
Chatuphalatika, a Thai herbal formula, ameliorate obesity and ...

y 186 (2007) 94–103www.elsevier.com/locate/jneuroim
Journal of Neuroimmunolog
COX-2 inhibitors ameliorate experimental autoimmune encephalomyelitisthrough modulating IFN-γ and IL-10 production by
inhibiting T-bet expression☆
Jia Ni a, Ying-Yi Shu b, Yi-Na Zhu a, Yun-Feng Fu a, Wei Tang a, Xiang-Gen Zhong a, Hui Wang c,Yi-Fu Yang a, Jin Ren c, Ming-Wei Wang b,⁎, Jian-Ping Zuo a,d,⁎
a State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences,Shanghai 201203, People's Republic of China
b The National Center for Drug Screening, Shanghai Institute of Materia Medica, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences,Shanghai 201203, People's Republic of China
c Center for Drug Safety Evaluation and Research, Shanghai Institute of Materia Medica, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences,Shanghai 201203, People's Republic of China
d Laboratory of Immunology and Virology, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, People's Republic of China
Received 8 February 2007; received in revised form 6 March 2007; accepted 7 March 2007
Abstract
The COX-2 inhibitors Rofecoxib (Rof) and Lumiracoxib (Lum) were evaluated in experimental autoimmune encephalomyelitis (EAE), themodel of multiple sclerosis (MS). Administration of Rof and Lum significantly reduced the incidence and severity of EAE, which was associatedwith the inhibition of MOG 35-55 lymphocyte recall response, anti-MOG 35-55 T cell responses, and modulation of cytokines production. In vitroRof and Lum inhibited primary T cells proliferation and modulated cytokine production. These findings highlight the fact that Rof and Lum likelyprevents EAE by modulating Th1/Th2 response, and suggest its utility in the treatment of MS and other autoimmune diseases.© 2007 Elsevier B.V. All rights reserved.
Keywords: Rofecoxib; Lumiracoxib; Experimental autoimmune encephalomyelitis; T cell; Cytokine
1. Introduction
Multiple sclerosis (MS) is the most common central nervoussystem (CNS) demyelinating disease, affecting approximately1,000,000 individuals worldwide (Steinman, 2001). Experi-mental autoimmune encephalomyelitis (EAE) is a T cell-
☆ This study was supported in part by grants from the Ministry of Science andTechnology of China (2004CB518902) and Shanghai Science and TechnologyDevelopment Fund (05dz22914).⁎ Corresponding authors. Zuo is to be contacted at Laboratory of
Immunopharmacology, Shanghai Institute of Materia Medica, Chinese Acad-emy of Sciences, 555 Zuchongzhi Road, Zhangjiang Hi-Tech Park, Shanghai201203, People's Republic of China. Tel./fax: +86 21 50806701. Wang, TheNational Center for Drug Screening, Shanghai Institute of Materia Medica,Chinese Academy of Sciences, 189 Guoshoujing Road, Shanghai 201203,People's Republic of China. Tel.: +86 21 5080 0598; fax: +86 21 5080 0721.
E-mail addresses: [email protected] (M.-W. Wang),[email protected] (J.-P. Zuo).
0165-5728/$ - see front matter © 2007 Elsevier B.V. All rights reserved.doi:10.1016/j.jneuroim.2007.03.012
mediated autoimmune disease, and serves as an animal modelfor human MS (Zamvil and Steinman, 1990). Similaritiesbetween Experimental autoimmune encephalomyelitis (EAE)and MS are present on many levels. In both diseases, CD4+ andCD8+ T cells can be found in lesions, including evidence ofpopulations of clonally derived T cells, some reactive to myelinproteins. EAE and MS are characterized by damage to themyelin sheath and in both the animal models and in MS there isevidence for axonal degeneration (Steinman and Zamvil, 2005).
In EAE, imbalance in T cell activation contributes to thepathogenesis of autoimmune diseases evidenced by release of pro-inflammatory cytokines, IL-2 and IFN-γ, from Th1 cells (Chitnisand Khoury, 2003; Gor et al., 2003; Sredni-Kenigsbuch, 2002). Onthe other hand, it is known that anti-inflammatory cytokines such asIL-10 produced by Th2 cells are associated with remissions of thedisease (Kennedy et al., 1992). IL-10 is crucial in inflammationwith potent anti-inflammatory and immunosuppressive activities

95J. Ni et al. / Journal of Neuroimmunology 186 (2007) 94–103
(Anderson et al., 2004). Some reports have shown that IL-10 is akey regulatory cytokine in EAE, and IL-10-deficient mice arehighly susceptible to EAE (Bettelli et al., 1998; Segal et al., 1998;Samoilova et al., 1998). In addition, administration of IL-10 couldmitigate the severity of EAE (Nagelkerken et al., 1997; Rott et al.,1994; Slavin et al., 2001).
Previous works described in the literature suggest that T-betplays a critical role in the progression of EAE as it can promote Th1development and IFN-γ production (Szabo et al., 2000). In vitrosuppression of T-bet during myelin-specific T cell differentiationand in vivo administration of T-bet-specific anti-sense oligonucle-otide or siRNA inhibited EAE (Lovett-Racke et al., 2004). T-bet-deficient mice immunized with myelin oligodendrocyte glycopro-tein (MOG) are resistant to the development of EAE, with minimalinflammatory infiltrates in the CNS (Bettelli et al., 2004). Recentlystudies have shown that the treatment of COX-2 inhibitorsprevented EAE (Moon et al., 2004; Miyamoto et al., 2006;Muthian et al., 2006), but the underlying mechanisms of action arenot fully understood. In this paper, we demonstrate that COX-2inhibitors could attenuate CNS inflammation and demyelination inEAE by inhibiting lymphocyte proliferation and IL-2/IFN-γproduction. This process was accompanied by an increase in IL-10 production by auto-reactive Tcells resulted at least in part from asuppression of T-bet expression.
IL-17 is a pro-inflammatory cytokine that activates T cells andother immune cells to produce a variety of cytokines, chemokines,and cell adhesion molecules. (Komiyama et al., 2006). Recentreports illustrate that IL-17 is crucially involved in the cytokinenetwork as an effector cytokine in EAE. Strong MOG-specific IL-17 production could be detected both in the spleen representing theimmune periphery with the induction phase and the CNS as thetarget organ of the autoimmune T-cell response in acute EAE.Neutralization of IL-17 with a monoclonal antibody alsoameliorated the disease course. (Hofstetter et al., 2005). Thedevelopment of EAE was significantly suppressed in IL-17(−/−)mice, and adoptive transfer of IL-17(−/−) CD4+ Tcells inefficientlyinduced EAE in recipient mice (Komiyama et al., 2006).
In the present study, we immunized C57BL/6 mice withMOG 35-55 to induce to EAE, a T-cell-mediated experimentmodel of chronic and non-relapsing EAE, and examined theeffects of Rofecoxib (Rof) and Lumiracoxib (Lum) on EAE.Our results demonstrate that Rof and Lum significantly reducedthe incidence and severity of the disease, which was associatedwith direct inhibition of T cell proliferation, modulating Th1/Th2 response and suppressing IL-17 expression.
2. Materials and methods
2.1. Reagents
Rofecoxib (Rof) and Lumiracoxib (Lum) were provided bythe National Center for Drug Screening. Peptide MOG 35-55(MEVGWYRSPFSRVVHLYRNGK) was synthesized by San-gon Biological Engineering Technology and Service Co., Ltd.(Shanghai, China). The sequence was confirmed by amino acidanalysis and mass spectroscopy, and the purity was greater than95%. Complete Freund's adjuvant CFA and Mycobacterium
tuberculosis H37Ra were purchased from Difco (Detroit, MI,USA). Bordetella pertussis toxin (PTX) and 3, 3′, 5, 5′-tetramethylbenzidine were supplied by Sigma-Aldrich. (St.Louis, MO, USA). RPMI 1640 was bought from GIBCO/LifeTechnologies Inc. (Gaithersburg, MD, USA), and fetal calfserum (FCS) obtained from Hyclone Laboratories (Logan,Utah, USA). [3H] thymidine was provided by Shanghai Instituteof Applied Physics, Chinese Academy of Science (Shanghai,PR China). The ELISA kits for IL-2, INF-γ and IL-10 wereprocured from PharMingen (San Diego, CA, USA).
2.2. Induction, treatment, and clinical evaluation of EAE
Female C57BL/6 mice, aged between 6 and 8 weeks, werepurchased from Shanghai Experimental Animal Center, Chi-nese Academy of Sciences. The animals were housed in aspecific pathogen-free environment. Experiments were carriedout according to the National Institutes of Health Guide for Careand Use of Laboratory Animals, and were approved by theBioethics Committee of the Shanghai Institute of MateriaMedica. Murine active EAE model was produced as describedpreviously (Fu et al., in press). Briefly, C57BL/6 mice wereimmunized on day 0 by s.c. injection with 100 μl of an emulsionof MOG 35-55 peptide in CFA. Each mouse additionallyreceived PTX via i.p. injection on day 0 and day 3 post-immunization (p.i.). Rof or Lum (50 mg/kg) was dissolved inPBS containing 0.5% sodium CM-cellulose and daily admin-istered orally following the immunization and continuedthroughout the study (n=10 mice). To examine the therapeuticefficacy, Rof or Lum (50 mg/kg) was administered from days 14to 30 p.i. The dose of Rof or Lum was chosen on the basis ofpreliminary experiments. Control mice received orally an equalvolume of PBS containing 0.5% CM-cellulose (n=10).
2.3. Histopathological analysis
To assess the degree of CNS inflammation and demyelination,mice in vehicle, Rof or Lum treated groups were anaesthetized byi.p. injection of sodium pentobarbital (30mg/kg) on day 21 (at thepeak of the disease) and perfused with 20 ml cold PBS. Spinalcords were dissected and fixed in 10% formalin. Five-micrometerthick transverse sections were taken from cervical, upper thoracic,lower thoracic, and lumbar regions of the spinal cord (foursections per mouse). The sections were stained with H&E toexamine inflammation. Signs of inflammation in the anterior,posterior, and two lateral columns (four quadrants) of the sectionwere scored under microscope by the standard describedpreviously (Fu et al., in press).
2.4. Splenocyte response assay
The in vivo and in vitro effects of COX-2 inhibitors onneural antigen specific T cell proliferation were measured by[3H] thymidine incorporation assay. Spleens (n=10) wereremoved from MOG-immunized mice treated with vehicle orCOX-2 inhibitors on day 14 p.i. Splenocyte (5×105 cells/well)suspensions were prepared from various treatment groups as
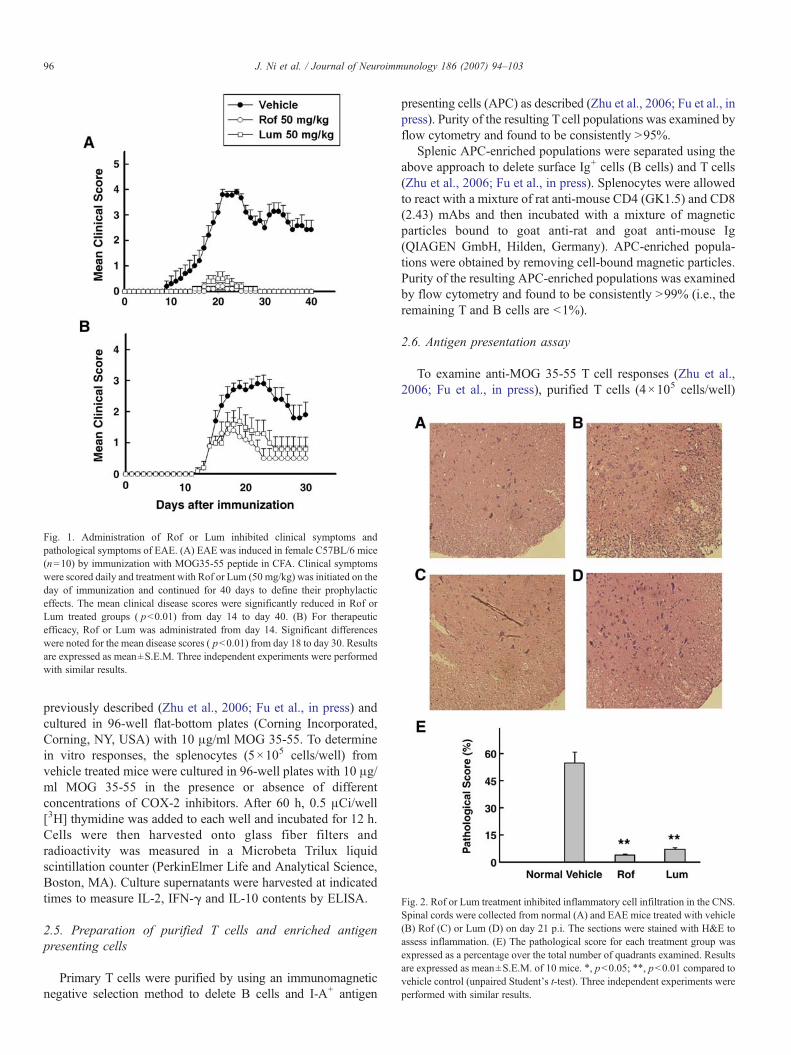
Fig. 1. Administration of Rof or Lum inhibited clinical symptoms andpathological symptoms of EAE. (A) EAE was induced in female C57BL/6 mice(n=10) by immunization with MOG35-55 peptide in CFA. Clinical symptomswere scored daily and treatment with Rof or Lum (50 mg/kg) was initiated on theday of immunization and continued for 40 days to define their prophylacticeffects. The mean clinical disease scores were significantly reduced in Rof orLum treated groups ( pb0.01) from day 14 to day 40. (B) For therapeuticefficacy, Rof or Lum was administrated from day 14. Significant differenceswere noted for the mean disease scores ( pb0.01) from day 18 to day 30. Resultsare expressed as mean±S.E.M. Three independent experiments were performedwith similar results.
Fig. 2. Rof or Lum treatment inhibited inflammatory cell infiltration in the CNS.Spinal cords were collected from normal (A) and EAE mice treated with vehicle(B) Rof (C) or Lum (D) on day 21 p.i. The sections were stained with H&E toassess inflammation. (E) The pathological score for each treatment group wasexpressed as a percentage over the total number of quadrants examined. Resultsare expressed as mean±S.E.M. of 10 mice. ⁎, pb0.05; ⁎⁎, pb0.01 compared tovehicle control (unpaired Student's t-test). Three independent experiments wereperformed with similar results.
96 J. Ni et al. / Journal of Neuroimmunology 186 (2007) 94–103
previously described (Zhu et al., 2006; Fu et al., in press) andcultured in 96-well flat-bottom plates (Corning Incorporated,Corning, NY, USA) with 10 μg/ml MOG 35-55. To determinein vitro responses, the splenocytes (5×105 cells/well) fromvehicle treated mice were cultured in 96-well plates with 10 μg/ml MOG 35-55 in the presence or absence of differentconcentrations of COX-2 inhibitors. After 60 h, 0.5 μCi/well[3H] thymidine was added to each well and incubated for 12 h.Cells were then harvested onto glass fiber filters andradioactivity was measured in a Microbeta Trilux liquidscintillation counter (PerkinElmer Life and Analytical Science,Boston, MA). Culture supernatants were harvested at indicatedtimes to measure IL-2, IFN-γ and IL-10 contents by ELISA.
2.5. Preparation of purified T cells and enriched antigenpresenting cells
Primary T cells were purified by using an immunomagneticnegative selection method to delete B cells and I-A+ antigen
presenting cells (APC) as described (Zhu et al., 2006; Fu et al., inpress). Purity of the resulting T cell populations was examined byflow cytometry and found to be consistently N95%.
Splenic APC-enriched populations were separated using theabove approach to delete surface Ig+ cells (B cells) and T cells(Zhu et al., 2006; Fu et al., in press). Splenocytes were allowedto react with a mixture of rat anti-mouse CD4 (GK1.5) and CD8(2.43) mAbs and then incubated with a mixture of magneticparticles bound to goat anti-rat and goat anti-mouse Ig(QIAGEN GmbH, Hilden, Germany). APC-enriched popula-tions were obtained by removing cell-bound magnetic particles.Purity of the resulting APC-enriched populations was examinedby flow cytometry and found to be consistently N99% (i.e., theremaining T and B cells are b1%).
2.6. Antigen presentation assay
To examine anti-MOG 35-55 T cell responses (Zhu et al.,2006; Fu et al., in press), purified T cells (4×105 cells/well)
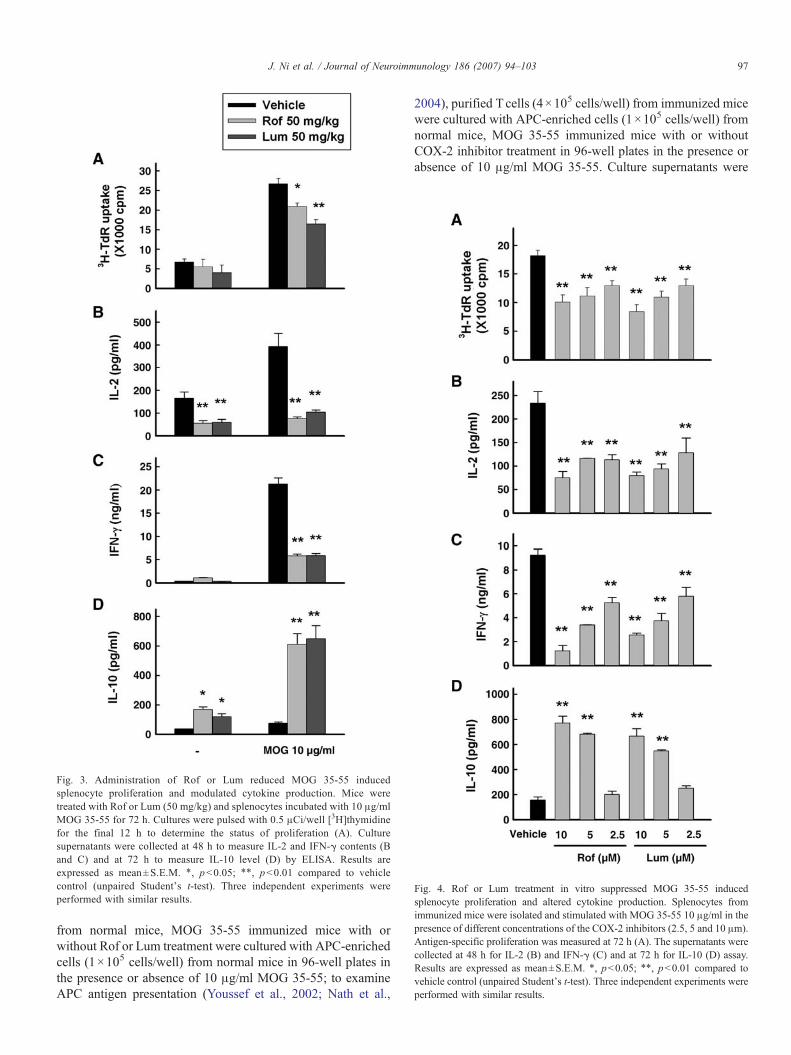
Fig. 3. Administration of Rof or Lum reduced MOG 35-55 inducedsplenocyte proliferation and modulated cytokine production. Mice weretreated with Rof or Lum (50 mg/kg) and splenocytes incubated with 10 μg/mlMOG 35-55 for 72 h. Cultures were pulsed with 0.5 μCi/well [3H]thymidinefor the final 12 h to determine the status of proliferation (A). Culturesupernatants were collected at 48 h to measure IL-2 and IFN-γ contents (Band C) and at 72 h to measure IL-10 level (D) by ELISA. Results areexpressed as mean±S.E.M. ⁎, pb0.05; ⁎⁎, pb0.01 compared to vehiclecontrol (unpaired Student's t-test). Three independent experiments wereperformed with similar results.
Fig. 4. Rof or Lum treatment in vitro suppressed MOG 35-55 inducedsplenocyte proliferation and altered cytokine production. Splenocytes fromimmunized mice were isolated and stimulated with MOG 35-55 10 μg/ml in thepresence of different concentrations of the COX-2 inhibitors (2.5, 5 and 10 μm).Antigen-specific proliferation was measured at 72 h (A). The supernatants werecollected at 48 h for IL-2 (B) and IFN-γ (C) and at 72 h for IL-10 (D) assay.Results are expressed as mean±S.E.M. ⁎, pb0.05; ⁎⁎, pb0.01 compared tovehicle control (unpaired Student's t-test). Three independent experiments wereperformed with similar results.
97J. Ni et al. / Journal of Neuroimmunology 186 (2007) 94–103
from normal mice, MOG 35-55 immunized mice with orwithout Rof or Lum treatment were cultured with APC-enrichedcells (1×105 cells/well) from normal mice in 96-well plates inthe presence or absence of 10 μg/ml MOG 35-55; to examineAPC antigen presentation (Youssef et al., 2002; Nath et al.,
2004), purified T cells (4×105 cells/well) from immunized micewere cultured with APC-enriched cells (1×105 cells/well) fromnormal mice, MOG 35-55 immunized mice with or withoutCOX-2 inhibitor treatment in 96-well plates in the presence orabsence of 10 μg/ml MOG 35-55. Culture supernatants were
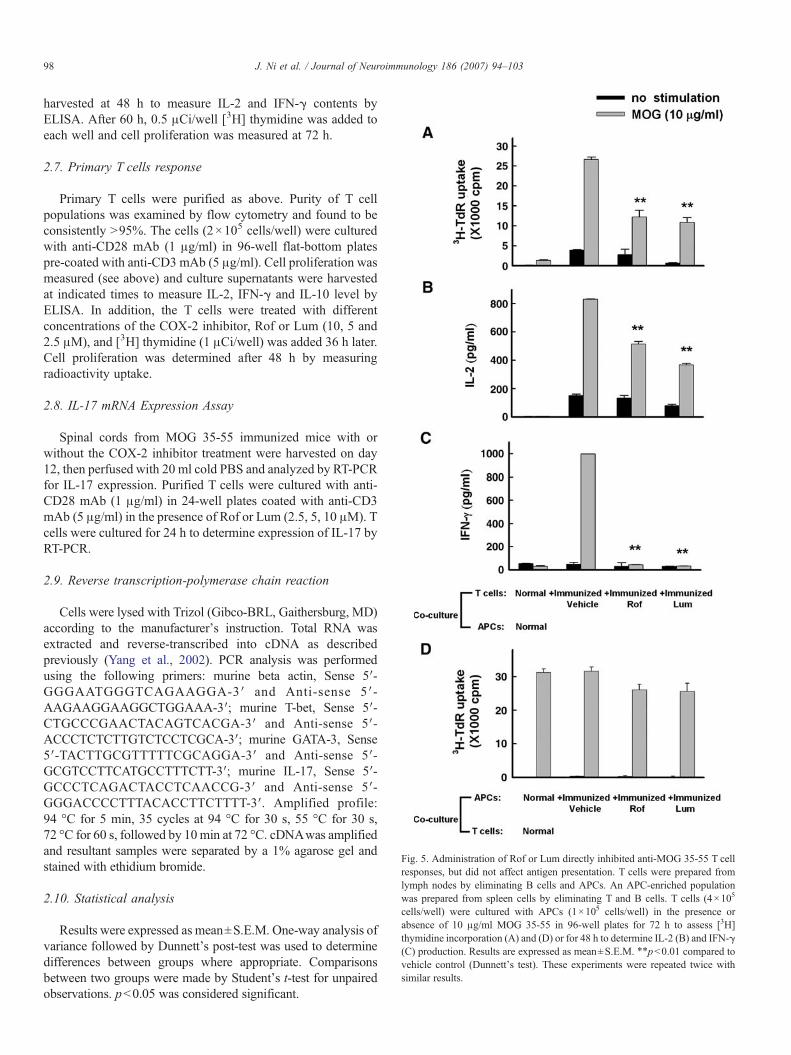
Fig. 5. Administration of Rof or Lum directly inhibited anti-MOG 35-55 T cellresponses, but did not affect antigen presentation. T cells were prepared fromlymph nodes by eliminating B cells and APCs. An APC-enriched populationwas prepared from spleen cells by eliminating T and B cells. T cells (4×105
cells/well) were cultured with APCs (1×105 cells/well) in the presence orabsence of 10 μg/ml MOG 35-55 in 96-well plates for 72 h to assess [3H]thymidine incorporation (A) and (D) or for 48 h to determine IL-2 (B) and IFN-γ(C) production. Results are expressed as mean±S.E.M. ⁎⁎pb0.01 compared tovehicle control (Dunnett's test). These experiments were repeated twice withsimilar results.
98 J. Ni et al. / Journal of Neuroimmunology 186 (2007) 94–103
harvested at 48 h to measure IL-2 and IFN-γ contents byELISA. After 60 h, 0.5 μCi/well [3H] thymidine was added toeach well and cell proliferation was measured at 72 h.
2.7. Primary T cells response
Primary T cells were purified as above. Purity of T cellpopulations was examined by flow cytometry and found to beconsistently N95%. The cells (2×105 cells/well) were culturedwith anti-CD28 mAb (1 μg/ml) in 96-well flat-bottom platespre-coated with anti-CD3 mAb (5 μg/ml). Cell proliferation wasmeasured (see above) and culture supernatants were harvestedat indicated times to measure IL-2, IFN-γ and IL-10 level byELISA. In addition, the T cells were treated with differentconcentrations of the COX-2 inhibitor, Rof or Lum (10, 5 and2.5 μM), and [3H] thymidine (1 μCi/well) was added 36 h later.Cell proliferation was determined after 48 h by measuringradioactivity uptake.
2.8. IL-17 mRNA Expression Assay
Spinal cords from MOG 35-55 immunized mice with orwithout the COX-2 inhibitor treatment were harvested on day12, then perfused with 20 ml cold PBS and analyzed by RT-PCRfor IL-17 expression. Purified T cells were cultured with anti-CD28 mAb (1 μg/ml) in 24-well plates coated with anti-CD3mAb (5 μg/ml) in the presence of Rof or Lum (2.5, 5, 10 μM). Tcells were cultured for 24 h to determine expression of IL-17 byRT-PCR.
2.9. Reverse transcription-polymerase chain reaction
Cells were lysed with Trizol (Gibco-BRL, Gaithersburg, MD)according to the manufacturer's instruction. Total RNA wasextracted and reverse-transcribed into cDNA as describedpreviously (Yang et al., 2002). PCR analysis was performedusing the following primers: murine beta actin, Sense 5′-GGGAATGGGTCAGAAGGA-3′ and Anti-sense 5′-AAGAAGGAAGGCTGGAAA-3′; murine T-bet, Sense 5′-CTGCCCGAACTACAGTCACGA-3′ and Anti-sense 5′-ACCCTCTCTTGTCTCCTCGCA-3′; murine GATA-3, Sense5′-TACTTGCGTTTTTCGCAGGA-3′ and Anti-sense 5′-GCGTCCTTCATGCCTTTCTT-3′; murine IL-17, Sense 5′-GCCCTCAGACTACCTCAACCG-3′ and Anti-sense 5′-GGGACCCCTTTACACCTTCTTTT-3′. Amplified profile:94 °C for 5 min, 35 cycles at 94 °C for 30 s, 55 °C for 30 s,72 °C for 60 s, followed by 10min at 72 °C. cDNAwas amplifiedand resultant samples were separated by a 1% agarose gel andstained with ethidium bromide.
2.10. Statistical analysis
Results were expressed as mean±S.E.M. One-way analysis ofvariance followed by Dunnett's post-test was used to determinedifferences between groups where appropriate. Comparisonsbetween two groups were made by Student's t-test for unpairedobservations. pb0.05 was considered significant.
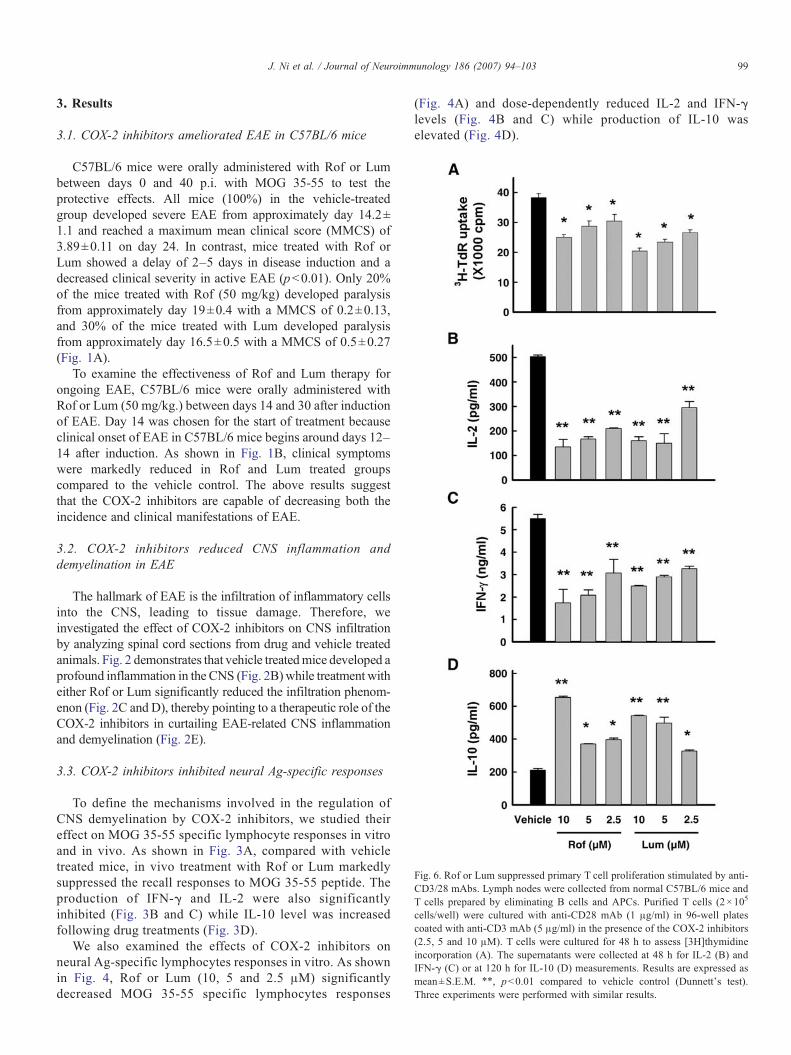
Fig. 6. Rof or Lum suppressed primary T cell proliferation stimulated by anti-CD3/28 mAbs. Lymph nodes were collected from normal C57BL/6 mice andT cells prepared by eliminating B cells and APCs. Purified T cells (2×105
cells/well) were cultured with anti-CD28 mAb (1 μg/ml) in 96-well platescoated with anti-CD3 mAb (5 μg/ml) in the presence of the COX-2 inhibitors(2.5, 5 and 10 μM). T cells were cultured for 48 h to assess [3H]thymidineincorporation (A). The supernatants were collected at 48 h for IL-2 (B) andIFN-γ (C) or at 120 h for IL-10 (D) measurements. Results are expressed asmean±S.E.M. ⁎⁎, pb0.01 compared to vehicle control (Dunnett's test).Three experiments were performed with similar results.
99J. Ni et al. / Journal of Neuroimmunology 186 (2007) 94–103
3. Results
3.1. COX-2 inhibitors ameliorated EAE in C57BL/6 mice
C57BL/6 mice were orally administered with Rof or Lumbetween days 0 and 40 p.i. with MOG 35-55 to test theprotective effects. All mice (100%) in the vehicle-treatedgroup developed severe EAE from approximately day 14.2±1.1 and reached a maximum mean clinical score (MMCS) of3.89±0.11 on day 24. In contrast, mice treated with Rof orLum showed a delay of 2–5 days in disease induction and adecreased clinical severity in active EAE (pb0.01). Only 20%of the mice treated with Rof (50 mg/kg) developed paralysisfrom approximately day 19±0.4 with a MMCS of 0.2±0.13,and 30% of the mice treated with Lum developed paralysisfrom approximately day 16.5±0.5 with a MMCS of 0.5±0.27(Fig. 1A).
To examine the effectiveness of Rof and Lum therapy forongoing EAE, C57BL/6 mice were orally administered withRof or Lum (50 mg/kg.) between days 14 and 30 after inductionof EAE. Day 14 was chosen for the start of treatment becauseclinical onset of EAE in C57BL/6 mice begins around days 12–14 after induction. As shown in Fig. 1B, clinical symptomswere markedly reduced in Rof and Lum treated groupscompared to the vehicle control. The above results suggestthat the COX-2 inhibitors are capable of decreasing both theincidence and clinical manifestations of EAE.
3.2. COX-2 inhibitors reduced CNS inflammation anddemyelination in EAE
The hallmark of EAE is the infiltration of inflammatory cellsinto the CNS, leading to tissue damage. Therefore, weinvestigated the effect of COX-2 inhibitors on CNS infiltrationby analyzing spinal cord sections from drug and vehicle treatedanimals. Fig. 2 demonstrates that vehicle treatedmice developed aprofound inflammation in the CNS (Fig. 2B) while treatment witheither Rof or Lum significantly reduced the infiltration phenom-enon (Fig. 2C and D), thereby pointing to a therapeutic role of theCOX-2 inhibitors in curtailing EAE-related CNS inflammationand demyelination (Fig. 2E).
3.3. COX-2 inhibitors inhibited neural Ag-specific responses
To define the mechanisms involved in the regulation ofCNS demyelination by COX-2 inhibitors, we studied theireffect on MOG 35-55 specific lymphocyte responses in vitroand in vivo. As shown in Fig. 3A, compared with vehicletreated mice, in vivo treatment with Rof or Lum markedlysuppressed the recall responses to MOG 35-55 peptide. Theproduction of IFN-γ and IL-2 were also significantlyinhibited (Fig. 3B and C) while IL-10 level was increasedfollowing drug treatments (Fig. 3D).
We also examined the effects of COX-2 inhibitors onneural Ag-specific lymphocytes responses in vitro. As shownin Fig. 4, Rof or Lum (10, 5 and 2.5 μM) significantlydecreased MOG 35-55 specific lymphocytes responses
(Fig. 4A) and dose-dependently reduced IL-2 and IFN-γlevels (Fig. 4B and C) while production of IL-10 waselevated (Fig. 4D).
100 J. Ni et al. / Journal of Neuroimmunology 186 (2007) 94–103
3.4. COX-2 inhibitors directly suppressed anti-MOG 35-55 Tcell responses, but did not affect antigen presentation
We used the antigen presenting assay to determine whetherthe inhibitory effects of the COX-2 inhibitors on T cellactivation reflect an alteration in T cell functions. Purified Tcells prepared from normal mice, MOG 35-55 immunizedmice with or without COX-2 inhibitor treatment werestimulated with MOG 35-55 (10 μg/ml) in the presence ofAPC-enriched cells obtained from normal animals. Fig. 5Adepicts that COX-2 inhibitor treatment significantly inhibitedT cell proliferation; similarly, treated cells produced aconsiderably less amount of IL-2 and IFN-γ upon MOG 35-55 stimulation (Fig. 5B and C).
To further examine antigen-presenting function, APC-enriched preparation from normal mice, MOG 35-55 immu-nized mice with or without COX-2 inhibitor treatment wascultured with purified T cells from immunized animals in thepresence of 10 μg/ml of MOG 35-55. There was no substantialdifference in T cell proliferation activated by APC-enrichedcells (Fig. 5D). The data indicate that administration of COX-2inhibitors could elicit a direct suppression on anti-MOG 35-55T cell responses, but had no effect on APC function.
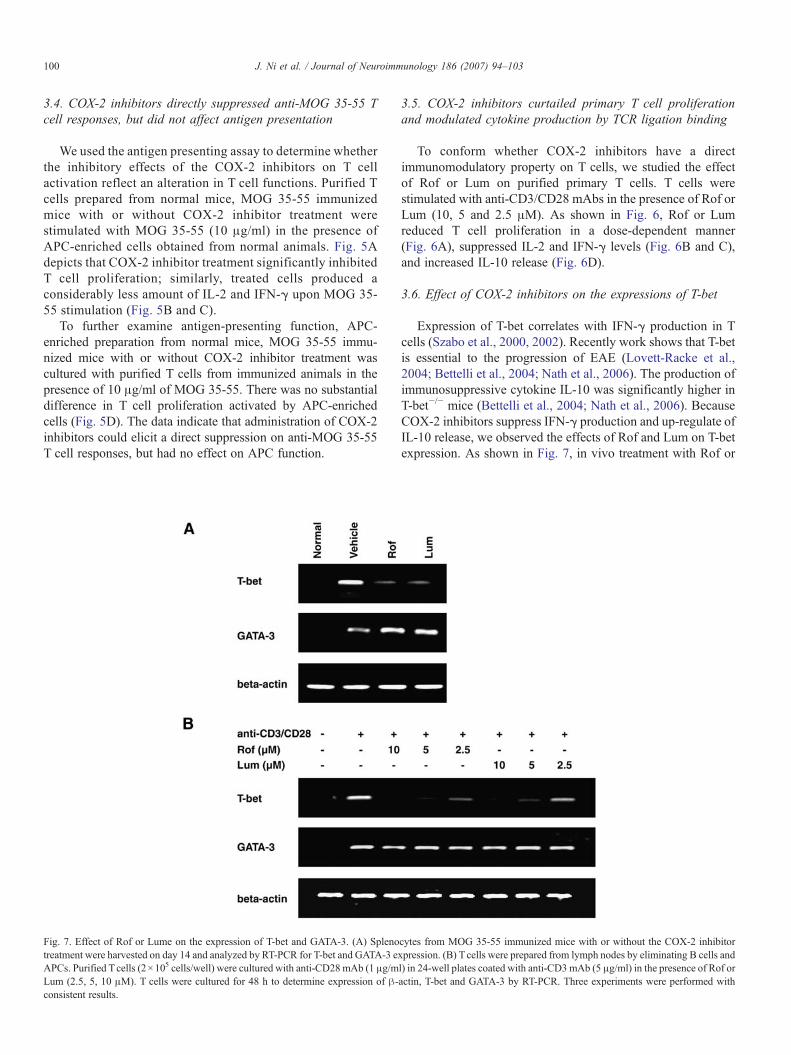
Fig. 7. Effect of Rof or Lume on the expression of T-bet and GATA-3. (A) Splenotreatment were harvested on day 14 and analyzed by RT-PCR for T-bet and GATA-3 eAPCs. Purified Tcells (2×105 cells/well) were cultured with anti-CD28mAb (1 μg/mLum (2.5, 5, 10 μM). T cells were cultured for 48 h to determine expression of β-consistent results.
3.5. COX-2 inhibitors curtailed primary T cell proliferationand modulated cytokine production by TCR ligation binding
To conform whether COX-2 inhibitors have a directimmunomodulatory property on T cells, we studied the effectof Rof or Lum on purified primary T cells. T cells werestimulated with anti-CD3/CD28 mAbs in the presence of Rof orLum (10, 5 and 2.5 μM). As shown in Fig. 6, Rof or Lumreduced T cell proliferation in a dose-dependent manner(Fig. 6A), suppressed IL-2 and IFN-γ levels (Fig. 6B and C),and increased IL-10 release (Fig. 6D).
3.6. Effect of COX-2 inhibitors on the expressions of T-bet
Expression of T-bet correlates with IFN-γ production in Tcells (Szabo et al., 2000, 2002). Recently work shows that T-betis essential to the progression of EAE (Lovett-Racke et al.,2004; Bettelli et al., 2004; Nath et al., 2006). The production ofimmunosuppressive cytokine IL-10 was significantly higher inT-bet−/− mice (Bettelli et al., 2004; Nath et al., 2006). BecauseCOX-2 inhibitors suppress IFN-γ production and up-regulate ofIL-10 release, we observed the effects of Rof and Lum on T-betexpression. As shown in Fig. 7, in vivo treatment with Rof or
cytes from MOG 35-55 immunized mice with or without the COX-2 inhibitorxpression. (B) Tcells were prepared from lymph nodes by eliminating B cells andl) in 24-well plates coated with anti-CD3mAb (5 μg/ml) in the presence of Rof oractin, T-bet and GATA-3 by RT-PCR. Three experiments were performed with

Fig. 8. Effect of Rof or Lum on the expression of IL-17. (A) Spinal cords from MOG 35-55 immunized mice with or without the COX-2 inhibitor treatment wereharvested on day 12 and analyzed by RT-PCR for IL-17 expression. (B) Purified T cells were cultured with anti-CD28 mAb (1 μg/ml) in 24-well plates coated withanti-CD3 mAb (5 μg/ml) in the presence of Rof or Lum (2.5, 5, 10 μM). T cells were cultured for 24 h to determine expression of IL-17 by RT-PCR. Two experimentswere performed with consistent results.
101J. Ni et al. / Journal of Neuroimmunology 186 (2007) 94–103
Lum (50 mg/kg) univocally suppressed T-bet expressionwithout affecting the expression of GATA3 (Fig. 7A). Inprimary T cells stimulated with anti-CD3/CD28 mAbs, COX-2inhibitors (10, 5 and 2.5 μM) reduced the expression of T-betsignificantly, whereas GATA3 was unaffected (Fig. 7B).
3.7. Effect of COX-2 inhibitors on the expressions of IL-17
To investigated whether COX-2 inhibitors can affect theexpression of IL-17, we examined the IL-17 mRNA expressionin CNS and primary T cells stimulated by CD3/CD28. Asshown in Fig. 8, in vivo treatment with Rof or Lum (50 mg/kg)obviously suppressed IL-17 expression (Fig. 8A). In primary Tcells stimulated with anti-CD3/CD28 mAbs, COX-2 inhibitors(10, 5 and 2.5 μM) reduced the expression of IL-17 significantly(Fig. 8B).
4. Discussion
In the present study, we demonstrated that administration ofCOX inhibitors, namely Rof or Lum, before and after diseaseonset prevented the clinical manifestations of EAE. The resultsshowed that Rof or Lum suppressed lymphocyte recall responseand modulated cytokine production through direct inhibition onanti-MOG T cell functions. Rof or Lum curtailed anti-CD3/28mAbs induced primary T cell proliferation and altered cytokinerelease profile. And we observed that Rof or Lum inhibited T-bet expression without affecting GATA-3 expression. We alsofound that Rof or Lum could suppress IL-17 expression both invivo and in vitro. It is therefore postulated that the COX-2inhibitors (Rof or Lum) are capable of preventing EAE viainhibition of antigen-specific T cell proliferation and modula-
tion of cytokine production by suppressing T-bet expression,and decreasing expression of IL-17.
Previous studies illuminated that COX inhibitors, NS398 andcelecoxib, had preventive effects on EAE (Muthian et al., 2006;Miyamoto et al., 2006). In this paper, we not only confirmedtheir observation using two different COX inhibitors, Rof orLum, but also established that they are equally efficacious intreating EAE.
The preventive action of NS398 on EAE was related to IL-12-induced JAK-STAT signaling pathway in activated T cells(Muthian et al., 2006), whereas that of celecoxib was shown tocorrelate with the expression of adhesion molecules andchemokines (Miyamoto et al., 2006). NS398 also possessesinhibitory properties on anti-CD3/28 mAbs-induced T cellsactivation, proliferation and cytokines production (Iniguezet al., 1999). However, whether the preventive effect of COXinhibitors on EAE is related to an alteration in primary T cellactivation is unknown, and this was one of the questionsaddressed in the study.
First, in vivo treatment with Rof or Lum reduced the MOG35-55 specific T cell proliferation compared to vehicle treatedmice, an outcome consistent with a published study displayingthe effect of another COX-2 inhibitor, NS398, on MOG 35-55specific splenocytes proliferation (Muthian et al., 2006).Inhibition of T cell proliferation could be partially attributedto a decrease in IL-2 production in these cells.
Second, Rof or Lum suppressed bothMOG35-55 induced andanti-CD3/28 mAbs mediated IFN-γ production. Previous reportsexhibited that, IFN-γ−/− and IFN-γ receptor−/− mice developedsevere diseases (Ferber et al., 1996; Willenborg et al., 1996; Chuet al., 2000). Although these observations suggested that IFN-γwas not essential to the induction of EAE, they did not negate the

102 J. Ni et al. / Journal of Neuroimmunology 186 (2007) 94–103
fact that encephalitogenic T cells generated in vivo in wild-typemice produced significant amounts of IFN-γ, or the fact that invitro suppression of IFN-γ production during stimulation ofmyelin specific T cells reduced the encephalitogenic capacity ofthese cells (Olsson, 1992). Taken together, these studies wouldimply that suppressing IFN-γ production in encephalitogenicT cells might provide therapeutic benefits.
In addition to inhibition of IFN-γ production, we alsodemonstrated that in vivo treatment with COX-2 inhibitorsenhanced the production of IL-10. IL-10 is associated withdisease remission in EAE. Recovery from EAE was correlatedwith an increased level of IL-10 in the CNS (Kennedy et al.,1992). IL-10 appeared to have a dominant role in the regulationof EAE since IL-10−/− mice suffered a severe non-remittingdisease (Bettelli et al., 1998) while mice that were transgenic forIL-10 were protected (Bettelli et al., 1998; Cua et al., 1999).Furthermore, administration of IL-10 was shown to suppressTh1 responses and incidences of EAE (Rott et al., 1994;Nagelkerken et al., 1997; Slavin et al., 2001). The observedincreased production of IL-10 throughout this study might bethe main driving factor in disease attenuation.
Another question is the cellular source of IL-10.Many types ofcells can produce IL-10 including antigen-presenting populationsuch as macrophages (Strassmann et al., 1994). So we testedwhether COX-2 inhibitors affected production of IL-10 onantigen-presenting cells. Administration of Rof or Lum didnot affect APCs to produce and induce more IL-10production in APC and T cells co-culture system (Data notshown). On the other hand, treatment with COX-2 inhibitorsin vivo or in vitro could enhance the production of IL-10. Webelieve that the increasing production of IL-10 was due toT cells.
Recent experiments have established that T-bet is requiredfor EAE induction, as T-bet-deficient mice immunized withMOG were resistant to EAE development (Bettelli et al., 2004;Nath et al., 2006). In vitro silencing T-bet in myelin-specific Tcells suppressed IFN-γ production and diminished the capacityof these cells to cause EAE when transferred into naive mice(Lovett-Racke et al., 2004). In vivo administration of a T-bet-specific anti-sense oligonucleotide or siRNA inhibited EAE,and there was no antigen-induced IFN-γ production in thesplenocytes of these mice (Lovett-Racke et al., 2004). Theproduction of regulatory cytokine IL-10 was significantlyhigher in the T-bet−/− mice (Bettelli et al., 2004; Nath et al.,2006). In this study, Rof or Lum curtailed the expression of T-bet to a considerably lower level (Fig. 7) suggesting thatinhibition of T-bet expression by Rof or Lum might contributeto the modulation on IFN-γ and IL-10 production.
Recent studies have demonstrated that the pro-inflammatorycytokine IL-17 has a significant role in the development ofpathological changes that accompany central nervous system(CNS) inflammation in experimental autoimmune encephalitis(EAE). IL-17−/− mice have a delayed disease onset and reduceddisease severity with early recovery (Komiyama et al., 2006),while administration of neutralizing anti-IL-17 antibodiesimmediately prior to the expected onset of actively inducedclinical disease in wild type animals reduces both disease
incidence and severity (Langrish et al., 2005; Park et al., 2005).In our study, Rof or Lum decreased the expression of IL-17 inCNS and in primary T cells stimulated by CD3/CD28 (Fig. 8).This data indicated that the amelioration of EAE by these twoCOX-2 inhibitors may partly through down regulation theexpression of IL-17.
Finally, a latest study demonstrated that the inhibitory effecton EAE by another COX-2 inhibitor (celecoxib) was evident inCOX-2-deficient mice, indicating that celecoxib suppressedEAE via a COX-2-independent mechanism (Miyamoto et al.,2006). Similarly, the up-regulation of IL-10 observed in oursystem also suggests that Rof and Lum may not act throughCOX-2-dependent pathway. In conclusion, the COX-2 inhibi-tors (Rof and Lum) showed potent therapeutic effects on EAEby modulating IFN-γ and IL-10 production through a COX-2independent mechanism, i.e., suppressing T-bet expression. AsIL-17 play an important role in EAE, the suppression of IL-17expression may also be an important mechanism. The datacollectively point to a potential application for MS treatmentusing COX-2 inhibitors.
Acknowledgements
We are indebted to Dr. G. X. Wang and Mr. J. S. Xie foradvice, and to Mr. Y. Zhang for technical assistance.
References
Anderson, A.C., Reddy, J., Nazareno, R., Sobel, R.A., Nicholson, L.B., Kuchroo,V.K., 2004. IL-10 plays an important role in the homeostatic regulation of theautoreactive repertoire in naive mice. J. Immunol. 173, 828–834.
Bettelli, E., Das, M.P., Howard, E.D., Weiner, H.L., Sobel, R.A., Kuchroo, V.K.,1998. IL-10 is critical in the regulation of autoimmune encephalomyelitis asdemonstrated by studies of IL-10-and IL-4-deficient and transgenic mice.J. Immunol. 161, 3299–3306.
Bettelli, E., Sullivan, B., Szabo, S.J., Sobel, R.A., Glimcher, L.H., Kuchroo, V.K.,2004. Loss of T-bet, but not STAT1, prevents the development of experimentalautoimmune encephalomyelitis. J. Exp. Med. 200, 79–87.
Chitnis, T., Khoury, S.J., 2003. Cytokine shifts and tolerance in experimentalautoimmune encephalomyelitis. Immunol. Res. 28, 223–239.
Chu, C.Q., Wittmer, S., Dalton, D.K., 2000. Failure to suppress the expansion ofthe activated CD4 T cell population in interferon gamma-deficient miceleads to exacerbation of experimental autoimmune encephalomyelitis.J. Exp. Med. 192, 123–128.
Cua, D.J., Groux, H., Hinton, D.R., Stohlman, S.A., Coffman, R.L., 1999.Transgenic interleukin 10 prevents induction of experimental autoimmuneencephalomyelitis. J. Exp. Med. 189, 1005–1010.
Ferber, I.A., Brocke, S., Taylor-Edwards, C., Ridgway, W., Dinisco, C., Steinman,L., Dalton, D., Fathman, C.G., 1996. Mice with a disrupted IFN-gamma geneare susceptible to the induction of experimental autoimmune encephalomyelitis(EAE). J. Immunol. 156, 5–7.
Fu, Y.F., Zhu, Y.N., Ni, J., Zhong, X.G., Tang, W., Ren, Y.D., Shi, L.P., Wan, J.,Yang, Y.F., Yuan, C., Nan, F.J., Lawson, B.R., and Zuo, J.P., in press. Areversible S-adenosyl-L-homocysteine hydrolase inhibitor amelioratesexperimental autoimmune encephalomyelitis by inhibiting T cell activation.J. Pharmacol. Exp. Ther.
Gor, D.O., Rose, N.R., Greenspan, N.S., 2003. TH1–TH2: a procrusteanparadigm. Nat. Immunol. 4, 503–505.
Hofstetter, H.H., Ibrahim, S.M., Koczan, D., Kruse, N., Weishaupt, A., Toyka,K.V., Gold, R., 2005. Therapeutic efficacy of IL-17 neutralization in murineexperimental autoimmune encephalomyelitis. Cell. Immunol. 237 (2),123–130 (Oct).

103J. Ni et al. / Journal of Neuroimmunology 186 (2007) 94–103
Iniguez, M.A., Punzon, C., Fresno, M., 1999. Induction of cyclooxygenase-2 onactivated T lymphocytes: regulation of T cell activation by cyclooxygenase-2 inhibitors. J. Immunol. 163, 111–119.
Kennedy, M.K., Torrance, D.S., Picha, K.S., Mohler, K.M., 1992. Analysis ofcytokine mRNA expression in the central nervous system of mice withexperimental autoimmune encephalomyelitis reveals that IL-10 mRNAexpression correlates with recovery. J. Immunol. 149, 2496–2505.
Komiyama, Y., Nakae, S., Matsuki, T., Nambum, A., Ishigame, H., Kakuta, S.,Sudo, K., Iwakura, Y., 2006. IL-17 plays an important role in thedevelopment of experimental autoimmune encephalomyelitis. J. Immunol.177 (1), 566–573 (Jul 1).
Langrish, C.L., Chen, Y., Blumenschein, W.M., Mattson, J., Basham, B.,Sedgwick, J.D., McClanahan, T., Kastelein, R.A., Cua, D.J., 2005. IL-23drives a pathogenic T cell population that induces autoimmune inflamma-tion. J. Exp. Med. 201, 233–240.
Lovett-Racke, A.E., Rocchini, A.E., Choy, J., Northrop, S.C., Hussain, R.Z.,Ratts, R.B., Sikder, D., Racke, M.K., 2004. Silencing T-bet defines a criticalrole in the differentiation of autoreactive T lymphocytes. Immunity 21,719–731.
Miyamoto, K., Miyake, S., Mizuno, M., Oka, N., Kusunoki, S., Yamamura, T.,2006. Selective COX-2 inhibitor celecoxib prevents experimental autoim-mune encephalomyelitis through COX-2-independent pathway. Brain 129,1984–1992.
Moon, C., Ahn, M., Jee, Y., Heo, S., Kim, S., Kim, H., Sim, K.B., Koh, C.S.,Shin, Y.G., Shin, T., 2004. Sodium salicylate-induced amelioration ofexperimental autoimmune encephalomyelitis in Lewis rats is associated withthe suppression of inducible nitric oxide synthase and cyclooxygenases.Neurosci. Lett. 356, 123–126.
Muthian, G., Raikwar, H.P., Johnson, C., Rajasingh, J., Kalgutkar, A., Marnett,L.J., Bright, J.J., 2006. COX-2 inhibitors modulate IL-12 signaling throughJAK-STAT pathway leading to Th1 response in experimental allergicencephalomyelitis. J. Clin. Immunol. 26, 73–85.
Nagelkerken, L., Blauw, B., Tielemans, M.I.M., 1997. IL-4 abrogates theinhibitory effect of IL-10 on the development of experimental allergicencephalomyelitis in SJL mice. Int. Immunol. 9, 1243–1251.
Nath, N., Giri, S., Prasad, R., Singh, A.K., Singh, I., 2004. Potential targets of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor for multiplesclerosis therapy. J. Immunol. 172, 1273–1286.
Nath, N., Prasad, R., Giri, S., Singh, A.K., Singh, I., 2006. T-bet is essential forthe progression of experimental autoimmune encephalomyelitis. Immunol-ogy 118, 384–391.
Olsson, T., 1992. Cytokines in neuroinflammatory disease: role of myelinautoreactive T cell production of interferon-gamma. J. Neuroimmunol. 40,211–218.
Park, H., Li, Z., Yang, X.O., Chang, S.H., Nurieva, R., Wang, Y.H., Wang, Y.,Hood, L., Zhu, Z., Tian, Q., Dong, C., 2005. A distinct lineage of CD4 Tcells regulates tissue inflammation by producing interleukin 17. Nat.Immunol. 6 (11), 1133–1141 (Nov).
Rott, O., Fleischer, B., Cash, E., 1994. Interleukin-10 prevents experimentalallergic encephalomyelitis in rats. Eur. J. Immunol. 24, 1434–1440.
Samoilova, E.B., Horton, J.L., Chen, Y., 1998. Acceleration of experimentalautoimmune encephalomyelitis in interleukin-10-deficient mice: roles ofinterleukin-10 in disease progression and recovery. Cell. Immunol. 188,118–124.
Segal, B.M., Dwyer, B.K., Shevach, E.M., 1998. An interleukin (IL)-10/IL-12immunoregulatory circuit controls susceptibility to autoimmune disease.J. Exp. Med. 187, 537–546.
Slavin, A.J., Maron, R., Weiner, H.L., 2001. Mucosal administration of IL-10enhances oral tolerance in autoimmune encephalomyelitis and diabetes. Int.Immunol. 13, 825–833.
Sredni-Kenigsbuch, D., 2002. TH1/TH2 cytokines in the central nervoussystem. Int. J. Neurosci. 112, 665–703.
Steinman, L., 2001. Multiple sclerosis: a two-stage disease. Nat. Immunol. 2,762–764.
Steinman, L., Zamvil, S.S., 2005. Virtues and pitfalls of EAE for thedevelopment of therapies for multiple sclerosis. Trends Immunol. 11,565–571.
Strassmann, G., Patil-Koota, V., Finkelman, F., Fong, M., Kambayashi, T., 1994.Evidence for the involvement of interleukin 10 in the differentialdeactivation of murine peritoneal macrophages by prostaglandin E2.J. Exp. Med. 180 (6), 2365–2370 (Dec 1).
Szabo, S.J., Kim, S.T., Costa, G.L., Zhang, X., Fathman, C.G., Glimcher, L.H.,2000. A novel transcription factor, T-bet, directs Th1 lineage commitment.Cell 100, 655–669.
Szabo, S.J., Sullivan, B.M., Stemmann, C., Satoskar, A.R., Sleckman, B.P.,Glimcher, L.H., 2002. Distinct effects of T-bet in TH1 lineage commitmentand IFN-gamma production in CD4 and CD8 T cells. Science 295 (5553),338–342 (Jan 11).
Willenborg, D.O., Fordham, S., Bernard, C.C., Cowden, W.B., Ramshaw, I.A.,1996. IFN-gamma plays a critical down-regulatory role in the induction andeffector phase of myelin oligodendrocyte glycoprotein-induced autoimmuneencephalomyelitis. J. Immunol. 157, 3223–3227.
Yang, Y.F., Mukai, T., Gao, P., Yamaguchi, N., Ono, S., Iwaki, H., Obika, S.,Imanishi, T., Tsujimura, T., Hamaoka, T., Fujiwara, H., 2002. A non-peptideCCR5 antagonist inhibits collagen-induced arthritis by modulating T cellmigration without affecting anti-collagen T cell responses. Eur. J. Immunol.32, 2124–2132.
Youssef, S., Stuve, O., Patarroyo, J.C., Ruiz, P.J., Radosevich, J.L., Hur, E.M.,Bravo, M., Mitchell, D.J., Sobel, R.A., Steinman, L., Zamvil, S.S., 2002.The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias andreverses paralysis in central nervous system autoimmune disease. Nature420, 78–84.
Zamvil, S.S., Steinman, L., 1990. The T lymphocyte in experimental allergicencephalomyelitis. Annu. Rev. Immunol. 8, 579–621.
Zhu, Y.N., Zhong, X.G., Feng, J.Q., Yang, Y.F., Fu, Y.F., Ni, J., Liu, Q.F., Tang,W., Zhao, W.M., Zuo, J.P., 2006. Periplocoside E inhibits experimentalallergic encephalomyelitis by suppressing interleukin 12-dependent CCR5expression and interferon-gamma-dependent CXCR3 expression in Tlymphocytes. J. Pharmacol. Exp. Ther. 318, 1153–1162.
Copyright © 2022 FDOKUMEN




![Design, synthesis and pharmacological evaluation of 4-[2-alkylthio-5(4)-(4-substitutedphenyl)imidazole-4(5)yl]benzenesulfonamides as selective COX2 inhibitors](https://static.fdokumen.com/doc/165x107/6316e643d16b3722ff0d1bc7/design-synthesis-and-pharmacological-evaluation-of-4-2-alkylthio-54-4-substitutedphenylimidazole-45ylbenzenesulfonamides.jpg)















