
Eosinophil-mediated signalling attenuates inflammatory responses in experimental colitis
Upload
independentCategory
view
1download
0
Rapid Development of Colitis in NSAID-TreatedIL-10–Deficient Mice
DANIEL J. BERG,* JUAN ZHANG,* JOEL V. WEINSTOCK,* HANAN F. ISMAIL,* KEITH A. EARLE,‡
HECTOR ALILA,‡ RIFAT PAMUKCU,‡ STEVEN MOORE,§ and RICHARD G. LYNCH§
*Department of Internal Medicine, and §Department of Pathology, University of Iowa College of Medicine, Iowa City, Iowa; ‡Cell Pathways,Horsham, Pennsylvania
Background & Aims: Interleukin (IL)-10 is an anti-inflam-matory and immune regulatory cytokine. IL-10–defi-cient mice (IL-10�/�) develop chronic inflammatorybowel disease (IBD), indicating that endogenous IL-10 isa central regulator of the mucosal immune response.Prostaglandins are lipid mediators that may be impor-tant mediators of intestinal inflammation. In this studywe assessed the role of prostaglandins in the regulationof mucosal inflammation in the IL-10�/� mouse modelof IBD. Methods: Prostaglandin (PG) synthesis was in-hibited with nonselective or cyclooxygenase (COX)-iso-form selective inhibitors. Severity of inflammation wasassessed histologically. Cytokine production was as-sessed by ribonuclease protection analysis and enzyme-linked immunosorbent assay. PGE2 levels were as-sessed by enzyme immunoassay. COX-1 and COX-2expression was assessed by Western blot analysis.Results: Nonsteroidal anti-inflammatory drug (NSAID)treatment of wild-type mice had minimal effect on thecolon. In contrast, NSAID treatment of 4-week-old IL-10�/� mice resulted in rapid development of colitischaracterized by infiltration of the lamina propria withmacrophages and interferon gamma–producing CD4� Tcells. Colitis persisted after withdrawal of the NSAID.NSAID treatment decreased colonic PGE2 levels by 75%.Treatment of IL-10�/� mice with sulindac sulfone (whichdoes not inhibit PG production) did not induce colitiswhereas the NSAID sulindac induced severe colitis.COX-1– or COX-2–selective inhibitors used alone did notinduce IBD in IL-10�/� mice. However, the combinationof COX-1– and COX-2–selective inhibitors did inducecolitis. Conclusions: NSAID treatment of IL-10�/� miceresults in the rapid development of severe, chronic IBD.Endogenous PGs are important inhibitors of the devel-opment of intestinal inflammation in IL-10�/� mice.
Prostaglandins (PGs) are lipid mediators that have animportant role in multiple physiologic processes in-
cluding blood clotting, ovulation, initiation of labor,wound healing, and kidney function.1 PGs also have beenimplicated as key mediators of acute inflammatory re-
sponses2,3 and chronic inflammatory states such as arthri-tis4,5 and inflammatory bowel disease (IBD).6
PG biosynthesis is tightly controlled. The key regu-latory enzyme of the PG biosynthesis pathway, PG syn-thase (EC 1.14.99.1), also known as cyclooxygenase(COX), is the first enzyme in the biosynthetic pathwayleading to PGs, thromboxanes, and prostacyclins. TheCOX enzyme exists in 2 isoforms: COX-1, a constitutiveform that is expressed in multiple cell types and isthought to produce PGs central to physiologic homeosta-sis,7 and COX-2, an inducible form that is rapidly up-regulated in response to lipopolysaccharides (LPSs), cy-tokines, and mitogens.7–9 PG production by bothisoforms is inhibited by nonsteroidal anti-inflammatorydrugs (NSAIDs), for example, piroxicam and sulin-dac.10–12
There is clear evidence that PG production is increasedin IBD. Increased PG levels have been documented instool, venous blood, and rectal mucosa in IBD patients.6
Biopsy specimens from patients with IBD also have beenfound to contain increased amounts of PGE2 and throm-boxane.13 In vivo estimation of PG production has beenperformed in multiple studies by using rectal dialysis,which has confirmed that PG production in the colon isincreased in patients with active disease.6 PG levelscorrelate with disease activity and decrease with success-ful medical intervention.Although PGs can mediate inflammation and the
production of PGs is clearly increased in IBD, there isgood evidence that PGs are protective in colitis. Inhibi-tion of PG production is therapeutic in a number ofinflammatory diseases (e.g., rheumatoid arthritis), how-ever, there is no role for NSAIDS in the treatment of
Abbreviations used in this paper: COX, cyclooxygenase; DMSO, di-methyl sulfoxide; ELISA, enzyme-linked immunosorbent assay; IFN�,interferon gamma; IL, interleukin; LPS, lipopolysaccharide; RNase,ribonuclease; Th1, T helper 1.
© 2002 by the American Gastroenterological Association0016-5085/02/$35.00
doi:10.1053/gast.
GASTROENTEROLOGY 2002;123:1527–1542

IBD. In fact, there is evidence that COX inhibitors canaggravate or reactivate IBD.10,14,15 The mechanism forthis detrimental effect is unknown; however, PGs haveimportant immunoregulatory effects. For example, PGE2
is a potent inhibitor of LPS-induced tumor necrosisfactor � production16 and nitric oxide synthesis.17 Inaddition, exogenously added PGE2 induces the produc-tion of interleukin (IL)-10,18,19 a key anti-inflammatorycytokine.IL-10 is a cytokine with potent anti-inflammatory
activity. IL-10 is a potent macrophage deactivator,blocking the induced synthesis of multiple inflammatorycytokines, including tumor necrosis factor �, IL-1, IL-6,IL-8, and granulocyte-macrophage colony-stimulatingfactor.20,21 IL-10 also indirectly suppresses the synthesisof interferon (IFN)� by helper T cells22 and natural killercells.23 Interestingly, mice with a targeted disruption ofthe IL-10 gene (IL-10�/�)24 develop a spontaneouschronic IBD characterized by increased inflammatorycytokine production as well as IFN� from T-helper 1(Th1) CD4� T cells.24,25 These studies indicated thatendogenous IL-10 is a central regulator of the mucosalimmune and inflammatory response in vivo. Impor-tantly, the spontaneous IBD in IL-10�/� mice may takemonths to develop, suggesting the presence of otherregulatory factors that can delay the development of IBDin this model. In the present study, we have usedNSAID-induced inhibition of PG production to studythe role of endogenous PGs in the initiation of inflam-mation in the IL-10�/� mouse model of IBD.
Materials and Methods
Animals
Healthy 4-week-old IL-10�/� mice on a 129/SvEv orC57BL/6 background were used for this study.25 Wild-type129/SvEv mice were obtained from Taconic Farms (German-town, NY). Wild-type C57BL/6 mice were obtained fromJackson Laboratories (Bar Harbor, ME). Mice were maintainedin microisolator cages under specific pathogen-free, Helicobacter-free conditions at the animal care facility at the University ofIowa.
Reagents
Piroxicam and Sulindac were obtained from Sigma (St.Louis, MO). NS-398 was obtained from Cayman ChemicalCompany (Ann Arbor, MI). SC-560 and SC-58236 werekindly provided by J. Portanova (Pharmacia, St. Louis, MO).Sulindac sulfone (Exisulind), 1000 ppm in AIN-93G diet wasprovided by Cell Pathways (Horsham, PA). 16,16-dimethyl-PGE2 and 17-phenyl-trinor-PGE2 were obtained from CaymanChemical Company. Rabbit polyclonal anti-murine COX-2
was obtained from Cayman Chemical Company; rabbit poly-clonal anti–COX-1 was obtained from Santa Cruz Biotechnol-ogy (Santa Cruz, CA). Hamster anti-CD3 (clone: 2C11) was akind gift from J. Bluestone (University of California, SanFrancisco, CA). LPS from Escherichia coli (serotype 0111:B4)was obtained from Sigma. Tissue culture grade dimethylsulf-oxide (DMSO) was obtained from American Type CultureCollection (Manassas, VA).
Experimental Protocol for NonsteroidalAnti-Inflammatory Drug Treatment
To test the role of PGs in the development of IBD,nonselective NSAIDs that inhibit both COX isoforms (piroxi-cam, 200 ppm in the diet; or Sulindac, 300–500 ppm in thediet) were fed to weanling mice (4 weeks of age) for 2 weeks ata dose of 200 ppm piroxicam in the diet. The NSAID wasmixed with rodent chow (NIH-31M) by using the techniqueof geometric dilution26 to ensure uniform distribution of theNSAID in the diet. Briefly, an equivalent amount of mousediet was added to the NSAID and then mixed thoroughly.Successive equivalent amounts of the mouse diet were added,mixing well after each dilution, until the entire quantity of themouse diet was incorporated. After the 2-week feeding period,mice were euthanized and colons were removed for furtherstudies.To test whether the NSAID-induced colitis became chronic,
after the 2-week feeding period the mice were subsequentlyplaced on standard sterile rodent chow for 4–10 weeks beforethey were assessed for colitis.To test the role of PG inhibition in NSAID-induced colitis,
4-week-old IL-10�/� mice were fed for 2 weeks with a dietcontaining either Sulindac sulfone (Exisulind, 1000 ppm),which does not inhibit PG production, Sulindac (300 or 500ppm), or control diet. In this series of experiments, the dietused was AIN-93G (Dyets, Bethlehem, PA). After the 2-weekfeeding period the colons were removed for histologic analysisof inflammation.
To assess the role of COX-1 or COX-2, mice were treatedfor 2 weeks with the following NSAIDs: SC-560 (COX-1inhibitor) 10 mg/kg by gavage, daily for 14 days; SC-58236(COX-2 inhibitor), 3 mg/kg by gavage, daily for 14 days;NS-398 (COX-2 inhibitor) 5 mg/kg by gavage, twice daily for14 days. Test drugs were solubilized in DMSO and diluted in0.5% carboxymethylcellulose (final concentration of 5%DMSO). In some experiments mice received both SC-560 andSC-58236. Control mice received vehicle only (0.5% car-boxymethylcellulose with 5% DMSO). After the 2-week treat-ment period the colons were removed for histologic analysis ofinflammation.
To assess the effect of PG supplementation, piroxicam-treated IL-10�/� mice were treated simultaneously with thePGE-receptor antagonists. The PG agonists 16,16-dimethyl-PGE2 and 17-phenyl-trinor-PGE2, 10 mg each in sterile phos-phate-buffered saline (PBS) were administered in 2 daily in-jections (8 AM and 5 PM) and once daily via gavage feeding (12
1528 BERG ET AL. GASTROENTEROLOGY Vol. 123, No. 5

PM) over a 7-day period.27 Mice were euthanized after 7 days ofpiroxicam treatment and the colons were assessed histologi-cally for evidence of inflammation.
Histologic Analysis
Tissues from wild-type and IL-10�/� mice were fixedin 10% neutral buffered formalin, routinely processed, sec-tioned at 6 �, and stained with H&E for light microscopicexamination. Samples from the entire gastrointestinal tractwere examined by the same pathologist (R.G.L.) withoutknowledge of which treatment group the samples were from.Because intestinal lesions were multifocal and of variable se-verity, the grades given to any section of intestine took intoaccount the number of lesions as well as their severity. A scorefrom 0 to 4 was based on the following criteria: grade 0, nochange from normal tissue; grade 1, one or a few multifocalmononuclear cell infiltrates in the lamina propria accompaniedby minimal epithelial hyperplasia and slight to no depletion ofmucus from goblet cells; grade 2, lesions were more frequentand typical changes included several multifocal, mild inflam-matory cell infiltrates in the lamina propria primarily com-posed of mononuclear cells with a few neutrophils. Mildepithelial hyperplasia and mucin depletion also were seen.Small epithelial erosions were occasionally present and inflam-mation rarely involved the submucosa; grade 3, lesions in-volved a large area of the mucosa or were more frequent thangrade 2 lesions. Inflammation was moderate and often involvedthe submucosa but was rarely transmural. Inflammatory cellswere a mixture of mononuclear cells as well as neutrophils, andcrypt abscesses were sometimes observed. Moderate epithelialhyperplasia and mucin depletion were seen. Ulcers were occa-sionally observed; grade 4, lesions usually involved most of theintestinal section and were more severe than grade 3 lesions.Inflammation was intense, including mononuclear cells andneutrophils, and was sometimes transmural. Epithelial hyper-plasia was marked with crowding of epithelial cells in elon-gated glands. Few mucin-containing cells were seen. Cryptabscesses and ulcers were present and foci of fibrinoid necrosiswere present in the submucosa contiguous to ulcerations andcrypt abscesses.
Immunohistochemical Analysis
Colons from control and NSAID-treated wild-type andIL-10�/� mice used for immunohistochemistry were fixed in azinc-based fixative (0.1 mol/L Tris, pH 7.4 containing 0.5 g Caacetate, 5 g Zn acetate, 5 g Zn chloride),28,29 processed byusing low melting point paraffin, and sectioned at a thicknessof 6 �. Standard immunohistochemical procedures were used.Slides were deparaffinized by incubation in xylene and gradedalcohols. Endogenous peroxidase was inhibited by incubatingtissue in buffer containing 0.3% H2O2. All monoclonal anti-bodies were diluted in 1% (vol/vol) fetal calf serum in PBS.After incubation with the primary, sections were incubatedwith biotinylated goat anti-rat immunoglobulin G (1:200,Vector Laboratories, South San Francisco, CA). Staining wasenhanced by using biotin-tyramide (TSA Immunohistochem-
istry kit; New England Nuclear, Boston, MA) exactly as perthe manufacturer’s instructions. 3,3�-diaminobenzidine tetra-hydrocholoride was used to visualize stained cells and sectionswere counterstained lightly with hematoxylin. Slides weredehydrated with graded alcohol and xylene and subsequentlymounted with Permount (Fisher Scientific, Fair Lawn, NJ)under glass coverslips. Ab used for section staining: purifiedrat anti-mouse CD4 (clone L3/T4; 10 �g/mL, Caltag, SouthSan Francisco, CA); purified rat anti-mouse F4/80 tissue cul-ture supernatant (clone A3-1; 10 �g/mL, Caltag); rat anti-mouse neutrophil (clone 8C5, 10 �g/mL, Caltag); purified ratanti-mouse CD45R (B220) (clone RA3-6B2, 10 �g/mL,Caltag) and rat anti-mouse CD8� (clone CT-CD8�, 10 �g/mL, Caltag). Rat immunoglobulin isotype control antibodieswere obtained from BD Pharmingen (San Diego, CA).
Quantification of Prostaglandin E2Colon tissue PGE2 levels were determined by using the
PGE2 enzyme immunoassay kit from Cayman Chemical Com-pany, as per the manufacturer’s instructions. Colons weresnap-frozen in liquid N2 and then homogenized in PBS con-taining 1 �g/mL aprotinin, 1 �g/mL leupeptin, 100 �g/mLphenylmethylsulfonyl fluoride, and 100 �mol/L piroxicam (toinhibit PG production during sample preparation). Debris wasremoved by centrifugation at 15,000 rpm for 20 minutes, at4°C. The supernatant was removed for determination of PGE2
concentration. Protein concentration in the supernatant wasmeasured by using a commercial reagent based on bicincho-ninic acid staining (Pierce, Rockford, IL), using bovine serumalbumin as an internal standard.
Ribonuclease Protection Assay
RNA from colons was prepared by using RNA-STAT-60 (Tel-Test, Friendswood, TX) exactly as per themanufacturer’s instructions. Cytokine messenger RNA(mRNA) was measured by using multiprobe mouse cytokineribonuclease (RNase) protection kits (mcCK-1 and mCK-2b)exactly as per the manufacturer’s instructions (RiboQuantMultiprobe RNase Protection Assay System, BD Pharmingen).Briefly, by an in vitro transcription, 32P-labeled RNA probeswere synthesized by using the mCK-2b set of cytokine com-plementary DNA templates. The synthesized probes werepurified by using phenol:chloroform extraction. Hybridizationreactions with 20 �g of total RNA were performed overnightat 56°C. After RNase digestion, the RNA duplexes wereisolated by electrophoresis and resolved on 5% polyacrylamidegels, dried, and visualized by autoradiography. Dried gels wereplaced on Kodak BMR film and were exposed at �70°C.
Western Blotting
Protein was isolated from cultured spleen cells byresuspending in lysis buffer (50 mmol/L Tris [hydroxymethyl]aminomethane, pH 7.5, 150 mmol/L NaCl, 100 �g/mL phe-nylmethylsulfonyl fluoride, 1 �g/mL aprotinin, 1 �g/mL leu-
November 2002 NSAID–INDUCED COLITIS IN IL–10�/� MICE 1529

peptin, 1 mmol/L diethyldithiocarbamic acid, 1% NonidetP-40, and 1% sodium deoxycholate). Cells were lysed bysonication (20 s, 4°C). Debris was eliminated by centrifugation(15 min, 1000g). Protein concentration was measured by usinga commercial reagent based on bicinchoninic acid staining(Pierce) by using bovine serum albumin as an internal stan-dard. Equal amounts of cellular protein were loaded onto a10% polyacrylamide gel and separated by electrophoresis (200V for 45 min). Proteins were then transferred to nitrocellulose(100 V for 1 h), and the membrane was blocked with 5%nonfat dry milk. The nitrocellulose was then incubated with arabbit polyclonal primary antibody (anti–COX-2, Cayman,1:1000 or anti–COX-1, 1:1000, Santa Cruz) overnight at25°C. Antibody labeling was detected by using enhancedchemiluminescence (Amersham, Buckinghamshire, England)as per the manufacturer’s instructions. Specificity of the anti-COX antibody was confirmed with the use of ram seminalvesicle COX-1 (Oxford, Oxford, MI) and sheep placentaCOX-2 (Cayman).Films and photographs of Western blots and RNase protec-
tion assays were scanned in at 600 dots per inch by using anEpson Expression 1600 scanner (Epson, Long Beach, CA).Densitometric analysis was performed by using NIH ImageJsoftware. Average and integrated optical density measure-ments were made on user-selected regions.
Preparation of Colon Lamina PropriaLymphocytes
Gut lymphocytes were isolated from IL-10�/� or wild-type mice according to a modified protocol of Lefrancois.30
Intestinal tissue was trimmed of fat and connective tissue. Theintestinal tissue was washed extensively in PBS, chopped, andthen incubated in 2 mmol/L ethylenediaminetetraacetic acid inPBS for 30 minutes at 37°C with stirring to release intraepi-thelial lymphocytes and epithelial cells. Lamina propria lym-phocytes were isolated from the remaining tissue fragments byincubation in 0.2 mg/mL collagenase/dispase (BoehringerMannheim, Indianapolis, IN) for 30 minutes at 37°C withstirring. The lamina propria lymphocyte preparations werethen washed and sieved twice through nylon gauze (pore size100 � and twice through 70 � Falcon cell filters; BectonDickinson). Lymphocytes were enriched by centrifugationwithin 40% (vol/vol) Percoll (Pharmacia LKB, Uppsala, Swe-den) overlaid on 75% Percoll for 20 minutes at 750g.
T-Cell Culture
T cells were enriched by depletion of B cells, neutro-phils, and macrophages. Cell preparations were incubated withanti-B220, anti-GR1 (8C5), and anti-Mac 1 Ab, respectively,and Ab-reactive cells were removed in a magnetic field byusing a combination of goat anti-rat immunoglobulin G (Fc)and goat anti-rat immunoglobulin G (H � L)–coated mag-netic beads (Advanced Magnetics, Cambridge, MA). The re-maining cells were stained with anti-CD4 for 20 minutes onice. Populations were �98% pure on reanalysis. Purified CD4T cells were incubated at a concentration of 1 � 106 cells/mL
in 96-well plates coated with anti-CD3. Culture supernatantswere collected after 24 hours of stimulation and frozen at�70°C until analysis of cytokine concentration by enzyme-linked immunosorbent assay (ELISA).
Colon Organ Culture
Colon organ cultures were prepared from age-matchedcontrol or piroxicam-treated wild-type or IL-10�/� mice in thefollowing manner. Colons, not including the cecum, weredissected from mice and flushed with cold PBS to remove fecalmatter. Each colon was then cut into 2-mm squares, washed ina large volume of PBS plus 5% fetal calf serum, and resus-pended in RPMI-1640 supplemented with 10% fetal calfserum, 50 mmol/L 2-ME, penicillin (100 U/mL), streptomycin(100 U/mL), and ciprofloxacin (100 �g/mL). Equivalentamounts of tissue (200 mg) were distributed into tissueculture plates (Falcon 3046; Becton Dickinson Labware) con-taining 5 mL of media alone or media supplemented with LPS(10 �g/mL) or Con A (5 �g/mL). Cultures were incubated at37°C in 5% CO2. Supernatants were harvested after 24 hoursand stored at �70°C before analysis of cytokine levels byELISA.
Determination of Cytokine Levels
IL-12 and IFN� concentrations in cell culture super-natants and colon explant culture supernatants were measuredby using ELISA kits purchased from BD Pharmingen accord-ing to the manufacturer’s directions.
ResultsRapid Induction of Colitis in NonsteroidalAnti-Inflammatory Drug–TreatedInterleukin-10–Deficient Mice
To assess the role of endogenous PGs in theregulation of the immune and inflammatory responses inthe colon, 4-week-old wild-type and IL-10�/� mice weretreated for 2 weeks with piroxicam, a nonselectiveNSAID. As expected, untreated control wild-type micehad no evidence of colonic inflammation (Table 1). Themajority (75%) of NSAID-treated wild-type mice alsowere completely normal. A very mild colitis (averagecolitis disease score 0.37 0.74, scale 0–4, with 0 � nocolitis and 4 � severe colitis) could be seen in 25% ofwild-type mice treated with piroxicam (Table 1). Thelamina propria in 75% of the piroxicam-treated wild-type mice was completely normal. In the NSAID-treatedwild-type mice with colitis, the lamina propria containedsmall focal infiltrates of mononuclear cells. The colonicepithelium of NSAID-treated wild-type mice was nor-mal, with no evidence of hyperplasia or ulcerations.Before initiation of NSAID treatment, we evaluated
the level of colonic inflammation in IL-10�/� mice from
1530 BERG ET AL. GASTROENTEROLOGY Vol. 123, No. 5

our colony. Untreated IL-10�/� mice at 4 weeks of agewere healthy and had no inflammatory infiltrates (Figure1A, Table 1). Although IL-10�/� mice develop a spon-taneous IBD,25 in our colony colonic inflammation wasnot present until mice are 3 months of age or older. Incontrast to wild-type mice, NSAID treatment of wean-ling IL-10�/� mice resulted in the development of mod-erate to severe colitis within 2 weeks (Figures 1B–1F,Table 1). Microscopic examination revealed markedchanges in the colonic mucosa of NSAID-treated IL-10�/� mice. Severe inflammatory infiltrates were foundmost commonly in the proximal colon (including thececum). In some instances, the mice had a pan-colitis.The cellular infiltrate predominantly involved the laminapropria. In some cases infiltrates were present in both themucosa and submucosa with areas of transmural inflam-mation. The inflammatory infiltrates were composed pre-dominately of mononuclear cells, although small num-bers of neutrophils and eosinophils also could be seen,usually in association with ulceration. Epithelial changeswere also noted in NSAID-treated IL-10�/� mice. Epi-thelial hyperplasia was common in areas with inflamma-tion. Ulcerations of the epithelium were present, as wererare crypt abscesses. Mesenteric lymph nodes from theNSAID-treated IL-10�/� mice were markedly enlargedas compared with mesenteric lymph nodes from wild-type and control IL-10�/� mice. Treatment of IL-10�/�
mice with chemically distinct nonselective NSAIDs, ei-ther piroxicam (Figures 1C and 1D) or sulindac (Figures1B, 1E, and 1F; see Table 3), resulted in similar levels ofcolitis. Histologic scores are shown in Table 1.
A Longitudinal Study of Nonsteroidal Anti-Inflammatory Drug–Treated Interleukin-10–Deficient Mice Was Performed to Studythe Progression of the NonsteroidalAnti-Inflammatory Drug–Induced Colitis
Pathologic changes were evident in piroxicam-treated mice as early as 3 days after initiation of treat-ment (Table 2). These changes consisted of small, mul-tifocal infiltrates located in the lamina propria of theproximal colon. The infiltrates consisted mainly of lym-phocytes and a small number of neutrophils. At this earlytime point there was no evidence of epithelial hyperplasiaor ulcerations. After 7 days of piroxicam treatment therewas a significant increase in lesion frequency and severityin the colon. These multifocal lesions were seen mainlyin the proximal colon and consisted of mononuclearinfiltrates with rare neutrophils. Accompanying the in-filtrates were occasional ulcerations in the epithelium aswell as epithelial hyperplasia. Colitis was observed in100% of the mice treated with piroxicam after 7 days ofNSAID treatment. The lesion severity in the micetreated for 7 days was not significantly different from thelesions noted after 14 days of NSAID-treatment; thus,the colitis scores are similar at both time points (seeTable 1). The frequency of the lesions, however, isgreater in the mice treated with NSAIDs for 14 days ascompared with those treated for only 7 days. Controluntreated IL-10�/� mice did not have colonic inflamma-tion at the tested time points.
Colitis Persists After Discontinuation ofNonsteroidal Anti-Inflammatory Drugs
Our previous studies25 showed that IL-10�/�
mice develop a spontaneous IBD that is chronic in na-ture. To determine whether NSAID-induced colitiswould persist, we treated 4-week-old IL-10�/� micewith piroxicam for 2 weeks and then placed the miceback on the standard mouse diet for 4–6 weeks. After
Table 1. Gastrointestinal Inflammation in NSAID-TreatedMice
Genetic background n Treatment% Of micewith colitis
Colitisdiseasescore per
group (0–4)a
129/SvEv/wild type 5 Control 0 0129/SvEv/wild type 18 Piroxicam 25 0.37 0.74b
129/SvEv/IL-10�/� 14 Control 0 0129/SvEv/IL-10�/� 21 Piroxicam 100 3.82 0.41b
C57BI/6J/IL-10�/� 6 Control 0 0C57BI/6J/IL-10�/� 12 Piroxicam 100 3.80 0.20b
NOTE. Gastrointestinal inflammation in NSAID-treated wild-type andIL-10�/� mice. Four-week-old mice were fed either control diet or dietcontaining piroxicam, 200 ppm, for 2 weeks. The gastrointestinaltract was subsequently evaluated histologically. n indicates the num-ber of mice per group.aColitis was graded on a scale of 0–4, with 0 � no colitis and 4 �severe colitis (see Materials and Methods section for further Details).Data are reported as mean SD per group.bP � 0.001, piroxicam-treated IL-10�/� vs. control mice.
Table 2. Longitudinal Study of NSAID-Induced Colitis inIL-10�/� Mice
Duration oftreatment n
% Of micewith colitis
Colitis disease scoreper group (0–4)a
3 days 5 80 1.40 0.897 days 11 100 3.56 0.88b
NOTE. IL-10�/� mice were fed control diet or diet containing piroxi-cam, 200 ppm, for 3 or 7 days. The gastrointestinal tract was sub-sequently evaluated histologically. n indicates the number of mice pergroup.aData reported as mean SD per group.bP � 0.001, 7 days of piroxicam treatment vs. 3 days of piroxicamtreatment.
November 2002 NSAID–INDUCED COLITIS IN IL–10�/� MICE 1531
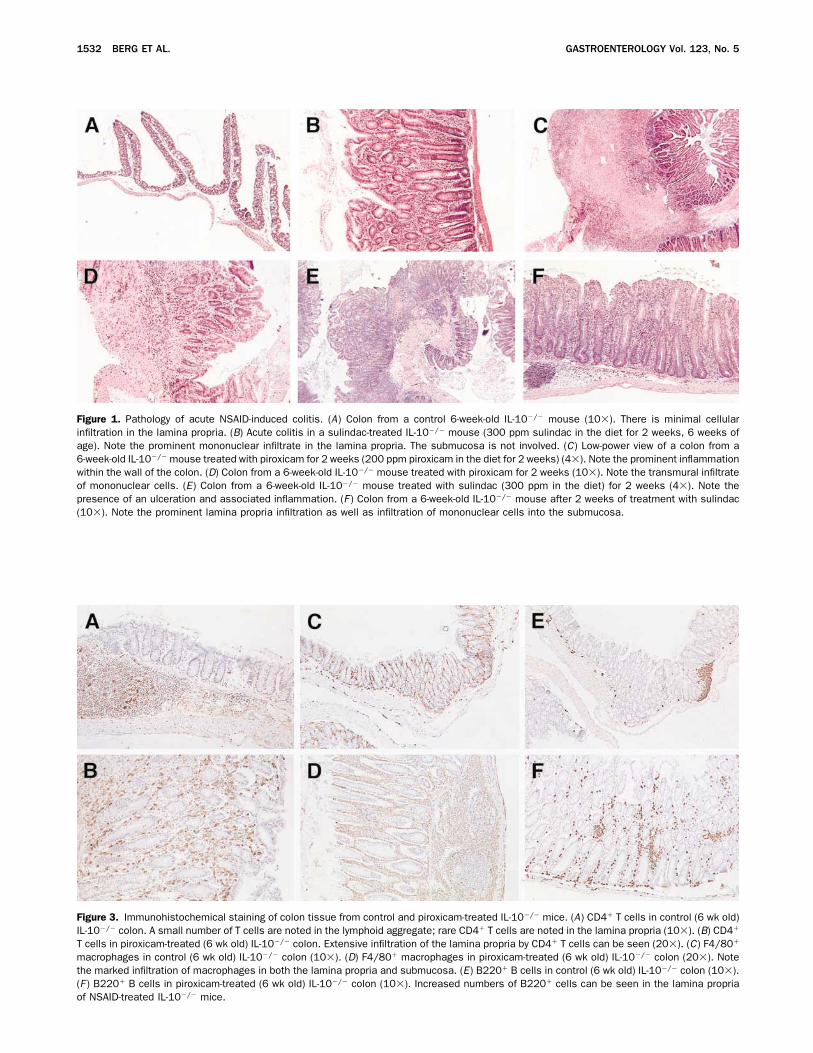
Figure 1. Pathology of acute NSAID-induced colitis. (A) Colon from a control 6-week-old IL-10�/� mouse (10�). There is minimal cellularinfiltration in the lamina propria. (B) Acute colitis in a sulindac-treated IL-10�/� mouse (300 ppm sulindac in the diet for 2 weeks, 6 weeks ofage). Note the prominent mononuclear infiltrate in the lamina propria. The submucosa is not involved. (C) Low-power view of a colon from a6-week-old IL-10�/� mouse treated with piroxicam for 2 weeks (200 ppm piroxicam in the diet for 2 weeks) (4�). Note the prominent inflammationwithin the wall of the colon. (D) Colon from a 6-week-old IL-10�/� mouse treated with piroxicam for 2 weeks (10�). Note the transmural infiltrateof mononuclear cells. (E) Colon from a 6-week-old IL-10�/� mouse treated with sulindac (300 ppm in the diet) for 2 weeks (4�). Note thepresence of an ulceration and associated inflammation. (F ) Colon from a 6-week-old IL-10�/� mouse after 2 weeks of treatment with sulindac(10�). Note the prominent lamina propria infiltration as well as infiltration of mononuclear cells into the submucosa.
Figure 3. Immunohistochemical staining of colon tissue from control and piroxicam-treated IL-10�/� mice. (A) CD4� T cells in control (6 wk old)IL-10�/� colon. A small number of T cells are noted in the lymphoid aggregate; rare CD4� T cells are noted in the lamina propria (10�). (B) CD4�
T cells in piroxicam-treated (6 wk old) IL-10�/� colon. Extensive infiltration of the lamina propria by CD4� T cells can be seen (20�). (C) F4/80�
macrophages in control (6 wk old) IL-10�/� colon (10�). (D) F4/80� macrophages in piroxicam-treated (6 wk old) IL-10�/� colon (20�). Notethe marked infiltration of macrophages in both the lamina propria and submucosa. (E) B220� B cells in control (6 wk old) IL-10�/� colon (10�).(F ) B220� B cells in piroxicam-treated (6 wk old) IL-10�/� colon (10�). Increased numbers of B220� cells can be seen in the lamina propriaof NSAID-treated IL-10�/� mice.
1532 BERG ET AL. GASTROENTEROLOGY Vol. 123, No. 5

this period, the mice were evaluated for the level ofcolonic inflammation. Control IL-10�/� mice receivedthe standard mouse diet during the entire study. All ofthe IL-10�/� mice treated with NSAIDs had colitis 4–6weeks after termination of the NSAID treatment. Thecolitis disease score for these mice was 3.70 0.67 (n �10), which was significantly different from the colitisdisease score of age-matched control IL-10�/� mice (av-erage colitis disease score, 0.6 0.69, mean SD, n �10, P � 0.05). There was no difference in the severity orcharacter of the colitis at 4 or 6 weeks after NSAIDtreatment. These studies show that withdrawal of theNSAID does not result in resolution of the colitis; rather,the colitis persisted. These data clearly indicate thatmaintenance of the inflammation at the assessed timepoints did not require continual treatment with theNSAID.Although the colitis disease score was similar in both
acute colitis and at 4–6 weeks after NSAID treatment,the histologic appearance of the colitis 4–6 weeks afterNSAID treatment differed from that of acute colitis. Themost significant differences were noted in the epithe-lium. Areas of epithelial cell atypia were evident inchronic colitis (Figure 2A ) but were not seen in controlmice or mice with acute colitis. Invasive colonic epithe-lium was noted in 30% of mice with chronic colitis(Figures 2B and 2C ), whereas none of the control mice ormice with acute colitis had evidence of invasion of epi-thelial cells into the lamina propria. The invasive epi-thelium was noted to have mild cytologic and nuclearpleomorphism, with a high nuclear:cytoplasmic ratio andfrequent loss of the basal orientation of the nuclei. Inva-sive glands also were surrounded by a prominent desmo-plastic response. Ulcerations of the epithelium, thoughpresent, were less prominent in chronic colitis as com-pared with acute colitis. There was more involvement of
the distal colon in the mice with chronic colitis ascompared with acute colitis.
The Effect of Genetic Background on theDevelopment of Nonsteroidal Anti-Inflammatory Drug–Induced Colitis inInterleukin-10–Deficient Mice
The contribution of heritable factors to the devel-opment of NSAID-induced intestinal inflammation wasevaluated by NSAID treatment of IL-10�/� mice oneither the C57BL/6J or the 129/SvEv genetic back-grounds. IL-10�/� mice were evaluated at 6 weeks of ageafter 2 weeks of piroxicam treatment. Of IL-10�/� miceof both the 129/SvEv and C57BL/6J strain backgrounds,100% developed colitis when treated with the NSAIDpiroxicam (Table 1). The disease severity in IL-10�/�
mice on the C57Bl/6 background was similar to that seenon the 129/SvEv mice and the characteristics of theinflammatory infiltrate did not differ between the 2strains.
Phenotypic Analysis of Infiltrating Cells
Immunohistochemical studies were performed tocharacterize the cellular infiltrate that develops inNSAID-treated IL-10�/� mice. Review of the H&E tis-sue sections showed that the infiltrate consisted primarilyof mononuclear cells, although neutrophils were present,generally in association with ulcerations (data notshown). A striking finding was the large increase inCD4� T cells within the lamina propria of piroxicam-treated IL-10�/� mice. Large aggregates of CD4� T cellswere noted throughout the lamina propria, as well as inassociation with ulcerations (Figures 3A and 3B).NSAID-treated IL-10�/� mice also had a marked in-crease in lamina propria macrophages (Figures 3C and3D). Increased numbers of B220� lymphocytes were also
Figure 2. Pathology of colitis 4–6 weeks after discontinuation of NSAID treatment. Four-week-old IL-10�/� mice were treated with NSAID(piroxicam 200 ppm in the diet) for 2 weeks. The animals subsequently were placed on standard rodent diet for 4–6 weeks before assessmentof colitis. (A) Cellular atypia in an epithelial gland from an IL-10�/� mouse 4–6 weeks after discontinuation of NSAID treatment (40�). Mildnuclear and cytologic pleomorphism are present as well as loss of basal orientation of the epithelial nuclei. (B) Invasive epithelium in an IL-10�/�
mouse 4–6 weeks after discontinuation of NSAID treatment (4�). The epithelium extends almost to the serosa. (C) Extensive area of invasiveepithelium in an IL-10�/� mouse 4–6 weeks after discontinuation of NSAID treatment (10�).
November 2002 NSAID–INDUCED COLITIS IN IL–10�/� MICE 1533

noted, mainly located at the base of the lamina propria(Figures 3E and 3F ). A minimal increase in CD8�
lymphocytes was noted in the lamina propria and intra-epithelial compartment of NSAID-treated IL-10�/�
mice (data not shown). There were no differences seen inthe cellular infiltrates of wild-type mice treated withpiroxicam as compared with control wild-type mice.
Nonsteroidal Anti-InflammatoryDrug–Treated Interleukin-10–Deficient MiceHave Increased Production of InflammatoryCytokines
Spontaneous IBD in IL-10�/� mice is character-ized by increased production of inflammatory cytokinesin the colon.25 Therefore, we assessed the effect ofNSAID treatment on cytokine mRNA expression in thecolon of control and NSAID-treated wild-type and IL-10�/� mice by using RNase protection assays. NSAIDtreatment of IL-10�/� mice resulted in marked increasesin mRNA expression of multiple inflammatory cytokines(Figures 4A and 4B). Small increases were noted inmRNA expression of IFN� and macrophage migrationinhibitory factor (Figure 4B). Large increases in mRNAexpression for IL-1� and IL-1 were consistently found(Figure 4B). Interestingly, there was no change in IL-18mRNA in the colons of NSAID-treated IL-10�/� mice.No expression of IL-4 or IL-13 mRNA was detected inwild-type or IL-10�/� mice either at baseline or after 2weeks of NSAID treatment (data not shown). There wasa low level of IL-10 mRNA expression detected in co-lonic mRNA from 3 of 10 wild-type mice; no expressionof IL-10 mRNA was detected in mRNA from IL-10�/�
mice. Unlike IL-10�/� mice, NSAID treatment of wild-type mice did not lead to increased expression of IL-12p40, IL-1�, IL-1 , or macrophage migration inhibi-tory factor mRNA (data not shown).To further define colonic cytokine production in
NSAID-induced colitis in IL-10�/� mice we measuredspontaneous production of IL-12 and IFN� in colonexplant cultures. Colonic tissue from NSAID-treatedIL-10�/� mice spontaneously produced larger amounts ofIL-12 and IFN� as compared with control IL-10�/�
colons (Figure 5A ).
Colonic T Cells From Nonsteroidal Anti-Inflammatory Drug–Treated Interleukin-10–Deficient Mice Produce Interferon�
The spontaneous IBD in IL-10�/� mice is causedby the development of IFN�-producing Th1-type Tcells.25,31 Therefore, we next assessed the phenotype ofthe lamina propria T cells from the colons of NSAID-treated IL-10�/� mice. Lamina propria T cells from
Figure 4. (A) RNase protection analysis of the effect of NSAID treat-ment on cytokine mRNA expression in the colon. Four-week-old IL-10�/� mice were maintained on a standard diet (control) or placed ona diet containing piroxicam (200 ppm in the diet). After 2 weeks, totalcolon mRNA was isolated and cytokine mRNA expression was ana-lyzed by RNase protection by using specific probes for murine cyto-kines. Lanes 1–4, colon RNA from control IL-10�/� mice. Lanes5–14, colon RNA from piroxicam-treated IL-10�/� mice. Data arerepresentative of 3 independent experiments. (B) Quantitation ofcytokine mRNA expression was assessed by image analysis. CytokinemRNA levels were normalized to the level of mRNA expression of thehousekeeping gene, L32. Data are representative of 3 independentexperiments. Mean SD, *P � 0.05, piroxicam-treated IL-10�/�
mice vs. control IL-10�/� mice. �, Control IL-10�/�; ■ , NSAID-treatedIL-10�/�.
1534 BERG ET AL. GASTROENTEROLOGY Vol. 123, No. 5

NSAID-treated IL-10�/� mice were isolated and cul-tured with plate-bound anti-CD3/anti-CD28 and thesupernatant was analyzed for cytokine content. As can beseen in Figure 5B, high levels of IFN� were producedfrom anti-CD3–treated unfractionated lamina propriacells as well as from lamina propria T cells from NSAID-treated IL-10�/� mice. No IFN� was produced fromlamina propria cells that were depleted of T cells norfrom lamina propria cells of healthy 5-week-old controlIL-10�/� mice. Moreover, no IL-4 or IL-5 productionwas detected from any of the anti-CD3–stimulated lam-ina propria cell preparations, indicating that the infil-trating T cells in NSAID-induced colitis have a Th1-type phenotype.
Basal Prostaglandin E2 Concentration inthe Colon Is Decreased in Mice TreatedWith Nonsteroidal Anti-Inflammatory Drugs
We next turned to a series of experiments todetermine the underlying mechanism for the inductionof IBD in NSAID-treated IL-10�/� mice. Piroxicam andsulindac are NSAIDs that inhibit PG production fromthe COX enzymes. Therefore, we assessed PGE2 levels incolonic tissues from NSAID-treated wild-type and IL-10�/� mice to determine if the NSAIDs given in the dietwere able to inhibit PG production in the colon. Wild-type and IL-10�/� mice fed for 5 days with a dietcontaining piroxicam at 200 ppm had approximately a75% decrease in colonic PGE2 levels (Figure 6). Thebasal levels of PGE2 concentration in untreated controlwild-type and 4-week-old IL-10�/� mice were not sig-nificantly different. Similar levels of suppression of co-lonic PGE2 were found in wild-type mice treated for 5days with SC-560, a specific inhibitor of COX-1 (datanot shown).Because we found that colons from wild-type and
IL-10�/� mice contained high levels of PGE2 that wasinhibited by the NSAID treatment, we next evaluatedthe expression of the COX enzymes to determine thesource(s) of the colonic PG production. By Western blotanalysis, the dominant colonic COX isoform was COX-1(Figure 7A ) in both wild-type and IL-10�/� mice. West-ern blot analysis also revealed a small amount of COX-2protein expression in 4-week-old IL-10�/� mice (Figure7B). Reverse-transcription polymerase chain reactionanalysis of colonic RNA showed the presence of COX-2mRNA in both wild-type and 4-week-old IL-10�/� mice
Figure 5. (A) Colonic cultures of piroxicam-treated IL-10�/� miceproduce increased amounts of IL-12 and IFN�. Colon explants fromcontrol IL-10�/� mice or piroxicam-treated IL-10�/� mice were cul-tured in medium for 36 hours. Supernatants were subsequently har-vested and analyzed for cytokine content by ELISA. Ten mice per groupwere analyzed. Data are representative of 2 independent experi-ments. Mean SD, *P � 0.05, piroxicam-treated IL-10�/� mice vs.control IL-10�/� mice. (B) Purified lamina propria T cells from thecolons of piroxicam-treated IL-10�/� mice produce IFN�. T cells wereplated at 1 � 106 cells/mL in anti-CD3 monoclonal antibody–coatedplates as described in the Materials and Methods section. After 24hours of culture, supernatants were harvested and analyzed for cyto-kine content (IFN� and IL-4) by ELISA. No IL-4 or IL-5 was detected.
Figure 6. PGE2 concentration in colonic tissues from 4-week-old wild-type or IL-10�/� mice maintained on either control diet or diet con-taining piroxicam (200 ppm) for 5 days. Tissue PGE2 concentrationswere measured by enzyme immunoassay in snap-frozen tissues ho-mogenized in indomethacin-containing buffer. Mean SD, n �5/group. *P � 0.05 control vs. piroxicam-treated mice.
November 2002 NSAID–INDUCED COLITIS IN IL–10�/� MICE 1535

(data not shown). Western blot analysis, however,showed that COX-2 expression dramatically increased inthe colon of IL-10�/� mice with NSAID-induced colitis(Figure 7B), whereas COX-1 expression was unchangedby NSAID-induced colitis (Figure 7C ). Taken together,these data indicate that both COX isoforms are presentin the normal mouse colon, but with the development ofNSAID-induced colitis, there is a dramatic increase inCOX-2 expression in the IL-10�/� mouse colon.
Effect of Sulindac, Sulindac Sulfone, andProstaglandin E–Receptor Agonists onNonsteroidal Anti-Inflammatory–InducedColitis
To further assess the role of PGs in the initiationof colitis, IL-10�/� mice were treated with Sulindac (a
nonselective NSAID) or Exisulind, the sulfone metabo-lite of Sulindac. Sulindac is a prodrug that is metabolizedin vivo to either the sulindac sulfide or sulindac sulfone(Exisulind) derivative.32 The sulindac sulfide derivativehas potent nonselective COX-inhibitory activity and isresponsible for the anti-inflammatory effects of Sulin-dac.32 Exisulind is the sulfone derivative of sulindac anddoes not have COX-inhibitory activity,33 yet retains theparent compound’s ability to inhibit chemically inducedcarcinogenesis.34 Weanling IL-10�/� mice were fed dietscontaining Sulindac or Exisulind for 2 weeks and sub-sequently were assessed for the presence of colonic in-flammation. All the sulindac-treated IL-10�/� miceuniformly developed colitis (Table 3). In contrast, allIL-10�/� mice treated with Exisulind (at 1000 ppm)survived and had no evidence of colitis or other toxiceffects (Table 3), indicating that inhibition of PG pro-duction was central to the development of NSAID-induced colitis.To determine if PG supplementation would prevent
disease, IL-10�/� mice were simultaneously treated withPGE-receptor agonists and piroxicam. After 7 days ofpiroxicam treatment, control (non-PGE2 treated) IL-10�/� mice had developed significant colitis (colitis dis-ease score 3.33 0.51, mean � SD, n � 6) whereasconcomitant PGE-receptor agonist treatment signifi-cantly protected against the development of NSAID-induced IBD (colitis disease score 1.33 0.6, mean SD, n � 3, P � 0.01, PGE2 treatment vs. piroxicamalone).
Functional Role of COX-1– andCOX-2–Derived Prostaglandins in theInitiation of Nonsteroidal Anti-InflammatoryDrug–Induced Colitis
To assess the relative contributions of PGs derivedfrom the COX-1 and COX-2 isoforms to the initiation of
Figure 7. Western blot analysis of COX expression. (A) COX-1 is ex-pressed in colons of wild-type and control IL-10�/� mice. Western blotanalysis was performed on colonic protein isolated from 4-week-oldwild-type or IL-10�/� mice. Equivalent amounts of protein (25 �g/lane)were separated on 10% sodium dodecyl sulfate–polyacrylamide gel elec-trophoresis, and COX-1 expression was assessed by Western blot anal-ysis. C, control protein from COX-1 expressing 3T3 fibroblast cell line. (B)COX-2 is expressed in control and piroxicam-treated IL-10�/� mice. West-ern blot analysis was performed on colonic protein isolated from 4-week-old IL-10�/� mice or piroxicam-treated IL-10�/� mice. Equivalentamounts of protein (25�g/lane) were separated on 10% sodium dodecylsulfate–polyacrylamide gel electrophoresis, and COX-2 expression wasassessed by Western blot analysis. C, control protein from LPS-stimu-lated RAW 264.7 macrophage cell line. Data are representative of 3independent experiments. (C) COX-1 expression is unchanged in controland piroxicam-treated IL-10�/� mice. Western blot analysis was per-formed on colonic protein isolated from 4-week-old IL-10�/� mice or6-week-old piroxicam-treated IL-10�/� mice. Equivalent amounts of pro-tein (25 �g/lane) were separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and COX-1 expression was assessedby Western blot analysis. Data are representative of 2 independent experi-ments. C, control protein from COX-1 expressing 3T3 fibroblast cell line.
Table 3. Gastrointestinal Inflammation in Sulindac- andExisulind-Treated IL-10�/� Mice
Treatment n% Of micewith colitis
Colitis disease scoreper group (0–4)a
Control Diet 9 22 0.44 1.0Sulindac 8 100 3.38 5b
Exisulind 11 9 0.09 0.3
NOTE. Colonic inflammation in IL-10�/� mice treated with Sulindac orExisulind. Four-week-old mice were fed for 2 weeks with control diet ordiet containing Sulindac (300 ppm) or Exisulind (1000 ppm). Thecolon was subsequently evaluated histologically. n indicates the num-ber of mice per group.aData reported as mean SD per group.bP � 0.001, Sulindac-containing diet vs. control diet. Data are repre-sentative of 2 independent experiments.
1536 BERG ET AL. GASTROENTEROLOGY Vol. 123, No. 5

colitis in NSAID-treated IL-10�/� mice we treated4-week-old IL-10�/� mice with selective COX inhibi-tors. Treatment of IL-10�/� mice with selective COX-2inhibitors (NS-398 or SC-58236) did not induce colitis(Table 3). As noted earlier, weanling IL-10�/� miceexpress low levels of the target isoform (COX-2) in thecolon at this time point. Interestingly, treatment ofIL-10�/� mice with the selected COX-1 inhibitor SC-560 also did not induce colitis (Table 4). However,treatment of IL-10�/� mice with both COX selectiveinhibitors (COX-1 inhibitor, SC-560, and the COX-2inhibitor, SC-58236) resulted in the development ofcolitis in all the IL-10�/� mice so treated (Table 4).These data suggest that PGs from both COX isoformsplay a role in the regulation of the colonic immune andinflammatory response.
DiscussionIn this study we have used wild-type and IL-
10�/� mice to assess the effect of inhibition of PGproduction on the mucosal immune and inflammatoryresponse. We found that wild-type mice treated withNSAIDs developed little or no colitis whereas NSAID-treated IL-10�/� mice developed severe colonic inflam-mation. A major finding was that, within 2 weeks ofNSAID treatment, IL-10�/� mice developed a signifi-cant cellular infiltrate in the lamina propria, which wassimilar to the spontaneous IBD that develops after 3months of age in our colony of IL-10�/� mice. Ourfinding that NSAID-treated IL-10�/� mice develop asevere IBD strongly indicates that PGs have an impor-tant role in the regulation of the mucosal immune sys-tem.NSAID-induced colitis is histologically quite similar
to the spontaneous colitis in IL-10�/� mice. Both models
are characterized by a predominantly mononuclear infil-trate in the lamina propria with areas of transmuralinflammation. Epithelial changes, including hyperplasiaand ulceration, can be seen in both spontaneous andNSAID-induced IBD. Similar to the spontaneous modelof IBD, NSAID-treatment did not affect the jejunum orileum. However, NSAID treatment is known to be acause of gastric ulcerations35 and, not unexpectedly, 2weeks of NSAID treatment did result in ulcerations atthe gastroduodenal junction in both wild-type and IL-10�/� mice. The severity of the gastroduodenal ulcer-ations was equivalent in the wild-type and IL-10�/�
mice, but ulcerations were more frequent in wild-typemice (37%) than IL-10�/� mice (12%). In contrast,spontaneous gastric involvement in IL-10�/� mice isuncommon, and when it occurs it is characterized by amild mononuclear infiltrate in the lamina propria ratherthan ulceration.NSAID-induced colitis in IL-10�/� mice has signifi-
cant immunologic similarities to spontaneous colitis inIL-10�/� mice. Both spontaneous and NSAID-inducedcolitis in IL-10�/� mice are characterized by markedinfiltration of the colon with CD4� T cells and macro-phages. In the spontaneous colitis model we showed anincreased production of inflammatory mediators such asIL-1�.25 Similarly, we found increased mRNA expres-sion of multiple inflammatory cytokines, includingIL-1� and IL-1 in colons from NSAID-treated IL-10�/� mice. Colon explant cultures also showed in-creased levels of IL-12 and IFN�, showing that theinflammatory cytokine production in NSAID-inducedcolitis is quite similar to that of the spontaneous IBDmodel.In previous studies, we showed that spontaneous IBD
in IL-10�/� mice is dependent on the development ofIFN�-producing Th1 T cells.25,31 IL-12 has been shownto drive Th1 differentiation and IFN� production.36,37
The generation of pathogenic Th1 T cells in spontaneousIBD in IL-10�/� mice is dependent on IL-12.38 In thisstudy, we showed increased colonic IL-12 mRNA pro-duction from NSAID-treated IL-10�/� mice. We alsofound that NSAID-induced colitis in IL-10�/� mice isassociated with infiltration of the colon by CD4� T cells.Moreover, lamina propria T cells from NSAID-treatedIL-10�/� mice stimulated with anti-CD3 producedIFN�, but not IL-4, indicating that the infiltrating cellshave a Th1-type phenotype. Studies of the pathogenicityof these CD4� T cells are ongoing.Ethnic and familial studies in humans have suggested
that certain individuals are genetically predisposed todevelop IBD. A difficulty in these studies, however, is
Table 4. Effect of Selective COX Inhibitors on theDevelopment of Colitis in IL-10�/� Mice
TreatmentCox isoforminhibited n
% Of micewith colitis
Colitis disease scoreper group (0–4)a
Control diet NA 19 21 0.42 0.9NS 398 COX-2 12 8 0.16 0.58SC58236 COX-2 9 0 0SC560 COX-1 16 0 0SC 560 �SC 58236 COX-1 � COX-2 8 100 3.5 0.7b
NOTE. Four-week-old mice were treated with either NS 398 (10 mg/kg/day) or SC 58236 (10 mg/kg/day). SC560 (5 mg/kg/day) or thecombination of SC58236 and SC560. The colon was subsequentlyevaluated histologically. n indicates the number of mice per group.aData reported as mean SD per group.bP � 0.001, SC 560- and SC 58236-containing diet vs. control diet.Data is pooled from 8 independent experiments.
November 2002 NSAID–INDUCED COLITIS IN IL–10�/� MICE 1537

that they have failed to show a simple type of inheritanceowing to dominant or recessive gene. In our previousstudy, we evaluated the development of spontaneous IBDin IL-10�/� mice on 3 different genetic backgrounds:C57BL/6J, 129/SvEv, and BALB/c.25 We found that the129/SvEv strain and BALB/c strain were permissive forthe development of IBD, whereas the C57BL/6 strainwas relatively resistant. These findings suggested that,similar to human IBD, there are genetic factors thatmodify the development of colitis in the IL-10�/� mice.Genetic contributions to the susceptibility to chronicintestinal inflammation have been reported in multipleexperimental animal models of colitis.39–41 In contrast tothe previous study, we show that NSAID-induced colitisdevelops rapidly in both the susceptible 129/SvEv strainand the more resistant C57BL/6J strain. The severity ofNSAID-induced colitis in the C57BL/6J strain is quitesimilar to that of the 129/SvEv strain whereas the straindifferences in the spontaneous IBD are much greater.These data indicate that the mechanism(s) underlyingNSAID-induced development of IBD can overcome theeffect of genetic modifiers that may serve to inhibitdisease development in the C57Bl/6 mouse strain.The most significant difference between NSAID-in-
duced colitis and spontaneous colitis was the rapiditywith which NSAID-induced colitis developed. In starkcontrast to the spontaneous model, NSAID treatmentresults in moderate to severe colitis within 2 weekswhereas spontaneous colitis requires at least 3 months ormore before significant lesions develop. An interestingfinding of the disease progression studies was that smallinflammatory lesions were seen within 3 days of piroxi-cam treatment of IL-10�/� mice. Importantly, theselesions consisted of increased lamina propria mononu-clear cells in the absence of any evidence of epithelialdamage. By 7 days of piroxicam treatment the lesionshad progressed and included a strong mononuclear infil-trate within the lamina propia as well as ulcerations.Continued NSAID treatment was not necessary for themaintenance of disease. When the NSAID treatment wasdiscontinued and the mice were placed on normal food,the disease continued and became chronic, suggestingthat the mechanisms of NSAID induction of IBD maydiffer from those that maintain the intestinal inflamma-tion.Several lines of evidence indicate that inhibition of PG
production is central to the development of colitis inIL-10�/� mice. Although the NSAID dose used in ourstudies was relatively low, PG production was effectivelyinhibited (by about 75%) in the colon of both wild-typeand IL-10�/� mice. Moreover, several chemically distinct
drugs (piroxicam, sulindac, and the combination of SC-560 and SC-58236), which all inhibit PG production,induced the same phenotype (colitis) in the IL-10�/�
mice whereas administration of PGE2-receptor agonistssimultaneously with NSAID markedly reduced the de-velopment of colitis. Finally, the lack of inflammation inExisulind-treated IL-10�/� mice supports the hypothesisthat inhibition of PG production is important for thedevelopment of colitis in IL-10�/� mice. Sulindac is aprodrug that is metabolized in vivo to sulindac sulfide,the moiety that inhibits COX enzymes, and sulindacsulfone, which has no COX-inhibitory activity. Sulindacinduced severe colitis at a dose of 300 ppm. In contrast,IL-10�/� mice treated with Exisulind at a dose of 1000ppm in the diet did not develop colitis and no otheradverse effects were noted. Taken together, these datastrongly suggest that inhibition of PG production iscentral to the development of colitis, and, conversely,suggest that endogenous PGs are important regulators ofthe mucosal immune response in IL-10�/� mice.Our data indicate that both COX isoforms have an
important role in the regulation of the mucosal immuneand inflammatory response. We found that wild-typeand healthy, weanling IL-10�/� mice (which do not havesignificant inflammation) have significant levels of PGE2
in the colon. The levels of PGE2 did not differ betweenthe wild-type and IL-10�/� mice at this age. The PGsappear to be derived from COX-1 based on the high levelof COX-1 protein expression in the colon (coincidentwith minimal COX-2 expression). Similarly, Morteau etal.42 found that COX-1–deficient mice have minimalPGE2 levels in the colon, indicating that the COX-1isoform produces most of the PGs in the normal, non-inflamed colon. Interestingly, COX-1–deficient micehave minimal mucosal PG levels yet have no evidence forintestinal inflammation.43 However, COX-1–deficientmice challenged with dextran sodium sulfate had in-creased susceptibility to injury, suggesting that COX-1–derived prostaglandins had a protective effect.42
Our data indicate that COX-2–derived PGs also playan important role in the regulation of colonic inflamma-tion in IL-10�/� mice. Although COX-2 expression ismarkedly less than that of COX-1 in the normal colon,protein and mRNA for COX-2 is present in the colon ofboth wild-type and IL-10�/� mice. Moreover, treatmentof IL-10�/� mice with selective COX-1 or COX-2 in-hibitors alone was not sufficient to induce colitis inIL-10�/� mice. Only nonselective inhibitors or the com-bination of selective COX-1 and COX-2 inhibitors wereable to induce colitis in the IL-10�/� mice. These datasuggest the possibility that PGs derived from one iso-
1538 BERG ET AL. GASTROENTEROLOGY Vol. 123, No. 5

form can compensate for the absence of PGs from theother. Indeed, examples of compensation in COX-defi-cient mice have been reported.44,45 Multiple studies haveshown the importance of COX-2–derived PGs in thegastrointestinal tract. Although COX-2–deficient micedo not have any evidence of colonic inflammation, thesemice do have an increased incidence of peritonitis ofunknown cause.46 In a model of dextran sodium sulfate–induced colitis, selective COX-2 deletion resulted in amarked enhancement of inflammation,42 indicating thatCOX-2–derived PG can down-regulate an inflammatoryresponse in the colon. Similarly, in a study of experimen-tal gastric ulcerations it was found that COX-2 is rapidlyup-regulated. Inhibition of COX-2 with a selectiveNSAID enhanced the pathology, indicating that COX-2–derived PGs were important for the repair of gastricmucosal lesions.47 A key role for COX-2–derived PGs inthe regulation of the mucosal immune response has beenshown in studies of oral tolerance. In an elegant study ofT-cell transgenic mice that recognized hen egg-whitelysozyme, Newberry et al.48 showed that administrationof hen egg-white lysozyme in the absence of COX-2–derived PGs resulted in loss of oral tolerance. Takentogether, these results support a protective role for COX-2–derived PGs in the regulation of the mucosal immuneresponse as well as in the maintenance of epithelialintegrity.How might PGs regulate the mucosal immune and
inflammatory response? PGs perform a myriad of func-tions that are dependent on the cell of origin and thephysiologic context. Inhibition of PG production mayhave promoted the development of IBD in IL-10�/�
mice by altering the immune and inflammatory re-sponses. Extensive literature indicates that PGs haveimportant immunoregulatory effects. PGs have signifi-cant effects on inflammatory cytokine production. Sim-ilar to IL-10, PGE2 is a potent inhibitor of LPS-inducedtumor necrosis factor � production16 and nitric oxidesynthesis.17 Absence of IL-10 and PGs may have resultedin increased inflammatory cytokine production; indeed,we have found that NSAID treatment induces a 2-foldincrease in LPS-induced IFN� in spleen cells from IL-10�/� mice (Berg DJ, data not shown). Similar to IL-10,49,50 exogenous PGs have been shown to decreasechemokine production and chemokine receptor expres-sion.51–54 The absence of both IL-10 and PGs may haveresulted in increased chemokine production, increasedtrafficking of immune and inflammatory cells into thecolon, and increased inflammatory cytokine production,thus, promoting the development of the intestinal in-flammation.
In addition to their effects on cytokine production,PGs may have important effects on dendritic cell func-tions and T-cell differentiation. The production of IL-12,a cytokine central to the development of Th1-type CD4�
T cells, is inhibited by exogenous PGE2.19,55 In addition,dendritic cells cultured in the presence of exogenousPGE2 skewed T cells to a Th2 phenotype whereas controldendritic cells generated Th0 cells.56,57 IL-10 is knownto have potent inhibitory effects on multiple dendriticcell functions.58–60 The absence of 2 potent regulators ofdendritic cell function (IL-10 and PGE2) may have rap-idly accelerated the development of pathogenic Th1 Tcells in the NSAID-treated IL-10�/� mice.PGs regulate epithelial growth and function and their
absence may lead to impaired function of the epithelialbarrier. PGs are potent stimulators of intestinal epithe-lial growth.61 COX-1–derived PGs have an importantrole in maintenance of the epithelial barrier. In a study ofthe effect of radiation injury on the colon, it was foundthat indomethacin (a nonselective COX inhibitor) in-creased the effects of radiation injury.62 Exogenous PGE2
reversed this effect whereas COX-2 selective inhibitorshad no effect, implicating COX-1–derived PGs as im-portant protectors of the epithelial barrier.62 Indeed,intestinal radiation injury in COX-1–deficient mice re-sulted in increased crypt epithelial cell apoptosis anddecreased clonogenic stem cell survival as compared withwild-type and COX-2–deficient mice,63 further showingan important role for COX-1–derived PGs in maintain-ing epithelial integrity in response to injury. In additionto their role in gastric protection,47 inhibition of COX-2–derived PGs resulted in increased colonic injury usingthe model of TNBS-induced colitis.64 PGs also regulateepithelial function and NSAID treatment has been doc-umented to increase epithelial permeability in humans.65
Altered epithelial barrier function has been implicated asa mechanism for the development of IBD in animalmodels, including a transgenic mouse model that ex-presses a dominant-negative N-cadherin mutant pro-tein66 and the MDR-1–deficient mouse.67 It has beenproposed that increased epithelial permeability contrib-utes to the development of spontaneous colitis in IL-10�/� mice.68 Interestingly, NSAID treatment of a sub-set of first-degree relatives of patients with Crohn’scolitis resulted in an exaggerated increase in intestinalpermeability, suggesting a genetic link between abnor-mal intestinal permeability and Crohn’s disease and po-tentially with the phenomenon of NSAID exacerbationof colitis.69,70 NSAID treatment may therefore increaseepithelial permeability directly by effects on epithelialcell functions or indirectly via inhibition of repair of
November 2002 NSAID–INDUCED COLITIS IN IL–10�/� MICE 1539

epithelial damage, thus, allowing bacterial antigens andbacterial products to enter the lamina propria. The ab-sence of IL-10 could then lead to an excessive inflamma-tory response in response to LPS (and other bacterialproducts such as CpG DNA), as suggested by the findingthat germ-free IL-10�/� mice do not develop colitis.71
The increased inflammatory response could then promotethe development of a cell-mediated immune response tobacterial proteins leading to the development of patho-genic Th1 T cells and chronic colitis.In summary, our studies indicate that PGs have a
pivotal role in the regulation of the immune and inflam-matory response in the colon of IL-10�/� mice. Wefound that NSAID treatment of IL-10�/� mice results inmarked cellular infiltration of the colon, increased in-flammatory cytokine production, and colonic epithelialproliferation. These changes occur within 2 weeks oftreatment in young IL-10�/� mice, whereas manymonths are required for the development of the sponta-neous IBD in IL-10�/� mice. NSAID-induced colitis inthe IL-10�/� mice also becomes chronic after removal ofthe drug. This novel model will allow us to study themechanisms involved in NSAID-induced exacerbation ofcolitis and potentially show the physiologic role of PGsin the regulation of the mucosal immune response. More-over, the rapid development of colitis in NSAID-treatedIL-10�/� mice affords us a model that will be useful forthe study of the mechanisms underlying the initiation ofIBD.
References1. Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van
De Putte LB, Lipsky PE. Cyclooxygenase in biology and disease.FASEB J 1998;12:1063–1073.
2. Needleman P, Isakson PC. The discovery and function of COX-2.J Rheumatol 1997;24(suppl 49):6–8.
3. Portanova JP, Zhang Y, Anderson GD, Hauser SD, Masferrer JL,Seibert K, Gregory SA, Isakson PC. Selective neutralization ofprostaglandin E2 blocks inflammation, hyperalgesia, and interleu-kin 6 production in vivo. J Exp Med 1996;184:883–891.
4. Anderson GD, Hauser SD, McGarity KL, Bremer ME, Isakson PC,Gregory SA. Selective inhibition of cyclooxygenase (COX)-2 re-verses inflammation and expression of COX-2 and interleukin 6 inrat adjuvant arthritis. J Clin Invest 1996;97:2672–2679.
5. Amin AR, Attur M, Abramson SB. Nitric oxide synthase and cy-clooxygenases: distribution, regulation, and intervention in arthri-tis. Curr Opin Rheumatol 1999;11:202–209.
6. MacDermott RP. Alterations in the mucosal immune system inulcerative colitis and Crohn’s disease. Med Clin North Am 1994;78:1207–1231.
7. Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide Hsynthases (cyclooxygenases)-1 and -2. J Biol Chem 1996;271:33157–33160.
8. Endo T, Ogushi F, Sone S. LPS-dependent cyclooxygenase-2 in-duction in human monocytes is down-regulated by IL-13, but notby IFN-gamma. J Immunol 1996;156:2240–2246.
9. DuBois RN, Tsujii M, Bishop P, Awad JA, Makita K, Lanahan A.Cloning and characterization of a growth factor-inducible cycloox-
ygenase gene from rat intestinal epithelial cells. Am J Physiol1994;266:G822–G827.
10. Aabakken L. Review article: non-steroidal, anti-inflammatorydrugs—the extending scope of gastrointestinal side effects. Ali-ment Pharmacol Ther 1992;6:143–162.
11. Giardiello FM, Spannhake EW, DuBois RN, Hylind LM, RobinsonCR, Hubbard WC, Hamilton SR, Yang VW. Prostaglandin levels inhuman colorectal mucosa: effects of sulindac in patients withfamilial adenomatous polyposis. Dig Dis Sci 1998;43:311–316.
12. Masferrer JL, Isakson PC, Seibert K. Cyclooxygenase-2 inhibitors:a new class of anti-inflammatory agents that spare the gastroin-testinal tract. Gastroenterol Clin North Am 1996;25:363–372.
13. Gould SR, Brash AR, Conolly ME, Lennard-Jones JE. Studies ofprostaglandins and sulphasalazine in ulcerative colitis. Prosta-glandins Med 1981;6:165–182.
14. Kaufmann HJ, Taubin HL. Nonsteroidal anti-inflammatory drugsactivate quiescent inflammatory bowel disease. Ann Intern Med1987;107:513–516.
15. Bjarnason I, Hayllar J, MacPherson AJ, Russell AS. Side effects ofnonsteroidal anti-inflammatory drugs on the small and large in-testine in humans. Gastroenterology 1993;104:1832–1847.
16. Kunkel SL, Spengler M, May MA, Spengler R, Larrick J, Remick D.Prostaglandin E2 regulates macrophage-derived tumor necrosisfactor gene expression. J Biol Chem 1988;263:5380–5384.
17. Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase induc-tion by suppressors of nitric oxide synthesis (IL-4, IL-10 andPGE2) in murine bone-marrow-derived macrophages. BiochemBiophys Res Commun 1995;206:667–673.
18. Strassmann G, Patil-Koota V, Finkelman F, Fong M, KambayashiT. Evidence for the involvement of interleukin 10 in the differen-tial deactivation of murine peritoneal macrophages by prosta-glandin E2. J Exp Med 1994;180:2365–2370.
19. van der Pouw Kraan TC, Boeije LC, Snijders A, Smeenk RJ,Wijdenes J, Aarden LA. Regulation of IL-12 production by humanmonocytes and the influence of prostaglandin E2. Ann N Y AcadSci 1996;795:147–157.
20. de Wall-Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE.Interleukin-10 inhibits cytokine synthesis by human monocytes:an autoregulatory role of IL-10 produced by monocytes. J ExpMed 1991;174:1209–1220.
21. Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O’Garra A.IL-10 inhibits cytokine production by activated macrophages.J Immunol 1991;147:3815–3822.
22. Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, MooreKW, O’Garra A. IL-10 acts on the antigen-presenting cell to inhibitcytokine production by Th1 cells. J Immunol 1991;146:3444–3451.
23. Hsu DH, Moore KW, Spits H. Differential effects of IL-4 and IL-10on IL-2-induced IFN-gamma synthesis and lymphokine-activatedkiller activity. Int Immunol 1992;4:563–569.
24. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993;75:263–274.
25. Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G,Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis andcolon cancer in interleukin-10-deficient mice are associated withaberrant cytokine production and CD4(�) TH1-like responses.J Clin Invest 1996;98:1010–1020.
26. Gennaro AR. Remington’s Pharmaceutical Sciences. 18th ed.Easton, PA: Mack Publishing Co, 1990:1632.
27. Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, WhelanJ. Prostaglandin E(2) protects intestinal tumors from nonsteroi-dal anti-inflammatory drug-induced regression in Apc(Min/�)mice. Cancer Res 2002;62:403–408.
28. Beckstead JH. A simple technique for preservation of fixation-sensitive antigens in paraffin-embedded tissues: addendum.J Histochem Cytochem 1995;43:345.
1540 BERG ET AL. GASTROENTEROLOGY Vol. 123, No. 5

29. Beckstead JH. A simple technique for preservation of fixation-sensitive antigens in paraffin-embedded tissues. J HistochemCytochem 1994;42:1127–1134.
30. Lefrancois L. Isolation of mouse small intestine intraepitheliallymphocytes. In: Coligan JE, Kruisbeek AM, Margulies OH, She-vach EM, Strober W, eds. Current protocols in immunology. Vol.1. New York: John Wiley and Sons, 1993:3.19.1–3.19.7.
31. Davidson NJ, Leach MW, Fort MM, Thompson-Snipes L, Kuhn R,Muller W, Berg DJ, Rennick DM. T helper cell 1-type CD4� T cells,but not B cells, mediate colitis in interleukin 10-deficient mice. JExp Med 1996;184:241–251.
32. Duggan DE, Hooke KF, Risley EA, Shen TY, Arman CG. Identifica-tion of the biologically active form of sulindac. J Pharmacol ExpTher 1977;201:8–13.
33. Piazza GA, Rahm AL, Krutzsch M, Sperl G, Paranka NS, Gross PH,Brendel K, Burt RW, Alberts DS, Pamukcu R. Antineoplastic drugssulindac sulfide and sulfone inhibit cell growth by inducing apo-ptosis. Cancer Res 1995;55:3110–3116.
34. Reddy BS, Kawamori T, Lubet RA, Steele VE, Kelloff GJ, Rao CV.Chemopreventive efficacy of sulindac sulfone against colon can-cer depends on time of administration during carcinogenic pro-cess. Cancer Res 1999;59:3387–3391.
35. Sung J, Russell RI, Nyeomans, Chan FK, Chen S, Fock K, Goh KL,Kullavanijaya P, Kimura K, Lau C, Louw J, Sollano J, Triadiafalo-poulos G, Xiao S, Brooks P. Non-steroidal anti-inflammatory drugtoxicity in the upper gastrointestinal tract. J Gastroenterol Hepa-tol 2000;15(Suppl):G58–G68.
36. Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY,Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK.IL-12-deficient mice are defective in IFN gamma production andtype 1 cytokine responses. Immunity 1996;4:471–481.
37. Gately MK, Renzetti LM, Magram J, Stern AS, Adorini L, Gubler U,Presky DH. The interleukin-12/interleukin-12-receptor system:role in normal and pathologic immune responses. Ann Rev Im-munol 1998;16:495–521.
38. Davidson NJ, Hudak SA, Lesley RE, Menon S, Leach MW, RennickDM. IL-12, but not IFN-gamma, plays a major role in sustainingthe chronic phase of colitis in IL-10-deficient mice. J Immunol1998;161:3143–3149.
39. Mahler M, Bristol IJ, Sundberg JP, Churchill GA, Birkenmeier EH,Elson CO, Leiter EH. Genetic analysis of susceptibility to dextransulfate sodium-induced colitis in mice. Genomics 1999;55:147–156.
40. Mahler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH,Elson CO, Sundberg JP. Differential susceptibility of inbredmouse strains to dextran sulfate sodium-induced colitis. Am JPhysiol 1998;274:G544–G551.
41. Sundberg JP, Elson CO, Bedigian H, Birkenmeier EH. Spontane-ous, heritable colitis in a new substrain of C3H/HeJ mice. Gas-troenterology 1994;107:1726–1735.
42. Morteau O, Morham SG, Sellon R, Dieleman LA, Langenbach R,Smithies O, Sartor RB. Impaired mucosal defense to acute co-lonic injury in mice lacking cyclooxygenase-1 or cyclooxygen-ase-2. J Clin Invest 2000;105:469–478.
43. Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI,Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, et al.Prostaglandin synthase 1 gene disruption in mice reduces ara-chidonic acid-induced inflammation and indomethacin-inducedgastric ulceration. Cell 1995;83:483–492.
44. Reese J, Brown N, Paria BC, Morrow J, Dey SK. COX-2 compen-sation in the uterus of COX-1 deficient mice during the pre-implantation period. Mol Cell Endocrinol 1999;150:23–31.
45. Ballou LR, Botting RM, Goorha S, Zhang J, Vane JR. Nociceptionin cyclooxygenase isozyme-deficient mice. Proc Natl Acad SciU S A 2000;97:10272–10276.
46. Morham SG, Langenbach R, Loftin CD, Tiano HF, VouloumanosN, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, et al.
Prostaglandin synthase 2 gene disruption causes severe renalpathology in the mouse. Cell 1995;83:473–482.
47. Mizuno H, Sakamoto C, Matsuda K, Wada K, Uchida T, NoguchiH, Akamatsu T, Kasuga M. Induction of cyclooxygenase 2 ingastric mucosal lesions and its inhibition by the specific antago-nist delays healing in mice. Gastroenterology 1997;112:387–397.
48. Newberry RD, Stenson WF, Lorenz RG. Cyclooxygenase-2-depen-dent arachidonic acid metabolites are essential modulators ofthe intestinal immune response to dietary antigen. Nat Med1999;5:900–906.
49. Marfaing-Koka A, Maravic M, Humbert M, Galanaud P, Emilie D.Contrasting effects of IL-4, IL-10 and corticosteroids on RANTESproduction by human monocytes. Int Immunol 1996;8:1587–1594.
50. Kopydlowski KM, Salkowski CA, Cody MJ, van Rooijen N, Major J,Hamilton TA, Vogel SN. Regulation of macrophage chemokineexpression by lipopolysaccharide in vitro and in vivo. J Immunol1999;163:1537–1544.
51. Takahashi S, Fujita T, Yamamoto A. Role of cyclooxygenase-2 inHelicobacter pylori-induced gastritis in Mongolian gerbils. Am JPhysiol 2000;279:G791–G798.
52. Zeidler R, Csanady M, Gires O, Lang S, Schmitt B, Wollenberg B.Tumor cell-derived prostaglandin E2 inhibits monocyte functionby interfering with CCR5 and Mac-1. FASEB J 2000;14:661–668.
53. Janabi N, Hau I, Tardieu M. Negative feedback between prosta-glandin and alpha- and beta-chemokine synthesis in human mi-croglial cells and astrocytes. J Immunol 1999;162:1701–1706.
54. Thivierge M, Le Gouill C, Tremblay MJ, Stankova J, Rola-Pleszc-zynski M. Prostaglandin E2 induces resistance to human immu-nodeficiency virus-1 infection in monocyte-derived macrophages:downregulation of CCR5 expression by cyclic adenosine mono-phosphate. Blood 1998;92:40–45.
55. van der Pouw Kraan TC, Boeije LC, Smeenk RJ, Wijdenes J,Aarden LA. Prostaglandin-E2 is a potent inhibitor of human inter-leukin 12 production. J Exp Med 1995;181:775–779.
56. Kalinski P, Hilkens CM, Snijders A, Snijdewint FG, KapsenbergML. Dendritic cells, obtained from peripheral blood precursors inthe presence of PGE2, promote Th2 responses. Adv Exp Med Biol1997;417:363–367.
57. Kalinski P, Hilkens CM, Snijders A, Snijdewint FG, KapsenbergML. IL-12-deficient dendritic cells, generated in the presence ofprostaglandin E2, promote type 2 cytokine production in maturinghuman naive T helper cells. J Immunol 1997;159:28–35.
58. Macatonia SE, Doherty TM, Knight SC, O’Garra A. Differentialeffect of IL-10 on dendritic cell-induced T cell proliferation andIFN-gamma production. J Immunol 1993;150:3755–3765.
59. Corinti S, Albanesi C, la Sala A, Pastore S, Girolomoni G. Regu-latory activity of autocrine IL-10 on dendritic cell functions. J Im-munol 2001;166:4312–4318.
60. D’Amico G, Frascaroli G, Bianchi G, Transidico P, Doni A, VecchiA, Sozzani S, Allavena P, Mantovani A. Uncoupling of inflamma-tory chemokine receptors by IL-10: generation of functional de-coys. Nat Immunol 2000;1:387–391.
61. Tessner TG, Cohn SM, Schloemann S, Stenson WF. Prostaglan-dins prevent decreased epithelial cell proliferation associatedwith dextran sodium sulfate injury in mice. Gastroenterology1998;115:874–882.
62. Cohn SM, Schloemann S, Tessner T, Seibert K, Stenson WF.Crypt stem cell survival in the mouse intestinal epithelium isregulated by prostaglandins synthesized through cyclooxygen-ase-1. J Clin Invest 1997;99:1367–1379.
63. Houchen CW, Stenson WF, Cohn SM. Disruption of cyclooxygen-ase-1 gene results in an impaired response to radiation injury.Am J Physiol 2000;279:G858–G865.
64. Reuter BK, Asfaha S, Buret A, Sharkey KA, Wallace JL. Exacer-bation of inflammation-associated colonic injury in rat through
November 2002 NSAID–INDUCED COLITIS IN IL–10�/� MICE 1541

inhibition of cyclooxygenase-2. J Clin Invest 1996;98:2076–2085.
65. Bjarnason I, Zanelli G, Prouse P, Williams P, Gumpel MJ, Levi AJ.Effect of non-steroidal anti-inflammatory drugs on the humansmall intestine. Drugs 1986;32(Suppl 1):35–41.
66. Hermiston ML, Gordon JI. Inflammatory bowel disease and ade-nomas in mice expressing a dominant negative N-cadherin. Sci-ence 1995;270:1203–1207.
67. Panwala CM, Jones JC, Viney JL. A novel model of inflammatorybowel disease: mice deficient for the multiple drug resistancegene, mdr1a, spontaneously develop colitis. J Immunol 1998;161:5733–5744.
68. Madsen KL, Malfair D, Gray D, Doyle JS, Jewell LD, Fedorak RN.Interleukin-10 gene-deficient mice develop a primary intestinalpermeability defect in response to enteric microflora. InflammBowel Dis 1999;5:262–270.
69. Zamora SA, Hilsden RJ, Meddings JB, Butzner JD, Scott RB,Sutherland LR. Intestinal permeability before and after ibuprofenin families of children with Crohn’s disease. Can J Gastroenterol1999;13:31–36.
70. Hilsden RJ, Meddings JB, Sutherland LR. Intestinal permeability
changes in response to acetylsalicylic acid in relatives of patientswith Crohn’s disease. Gastroenterology 1996;110:1395–1403.
71. Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W,Balish E, Rennick DM, Sartor RB. Resident enteric bacteria arenecessary for development of spontaneous colitis and immunesystem activation in interleukin-10-deficient mice. Infect Immun1998;66:5224–5231.
Received December 7, 2001. Accepted July 18, 2002.Address requests for reprints to: Daniel J. Berg, M.D., Department of
Internal Medicine, C32-GH, University of Iowa Hospitals, 200 HawkinsDrive, Iowa City, IA 52242. e-mail: [email protected]; fax: (319)353-8383.Supported by the Crohn’s and Colitis Foundation of America, First
Award (to D.J.B.).Dr. Earle is a Cell Pathways, Inc. employee. Dr. Pamukcu is the
Founder, Director, and Chief Scientific Officer at Cell Pathways, Inc.and has significant stock ownership and equity interests in the com-pany. Dr. Alila is also an employee at Cell Pathways, Inc. with stockownership in the company.
1542 BERG ET AL. GASTROENTEROLOGY Vol. 123, No. 5
Copyright © 2022 FDOKUMEN




















