
Calcium microdomains and oxidative stress
14
Cell Calcium 40 (2006) 561–574 Calcium microdomains and oxidative stress Sean M. Davidson a,∗ , Michael R. Duchen b a The Hatter Cardiovascular Institute, Royal Free and University College Medical School, London, Department of Medicine, 67 Chenies Mews, London WC1E 6HX, United Kingdom b Department of Physiology and Mitochondrial Biology Group, University College London, London, United Kingdom Received 1 August 2006; accepted 23 August 2006 Available online 17 October 2006 Abstract The phenomenon of calcium microdomains is firmly established in the field of subcellular physiology. These regions of localized, transient calcium increase are exemplified by the spontaneous ‘sparks’ released through the ryanodine receptor in myocytes, but include subplasmalem- mal microdomains, focal calcium oscillations and microdomains enclosed within organelles, such as the endoplasmic reticulum, golgi and mitochondria. Increasing evidence suggests that oxidative stress regulates both the formation and disappearance of microdomains. Calcium release channels and transporters are all modulated by redox state, while several mechanisms that generate oxidative or nitrosative stress are regulated by calcium. Here, we discuss the evidence for the regulation of calcium microdomains by redox state, and, by way of example, demonstrate that the frequency of calcium sparks in cardiomyocytes is increased in response to oxidative stress. We consider the evidence for the existence of analogous microdomains of reactive oxygen and nitrogen species and suggest that the refinement of imaging techniques for these species might lead to similar concepts. The interaction between Ca 2+ microdomains and proteins that modulate their formation results in a complex and dynamic, spatial signaling mechanism, which is likely to be broadly applicable to different cell types, adding new dimensions to the calcium signaling ‘toolkit’. © 2006 Elsevier Ltd. All rights reserved. Keywords: Calcium; Oxidative stress; ROS 1. Introduction Mammalian cells are bathed in interstitial fluid that con- tains greater than 1 mM Ca 2+ . Only by functioning continu- ously are extrusion mechanisms able to counter the inward leak of this calcium, and maintain cytosolic free calcium at ∼100 nM. These include the plasma membrane Ca 2+ ATPase (PMCA) and Na + /Ca 2+ exchangers (NCX). Although the means by which calcium slowly leaks into cells is not under- stood, there are a number of routes by which calcium can enter which are much more well-defined. These include Ca 2+ permeant channels, such as transient receptor potential (TRP) channels (activated by a range of extracellular signals), NMDA gated channels in the CNS and, of course, a wide range of voltage-operated Ca 2+ channels (VOC), which ini- tiate the calcium waves that initiate contraction in cardiomy- ∗ Corresponding author. Tel.: +44 207 380 9683. E-mail address: [email protected] (S.M. Davidson). ocytes [1,2]. Any sudden influx of Ca 2+ through a small local- ized pathway via these mechanisms must inevitably lead to a local, transient increase in [Ca 2+ ] c beneath the plasmalemma. The high calcium buffering capacity of the cytosol limits cal- cium diffusion [3], and so such increases may remain spatially restricted, requiring other mechanisms for amplification and propagation. Such localized increases in [Ca 2+ ] c have been referred to as “Ca 2+ microdomains” (for reviews, see [2], and other articles in this issue of Cell Calcium). However, a discussion of calcium microdomains is not limited to the transient increases that occur beneath the plasma membrane. Both the ER and golgi are microenviron- ments within the cell which contain a concentration of Ca 2+ which is far higher than that within in the cytosol, and so any release of Ca 2+ from these organelles must necessarily also result in a localized increase in [Ca 2+ ] c . Such increases have been defined operationally as “Ca 2+ puffs” (originating from IP3R in the ER) [4], “Ca 2+ sparks” (originating from RyR in the SR) [5], “Ca 2+ marks” (transient increases in mitochon- 0143-4160/$ – see front matter © 2006 Elsevier Ltd. All rights reserved. doi:10.1016/j.ceca.2006.08.017
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Calcium microdomains and oxidative stress

A
cmmrrdttat©
K
1
tol∼(mseC(Nrt
0d
Cell Calcium 40 (2006) 561–574
Calcium microdomains and oxidative stress
Sean M. Davidson a,∗, Michael R. Duchen b
a The Hatter Cardiovascular Institute, Royal Free and University College Medical School, London, Department of Medicine,67 Chenies Mews, London WC1E 6HX, United Kingdom
b Department of Physiology and Mitochondrial Biology Group, University College London, London, United Kingdom
Received 1 August 2006; accepted 23 August 2006Available online 17 October 2006
bstract
The phenomenon of calcium microdomains is firmly established in the field of subcellular physiology. These regions of localized, transientalcium increase are exemplified by the spontaneous ‘sparks’ released through the ryanodine receptor in myocytes, but include subplasmalem-al microdomains, focal calcium oscillations and microdomains enclosed within organelles, such as the endoplasmic reticulum, golgi anditochondria. Increasing evidence suggests that oxidative stress regulates both the formation and disappearance of microdomains. Calcium
elease channels and transporters are all modulated by redox state, while several mechanisms that generate oxidative or nitrosative stress areegulated by calcium. Here, we discuss the evidence for the regulation of calcium microdomains by redox state, and, by way of example,emonstrate that the frequency of calcium sparks in cardiomyocytes is increased in response to oxidative stress. We consider the evidence forhe existence of analogous microdomains of reactive oxygen and nitrogen species and suggest that the refinement of imaging techniques for
hese species might lead to similar concepts. The interaction between Ca2+ microdomains and proteins that modulate their formation results incomplex and dynamic, spatial signaling mechanism, which is likely to be broadly applicable to different cell types, adding new dimensionso the calcium signaling ‘toolkit’.2006 Elsevier Ltd. All rights reserved.
oilTcrpro
lpm
eywords: Calcium; Oxidative stress; ROS
. Introduction
Mammalian cells are bathed in interstitial fluid that con-ains greater than 1 mM Ca2+. Only by functioning continu-usly are extrusion mechanisms able to counter the inwardeak of this calcium, and maintain cytosolic free calcium at
100 nM. These include the plasma membrane Ca2+ ATPasePMCA) and Na+/Ca2+ exchangers (NCX). Although theeans by which calcium slowly leaks into cells is not under-
tood, there are a number of routes by which calcium cannter which are much more well-defined. These includea2+ permeant channels, such as transient receptor potential
TRP) channels (activated by a range of extracellular signals),
MDA gated channels in the CNS and, of course, a wideange of voltage-operated Ca2+ channels (VOC), which ini-iate the calcium waves that initiate contraction in cardiomy-
∗ Corresponding author. Tel.: +44 207 380 9683.E-mail address: [email protected] (S.M. Davidson).
wrrbIt
143-4160/$ – see front matter © 2006 Elsevier Ltd. All rights reserved.oi:10.1016/j.ceca.2006.08.017
cytes [1,2]. Any sudden influx of Ca2+ through a small local-zed pathway via these mechanisms must inevitably lead to aocal, transient increase in [Ca2+]c beneath the plasmalemma.he high calcium buffering capacity of the cytosol limits cal-ium diffusion [3], and so such increases may remain spatiallyestricted, requiring other mechanisms for amplification andropagation. Such localized increases in [Ca2+]c have beeneferred to as “Ca2+ microdomains” (for reviews, see [2], andther articles in this issue of Cell Calcium).
However, a discussion of calcium microdomains is notimited to the transient increases that occur beneath thelasma membrane. Both the ER and golgi are microenviron-ents within the cell which contain a concentration of Ca2+
hich is far higher than that within in the cytosol, and so anyelease of Ca2+ from these organelles must necessarily also
esult in a localized increase in [Ca2+]c. Such increases haveeen defined operationally as “Ca2+ puffs” (originating fromP3R in the ER) [4], “Ca2+ sparks” (originating from RyR inhe SR) [5], “Ca2+ marks” (transient increases in mitochon-
5 / Cell
dCg
tachwrmtcpsrbeittlb
evnpnabcmttbtcm
2
Eubitaccnmtd
usRstsrtm
mbafwipdoRsaCwtieTaC
watmafovasfn
it(Noots
62 S.M. Davidson, M.R. Duchen
rial Ca2+ which occur as a consequence of mitochondriala2+ uptake from nearby cytoplasmic sparks) [6], or, moreenerally, as [Ca2+]c “hot-spots”.
The concept of microdomains has helped in many caseso explain how [Ca2+]c signals may be generated withoutny measurable change in net [Ca2+]c levels over the wholeell. For instance, in the example above, PMCA transportersave a high affinity for calcium, so their activity is increasedithin the subplasmalemmal microdomain, leading to rapid
estoration of the resting [Ca2+]c. The kinetics of the for-ation and disappearance of the [Ca2+]c microdomains or
ransients will vary depending on the rates of the relevanthannels and transporters. The characteristics of the trans-orters are therefore fundamental to the dynamics of [Ca2+]cignaling, and are actively modified, either by wholesaleeplacement of different isoforms in different cell types [7], ory post-translational modifications. These modifications mayntail phosphorylation by protein kinases, protein–proteinnteractions or, as highlighted in this paper, redox regula-ion. Exposed sulfhydryl groups are particularly susceptibleo redox modulation, and they have been assigned a physio-ogical role in channel regulation in several cases, which wille discussed later.
As a consequence of increased [Ca2+]c, calcium-sensitivenzymes localized to the region of the increase will be acti-ated, and so the focal [Ca2+]c signal can serve as a mecha-ism to achieve selective local activation of Ca2+ dependentrocesses. These may include both the neuronal isoform ofitric oxide synthase (nNOS) [8] and the NADPH oxidase,n enzyme originally thought to be restricted to phagocytes,ut which is now known to generate oxidative stress in manyells types. Similarly, increased levels of calcium within theitochondria enhance respiratory activity [9], and may lead
o increased production of reactive oxygen species (ROS) byhe respiratory chain [10]. Thus, [Ca2+]c microdomains cane strongly linked to mechanisms that generate both oxida-ive and nitrosative stress, and there exists the potential for aomplex and interdependent organization of Ca2+ and ROSicrodomains in time and space.
. Sources of oxidative stress
Mitochondria are a major site of ROS production [11–13].ven under resting conditions between 2% and 5% of molec-lar oxygen consumed by mitochondria is partially reducedy the electron transport chain to form superoxide, whichs subsequently dismutated to hydrogen peroxide. Althoughhe production of most mitochondrial ROS is countered byn armory of antioxidant proteins and molecules within mito-hondria [11,12], it is possible that some mitochondrial ROSan exit the mitochondria and contribute to intracellular sig-
aling pathways. Hydrogen peroxide can diffuse across theitochondrial membrane, but superoxide cannot, leading tohe suggestion that it may exit via anion channels [11]. Asiscussed in a recent review [11], it has been difficult to
fic[
Calcium 40 (2006) 561–574
nequivocally establish whether ROS exit mitochondria initu, in normal cells, because most experiments in whichOS have been detected have been performed on suspen-ions of isolated mitochondria. However, it does seem clearhat under certain pathological situations, such as oxidativetress, antioxidant defenses are depleted or overwhelmed,esulting in the transit of mitochondrially produced ROS intohe cytoplasm where they may influence [Ca2+]c signaling
achinery and the formation of microdomains.This is not the place for a more detailed description of
echanisms and pathways of mitochondrial ROS generation,ut these have been discussed at length in reference [11] andlso see recent papers [14–16]. It has been suggested that ROSrom complex I are released into the mitochondrial matrix,hile ROS from complex III can also be released into the
ntermembrane space [16]. The rate of mitochondrial ROSroduction is thought to be highly dependent on mitochon-rial membrane potential and is affected by changes in the ratef oxidative phosphorylation. Consequently, mitochondrialOS production may vary greatly in different pathologicalettings, and may also be influenced by other factors thatffect the rate of respiration, including the mitochondriala2+ concentration [10]. Although the Ca2+ uptake path-ays of mitochondria are relatively low affinity, in many cell
ypes, the physical location of mitochondria, directly appos-ng the Ca2+ release units of ER/SR, means that they arexposed effectively to microdomains of high [Ca2+]c [17].he mitochondrial matrix buffers Ca2+ very effectively, withn effective Ca2+-binding ratio that is about 40 times thea2+-binding ratio of the cytoplasm [18].
The NADPH oxidase consists of a complex of proteinshich localize to the plasma membrane, and which function
s a simple electron transport chain transferring an electrono molecular oxygen. Although originally understood as the
echanism by which neutrophils generate antibacterial ROS,much more widespread regulatory role is being uncovered
or NADPH oxidase in various cell types including cardiomy-cytes [19], microglia [20] and astrocytes [21], which expressarious isoforms of the enzyme system at significant levels,lbeit at lower levels than found in neutrophils. It has to beaid that even though the enzyme is widely expressed in oneorm or another, its physiological and pathophysiological sig-ificance still need to be clarified [22].
Xanthine dehydrogenase (XDH) is a hydroxylase involvedn the oxidative metabolism of purines. Sulfhydryl oxida-ion readily and reversibly converts XDH to xanthine oxidaseXOR). Whereas the XDH form of this enzyme generally usesAD(+) as an electron acceptor, XOR can generate super-xide anion or hydrogen peroxide using molecular oxygen,r can generate nitric oxide via nitrate and nitrite reduc-ase activities [23]. XOR activity has been implicated ineveral pathological conditions, particularly endothelial dys-
unction, hypertension and heart failure [23]. Recent exper-ments demonstrated that XOR is localized to the SR ofardiac myocytes, and interacts with nitric oxide synthase24].
/ Cell
esibshfrtcrppR
3m
srtcTpmnsiopwadwwpie[
irumdrsoreitd
3
crdrcitcomottaspstret
cisnCStoikFsc[
erplahudaoob
S.M. Davidson, M.R. Duchen
It is difficult to discuss oxidative stress without consid-ring the contribution of nitric oxide (NO) and nitrosativetress. Both ROS and NO are highly reactive, and they cannteract, limiting each other’s effective diffusion distanceut also, in the process, producing highly reactive nitritepecies [25]. Reactive oxygen and nitrogen species (RNS)ave certain characteristics in common with calcium, whichavors their roles as signaling molecules. In particular, theirelatively short diffusion distances due to intracellular spa-ial buffering or reactivity will result in local microdomainsontaining high concentrations of reactive species. We willeturn later to the issue of whether ROS and RNS might beresent as microdomains, and will next describe some exam-les where interaction between calcium microdomains andOS, or RNS, seems likely to be important.
. Evidence for redox regulation of calciumachinery
There has been evidence for many years suggesting thateveral calcium signaling pathways may be regulated byedox state. The properties of ryanodine receptors, IP3 recep-ors, NMDA glutamate receptors and probably voltage gatedalcium channels are all modulated by redox agents [26,27].he key questions must concern the physiological or patho-hysiological relevance of these pathways. Do they serve toodulate calcium signals during routine matters of cell sig-
aling or are they only significant at the extremes of redoxtate associated with pathology? The term “oxidative stress”s used loosely and seems somewhat vague, since reactivexygen species (ROS) including superoxide and hydrogeneroxide are continually produced even in resting cells. Aorking definition is: “An imbalance between oxidants and
ntioxidants in favor of the oxidants, potentially leading toamage,” [28]. We will therefore describe several exampleshere calcium machinery is regulated by redox state, andhich therefore has the potential to be modulated duringeriods of oxidative stress, such as occur during exposure toschemia and reperfusion, or, as has recently become appar-nt, within neurons in response to the peptide �-amyloid86].
The redox sensitivity of calcium transporters and channelss commonly conferred by sulfhydryl (SH) groups of cysteineesidues, hence, sensitivity to redox modulation dependspon the number of exposed sulfhydryl groups. Oxidationay result in either an increase in activity, or a decrease,
epending on the particular channel. While in many casesedox regulation appears to be exquisitely sensitive, exces-ive oxidation, such as may occur in oxidative stress, canften lead to irreversible inactivation [26]. The potential foredox regulation of many Ca2+ channels essential to cardiac
xcitation–contraction coupling has recently been reviewedn detail [26], so only those points particularly pertinent tohe formation and regulation of calcium microdomains areescribed below.am
i
Calcium 40 (2006) 561–574 563
.1. Ryanodine receptor
In muscle, the ryanodine receptor (RyR) Ca2+ releasehannels in the SR are responsible for the bulk of the Ca2+
eleased into the cytoplasm in response to activation of theihydropyridine-sensitive L-type Ca2+ channels (dihydropy-idine receptor or DHPR) in the plasma membrane. In thease of cardiac muscle, Ca2+ release is induced by Ca2+
nflux and Ca2+ induced Ca2+ release (CICR), while in skele-al muscle, Ca2+ is released in response to an electrome-hanical interaction between the RyR and DHPR. Activityf the RyR can be modulated by various post-translationalodifications, such as phosphorylation and interaction with
ther proteins, as well as thiol oxidation of critical cys-eines [26,29,30]. The RyR contains on the order of 80 cys-eine residues per monomer, although only a few of theseppear to confer redox sensitivity [31]. These hyperreactiveulfhydryl groups were shown to have a well-defined redoxotential in skeletal muscle SR. Thus, under mild oxidativetress, relatively small changes in the cellular redox poten-ial can contribute to significant stimulation of the ryanodineeceptor [32]. As one might expect, however, exposure toxcessive levels of ROS irreversibly destroy channel func-ion [33].
As well as directly modifying the open probability of thehannel, redox modification of the RyR can alter its sensitiv-ty to cytosolic Ca2+ [34]. Incubation of hamster eggs with theulfhydryl reagent thimerosal lowers the threshold of Ca2+
eeded to stimulate Ca2+ induced Ca2+ release (CICR) [35].alsequestrin, the principal lumenal Ca2+-binding protein inR, is concentrated in the junctional region of the SR. Here,
riadin anchors calsequestrin in place, and redox modulationf the RyR can alter its interaction with triadin, thus alteringts sensitivity to luminal Ca2+ [31]. It is also important toeep in mind that ROS may modify RyR activity indirectly.or example, superoxide has been found to stimulate conver-ion of NAD+ into cADPR in heart homogenates [36], andADPR is believed to increase the Ca2+ sensitivity of RyRs1].
Many of the regulatory components of the RyR are teth-red in the immediate vicinity, permitting rapid and preciseegulation. For example, PKA, a protein kinase that phos-horylates and activates RyR in response to elevated cAMPevels, is attached to RyR in skeletal muscle via A kinase-nchoring protein (mAKAP) [37]. There is also evidence thatighly localized production of ROS is involved in the mod-lation of RyR activity. For example, superoxide productionetected in the sarcoplasmic reticulum isolated from coronaryrtery smooth muscle has been attributed to a local NAD(P)Hxidase system, and this appears to be able to increase RyRpen probability [38]. NAD(P)H oxidase activity has alsoeen detected in cardiac sarcoplasmic reticulum fractions,
nd, by enhancing the redox modification of RyR, seems toediate the electrical induction of tachycardia [39].As mentioned, it is becoming increasingly evident that,n many cases, oxidative stress tells only half of the story,
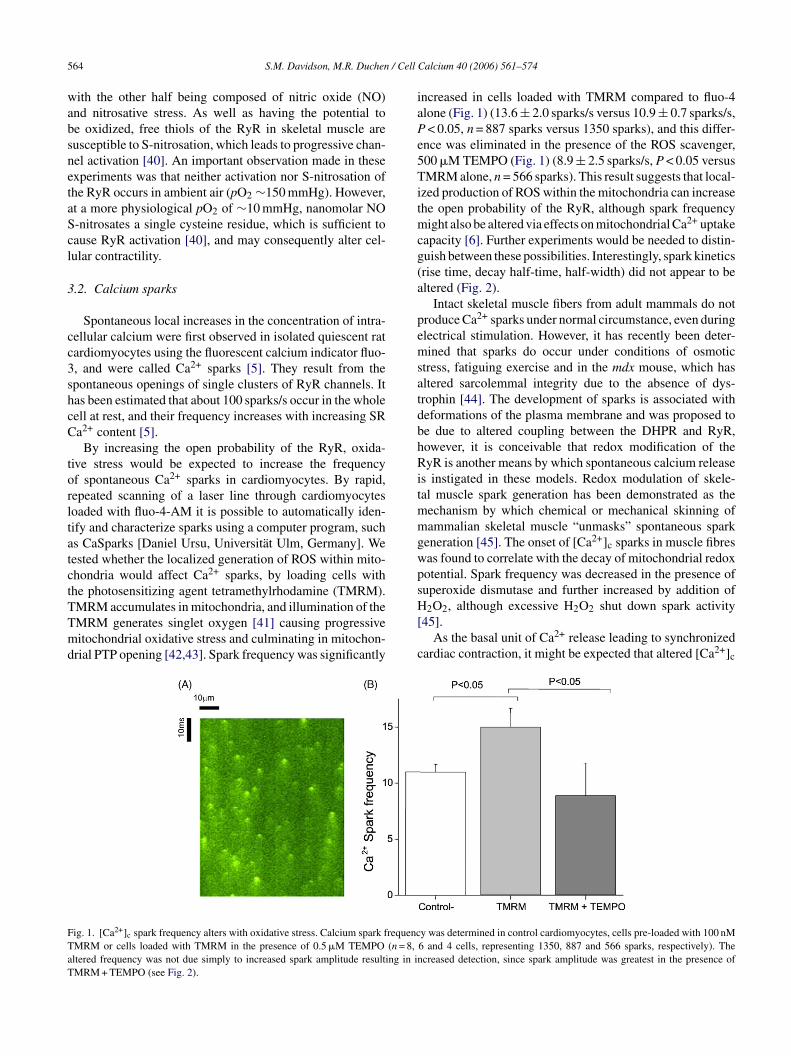
5 / Cell
wabsnetaScl
3
cc3shcC
torltatctTTmd
iaPe5Titmcg(a
pemsatdbhRitmmgwps
FTaT
64 S.M. Davidson, M.R. Duchen
ith the other half being composed of nitric oxide (NO)nd nitrosative stress. As well as having the potential toe oxidized, free thiols of the RyR in skeletal muscle areusceptible to S-nitrosation, which leads to progressive chan-el activation [40]. An important observation made in thesexperiments was that neither activation nor S-nitrosation ofhe RyR occurs in ambient air (pO2 ∼150 mmHg). However,t a more physiological pO2 of ∼10 mmHg, nanomolar NO-nitrosates a single cysteine residue, which is sufficient toause RyR activation [40], and may consequently alter cel-ular contractility.
.2. Calcium sparks
Spontaneous local increases in the concentration of intra-ellular calcium were first observed in isolated quiescent ratardiomyocytes using the fluorescent calcium indicator fluo-, and were called Ca2+ sparks [5]. They result from thepontaneous openings of single clusters of RyR channels. Itas been estimated that about 100 sparks/s occur in the wholeell at rest, and their frequency increases with increasing SRa2+ content [5].
By increasing the open probability of the RyR, oxida-ive stress would be expected to increase the frequencyf spontaneous Ca2+ sparks in cardiomyocytes. By rapid,epeated scanning of a laser line through cardiomyocytesoaded with fluo-4-AM it is possible to automatically iden-ify and characterize sparks using a computer program, suchs CaSparks [Daniel Ursu, Universitat Ulm, Germany]. Weested whether the localized generation of ROS within mito-hondria would affect Ca2+ sparks, by loading cells withhe photosensitizing agent tetramethylrhodamine (TMRM).
MRM accumulates in mitochondria, and illumination of theMRM generates singlet oxygen [41] causing progressiveitochondrial oxidative stress and culminating in mitochon-rial PTP opening [42,43]. Spark frequency was significantly
H[
c
ig. 1. [Ca2+]c spark frequency alters with oxidative stress. Calcium spark frequencMRM or cells loaded with TMRM in the presence of 0.5 �M TEMPO (n = 8,ltered frequency was not due simply to increased spark amplitude resulting in inMRM + TEMPO (see Fig. 2).
Calcium 40 (2006) 561–574
ncreased in cells loaded with TMRM compared to fluo-4lone (Fig. 1) (13.6 ± 2.0 sparks/s versus 10.9 ± 0.7 sparks/s,< 0.05, n = 887 sparks versus 1350 sparks), and this differ-
nce was eliminated in the presence of the ROS scavenger,00 �M TEMPO (Fig. 1) (8.9 ± 2.5 sparks/s, P < 0.05 versusMRM alone, n = 566 sparks). This result suggests that local-
zed production of ROS within the mitochondria can increasehe open probability of the RyR, although spark frequency
ight also be altered via effects on mitochondrial Ca2+ uptakeapacity [6]. Further experiments would be needed to distin-uish between these possibilities. Interestingly, spark kineticsrise time, decay half-time, half-width) did not appear to beltered (Fig. 2).
Intact skeletal muscle fibers from adult mammals do notroduce Ca2+ sparks under normal circumstance, even duringlectrical stimulation. However, it has recently been deter-ined that sparks do occur under conditions of osmotic
tress, fatiguing exercise and in the mdx mouse, which hasltered sarcolemmal integrity due to the absence of dys-rophin [44]. The development of sparks is associated witheformations of the plasma membrane and was proposed toe due to altered coupling between the DHPR and RyR,owever, it is conceivable that redox modification of theyR is another means by which spontaneous calcium release
s instigated in these models. Redox modulation of skele-al muscle spark generation has been demonstrated as the
echanism by which chemical or mechanical skinning ofammalian skeletal muscle “unmasks” spontaneous spark
eneration [45]. The onset of [Ca2+]c sparks in muscle fibresas found to correlate with the decay of mitochondrial redoxotential. Spark frequency was decreased in the presence ofuperoxide dismutase and further increased by addition of
2O2, although excessive H2O2 shut down spark activity45].As the basal unit of Ca2+ release leading to synchronized
ardiac contraction, it might be expected that altered [Ca2+]c
y was determined in control cardiomyocytes, cells pre-loaded with 100 nM6 and 4 cells, representing 1350, 887 and 566 sparks, respectively). Thecreased detection, since spark amplitude was greatest in the presence of

S.M. Davidson, M.R. Duchen / Cell Calcium 40 (2006) 561–574 565
Fig. 2. The average profile of spontaneous [Ca2+]c sparks in cardiomyocytes is not altered by oxidative stress. The average profile of [Ca2+]c sparks wascomputed using the same data analyzed for Fig. 1. For control cells the results were: average rise time 11.0 ± 0.2 ms; amplitude 0.71 ± 0.02 (�F/F ); width ath s comps + 0.5 mt resence
sccqieamTmalc
mqirewmd
alf maximum 2.27 ± 0.05 �m; half-decay time 27.3 ± 0.2 ms. These valueignificantly different for cells in the presence of 100 nM TMRM or TMRMhe presence of TMRM, and significantly (20%, P < 0.01) increased in the p
park frequency or dynamics would have serious pathologicalonsequences. Indeed, atrial fibrillation is the most commonardiac arrhythmia in humans, and a significantly higher fre-uency of [Ca2+]c sparks has been detected in atrial myocytessolated from patients with atrial fibrillation, despite an appar-ntly normal SR Ca2+ content [46]. This was associated withgreater frequency of spontaneous [Ca2+]c waves in theyocytes, and might thereby explain the arrhythmia [46].he authors hypothesized that the increase in spark frequency
ight be due to an increase in the SR Ca2+ release channelctivity. Based on the evidence presented thus far, an under-ying pathology leading to increased oxidative stress mightonceivably have led to this increase in channel activity.
omaa
0
are well with published values for cardiomyocytes [114]. Values were notM TEMPO, although the average amplitude was slightly increased (6%) inof TMRM + TEMPO.
Discounting altered SR Ca2+ content, the most obviousechanism by which oxidative stress could affect the fre-
uency of spark formation is by a direct effect on RyR,ncreasing the probability of opening by oxidation of criticalesidues. However, indirect pathways should also be consid-red. For example, activation of kinases, such as PKA or PKC,hich phosphorylate and modulate RyR/phospholamban,ay affect spark frequency or kinetics. A third possibility
erives from the interesting observation that the activation
f IP3R, which co-localizes with some of the RyR in atrialyocytes, can lower the threshold for SR Ca2+ release [47],lthough it is not known whether this mechanism can oper-te in ventricular myocytes. Lastly, decreased mitochondrial

5 / Cell
pc[
3
nkHlfmocth
dtcimitcovmdcpmrd
4
intcte[usecC[rHI
srabbcrb
ulmdssovaoaedfooonmd
5(
uSoSR[SprSapap
66 S.M. Davidson, M.R. Duchen
otential, hence reduced mitochondrial calcium bufferingapacity, has also been found to increase spark frequency48].
.3. BK channels
Large-conductance Ca2+-activated potassium (BK) chan-els in the sarcolemma of arterial smooth muscle have beennown for some time to play an important role in vasodilation.owever, most vasodilators barely influence global intracel-
ular [Ca2+] in smooth muscle, and the affinity of BK channelsor Ca2+ is relatively low (KD ∼20 �M [reviewed in Well-an and Nelson [49]]). This is explained by the generation
f subplasmalemmal Ca2+ sparks which result in local Ca2+
oncentrations estimated at 10–30 �M [49]—greatly poten-iating coupled BK channel opening and causing a membraneyperpolarization of up to 20 mV per spark [50].
Recent data suggests that ROS generated by mitochon-ria increase Ca2+ spark generation in arterial smooth muscle,hereby increasing BK activation, membrane polarization andonsequently vasodilation [51]. The ROS species involveds likely to be H2O2, since it can freely diffuse across the
itochondrial membrane, but could also be superoxide, exit-ng mitochondria via anion channels. Interestingly, it appearshat mitochondrial ROS production induced by mild mito-hondrial uncoupling (in response to application of FCCPr the KATP channel opener diazoxide), is sufficient to acti-ate this pathway of vasodilation [51]—despite the fact thatild uncoupling is usually described as a mechanism of
ecreasing ROS production [52]. This pathway linking mito-hondrial ROS and vasodilation may turn out to have broadhysiological relevance. For example, H2O2 generated withinitochondria is known to mediate arterial vasodilation in
esponse to shear stress, though the mechanism has not beenefined [53].
. Ins (1,4,5)P3 receptor
The second major class of channel responsible for calciumnduced calcium release from ER/SR stores, particularly inon-excitable cells, is the Ins (1,4,5)P3 receptor (IP3R). Ashe name suggests, binding of Ins (1,4,5)P3 sensitizes thehannel to activation by Ca2+ [1]. Recent studies indicatehat a set of highly reactive cysteine residues are differ-ntially accessible in two splice variants of type 1 IP3R54], and sulfhydryl modification has been shown to mod-late IP3R activity in various cell types. For example, theulfhydryl reagent thimerosal sensitizes the IP3R to IP3, andvokes spontaneous Ca2+ spikes in hepatocytes and HeLaells [55,56]. Similarly, thimerosal dramatically increasesa2+ release by IP3R activation in cultured rat astrocytes
57]. In vascular smooth muscle, superoxide sensitizes Ca2+
elease stimulated by IP3 [58]. Oxidized glutathione [59] and2O2 [60] have also been shown to induce Ca2+ release from
P3-mediated stores in endothelial cells.
6
n
Calcium 40 (2006) 561–574
Hypoxic mobilization of Ca2+ in neuronal cells has beenhown to be directly mediated by stimulation of IP3R Ca2+
elease by increased NADH levels [61]. Interestingly, thisppears to be mediated by local domains of NADH generatedy GAPDH bound to the IP3R [62]. Nitric oxide has alsoeen suggested to directly modify the IP3R under hypoxiconditions [63], although the possibility that it acts via indi-ect effects on mitochondrial metabolism, etc., should alsoe considered.
[Ca2+]c oscillations are observed in many types of cellnder stimulation, and the actual frequency of Ca2+ oscil-ation regulates the activity of various Ca2+-dependentolecules such as NFAT [64], Ca2+- and calmodulin-
ependent kinase II [65] and PKC [66]. A recent eleganttudy in HeLa cells suggests that, while the initial [Ca2+]cpike is due to Ca2+ release from the ER via IP3R, subsequentscillations depend on the release of mitochondrial Ca2+
ia Na+/Ca2+ exchangers (NCX), which induces a regener-tive ER Ca2+ release [67]. As a consequence, the rate ofscillation is controlled by Ca2+ shuttling between the ERnd mitochondria in phase with the oscillations [67]. Cellsxpressing mutant IP3R with a reduced sensitivity to Ca2+
o not generate [Ca2+]c oscillations [68]. One might there-ore expect an additional consequence of redox modificationf IP3R and NCX exchangers to be altered characteristicsf [Ca2+]c oscillations. Indeed, an interesting recent studybserved that [Ca2+]c oscillations induced in pancreatic aci-ar cells by cholecystokinin are associated with low levels ofitochondrial ROS production and are inhibited by antioxi-
ants targeted to the mitochondria [69].
. Sarco-/endo-plasmic reticulum Ca2+ ATPaseSERCA)
Ca2+ is driven into the sarcoplasmic or endoplasmic retic-lum by SERCA, utilizing the energy of ATP hydrolysis.ERCA maintains [Ca2+]ER at ∼300 �M, three to four ordersf magnitude greater than [Ca2+]c [70]. The activity ofERCA is modulated by redox state, but in contrast to theyR and IP3R, its activity is inhibited by thiol oxidation
26]. Recent evidence suggests that redox modulation ofERCA activity can affect [Ca2+]c microdomains. In Xeno-us oocytes, [Ca2+]c oscillations are triggered by cyclic Ca2+
elease via IP3R and subsequent reuptake into the ER byERCA, and mutation of critical cysteine residues in SERCAlters the frequency of this oscillation [71]. Interestingly, ERrotein 57, a ubiquitous ER thiol-dependent oxidoreductase,ppears to modulate the redox state of these ER-facing thiols,roviding dynamic control of ER Ca2+ homeostasis [71].
. Other channels
It is certainly the case that the activity of other Ca2+ chan-els can also be modulated by redox status, either directly

/ Cell
omwmmcibrimtcda[shtui
6
cbtlrvddg“c[iaustphatcsmimpttd[
tmSddRstttwtcaiAosoiwelaiwo
aomtAom(flutempbRApiadti
S.M. Davidson, M.R. Duchen
r indirectly, although the importance of this in regards toicrodomains is generally less well defined. For example, asell as inhibiting SERCA activity, ROS can inhibit otherembers of the P-type ATPase family, such as the plas-alemmal Ca2+ ATPase (PMCA). Stimulation of the sar-
olemmal Na+–Ca2+ exchanger (NCX) by ROS can resultn the accumulation of intracellular calcium, which maye particularly important in pathological situations, such aseperfusion after ischemia (reviewed in reference [26]). Theres also evidence that the L-type Ca2+ channel of cardiac
yocytes is modulated by redox state [72,73], although inhis case oxidation of thiols by thimerosal causes irreversiblehannel inhibition [74]. Redox modulation of the N-methyl--aspartate (NMDA) receptor is physiologically important,nd thiol oxidizing agents decrease NMDA-evoked currents27]. Interestingly, antioxidants have also been found to tran-iently inhibit the NMDA-evoked current in cultured ratippocampal neurones [75]. A single cysteine residue inhe NMDA receptor has been identified as the target of NOnder physiological conditions, and modulates channel activ-ty [76].
.1. Mitochondrial permeability transition pore
A channel which deserves special mention is the mito-hondrial permeability transition pore (mPTP), since it haseen suggested to be involved in the regulation of specificypes of Ca2+ and ROS domains (below). The mPTP is aarge, voltage-dependent, non-selective pore, which opens inesponse to high levels of Ca2+ and oxidative stress. Irre-ersible opening of the mPTP causes rapid mitochondrialepolarization and leads inevitably to cell death due to ATPepletion. Transient opening of the mPTP has been sug-ested to have a role in normal physiology, perhaps as arelease valve”, allowing excess accumulated mitochondrialalcium to escape into the cytoplasm (discussed in reference77]). In astrocytes, the frequency of transient mPTP open-ng is increased by the interplay between mitochondrial ROS,nd microdomains of Ca2+ released from the ER and takenp by the mitochondria [43]. Although the physiologicalignificance of transient mPTP opening remains unknown,here is no doubt that irreversible opening occurs in manyathological situations. For example, opening of the mPTPas been detected during the initial minutes of reperfusionfter ischemia [78]. Although, it has widely been assumedhat mitochondrial Ca2+ overload is the critical pathologi-al feature leading to mPTP opening, accumulating evidenceuggests that oxidative stress is fundamentally important anday sensitize mPTP opening in response to Ca2+ [79]. In fact,
t was suggested nearly 20 years ago that oxidation of criticalitochondrial thiol groups may trigger mPTP opening [80],
ossibly by modifying thiol groups on the adenine nucleotide
ranslocase [81], a component of the pore. A second site inhe mPTP confers sensitivity to redox state of the mitochon-rial NAD(P)H pool, possibly by an allosteric mechanism82].ttda
Calcium 40 (2006) 561–574 567
The generation of ROS within mitochondria by photoac-ivation of TMRM, as described above, leads eventually to
PTP opening and mitochondrial depolarization [42,83].ubsequent to mPTP opening, a burst of localized ROS pro-uction produced by the electron transport chain can beetected using fluorescent substrates. This has been termedOS induced ROS release (RIRR) [83]. RIRR has beenhown to correlate with a wave of mitochondrial depolariza-ion along the length of the cardiomyocytes [84], implyinghe propagation of a ROS microdomain or wave front throughhe cytosol. This wave was initiated by localized oxidationithin a single cell, and moved at 5 �m/min—much slower
han the Ca2+ induced Ca2+ release wave responsible forell contraction which moves at 10–50 �m/s, but similar toredox wave propagating at 2 �m/s that has been observed
n cardiomyocytes cultured in a glucose-free medium [85].s recognized by Brady et al., considering the short half-lifef ROS, the progression of such a slow wave must involveome regenerative mechanism. However, this wave was onlybserved in roughly 50% of cells [84]. In our hands, local-zed oxidation within a single myocyte did not generate aave of RIRR without an additional “boost” of ROS gen-
rated by photo-oxidation of the whole cell, suggesting thatocal antioxidant defences may be sufficient to restrict therea of oxidative damage. For example, 5 min after localizednduction of mPTP opening by photo-oxidation of TMRM,e could detect no expansion in the area of depolarization,r of ROS (indicated by DCF-DA fluorescence) (Fig. 3).
Considering the evidence for redox regulation of RyRctivity, we wondered whether the increase in ROS at mPTPpening would increase spark frequency. We used the aboveodel to generate ROS within mitochondria and examined
he effect of localized ROS production on spark production.s has previously been observed, rapid, repeated line scanf a region of an isolated adult rat cardiomyocytes causedPTP opening, detected as the loss of TMRM fluorescence
Fig. 4A). By simultaneously imaging the fluorescence ofuo-4 we were able to observe individual [Ca2+]c sparks. Wesed a simple procedure to estimate spark production overime at a single point along the line scan, and plotted thesevents alongside local mitochondrial potential (Fig. 4B). Inany cells, we noticed a sharp increase in the rate of spark
roduction when the mPTP opened (Fig. 4B and C). We incu-ated cells in the presence of TEMPO to determine whetherOS were responsible for the increase in spark frequency.s expected, mPTP opening was significantly delayed in theresence of the ROS scavenger. TEMPO also prevented thencrease in spark frequency at mPTP opening (Fig. 4D). Welso incubated cells in thimerosal to demonstrate that oxi-ation of thiols increases basal spark activity. This appearedo increase spark frequency to a maximum since no furtherncrease in spark activity was observed at mPTP opening in
he presence of thimerosal (Fig. 4E). These data illustratehat, upon mPTP opening, local spark frequency increasesue to higher ROS levels, probably due to enhanced RyRctivity.
568 S.M. Davidson, M.R. Duchen / Cell
Fig. 3. Oxidative stress can be spatially restricted within a cardiomyocyte.Opening of the mPTP can be induced in a region of a cardiomyocyte withoutleading to any spread of ROS induced ROS release. Adult rat cardiomy-ocytes were loaded with 100 nM TMRM (top panel), and mPTP opening wasinduced in a region of the cell by repeated linescan using a confocal micro-sit
7C
AiwNqpcooomTdmtist
nrmN
ra
8
rwnnansaoitacpeap
dtadRwcpa
tSdsse(maldaSrtpt
cope (middle panel). After 5 min, the cell was imaged again. The locallylluminated mitochondria remained depolarized, but no further increase inhe region of depolarized mitochondria could be observed (lower panel).
. Other evidence for interdependence of ROS anda2+ microdomains
Amyloid peptide beta (A�) accumulates in the CNS inlzheimer’s disease, and causes sporadic transient increases
n [Ca2+]c in astrocytes. These [Ca2+]c spikes are associatedith a Ca2+-dependent increase in the generation of ROS byADPH oxidase. ROS generated within astrocytes is subse-uently transmitted to neurons and causes their death [86],robably reflecting a failure of trophic support, as astro-ytes feed neurons with glutathione precursors [87]. Thexidative stress generated by the oxidase also has an impactn mitochondrial potential, causing a slow depolarizationf astrocyte mitochondria due to impaired substrate supply,ost likely by oxidative damage to glucose uptake pathways.hus, the local sublasmalemmal microdomains of ROS pro-uced by NADPH oxidase in response to [Ca2+]c spikes mayediate some of the toxic effects of A�. However, the oxida-
ive stress also sensitizes astrocyte mitochondria to the A�nduced [Ca2+]c transients, causing transient mPTP opening,uggesting that the ROS cannot be restricted closely underhe plasmalemma [86,88].
It is also important to keep in mind that many other sig-
aling proteins and transcription factors can be activated inesponse to oxidative stress, such as the small GTPase RAS,itogen activated proteins kinase (MAPK), p38, JNK, Ask1,F-kappa B and hypoxia inducible factor (HIF-1). Redoxcimt
Calcium 40 (2006) 561–574
egulation of some of these signaling pathways may wellffect the formation of Ca2+ microdomains.
. ROS microdomains
Theoretically, one means of generating a spatiallyestricted increase in Ca2+ is the localized generation of ROS,hich then activates local Ca2+ release channels by mecha-isms outlined above. Thus far, the “ROS microdomain” hasot really emerged as a distinct concept in the same ways it has for Ca2+ microdomains, although there are sig-ificant parallels with the Ca2+ regulatory network, whichuggest that it might be a useful concept. For example, there islready substantial evidence for highly localized productionf ROS (discussed above), there is a powerful ROS buffer-ng/scavenging system, which will control the decay of ROSransients [6,7], and ROS can diffuse through the cytoplasm tocertain extent. In some cases, a global increase in cytosolicalcium may result in localized ROS production. For exam-le, treatment of astrocytes with a calcium ionophore, orxposure to amyloid � peptide, or ATP, causes the gener-tion of ROS by NADPH oxidase, which is localized at thelasma membrane [21].
There are some conceptual differences between micro-omains of calcium and ROS or RNS. The term when appliedo calcium is usually reserved for transient fluctuations, suchs sparks, which have a half-life of 20 ms or so in car-iomyocytes (see Fig. 2). In contrast, the prevalent view ofOS/RNS production is of a continually produced species,ith the rate of production being altered (or ceasing), under
ertain conditions. One consequence of reactive species beingroduced continuously from a source point and then diffusingway, would be a sustained concentration gradient.
An important point here is to consider the species of ROShat might be able to generate significant local microdomains.uperoxide is the major immediate product of the mitochon-rial respiratory chain and of the NADPH oxidase. However,uperoxide is thought not to cross membranes readily, ando if generated in the matrix side of the mitochondrion mustither exit through a channel or after conversion to peroxideby mitochondrial superoxide dismutase), which does crossembranes freely. The NADPH oxidase transfers electrons
cross membranes to generate superoxide, but this also willikely be rapidly converted to peroxide. NOS requires tetrahy-robiopterin (THB) as a cofactor, and when levels of THBre reduced, also has the capacity to generate superoxide.ignificantly, microdomains of calcium have the potential toegulate highly localized isoforms of nitric oxide synthasehat are calcium-sensitive, such as nNOS or eNOS at thelasma membrane, and mtNOS within mitochondria. Fur-hermore, in some cases, ROS produced by NADPH oxidase
an activate eNOS [89]. These examples suggest the intrigu-ng possibility that microdomains of different moleculesay sustain and influence each other. The ramifications ofhese ROS/RNS “microdomains” largely remain unexplored,
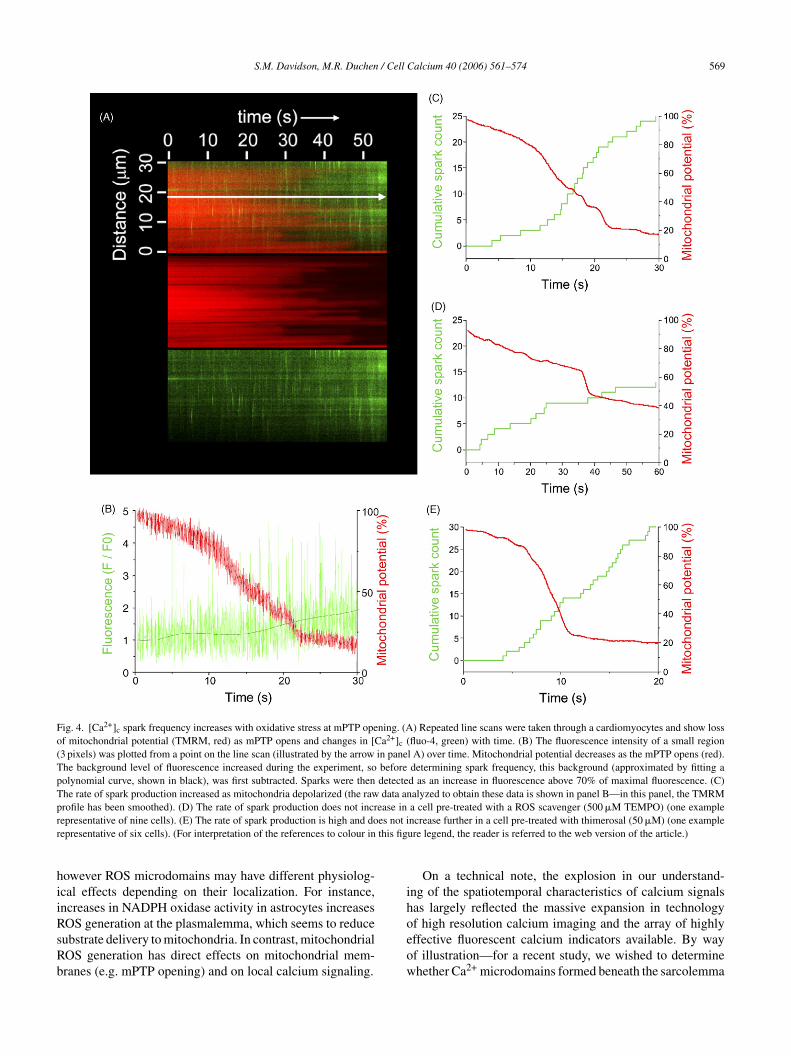
S.M. Davidson, M.R. Duchen / Cell Calcium 40 (2006) 561–574 569
Fig. 4. [Ca2+]c spark frequency increases with oxidative stress at mPTP opening. (A) Repeated line scans were taken through a cardiomyocytes and show lossof mitochondrial potential (TMRM, red) as mPTP opens and changes in [Ca2+]c (fluo-4, green) with time. (B) The fluorescence intensity of a small region(3 pixels) was plotted from a point on the line scan (illustrated by the arrow in panel A) over time. Mitochondrial potential decreases as the mPTP opens (red).The background level of fluorescence increased during the experiment, so before determining spark frequency, this background (approximated by fitting apolynomial curve, shown in black), was first subtracted. Sparks were then detected as an increase in fluorescence above 70% of maximal fluorescence. (C)The rate of spark production increased as mitochondria depolarized (the raw data analyzed to obtain these data is shown in panel B—in this panel, the TMRMprofile has been smoothed). (D) The rate of spark production does not increase in a cell pre-treated with a ROS scavenger (500 �M TEMPO) (one exampler es not ir this fig
hiiRsRb
ih
epresentative of nine cells). (E) The rate of spark production is high and doepresentative of six cells). (For interpretation of the references to colour in
owever ROS microdomains may have different physiolog-cal effects depending on their localization. For instance,ncreases in NADPH oxidase activity in astrocytes increases
OS generation at the plasmalemma, which seems to reduceubstrate delivery to mitochondria. In contrast, mitochondrialOS generation has direct effects on mitochondrial mem-ranes (e.g. mPTP opening) and on local calcium signaling.oeow
ncrease further in a cell pre-treated with thimerosal (50 �M) (one exampleure legend, the reader is referred to the web version of the article.)
On a technical note, the explosion in our understand-ng of the spatiotemporal characteristics of calcium signalsas largely reflected the massive expansion in technology
f high resolution calcium imaging and the array of highlyffective fluorescent calcium indicators available. By wayf illustration—for a recent study, we wished to determinehether Ca2+ microdomains formed beneath the sarcolemma
5 / Cell
owiuaNtlatcp
9
isathcdbjafdNpdCt
optcdptei
aifidctapho
lmaptiaatDtbbTacotsfs
itAsDtoiidmctwcactb
tmdlitcer
70 S.M. Davidson, M.R. Duchen
f neutrophils, but quantification of the changes we observedas complicated by the movement of the cells. The availabil-
ty of the ratiometric Ca2+-sensing dyes, indo-1, permittednequivocal detection of these Ca2+ microdomains, gener-ted in neutrophils and associated with the activation ofADPH oxidase [90]. In contrast, our knowledge of the spa-
iotemporal properties of ROS/RNS signals has been largelyimited by the relatively impoverished array of tools avail-ble to study them and the limitations of all imaging probeso provide meaningful spatial resolution. This is likely tohange with new generations of fluorescent protein basedrobes [91–93].
. Pathophysiological implications
Dysregulation of intracellular ROS handling has beenmplicated in the development of various disease states,uch as diabetes mellitus, sepsis, neurodegenerative diseases,therosclerosis, ischemia and reperfusion injury, and formshe basis of a theory on the mechanism of ageing whichas gained widespread support [94]. Of course, excessiveoncentrations of ROS are capable of causing widespreadamage to just about all components of the cell; from mem-rane lipids to proteins and DNA. But when one considersust how exquisitely sensitivity the Ca2+ handling proteinsre to redox modulation; how the Ca2+ concentration in dif-erent subcellular compartments is subject to continuous,elicate regulation; and the subtle interplay between ROS,O and Ca2+ networks, it is not difficult to imagine thatathologies that cause even minor oxidative stress might haveetrimental effects on critical signaling elements involvinga2+ microdomains and so influence the spatiotemporal pat-
erning of [Ca2+]c signals in cells and tissues.Probably the best evidence for a causal chain linking
xidative stress to alterations in [Ca2+]c microdomains toathological disturbances at an organism level comes fromhe cardiovascular field, since the “readout” of altered cal-ium dynamics is readily observable in disturbances in car-iac contractile function and vascular function. For exam-le, “myocardial stunning”, which is a type of contrac-ile dysfunction that is believed to be due to impairedxcitation–contraction coupling has been linked to increasesn cellular generation of ROS [95].
Experimental cardiac hypertrophy, either in response to �-drenergic agonists or pressure overload, causes an increasen the activity of NAD(P)H oxidase and may lead to heartailure [96–98]. There is also strong evidence that a defectn Ca2+ signaling via calcineurin is a causative factor in car-iac hypertrophy [99]. Interestingly, overexpression of cal-ineurin in the hearts of mice causes mitochondrial dysfunc-ion, elevated superoxide production and leads to hypertrophy
nd heart failure within 8–12 weeks [100,101]. One inter-retation is that in pathological situations, defects in Ca2+andling and oxidative stress can mutually reinforce eachther.
mafe
Calcium 40 (2006) 561–574
Another interesting clinical manifestation of the directink between pathological oxidative stress and [Ca2+]c
icrodomains may be the enhanced likelihood of cardiacrrhythmias with age, after myocardial infarction, and inatients with diabetes or hypertension [102,103]. As men-ioned previously, [Ca2+]c spark dynamics may be alteredn the presence of diabetes. Higher spontaneous Ca2+ sparkctivity has been observed in cardiac myocytes isolated fromged mice and rats, with altered spark kinetics suggesting thathere were alterations in RyR gating properties [104,105].ysregulated redox modulation of L-type Ca2+ channels or
he RyR can cause arrhythmias. In support of the possi-ility that oxidative stress is involved, oxidative injury haseen detected in human patients with atrial fibrillation [106].here is some tantalizing evidence that treatment with anntioxidant reduces the incidence of atrial fibrillation afterardiac bypass graft surgery [107]. Although a great dealf fundamental research remains to be done in this area,his result suggests the hugely attractive possibility that theimple administration of antioxidants can be used to success-ully treat complex pathologies that can result when oxidativetress results in defective Ca2+ handling.
Oxidative stress is believed to be fundamentally involvedn the development of a wide range of medically impor-ant conditions, including cardiovascular disease, diabetes,lzheimer’s disease and sepsis, and the influence of oxidative
tress on calcium microdomains may be potentially relevant.iabetes and cardiovascular disease are considered to be par-
icularly important in those countries that fund the majorityf medical research, but their rapidly increasing prevalencen developing countries is also predicted to have devastat-ng consequences on mortality and morbidity. The metabolicefects that define diabetes also increase the risk of arrhyth-ias and silent cardiac infarctions, with the results that
ardiovascular complications including myocardial infarc-ion are one of the major causes of death in individualsith diabetes [108]. In models of types I and II diabetes,
ontractility of isolated ventricular myocytes is impaired,s is the calcium decay rate, possibly through reduced SRalcium handling [109–111]. This suggests the possibilityhat [Ca2+]c microdomains would also be modified in dia-etes.
The characteristics of Ca2+ sparks have been examined inhe hearts of two different animal models of diabetes. db/db
ice lack the leptin receptor and develop obesity and severeiabetes, displaying hyperglycemia and insulin resistance,eading to cardiac failure as early as 12 weeks. [Ca2+]c sparksn cardiomyocytes isolated from db/db mice are less frequenthan in +/+ myocytes, partly because of a depression in sar-oplasmic reticulum Ca2+ load but also because of a reducedxpression of the RyR [112]. However, this contrasts withesults from rats harboring type I diabetes induced by treat-
ent with streptozotocin, where basal [Ca2+]c level as wells [Ca2+]c-spark frequency of isolated cardiomyocytes wasound to be significantly increased [113]. [Ca2+]c transientsxhibited significantly reduced amplitude and prolonged time

/ Cell
crphomit
qAmtptotcpip
1
lpoaaisistpCm
ioust
R
S.M. Davidson, M.R. Duchen
ourses as well as depressed Ca2+ loading of sarcoplasmiceticulum in diabetic rats. Western blotting using an anti-hospho-RyR2 antibody RyR was used to show that RyR wasyperphosphorylated in diabetic rat hearts [113], althoughther modifications were not tested for. Interestingly, treat-ent of isolated cardiomyocytes from diabetic animals with
nsulin for 4 h was sufficient to restore normal spark charac-eristics [113].
As yet, the role of oxidative stress in the altered spark fre-uency of diabetic cardiomyocytes has not been determined.s we have seen, the response of the RyR to oxidative stressay be biphasic, with sensitization at low stress levels leading
o inactivation by strong stresses. This raises the interestingossibility that different diabetic models have either sensi-ization or resistance of RyR activity, and, hence, increasedr decreased spark activity, depending on the level of oxida-ive stress. However, it may be challenging to isolate a singleause in an animal model with alterations in various com-onents of the [Ca2+]c signaling pathway, not to mentionnterdependent pathways of oxidative stress and nitric oxideroduction.
0. Conclusion
The concept of the cell cytoplasm as a linear network ofogically ordered signaling molecules swimming in a cyto-lasmic pool is being superseded by a much more spatiallyrientated model whereby transient microdomains of calciumnd ROS/RNS interact and transmit signals to moleculesnchored in membranes and scaffolding structures. The activ-ty of all the major Ca2+ channels and transporters has beenhown to be modulated by redox state, both directly, andn some cases indirectly via NADH levels or other redox-ensitive proteins. Although the significance of this is difficulto establish in normal cells, there is strong evidence thatathological levels of oxidative stress cause disturbances ina2+ microdomain formation and transmission, and that thisay, in some cases, lead to disease.The evolution of imaging technologies enabling the local-
zation of areas of ROS production with high resolutionver time and space will doubtless lead to a much improvednderstanding of how ROS domains influence intracellularignaling, much as Ca2+ imaging technology has done forhat speciality.
eferences
[1] M.J. Berridge, M.D. Bootman, H.L. Roderick, Calcium signalling:dynamics, homeostasis and remodelling, Nat. Rev. Mol. Cell Biol. 4(2003) 517–529.
[2] R. Rizzuto, T. Pozzan, Microdomains of intracellular Ca2+: molecular
determinants and functional consequences, Physiol. Rev. 86 (2006)369–408.[3] N.L. Allbritton, T. Meyer, L. Stryer, Range of messenger action ofcalcium ion and inositol 1,4,5-trisphosphate, Science 258 (1992)1812–1815.
Calcium 40 (2006) 561–574 571
[4] Y. Yao, J. Choi, I. Parker, Quantal puffs of intracellular Ca2+ evokedby inositol trisphosphate in Xenopus oocytes, J. Physiol. 482 (Pt 3)(1995) 533–553.
[5] H. Cheng, W.J. Lederer, M.B. Cannell, Calcium sparks: elementaryevents underlying excitation-contraction coupling in heart muscle,Science 262 (1993) 740–744.
[6] P. Pacher, A.P. Thomas, G. Hajnoczky, Ca2+ marks: miniature calciumsignals in single mitochondria driven by ryanodine receptors, Proc.Natl. Acad. Sci. U.S.A. 99 (2002) 2380–2385.
[7] A.J. Caride, A.G. Filoteo, A.R. Penheiter, K. Paszty, A. Enyedi, J.T.Penniston, Delayed activation of the plasma membrane calcium pumpby a sudden increase in Ca2+: fast pumps reside in fast cells, CellCalcium 30 (2001) 49–57.
[8] R. Sattler, Z. Xiong, W.Y. Lu, M. Hafner, J.F. MacDonald, M.Tymianski, Specific coupling of NMDA receptor activation to nitricoxide neurotoxicity by PSD-95 protein, Science 284 (1999) 1845–1848.
[9] M.R. Duchen, Mitochondria and calcium: from cell signalling to celldeath, J. Physiol. 529 (Pt 1) (2000) 57–68.
[10] P.S. Brookes, Y. Yoon, J.L. Robotham, M.W. Anders, S.S. Sheu,Calcium, ATP, and ROS: a mitochondrial love-hate triangle, Am. J.Physiol. Cell Physiol. 287 (2004) C817–C833.
[11] A.Y. Andreyev, Y.E. Kushnareva, A.A. Starkov, Mitochondrialmetabolism of reactive oxygen species, Biochemistry (Moscow) 70(2005) 200–214.
[12] J.F. Turrens, Mitochondrial formation of reactive oxygen species, J.Physiol. 552 (2003) 335–344.
[13] G. Lenaz, The mitochondrial production of reactive oxygen species:mechanisms and implications in human pathology, IUBMB Life 52(2001) 159–164.
[14] C. Chinopoulos, V. dam-Vizi, Calcium, mitochondria and oxidativestress in neuronal pathology. Novel aspects of an enduring theme,FEBS J. 273 (2006) 433–450.
[15] G.B. Waypa, P.T. Schumacker, Role for mitochondrial reactive oxy-gen species in hypoxic pulmonary vasoconstriction, Novartis FoundSymp. 272 (2006) 176–192.
[16] F.L. Muller, Y. Liu, R.H. Van, Complex III releases superoxide toboth sides of the inner mitochondrial membrane, J. Biol. Chem. 279(2004) 49064–49073.
[17] R. Rizzuto, M.R. Duchen, T. Pozzan, Flirting in little space: theER/mitochondria Ca2+ liaison, Sci. STKE 2004 (2004) re1.
[18] D.F. Babcock, J. Herrington, P.C. Goodwin, Y.B. Park, B. Hille, Mito-chondrial participation in the intracellular Ca2+ network, J. Cell Biol.136 (1997) 833–844.
[19] K.K. Griendling, Novel NAD(P)H oxidases in the cardiovascular sys-tem, Heart 90 (2004) 491–493.
[20] C.A. Colton, Microglial oxyradical production: causes and conse-quences, Neuropathol. Appl. Neurobiol. 20 (1994) 208–209.
[21] A.Y. Abramov, J. Jacobson, F. Wientjes, J. Hothersall, L. Canevari,M.R. Duchen, Expression and modulation of an NADPH oxidase inmammalian astrocytes, J. Neurosci. 25 (2005) 9176–9184.
[22] J.D. Lambeth, NOX enzymes and the biology of reactive oxygen, Nat.Rev. Immunol. 4 (2004) 181–189.
[23] C.E. Berry, J.M. Hare, Xanthine oxidoreductase and cardiovasculardisease: molecular mechanisms and pathophysiological implications,J. Physiol. 555 (2004) 589–606.
[24] S.A. Khan, K. Lee, K.M. Minhas, et al., Neuronal nitric oxide synthasenegatively regulates xanthine oxidoreductase inhibition of cardiacexcitation-contraction coupling, Proc. Natl. Acad. Sci. U.S.A. 101(2004) 15944–15948.
[25] G.C. Brown, V. Borutaite, Inhibition of mitochondrial respiratorycomplex I by nitric oxide, peroxynitrite and S-nitrosothiols, Biochim.
Biophys. Acta 1658 (2004) 44–49.[26] A.V. Zima, L.A. Blatter, Redox regulation of cardiac calcium channelsand transporters, Cardiovasc. Res. (2006).
[27] Y.B. Choi, S.A. Lipton, Redox modulation of the NMDA receptor,Cell Mol. Life Sci. 57 (2000) 1535–1541.

5 / Cell
72 S.M. Davidson, M.R. Duchen[28] H. Sies, Oxidative stress: oxidants and antioxidants, Exp. Physiol. 82(1997) 291–295.
[29] H. Xiong, E. Buck, J. Stuart, I.N. Pessah, G. Salama, J.J. Abram-son, Rose bengal activates the Ca2+ release channel from skeletalmuscle sarcoplasmic reticulum, Arch. Biochem. Biophys. 292 (1992)522–528.
[30] J.I. Kourie, Interaction of reactive oxygen species with ion transportmechanisms, Am J. Physiol. 275 (1998) C1–C24.
[31] G. Liu, J.J. Abramson, A.C. Zable, I.N. Pessah, Direct evidencefor the existence and functional role of hyperreactive sulfhydrylson the ryanodine receptor-triadin complex selectively labeled bythe coumarin maleimide 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin, Mol. Pharmacol. 45 (1994) 189–200.
[32] R. Xia, T. Stangler, J.J. Abramson, Skeletal muscle ryanodine receptoris a redox sensor with a well defined redox potential that is sensitiveto channel modulators, J. Biol. Chem. 275 (2000) 36556–36561.
[33] S.R. Holmberg, D.V. Cumming, Y. Kusama, et al., Reactive oxygenspecies modify the structure and function of the cardiac sarcoplasmicreticulum calcium-release channel, Cardioscience 2 (1991) 19–25.
[34] J.J. Marengo, C. Hidalgo, R. Bull, Sulfhydryl oxidation modifiesthe calcium dependence of ryanodine-sensitive calcium channels ofexcitable cells, Biophys. J. 74 (1998) 1263–1277.
[35] K. Swann, Thimerosal causes calcium oscillations and sensitizescalcium-induced calcium release in unfertilized hamster eggs, FEBSLett. 278 (1991) 175–178.
[36] S. Kumasaka, H. Shoji, E. Okabe, Novel mechanisms involvedin superoxide anion radical-triggered Ca2+ release from cardiacsarcoplasmic reticulum linked to cyclic ADP-ribose stimulation,Antioxid Redox Signal. 1 (1999) 55–69.
[37] M.L. Ruehr, M.A. Russell, D.G. Ferguson, et al., Targeting of proteinkinase A by muscle A kinase-anchoring protein (mAKAP) regulatesphosphorylation and function of the skeletal muscle ryanodine recep-tor, J. Biol. Chem. 278 (2003) 24831–24836.
[38] X.Y. Yi, V.X. Li, F. Zhang, et al., Characteristics and actions ofNAD(P)H oxidase on the sarcoplasmic reticulum of coronary arterysmooth muscle, Am J. Physiol. Heart Circ. Physiol. 290 (2006)H1136–H1144.
[39] G. Sanchez, Z. Pedrozo, R.J. Domenech, C. Hidalgo, P. Donoso,Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle, J. Mol. Cell Cardiol. 39(2005) 982–991.
[40] J.P. Eu, J. Sun, L. Xu, J.S. Stamler, G. Meissner, The skeletal mus-cle calcium release channel: coupled O2 sensor and NO signalingfunctions, Cell 102 (2000) 499–509.
[41] J.R. Bunting, A test of the singlet oxygen mechanism of cationic dyephotosensitization of mitochondrial damage, Photochem. Photobiol.55 (1992) 81–87.
[42] M.R. Duchen, Mitochondria and Ca(2+)in cell physiology and patho-physiology, Cell Calcium 28 (2000) 339–348.
[43] J. Jacobson, M.R. Duchen, Mitochondrial oxidative stress and celldeath in astrocytes—requirement for stored Ca2+ and sustained open-ing of the permeability transition pore, J. Cell Sci. 115 (2002)1175–1188.
[44] X. Wang, N. Weisleder, C. Collet, et al., Uncontrolled calcium sparksact as a dystrophic signal for mammalian skeletal muscle, Nat. CellBiol. 7 (2005) 525–530.
[45] E.V. Isaeva, V.M. Shkryl, N. Shirokova, Mitochondrial redox state andCa2+ sparks in permeabilized mammalian skeletal muscle, J. Physiol.565 (2005) 855–872.
[46] L. Hove-Madsen, A. Llach, A. Bayes-Genis, et al., Atrial fibrillationis associated with increased spontaneous calcium release from thesarcoplasmic reticulum in human atrial myocytes, Circulation 110
(2004) 1358–1363.[47] L. Mackenzie, M.D. Bootman, M. Laine, et al., The role of inositol1,4,5-trisphosphate receptors in Ca(2+) signalling and the generationof arrhythmias in rat atrial myocytes, J. Physiol. 541 (2002) 395–409.
Calcium 40 (2006) 561–574
[48] D.N. Bowser, T. Minamikawa, P. Nagley, D.A. Williams, Role of mito-chondria in calcium regulation of spontaneously contracting cardiacmuscle cells, Biophys. J. 75 (1998) 2004–2014.
[49] G.C. Wellman, M.T. Nelson, Signaling between SR and plasmalemmain smooth muscle: sparks and the activation of Ca2+-sensitive ionchannels, Cell Calcium 34 (2003) 211–229.
[50] M.T. Nelson, H. Cheng, M. Rubart, et al., Relaxation of arterialsmooth muscle by calcium sparks, Science 270 (1995) 633–637.
[51] Q. Xi, S.Y. Cheranov, J.H. Jaggar, Mitochondria-derived reactive oxy-gen species dilate cerebral arteries by activating Ca2+ sparks, Circ.Res. 97 (2005) 354–362.
[52] V.P. Skulachev, Role of uncoupled and non-coupled oxidations inmaintenance of safely low levels of oxygen and its one-electron reduc-tants, Q. Rev. Biophys. 29 (1996) 169–202.
[53] Y. Liu, H. Zhao, H. Li, B. Kalyanaraman, A.C. Nicolosi, D.D. Gut-terman, Mitochondrial sources of H2O2 generation play a key role inflow-mediated dilation in human coronary resistance arteries, Circ.Res. 93 (2003) 573–580.
[54] S.K. Joseph, S.K. Nakao, S. Sukumvanich, Reactivity of free thiolgroups in type-I inositol trisphosphate receptors, Biochem. J. 393(2006) 575–582.
[55] L. Missiaen, C.W. Taylor, M.J. Berridge, Spontaneous calcium releasefrom inositol trisphosphate-sensitive calcium stores, Nature 352(1991) 241–244.
[56] M.D. Bootman, C.W. Taylor, M.J. Berridge, The thiol reagent,thimerosal, evokes Ca2+ spikes in HeLa cells by sensitizing the inos-itol 1,4,5-trisphosphate receptor, J. Biol. Chem. 267 (1992) 25113–25119.
[57] S. Peuchen, J.B. Clark, M.R. Duchen, Mechanisms of intracellu-lar calcium regulation in adult astrocytes, Neuroscience 71 (1996)871–883.
[58] Y.J. Suzuki, G.D. Ford, Superoxide stimulates IP3-induced Ca2+
release from vascular smooth muscle sarcoplasmic reticulum, Am.J. Physiol. 262 (1992) H114–H116.
[59] P.N. Henschke, S.J. Elliott, Oxidized glutathione decreases luminalCa2+ content of the endothelial cell Ins (1,4,5)P3-sensitive Ca2+ store,Biochem. J. 312 (Pt 2) (1995) 485–489.
[60] D.E. Wesson, S.J. Elliott, The H2O2-generating enzyme, xanthineoxidase, decreases luminal Ca2+ content of the IP3-sensitive Ca2+
store in vascular endothelial cells, Microcirculation 2 (1995) 195–203.
[61] A.I. Kaplin, S.H. Snyder, D.J. Linden, Reduced nicotinamide ade-nine dinucleotide-selective stimulation of inositol 1,4,5-trisphosphatereceptors mediates hypoxic mobilization of calcium, J. Neurosci. 16(1996) 2002–2011.
[62] R.L. Patterson, D.B. van Rossum, A.I. Kaplin, R.K. Barrow, S.H.Snyder, Inositol 1,4,5-trisphosphate receptor/GAPDH complex aug-ments Ca2+ release via locally derived NADH, Proc. Natl. Acad. Sci.U.S.A. 102 (2005) 1357–1359.
[63] O.P. Mishra, I. Qayyum, M. ivoria-Papadopoulos, Hypoxia-inducedmodification of the inositol triphosphate receptor in neuronal nucleiof newborn piglets: role of nitric oxide, J. Neurosci. Res. 74 (2003)333–338.
[64] T. Tomida, K. Hirose, A. Takizawa, F. Shibasaki, M. Iino, NFATfunctions as a working memory of Ca2+ signals in decoding Ca2+
oscillation, EMBO J. 22 (2003) 3825–3832.[65] K.P. De, H. Schulman, Sensitivity of CaM kinase II to the frequency
of Ca2+ oscillations, Science 279 (1998) 227–230.[66] E. Oancea, T. Meyer, Protein kinase C as a molecular machine for
decoding calcium and diacylglycerol signals, Cell 95 (1998) 307–318.
[67] K. Ishii, K. Hirose, M. Iino, Ca2+ shuttling between endoplasmic retic-
ulum and mitochondria underlying Ca2+ oscillations, EMBO Rep. 7(2006) 390–396.[68] T. Miyakawa, A. Mizushima, K. Hirose, et al., Ca(2+)-sensor regionof IP(3) receptor controls intracellular Ca(2+) signalling, EMBO J.20 (2001) 1674–1680.

/ Cell
S.M. Davidson, M.R. Duchen[69] M.C. Camello-Almaraz, M.J. Pozo, M.P. Murphy, P.J. Camello, Mito-chondrial production of oxidants is necessary for physiological cal-cium oscillations, J. Cell Physiol. 206 (2006) 487–494.
[70] T. Pozzan, R. Rizzuto, P. Volpe, J. Meldolesi, Molecular and cellularphysiology of intracellular calcium stores, Physiol. Rev. 74 (1994)595–636.
[71] Y. Li, P. Camacho, Ca2+-dependent redox modulation of SERCA 2bby ERp57, J. Cell Biol. 164 (2004) 35–46.
[72] L.C. Hool, Reactive oxygen species in cardiac signalling: from mito-chondria to plasma membrane ion channels, Clin. Exp. Pharmacol.Physiol. 33 (2006) 146–151.
[73] D.L. Campbell, J.S. Stamler, H.C. Strauss, Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanismregulation by nitric oxide and S-nitrosothiols, J. Gen. Physiol. 108(1996) 277–293.
[74] I.M. Fearon, A.C. Palmer, A.J. Balmforth, S.G. Ball, G. Varadi, C.Peers, Modulation of recombinant human cardiac L-type Ca2+ chan-nel alpha1C subunits by redox agents and hypoxia, J. Physiol. 514 (Pt3) (1999) 629–637.
[75] O. Vergun, A.I. Sobolevsky, M.V. Yelshansky, J. Keelan, B.I.Khodorov, M.R. Duchen, Exploration of the role of reactive oxy-gen species in glutamate neurotoxicity in rat hippocampal neuronesin culture, J. Physiol. 531 (2001) 147–163.
[76] Y.B. Choi, L. Tenneti, D.A. Le, et al., Molecular basis of NMDAreceptor-coupled ion channel modulation by S-nitrosylation, Nat.Neurosci. 3 (2000) 15–21.
[77] T.E. Gunter, D.R. Pfeiffer, Mechanisms by which mitochondria trans-port calcium, Am. J. Physiol. 258 (1990) C755–C786.
[78] E.J. Griffiths, A.P. Halestrap, Mitochondrial non-specific poresremain closed during cardiac ischaemia, but open upon reperfusion,Biochem. J. 307 (Pt 1) (1995) 93–98.
[79] J.S. Kim, Y. Jin, J.J. Lemasters, Reactive oxygen species, butnot Ca2+ overloading, trigger pH- and mitochondrial permeabilitytransition-dependent death of adult rat myocytes after ischemia-reperfusion, Am. J. Physiol. Heart Circ. Physiol. 290 (2006) H2024–H2034.
[80] M. Crompton, A. Costi, Kinetic evidence for a heart mitochondrialpore activated by Ca2+, inorganic phosphate and oxidative stress. Apotential mechanism for mitochondrial dysfunction during cellularCa2+ overload, Eur. J. Biochem. 178 (1988) 489–501.
[81] A.P. Halestrap, K.Y. Woodfield, C.P. Connern, Oxidative stress,thiol reagents, and membrane potential modulate the mitochon-drial permeability transition by affecting nucleotide binding to theadenine nucleotide translocase, J. Biol. Chem. 272 (1997) 3346–3354.
[82] P. Costantini, B.V. Chernyak, V. Petronilli, P. Bernardi, Modulation ofthe mitochondrial permeability transition pore by pyridine nucleotidesand dithiol oxidation at two separate sites, J. Biol. Chem. 271 (1996)6746–6751.
[83] D.B. Zorov, C.R. Filburn, L.O. Klotz, J.L. Zweier, S.J. Sollott,Reactive oxygen species (ROS)-induced ROS release: a new phe-nomenon accompanying induction of the mitochondrial permeabil-ity transition in cardiac myocytes, J. Exp. Med. 192 (2000) 1001–1014.
[84] N.R. Brady, S.P. Elmore, J.J. van Beek, et al., Coordinated behaviorof mitochondria in both space and time: a reactive oxygen species-activated wave of mitochondrial depolarisation, Biophys. J. 87 (2004)2022–2034.
[85] D.N. Romashko, E. Marban, B. O’Rourke, Subcellular metabolic tran-sients and mitochondrial redox waves in heart cells, Proc. Natl. Acad.Sci. U.S.A. 95 (1998) 1618–1623.
[86] A.Y. Abramov, L. Canevari, M.R. Duchen, Beta-amyloid peptides
induce mitochondrial dysfunction and oxidative stress in astrocytesand death of neurons through activation of NADPH oxidase, J. Neu-rosci. 24 (2004) 565–575.[87] J. Keelan, N.J. Allen, D. Antcliffe, S. Pal, M.R. Duchen, Quanti-tative imaging of glutathione in hippocampal neurons and glia in
Calcium 40 (2006) 561–574 573
culture using monochlorobimane, J. Neurosci. Res. 66 (2001) 873–884.
[88] A.Y. Abramov, L. Canevari, M.R. Duchen, Calcium signals inducedby amyloid beta peptide and their consequences in neurons and astro-cytes in culture, Biochim. Biophys. Acta 1742 (2004) 81–87.
[89] H. Cai, Z. Li, S. Dikalov, et al., NAD(P)H oxidase-derived hydrogenperoxide mediates endothelial nitric oxide production in response toangiotensin II, J. Biol. Chem. 277 (2002) 48311–48317.
[90] J. Ahluwalia, A. Tinker, L.H. Clapp, et al., The large-conductanceCa2+-activated K+ channel is essential for innate immunity, Nature427 (2004) 853–858.
[91] N. Demaurex, Calcium measurements in organelles with Ca2+-sensitive fluorescent proteins, Cell Calcium 38 (2005) 213–222.
[92] R. Rudolf, M. Mongillo, R. Rizzuto, T. Pozzan, Looking forward toseeing calcium, Nat. Rev. Mol. Cell Biol. 4 (2003) 579–586.
[93] V.V. Belousov, A.F. Fradkov, K.A. Lukyanov, et al., Geneticallyencoded fluorescent indicator for intracellular hydrogen peroxide,Nat. Methods 3 (2006) 281–286.
[94] W. Droge, Free radicals in the physiological control of cell function,Physiol. Rev. 82 (2002) 47–95.
[95] M.J. Shattock, Do we know the mechanism of myocardial stunning?Basic Res. Cardiol. 93 (1998) 145–149.
[96] D. Sorescu, K.K. Griendling, Reactive oxygen species, mitochondria,and NAD(P)H oxidases in the development and progression of heartfailure, Congest. Heart Fail 8 (2002) 132–140.
[97] J.M. Li, N.P. Gall, D.J. Grieve, M. Chen, A.M. Shah, Activation ofNADPH oxidase during progression of cardiac hypertrophy to failure,Hypertension 40 (2002) 477–484.
[98] C.E. Murdoch, D.J. Grieve, A.C. Cave, Y.H. Looi, A.M. Shah,NADPH oxidase and heart failure, Curr. Opin. Pharmacol. 6 (2006)148–153.
[99] B.J. Wilkins, J.D. Molkentin, Calcium-calcineurin signaling in theregulation of cardiac hypertrophy, Biochem. Biophys. Res. Commun.322 (2004) 1178–1191.
[100] J.D. Molkentin, J.R. Lu, C.L. Antos, et al., A calcineurin-dependenttranscriptional pathway for cardiac hypertrophy, Cell 93 (1998)215–228.
[101] M.R. Sayen, A.B. Gustafsson, M.A. Sussman, J.D. Molkentin, R.A.Gottlieb, Calcineurin transgenic mice have mitochondrial dysfunctionand elevated superoxide production, Am. J. Physiol. Cell Physiol. 284(2003) C562–C570.
[102] M. Leonardi, J. Bissett, Prevention of atrial fibrillation, Curr. Opin.Cardiol. 20 (2005) 417–423.
[103] M. Andrews, B.P. Nelson, Atrial fibrillation, Mt. Sinai. J. Med. 73(2006) 482–492.
[104] S.E. Howlett, S.A. Grandy, G.R. Ferrier, Calcium spark properties inventricular myocytes are altered in aged mice, Am J. Physiol. HeartCirc. Physiol. 290 (2006) H1566–H1574.
[105] X. Zhu, B.A. Altschafl, R.J. Hajjar, H.H. Valdivia, U. Schmidt, AlteredCa2+ sparks and gating properties of ryanodine receptors in agingcardiomyocytes, Cell Calcium 37 (2005) 583–591.
[106] M.J. Mihm, F. Yu, C.A. Carnes, et al., Impaired myofibrillar energeticsand oxidative injury during human atrial fibrillation, Circulation 104(2001) 174–180.
[107] C.A. Carnes, M.K. Chung, T. Nakayama, et al., Ascorbate attenuatesatrial pacing-induced peroxynitrite formation and electrical remod-eling and decreases the incidence of postoperative atrial fibrillation,Circ. Res. 89 (2001) E32–E38.
[108] R.W. Nesto, Correlation between cardiovascular disease and dia-betes mellitus: current concepts, Am. J. Med. 116 (Suppl. 5A) (2004)11S–22S.
[109] D. Lagadic-Gossmann, K.J. Buckler, P.K. Le, D. Feuvray, Altered
Ca2+ handling in ventricular myocytes isolated from diabetic rats,Am. J. Physiol. 270 (1996) H1529–H1537.[110] S. Penpargkul, F. Fein, E.H. Sonnenblick, J. Scheuer, Depressed car-diac sarcoplasmic reticular function from diabetic rats, J. Mol. CellCardiol. 13 (1981) 303–309.

5 / Cell
74 S.M. Davidson, M.R. Duchen[111] D.D. Belke, E.A. Swanson, W.H. Dillmann, Decreased sarcoplasmic
reticulum activity and contractility in diabetic db/db mouse heart,Diabetes 53 (2004) 3201–3208.[112] L. Pereira, J. Matthes, I. Schuster, et al., Mechanisms of [Ca2+]i
transient decrease in cardiomyopathy of db/db type 2 diabetic mice,Diabetes 55 (2006) 608–615.
Calcium 40 (2006) 561–574
[113] N. Yaras, M. Ugur, S. Ozdemir, et al., Effects of diabetes on ryanodine
receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart,Diabetes 54 (2005) 3082–3088.[114] J.H. Jaggar, V.A. Porter, W.J. Lederer, M.T. Nelson, Calcium sparksin smooth muscle, Am. J. Physiol. Cell Physiol. 278 (2000) C235–C256.




















