
Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition of nuclear factor...
16
Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition of nuclear factor kappaB and induction of apoptosis Sanjay Gupta 1,4 , Kedar Hastak 2,4 , Farrukh Afaq 3 , Nihal Ahmad 3 and Hasan Mukhtar* ,3 1 Department of Urology, Case Western Reserve University, Cleveland, OH 44106, USA; 2 Department of Environmental Health Sciences, Case Western Reserve University, Cleveland, OH 44106, USA; and 3 Department of Dermatology, Medical Sciences Center, 1300 University Avenue, University of Wisconsin, Madison, WI 53706, USA Green tea constituent () epigallocatechin-3-gallate (EGCG) has shown remarkable cancer-preventive and some cancer-therapeutic effects. This is partially because of its ability to induce apoptosis in cancer cells without affecting normal cells. Previous studies from our labora- tory have shown the involvement of NF-jB pathway in EGCG-mediated cell-cycle deregulation and apoptosis of human epidermoid carcinoma A431 cells. Here we show the essential role of caspases in EGCG-mediated inhibi- tion of NF-jB and its subsequent apoptosis. Treatment of A431 cells with EGCG (10–40 lg/ml) resulted in dose- dependent inhibition of NF-jB/p65, induction of DNA breaks, cleavage of poly(ADP-ribose) polymerase (PARP) and morphological changes consistent with apoptosis. EGCG treatment of cells also resulted in significant activation of caspases, as shown by the dose- and time-dependent increase in DEVDase activity, and protein expression of caspase-3, -8 and -9. EGCG- mediated caspase activation induces proteolytic cleavage of NF-jB/p65 subunit, leading to the loss of transactiva- tion domains, and driving the cells towards apoptosis. EGCG-mediated induction of apoptosis was significantly blocked by the caspase inhibitor N-benzyloxycarbonyl- Val-Ala-Asp(OMe)-fluoromethylketone (Z-VAD-FMK), and moderately blocked by the specific caspase-3 inhibitor Z-DEVD-FMK. Further, pretreatment of cells with Z- VAD-FMK was found to suppress the cleavage of NF-jB/ p65 subunit, thereby increasing nuclear translocation, DNA binding and transcriptional activity, thus protecting the cells from EGCG-induced apoptosis. Taken together, these studies for the first time demonstrate that EGCG- mediated activation of caspases is critical, at least in part, for inhibition of NF-jB and subsequent apoptosis. Oncogene (2004) 23, 2507–2522. doi:10.1038/sj.onc.1207353 Published online 15 December 2003 Keywords: green tea; epigallocatechin-3-gallate; cas- pases; apoptosis; nuclear factor-kappa B Introduction Epigallocatechin-3-gallate (EGCG), the major polyphe- nolic constituent present in green tea, has received much attention over the last few years, as a potential cancer- chemopreventive and cancer-chemotherapeutic agent, possibly because of its wide range of effects on a number of cellular processes and efficacy in many tumor model systems (Singh et al., 2002; Chung et al., 2003; Gupta et al., 2003; Hastak et al., 2003). Our previous studies have shown that EGCG, at pharmacologically attain- able concentrations, results in inhibition of cell growth, arrest of the cell cycle in G0/G1 phase and induction of apoptosis in some human carcinoma cells (Ahmad et al., 1997). In subsequent studies, many other laboratories reported similar findings in many other cancer cell types (Yang et al., 1998; Chung et al., 2001; Masuda et al., 2001). The mechanism of EGCG-mediated apoptosis of cancer cells is incompletely understood. Such an under- standing could be critical for developing EGCG and related compounds as agents for prevention and/or therapy of cancer. The transcription factor nuclear factor-kappaB (NF- kB) plays a critical role in the regulation of genes related to cell survival, proliferation and apoptosis (Schwartz et al., 1999; Joyce et al., 2001; Karin and Lin, 2002). NF-kB consists of multiple members of the Rel family of proteins that include NF-kB1 (p105/p50), NF-kB2 (p100/p52), RelA (p65), RelB and c-Rel (Bours et al., 2000). The NF-kB dimers are present in cytoplasm in an inactive form bound to inhibitory subunits, IkB (Bours et al., 2000). Upon activation, IkB is phosphorylated, which marks the inhibitor for ubiquitination and degradation by the proteasome-dependent pathway (Aggarwal, 2000; Bours et al., 2000). This process allows translocation of active NF-kB complexes into the nucleus, where they bind to specific DNA motifs in the promoter/enhancer regions of target genes and activate their transcription. Modulation in the transac- tivation domains and/or nuclear translocation of NF-kB by a variety of stimuli leads to cell cycle arrest and apoptosis (Bours et al., 2000; Joyce et al., 2001; Karin and Lin, 2002). In recent years, considerable attention has been focused on NF-kB inhibition as a target for developing agents for cancer prevention and therapy (Yamamoto and Gaynor, 2001). Received 22 October 2003; revised 11 November 2003; accepted 11 November 2003 *Correspondence: H Mukhtar; E-mail: [email protected] 4 Contributed equally to this work Oncogene (2004) 23, 2507–2522 & 2004 Nature Publishing Group All rights reserved 0950-9232/04 $25.00 www.nature.com/onc
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition of nuclear factor...

Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition
of nuclear factor kappaB and induction of apoptosis
Sanjay Gupta1,4, Kedar Hastak2,4, Farrukh Afaq3, Nihal Ahmad3 and Hasan Mukhtar*,3
1Department of Urology, Case Western Reserve University, Cleveland, OH 44106, USA; 2Department of Environmental HealthSciences, Case Western Reserve University, Cleveland, OH 44106, USA; and 3Department of Dermatology, Medical Sciences Center,1300 University Avenue, University of Wisconsin, Madison, WI 53706, USA
Green tea constituent (�) epigallocatechin-3-gallate(EGCG) has shown remarkable cancer-preventive andsome cancer-therapeutic effects. This is partially becauseof its ability to induce apoptosis in cancer cells withoutaffecting normal cells. Previous studies from our labora-tory have shown the involvement of NF-jB pathway inEGCG-mediated cell-cycle deregulation and apoptosis ofhuman epidermoid carcinoma A431 cells. Here we showthe essential role of caspases in EGCG-mediated inhibi-tion of NF-jB and its subsequent apoptosis. Treatment ofA431 cells with EGCG (10–40 lg/ml) resulted in dose-dependent inhibition of NF-jB/p65, induction of DNAbreaks, cleavage of poly(ADP-ribose) polymerase(PARP) and morphological changes consistent withapoptosis. EGCG treatment of cells also resulted insignificant activation of caspases, as shown by the dose-and time-dependent increase in DEVDase activity, andprotein expression of caspase-3, -8 and -9. EGCG-mediated caspase activation induces proteolytic cleavageof NF-jB/p65 subunit, leading to the loss of transactiva-tion domains, and driving the cells towards apoptosis.EGCG-mediated induction of apoptosis was significantlyblocked by the caspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone (Z-VAD-FMK),and moderately blocked by the specific caspase-3 inhibitorZ-DEVD-FMK. Further, pretreatment of cells with Z-VAD-FMK was found to suppress the cleavage of NF-jB/p65 subunit, thereby increasing nuclear translocation,DNA binding and transcriptional activity, thus protectingthe cells from EGCG-induced apoptosis. Taken together,these studies for the first time demonstrate that EGCG-mediated activation of caspases is critical, at least in part,for inhibition of NF-jB and subsequent apoptosis.Oncogene (2004) 23, 2507–2522. doi:10.1038/sj.onc.1207353Published online 15 December 2003
Keywords: green tea; epigallocatechin-3-gallate; cas-pases; apoptosis; nuclear factor-kappa B
Introduction
Epigallocatechin-3-gallate (EGCG), the major polyphe-nolic constituent present in green tea, has received muchattention over the last few years, as a potential cancer-chemopreventive and cancer-chemotherapeutic agent,possibly because of its wide range of effects on a numberof cellular processes and efficacy in many tumor modelsystems (Singh et al., 2002; Chung et al., 2003; Guptaet al., 2003; Hastak et al., 2003). Our previous studieshave shown that EGCG, at pharmacologically attain-able concentrations, results in inhibition of cell growth,arrest of the cell cycle in G0/G1 phase and induction ofapoptosis in some human carcinoma cells (Ahmad et al.,1997). In subsequent studies, many other laboratoriesreported similar findings in many other cancer cell types(Yang et al., 1998; Chung et al., 2001; Masuda et al.,2001). The mechanism of EGCG-mediated apoptosis ofcancer cells is incompletely understood. Such an under-standing could be critical for developing EGCG andrelated compounds as agents for prevention and/ortherapy of cancer.
The transcription factor nuclear factor-kappaB (NF-kB) plays a critical role in the regulation of genes relatedto cell survival, proliferation and apoptosis (Schwartzet al., 1999; Joyce et al., 2001; Karin and Lin, 2002).NF-kB consists of multiple members of the Rel family ofproteins that include NF-kB1 (p105/p50), NF-kB2(p100/p52), RelA (p65), RelB and c-Rel (Bours et al.,2000). The NF-kB dimers are present in cytoplasm in aninactive form bound to inhibitory subunits, IkB (Bourset al., 2000). Upon activation, IkB is phosphorylated,which marks the inhibitor for ubiquitination anddegradation by the proteasome-dependent pathway(Aggarwal, 2000; Bours et al., 2000). This processallows translocation of active NF-kB complexes intothe nucleus, where they bind to specific DNA motifs inthe promoter/enhancer regions of target genes andactivate their transcription. Modulation in the transac-tivation domains and/or nuclear translocation of NF-kBby a variety of stimuli leads to cell cycle arrest andapoptosis (Bours et al., 2000; Joyce et al., 2001; Karinand Lin, 2002). In recent years, considerable attentionhas been focused on NF-kB inhibition as a target fordeveloping agents for cancer prevention and therapy(Yamamoto and Gaynor, 2001).
Received 22 October 2003; revised 11 November 2003; accepted 11November 2003
*Correspondence: H Mukhtar; E-mail: [email protected] equally to this work
Oncogene (2004) 23, 2507–2522& 2004 Nature Publishing Group All rights reserved 0950-9232/04 $25.00
www.nature.com/onc

Several recent studies have identified the involvement ofmultiple caspases in the proteolytic cascade of apoptosis(Grutter, 2000; Zimmermann et al., 2001). Caspases aresynthesized as zymogens that require proteolytic cleavageto generate active enzyme subunits (Nicholson, 1999;Grutter, 2000). These activating cleavage events areconducted by other caspases, and are thought to representa major regulatory step in the apoptotic pathway(Nicholson, 1999; Stennicke and Salvesen, 2000). Acti-vated caspases cleave several target proteins includingpoly(ADP-ribose) polymerase (PARP) (Tewari et al.,1995), retinoblastoma protein (Janicke et al., 1996),cytoskeletal proteins (Lazebnik et al., 1995), Bcl2 andBcl-xL (Cheng et al., 1997; Clem et al., 1998). Studies have
a
b
c
d
2
0
4
6
Num
ber
of c
ells
X 1
06
0 10 20 40
0 10 20 40
EGCG (µg/ml); 24 h
EGCG (µg/ml); 48 h
EGCG (µg/ml); 48 h
EGCG (µg/ml); 24 h
Enr
ichm
ent F
acto
r
2
1.5
0.5
1
0
0
0
10 20 40
10 20 40
**
*
PARP 116 KD85 KD
*
Figure 1 Dose-dependent effect of EGCG on cell growthinhibition and induction of apoptosis in A431 cells. (a) Cell growthinhibition was ascertained by trypan blue exclusion assay. The datashown are means7s.e. of three independent experiments. Statis-tical analysis was performed by Student’s t-test, and Po0.05 wasconsidered significant. (b) Apoptosis was determined by cell-deathELISAPLUS, as per the vendor’s protocol. Data are expressed asenrichment factors. Statistical analysis was performed by Student’st-test, and Po0.05 was considered significant. (c) DNA ladderformation. Cells were treated with the indicated dose of EGCG for48 h, as described under ‘Materials and methods’; DNA wasisolated and subjected to gel electrophoresis, followed by visualiza-tion of bands. (d) Immunoblot analysis of PARP cleavage. The celllysate was prepared following treatment, and 50 mg of protein wassubjected to 12% Tris-glycine gel electrophoresis, followed byimmunoblot analysis and chemiluminescence detection, as de-scribed under ‘Materials and methods’. The data shown here arerepresentative of three independent experiments with similar results
a
b
c
d
2
0
4
6
Num
ber
of c
ells
X 1
06
0 12 24 48
0 12 24 48Time (h); E 20 µg/ml
Time (h); E 20 µg/ml
Time (h); E 20 µg/ml
Time (h); E 20 µg/ml
Enr
ichm
ent F
acto
r
2
1.5
0.5
1
0
0 12 24 48
0 12 24 48
116 KD
85 KDPARP
*
*
*
**
Role of caspases in EGCG-induced apoptosisS Gupta et al
2508
Oncogene

also identified RelA and p50 as a substrate for activatedcaspase (Levkau et al., 1999). Further, cleavage of the45kDa subunit of DNA fragmentation factor of genomicDNA into nucleosomal fragments is the hallmark ofapoptosis (Tang and Kidd, 1998).
Studies from our laboratory and elsewhere haveshown that EGCG is capable of inhibiting NF-kBactivation in cancer cells (Ahmad et al., 2000; Surh et al.,2001). Recently, EGCG has been shown to modulatecaspase activation (Islam et al., 2000; Hayakawa et al.,2001). However, the relationship between caspaseactivation and NF-kB inhibition by EGCG is comple-tely unknown. Here we demonstrate the essential role ofcaspases in EGCG-mediated inhibition of NF-kB andinduction of apoptosis. The mechanism responsible forEGCG-mediated inhibition of NF-kB is the cleavage ofNF-kB/p65 subunit by active caspases, and it providesevidence that this cleavage contributes to the apoptosisof human epidermoid carcinoma A431 cells.
Results
EGCG-mediated growth inhibition and apoptosis in A431cells
Employing A431 cells, we first evaluated the effect ofEGCG on cell growth by trypan blue exclusion assay.
Figure 2 Time-dependent effect of EGCG on cell growthinhibition and induction of apoptosis in A431 cells. (a) Cell growthinhibition was ascertained by trypan blue exclusion assay. The datashown are means7s.e. of three independent experiments. Statis-tical analysis was performed by Student’s t-test, and Po0.05 wasconsidered significant. (b) Apoptosis was determined by cell-deathELISAPLUS as per vendor’s protocol. Data are expressed asenrichment factors. The data shown are means7s.e. of threeindependent experiments, statistical analysis was performed byStudent’s t-test, and Po0.05 was considered significant. (c) DNAladder formation. Cells were treated with 20-mg/ml dose of EGCGfor the indicated time interval, as described under ‘Materials andmethods’; DNA was isolated and subjected to gel electrophoresis,followed by visualization of bands. (d) Immunoblot analysis ofPARP cleavage. The cell lysate was prepared following treatment,and 50 mg of protein was subjected to 12% Tris-glycine gelelectrophoresis followed by immunoblot analysis and chemilumi-nescence detection, as described under ‘Materials and methods’.The data shown here are representative of three independentexperiments with similar results
Figure 3 Effect of EGCG on cell morphology and quantification of apoptosis in A431 cells. (a) Dose-dependent and (b) time-dependent effect of EGCG on morphological changes in A431 cells, as evidenced by fluorescence microscopy. Apoptotic cells arestained with annexin V (green fluorescence) and the necrotic cells are stained with PI (red fluorescence). Details are described under‘Materials and methods’ section. (c) Dose-dependent and (d) time-dependent quantification of apoptosis in A431 cells by flowcytometry. Cells were treated at the indicated dose and times with EGCG, labeled with deoxyuridine triphosphate using terminaldeoxynucleotide transferase and PI, by using an apoptosis kit followed by flow cytometry. Cells showing deoxyuridine triphosphatefluorescence above that of control population, as indicated by the line in each histogram, are considered as apoptotic cells, and theirpercentage population is shown in each box. Details are described under ‘Materials and methods’ section. Data shown here are from arepresentative experiment repeated three times with similar results
Role of caspases in EGCG-induced apoptosisS Gupta et al
2509
Oncogene

As shown in Figure 1a, EGCG treatment (10–40 mg/mlfor 24 h) of A431 cells resulted in a dose-dependentinhibition of cell growth, which was more pronounced in20 and 40 mg/ml of EGCG treatment. To evaluate theinduction of apoptosis by EGCG, we performedenzyme-linked immunoabsorbent assay (ELISA)(Figure 1b). Compared to control, EGCG treatmentresulted in significant apoptosis in A431 cells (20 and40 mg/ml), in a dose-dependent fashion. The induction of
Figure 4 Effect of EGCG on DEVDase activity and proteinexpression of caspases in A431 cells. (a) Dose-dependent and (b)time-dependent effect of EGCG on DEVDase activity assay.Caspase activity was measured by AFC-DEVDase assay, asdescribed under ‘Materials and methods’. Data represents mean-s7s.e. of three independent experiments. Statistical analysis wasperformed by Student’s t-test, and Po0.05 was consideredsignificant. (c) Dose-dependent and (d) time-dependent effect ofEGCG on protein expression of the pro- and active forms ofcaspase-3 and -8. For caspase-9, the antibody detects only the activeform. The cell lysate was prepared following treatment, and 50 mgof protein was subjected to 12% Tris-glycine gel electrophoresisfollowed by immunoblot analysis and chemiluminescence detection,as described under ‘Materials and methods’. The data shown hereare representative of three independent experiments with similarresults
9
12
6
3
0
0 10 20 40
0 10 20 40
EGCG (µg/ml); 24 h
Time (h); E 20 µg/ml
Time (h); E 20 µg/ml
0 12 24 48
0 12 24 48
EGCG (µg/ml); 24 h
Spe
cific
Act
ivity
pmol
es/m
in/m
g pr
otei
n
9
12
6
3
0
Spe
cific
Act
ivity
pmol
es/m
in/m
g pr
otei
n
*
*
**
Caspase 3
Caspase 8
Caspase 9
Caspase 3
Caspase 8
Caspase 9
32 KD (pro)17 KD (active)
55 KD (pro)43 KD (active)
18 KD (active)
35 KD (active)
32 KD (pro)17 KD (active)
55 KD (pro)43 KD (active)
18 KD (active)
35 KD (active)
a
b
c
d
a
b
c
d
6
4
2
00 0 10 10 20 20 40 40
- - - -+ + + +
0 0 10 10 20 20 40 40
- - - -+ + + +
2
1
0
Num
ber
of c
ells
X 1
06EGCG (µg/ml)
Z-VAD-FMK (10µm)
EGCG (µg/ml)
Z-VAD-FMK (10µm)
EGCG (µg/ml)
Z-VAD-FMK (10µm)
0 0 10 10 20 20 40 40- - - -+ + + +
0 0 10 10 20 20 40 40
- - - -+ + + +EGCG (µg/ml)Z-VAD-FMK (10µm)
Enr
ichm
ent F
acto
r
15
12
9
6
3
0
Spe
cific
Act
ivity
pmol
es/m
in/m
g pr
otei
n
Caspase 3
Caspase 8
Caspase 9
32 KD (pro)17 KD (active)
55 KD (pro)43 KD (active)
18 KD (active)
35 KD (active)
**
*
**
**
Role of caspases in EGCG-induced apoptosisS Gupta et al
2510
Oncogene

apoptosis was further assessed by the formation ofDNA ladder. As shown in Figure 1c, compared tocontrol, EGCG treatment (20 and 40 mg/ml for 48 h)resulted in the formation of DNA fragments in A431cells. Additionally, PARP cleavage analysis showed thatthe full-size PARP (116 kDa) protein was cleaved toyield an 85 kDa fragment after treatment of cells withEGCG at 20 and 40 mg/ml concentrations (Figure 1d).
In a time-dependent study, we found that a 20 mg/mldose of EGCG resulted in a cell inhibitory response,which was significant as early as 12 h post-EGCGtreatment, with an increasing trend up to 48 h ofobservation, as shown by trypan blue exclusion assay(Figure 2a). The induction of apoptosis was observed byELISA assay that showed significant apoptosis byEGCG (20 mg/ml) at 24 and 48 h post-EGCG treatment(Figure 2b). Similarly, treatment with 20 mg/ml concen-tration of EGCG resulted in the formation of DNAladder at 48 h post-treatment (Figure 2c). The time-dependent effect of EGCG treatment was similar to thatof DNA fragmentation, where a 20 mg/ml concentrationof EGCG resulted in the cleavage of PARP at 48 hpost-treatment (Figure 1d).
The induction of apoptosis by EGCG was alsoevident from the morphology of cells, as assessed byfluorescence microscopy, after labeling the cells with anapoptosis detection kit containing annexin V andpropidium iodide (PI) (Figure 3a, b). We used thismethod because it identifies the apoptotic (greenfluorescence) as well as necrotic (red fluorescence) cells.As shown by the data in Figure 3a, EGCG treatmentresulted in a dose-dependent apoptosis in A431 cells.The time-dependent effect of EGCG treatment wasidentical to that of other apoptosis parameters examined(Figure 3b). These data indicated that EGCG treatmentalso resulted in necrosis of these cells, which may be asecondary event in the apoptotic process.
We next quantified the extent of apoptosis by flow-cytometric analysis of EGCG-treated A431 cells labeledwith bromo-deoxyuridine triphosphate and PI. Asshown by the data in Figure 3c, compared to control(1.5%), EGCG treatment resulted in 8.0, 12.5 and16.8% of TUNEL-positive cells at 10, 20 and 40 mg/mldose. The time-dependent assay for apoptosis (20 mg/mldose) exhibited a significant increase in TUNEL-positivecells (control 1.6 versus 6.5, 12.8 and 18.4%) at 12, 24and 48 h post-EGCG treatment, respectively (Figure 3d).Based on these data, we selected a dose of 20 mg/ml and24 h post-EGCG treatment for further studies.
EGCG-induced caspase activation and increase in proteinexpression of caspase-3, -8 and -9 in A431 cells
To investigate the involvement of caspase(s) in EGCG-mediated apoptosis, we first evaluated the caspaseactivity by incubating the fluorogenic peptideAc-DEVD-AFC with the cell lysates undergoingapoptosis. The release of 7-amino-4-trifluoromethyl-coumarin (AFC) was measured with a fluorescencespectrophotometer at 400-nm excitation and 505-nmemission. Treatment of A431 cells with EGCG leadsto the dose-dependent activation of caspases with 1.2-,1.7- and 1.8-fold induction at 10, 20 and 40 mg/ml,respectively (Figure 4a). The time-dependent EGCG-mediated caspase activation of A431 cells (20 mg/mldose) was maximum (1.9-fold) at 24 h of treatment(Figure 4b).
We next performed immunoblot analysis of themembers of group II and III caspases in the cell lysateobtained from EGCG-treated A431 cells. We have usedantibodies that specifically recognize the pro- and activeforms of caspases (for caspase-3, and -8). For caspase-9,we have used the antibody that recognizes the cleaved(active) product at 35 kDa. EGCG treatment of cellsresulted in a dose-dependent increase in the cleavedproduct of casapse-3 (17 kDa), with concomitantdecrease in the pro-form (32 kDa). Similarly, a dose-dependent increase in the active form of caspase-8 (43and 18 kDa) and caspase-9 (35 kDa) was observed afterEGCG treatment (Figure 4c). Further, EGCG treat-ment also resulted in a significant time-dependentincrease in the active products of caspase-3, -8 and –9,respectively (Figure 4d).
Inhibition of caspases increases cell growth and inhibitsEGCG-mediated apoptosis in A431 cells
The above results indicate that EGCG treatment ofA431 cells results in apoptosis via activation of caspases.Since Z-VAD-FMK inhibits caspase activation, we nextasked whether Z-VAD-FMK could block EGCG-mediated apoptosis. Compared to EGCG treatment(10, 20 and 40 mg/ml), pre-incubation of A431 cells withZ-VAD-FMK (10 mM) 2 h before EGCG treatmentresulted increases of 14.5, 18 and 12% in cell growth(Figure 5a). These results were further confirmed byELISA assay, where incubation of cells with Z-VAD-FMK with EGCG inhibited cell death by 16%
Figure 5 EGCG-mediated cell growth inhibition and induction ofapoptosis is blocked by caspase inhibitor in A431 cells. (a) Cellgrowth inhibition, as ascertained by trypan blue exclusion assay.Cells were incubated with 10mM concentration of the generalcaspase inhibitor Z-VAD-FMK for 2 h, followed by addition of theindicated doses of EGCG. The data shown are means7s.e. of threeindependent experiments; statistical analysis was performed byStudent’s t-test, and Po0.05 was considered significant. (b)Apoptosis as determined by cell-death ELISAPLUS kit accordingto the vendor’s protocol. Data are expressed as enrichment factors.The data shown are means7s.e. of three independent experiments.Statistical analysis was performed by Student’s t-test, and Po0.05was considered significant. (c) DEVDase activity assay. Caspaseactivity was measured by AFC-DEVDase assay, as described under‘Materials and methods’. Data represent means7s.e. of threeindependent experiments. Statistical analysis was performed byStudent’s t-test, and Po0.05 was considered significant. (d)Immunoblot analysis for protein expression of the pro- and activeforms of caspase-3 and -8. For caspase-9, the antibody detects onlythe active form. The cell lysate was prepared following treatment,and 50 mg of protein was subjected to 12% Tris-glycine gelelectrophoresis, followed by immunoblot analysis and chemilumi-nescence detection, as described under ‘Materials and methods’.The data shown here are representative of three independentexperiments with similar results
Role of caspases in EGCG-induced apoptosisS Gupta et al
2511
Oncogene

at 10 mg/ml, 32% at 20 mg/ml and 18% at 40 mg/ml, ascompared to EGCG alone treatment (Figure 5b).
We next investigated the effect of caspase inhibitorand EGCG on the extent of caspase activation. Pre-incubation of cells with Z-VAD-FMK resulted insignificant decrease in caspase activity. As observed bya comparison to the corresponding control (10-, 20- and40-mg/ml EGCG concentration), a significant decrease(1.5-, 1.7- and 1.6-fold) in caspase activity was observedwith Z-VAD-FMK treatment (Figure 5c). Immunoblotanalysis further confirmed the results, where a signifi-cant decrease in the protein expression of the activeproducts of caspase-3, -8 and -9 was observed by thepre-incubation of A431 cells with the Z-VAD-FMKcaspase inhibitor. No significant modulation in theprotein expression of caspase-3, -8 and -9 was observedin the Z-VAD-FMK treatment group (Figure 5d). Inaddition to Z-VAD-FMK, we also used the caspase-3-specific inhibitor Z-DVED-FMK to assess the invol-vement of caspase-3 during EGCG-mediated apoptosis.Compared to control, pre-incubation of A431 cells withZ-DVED-FMK exhibited increased cell growth andinhibition of apoptosis, but the effects were moderate incomparison to the general caspase inhibitor Z-VAD-FMK (data not shown).
We next quantified the extent of apoptosis by flowcytometry, exhibiting 13 and 17% of TUNEL-positivecells at 20- and 40-mg/ml concentrations of EGCG,which were considerably reduced to 4.5 and 9.3%, as aresult of the treatment of cells with Z-VAD-FMK(Figure 6a). The extent of apoptosis was furtherestimated by fluorescence microscopy using annexin-V- and PI-stained cells. As shown by the representativepictures (Figure 6b), EGCG (20 and 40 mg/ml) treatmentof A431 cells resulted in apoptosis at both theconcentrations, as evidenced by chromatin condensation
and nuclear condensation (20 mg/ml). The cells pre-incubated with Z-VAD-FMK exhibited few apoptoticcells at 20 and 40 mg/ml of EGCG. No apoptotic cellswere observed in control and Z-VAD-FMK-treatedgroups (data not shown).
Inhibition of NF-kB increases EGCG-mediated apoptosisin A431 cells
In the next series of experiments, we investigated theeffect of EGCG on the constitutive expression of NF-kB/p65, NF-kB/p50 and IkBa in A431 cells. As shownby immunoblot analysis, EGCG treatment resulted in adose- and time-dependent decrease in nuclear transloca-tion of NF-kB/p65 protein expression, but not of theNF-kB/p50, as estimated in the nuclear fraction of thesecells (Figure 7a). A similar effect was observed incytosolic fraction, where EGCG treatment of cells leadsto inhibition in the protein expression of NF-kB/p65 butnot of NF-kB/p50 isomer (data not shown). Theinhibition of NF-kB/p65 in the nuclear fractioncorrelated with the increase in the protein expressionof IkBa in the cytosol, both in dose- and time-dependentfashion (Figure 7a), which was further confirmed by amore sensitive method that can detect NF-kB/p65 andrecognize an epitope on p65 by ELISA. EGCG-mediated inhibition of NF-kB/p65 expression wasfurther correlated with increasing apoptosis in a dose-dependent fashion (Figure 7b). Similar results wereobserved by treatment of cells with 20 mg/ml ofEGCG for different time intervals (Figure 7c). Theseobservations suggest the existence of two intriguingpossibilities for the mechanism of EGCG-mediated NF-kB inactivation and apoptosis: (i) EGCG may causephosphorylative degradation of IkBa, thereby inhibitingthe constitutive NF-kB expression, and (ii) modulation
Figure 6 EGCG-mediated apoptosis is blocked by caspase inhibitor in A431 cells. (a) Quantification of apoptosis in A431 cells byflow cytometry. Cells were incubated with 10mM concentration of the general caspase inhibitor Z-VAD-FMK for 2 h, followed byaddition of the indicated doses of EGCG, labeled with deoxyuridine triphosphate using terminal deoxynucleotide transferase and PI,by using the apoptosis kit followed by flow cytometry. Cells showing deoxyuridine triphosphate fluorescence above that of the controlpopulation, as indicated by the line in each histogram, are considered as apoptotic cells, and their percentage population is shown ineach box. Details are described under ‘Materials and methods’ section. Data shown here are from a representative experiment repeatedthree times with similar results. (b) Morphological changes as evidenced by fluorescence microscopy. Apoptotic cells are stained withannexin V (green fluorescence) and the necrotic cells are stained with PI (red fluorescence). Details are described under ‘Materials andmethods’ section
Role of caspases in EGCG-induced apoptosisS Gupta et al
2512
Oncogene

of the transactivation domains of NF-kB, critical for cellsurvival.
NF-kB DNA-binding activity and transcription is alteredin EGCG-mediated apoptosis of A431 cells
Previous experiments have demonstrated that treatmentof A431 cells with EGCG resulted in caspase activation,inhibition of NF-kB, and exhibit typical morphologicaland biochemical criteria for apoptosis. In the next seriesof experiment, we performed gel shift assays in controland EGCG-treated groups. As shown in Figure 8, thecontrol group exhibited binding of NF-kB/p65 to DNA,which was significantly increased by challenging the cellswith TNF-a (positive control). In contrast, EGCG
treatment to A431 cells exhibited a dose- and time-dependent decrease in NF-kB/p65 DNA-binding activ-ity (Figure 8a, b). To determine the nature of differencein NF-kB complexes between control and EGCG-treated groups, we next performed supershift assayswith antibodies against the NF-kB/p65 and NF-kB/p50subunits of NF-kB. The EGCG-mediated responses inNF-kB complexes were mainly due to the NF-kB/p65subunit (data not shown). These observations indicatethat a molecular alteration of NF-kB/p65 may beresponsible for EGCG-mediated apoptosis of A431cells.
To test whether the induction of NF-kB-DNA-binding activity corresponds to an increase inNF-kB-dependent transcription, we transfected the cellswith ELAM-specific luciferase (ELAM-Luc) reporterconstruct, having NF-kB- and AP-1-binding sites,and measured the increase in luciferase activity incontrol cells and after EGCG treatment. We usedTNF-a-stimulated cells at a dose of 25 ng/ml as apositive control throughout the studies. AlthoughELAM-specific transcription is low in control cells,EGCG-treated cells exhibited a significant dose- andtime-dependent decrease in transcriptional activity(Figure 8c, d).
NF-kB/p65 is cleaved by caspases during EGCG-mediatedapoptosis of A431 cells
To characterize the molecular changes occurring inthe NF-kB complex during EGCG-mediated apoptosis,we analysed the NF-kB/p65 subunit. Since previousstudies have shown RelA/p65 as a substratefor activated caspase that leads to the loss ofcarboxy-terminal transactivation domain of activeNF-kB/p65 (Levkau et al., 1999), we asked whetherEGCG treatment leads to caspase-mediated cleavage ofNF-kB/p65 subunit in A431 cells. For this, we haveused a commercially available anti-p65 antibodythat crossreacts with a protein of approximatelyB54 kDa specific to detect the cleavage of NF-kB/p65at the carboxy-terminal transactivation domains.Studies have shown that B54 kDa protein (DN p65) isrecognized by this antibody raised against aminoacids 3–19 at the amino terminus of p65; therefore, itis likely that this protein is a C-terminally truncatedp65 molecule, specifically observed in apoptotic cells(Levkau et al., 1999). As shown by immunoblotanalysis, EGCG treatment resulted in a dose- andtime-dependent increase in the B54 kDa (DN p65)protein in the cytosolic fraction, suggesting thatNF-kB/p65 may be cleaved during EGCG-mediatedcaspase activation and subsequent apoptosis of A431cells (Figure 9a, b). This cleavage leads to the loss ofcarboxy-terminal transactivation domains of NF-kB/p65, and this may act as a dominant-negative inhibitorof NF-kB, promoting EGCG-mediated apoptosis.EGCG treatment resulted in dose-dependent increasein caspase activation which correlated with DN p65protein expression, the cleaved product of NF-kB/p65(Figure 9c). Similar results were observed by treatment
NF-κB/p65
NF-κB/p50
1κBα
0 010 20 40
10 20 40
12 24 48
EGCG (µg/ml); 24 h Time (h); E 20 µg/ml
0 12 24 48 h; E 20 µg/ml
120
90
60
30
00 �g/ml
20
15
10
5
0
EGCG
NF
-κB
/65
activ
atio
n (O
D 4
50 n
m)
% C
hang
e
120
90
60
30
0NF
-κB
/65
activ
atio
n (O
D 4
50 n
m)
% C
hang
e
Apoptotic cells; (%
)
20
15
10
5
0
Apoptotic cells; (%
)
Time
Nuclear Extract
Cytoplasmic Extract
NF-κBApoptosis
NF-κBApoptosis
a
b
c
Figure 7 Effect of EGCG on constitutive NF-kB expression inA431 cells. (a) Immunoblot analysis of nuclear translocation ofNF-kB/p65, NF-kB/p50 and cytoplasmic levels of IkBa. Cells weretreated with at indicated dose and times with EGCG. The detailsare described in ‘Materials and methods’ section. (b) Dose-dependent and (c) time-dependent downmodulation of constitutiveNF-kB/p65 induces apoptosis. Cells were treated with differentconcentrations of EGCG for 24 h, and then labeled with dUTPusing terminal deoxynucleotide transferase and PI, and TUNEL-positive (apoptotic) cells were analysed by flow cytometry. Thelevel of NF-kB/p65 was estimated by ELISA. The details aredescribed in ‘Materials and methods’ section. The data shown hereare representative of three independent experiments with similarresults
Role of caspases in EGCG-induced apoptosisS Gupta et al
2513
Oncogene

of cells with 20 mg/ml of EGCG for different timeintervals (Figure 9d).
Inhibition of caspases increases EGCG-mediatedconstitutive expression, and nuclear localization of NF-kB/p65 and IkBa in A431 cells
To further examine the effect of EGCG-mediated NF-kB inhibition and induction of caspase-dependent
apoptosis, A431 cells were incubated with Z-VAD-FMK prior to EGCG treatment. As shown in Figure10a, pre-incubation of cells with Z-VAD-FMK resultedin a significant increase in the protein expression ofNF-kB/p65 in the nuclear extract, as observed byimmunoblot analysis. Further, increase of NF-kB/p65expression in the nuclear fraction of A431 cells pre-incubated with Z-VAD-FMK correlated with a decreasein the protein expression of IkBa in the cytosol (Figure10a). These results were further confirmed by moresensitive NF-kB/p65 ELISA in the nuclear extract(Figure 10b). In the next series of experiments, weexamined the dose-dependent effect of the caspaseinhibitor on EGCG-mediated NF-kB/p65 inhibition ofA431 cells. As shown in Figure 10c, pre-incubation of Z-VAD-FMK (10–40 mM) resulted in a dose-dependentincrease in the protein expression of NF-kB/p65 in thenuclear fraction of EGCG (40-mg/ml)-treated A431 cells.The increase of NF-kB/p65 expression in the nuclearfraction of A431 cells pre-incubated with Z-VAD-FMKcorrelated with a dose-dependent decrease in the proteinexpression of IkBa in the cytosol. These results werefurther confirmed by more sensitive NF-kB/p65 ELISAin the nuclear extract (Figure 10d).
Inhibition of caspases increases EGCG-mediated DNA-binding activity and transcription of NF-kB in A431 cells
We next investigated the effect of EGCG-mediated NF-kB DNA-binding activity and its effect on caspaseinhibition. As shown in Figure 11a, pre-incubation ofcells with 10 mM concentration of Z-VAD-FMK resultedin a modest increase in the DNA-binding activitycompared to the corresponding EGCG-treated group.
a
b
c
d
NF-κB
NF-κB
TNF α (25 ng/ml)
0 0 10 20 40 �g/mlEGCG
TNF α (25 ng/ml)
EGCG
TNF α (25 ng/ml)
Time
TNF α (25 ng/ml)
Time
- - - -+
0 0 10 20 40
0 0 12 24 48
�g/ml
h; E 20�g/ml
0 0 12 24 48 h; E 20�g/ml
- - - -+
- - - -+
- - - -+
4
3
2
1
0
Fol
d In
duct
ion
4
3
2
1
0
Fol
d In
duct
ion
* * *
***
Figure 9 EGCG-mediated cleavage of NF-kB/p65 in A431 cells.(a) Dose-dependent and (b) time-dependent effect of EGCG oncleavage of NF-kB/p65, as observed by the cleaved product atB54 kDa, raised against amino acids 3–19 at the amino terminus ofNF-kB/p65. Cells were treated at the indicated dose and times withEGCG. Negative control: Cells were stimulated with 25 ng/ml ofTNF-a for 4 h. Positive control: Cells were treated with camp-tothecin with 5mg/ml for 12 h. The details are described in‘Materials and methods’ section. (c) Dose-dependent and (d)time-dependent increase in EGCG-mediated caspase activationcauses cleavage of the NF-kB/p65 subunit. The data are comparedbetween the fold increase in caspase activity, as shown in a previousexperiment, versus increase in protein expression of DN p65, thecleaved product at B54 kDa, raised against amino acids 3—19, atthe amino terminus of NF-kB/p65
Figure 8 EGCG-mediated NF-kB DNA binding and transcrip-tion is inhibited in A431 cells. (a) Dose-dependent and (b) time-dependent effect of EGCG on NF-kB DNA-binding activity, asperformed by EMSA. Cells were treated at the indicated dose andtimes with EGCG. (c) Dose-dependent and (d) time-dependentreporter assay in A431 cells transfected with ELAM-Luc construct.Luciferase activity normalized to b-galactosidase activity wasdetermined after EGCG treatment at the indicated doses andtimes, as described under ‘Materials and methods’. Values arerepresented as fold induction. Positive control: Cells werestimulated with 25 ng/ml of TNF-a for 4 h. Statistical analysiswas performed by Student’s t-test, and Po0.05 was consideredsignificant
Role of caspases in EGCG-induced apoptosisS Gupta et al
2514
Oncogene
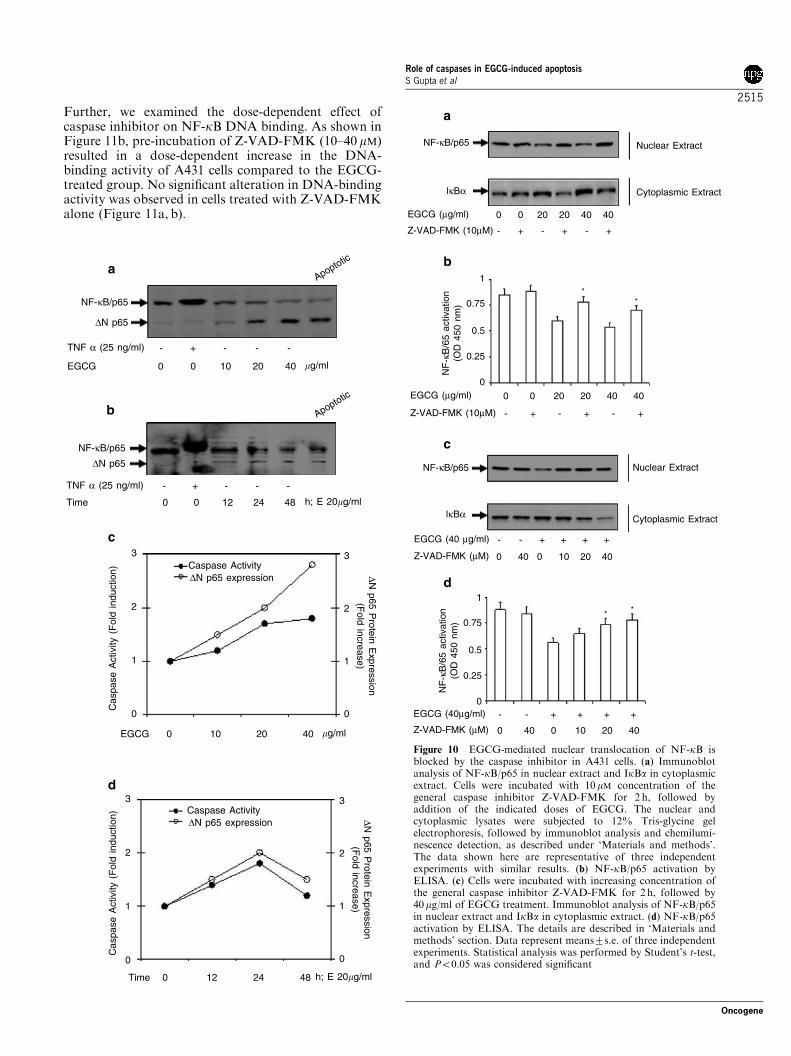
Further, we examined the dose-dependent effect ofcaspase inhibitor on NF-kB DNA binding. As shown inFigure 11b, pre-incubation of Z-VAD-FMK (10–40 mM)resulted in a dose-dependent increase in the DNA-binding activity of A431 cells compared to the EGCG-treated group. No significant alteration in DNA-bindingactivity was observed in cells treated with Z-VAD-FMKalone (Figure 11a, b).
a
c
d
b
NF-κB/p65
∆N p65
EGCG
Time
- - - -+
- - - -+
TNF α (25 ng/ml)
NF-κB/p65
∆N p65
TNF α (25 ng/ml)
0 0 10 20 40 �g/ml
EGCG 0 10 20 40 �g/ml
0 0 12 24 48 h; E 20�g/ml
Time 0 12 24 48 h; E 20�g/ml
Apoptotic
Apoptotic
3
2
1
0
3
2
1
0
3
2
1
0
Cas
pase
Act
ivity
(F
old
indu
ctio
n)C
aspa
se A
ctiv
ity (
Fol
d in
duct
ion)
∆N
p65 Protein E
xpression(F
old increase)
3
2
1
0
∆N
p65 Protein E
xpression(F
old increase)
Caspase Activity∆N p65 expression
Caspase Activity∆N p65 expression
a
c
d
b
NF-κB/p65
lκBα
EGCG (µg/ml)
Z-VAD-FMK (10µM)
EGCG (µg/ml)
Z-VAD-FMK (10µM)
EGCG (40µg/ml)
Z-VAD-FMK (µM)
0 0 20 20 40 40
- - - +++
0 0 20 20 40 40
- - - +++
Nuclear Extract
Cytoplasmic Extract
NF-κB/p65
lκBα
EGCG (40 µg/ml)
Z-VAD-FMK (µM) 0 40 0 10 20 40
- + ++ +-
0 40 0 10 20 40
- + ++ +-
Nuclear Extract
Cytoplasmic Extract
1
0.75
0.5
0.25
0
1
0.75
0.5
0.25
0
NF
-κB
/65
activ
atio
n(O
D 4
50 n
m)
NF
-κB
/65
activ
atio
n(O
D 4
50 n
m)
**
**
Figure 10 EGCG-mediated nuclear translocation of NF-kB isblocked by the caspase inhibitor in A431 cells. (a) Immunoblotanalysis of NF-kB/p65 in nuclear extract and IkBa in cytoplasmicextract. Cells were incubated with 10 mM concentration of thegeneral caspase inhibitor Z-VAD-FMK for 2 h, followed byaddition of the indicated doses of EGCG. The nuclear andcytoplasmic lysates were subjected to 12% Tris-glycine gelelectrophoresis, followed by immunoblot analysis and chemilumi-nescence detection, as described under ‘Materials and methods’.The data shown here are representative of three independentexperiments with similar results. (b) NF-kB/p65 activation byELISA. (c) Cells were incubated with increasing concentration ofthe general caspase inhibitor Z-VAD-FMK for 2 h, followed by40mg/ml of EGCG treatment. Immunoblot analysis of NF-kB/p65in nuclear extract and IkBa in cytoplasmic extract. (d) NF-kB/p65activation by ELISA. The details are described in ‘Materials andmethods’ section. Data represent means7s.e. of three independentexperiments. Statistical analysis was performed by Student’s t-test,and Po0.05 was considered significant
Role of caspases in EGCG-induced apoptosisS Gupta et al
2515
Oncogene

We initially tested the effect of the caspase inhibitorZ-VAD-FMK on the constitutive expression of NF-kB/p65, nuclear localization and DNA-binding activityfollowing EGCG treatment. Further, we wanted toknow if the caspase inhibition leads to survival of cellsfrom EGCG-mediated apoptosis, which is linked to NF-kB. To address this issue, the A431 cells were transfectedwith ELAM-Luc plasmids and were incubated with Z-VAD-FMK prior to EGCG treatment. Treatment ofA431 cells with EGCG resulted in significant decrease inELAM-specific luciferase activity, suggesting the role ofNF-kB in cell survival. Further, pre-incubation of cellswith Z-VAD-FMK (10 mM) resulted in 1.2- and 1.6-foldinduction in ELAM-specific luciferase activity at 20 and40 mg/ml of EGCG (Figure 11c). The dose-dependenteffect of caspase inhibitor (10–40 mM) resulted in asignificant induction (1.6-fold at 10 mM, 1.9-fold at20 mM and 3.2-fold at 40 mM Z-VAD-FMK) in EGCG-mediated ELAM-specific luciferase activity (Figure11d), which seems to play a role in cell survival. Thiseffect was of a magnitude almost similar to that elicitedby TNF-a, a well-characterized activator of NF-kB.
Inhibition of caspases decreases cleavage of NF-kB/p65and increases survival of A431 cells
We initially tested the effect of caspase inhibitor Z-VAD-FMK on the constitutive expression of NF-kB, nuclearlocalization, DNA-binding activity and transcriptionfollowing EGCG treatment. Further, we wanted to
a
b
c
d
NF-κB
NF-κB
Control
EGCG (40µg/ml)
Z-VAD-FMK (10µM)
Control
EGCG (µg/ml)Z-VAD-FMK (10µM)
EGCG (µg/ml)
Z-VAD-FMK (10µM)
EGCG (40µg/ml)
Z-VAD-FMK (µM)
+
+ + +-----
-
+
+ + + +
-
- - - -
++
++
+ + +--
- - -
- - - - -
- - -
+
+ + +
+ - - - - - -
-
0 0 0 0 20 20 40 40
40 4020100000
40
40 40
4020
20 20 20
100000
0 0 0
TNFα (25ng/ml)
TNFα (25ng/ml)
3
2
1
0
Fol
d In
duct
ion
3
2
1
0
Fol
d In
duct
ion
Figure 11 EGCG-mediated NF-kB DNA binding in the nucleus isincreased by caspase inhibitor in A431 cells. (a) Immunoblotanalysis of NF-kB DNA binding in nuclear extract by EMSA. Cellswere incubated with 10 mM concentration of the general caspaseinhibitor Z-VAD-FMK for 2 h, followed by addition of theindicated doses of EGCG, as described under ‘Materials andmethods’. The data shown here are from a representativeexperiment repeated three times with similar results. (b) Immuno-blot analysis of NF-kB DNA binding in the nuclear extract byEMSA, with increasing concentration of caspase inhibitor. Cellswere incubated with increasing concentrations of the generalcaspase inhibitor Z-VAD-FMK for 2 h, followed by 40mg/ml ofEGCG treatment, as described under ‘Materials and methods’.Positive control: biotin end-labeled target DNA with extract,negative control: biotin end-labeled target DNA with extract and a200-fold molar excess of unlabeled DNA. (c) Reporter assay inA431 cells transfected with NF-kB-dependent ELAM-Luc con-struct having NF-kB- and AP-1-binding sites. Cells were incubatedwith 10mM concentration of the general caspase inhibitor Z-VAD-FMK for 2 h, followed by addition of the indicated doses ofEGCG. Luciferase activity normalized to b-galactosidase activitywas determined 24 h after EGCG treatment, as described under‘Materials and methods’. Values are represented as fold induction.(d) Reporter assay in A431 cells transfected with NF-kB-dependentELAM-Luc construct with increasing concentrations of caspaseinhibitor. Cells were incubated with increasing concentration of thegeneral caspase inhibitor Z-VAD-FMK for 2 h, followed by 40 mg/ml of EGCG treatment. Luciferase activity normalized to b-galactosidase activity was determined 24 h after EGCG treatment,as described under ‘Materials and methods’. Values are representedas fold induction. Control: Cells were stimulated with 25 ng/ml ofTNF-a for 4 h with or without addition of EGCG. The datarepresent means7s.e. of three independent experiments. Statisticalanalysis was performed by Student’s t-test, and Po0.05 wasconsidered significant
Role of caspases in EGCG-induced apoptosisS Gupta et al
2516
Oncogene

know whether caspase activation is related to NF-kBinhibition during EGCG-mediated apoptosis of A431cells. To address this issue, the cells were incubated withcaspase inhibitor Z-VAD-FMK prior to EGCG treat-ment, and probed for the cleaved product of NF-kB/p65subunit. As shown by immunoblot analysis, pre-incuba-tion of cells with Z-VAD-FMK resulted in significantdecrease in DN p65, the cleaved B54 kDa proteinexpression in the cytosolic fraction compared with theEGCG-treated groups (Figure 12a). The dose-dependenteffect of caspase inhibitor (10–40mM) resulted in asignificant decrease in DN p65 protein expressioncompared to the EGCG-treated group (Figure 12b).These observations suggest that decrease in EGCG-mediated caspase activation leads to inhibition ofproteolytic cleavage of NF-kB/p65 and subsequentapoptosis. These results could be partially explained bythe gelshift and transcription assays, where a modestincrease in DNA-binding activity and transcription wasobserved after treatment with caspase inhibitor.
We next asked whether modulation in NF-kB/p65activation and cleavage by caspase inhibition leads tosurvival of cells from EGCG-mediated apoptosis. Toexamine this effect, we incubated A431 cells with Z-VAD-FMK with or without EGCG, and determined itseffect on cell survival by trypan blue exclusion assay andapoptosis by ELISA, as previously described. Pre-incubation of cells with Z-VAD-FMK prior to EGCGtreatment resulted in increase in the number of survivalcells with concomitant decrease in apoptosis (data notshown). Further, a dose-dependent effect was observedwith caspase inhibitor Z-VAD-FMK (data not shown).Next, we performed PARP cleavage assay in A431 cellsincubated with Z-VAD-FMK prior to EGCG treat-ment. As shown in Figure 12c, pre-incubation of cellswith Z-VAD-FMK resulted in decrease in cleavage ofPARP compared to EGCG treatment. Further, a dose-dependent decrease in PARP cleavage was observed incells pre-incubated with 10–40 mM concentrations of Z-VAD-FMK (Figure 12d). No cleavage of PARP wasobserved in cells treated with Z-VAD-FMK alone(Figure 12c, d). These results indicate that cells couldbe rescued from apoptosis by pretreatment with caspaseinhibitor Z-VAD-FMK, conferring an essential role ofcaspases in EGCG-mediated inhibition of NF-kB andinduction of apoptosis.
Discussion
In recent years, green tea, specifically its majorconstituent EGCG, has gained much attention due toits cancer chemopreventive properties. A significantreason for the great interest in EGCG is because of itsremarkable effects in many in vitro and in vivo tumormodel systems (Gupta et al. 2000; Singh et al., 2002;Chung et al., 2003; Gupta et al., 2003; Hastak et al.,2003). Part of this effect of EGCG appears to bebecause, at physiologically attainable concentrations, itinduces apoptosis in cancer cells without affectingnormal cells (Ahmad et al., 1997, 2000; Chen et al.,
1998). Our earlier studies have shown that EGCGtreatment of cancer cells resulted in a dose-dependent (i)inhibition of cell growth, (ii) G0/G1 phase arrest of thecell cycle and (iii) induction of apoptosis (Ahmad et al.,1997; Gupta et al., 2003). Apoptosis represents auniversal and exquisitely efficient suicide pathway, anideal way for elimination of damaged cells (Ferreiraet al., 2002). The apoptotic process may conceptually be
a
c
d
b
NF-κB/p65∆N p65
EGCG (µg/ml)
Z-VAD-FMK (10µM)
EGCG (µg/ml)
Z-VAD-FMK (10µM)
NF-κB/p65∆N p65
EGCG (40µg/ml)
Z-VAD-FMK (µM)
0 0 20 20 40 40
0 0 10 2040 40
- - -
- -
+ + +
0 0 20 20 40 40
-- -+ + +
+ + + +
0 0 10 2040 40
- - + + + +
PARP
EGCG (40µg/ml)
Z-VAD-FMK (µM)
PARP
116 KD85 KD
116 KD85 KD
Figure 12 EGCG-mediated cleavage of NF-kB/p65 and apoptosisis decreased by the caspase inhibitor in A431 cells. (a) Immunoblotanalysis of NF-kB/p65 and DN p65, the cleaved product atB54 kDa, raised against amino acids 3–19 at the amino terminus ofNF-kB/p65. Cells were incubated with 10 mM concentration of thegeneral caspase inhibitor Z-VAD-FMK for 2 h, followed byaddition of the indicated doses of EGCG, as described under‘Materials and methods’. (b) Immunoblot analysis of NF-kB/p65and DN p65, the cleaved product atB54 kDa, raised against aminoacids 3–19 at the amino terminus of NF-kB/p65. Cells wereincubated with increasing concentration of the general caspaseinhibitor Z-VAD-FMK for 2 h, followed by 40 mg/ml of EGCGtreatment, as described under ‘Materials and methods’. (c)Immunoblot analysis of PARP cleavage. Cells were incubated with10mM concentration of general caspase inhibitor Z-VAD-FMK for2 h, followed by addition of the indicated doses of EGCG. The celllysates were prepared following treatment, and 50 mg of protein wassubjected to 12% Tris-glycine gel electrophoresis, followed byimmunoblot analysis and chemiluminescence detection, as de-scribed under ‘Materials and methods’. The data shown here arerepresentative of three independent experiments with similarresults. (d) Immunoblot analysis of PARP cleavage with increasingconcentration of caspase inhibitor. Cells were incubated withincreasing concentration of the general caspase inhibitor Z-VAD-FMK for 2 h, followed by 40 mg/ml of EGCG treatment. The celllysates were prepared following treatment, and 50 mg of protein wassubjected to 12% Tris-glycine gel electrophoresis, followed byimmunoblot analysis and chemiluminescence detection, as de-scribed under ‘Materials and methods’. The data shown here arerepresentative of three independent experiments with similar results
Role of caspases in EGCG-induced apoptosisS Gupta et al
2517
Oncogene

divided into three phases: an induction phase, the natureof which depends on the specific death-inducing signals,an effector phase, during which the central executioneris activated, and a degradation phase that leads tomorphological features of end-stage apoptosis (Nichol-son, 1999; Grutter, 2000; Zimmermann et al., 2001).Since the execution machinery of apoptosis appears tobe evolutionarily conserved, it seems possible that itplays an especially important role in governing both theaccuracy and efficiency of the apoptotic pathway. Theexecution phase occurs through the activation andfunction of caspases, which cleave certain critical targetsleading to apoptosis (Lazebnik et al., 1995; Tewari et al.,1995; Janicke et al., 1996; Cheng et al., 1997; Clem et al.,1998; Tang and Kidd, 1998; Levkau et al., 1999;Stennicke and Salvesen, 2000). Many investigators havestudied the signaling pathway in the initiation andpropagation of various chemopreventive agents; how-ever, the execution phase has not been well studied(Clement et al., 1998; Yu et al., 1998). Our present studyprovides an essential role of caspases as the executionaryphase in EGCG-mediated apoptosis.
Caspases are part of a growing family of cysteineproteases that have been shown to be involved in manyforms of apoptosis (Nicholson, 1999; Grutter, 2000;Zimmermann et al., 2001). Activation of caspaseproteases was previously shown to be required for theinduction of apoptosis in different cell types (Islam et al.,2000; Hayakawa et al., 2001). Caspases are activated asa consequence of cell membrane signaling events thatcan be classified as initiating or upstream caspases.Several caspases (caspase-2, -8 and -10) have large pro-domain-containing motifs for protein–protein interac-tions, allowing for their interaction with activation byproteins associated with cytoplasmic domains of thefamily of death receptors. Recent studies on receptor-mediated apoptosis and activation of caspase-8 is theearliest event, which is synthesized as inactive proen-zymes activated by proteolytic cleavage after specificaspartate residue, and is capable of activating down-stream caspases (Kim et al., 2001). The effector, ordownstream caspase (caspase-3, -6 and -7), is activatedlater than the upstream caspases. These effectors ordownstream caspases have been defined as a keyexecutioner involved in the apoptosis induced by variousstimuli, and is also necessary for nuclear changesassociated with apoptosis, such as chromatin condensa-tion and PARP cleavage (Nicholson, 1999; Grutter,2000; Stennicke and Salvesen, 2000). In the presentstudy, we have shown that caspase-3, -8 and -9 areactivated during EGCG-mediated apoptosis. Ourstudies have further shown that addition of Z-VAD-FMK, a general caspase inhibitor, significantlyreduces EGCG-mediated apoptosis, while inhibition ofcaspase-3 by Z-DVED-FMK resulted in only partialprevention of apoptosis. Inhibitory effects of Z-VAD-FMK and Z-DVED-FMK provide evidence thatEGCG-mediated apoptosis in A431 cells is caspasedependent.
We have previously demonstrated the involvementof NF-kB in regulating EGCG-mediated apoptotic
response of cancer cells (Ahmad et al., 2000). NF-kBhas been implicated in a number of cellular functions,including cell survival, proliferation and apoptosis.Numerous studies have shown that NF-kB regulatessusceptibility of cells to apoptosis through the transcrip-tional control of genes (Bours et al., 2000; Joyce et al.,2001; Karin and Lin, 2002). Inhibition of NF-kB andnuclear translocation has been shown to increase thesusceptibility of cells to undergo apoptosis induced byTNF-a, LPS, ionizing radiation and anticancer agents(Wang et al., 1996; Bours et al., 2000; Joyce et al., 2001;Karin and Lin, 2002). Recent studies have demonstratedthat suppression of NF-kB activation by proteaseinhibitors could block IkBa degradation-induced apop-tosis (Wu et al., 1996). Further, ectopic expression of c-Rel was found to ablate the induction of apoptosisfollowing suppression of NF-kB activation by inhibitorsof IkBa degradation (Wu et al., 1996). These findingspoint to a critical role of NF-kB and essential role ofcaspases in apoptotic cell death. In the present study, wehave demonstrated that EGCG-mediated caspase acti-vation leads to constitutive NF-kB inhibition andsubsequent apoptosis of human epidermoid carcinomaA431 cells. The maintenance of NF-kB activity by thecells is required for cell survival and, conversely,inhibition in the constitutive activity directs the cellstowards apoptosis. The inhibition of NF-kB transcrip-tional activity observed during EGCG-mediated apop-tosis could be the result of two distinct inhibitorymechanisms: first, blockade of IkB degradation, andsecond, cleavage of NF-kB subunit that may disable thetransactivation capacity. Our studies have identified theNF-kB/p65 component of the NF-kB complex as atarget for specific cleavage by caspases during EGCG-mediated apoptosis. This cleavage leads to the loss ofcarboxy-terminal transactivation domains of NF-kB/p65, resulting in truncated p65 and promoting apopto-sis. Further, pre-incubation of Z-VAD-FMK prior toEGCG treatment was found to result in the increase incell survival, as observed by modest increase inconstitutive NF-kB expression, DNA binding andtranscriptional activity. This observation leads to theconfirmation that, as long as the cells are capable ofmaintaining transcriptionally active NF-kB, they areable to resist EGCG-mediated apoptotic cell death. Thefact is that EGCG-induced caspase activation leads tocleavage of NF-kB/p65 and subsequently decreases celltranscription that may be aborted by pre-incubation ofA431 cells with the general caspase inhibitor Z-VAD-FMK. Whether suppression of NF-kB activation isresponsible for initiation of EGCG-mediated apoptosisin A431 cells is further supported by the observationthat transfection of cells with a NF-kB-dependentreporter construct (ELAM-Luc) having NF-kB- andAP-1-binding sites resulted in significant decrease inELAM-specific luciferase activity. Importantly, pre-incubation of Z-VAD-FMK was found to result in theincrease in EGCG-mediated ELAM-specific luciferaseactivity. Similar effects were observed on humanepithelial prostate carcinoma LNCaP cells (data notshown). These findings point to a critical role of
Role of caspases in EGCG-induced apoptosisS Gupta et al
2518
Oncogene

caspases in modulating NF-kB family members in cellsurvival and cell death, which is not cell type specific.These data further suggest that the apoptotic programelicited by EGCG in cancer cells can destroy NF-kBthrough an elegant and efficient caspase-mediatedswitch-off mechanism that commits the cells towardsapoptosis. The schematic representation of the action ofEGCG is depicted in Figure 13.
A number of pathways have been suggested that maylead to program cell death by chemopreventive agents(Kong et al., 2001). In this aspect, two central pathwayshave been shown to be involved in the process ofapoptotic cell death: one shows the direct involvementof caspase proteases (especially caspase-8) and, theother, the mitochondrial pathway where caspases areactivated as downstream events in apoptosis (Green andKroemer, 1998). The molecular ordering of caspases hasbeen upheld in several cases, and is best exemplified bycaspase-8-mediated activation of caspase-3 and -7 in theCD95/Fas/APO-1 system, and caspase-9-mediated acti-vation of caspase-3 in the Apaf-1/cytochrome c pathway(Nicholson, 1999). Recent studies from our group havedemonstrated the role of EGCG in disruption ofmitochondrial pathway and induction of apoptosis(Hastak et al., 2003). The role of EGCG in theactivation of death receptor Fas/APO-1 has beenrecently demonstrated by another group (Kuo andLin, 2003). However, this study and the pathways thatlead to EGCG-induced apoptosis described herein arenovel and have not been elucidated. Experimentalevidence has further shown CD95/Fas-induced cas-pase-mediated proteolysis of NF-kB, suggesting a linkbetween NF-kB and programmed cell death or apopto-sis (Ravi et al., 1998). This phenomenon leads to the
existence of multiple distinct apoptotic mechanisms forthe inactivation of NF-kB, and further support the ideathat its inactivation has a central role in the commitmentto apoptosis. Additional studies are needed to identifydifferent caspase cleavage sites on NF-kB that areinvolved in EGCG-mediated apoptosis. It is importantto emphasize here that caspase activity varies accordingto the cell type and apoptotic stimuli. Since induction ofapoptosis is considered an important mechanism ofprevention/treatment of cancer by chemopreventiveagent(s), further elucidation of the mechanism viadifferent signaling pathways during EGCG-mediatedapoptosis would provide useful information concerningthe basis for its use as a potential therapeutic agent.
Materials and methods
Materials
A purified preparation of EGCG (498% pure) was kindlyprovided by Dr Yukihiko Hara of Mitsui Norin Co., Ltd(Shizuoka, Japan). Antibody for casapase-3 was purchasedfrom Upstate Biotechnology (Lake Placid, NY, USA),caspase-8 from Biosource International (Camarillo, CA,USA) and caspase-9 from Santa Cruz Biotechnology, Inc.(Santa Cruz, CA, USA). NF-kB/p65 and NF-kB/p50 anti-bodies were obtained from Geneka Biotechnology Inc.(Montreal, Canada). Another NF-kB/p65 antibody used forthe studies was obtained from Santa Cruz Biotechnology, Inc.that recognizes both NF-kB/p65 subunit and the cleavedproduct at B54 kDa, raised against amino acids 3–19 at theamino terminus of NF-kB/p65. The antibody for IkBa waspurchased from New England Biolabs, Inc. (Beverly, MA,USA), while NF-kB/p65 Trans-AM kit was obtained fromActive Motif North America (Carlsbad, CA, USA). The
EGCG
A431 cells
cytoplasm
p50
p50
p65
p65nucleus
IκBα super-repressor / inhibitor
caspaseinhibitor
Less DNATranscription/Survival
APOPTOSIS SURVIVAL
PARP CLEAVAGE
Active caspase
Procaspase
NF-κB inhibitor
Ub-Ub-UbIκB
P
Figure 13 Schematic representation for caspase activation, inhibition of NF-kB and induction of apoptosis by EGCG. k representsinhibition
Role of caspases in EGCG-induced apoptosisS Gupta et al
2519
Oncogene

general caspase inhibitor Z-VAD-FMK was purchased fromR&D Systems, Inc. (Minneapolis, MN, USA), while caspase-3-specific inhibitor Z-DVED-FMK was purchased fromCalbiochem Labs (San Diego, CA, USA). The ELISA CellDeath DetectionPLUS kit and Annexin-V-FLUOS staining kitswere purchased from Roche Diagnostic Corporation (India-napolis, IN, USA). The APO-BRDU kit for quantitation ofapoptosis by flow cytometry was purchased from PhoenixFlow Systems, Inc. (San Diego, CA, USA).
Cell culture
The human epidermoid carcinoma (A431) and androgen-sensitive human prostate carcinoma LNCaP cells wereobtained from American Type Culture Collection (ATCC)(Rockville, MD, USA). The cells were cultured under standardculture condition in Dulbecco’s modified Eagle’s medium(DMEM) (Life Technologies, Grand Island, NY, USA)supplemented with 10% fetal bovine serum and 1% penicil-lin-streptomycin. The cells were maintained at 371C and 5%CO2 in a humid environment.
Treatment of cells
EGCG was dissolved in PBS (pH 7.4) and employed for thetreatment of cells. The cells were treated with 10-, 20- and 40-mg/ml concentrations for 24 h. For DNA fragmentation andPARP cleavage, the cells were treated with the desired doses ofEGCG for 48 h. For time-dependent assay, the cells weretreated with 20mg/ml of EGCG for 12, 24 and 48 h,respectively. For caspase-inhibition experiments, the cells wereincubated with Z-VAD-FMK or Z-DEVD-FMK at thedesired concentration for 2 h before addition of EGCG. At24 h after the addition of EGCG, cells were harvested andprocessed for trypan blue exclusion assay, ELISA andTUNEL assay or immunoblot analysis.
Cell viability
The cells were grown to 70% confluence and treated withEGCG (10, 20, and 40mg/ml) for 24 h, and the cell viabilitywas determined by trypan blue exculsion assay.
Apoptosis by ELISA
Following EGCG treatment, the extent of apoptosis wasdetermined by Cell Death Detection ELISAPLUS assay,according to the manufacturer’s protocol. Briefly, the cellswere harvested after EGCG treatment at 10-, 20- and 40-mg/mldoses and 12, 24 and 48 h time intervals, and incubated on icefor 30min in Tris lysis buffer (50mM Tris-HCl, pH 7.5,150mM NaCl, 1mM EDTA, 20mM NaF, 0.5% NP-40, 1%Triton X-100) containing fresh protease inhibitors (5 mg/ml,aprotinin, 10mg/ml phenylmethylsulfonyl fluoride and 10 mg/ml sodium vanadate), and then centrifuged at 12 000 r.p.m. for10min at 41C. The total cell lysate was used for proteindetermination by the DC BioRads protein assay. The lysate(30mg of total protein) was added to the lysis buffer andpipetted on a streptavidin-coated 96-well microtiter plate towhich the immunoreagent mix was added and incubated for2 h at room temperature, with continuous shaking at500 r.p.m. The wells were then washed with washing buffer,the substrate solution added, the color developed (10–20min)was read at 405 nm against the blank, reference wavelength of490 nm. The enrichment factor (total amount of apoptosis)was calculated by dividing the absorbance of the sample(A405 nm) by the absorbance of the controls withouttreatment (A490 nm).
DNA fragmentation assay
Following treatment of cells as described above, the cells werewashed twice with PBS (pH 7.4), incubated with DNA lysisbuffer (10mM Tris, pH 7.5, 400mM NaCl, 1mM EDTA and1% Triton X-100) for 30min on ice and then centrifuged at14 000� g at 41C. The supernatant obtained was incubatedovernight with RNAse (0.2mg/ml) at room temperature andthen with Proteinase K (0.1mg/ml) for 2 h at 371C. DNA wasextracted using phenol : chloroform (1 : 1) and precipitatedwith 95% ethanol for 2 h at �801C. The DNA precipitate wascentrifuged at 14 000 g at 41C for 15min and the pellet was air-dried and dissolved in 20 ml of Tris-EDTA buffer (10mM Tris-HCl, pH 8.0 and 1mM EDTA). The total amount of DNA wasresolved over 1.5% agarose gel, containing 0.3 mg/ml ethidiumbromide in 1� TBE buffer (pH 8.3, 89mM Tris, 89mM boricacid and 2mM EDTA) (Bio Wittaker, Inc., Walkersville, MD,USA). The bands were visualized under a UV transilluminator(Model # TM-36, UVP Inc., San Gabriel, CA, USA) followedby polaroid photography (MP-4 Photographic System, Foto-dyne Inc., Hartland, WI, USA).
Apoptosis by flow cytometry
The cells were grown at a density of 1� 106 cells in 100-mmculture dish, and were treated with EGCG (10-, 20- and 40-mg/ml concentration) for 24 h and 20 mg/ml EGCG for 12–48 h.The cells were trypsinized, washed with PBS and processed forlabeling with fluorescein-tagged bromodeoxyuridine tripho-sphate (Br-dUTP) and propidium iodide, using the APO-BRDU apoptosis kit, as per the manufacturer’s protocol. Thelabeled cells were then analysed by flow cytometry.
Caspase activity assay
The activity of caspases was measured by the fluorometricassay, following the manufacturer’s protocol. Briefly, cellstreated with EGCG and/or caspase inhibitors followed theirlysis using lysis buffer (0.1M HEPES buffer, pH 7.4, contain-ing 2mM DTT, 0.1% Chaps and 1% sucrose). Cell lysateswere incubated with 10ml of 100 mM fluorogenic substrates, Ac-DEVD-AFC (Ac-Asp-Glu-Val-Asp-AFC), purchased fromCalbiochem (San Diego, CA, USA) for 30min at 301C. Thisis a fluorogenic caspase-3 substrate, and also acts as a substratefor granzymeB-processed ICE-LAP3, caspase-6, -7, -8 and -10.The release of AFC was measured with a fluorescencespectrophotometer at 400-nm excitation and 505-nm emission.
Protein extraction and immunoblot analysis
Following the treatment of cells as described above, the mediawas aspirated, the cells were washed with cold PBS (pH 7.4),and ice-cold lysis buffer (50mM Tris-HCl, 150mM NaCl, 1mM
EGTA, 1mM EDTA, 20mM NaF, 100mM Na3VO4, 0.5%NP-40, 1% Triton X-100, 1mM PMSF (pH 7.4)) with freshlyadded protease inhibitor cocktail (Protease Inhibitor CocktailSet III, Calbiochem, La Jolla, CA, USA) was added and it wasincubated over ice for 30min. The cells were scraped, the lysatewas collected in a microfuge tube and passed through a 211
2G
needle to break up the cell aggregates. The lysate was clearedby centrifugation at 14 000 g for 15min at 41C, and thesupernatant (total cell lysate) was used or immediately storedat �801C. For NF-kB/p65 assay, the cells were processed fornuclear and postnuclear fractions, as described previously(Ahmad et al., 2000). The protein concentration was deter-mined by DC Bio-Rad assay using the manufacturer’sprotocol (Bio Rad Laboratories, Hercules, CA, USA).
Role of caspases in EGCG-induced apoptosisS Gupta et al
2520
Oncogene

For immunoblot analysis, an appropriate amount of celllysates (25–50mg protein) was resolved over 12% Tris-Glycinepolyacrylamide gel and then transferred onto the nitrocellulosemembrane. The blots were blocked using 5% non-fat dry milkand probed using appropriate primary antibodies in blockingbuffer overnight at 41C. The membrane was then incubatedwith the appropriate secondary antibody horseradish perox-idase (HRP) conjugate (Amersham Life Sciences Inc., Arling-ton Heights, IL, USA), followed by detection using achemiluminescence ECL kit (Amersham Life Sciences Inc.,Arlington Heights, IL, USA). For equal loading of protein, themembrane was stripped and reprobed with an anti-b-actinantibody.
ELISA assay for NF-kB activity
ELISA was performed for NF-kB/p65 activity, according tothe vendor’s protocol. This assay uses an oligonucleotidecontaining NF-kB consensus site (50-GGGACTTTCC-30) thatbinds to the cell extract and can detect NF-kB, which canrecognize an epitope on p65 activated and bound to its targetDNA. This assay is specific for NF-kB/p65 activation, and ishighly sensitive when compared with the gel-retardationtechnique.
Electrophoretic mobility shift assay (EMSA) and Supershiftassay
EMSA for NF-kB was performed using lightshiftt chemilu-miniscent EMSA kit (Pierce, Rockford, IL, USA), followingthe manufacturer’s protocol. Briefly, DNA was biotin labeledusing the biotin 30 end-labeling kit (Pierce, Rockford, IL,USA) in a 50ml reaction buffer, 5 pmol of double-strandedNF-kB oligonucleotide (50-AGTTGAGGGGACTTTCCCAGGC-30 and 30-TCAACTCCCCTGAAAGGGTCCG-50)incubated in a microfuge tube with 10ml of 5� terminaldeoxynucleotidyl transferase (TdT) buffer, 5 ml of 5mM biotin-N4-CTP, 10U of diluted TdT and 25 ml of ultrapure water at371C for 30min. The reaction was stopped with 2.5 ml of 0.2M
EDTA. To extract labeled DNA, 50ml of chloroform : isoamylalcohol (24 : 1) was added to each tube and centrifuged at13 000 g. The top aqueous phase containing the labeled DNAwas further used for binding reactions. Each binding reactioncontained 1� binding buffer (100mM Tris, 500mM KCl,10mM dithiothreitol, pH 7.5), 2.5% glycerol, 5mM MgCl2,50 ng/ml poly (dI-dC), 0.05% NP-40, 2.5 mg of nuclear extractand 20–50 fm of biotin end-labeled target DNA. The contentswere incubated at room temperature for 20min. To thisreaction mixture, 5ml of 5� loading buffer was added,subjected to gel electrophoresis on a native polyacrylamidegel and transferred to a nylon membrane. After transferwas completed, DNA was crosslinked to the membrane at120mJ/cm2 using a UV crosslinker equipped with 254-nmbulb. The biotin end-labeled DNA was detected usingstreptavidin–horseradish peroxidase conjugate and a chemilu-miniscent substrate. The membrane was exposed to an X-rayfilm (XAR-5 Amersham Life Science Inc., Arlington Height,IL, USA) and developed using a Kodak film processor. Forsupershift assay, antibodies against NF-kB/p65 and NF-kB/p50 were added 30min after beginning of the reaction, and
incubation was continued for an additional 30–45min. DNAprotein complexes were analysed as described earlier.
Fluorescence microscopy
The cells were treated as described earlier, and washed withPBS and resuspended and incubated with 100 ml of stainingsolution for 10–15min at room temperature. The mixture wasthen placed on the stage of Zeiss Axiovert 100 microscope. Thesamples were excited at 330–380 nm, and the image wasobserved and photographed under a combination of a 400-nmdichoric mirror and then a 420-nm high-pass filter. Apoptoticand necrotic cells were scored under the microscope. Cells withgreen, condensed chromatin pattern were considered apopto-tic, whereas those with orange nuclei without nuclearcondensation were considered necrotic.
Transfection and reporter assay
The NF-kB-dependent reporter plasmid (ELAM-Luc) havingNF-kB- and AP-1-binding sites, and CMV-b-gal plasmid, wereobtained as a kind gift from Dr Munna L Agarwal (ClevelandClinic Foundation, Cleveland, OH, USA). The A431 cells weretransiently transfected using Fugene 6 reagent with 1.5 mgELAM-Luc and 1mg of CMV-b-gal plasmid. After 24 h, themedia was changed and cells were treated with the desiredconcentration of Z-VAD-FMK 2h, prior to EGCG addition.For positive control, the cells were stimulated with 25 ng/ml ofTNF-a (Roche, Indianapolis, IN, USA) for 4 h with or withoutaddition of EGCG. After treatment with EGCG for 24 h,luciferase and galactosidase activity was determined with theluciferase assay system and b-galactosidase assay system(Promega, Madison, WI, USA), according to the vendor’sprotocol. Luciferase activity was normalized to b-galactosidaseactivity to control for transfection efficiency.
Statistical analysis
The enzyme activity and other values are expressed asmean7s.e. The significance values between the control andtreated groups were calculated by the Student’s ‘t’-test, and P-values less than 0.05 were taken as significant in all theexperiments.
Abbreviations
EGCG, (�)-epigallocatechin-3-gallate; NF-kB, nuclear factor-kappaB; PARP, poly(ADP-ribose) polymerase; Z-VAD-FMK,N-benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone;Z-DEVD-FMK, CPP32/apopain inhibitor.
Acknowledgements
This work was supported by grants from United States PublicHealth Service (RO1CA 78809), American Institute for CancerResearch (00A030) and Department of Defense (DAMD 17-00-1-0527). Sanjay Gupta, PhD is grateful to the ‘CancerResearch Foundation of America’ and ‘The O-CHA (Tea)Pioneer Academic Research Grant Program’, Japan, for apartial support of funds.
References
Aggarwal BB. (2000). Biochem. Pharmacol., 60, 1033–1039.Ahmad N, Feyes DK, Nieminen AL, Agarwal R and MukhtarH. (1997). J. Natl. Cancer Inst., 89, 1881–1886.
Ahmad N, Gupta S and Mukhtar H. (2000). Arch. Biochem.Biophys., 376, 338–346.
Bours V, Bentires-Alj M, Hellin AC, Viatour P, Robe P,Delhalle S, Benoit V and Merville MP. (2000). Biochem.Pharmacol., 60, 1085–1089.
Chen ZP, Schell JB, Ho CT and Chen KY. (1998). CancerLett., 129, 173–179.
Role of caspases in EGCG-induced apoptosisS Gupta et al
2521
Oncogene

Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A,Ueno K and Hardwick JM. (1997). Science, 278, 1966–1968.
Chung JH, Han JH, Hwang EJ, Seo JY, Cho KH, Kim KH,Youn JI and Eun HC. (2003). FASEB J., 17, 1913–1915.
Chung LY, Cheung TC, Kong SK, Fung KP, Choy YM, ChanZY and Kwok TT. (2001). Life Sci., 68, 1207–1214.
Clem RJ, Cheng EH, Karp CL, Kirsch DG, Ueno K,Takahashi A, Kastan MB, Griffin DE, Earnshaw WC,Veliuona MA and Hardwick JM. (1998). Proc. Natl. Acad.Sci. USA, 95, 554–559.
Clement MV, Hirpara JL, Chawdhury SH and Pervaiz S.(1998). Blood, 92, 996–1002.
Ferreira CG, Epping M, Kruyt FA and Giaccone G. (2002).Clin. Cancer Res., 8, 2024–2034.
Green D and Kroemer G. (1998). Trends Cell Biol., 8, 267–271.Grutter MG. (2000). Curr. Opin. Struct. Biol., 10, 649–655.Gupta S, Ahmad N, Nieminen AL and Mukhtar H. (2000).
Toxicol. Appl. Pharmacol., 164, 82–90.Gupta S, Hussain T and Mukhtar H. (2003). Arch. Biochem.
Biophys., 410, 177–185.Hastak K, Gupta S, Ahmad N, Agarwal MK, Agarwal MLand Mukhtar H. (2003). Oncogene, 22, 4851–4859.
Hayakawa S, Saeki K, Sazuka M, Suzuki Y, Shoji Y, Ohta T,Kaji K, You A and Isemura M. (2001). Biochem. Biophys.Res. Commun., 285, 1102–1106.
Islam S, Islam N, Kermode T, Johnstone B, Mukhtar H,Moskowitz RW, Goldberg VM, Malemud CJ and HaqqiTM. (2000). Biochem. Biophys. Res. Commun., 270, 793–797.
Janicke RU, Walker PA, Lin XY and Porter AG. (1996).EMBO J., 15, 6969–6978.
Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B and PestellRG. (2001). Cytokine Growth Factor Rev., 12, 73–90.
Karin M and Lin A. (2002). Nat. Immunol., 3, 221–227.Kim PK, Mahidhara R and Seol DW. (2001). Drug Resist.
Updat., 4, 293–296.Kong AN, Yu R, Hebbar V, Chen C, Owuor E, Hu R, Ee Rand Mandlekar S. (2001). Mutat. Res., 480–481, 231–241.
Kuo PL and Lin CC. (2003). J. Biomed. Sci., 10, 219–227.
Lazebnik YA, Takahashi A, Moir RD, Goldman RD, PoirierGG, Kaufmann SH and Earnshaw WC. (1995). Proc. Natl.Acad. Sci. USA, 92, 9042–9046.
Levkau B, Scatena M, Giachelli CM, Ross R and Raines EW.(1999). Nat. Cell Biol., 1, 227–233.
Masuda M, Suzui M and Weinstein IB. (2001). Clin. CancerRes., 7, 4220–4229.
Nicholson DW. (1999). Cell Death Differ., 6, 1028–1042.Ravi R, Bedi A, Fuchs EJ and Bedi A. (1998). Cancer Res., 58,882–886.
Schwartz SA, Hernandez A and Mark Evers B. (1999). Surg.Oncol., 8, 143–153.
Singh AK, Seth P, Anthony P, Husain MM, Madhavan S,Mukhtar H and Maheshwari RK. (2002). Arch. Biochem.Biophys., 401, 29–37.
Stennicke HR and Salvesen GS. (2000). Biochim. Biophys.Acta, 1477, 299–306.
Surh YJ, Chun KS, Cha HH, Han SS, Keum YS, Park KKand Lee SS. (2001). Mutat. Res., 480–481, 243–268.
Tang D and Kidd VJ. (1998). J. Biol. Chem., 273,28549–28552.
Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z,Beidler DR, Poirier GG, Salvesen GS and Dixit VM. (1995).Cell, 81, 801–809.
Wang C-Y, Mayo MW and Baldwin Jr AS. (1996). Science,274, 784.
Wu M, Lee H, Bellas RE, Schauer SL, Arsura M, Katz D,FitzGerald MJ, Rothstein TL, Sherr DH and SonensheinGE. (1996). EMBO J., 15, 4682–4690.
Yamamoto Y and Gaynor RB. (2001). J. Clin. Invest., 107,135–142.
Yang GY, Liao J, Kim K, Yurkow EJ and Yang CS. (1998).Carcinogenesis, 19, 611–616.
Yu R, Mandlekar S, Harvey KJ, Ucker DS and Kong AN.(1998). Cancer Res., 58, 402–408.
Zimmermann KC, Bonzon C and Green DR. (2001).Pharmacol. Ther., 92, 57–70.
Role of caspases in EGCG-induced apoptosisS Gupta et al
2522
Oncogene




















