
Early Low Protein Diet Aggravates Unbalance between Antioxidant Enzymes Leading to Islet Dysfunction
Upload
independentCategory
view
0download
0
LIVER FAILURE/CIRRHOSIS/PORTAL HYPERTENSION
NOX1/Nicotinamide Adenine Dinucleotide Phosphate,Reduced Form (NADPH) Oxidase Promotes Proliferationof Stellate Cells and Aggravates Liver Fibrosis Induced
by Bile Duct LigationWenhao Cui,1 Kuniharu Matsuno,1 Kazumi Iwata,1 Masakazu Ibi,1 Misaki Matsumoto,1 Jia Zhang,1 Kai Zhu,1
Masato Katsuyama,2 Natalie J. Torok,3 and Chihiro Yabe-Nishimura1
Among multiple isoforms of nicotinamide adenine dinucleotide phosphate, reduced form(NADPH) oxidase expressed in the liver, the phagocytic NOX2 isoform in hepatic stellate cells(HSCs) has been demonstrated to play a key role in liver fibrogenesis. The aim of this study wasto clarify the role of NOX1, a nonphagocytic form of NADPH oxidase, in the development of fi-brosis using Nox1-deficient mice (Nox1KO). Liver injury and fibrosis were induced by bile ductligation (BDL) and carbon tetrachloride in Nox1KO and wildtype littermate mice (WT). Pri-mary HSCs were isolated to characterize the NOX1-induced signaling cascade involved in liverfibrogenesis. Following BDL, a time-dependent increase in NOX1 messenger RNA (mRNA) wasdemonstrated in WT liver. Compared with those in WT, levels of collagen-1a mRNA andhydroxyproline were significantly suppressed in Nox1KO with a reduced number of activatedHSCs and less severe fibrotic lesions. The expression levels of a-smooth muscle actin, a markerof HSCs activation, were similar in cultured HSCs isolated from both genotypes. However, cellproliferation was significantly attenuated in HSCs isolated from Nox1KO. In these cells, theexpression of p27kip1, a cell cycle suppressor, was significantly up-regulated. Concomitantly, asignificant reduction in phosphorylated forms of Akt and forkhead box O (FOXO) 4, a down-stream effector of Akt that regulates the transcription of p27kip1 gene, was demonstrated inNox1KO. Finally, the level of the oxidized inactivated form of phosphatase and tensin homolog(PTEN), a negative regulator of PI3K/Akt pathway, was significantly attenuated in HSCs ofNox1KO. Conclusion: These findings indicate that reactive oxygen species derived fromNOX1/NADPH oxidase oxidize and inactivate PTEN to positively regulate the Akt/FOXO4/p27kip1 signaling pathway. NOX1 may thus promote proliferation of HSCs and accelerate thedevelopment of fibrosis following BDL-induced liver injury. (HEPATOLOGY 2011;54:949-958)
Liver fibrosis, characterized by accumulation ofextracellular matrix (ECM), is a wound-healingresponse to acute or chronic liver injury.
Chronic hepatitis viral infection, alcohol abuse, and
nonalcoholic steatohepatitis (NASH) are among theetiological factors documented in the development ofliver fibrosis.1,2 Upon acute or chronic injury, the liverundergoes an injury/repair process accompanied by an
Abbreviations: a-SMA, a-smooth muscle actin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BDL, bile duct ligation; CCl4, carbontetrachloride; CDK, cyclin-dependent kinase; col-1a, collagen 1a; DHE, dihydroethidium; ECM, extracellular matrix; FOXO, forkhead box O; FP, PGF2areceptor; HSCs, hepatic stellate cells; NADPH, nicotinamide adenine dinucleotide phosphate, reduced form; NASH, nonalcoholic steatohepatitis; PBC, primarybiliary cirrhosis; PDGF, platelet-derived growth factor; PTEN, phosphatase and tensin homolog; ROS, reactive oxygen species; TGF, transforming growth factor.From the 1Department of Pharmacology, Kyoto Prefectural University of Medicine, Kyoto, Japan; 2Radioisotope Center, Kyoto Prefectural University of Medicine,
Kyoto, Japan; 3Division of Gastroenterology and Hepatology, Department of Internal Medicine, UC Davis Medical Center, Sacramento, CA.Received December 14, 2010; accepted May 11, 2011.Supported by a Grant-in-Aid for Young Scientists (B) 21790246 (to K.M.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.Address reprint requests to: Dr. C. Yabe-Nishimura, Department of Pharmacology, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan. E-mail:
[email protected]; fax: þ81-75-251-5348.CopyrightVC 2011 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.24465Potential conflict of interest: Dr. Yabe-Nishimura owns stock in Genkyotex.Additional Supporting Information may be found in the online version of this article.
949

inflammatory response and deposition of ECM. Dur-ing the progression of fibrosis, apoptosis is promotedand inflammatory cells are increasingly recruited,which can further worsen the liver function and tissuedamage.In the fibrotic process, hepatic stellate cells (HSCs)
are the major source of ECM. In the normal liver, qui-escent HSCs, located in the space of Disse, store fatand lipid-soluble vitamins including vitamin A. Inresponse to liver injury, HSCs undergo an activationprocess and transform into myofibroblast-like cells, aprocess characterized by loss of vitamin A and expres-sion of a-smooth muscle actin (a-SMA).3,4 ActivatedHSCs promote collagen synthesis and secretion. Inaddition, these cells produce a number of cytokinessuch as transforming growth factor-b (TGF-b) andplatelet-derived growth factor (PDGF), which play im-portant roles in the development of liver fibrosis.5,6
The involvement of reactive oxygen species (ROS)in liver fibrogenesis has been documented. The activa-tion and expression of various mediators are regulatedby ROS by way of the redox-sensitive protein kinasesand transcription factors in HSCs.7,8 Recently, nicotin-amide adenine dinucleotide phosphate, reduced form(NADPH) oxidase, originally identified as a majorsource of ROS in phagocytes, was shown to play a keyrole in cellular signaling. NOX is a catalytic subunit ofNADPH oxidase, a multisubunit enzyme composed ofmembrane-bound NOX and p22phox, associated withseveral cytosolic regulatory subunits including p47phox.In recent years, several isoforms of NOX have beenidentified.9 NOX2 (gp91phox) is the phagocytic formof NOX, whereas NOX1 is a newly identified formthat has been implicated in the pathogenesis of hyper-tension and inflammatory pain.10,11
Previously, it was reported that mice lackingp47phox, a cytosolic subunit of NADPH oxidase, dem-onstrated reduced liver injury and fibrosis after bileduct ligation (BDL).12 In transgenic mice overexpress-ing Rac1, another cytosolic subunit necessary forenzyme activation, greater numbers of activated HSCs,increased liver damage, and fibrosis were demonstratedfollowing treatment with carbon tetrachloride(CCl4).
13 In line with these findings, reduced liver fi-brosis was demonstrated in mice lacking NOX2treated with CCl4.
14 Furthermore, NOX2 was recentlyshown to play a key role in activation of HSCs follow-ing phagocytosis of apoptotic hepatocytes.15 A consid-erable amount of evidence thus suggests a critical rolefor the NOX2 isoform of NADPH oxidase in liverfibrogenesis. On the other hand, little is known aboutthe role of NOX1 in the pathogenesis of liver fibrosis,
although induction of NOX1 mRNA in the activatedHSCs was reported.12 This led us to investigatewhether the NOX1 isoform of NADPH oxidase isinvolved in the development of liver fibrosis usingmice deficient in the Nox1 gene. We here report thatNOX1 promotes the proliferation of HSCs and liverfibrogenesis by inactivating phosphatase and tensinhomolog (PTEN). NOX1 thus positively regulates theAkt/FOXO4 pathway to reduce a cell cycle suppressor,p27kip1.
Materials and Methods
Animal Model. Liver fibrosis was induced by BDLor injection of carbon tetrachloride (CCL4).Details of the methods are described in the Support-
ing Materials and Methods.Isolation and Culture of HSCs and Hepatocy-
tes. HSCs and hepatocytes were isolated by in situ col-lagenase perfusion and density gradient centrifugationon OptiPrep (Sigma), as described.16 Refer to the Sup-porting Materials and Methods section for details.Real-Time Polymerase Chain Reaction (PCR). Real-
time PCR was performed with SYBR green dye.Details of the methods are described in the SupportingMaterials and Methods. Primers used for real-timePCR are shown in Table 1.Biochemical Measurements. The analyses of serum
alanine aminotransferase (ALT) and aspartate amino-transferase (AST) were performed as described.13 Thelevel of hydroxyproline was quantified colorimetrically.Refer to the Supporting Materials and Methods sectionfor details.Detection of Superoxide Production. Superoxide
production was measured by the lucigenin chemilumi-nescence method as described.17 The lucigenin
Table 1.
Sense Antisense
Nox1 CTGACAAGTACTATTACACGAGAG CATATATGCCACCAGCTTATGGAAG
Nox2 AACTGTATGCTGATCCTGCTGC GTTCTCATTGTCACCGATGTCAG
Nox4 TGAGGAGTCACTGAACTA
TGAAGTTAATC
TGACTGAGGTACAGCTGGA
TGTTCACA
p27kip1 CGTGAGAGTGTCTAACGGG CGAGTCAGGCATTTGGTCC
PDGF-B AATGCTGAGCGACCACTCCAT CGGCATTGCACATTGCGGTTATT
TGF-b1 GCAACATGTGGAACTCTACCAG GACGTCAAAAGACAGCCACTCA
p21cip1 GTCTGAGCGGCCTGAAGATT TCCTGACCCACAGCAGAAGA
col-1a AAGGTTCTCCTGGTGAAGCTGGT CTGAGCTCCAGCTTCTCCATCTT
a-SMA GAAGTATCCGATAGAACACGGCATC CCAGCACAATACCAGTTGTACGTC
MCP1 TTCTGGGCCTGCTGTTCAC TCCAGCCTACTCATTGGGATC
RANTES CTGCTGCTTTGCCTACCTCT TTGAACCCACTTCTTCTCTGG
TNFa AGAACTCCAGGCGGTGCCTAT TTGAGATCCATGCCGTTGGC
GADPH TCAACGGGAAACCCATCACCAT GAACACGGAAGGCCATGCCAGT
950 CUI ET AL. HEPATOLOGY, September 2011

chemiluminescence is expressed as relative light units(RLU) per milligram of protein per minute.Dihydroethidium (DHE) Staining. After one pas-
sage, HSCs were plated in Lab-Tek II chamber slides(Thermo Fisher Scientific, Waltham, MA) and cul-tured for another 5 days. Cells were washed with phos-phate-buffered saline (PBS) twice and DHE (10 lmol/L, Molecular Probes) was topically applied. Slides wereincubated at 37�C for 30 minutes in a light-protectedhumidified chamber and ethidium bromide wasdetected under a 543-nm He-Ne laser.Western Blot Analysis. Proteins separated by so-
dium dodecyl sulfate-polyacrylamide gel electrophore-sis (SDS-PAGE) were subjected to western blotting asdescribed.17 Refer to the Supporting Materials andMethods section for details.Flow Cytometry (FACS). Cells were trypsinized and
fixed in 75% ethanol overnight at �20�C. After wash-ing with PBS, cells were incubated with 0.01% RNasefor 30 minutes at 25�C. Propidium iodide (PI; 500lg/mL) was added just before the measurement andDNA content was determined using a FACS Caliburflow cytometer with Cell Quest software (BD Bio-sciences, NJ). For all assays, 10,000 cells werecounted.Histological Analysis. Paraffin-embedded sections
were prepared as described.17 For Sirius red staining,sections were stained with 0.1% Sirius red and fastgreen in saturated picric acid for 1 hour, followed bywashing with 0.01 N HCl. For immunohistochemicalanalysis, an antibody against a-SMA (Sigma, St. Louis,MO) was used. Signal was detected using VectastainABC Kit (Vector Laboratories, Burlingame, CA).Statistics. Results are expressed as mean 6 standard
error of the mean (SEM). The statistical analyses wereperformed with two-way analysis of variance(ANOVA). For multiple treatment groups, post-hocmultiple comparison (Dunnett’s test) was applied.
Results
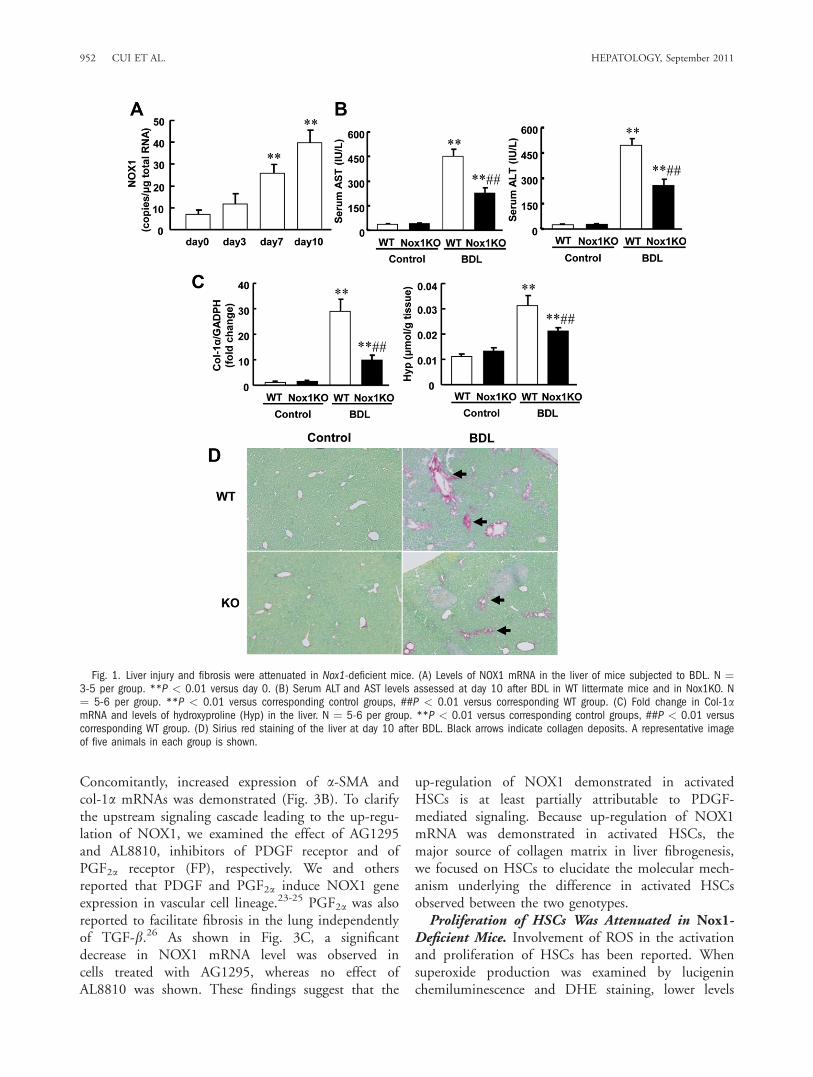
Liver Injury and Fibrosis Were Attenuated inNox1-Deficient Mice. A marked increase in the levelof NOX1 mRNA was demonstrated in WT liver atday 3 and reached up to 6-fold at day 10 after BDL(Fig. 1A). In both genotypes the levels of NOX2mRNA were similarly increased at day 10 after BDL.Meanwhile, the expression of NOX4 mRNA wasunchanged (Supporting Fig. 1).To examine the effect of Nox1 deficiency on liver
injury, serum ALT and AST levels were measured. Tendays after BDL, both parameters were significantly
higher in WT than those in Nox1-deficient mice(Nox1KO) (Fig. 1B).Furthermore, increased levels of collagen 1a (col-1a)
mRNA and hydroxyproline, an amino acid specificallycontained in collagen, were significantly suppressed inthe liver of Nox1KO after BDL (Fig. 1C). These find-ings suggest that liver injury and fibrosis were attenu-ated in the absence of NOX1. In line with these find-ings, histological analyses demonstrated markedcollagen deposition in WT liver, whereas less deposi-tion was observed in Nox1KO after BDL (Fig. 1D).Activated HSCs Were Reduced in Nox1-Deficient
Mice. As activated HSCs are the major source of colla-gen matrix, expression of a-SMA, a marker of acti-vated HSCs, was examined at 10 days after BDL. Amarked increase in mRNA and protein levels of a-SMA was observed in WT, whereas the levels were sig-nificantly suppressed in Nox1KO (Fig. 2A,B). Histo-logical analyses also illustrated that a-SMA-positiveHSCs were less apparent in Nox1KO (Fig. 2C).Several cytokines, such as PDGF, TGF-b1, tumor
necrosis factor alpha (TNF-a), and macrophage che-moattractant protein-1 (MCP1) are known to takepart in the development of liver fibrosis.1,18-20 Follow-ing BDL, levels of PDGF, TGF-b1, TNFa, andMCP1 mRNAs in the liver were markedly increased,but no difference was observed between WT andNox1KO (Supporting Fig. 2).We next investigated whether similar findings are
observed in a different model of liver fibrosis.Repeated administration of the chemical hepatotoxinCCl4 elicited a significant increase in NOX1 mRNAin the liver. In contrast to the BDL model, however,the levels of col-1a and a-SMA protein were unaf-fected by Nox1 deficiency after 8 weeks of CCL4 treat-ment (Supporting Fig. 3).NOX1 Was Up-regulated in Cultured HSCs. In
the liver, expression of NOX1 was reported in hepato-cytes and HSCs, but not in Kupffer cells.21 We there-fore isolated hepatocytes and treated them with cheno-deoxycholic acid, one of the key components involvedin the pathogenesis of BDL-induced fibrosis. Nochange in the level of NOX1 mRNA was observed,whereas activation of caspase-3 induced by bile acidwas significantly diminished in hepatocytes isolatedfrom Nox1KO (Supporting Fig. 4A,B).We next isolated HSCs and the expression of
NOX1 mRNA was assessed in primary culture. It isknown that HSCs in culture are spontaneously acti-vated.22 As shown in Fig. 3A, a time-dependentincrease in NOX1 mRNA, which was undetectableuntil day 3, was demonstrated in cultured HSCs.
HEPATOLOGY, Vol. 54, No. 3, 2011 CUI ET AL. 951
Concomitantly, increased expression of a-SMA andcol-1a mRNAs was demonstrated (Fig. 3B). To clarifythe upstream signaling cascade leading to the up-regu-lation of NOX1, we examined the effect of AG1295and AL8810, inhibitors of PDGF receptor and ofPGF2a receptor (FP), respectively. We and othersreported that PDGF and PGF2a induce NOX1 geneexpression in vascular cell lineage.23-25 PGF2a was alsoreported to facilitate fibrosis in the lung independentlyof TGF-b.26 As shown in Fig. 3C, a significantdecrease in NOX1 mRNA level was observed incells treated with AG1295, whereas no effect ofAL8810 was shown. These findings suggest that the
up-regulation of NOX1 demonstrated in activatedHSCs is at least partially attributable to PDGF-mediated signaling. Because up-regulation of NOX1mRNA was demonstrated in activated HSCs, themajor source of collagen matrix in liver fibrogenesis,we focused on HSCs to elucidate the molecular mech-anism underlying the difference in activated HSCsobserved between the two genotypes.Proliferation of HSCs Was Attenuated in Nox1-
Deficient Mice. Involvement of ROS in the activationand proliferation of HSCs has been reported. Whensuperoxide production was examined by lucigeninchemiluminescence and DHE staining, lower levels
Fig. 1. Liver injury and fibrosis were attenuated in Nox1-deficient mice. (A) Levels of NOX1 mRNA in the liver of mice subjected to BDL. N ¼3-5 per group. **P < 0.01 versus day 0. (B) Serum ALT and AST levels assessed at day 10 after BDL in WT littermate mice and in Nox1KO. N¼ 5-6 per group. **P < 0.01 versus corresponding control groups, ##P < 0.01 versus corresponding WT group. (C) Fold change in Col-1amRNA and levels of hydroxyproline (Hyp) in the liver. N ¼ 5-6 per group. **P < 0.01 versus corresponding control groups, ##P < 0.01 versuscorresponding WT group. (D) Sirius red staining of the liver at day 10 after BDL. Black arrows indicate collagen deposits. A representative imageof five animals in each group is shown.
952 CUI ET AL. HEPATOLOGY, September 2011

Fig. 2. Activated HSCs were reduced inNox1KO mice. (A) Levels of a-SMA mRNAin the liver at day 10 after BDL. N ¼ 6-9per group. *P < 0.05, **P < 0.01 ver-sus corresponding control groups, ##P <0.01 versus corresponding WT group. (B)Representative immunoblots and quantita-tive densitometric analysis of a-SMA inthe liver at day 10 after BDL. N ¼ 4-5per group. **P < 0.01 versus corre-sponding control groups, #P < 0.05 ver-sus corresponding WT group. (C)Immunohistochemical staining of a-SMAin the liver at day 10. Black arrows indi-cate a-SMA-positive cells. A representativeimage of five animals in each group isshown.
Fig. 3. NOX1 is up-regulated in activated HSCs in culture. (A) A time-dependent increase in NOX1 mRNA in primary HSCs isolated from theliver. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). (B) Levels of a-SMA and collagen-1a mRNAs in HSCs. N ¼ 4-6. *P < 0.05, **P < 0.01 versus day 3. (C) Effects of inhibitors of PDGF and PGF2a receptorson NOX1 mRNA expression. Cells were treated with AG1295 (10 lM) or AL8810 (10 lM) on day 5. On day 7, total RNA was extracted andNOX1 mRNA was measured by real-time PCR. N ¼ 3-5. *P < 0.05 versus control.
HEPATOLOGY, Vol. 54, No. 3, 2011 CUI ET AL. 953

were observed in cells isolated from Nox1KO (Fig.4A,B). When mRNA levels of col-1a and a-SMA wereevaluated, no difference was observed in HSCs isolatedfrom either genotype. Furthermore, no difference inthe levels of RANTES and MCP1, proinflammatorycytokines released from HSCs, was observed in cellsisolated from either genotype (Supporting Fig. 5).Accordingly, activation of HSCs was not affected byNox1 deficiency.On the other hand, proliferation of HSCs isolated
from Nox1KO was significantly suppressed comparedwith that from WT (Fig. 4C,D). Because flow cyto-metric analyses indicated similar amounts of sub-G1
DNA, an indicator of apoptosis (Fig. 4D), the findingwas verified by measuring the activity of caspase-3. Asshown in Fig. 4E, no difference was observed in cellsisolated from either genotype. These findings suggestthe involvement of NOX1 in cell cycle progression,but not in apoptosis or activation of HSCs in thecourse of liver fibrosis.NOX1 Regulated PTEN/Akt/p27kip1 Pathway in
HSCs. We then investigated the role of NOX1 in cellcycle regulation. p27kip1, a cyclin-dependent kinase(CDK) inhibitor, is a key regulator of the cell cycle. InHSCs isolated from Nox1KO, the expression ofp27kip1 was significantly increased at both the mRNA
and protein levels. In contrast, no change in anothercell cycle inhibitor, p21cip1, was indicated (Fig. 5A,B).The phosphorylation of forkhead box O (FOXO)
transcription factors by Akt leads to inactivation oftheir transcriptional activities. Because p27kip1 is regu-lated by the Akt/FOXO pathway,27 we examined thephosphorylation of Akt and FOXO transcription fac-tors. In HSCs isolated from Nox1KO, the levels ofphosphorylated Akt and FOXO4 were significantlydecreased, whereas no difference in the level of phos-phorylated PI3K was observed (Fig. 6A,B). When cellsisolated from WT were cultured in the presence of N-acetylcysteine, a free radical scavenger and glutathioneprecursor, the level of phosphorylated Akt was signifi-cantly suppressed (Fig. 6C). These data suggest thatthe Akt/FOXO4 pathway downstream of PI3K plays acrucial role in the increased expression of p27kip1 inHSCs of Nox1KO.Phosphatase and tensin homolog (PTEN) is a dual
phosphatase that negatively regulates the PI3K/Aktpathway.28 The activity of PTEN is regulated by phos-phorylation and oxidation. Phosphorylation of PTENinfluences protein stability,29 whereas oxidation ofPTEN negatively regulates enzyme activity.30 BecauseROS derived from NADPH oxidase could oxidize andinactivate PTEN,31 we examined the potential role of
Fig. 4. Proliferation was attenuated in HSCs isolated from Nox1KO mice. (A) Production of ROS in primary cultured HSCs determined by luci-genin chemiluminescence. N ¼ 10. *P < 0.05 versus WT. (B) DHE staining of primary cultured HSCs. (C) Proliferation of HSCs in culture. Num-ber of HSCs was determined at indicated hours after passage at day 7. Averages were obtained from five random fields. **P < 0.01 versus WT.(D) FACS analyses of cell cycle distribution in HSCs. The analysis was performed at day 14. N ¼ 6. *P < 0.05 versus WT. (E) The activity ofcaspase-3 in HSCs. The assay was performed at day 14. N ¼ 5.
954 CUI ET AL. HEPATOLOGY, September 2011

PTEN in NOX1-mediated regulation of cell prolifera-tion. As shown in Fig. 7A, a significant decrease in theoxidized form of PTEN was demonstrated in HSCsisolated from Nox1KO. In accordance with these find-ings, a marked decrease in the oxidized form of PTENwas demonstrated in WT cells treated with N-acetyl-cysteine (Fig. 7B). On the other hand, no difference inthe level of phosphorylated PTEN was observedbetween the two genotypes (Fig. 7C). These findingssuggest that ROS derived from NOX1 inactivatePTEN to enhance the Akt signaling pathway.
Discussion
In this study, NOX1/NADPH oxidase was demon-strated to play an essential role in liver injury and fi-brosis. The principal findings obtained include the fol-lowing: (1) NOX1 was up-regulated in the liver ofmice subjected to BDL. (2) Increased parameters indi-cating liver injury and collagen synthesis induced byBDL were significantly attenuated in Nox1KO. (3)Activated HSCs were reduced in Nox1KO liver afterBDL. (4) Proliferation of cultured HSCs was attenu-ated in Nox1KO. (5) Increased expression of p27kip1
was accompanied by decreased levels of phosphorylatedAkt/FOXO4 and of the oxidized form of PTEN inHSCs isolated from Nox1KO. These results suggestthat ROS derived from NOX1 inactivate PTEN andpromote proliferation of HSCs by way of the Akt/FOXO4/p27kip1 signaling pathway, which leads to thedevelopment of fibrosis following liver injury (Fig. 8).Tyrphostin AG1295, a selective inhibitor of PDGF-
receptor kinase, partially but significantly suppressedthe induction of NOX1 in cultured HSCs. As PDGFis known to induce NOX1 in vascular smooth musclecells,24,25 up-regulation of NOX1 may be at leastpartly attributable to augmented PDGF signaling fol-lowing liver injury. In fact, the level of PDGF mRNAwas significantly elevated in the liver of BDL mice.Recently, NOX1 was reported to control autocrine cellgrowth of liver tumor cells through regulation of theepidermal growth factor receptor (EGFR) pathway.Targeted knockdown of NOX1 attenuated autocrinecell growth along with decreased EGFR expression andsignaling.32 In our primary cultured HSCs, increasedexpression of NOX1 mRNA was observed under se-rum starvation (Supporting Fig. 6A). However, therewas no difference in the level of EGFR between WTand Nox1KO HSCs (Supporting Fig. 6B). Accord-ingly, the EGFR pathway does not appear to take partin the increased expression of NOX1 and proliferationof HSCs.A significant increase in NOX2 mRNA was
observed in the liver of both genotypes by BDL. Inmice with NOX2 deficiency, chronic administration ofCCl4 elicited increased inflammation and hepatic ne-crosis, whereas fewer HSCs and less fibrosis were dem-onstrated compared with those in WT.14 Furthermore,ROS derived from NOX2 were recently reported toinduce collagen expression at the transcriptionallevel.15 In contrast, expression of collagen was unaf-fected in HSCs isolated from Nox1KO. Instead,NOX1-derived ROS augmented liver fibrosis by pro-moting the proliferation of activated HSCs. Thesefindings clearly indicated distinct roles for NOX1 andNOX2 in the pathogenesis of liver fibrosis. Why ROSderived from different NOX isoforms have diversefunctions remains to be determined.In the liver, NOX1 was not only expressed in
HSCs, but also in hepatocytes. To examine whetherthe expression of NOX1 was also affected in parenchy-mal cells in the liver, we exposed primary cultured he-patocytes to endogenous factors involved in the patho-genesis of BDL-induced liver injury. Although thelevel of NOX1 mRNA was unchanged in cells treatedwith bile acid, activation of caspase-3 induced by bile
Fig. 5. A cyclin-dependent kinase inhibitor, p27kip1, was increasedin HSCs isolated from Nox1KO. (A) Levels of p27kip1 and p21cip1
mRNAs in HSCs isolated from WT and Nox1KO. HSCs at day 14 in cul-ture were analyzed. N ¼ 5-8. *P < 0.05, versus WT. (B) Representa-tive immunoblots and quantitative densitometric analysis of p27kip1 inHSCs at day 14. N ¼ 4-5. **P < 0.01 versus WT.
HEPATOLOGY, Vol. 54, No. 3, 2011 CUI ET AL. 955

Fig. 7. The oxidized form of PTEN was decreased in HSCs isolated from Nox1KO. (A) Representative immunoblots and quantitative analysis ofthe oxidized form of PTEN. Protein lysate prepared in buffer containing iodoacetamide in the absence of b-mercaptoethanol and dithiothreitol(DTT) was fractionated by nonreducing SDS/PAGE. DTT was added to verify the specificity of the oxidized form. Bars represent fold change relativeto WT. N ¼ 4-6. *P < 0.05 versus WT. (B) Effects of the antioxidant N-acetylcysteine (NAC) on the oxidized form of PTEN in HSCs. After passageat day 7, cells were cultured in the medium containing NAC (5 mM). On day 14, cells were harvested for immunoblot analysis. Representativeimmunoblots and quantitative analysis are demonstrated. Bars represent fold change relative to control. N ¼ 3-4. **P < 0.01 versus WT group.(C) Representative immunoblots and quantitative analysis of phospho-PTEN. HSCs at day 14 in culture were analyzed. N ¼ 4-5 per group.
Fig. 6. The Akt/FOXO pathwaywas attenuated in HSCs isolatedfrom Nox1KO. (A) Representativeimmunoblots and quantitative anal-ysis of phospho-Akt, total Akt, andphospho-PI3K. (B) Representativeimmunoblots and quantitative anal-ysis of phospho-FOXO4 and totalFOXO4. HSCs at day 14 in culturewere analyzed. Bars represent foldchange relative to WT. N ¼ 4-5.**P < 0.01 versus WT. (C) Levelsof phospho-Akt and total Akt inHSCs cultured in the presence ofN-acetylcysteine (NAC). After pas-sage at day 7, cells were culturedin the medium containing NAC (5mM). On day 14, cells were har-vested for immunoblot analysis.Representative immunoblots andquantitative analysis are demon-strated. Bars represent fold changerelative to controls. N ¼ 3-4. *P< 0.05 versus WT.

acid was significantly suppressed by Nox1 deficiency.The data corroborated our in vivo findings, in that se-rum ALT and AST levels were significantly lower inNox1KO than those in WT after BDL. Because apo-ptosis of hepatocytes could directly activate HSCs,15
decreased apoptosis by Nox1 deficiency may possiblycontribute to the attenuation of fibrotic responses. Inthis context, the crosstalk between HSCs and hepato-cytes may be causally related to the development ofBDL-induced liver fibrosis.FOXO transcription factors, including FOXO1,
FOXO3, and FOXO4, are known to take part in cel-lular metabolism, proliferation, and survival.33
FOXO4, also known as AFX, directly binds to thepromoter region of the p27kip1 gene and stimulates itstranscription.27 In this study we demonstrated that thelevel of the phosphorylated, inactive form of FOXO4was significantly reduced in HSCs isolated fromNox1KO. This finding was coupled with the increasedexpression of p27kip1 in Nox1KO HSCs. Moreover,phosphorylation of Akt, which directly phosphorylatesFOXO transcription factors, was suppressed inNox1KO. These results suggested that Akt-mediatedphosphorylation of FOXO4 was instrumental in theregulation of the cell cycle in HSCs. On the otherhand, a previous study demonstrated that FOXO1 wasessential for the proliferation of HSCs.34 In HSCsused in this study, however, we could not detect thephosphorylated form of FOXO1 (data not shown).This may be due to the difference in culture
conditions or in species, because rat HSCs were usedin the previous study.PTEN, a tumor suppressor, is an inhibitor of PI3K/
Akt signaling by converting PIP3 to PIP2. The activityof PTEN is regulated by phosphorylation and oxida-tion. Phosphorylation of PTEN was reported to affectthe stability of protein, thus influencing its activity.29
Meanwhile, PTEN could be oxidized by ROS and forma stable intramolecular disulfide bond in the active site,which could negatively regulate the activity.30 Oxidized/inactivated PTEN could increase PIP3 levels, whichcould then recruit Akt and PDK1 to the plasma mem-brane.28 Once recruited to the plasma membrane, Aktis phosphorylated and activated by PDK1.35 In thepresent study the oxidized form of PTEN was signifi-cantly reduced, whereas no change in the phosphoryl-ated form was observed in HSCs isolated fromNox1KO. A recent study showed that oxidized PTENwas also decreased in colonic tissues of Nox1-deficientmice. In contrast to our findings, however, the phos-phorylated form of PTEN was concomitantly reducedin the colon of these mice.31 In any event, these dataendorse the concept that NOX1-derived ROS inactivatePTEN, thus enhancing the PI3K/Akt signaling cascadeto increase cell proliferation in different cell lineages.During the course of this study the involvement of
NOX1 in not only BDL-induced, but also CCL4-induced liver fibrosis was reported using Nox1-defi-cient mice of different origin.21 Increased mRNAexpression of a-SMA and col-1a induced by CCL4was significantly blunted by Nox1-deficiency. In con-trast, the levels of a-SMA and col-1a in the liver weresimilarly up-regulated in our Nox1KO treated withCCL4. The reason for such a discrepancy is presentlyunclear. It might be due to different sources of geneti-cally modified mice and/or experimental conditions. InPaik’s CCL4 model,21 more robust induction of a-SMA and col-1a mRNAs was demonstrated comparedwith our mice. Notably, the levels of a-SMA and col-1a were still markedly elevated in Nox1-deficient micetreated with CCL4 relative to those in vehicle-treatedcounterparts.21 Of interest in this regard, treatmentwith CCL4 consistently up-regulated a-SMA and col-1a mRNAs in Nox1-deficient mice of both origins. Inthe BDL model, accumulation of bile acid and mobili-zation of intestinal bacteria are the events initiatingfibrogenesis. Because bacterial endotoxin lipopolysac-charide (LPS) induces expression of NOX1 in macro-phage36 and in hepatocytes (data not shown), earlystimulation with LPS, together with PDGF, may up-regulate NOX1 and enhance liver injury and fibrosis.Portal myofibroblasts originated from HSCs and other
Fig. 8. A schematic model proposed for the role of NOX1/NADPHoxidase in liver fibrosis. Increased PDGF up-regulates NOX1 expres-sion. ROS derived from NOX1 oxidize and inactivate PTEN, whichresults in activation of the Akt/FOXO4 pathway to down-regulatep27kip1, a cell cycle suppressor. This cascade of events leads to prolif-eration of HSCs and ensuing fibrogenesis following liver injury.
HEPATOLOGY, Vol. 54, No. 3, 2011 CUI ET AL. 957

precursor cells, including vascular smooth muscle cells,have been documented to take part in the developmentof biliary fibrosis.37 The fact that NOX1 is expressed invascular smooth muscle cells9,23 further supports the rolefor NOX1 in the development of BDL-induced fibrosis.In conclusion, our findings highlight the importance
of NOX1 in the pathogenesis of BDL-induced liver fi-brosis by regulating the proliferation of HSCs. ROSderived from NOX1 inactivate PTEN to positively reg-ulate the Akt/FOXO4/p27kip1 signaling pathway,thereby accelerating the development of fibrosis follow-ing liver injury. Thus, NOX1 may become a novel ther-apeutic target for the treatment of chronic liver diseases.
References1. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:
209-218.
2. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology2008;134:1655-1669.
3. Burt AD. Pathobiology of hepatic stellate cells. J Gastroenterol 1999;34:299-304.
4. Friedman SL. The virtuosity of hepatic stellate cells. Gastroenterology1999;117:1244-1246.
5. Czaja MJ, Weiner FR, Flanders KC, Giambrone MA, Wind R, Biemp-ica L, et al. In vitro and in vivo association of transforming growth fac-tor-beta 1 with hepatic fibrosis. J Cell Biol 1989;108:2477-2482.
6. Friedman SL, Arthur MJ. Activation of cultured rat hepatic lipocytesby Kupffer cell conditioned medium. Direct enhancement of matrixsynthesis and stimulation of cell proliferation via induction of platelet-derived growth factor receptors. J Clin Invest 1989;84:1780-1785.
7. De Minicis S, Brenner DA. NOX in liver fibrosis. Arch Biochem Bio-phys 2007;462:266-272.
8. Loguercio C, Federico A. Oxidative stress in viral and alcoholic hepati-tis. Free Radic Biol Med 2003;34:1-10.
9. Bedard K, Krause KH. The NOX family of ROS-generating NADPH ox-idases: physiology and pathophysiology. Physiol Rev 2007;87:245-313.
10. Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M,et al. Nox1 is involved in angiotensin II-mediated hypertension: a studyin Nox1-deficient mice. Circulation 2005;112:2677-2685.
11. Ibi M, Matsuno K, Shiba D, Katsuyama M, Iwata K, Kakehi T, et al.Reactive oxygen species derived from NOX1/NADPH oxidase enhanceinflammatory pain. J Neurosci 2008;28:9486-9494.
12. Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, et al.NADPH oxidase signal transduces angiotensin II in hepatic stellate cellsand is critical in hepatic fibrosis. J Clin Invest 2003;112:1383-1394.
13. Choi SS, Sicklick JK, Ma Q, Yang L, Huang J, Qi Y, et al. Sustainedactivation of Rac1 in hepatic stellate cells promotes liver injury andfibrosis in mice. HEPATOLOGY 2006;44:1267-1277.
14. Aram G, Potter JJ, Liu X, Wang L, Torbenson MS, Mezey E. Deficiencyof nicotinamide adenine dinucleotide phosphate, reduced form oxidaseenhances hepatocellular injury but attenuates fibrosis after chronic car-bon tetrachloride administration. HEPATOLOGY 2009;49:911-919.
15. Jiang JX, Venugopal S, Serizawa N, Chen X, Scott F, Li Y, et al. Nico-tinamide adenine dinucleotide phosphate reduced oxidase 2 plays a keyrole in stellate cell activation and liver fibrogenesis in vivo. Gastroenter-ology 2010;139:1375-1384.
16. Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis inmice by inhibiting stellate cell proliferation and stimulating NK cellcytotoxicity. HEPATOLOGY 2006;44:1441-1451.
17. Cui W, Matsuno K, Iwata K, Ibi M, Katsuyama M, Kakehi T, et al.NADPH oxidase isoforms and anti-hypertensive effects of atorvastatin
demonstrated in two animal models. J Pharmacol Sci 2009;111:260-268.
18. Friedman SL. Molecular regulation of hepatic fibrosis, an integratedcellular response to tissue injury. J Biol Chem 2000;275:2247-2250.
19. Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, et al.Tumour necrosis factor alpha signalling through activation of Kupffercells plays an essential role in liver fibrosis of non-alcoholic steatohepa-titis in mice. Gut 2006;55:415-424.
20. Marra F, Romanelli RG, Giannini C, Failli P, Pastacaldi S, ArrighiMC, et al. Monocyte chemotactic protein-1 as a chemoattractant forhuman hepatic stellate cells. HEPATOLOGY 1999;29:140-148.
21. Paik YH, Iwaisako K, Seki E, Inokuchi S, Schnabl B, Osterreicher CH,et al. The nicotinamide adenine dinucleotide phosphate oxidase homo-logues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice.HEPATOLOGY 2011;53:1730-1741.
22. Friedman SL, Roll FJ. Isolation and culture of hepatic lipocytes,Kupffer cells, and sinusoidal endothelial cells by density gradient cen-trifugation with Stractan. Anal Biochem 1987;161:207-218.
23. Katsuyama M, Fan C, Yabe-Nishimura C. NADPH oxidase is involved inprostaglandin F2alpha-induced hypertrophy of vascular smooth muscle cells:induction of NOX1 by PGF2alpha. J Biol Chem 2002;277:13438-13442.
24. Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, et al.Novel gp91(phox) homologues in vascular smooth muscle cells: nox1mediates angiotensin II-induced superoxide formation and redox-sensi-tive signaling pathways. Circ Res 2001;88:888-894.
25. Katsuyama M, Fan C, Arakawa N, Nishinaka T, Miyagishi M, Taira K,et al. Essential role of ATF-1 in induction of NOX1, a catalytic subu-nit of NADPH oxidase: involvement of mitochondrial respiratorychain. Biochem J 2005;386:255-261.
26. Oga T, Matsuoka T, Yao C, Nonomura K, Kitaoka S, Sakata D, et al.Prostaglandin F(2alpha) receptor signaling facilitates bleomycin-inducedpulmonary fibrosis independently of transforming growth factor-beta.Nat Med 2009;15:1426-1430.
27. Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkheadtranscription factors mediate cell-cycle regulation by Ras and PKBthrough p27kip1. Nature 2000;404:782-787.
28. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1,dephosphorylates the lipid second messenger, phosphatidylinositol3,4,5-trisphosphate. J Biol Chem 1998;273:13375-13378.
29. Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylationof the PTEN tail regulates protein stability and function. Mol CellBiol 2000;20:5010-5018.
30. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inac-tivation of the tumor suppressor PTEN by H2O2. J Biol Chem 2002;277:20336-20342.
31. Coant N, Ben Mkaddem S, Pedruzzi E, Guichard C, Treton X, DucrocR, et al. NADPH oxidase 1 modulates WNT and NOTCH1 signalingto control the fate of proliferative progenitor cells in the colon. MolCell Biol 2010;30:2636-2650.
32. Sancho P, Fabregat I. NADPH oxidase NOX1 controls autocrinegrowth of liver tumor cells through up-regulation of the epidermalgrowth factor receptor pathway. J Biol Chem 2010;285:24815-24824.
33. Maiese K, Chong ZZ, Shang YC, Hou J. A ‘‘FOXO’’ in sight: target-ing Foxo proteins from conception to cancer. Med Res Rev 2009;29:395-418.
34. Adachi M, Osawa Y, Uchinami H, Kitamura T, Accili D, Brenner DA.The forkhead transcription factor FoxO1 regulates proliferation andtransdifferentiation of hepatic stellate cells. Gastroenterology 2007;132:1434-1446.
35. Downward J. Mechanisms and consequences of activation of proteinkinase B/Akt. Curr Opin Cell Biol 1998;10:262-267.
36. Kim JS, Yeo S, Shin DG, Bae YS, Lee JJ, Chin BR, et al. Glycogensynthase kinase 3beta and beta-catenin pathway is involved in toll-likereceptor 4-mediated NADPH oxidase 1 expression in macrophages.FEBS J 2010;277:2830-2837.
37. Dranoff JA, Wells RG. Portal fibroblasts: underappreciated mediatorsof biliary fibrosis. HEPATOLOGY 2010;51:1438-1444.
958 CUI ET AL. HEPATOLOGY, September 2011
Copyright © 2022 FDOKUMEN




















