
NADPH oxidase-dependent redox signaling in human heart failure: Relationship between the left and...
9
Original article NADPH oxidase-dependent redox signaling in human heart failure: Relationship between the left and right ventricle Chiara Nediani a, ⁎ , Elisabetta Borchi a , Carla Giordano c , Serena Baruzzo a , Vanessa Ponziani a , Mariangela Sebastiani c , Paolo Nassi a , Alessandro Mugelli b , Giulia d'Amati c , Elisabetta Cerbai b a Department of Biochemical Sciences, University of Florence, Viale Morgagni, 50, 50134 Florence, Italy b Department of Preclinical and Clinical Pharmacology, University of Florence, Florence, Italy c Department of Experimental Medicine and Pathology, La Sapienza University, Rome, Italy Received 14 December 2006; received in revised form 16 January 2007; accepted 18 January 2007 Available online 31 January 2007 Abstract Oxidative stress resulting from increased superoxide generation by NADPH oxidase is implicated in the pathophysiology of human heart failure. Downstream targets of NADPH oxidase remain undefined and available information is restricted to the left ventricle (LV). Thus, we aimed to assess the cascade of events triggered by increased NADPH oxidase activity (lipid peroxidation and activation of mitogen-activated protein kinases ERK1/2, JNK and p38) and their mutual relationship in right (RV) and (LV) of end-stage failing human hearts. When compared to control ventricles, diseased RV and LV showed a significant increase in NADPH oxidase superoxide production that positively correlated with p47 phox membrane translocation (RV: r = 0.76, P <0.001; LV: r = 0.79, P < 0.001). MDA content, a marker of lipid peroxidation, was also enhanced and ERK and p38, but not JNK, were activated. For all these relevant steps of the oxidative stress pathway, a significant correlation was observed between LVand RV from the same heart (NADPH-dependent superoxide production: r = 0.678, P < 0.0055; MDA: r = 0.95, P < 0.0001; p-p38/p38 ratio: r = 0.926, P < 0.0001; p-ERK/ERK ratio: r = 0.913, P <0.0001). We concluded that in human heart failure, both ventricles are targets of NADPH oxidase superoxide generation which in turn may trigger the coordinated activation of downstream signaling components. This pathway may contribute to adverse remodeling of the LV and RV and subsequent progression toward end-stage heart failure, suggestive of new therapeutic targeting strategy. © 2007 Elsevier Inc. All rights reserved. Keywords: Humans; Heart failure; Oxidative stress; NADPH oxidase; MAPKs 1. Introduction Oxidative stress resulting from increased cardiac generation of reactive oxygen species (ROS) plays a major part in the pathophysiology of human heart failure [1]. Excess ROS production results in the oxidation of many macromolecules contributing to cell damage and promoting cell death through apoptosis and necrosis [2]. Such effects, especially important in advanced heart failure, are also implicated in critical steps of cardiac remodeling, such as cellular hypertrophy and contractile dysfunction [2,3]. The NADPH oxidase enzymes, the major source of ROS in the cardiovascular system [4], are heteromeric cytochromes containing a catalytic Nox subunit and a lower molecular weight subunit, p22 phox . Of the five Nox isoforms identified so far, the main isoforms expressed in cardiac cells seem to be Nox2 and Nox4. Activation of Nox2 (also known as gp91 phox ) oxidase, the most common isoform in the human myocardium [5], requires the association of cytosolic regulatory units (p47 phox , p67 phox , p40 phox and rac1) with the cytochrome in order to activate superoxide (O 2 - ) production, while Nox 4 activation does not [6]. NADPH oxidase activity is enhanced by several stimuli that are relevant to left ventricular hypertrophy and heart failure [7–9]. The prerequisite for oxidase activation and ROS produc- tion seems to be the increase in subunit protein expression and Journal of Molecular and Cellular Cardiology 42 (2007) 826 – 834 www.elsevier.com/locate/yjmcc ⁎ Corresponding author. Tel.: +39 0554598321; fax: +39 0554598905. E-mail address: [email protected] (C. Nediani). 0022-2828/$ - see front matter © 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.yjmcc.2007.01.009
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of NADPH oxidase-dependent redox signaling in human heart failure: Relationship between the left and...

Journal of Molecular and Cellular Cardiology 42 (2007) 826–834www.elsevier.com/locate/yjmcc
Original article
NADPH oxidase-dependent redox signaling in human heart failure:Relationship between the left and right ventricle
Chiara Nediani a,⁎, Elisabetta Borchi a, Carla Giordano c, Serena Baruzzo a, Vanessa Ponziani a,Mariangela Sebastiani c, Paolo Nassi a, Alessandro Mugelli b, Giulia d'Amati c, Elisabetta Cerbai b
a Department of Biochemical Sciences, University of Florence, Viale Morgagni, 50, 50134 Florence, Italyb Department of Preclinical and Clinical Pharmacology, University of Florence, Florence, Italyc Department of Experimental Medicine and Pathology, La Sapienza University, Rome, Italy
Received 14 December 2006; received in revised form 16 January 2007; accepted 18 January 2007Available online 31 January 2007
Abstract
Oxidative stress resulting from increased superoxide generation by NADPH oxidase is implicated in the pathophysiology of human heartfailure. Downstream targets of NADPH oxidase remain undefined and available information is restricted to the left ventricle (LV). Thus, we aimedto assess the cascade of events triggered by increased NADPH oxidase activity (lipid peroxidation and activation of mitogen-activated proteinkinases ERK1/2, JNK and p38) and their mutual relationship in right (RV) and (LV) of end-stage failing human hearts. When compared to controlventricles, diseased RV and LV showed a significant increase in NADPH oxidase superoxide production that positively correlated with p47phox
membrane translocation (RV: r=0.76, P<0.001; LV: r=0.79, P<0.001). MDA content, a marker of lipid peroxidation, was also enhanced andERK and p38, but not JNK, were activated. For all these relevant steps of the oxidative stress pathway, a significant correlation was observedbetween LV and RV from the same heart (NADPH-dependent superoxide production: r=0.678, P<0.0055; MDA: r=0.95, P<0.0001; p-p38/p38ratio: r=0.926, P<0.0001; p-ERK/ERK ratio: r=0.913, P<0.0001). We concluded that in human heart failure, both ventricles are targets ofNADPH oxidase superoxide generation which in turn may trigger the coordinated activation of downstream signaling components. This pathwaymay contribute to adverse remodeling of the LV and RVand subsequent progression toward end-stage heart failure, suggestive of new therapeutictargeting strategy.© 2007 Elsevier Inc. All rights reserved.
Keywords: Humans; Heart failure; Oxidative stress; NADPH oxidase; MAPKs
1. Introduction
Oxidative stress resulting from increased cardiac generationof reactive oxygen species (ROS) plays a major part in thepathophysiology of human heart failure [1]. Excess ROSproduction results in the oxidation of many macromoleculescontributing to cell damage and promoting cell death throughapoptosis and necrosis [2]. Such effects, especially important inadvanced heart failure, are also implicated in critical steps ofcardiac remodeling, such as cellular hypertrophy and contractiledysfunction [2,3].
⁎ Corresponding author. Tel.: +39 0554598321; fax: +39 0554598905.E-mail address: [email protected] (C. Nediani).
0022-2828/$ - see front matter © 2007 Elsevier Inc. All rights reserved.doi:10.1016/j.yjmcc.2007.01.009
The NADPH oxidase enzymes, the major source of ROS inthe cardiovascular system [4], are heteromeric cytochromescontaining a catalytic Nox subunit and a lower molecularweight subunit, p22phox. Of the five Nox isoforms identified sofar, the main isoforms expressed in cardiac cells seem to beNox2 and Nox4. Activation of Nox2 (also known as gp91phox)oxidase, the most common isoform in the human myocardium[5], requires the association of cytosolic regulatory units(p47phox, p67phox, p40phox and rac1) with the cytochrome inorder to activate superoxide (O2
−) production, while Nox 4activation does not [6].
NADPH oxidase activity is enhanced by several stimuli thatare relevant to left ventricular hypertrophy and heart failure[7–9]. The prerequisite for oxidase activation and ROS produc-tion seems to be the increase in subunit protein expression and

Table 1Clinical characteristics of the patients
NF IHD DCM
Total number 6 7 7Sex (M/F) 4/2 6/1 4/3Age (range) 44 (38–52) 52 (41–67) 45 (33–62)LVEF (%) 26±1.89 17±0.38CI (l/min/m2) 2.37±0.23 1.8±0.07PAP (mm Hg) 39.2±4.87 29.2±5.36PCWP (mm Hg) 25.2±2.92 21±5.36RAP (mm Hg) 17±5.79 10.6±4.78NYHA IV IVMedicationDiuretics – 7 7Digoxin – 0 0Antiarrhythmics – 3 2ACE-I – 7 7β-Blockers – 7 7Nitrates – 4 0HMGCoA-Inh – – –ANF/rRNA 18S 1.04 ± 0.013 10.83 ±1.06 11.93 ±1.1MCH-α/rRNA 18S 1.30 ± 0.04 0.057 ±0.004 0.03 ±0.007MCH-β/rRNA 18S 1.36 ± 0.12 0.04 ±0.007 0.46 ±0.03
LVEF=left ventricular ejection fraction; CI=cardiac index; ACE-I=angiotensinconverting enzyme inhibitors; HMGCoA inhibitors (statins); ANF=atrialnatriuretic peptide; MCH-α/β=myosin heavy chain-α/β; PAP=pulmonaryartery pressure; PCWP=pulmonary capillary wedge pressure; RAP=rightatrium pressure.
827C. Nediani et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 826–834
the translocation of regulatory subunits from cytosol to mem-brane, in particular of p47phox [5,7,8,10]. NADPH oxidase-derived ROS likely act through specific redox-regulatedsignaling pathways, which play important roles in the remo-deling of the failing heart and in the promotion of cardiomyo-cyte hypertrophy and interstitial fibrosis [11]. However, thenature of the downstream pathways remains poorly defined.Among these, potential downstream targets of NADPH oxidaseROS production are the redox-sensitive mitogen-activatedprotein kinases (MAPKs), including extracellular signal-regulated kinase (ERK1/2), p38 and c-jun N-terminal kinase(JNK) [3,7,12]. The modulation of this detrimental response is apromising strategy for the prevention and treatment of myo-cardial dysfunction and heart failure.
Notwithstanding this premise, few data are available on thediseased human heart, and they are mostly limited to singleand specific aspects of the complex oxidative stress pathway[10,13,14]. Thus, the sequence of events arising fromincreased NADPH oxidase activity remains undefined [15].In addition, all information is restricted to the left ventricle(LV). The LV and the right ventricle (RV) are interdependent,and changes in the performance of one may affect the other. Inhuman heart failure, the structural and functional RVremodeling reproduces what occurs in the LV [16,17] andmay adversely affect the clinical outcome [18–20]. On thisbasis, defining the oxidative stress pathway in the RV mayhelp to clarify the mechanisms responsible for the progressiontoward end-stage heart failure.
The aim of this study was twofold: first, to assess the cascadeof events triggered by increased NADPH oxidase activity inhuman heart failure and their mutual relationship; second, toassess whether these mechanisms are equally effective in boththe left and the right ventricles. To this purpose, NADPHoxidase activity, its regulation through p47phox, lipid peroxida-tion and the activation of redox-sensitive MAPKs wereinvestigated in RV and LV myocardium from end-stageischemic (IHD) and dilated (DCM) cardiomyopathy, and innon-failing donor hearts.
2. Methods
2.1. Patients
All studies conformed to the Declaration of Helsinki andinstitutional ethical regulations. Explanted failing hearts wereobtained from patients undergoing transplantation for end-stageheart failure secondary to idiopathic dilated cardiomyopathy(n=4 males, n=3 females; mean age 45±3.6 years) or ischemicheart disease (n=6 males, n=1 females; mean age 52±6.3years). All patients had New York Heart Association class IVheart failure, with a mean LVejection fraction of 21.5±1.14 anda mean pulmonary artery pressure of 34.2±3.8. Non-failing(NF) donor hearts (n=6; mean age 44±2.1 years; n=4 males,n=2 females; no cardiac medication) which were unsuitablefor transplantation for technical reasons, were used as controls.Clinical characteristics of the three groups are shown in Table 1.Immediately after explant, myocardial tissue samples were
snap-frozen in liquid nitrogen-chilled isopentane for protein,RNA analysis, and biochemistry. All tissues were stored at−80 °C. Sections stained with hematoxylin–eosin and MassonTrichrome stain, were obtained from each sample for morpho-logical examination prior to each experiment. Histologic slidesfrom both failing and non-failing hearts were observed underlight microscopy. Myocyte hypertrophy was a common findingin all failing heart, associated with variable degrees ofinterstitial fibrosis, graded from mild (+1) to severe (+3) on asemi-quantitative basis. According to the results of histologicexamination, myocardial samples with minimal amounts offibrosis and devoid of inflammatory infiltrates were selected formolecular and biochemical studies. In addition, atrial natriureticfactor (ANF), and myosin heavy chain (MHC)-α and -βisoforms mRNA expression, molecular markers of cardiachypertrophy, were evaluated quantitatively both on NF andfailing hearts by RT-PCR using the Platinum SYBR GreenqPCR Super Mix-UDG (Invitrogen).
2.2. Sample preparation
LVand RV samples from non-failing and failing hearts werehomogenized in ice-cold 20 mM Tris–HCl, pH 7.4, containing10 μg/ml leupeptin, 10 μg/ml aprotinin, 0.2 mM PMSF andphosphatase inhibitor cocktail (Sigma). After spinning for10 min, 3000 rpm at 4 °C, total heart homogenate samples wereused for Western blotting, lipid peroxidation assay andmyocardial ROS production. Heart homogenate aliquots werealso separated into soluble (cytosolic) and membrane fractionsby ultracentrifugation for 30 min at 30,000 rpm at 4 °C in aBeckman centrifuge, and used for immunoblotting to detect

828 C. Nediani et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 826–834
p47phox translocation. Protein concentration was measured bybicinchoninic acid (BCA) protein assay (Pierce).
2.3. Lucigenin chemiluminescence
Myocardial O2− production was assessed by lucigenin-
enhanced chemiluminescence [5]. Experiments were per-formed on a luminometer (Lumat LB 9507 EG&G Berthold)using 100 μg protein/tube, and a non-redox cycling 5 μMlucigenin concentration [5,21,22]. Superoxide generation wasmeasured in the absence and in the presence of specific oxidasesubstrates (300 μM NADPH for NADPH oxidase, 5 mMsuccinate for mitochondrial oxidase complex I, 1 mM xanthinefor xanthine oxidase), at room temperature. Further measure-ments were taken after the addition of specific inhibitors ofoxidase systems: diphenyleneiodonium (DPI, 20 μM), aNADPH oxidase flavoprotein inhibitor, Nω-nitro-L-argininemethyl ester hydrochloride (L-NAME, 100 μM), a nitric oxidesynthase inhibitor; rotenone (50 μM), a mitochondrial oxidasecomplex I inhibitor; oxypurinol (100 μM), a xanthine oxidaseinhibitor. A cell-permeable superoxide scavenger 4,5-dihy-droxy-1,3-benzene-disulfonic acid (Tiron, 20 mM) was alsoused. A buffer blank was subtracted from each reading.Superoxide production was expressed as an arbitrary light unitover 10 min.
2.4. Lipid peroxidation assay
To assess the level of oxidative stress, malonaldehyde(MDA) concentration was determined, as a marker of lipidperoxidation, using a Bioexytech LPO-586 kit (Oxis Interna-tional Inc.).
2.5. Western blotting
Equal amounts (60 μg/sample) of ventricular homogenates,membrane or cytosolic proteins were separated on 12% SDS-PAGE gels and transferred to polyvinylidine difluoride (PVDF)membranes. Membranes were then incubated with the follow-ing antibodies: polyclonal anti-gp91phox (1:500; sc-20782),polyclonal anti-p47phox (1:2000 sc-14015), anti-JNK (1:1000;sc-571), anti-ERK (1:1000; sc-93), and anti-p38 (1:1000; sc-535) (Santa Cruz Biotech, Santa Cruz, California), monoclonalanti-p-JNK, anti-p-ERK (Thr202/Tyr204), anti-p-p38 (1:1000;#9938), Cell Signaling Technology, Inc.), and anti- (Santa CruzBiotech). Anti-mouse (sc-2005; 1:5000) or -rabbit (sc-2004;1:10000) horseradish peroxidase-conjugated secondary anti-bodies (Santa Cruz Biotech) were used. Immunoreactive bandswere detected by chemiluminescence SuperSignal West Durasolution (Pierce) and quantified by densitometry analysis usinga Chemi Doc system and Quantity One software (Bio-RADLaboratories). β-Tubulin (1:1000; sc-5274, Santa Cruz Biotech)was used to control for equal protein loading of gp91phox andp47phox.
Activation of MAPKs was assessed by Western blotting withantibodies that specifically recognize the phosphorylated activeforms of these kinases. Results are expressed relative to their
total protein abundance obtained with primary antibody used todetect JNK, ERK, and p38 total protein levels.
2.6. Statistical analysis
All values are expressed as mean±SEM. Comparisons wereperformed using ANOVA followed by a Tukey–Kramermultiple comparison test. Correlation coefficient r was obtainedusing a linear (Pearson) correlation test. Probabilities ofP<0.05 were considered statistically significant.
3. Results
3.1. Increased NADPH dependent O2− production in failing
right and left ventricles
O2− production measured by lucigenin chemiluminescence in
the absence of substrates or in the presence of succinate orxanthine was minimal in the RVand LV homogenate and did notdiffer between NF and failing hearts (data not shown). In thepresence of NADPH, significantly higher O2
− generation wasdetected in NF LV compared to NF RV (240.14±8.1 MLU/s/mgprotein vs 160±10.6 MLU/s/mg protein, respectively). Dis-eased LVand RV specimens exhibited a marked increase in O2
−
production compared to NF controls. In particular, the increasein O2
− generation was less marked, although statisticallysignificant in failing LVs (IHD 1.38-fold, P<0.001; DCM1.23-fold, P<0.05; ICM and DCM vs NF), while a 2-fold and1.65-fold increase in NADPH-dependent O2
− production wasobserved in the RVs from IHD and DCM hearts (P<0.001 vsNF) (Fig. 1A). Interestingly, in RV samples from diseasedhearts, O2
− production, attributable to NADPH oxidase activitywas positively related to the respective pulmonary arterialpressure (PAP; r=0.86, P=0.0014) (Fig. 1B). To investigatethe sources of O2
− production, experiments were performed inthe presence of potential ROS-generating system inhibitors.NADPH-dependent O2
− production was virtually abolished bythe O2
− scavenger Tiron, thus confirming O2− as the major source
of ROS detected in our experimental conditions. In both RVandLV samples from failing hearts, NADPH-dependent O2
−
generation was significantly decreased by diphenyleneiodo-nium, an inhibitor of NADPH oxidase, but was unaltered byNOS inhibitor (L-NAME), or by the inhibitor of the mitochon-drial electron transport chain, rotenone, or by the xanthineoxidase inhibitor, oxypurinol, supporting a phagocyte-typeNADPH oxidase as the likely major source of O2
− in the humanheart (Figs. 1C, D).
3.2. Protein expression and membrane translocation of p47phox
To elucidate the mechanisms underlying the increasedNADPH oxidase activity in the failing myocardium, experi-ments were focused on the Nox2 (gp91phox), i.e., the subunitresponsible for catalytic activity of the oxidase, and itsregulatory subunit p47phox, which is likely to modulate oxidaseactivation following translocation to the myocyte membrane[5,7,8,23]. Nox2 protein expression, assessed by Western
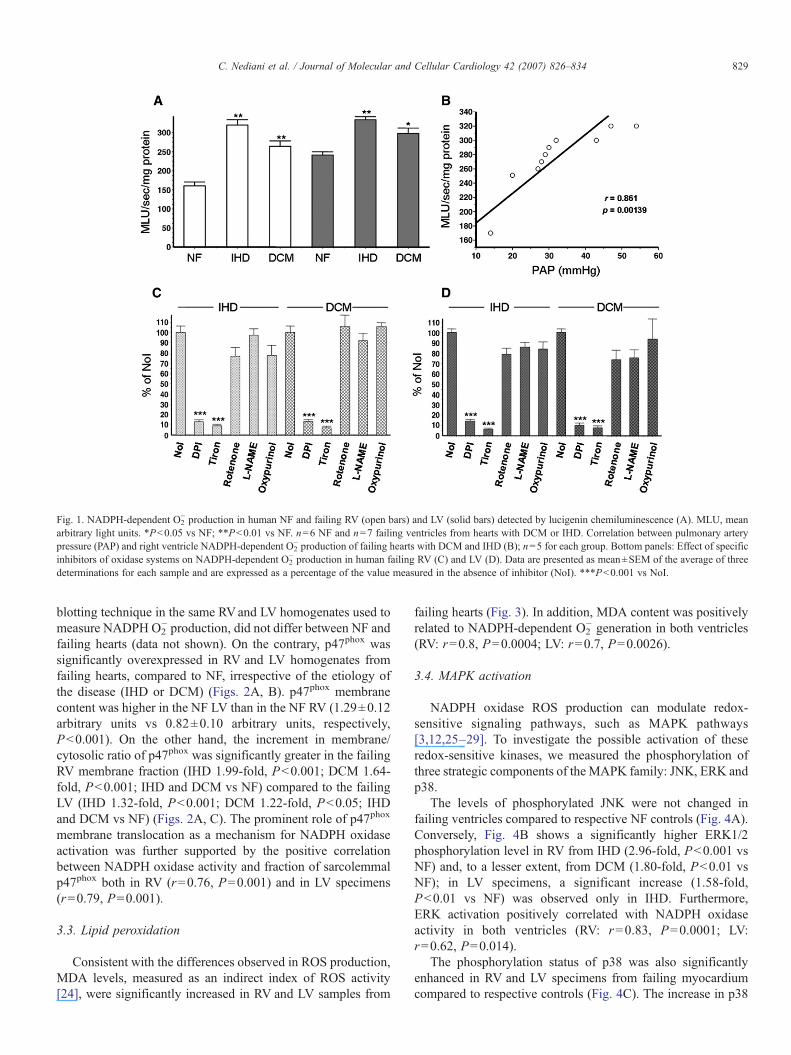
Fig. 1. NADPH-dependent O2− production in human NF and failing RV (open bars) and LV (solid bars) detected by lucigenin chemiluminescence (A). MLU, mean
arbitrary light units. *P<0.05 vs NF; **P<0.01 vs NF. n=6 NF and n=7 failing ventricles from hearts with DCM or IHD. Correlation between pulmonary arterypressure (PAP) and right ventricle NADPH-dependent O2
− production of failing hearts with DCM and IHD (B); n=5 for each group. Bottom panels: Effect of specificinhibitors of oxidase systems on NADPH-dependent O2
− production in human failing RV (C) and LV (D). Data are presented as mean±SEM of the average of threedeterminations for each sample and are expressed as a percentage of the value measured in the absence of inhibitor (NoI). ***P<0.001 vs NoI.
829C. Nediani et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 826–834
blotting technique in the same RVand LV homogenates used tomeasure NADPH O2
− production, did not differ between NF andfailing hearts (data not shown). On the contrary, p47phox wassignificantly overexpressed in RV and LV homogenates fromfailing hearts, compared to NF, irrespective of the etiology ofthe disease (IHD or DCM) (Figs. 2A, B). p47phox membranecontent was higher in the NF LV than in the NF RV (1.29±0.12arbitrary units vs 0.82±0.10 arbitrary units, respectively,P<0.001). On the other hand, the increment in membrane/cytosolic ratio of p47phox was significantly greater in the failingRV membrane fraction (IHD 1.99-fold, P<0.001; DCM 1.64-fold, P<0.001; IHD and DCM vs NF) compared to the failingLV (IHD 1.32-fold, P<0.001; DCM 1.22-fold, P<0.05; IHDand DCM vs NF) (Figs. 2A, C). The prominent role of p47phox
membrane translocation as a mechanism for NADPH oxidaseactivation was further supported by the positive correlationbetween NADPH oxidase activity and fraction of sarcolemmalp47phox both in RV (r=0.76, P=0.001) and in LV specimens(r=0.79, P=0.001).
3.3. Lipid peroxidation
Consistent with the differences observed in ROS production,MDA levels, measured as an indirect index of ROS activity[24], were significantly increased in RV and LV samples from
failing hearts (Fig. 3). In addition, MDA content was positivelyrelated to NADPH-dependent O2
− generation in both ventricles(RV: r=0.8, P=0.0004; LV: r=0.7, P=0.0026).
3.4. MAPK activation
NADPH oxidase ROS production can modulate redox-sensitive signaling pathways, such as MAPK pathways[3,12,25–29]. To investigate the possible activation of theseredox-sensitive kinases, we measured the phosphorylation ofthree strategic components of the MAPK family: JNK, ERK andp38.
The levels of phosphorylated JNK were not changed infailing ventricles compared to respective NF controls (Fig. 4A).Conversely, Fig. 4B shows a significantly higher ERK1/2phosphorylation level in RV from IHD (2.96-fold, P<0.001 vsNF) and, to a lesser extent, from DCM (1.80-fold, P<0.01 vsNF); in LV specimens, a significant increase (1.58-fold,P<0.01 vs NF) was observed only in IHD. Furthermore,ERK activation positively correlated with NADPH oxidaseactivity in both ventricles (RV: r=0.83, P=0.0001; LV:r=0.62, P=0.014).
The phosphorylation status of p38 was also significantlyenhanced in RV and LV specimens from failing myocardiumcompared to respective controls (Fig. 4C). The increase in p38
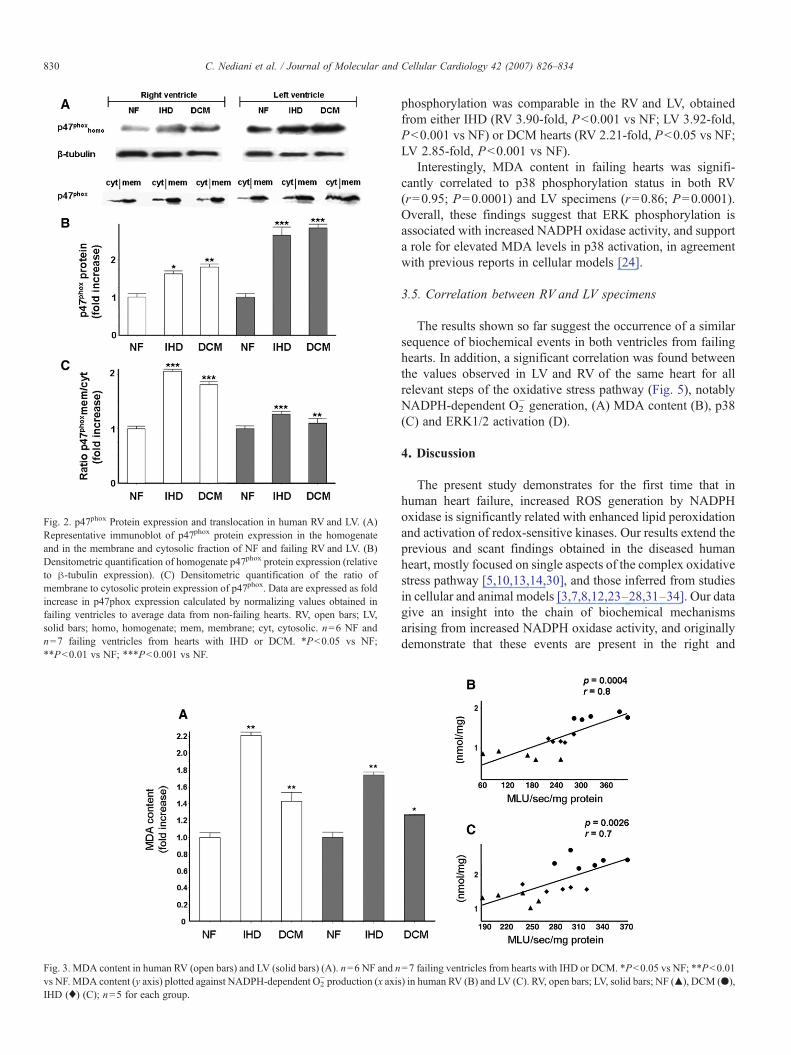
Fig. 3. MDA content in human RV (open bars) and LV (solid bars) (A). n=6 NF and nvs NF. MDA content (y axis) plotted against NADPH-dependent O2
− production (x axisIHD (♦) (C); n=5 for each group.
Fig. 2. p47phox Protein expression and translocation in human RV and LV. (A)Representative immunoblot of p47phox protein expression in the homogenateand in the membrane and cytosolic fraction of NF and failing RV and LV. (B)Densitometric quantification of homogenate p47phox protein expression (relativeto β-tubulin expression). (C) Densitometric quantification of the ratio ofmembrane to cytosolic protein expression of p47phox. Data are expressed as foldincrease in p47phox expression calculated by normalizing values obtained infailing ventricles to average data from non-failing hearts. RV, open bars; LV,solid bars; homo, homogenate; mem, membrane; cyt, cytosolic. n=6 NF andn=7 failing ventricles from hearts with IHD or DCM. *P<0.05 vs NF;**P<0.01 vs NF; ***P<0.001 vs NF.
830 C. Nediani et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 826–834
phosphorylation was comparable in the RV and LV, obtainedfrom either IHD (RV 3.90-fold, P<0.001 vs NF; LV 3.92-fold,P<0.001 vs NF) or DCM hearts (RV 2.21-fold, P<0.05 vs NF;LV 2.85-fold, P<0.001 vs NF).
Interestingly, MDA content in failing hearts was signifi-cantly correlated to p38 phosphorylation status in both RV(r=0.95; P=0.0001) and LV specimens (r=0.86; P=0.0001).Overall, these findings suggest that ERK phosphorylation isassociated with increased NADPH oxidase activity, and supporta role for elevated MDA levels in p38 activation, in agreementwith previous reports in cellular models [24].
3.5. Correlation between RV and LV specimens
The results shown so far suggest the occurrence of a similarsequence of biochemical events in both ventricles from failinghearts. In addition, a significant correlation was found betweenthe values observed in LV and RV of the same heart for allrelevant steps of the oxidative stress pathway (Fig. 5), notablyNADPH-dependent O2
− generation, (A) MDA content (B), p38(C) and ERK1/2 activation (D).
4. Discussion
The present study demonstrates for the first time that inhuman heart failure, increased ROS generation by NADPHoxidase is significantly related with enhanced lipid peroxidationand activation of redox-sensitive kinases. Our results extend theprevious and scant findings obtained in the diseased humanheart, mostly focused on single aspects of the complex oxidativestress pathway [5,10,13,14,30], and those inferred from studiesin cellular and animal models [3,7,8,12,23–28,31–34]. Our datagive an insight into the chain of biochemical mechanismsarising from increased NADPH oxidase activity, and originallydemonstrate that these events are present in the right and
=7 failing ventricles from hearts with IHD or DCM. *P<0.05 vs NF; **P<0.01) in human RV (B) and LV (C). RV, open bars; LV, solid bars; NF (▴), DCM (●),
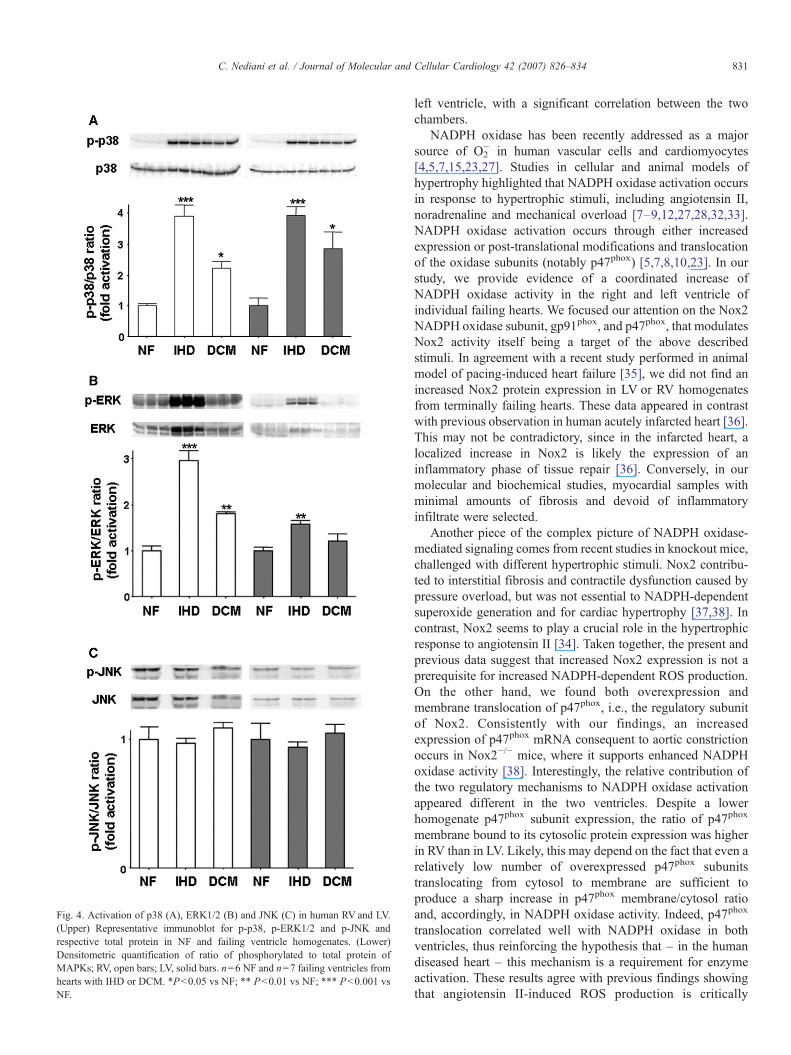
Fig. 4. Activation of p38 (A), ERK1/2 (B) and JNK (C) in human RV and LV.(Upper) Representative immunoblot for p-p38, p-ERK1/2 and p-JNK andrespective total protein in NF and failing ventricle homogenates. (Lower)Densitometric quantification of ratio of phosphorylated to total protein ofMAPKs; RV, open bars; LV, solid bars. n=6 NF and n=7 failing ventricles fromhearts with IHD or DCM. *P<0.05 vs NF; ** P<0.01 vs NF; *** P<0.001 vsNF.
831C. Nediani et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 826–834
left ventricle, with a significant correlation between the twochambers.
NADPH oxidase has been recently addressed as a majorsource of O2
− in human vascular cells and cardiomyocytes[4,5,7,15,23,27]. Studies in cellular and animal models ofhypertrophy highlighted that NADPH oxidase activation occursin response to hypertrophic stimuli, including angiotensin II,noradrenaline and mechanical overload [7–9,12,27,28,32,33].NADPH oxidase activation occurs through either increasedexpression or post-translational modifications and translocationof the oxidase subunits (notably p47phox) [5,7,8,10,23]. In ourstudy, we provide evidence of a coordinated increase ofNADPH oxidase activity in the right and left ventricle ofindividual failing hearts. We focused our attention on the Nox2NADPH oxidase subunit, gp91phox, and p47phox, that modulatesNox2 activity itself being a target of the above describedstimuli. In agreement with a recent study performed in animalmodel of pacing-induced heart failure [35], we did not find anincreased Nox2 protein expression in LV or RV homogenatesfrom terminally failing hearts. These data appeared in contrastwith previous observation in human acutely infarcted heart [36].This may not be contradictory, since in the infarcted heart, alocalized increase in Nox2 is likely the expression of aninflammatory phase of tissue repair [36]. Conversely, in ourmolecular and biochemical studies, myocardial samples withminimal amounts of fibrosis and devoid of inflammatoryinfiltrate were selected.
Another piece of the complex picture of NADPH oxidase-mediated signaling comes from recent studies in knockout mice,challenged with different hypertrophic stimuli. Nox2 contribu-ted to interstitial fibrosis and contractile dysfunction caused bypressure overload, but was not essential to NADPH-dependentsuperoxide generation and for cardiac hypertrophy [37,38]. Incontrast, Nox2 seems to play a crucial role in the hypertrophicresponse to angiotensin II [34]. Taken together, the present andprevious data suggest that increased Nox2 expression is not aprerequisite for increased NADPH-dependent ROS production.On the other hand, we found both overexpression andmembrane translocation of p47phox, i.e., the regulatory subunitof Nox2. Consistently with our findings, an increasedexpression of p47phox mRNA consequent to aortic constrictionoccurs in Nox2−/− mice, where it supports enhanced NADPHoxidase activity [38]. Interestingly, the relative contribution ofthe two regulatory mechanisms to NADPH oxidase activationappeared different in the two ventricles. Despite a lowerhomogenate p47phox subunit expression, the ratio of p47phox
membrane bound to its cytosolic protein expression was higherin RV than in LV. Likely, this may depend on the fact that even arelatively low number of overexpressed p47phox subunitstranslocating from cytosol to membrane are sufficient toproduce a sharp increase in p47phox membrane/cytosol ratioand, accordingly, in NADPH oxidase activity. Indeed, p47phox
translocation correlated well with NADPH oxidase in bothventricles, thus reinforcing the hypothesis that – in the humandiseased heart – this mechanism is a requirement for enzymeactivation. These results agree with previous findings showingthat angiotensin II-induced ROS production is critically
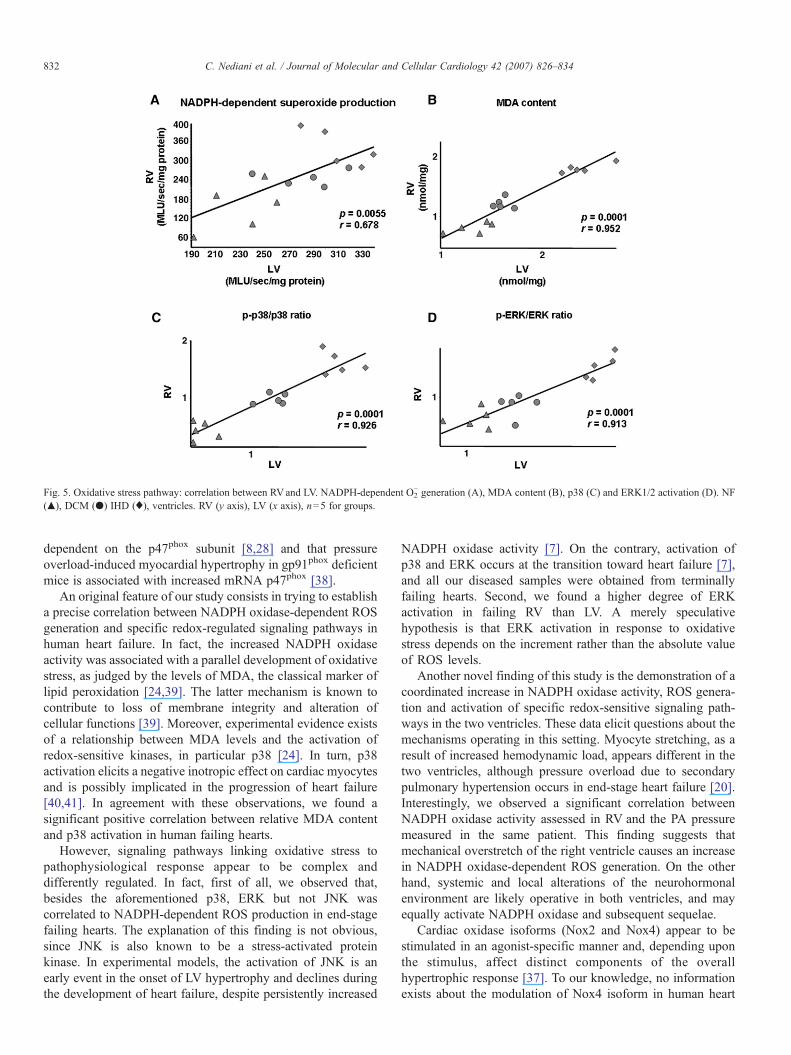
Fig. 5. Oxidative stress pathway: correlation between RVand LV. NADPH-dependent O2− generation (A), MDA content (B), p38 (C) and ERK1/2 activation (D). NF
(▴), DCM (●) IHD (♦), ventricles. RV (y axis), LV (x axis), n=5 for groups.
832 C. Nediani et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 826–834
dependent on the p47phox subunit [8,28] and that pressureoverload-induced myocardial hypertrophy in gp91phox deficientmice is associated with increased mRNA p47phox [38].
An original feature of our study consists in trying to establisha precise correlation between NADPH oxidase-dependent ROSgeneration and specific redox-regulated signaling pathways inhuman heart failure. In fact, the increased NADPH oxidaseactivity was associated with a parallel development of oxidativestress, as judged by the levels of MDA, the classical marker oflipid peroxidation [24,39]. The latter mechanism is known tocontribute to loss of membrane integrity and alteration ofcellular functions [39]. Moreover, experimental evidence existsof a relationship between MDA levels and the activation ofredox-sensitive kinases, in particular p38 [24]. In turn, p38activation elicits a negative inotropic effect on cardiac myocytesand is possibly implicated in the progression of heart failure[40,41]. In agreement with these observations, we found asignificant positive correlation between relative MDA contentand p38 activation in human failing hearts.
However, signaling pathways linking oxidative stress topathophysiological response appear to be complex anddifferently regulated. In fact, first of all, we observed that,besides the aforementioned p38, ERK but not JNK wascorrelated to NADPH-dependent ROS production in end-stagefailing hearts. The explanation of this finding is not obvious,since JNK is also known to be a stress-activated proteinkinase. In experimental models, the activation of JNK is anearly event in the onset of LV hypertrophy and declines duringthe development of heart failure, despite persistently increased
NADPH oxidase activity [7]. On the contrary, activation ofp38 and ERK occurs at the transition toward heart failure [7],and all our diseased samples were obtained from terminallyfailing hearts. Second, we found a higher degree of ERKactivation in failing RV than LV. A merely speculativehypothesis is that ERK activation in response to oxidativestress depends on the increment rather than the absolute valueof ROS levels.
Another novel finding of this study is the demonstration of acoordinated increase in NADPH oxidase activity, ROS genera-tion and activation of specific redox-sensitive signaling path-ways in the two ventricles. These data elicit questions about themechanisms operating in this setting. Myocyte stretching, as aresult of increased hemodynamic load, appears different in thetwo ventricles, although pressure overload due to secondarypulmonary hypertension occurs in end-stage heart failure [20].Interestingly, we observed a significant correlation betweenNADPH oxidase activity assessed in RV and the PA pressuremeasured in the same patient. This finding suggests thatmechanical overstretch of the right ventricle causes an increasein NADPH oxidase-dependent ROS generation. On the otherhand, systemic and local alterations of the neurohormonalenvironment are likely operative in both ventricles, and mayequally activate NADPH oxidase and subsequent sequelae.
Cardiac oxidase isoforms (Nox2 and Nox4) appear to bestimulated in an agonist-specific manner and, depending uponthe stimulus, affect distinct components of the overallhypertrophic response [37]. To our knowledge, no informationexists about the modulation of Nox4 isoform in human heart

833C. Nediani et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 826–834
failure. Thus, the possibility that increased ROS production inthe human right and left ventricles relies upon the activation ofdistinct Nox isoforms remains an intriguing hypothesis whichwarrants further investigation.
The findings of our study should be interpreted in the contestof their limitations. A mechanistic interpretation of the eventsdownstream to NADPH oxidase activation is prevented inhuman end-stage hearts and therefore our results cannot providea clear evidence for a cause–effect relationship. However, thehighly significant correlations among ROS production, p47phox
translocation and phosphorylation of specific MAP kinases aresuggestive of a close association among these factors.
Another insight provided by the present investigation is thecorrelation between the left and right ventricle from the samepatient, a report of scientific and prospectively clinicalrelevance. A rigorous statistical interpretation of such acorrelation is difficult, since control, IHD or DCM valuestend to cluster. This might indicate that, in our conditions, theetiology rather than the severity of the disease is crucial for thetested parameters; however, all diseased samples came frompatients with end-stage heart failure. Studies in animal modelswith different degree of cardiac disease will hopefully allow adeeper understanding of the mutual relationship between leftand right ventricular remodeling and causal mechanisms.Notwithstanding these limitations, as recently pointed out[42], advancing knowledge about molecular and functionalcharacteristics of the right ventricle might lead to progress in thetreatment of cardiomyopathy.
In conclusion, notwithstanding the relative contribution ofsystemic and hemodynamic factors operative in human heartfailure, that could differently activate distinct ROS sources andrelated redox signaling, our data strongly suggest that oxidativestress may contribute to harmful RV (besides LV) remodeling.Given that RV function is a predictor of survival in heart failurepatients [18–20], this observation provides new insights into thepathophysiological processes involved in the progressiontoward end-stage heart failure and supports the therapeutictargeting of ROS-dependent processes.
Acknowledgments
The financial support from Ministero dell' Istruzione, dell'Università e della Ricerca (MIUR) and Telethon–Italy (GrantNo GGP05093) is gratefully acknowledged.
References
[1] Byrne JA, Grieve DJ, Cave AC, Shah AM. Oxidative stress and heartfailure. Arch Mal Coeur Vaiss 2003;96:214–21.
[2] Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J ClinInvest 2005;115:500–8.
[3] Qin F, Shite J, Liang C-S. Antioxidants attenuate myocyte apoptosis andimprove cardiac function in CHF: association with changes in MAPKpathways. Am J Physiol: Heart Circ Physiol 2003;285:H822–32.
[4] Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P) H oxidase: role incardiovascular biology and disease. Circ Res 2000;86:494–501.
[5] Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G,et al. Increased myocardial NADPH oxidase activity in human heartfailure. J Am Coll Cardiol 2003;41:2164–71.
[6] Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat RevImmunol 2004;4:181–9.
[7] Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPHoxidase during progression of cardiac hypertrophy to failure. Hypertension2002;40:477–84.
[8] Li JM, Shah AM. Mechanism of endothelial cell NADPH oxidaseactivation by angiotensin II. Role of the p47phox subunit. J Biol Chem2003;278:12094–100.
[9] Li JM, Fan LM, Christie MR, Shah AM. Acute tumor necrosis factor alphasignaling via NADPH oxidase in microvascular endothelial cells: role ofp47phox phosphorylation and binding to TRAF4. Mol Cell Biol 2005;25:2320–30.
[10] Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M, et al.Oxygen free radical release in human failing myocardium is associatedwith increased activity of Rac1-GTPase and represents a target for statintreatment. Circulation 2003;108:1567–74.
[11] Murdoch CE, Zhang M, Cave AC, Shah AM. ADPH oxidase-dependentredox signaling in cardiac hypertrophy, remodelling and failure.Cardiovasc Res 2006;71:208–15.
[12] Zhang G-X, Kimura S, Nishiyama A, Shokoji T, Rahman M, Abe Y. ROSduring the acute phase of Ang II hypertension participates in cardiovas-cular MAPK activation but not vasoconstriction. Hypertension 2004;43:117–24.
[13] Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, et al.Differential activation of signal transduction pathways in human hearts withhypertrophy versus advanced heart failure. Circulation 2001;103:670–7.
[14] Cook SA, Sugden PH, Clerk A. Activation of c-Jun N-terminal kinases andp38-mitogen-activated protein kinases in human heart failure secondary toischaemic heart disease. J Mol Cell Cardiol 1999;31:1429–34.
[15] Murdoch CE, Grieve DJ, Cave AC, Looi YH, Shah AM. NADPH oxidaseand heart failure. Curr Opin Pharmacol 2006;6:1–6.
[16] Kucuker. Evidence of improved right ventricular structure after LVADsupport in patients with end-stage cardiomyopathy. Heart Lung Transplant2004;23:28–35.
[17] Li GR, Lau CP, Nattel S. Ionic current abnormalities associated withprolonged action potentials in cardiomyocytes from diseased human rightventricles. Heart Rhythm 2004;1:460–8.
[18] Di Salvo TG, Mathier M, Semigrain MJ, Dec GW. Preserved rightventricular ejection fraction predicts exercise capacity and survival inadvanced heart failure. J Am Coll Cardiol 1995;25:1143–53.
[19] Gavazzi A, Berzuini C, Campana C, Inserra C, Ponzetta M, Sebastiani R,et al. Value of right ventricular ejection fraction in predicting short-termprognosis of patients with severe chronic heart failure. J Heart LungTransplant 1997;16:774–85.
[20] Brieke A, De Nofrio D. Right ventricular dysfunction in chronic dilatedcardiomyopathy and heart failure. Coron Artery Dis 2005;16:5–11.
[21] Skatchkov MP, Sperling D, Hink U, Mulsch A, Harrison DG, SindermannI, et al. Validation of lucigenin as a chemiluminescent probe to monitorvascular superoxide as well as basal vascular nitric oxide production.Biochem Biophys Res Commun 1999;254:319–24.
[22] Li JM, Shah AM. Differential NADPH-versus NADH-dependent super-oxide production by phagocyte-type endothelial cell NADPH oxidase.Cardiovasc Res 2001;52:477–86.
[23] MacCarthy PA, Grieve DJ, Li J-M, Dunster C, Kelly FJ, Shah AM.Impaired endothelial regulation of ventricular relaxation in cardiachypertrophy: role of reactive oxygen species and NADPH oxidase.Circulation 2001;104:2967–74.
[24] Folden DV, Gupta A, Sharma AC, Li SY, Saari JT, Ren J. Malondialdehydeinhibits cardiac contractile function in ventricular myocytes via a p38mitogen-activated protein kinase-dependent mechanism. Br J Pharmacol2003;139:1310–6.
[25] Widder J, Behr T, Fraccarollo D, Hu K, Galuppo P, Tas P, et al. Vascularendothelial dysfunction and superoxide anion production in heart failureare p38 MAP kinase-dependent. Cardiovasc Res 2004;63:161–7.
[26] Fiorillo C, Nediani C, Ponziani V, Giannini L, Celli A, Nassi N, et al.Cardiac volume overload rapidly induces oxidative stress-mediatedmyocyte apoptosis and hypertrophy. Biochim Biophys Acta 2005;1741:173–82.

834 C. Nediani et al. / Journal of Molecular and Cellular Cardiology 42 (2007) 826–834
[27] Qin F, Patel R, Yan C, Liu W. NADPH oxidase is involved in angiotensinII-induced apoptosis in H9C2 cardiac muscle cells: effect of apocynin. FreeRadical Biol Med 2006;40:236–46.
[28] Li JM, Wheatcroft S, Fan LM, Kearney MT, Shah AM. Opposing rolesof p47phox in basal versus angiotensin II-stimulated alterations in vascularO2− production, vascular tone, and mitogen-activated protein kinase
activation. Circulation 2004;109:1307–13.[29] Sumbayev VV, Yasinka IM. Regulation of MAP kinase-dependent
apoptotic pathway: implication of reactive oxygen and nitrogen species.Arch Biochem Biophys 2005;436:406–12.
[30] Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C,et al. A myocardial Nox2 containing NADPH Oxidase contributes tooxidative stress in human atrial fibrillation. Circ Res 2005;97:629–36.
[31] Li JM, Shah AM. Endothelial cell superoxide generation: regulation andrelevance for cardiovascular pathophysiology. Am J Physiol, Regul IntegrComp Physiol 2004;287:R1014–30.
[32] Li JM, Mullen AM, Yun S, Wientjes F, Brouns GY, Thrasher AJ, et al.Essential role of the NADPH oxidase subunit p47phox in endothelial cellsuperoxide production in response to phorbol ester and tumor necrosisfactor-α. Circ Res 2002;90:143–50.
[33] Xiao L, Pimentel DR, Wang J, Singh K, Colucci WS, Sawyer DB. Role ofreactive oxygen species and NAD(P) H oxidase in alpha(1)-adrenoceptorsignaling in adult rat cardiac myocytes. Am J Physiol: Cell Physiol2002;282:C926–34.
[34] Bendall JK, Cave CA, Heymes C, Gall N, Shah AM. Pivotal role of agp91phox-containing NADPH oxidase in angiotensin II-induced cardiachypertrophy in mice. Circulation 2005;105:293–6.
[35] Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinsky V,et al. Glucose-6-phosphate dehydrogenase-derived NADPH fuelssuperoxide production in the failing heart. J Mol Cell Cardiol 2006;41:340–9.
[36] Krijnen PAJ, Meischl C, Hack CE, Meijer CJ, Visser CA, Roos D, et al.Increased Nox2 expression in human cardiomyocyte after acute myocar-dial infarction. J Clin Pathol 2003;56:194–9.
[37] Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC, et al.Involvement of the nicotinamide adenosine dinucleotide phosphateoxidase isoform Nox2 in cardiac contractile dysfunction occurring inresponse to pressure overload. J Am Coll Cardiol 2006;47:817–26.
[38] Maytin M, Siwik DA, Ito M, Xiao L, Sawyer D, Ronglih L, et al. Pressureoverload-induced myocardial hypertrophy in mice does not requiregp91phox. Circulation 2004;109:1168–71.
[39] Porter NA, Caldweel SE, Mills KA. Mechanisms of free radical oxidationof unsaturated lipids. Lipids 1995;30:277–90.
[40] Liao P, Wang SQ, Wang S, Zheng M, Zheng M, Zhang SJ, et al. p38Mitogen-activated protein kinase mediates a negative inotropic effect incardiac myocytes. Circ Res 2002;90:190–6.
[41] Ma XL, Kumar S, Gao F, Louden CS, Lopez BL, Christopher TA, et al.Inhibition of p38 mitogen-activated protein kinase decreases cardiomyo-cyte apoptosis and improves cardiac function after myocardial ischemiaand reperfusion. Circulation 1999;99:1685–91.
[42] Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, MeldrumDR, et al. Right ventricular function and failure: report of a National Heart,Lung, and Blood Institute working group on cellular and molecularmechanisms of right heart failure. Circulation 2006;114:1883–91.




















