
Phospholipase D1 is associated with amyloid precursor protein in Alzheimer's disease
13
Neurobiology of Aging 28 (2007) 1015–1027 Phospholipase D1 is associated with amyloid precursor protein in Alzheimer’s disease Jae-Kwang Jin a , Bong-Hyun Ahn b , Yeo-Jung Na a , Jae-Il Kim c , Yong-Sun Kim a , Eun-Kyoung Choi a , Young-Gyu Ko d , Kwang Chul Chung e , Piotr B. Kozlowski c , Do Sik Min f,∗ a Ilsong Institute of Life Science, Hallym University, Kwanyang-dong, Dongan-gu, Anyang, Kyonggi-do 431-060, Republic of Korea b Cardiovascular Branch National Heart, Lung and Blood Institute (NHLBI), NIH Building 10/CRC 5-3288 10 Center Drive, Bethesda, MD 20892, USA c New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY 10314, USA d College of Life Science and Biotechnology, Korea University, Anam-dong, Sungbook-ku, Seoul 136-701, Republic of Korea e Department of Biology, College of Sciences, Yonsei University, Sinchon-dong, Seodaemun-ku, Seoul 120-752, Republic of Korea. f Department of Molecular Biology, College of Natural Science, Pusan National University, 30 Jangjeon dong, Geumjeong gu, Busan 609-735, Republic of Korea Received 23 January 2006; received in revised form 8 May 2006; accepted 11 May 2006 Available online 23 June 2006 Abstract Amyloid precursor protein (APP) is a widely expressed transmembrane protein of unknown function that is involved in the pathogenesis of Alzheimer’s disease (AD). We investigated the involvement of phospholipase D (PLD) in the pathophysiology of AD. We showed dramatic upregulation of PLD1 immunoreactivity in reactive astroglial cells in brain tissue sections from authentic AD patients. Expression and activity of PLD1 were up-regulated in brain tissues from AD patients, especially caveolae membrane fraction, compared with those of control brains. Interestingly, PLD1 physically interacts and colocalizes with APP and caveolin-3. We found that APP was associated with the pleckstrin homology domain of PLD1, and the amyloid region of APP interacted with PLD. Elevated expression of APP stimulated PLD activity in human astroglioma cells. These results suggest that up-regulation of PLD might have a role in the neuronal pathology associated with AD. © 2006 Elsevier Inc. All rights reserved. Keywords: Alzheimer’s disease; Amyloid precursor protein; -Amyloid; Phospholipase D; Reactive astrocytes; Caveolin 1. Introduction Alzheimer’s disease (AD), the most common cause of dementia in elderly patients, is a complex disorder of the Abbreviations: AD, Alzheimer’s disease; FAD, familial Alzheimer’s dis- ease; APP, amyloid precursor protein; A, -amyloid; NFT, neurofibrillary tangles; PLD, phospholipase D; CEM, caveolin-enriched membrane; ARF, ADP-ribosylation factor; PC, phosphatidylcholine; PA, phosphatidic acid; PKC, protein kinase C; DMEM, Dulbecco’s modified Eagles’s medium; GFAP, glial fibrillary acidic protein; PBS, phosphate-buffered saline; TBS, tris-buffered saline; MBS, MES-buffered saline; PMSF, phenylmethylsul- fonyl fluoride; PtdBut, phosphatidylbutanol; PH, pleckstrin homology; PX, N-terminal phox ∗ Corresponding author. Tel.: +82 51 510 3682; fax: +82 51 513 9258. E-mail address: [email protected] (D.S. Min). central nervous system clinically characterized by a pro- gressive loss of cognitive abilities. Pathological hallmarks of AD include widespread neuronal degeneration, neurite plaques containing -amyloid (A), and intracellular neu- rofibrillary tangles [24,35]. The A is the major component of senile plaques and is derived from the amyloid precur- sor protein (APP) by proteolytic cleavage [36]. Although accumulated evidence suggests that A is a key causative agent of AD [11,33], the exact mechanism of neuronal degeneration in AD has not been clear. It is likely that multiple factors are involved in the development of the disease. Kanfer et al. [17] have suggested that neuronal degen- eration in AD might be related to an alteration in tissue phospholipids with a corresponding increase in the catabolic 0197-4580/$ – see front matter © 2006 Elsevier Inc. All rights reserved. doi:10.1016/j.neurobiolaging.2006.05.022
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Phospholipase D1 is associated with amyloid precursor protein in Alzheimer's disease

A
AuoIhh©
K
1
d
etAPGtfN
0d
Neurobiology of Aging 28 (2007) 1015–1027
Phospholipase D1 is associated with amyloid precursorprotein in Alzheimer’s disease
Jae-Kwang Jin a, Bong-Hyun Ahn b, Yeo-Jung Na a, Jae-Il Kim c, Yong-Sun Kim a,Eun-Kyoung Choi a, Young-Gyu Ko d, Kwang Chul Chung e,
Piotr B. Kozlowski c, Do Sik Min f,∗a Ilsong Institute of Life Science, Hallym University, Kwanyang-dong, Dongan-gu, Anyang, Kyonggi-do 431-060, Republic of Korea
b Cardiovascular Branch National Heart, Lung and Blood Institute (NHLBI), NIH Building 10/CRC 5-3288 10 Center Drive, Bethesda, MD 20892, USAc New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY 10314, USA
d College of Life Science and Biotechnology, Korea University, Anam-dong, Sungbook-ku, Seoul 136-701, Republic of Koreae Department of Biology, College of Sciences, Yonsei University, Sinchon-dong, Seodaemun-ku, Seoul 120-752, Republic of Korea.
f Department of Molecular Biology, College of Natural Science, Pusan National University, 30 Jangjeon dong,Geumjeong gu, Busan 609-735, Republic of Korea
Received 23 January 2006; received in revised form 8 May 2006; accepted 11 May 2006Available online 23 June 2006
bstract
Amyloid precursor protein (APP) is a widely expressed transmembrane protein of unknown function that is involved in the pathogenesis oflzheimer’s disease (AD). We investigated the involvement of phospholipase D (PLD) in the pathophysiology of AD. We showed dramaticpregulation of PLD1 immunoreactivity in reactive astroglial cells in brain tissue sections from authentic AD patients. Expression and activityf PLD1 were up-regulated in brain tissues from AD patients, especially caveolae membrane fraction, compared with those of control brains.
nterestingly, PLD1 physically interacts and colocalizes with APP and caveolin-3. We found that APP was associated with the pleckstrinomology domain of PLD1, and the amyloid region of APP interacted with PLD. Elevated expression of APP stimulated PLD activity inuman astroglioma cells. These results suggest that up-regulation of PLD might have a role in the neuronal pathology associated with AD.2006 Elsevier Inc. All rights reserved.
hosphol
c
eywords: Alzheimer’s disease; Amyloid precursor protein; �-Amyloid; P
. Introduction
Alzheimer’s disease (AD), the most common cause ofementia in elderly patients, is a complex disorder of the
Abbreviations: AD, Alzheimer’s disease; FAD, familial Alzheimer’s dis-ase; APP, amyloid precursor protein; A�, �-amyloid; NFT, neurofibrillaryangles; PLD, phospholipase D; CEM, caveolin-enriched membrane; ARF,DP-ribosylation factor; PC, phosphatidylcholine; PA, phosphatidic acid;KC, protein kinase C; DMEM, Dulbecco’s modified Eagles’s medium;FAP, glial fibrillary acidic protein; PBS, phosphate-buffered saline; TBS,
ris-buffered saline; MBS, MES-buffered saline; PMSF, phenylmethylsul-onyl fluoride; PtdBut, phosphatidylbutanol; PH, pleckstrin homology; PX,-terminal phox∗ Corresponding author. Tel.: +82 51 510 3682; fax: +82 51 513 9258.
E-mail address: [email protected] (D.S. Min).
goprosaadmd
ep
197-4580/$ – see front matter © 2006 Elsevier Inc. All rights reserved.oi:10.1016/j.neurobiolaging.2006.05.022
ipase D; Reactive astrocytes; Caveolin
entral nervous system clinically characterized by a pro-ressive loss of cognitive abilities. Pathological hallmarksf AD include widespread neuronal degeneration, neuritelaques containing �-amyloid (A�), and intracellular neu-ofibrillary tangles [24,35]. The A� is the major componentf senile plaques and is derived from the amyloid precur-or protein (APP) by proteolytic cleavage [36]. Althoughccumulated evidence suggests that A� is a key causativegent of AD [11,33], the exact mechanism of neuronalegeneration in AD has not been clear. It is likely thatultiple factors are involved in the development of the
isease.Kanfer et al. [17] have suggested that neuronal degen-
ration in AD might be related to an alteration in tissuehospholipids with a corresponding increase in the catabolic

1 gy of Ag
ptiisaaDw
(pPacPrgsitPrbn
ipuaw
2
2
dfarAsst
2
NaffwAms
2
sHoL(CoNst
2cc
iparCwwshob
TD
N
123456
016 J.-K. Jin et al. / Neurobiolo
roducts of phospholipids metabolism. A� is known to affecthe activity of several lipid-metabolizing enzymes, includ-ng the phospholipases A2, C and D [16,31,32]. However,t is unknown whether these phospholipids alternations areufficient and necessary for the etiology of AD or if theyre secondary events in the pathogenesis of neurodegener-tion. Recently, phospholipids breakdown by phospholipase(PLD) has been recognized as an important signaling path-ay in the nervous system.PLD catalyzes the hydrolysis of phosphatidylcholine
PC), the major membrane phospholipid, to produce theleiotropic signaling lipid messenger phosphatidic acid (PA).A is generally recognized as the signaling product of PLDnd functions as an effector in multiple physiological pro-esses [21]. In mammals, two isoforms of PLD, PLD1 andLD2, have been cloned and are being characterized foregulation and cellular function [21]. However, the segre-ated roles of the two PLD isoforms in cellular responses aretill poorly understood. Activation of PLD occurs throughnteractions of the ARF and Rho families as well as with pro-ein kinase C (PKC) [21]. Recently, we have reported thatLD1 immunoreactivity and its enzymatic activity were up-egulated in the activated astrocytes of scrapie-infected micerain [14]. However, the expression of PLD in AD brains hasot been investigated so far.
These studies have raised the possibility that PLD might benvolved in the neuronal pathology associated with AD. In theresent study, we demonstrate for the first time that PLD1 isp-regulated in the caveolae membrane fraction and reactivestrocytes from AD brains, and co-localized and associatedith APP.
. Materials and methods
.1. Brain tissues
A total of six post-mortem brain specimens of Alzheimer’sisease patients and age-matched controls were obtainedrom the IBR brain bank (Dr. Piotr B. Kozlowski). The char-cteristics of the brain specimens with post-mortem intervalsanging from 0.5 to 14.5 h are shown in Table 1. Cases of
lzheimer’s disease and age-matched controls were clas-ified based on quantitative pathological features includingenile plaques and neurofibrillary tangles (NFT) accordingo Braak and Braak [1].
5drc
able 1emographic detail of control and Alzheimer’s disease post-mortem brain specime
umber Case type Age/sex Post-mortem interva
Control 68/F 2.5Control 68/F 5.0Control 67/F 4.0AD 74/F 14.5AD 79/F 5.0AD 73/F 0.5
ing 28 (2007) 1015–1027
.2. Cell culture and transfections
COS-7 cells were transfected with human PLD1, PLD2,-terminally deleted mutants of PLD1 �209 (210–1036)
nd PLD1 �329 (330–1036) expression vectors using Lipo-ectAMINE. Human U87 astrocytoma cells were purchasedrom the American Type Culture Collection and transfectedith vector or myc-tagged APP695 and stimulated with PMA.ll cells were maintained in Dulbecco’s modified Eagle’sedium (DMEM) supplemented with 10% fetal bovine
erum.
.3. Antibodies
The following monoclonal antibodies and polyclonal anti-era were used: mouse anti-4G8 (SENETEK, Marylandeights, MO); mouse anti-caveolin-1/3 (Transduction Lab-ratories, Lexington, KY); rabbit anti-APP (Sigma, Saintouis, MO); mouse anti-glial fibrillary acidic protein (GFAP)
Chemicon, Temecula, CA) and rabbit anti-GFAP (Dako,openhagen, Denmark). Rabbit polyclonal antibody that rec-gnizes PLD1 was generated as described previously [23].on-immune serum and secondary antibodies used in this
tudy were obtained from Jackson ImmunoResarch Labora-ories (West Grove, PA).
.4. Preparation of total membrane andaveolin-enriched membrane (CEM) fractions fromontrol and Alzheimer’s brains
Total membrane fractions were obtained by homogeniz-ng brains in TBS (10 mM Tris, pH 7.4, 150 mM NaCl) in theresence of protease inhibitors (10 �g/ml leupeptin, 2 �g/mlprotinin, 1 mM phenylmethylsulfonyl fluoride (PMSF)),emoval of nuclei, and centrifugation at 100,000 × g for 1 h.EM fractions were prepared as described previously [10]ith some modification. In brief, brains were homogenizedith 100 strokes of Dounce homogenizer in MES-buffered
aline (MBS, 25 mM MES, pH 6.5, 150 mM NaCl). Theomogenate was adjusted to 40% sucrose by 1:1 additionf 80% sucrose prepared in MBS buffer and placed in theottom of a tube. Four-milliliters of 30% and then 4 ml of
% sucrose in MBS buffer were layered on top, and the gra-ient was centrifuged at 39,000 rpm for 18 h in an SW41otor (Beckman Instruments). One-milliliter fractions wereollected from the top, yielding a total of 12 fractions.ns
l (h) Brain weight (g) Braak stage of AD brains [1]
1240 None1200 None1400 None1150 III/IV1090 III/IV1210 III/IV

gy of Ag
2
pFp1Pf2t(aw[w(ttfop
2
t1prwtwacabl(N4wpebaboe
2
Ppi
gelbpPpfa
2
ttta0bba(srpspw
t11cl(Rsam4T(pcttCpasc
J.-K. Jin et al. / Neurobiolo
.5. Measurement of PLD activity
PLD activity was measured by the formation of phos-hatidylbutanol (PtdBut) as described by Brown et al. [2].or in vitro assay, the total membrane fractions were resus-ended in homogenization buffer (300 mM HEPES, pH 7.2,8 mM EGTA, 18 mM CaCl2, 18 mM MgCl2, 480 mM KCl).LD1 activity was measured by the formation of [3H] PtdButrom [3H-palmitoyl] PC in the presence of 100 �M GTP�S,�M ARF3 and 1% butanol. Phospholipid vesicles con-
aining PE/phosphatidylinositol 4,5-bisphosphate (PIP2)/PC16:1.4:1 v/v/v) were used as substrate. For in vivo PLD assay,fter 20 h transfection with LipofectAMINE, cells in six-ell plates were serum–starved in the presence of 1 �Ci/ml
3H]myristic acid. After overnight starvation, the cells wereashed three times with 5 ml of phosphate-buffered saline
PBS) and preequilibrated in serum-free DMEM for 1 h. Forhe final 10 min of preincubation, 0.3% butanol was added. Athe end of the preincubation, cells were treated with agonistsor the indicated times. The extraction and characterizationf lipids by thin-layer chromatography were performed asreviously described [23].
.6. Immunoprecipitation and Western blot analysis
Isolated fractions were lysed with radio immunoprecipita-ion assay buffer (20 mM HEPES, pH 7.2, 1% Triton X-100,% deoxycholate, 0.1% SDS, 150 mM NaCl, 10 �g/ml leu-eptin, 10 �g/ml aprotinin, 1 mM phenylmethylsulfonyl fluo-ide) by homogenization. For immunoprecipitation, the lysateas then precleared for 30 min with preimmune IgG and pro-
ein A sepharose at 4 ◦C with rocking. Protein concentrationsere determined using Bradford method with bovine serum
lbumin as a standard [26]. Equal protein aliquots of pre-leared lysates (1–20 mg) were incubated with the indicatedntibodies and 40 �l of a 1:1 slurry of protein A sepharoseeads for 4 h at 4 ◦C. The immune complexes were col-ected by centrifugation and washed five times with a buffer20 mM Tris, pH 7.5, 1 mM EDTA, 1 mM EGTA, 150 mMaCl, 2 mM Na3VO4, 10% glycerol and 1% Nonidet P-0) and resuspended in sample buffer. Immune complexesere boiled in SDS-sample buffer. For Western blotting,rotein samples were analyzed by SDS-polyacrylamide gellectrophoresis and were transferred to a nitrocellulose mem-rane. The blots were then blocked with 5% non-fat milknd incubated with appropriate primary antibodies followedy incubation with horseradish peroxidase-conjugated sec-ndary antibody. Immunoreactive bands were detected usingnhanced chemiluminescence.
.7. In vitro binding assay
Using human U87 astrocytoma cells transfected withLD1 and PLD2 and brain tissues from AD patients, GSTull-down assay was performed. Clarified lysates (1 mg) werencubated with 2 �g of GST-fusion proteins immobilized on
2
d
ing 28 (2007) 1015–1027 1017
lutathione-sepharose beads in a final volume of 500 �l of thextraction buffer for 1 h at 4 ◦C. Protein complexes were col-ected by centrifugation and washed four times with washinguffer (1% Triton X-100, 150 mM NaCl, 20 mM Tris–HCl,H 8.0, 20 mM NaF, 200 �M sodium orthovanadate, 1 mMMSF, 10 �g/ml leupeptin, 10 �g/ml aprotinin). Associatedrotein complexes were resolved by SDS-PAGE and trans-erred to a nitrocellulose membrane, followed by immunoblotnalysis.
.8. Immunohistochemistry
Immunohistochemical procedures were carried out usinghe ABC kit (Vector, Burlingame, CA) by a modifica-ion of the avidin–biotin-peroxidase method. Briefly, sec-ions (6 �m) of the brain were deparaffinized with xylenend hydrated with graded ethanol, and then treated with.3% hydrogen peroxide in methyl alcohol for 20 min tolock endogenous peroxidase. After three washes in PBSuffer, the sections were exposed to normal donkey serum,nd then incubated with polyclonal antibody for PLD1diluted in 1:200) overnight at 4 ◦C. After washing, theections were sequentially treated with biotinylated anti-abbit immunoglobulin and avidin–biotin-peroxidase com-lex, developed with diaminobenzidine–hydrogen peroxideolution (0.003% 3,3-diaminobenzidine and 0.03% hydrogeneroxide in 0.05 M Tris-buffer), and finally counterstainedith hematoxylin.For double staining, the sections were incubated in
he following order: 10% normal donkey serum/PBS forh; monoclonal/polyclonal anti-PLD1 antibody (diluted in:100); anti-APP (diluted in 1:1000) or monoclonal anti-aveolin-3 antibody (diluted in 1:200) overnight at 4 ◦C;issamine rhodamine (LRSC)/fluorescein isothiocyanateFITC)-conjugated donkey anti-mouse/rabbit IgG for 1 h atT and washed, and then blocked in 10% normal rabbit/goaterum for 1 h at RT; incubated with monoclonal/polyclonalnti-GFAP (diluted in 1:200); anti-APP (diluted in 1:1000) oronoclonal anti-caveolin-3 antibody (diluted in 1:200); anti-
G8 (diluted in 1:200) overnight at 4 ◦C and then washed.he final step was incubation with fluorescein isothiocyanate
FITC)/lissamine rhodamine (LRSC)-conjugated affinityurified rabbit/goat anti-mouse IgG. After immunofluores-ence, to reduce or eliminate lipofuscin autofluorescence,he sections were washed in PBS (three times for 1 h) at RT,hen dipped briefly in distilled H2O, and treated with 10 mMuSO4 in ammonium acetate buffer (50 mM CH3COONH4,H 5.0) for 20 min, dipped briefly again in distilled H2O,nd finally returned to PBS [30]. The immunofluorescence-tained specimens were examined under a LSM 510 laseronfocal microscope (Zeiss, Oberkochen, Germany).
.9. Statistical analysis
Quantitative results are expressed as mean ± standardeviation (S.D.). Comparisons of expression levels and enzy-

1 gy of Ag
mh
3
3b
asecdsis
tafch
pwHpacit
FrTcb
018 J.-K. Jin et al. / Neurobiolo
atic activity of PLD1 in control and Alzheimer’s brainomogenates were made by Student’s t-test.
. Results
.1. PLD1 is up-regulated in reactive astrocytes of therains of AD patients
To investigate the possibility that PLD expression isltered in AD brain tissues, we immunostained AD brainections from patients using anti-PLD antibody. We observednhanced PLD immunoreactivity in the brains of AD patientsompared to that of elderly controls (Fig. 1A and B). Barely
etectable levels were found in normal human hippocampusection (Fig. 1A). We detected strong PLD immunoreactiv-ty in the hippocampal sections from AD patients via DABtaining (Fig. 1B). The cells that stained positive for PLD hadpsai
ig. 1. PLD1 is up-regulated in the reactive astrocytes associated with senile plaqeactive astrocytes (B) in AD brains, but not in controls (A). Immunofluorescent co-he sections were incubated with antibodies to PLD1 (C) and GFAP (D), and were exells were also positive for GFAP (E). Confocal image of double immunostaining oy a senile plaque. The merged image is shown (H). Scale bars, 10 �m. Data are re
ing 28 (2007) 1015–1027
he morphological and anatomical characteristics of reactivestrocytes, because they displayed spikes on their cell sur-aces. Double immunostaining revealed that PLD-positiveells were indeed activated astrocytes, identified by theireavy GFAP labeling (Fig. 1C–E).
A �-amyloid antibody was next used to detect amyloidlaques (Fig. 1G) and PLD1 was found to be co-localizedith A� in reactive astrocyte, but not in plaques (Fig. 1F and). Essentially identical results were obtained in three inde-endent AD brain tissue sections. Double labeling of PLD1nd �-amyloid peptide revealed that PLD1-positive astro-ytes were surrounded by senile plaques (Fig. 1H). PLD1mmunoreactivity in astrocytes was eliminated when the sec-ions were incubated with anti-PLD1 antibody that had been
reabsorbed with an excess amount of the antigen (data nothown). These results suggest that the reactive astrocytesssociated with senile plaques specifically overexpress PLD1n the brains of AD patients.ue of the brains of AD patients. PLD1 immunoreactivity was observed inlocalization of PLD1 with GFAP in hippocampus sections from AD brains.amined using confocal laser scanning microscopy. Note that PLD1-positivef PLD1 (F) and A� (G) showed that PLD1-positive cells were surrounded
presentative of three experiments.

gy of Ag
3i
bsaGPPfcastsudpPsbiP
FbfRlbWtPl
3fA
pdaraeccsacabrains compared with that of age-matched control subjects. Inaddition, caveolin-3 was not detected in caveolae membranesfrom normal human brain tissues, but was up-regulated incaveolae membranes from AD brain tissues. These results are
J.-K. Jin et al. / Neurobiolo
.2. Expression and enzymatic activity of PLD1 arencreased in the brains of AD patients
To investigate whether PLD activity is altered in therains of AD patients, PLD activity was measured usingubstrate vesicles containing PIP2 in the presence of PEnd PC. The assays also contained GTP�S and the small
protein ARF, as this greatly enhances the activity ofLD. As shown in Fig. 2A, the activity of ARF-dependentLD was significantly increased in the particulate fractionsrom the brains of AD patients, compared with that ofontrol brains. To examine whether the increase in PLDctivity is a result of changes in the PLD protein expres-ion level, we analyzed the level of PLD protein from con-rol and AD patients’ brains. Western blot analysis demon-trated that the level of PLD1 expression was significantlyp-regulated in AD brains (Fig. 2B), but PLD2 was notetected in either control or AD brain tissues. This expressionattern was correlated with the activity of ARF-dependentLD. It is possible that human brains may express a verymall amount of PLD2, and so PLD2 was not detectabley the anti-PLD antibody, indicating that the PLD activ-
ty shown in these brain tissues may be mainly due toLD1.ig. 2. The expression and activity of PLD1 are up-regulated in Alzheimer’srains. (A) The membrane fractions of brains were prepared and assayedor PLD activity in the presence of 100 �M GTP�S and 2 mM ARF3.esults show means ± S.D. of three independent experiments. (B) The
evel of PLD1 expression is up-regulated in the AD brains. The mem-rane fractions were prepared as described Section 2 and analyzed byestern blot using anti-PLD1 or actin antibody. A: AD brains; C: con-
rol brains, data are representative of three experiments. The levels ofLD-1 were determined by densitometer analysis. *p < 0.05 vs. control
evels.
Fa(famTr
ing 28 (2007) 1015–1027 1019
.3. Expression of PLD1 is increased in caveolarraction of AD brain and forms a physical complex withPP and caveolin-3
There is increasing evidence that caveolae may have aivotal role in the initiation and progression of Alzheimer’sisease. Caveolin-1 and 3 have been shown to physicallyssociate with APP [13,25]. Caveolin-3 is known to be up-egulated in reactive astrocyte in brain tissue sections fromuthentic AD patients [25]. We next examined the hypoth-sis that APP, PLD1 and caveolin are co-enriched withinaveolae membranes in normal and AD brains. We purifiedaveolae microdomains using an established detergent-freeucrose density gradient approach [10]. As shown in Fig. 3,ntibody specific for caveolin-1 as an internal markers foraveolar fractions recognized caveolin-1 in the CEM fractionnd the expression level of caveolin-1 was increased in AD
ig. 3. PLD1, APP and caveolin are co-enriched within caveolae membranesnd up-regulated in AD brain. CEM fractions from control (A) and ADB) brains were prepared as described in Section 2. Equal sucrose gradientraction volume (30 �l) was analyzed by Western blot using the indicatedntibody. Antibodies specific for caveolin-1 or caveolin-3 as an internalarkers for caveolar fractions recognized caveolin-1/3 in the CEM fraction.hree experiments were performed with three different brains, and similar
esults were obtained in each experiment.

1 gy of Ag
c1dbaP
lb
Fin(
020 J.-K. Jin et al. / Neurobiolo
onsistent with recent reports that protein level of caveolin-is increased in AD brain [9], and caveolin-3 is barely
etected in normal human brain, but strongly detected in ADrain [25]. The immunoreactivity of PLD1 and APP werelso recovered in the CEM fraction (Fig. 3), suggesting thatLD1 and APP are localized within caveolin-positive caveo-
cwil
ig. 4. PLD1 physically interacts with APP and caveolin-3 in caveolae membrane frammunoprecipitated with normal IgG and anti-APP, and immunoblotted with anti-Pormal IgG, anti-PLD1, anti-APP and anti-caveolin-3 antibodies. The immunoprecBlot) with anti-PLD1, anti-APP or anti-caveolin-3 antibody. Data are representativ
ing 28 (2007) 1015–1027
ae membrane domains. PLD1 is mainly detected in CEM inoth normal and AD brain. Interestingly, PLD1 was signifi-
antly increased in CEM fractions from AD brains comparedith that of control brains. A relatively small part of APPs detected in the lipid raft fractions and neither expressionevels of APP in normal brain or those in AD brain change
ction of AD. (A) Total lysates from normal brain (C) and AD brain (A) wereLD1 antibody. (B) The CEM fractions were immunoprecipitated (IP) withipitated proteins were subjected to 8–15% SDS-PAGE and immunoblottede results from three experiments.

J.-K. Jin et al. / Neurobiology of Aging 28 (2007) 1015–1027 1021
Fig. 5. PLD1 is co-localized with APP and caveolin-3 in reactive astrocytes from AD brain tissue. The sections of hippocampus from AD brains were incubatedwith antibodies to PLD1, APP and caveolin-3. Confocal image of double immunostaining of PLD1 and APP showed that PLD1-positive cells (A: green) wereAPP-positive cells (B: red). The merged image is shown in (C). Confocal image of double immunostaining of PLD1 and caveolin-3 showed that PLD1-positivecells (D: green) were in reactive astrocyte expressing caveolin-3 (E: red). The merged image is shown in (F). Confocal image of double immunostaining of APPand caveolin-3 showed that APP-positive cells (G: green) were caveolin-3-positive cells (H: red). The merged image is shown in I. The sections were incubatedwith antibodies to caveolin-3 (J), APP (M) and GFAP (K and N), and were examined using confocal laser scanning microscopy. Note that both caveolin-3 andAPP-positive cells were also positive for GFAP (L and O). Scale bars: 10 �m. Data are representative of three experiments.

1 gy of Ag
scAwtslstBtpatpIs(ApbwstTenTA
3r
Aa3waoattCtDA(3t
3i
FAPar
022 J.-K. Jin et al. / Neurobiolo
o much. We examined the expression of actin as an internalontrol whose level is equal in both control and AD brains.s shown in Fig. 3, the amount of actin in control brainas similar with that of AD brain. And then, we examined
he interaction of PLD1 with APP in the normal brain. Ashown in Fig. 4A, PLD1 was associated with APP in bothysates from normal brain and AD brain. As shown in Fig. 3A,ince caveolin-3 was not detected in normal brain, it seemshat PLD does not interact with caveolin-3 in normal brain.ecause caveolin provides a physical means for the associa-
ion of APP or PLD with caveolae, we next investigated theossibility that PLD1 forms a physical complex with APPnd caveolin-3 in AD brain tissues. We prepared CEM frac-ions from AD brain tissue and co-immunoprecipitation waserformed using anti-PLD1 and anti-caveolin-3 antibodies.mmunoblot analysis of these immunoprecipitates demon-trated that PLD1 forms a physical complex with caveolin-3Fig. 4). Caveolin-3 is also known to associate with APP [25].s shown in Fig. 4, caveolin-3 was detected after immuno-recipitation of APP in caveolae-enriched fractions from ADrain tissue. In addition, we also examined whether PLD1as associated with APP. CEM fractions from AD brain tis-
ues were immunoprecipitated with anti-PLD antibody andhe precipitates were probed with anti-APP antibody (Fig. 4).he presence of APP in the PLD immune complex was appar-
nt. Significantly, immunoprecipitation with normal IgG didot co-immunoprecipitate either PLD1, APP or caveolin-3.hese data suggest that PLD1 forms a physical complex withPP and caveolin-3 in CEM fractions from AD brain tissues.bCaF
ig. 6. �-Amyloid region of APP is involved in the interaction with PLD1. (A) FoPP and GST-A� were used in in vitro binding experiments as described in SectionLD1 from AD brain lysates. (B) The lysates of U87 astroglioma transfected wind immunoblotted with antibody to PLD. The amount of the GST fusion proteinepresentative of three experiments.
ing 28 (2007) 1015–1027
.4. PLD1, APP and caveolin-3 are co-localized ineactive astrocytes from AD brain tissue
We tried to further check whether PLD1 co-localized withPP and caveolin-3. Astrogliosis is known to be associ-
ted with increased expression of PLD1, APP and caveolin-in reactive astrocytes. Therefore, AD brain sections
ere double-immunolabeled using anti-PLD1, anti-APP, andnti-caveolin-3 antibodies to identify the co-localizationf APP and caveolin-3 with PLD1 in the same reactivestrocytes. Examination by confocal microscopy revealedhat PLD1-immunoreactive astrocytes in hippocampus sec-ion co-localized with APP and caveolin-3 (Fig. 5A–F).aveolin-3 immunoreactivity was also detected in reac-
ive astrocytes showing APP immunoreactivity (Fig. 5G–I).ouble immunostaining demonstrated that caveolin-3 andPP-positive cells were indeed GFAP-positive astrocytes
Fig. 5J–O). These results and PLD1, APP and caveolin-are co-localized in reactive astrocytes from AD brain
issues.
.5. β-Amyloid region of APP is involved in thenteraction with PLD1
To map the region on the APP protein that is responsi-
le for the interaction with PLD1, we prepared GST-fused-terminal 100 amino acids (C100) of APP and GST-A�,nd used them in vitro binding experiments. As shown inig. 6A, the GST-C100 and GST-A� precipitated PLD1 fromr GST pull down assay, GST-fused C-terminal 100 amino acids (C100) of2. Both GST-C100 and GST-A� fusion proteins but not GST precipitated
th PLD1 or 2 were incubated in 2 �g of GST or GST-A� fusion proteinss was identified by immunoblotting using an anti-GST antibody. Data are

gy of Ag
AaUTaccra
3P
Ptt
oauwtaCiPaPpp
F4laebt3tw
J.-K. Jin et al. / Neurobiolo
D brain lysates. Furthermore, to examine whether A� inter-cts with PLD1 and PLD2, we used the lysates from human87 MG astroglioma cells overexpressing PLD1 and PLD2.he physical interaction of A� with PLD1 and PLD2 waslso observed in astrocytoma cells (Fig. 6B). Importantly,ontrol precipitates with GST-glutathione-sepharose did noto-precipitate PLD. Taken together, these results suggest thategulation of PLD by APP might occur through in vivo inter-ction.
.6. The N-terminal pleckstrin homology domain ofLD is required for its interaction with APP
We examined further the physical interaction betweenLD isozymes and APP using a heterologous expression sys-
em, i.e., transient transfection of COS-7 cells. We transientlyransfected the cDNA encoding myc-tagged APP and PLD1
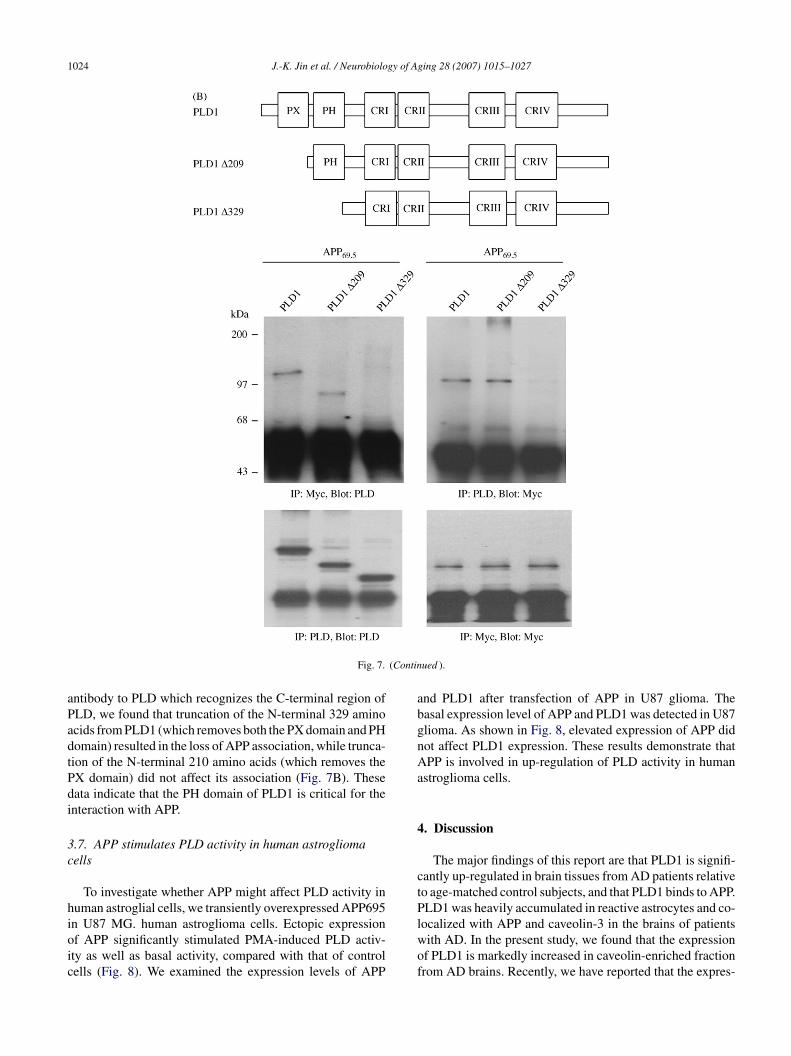
ft(
ig. 7. The N-terminal pleckstrin homology domain of PLD is required for its i0 h with PLD1 or PLD2 plus myc-tagged APP695 expression vectors using Lipofysates were immunoprecipitated with anti-PLD or anti-myc antibodies and thenti-PLD antibodies, respectively. Expression of PLDs and APP was determined byxperiments. (B) Upper panel: schematic representation of PLD1. The boxes indiceen proposed or demonstrated in Ref. [33]. PX: phox domain; PH: pleckstrin homransiently co-transfected with plasmids encoding PLD1 and APP, or N-terminal 229-amino acid-truncated PLD2 �329 (330–1036) and APP. Cell lysates were subhe amount of co-immunoprecipitated APP or PLD was determined by immunobloas determined using anti-PLD or anti-myc antibodies.
ing 28 (2007) 1015–1027 1023
r PLD2 into COS-7 cells. Next, cell lysates were prepared,nd PLD isozymes or APP were co-immunoprecipitatedsing antibodies to PLD and APP. These immunoprecipitatesere then subjected to immunoblot analysis with antibodies
o APP or PLD. As shown in Fig. 7A, PLD1 and PLD2 formphysical complex with APP during transient expression inOS-7 cells. PLD contains a number of functional domains,
ncluding conserved catalytic regions (I, II, III and IV), aIP2-binding site, a pleckstrin homology (PH) domain andn N-terminal phox (PX) domain (Fig. 7B). The N-terminalX domain has been proposed to mediate a wide variety ofrotein–protein interactions as well as the binding of phos-holipids [8,12,39].
Next, to determine the domain(s) of PLD1 requiredor APP695 interaction, cells were co-transfected with N-erminally deleted mutant construct of PLD1, namely �209210–1036) or �329 (330–1036), plus APP695. Using an
nteraction with APP. (A) COS-7 cells were transiently co-transfected forectAMINE (Invitrogen) according to the manufacturer’s instructions. Cellimmune complexes were subjected to immunoblotting using anti-myc orimmunoprecipitation and immunoblotting. Data are representative of threeate highly conserved sequences of PLD, and their possible functions haveology-like domain; CR: conserved region. Lower panel: COS-7 cells were09-amino acid-truncated PLD2 �209 (210–1036) and APP, or N-terminaljected to immunoprecipitation using anti-PLD or anti-myc antibodies, andtting with anti-myc or anti-PLD antibodies. Expression of PLD1 and APP
1024 J.-K. Jin et al. / Neurobiology of Aging 28 (2007) 1015–1027
(Contin
aPadtPdi
3c
hioic
abgnAa
4
ctP
Fig. 7.
ntibody to PLD which recognizes the C-terminal region ofLD, we found that truncation of the N-terminal 329 aminocids from PLD1 (which removes both the PX domain and PHomain) resulted in the loss of APP association, while trunca-ion of the N-terminal 210 amino acids (which removes theX domain) did not affect its association (Fig. 7B). Theseata indicate that the PH domain of PLD1 is critical for thenteraction with APP.
.7. APP stimulates PLD activity in human astrogliomaells
To investigate whether APP might affect PLD activity inuman astroglial cells, we transiently overexpressed APP695
n U87 MG. human astroglioma cells. Ectopic expressionf APP significantly stimulated PMA-induced PLD activ-ty as well as basal activity, compared with that of controlells (Fig. 8). We examined the expression levels of APPlwof
ued ).
nd PLD1 after transfection of APP in U87 glioma. Theasal expression level of APP and PLD1 was detected in U87lioma. As shown in Fig. 8, elevated expression of APP didot affect PLD1 expression. These results demonstrate thatPP is involved in up-regulation of PLD activity in human
stroglioma cells.
. Discussion
The major findings of this report are that PLD1 is signifi-antly up-regulated in brain tissues from AD patients relativeo age-matched control subjects, and that PLD1 binds to APP.LD1 was heavily accumulated in reactive astrocytes and co-
ocalized with APP and caveolin-3 in the brains of patientsith AD. In the present study, we found that the expressionf PLD1 is markedly increased in caveolin-enriched fractionrom AD brains. Recently, we have reported that the expres-

J.-K. Jin et al. / Neurobiology of Ag
Fig. 8. APP stimulates PLD activity in human astroglioma cells. U87astroglioma cells were transiently transfected with empty vector or APP andlabeled with [3H]myristic acid for 18 h. The cells were treated with or without50 nM PMA for 30 min in the presence of 0.3% 1-butanol. The expression ofAPP and PLD1 was detected by antibodies to APP and PLD1. Radioactivityi2
sgotmtffArrIaapPradoopp
ctPeAtc
cmIrmuhavtf[AlAt[woCmpAbosnTcel
raAwpppirlstr�gootAc
ncorporated into phosphatidylbutanol was measured as described in Section. Results show means ± S.D. of three independent experiments.
ion of PLD1 in activated astrocytes may be involved in theeneration of phospholipids products and the developmentf neurodegeneration in scrapie [14]. It was reported thathe altered metabolic lipid products can give rise to the for-
ation of oxygen free radicals and lipid peroxides and, inurn, induce tissue damage [28]. Thus, it can be speculatedrom these findings that altered lipids are one of the criticalactors in the pathogenic mechanisms of the aggregation of� seen in AD brains. In addition, we and our collaborators
eported the possibility that the activation of PLD1 is closelyelated to repair and remodeling after an ischemic injury [20].ncreased PLD expression in astrocytes was also seen aftercompression injury of rat spinal cord [15], cryoinjury inrat cortex [18] and kainate-induced seizures in a rat hip-
ocampus [19]. Taken together, it appears that increases ofLD activities during neurodegeneration may be due to up-egulation of astroglial PLD during inflammatory processesnd glial proliferation (astrogliosis). In view of these contra-ictory findings, it is currently unclear whether the activationf PLD1 leading to PA formation is involved in protectiver damaging mechanisms in AD. Further study needs to beerformed to clarify the specific role of PLD in the patho-hysiology of AD.
Several studies have shown that APP and PLD are asso-iated with lipid rafts or caveolae [22,25]. It is knownhat APP interacts with caveolin [22], and caveolin bindsLD [10,25]. Our finding demonstrates that PLD1 is highly
nriched in caveolae and forms physical complexes withPP and caveolin-3. Our findings also provide evidencehat PLD is up-regulated within caveolin-3-positive astro-ytes and APP-positive astrocytes. Caveolin as a principal
paci
ing 28 (2007) 1015–1027 1025
omponent of caveolae membrane is a cholesterol-bindingembrane protein involved in cellular cholesterol transport.
ncreasing evidence suggests that cholesterol plays a centralole in the pathophysiology of AD. Although the protectiveechanism of cholesterol-lowering drugs remains largely
nknown, it is postulated that alterations in brain cholesterolomeostasis could explain both the reduced risk and alter-tion in �-amyloid metabolism observed in AD [7,34]. Thisiew is consistent with a study showing increased choles-erol and caveolin in the caveolae-enriched fractions isolatedrom AD patient when compared to age-matched controls5]. Moreover, recent observations connecting caveolin toPP metabolism via cholesterol regulation serve to high-
ight the importance of this finding in the pathophysiology ofD. It is well known that aging also leads to distinct varia-
ions in the properties and composition of plasma membrane37,38]. Recently, it has been reported that PLD activitiesere increased in the mitochondrial-synaptosomal fractionf rat cerebellum and cerebral cortex during aging [27,29].aveolin expression is also increased in the brain of agedice as compared to young animals [6,38]. Changes in phos-
holipids or cholesterol homeostasis associated with aging orD may alter the structure and functional integrity of mem-rane microdomains. The up-regulation of PLD and caveolinbserved in the brains of the aged and in AD patients’ brainsupports this assumption. The expression of PLD1 was sig-ificantly enhanced in the reactive astrocytes of AD brains.he same phenomena were observed in the reactive astro-ytes of scrapie-infected brains [14]. Interestingly, enhancedxpression of PLD1 is not seen in activated astrocytes fol-owing LPS stimulation [14].
From this view, PLD1 may contribute to the glioticesponse in AD and scrapie. PLD is co-localized with APPnd A� in reactive astrocytes, and associated with APP.� or C100-terminal amino acids (C100) of APP interactith PLD and APP binds to a region of PLD1 containingleckstrin homology (PH), which is known to be involved inrotein–protein interactions as well as the binding of phos-holipids [39]. Although the molecular mechanisms involvedn PLD activation by APP remain to be determined, theseesults suggest that APP may affect the membrane phospho-ipids’ composition and/or stimulate the formation of lipidecondary messengers via PLD activation. Recently, the rela-ionship between presenilin-1 and PLD1 in neuron has beeneported [3,4]. Presenilin-1, which is major component of-secretase, the activity that mediates proteolysis of APP toenerate A� presenilin-1 binds to PLD1, and overexpressionf PLD1 decreases �-secretase activity and down-regulationf PLD1 increases �-secretase activity. Moreover, it is knownhat caveolin-3 form a physical complex with presenilin-1 andPP, and elevated expression of caveolin-3 in reactive astro-
ytes increase beta-secretase-mediated processing of APP
rocessing [25]. In our study, PLD1 associates with APPnd caveolin-3. Taken together, it PLD1 might form multipleomplexes with APP, caveolin-3 and presenilin-1. Therefore,t must be considered that the association of PLD1 with APP
1 gy of Ag
am
bAioss
alatcg
A
1KnfaHopIYwp
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
026 J.-K. Jin et al. / Neurobiolo
s well as interaction of PLD1 with presenilin or caveolin-3ight somehow affect the pathology of AD.PLD1 associates with APP in normal brain as well as AD
rain. Although the high level of PLD1 in the brain tissue ofD patients might provide a clue to the mechanism underly-
ng the pathophysiology of AD, the issue of how associationf PLD1 with APP affect AD brain might be explained andpeculated through future studies using experimental toolsuch as PLD1 knock-out mice and AD animal model.
To clarify how the association between PLD1 and APPffects the processing of APP will be examined in the fol-owing study including fine mapping and mutation of inter-ction site of PLD1 with APP. Further studies are requiredo fully understand the significance of the interaction ando-accumulation of PLD with caveolin and APP in the patho-enesis of AD.
cknowledgements
This study was supported by grant No. R01-2006-000-0521-0 (2006) from the Basic Research Program of theorea Science & Engineering Foundation and by the Biotech-ology Development Program (grant number 2005-00115)rom Ministry of Science and Technology (MOST), Koreand grant of the Korea Health 21 R&D project, Ministry ofealth and Welfare, Republic of Korea (A020007). A totalf six post-mortem brain specimens of Alzheimer’s diseaseatients and age-matched controls were obtained from theBR Brain Bank (Dr. Piotr B. Kozlowski, who is head of Nework IBR brain bank and co-author of this manuscript). So,e used post-mortem brain specimens of Alzheimer’s diseaseatients according to appropriate approval and procedure.
eferences
[1] Braak H, Braak E. Neuropathological stageing of Alzheimer-relatedchanges. Acta Neuropathol 1991;82:239–59.
[2] Brown HA, Gutowski S, Moomaw CR, Slaughter C, Sternweis PC.ADP-ribosylation factor, a small GTP-dependent regulatory protein,stimulates phospholipase D activity. Cell 1993;75:1137–44.
[3] Cai D, Netzer WJ, Zhong M, Lin Y, Du G, Frohman M, et al. Presenilin-1 uses phospholipase D1 as a negative regulator of b-amyloid formation.Proc Natl Acad Sci USA 2006;103:1941–6.
[4] Cai D, Zhong M, Netzer WJ, Shields D, Zheng H, Sisodia SS, et al.Phospholipase D1 corrects impaired betaAPP trafficking and neuriteoutgrowth in familial Alzheimer’s disease-linked presenilin-1 mutantneurons. Proc Natl Acad Sci USA 2006;103:1936–40.
[5] Dufour F, Zhao W, Ravindranath L, Alkon DL. Abnormal cholesterolprocessing in Alzheimer’s disease patient’s fibroblasts. Neurobiol Lipid2003;7:7.
[6] Eckert GP, Wood WG, Muller WE. Effects of aging and beta-amyloidon the properties of brain synaptic and mitochondrial membranes. JNeural Transm 2001;108:1051–64.
[7] Fassbender K, Simon M, Bergmann C, Stroick M, Lutjohann D, Keller
P. Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. ProcNatl Acad Sci USA 2001;98:5856–61.[8] Frohman MA, Morris AJ. Phospholipase D structure and regulation.Chem Phys Lipids 1999;98:127–40.
[
ing 28 (2007) 1015–1027
[9] Gaudreault SB, Dea D, Poirier J. Increased caveolin-1 expression inAlzheimer’s disease brain. Neurobiol Aging 2004;25:753–9.
10] Han JM, Kim Y, Lee JS, Lee CS, Lee BD, Ohba M, et al. Localizationof phospholipase D1 to caveolin-enriched membrane via palmitoyla-tion: implications for epidermal growth factor signaling. Mol Biol Cell2002;13:3976–88.
11] Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s dis-ease: progress and problems on the road to therapeutics. Science2002;297:353–6.
12] Houle MG, Bourgoin S. Regulation of phospholipase D by phos-phorylation-dependent mechanisms. Biochim Biophys Acta 1999;1439:135–49.
13] Ikezu T, Trapp BD, Song KS, Schlegel A, Lisanti MP, OkamotoT. Caveolae, plasma membrane microdomains for alpha-secretase-mediated processing of the amyloid precursor protein. J Biol Chem1998;273:10485–95.
14] Jin JK, Kim NH, Min DS, Kim JI, Choi JK, Jeong BH, et al. Increasedexpression of phospholipase D1 in the brains of scrapie-infected mice.J Neurochem 2005;92:452–61.
15] Jung K, Min DS, Sim KB, Ahn M, Kim H, Cheong J, et al. Upregulationof phospholipase D1 in the spinal cords of rats with clip compressioninjury. Neurosci Lett 2003;336:126–30.
16] Kanfer JN, Singh IN, Pettegrew JW, McCartney DG, Sorrentino G.Phospholipid metabolism in Alzheimer’s disease and in a humancholinergic cell. J Lipid Mediat Cell Signal 1996;14:361–3.
17] Kanfer JN, Sorrentino G, Sitar DS. Phospholipases as mediators ofamyloid beta peptide neurotoxicity: an early event contributing to neu-rodegeneration characteristic of Alzheimer’s disease. Neurosci Lett1998;257:93–6.
18] Kim MD, Min DS, Sim KB, Cho HJ, Shin T. Expression and potentialrole of phospholipase D1 in cryoinjured cerebral cortex of rats. HistolHistopathol 2004;19:1015–9.
19] Kim SY, Min DS, Choi JS, Choi YS, Park HJ, Sung KW, et al. Differ-ential expression of phospholipase D isozymes in the hippocampusfollowing kainic acid-induced seizures. J Neuropathol Exp Neurol2004;63:812–20.
20] Lee MY, Kim SY, Min DS, Choi YS, Sin SL, Chun MH, et al. Upregula-tion of phospholipase D in astrocytes in response to transient forebrainischemia. Glia 2000;30:311–7.
21] McDermott M, Wakelam MJ, Morris AJ, Phospholipase D. BiochemCell Biol 2004;82:225–53.
22] Meacci E, Donati C, Cencetti F, Romiti E, Farnararo M, Bruni P.Receptor-activated phospholipase D is present in caveolin-3-enrichedlight membranes of C2C12 myotubes. FEBS Lett 2000;473:10–4.
23] Min DS, Ahn BH, Rhie DJ, Yoon SH, Hahn SJ, Kim MS, et al. Expres-sion and regulation of phospholipase D during neuronal differentiationof PC12 cells. Neuropharmacology 2001;41:384–91.
24] Mudher A, Lovestone S. Alzheimer’s disease-do tauists and baptistsfinally shake hands? Trends Neurosci 2002;25:22–6.
25] Nishiyama K, Trapp BD, Ikezu T, Ransohoff RM, Tomita T, Iwatsubo T,et al. Caveolin-3 upregulation activates beta-secretase-mediated cleav-age of the amyloid precursor protein in Alzheimer’s disease. J Neurosci1999;19:6538–48.
26] O’Banion MK, Miller JC, Chang JW, Kaplan MD, Coleman PD.Interleukin-1 beta induces prostaglandin G/H synthase-2 (cyclooxy-genase-2) in primary murine astrocyte cultures. J Neurochem 1996;66:2532–40.
27] Pasquare SJ, Salvador GA, Giusto NM. Phospholipase D and phos-phatidate phosphohydrolase activities in rat cerebellum during aging.Lipids 2004;39:553–60.
28] Ryu SB, Wang X. Activation of phospholipase D and the possible mech-anism of activation in wound-induced lipid hydrolysis in castor bean
leaves. Biochim Biophys Acta 1996;1303:243–50.29] Salvador GA, Boschero ID, Pasquare SJ, Giusto NM. Phosphatidicacid and diacylglycerol generation is regulated by insulin in cere-bral cortex synaptosomes from adult and aged rats. J Neurosci Res2005;81:244–52.

gy of Ag
[
[
[
[
[
[
[
[
[position of cortical synaptosomes from different age groups of mice.
J.-K. Jin et al. / Neurobiolo
30] Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-likeautofluorescence in fluorescently labeled tissue. J Histochem Cytochem1999;47:719–30.
31] Singh IN, McCartney DG, Kanfer JN. Amyloid beta protein (25–35)stimulation of phospholipases A, C and D activities of LA-N-2 cells.FEBS Lett 1995;365:125–8.
32] Singh IN, Sorrentino G, Kanfer JN. Activation of LA-N-2 cellphospholipase D by amyloid beta protein (25–35). Neurochem Res1998;23:1225–32.
33] Sommer B. Alzheimer’s disease and the amyloid cascade hypothesis:
10 years on. Curr Opin Pharmacol 2002;2:87–92.34] Sparks DL. Intraneuronal beta-amyloid immunoreactivity in the CNS.Neurobiol Aging 1996;17:291–9.
35] Suh YH, Checler F. Amyloid precursor protein, presenilins, andalpha-synuclein: molecular pathogenesis and pharmacological appli-
[
ing 28 (2007) 1015–1027 1027
cations in Alzheimer’s disease. Pharmacol Rev 2002;54:469–525.
36] Vassar R, Citron M. Abeta-generating enzymes: recent advancesin beta- and gamma-secretase research. Neuron 2000;27:419–22.
37] Viani P, Cervato G, Fiorilli A, Cestaro B. Age-related differences insynaptosomal peroxidative damage and membrane properties. J Neu-rochem 1991;56:253–8.
38] Wood WG, Strong R, Williamson LS, Wise RW. Changes in lipid com-
Life Sci 1984;35:1947–52.39] Xu Y, Seet LF, Hanson B, Hong W. The Phox homology (PX)
domain, a new player in phosphoinositide signalling. Biochem J2001;360:513–30.




















