
Role of Bcl2 family proteins and caspases in the regulation of apoptosis
18
Role of Bcl-2 family proteins and caspases in the regulation of apoptosis Mohammad Shamsul Ola • Mohd. Nawaz • Haseeb Ahsan Received: 29 September 2010 / Accepted: 13 December 2010 / Published online: 6 January 2011 Ó Springer Science+Business Media, LLC. 2011 Abstract Apoptosis, or programmed cell death, plays a pivotal role in the elimination of unwanted, damaged, or infected cells in multicellular organisms and also in diverse biological processes, including development, cell differ- entiation, and proliferation. Apoptosis is a highly regulated form of cell death, and dysregulation of apoptosis results in pathological conditions including cancer, autoimmune and neurodegenerative diseases. The Bcl-2 family proteins are key regulators of apoptosis, which include both anti- and pro-apoptotic proteins, and a slight change in the dynamic balance of these proteins may result either in inhibition or promotion of cell death. Execution of apoptosis by various stimuli is initiated by activating either intrinsic or extrinsic pathways which lead to a series of downstream cascade of events, releasing of various apoptotic mediators from mitochondria and activation of caspases, important for the cell fate. In view of recent research advances about underlying mechanism of apoptosis, this review highlights the basics concept of apoptosis and its regulation by Bcl-2 family of protein. Furthermore, this review discusses the interplay of various apoptotic mediators and caspases to decide the fate of the cell. We expect that this review will add to the pool of basic information necessary to under- stand the mechanism of apoptosis which may implicate in designing better strategy to develop biomedical therapy to control apoptosis. Keywords Apoptosis Á Bcl-2 Á BH-3 only proteins Á Caspases Á Mitochondrial proteins Á Programmed cell death Abbreviations PCD Programmed cell death Bcl-2 B cell lymphoma-2 protein Bax Bcl-2 associated X protein Bid Bcl-2 interacting domain death agonist Bad Bcl-2 antagonist of cell death Bcl-xl Bcl-extra long Bim Bcl-2 interacting mediator of cell death Bik Bcl-2 interacting killer Bmf Bcl-2 modifying factor Boo Bcl-2 homolog of ovary Bcl-xs Bcl-extra short Bak Bcl-2 antagonistic killer Bok Bcl-2 related ovarian killer Apaf-1 Apoptosis protease-activating factor-1 Diablo Direct IAP binding Protein with low pI FADD Fas-associated death domain protein TNF-R Tumor necrosis factor receptor Fas-L Fas ligand HtrA High-temperature requirement IAP Inhibitor of apoptosis protein IMM Inner mitochondrial membrane Omi/HtrA2 Mammalian serine protease SMAC Second mitochondrial activator of caspase TNF-a Tumor Necrosis Factor alpha TRADD TNF-receptor-1 associated death domain protein VDAC Voltage-dependent anion channel Cyt c Cytochrome c PIDDosome p53-Inducible death domain containing protein complex DISC Death-inducing signaling complex M. S. Ola Á Mohd. Nawaz Department of Ophthalmology, College of Medicine, King Saud University, Riyadh 11411, KSA H. Ahsan (&) Department of Biochemistry, Faculty of Dentistry, Jamia Millia Islamia (A Central University), New Delhi 110025, India e-mail: [email protected] 123 Mol Cell Biochem (2011) 351:41–58 DOI 10.1007/s11010-010-0709-x
-
Upload
ucsc-chile -
Category
Documents
-
view
0 -
download
0
Transcript of Role of Bcl2 family proteins and caspases in the regulation of apoptosis

Role of Bcl-2 family proteins and caspases in the regulationof apoptosis
Mohammad Shamsul Ola • Mohd. Nawaz •
Haseeb Ahsan
Received: 29 September 2010 / Accepted: 13 December 2010 / Published online: 6 January 2011
� Springer Science+Business Media, LLC. 2011
Abstract Apoptosis, or programmed cell death, plays a
pivotal role in the elimination of unwanted, damaged, or
infected cells in multicellular organisms and also in diverse
biological processes, including development, cell differ-
entiation, and proliferation. Apoptosis is a highly regulated
form of cell death, and dysregulation of apoptosis results in
pathological conditions including cancer, autoimmune and
neurodegenerative diseases. The Bcl-2 family proteins are
key regulators of apoptosis, which include both anti- and
pro-apoptotic proteins, and a slight change in the dynamic
balance of these proteins may result either in inhibition or
promotion of cell death. Execution of apoptosis by various
stimuli is initiated by activating either intrinsic or extrinsic
pathways which lead to a series of downstream cascade of
events, releasing of various apoptotic mediators from
mitochondria and activation of caspases, important for the
cell fate. In view of recent research advances about
underlying mechanism of apoptosis, this review highlights
the basics concept of apoptosis and its regulation by Bcl-2
family of protein. Furthermore, this review discusses the
interplay of various apoptotic mediators and caspases to
decide the fate of the cell. We expect that this review will
add to the pool of basic information necessary to under-
stand the mechanism of apoptosis which may implicate in
designing better strategy to develop biomedical therapy to
control apoptosis.
Keywords Apoptosis � Bcl-2 � BH-3 only proteins �Caspases � Mitochondrial proteins � Programmed cell death
Abbreviations
PCD Programmed cell death
Bcl-2 B cell lymphoma-2 protein
Bax Bcl-2 associated X protein
Bid Bcl-2 interacting domain death agonist
Bad Bcl-2 antagonist of cell death
Bcl-xl Bcl-extra long
Bim Bcl-2 interacting mediator of cell death
Bik Bcl-2 interacting killer
Bmf Bcl-2 modifying factor
Boo Bcl-2 homolog of ovary
Bcl-xs Bcl-extra short
Bak Bcl-2 antagonistic killer
Bok Bcl-2 related ovarian killer
Apaf-1 Apoptosis protease-activating factor-1
Diablo Direct IAP binding Protein with low pI
FADD Fas-associated death domain protein
TNF-R Tumor necrosis factor receptor
Fas-L Fas ligand
HtrA High-temperature requirement
IAP Inhibitor of apoptosis protein
IMM Inner mitochondrial membrane
Omi/HtrA2 Mammalian serine protease
SMAC Second mitochondrial activator of caspase
TNF-a Tumor Necrosis Factor alpha
TRADD TNF-receptor-1 associated death domain
protein
VDAC Voltage-dependent anion channel
Cyt c Cytochrome c
PIDDosome p53-Inducible death domain containing
protein complex
DISC Death-inducing signaling complex
M. S. Ola � Mohd. Nawaz
Department of Ophthalmology, College of Medicine, King Saud
University, Riyadh 11411, KSA
H. Ahsan (&)
Department of Biochemistry, Faculty of Dentistry, Jamia Millia
Islamia (A Central University), New Delhi 110025, India
e-mail: [email protected]
123
Mol Cell Biochem (2011) 351:41–58
DOI 10.1007/s11010-010-0709-x

Introduction
Apoptosis is an ancient Greek word which means ‘‘the
falling of leaves from a tree’’. John Kerr first introduced the
name which refers to the morphological feature of forma-
tion of ‘‘apoptotic bodies’’ from a cell [1]. Apoptosis, also
known as programmed cell death (PCD), plays an integral
role in a variety of biological events including morpho-
genesis, tissue homeostasis, aging, and removal of
unwanted harmful cells [2]. Changes typical for apoptosis
include condensation of the nuclei, DNA fragmentation,
chromatin condensation, generation of envoluted mem-
brane segments, cellular shrinkage, and disintegration of
mitochondria [3, 4]. Apoptosis can be induced by a variety
of physiological and pathophysiological stimuli, such as
specific receptor molecules-CD95 [5, 6], tumor necrosis
factor (TNFa) [7], growth factors [8], ultraviolet light [9,
10], irradiation [11], heat shock [12], cytotoxic drugs [13,
14], oxidative stress [15], ceramide treatment [16], and
bacteria [17]. Dysfunction of apoptotic pathways result in
various pathological conditions including cancer, autoim-
mune, and neurodegenerative diseases [18–21].
A significant progress has been made toward under-
standing the mechanism of apoptosis and other apoptosis
related dysfunction, but it needs more intensive research to
better understand the whole cascade of events involved in
apoptosis and also the mechanism of their regulatory
pathways [22–24]. The nematode, Caenorhabditis elegans
(C. elegans), has been widely used to elucidate the
molecular pathways implicated in the regulation of PCD
[25–27]. For elucidating the underlying genetics and bio-
chemical pathways in PCD, three scientists, namely,
S. Brenner, J. Sulston, and R. Horvitz were awarded the
Noble Prize in medicine (2002).
The Bcl-2 and apoptosis
The underlying mechanism and genetics of apoptosis
began a new approach of research after the landmark dis-
covery by Horvitz, 1999 [28]. The protein expression of
two C. elegans cell death genes (CED-3 and CED-4) is
necessary for PCD during development, and CED-9 is the
functional homolog to mammalian protein Bcl-2, which is
shown to correct the phenotype of C. elegans by its anti-
apoptotic function [29, 30]. CED-9 was found to act
upstream of aspartate-directed cysteine protease (CED-3
and CED-4) known as caspases [31]. Previously, it was
revealed that proteins that are encoded by the mutant genes
discovered in C. elegans shared homology with mamma-
lian proteins, including Bcl-2 [32].
The role of Bcl-2 (B cell lymphoma 2), a founder
member of the Bcl-2 family of apoptosis regulator proteins,
has been elucidated in tumor development by dysfunction
in apoptotic pathways [20, 30]. In mammals, there is an
extrinsic apoptotic pathway that involves death receptors
and an intrinsic apoptotic pathway that involves the mito-
chondria. Mammalian mitochondria control apoptosis by
releasing many apoptogenic mediators from intermem-
brane space [33] and that further leads to a cascade of
reaction with the assistance of caspases, proteases that
cleave key cellular proteins [34, 35]. A major difference
between apoptosis in worm and mammals is the greater
role of mitochondria in the mammals but not in the worm.
Bcl-2 expression specifically blocks the morphologic fea-
tures of apoptosis, including the plasma membrane bleb-
bing, DNA cleavage, and nuclear condensation. Bcl-2
plays a role in cell survival and also inhibits cell death
induced by various stimuli such as chemotherapeutic
agents, ethanol and heat shock, indicating Bcl-2 as a neg-
ative regulator of cell death. The anti-death role is also
demonstrated in vivo by generation of mice lacking the
bcl-2 gene, which shows a variety of abnormalities
including excessive cell death.
Members and structural classification of Bcl-2 proteins
On the basis of various structural and functional charac-
teristics, the Bcl-2 family is divided into anti-apoptotic and
pro-apoptotic proteins [36–38]. All of the members of Bcl-
2 family have one or more homology domains labeled as
Bcl-2 homology (BH1, -2, -3, and -4) which are important
for heterodimeric interaction among members of the Bcl-2
family [27] (Fig. 1). Anti-apoptotic Bcl-2 family multido-
main proteins include Bcl-2, Bcl-xL (Bcl-extra long), A1,
Bcl-w, and Boo (Bcl-2 homolog of ovary) contain BH-
(1-4) domains. The Bcl-xL and Bcl-2 have a carboxy-ter-
minal hydrophobic transmembrane tail domain which helps
in localization of protein in outer mitochondrial membrane
(OMM) with the exception that Bcl-2 also resides in the
nuclear and endoplasmic reticulum membrane and is
translocated to OMM upon an apoptotic signal. Myeloid
cell leukemia factor-1 (Mcl-1) is the only antiapoptotic
Bcl-2 protein with three BH domains (BH-1, -2, and -3).
Pro-apoptotic Bcl-2 family of proteins are divided into
two subgroups according to the number of BH domains
(e.g., Bax, Bak, and Box) or those proteins that have only
the BH3 domain (e.g., Bid, Bim, and Bad) with the
exception of Bcl-xS which has only BH3 and BH4 domains
(Fig. 1). There are eight members of BH3 only protein,
which include hara-kiri (Hrk), BH3 interacting domain
death agonist (Bid), Bcl-2 interacting mediator of cell
death (Bim), Bcl-2 modifying factor (Bmf), p53, promoter-
upregulated modulator of apoptosis (Puma), Noxa (named
for ‘‘damage’’), Bcl-2 antagonist of cell death (Bad), and
42 Mol Cell Biochem (2011) 351:41–58
123

Bcl-2 interacting killer (Bik). Bid, Bad, and Bim have
cytosolic location with respect to mitochondria. Following
death signal, BH3 domain-only proteins also known as
minimal death domain can neutralize or depress antiapo-
ptotic Bcl-2 proteins allowing pro-apoptotic proteins such
as Bax/Bak-like proteins to induce apoptosis [39, 40].
A restricted subset of these BH3 proteins undergoes
post-translation modification such as cleavage, phosphor-
ylation, transcriptional upregulation, and dissociation. Such
modifications result in their activation and translocation to
outer mitochondrial membrane (OMM) which helps it to
interact with multidomain pro-apototic members, Bax/Bak-
like proteins, leading to their oligomerization and forma-
tion of pore. Such pore may further lead to release of other
pro-apoptotic protein on the intermembrane space (IMS)
that enhances the processes of apoptosis [41]. Thus the
regulation of expression and function of BH3 domain-only
proteins is critical in mediating cell death and under-
standing their expression and function necessary for cyto-
protective strategies. In general, the relative ratio of pro-
survival (Bcl-2) and pro-apoptotic (Bax and BH3-only)
proteins seems to determine the cell sensitivity or resis-
tance to the apoptotic stimuli.
Mechanism of action and role of Bcl-2 family members
in the regulation of mitochondrial apoptosis
Antiapoptotic or pro-survival proteins
It is now becoming evident that almost every nucleated cell
requires protection by at least one of the member Bcl-2
homolog, and that regulates homeostasis in mammalian cell
and tissues. Over-expression of Bcl-2 protein in hemato-
poietic lineages yields excess T, B, and myeloid cells and
enhance the survival of these progenitor cells that are hard to
avoid cytotoxic insults [29, 42]. Alternatively, inactivation
of the Bcl-2 gene increases apoptosis in specific cell types,
presumably because the concentrations of other Bcl-2
homolog are too low to compensate. Bcl-2 itself is required
for the survival of kidney, mature lymphocytes and mela-
nocyte stem cells [43], Bcl-w for sperm progenitors in adult
mice [44], an A1 gene for neutrophils [45], Mcl-1 for zygote
implantation [46], and Bcl-xL for erythroid and neuronal
cells [47]. The hydrophobic carboxy-terminal domain of
these antiapoptotic proteins help to anchor themselves to the
cytoplasmic face of three intracellular membranes: the outer
mitochondrial membrane, the nuclear envelope, and the
endoplasmic reticulum (ER). Bcl-2 is an integral membrane
protein, whereas Bcl-xL and Bcl-w only become tightly
associated with the membrane after cytotoxic signals which
induce conformational changes to protect cell. The antiap-
optotic member of Bcl-2 family, including Bcl-2 itself,
decreases apoptosis mainly by preventing mitochondrial
outer membrane permeabilization (MOMP) either by neu-
tralizing the activity of both pro-apoptotic members (BH3
only protein and multidomain members) [48]. If Bcl-2 or
Bcl-xl cannot exert their antiapoptotic property, Bax oligo-
merization takes place and apoptosis is promoted.
Pro-apoptotic or anti-survival protein
It includes both multi-domain and BH3-only protein
members.
Multi-domain pro-apoptotic member
Antiapoptotic members such as Bcl-2 interact with Bak and
Bax and inhibit their oligomerization and/or bind to BH3
only proteins to block apoptosis [48]. Bak interacts with
two mitochondrial fusion proteins (Mfn1 and Mfn2). After
receiving apoptotic signal, Bak dissociates from Mfn2 to
become associated with Mfn1. It has been shown that
reconstitution of Bak into Bax/Bak double knockout cells
Fig. 1 According to conserve
domain (BH) and its anti or pro-
apoptotic action, the Bcl-2
super-family can be divided into
three sub-families: Anti-
apoptotic or Pro-survival multi-
domain, pro-apoptotic multi-
domain and pro-apoptotic BH3
only. Few examples of protein
from each group of the family
are given in parenthesis. TM
(transmembrane domain)
Mol Cell Biochem (2011) 351:41–58 43
123

restores mitochondrial fragmentation and Bak in associa-
tion with Bax permeabilize the outer membrane of mito-
chondria rendering apoptotic cascade [49]. Bak may
contribute to early mitochondrial fragmentation while Bax
is probably more important for subsequent pores develop-
ment and degeneration in the outer membrane [49]. A
number of studies have shown that insertion of activated
oligomerized Bax and/or Bak into the OMM exerts stress
on the membrane, leading to supramolecular pores that
include lipids (lipidic pores) in the OMM [50–52].
In healthy cells, Bak is inactive in the OMM through its
association with Voltage-dependent Anion channel
(VDAC-2) [48], whereas Bax is dormant in the cytosol
through interactions with several proteins, including
Ku-70, 14-3-3, and the humanin peptide. Many apoptotic
signals can trigger BH3-only-protein-dependent transloca-
tion of Bax, followed by its insertion into the OMM and the
formation of Bak or Bax homo-oligomers. Several models
have been proposed for how Bcl-2 anti-apoptotic proteins
antagonize the functions of Bax, Bak, and BH3-only pro-
teins. Activation of Bax and Bak leads to a conformational
change that exposes the N-terminus of the proteins, which is
otherwise hidden in the inactive state [53]. After Bak and
Bax form homo-oligomers, they participate in forming pores
in OMM and cause permeabilization leading to the release of
the contents of the mitochondrial intermembrane space
(IMS) including Smac and cytochrome c into the cytosol
[54, 55]. The released mitochondrial protein then activates
caspases, which causes a series of cascade reactions that
cleave essential proteins complement throughout the cell.
Two models of Bax and Bak activation, exist the direct
and indirect models. According to the direct model various
BH3 pro-apoptotic proteins called as activator proteins (Bid,
Bim, Puma, and p53) directly interact and induce confor-
mational changes in Bak and Bax [56]. Members of anti-
apoptotic proteins bind and sequester such activator, and
also bind to activated Bak and Bax proteins that might be
present and thus prevent apoptosis. BH3-only proteins are
further divided into activator and sensitizer categories [57].
Sensitizers bind to anti-apoptotic proteins and cause the
release of activator BH3-only proteins leading to activation
of Bak and Bax. However, it is likely that additional factors
other than Bid and Bim can act as activators such as Puma,
p53, and heat as activators of Bax and Bak [58–60], and
possibly others. Gavathiotis and colleagues have demon-
strated that stabilized alpha-helix of Bcl-2 domains (SA-
HBs) directly initiates Bax-mediated mitochondrial
apoptosis [61].
According to the indirect model, anti-apoptotic proteins
always bind to Bak and Bax and prevent their activation,
whereas in response to death signal, BH3-only proteins
bind to antiapoptotic proteins causing its release and ini-
tiating death signal through activation of Bax and Bak [62].
The sequestered forms of Bak and Bax are those fractions
of the total Bak and Bax population that are already
activated either spontaneously, or by other unspecified
means [63]. Bax and Bak can also alternatively interact
with proteins that remove the requirement for sequestra-
tion by anti-apoptotic proteins [64]. Activated population
of Bak and Bax are needed to kill, and that should be
sequestered by anti-apoptotic proteins to maintain survival
of the cell. Activated Bak and Bax are responsible for the
permeabilization of membranes. The activation is
achieved, either by interacting with activator proteins or
through some agents, while the anti-apoptotic proteins
inhibit death by sequestering activated Bak and Bax or
activator proteins.
BH3-only pro-apoptotic members
BH3-only pro-apoptotic proteins are involved in main-
taining the functionality and cellular integrity of the cell in
which Bim acts as sensor for cytoskeleton integrity, Bid
acts as sensor for death domain receptor signaling, and Bad
acts as an inhibitor for growth factors withdrawal. Indi-
vidual BH3-only proteins are normally controlled by
diverse mechanisms.
Cleavage of Bid Activation of cell surface receptors such
as tumor necrosis factor receptor (TNF-R) activates cas-
pase-8, which leads to the cleavage of Bid, and then
truncated Bid (active Bid or tBid) translocates from cytosol
to OMM and induces cytochrome c release [65–67]. Oh
and his group have demonstrated that at nanomolar con-
centrations of a synthetic Bid activates Bax almost as
efficiently as tBid itself thus tBid engages Bax to trigger its
pro-apoptotic activity [68]. It is now well established that
tBid leads to oligomerization of Bax and Bak, which forms
pores in OMM which in turn permits the release of IMS
proteins into the cytosol [54, 69]. During apoptosis, Bid
induces the mobilization of cyt c by remodeling mito-
chondrial cristae by interacting Bid with cardiolipin [70,
71]. Addition of tBid to permeabilized cells leads to
hydrolysis of cardiolipin molecules thereby decreasing the
cardiolipin levels [72, 73]. But tBid alone does not cause
mitochondrial outer membrane permeabilization [74].
Phosphorylation of Bad In the absence of various sur-
vival factors, Bad is activated by dephosphorylation [75].
The BH3 domain of Bad binds and inactivates Bcl-2 and/or
Bcl-xL at the OMM, thereby promoting cell death. Con-
versely, in the presence of survival factors, Bad is phos-
phorylated, making it to dissociate from Bcl-2 and/or Bcl-
xl, permitting, and survival promotion. There are several
phosphatases that dephosphorylate Bad in vitro [75, 76].
44 Mol Cell Biochem (2011) 351:41–58
123

However, a report by Datta and his group showed that Akt
phosphorylates Bad, both in vitro and in vivo, and blocks
the Bad-induced death of primary neurons [77]. Further-
more, several studies elucidate the importance of Bad
phosphorylation and underlying mechanism of cell death
[78–80].
Dissociation of Bim and Bmf In response to several
apoptotic stimuli, such as detachment of adherent cells
from their substratum (anoikis) or ultraviolet irradiation,
Bmf is released from the myosin V motor complex,
translocates and binds to antiapoptotic family proteins,
such as Bcl-2, Bcl-xl, and Bcl-w, but does not interact with
the pro-apoptotic family protein, such as Bax, Bid, and Bad
[81]. However, c-Jun N-terminal protein kinase (JNK) that
causes phosphorylation of Bim is involved in ischemia-
induced neuronal apoptosis through activation and trans-
location of Bax [82, 83]. Bim is the main regulator of
hematopoietic homeostasis [84], essential for the elimina-
tion of autoreactive lymphocytes [85] and plays vital role
in neuronal death [86]. Recently, it has been shown that
Bim displaces Bcl-xl in the mitochondria and promotes
Bax translocation during intrinsic pathways assisted by
TNFa [87] or UV-induced [88] apoptosis. Histone deace-
tylase (HDAC) inhibitors increase ionizing radiation-
induced apoptosis in several cancer cells via activation of
Bmf transcription [89, 90]. In TGF beta-induced cell death,
there is upregulation of Bmf and Bim, and thus, inhibition
of the TGFb provides an important therapeutic and pro-
tection of cells from apoptosis [91].
Transcriptional regulation of BH3-only proteins BH3-
only proteins are also transcriptionally regulated as in DNA
damage-induced apoptosis which requires synthesis of new
protein. Transcription of Noxa and Puma, the BH3-only
group members, is induced by p53 [92–94]. Apoptosis in
fibroblast in response to DNA damage is decreased in mice
knocked out in Noxa/puma whereas mutations of the BH3
domain suppress the pro-apoptotic activity of Noxa [94],
and Noxa-induced apoptosis is inhibited by Bcl-xl and
Bcl-2 [95]. Recently, it has been reported that Puma
expressed independent of p53 regulation [96, 97] and ini-
tiates apoptosis by dissociating Bcl-xl and Bax, promoting
Bax multimerization and mitochondrial translocation
[9, 96]. Puma rapidly induces apoptosis in cells lacking not
only the BH3-only proteins but in the absence of Bid and
Bim [98]. Puma induced apoptosis is associated by
regeneration of superoxide and H2O2 which is Bax-
dependent and that can be confirmed by the presence of
antioxidants that prevent Puma-dependent apoptosis [99].
Whereas Bax inactivation confers a resistance to Puma-
dependent apoptosis [100].
Apoptotic pathways
Apoptosis or PCD has conserved genetic and biochemical
pathways [27, 101]. In vertebrates, caspase-dependent
apoptosis occurs through two main interconnected path-
ways which are intrinsic and extrinsic pathways [102]. The
intrinsic pathway or intracellular path is mediated by Bcl-2
family, whereas the death receptor or extrinsic pathway is
activated by signal from other cells [7, 103, 104].
Intrinsic pathway
The intrinsic pathways also known as mitochondrial path-
ways or stress pathways are activated by a diverse array of
death stress, genomic stress, metabolic stress, presence of
unfolded proteins, and other stimuli that lead to perme-
abilization of OMM and release of apoptotic proteins into
the cytosol (Fig. 2). Several of these proteins including
cytochrome c (cyt c) initiate or regulate caspase activation.
The cyt c plays the main role in this pathway which is
activated after its interaction with apoptotic protease-acti-
vating factor (Apaf1) and deoxyadenosine triphosphate
(dATP) to form apoptosome [102, 105]. The apoptosome
creates a platform to bring together molecules of the ini-
tiator caspase of the intrinsic pathway. Progression through
the pathway usually leads to activation of caspase-9,
enabling their auto-activation. Caspase-8 also has a major
role to play by activating the Bid leading to the formation
of activated truncated Bid (tBid), which translocates to
mitochondria and releases cyt c. The activated caspase-8
then cleaves procaspase-3, giving activated caspase-3,
which acts as an executioner, by cleaving multiple of other
substrate within the cells [34, 106].
Extrinsic pathway
The extrinsic pathway involves the association of receptor-
mediated transmembrane death receptor (FAS and TNF-a)
and its extracellular ligand (FAS-L and TNF-aL) [107–
109]. For association the receptor trimerizes and death
adapter molecules are recruited on the cytoplasmic side of
the mitochondrial membrane. Fas recruits Fas-associated
death domain protein (FADD) whereas TNF-R recruits
TNF-R1-associated death domain protein (TRADD) which
again recruits FADD [7]. Such association leads to for-
mation of death-inducing signaling complex (DISC) con-
sisting of a complex of receptor, its ligand, the initiator
procaspase-8 (or procaspase-10), and some other regulators
and co-factor [110]. The complex helps in recruiting more
of procaspase-8 (or procaspase-10) and enables their
autoactivation (Fig. 2).
Mol Cell Biochem (2011) 351:41–58 45
123
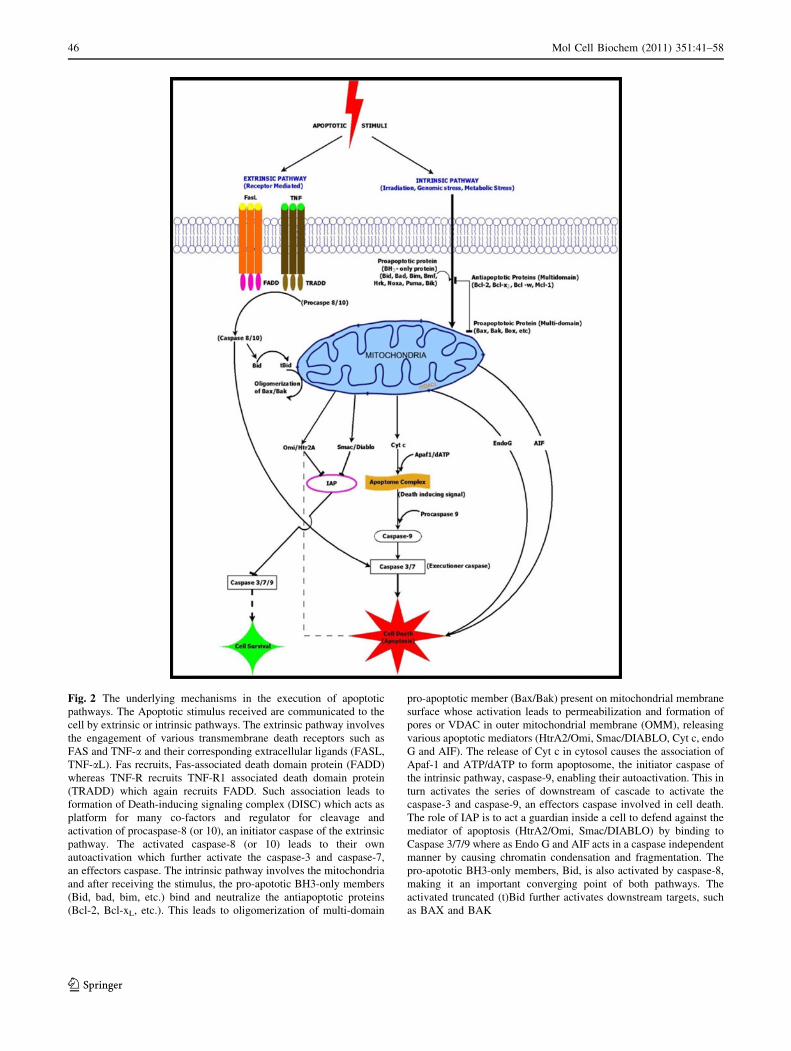
Fig. 2 The underlying mechanisms in the execution of apoptotic
pathways. The Apoptotic stimulus received are communicated to the
cell by extrinsic or intrinsic pathways. The extrinsic pathway involves
the engagement of various transmembrane death receptors such as
FAS and TNF-a and their corresponding extracellular ligands (FASL,
TNF-aL). Fas recruits, Fas-associated death domain protein (FADD)
whereas TNF-R recruits TNF-R1 associated death domain protein
(TRADD) which again recruits FADD. Such association leads to
formation of Death-inducing signaling complex (DISC) which acts as
platform for many co-factors and regulator for cleavage and
activation of procaspase-8 (or 10), an initiator caspase of the extrinsic
pathway. The activated caspase-8 (or 10) leads to their own
autoactivation which further activate the caspase-3 and caspase-7,
an effectors caspase. The intrinsic pathway involves the mitochondria
and after receiving the stimulus, the pro-apototic BH3-only members
(Bid, bad, bim, etc.) bind and neutralize the antiapoptotic proteins
(Bcl-2, Bcl-xL, etc.). This leads to oligomerization of multi-domain
pro-apoptotic member (Bax/Bak) present on mitochondrial membrane
surface whose activation leads to permeabilization and formation of
pores or VDAC in outer mitochondrial membrane (OMM), releasing
various apoptotic mediators (HtrA2/Omi, Smac/DIABLO, Cyt c, endo
G and AIF). The release of Cyt c in cytosol causes the association of
Apaf-1 and ATP/dATP to form apoptosome, the initiator caspase of
the intrinsic pathway, caspase-9, enabling their autoactivation. This in
turn activates the series of downstream of cascade to activate the
caspase-3 and caspase-9, an effectors caspase involved in cell death.
The role of IAP is to act a guardian inside a cell to defend against the
mediator of apoptosis (HtrA2/Omi, Smac/DIABLO) by binding to
Caspase 3/7/9 where as Endo G and AIF acts in a caspase independent
manner by causing chromatin condensation and fragmentation. The
pro-apototic BH3-only members, Bid, is also activated by caspase-8,
making it an important converging point of both pathways. The
activated truncated (t)Bid further activates downstream targets, such
as BAX and BAK
46 Mol Cell Biochem (2011) 351:41–58
123

Apoptotic mediators
Permeabilization of the outer mitochondrial membrane
allows the leakage of at least five apoptotic mediators
(apoptogenic proteins) from the mitochondrial intermem-
brane space, such as cyt c, second mitochondrial activator
of caspases/Direct IAP protein with low pI (Smac/DIA-
BLO), HtrA2/Omi, apoptosis-inducing factors (AIF), and
endonuclease G [32, 37]. These proteins induce apoptosis
in different ways. Smac/DIABLO and HtrA2/Omi suppress
the ability of IAPs (inhibitors of apoptosis proteins) to
inhibit caspases. Endonuclease G and AIF are involved in
DNA fragmentation, and AIF is also involved in chromatin
condensation (Fig. 2). The release of these apoptotic
mediators from mitochondria is known to be regulated by
Bcl-2 family of proteins [55, 111]. However, caspase-
independent mitochondrial cell death results from loss of
respiration and not from the release of cytotoxic apoptotic
mediators [112].
Release of cytochrome c
Cyt c is a water soluble 13 kDa protein encoded by nuclear
gene that is translated in cytosol to be finally imported into
mitochondria. It normally resides in the spaces within
cristae of the inner mitochondrial membrane (IMM) and at
narrow cristae junctions [113]. The role of cyt c in the
intrinsic pathways in mammalian cells is well known.
Addition of dATP to cytosolic extract induces caspase
activity [114] and depletion of cyt c in cell extract inhibits
its apoptotic potential and also microinjection of cyt c in
various cell types enhances the apoptotic pathways [115,
116]. Thus, cyt c is the main mediator of apoptosis [117,
118], and the release of cyt c occurs due to DNA damage
[119, 120]. Over-expression of Bcl-2 blocks the release of
cyt c from mitochondria and inhibits the initiation of
apoptosis [121].
Release of endonuclease G (EndoG)
EndoG is a 30 kDa nuclease protein located in mitochon-
drial intermembrane space [122, 123]. The release of
EndoG after apoptotic signal leads to DNA fragmentation
as found in inhibitor of caspase-activated DNase (ICAD)-
deficient cells after induction of apoptosis by TNF treat-
ment and UV-irradiation [123, 124]. Once released, EndoG
participates in DNA fragmentation but without assistance
of caspases [125–127].
Release of apoptosis-inducing factors (AIF)
Apoptosis-inducing factor (AIF), which resembles bacterial
oxidoreductase, is a 57 kDa flavoprotein present in the
mitochondrial intermembrane space [128]. Upon induction
of apoptosis, AIF translocates from the mitochondria to the
nucleus and causes DNA fragmentation and chromatin
condensation [128]. These effects are independent of its
oxidoreductase and caspases activity [129, 130]. Disruption
of AIF in mice prevents normal apoptosis necessary for the
activation of embryoid bodies in the embryo [131]. In
addition, AIF is required for specific cell death pathways
including lethal responses to excitotoxins such as gluta-
mate and N-methyl-D-aspartate (NMDA), DNA-alkylating
agents, hypoxia–ischemia, or growth factor deprivation
[132]. Recently, Schulthess and coworkers have shown a
protective role of AIF on b-cell turnover and the loss of
AIF increases b-cell apoptosis. AIF is essential for main-
taining b-cell mass and oxidative stress response [133].
The mechanism of AIF-induced large-scale chromatin
condensation and DNA fragmentation is still not clear
[128, 131, 134–136]. More recently, the role of AIF in
12/15-lipoxygenase (LOX)-dependent organelle damage
pathway has been reported showing that AIF and 12/15-
LOX are important mediators in a common cell death
pathway in stroke-induced brain damage [137].
Release of Smac/DIABLO
SMAC (Second Mitochondrial Activator of Caspases) or
DIABLO (Direct IAP Binding Protein with Low pI) are a
25 kDa, pro-apoptotic protein released from the inter-
membrane space that neutralizes the inhibitory activity of
IAP leading to activation of caspases and apoptosis [138–
140]. During reovirus-induced apoptosis, Smac/DIABLO
are released that decreases the level of IAP’s and thus
activates apoptosis [139]. Sphingosine 1-phosphate inhibits
the release of Smac/DIABLO from mitochondria and
antagonizes apoptosis of human leukemia cells. Smac/
DIABLO is involved in many cancer manifestation and
progression [141] such as cervical cancer [142], colon
cancer [143], and hepatocellular carcinoma [144].
Release of Omi/HtrA2
The mammalian serine protease Omi/HtrA2 (high-tem-
perature requirement) is a 49 kD protein, homologous to
the bacterial endoprotease also known as DegP [145].
Unlike Smac/DIABLO, the pro-apoptotic activity of Omi/
HtrA2 involves both IAP binding and serine protease
activity. Omi/HtrA2 has a dual function, when residing
inside the mitochondria it promotes cell survival, but when
released into the cytoplasm it participates in both caspase-
dependent and -independent cell death. It prevents the
IAP’s action via amino-terminal reaper-related motif which
induces caspase activity [99, 146–148]. It also mediates
caspase-independent cell death through its own protease
Mol Cell Biochem (2011) 351:41–58 47
123

activity, by the fact that simultaneous deletion of the other
IAP binding protein, Smac/DIABLO, does not alter the
phenotype of Omi/HtrA2 knockout mice or cells derived
from them [149]. The caspase-independent role of Omi/
HtrA2 in apoptosis is evident in human cardiac-specific
inhibitor of cell cycle protein, Thanatos-Associated Protein
5 (THAP5) that are cleaved by pro-apoptotic Omi/HtrA2
during cardiomyocytic cell death [150]. The role of Omi/
HtrA2 in colon cancer [151] and oxidative stress of pig-
ment epithelial cell [152] further supports its application of
mediating apoptosis in cells. The role of Omi/HtrA2 in
promoting cell death by binding and degrading ped/pea-15,
an anti-apoptotic protein, establishes the pro-apoptotic
effect of Omi/HtrA2 [153] and its role in the apoptosis of
prostate cancer cell, PC-3 [154]. However, a contradictory
role of mitochondrial Omi/HtrA2 has been reported. In
response to some extracellular inducers of mitochondrial
stress, Omi/HtrA2 stabilizes mitochondrial membrane
potential and inhibits mitochondrial superoxide generation
and hence controls apoptosis [155].
Regulation of apoptosis by IAPs
Inhibitor of apoptosis (IAPs) is a family of antiapoptotic
proteins that associate with caspases in response to diverse
stimuli. IAP has been discovered both in invertebrate and
vertebrates. So far, eight human IAP homologs have been
identified which includes X-chromosome-linked IAP
(XIAP, also known as hILP, MIHA, or BIRC4), survivin
(also known as TIAP or BIRC5), cellular IAP1 (c-IAP1, also
known as HIAP2, MIHB or BIRC2), c-IAP2 (also known as
HIAP1, MIHC, or BIRC3), neuronal apoptosis inhibitory
protein (also known as BIRC1), IAP-like protein 2 (also
known as BIRC8, or Ts-IAP), apollon (also known as Bruce
or BIRC6), melanoma IAP (ML-IAP, also known as KIAP,
livin, or BIRC7) [156, 157]. The best characterized IAPs
such as XIAP, c-IAP1, and c-IAP2, bind caspase-3, caspase-
7, and caspase-9, thereby inhibiting their activation and
preventing apoptosis (Table 1) [156, 158–160]. Also, cIAP1
and cIAP2 have been shown to bind caspases, although how
they inhibit apoptosis at the molecular level is not com-
pletely understood [161]. Direct inhibition of caspase
activity by c-IAPs is an important means of regulation in
order to protect cell death. Activity of XIAP is blocked by
binding to Omi/Htr2A and Smac/DIABLO proteins released
from mitochondria after pro-apoptotic stimuli. Thus, Smac/
DIABLO is a negative regulator of IAPs [140, 162]. Simi-
larly, Omi/HtrA2 also inhibits the XIAP through a reaper-
like motif [148] and has a prognostic significance in hepa-
tocellular [144] and renal cell carcinoma [163]. Inhibition of
apoptosis increases the survival rate of cancer cells and
facilitates their escape from cytotoxic therapies and immune
surveillance [144, 164, 165].
Caspases and underlying mechanism in controlling
cellular apoptosis
Caspases are not only involved in the process of apoptosis
but also needed for the development and maturation of
cytokines leading to cell growth and differentiation [166].
Apoptotic cell death is dependent on a family of aspartate-
specific cysteine proteases (caspases) that cleave certain
vital structural proteins (e.g., lamins, gelsolin) and pro-
teolytically activate latent enzymes (e.g., nucleases) that
contribute to cell death. These enzymes exist in most cells
as inactive precursors (procaspases) that are converted into
their active forms by proteolytic cleavage at internal
aspartic acid residues, which separates the caspase into
small and large subunits [167] and then they become
activated by autoproteolysis.
Through genetic analysis of cell-death defective (CED)
mutants, it was found that the product of the aspartate-
directed cysteine protease (CED-3) is required for all
developmental-related programmed cell deaths in the
worm [168]. In this multicellular model, various genes that
encode for proteins essential for the regulation and exe-
cution of apoptosis were identified and their mammalian
homologs were described [33, 76]. For example, ced-9 and
Bcl-2 encode proteins that inhibit apoptosis, Apaf-1 and
ced-4 encode an adaptor protein that permits the interaction
between initiator proteins ced-3 and caspase-9. Ced-3
encodes a protein homologous to caspase-9 which is
responsible for the apoptotic initiation process. Fifteen
mammalian caspases have been identified, with caspase-11
and 12 identified only in mouse as shown in Table 1.
All caspases are synthesized as zymogens sharing a com-
mon domain structure consisting of a large (p10) and a small
(p20) catalytic subunit. Among two fundamentally different
groups, the first group, termed as initiator caspases, is char-
acterized by a long prodomain which provides a protein–
protein interaction platform [167, 169]. Prodomains allow the
recruitment of procaspases into an activating protein com-
plex. Long prodomain caspases are caspase-1, -2, -4, -5, -9,
-11, and -12 with an N-terminal caspase-activating recruit-
ment domain (CARD), and caspase-8 and -10 with an
N-terminal death effector domain (DED) (Table 1). In con-
trast to the initiator caspases, the second group consisting of
executioner caspases-3, -6, and -7 lack the large N-terminal
non-enzymatic domain and they are responsible for the
majority of cellular destruction during apoptosis [170]. When
procaspase-8 or -10 recruited to ligate death receptors by Fas-
Associated Death Domain (FADD), they undergo autocatal-
ysis, releasing the p10 and p20 subunits that form the active
(tetrameric) enzyme. Caspase-9 is activated in the presence of
ATP and cyt c by an allosteric change on a heptameric scaf-
fold of apoptotic protease-activating factor 1 (Apaf1) proteins
termed as apoptosome. Caspase-9 processing occurs
48 Mol Cell Biochem (2011) 351:41–58
123
secondary to caspase-3 activation during Smac-induced
apoptosis. In a heat shock-induced death, Caspase-2 induces
apoptosis via cleavage of Bid [171].
Interdependence of caspases and Bcl-2 in the regulation
of apoptosis
Numerous studies suggest that many apoptotic signaling
pathways converge at the mitochondria, where signals are
processed through a series of molecular events culminating
in the release of potent death factors that trigger either
through the extrinsic or the intrinsic pathway. Basically,
the release of the pro-apoptotic proteins from the inter-
membrane space triggers apoptosis, in a caspase-dependent
(through cyt c, Omi/HtrA2, and SMAC) as well as in a
caspase-independent form (through AIF and Endo G).
In mammalian cells, caspases-9, -8, and -2 rely on the
formation of apoptosome (Apaf-1), death-inducing signal-
ing complex (DISC), and PIDDosome, respectively, for
activation of apoptotic signals. Apoptosome is composed of
seven molecules (heptamer) of Apaf-1 bound to cyt c in the
presence of ATP/dATP, Fig. 2. DISC is assembled follow-
ing binding of death ligand to its receptor and contains
FADD and caspase-8 (or –10) whereas PIDDosome contains
at least three components, PIDD, RAIDD, and caspase-2.
Apoptosome complex
Apoptosome is a multimeric protein complex that mediates
activation of an initiator caspase at the onset of apoptosis.
Biochemical and structural investigations revealed
insights into the assembly and function of the various
apoptosomes from fruit fly (Drosophilia melanogaster),
worm (C. elgans) and mammals [172, 173]. The assembly
of the mammalian apoptosome which is responsible for the
activation of caspase-9 requires the binding of Apaf-1, cyt
c, and ATP/dATP. The apoptosome is an oligomeric sig-
naling platform that has a core of seven apoptotic Apaf1.
Each Apaf-1 monomer contains an N-terminal caspase
recruitment domain (CARD), a nucleotide-binding and
oligomerization domain (NOD), and a string of WD40
(tryptophan-aspartic acid) repeats at the C terminus
(Fig. 2). The WD40 repeats are thought to be the site of cyt
c binding [102, 174].
Death-inducing signaling complex (DISC)
DISC consist of complex of the death receptor (FAS), the
adaptor FAS-associated death domain protein (FADD), the
initiator caspase procaspase-8 (or procaspase-10), and
possibly other co-factors and regulators [175]. Kischkel
and coworkers reported the formation of a protein complex
in the dying cell and named it as DISC. Upon receiving the
death signal the activated death ligands homo-trimerizes,
which in turn, induces oligomerization of the Fas death
receptors (Fig. 2) [176, 177]. Dimerization of caspase-8 is
a crucial factor for activation and suggests that DISC may
facilitate the activation of caspase-8 through dimerization
[178]. The DISC creates a platform that brings together
molecules of the initiator caspase-8 (or caspase-10) and
other co-factors and regulators for execution of extrinsic
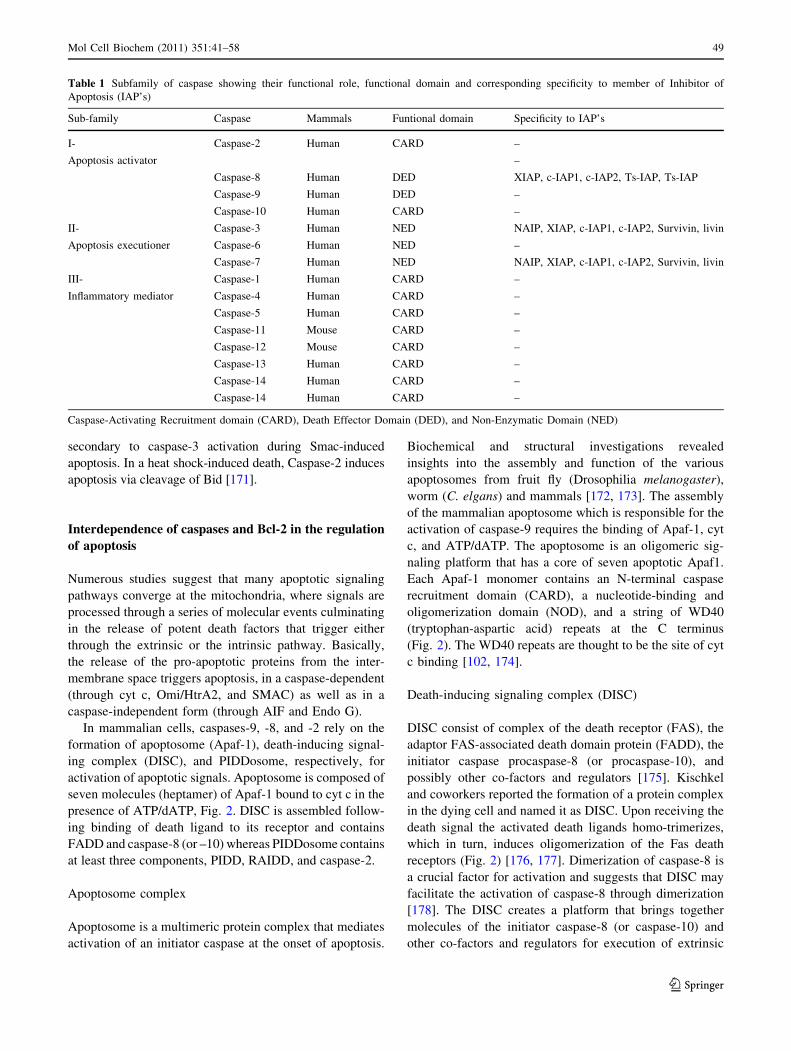
Table 1 Subfamily of caspase showing their functional role, functional domain and corresponding specificity to member of Inhibitor of
Apoptosis (IAP’s)
Sub-family Caspase Mammals Funtional domain Specificity to IAP’s
I- Caspase-2 Human CARD –
Apoptosis activator –
Caspase-8 Human DED XIAP, c-IAP1, c-IAP2, Ts-IAP, Ts-IAP
Caspase-9 Human DED –
Caspase-10 Human CARD –
II- Caspase-3 Human NED NAIP, XIAP, c-IAP1, c-IAP2, Survivin, livin
Apoptosis executioner Caspase-6 Human NED –
Caspase-7 Human NED NAIP, XIAP, c-IAP1, c-IAP2, Survivin, livin
III- Caspase-1 Human CARD –
Inflammatory mediator Caspase-4 Human CARD –
Caspase-5 Human CARD –
Caspase-11 Mouse CARD –
Caspase-12 Mouse CARD –
Caspase-13 Human CARD –
Caspase-14 Human CARD –
Caspase-14 Human CARD –
Caspase-Activating Recruitment domain (CARD), Death Effector Domain (DED), and Non-Enzymatic Domain (NED)
Mol Cell Biochem (2011) 351:41–58 49
123

apoptotic pathway which finally leads to their auto-acti-
vation [179, 180].
p53-Inducible death domain containing protein
complex (PIDDosome)
The PIDDosome complex under physiological conditions
contains PIDD and associate with the activation of another
initiator caspase, caspase-2 [181]. Although, the role of the
PIDDosome in apoptosis remains controversial, its
expression is inducible upon DNA damage [173, 181, 182].
However, PIDD-deficient mice undergo apoptosis not only
in response to DNA damage, but also in response to various
p53-independent stress signals and to death receptor
engagement. In the absence of PIDD, both caspase-2 pro-
cessing and activation occur in response to DNA damage
indicating that PIDD does not play an essential role for all
p53-mediated or p53-independent apoptotic pathways
[182]. The role of caspase-2 in the mitochondrial pathway
is now widely accepted [183–185]. Thus, the initial stage
of DNA damage facilitated by p53-mediated apoptosis
occurs by a PIDD and caspase 2-dependent mechanism.
For events that are downstream of cyt c release, p53’s full
transcriptional regulatory functions are required [186].
Apoptosis and the caspase-independent pathway
There also exists a caspase-independent apoptotic pathway
that is associated to AIF [131, 134], endonuclease G [187],
as well as Omi/HtrA2 [150–152]. As already discussed
above in this review, cells after receiving the apoptotic
signals release the nuclear AIF molecule and endonuclease
G protein which are translocated to the nucleus causing a
large-scale chromatin condensation and DNA fragmenta-
tion independently of caspase activation. Similarly Omi/
HtrA2 mediates caspase-independent cell death through its
own protease activity as can be observed during apoptosis.
A specific Omi/HtrA2 inhibitor can stop degradation of
THAP5 protein (THAP family of proteins), which leads to
reduced cell death [150]. A few studies have focused the
role of Heat Shock Proteins (HSPs) that are either consti-
tutively expressed or expressed under variety of stresses
stimuli [188], have shown their role in apoptosis via cas-
pase-independent pathway [189]. Members of HSP protein
such as HSP27 and HSP70 participate in oncogenesis,
probably by interfering apoptotic pathways. First, they act
as chaperones and play a role in proteasome-mediated
degradation of apoptosis-regulatory proteins. Second, they
inhibit key effectors of the apoptotic machinery including
the apoptosome and apoptosis-inducing factor [190].
However recent work on caspase-independent apoptotic
pathway has lead to the discovery of various other
molecules which has a major role in the pathway. For
example, the work by Yuan and his group have shown that
Ste20-like protein kinase 3 respond to apoptosis of HeLa
cells to trigger the caspase-independent apoptotic pathway
[191]. Similarly activated analog of CY, 4-hydroperoxy-
cyclophosphamide (4-OOH-CY) is being used for the
therapy for hematological malignancies and autoimmune
disorders through caspase-independent T-cell apoptosis
[192].
Therapeutic implication in the control of apoptosis
Lack of the phenomena of apoptosis results in excessive
increase of cell number and has implications in autoim-
munity and tumorigenesis. On the other hand, excessive
apoptosis decreases the cell population, which is linked to
many neurodegenerative disorders such as Parkinson’s
disease, Alzheimer’s disease, Huntington’s disease, and
spinal muscular atrophy. Alzheimer’s disease is a complex
neurological disorder in which the beta-amyloid peptides
are formed in the brain. In this, the Bcl-2 is down-regulated
and Bax is up-regulated [193, 194]. With respect to the role
of Bcl-2 proteins and its role in Parkinson’s disease, it has
been anticipated that pro-apoptotic family members par-
ticipate in neuronal death in a variety of Parkinson’s dis-
ease models [195]. Numerous studies show that activation
of apoptosis has also been found to be involved in the
pathogenesis of other human diseases such as chronic heart
failure [196], diabetes [197, 198], and atherosclerosis
[199]. The increased expression of Bcl-2 in the vascular
endothelium inhibits the diabetes-induced degeneration of
retinal capillaries and superoxide generation [198, 200].
An imbalance among the Bcl-2 family of proteins, in
favor of the anti-apoptotic members, is a phenomenon that
naturally, and frequently occurs in cancer cells [201–203].
Over-expression of anti-apoptotic Bcl-2 or Bcl-xl probably
occurs in more than half of all cancers. Moreover, loss of
expression of Bax is also found in some colorectal cancers
and in hematopoietic malignancies, whereas the expression
of a highly apoptogenic variant of Bax (Baxw) is correlated
with an increased survival of patients with glioblastoma
multiforme, an aggressive form of brain tumors [204]. The
defects may arise from the fact that neoplastic cells are
under strong selective pressure to stabilize their mito-
chondrial permeability, even if they harbor alterations in
the p53 tumor suppressor pathway. c-Myc, for instance,
can induce mitochondrial damage independently from the
transcriptional activity of p53. Since p53 also activates the
mitochondrial death pathway, the mitochondrion appears to
integrate the diverse pro-apoptotic mechanisms induced by
oncogenes. Studies in transgenic mice have revealed that
Bcl-2 (and/or Bcl-xl) over-expression and p53 mutations
50 Mol Cell Biochem (2011) 351:41–58
123

(or ARF loss) are selected independently during Myc-
induced lymphomagenesis [205–207].
The emerging knowledge about proteins that are
involved in apoptosis, including their 3-D structures and
biochemical mechanisms, has provided therapeutic ave-
nues by discovering molecules or targets which may
modulate apoptosis. A number of therapeutic approaches
are undergoing using an antisense RNA [208], various
small molecules [209–218, 222, 223], and peptidic com-
pounds [219–221, 224, 225] classified as potential thera-
peutics to target the pathway of apoptosis, as briefly
summarized in Table 2. Most of these therapeutics involve
the targeting of structurally defined multidomain of the
member of Bcl-2 family of protein. Some of these mole-
cules/agents are in clinical trials.
The combination of IAP antagonists with drugs that
target ErbB receptors promotes apoptosis thereby reduces
the cell turnover of breast cancer cells [226]. The apoptotic
response to most chemotherapeutic drugs in mammalian
cells involves the induction of mitochondrial pathway in
which mitochondrial membrane permeabilization con-
trolled by the Bcl-2 protein family, is induced. Other
strategies include the IAP proteins as therapeutic targets
that are expressed in the majority of human tumor through
the inhibition of cellular death and participation in sig-
naling pathways associated with malignancies [157, 227].
Further more, Table 2 has summarized the list of antisense
[228], small molecules [229–235], and peptidic compounds
[234, 236, 237] that are under investigation to develop
potential therapeutics against IAPs protein which are
involved in cancer.
Acknowledgments MSO and MN thank Medical Research Chair in
Ophthalmology funded by Dr. Nasser Al-Rasheed, College of Med-
icine, Kind Saud University for support. HA would like to thank Dr.
Nihal Ahmad, School of Medicine and Public Health, University of
Wiscosnsin, Madison for a cancer cell biology research fellowship.
The authors would also like to thank Ms. Crisalis Longanilla-Bautista
and Mr. Miaraj Siddiquei in helping with figures and proof reading.
References
1. Kerr JFR, Wyllie AH, Currie AR (1972) Apoptosis: a basic
biological phenomenon with wide-ranging implications in tissue
kinetics. Br J Cancer 26:239–257
2. Adams JM (2003) Ways of dying: multiple pathways to apop-
tosis. Genes Dev 17:2481–2495
3. Saikumar P, Dong Z, Mikhailov V, Denton M, Weinberg JM,
Venkatachalam MA (1999) Apoptosis: definition, mechanisms,
and relevance to disease. Am J Med 107:489–506
4. Gulbins E, Jekle A, Ferlinz K, Grassme H, Lang F (2000)
Physiology of apoptosis. Am J Physiol Renal Physiol 279:
605–615
5. Janssen O, Qian J, Linkermann A, Kabelitz D (2003) CD95
ligand—death factor and costimulatory molecule? Cell Death
Differ 10:1215–1225
6. Schutze S, Tchikov V, Schneider-Brachert W (2008) Regulation
of TNFR1 and CD95 signalling by receptor compartmentaliza-
tion. Nat Rev Mol Cell Biol 9:655–662
7. Ashkenazi A (2002) Targeting death and decoy receptors of the
tumour-necrosis factor superfamily. Nat Rev Cancer 2:420–430
8. Letai A (2006) Growth factor withdrawal and apoptosis: the
middle game. Mol Cell 21:728–730
Table 2 Compounds on high clinical development against the Bcl-2 and IAP’s family of proteins involved in the mitochondria-mediated
apoptosis pathway
Compound Class Mechanism (target) Reference
Targets for antiapoptotic Bcl-2 family of protein
Genasense (Bcl-2 antisense) Antisense Anti-apoptotic mRNA (Bcl-2) [208]
BH3I-1/BH3I-2; antimycin A3; compound 6;
apogossypol; theaflavanin; GX15-070; AT101: (-)
gossypol; polyphenol E; A-779024 (ABT 737);
HA14-1 analogs
Small molecules Anti-apoptotic inhibition (Bcl-2) [209–218]
CPM-1285 analogs; terphenyl derivative; SAHBs Peptide Anti-apoptotic inhibition (Bcl-2) [219–221]
Targets for pro-apoptotic Bcl-2 family of protein
3,6-dibromocarbazole piperazine derivatives of
2-propanol; 4-phenylsulfanyl-phenylamine derivatives
Small molecules Pro-apoptotic inhibition (BAX)/(BID) [222, 223]
Humanin peptides; Ku70 peptides Peptides Pro-apoptotic inhibition (BAX) [224, 225]
Targets for IAP’s family of protein
XIAP antisense (AEG35156/GEM640) Antisense Inhibition of XIAP [228]
Benzenesulfonamide derivatives; di/triphenylureas
(1396-11,12,34); embeline; compound 3/8/11; SM-
164; LBW242
Small molecules Inhibition of IAP [229–235]
Capped tripeptides containing unnatural amino acids;
Smac/penetratin fusion peptides
Peptide Inhibition of IAP [234, 236, 237]
Mol Cell Biochem (2011) 351:41–58 51
123

9. Zhang Y, Xing D, Liu L (2009) PUMA promotes Bax translo-
cation by both directly interacting with Bax and by competitive
binding to Bcl-X L during UV-induced apoptosis. Mol Biol Cell
20:3077–3087
10. Gentile M, Latonen L, Laiho M (2003) Cell cycle arrest and
apoptosis provoked by UV radiation-induced DNA damage are
transcriptionally highly divergent responses. Nucleic Acids Res
31:4779–4790
11. Stevenson MA, Pollock SS, Coleman CN, Calderwood SK
(1994) X-irradiation, phorbol esters, and H2O2 stimulate mito-
gen-activated protein kinase activity in NIH-3T3 cells through
the formation of reactive oxygen intermediates. Cancer Res
54:12–15
12. Dudeja V, Mujumdar N, Phillips P, Chugh R, Borja-Cacho D,
Dawra RK, Vickers SM, Saluja AK (2009) Heat shock pro-
tein 70 inhibits apoptosis in cancer cells through simultaneous
and independent mechanisms. Gastroenterology 136:1772–
1782
13. Solary E, Droin N, Bettaieb A, Corcos L, Dimanche-Boitrel MT,
Garrido C (2000) Positive and negative regulation of apoptotic
pathways by cytotoxic agents in hematologica. Leukemia 14:
1833–1849
14. Ahsan H, Reagan-Shaw S, Breur J, Ahmad N (2007) Sanguin-
arine induces apoptosis of human pancreatic carcinoma AsPC-1
and BxPC-3 cells via modulations in Bcl-2 family proteins.
Cancer Lett 249:198–208
15. Meng SJ, Yu LJ (2010) Oxidative stress, molecular inflamma-
tion and sarcopenia. Int J Mol Sci 11:1509–1526
16. Gu X, Song X, Dong Y, Cai H, Walters E, Zhang R, Pang X, Xie
T, Guo Y, Sridhar R, Califano JA (2008) Vitamin E succinate
induces ceramide-mediated apoptosis in head and neck squa-
mous cell carcinoma in vitro and in vivo. Clin Cancer Res
14:1840–1848
17. Lancellotti M, Pereira RF, Cury GG, Hollanda LM (2009)
Pathogenic and opportunistic respiratory bacteria-induced
apoptosis. Braz J Infect Dis 13:226–231
18. Sorensen CM (2004) Bcl-2 family members and disease. Bio-
chim Biophys Acta 1644:179–188
19. Thompson CB (1995) Apoptosis in the pathogenesis and treat-
ment of disease. Science 267:1456–1462
20. Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell
100:57–70
21. Bidere N, Su HC, Lenardo MJ (2006) Genetic disorders of
programmed cell death in the immune system. Annu Rev
Immunol 24:321–352
22. Mersich S, Gadaleta P (2003) Nuevas estrategias terapeuticas
basadas en apoptosis y virus. Acta Bioquımica Clınica Latino-
americana 37:13–21
23. Fisher DE (1994) Apoptosis in cancer therapy: crossing the
threshold. Cell 78:539–542
24. Johnstone RW, Ruefli AA, Lowe SW (2002) Apoptosis:
a link between cancer genetics and chemotherapy. Cell 108:
153–164
25. Pecina-Slaus N (2009) Genetic and molecular insights into
apoptosis. Acta Med Croatica 63(Suppl 2):13–19
26. Schaffitzel E, Hertweck M (2006) Recent aging research in
Caenorhabditis elegans. Exp Gerontol 41:557–563
27. Danial NN, Korsmeyer SJ (2004) Cell death: critical control
points. Cell 116(2):205–219
28. Horvitz HR (1999) Genetic control of programmed cell death in
the nematode Caenorhabditis elegans. Cancer Res 59:1701S–
1706S
29. McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U,
McKearn JP, Korsmeyer SJ (1989) Bcl-2-immunoglobulin
transgenic mice demonstrate extended B cell survival and fol-
licular lymphoproliferation. Cell 57:79–88
30. Vaux DL, Cory S, Adams JM (1988) Bcl-2 gene promotes
haemopoietic cell survival and cooperates with c-myc to
immortalize pre-B cells. Nature 335:440–442
31. Vaux DL, Weissman IL, Kim SK (1992) Prevention of pro-
grammed cell death in Caenorhabditis elegans by human bcl-2.
Science 258:1955–1957
32. Hengartner MO, Horvitz HR (1994) C. elegans cell survival
gene ced-9 encodes a functional homolog of the mammalian
proto-oncogene bcl-2. Cell 76:665–676
33. Mohamad N, Gutierrez A, Nunez M, Cocca C, Martın G, Cricco
G, Medina V, Rivera E, Bergoc R (2005) Mitochondrial apop-
totic pathways. Biocell 29:149–161
34. Fan TJ, Han LH, Cong RS, Liang J (2005) Caspase family
proteases and apoptosis. Acta Biochimica et Biophysica Sinica
37:719–727
35. Li J, Yuan J (2008) Caspases in apoptosis and beyond. Onco-
gene 27(48):6194–6206
36. Petros AM, Olejniczak ET, Fesik SW (2004) Structural biology
of the Bcl-2 family of proteins. Biochim Biophys Acta
1644:83–94
37. Germain M, Shore GC (2003) Cellular distribution of Bcl-2
family proteins. Sci STKE 173:pe10
38. Budd R (2001) Activation-induced cell death. Curr Opin
Immunol 13:356–362
39. Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ
(1996) BID: a novel BH3 domain-only death agonist. Genes
Dev 10:2859–2869
40. Brunelle JK, Letai A (2009) Control of mitochondrial apoptosis
by the Bcl-2 family. J Cell Sci 122:437–441
41. Puthalakath H, Strasser A (2002) Keeping fillers on a tight leash:
transcriptional and post-translational control of the proapoptotic
activity of BH3-only proteins. Cell Death Differ 9:505–512
42. Ogilvy S, Metcalf D, Print CG, Bath ML, Harris AW, Adams
JM (1999) Constitutive Bcl-2 expression throughout the hema-
topoietic compartment affects multiple lineages and enhances
progenitor cell survival. Proc Natl Acad Sci USA 96:
14943–14948
43. Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ (1993) Bcl-2-
deficient mice demonstrate fulminant lymphoid apoptosis,
polycystic kidneys, and hypopigmented hair. Cell 75:229–240
44. Ross AJ, Waymire KG, Moss JE, Parlow AF, Skinner MK,
Russell LD, MacGregor GR (1998) Testicular degeneration in
Bcl-w-deficient mice. Nat Genet 18:251–256
45. Hamasaki A, Sendo F, Nakayama K, Ishida N, Negishi I,
Nakayama K, Hatakeyama S (1998) Accelerated neutrophil
apoptosis in mice lacking A1-a, a subtype of the Bcl-2-related
A1 gene. J Exp Med 188:1985–1992
46. Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ
(2000) Mcl-1 deficiency results in peri-implantation embryonic
lethality. Genes Dev 14:23–27
47. Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K,
Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S (1995)
Massive cell death of immature hematopoietic cells and neurons
in Bcl-x deficient mice. Science 267:1506–1510
48. Youle RJ, Strasser A (2008) The BCL-2 protein family:
opposing activities that mediate cell death. Nat Rev Mol Cell
Biol 9:47–59
49. Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie ZJ, Dong
Z (2007) Bak regulates mitochondrial morphology and pathol-
ogy during apoptosis by interacting with mitofusins. Proc Natl
Acad Sci USA 104:11649–11654
50. Mikhailov V et al (2003) Association of Bax and Bak homoo-
ligomers in mitochondria. Bax requirement for Bak reorgani-
zation and cytochrome c release. J Biol Chem 278:5367–5376
51. Basanez G, Sharpe JC, Galanis J, Brandt TB, Hardwick JM,
Zimmerberg J (2002) Bax-type apoptotic proteins porate pure
52 Mol Cell Biochem (2011) 351:41–58
123

lipid bilayers through a mechanism sensitive to intrinsic
monolayer curvature. J Biol Chem 277:49360–49365
52. Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M,
Schneiter R, Green DR, Newmeyer DD (2002) Bid, Bax, and
lipids cooperate to form supramolecular openings in the outer
mitochondrial membrane. Cell 111:331–342
53. Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW
(2003) Interaction with a membrane surface triggers a reversible
conformational change in Bax normally associated with induc-
tion of apoptosis. J Biol Chem 278:48935–48941
54. Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou
V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Kors-
meyer SJ (2001) Proapoptotic BAX and BAK: a requisite
gateway to mitochondrial dysfunction and death. Science
292:727–730
55. Wang X (2001) The expanding role of mitochondria in apop-
tosis. Genes Dev 15:922–2933
56. Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber
B, Andrews DW (2008) Membrane binding by tBid initiates an
ordered series of events culminating in membrane permeabili-
zation by Bax. Cell 135(6):1074–1084
57. Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S,
Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or
activate mitochondrial apoptosis, serving as prototype cancer
therapeutics. Cancer Cell 2:183–192
58. Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, New-
meyer DD, Schuler M, Green DR (2004) Direct activation of
Bax by p53 mediates mitochondrial membrane permeabilization
and apoptosis. Science 303:1010–1014
59. Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP,
Hsieh JJ, Cheng EH (2006) Hierarchical regulation of mito-
chondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell
Biol 8:1348–1358
60. Pagliari LJ, Kuwana T, Bonzon C, Newmeyer DD, Tu S, Beere
HM, Green DR (2005) The multidomain proapoptotic molecules
Bax and Bak are directly activated by heat. Proc Natl Acad Sci
USA 102:17975–17980
61. Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz
SG, Tu HC, Kim H, Cheng EH, Tjandra N, Walensky LD (2008)
BAX activation is initiated at a novel interaction site. Nature
455:1076–1081
62. Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L,
Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser
A, Kluck RM, Adams JM, Huang DC (2007) Apoptosis initiated
when BH3 ligands engage multiple Bcl-2 homologs, not Bax or
Bak. Science 315:856–859
63. Fletcher JI, Meusburger S, Hawkins CJ, Riglar DT, Lee EF,
Fairlie WD, Huang DC, Adams JM (2008) Apoptosis is trig-
gered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc
Natl Acad Sci USA 105:18081–18087
64. Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ
(2003) VDAC2 inhibits BAK activation and mitochondrial
apoptosis. Science 301:513–517
65. Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase
8 mediates the mitochondrial damage in the Fas pathway of
apoptosis. Cell 94:491–501
66. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a
Bcl2 interacting protein, mediates cytochrome c release from
mitochondria in response to activation of cell surface death
receptors. Cell 94:481–490
67. Billen LP, Shamas-Din A, Andrews DW (2008) Bid: a Bax-like
BH3 protein. Oncogene 27(Suppl 1):S93–S104
68. Oh KJ, Barbuto S, Pitter K, Morash J, Walensky LD, Korsmeyer
SJ (2006) A membrane-targeted BID BCL-2 homology 3 pep-
tide is sufficient for high potency activation of BAX in vitro.
J Biol Chem 281:36999–37008
69. Eskes R, Desagher S, Antonsson B, Martinou JC (2000) Bid
induces the oligomerization and insertion of Bax into the outer
mitochondrial membrane. Mol Cell Biol 20:929–935
70. Balakrishnan G, Hu Y, Oyerinde OF, Su J, Groves JT, Spiro TG
(2007) A conformational switch to b-sheet structure in cyto-
chrome c leads to heme exposure. Implications for cardiolipin
peroxidation and apoptosis. J Am Chem Soc 129:504–505
71. Kim TH et al (2004) Bid–cardiolipin interaction at mitochon-
drial contact site contributes to mitochondrial cristae reorgani-
zation and cytochrome c release. Mol Biol Cell 15:3061–3072
72. Giordano A et al (2005) tBid induces alterations of mitochon-
drial fatty acid oxidation flux by malonyl-CoA-independent
inhibition of carnitine palmitoyltransferase-1. Cell Death Differ
12:603–613
73. Tyurin VA et al (2007) Interactions of cardiolipin andlyso-car-
diolipins with cytochrome c and tBid: conflict or assistance in
apoptosis. Cell Death Differ 14:872–875
74. Orrenius S, Gogvadze V, Zhivotovsky B (2007) Mitochondrial
oxidative stress: implications for cell death. Annu Rev Phar-
macol Toxicol 47:143–183
75. Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ (1996) Serine
phosphorylation of death agonist BAD in response to survival
factor results in binding to 14-3-3 not BCL-X(L). Cell 87:
619–628
76. Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D,
Darnell JE, Freeman WH (2000) Molecular cell biology, 4th
edn. W. H. Freeman & Co, New York, Chapter 23–28
77. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y,
Greenberg ME (1997) Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell 91:
231–241
78. Tommasini I, Cerioni L, Palomba L, Cantoni O (2008) Prosta-
glandin E2 signals monocyte/macrophage survival to perox-
ynitrite via protein kinase A converging in bad phosphorylation
with the protein kinase C alpha-dependent pathway driven by
5-hydroxyeicosatetraenoic acid. J Immunol 181:5637–5645
79. Grund K, Ahmadi R, Jung F, Funke V, Gdynia G, Benner A,
Sykora J, Walczak H, Joos S, Felsberg J, Reifenberger G,
Wiestler OD, Herold-Mende C, Roth W (2008) Troglitazone-
mediated sensitization to TRAIL-induced apoptosis is regulated
by proteasome-dependent degradation of FLIP and ERK1/2-
dependent phosphorylation of BAD. Cancer Biol Ther
7:1982–1990
80. Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ (2009) {Beta}-
arrestin-2 mediates anti-apoptotic signaling through regulation
of BAD phosphorylation. J Biol Chem 284:8855–8865
81. Puthalakath H, Villunger A, O’Reilly LA, Beaumont JG,
Coultas L, Cheney RE, Huang DC, Strasser A (2001) Bmf: a
pro-apoptotic BH3-only protein regulated by interaction with the
myosin V actin motor complex, activated by anoikis. Science
293:1829–1832
82. Lei K, Davis RJ (2003) JNK phosphorylation of Bim-related
members of the Bcl2 family induces Bax-dependent apoptosis.
Proc Natl Acad Sci USA 100:2432–2437
83. Okuno S, Saito A, Hayashi T, Chan PH (2004) The c-Jun
N-terminal protein kinase signaling pathway mediates Bax
activation and subsequent neuronal apoptosis through interac-
tion with Bim after transient focal cerebral ischemia. J Neurosci
24:7879–7887
84. Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW,
Kontgen F, Adams JM, Strasser A (1999) Proapoptotic Bcl-2
relative Bim required for certain apoptotic responses, leukocyte
homeostasis, and to preclude autoimmunity. Science 286:
1735–1738
85. Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L,
Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A
Mol Cell Biochem (2011) 351:41–58 53
123

(2002) BH3-only Bcl-2 family member Bim is required for
apoptosis of autoreactive thymocytes. Nature 415:922–926
86. Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA,
Strasser A, Johnson EM (2001) Induction of Bim, a proapoptotic
BH3- only Bcl-2 family member, is critical for neuronal apop-
tosis. Neuron 29:615–628
87. Zhang L, Xing D, Chen M (2008) Bim(L) displacing Bcl-
x(L) promotes Bax translocation during TNFalpha-induced
apoptosis. Apoptosis 13:950–958
88. Wang X, Xing D, Liu L, Chen WR (2009) BimL directly neu-
tralizes Bcl-xL to promote Bax activation during UV-induced
apoptosis. FEBS Lett 583:1873–1879
89. Zhang Y, Adachi M, Kawamura R, Zou HC, Imai K, Hareyama
M, Shinomura Y (2006) Bmf contributes to histone deacetylase
inhibitor-mediated enhancing effects on apoptosis after ionizing
radiation. Apoptosis 11:1349–1357
90. Zhang Y, Adachi M, Kawamura R, Imai K (2006) Bmf is a
possible mediator in histone deacetylase inhibitors FK228 and
CBHA-induced apoptosis. Cell Death Differ 13:129–140
91. Ramjaun AR, Tomlinson S, Eddaoudi A, Downward J (2007)
Upregulation of two BH3-only proteins, Bmf and Bim, during
TGF beta-induced apoptosis. Oncogene 26:970–981
92. Yakovlev AG, Giovanni SD, Wang G et al (2004) Bok and Noxa
are essential mediators of p53-dependent apoptosis. J Biol Chem
279:28367–28374
93. Oda E, Ohki R, Murasawa H et al (2000) Noxa, a BH3- only
member of the Bcl-2 family and candidate mediator of p53-
induced apoptosis. Science 288:1053–1058
94. Villunger A, Michalak EM, Coultas L et al (2003) p53- and
drug-induced apoptotic responses mediated by BH3-only pro-
teins Puma and Noxa. Science 302:1036–1038
95. Karst AM, Li G (2007) BH3-only proteins in tumorigenesis and
malignant melanoma. Cell Mol Life Sci 64:318–330
96. Ming L, Wang P, Bank A et al (2006) Puma dissociates Bax and
Bcl-XL to induce apoptosis in colon cancer cells. J Biol Chem
281:16034–16042
97. Wyttenbach A, Tolkovsky AM (2006) The BH3-only protein
Puma is both necessary and sufficient for neuronal apoptosis
induced by DNA damage in sympathetic neurons. J Neurochem
96:1213–1226
98. Jabbour AM, Heraud JE, Daunt CP, Kaufmann T, Sandow J,
O’Reilly LA, Callus BA, Lopez A, Strasser A, Vaux DL, Ekert
PG (2009) Puma indirectly activates Bax to cause apoptosis in
the absence of Bid or Bim. Cell Death Differ 16:555–563
99. Liu Z, Lu H, Shi H et al (2005) Puma overexpression induces
reactive oxygen species generation and proteasome- mediated
stathmin degradation in colorectal cancer cells. Cancer Res
65:1647–1654
100. Hemann MT, Zilfou JT, Zhao Z et al (2004) Suppression of
tumorigenesis by the p53 target Puma. Proc Natl Acad Sci USA
101:9333–9338
101. Adams JM, Cory S (2007) The Bcl-2 apoptotic switch in cancer
development and therapy. Oncogene 26:1324–1337
102. Riedl SJ, Salvesen GS (2007) The apoptosome: signalling
platform of cell death. Nat Rev Mol Cell Biol 8:405–413
103. Strasser A, O’Connor L, Dixit VM (2000) Apoptosis signaling.
Annu Rev Biochem 69:217–245
104. Strasser A, Harris AW, Huang DCS, Krammer PH, Cory S
(1995) Bcl-2 and Fas/APO-1 regulate distinct pathways to
lymphocyte apoptosis. EMBO J 14:6136–6147
105. Li P et al (1997) Cytochrome c and dATP-dependent formation
of Apaf-1/caspase-9 complex initiates an apoptotic protease
cascade. Cell 91:479–489
106. Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W,
Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, Los
M (2009) Apoptosis and cancer: mutations within caspase
genes. J Med Genet 46:497–510
107. Locksley RM, Killeen N, Lenardo MJ (2001) The TNF and TNF
receptor superfamilies: integrating mammalian biology. Cell
104:487–501
108. Wajant H (2002) The Fas signaling pathway: more than a par-
adigm. Science 296:1635–1636
109. Chen G, Goeddel D (2002) TNF-1 signaling: a beautiful path-
way. Science 296:1634–1635
110. Yang JK, Wang L, Zheng L, Wan F, Ahmed M, Lenardo MJ,
Wu H (2005) Crystal structure of MC159 reveals molecular
mechanism of DISC assembly and FLIP inhibition. Mol Cell
20:939–949
111. Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger
PH (2000) Pro-apoptotic cascade activates BID, which oligo-
merizes BAK or BAX into pores that result in the release of
cytochrome c. Cell Death Differ 7:1166–1173
112. Lartigue L, Kushnareva Y, Seong Y, Lin H, Faustin B,
Newmeyer DD (2009) Caspase-independent mitochondrial cell
death results from loss of respiration, not cytotoxic protein
release. Mol Biol Cell 20:4871–4884
113. Pellegrini L, Scorrano L (2007) A cut short to death: Parl and
Opa1 in the regulation of mitochondrial morphology and
apoptosis. Cell Death Differ 14:1275–1284
114. Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996)
Induction of apoptotic program in cell-free extracts: requirement
for dATP and cytochrome c. Cell 86:147–157
115. Li F et al (1997) Cell-specific induction of apoptosis by
microinjection of cytochrome c. Bcl-xL has activity independent
of cytochrome c release. J Biol Chem 272:30299–30305
116. Zhivotovsky B, Orrenius S, Brustugun OT, Doskeland SO
(1998) Injected cytochrome c induces apoptosis. Nature
391:449–450
117. Newmeyer DD, Farschon DM, Reed JC (1994) Cell-free apop-
tosis in Xenopus egg extracts: inhibition by Bcl-2 and require-
ment for an organelle fraction enriched in mitochondria. Cell
79:353–364
118. Kluck RM, Bossy-Wetzel E, Green DR, New meyer DD (1997)
The release of cytochrome c from mitochondria: a primary site
for Bcl-2 regulation of apoptosis. Science 275:1132–1136
119. Glazunova VA, Shtil AA (2008) Mitochondrial mechanisms of
apoptosis in response to DNA damage. Mol Biol (Mosk)
42:765–771
120. Ravi D, Das KC (2004) Redox-cycling of anthracyclines by
thioredoxin system: increased superoxide generation and DNA
damage. Cancer Chemother Pharmacol 54:449–458
121. Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI,
Jones DP, Wang X (1997) Prevention of apoptosis by Bcl-2:
release of cytochrome c from mitochondria blocked. Science
275:1129–1132
122. Bajt ML, Cover C, Lemasters JJ, Jaeschke H (2006) Nuclear
translocation of endonuclease G and apoptosis-inducing factor
during acetaminophen-induced liver cell injury. Toxicol Sci
94:217–225
123. Li LY, Luo X, Wang X (2001) Endonuclease G (EndoG) is an
apoptotic DNAse when released from mitochondria. Nature
412:95–99
124. Zhang J, Liu X, Scherer DC, van Kaer L, Wang X, Xu M (1998)
Resistance to DNA fragmentation and chromatin condensation
in mice lacking the DNA fragmentation factor. Proc Natl Acad
Sci 95:12480–12485
125. Ohsato T, Ishihara N, Muta T, Umeda S, Ikeda S, Mihara K,
Hamasaki N, Kang D (2002) Mammalian mitochondrial endo-
nuclease G digestion of R-loops and localization in intermem-
brane space. Eur J Biochem 269:5765–5770
54 Mol Cell Biochem (2011) 351:41–58
123

126. van Loo G, Schotte P, van Gurp M, Demol H, Hoorelbeke B,
Gevaert K, Rodriguez I, Ruiz-Carrillo A, Vandekerckhove J,
Declercq W, Beyaert R, Vandenabeele P (2001) Endonuclease
G: a mitochondrial protein released in apoptosis and involved in
caspase-independent DNA degradation. Cell Death Differ
8:1136–1142
127. Widlak P, Li LY, Wang X, Garrard WT (2001) Action of
recombinant human apoptotic endonuclease G on naked DNA
and chromatin substrates: cooperation with exonuclease and
DNase. I. J Biol Chem 276:48404–48409
128. Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE,
Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M,
Larochette N, Goodlett DR, Aebersold R, Siderovski DP,
Penninger JM, Kroemer G (1999) Molecular characterization of
mitochondrial apoptosis-inducing factor. Nature 397:441–446
129. Kondo K, Obitsu S, Ohta S, Matsunami K, Otsuka H, Teshima R
(2010) Poly(ADP-ribose) polymerase (PARP)-1-independent
apoptosis-inducing factor (AIF) release and cell death are
induced by eleostearic acid and blocked by a-tocopherol and
MEK inhibition. J Biol Chem 285:13079–13091
130. Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D,
Brothers G, Penninger JM, Peleato ML, Kroemer G, Susin SA
(2001) NADH oxidase activity of mitochondrial apoptosis-
inducing factor. J Biol Chem 276:16391–16398
131. Joza N, Pospisilik JA, Hangen E, Hanada T, Modjtahedi N,
Penninger JM, Kroemer G (2009) AIF: not just an apoptosis-
inducing factor. Ann N Y Acad Sci 1171:2–11
132. Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY,
Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami
N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW,
Zuniga-Pflucker JC, Kroemer G, Penninger JM (2001) Essential
role of the mitochondrial apoptosis-inducing factor in pro-
grammed cell death. Nature 410:549–554
133. Schulthess FT, Katz S, Ardestani A, Kawahira H, Georgia S,
Bosco D, Bhushan A, Maedler K (2009) Deletion of the mito-
chondrial flavoprotein apoptosis inducing factor (AIF) induces
b-cell apoptosis and impairs b-cell mass. PLoS One 4:4394
134. Cande C, Cecconi F, Dessen P, Kroemer G (2002) Apoptosis-
inducing factor (AIF): key to the conserved caspase-independent
pathways of cell death? J Cell Sci 115:4727–4734
135. Yu W, Gubkina O, Mechawar N, Elwell D, Quirion R, Krantic S
(2009) Expression of apoptosis-inducing factor (AIF) in the
aged rat brain. Neurobiol Aging 32(1):179–180
136. Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N,
Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prevost MC,
Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G
(2000) Two distinct pathways leading to nuclear apoptosis.
J Exp Med 192:571–580
137. Pallast S, Arai K, Pekcec A, Yigitkanli K, Yu Z, Wang X, Lo
EH, Leyen KV (2010) Increased nuclear apoptosis-inducing
factor after transient focal ischemia: a 12/15-lipoxygenase-
dependent organelle damage pathway. J Cereb Blood Flow
Metab 30:1157–1167
138. Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM,
Reid GE, Moritz RL, Simpson RJ, Vaux DL (2000) Identifica-
tion of DIABLO, a mammalian protein that promotes apoptosis
by binding to and antagonizing IAP proteins. Cell 102:43–53
139. Kominsky DJ, Bickel RJ, Tyler KL (2002) Reovirus-induced
apoptosis requires mitochondrial release of Smac/DIABLO and
involves reduction of cellular inhibitor of apoptosis protein
levels. J Virol 76:11414–11424
140. Wilkinson JC, Wilkinson AS, Scott FL, Csomos RA, Salvesen
GS, Duckett CS (2004) Neutralization of Smac/Diablo by
inhibitors of apoptosis (IAPs). A caspase-independent mecha-
nism for apoptotic inhibition. J Biol Chem 279:51082–51090
141. Martinez-Ruiz G, Maldonado V, Ceballos-Cancino G, Grajeda
JP, Melendez-Zajgla J (2008) Role of Smac/DIABLO in cancer
progression. J Exp Clin Cancer Res 26:48
142. Arellano-Llamas A, Garcia FJ, Perez D, Cantu D, Espinosa M,
De la Garza JG, Maldonado V, Melendez-Zajgla J (2006) High
Smac/DIABLO expression is associated with early local recur-
rence of cervical cancer. BMC Cancer 6:256
143. Kohli M, Yu J, Seaman C, Bardelli A, Kinzler KW, Vogelstein
B, Lengauer C, Zhang L (2004) SMAC/Diablo-dependent
apoptosis induced by nonsteroidal antiinflammatory drugs
(NSAIDs) in colon cancer cells. Proc Natl Acad Sci USA
101:16897–16902
144. Augello C, Caruso L, Maggioni M, Donadon M, Montorsi M,
Santambrogio R, Torzilli G, Vaira V, Pellegrini C, Roncalli M,
Coggi G, Bosari S (2009) Inhibitors of apoptosis proteins (IAPs)
expression and their prognostic significance in hepatocellular
carcinoma. BMC Cancer 9:125
145. Gray CW, Ward RV, Karran E, Turconi S, Rowles A,
Viglienghi D, Southan C, Barton A, Fantom KG, West A,
Savopoulos J, Hassan NJ, Clinkenbeard H, Hanning C, Ame-
gadzie B, Davis JB, Dingwall C, Livi GP, Creasy CL (2000)
Characterization of human HtrA2, a novel serine protease
involved in the mammalian cellular stress response. Eur J Bio-
chem 267:5699–5710
146. Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K,
Takahashi R (2001) A serine protease, HtrA2, is released from
the mitochondria and interacts with XIAP, inducing cell death.
Mol Cell 8:613–621
147. Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H,
Connolly LM, Day CL, Tikoo A, Burke R, Wrobel C, Moritz
RL, Simpson RJ, Vaux DL (2002) HtrA2 promotes cell death
through its serine protease activity and its ability to antagonize
inhibitor of apoptosis proteins. J Biol Chem 277:445–454
148. Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF,
Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C,
Downward J (2002) The serine protease Omi/HtrA2 regulates
apoptosis by binding XIAP through a repear-like motif. J Biol
Chem 277:439–444
149. Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N,
Teismann P, Abuin A, Grau E, Geppert M, Livi GP, Creasy CL,
Martin A, Hargreaves I, Heales SJ, Okada H, Brandner S, Schulz
JB, Mak T, Downward J (2004) Neuroprotective role of the
reaper-related serine protease HtrA2/Omi revealed by targeted
deletion in mice. Mol Cell Biol 24:9848–9862
150. Balakrishnan MP, Cilenti L, Mashak Z, Popat P, Alnemri ES,
Zervos AS (2009) THAP5 is a human cardiac-specific inhibitor
of cell cycle that is cleaved by the proapoptotic Omi/HtrA2
protease during cell death. Am J Physiol Heart Circ Physiol
297:H643–H653
151. Pruefer FG, Lizarraga F, Maldonado V, Melendez-Zajgla J
(2008) Participation of Omi Htra2 serine-protease activity in the
apoptosis induced by cisplatin on SW480 colon cancer cells.
J Chemother 20:348–354
152. Ding X, Patel M, Shen D, Herzlich AA, Cao X, Villasmil R,
Klupsch K, Tuo J, Downward J, Chan CC (2009) Enhanced
HtrA2/Omi expression in oxidative injury to retinal pigment
epithelial cells and murine models of neurodegeneration. Invest
Ophthalmol Vis Sci 50:4957–4966
153. Trencia A, Fiory F, Maitan MA, Vito P, Barbagallo AP, Perfetti
A, Miele C, Ungaro P, Oriente F, Cilenti L, Zervos AS,
Formisano P, Beguinot F (2004) Omi/HtrA2 promotes cell death
by binding and degrading the anti-apoptotic protein ped/pea-15.
J Biol Chem 279:46566–46572
154. Hu XY, Chen XC, Zhu ZH, Chen CH, Zeng FQ, Lu GC (2006)Effects of Omi/HtrA2 on expression of anti-apoptotic protein
Mol Cell Biochem (2011) 351:41–58 55
123

PED/PEA-15 and apoptosis of prostate cancer cell line PC-3. Ai
Zheng 25:677–682
155. Krick S, Shi S, Ju W, Faul C, Tsai SY, Mundel P, Bottinger EP
(2008) Mpv17l protects against mitochondrial oxidative stress
and apoptosis by activation of Omi/HtrA2 protease. Proc Natl
Aca. Sci USA 105:14106–14111
156. Salvesen GS, Duckett CS (2002) IAP proteins: blocking the road
to death’s door. Nat Rev Mol Cell Biol 3:401–410
157. Vucic D, Fairbrother WJ (2007) The inhibitor of apoptosis
proteins as therapeutic targets in cancer. Clin Cancer Res
13:5995–6000
158. Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J,
Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES
(2001) A conserved XIAP-interaction motif in caspase-9 and
Smac/DIABLO regulates caspase activity and apoptosis. Nature
410:112–116
159. Srinivasula SM, Gupta S, Datta P, Zhang Z, Hegde R, Cheong
N, Fernandes-Alnemri T, Alnemri ES (2003) Inhibitor of
apoptosis proteins are substrates for the mitochondrial serine
protease Omi/HtrA2. J Biol Chem 278:31469–31472
160. Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H
(2001) Structural basis of caspase inhibition by XIAP: differ-
ential roles of the linker versus the BIR domain. Cell 104:81–90
161. Eckelman BP, Salvesen GS (2006) The human antiapoptotic
proteins cIAP1 and cIAP2 bind but do not inhibit caspases.
J Biol Chem 281:3254–3260
162. Zobel K, Wang L, Varfolomeev E, Franklin MC, Elliott LO,
Wallweber HJ, Okawa DC, Flygare JA, Vucic D, Fairbrother
WJ, Deshayes K (2006) Design, synthesis, and biological
activity of apotent Smac mimetic that sensitizes cancer cells to
apoptosis by antagonizing IAPs. ACSChemBiol 1:525–533
163. Kempkensteffen C, Hinz S, Christoph F, Krause H, Magheli A,
Schrader M, Schostak M, Miller K, Weikert S (2008) Expression
levels of the mitochondrial IAP antagonists Smac/DIABLO and
Omi/HtrA2 in clear-cell renal cell carcinomas and their prog-
nostic value. J Cancer Res Clin Oncol 134:543–550
164. Wagener N, Crnkovic-Mertens I, Vetter C, Macher-Goppinger
S, Bedke J, Grone EF, Zentgraf H, Pritsch M, Hoppe-Seyler K,
Buse S, Haferkamp A, Autschbach F, Hohenfellner M, Hoppe-
Seyler F (2007) Expression of inhibitor of apoptosis protein
Livin in renal cell carcinoma and non-tumorous adult kidney. Br
J Cancer 97:1271–1276
165. Fulda S (2008) Targeting inhibitor of apoptosis proteins (IAPs)
for cancer therapy. Anticancer Agents Med Chem 8:533–539
166. Wang J, Lenardo MJ (2000) Roles of caspases in apoptosis,
development, and cytokine maturation revealed by homozygous
gene deficiencies. J Cell Sci 113(Pt 5):753–757
167. Shi Y (2002) Mechanisms of caspase activation and inhibition
during apoptosis. Mol. Cell 9:459–470
168. Los M, van de Craen M, Penning CL, Schenk H, Westendorp M,
Baeuerle PA, Droge W, Krammer PH, Fiers W, Schulze-Osthoff
K (1995) Requirement of an ICE/Ced-3 protease for Fas/Apo-
1–1mediated apoptosis. Nature 375:81–83
169. Thornberry NA, Lazebnik Y (1998) Caspases: enemies within.
Science 281:1312–1316
170. Sprick MR and Walczak H (2004) The interplay between the
Bcl-2 family and death receptor-mediated apoptosis. Biochim
Biophys Acta 1644:125–132
171. Bonzon C, Bouchier-Hayes L, Pagliari LJ, Green DR,
Newmeyer DD (2006) Caspase-2-induced apoptosis requires bid
cleavage: a physiological role for bid in heat shock-induced
death. Mol Biol Cell 17:2150–2157
172. Shi Y (2008) Apoptosome assembly Methods. Enzymol
442:141–156
173. Bao Q, Shi Y (2007) Apoptosome: a platform for the activation
of initiator caspases. Cell Death Differ 14:56–65
174. Ow YP, Green DR, Hao Z, Mak TW (2008) Cytochrome c:
functions beyond respiration. Nat Rev Mol Cell Biol 9:532–542
175. Peter ME, Krammer PH (2003) The CD95(APO-1/Fas) DISC
and beyond. Cell Death Differ 10:26–35
176. Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M,
Krammer PH, peter ME (1995) Cytotoxicity-dependent APO-1
(Fas/CD95)-associated proteins form a death-inducing signaling
complex (DISC) with the receptor. EMBO J 14:5579–5588
177. Yan N, Shi Y (2005) Mechanisms of apoptosis through struc-
tural biology. Annu Rev Cell Dev Biol 21:35–56
178. Donepudi M, Mac Sweeney A, Briand C, Grutter MG (2003)
Insights into the regulatory mechanism for caspase-8 activation.
Mol Cell 11:543–549
179. Davis AR, Lotocki G, Marcillo AE, Dietrich WD, Keane RW
(2007) FasL, Fas, and death-inducing signaling complex (DISC)
proteins are recruited to membrane rafts after spinal cord injury.
J Neurotrauma 24:823–834
180. Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS
(2004) Activation of caspases-8 and -10 by FLIP(L). Biochem J
382:651–657
181. O’Reilly LA, Ekert P, Harvey N et al (2002) Caspase-2 is not
required for thymocyte or neuronal apoptosis even thoughclea-
vage of caspase-2 is dependent on both Apaf-1 and caspase 9.
Cell Death Differ 9:832–841
182. Kim IR, Murakami K, Chen NJ, Saibil SD, Matysiak-Zablocki
E, Elford AR, Bonnard M, Benchimol S, Jurisicova A, Yeh WC,
Ohashi PS (2009) DNA damage- and stress-induced apoptosis
occurs independently of PIDD. Apoptosis 14:1039–1049
183. Ho LH, Read SH, Dorstyn L, Lambrusco L, Kumar S (2008)
Caspase-2 is required for cell death induced by cytoskeletal
disruption. Oncogene 27:3393–3404
184. Park MS, Kim BS, Devarajan P (2007) Hypoxia/reoxygenation
injury induces apoptosis of LLC-PK1 cells by activation of
caspase-2. Pediatr Nephrol 22:202–208
185. Gogvadze V, Orrenius S, Zhivotovsky B (2006) Multiple path-
ways of cytochrome c release from mitochondria in apoptosis.
Biochim Biophys Acta 1757:639–647
186. Baptiste-Okoh N, Barsotti AM, Prives C (2008) A role for
caspase 2 and PIDD in the process of p53-mediated apoptosis.
Proc Natl Acad Sci USA 105:1937–1942
187. Comelli M, Genero N, Mavelli I (2009) Caspase-independent
apoptosis in Friend’s erythroleukemia cells: role of mitochondrial
ATP synthesis impairment in relocation of apoptosis-inducing
factor and endonuclease G. J Bioenerg Biomembr 41:49–59
188. Parcellier A, Gurbuxani S, Schmitt E, Solary E, Garrido C
(2003) Heat shock proteins, cellular chaperones that modulate
mitochondrial cell death pathways. Biochem Biophys Res
Commun 304:505–512
189. Laudanski K, Wyczechowska D (2006) The distinctive role ofsmall heat shock proteins in oncogenesis. Arch Immunol Ther
Exp (Warsz) 54:103–111
190. Garrido C, Schmitt E, Cande C, Vahsen N, Parcellier A,
Kroemer G (2003) HSP27 and HSP70: potentially oncogenic
apoptosis inhibitors. Cell Cycle 2:579–584
191. Lin CY, Wu HY, Wang PL, Yuan CJ (2010) Mammalian Ste20-
like protein kinase 3 induces a caspase-independent apoptotic
pathway. Int J Biochem Cell Biol 42:98–105
192. Strauss G, Westhoff MA, Fischer-Posovszky P, Fulda S,
Schanbacher M, Eckhoff SM, Stahnke K, Vahsen N, Kroemer
G, Debatin KM (2008) 4-hydroperoxy-cyclophosphamide
mediates caspase-independent T-cell apoptosis involving oxi-
dative stress-induced nuclear relocation of mitochondrial apop-
togenic factors AIF and EndoG. Cell Death Differ 15:332–343
193. Satou T, Cummings BJ, Cotman CW (1995) Immunoreactivity
for Bcl-2 protein within neurons in the Alzheimer’s disease
brain increases with disease severity. Brain Re 697:35–43
56 Mol Cell Biochem (2011) 351:41–58
123

194. Jarskog LF, Gilmore JH (2000) Developmental expression of
Bcl-2 protein in human cortex. Brain Res Dev Brain Res
119:225–230
195. Levy OA, Malagelada C, Greene LA (2009) Cell death path-
ways in Parkinson’s disease: proximal triggers, distal effectors,
and final steps. Apoptosis 14:478–500
196. Morissette MR, Rosenzweig A (2005) Targeting survival sig-
naling in heart failure. Curr Opin Pharmacol 5:165–170
197. Donath MY, Halban PA (2004) Decreased beta-cell mass in
diabetes: significance, mechanisms and therapeutic implications.
Diabetologia 47:581–589
198. Kern TS, Du Y, Miller CM, Hatala DA, Levin LA (2010)
Overexpression of Bcl-2 in vascular endothelium inhibits the
microvascular lesions of diabetic retinopathy. Am J Pathol
176:2550–2558
199. Littlewood TD, Bennett MR (2003) Apoptotic cell death in
atherosclerosis. Curr Opin Lipidol 14:469–475
200. Susnow N, Zeng L, Margineantu D, Hockenbery DM (2009)
Bcl-2 family proteins as regulators of oxidative stress. Semin
Cancer Biol 9:42–49
201. Hajra KM, Liu JR (2004) Apoptosome dysfunction in human
cancer. Apoptosis 9:691–704
202. Yip KW, Reed JC (2008) Bcl-2 family proteins and cancer.
Oncogene 27:6398–6406
203. Reagan-Shaw S, Nihal M, Ahsan H, Mukhtar H, Ahmad N
(2008) Combination of vitamin E and delenium causes an
induction of apoptosis of human prostate cancer cells by
enhancing the Bax/Bcl-2 ration. Prostate 68:1624–1634
204. Cartron PF, Oliver L, Martin S, Moreau C, LeCabellec MT,
Jezequel P, Meflah K, Vallette FM (2002) The expression of a
new variant of the pro-apoptotic molecule Bax, Baxw, is cor-
related with an increased survival of glioblastoma multiforme
patients. Hum Mol Genet 11:675–687
205. Kirkin V, Joos S, Zornig M (2004) The role of Bcl-2 family
members in tumorigenesis. Biochim Biophys Acta 1644:229–249
206. O’Neill J, Manion M, Schwartz P, Hockenbery DM (2004)
Promises and challenges of targeting Bcl-2 anti-apoptotic pro-
teins for cancer therapy. Biochim Biophys Acta 1705:43–51
207. Juin P, Geneste O, Raimbaud E, Hickman JA (2004) Shooting at
survivors: Bcl-2 family members as drug targets for cancer.
Biochim Biophys Acta 1644:251–260
208. Frankel SR (2003) Oblimersen sodium (G3139 Bcl-2 antisense
oligonucleotide) therapy in Waldenstrom’s macroglobulinemia: a
targeted approach to enhance apoptosis. Semin Oncol 30:
300–304
209. Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G,
Mitchison T, Yuan J (2001) Identification of small-molecule
inhibitors of interaction between the BH3 domain and Bcl-xL.
Nat Cell Biol 3:173–182
210. Tzung SP, Kim KM, Basanez G, Giedt CD, Simon J, Zimmer-
berg J, Zhang KY, Hockenbery DM (2001) Antimycin A mimics
a cell-death-inducing Bcl-2 homology domain 3. Nat Cell Biol
3:183–191
211. Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu X, Cao Y, Guo R,
Li B, Zhu X, Huang Y, Long YQ, Roller PP, Yang D,
Wang S (2001) Discovery of small-molecule inhibitors of Bcl-2
through structure-based computer screening. J Med Chem
44:4313–4324
212. Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M
(2003) Discovery, characterization, and structure-activity rela-
tionships studies of proapoptotic polyphenols targeting B-cell
lymphocyte/leukemia-2 proteins. J Med Chem 46:4259–4264
213. Becattini B, Kitada S, Leone M, Monosov E, Chandler S, Zhai
D, Kipps TJ, Reed JC, Pellecchia M (2004) Rational design and
real time, in-cell detection of the proapoptotic activity of a novel
compound targeting Bcl-X(L). Chem Biol 11:389–395
214. Li J, Viallet J, Haura EB (2008) A small molecule pan-Bcl-2
family inhibitor, GX15–070, induces apoptosis and enhances
cisplatin-induced apoptosis in non-small cell lung cancer cells.
Cancer Chemother Pharmacol 61:525–534
215. McGregor N, Patel L, Craig M, Weidner S, Wang S, Pienta KJ
(2010) AT-101 (R-(-)-gossypol acetic acid) enhances the
effectiveness of androgen deprivation therapy in the VCaP
prostate cancer model. J Cell Biochem 110:1187–1194
216. Ferrer P, Asensi M, Priego S, Benlloch M, Mena S, Ortega A,
Obrador E, Esteve JM, Estrela JM (2007) Nitric oxide mediates
natural polyphenol-induced Bcl-2 down-regulation and activa-
tion of cell death in metastatic B16 melanoma. J Biol Chem
282:2880–2890
217. Muilenburg DJ, Coates JM, Virudachalam S, Bold RJ (2010)
Targeting Bcl-2-mediated cell death as a novel therapy in pan-
creatic cancer. J Surg Res 163(2):276–281
218. An J, Chen Y, Huang Z (2004) Critical upstream signals of
cytochrome C release induced by a novel Bcl-2 inhibitor. J Biol
Chem 279:19133–19140
219. Wang JL, Zhang ZJ, Choksi S, Shan S, Lu Z, Croce CM,
Alnemri ES, Korngold R, Huang Z (2000) Cell permeable Bcl-2
binding peptides: a chemical approach to apoptosis induction in
tumor cells. Cancer Res 60:1498–1502
220. Yin H, Lee GI, Sedey KA, Kutzki O, Park HS, Orner BP, Ernst
JT, Wang HG, Sebti SM, Hamilton AD (2005) Terphenyl-Based
Bak BH3 alpha-helical proteomimetics as low-molecular-weight
antagonists of Bcl-xL. J Am Chem Soc 127:10191–10196
221. Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright
RD, Wagner G, Verdine GL, Korsmeyer SJ (2004) Activation of
apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science
305:1466–1470
222. Bombrun A, Gerber P, Casi G, Terradillos O, Antonsson B,
Halazy S (2003) 3, 6-dibromocarbazole piperazine derivatives
of 2-propanol as first inhibitors of cytochrome c release via Bax
channel modulation. J Med Chem 46:4365–4368
223. Becattini B, Sareth S, Zhai D, Crowell KJ, Leone M, Reed JC,
Pellecchia M (2004) Targeting apoptosis via chemical design:
inhibition of bid-induced cell death by small organic molecules.
Chem Biol 11:1107–1117
224. Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait
AC, Reed JC (2003) Humanin peptide suppresses apoptosis by
interfering with Bax activation. Nature 423:456–461
225. Sawada M, Sun W, Hayes P, Leskov K, Boothman DA,
Matsuyama S (2003) Ku70 suppresses the apoptotic transloca-
tion of Bax to mitochondria. Nat Cell Biol 5:320–329
226. Foster FM, Owens TW, Tanianis-Hughes J, Clarke RB, Brennan
K, Bundred NJ, Streuli CH (2009) Targeting inhibitor of
apoptosis proteins in combination with ErbB antagonists in
breast cancer. Breast Cancer Res 1:R41
227. Liston P, Fong WG, Korneluk RG (2003) The inhibitors of
apoptosis: there is more to life than Bcl2. Oncogene 22:8568–
8580
228. LaCasse EC, Cherton-Horvat GG, Hewitt KE, Jerome LJ,
Morris SJ, Kandimalla ER, Yu D, Wang H, Wang W, Zhang R,
Agrawal S, Gillard JW, Durkin JP (2006) Preclinical charac-
terization of AEG35156/GEM 640, a second-generation anti-
sense oligonucleotide targeting X-linked inhibitor of apoptosis.
Clin Cancer Res 12:5231–5241
229. Wu TY, Wagner KW, Bursulaya B, Schultz PG, Deveraux QL
(2003) Development and characterization of nonpeptidic small
molecule inhibitors of the XIAP/caspase-3 interaction. Chem
Biol 10:759–767
230. Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M,
Bonneau MJ, Pedersen IM, Kitada S, Scott FL, Bailly-Maitre B,
Glinsky G, Scudiero D, Sausville E, Salvesen G, Nefzi A,
Ostresh JM, Houghten RA, Reed JC (2004) Small-molecule
Mol Cell Biochem (2011) 351:41–58 57
123

antagonists of apoptosis suppressor XIAP exhibit broad antitu-
mor activity. Cancer Cell 5:25–35
231. Chen J, Nikolovska-Coleska Z, Wang G, Qiu S, Wang S (2006)
Design, synthesis, and characterization of new embelin deriva-
tives as potent inhibitors of X-linked inhibitor of apoptosis
protein. Bioorg Med Chem Lett 16:5805–5808
232. Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X,
Harran PG (2004) A small molecule Smac mimic potentiates
TRAIL- and TNFalpha-mediated cell death. Science 305:
1471–1474
233. Zobel K, Wang L, Varfolomeev E, Franklin MC, Elliott LO,
Wallweber HJ, Okawa DC, Flygare JA, Vucic D, Fairbrother
WJ, Deshayes K (2006) Design, synthesis, and biological
activity of a potent Smac mimetic that sensitizes cancer cells to
apoptosis by antagonizing IAPs. ACS Chem Biol 1:525–533
234. Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF,
Deckwerth TL, Ding H, Elmore SW, Meadows RP, Olejniczak
ET, Oleksijew A, Oltersdorf T, Rosenberg SH, Shoemaker AR,
Tomaselli KJ, Zou H, Fesik SW (2004) Discovery of potent
antagonists of the antiapoptotic protein XIAP for the treatment
of cancer. J Med Chem 47:4417–4426
235. Lu J, Bai L, Sun H, Nikolovska-Coleska Z, McEachern D, Qiu
S, Miller RS, Yi H, Shangary S, Sun Y, Meagher JL, Stuckey
JA, Wang S (2008) SM-164: a novel, bivalent Smac mimetic
that induces apoptosis and tumor regression by concurrent
removal of the blockade of cIAP-1/2 and XIAP. Cancer Res
68:9384–9393
236. Chauhan D, Neri P, Velankar M, Podar K, Hideshima T, Fulc-
initi M, Tassone P, Raje N, Mitsiades C, Mitsiades N, Rich-
ardson P, Zawel L, Tran M, Munshi N, Anderson KC (2007)
Targeting mitochondrial factor Smac/DIABLO as therapy for
multiple myeloma (MM). Blood 109:1220–1227
237. Arnt CR, Chiorean MV, Heldebrant MP, Gores GJ, Kaufmann
SH (2002) Synthetic Smac/DIABLO peptides enhance the
effects of chemotherapeutic agents by binding XIAP and cIAP1
in situ. J Biol Chem 277:44236–44243
58 Mol Cell Biochem (2011) 351:41–58
123




















