 0 0 0 25Fusidie acid 50 40 3 0 0 7 0 6Novobiocin 21 82 2 0 1 0 0 0Pristinamycin 14 46 36 9 1 0 0 0Ciprofloxacin* 1 8 53 26 4 9 0 5Mupirocin 0 84 22 0 0 0 0 0Ramoplanin 0 25 74 7 0 0 0 0Vancomycin 0 1 4 90 11 0 0 0Teicoplanin 0 6 32 68 0 0 0 0
*As trimethoprim and ciprofloxacin are synthetic they should be called “antimicrobials” but we have included them as antibiotics for convenience.
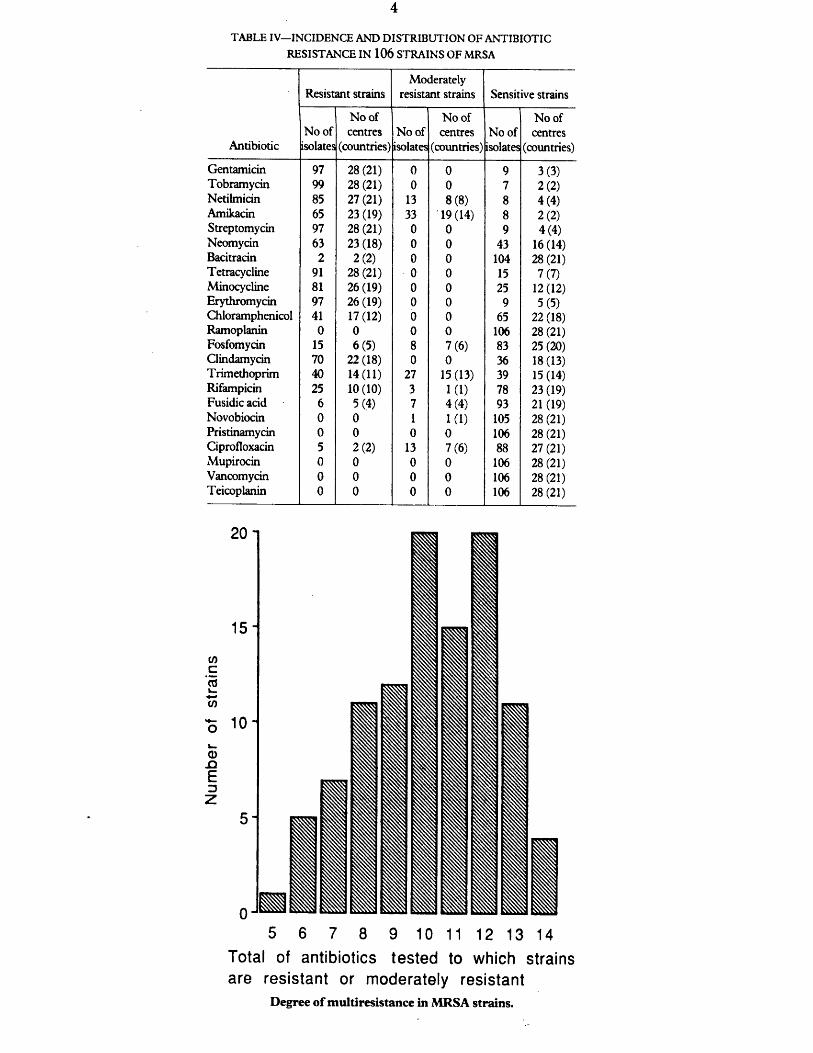
4TABLE IV—INCIDENCE AND DISTRIBUTION OF ANTIBIOTIC
RESISTANCE IN 106 STRAINS OF MRSA
Antibiotic
Resistant strainsModerately
resistant strains Sensitive strains
No of isolates
No of centres
(countries)No of isolates
No of centres
(countries)No of isolates
No of centres
(countries)
Gentamicin 97 28(21) 0 0 9 3(3)Tobramycin 99 28 (21) 0 0 7 2(2)Netilmicin 85 27(21) 13 8(8) 8 4(4)Amikacin 65 23(19) 33 19 (14) 8 2(2)Streptomycin 97 28(21) 0 0 9 4(4)Neomycin 63 23 (18) 0 0 43 16(14)Bacitracin 2 2(2) 0 0 104 28(21)Tetracycline 91 28(21) 0 0 15 7(7)Minocycline 81 26 (19) 0 0 25 12(12)Erythromycin 97 26 (19) 0 0 9 5(5)Chloramphenicol 41 17(12) 0 0 65 22 (18)Ramoplanin 0 0 0 0 106 28 (21)Fosfomycin 15 6(5) 8 7(6) 83 25 (20)Clindamycin 70 22 (18) 0 0 36 18(13)Trimethoprim 40 14(11) 27 15(13) 39 15(14)Rifampicin 25 10(10) 3 1(1) 78 23(19)Fusidie acid 6 5(4) 7 4(4) 93 21 (19)Novobiocin 0 0 1 1(1) 105 28 (21)Pristinamycin 0 0 0 0 106 28 (21)Ciprofloxacin 5 2(2) 13 7(6) 88 27 (21)Mupirocin 0 0 0 0 106 28(21)Vancomycin 0 0 0 0 106 28 (21)Teicoplanin 0 0 0 0 106 28 (21)
5 6 7 8 9 10 11 12 13 14Total of antibiotics tested to which strains are resistant or moderately resistant
Degree of multiresistance in MRSA strains.

streptomycin), and resistance to clindamycin, erythromycin, tetracycline, and minocycline was widespread. By contrast, all MRSA were sensitive to mupirocin, pristinamycin, ramoplanin, teicoplanin, and vancomycin. The strains that were resistant to ciprofloxacin (MIC 16 mg/1) were of French and German origin whereas those that were moderately resistant (MIC=2 or 4 mg/1) originated from various centres in Europe, USA, and Asia. MRSA that were resistant/moderately resistant to fosfomycin came from France, Germany, Italy, Turkey, and Brazil. 2 strains (1 from Switzerland, 1 from Brazil) were resistant to bacitracin. For 1 strain (from Ireland) the MIC of novobiocin was high (2 mg/1).
12 of the 15 strains resistant/moderately resistant either to thirteen or to fourteen antibiotics (figure) came from France, Turkey, and Brazil. The other 3 strains were from the USA, Kuwait, and Switzerland. 2 of the 15 strains were sensitive to rifampicin, 3 to fosfomycin, and 11 to fusidie acid and ciprofloxacin.
DISCUSSION
Gentamicin resistance in MRSA is encountered world wide; resistance can be chromosomally or plasmid mediated.7 Various patterns of aminoglycoside resistance have been recorded; for example, Rimland8 has reported tobramycin-resistant, gentamicin-sensitive strains, and Schaefler et al9 observed MRSA that were resistant to gentamicin, tobramycin, and amikacin. Even when the organisms are sensitive, the clinical use of aminoglycosides, either alone10 or with P-lactams,11 has proved to be less than satisfactory for the treatment of staphylococcal infections.
Resistance to antibiotics of the “macrolide, lincosamide, and streptogramin” (MLS) group—represented in the present study by erythromycin, clindamycin, and pristinamycin—is due either to methylation of ribosomal RNA, or to enzymic modification of the antibiotic. The first type of MLS resistance may be either constitutive (strains are resistant to erythromycin and clindamycin, but remain sensitive to pristinamycin), or inducible (strains are resistant to erythromycin only, but clindamycin resistance can develop in the presence of subinhibitory concentrations of erythromycin). Many of the MRSA that were sensitive to clindamycin showed inducible clindamycin resistance. None of the strains in the present study was resistant to pristinamycin, although resistance has been reported as a rare event. Our findings concerning MLS resistance are similar to those of Duval.12
Where the MRSA strains were trimethoprim-sensitive— commonly in the USA13—co-trimoxazole (trimethoprim/ sulphamethoxazole) has been used successfully.14 MRSA from the UK and Australia were resistant to trimethoprim, whereas strains from many European centres were sensitive.

Resistance to rifampicin and to fusidie acid is due to chromosomal mutations followed by selection. Selection is less likely if these agents are used in appropriate combinations.1516 We are concerned that as many as 28 of the 106 strains studied were not sensitive to rifampicin, particularly those from France, Turkey, and Brazil.
There are few clinical reports on the use of novobiocin, fosfomycin, and ciprofloxacin for treatment of infections caused by MRSA. We have found widespread resistance to ciprofloxacin: this is disconcerting because the antibiotic has a novel structure and has only recently entered clinical use. Our concern is heightened by reports from the USA of ciprofloxacin resistance in MRSA after treatment.1718 There have, however, been encouraging reports about the use of fosfomycin for MRSA infections.19 Development of resistance during treatment can occur, but does not seem to arise if fosfomycin is not used alone.20
Only 1 of our strains was resistant to novobiocin: in vitro, there is promising activity with this compound and the related compound, coumermycin.2U2 None of the strains in the present study was resistant to vancomycin or to the structurally similar teicoplanin. S aureus has remained sensitive to vancomycin, although reports of resistance by 5 epidermidis,23 and, more recently, of plasmid mediated resistance by enterococci4 warn against complacency. Teicoplanin, which is currently under clinical evaluation,24 is less toxic and is easier to administer than vancomycin.
Opinions vary on the efficacy of topical antibiotics for eradicating MRSA carriage. Like many others25 we believe that certain topical agents should only be regarded as an adjunct to rigorous measures of hospital control of infection: Undoubtedly, scrupulous handwashing and access to isolation facilities are much more effective in controlling the spread of MRSA. Bacitracin is ineffective,26 and many strains are resistant to neomycin and tetracycline. There are encouraging reports about the efficacy of mupirocin,27 but even with this drug, resistance has been encountered.28 The novel lipoglycodepsipeptide antibiotic, ramoplanin29 (formerly called A-16686), may have an important future role as a topical agent.
In the present study, numerous patterns of resistance to antibiotics were recorded. Although the severity of multiresistance ranged from 5 to 14 antibiotics, the choice of effective recognised chemotherapy in many instances was limited to rifampicin, to fusidie acid, or to vancomycin. For the highly multiresistant strains—ie, those resistant to 13 or 14 antibiotics—vancomycin was often the only antibiotic so far approved by the regulatory bodies to which the strains were sensitive. A thorough in-vitro and in-vivo assessment of fosfomycin, novobiocin (or the related compound coumermycin), and pristinamycin, in terms of efficacy and development of resistance, is essential if we are to increase therapeutic options. In view of the prevalence of

ciprofloxacin resistance found in the present study and by others17 we feel that the use of fluoroquinolones against M RSA should be re-evaluated.
12 of the 15 highly multiresistant strains came from France, Turkey, and Brazil: we have yet to determine whether this multiresistance is a consequence o f strains with enhanced genetic adaptability, or of selection caused by favourable environmental factors. Additional resistance can be prevented: first, it is essential that antibiotics that can be used systemically should not also be used topically; and, second, for antibiotics where m utation to resistance is likely (eg, ciprofloxacin, fosfomycin, fusidie acid, and rifampicin), use of appropriate combinations will minimise development of resistance.
Finally, because of the possibility of spread o f resistant strains from one country to another, close international cooperation is the key to limiting occurrence and spread.
This study has been aided by the Special Trustees o f the Royal Free Hospital. A fellowship was made available to J. M . T . H -M . by th e - Leverhulme Trust.
W e are indebted to the following, who supplied the strains used in this study: J. F. Acar, O. Ang, G. F. Araj, C. A. Bartzokas, E. Bergogne-Berezin, J. Borowski, M . T . Cafferkey, P. Y. Chau, J. Cooper, S. D ixson, A. A. Forder, A. Georgopoulos, H . Giamarellou, F. W . Goldstein, W. Graninger, G. G. Grassi, D . Hanslo, C. H ohne, F. H . Kayser, C. T . Keane, C. C. Linnemann, R. C. Moellering, E. C. M oorhouse, H . C. N eu , C. E. O. Pires de Campos, G. W . Smith, K. H. Spitzy, J. L. Staneck, A. Torres-Pereira, E. P. Trallero, W. H . Traub, K. Ubukata, C. Watanakunakom, R. P. Wenzel, E. Yourassowsky.
Correspondence should be addressed to W . B.
REFERENCES
1. Shanson D C, Kensit JG , Duke R. Outbreak of hospital infection with a strain ofStaphylococcus aureus resistant to gentamicin and methicillin. Lancet 1976; ii: 1347—48.
2. Soussy CJ, Dublanchet A, Cormier M , et al. Nouvelles resistances piasmidiques deStaphylococcus aureus aux aminosides (gentamicine, tobramycine, amikacine). Nouv Presse Med 1976; 5: 2599-602.
3. Milatovic D. Vancomycin for treatment of infections with methicillin-resistantStaphylococcus aureus: are there alternatives? E urJ Clin Microbiol 1986; 5:689-92.
4. LeClercq R, Derlot E, Duval J, Courvalin P. Plasmid mediated resistance tovancomycin and teicoplanin in Enterococcus faecium. N Engl J M ed 1988; 319: 157-61.
5. British Society for Antimicrobial Chemotherapy. Break-points in in-vitro antibioticsensitivity testing. J Antimicrob Chemoiher 1988; 21: 701-10.
6. Andrews JM , Baquero F, Beltran JM , et al. International collaborative study onstandardization of bacterial sensitivity to fosfomycin. J Antimicrob Chemoiher 1983; 12: 357-61.
7. Lyon BR, Skurray R. Antimicrobial resistance of Staphylococcus aureus: genetic basis.Microbiol Rev 1987; 51: 88-134.
8. Rimland D. Nosocomial infections with methicillin and tobramycin resistantStaphylococcus aureus: Implications of physiotherapy in hospital-widedissemination. Am J M ed Sci 1987; 290: 91-97.
9. Schaefler S, Jones D, Perry W, et al. Emergence of gentamicin and methicillin resistantStaphylococcus aureus strains in New York City hospitals. J Clin Microbiol 1981; 13: 754-59.
10. Cafferkey M T , Hone R, Keane CT. Antimicrobial chemotherapy of septicaemia dueto methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 1985; 28: 819-23.
11. Acar JF , Courvalin F, Chabbert YA. Methicillin-resistant staphylococcaemia:bacteriological failure of treatment with cephalosporins. Antimicrob Agents Chemoiher!1970. American Society for Microbiology, 1971: 280-85.

8
12. Duval J. Evolution and epidemiology of MLS resistance. J Antimicrob Chemother1985; 16 (suppl A): 137-49.
13. Elwell LP, Wilson RH, Knick VB, Keith BR. In vitro and in vivo efficacy of thecombination trimethoprim-sulfamethoxazole against clinical isolates of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 1986; 29: 1092-94.
14. Markowitz N, Saravolatz L, Pohlod D, et al. Comparative efficacy and toxicity oftrimethoprim-sulfamethoxazole versus vancomycin in the therapy of seriousS. aureus infections. In: 23rd Interscience Conference Antimicrobial Agents Chemotherapy 1983; Abs No 903:201. Washington DC: American Society for Microbiology, 1983.
15. Kapusnik JE, Parenti F, Sande MA. The use of rifampicin in staphylococcalinfections—a review. J Antimicrob Chemother 1984; 13 (suppl C): 61-66.
16. Jensen K. Methicillin-resistant staphylococci. Lancet 1968; ii: 1078.17. Piercy EA, Barbara D, Luby JP, Mackowiak PA. Ciprofloxacin for methicillin-
resistant Staphylococcm aureus infections. Antimicrob Agents Chemother 1989; 33: 128-30.
18. Isaacs RD, Kunke PJ, Cohen RL, Smith JW. Ciprofloxacin resistance in epidemicmethicillin-resistant Staphylococcia aureus. Lancet 1988; ii 843.
19. Lau WY, Teoh-Chan CH, Fan ST, Lau KF. In vitro and in vivo study of fosfomycinin methicillin resistant Staphylococcus aureus. J Hyg t Camb) 1986; 96:419-23.
20. Baron D, Drugeon H, Courtieu AL, Nicolas F. Septicemies et infections graves agermes multiresistants. Resultats du traitement par la fosfomycine. Med Malad Infect 1981; 11:255-61.
21. Walsh TC, Hansen SL, Tatem BA, Auger F, Standiford HC. Activity of novobiocinagainst methicillin resistant Staphylococcus aureus. J Antimicrob Chemoiher 1985; 15:435-40.
22. Unowsky J, Chandrasekar PH, De Lorenzo W, Levine DP. In vitro and in vivo activityof coumermycin and other antibacterial agents against methicillin resistant strains of Staphylococcus aureus. Chemotherapy 1986; 32:499-505.
23. Schwalbe RS, Stapleton JT, Gilligan PH. Emergence of vancomycin-resistance incoagulase negative staphylococci. N EnglJ Med 1987; 316:927-31.
24. Stille E, Sietzen W, Dieterich HA, Fell JJ. Clinical efficacy and safety of teicoplanin.J Antimcrob Chemother 1988; 21 (suppl A): 69-79.
25. Editorial. Staphylococci resistant to neomycin and bacitracin. Lancet 1965; ii: 421-22.26. McNally T, Lewis MR, Brown DR. Effect of rifampicin and bacitracin on nasal
carriers of Staphylococcus aureus. Antimicrob Agents Chemother 1984; 25:422-26.27. Casewell MW, Hill RLR. Mupirocin (“pseudomonic acid”)—a promising new topical
antimicrobial agent. J Antimicrob Chemother 1987; 19:1-5.28. Baird D, Coia J. Mupirocin-resistant Staphylococcta aureus. Lancet 1987; ii: 387-88.29. Pallanza R, Scotti R, Beretta G, Cavalleri B, Arioli V. In vitro activity of A-16686, a
potential antiplaque agent. Antimicrob Agents Chemother 1984; 26:462-65.
Printed in Great Britain © 1989 The Lancet, by Robquest Ashford Kent 46 Bedford Square London W C 1 B 3SL

Journal oj Antimicrobial Chemotherapy (1989) 23, 517-525
Comparative in-vitro activity of vancomycin, teicoplanin, ramoplanin (formerly A16686), paldimycin, DuP 721 and DuP 105 against methicillin and gentamicin resistant Staphylococcus aureus
P. A. C. Maple, J. M. T. Hamilton-Miller* and W. Brumfitt
Department o f Medical Microbiology, Royal Free Hospital School o f Medicine, PondStreet, Hampstead, London NW3 2QG, UK
The in-vitro activities of five anti-staphylococcal agents, teicoplanin, ramoplanin, paldimycin, D u P 721 and D u P 105 have been compared to vancomycin. Minimum inhibitory concentrations (MICs) and minimum bactericidal concentrations (MBGs) have been determined for a collection of methicillin and gentamicin resistant Staphylococcus aureus (MGRSA), comprising 75 strains obtained from 22 centres.
In terms of geometric mean MICs (inoculum size 105 cfu) paldimycin was the most active agent (0-4 mg/1) followed by ramoplanin (0-75 mg/1), teicoplanin (1*0 mg/1),D u P 721 and vancomycin (2-0 mg/1) and D u P 105 (6-8 mg/1).
Ramoplanin was bactericidal within six hours to all strains at a concentration of 1 0 mg/1. The M B C 90s for vancomycin and teicoplanin were > 32 mg/1 after 22 h exposure to antibiotic and 2-5 and 4 0 mg/1 respectively after 26 h exposure. Paldimycin was bactericidal against only some strains, while D u P 721 and D u P 105 were not bactericidal.
Ramoplanin is the most interesting of the new antibiotics, on account of its rapid and consistent bactericidal activity.
Introduction
Before 1975, strains of Staphylococcus aureus resistant to gentamicin were rarely isolated (Soussy et al., 1975; Porthouse et al., 1976). Outbreaks of hospital infection with strains of S. aureus resistant to methicillin and gentamicin (MGRSA) were first reported in 1976 (Shanson, Kensit & Duke, 1976; Soussy et al., 1976). Subsequently, such strains have shown increasing prevalence worldwide (Leading article, 1981; Thompson & Wenzel, 1982; Townsend et al., 1987).
Vancomycin is currently the antibiotic of choice for treating systemic infections caused by methicillin resistant staphylococci (Watanakunakorn, 1982; Kucers, 1984). Concern that the now widespread use of vancomycin will lead to the emergence of resistance has prompted the search for new agents. The toxicity of vancomycin, its inconvenient administration and its cost, are reasons for contemplating alternative antimicrobial therapy.
Recently, a number of new compounds with activity predominantly against Gram- positive species have been reported. We have investigated five such compounds with characteristics which make them potential alternatives to vancomycin.
♦Corresponding author.
5170305-7453/89/040517 + 09 $02.00/0 © 1989 The British Society for Antimicrobial Chemotherapy

1988). Ramoplanin (formerly called A 16686 and M D L 62198) and paldimycin also have good anti-staphylococcal activity (Neu & Neu, 1986; Pfaller, Bale & Barrett, 1987). The latter two compounds are structurally novel, paldimycin being derived from the paulomycins (Argoudelis et al., 1987a) while ramoplanin is a lipoglycodepsipep- tide, resembling but distinct from daptomycin. D uP 721 and DuP 105 are entirely new chemical entities, substituted oxazolidinones (Slee et al., 1987).
In this study, we report the activity o f these antimicrobial agents against a collection o f M GRSA comprising 75 strains from 22 centres in 17 countries distributed over five continents.
Materials and methods
Media
Iso-Sensitest Agar (ISA), Nutrient Agar (NA) and Antibiotic Medium No. 2 (AM) were from Oxoid Ltd. Basingstoke.
Bacterial strains
The 75 strains o f M GRSA were kindly supplied from Liverpool, UK; Crewe, UK; London, UK; Dublin, Eire; Paris, France; Brussels, Belgium; Hamburg, FGR; Vienna, Austria; Zurich, Switzerland; Lisbon, Portugal; Athens, Greece; Pavia, Italy; Istanbul, Turkey; Hong-Kong; Melbourne, Australia; Tokyo, Japan; Ohio, USA; Botucatu, Brazil; Cape Town, South Africa.
Twenty strains o f methicillin sensitive S. aureus (MSSA) from the routine microbiology laboratory at The Royal Free Hospital were used for additional studies on the bactericidal activity o f vancomycin and teicoplanin.
The antimicrobial agents used were: vancomycin hydrochloride (Sigma Chemicals, St Louis, Missouri, USA), gentamicin sulphate (Roussel Laboratories, London, UK), teicoplanin and ramoplanin (Merrell Dow, Lepetit Research Centre, Milan), paldimycin (Upjohn Co., Kalamazoo, Michigan, USA), D uP 721 and DuP 105 (Du Pont de Nemours, Geneva, Switzerland).
Microbiological methods
The strains of S. aureus were confirmed as tube coagulase-positive, catalase-positive, Gram-positive cocci.
To test for methicillin resistance, strains and appropriate controls were streaked on to NA, and then paper strips containing 25 jug methicillin (M ast Laboratories, Merseyside) were laid on the agar at right angles to the inocula. Plates were read after incubation for 40 h at 30°C.
Minimum inhibitory concentrations (MICs) were determined by incorporating doubling dilutions o f antimicrobial agents into agar. For paldimycin, because of its better stability at pH 6*5, AM was used, while ISA was used for all the other agents. Using a multi-point inoculator (Denley Instruments, Billingshurst, Sussex) all plates
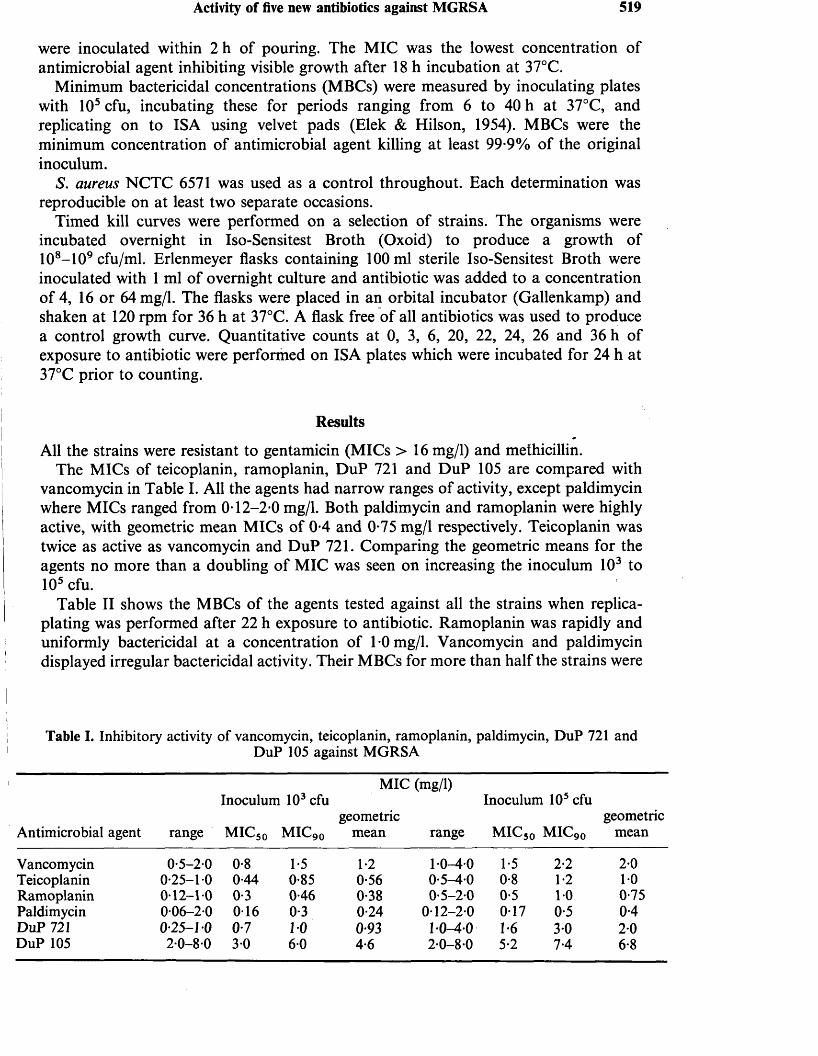
Activity of five new antibiotics against MGRSA 519
were inoculated within 2 h of pouring. The MIC was the lowest concentration of antimicrobial agent inhibiting visible growth after 18 h incubation at 37°C.
Minimum bactericidal concentrations (MBCs) were measured by inoculating plates with 105 cfu, incubating these for periods ranging from 6 to 40 h at 37°C, and replicating on to ISA using velvet pads (Elek & Hilson, 1954). MBCs were the minimum concentration of antimicrobial agent killing at least 99*9% of the original inoculum.
S. aureus NCTC 6571 was used as a control throughout. Each determination was reproducible on at least two separate occasions.
Timed kill curves were performed on a selection of strains. The organisms were incubated overnight in Iso-Sensitest Broth (Oxoid) to produce a growth of 108-109 cfu/ml. Erlenmeyer flasks containing 100 ml sterile Iso-Sensitest Broth were inoculated with 1 ml of overnight culture and antibiotic was added to a concentration of 4, 16 or 64 mg/1. The flasks were placed in an orbital incubator (Gallenkamp) and shaken at 120 rpm for 36 h at 37°C. A flask free of all antibiotics was used to produce a control growth curve. Quantitative counts at 0, 3, 6, 20, 22, 24, 26 and 36 h of exposure to antibiotic were performed on ISA plates which were incubated for 24 h at 37°C prior to counting.
Results
All the strains were resistant to gentamicin (MICs > 1 6 mg/1) and methicillin.The MICs of teicoplanin, ramoplanin, DuP 721 and DuP 105 are compared with
vancomycin in Table I. All the agents had narrow ranges of activity, except paldimycin where MICs ranged from 0*12-2*0 mg/1. Both paldimycin and ramoplanin were highly active, with geometric mean MICs of 0*4 and 0*75 mg/1 respectively. Teicoplanin was twice as active as vancomycin and DuP 721. Comparing the geometric means for the agents no more than a doubling of MIC was seen on increasing the inoculum 103 to 105 cfu.
Table II shows the MBCs of the agents tested against all the strains when replica- plating was performed after 22 h exposure to antibiotic. Ramoplanin was rapidly and uniformly bactericidal at a concentration of 1*0 mg/1. Vancomycin and paldimycin displayed irregular bactericidal activity. Their MBCs for more than half the strains were
Table I. Inhibitory activity of vancomycin, teicoplanin, ramoplanin, paldimycin, D u P 721 andD u P 105 against M G R S A
M I C (mg/1)Inoculum 103 cfu Inoculum 105 cfu
geometric geometricAntimicrobial agent range m i c 50 m i c 90 mean range m i c 50 MIC90 mean
Vancomycin 0*5-20 0*8 1*5 1*2 1 0-4*0 1*5 2*2 2*0Teicoplanin 0*25-1*0 0*44 0*85 0*56 0*5-40 0*8 1*2 1*0Ramoplanin 0*12-1*0 0*3 0*46 0*38 0*5-20 0*5 1*0 0*75Paldimycin 0*06-2*0 0-16 0*3 0*24 0*12-2*0 0*17 0*5 0*4D u P 721 0*25-1*0 0*7 10 093 1 *0-4*0 1*6 3*0 2*0D u P 105 2*0-80 3*0 6*0 4*6 2*0-8 *0 5*2 7*4 6*8
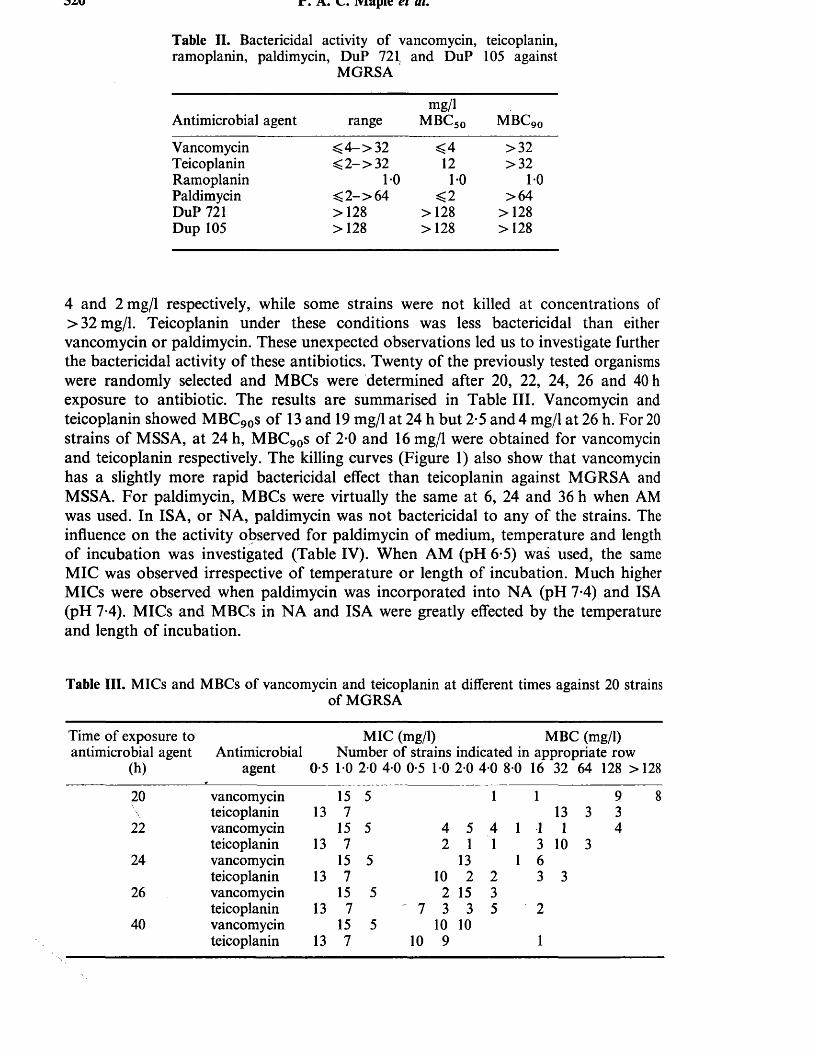
DZu jr. a. l. iviapie ei at.
Table II. Bactericidal activity of vancomycin, teicoplanin, ramoplanin, paldimycin, D u P 721( and D u P 105 against
M G R S A
Antimicrobial agent rangemg/l
m b c 50 m b c 90Vancomycin < 4 - > 3 2 <4 >32Teicoplanin < 2- > 32 12 >32Ramoplanin 10 10 10Paldimycin < 2 - > 6 4 <2 >6 4D u P 721 >128 >128 >128D u p 105 >128 >128 >128
4 and 2 mg/1 respectively, while some strains were not killed at concentrations of >32 mg/1. Teicoplanin under these conditions was less bactericidal than either vancomycin or paldimycin. These unexpected observations led us to investigate further the bactericidal activity of these antibiotics. Twenty of the previously tested organisms were randomly selected and MBCs were determined after 20, 22, 24, 26 and 40 h exposure to antibiotic. The results are summarised in Table III. Vancomycin and teicoplanin showed MBC90s of 13 and 19 mg/1 at 24 h but 2*5 and 4 mg/1 at 26 h. For 20 strains of MSSA, at 24 h, MBC90s of 2-0 and 16 mg/1 were obtained for vancomycin and teicoplanin respectively. The killing curves (Figure 1) also show that vancomycin has a slightly more rapid bactericidal effect than teicoplanin against MGRSA and MSSA. For paldimycin, MBCs were virtually the same at 6, 24 and 36 h when AM was used. In ISA, or NA, paldimycin was not bactericidal to any of the strains. The influence on the activity observed for paldimycin of medium, temperature and length of incubation was investigated (Table IV). When AM (pH 6*5) was used, the same MIC was observed irrespective of temperature or length of incubation. Much higher MICs were observed when paldimycin was incorporated into NA (pH 7*4) and ISA (pH 7-4). MICs and MBCs in NA and ISA were greatly effected by the temperature and length of incubation.
Table III. MICs and M B C s of vancomycin and teicoplanin at different times against 20 strainsof M G R S A
Time of exposure to M I C (mg/1) M B C (mg/1)antimicrobial agent Antimicrobial Number of strains indicated in appropriate row
(h) agent 0-5 1 0 2 0 4 0 0-5 1 0 2 0 4 0 80 16 32 64 128 >128
20 vancomycinteicoplanin 13
157
5 1 113 3
9 8 3
22 vancomycin 15 5 4 5 4 1 1 1 4teicoplanin 13 7 2 1 1 3 10 3
24 vancomycin 15 5 13 1 6teicoplanin 13 7 10 2 2 3 3
26 vancomycin 15 5 2 15 3teicoplanin 13 7 ' 7 3 3 5 2
40 vancomycinteicoplanin 13
157
510
109
101
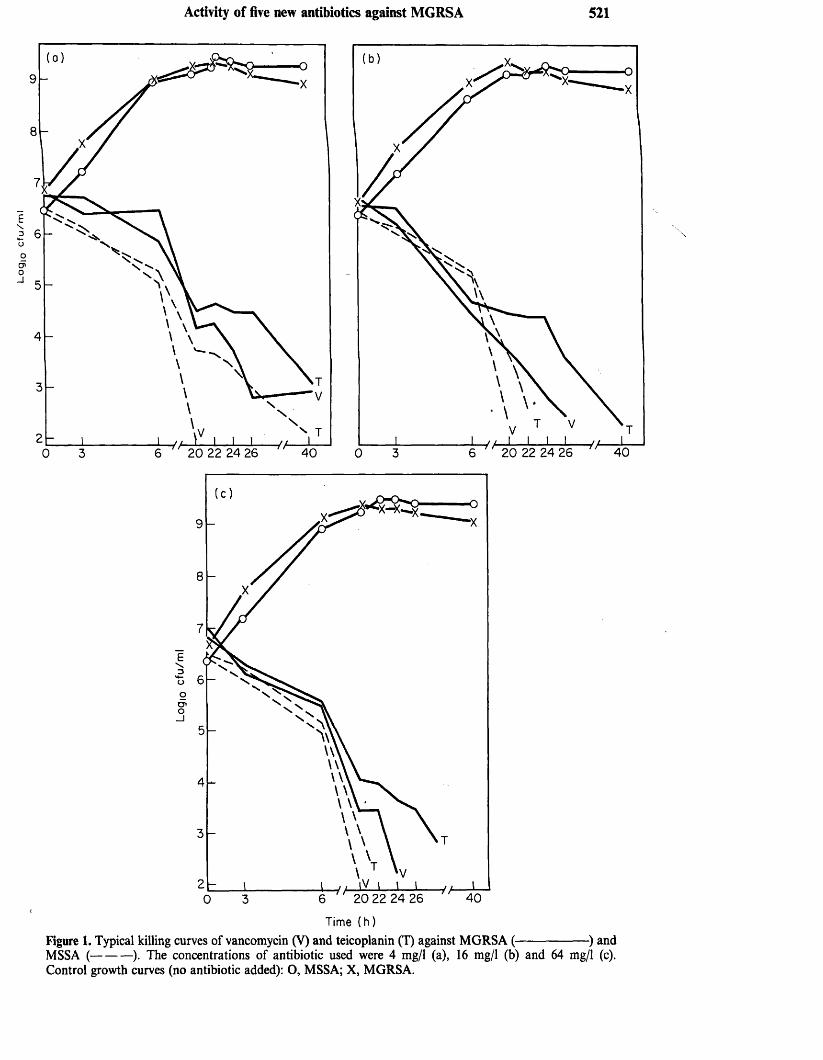
Logi
c c
fu/m
lActivity of five new antibiotics against MGRSA 521
Es3UoO'o_ l
4020 22 24 26
T i m e ( h )
Figure 1. Typical killing curves of vancomycin (V) and teicoplanin (T) against MGRSA (------------------ ) andMSSA (-----------). The concentrations of antibiotic used were 4 mg/1 (a), 16 mg/1 (b) and 64 mg/1 (c).Control growth curves (no antibiotic added): O, MSSA; X, MGRSA.
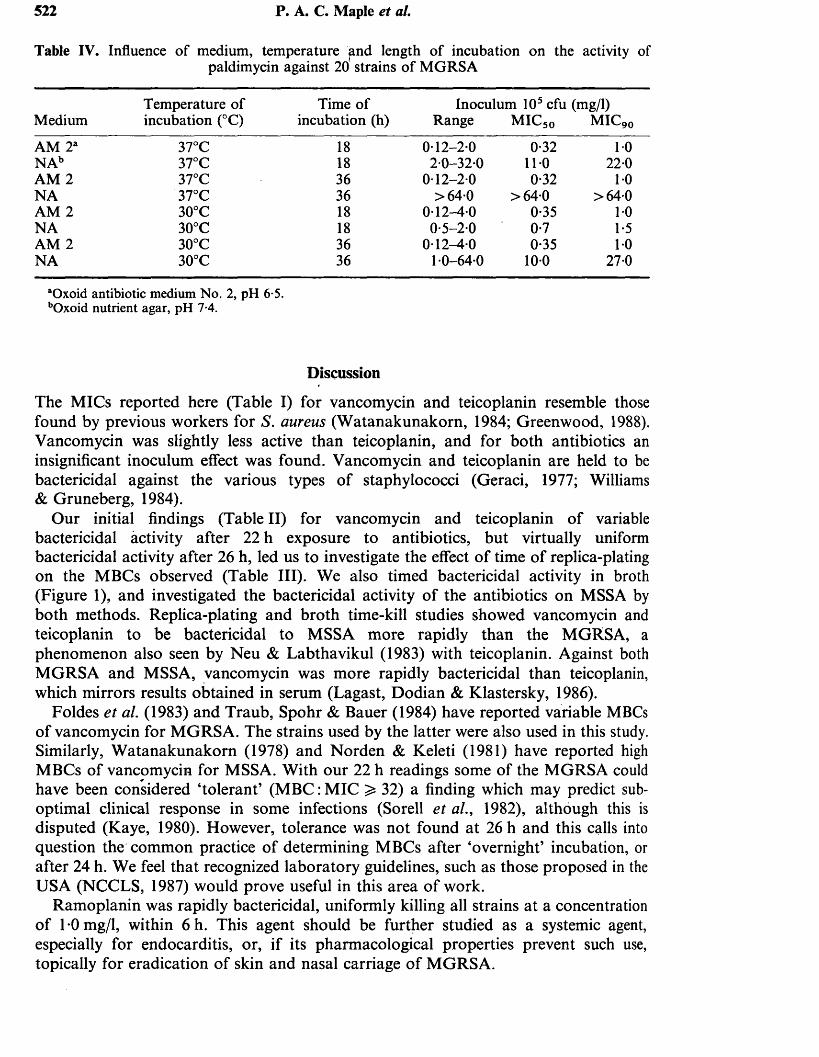
522 P. A. C. Maple et al.
Table IV. Influence of medium, temperature and length of incubation on the activity ofpaldimycin against 20 strains of M G R S A
M e d i u mTemperature of incubation (°C)
Time of incubation (h)
Inoculum 105 cfu (mg/1) Range M I C 50 M I C 90
A M 2a 37°C 18 0-12-2*0 0*32 1*0N A b 37°C 18 2*0-32*0 11-0 22*0A M 2 37°C 36 0*12-2*0 0*32 1*0N A 37°C 36 >64*0 >64*0 >64*0A M 2 30°C 18 0*12-4*0 0*35 1*0N A 30°C 18 0*5-20 0*7 1*5A M 2 30°C 36 0*12-4*0 0*35 1*0N A 30°C 36 1*0-64*0 10*0 27*0
“Oxoid antibiotic medium N o. 2, pH 6*5. bOxoid nutrient agar, pH 7-4.
Discussion
The MICs reported here (Table I) for vancomycin and teicoplanin resemble those found by previous workers for S. aureus (W atanakunakorn, 1984; Greenwood, 1988). Vancomycin was slightly less active than teicoplanin, and for both antibiotics an insignificant inoculum effect was found. Vancomycin and teicoplanin are held to be bactericidal against the various types of staphylococci (Geraci, 1977; Williams & Gruneberg, 1984).
Our initial findings (Table II) for vancomycin and teicoplanin of variable bactericidal activity after 22 h exposure to antibiotics, but virtually uniform bactericidal activity after 26 h, led us to investigate the effect of time of replica-plating on the MBCs observed (Table III). We also timed bactericidal activity in broth (Figure 1), and investigated the bactericidal activity of the antibiotics on MSSA by both methods. Replica-plating and broth time-kill studies showed vancomycin and teicoplanin to be bactericidal to MSSA more rapidly than the MGRSA, a phenomenon also seen by Neu & Labthavikul (1983) with teicoplanin. Against both MGRSA and MSSA, vancomycin was more rapidly bactericidal than teicoplanin, which mirrors results obtained in serum (Lagast, Dodian & Klastersky, 1986).
Foldes et al. (1983) and Traub, Spohr & Bauer (1984) have reported variable MBCs of vancomycin for MGRSA. The strains used by the latter were also used in this study. Similarly, W atanakunakorn (1978) and Norden & Keleti (1981) have reported high MBCs of vancomycin for MSSA. With our 22 h readings some of the MGRSA could have been considered ‘tolerant’ (M BC: MIC ^ 32) a finding which may predict sub- optimal clinical response in some infections (Sorell et al., 1982), although this is disputed (Kaye, 1980). However, tolerance was not found at 26 h and this calls into question the common practice of determining MBCs after ‘overnight’ incubation, or after 24 h. We feel that recognized laboratory guidelines, such as those proposed in the USA (NCCLS, 1987) would prove useful in this area of work.
Ramoplanin was rapidly bactericidal, uniformly killing all strains at a concentration of 1 *0 mg/1, within 6 h. This agent should be further studied as a systemic agent, especially for endocarditis, or, if its pharmacological properties prevent such use, topically for eradication of skin and nasal carriage of MGRSA.

Paldimycins, like the corresponding paulomycins, are sensitive to heat and acidic or alkaline environments (Argoudelis et al., 19876). Rolston et a l, (1987) also found the activity of paldimycin to be medium dependent. There was significant loss of activity when paldimycin was incorporated in NA (pH 7-4), influenced both by the nature of the medium, and temperature. We have found paldimycin to be stable in AM (pH 6-8) over 36 h, irrespective of temperature (Table IV).
DuP 721 and DuP 105 are synthetic agents containing a novel oxazolidinone functional grouping. Like other workers, we havfc found DuP 721 to be more active than DuP 105 against S. aureus (Neu et al., 1988), and that both compounds are bacteriostatic (Daly et al., 1988). 1
In conclusion, all the antimicrobial agents except DuP 105, showed quantitatively superior or similar in-vitro activity to vancomycin in terms of MIC. Further work is required with ramoplanin, DuP 721 and paldimycin to establish their pharmacokinetic profiles and in-vivo efficacy. Paldimycin augments serum killing of S. aureus (Cialdella, Ulrich & Marshall, 1988). However, until detailed published studies correlating its activity in vitro with that in vivo are available, a state of confusion over conditions for susceptibility testing will exist.
We believe ramoplanin to be a most promising antibiotic, on account of its rapid, uniform bactericidal activity against MGRSA, and also because of its reported good activity against streptococci, especially faecal types (Pallanza et al., 1984). Teicoplanin is undergoing clinical trials now, and should it have similar clinical efficacy to vancomycin, offers the prospect of more easily administered and better tolerated treatment.
Finally, our MBC studies showed teicoplanin to be less rapidly bactericidal than vancomycin for MGRSA and MSSA. Whether this observation is clinically relevant is debatable, especially when considering the influence of technical variableson the observation of increased MBC (Pelletier, 1984).
Acknowledgement
This work was sponsored entirely by grants from The Special Trustees of The Royal Free Hospital and from The Leverhulme Trust. We are grateful to the following for supplying strains of S. aureus: C. A. Bartzokas, J. Cooper, G. W. Smith, E. C.Moorhouse, C. T. Keane, J. F. Acar, E. Yourassowsky, W. H. Traub, K. H. Spitzy,F. H. Kayser, A. Torres Pereira, H. Giamarellou, G. G. Grassi, O. Ang, P. Y. Chau, S. Dixson, K. Ubukata, C. W atanakunakorn, C. Linnemann, C. E. O. Pires de Campos, A. A. Forder and D. Hanslo.
References
Argoudelis, A. D ., Baczynskyj, L., Buege, J. A., Marshall, V. P., Mizak, S. A. & Wiley, P. F. (1987a). Paulomycin — related antibiotics: paldimycins and antibiotics 273a2. Isolation and characterisation. Journal o f Antibiotics 40, 408-18.
Argoudelis, A. D., Baczynskyj, L., Mizak, S. A., Shilliday, F. B., Spinelli, P. A. & DeZwaan, J. (19876). Paldimycins A and B and antibiotics 273a2a and 273a2/!. Synthesis and characterisation. Journal o f Antibiotics 40, 419-36.
Cialdella, J. I., Ulrich, R. G. & Marshall, V. P. (1988). Augmentation o f serum bactericidal activity by paldimycin. Journal o f Antibiotics 41, 660-66.
Daly, S. J., Eliopoulos, G. M., Reiszner, E. & Moellering, R. C. (1988). Activity and mechanism

524 P. A. C. Maple et al.
of action of D u P 105 and D u P 721, new oxazolidinone compounds. Journal of Antimicrobial Chemotherapy 21, 721-30.
Elek, S. D. & Hilson, G. R. F. (1954). Combined agar diffusion and replica plating techniques in the study of antibacterial substances. Journal o f Clinical Pathology 7, 37-44.
Foldes, M., Munro, R., Sorrell, T. C., Shanker, S. & Toohey, M. (1983). In-vitro effects of vancomycin, rifampicin, and fusidie acid, alone and in combination, against methicillin- resistant Staphylococcus aureus. Journal o f Antimicrobial Chemotherapy 11, 21-6.
Geraci, J. E. (1977). Vancomycin. Mayo Clinic Proceedings 52, 631-64.Greenwood, D. (1988). Microbiological properties of teicoplanin. Journal o f Antimicrobial
Chemotherapy 21, Suppl. A, 1-13.Kaye, D. (1980). The clinical significance of tolerance of Staphylococcus aureus. Annals of
Internal Medicine 93, 924-96.Kucers, A. (1984). Vancomycin. Journal o f Antimicrobial Chemotherapy 14, 564— 7.Lagast H., Dodian, P. & Klastersky, J. (1986). Comparison of pharmacokinetics and
bactericidal activity of teicoplanin and vancomycin. Journal o f Antimicrobial Chemotherapy 18, 513-20.
Leading article. (1981). Of gentamicin and staphylococci. Lancet ii, 127-8.National Committee for Clinical Laboratory Standards (1987). Methods for determining
bactericidal activity of antimicrobial agents. Proposed Guideline M26-P. NCCLS, Villanova, PA.
Neu, H. C. & Labthavikul, P. (1983). In vitro activity of teichomycin compared with those of other antibiotics. Antimicrobial Agents and Chemotherapy 24, 425-8.
Neu, H. C. & Neu, N. M. (1986). In vitro activity of A16686, a new glycopeptide. Chemotherapy 32, 453-457.
Neu, H. C, Novelli, A., Saha, G. & Chin, N.-X. (1988). In vitro activities of two oxazolidinone antimicrobial agents, D u P 721 and D u P 105. Antimicrobial Agents and Chemotherapy 32, 580-3.
Norden, C. W., & Keleti, E. (1981). Antibiotic tolerance in strains of Staphylococcus aureus. Journal o f Antimicrobial Chemotherapy 7, 599-605.
Pallanza, R., Berti, M., Scotti, R., Randisi, E. & Arioli, V. (1984). A-16686, a new antibiotic from Actinoplanes. II Biological properties. Journal o f Antibiotics 37, 318-24.
Pelletier, L. L. (1984). Lack of reproducibility of macrodilution M B C ’s for Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 26, 815-8.
Pfaller, M. A., Bale, M. & Barrett, M. (1987). In-vitro activity of paldimycin against methicillin- resistant and susceptible isolates of Staphylococcus aureus and S. epidermidis. Journal o f Antimicrobial Chemotherapy 20, 286-8.
Porthouse, A., Brown, D. F. J., Graeme Smith, R. & Rogers, T. (1976). Gentamicin resistance in Staphylococcus aureus. Lancet i, 20-1.
Rolston, K. V. I., LeBlanc, B., Ho, D. H. & Bodey, G. P. (1987). In vitro activity of paldimycin (U-70138F) against Gram-positive bacteria isolated from patients with cancer. Antimicrobial Agents and Chemotherapy 31, 650-2.
Shanson, D. C., Kensit, J. G. & Duke, R (1976). Outbreak of hospital infection with a strain of Staphylococcus aureus resistant to gentamicin and methicillin. Lancet ii, 1347-8.
Slee, A. M., Wuonola, M. A., McRipley, R. J., Zajac, I., Zawada, M. J., Bartholomew, P. T. et al. (1987). OxazPlidinones, a new class of synthetic antibacterial agents: in vitro and in vivo activities of D u P 105 and D u P 721. Antimicrobial Agents and Chemotherapy 31, 1791-7.
Sorrell, T. C., Packham, D. R., Shanker, S., Foldes, M. & Munro, R. (1982). Vancomycin therapy for methicillin-resistant Staphylococcus aureus. Annals o f Internal Medicine 97, 344-50.
Soussy, C. J., Bouanchaud, D. H., Fouace, J., Dublanchet, A. & Duval, J. (1975). A gentamicin resistance plasmid in Staphylococcus aureus. Annales de Microbiologie 126B, 91-4.
Soussy, C. J., Dublanchet, A., Cormier, M., Bismuth, R., Nizon, F., Chardon, F. et al. (1976). Nouvelles resistances plasmidiques de Staphylococcus aureus aux aminosides (gentamicine, tobramycine, amikacine). Nouvelle Presse Medicale 5, 2599-602.
Stille, W., Sietzen, W., Dieterich, H.-A. & Fell, J. J. (1988). Clinical efficacy and safety of teicoplanin. Journal o f Antimicrobial Chemotherapy 21, Suppl. A, 69-79.

Activity ot live new antibiotics against MGK5A 525
Thompson, R. L. & Wenzel, R. P. (1982). International recognition of methicillin-resistant strains of Staphylococcus aureus. Annals o f Internal Medicine 97, 925-6.
Townsend, D. E., Ashdown, N., Bolton, S., Bradley, J., Duckworth, G., Moorhouse, E. C. et al. (1987). The international spread of methicillin resistant Staphylococcus aureus. Journal o f Hospital Infection 9, 60-71.
Traub, W. H., Spohr, M. & Bauer, D. (1984). Gentamicin and methicillin resistant, clinical isolates of Staphylococcus aureus: comparative in vitro and in vivo efficacy of alternative antimicrobial drugs. Chemotherapy 30, 102-12.
Watanakunakorn, C. (1978). Antibiotic-tolerant Staphylococcus aureus. Journal o f Antimicrobial Chemotherapy 4, 561-8.
Watanakunakorn, C. (1982). Treatment of infections due to methicillin-resistant Staphylococcus aureus. Annals o f Internal Medicine 97, 370-8.
Watanakunakorn, C. (1984). Mode of action and in-vitro activity of vancomycin. Journal o f Antimicrobial Chemotherapy 14, Suppl. D, 7-18.
Williams, A. H. & Gruneberg, R. N. (1984). Teicoplanin. Journal o f Antimicrobial Chemotherapy 14, 441-8.
{Received 11 August 1988; revised version accepted 20 December 1988)

622 Eur. J. Clin. Microbiol. Infect. Dis.
more, for detection o f catheter-related bacteremia, this combination had 100% sensitivity and a positive predictive value similar to the actual rate o f catheter- related bacteremia.
References
1. Vanherweghem, J. L., Cabolet, P., Dhaene, M., Goldman, M., Stolear, J. C., Sabot, J. P., Waterlot, Y., Marchal,M.: Complications related to subclavian catheters for hemodialysis. American Journal o f Nephrology 1986, 6: 3 39 -345 .
2. Qeri, J., Corrado, M. L., Seligman, S. J.: Quantitative culture o f intravenous catheters and other intravascular inserts. Journal of Infectious Diseases 1980, 141: 781— 786.
3. Maki, D. G., Weise, C. E., Sarafin, H. W.: A semiquan- titative culture method for identifying intravenous- catheter related infection. New England Journal of Medicine 1977, 296: 1305-1309 .
4. Liffares, J., Sitges-Serra, A., Garau, J., Perez, J. L., Martin, R.: Pathogenesis of catheter sepsis: a prospective study with quantitative and semiquantitative cultures of catheter hub and segments. Journal of Clinical Microbiology 1985, 21: 357—360.
5. Mausner, J. S., Bahn, A. K.: Epidemiology. Saunders, Philadelphia, 1974, p. 243—252.
6. Norwood, M. A. G., Civetta, J. M.: Evaluating sepsis in critically ill patients. Chest 1987, 92: 137—144.
7. Collignon, P. J., Soni, P., Pearson, I. Y., Woods, W. P., Munro, R., Sorrell, T. C.: Is semiquantitative culture of central vein catheter tips useful in the diagnosis of catheter-associated bacteremia? Journal o f Clinical Microbiology 1986, 24: 5 3 2 -5 3 5 .
8. Bozzetti, F., Terno, G., Camerini, E., Baticci, F., Scarpa, D., Pupa, A.: Pathogenesis and predictability of central venous catheter sepsis. Surgery 1982, 91: 3 8 3 -3 8 9 .
9. Moyer, M. A., Edwards, L. D., Farley, L.: Comparative culture methods on 101 intravenous catheters: routine, semiquantitative and blood cultures. Archives of Internal Medicine 1983, 143: 6 6 -6 9 .
10. Sitges-Serra, A., Puig, P., Jaurrieta, E., Garau, J., Alastrue, A., Sitges-Creus, A.: Catheter sepsis due to Staphylococcus epidermidis during parenteral nutrition. Surgery, Gynecology and Obstetrics 1980, 151: 481—483.
Ciprofloxacin Resistance in Methicillin- and Gentamicin-Resistant Staphylococcus aureus
P. Maple*, J. Hamilton-Miller, W. Brumfitt
A total of 112 Staphylococcus aureus strains resistant to methicillin and gentamicin were collected from 31 centres in 22 countries worldwide. Many strains
Department o f Medical Microbiology, The Royal Free Hospital School o f Medicine, Pond Street, Hampstead, London NW3 2QG, UK.
were multi-resistant. In tests to determine the susceptibility of the organisms to ciprofloxacin 16 strains (14.3 %), originating from France, the FRG, Israel and Italy, were shown to be resistant to this agent. To limit ciprofloxacin resistance, a reappraisal is necessary of fluoroquinolone usage against methicillin- and gentamicin-resistant Staphylococcus aureus.
In recent years, there have been a growing number o f reports o f infection or colonization due to methicillin - and gentamicin-resistant Staphylococcus aureus (MGRSA). As many o f these strains are also resistant to numerous other^ antibiotics besides the beta- lactams and gentamicin (1), in certain instances the choice o f effective antistaphylococcal chemotherapy may be severely restricted. Ciprofloxacin, on account o f its structural novelty and reported in vitro activity against MRSA (2), has found favour with some clinicians for treatment o f infection or colonization with these organisms.
The first specific report o f ciprofloxacin resistant MRSA appeared in October 1988 (3). Subsequent accounts show that resistance to fluoroquinolones in these organisms can become common (4 , 5). In order to determine how widespread this phenomenon is, we tested a total o f 112 strains o f MGRSA from 31 centres in 22 countries for susceptibility to ciprofloxacin.
Materials and M ethods. Strains o f MGRSA have been collected from 31 centres worldwide over the last three years. For this study, a total o f 112 strains were used. No more than four and no fewer than two strains from any one centre were represented in the study collection. For each centre, only strains with different phage-types or biotypes were used. The sources o f the strains were: Australia (S. Dixson), Austria (K. H. Spitzy), Belgium (E. Yourassowsky), Brazil (C. E. O. Pires de Campos), UK (J. Cooper, W. Brumfitt, C. A. Bartzokas), Federal Republic o f Germany (W. H. Traub), France (J. F. Acar, E. Bergogne Berezin), German Democratic Republic (C. Hohne), Greece (H. Giamarellou), Hong Kong (P. Y. Chau), Republic o f Ireland (C. T. Keane, E. C. Moorhouse), Israel (D. Merzbach), Italy (G .G . Grassi), Japan (K. Ubukata), Kuwait (G. F.Araj), Poland (J. Borowski), Portugal (A. Torres Pereira), South Africa (A. A. Forder, D. Hanslo), Spain (E. P. Trallero), Switzerland (F. H. Kayser), Turkey (O. Ang), USA (R. C. Moellering, R. P. Wenzel, C.C. Linnemann, H. C. Neu, C. Watanakunakorn).
All the strains were coagulase-positive (tube test), catalase-positive, gram-positive cocci which grew up to the edge o f paper strips containing 25 p% o f methicillin (Mast laboratories, UK) after incubation for 40 h at 30 °C on nutrient agar. All strains were gentamicin-resistant (MIC > 1 6 mg/1).
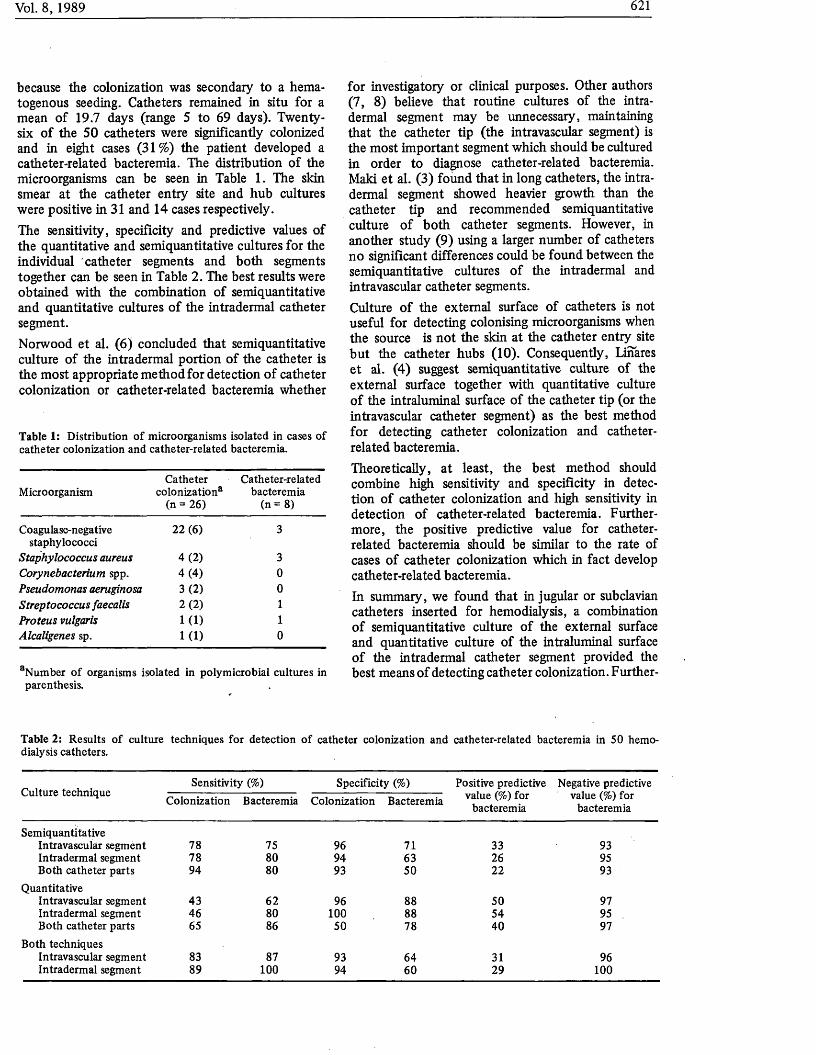
because the colonization was secondary to a hematogenous seeding. Catheters remained in situ for a mean of 19.7 days (range 5 to 69 days). Twenty- six of the 50 catheters were significantly colonized and in eight cases (31%) the patient developed a catheter-related bacteremia. The distribution of the microorganisms can be seen in Table 1. The skin smear at the catheter entry site and hub cultures were positive in 31 and 14 cases respectively.The sensitivity, specificity and predictive values of the quantitative and semiquantitative cultures for the individual catheter segments and both segments together can be seen in Table 2. The best results were obtained with the combination of semiquantitative and quantitative cultures of the intradermal catheter segment.Norwood et al. (6) concluded that semiquantitative culture of the intradermal portion of the catheter is the most appropriate method for detection of catheter colonization or catheter-related bacteremia whether
Table 1: Distribution o f microorganisms isolated in cases of catheter colonization and catheter-related bacteremia.
MicroorganismCatheter
colonization® (n = 26)
Catheter-related bacteremia
(n = 8)
Coagulase-negativestaphylococci
22 (6) 3
Staphylococcus aureus 4 (2 ) 3Corynebacterium spp. 4 (4 ) 0Pseudomonas aeruginosa 3 (2 ) 0Streptococcus faecalis 2 (2 ) 1Proteus vulgaris 1 (1 ) 1Alcaligenes sp. 1 (1 ) 0
aNumber o f organisms isolated in polymicrobial cultures in parenthesis.
for investigatory or clinical purposes. Other authors (7, 8) believe that routine cultures of the intradermal segment may be unnecessary, maintaining that the catheter tip (the intravascular segment) is the most important segment which should be cultured in order to diagnose catheter-related bacteremia. Maki et al. (3) found that in long catheters, the intradermal segment showed heavier growth than the catheter tip and recommended semiquantitative culture of both catheter segments. However, in another study (9) using a larger number of catheters no significant differences could be found between the semiquantitative cultures of the intradermal and intravascular catheter segments.Culture of the external surface of catheters is not useful for detecting colonising microorganisms when the source is not the skin at the catheter entry site but the catheter hubs (10). Consequently, Linares et al. (4) suggest semiquantitative culture of the external surface together with quantitative culture of the intraluminal surface of the catheter tip (or the intravascular catheter segment) as the best method for detecting catheter colonization and catheter- related bacteremia.Theoretically, at least, the best method should combine high sensitivity and specificity in detection of catheter colonization and high sensitivity in detection of catheter-related bacteremia. Furthermore, the positive predictive value for catheter- related bacteremia should be similar to the rate of cases of catheter colonization which in fact develop catheter-related bacteremia.In summary, we found that in jugular or subclavian catheters inserted for hemodialysis, a combination of semiquantitative culture of the external surface and quantitative culture of the intraluminal surface of the intradermal catheter segment provided the best means of detecting catheter colonization. Further-
Table 2: Results o f culture techniques for detection o f catheter colonization and catheter-related bacteremia in 50 hemodialysis catheters.
Sensitivity (%) Specificity (%) Positive predictive Negative predictiveCulture technique Colonization Bacteremia Colonization Bacteremia ™lue «*) for value (%) for
bacteremia bacteremia
SemiquantitativeIntravascular segment 78 75 96 71 33 93Intradermal segment 78 80 94 63 26 95Both catheter parts 94 80 93 50 22 93
QuantitativeIntravascular segment 43 62 96 88 50 97Intradermal segment 46 80 100 88 54 95Both catheter parts 65 86 50 78 40 97
Both techniquesIntravascular segment 83 87 93 64 31 96Intradermal segment 89 100 94 60 29 100

MICs of gentamicin and ciprofloxacin were determined by the agar dilution method. Doubling dilutions of antibiotics were incorporated into molten Iso-Sensitest agar (Oxoid, UK) held at 50 °C. Inocula of approximately 5 X 105 C F U from 24 h-old peptone water cultures grown at 37 °C were delivered onto the plates using a multi-point inocula- tor (Denley Instruments, UK). MICs were read after incubation for 24 h at 37 °C. MBCs were determined after exposure for 24 h to antibiotic at 37 °C by replica-plating onto fresh Iso-Sensitest agar plates which were incubated for 24 h at 37 °C. MBCs were defined as the minimum concentration of antibiotic killing at least 99.9 % of the original inoculum.Phage typing was performed with the International Set at routine test dilution (RTD) and 100 x RTD. Biotyping was performed by a modification of the method of Putland and Guinness (6), which detects haemolysis (sheep blood agar), lipolysis (egg yolk agar) and pigmentation (milk agar). Antibiotic sensitivity profiles were also determined by means of the disc method using DST Agar (Oxoid, UK) supplemented with 7 % lysed horse blood. The discs contained as follows (content in parenthesis): netilmicin (10 jtrg), amikacin (10 Mg), neomycin (10 Mg), tetracycline (10 Mg), clindamycin (2 Mg), chloramphenicol (10 Mg), trimethoprim (1.25 Mg) and rifampicin (5 Mg)- An inoculum of approximately 5 X 10s C F U per plate was used. Sensitivity results were read after incubation for 20 h at 37 °C. Strains were classed as resistant if the diameter of the inhibition zone (total diameter minus disc diameter) was < 6 mm, and sensitive if the diameter was > 6 mm.
Results and Discussion. The following MIC breakpoints of ciprofloxacin were adopted after consultation with Bayer, UK: sensitivity < 1 mg/1, intermediate sensitivity > 1 mg/1 but < 4 mg/1, and resistance > 4 mg/1. Applying these criteria 94 strains were classified as sensitive, two as intermediate and 16 as resistant (Table 1). The ciprofloxacin-resistant strains originated from the FRG. France, Israel and Italy. Ciprofloxacin MICs of > 16 mg/1 and MBCs of > 32 mg/1 were found for M G R S A from France and Israel.Until now, fluoroquinolone (ciprofloxacin or pefloxa- cin) resistant strains of M R S A have been reported from the U S A (3,5), France (4) and the U K (7).
All these strains were found in hospitals where either ciprofloxacin or pefloxacin had been used to treat infections or colonization. As we had made no attempt to collect M G R S A specifically resistant to ciprofloxacin for this study, the high incidence of ciprofloxacin resistance is cause for concern.Ciprofloxacin has been reported to have good in vitro activity against MRSA, and has been suggested as an important new alternative for treatment of M R S A colonization (8). In common with other investigators (9), we found ciprofloxacin to be bactericidal: many of the sensitive strains tested were killed by a concentration of 1 mg/1 (Table 1).The finding of an incidence of 14.3 % ciprofloxacin resistance in this collection of organisms contradicts earlier claims that such resistance would rarely be encountered (10). Our ciprofloxacin-resistant M G R S A strains were also resistant to enoxacin, norfloxacin, ofloxacin and pefloxacin. Thus we would urge caution before fluoroquinolones are used for the management of patients with MGRSA. It would be unfortunate if ciprofloxacin-resistant strains were to emerge due to inappropriate use of fluoroquinolones, especially as the antimicrobial agents available for treatment of infections caused by M G R S A are already so limited. We advocate that ciprofloxacin (or any other fluoroquinolone) should not be used for the eradication of carriage. Furthermore, if quinolones are required for treatment, then their use in appropriate combination should be seriously considered in order to reduce the chance of selecting for resistance.
References
1. Maple, P. A . C., Hamilton-Miller, J. M. T., Brumfitt, W.:World-wide antibiotic resistance in methicillin-resistant Staphylococcus aureus. Lancet 1989, i: 53 7 —540.
2. Smith, S. M., Eng, R. H. K.: Activity o f ciprofloxacin against methicillin-resistant Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 1985, 27: 6 8 8 -6 9 1 .
3. Isaacs, R. D ., Kunke, P. J., Cohen, R. L., Smith, J. W.:Ciprofloxacin resistance in epidemic methicillin-resistant Staphylococcus aureus. Lancet 1988, ii: 843.
4. Jean-Pierre, H., Boyer, G., Darbas, H.: Evolution de la resistance des Staphylococcus aureus a la pefloxacin. Etude portant sur 782 souches isole'es en 1985 et 1986. Pathologie Biologie 1988, 36: 9 5 6 -9 5 8 .
Table 1: MICs and MBCs o f ciprofloxacin for 112 strains o f methicillin- and gentamicin-resistant Staphylococcus aureus collected worldwide.
Concentration o f ciprofloxacin (mg/1)
< 0 .2 5 0.5 1 2 4 8 16 32 > 3 2
No. o f strains inhibited 10 52 32 2 9 0 3 4 0No. of strains killed 2 5 70 15 10 3 0 0 7

5. Schaefler, S.: Methicillin-resistant strains o f Staphylococcus aureus resistant to quinolones. Antimicrobial Agents and Chemotherapy 1989, 27: 335—336.
6. Putland, R. A ., Guinness, M. D. G.: Autobac susceptibility testing o f methicillin-resistant Staphylococcus aureus isolated in an Australian hospital. Journal o f Clinical Microbiology 1985, 22: 8 2 2 -8 2 7 .
7. Milne, F. M., Faiers, M. C.: Ciprofloxacin resistance in epidemic methicillin-resistant Staphylococcus aureus. Lancet 1988, ii: 843.
8. Mulligan, M. E., Ruane, P. J., Johnston, L., Wong, P., Wheelock, J. P., MacDonald, K., Reinhardt, J. F., Johnson, C. C., Statner, B. B., Blomquist, I., McCarthy, J., O’Brien, W., Gardner, S., Hammer, L., Citron, D. M.: Ciprofloxacin for eradication o f methicillin-resistant Staphylococcus aureus colonization. American Journal o f Medicine 1987, 82, Supplement 4A: 215—219.
9. Smith, S. M., Eng, R. H. K., Berman, E.: The effect of ciprofloxacin on methicillin-resistant Staphylococcus aureus. Journal o f Antimicrobial Chemotherapy 1986, 17: 2 8 7 -2 9 5 .
10. Smith, J. T.: Mode o f action o f the 4-quinolone antibacterial agents. In: Ciprofloxacin product monograph. ADIS Press, Auckland, New Zealand, 1986, p. 1 9 -3 1 .
In Vitro Activity of Cefoperazone- Sulbactam Combinations against Cefoperazone-Resistant Clinical Bacterial Isolates
G. M. Eliopoulos1,3*, K. Klimm1,M. J. Ferraro2,3, R.C. Moellering, Jr.1,3*
From July 1987 to January 1988,452 cefoperazone- resistant bacterial isolates were identified among strains subjected to routine susceptibility testing in a clinical microbiology laboratory. The 452 isolates were tested for susceptibility to cefoperazone, sulbactam, and a 2:1 combination of these drugs by agar dilution techniques. The greatest benefit of the cefo- perazone-sulbactam combination was noted against Bacteroides spp. and Acinetobacter spp. The combination demonstrated clinically significant synergism against approximately 20% of strains of Pseudomonas aeruginosa.
Department o f Medicine, New England Deaconess Hospital, 185 Pilgrim Road, Boston, Massachusetts 02215, USA. Departments o f Medicine and Microbiology, Massachusetts General Hospital, Fruit Street, Boston, Massachusetts 02114, USA.Harvard Medical School, 25 Shattuck Street, Boston, Massachusetts 02115, USA.
The beta-lactamase inhibitors sulbactam, clavulanic acid and tazobactam effectively inhibit a number of commonly encountered beta-lactamases of either plasmid or chromosomal origin (1). Although ex- panded-spectrum cephalosporins such as cefoperazone are relatively resistant to hydrolysis by many of these enzymes, occasional bacterial isolates demonstrate substantially greater susceptibility to cefo- perazone-sulbactam combinations than to the cephem alone (2). The purpose of this study was to examine the activity of cefoperazone-sulbactam combinations against the cefoperazone-resistant bacteria encountered in a busy clinical microbiology laboratory over a period of seven consecutive months.
Materials and Methods. During the period of July 1987 through January 1988, all routine clinical isolates subjected to antimicrobial susceptibility testing at the Massachusetts General Hospital, Boston, Massachusetts, were examined for susceptibility to cefoperazone. Aerobic and facultative bacteria resistant to cefoperazone as tested by the disk diffusion method (zone diameter < 15 m m ) and anaerobic isolates resistant as tested by the disk elution method (growth in 30Mg/ml) were referred for subsequent testing by agar dilution reference methods (3). Susceptibility to cefoperazone and sulbactam (Pfizer, USA), alone and in a 2:1 combination, was determined (2). Antibiotics were incorporated into Mueller- Hinton agar (BBL Microbiology Systems, USA) for testing aerobic organisms. Inocula of approximately 104 CFU/spot were applied to plates using a multiprong inoculating device. Anaerobes were tested on Wilkins-Chalgren agar (Oxoid, UK) using inocula of about 105/spot. Plates were incubated in air at room temperature for 18h or in an anaerobic atmosphere (GasPak, BBL) for 48h at 35 °C.
Results and Discussion. During the study period, 452 organisms were referred for further testing based on resistance to cefoperazone as determined by aforementioned criteria (Table 1). Cefoperazone- resistant organisms accounted for 6 % of routine clinical isolates of gram-negative bacteria screened during a representative three-month interval. Rates of resistance to cefoperazone varied widely between species, ranging from 0.2% of Escherichia coli to 42% of Acinetobacter anitratus. Resistance to cefoperazone based on disk diffusion testing predicted resistance by dilution testing (MIC > 64Mg/ml) (2) in 97.6% of aerobic isolates. Of the 369 facultative and aerobic organisms determined to be cefoperazone-resistant by disk diffusion, only eight were inhibited by a cefoperazone concentration of 32Mg/ml, while one was inhibited at 16 pglmt. Among the Bacteroides spp., 18 of 83 (21.7%) judged to be resistant by the elution method demonstrated intermediate levels of susceptibility (MIC = 32 /Ltg/ml) by




















