
Activation of apoptosis, but not necrosis, during Mycobacterium tuberculosis infection correlated...
15
This article was published in an Elsevier journal. The attached copy is furnished to the author for non-commercial research and education use, including for instruction at the author’s institution, sharing with colleagues and providing to institution administration. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
Transcript of Activation of apoptosis, but not necrosis, during Mycobacterium tuberculosis infection correlated...

This article was published in an Elsevier journal. The attached copyis furnished to the author for non-commercial research and
education use, including for instruction at the author’s institution,sharing with colleagues and providing to institution administration.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Activation of apoptosis, but not necrosis, duringMycobacterium tuberculosis infection correlated with decreased
bacterial growth: Role of TNF-a, IL-10, caspasesand phospholipase A2 q
Mary Luz Arcila a,1, Marıa Dulfary Sanchez a,1, Blair Ortiz a, Luis Fernando Barrera a,Luis F. Garcıa a, Mauricio Rojas a,b,*
a Grupo de Inmunologıa Celular e Inmunogenetica, Facultad de Medicina, Universidad de Antioquia, Medellın, Colombiab Unidad de Citometrıa, Sede de Investigacion Universitaria, Universidad de Antioquia, Medellın, Colombia
Received 12 September 2007; accepted 15 November 2007Available online 26 December 2007
Abstract
Monocyte/macrophage cell death is an important event during mycobacterial infection. To get insights about the influence of mono-nuclear phagocyte maturation in this event we compared the response to Mycobacterium tuberculosis (Mtb) infection of fresh isolatedmonocytes and monocyte-derived macrophages (MDM) from healthy tuberculin positive individuals. Both monocytes and MDM under-went apoptosis, however, there was a higher numbers of apoptotic macrophages with active Caspases 8 and 9. We also compared Mtb-induced cell death in U937 pro-monocytes and PMA-differentiated cells (U937D). In response to Mtb infection, U937D cells underwentapoptosis and promonocytes both apoptosis and necrosis. There were high number of U937D cells producing TNF-a and high numberof IL-10+ promonocytes. These evidences suggest that U937 could be a valid model to study the mechanisms that rule Mtb-induced celldeath. Experiments with the cell line and fresh isolated mononuclear cells with pharmacological inhibitors showed that induction ofnecrosis involved calcium and cAMP signals resulting in IL-10 production. Necrosis also correlated with Caspase 3, PLA2 activityand bacterial growth. In U937D cells and monocytes from healthy donors there was activation of calcium, TNF-a and Caspase 8 acti-vation and decreased bacterial load. Understanding the mechanisms that control the dichotomy events between apoptosis and necrosis/oncosis associated with cell maturity might open new strategies to better control the course of mycobacterial infections.� 2007 Elsevier Inc. All rights reserved.
Keywords: Monocytes/macrophages; Necrosis; Apoptosis; Mycobacterial infection; TNF-a; IL-10 and phospholipase A2
1. Introduction
It is estimated that one-third of the world population isinfected with Mycobacterium tuberculosis (Mtb) [1]; how-
ever, most of the infected individuals never develop activedisease, supporting the importance of the interplaybetween the host genetic background and the immuneresponse that maintains the infection under control. How-ever, the increased incidence of tuberculosis over the lastdecades has made more urgent to further understand thehost factors that control susceptibility to tuberculosis.
Inhaled Mtb particles are readily phagocytosed by alve-olar macrophages [2], but the establishment of infectiondepends on the ability of the mycobacteria to survive andmultiply within the macrophages. However, infection ofmacrophages with Mtb also leads to the activation of
0008-8749/$ - see front matter � 2007 Elsevier Inc. All rights reserved.
doi:10.1016/j.cellimm.2007.11.006
q Supported by COLCIENCIAS, Bogota, Colombia. Grant No. 1115-04-11957.
* Corresponding author. Address: Grupo de Inmunologıa Celular eInmunogenetica, Facultad de Medicina, Sede de Investigaciones Univers-itaria, Universidad de Antioquia, Cra 53 No. 61-30 Lab 420, Medellın,Colombia. Fax: +57 4 210 6450.
E-mail address: [email protected] (M. Rojas).1 These authors had an equal contribution to this paper.
www.elsevier.com/locate/ycimm
Available online at www.sciencedirect.com
Cellular Immunology 249 (2007) 80–93

Author's personal copy
multiple anti-mycobacterial mechanisms by the infectedphagocyte, the induction of the T-cell dependent acquiredcellular responses and the granuloma formation. Mycobac-teria maybe contained within the granulomas resulting in alatent infection, but it can also multiply and disseminate,leading to active TB disease [3]. Tissue macrophages, aswell newly recruited monocytes, are important cellularcomponents of the granumolatous reaction. Studies onthe mononuclear phagocyte distribution within the granu-loma evidenced differences in cell maturity that correlatewith bacterial burden. Cells located at the center of thegranuloma are less mature and have a high bacillary load,whereas the cells at the periphery have a more mature phe-notype and few bacilli [4].
Our group has provided evidence that circulating mono-cytes from TB patients are phenotypically and functionallydifferent from monocytes from healthy controls, and thesedifferences are reversed during anti-TB treatment [5]. Also,we have previously reported that upon in vitro infectionwith Mtb, or stimulation with PPD, monocytes from tuber-culin skin test positive individuals undergo apoptosis, whilemonocytes from patients with pulmonary TB undergo bothapoptosis and necrosis/oncosis [5,6]. These results sup-ported our observations with murine macrophages [7–9]suggesting that mechanisms of cell death may correlatewith macrophage innate capability to restrain Mtb intracel-lular growth, which may be also dependent on the matura-tion stage of the phagocyte.
In this study, we further investigated the mechanismsinvolved in the necrosis and apoptosis induced by virulentMtb in human monocytes and monocyte-derived macro-phages (MDM). These events were also studied in thehuman U937 promonocytic cell line and in PMA-differen-tiated cells (U937D). Results show that after infection withMtb, monocytes from TB patients and U937 promonocytesare more prone to undergo necrosis, compared to mono-cytes from healthy individuals, MDMs and U937D cellsthat preferentially underwent apoptosis, suggesting thatphagocyte maturity may play a critical role in Mtb-inducedcell death. The differences in cell death correlated with vari-ations in Caspases 8 and 9 activation, calcium mobiliza-tion, activation of phospholipase A2 (PLA2), productionof TNF-a, and IL-10 and the capacity to control mycobac-terial growth. These results provide new insights about therole of Mtb-induced macrophage cell death and providenew insights to improve bacterial control.
2. Materials and methods
2.1. Reagents and antibodies
Saponin, trypan blue and phorbol-12-myristate-13-ace-tate (PMA) were obtained from Sigma–Aldrich (SaintLouis, MO); bovine fetal serum (PHS) from HyClone(Logan, UT); RPMI-1640, phosphate buffered saline(PBS, ph 7.4) and Dulbecco’s modified Eagle medium(DMEM) from Gibco-BRL (Grand Island, NY); caspase
inhibitors and biotynilated substrates: YVAD (Caspase 1substrate), DEVD (Caspase 3 substrate) and IETD (Cas-pase 8 substrate) from Calbiochem (La Jolla, CA). Sub-strates for Caspase 8, FLICA (FAM-LETD-FMK), andCaspase 9, FLICA (FAM-LEHD-FMK) from Immuno-chemistry Technologies, LLC, Bloomington, MN;pentoxyfilline, BAPTA/AM (1,2 bis-[o-aminophenoxy]-ethane-N,N 0,N 0-tetra-acetic acid, tetra-acetoxymethylester), dBcAMP (dibutyryl-cAMP); etidium bromide(EB), DEDA (7,7-dimethyleicosadienoic acid), andNaH2PO4 from ICN (Cleveland, OH); bongkrekic acidfrom Biomol Research Laboratories (Plymouth Meeting,PA). pooled human serum, male, AB (PHS) and Lympho-cyte Separation Medium (LSM, density = 1.077) from Bio-Whittaker (Walkersville, MD); MDL-1 (cis-N-[2-phenyl-cyclopentyl]azacyclotride-1-en-z-amine, HCl) y 3H-uracil(27 mCi/mmol) from Amersham (Buckinghamshire, UK);Middlebrook 7H9 broth and Middlebrook 7H10 agar fromBecton Dickinson (Cockeysville, MD); DIOC6 (3,3 0-dihexyloxacarbocyanine-iodide) and propidium iodidefrom Molecular Probes (Eugene, OR); p-formaldehyde(PFA) from Fisher Scientific (Pittsburgh, PA); glycerolfrom Mallinckrodt Baker (Xalostoc, Edo. De Mex., Mex-ico); monoclonal antibodies anti-TNF-a-FITC (cloneMab11), anti-IL-10-PE (clone JES3-19F1), IgG1-FITCfrom Alexis (San Diego, CA), IgG2a-PE, (clone G155-178), IgG1, k-Cy-Chrome (clone G155-228) anti-CD14-PE (clone M5E2), anti-CD3-Cy-Chrome (clone UCHT1)from BD Pharmingen (San Diego, CA). All reagents andmedia were endoctoxin free.
2.2. Culture and maintenance of M. tuberculosis H37Rv
Mtb H37Rv was grown in Middlebrook 7H9 supple-mented with 10% oleic acid dextrose. To disrupt bacterialclumps, bacteria were resuspended in PBS 1·, sonicatedat 25 W for 3 min at 4 �C (CV33 Sonics Vibra Cell, New-town, CT), centrifuged for 5 min at 400g, in order to pre-cipitate mycobacterial clumps. The supernatant wasdiluted in PBS 1· adjusted to final concentration of 0.1(OD550). The number of CFU was determined by plating5 ll of serial dilutions onto petri dishes containing Middle-brook 7H10 agar, supplemented with glycerol and oleicacid-albumin-dextrose pH 7.2. Bacteria (1.8 · 108 UFC/ml) were frozen at �70 �C in DMEM containing 30% glyc-erin and 10% heat-inactivated PHS, until use.
2.3. Patients and control subjects
Thirteen patients with pulmonary TB, clinically andbacteriologically diagnosed were recruited from differentHospitals and Health Centers from Medellın, Colombia.Bacteriological confirmation of TB was done by sputumacid-fast staining and culture. All participants were HIVnegative, they were not receiving any immunosuppressivedrug, and patients with cancer and diabetes and otherrespiratory diseases were excluded from this study. There
M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93 81

Author's personal copy
were six males and seven females whose ages ranged from18 to 55 years old. All patients were studied within 2 weeksof diagnosis. As control, tuberculin skin test positivehealthy donors (n = 13, with same range of age that TBpatients) were studied (PPD RT 23, Statens Serum InstitutCopenhagen, Denmark). Individuals included in this studydid not previously received anti-TB treatment. All subjectsstudied signed an informed consent previously approved bythe Ethics Committee of the Centro de InvestigacionesMedicas, Facultad de Medicina, Universidad de Antio-quia, Medellın, Colombia.
2.4. Isolation of mononuclear cells (PBMC)
Thirty five milliliter samples of venous blood werepoured into Erlenmeyer flasks containing 25 sterile glass-beads (5-mm diameter). Samples were gentle rotated for10 min until a fibrin-clot appeared. Defibrinated bloodwas collected, diluted three times with PBS and separatedon a density gradient (Lymphocyte Separation Medium,density 1.077 g/ml) at 400g for 25 min at room tempera-ture. The fraction containing the PBMC was recoveredand washed twice with PBS. Cells were resuspended inRPMI-1640 supplemented with 2 mM L-glutamine and0.5% PHS without antibiotics (medium of adhesion). Cellviability was determined by trypan blue exclusion and itwas P95%.
2.5. Monocytes and monocyte-derived macrophages
(MDM) monolayers infection with Mtb
Monolayers were obtained after determination by flowcytometry of the percentage CD14+ cells contained in thePBMC fraction. Five hundred thousand CD14+ cells werecultured in 24 wells plates (BD Falcon, Bedford, MA), in1 ml of RPMI-1640 supplemented with 0.5% PHS at37 �C, 5% CO2. Two hours later, nonadherent cells wereremoved by extensive washings with prewarmed PBS con-taining 0.5% PHS. Thereafter, adherent cells were incu-bated in 1 ml of RPMI-1640 supplemented with 10%PHS and 2 mM L-glutamine without antibiotics and cul-tured under conditions described below for specific experi-ments. To determine the number of adhered cells extrawells were used for the nuclear staining according to Naka-gawara and Nathan (0.05% de naphtol blue black, citricacid 0.1, 1% Triton X-100, pH 2.2) [10]. The percentageof CD3+ cells in monolayers was always <5%, as estimatedby flow cytometry. Monocytes were cultured during 5 daysin order to obtain MDM.
2.6. Culture and differentiation of human promonocytic U937
cell line
The human promonocytic cell line U937 was cultured inRPMI-1640 supplemented with 10% PHS and 2 mM L-glu-tamine. The conditions for PMA-induced cell differentia-tion were established by kinetics and dose-response
experiments measuring CD68 expression [11–13]. U937promonocytes, 1 · 106, were plated in 6 wells plates (Corn-ing, Elmira, NY) and stimulated with 10 ng/ml of PMA for0–120 h, or treated with 0.1–10 ng/ml of PMA for 72 h at37 �C and 5% CO2. Nonadherent cells were removed bywashing with pre-warmed PBS. Adherent cells were perme-abilized with 200 ll 0.5% Tween 20, 0.2% BSA in PBS for30 min at room temperature, stained with anti-CD68-PE orthe respective isotype control, and analyzed by flowcytometry.
2.7. Cell infection with Mtb H37Rv
U937 cells, human monocyte and MDM monolayers(5 · 106 cells/well) were cultured by duplicates in RPMI-1640 supplemented with 10% PHS in flat-bottom 24-wellculture plates (Corning, Elmira, NY), infected or not withMtb H37Rv macrophage (5:1) and incubated at 37 �C, 5%CO2, during variable periods of time, depending on theexperimental conditions.
2.8. Determination of cell death by flow cytometry
2.8.1. Mitochondrial permeability transition (MPT)
alterations and cytoplasm membrane damage
To determine variations of MPT, as indicative of apop-tosis, and cytoplasmic membrane damage, indicative ofnecrosis [6], cells were resuspended in 2 ml of PBS contain-ing 10 ll of DIOC6 (7 lM) and 25 ll of ethidium bromide(EB, 1.3 mg/ml). Cell cultures were incubated at room tem-perature and darkness during 20 min, washed, scraped witha rubber policeman, resuspended in PBS and analyzed byflow cytometry (Coulter EPICS XL, Hialeah, FL, USA).Cells with high fluorescence intensity for DIOC6 and EBnegative were considered as living cells. Cells with low fluo-rescence intensity for DIOC6 and EB negative were consid-ered apoptotic cells, and cells with high fluorescenceintensity for EB were considered necrotic cells [6].
2.8.2. Annexin V—propidium iodide (PI)
Exposure of phosphatidyl serine on the outer surfacemembrane is one of earliest hallmarks of apoptosis thatcan be detected using annexin V staining [14]. Cells (1–2 · 106 cells/ml) were washed with annexin-binding buffer(10 mM Hepes, 0.14 M NaCl; 2.5 mM CaCl2; pH 7.4). Fivemicroliters of annexin V-FITC and 5 lg/ml propidiumiodide were added to 1 · 106 cells, gently mixed and incu-bate for 15 min at room temperature in darkness. Thereaf-ter, the cells were washed and analyzed by flow cytometry.annexin V�PI� were considered viable, annexin+PI� earlyapoptotic, annexin V+PI+ late apoptotic cells and annexinV�PI+ were considered necrotic cells.
2.8.3. Measurement of activated Caspases 1, 3, 8 and 9
Active Caspases 1 and 3 were studied by flow cytometryas previously reported [9]. Cells were washed with PBSand fixed with 300 ll of 2% PFA at different time points
82 M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93

Author's personal copy
post-infection. Cells were permeabilized with 200 ll 0.5%Tween 20, 0.2% BSA in PBS for 30 min at room tempera-ture and then incubated with 20 mM biotin-YVAD-CMK(Caspase 1 substrate) or 20 mM biotin-DEVD-CHO (Cas-pase 3 substrate) for 1 h at 37 �C and 5% CO2. Thereafter,cells were washed with PBS and incubated with 0.5 lg/mlStreptavidin-FITC 30 min at room temperature in dark-ness. The excess avidin-FITC was removed by washingwith PBS. Cells were resuspended in 300 ll of PBS andanalyzed by flow cytometry. Active Caspases 8 and 9 weredetected incubating the cells with cell permeable peptidesFLICA (FAM-LETD-FMK) and FLICA (FAM-LEHD-FMK), respectively [15] for 1 h at 37 �C under 5% CO2,protecting the tubes from light. Cells were washed, gentlymixed to disrupt any cell-to-cell clumping and resuspendin 0.5 mL wash buffer and analyzed by flow cytometry.
2.8.4. Determination of intracellular TNF-a and IL-10
Infected cells were cultured for 18 h at 37 �C and 5%CO2, at this time 1 lg/ml brefeldin A was added and thecultures were further incubated for 6 h. Cells were washedtwice with PBS, fixed with 2% PFA in 0.1 M de NaH2PO4
during 20 min at room temperature and harvested. Then,cells were washed once with permeabilization buffer (PBS1· containing 1% PHS, 1% BSA, 0.1% sodium azide and0.1% saponin, pH 7.4), and incubated with 200 ll of per-meabilization buffer during 30 min at 4 �C containing2.5 lg mAb anti-TNF-a-FITC and 2.5 lg anti-IL-10-PE,or IgG2a-FITC and IgG2b-PE as isotype controls during30 min at 4 �C. Cells were washed three times with PBScontaining 1% PHS, 1% BSA and 0.1% sodium azide, pH7.4. As control for the intracellular signal, non-permeabili-zed cells were stained with specific antibodies. Thereaftercells were analyzed by flow cytometry.
2.8.5. TNF-a bioassay
TNF-a bioactivity was determined by a modified cyto-toxicity assay using L929 cells [9]. Briefly, L929 cell mono-layers, cultured in RPMI-1640 plus 10% FBS and 1 lg/mlof actinomycin D for 48 h on flat-bottom 24-well plates(1 · 106 cells/well), were overlaid with 1 ml of twofoldserial dilutions of supernatants from U937 and U937Dcells infected or not with Mtb. As control growing concen-trations of hrTNF-a (1–100 U/ml) were added. After incu-bation at 37 �C for 24 h, the cells were resuspended in1.5 ml of PBS containing 50 lg/ml of PI and incubatedfor 5 min at room temperature in darkness. Thereafter,cells were washed two times with cold PBS and the percent-age of PI fluorescent cells was determined by flowcytometry.
2.8.6. Determination of mycobacterial growth
Intracellular replication of Mtb was determined asdescribed [16]. Fifty thousand cells were cultured in 96-flat-bottom wells culture plates (BD Falcon, Bedford,MA) and infected with Mtb H37Rv at different multiplicityof infections. At indicated time-points cells were treated
with RPMI-1640 containing 10% PHS and 2% saponin torelease the intracellular bacteria, and supplemented with1.6% L-asparagine, 1.6% sodium glutamate, 0.04% ferricammonium citrate and 0.5 lCi/well of 3H-UdR for 8 d toevaluate bacterial growth. Thereafter, cultures were har-vested onto glass filters (Inotech, Byosistems Internacional.Lasing, USA). 3H-Uracil incorporation was measured in aliquid scintillation counter (Beckman LS6500, Albertville,MN).
2.8.7. Inhibitors
Cell cultures were treated with different inhibitors 1 hbefore Mtb infection: MDL-1 1 nM, to block of adenilatecyclase activity; 1 lg/ml dBcAMP, a cAMP analogue;20 lg/ml pentoxyfilline (PTX, an analogue of endogen pur-ines), in order to inhibit phosphodiesterase activity; 56 lMDEDA, to inhibit phospholipase A2 (PLA2) activity and20 lM BAPTA/AM, to chelate calcium and 2.5 lg/ml,bongkrekic acid 100 lM, anti-TNF-a and anti-IL-10.Z-YVAD-FMK for Caspase 1; Ac-DEVD-CMK, for Cas-pase 3; and Z-IETD-FMK, for Caspase 8. Cells were cul-tured with these inhibitors 1 h before of Mtb infection(5:1, 24 h). Thereafter, the induction of cell death usingDIOC6 and EB was determined.
2.8.8. Statistical analysis
All experiments were done in triplicate and indepen-dently repeated at least three or four times. Data were ana-lyzed by ANOVA-Type III of square sum. Interactionsabove the second level were excluded. Statistical signifi-cance was tested at p < 0.05 as critical value, calculatedby the interactions between the factors. Data are presentedas the mean ± 95% confidence interval for mean (95% CI).For all analysis we used Statgrafphics Plus, release 2, 1996(Statgraphics Corp., Rockville, MD).
3. Results
3.1. Macrophages are more prone than monocytes to undergoMtb-induced apoptosis
To determine whether macrophage differentiation mightaffect the Mtb-induced cell death, freshly isolated mono-cytes and MDM obtained from healthy donors wereinfected after 2 h of adherence or 5 days of in vitro differen-tiation, respectively. The cells were infected for 48 h andthereafter stained with annexin V and PI and with cell per-meable peptides to detect active Caspases 8 and 9. Non-infected monocytes and macrophages had less than 2% ofdead cells (not shown). After Mtb infection there wasabout 16% of annexin V+ monocytes and 33% annexinV+ MDM (p < 0.001) (Fig. 1). The percentage of mono-cytes with active Caspases 9 and 8 (18% and 25%, respec-tively) was lower than the percentage of MDM (88% and55%, respectively) (p < 0.001). In addition, the meanfluorescence intensity was also higher in macrophagescompared to monocytes, indicating that the amount of
M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93 83

Author's personal copy
active caspase per cell was augmented upon celldifferentiation.
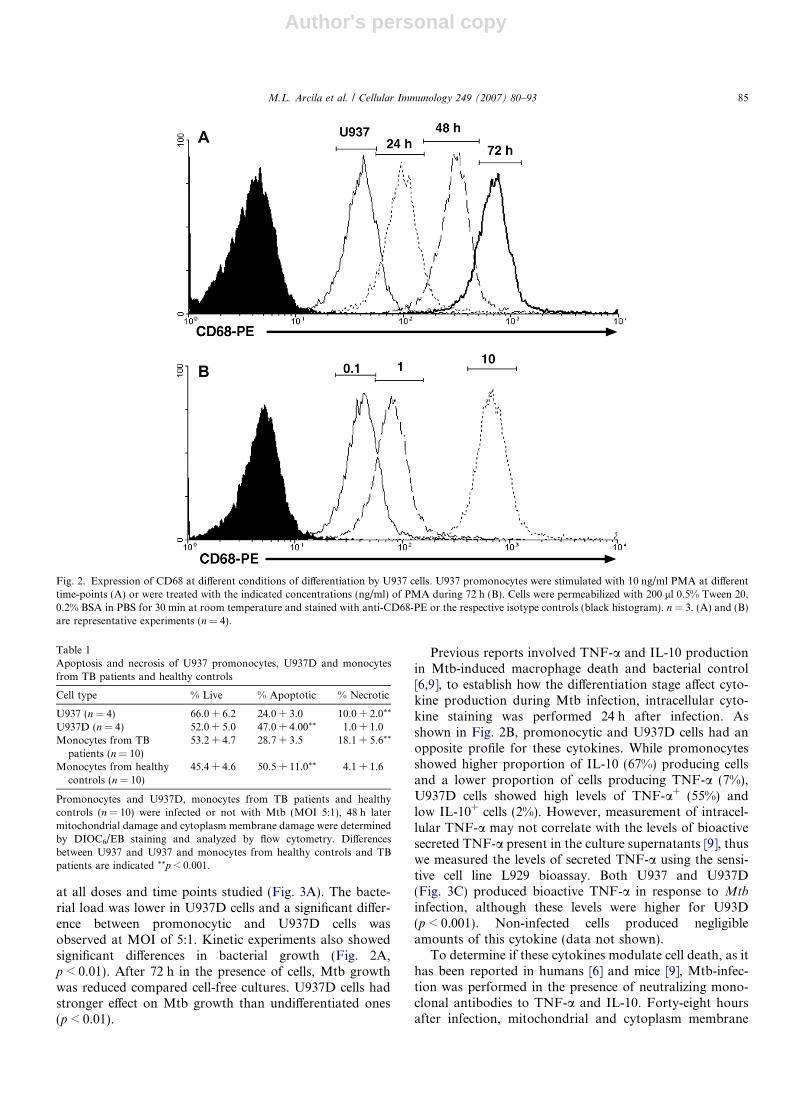
The influence of cell differentiation on Mtb-induced celldeath was also studied in U937 promonocytes differenti-ated or not with PMA [17] and monocytes from healthydonors and patients with pulmonary TB. The effect ofPMA on U937 cell differentiation was initially assessedby the expression CD68 at different time-points usingincreasing concentrations of PMA. As shown in Fig. 2A,10 ng/ml of PMA induced a time-dependent increase inCD68 expression. Fig. 2B shows that at 72 h, the expres-sion of CD68 increased in a PMA-concentration-depen-dent manner (Fig. 2B). CD68 expression was stable atlater time-points but concentrations higher than 10 ng/mldecreased cell adherence and viability (data not shown),thus for the following experiments U937 cells were differen-tiate with 10 ng/ml of PMA during 72 h.
Non-infected cells did not undergo apoptosis or necrosis(data not shown). Mtb-infected promonocytic U937 cellsshowed 24 ± 3% apoptotic cells and 10 ± 2% necrotic cells(Table 1). In U937D cells, Mtb induced mitochondrialdamage compatible with apoptosis in 47.0 ± 4% of the cells
with a negligible percentage of cells with cytoplasm mem-brane damage compatible with necrosis (1 ± 1%). Mono-cytes from patients with TB showed apoptotic(28.7 ± 3.5) and necrotic cells (18.1 ± 5.6), compared withtuberculin-skin test positive healthy controls that exhibited50.5 + 11.0 of apoptotic cells with low percentage of necro-tic cells (4.1 ± 1.6) in response to Mtb infection, as previ-ously reported [6]. These results suggest that themechanisms of cell death could be related with the statusof mononuclear phagocyte differentiation and that U937cells are a feasible model to study Mtb-induced apoptosisand necrosis.
To establish whether U937 and U937D cells differ intheir ability to control the intracellular growth of Mtb,cells were infected at different MOI and collected at differ-ent time-points to evaluate uptake of 3HUdR. BecauseU937 cells grow in suspension, extra cellular bacteria werenot removed and the bacterial growth in these wells wascompared with control cultures without cells. As shownin Fig. 3A there was a MOI- and time-depending increasein bacterial load as determined by uptake of 3HUdR.Incorporation of 3HUdR was higher in cell-free cultures
Annexin V
PI
Monocytes + Mtb MDM + Mtb
Caspase 8 (FAM-LEDH)
Caspase 9 (FAM-LETD)
PI
25% 55%
18% 88%
0.5 0.5
85 16
0.5 5
28
0
25
50
75
100Non-InfectedMtb
%A
nn
exin
V+
cells
0
25
50
75
100
%C
asp
ase
9+ c
ells
Monocytes MDM
0
25
50
75
100
%C
asp
ase
8+ c
ells
**
**
**
Fig. 1. Mononuclear phagocytes activate different pathways of Mtb-induced cell death depending on maturation status. Monocytes (Left) and monocyte-derived macrophages (MDM, Right) were isolated healthy donors (n = 5). After 2 h of adherences monocytes were infected with Mtb (5:1) or allowed todifferentiate into macrophages by adherence to the plastic during 5 days before infection. Forty-eight hours after infection, cells were stained with annexinV and propidium iodide to determine apoptosis and necrosis by flow cytometry (A). Cells were stained with FLICA (FAM-LEHD-fmk) to detect Caspase9 and with FLICA (FAM-LETD-fmk) to detect Caspase 8. This figure shows one representative of five experiments (B).
84 M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93
Author's personal copy
at all doses and time points studied (Fig. 3A). The bacte-rial load was lower in U937D cells and a significant differ-ence between promonocytic and U937D cells wasobserved at MOI of 5:1. Kinetic experiments also showedsignificant differences in bacterial growth (Fig. 2A,p < 0.01). After 72 h in the presence of cells, Mtb growthwas reduced compared cell-free cultures. U937D cells hadstronger effect on Mtb growth than undifferentiated ones(p < 0.01).
Previous reports involved TNF-a and IL-10 productionin Mtb-induced macrophage death and bacterial control[6,9], to establish how the differentiation stage affect cyto-kine production during Mtb infection, intracellular cyto-kine staining was performed 24 h after infection. Asshown in Fig. 2B, promonocytic and U937D cells had anopposite profile for these cytokines. While promonocytesshowed higher proportion of IL-10 (67%) producing cellsand a lower proportion of cells producing TNF-a (7%),U937D cells showed high levels of TNF-a+ (55%) andlow IL-10+ cells (2%). However, measurement of intracel-lular TNF-a may not correlate with the levels of bioactivesecreted TNF-a present in the culture supernatants [9], thuswe measured the levels of secreted TNF-a using the sensi-tive cell line L929 bioassay. Both U937 and U937D(Fig. 3C) produced bioactive TNF-a in response to Mtb
infection, although these levels were higher for U93D(p < 0.001). Non-infected cells produced negligibleamounts of this cytokine (data not shown).
To determine if these cytokines modulate cell death, as ithas been reported in humans [6] and mice [9], Mtb-infec-tion was performed in the presence of neutralizing mono-clonal antibodies to TNF-a and IL-10. Forty-eight hoursafter infection, mitochondrial and cytoplasm membrane
Fig. 2. Expression of CD68 at different conditions of differentiation by U937 cells. U937 promonocytes were stimulated with 10 ng/ml PMA at differenttime-points (A) or were treated with the indicated concentrations (ng/ml) of PMA during 72 h (B). Cells were permeabilized with 200 ll 0.5% Tween 20,0.2% BSA in PBS for 30 min at room temperature and stained with anti-CD68-PE or the respective isotype controls (black histogram). n = 3. (A) and (B)are representative experiments (n = 4).
Table 1Apoptosis and necrosis of U937 promonocytes, U937D and monocytesfrom TB patients and healthy controls
Cell type % Live % Apoptotic % Necrotic
U937 (n = 4) 66.0 + 6.2 24.0 + 3.0 10.0 + 2.0**
U937D (n = 4) 52.0 + 5.0 47.0 + 4.00** 1.0 + 1.0Monocytes from TB
patients (n = 10)53.2 + 4.7 28.7 + 3.5 18.1 + 5.6**
Monocytes from healthycontrols (n = 10)
45.4 + 4.6 50.5 + 11.0** 4.1 + 1.6
Promonocytes and U937D, monocytes from TB patients and healthycontrols (n = 10) were infected or not with Mtb (MOI 5:1), 48 h latermitochondrial damage and cytoplasm membrane damage were determinedby DIOC6/EB staining and analyzed by flow cytometry. Differencesbetween U937 and U937 and monocytes from healthy controls and TBpatients are indicated **p < 0.001.
M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93 85

Author's personal copy
damage were studied. Compared with the isotype control,anti-TNF-a increased the number of EB+ promonocytesand had a negligible effect on the percentage of necroticU937D cells. Anti-TNF-a reduced the percentage of cellswith mitochondrial damage from 17% to 4% in U937 cellsand abolished apoptosis in apoptosis in U937D. Blockadewith anti-IL10 prevented induction of necrosis in promon-ocytic cells (Fig. 3D) and increased the percentage of apop-totic cells in U937D cells.
In addition to modulation of cell death, we also testedwhether anti-TNF-a and anti-IL10 treatments affect intra-cellular growth of Mtb. As shown in Fig. 3D, in the pres-ence of isotype antibodies there was a significantdifference between U937D cells and promonocytes(p < 0.0001). Treatment with anti-TNF-a increases bacte-rial growth (Fig. 1F, p < 0.01) in both cells, although thiseffect was more pronounced in U937D cells. In U937
promonocytes and U937D cells, blockade of IL-10 reducedbacterial load. These results strongly support a relationshipbetween the mechanisms to control Mtb and those associ-ated induction of cell death.
3.2. Chelation of intracellular calcium and stabilization of
permeability transition (PT) decreased apoptosis andnecrosis but enhanced bacterial growth
We have previously reported that macrophage infectionwith Mtb induced calcium influx and Ca+2-dependentevents seem to be responsible for PT alterations in murinemacrophages [18]. Thus, we explored how removal of cal-cium and stabilization of PT affected cell death, cytokineproduction and bacterial growth. For this purpose we trea-ted promonocytic U937 and U937D cells with BAPTA/AM, a calcium chelator, and Bongkrekic acid, an inhibitor
TNFα
IL-10
67% U937IL10+
55% U937D TNFα+
DIOC6
EB
U937
U937D
anti-IL-10
0:1 2.5:1 5:1 7.5:1 10:1
0
5000
10000
15000
20000Without cellsU937U937 D
*
MOI
H3 U
dR
(cp
m)
0 24 48 72 96 120
0
5000
10000
15000
20000Without cellsU937U937 D
**
Time
H3 U
dR
(cp
m)
U937 U937D
0
5000
10000
15000
20000
25000
**
***
Iso
typ
e
anti
-TN
Fα
anti
-IL
-10
Iso
typ
e
anti
-TN
Fα
anti
-IL
-10
H3U
dR
(cp
m)
Isotype control anti-TNFα
12%
17%
80%
4% 98%
1%
26%
4%
1% 55%45%
7% U937 TNFα+
U93
7
U93
7D
0
50
100
150
***
TN
F-α
U/m
l
U937 D
A B
C D E
Fig. 3. U937D cells are more prone to undergo apoptosis produce more TNF-a and control more efficiently Mtb-growth than U937 cells. (A) U937promonocytes and PMA-differentiated U937 cells were infected with Mtb at indicated MOI during 96 h (TOP), or at MOI 5:1 at indicated time-points(Bottom). Cell were lysed with Saponin and supplemented for bacterial growth and pulsed with 3HUdR. Seven days later, 3HUdR incorporation wasdetermined. (B) Promonocytes and PMA-differentiated were infected with Mtb (MOI 5:1). Eighteen hour later cells were treated with Brefeldin A, and24 h after initial infection cells were fixed and permeabilized for intracellular staining with anti-TNF-a (TOP) and anti-IL-10 (Bottom). (C) BioactiveTNF-a was determined in supernatants from Mtb infected cells. Promonocytes (U937) and PMA-differentiated (U937D) cells were infected with Mtb (5:1)and 48 h later mitochondrial damage and cytoplasm membrane damage were determined by DIOC6/EB staining and cells were analyzed by flow cytometry(D). Promonocytes and U937D cells were infected with Mtb (5:1) during 96 h in the presence of anti-TNF-a and anti-IL-10, Mtb growth was determinedas indicated above (E). (E) Comparisons were performed between isotype controls and treated cells, *p < 0.01; **p < 0.001, ***p < 0.0001, n = 4. (A) and(C) are representative experiments.
86 M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93

Author's personal copy
of mitochondrial PT, prior to Mtb-infection [19]. As shownin Fig. 4A, removal of calcium and stabilization of PTprevented both Mtb-induced necrosis and apoptosis inpromonocytes and differentiated U937 cells. In U937 cells,pre-treatment with BAPTA/AM and Bongkrekic acid didnot affect the percentage of TNF-a+ cells but these percent-ages were reduced in Mtb-infected U937D cells. BAPTA/AM and Bongkrekic acid significantly decreased IL-10+
promonocytes (Fig. 4B and C, p < 0.001) but they did notaffect U937D cells. Both treatments significantly increasedbacterial load in U937 and U937D cells (p < 0.01, Fig. 4D).
3.3. Increased cAMP levels decreased Mtb-induced
apoptosis, increased necrosis and enhanced bacterial growth
which correlates with increased percentage of IL-10+ cells
cAMP is a second messenger induced during infectionwith Mtb, involved in modulation of cell death by thenuclear activation of CREB and modulation of IL-10 pro-
duction [18]. cAMP seems to be activated as consequenceof calcium influx, since removal of calcium prevented manyof cAMP-mediated effects [18]. The effect of cAMP onMtb-induced death of U937 and U937D cells was testedby incubation with MDL-1, an inhibitor of cAMP produc-tion [20], dBcAMP, a cAMP agonist [21], and pentoxyfil-line (PTX), that increases intracellular cAMP levels bypreventing its enzymatic degradation [22]. Pretreatmentwith MDL-1 inhibited induction of necrosis and increasedapoptosis in pro-monocytes and U937D cells (Fig. 4A).Pretreatment with dBcAMP and pentoxyfilline increasedthe percentage of necrotic promonocytes, and almost com-pletely abolished the percentage apoptotic U937D cells.Interestingly, MDL-1 increased the proportion of promon-ocytic cells producing TNF-a (Fig. 5B) while significantlyreduced the proportion of IL-10+ promonocytes(Fig. 5C). In U937D, MDL-1 slightly increased thepercentage of TNF-a producing cells and did not affectIL-10+ cells. dBcAMP- and pentoxyfillline-pretreated
Fig. 4. Effect of PT-stabilization on Mtb-induced cell death and cells producing TNF-a and IL-10. U937 and U937D cells were treated or not withBAPTA/AM and Bongkrekic acid (BGRK) 1 h before infection with Mtb (5:1), 48 h later mitochondrial damage and cytoplasm membrane damage weredetermined by DIOC6/EB staining and cells were analyzed by flow cytometry (A). In parallel cultures, infected cells were treated with Brefeldin A, fixedand permeabilized to detect intracellular TNF-a (B) and IL-10 (C) after 24 h. Cells were treated as indicated in Section 2, and Mtb-intracellular growthwas determined by 3HUdR incorporation (D). Comparisons were performed between non-treated controls and treated cells, *p < 0.01; **p < 0.001, n = 4.(A) is a representative experiment.
M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93 87

Author's personal copy
promonocyte and U937D cells did not produce TNF-a butboth encompassed higher levels of IL-10+ cells. In parallelexperiments, pretreatment with MDL-1 significantlydecreased bacterial growth in promonocytes (p < 0.01) witha subtle effect on U937D cells; however, dBcAMP andpentoxyfilline significantly increased intracellular growthof Mtb in both promonocytes and U937D cells (Fig. 5D).
3.4. Blockade of phospholipase A2 and caspases had opposite
effects on mycobacterial intracellular growth
Several mechanisms are involved in regulating the effec-tor phase of different types of cell death: Caspase 3 [23] isa cornerstone during the effector phase of apoptosis, andCaspase 8 [23] is activated by different cell surface deathreceptors, Caspase 1 is involved in Mtb-induced cell death[9] and in a type of cell death known as pyroptosis [24],and PLA2 has been involved in both apoptosis and necro-sis [25]. Thus, we determined the activity of Caspases 1and 3 in promonocytic and U937D cells during infection
with Mtb. For this purpose we performed a kinetic exper-iment using fluorescent peptides able to bind active casp-ases. The activity of Caspases 1 and 3 in promonocyticcells is shown Fig. 6A and U937D cells in Fig. 5B. Atbasal level, time-point cero, there was not caspase activity.During the experimental time course, promonocytic cellsdisplayed high MFI for Caspase 3 and low MFI for Cas-pase 1; on the contrary, U937D cells had high MFI forCaspase 1 and no Caspase 3 activity, indicating that differ-ent effector mechanisms are controlling cell death upon-differentiation.
To further establish the participation of different execu-tioner mechanisms in apoptosis and necrosis induced byMtb, we pretreated cells with peptides able to block Cas-pase 1 (YVAD), Caspase 8 (IETED), Caspase 3 (DEVD),or with DEDA, a chemical able to block calcium-depend-ing secretory PLA2. Blockade of caspases inhibited apop-tosis in both promonocytes and U937D cells, whileDEDA inhibited apoptosis in U937 promonocytes butincreased it in U937D cells from 20% to 50%. Treatment
Fig. 5. Effect of cAMP and PLA2 inhibitors on Mtb-induced cell death and cells producing TNF-a and IL-10. Promonocytes (U937) and PMA-differentiated (U937D) cells were treated or not with MDL-1, dBcAMP and pentoxyfilline 1 h before infection with Mtb (5:1); 48 h later mitochondrialdamage and cytoplasm membrane damage were determined by DIOC6/EB staining (A). Cells were fixed and permeabilized for intracellular detection ofTNF-a (B) and IL-10 24 h (C) or to determine Mtb-intracellular growth after 96 h (D). Comparisons were performed between non-treated controls andtreated cells, *p < 0.01; **p < 0.001, n = 4. A is a representative experiment.
88 M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93

Author's personal copy
with DEVD and DEDA prevented induction of necrosis inU937 cells (Fig. 6C), suggesting that Caspase 3 and PLA-2are important for the regulation of both types of cell deathin non-differentiated cells. DEDA treatment also resultedin a decrease in both apoptosis and necrosis in U937.
Blockade of caspases had a negligible effect on the per-centage of TNF-a+ cells; however, DEDA increased theproportion of both U937 and U937D cells producingTNF-a (Fig. 5D, p < 0.001). Blockade of Caspases 1 and8 increased the proportion of IL-10+ U937D cells, withno effect on promonocytes, but DEDA significantlydecreased IL-10+ U937 cells (p < 0.001, Fig. 5E). In prom-onocytes, blockade of caspases did not have any significanteffect in 3HUdR uptake; however, in U937D cells, YVADand IETED (p < 0.001) increased in 3HUdR uptake com-pared with DEVD (p < 0.01). Treatment with DEDA sig-
nificantly decreased bacterial growth in bothpromonocytes and U937D cells (Fig. 6D, p < 0.01).
Infection with Mtb activates different mechanisms of celldeath in monocytes from TB patients and healthy controls,which are modulated by cAMP and PLA-2. To further ver-ify that regulation of cell death was not an intrinsic mech-anism associated with a differentiation of U937promonocytes, we isolated monocytes from TB patientsand healthy controls. These cells were pre-treated withdBcAMP, MDL-1 and DEDA, infected with Mtb and celldeath was determined 48 h later. As expected, only mono-cytes from TB patients underwent necrosis after Mtb infec-tion (Fig. 7A), dBcAMP increased the number of necrotic(annexin V�, PI+) monocytes from TB patients and healthycontrols, and prevented induction of apoptosis (annexinV+, PI�). MDL-1 and DEDA almost completely abrogated
Fig. 6. Differential activity of Caspases 1 and 3 in U937 and U937D during Mtb infection. Effect of phospholipase A2 and caspases inhibitors on celldeath, TNF-a, IL-10 and mycobacterial growth. U937 (A) and U937D (B) cells infected with Mtb (5:1) for different time points. Cells were fixed,permeabilized and incubated with fluorescent peptides able to bind active Caspases 1 and 3. U937 and U937D cells were treated or not with YVAD,DEVD, IETED and DEDA from 1 h before and during infection with Mtb (5:1), 48 h later mitochondrial damage and cytoplasm membrane damage weredetermined by DIOC6/EB staining (C). Cells were fixed and permeabilized to detect intracellular TNF-a (D) and anti-IL-10 after 24 h (E). or to determineMtb-intracellular growth after 96 h (F). Comparisons were performed between non-treated controls and treated cells, *p < 0.01; **p < 0.001, n = 4.
M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93 89

Author's personal copy
induction of necrosis in TB patients (annexin V� PI+
monocytes) and healthy control, but increased apoptosis(annexin V+ cells). Pentoxyfilline, BAPTA/AM, Bon-gkrekic acid and blockade of caspases with peptides hada similar effect to the observed in experiments describedbefore using promonocytic and U937D cells (data notshown).
Experiments with monocytes from healthy controls inthe absence of treatment, infected monocytes exhibit ahigher percentage of TNF-a+ cells (Fig. 7B). dBcAMPand pentoxyfilline significantly reduced the percentage ofTNF-a+ monocytes, from both healthy and TB patients.Contrariwise, treatment with MDL-1 increased the propor-tion of TNF-a+ cells. Untreated Mtb-Infected monocytesfrom TB patients showed a higher percentage of IL-10+
monocytes compared with healthy controls. dBcAMPand pentoxyfilline non-significantly increased the percent-age of L-10+ monocytes. Treatment with DEDA did nothave any effect. MLD-1 and DEDA decreased the percent-age of IL-10+ cells in both healthy controls and TB-patients (p < 0.001). In untreated Mtb-Infected monocytes
from healthy control, there was not IL-10+ cells. dBcAMPand pentoxyfilline significantly increased this percentage(p < 0.05, Fig. 7C).
We also compared activated Caspases 1 and 3 in mono-cytes isolated from healthy controls and TB patients. Asshown in Fig. 7D, in monocytes from TB patients infectionwith Mtb induced a higher proportion of cells with acti-vated Caspase 3 than Caspase 1. Monocytes from healthycontrols had an opposite profile since they presented ahigher proportion of cells with activated Caspase 1 thanwith activated of Caspase 3.
4. Discussion
Using different experimental systems we and other inves-tigators have previously reported induction of cell deathduring Mtb infection in murine [6–9,18,26,27] and human[5,6] mononuclear phagocytes and that induction ofapoptosis might be activated through different metabolicpathways (references). Although the evidence highlightsthat macrophage apoptosis is a common event during
Fig. 7. Mtb induced different events associated with cell death in monocytes isolated from TB patients and healthy controls. Monocytes isolated frompatients with pulmonary TB (TOP) and healthy controls (BOTTOM) were treated with dBcAMP, MDL-1 and DEDA 1 h before infection with Mtb (5:1).Forty-eight hours later, cells were stained with annexin V and propidium iodide to determine apoptosis and necrosis by flow cytometry (A). Cells weretreated with indicated inhibitors before infection with Mtb, fixed and permeabilized to detect intracellular TNF-a (B), IL-10 (C) and Caspases 1 and 3 (D).In (B) and (C) comparisons were performed between non-treated controls and treated cells, in (D) comparisons were performed between monocytesisolated from TB patients and healthy controls, *p < 0.01; **p < 0.001, n = 4 independent experiments.
90 M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93

Author's personal copy
Mtb-infection the role of cell death in the pathogenesis ofTB is clearly understood.
Previously, we reported that monocytes from healthycontrols undergo apoptosis, whereas monocytes frompatients with pulmonary TB might undergo apoptosisand necrosis/oncosis during in vitro infection with Mtb[6] and that the induction of necrosis is reversed with theanti-TB treatment [5]. There is extensive evidence for roleof apoptosis as a defense mechanisms to contain bacterialreplication, avoiding the inflammatory response observedin susceptible hosts is supported [7,28]). However, the rela-tionship between the mechanisms of cell death and bacte-rial control is still poorly understood. The resultspresented herein clearly indicated that after Mtb-infectionmonocytes and macrophages activate mechanisms of celldeath using different pathways that also influence theircapacity to control Mtb growth. Furthermore, our results,using cell lines, suggests that the status of cell differentia-tion might affect the time course of infection and the acti-vation of cell death; additionally, the results with freshmonocytes suggest that different circulating monocytessubsets could be found in TB patients and healthy controls.In this regard, we already reported differences on cell mark-ers associated with innate response in monocytes isolatedfrom TB patients and healthy controls [5], suggesting thatsystemic effects of Mtb-infection might affect mononuclearphagocytes differentiation and traffic.
In our experimental system, we tried to correlate activa-tion of different pathways of cell death with the capacity ofinfected cells to counteract mycobacterial infection. Forthis purpose we used anti-TNF-a and anti-IL-10 [29], cyto-kines known to have opposite effects on macrophage acti-vation, cell death and mycobacterial control. Blockade ofthese cytokines clearly showed an association betweenapoptosis and bacterial control and necrosis with bacterialproliferation. Whereas anti-TNF-a enhanced Mtb 3HUdRincorporation, and increased membrane damage, anti-IL-10 decreased Mtb 3HUdR incorporation, and increasedmitochondrial PT.
It is known that Mtb is able to induce early mitochon-drial damage in a calcium-depending way. In our experi-ments, removal of calcium and stabilization ofmitochondrial PT prevented induction of both apoptosisand necrosis, decreased the proportion of TNF-a- andIL-10-producing cells and enhanced bacterial growth.The inhibition of both necrosis and apoptosis by BAP-TA/AM and BGRK, suggests that these forms of cell deathmay be triggered by a common event. Other authors haveshown that treatment of Mtb-infected macrophages withthe calcium ionophore A23187 blocks necrosis, stabilizedPT, decreased cytochrome C release, Caspase 9 activation,and effectively controlled mycobacterial growth [30]. Mac-rophages treated with the mitochondrial calcium uniporterinhibitor ruthenium and infected with Mtb showed mito-chondrial swelling characteristic of necrosis, decreased PTand anti-mycobacterial activity. Our results also supportedthat Mtb-infected macrophages showing hallmarks of
apoptosis are effective in controlling mycobacterial growth,in contrast, macrophages undergoing necrosis are unableto constrain Mtb replication [30].
Previous evidence demonstrated an interplay betweenIL-10 and cAMP metabolism to modulate apoptosis [18].We also explored the modulation of cAMP using MDL-1,dBcAMP and pentoxyfilline. Our results show that cAMPdirectly affected induction of cell death and bacterialgrowth in a cytokine-dependent way. Blockade of cAMPincreased the proportion of TNF-a+ cells, decreasedIL-10+ cells and bacterial growth and prevented inductionnecrosis. Conversely, high levels of cAMP increased IL-10+
cells, necrosis and bacterial growth.We also studied the participation of caspases and PLA-2
in Mtb induced cell death. Caspase 1 is involved in Mtbinduced cell death [9,27,31] and pyroptosis which activelypromotes inflammation [24]. Caspase 8 participates inTNF-a-dependent pathway; and Caspase 3, activatedthrough the mitochondrial and TNF-a pathways, is adownstream executioner protease that is activated by theupstream caspases, it acts itself to cleave cellular targetsassociated with induction of both apoptosis and necrosis[30,32]. It is noteworthy that the initiator (Caspase 1) andthe executioner (Caspase 3) were differentially activatedin promonocytes and U937D cells, and that, in the sameway, the number of Caspase 3+ cells was higher in TBpatients while Caspase 1 was higher in healthy controls.Blockade of the initiator Caspases 1 and 8 prevented induc-tion of apoptosis with no effect on necrosis, suggesting thatCaspases 8 and 1 may have a common downstream medi-ator of cell death activated during Mtb-infection. Blockadeof Caspase 3 prevented both apoptosis and necrosis [33,34],while blockade of Caspases 1, 3 and 8 increased bacterialgrowth in U937D cells. In this context, our results are par-tially in agreement with the results reported by Ciaramellaet al. [31] observed that Caspase 1 inhibitor (YVAD), butnot Caspase 3 (DEVD), was able to inhibit Mtb-inducedapoptosis [31]. However, although in both systems apopto-sis was strongly dependent of TNF-a production, blockadeof caspases did not have any effect on TNF-a+ cells, andblockade of Caspases 1 and 8, but no Caspase 3, increasedthe proportion of U937D IL-10+ cells. Importantly, block-ade of the three caspases significantly increased bacterialgrowth in U937D cells, but in non-differentiated promono-cytes there was small decrease in bacterial growth afterblockade of Caspase 3, supporting that Caspase 3 isinvolved in both apoptosis and necrosis, and that the exe-cutioner necrotic pathway in promonocytic cells is notimplicated in bacterial control.
PLA2s belongs to a family of esterases that hydrolyzethe ester bond in phospholipids, releasing free fatty acidsand lysophospholipids that may act as second messengersor effector molecules. PLA2s are important in the signalingof several cellular processes during ischemia/reperfusion-induced cellular injury [35]. The role of PLA2s in apoptosisand necrosis/oncosis depends upon the PLA2 isoform, thecell type, and the agent of injury [33]. Recent evidence indi-
M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93 91

Author's personal copy
cates that the non-virulent M. tuberculosis H37Ra, whichinduces TNF-a-dependent apoptosis but not necrosis,induces release of arachidonic acid [36]. Apoptosis ofinfected macrophages was abrogated by group IV cPLA2inhibitors, but not by inhibitors of group VI Caþ2 -indepen-dent. Importantly, group IV cPLA2 inhibitors significantlyreduced macrophage antimycobacterial activity and addi-tion of arachidonic acid, the major product of group IVcPLA2, to infected macrophages treated with cPLA2 inhib-itors, completely restored the antimycobacterial activity,but addition of arachidonic acid alone significantly reducedthe mycobacterial burden. These authors proposed thatMtb induces macrophage apoptosis through at least twoindependent signaling pathways, TNF-a and cPLA2,which are both also critical for anti-mycobacterial hostdefense [36]. In addition, It has been previously reportedthat inhibition of PLA2 leads to an increase in Mtb multi-plication [37]. In our experiments, treatment with DEDA,which is a general inhibitor of PLA2s, decreased IL-10+
cells, increased both TNF-a+ and apoptosis and decreasedbacterial count in U937D cells, suggesting that one ofPLA2 isoforms mediates a critical step in the mycobacterialoutcome. These evidence, in contrast with evidencedescribed above [36], suggest that in the case of M. tubercu-
losis H37Rv PLA2 plays a critical role in modulation of celldeath.
Besides PLA2, another enzymes involve in lipid metab-olism might play a role in bacterial clearance. Sphingosine1-phosphate participates in several intracellular pathways,such as calcium mobilization, cell growth and differentia-tion. In vivo and in vitro S1P induces antimicrobial activityin human macrophages against non-pathogenic Mycobac-
terium smegmatis and pathogenic M. tuberculosis infec-tions. This activity is mediated by host phospholipase D,which favors the acidification of mycobacteria-containingphagosomes, reduced mycobacterial load and pulmonarytissue damage. In this context, reinforcement of endoge-nous pathways that increase macrophage effectors capabil-ities might also affect acquired immunity [38].
Our results clearly show that mechanisms of cell deathobserved in U937 promonocytes and U937D and mono-cytes from TB patients and healthy controls, respectivelyare strongly influenced by the stage of cell differentiation.Altogether, these observations allow us to propose thatafter infection with Mtb, the earliest events are modulatedby calcium. Calcium seems to be responsible for mitochon-drial damage as we previously reported [18]; however, atlater after infection, cytokines may provide a second signal.In addition, a positive feedback may take place becauseremoval of calcium significantly reduced the proportionof TNF-a+ and IL-10+ cells. Thus, induction of necrosiswould require calcium and IL-10. In the presence of IL-10, macrophages effector mechanisms are not enough tocontain bacterial replication, allowing a permissive envi-ronment that favors calcium overload, mitochondrial dam-age and activation of Caspase 3 and PLA2, which are theexecutioners of necrosis.
On the other hand, infected cells might produce TNF-a,which is responsible for the apical activation of Caspases 1and 8 and the induction of apoptosis. At this point, it is notclear what governs the switch or the balance TNF-a/IL-10.It is tempting to speculate that monocyte/macrophage dif-ferentiation status balances both cytokines during myco-bacterial infection. The observation that monocytes fromTB patients and healthy control have unbalanced propor-tion of Caspases 1/3 also suggest that probably in mono-cytes from TB patients there is an active apoptoticpathway; however, infection might decrease the ATP reser-voirs switching cells into necrosis.
We also inquired about the effect of intrinsic macro-phage differentiation on cell death. Is well established thatcirculating monocytes differentiate into a variety of tissueresident macrophages as well as into myeloid dendritic cells[39]. During infection monocytes are elicited and recruitedin high numbers into the granulomatous lesions where theybecome macrophages. It has been accepted, that in TBpatients there is a high number of immature circulatingmonocytes [5]. Thus, under this premise we decide to infectnon-differentiated monocytes and macrophages after 5days of differentiation on plastic adherence. Our resultsclearly show that macrophages are more prone to undergoapoptosis than monocytes upon infection with Mtb.
In different experiments we observed a higher correla-tion between annexin V and active Caspase 9, indicatingthat although apical pathway was initiated probablythrough TNF-a, the mitochondrial pathway prevailed inthe execution of cell death and that apical pathway goesthrough induction of the mitochondrial damage. In addi-tion, our evidence indicates that Mtb-induced apical path-way might reinforce mitochondrial pathway, sinceblockade of TNF-a also affected mitochondrial permeabil-ity transition. In this way, it will be interesting to determinewhether Mtb infection is able to induce activation of tBID,which is an established connection between the apical andmitochondrial pathways of cell death [40]. Our results indi-cate that macrophage differentiation may be a critical clueto understand the outcome of mycobacterial infection; theyseem to respond in different ways depending upon the stageof differentiation, affecting the mechanisms of cell deathand the bacterial control. This process may account for dif-ferent phenomena during active diseases in TB patientsthat, in response to the infection, may increase the numberof circulating immature precursors from the bone marrow,and they may have decreased capability to contain Mtbinfection compared with mature mononuclear phagocytes.
References
[1] AIDS Policy Law 21 (2006) 5.[2] L.S. Schlesinger, J. Invest. Med. 44 (1996) 312–323.[3] D.G. Russell, Nat. Rev. Microbiol. 5 (2007) 39–47.[4] A. Pedroza-Gonzalez, G.S. Garcia-Romo, D. Aguilar-Leon, J.
Calderon-Amador, R. Hurtado-Ortiz, H. Orozco-Estevez, B.N.Lambrecht, I. Estrada-Garcia, R. Hernandez-Pando, L. Flores-Romo, Int. J. Exp. Pathol. 85 (2004) 135–145.
92 M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93

Author's personal copy
[5] M.D. Sanchez, Y. Garcia, C. Montes, S.C. Paris, M. Rojas, L.F.Barrera, M.A. Arias, L.F. Garcia, Microb. Infect. 8 (2006) 2492–2500.
[6] D.P. Gil, L.G. Leon, L.I. Correa, J.R. Maya, S.C. Paris, L.F. Garcia,M. Rojas, J. Infect. Dis. 189 (2004) 2120–2128.
[7] H. Pan, B.S. Yan, M. Rojas, Y.V. Shebzukhov, H. Zhou, L. Kobzik,D.E. Higgins, M.J. Daly, B.R. Bloom, I. Kramnik, Nature 434 (2005)767–772.
[8] M. Rojas, L.F. Barrera, G. Puzo, L.F. Garcia, J. Immunol. 159(1997) 1352–1361.
[9] M. Rojas, M. Olivier, P. Gros, L.F. Barrera, L.F. Garcia, J.Immunol. 162 (1999) 6122–6131.
[10] A. Nakagawara, C.F. Nathan, J. Immunol. Methods 56 (1983) 261–268.
[11] H. Kurushima, M. Ramprasad, N. Kondratenko, D.M. Foster, O.Quehenberger, D. Steinberg, J. Leukoc. Biol. 67 (2000) 104–108.
[12] F. Pinet, A. Dupont, N. Bencherif, A.L. Guihot, B. Quatannens, P.Amouyel, Cell Mol. Biol. (Noisy.-le-grand) 49 (2003) 899–905.
[13] M.P. Ramprasad, V. Terpstra, N. Kondratenko, O. Quehenberger,D. Steinberg, Proc. Natl. Acad. Sci. USA 93 (1996) 14833–14838.
[14] G. Denecker, H. Dooms, G. Van Loo, D. Vercammen, J. Grooten,W. Fiers, W. Declercq, P. Vandenabeele, FEBS Lett. 465 (2000) 47–52.
[15] P. Smolewski, E. Bedner, L. Du, T.C. Hsieh, J.M. Wu, D.J. Phelps, Z.Darzynkiewicz, Cytometry 44 (2001) 73–82.
[16] M. Arias, M. Rojas, J. Zabaleta, J.I. Rodriguez, S.C. Paris, L.F.Barrera, L.F. Garcia, J. Infect. Dis. 176 (1997) 1552–1558.
[17] M.T. Chateau, H. Rabesandratana, R. Caravano, FEMS Immunol.Med. Microbiol. 13 (1996) 19–28.
[18] M. Rojas, L.F. Garcia, J. Nigou, G. Puzo, M. Olivier, J. Infect. Dis.182 (2000) 240–251.
[19] C.Y. Shin, J. Shin, B.M. Kim, M.H. Wang, J.H. Jang, Y.J. Surh, U.Oh, Mol. Cell Neurosci. 24 (2003) 57–68.
[20] A. Gadea, E. Lopez, A.M. Lopez-Colome, Brain Res. 838 (1999) 200–204.
[21] Y. Berthiaume, J. Appl. Physiol. 70 (1991) 2490–2497.
[22] P. Rieckmann, F. Weber, A. Gunther, S. Martin, A. Bitsch, A.Broocks, B. Kitze, T. Weber, T. Borner, S. Poser, J. Neuroimmunol.64 (1996) 193–200.
[23] A. Thorburn, Cell Signal. 16 (2004) 139–144.[24] S.L. Fink, B.T. Cookson, Infect. Immunol. 73 (2005) 1907–1916.[25] J.M. Gutierrez, V. Arce, F. Brenes, F. Chaves, Exp. Mol. Pathol. 52
(1990) 25–36.[26] M. Rojas, L.F. Barrera, L.F. Garcia, Biochem. Biophys. Res.
Commun. 247 (1998) 436–442.[27] M. Rojas, M. Olivier, L.F. Garcia, Cell Immunol. 217 (2002) 58–66.[28] A. Molloy, P. Laochumroonvorapong, G. Kaplan, J. Exp. Med. 180
(1994) 1499–1509.[29] Y. Lin, M. Zhang, F.M. Hofman, J. Gong, P.F. Barnes, Infect.
Immunol. 64 (1996) 1351–1356.[30] L. Duan, H. Gan, D.E. Golan, H.G. Remold, J. Immunol. 169 (2002)
5181–5187.[31] A. Ciaramella, A. Martino, R. Cicconi, V. Colizzi, M. Fraziano, Cell
Death Differ. 7 (2000) 1270–1272.[32] H. Jaeschke, M.A. Fisher, J.A. Lawson, C.A. Simmons, A. Farhood,
D.A. Jones, J. Immunol. 160 (1998) 3480–3486.[33] B.S. Cummings, J. McHowat, R.G. Schnellmann, J. Pharmacol. Exp.
Ther. 294 (2000) 793–799.[34] K. Hamahata, S. Adachi, H. Matsubara, M. Okada, T. Imai, K.
Watanabe, S.Y. Toyokuni, M. Ueno, S. Wakabayashi, Y. Katano-saka, S. Akiba, M. Kubota, T. Nakahata, Eur. J. Pharmacol. 516(2005) 187–196.
[35] Y.A. Bellido-Reyes, H. Akamatsu, K. Kojima, H. Arai, H. Tanaka,M. Sunamori, Transplantation 81 (2006) 1700–1707.
[36] L. Duan, H. Gan, J. Arm, H.G. Remold, J. Immunol. 166 (2001)7469–7476.
[37] K. Kanai, E. Kondo, Jpn. J. Med. Sci. Biol. 33 (1980) 87–101.[38] Z.A. Malik, C.R. Thompson, S. Hashimi, B. Porter, S.S. Iyer, D.J.
Kusner, J. Immunol. 170 (2003) 2811–2815.[39] D.A. Hume, Curr. Opin. Immunol. 18 (2006) 49–53.[40] M. Tafani, N.O. Karpinich, A. Serroni, M.A. Russo, J.L. Farber, J.
Cell Physiol. 208 (2006) 556–565.
M.L. Arcila et al. / Cellular Immunology 249 (2007) 80–93 93




















