
World Journal of Diabetes - BPG Management System
186
Published by Baishideng Publishing Group Inc World Journal of Diabetes World J Diabetes 2015 April 15; 6(3): 367-542 ISSN 1948-9358 (online)
-
Upload
khangminh22 -
Category
Documents
-
view
25 -
download
0
Transcript of World Journal of Diabetes - BPG Management System

Published by Baishideng Publishing Group Inc
World Journal of DiabetesWorld J Diabetes 2015 April 15; 6(3): 367-542
ISSN 1948-9358 (online)

EDITOR-IN-CHIEFLu Qi, BostonJingbo Zhao, Aalborg
STRATEGY ASSOCIATE EDITOR-IN-CHIEFUndurti Narasimha Das, Shaker HeightsMin Du, LaramieGregory I Liou, AugustaZhong-Cheng Luo, QuebecDemosthenes B Panagiotakos, Athens
GUEST EDITORIAL BOARD MEMBERSJuei-Tang Cheng, TainanChih-Hsung Chu, KaohsiungLow-Tone (Larry) Ho, TaipeiCheng-Cheng Hsiao, KeelungYung-Hsi Kao, TaoyuanChi Feng Liu, TaipeiShing-Hwa Liu, TaipeiWayne H-H Sheu, TaichungEing-Mei Tsai, KaohsiungChin-Hsiao Tseng, TaipeiYen Tzung-Hai, TaipeiChing-Shuang Wu, KaohsiungWei-Chung Vivian Yang, TaipeiWen-Chin Yang, Taipei
MEMBERS OF THE EDITORIAL BOARD
Argentina
Justo P Castaño, CordobaEduardo Spinedi, La Plata
Australia
Sof Andrikopoulos, Heidelberg HeightsHugh Russell Barrett, PerthBernhard T Baune, TownsvilleGrant Brinkworth, AdelaideLouise Janet Maple Brown, CasuarinaMelinda Therese Coughlan, MelbourneJosephine Maree Forbes, MelbournePaul A Fournier, PerthAngela Gialamas, Adelaide Mark Douglas Gorrell, NewtownGraeme Hankey, PerthAnandwardhan A Hardikar, MelbourneMichael Horowitz, AdelaideKarin Jandeleit-Dahm, MelbourneMartha Lappas, VictoriaPeter J Little, MelbourneXin Liu, BrisbaneDianna Josephine Magliano, CaufieldRobyn McDermott, AdelaideBeverly Sara Muhlhausler, AdelaideChristopher Nolan, CanberraLuciano Pirola, MelbourneMaryam Rashidi, VictoriaKarly Calliopi Sourris, VictoriaGreg Tesch, ClaytonJack Ronald Wall, PenrithOwen Llewellyn Woodman, Bundoora
AustriaChristian Heinz Anderwald, ViennaHelmuth Martin Borkenstein, GrazWalter Hermann Hörl, ViennaAlexandra Kautzky-Willer, Vienna
Friedrich Mittermayer, ViennaMarkus Paulmichl, SalzburgStefan Pilz, GrazGuntram Schernthaner, ViennaHarald Sourij, GrazThomas Michael Stulnig, ViennaLudwig Wagner, Vienna
Belgium
Giovanni Dapri, BrusselsChristophe De Block, AntwerpEkaterine Tskitishvili, LiegeF Andre Van Assche, LeuvenLuc F Van Gaal, Antwerp
Brazil
Monica Levy Andersen, Vila ClementinoClaudia RL Cardoso, Rio de JaneiroRicardo Vitor Cohen, São PauloMarcelo Correia, Rio de JaneiroCassyano Januario Correr, CuritibaMatheus Roriz Cruz, Porto AlegreCintia Chaves Curioni, Rio de JaneiroFreddy Goldberg Eliaschewitz, Rua GoiásRodrigo Jorge, Ribeirão PretoLuciana Ansaneli Naves, Asa NorteJúlio César Voltarelli, Ribeirão PretoBernardo L Wajchenberg, PinheirosJacqueline Nelisis Zanoni, Maringá
Canada
Jean-Luc Ardilouze, Sherbrooke
I
Editorial Board2011-2015
The World Journal of Diabetes Editorial Board now consists of 712 members, representing a team of worldwide experts in diabetes mellitus. They are from 56 countries, including Argentina (2), Australia (27), Austria (11), Belgium (5), Brazil (13), Canada (25), Chile (3), China (40), Cuba (1), Czech Republic (3), Denmark (16), Egypt (3), Finland (5), France (12), Germany (27), Greece (17), Hungary (4), India (28), Iran (8), Iraq (2), Ireland (3), Israel (10), Italy (56), Japan (30), Jordan (1), Kuwait (3), Lebanon (1), Malaysia (1), Malta (1), Mexico (4), Netherlands (9), New Zealand (3), Nigeria (2), Norway (2), Oman (3), Pakistan (2), Poland (7), Portugal (1), Qatar (1), Romania (2), Saudi Arabia (1), Singapore (4), Slovakia (1), South Africa (1), South Korea (15), Spain (24), Sweden (5), Switzerland (4), Thailand (4), Tunisia (1), Turkey (13), United Arab Emirates (3), United Kingdom (27), United States (213), Venezuela (1), and Yemen (1).
December 11, 2012WJD|www.wjgnet.com
World Journal of DiabetesW J D

Subrata Chakrabarti, LondonDavid Cherney, OntarioMervyn Deitel, TorontoJean-Pierre Després, QuebecDavid Joseph Hill, LondonTian-Ru Jin, TorontoArulmozhi D Kandasamy, EdmontonJennifer L Kuk, TorontoIsmail Laher, VancouverRoger S McIntyre, TorontoDavid Meyre, OntarioJoseph Fomusi Ndisang, SaskatoonRaj Padwal, AlbertaCiriaco A Piccirillo, MontrealRemi Rabasa-Lhoret, MontrealAM James Shapiro, EdmontonGuang Sun, St. John’sValerie Taylor, HamiltonCory Toth, CalgaryAndré Tremblay, MontréalVincent C Woo, WinnipegJames Roscoe Wright, CalgaryXi-Long Zheng, Calgary
Chile
Sebastian San Martin, ValparaisoArmando Rojas-Rubio, TalcaLuis Sobrevia, Santiago
China
Pang-Zeng Chang, QingdaoJie Chen, NanjingBernard Man Yung Cheung, Hong KongWilliam Chi-shing Cho, Hong KongTian-Pei Hong, BeijingQin Huang, ShanghaiPo Sing Leung, Hong KongChao Liu, NanjingJian-Kang Liu, Xi’anLie-Gang Liu, WuhanRonald Ching Wan Ma, Hong KongJin-Sheng Qi, ShijiazhuangWing Yee So, Hong KongCheuk Chun Szeto, Hong KongKathryn Tan, Hong KongCheng-Ming Wang, YangzhouCong-Yi Wang, WuhanYu Wang, Hong KongGuang-Da Xiang, WuhanBao-Feng Yang, HarbinShu-Yu Yang, FujianXi-Lin Yang, Hong KongZai-Qing Yang, WuhanShan-Dong Ye, HefeiShi-Sheng Zhou, DalianZhi-Guang Zhou, Changsha
Cuba
Luis Sarmiento-Pérez, Havana
Czech Republic
Martin Haluzik, Prague
Michal Krcma, PlzenTerezie Pelikanova, Prague
Denmark
Charlotte Brøns, GentofteJens Sandahl Christiansen, ArhusFlemming Dela, CopenhagenKristine Færch, GentofteErik L Grove, AarhusLouise Groth Grunnet, GentofteR Scott Heller, GentofteKurt Højlund, Odense CFilip K Knop, HellerupHelle Markholst, MåløvJens D Mikkelsen, CopenhagenOle Hartvig Mortensen, CopenhagenOluf Pedersen, CopenhagenEsben Thyssen Vestergaard, AarhusMilan Zdravkovic, Søborg
Egypt
Mamdouh Moawad Ali Hssan, CairoMoshira Abdel Hakim Rateb, CairoMona Farag Schaalan, Cairo
Finland
Siamak Bidel, HelsinkiGang Hu, HelsinkiThomas Kietzmann, OuluQing Qiao, HelsinkiKaroliina Wehkalampi, Helsinki
France
Jean Claude Ansquer, DijonBertrand Cariou, NantesSylvie Dejager, Rueil-MalmaisonNaim Akhtar Khan, DijonJean-Philippe Lavigne, NîmesMichel Marre, ParisMarie-Claude Morice, MassyRiccardo Perfetti, ParisGérard Said, ParisSophie Visvikis Siest, NancyDominique Simon, ParisDidier Vieau, Villeneuve d’Ascq
Germany
Ioanna Gouni Berthold, CologneChrista Buechler, RegensburgRoland Büttner, HeidelbergMichael Froehner, DresdenHammes Hans-Peter, MannheimNadj Herbach, MunichAndrea Icks, DüsseldorfThomas Jax, NeussUlrich Arthur Julius, DresdenMichael Kluge, MunichFlorian Lang, Tuebingen Matthias Laudes, KölnRalf Lobmann, Stuttgart
Rafael T Mikolajczyk, BremenAndreas Stefan Mueller, Halle (Saale)Karsten Müssig, TübingenNahid Parvizi, Neustadt am RübenbergeThomas Peter Reinehr, DattelnMichael Ristow, JenaSven Schinner, DuesseldorfPeter Egbert Hermann Schwarz, DresdenKonstantinos Stellos, TubingenOvidiu Alin Stirban, Bad OeynhausenDiego J Walther, BerlinSilvia Anette Wein, KielChristian Wrede, BerlinDan Ziegler, Düsseldorf
Greece
George P Chrousos, AthensMoses S Elisaf, IoanninaPanagiotis Georgoulias, LarissaNikolaos Kadoglou, ThessalonikiGerasimos E Krassas, KriniSpilios Manolakopoulos, AttikiNikolaos Papanas, AlexandroupolisDimitrios Papazoglou, AlexandroupolisSokratis Pastromas, AthensMelpomeni Peppa, AthensChristina Piperi, GoudiNicholas K Tentolouris, AthensKonstantinos A Toulis, SalonikaApostolos Tsapas, ThessalonikiKonstantinos Tziomalos, ThessalonikiElias Zintzaras, Thessaly
Hungary
Mária Bagyánszki, SzegedGyörgy Jermendy, BudapestKaroly Racz, BudapestGyula Soltesz, Pécs
India
Deepak Narayan Amrapurkar, MumbaiC V Anuradha, Tamil NaduSarika Arora, New DelhiPitchai Balakumar, SivakasiMuthuswamy Balasubramanyam, ChennaiSubhabrata Chakrabarti, HyderabadAbhay Sankar Chakraborti, KolkataTapan K Chaudhuri, New DelhiKanwaljit Chopra, ChandigarhMalabika Datta, DelhiDebidas Ghosh, West BengalRavinder Goswami, New DelhiPappachan M Joseph, KeralaJothydev Kesavadev, KeralaKVS Hari Kumar, LucknowAnoop Misra, New DelhiAnalava Mitra, KharagpurViswanathan Mohan, ChennaiS P Murthy, BangalorePallavi Panchu, GunturUsharani Pingali, HyderabadAmbady Ramachandran, Egmore ChennaiVadde Ramakrishna, Kadapa
II December 11, 2012WJD|www.wjgnet.com

III December 11, 2012WJD|www.wjgnet.com
Geetha Vani Rayasam, HaryanaRajat Sandhir, ChandigarhManju Sharma, New DelhiSuman Bala Sharma, DelhiTarun Sharma, Chennai
Iran
Mohammad Abdollahi, TehranMohammad Kazemi Arababadi, RafsanjanLeila Azadbakht, IsfahanHamid Baradaran, TehranBehrooz Broumand, TehranAhmad Esmaillzadeh, IsfahanMajid Ghayour-Mobarhan, Mashhad Mohsen Janghorbani, Isfahan
Iraq
Saad Abdul-Rahman Hussain, BaghdadAbbas Ali Mansour, Basrah
Ireland
Amar Agha, DublinMark Philip Hehir, DublinGerald H Tomkin, Dublin
Israel
Michael Aviram, HaifaGal Dubnov-Raz, Tel HashomerShimon Efrat, Tel AvivRaymond Elias Farah, SafedOren Froy, RehovotSaher Hamed, HaifaArid Nakhoul, HaifaOrit Pinhas-Hamiel, Tel HashomerHaim Werner, Tel AvivMarina Shargorodsky Zimlichman, Holon
Italy
Luigi Angrisani, NapoliMoschetta Antonio, BariAntonio Aversa, RomeRoberto Baldelli, RomeGiuseppe Barbaro, RomeAlessandro Bartolomucci, ParmaGiuseppina Basta, PisaSimona Bertoli, MilanoFederico Bilotta, RomeFabio Broglio, TorinoFrancesco G Chiarelli, ChietiSergio Coccheri, BolognaMassimo Collino, TorinoMarco Aristide Comaschi, GenoaRenzo Cordera, GenovaFrancesco Dotta, SienaGagliardini Elena, BergamoStefano Fiorucci, PerugiaMaurizio Galderisi, NaplesAmalia Gastaldelli, Pisa
Ezio Ghigo, TurinCarla Giordano, PalermoPaolo Gisondi, VeronaRiccarda Granata, TurinGiorgio Iervasi, PisaClaudia Kusmic, PisaCarmelo La Rosa, CataniaFrancesco Landi, RomeMonica Rosa Loizzo, Arcavacata RendePaolo Magni, MilanoMariano Malaguarnera, CataniaMelania Manco, RomePiero Marchetti, PisaMassimo Massi-Benedetti, PerugiaAntonio Nicolucci, ImbaroLucia Pacifico, RomeStefano Palomba, CatanzaroGiampaolo Papi, CarpiRenato Pasquali, BolognaPiermarco Piatti, MilanoDario Pitocco, RomeAntonio E Pontiroli, MilanoGiulio Marchesini Reggiani, BolognaGiuseppe Remuzzi, BergamoManfredi Rizzo, PalermoRaffaella Rosso, GenoaGiuseppe Schillaci, PerugiaLeonardo A Sechi, SassariImad Sheiban, TorinoCesare R Sirtori, MilanoGiovanni Tarantino, NaplesGiovanni Targher, VeronaDonadon Valter, PordenoneAlberto Verrotti, ChietiAndrea Viggiano, NapoliGianvincenzo Zuccotti, Milan
Japan
Masato Asahina, ChibaTakuya Awata, SaitamaYuichiro Eguchi, SagaGoji Hasegawa, KyotoSatoshi Inoue, TokyoEiji Ishimura, OsakaMasayuki Iwano, NaraTakashi Kadowaki, TokyoEisuke Kagawa, HiroshimaMasahito Katahira, AichiEiji Kawasaki, NagasakiNoriyuki Koibuchi, GunmaKazuhiko Kotani, TochigiDaisuke Koya, IshikawaNorikazu Maeda, OsakaTakayuki Masaki, OitaYuji Matsuzawa, OsakaKazuaki Nishio, TokyoKenji Okumura, NagoyaMotoaki Saito, YonagoToshiyasu Sasaoka, ToyamaMichio Shimabukuro, OkinawaKohzo Takebayashi, SaitamaHiroyuki Tamemoto, TochigiTakashi Togo, YokohamaJun Udagawa, IzumoYoshinari Uehara, FukuokaTakuya Watanabe, TokyoToshihiko Yada, Tochigi
Tohru Yorifuji, Osaka
Jordan
Yousef S Khader, Irbid
Kuwait
Kamal AA Sulaiman Al-Shoumer, KuwaitIbrahim Fadil Benter, SafatAbu Salim Mustafa, Kuwait
Lebanon
Ramzi F Sabra, Beirut
Malaysia
Mafauzy Mohamed, Kota Bharu
Malta
Charles Savona-Ventura, Msida
Mexico
Manuel González-Ortiz, GuadalajaraFernando Guerrero-Romero, DurangoJesus Alberto Olivares-Reyes, Mexico CityRocío Salceda, Mexico City
Netherlands
Sander Kersten, WageningenNanne Kleefstra, ZwolleEdwin Mariman, MaastrichtDon Poldermans, RotterdamFrançois Pouwer, TilburgHan Roelofsen, GroningenHendrik-Jan Schuurman, UtrechtSuat Simsek, AlkmaarMarcel Twickler, Bergen op Zoom
New Zealand
Paul Hofman, AucklandPeter E Lobie, AucklandElaine Rush, Auckland
Nigeria
Adejuwon A Adeneye, LagosAnthonia Okeoghene Ogbera, Lagos
Norway
Akhtar Hussain, OsloOdd Erik Johansen, Hovik
IV December 11, 2012WJD|www.wjgnet.com
Oman
Mohammed Al Shafaee, MuscatJumana S Saleh, MuscatRadha Shenoy, Muscat
Pakistan
Shahid Hameed, IslamabadJamil A Malik, Islamabad
Poland
Marcin Baranowski, BialystokJerzy Beltowski, LublinAlicia Hubalewska Dydejczyk, KrakowMaciej Owecki, PoznańEwa Pankowska, WarsawAgnieszka Piwowar, WroclawDorota Anna Zieba, Krakow
Portugal
M Graça Pereira, Braga
Qatar
Hong Ding, Doha
Romania
Elena Ganea, BucharestAdriana Georgescu, Bucharest
Saudi Arabia
J Fernando Arevalo, Caracas
Singapore
S Thameem Dheen, SingaporeYung Seng Lee, SingaporeDaniel Ng, SingaporeRob Martinus van Dam, Singapore
Slovakia
Katarína Šebeková, Bratislava
South Africa
Md Shahidul Islam, Durban
South Korea
Huneg-Sik Choi, GwangjuKyung Mook Choi, SeoulWon Mi Hwang, SeoulEui-Bae Jeung, Chungbuk
Ju-Hee Kang, IncheonSin Gon Kim, Seongbuk-GuSung-Jin Kim, SeoulYoung-Gyu Ko, SeoulKang-Beom Kwon, ChonbukMyung Gull Lee, BucheonSoo Lim, SeongnamByung-Hyun Park, JeonbukSeungjoon Park, SeoulKun-Ho Yoon, SeoulJeesuk Yu, Cheonan
Spain
Vivencio Barrios, MadridM Lusia Bonet, Palma de MallorcaManuel Vazquez Carrera, BarcelonaMaria Luz Martinez Chantar, DerioManuel Aguilar Diosdado, CádizJavier Espino, BadajozRicardo V García-Mayor, VigoJosé Manuel Gómez-Sáez, BarcelonaOreste Gualillo, Santiago de CompostelaJ Alfredo Martínez Hernández, PamplonaEmilio Herrera, MadridAmelia Marti, PamplonaMerce Miranda, TarragonaJF Navarro-González, Santa Cruz de TenerifeAlberto Ortiz, MadridMaria Javier Ramirez, PamplonaEugenia Resmini, BarcelonaPedro Romero-Aroca, ReusJordi Salas-Salvadó, ReusGines M Salido, CaceresVictor Sanchez-Margalet, SevilleHelmut Schröder, BarcelonaCarmen Segundo, CádizRafael Simó, Barcelona
Sweden
Joanna Hlebowicz, MalmöKaj S Stenlöf, GöteborgAnn-Britt Wiréhn, LinköpingWeili Xu, StockholmShao-Nian Yang, Stockholm
Switzerland
Kaspar Berneis, ZurichPascal Bovet, LausanneLuc Tappy, LausanneChristian Toso, Geneva
Thailand
Narattaphol Charoenphandhu, BangkokArthorn Riewpaiboon, BangkokRawee Teanpaisan, Hat-YaiViroj Wiwanitkit, Bangkok
Tunisia
Khaled Hamden, Sfax
Turkey
Ugur Cavlak, DenizliTeoman Dogru, AnkaraErsin Fadillioglu, AnkaraAbdurrahman Fatih Fidan, AfyonkarahisarMuammer Karadeniz, Bornova-IzmirCevdet Kaya, IstanbulFahrettin Kelestimur, KayseriAltan Onat, IstanbulSemir Ozdemir, AntalyaMustafa Şahin, AnkaraIlker Tasci, AnkaraBelma Turan, AnkaraSerap Yalın, Mersin
United Arab Emirates
Ernest Akingunola Adeghate, Al AinMukesh M Agarwal, Al AinSamir M Awadallah, Sharjah
United Kingdom
Nisreen Alwan, LeedsAmbika P Ashraf, BirminghamChen Bing, LiverpoolFay Crawford, EdinburghTim M Curtis, BelfastUmesh Dashora, HastingsGareth Davison, BelfastPeter Raymond Flatt, ColeraineKathleen M Gillespie, BristolPeter John Grant, LeedsLorna W Harries, ExeterNigel Hoggard, AberdeenNigel Irwin, ColeraineEdward Jude, LancashireAndreas F Kolb, AberdeenStefan Marciniak, CambridgeMoffat Joha Nyirenda, EdinburghJeetesh Patel, BirminghamSnorri Bjorn Rafnsson, EdinburghThozhukat Sathyapalan, YorkshireLatika Sibal, NewcastleRajagopalan Sriraman, LincolnRamasamyiyer Swaminathan, LondonAbd A Tahrani, BirminghamG Neil Thomas, BirminghamCecil Thompson, LondonPaul Henry Whiting, Leicester
United States
Varun Agrawal, SpringfieldMohamed Al-Shabrawey, AugustaPascale Alard, LouisvilleOmar Ali, MilwaukeeJudith Aponte, New YorkBalamurugan N Appakalai, MinneapolisHwyda A Arafat, PhiladelphiaCarl V Asche, Salt LakeSanford A Asher, PittsburghAnthony Atala, Winston-SalemSami Toufic Azar, Beirut

V December 11, 2012WJD|www.wjgnet.com
George Louis Bakris, ChicagoAlistair J Barber, HersheyDaniel C Batlle, ChicagoDavid SH Bell, BirminghamRita Bortell, WorcesterSebastien G Bouret, Los AngelesDavid Lloyd Brown, Stony BrookLu Cai, LouisvilleJack D Caldwell, ErieAnna C Calkin, Los AngelesRoberto A Calle, GrotonR Keith Campbell, PullmanCarlos Campos, New BraunfelsHeping Cao, New OrleansKrista Casazza, BirminghamAaron Brandon Caughey, PortlandEileen R Chasens, PittsburghMunmun Chattopadhyay, Ann ArborXiao-Li Chen, St PaulSheri Renee Colberg, NorfolkCraig Ian Coleman, HartfordRobert Russell Conley, IndianapolisColleen M Croniger, ClevelandDoyle M Cummings, GreenvilleWilliam C Cushman, MemphisPatricia Ann D’Amore, BostonPatricia Darbishire, West LafayetteGuillaume Darrasse-Jèze, New YorkRavi M Dasu, SacramentoMichael Harvey Davidson, ChicagoPrakash Deedwania, SanFranciscoHong-Wen Deng, Kansas CityTeresa P DiLorenzo, BronxScot E Dowd, LubbockSamuel C Durso, BaltimoreKrystal L Edwards, DallasAlexander M Efanov, IndianapolisAzza B El-Remessy, AugustaAmy Zhihong Fan, AtlantaMelissa Spezia Faulkner, TucsonGeorge S Ferzli, Staten IslandPaolo Fiorina, BostonJames Edward Foley, East HanoverSamuel N Forjuoh, TempleAlessia Fornoni, MiamiMartha M Funnell, Ann ArborTrudy Gaillard, ColumbusPietro Galassetti, IrvineClaudia Gragnoli, HersheyJennifer B Green, DurhamGary J Grover, PiscatawayAlok Kumar Gupta, Baton RougeWerner K Gurr, New HavenSamy L Habib, San AntonioAbdel Rahim Hamad, BaltimoreDaniel M Herron, New YorkTiffany Hilton, RochesterRaimund Hirschberg, TorranceMichael Francis Holick, BostonZhaoyong Hu, HoustonRachel Mary Hudacko, New BrunswickYasuo Ido, BostonBrian K Irons, LubbockPamela Itkin-Ansari, La JollaHieronim Jakubowski, NewarkHong-Lin Jiang, BlacksburgPing Jiao, ProvidenceShengkan Jin, PiscatawayArpita Kalla, St LouisRichard Evers Katholi, Springfield
Melina Rae Kibbe, ChicagoBhumsoo Kim, Ann ArborTomoshige Kino, BethesdaJulienne K Kirk, Winston-SalemRenu A Kowluru, DetroitLewis H Kuller, PittsburghRajesh Kumar, TempleBlandine Laferrère, New YorkSang Yeoup Lee, MayoCong-Jun Li, BeltsvilleChing-Shwun Lin, San FranciscoJulie Lin, BostonShuo Lin, Los AngelesPeter Lindgren, San DiegoJames F List, PrincetonDong-Min Liu, BlacksburgZhen-Qi Liu, CharlottesvilleGeorge William Lyerly, ConwayJian-Xing Ma, Oklahoma CityRong Ma, Fort WorthXin-Laing Ma, PhiladelphiaDavid Maggs, San DiegoKenneth Maiese, DetroitKevin C Maki, Glen EllynSridhar Mani, BronxSuresh Mathews, AuburnLauraar McCabe, East LansingSarah E Messiah, MiamiThomas O Metz, RichlandShannon A Miller, OrlandoMurielle Mimeault, OmahaRaghavendra G Mirmira, IndianapolisPrasun J Mishra, BethesdaReema Mody, GrayslakeArshag D Mooradian, JacksonvilleMohammad Reza Movahed, TucsonJames Mu, RahwayMuraleedharan G Nair, East LansingManuel F Navedo, SeattleCharles B Nemeroff, AtlantaJoshua J Neumiller, SpokaneSteven Nissen, ClevelandHirofumi Noguchi, Fort WorthCraig Nunemake, CharlottesvillePatrick J O’Connor, MinneapolisErin St Onge, ApopkaWei-Hong Pan, Baton RougeNaushira Pandya, Fort LauderdaleMichael R Peluso, CorvallisInga Peter, New YorkAxel Pflueger, RochesterGretchen A Piatt, PittsburghJohn D Piette, Ann ArborLeonid Poretsky, New YorkWalter J Pories, GreenvilleParviz M Pour, OmahaWei Qiao Qiu, BostonTeresa Quattrin, BuffaloCristina Rabadán-Diehl, BethesdaRajendra S Raghow, MemphisSwapnil Rajpathak, BronxArmin Rashidi, NorfolkMohammed S Razzaque, BostonBeverly A S Reyes, PhiladelphiaDavid Rodbard, PotomacHelena W Rodbard, RockvilleJune Hart Romeo, ClevelandRaul J Rosenthal, Fort LauderdaleJuan M Saavedra, BethesdaStephen W Schaffer, Mobile
Frank AJL Scheer, BostonRichard E Scranton, TivertonVallabh (Raj) O Shah, AlbuquerqueAziz Shaibani, HoustonJin-Xiong She, AugustaGuo-Ping Shi, BostonCarol Ann Shively, Winston-SalemAnders AF Sima, DetroitPramil N Singh, Loma LindaRajan Singh, Los AngelesJay S Skyler, MiamiDawn Smiley, AtlantaMatthew D Solomon, StanfordMark A Sperling, PittsburghRakesh K Srivastava, TylerBangyan Stiles, Los AngelesYu-Xiang Sun, HoustonSalim Surani, Corpus ChristiArthur L M Swislocki, MartinezYa-Xiong Tao, AuburnJohn A Tayek, TorranceJohn Gaylord Teeter, New HavenCarlos Marcelo Telleria, VermillionChristopher Gordon Thanos, ProvidenceRonald G Tilton, GalvestonSerena Tonstad, Loma LindaMichael Lawrence Traub, Staten IslandGuillermo E Umpierrez, AtlantaMargrit Urbanek, ChicagoVladimir N Uversky, IndianapolisGabriel I Uwaifo, Baton RougeVolker Vallon, San DiegoShambhu D Varma, BaltimoreMaria Virella, CharlestonHong-Jun Wang, BostonMark E Williams, BostonNathan D Wong, IrvineGuangyu Wu, New OrleansZhong-Jian Xie, San FrancisocoMing-Zhao Xing, BaltimoreHariom Yadav, BethesdaLijun Yang, GainesvilleRuojing Yang, RahwaySubhashini Yaturu, AlbanyJoseph Yeboah, CharlottesvilleDengping Yin, NashvilleYisang Yoon, RochesterYi-Hao Yu, New YorkKevin CJ Yuen, PortlandIan Stuart Zagon, HersheyRobert Yuk-Lung Zee, BostonCui-Lin Zhang, RockvilleJames Xuejie Zhang, RichmondSarah Zhang, OklahomaGuixiang Zhao, AtlantaYang Zhao, IndianapolisMing-Hui Zou, Oklahoma City
Venezuela
Fuad Lechin, Caracas
Yemen
Khaled Abdul-Aziz Ahmed, Ibb

-----=
FIELD OF VISION367 Ménage-à-trois of bariatric surgery, bile acids and the gut microbiome
Raghow R
REVIEW371 Telehealth interventions to reduce management complications in type 1 diabetes: A review
Balkhi AM, Reid AM, Westen SC, Olsen B, Janicke DM, Geffken GR
380 Type 1 diabetes: A predictable disease
Simmons KM, Michels AW
391 Nociception at the diabetic foot, an uncharted territory
Chantelau EA
403 Gut microbiota and Ma-Pi 2 macrobiotic diet in the treatment of type 2 diabetes
Fallucca F, Fontana L, Fallucca S, Pianesi M
412 Some of the experimental and clinical aspects of the effects of the maternal diabetes on developing
hippocampus
Hami J, Shojae F, Vafaee-Nezhad S, Lotfi N, Kheradmand H, Haghir H
423 Endothelial and platelet markers in diabetes mellitus type 2
Kubisz P, Stančiaková L, Staško J, Galajda P, Mokáň M
432 Diabetic neuropathic pain: Physiopathology and treatment
Schreiber AK, Nones CFM, Reis RC, Chichorro JG, Cunha JM
445 New-onset diabetes mellitus after kidney transplantation: Current status and future directions
Palepu S, Prasad GVR
456 Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus
Tangvarasittichai S
MINIREVIEWS481 Birth defects in pregestational diabetes: Defect range, glycemic threshold and pathogenesis
Gabbay-Benziv R, Reece EA, Wang F, Yang P
489 Diabetic retinopathy - ocular complications of diabetes mellitus
Nentwich MM, Ulbig MW
Contents Monthly Volume 6 Number 3 April 15, 2015
April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com I

500 Tyrosine isomers and hormonal signaling: A possible role for the hydroxyl free radical in insulin resistance
Molnár GA, Mikolás EZ, Szijártó IA, Kun S, Sélley E, Wittmann I
508 Interplay between Rab27a effectors in pancreatic β-cells
Yamaoka M, Ishizaki T, Kimura T
517 Eating disorders in adolescents with type 1 diabetes: Challenges in diagnosis and treatment
Pinhas-Hamiel O, Hamiel U, Levy-Shraga Y
ORIGINAL ARTICLE Prospective Study
527 Perception of difficulty and glucose control: Effects on academic performance in youth with type Ⅰ diabetes
Potts TM, Nguyen JL, Ghai K, Li K, Perlmuter L
SYSTEMATIC REVIEWS534 Effect of aerobic and anaerobic exercises on glycemic control in type 1 diabetic youths
Lukács A, Barkai L
ContentsWorld Journal of Diabetes
Volume 6 Number 3 April 15, 2015
April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com II

ContentsWorld Journal of Diabetes
Volume 6 Number 3 April 15, 2015
FLYLEAF
EDITORS FOR THIS ISSUE
Responsible Assistant Editor: Li Xiang Responsible Science Editor: Fang-Fang JiResponsible Electronic Editor: Su-Qing Liu Proofing Editorial Office Director: Xiu-Xia SongProofing Editor-in-Chief: Lian-Sheng Ma
NAME OF JOURNAL World Journal of Diabetes
ISSNISSN 1948-9358 (online)
LAUNCH DATEApril 15, 2010
FREQUENCY Monthly
EDITORS-IN-CHIEFLu Qi, MD, PhD, Assistant Professor, Department of Nutrition, Harvard School of Public Health, 665 Huntington Ave., Boston, MA 02115, United States
Jingbo Zhao, PhD, Associate Professor, Aalborg Hospital Science and Innovation Centre, Aalborg Hospital, Aarhus University Hospital, Aalborg 9000, Denmark
EDITORIAL OFFICEJin-Lei Wang, Director
Xiu-Xia Song, Vice DirectorWorld Journal of DiabetesRoom 903, Building D, Ocean International Center, No. 62 Dongsihuan Zhonglu, Chaoyang District, Beijing 100025, ChinaTelephone: +86-10-85381891Fax: +86-10-85381893E-mail: [email protected] Desk: http://www.wjgnet.com/esps/helpdesk.aspxhttp://www.wjgnet.com
PUBLISHERBaishideng Publishing Group Inc8226 Regency Drive, Pleasanton, CA 94588, USATelephone: +1-925-223-8242Fax: +1-925-223-8243E-mail: [email protected] Desk: http://www.wjgnet.com/esps/helpdesk.aspxhttp://www.wjgnet.com
PUBLICATION DATEApril 15, 2015
COPYRIGHT© 2015 Baishideng Publishing Group Inc. Articles published by this Open-Access journal are distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non-commercial and is otherwise in compliance with the license.
SPECIAL STATEMENT All articles published in journals owned by the Baishideng Publishing Group (BPG) represent the views and opin-ions of their authors, and not the views, opinions or policies of the BPG, except where otherwise explicitly indicated.
INSTRUCTIONS TO AUTHORSFull instructions are available online at http://www.wjgnet.com/1948-9358/g_info_20100107165233.htm
ONLINE SUBMISSION http://www.wjgnet.com/esps/
ABOUT COVER
April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com III
AIM AND SCOPE
Editorial Board Member of World Journal of Diabetes , Shan-Dong Ye, MD, Professor, Department of Endocrionology, Anhui Provincial Hospital, No. 17, Lujiang Road, Hefei 230001, Anhui Province, China
World Journal of Diabetes (World J Diabetes, WJD, online ISSN 1948-9358, DOI: 10.4239), is a peer-reviewed open access academic journal that aims to guide clinical practice and improve diagnostic and therapeutic skills of clinicians.
WJD covers topics concerning α, β, δ and PP cells of the pancreatic islet, the effect of insulin and insulinresistance, pancreatic islet transplantation, adipose cells and obesity.
We encourage authors to submit their manuscripts to WJD. We will give priority to manuscripts that are supported by major national and international foundations and those that are of great clinical significance.
World Journal of Diabetes is now indexed in PubMed Central, PubMed, Digital Object Identifier, and Directory of Open Access Journals.
I-V Editorial Board
INDEXING/ABSTRACTING

Rajendra Raghow
Rajendra Raghow, Department of Veterans Affairs Medical Center, Memphis, TN 38104, United StatesRajendra Raghow, Department of Pharmacology, University of Tennessee Health Science Center, Memphis, TN 38163, United StatesAuthor contributions: Raghow R solely wrote this paper.Conflict-of-interest: Rajendra Raghow declares that there is neither a conflict of interest with regard to the publication discussed in this FOV communication nor with respect to a commercial entity.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Rajendra Raghow, PhD, Professor, Department of Veterans Affairs Medical Center, 1030 Jefferson Avenue, Memphis, TN 38104, United States. [email protected]: +1-901-5238990 Fax: +1-901-5237274Received: June 13, 2014 Peer-review started: June 14, 2014 First decision: June 27, 2014 Revised: December 26, 2014 Accepted: January 9, 2015 Article in press: January 12, 2015Published online: April 15, 2015
AbstractBariatric surgeries have emerged as highly effective treatments for obesity associated type-2 diabetes mellitus. Evidently, the desired therapeutic endpoints such as rates of weight loss, lower levels of glycated hemoglobin and remission of diabetes are achieved more rapidly and last longer following bariatric surgery, as opposed to drug therapies alone. In light of these findings, it has been suspected that in addition to
causing weight loss dependent glucose intolerance, bariatric surgery induces other physiological changes that contribute to the alleviation of diabetes. However, the putative post-surgical neuro-hormonal pathways that underpin the therapeutic benefits of bariatric surgery remain undefined. In a recent report, Ryan and colleagues shed new light on the potential mechanisms that determine the salutary effects of bariatric surgery in mice. The authors demonstrated that the improved glucose tolerance and weight loss in mice after vertical sleeve gastrectomy (VSG) surgery were likely to be caused by post-surgical changes in circulating bile acids and farnesoid-X receptor (FXR) signaling, both of which were also mechanistically linked to changes in the microbial ecology of the gut. The authors arrived at this conclusion from a comparison of genome-wide, metabolic consequences of VSG surgery in obese wild type (WT) and FXR knockout mice. Gene expression in the distal small intestines of WT and FXR knockout mice revealed that the pathways regulating bile acid composition, nutrient metabolism and anti-oxidant defense were differentially altered by VSG surgery in WT and FXR-/- mice. Based on these data Ryan et al , hypothesized that bile acid homeostasis and FXR signaling were mechanistically linked to the gut microbiota that played a role in modulating post-surgical changes in total body mass and glucose tolerance. The authors’ data provide a plausible explanation for putative weight loss-independent benefits of bariatric surgery and its relationship with metabolism of bile acids.
Key words: Vertical sleeve gastrectomy; Bile acids; Farnesoid-X-receptor; Type-2 diabetes mellitus; Gut microbiome; Bariatric surgery
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: The report of Ryan et al , raises a number of questions with regard to the prevailing notion that
FIELD OF VISION
367 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Ménage-à-trois of bariatric surgery, bile acids and the gut microbiome
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.367
World J Diabetes 2015 April 15; 6(3): 367-370ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

mechanical restriction of the stomach and weight loss are the sole mechanisms that mediate the therapeutic effects of bariatric surgery. The authors showed that both lowering of body mass index and improved glucose tolerance after vertical sleeve gastrectomy (VSG) surgery were mechanistically linked to an altered composition of circulating bile acids and their ability to modulate farnesoid-X receptor (FXR) mediated signaling mechanisms. Additionally, it was observed that the wild type and FXR knockout mice, after receiving VSG surgery, were significantly different with respect to the make up of their gut microbiomes. Finally, the experiments revealed that the composition of gut microbiota in wild type VSG and FXR-/- VSG mice were highly correlated with their differential abilities to lose weight and acquire glucose tolerance.
Raghow R. Ménage-à-trois of bariatric surgery, bile acids and the gut microbiome. World J Diabetes 2015; 6(3): 367-370 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/367.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.367
COMMENTARY ON HOT TOPICSThe provocative mechanism proposed by Ryan et al[1], that mechanistically links bile acids, farnesoid-X receptor (FXR) nuclear receptor signaling and microbial ecology of the gut to therapeutic superiority of bariatric surgery over intensive drug therapies, opens up new avenues of clinical research that deserves serious consideration.
According to the World Health Organization (WHO), obesity associated type-2 diabetes mellitus (T2DM) and cardiovascular diseases represent a looming health-care crisis of the 21st century [WHO Global Infobase: data on overweight and obesity mean body mass index (BMI), healthy diets and physical inactivity; www.who.int/mediacenter/]. Traditionally, obesity, T2DM and their co-morbidities have been treated with anti-diabetic drugs combined with behavioral modification therapies (e.g., better nutrition and regular exercise). However, in recent years, a number of surgical interventions, that include Roux-en Y gastric bypass, vertical sleeve gastrectomy (VSG), laparoscopic adjustable gastric banding and bilio-pancreatic diversion, have emerged as highly effective treatments for obesity-associated diabetes[2].
There is a growing body of evidence to indicate that patients undergoing bariatric surgery, achieve desirable clinical endpoints more rapidly and for a longer duration compared with therapeutic benefits obtained with anti-diabetic drug therapy alone[2]. A recently completed, randomized clinical trial revealed that the superior post-surgical therapeutic benefits (e.g., improved glucose tolerance and lower BMI) of bariatric surgery over intensive drug treatment persisted for at least three years, when the study ended. Additionally, patients
receiving bariatric surgery not only achieved their main therapeutic goal of weight loss and alleviation of diabetes but also enjoyed a better quality of life, with fewer obesity-associated complications[3]. Similar beneficial effects of bariatric surgery have also been observed in rodent models of obesity[4,5]. The strikingly more effective therapeutic outcomes of surgical procedures over pharmacological and behavioral interventions in the treatment of obesity-associated diabetes represent an unsolved medical puzzle[6,7].
The hormonal and metabolic underpinnings of surgical interventions and how they differ from conven-tional drug therapy in shaping the anabolic and catabolic pathways are being actively pursued[6-8]. Obviously, much of this research is prompted by a desire to define the basic mechanisms involved in the clinical outcome of bariatric surgery. An additional motivation for such studies is the fact that bariatric surgery poses definite risks, and thus there is an urgent need for safer, less invasive therapies to assuage obesity associated diabetes and its complications. Investigations into the putative mechanisms that differentially regulate the therapeutic outcomes of bariatric surgery vs conventional drug therapies have led to somewhat conflicting findings[6,9,10]. According to the prevailing view, a reduced intake and absorption of macronutrients by surgically downsized stomach is responsible for the post-surgical insulin sensitivity and weight loss after bariatric surgery[2]. Although this scenario is generally supported by the data gathered from rodent models of obesity and in humans, this mechanism fails to explain the paradoxical finding that a substantial fraction of patients experienced remission of their diabetes long before they exhibited a significant weight loss[6,7]. This has led some investigators to posit that in addition to causing weight loss bariatric surgery also leads to other physiological changes in the gastrointestinal (GI) tract. One of the key pieces of evidence in support of this view includes post-surgical changes in circulating bile acids seen in both rodents and humans after bariatric surgery[11,12]. It has been suggested that simultaneous changes in the composition of bile acids and gut microbiota may be causally related to obesity and a loss of BMI[13,14]. A highly revealing study, carried out with human twins discordant for obesity, showed that obese and lean individuals had unique gut microbial ecology; transplantation of the fecal microbiota from the obese or lean patients conferred analogous BMI and adiposity to the germ-free mice[15]. Differences in body composition were correlated with differences in the metabolism of short chain fatty acids, branched chain amino acids, and altered bile acids and FXR signaling[15]. These tantalizing correlations notwithstanding, the underlying mechanism by which gut microbiota regulates total body weight and glucose tolerance remain to be fully elucidated.
The experiments of Ryan et al[1], reported in a recent issue of Nature, bring us a step closer to defining the molecular interactions that mediate the metabolic effects of VSG surgery in mice. Since bariatric surgery is known
Raghow R. Gut microbiome and bile acids
368 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

to enhance enterohepatic circulation of bile acids[16] that signal via nuclear FXR, Ryan et al[1] hypothesized that altered bile acid homeostasis and FXR signaling were mechanistically involved in the weight loss and glucose tolerance. To test this hypothesis, the authors compared the gene expression profiles in the distal small intestines of wild type (WT) and FXR knockout mice after VSG surgery. These analyses led to the discovery of “glutathione-mediated detoxification”, “nuclear receptors in lipid metabolism and toxicity”, “meta-pathway bio-transformation” and “type Ⅱ interferon signaling” pathways that were highly suggestive of a role of the gut microbiome in the metabolic changes elicited by VSG surgery.
The authors undertook an empirical investigation of this hypothesis. The WT and FXR-/- mice, kept on high fat diet to induce obesity, showed similar rates of body weight loss in the first week after surgery. However, the FXR-/- mice were compromised with respect to sustained (at 5 wk and after) losses of BMI and body fat. Interestingly, while the WT and FXR-/- mice consumed lower calories in the first week after surgery only the former continued to maintain their hypophagic behavior. The cumulative caloric intake by sham-operated and VSG FXR-/- mice were similar; both cohorts of animals consumed 15% higher than VSG WT mice. Thus, the authors concluded that the inability of FXR-/- animals for sustained loss of body weight and total body fat were a result of their feeding behavior. Since FXR-/- mice were also refractory to the metabolic benefits of VSG surgery, it became apparent that the metabolic outcomes of surgery were also influenced by the genotype of the mice. Nevertheless, VSG surgery had a more pronounced effect on the bacterial ecology of the gut than the absence of a functional FXR gene. Un-weighted UniFrac-based comparison of bacterial 16S rRNA data in sham-operated and VSG mice revealed that the population of Bacterioides was significantly reduced after VSG surgery in WT mice whose guts also showed greater abundance of Porphyromonadaceae and Roseburia. Genotype independent post-surgical changes in the populations of Lactobacilli and Lactococci in the guts were also observed. The abundance of Roseburia in the WT VSG correlated negatively with glucose intolerance. The abundance of Roseburia in the guts of sham operated and VSG FXR-/- was similar and was significantly lower than in WT VSG guts. Thus, the authors posited that a functional FXR signaling was involved in the mechanisms of VSG-induced weight loss, feeding behavior and insulin sensitivity.
The current study did not directly address the molecular basis of how FXR contributes to the meta-bolic consequences of VSG surgery. The authors acknowledged however[1], that in light of the inherent signaling complexity of bile acids that activate FXR as well as a G-protein coupled receptor, takeda G-protein-coupled receptor-5[16], the explanation of these data is unlikely to be straightforward. This sentiment is supported by the contradictory phenotype of FXR-/-
mice elicited in this study; thus, FXR knockout mice were resistant to some of the deleterious effects of high fat diet while remaining less responsive to the therapeutic benefits of VSG surgery. The apparently paradoxical phenotype of FXR-/- mice is likely to be caused by (1) unique tissue-specific mechanisms involved in the activation of FXR; and (2) a possible induction of compensatory mechanisms that result from a global knockout of FXR. The data contained in this paper also did not directly address the key question of how post-surgical changes in bile acids modulate the gut microbial ecology, and vice versa. Finally, a role of the gut microbiota in differentially regulating the metabolism and energetics of WT and FXR-/- mice could only be speculated upon in light of the limited information contained in this paper. It worth remembering however, that a number of earlier studies were driven by the hypothesis that obesity-associated microbiome was more efficient at extracting energy from lipids and carbohydrates[13,14]. The observed differences in the abundance of common fatty acids and related metabolites among the WT and FXR-/-
mice following VSG surgery were not sufficiently clear to warrant an unequivocal explanation. Nevertheless, the mechanism involving more efficient extraction of energy from macronutrients by the microbiota appears to be too simplistic in light of some recent data showing that changes in gut microbial ecology impinged on the signaling mechanisms underlying satiety and motility of the GI tract[17,18]. Despite these caveats, the experiments of Ryan et al[1], have broken a new ground in elucidating a functional connection of FXR signaling and microbial ecology with the metabolic consequences of bariatric surgery. These exciting findings deserve to be systematically validated and extended in humans with an aim to discover less invasive therapies to treat obesity and its complications.
REFERENCES1 Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P,
Myronovych A, Karns R, Wilson-Pérez HE, Sandoval DA, Kohli R, Bäckhed F, Seeley RJ. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 2014; 509: 183-188 [PMID: 24670636 DOI: 10.1038/nature13135]
2 Buchwald H, Estok R, Fahrbach K, Banel D, Jensen MD, Pories WJ, Bantle JP, Sledge I. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am J Med 2009; 122: 248-256.e5 [PMID: 19272486 DOI: 10.1016/j.amjmed.2008.09.041]
3 Schauer PR, Kashyap SR, Wolski K, Brethauer SA, Kirwan JP, Pothier CE, Thomas S, Abood B, Nissen SE, Bhatt DL. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N Engl J Med 2012; 366: 1567-1576 [PMID: 22449319 DOI: 10.1056/NEJMoa1200225]
4 Chambers AP, Jessen L, Ryan KK, Sisley S, Wilson-Pérez HE, Stefater MA, Gaitonde SG, Sorrell JE, Toure M, Berger J, D’Alessio DA, Woods SC, Seeley RJ, Sandoval DA. Weight-independent changes in blood glucose homeostasis after gastric bypass or vertical sleeve gastrectomy in rats. Gastroenterology 2011; 141: 950-958 [PMID: 21699789 DOI: 10.1053/j.gas-tro.2011.05.050]
5 Wilson-Pérez HE, Chambers AP, Ryan KK, Li B, Sandoval DA,
369 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Raghow R. Gut microbiome and bile acids

metabolism. Obesity (Silver Spring) 2009; 17: 1671-1677 [PMID: 19360006 DOI: 10.1038/oby.2009.102]
13 Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 2005; 102: 11070-11075 [PMID: 16033867 DOI: 10.1073/pnas.0504978102]
14 Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, Mehrabian M, Ursell LK, He A, Castellani LW, Zinker B, Kirby M, Drake TA, Drevon CA, Knight R, Gargalovic P, Kirchgessner T, Eskin E, Lusis AJ. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab 2013; 17: 141-152 [PMID: 23312289 DOI: 10.1016/j.cmet.2012.12.007]
15 Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013; 341: 1241214 [PMID: 24009397 DOI: 10.1126/science.1241214]
16 Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Mol Cell Endocrinol 2013; 368: 17-29 [PMID: 22609541 DOI: 10.1016/j.mce.2012.05.004]
17 Schéle E, Grahnemo L, Anesten F, Hallén A, Bäckhed F, Jansson JO. The gut microbiota reduces leptin sensitivity and the expression of the obesity-suppressing neuropeptides proglucagon (Gcg) and brain-derived neurotrophic factor (Bdnf) in the central nervous system. Endocrinology 2013; 154: 3643-3651 [PMID: 23892476 DOI: 10.1210/en.2012-2151]
18 Wichmann A, Allahyar A, Greiner TU, Plovier H, Lundén GÖ, Larsson T, Drucker DJ, Delzenne NM, Cani PD, Bäckhed F. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe 2013; 14: 582-590 [PMID: 24237703 DOI: 10.1016/j.chom.2013.09.012]
P- Reviewer: Bernhardt GA, Zhang ZM S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
Stoffers D, Drucker DJ, Pérez-Tilve D, Seeley RJ. Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like Peptide 1 receptor deficiency. Diabetes 2013; 62: 2380-2385 [PMID: 23434938 DOI: 10.2337/db12-1498]
6 Pories WJ. Bariatric surgery: risks and rewards. J Clin Endocrinol Metab 2008; 93: S89-S96 [PMID: 18987275 DOI: 10.1210/jc.2008-1641]
7 Rubino F, Cummings DE. Surgery: the coming of age of metabolic surgery. Nat Rev Endocrinol 2012; 8: 702-704 [PMID: 23147581 DOI: 10.1038/nrendo.2012.207]
8 Stefater MA, Wilson-Pérez HE, Chambers AP, Sandoval DA, Seeley RJ. All bariatric surgeries are not created equal: insights from mechanistic comparisons. Endocr Rev 2012; 33: 595-622 [PMID: 22550271 DOI: 10.1210/er.2011-1044]
9 Campos GM, Rabl C, Peeva S, Ciovica R, Rao M, Schwarz JM, Havel P, Schambelan M, Mulligan K. Improvement in peripheral glucose uptake after gastric bypass surgery is observed only after substantial weight loss has occurred and correlates with the magnitude of weight lost. J Gastrointest Surg 2010; 14: 15-23 [PMID: 19838759 DOI: 10.1007/s11605-009-1060-y]
10 Lima MM, Pareja JC, Alegre SM, Geloneze SR, Kahn SE, Astiarraga BD, Chaim EA, Geloneze B. Acute effect of roux-en-y gastric bypass on whole-body insulin sensitivity: a study with the euglycemic-hyperinsulinemic clamp. J Clin Endocrinol Metab 2010; 95: 3871-3875 [PMID: 20484482 DOI: 10.1210/jc.2010-0085]
11 Myronovych A, Kirby M, Ryan KK, Zhang W, Jha P, Setchell KD, Dexheimer PJ, Aronow B, Seeley RJ, Kohli R. Vertical sleeve gastrectomy reduces hepatic steatosis while increasing serum bile acids in a weight-loss-independent manner. Obesity (Silver Spring) 2014; 22: 390-400 [PMID: 23804416 DOI: 10.1002/oby.20548]
12 Patti ME, Houten SM, Bianco AC, Bernier R, Larsen PR, Holst JJ, Badman MK, Maratos-Flier E, Mun EC, Pihlajamaki J, Auwerx J, Goldfine AB. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid
370 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Raghow R. Gut microbiome and bile acids

Amanda M Balkhi, Adam M Reid, Sarah C Westen, Brian Olsen, David M Janicke, Gary R Geffken
Amanda M Balkhi, Adam M Reid, Sarah C Westen, David M Janicke, Gary R Geffken, Department of Clinical and Health Psychology, University of Florida, Gainesville, FL 32611, United StatesAmanda M Balkhi, Adam M Reid, Brian Olsen, Gary R Geffken, Division of Medical Psychology, Department of Psychiatry, University of Florida, Gainesville, FL 32611, United StatesGary R Geffken, Department of Pediatrics, University of Florida, Gainesville, FL 32611, United StatesAuthor contributions: All the authors equally contributed to this work.Conflict-of-interest: The authors have no conflict of interest to report.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Amanda M Balkhi, MS, Department of Clinical and Health Psychology, University of Florida, PO Box 100165, 1600 S Archer Rd, Gainesville, FL 32611, United States. [email protected]: +1-352-2658873Fax: +1-352-3766270Received: August 29, 2014Peer-review started: August 30, 2014First decision: November 27, 2014Revised: December 20, 2014Accepted: January 18, 2015 Article in press: January 20, 2015Published online: April 15, 2015
AbstractType 1 diabetes is a chronic illness with a high burden of care. While effective interventions and recommendations for diabetes care exist, the intensive nature of diabetes management makes compliance difficult. This is
especially true in children and adolescents as they have unique psychosocial and diabetes needs. Despite the development of effective in-person interventions targeting improving self-management and ameliorating psychosocial difficulties there are still a number of barriers to implementing these interventions, namely time, cost, and access. Telehealth interventions allow for the dissemination of these interventions to a broader audience. Self-management and psychosocial telehealth interventions are reviewed with a special emphasis on mobile phone and internet based technology use. While efficacy has been demonstrated in a number of telehealth interventions with improved cost effectiveness over in-person interventions, many challenges remain including high participant attrition and difficulties with receiving reimbursement for services rendered. These and other challenges are discussed with recommendations for researchers and telehealth providers provided.
Key words: Telehealth; Disease management; E-health interventions; Type 1 diabetes management; Type 1 diabetes
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Type 1 diabetes is a chronic illness with a high burden of care. Despite the development of effective in-person interventions, telehealth interventions are necessary to improve access to and engagement in interventions to improve diabetes management. Mobile phone and internet based interventions appear to have the most potential to enact change. Challenges and recommendations for these telehealth interventions are provided.
Balkhi AM, Reid AM, Westen SC, Olsen B, Janicke DM, Geffken GR. Telehealth interventions to reduce management complications in type 1 diabetes: A review. World J Diabetes 2015; 6(3): 371-379 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/371.htm DOI: http://dx.doi.
REVIEW
371 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Telehealth interventions to reduce management complications in type 1 diabetes: A review
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.371
World J Diabetes 2015 April 15; 6(3): 371-379ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

org/10.4239/wjd.v6.i3.371
INTRODUCTIONThe prevalence of chronic diseases, such as diabetes, cancer, cardiometabolic and respiratory conditions continues to pose a challenge for often overtaxed health care systems, requiring fundamental changes in the delivery and maintenance of patient care[1-4]. Telehealth (TH), defined as any medical activity involving an element of distance and use of a telecommunications strategy[5], represents an approach which may enable patients with chronic medical conditions to seek disease specific information and support[6-9], to be followed by clinicians more frequently and away from hospital settings[10-12], reduce healthcare costs[13], and to ultimately promote improved adherence to medical regimens resulting in improvement in health outcomes[14].
BRIEF HISTORY OF THWhile TH interventions began more than 50 years ago with closed-circuit television, research into TH interventions did not truly begin to accelerate until the 1990s with a dramatic increase in TH publications through the 2000s[15,16]. Initial TH interventions primarily emphasized providing the same care that would be provided in-person through an intermediary such as a closed circuit television or telephone. A majority of interventions that subsequently developed relied on direct telephone contact by nurses or skilled health care professionals or transmission of simple self-management data via a modem[15]. The primary strength of these early TH interventions was in providing care coordination with more frequent feedback and without an in-person visit, which resulted in cost savings and improved patient health[14]. As technology advanced, TH interventions did as well, moving to material presented through video phones, home computers, pre-programmed interactive problem solving programs, and mobile phone and internet based interventions[17,18]. The flexibility and cost effectiveness of TH makes it well suited to be used in the treatment of chronic illnesses such as diabetes.
Previous interventions have shown efficacy imple-menting TH for a variety of chronic conditions, including cancer, transplant recipients, heart failure, and chronic pulmonary disease[3,7-10,12]. These interventions have shown support for TH in providing condition specific education, social support, and self management assistance. In addition, this previous work has demonstrated the wide acceptability of TH and the ability for TH interventions to reach previously underserved populations.
NEED FOR TH IN TYPE 1 DIABETES Type 1 diabetes (T1D) is one of the most common chronic diseases in pediatrics in the United States and affects
more than 151000 youth under 20 years of age[19]. Poorlycontrolled diabetes poses many serious health com-plications thus optimal T1D management during childhood and adolescence is necessary to reduce negative health outcomes and improve life expectancy[19,20]. The mana-gement of T1D is a complex and challenging task that involves integration of daily medical tasks and lifestyle modifications. While demanding, the successful intensive management of T1D is associated with improved health outcomes and protections against complications that maintain for as many as 6-10 years following intensive management[21].
Children and adolescents with T1D have unique needs that dictate different standards of care than adults[22]. Despite parental involvement in diabetes management being common, non-adherence is especially high in the transition to and within adolescence, increasing the risk of immediate and future microvascular complications[23-28]. T1D management is further complicated by the social, emotional, and psychological demands of the disease[23]. Poor psychosocial wellbeing (e.g., depression, anxiety, stress) is related to poorer short and long term health outcomes due to suboptimal disease management[29-34]. Family functioning, parent wellbeing, and family cohesion have also been identified as an important contributor to diabetes control[35-44]. Therefore, when evaluating T1D management interventions, assessing and addressing the impact of patient and parent psychosocial wellbeing while being flexible and developmentally sensitive to the needs of the patient is essential to ensure that the intervention has a lasting impact.
TH IN T1D While previous in-person diabetes interventions have successfully targeted increasing patient knowledge[45-47], improving illness perception[48,49], fostering family communication and relationships[50,51], and advancing technological accuracy and ease of management devic-es[52-54], there are several remaining challenges to in-person interventions. Primary limitations of in-person interventions include poor ease of access for rural or underserved families, increased healthcare utilization costs, and poor attendance[55,56]. Additionally, individuals at greater risk for medical regimen nonadherence are likely to also be individuals who are at greater risk for not attending medical appointments[57], making traditional clinic based recruitment and interventions potentially ineffective. TH addresses many of the limitations of previous T1D interventions by providing a unique avenue for improving the management of T1D that is engaging, cost effective, and accessible[58].
SELF MONITORING AND EDUCATION INTERVENTIONSA hallmark of many TH interventions are to focus on providing education, improving self-monitoring through
Balkhi AM et al . Telehealth and management in diabetes
372 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

electronic check ins, and establishing more frequent communication with health care providers. While traditional phone interventions have demonstrated positive improvements in glycemic control and self-efficacy[59,60], the increased availability of smartphones and the internet facilitated further innovation and development. Deploying interventions on a mobile device, especially those compatible with text messaging, also proved effective in improving glycemic control in both adults and children[59,61-64]. These results suggestthat text messaging and intervention through mobile phones are a substantial area for outreach and inter-vention. However, despite the increased ownership of mobile phones among adults (91%) and adolescents (78%), there is still a substantial portion of individuals without a mobile phone or texting ability, especially among younger adolescents[65,66]. Additionally, some interventions using text messaging or smartphone applications in children or adolescents have not shown an ability to improve glycemic control, although secondary benefits such as increased adherence, communication or knowledge are generally noted[41,67-69]. As such, while mobile based interventions are promising, continued research into maximizing desired outcomes and cost-effectiveness is necessary.
Internet based interventions have the potential to overcome this limitation of mobile phone based interventions, because of the wide spread availability of the internet for adults (85%) and teens (95%) in the United States[66,70,71]. The internet may be especially appropriate for diabetes intervention, as one study suggests that 63.6% of parents of children with T1D use the internet to seek out diabetes information on their own[72]. For a child or adolescent with T1D, diabetes psychoeducation[73], problem solving vignettes[74], and physician monitoring of HbA1c and intervention[75-77], have all recently shown moderate to strong evidence of successfully improving glycemic control when imple-mented in an online environment. Similar results have been found with adults; however, most studies rely on adults with T2D[78-80]. Despite their demonstrated efficacy and the wide spread availability of the internet, the primary challenge that continues to plague internet based interventions is the decreased engagement and participation of users over time, with participant attrition rates of 11.5%-37% reported[74,78].
Notwithstanding the challenges in self-management interventions, these interventions have demonstrated effectiveness in multiple delivery modalities including voice calls, SMS/Text messaging, email, customized web portals, and video conferencing[61,81,82]. A systematic review revealed that telemedicine solutions for diabetes care are also feasible and acceptable to patients and providers[59]. Therefore, future research is necessary to integrate the previously proven delivery strategies with new technology that is engaging to users and cost-
effective.
PSYCHOSOCIAL AND SUPPORTIVE TH INTERVENTIONSOther TH interventions have strived to improve adherence by providing psychosocial support and decreasing family conflict around T1D management. One such method of intervention developed by Grey et al[73] bundled effective psychoeducation intervention with Coping Skills Training which improved glycemic control, quality of life, social acceptance, and self-efficacy which maintained for a year after beginning the online program (which was only 5 wk in duration). Self-efficacy has also been targeted as a potential area of psychosocial intervention, with online interventions demonstrating a significant positive impact on self-care activities[83]. Individual wellbeing has also been successfully addressed with web-based Cognitive Behavioral Therapy and peer mentoring[84,85]. Taken together, the existing psychosocial interventions for patients with T1D have shown success in engaging patients and improving psychological wellbeing, but are mixed on their abilities to minimize attrition and improve objective measures of glycemic control (i.e., HbA1c).
TH interventions have also been utilized to support the family and environment of patients with T1D. These interventions have successfully improved communication, improved HbA1c levels, and quality of life suggesting that targeting those supporting the individual with T1D (i.e., nurses, physicians, and family) may also be an effective way to improve T1D health outcomes[81]. There also appears to be awareness from family members and service providers of their need to find information and support regarding T1D care and a preference for online interventions[86]. One way to reach supporting individuals and patients may be to extend interventions to build on pre-existing online networks and supports. Social networks and forums for T1D have been qualitatively examined; despite concerns regarding the quality of the information presented on these sources, it is clear that patients and family members actively use these online sources (such as Facebook and online message boards) for diabetes information and social support[6,87-90]. Most notably, in one study 84.3% of caregivers that used online forums reported that their child’s care was impacted by information they encountered online[6]. Recent data also suggests that more than half of parents within a pediatric T1D clinic use the internet to seek out T1D information[72]. This identifies pre-existing internet sources as a potentially strong source for information dissemination but also as a potential venue for the unintended spread of misinformation. While prospective studies are needed to understand the association between parents’ use of online forums and their child’s glycemic control, these may be an appropriate area for
373 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Balkhi AM et al . Telehealth and management in diabetes

limited, federal, and reciprocal licenses, it is clear that the current system of licensure is hindering providers’ ability to reach out to patients who live in other states and steps need to be made to resolve these concerns[95].
PATIENT ENGAGEMENT AND ATTRITIONAttrition and noncompliance in TH interventions creates yet another barrier to successful establishment of TH interventions. As stated previously, attrition rates of 11.5%-37% among internet interventions have been reported [74,78]. However, patients who complete TH interventions may also not adhere adequately to the intervention, given the lack of in-person oversight. In a study conducted by Wangberg[83], participants were requested to repeatedly view and engage with online modules targeting self-efficacy and diabetes self-care, yet only 34% logged in more than twice to interact with the modules. Similarly, a review of TH adherence found a recurring theme in suboptimal frequency of uploading and submitting blood glucose values[96]. Given these relatively high drop-out rates and problems with noncompliance, TH programs should incorporate measures to improve adherence and keep the patients engaged in treatment.
To this end, some studies have shown improved adherence when the TH interventions are tailored toward the patients unique needs by using customized messages, programs, or personalized functions within the program[97]. For instance, a program may allow patients to use a data base of pictures of foods that have predetermined carbohydrate amounts instead of requiring the patient to estimate the carbohydrates for food they consume. Including features that communicated with patient’s preexisting diabetes technology (e.g., glucose monitors, insulin pumps) and automatically upload patients’ blood glucose levels in order for parents or providers to review and provide feedback may also be helpful. Overall, these programs should be easy to access and provide immediate feedback[98]. Programs should also take demographics into account. For instance, older individuals[99], women[100], and patients with higher self-efficacy[101,102], are more likely to adhere to internet interventions. Thus, providers who are developing or implementing TH interventions should work to determine which patients are going to be appropriate for the TH intervention and how to address those groups with a history of poor adherence to TH interventions[103].
PRIVACY AND SECURITYPossibly the most common TH concern relates to the ability for TH interventions to maintain patient privacy and security in a mobile or online environment. Appropriately managing personal health information (PHI) is an important piece to maintaining patients’ confidentiality. The federal laws provide regulations for protecting PHI under the Health Information Portability and Accountability Act[104] (HIPAA). Each state also has
future intervention.
ONGOING CHALLENGES IN THWhile research suggests that TH for patients with T1D can be a useful and effective method of improving glycemic control and overall adherence, there has been a significant delay in transitioning efficacious research interventions of T1D into community treatment settings. Chief among these issues are the financial feasibility and reimbursement for services delivered by skilled staff, creating and maintaining patient involvement in TH interventions while minimizing patient attrition, and ensuring patient safety, privacy, and legal accountability.
FINANCIAL CHALLENGESA key challenge that permeates across the literature in TH is the difficulty in obtaining reimbursement for services. The literature suggests that providers’ experience of receiving reimbursement varies signi-ficantly[91-93]. As of this publication only 15 states in the United States mandate coverage for TH services with 39 states providing at least some reimbursement for TH, although dramatically less so for behavioral TH despite behavioral TH’s appropriateness[94,95]. Additionally, recent studies have suggested that private third party reimbursement is improving across the board, though the trajectory of these improvements continues to be slow[93]. For example in 2005, 58% of TH programs received reimbursement for their services while in 2012, 45% of TH programs sought reimbursement and 81% of those reported receiving it[92,93]. Obtaining grant funding to offset these costs increases the institution’s ability to build a program[92]; however, if program personnel lack information about how to obtain reimbursement for their services from third party payers, the program may be discontinued after the grant funding has remitted[91,93]. This pattern of short term growth with long term discon-tinuation is concerning and hinders the growth of TH services.
The licensing and credentialing rules create another barrier to TH implementation. Similar to reimbursement regulations, there is a large discrepancy in state TH licensing laws. Current laws generally refer to the physical location of the patient as the place of service, regardless of the provider’s location. Moreover, state laws generally require that providers be licensed and credentialed at the place of service, making state lines a finite barrier to service delivery [91]. In 2011, Children’sMedical Services (CMS) began to allow institutions to accept the credentialing of the provider’s home institution instead of requiring the outside institution to put the provider through their credentialing process[91]. The CMS regulations show promise for expanding the credentialing requirements and may facilitate providers’ ability to reach patients who otherwise may not have been able to receive care. While efforts have been made to extend licensing adjustments by implementing
374 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Balkhi AM et al . Telehealth and management in diabetes

laws for managing PHI, which is not consistent from state to state and may be more or less stringent than HIPAA regulations[95]. When state privacy laws conflict with HIPAA, the general rule is that the provider follows the more stringent law[95]. TH providers who provide services across state lines must be aware of laws in both states and must work to resolve conflicts as they arise while being prepared for conflicting laws or regulations[105]. While managing these challenges across providers, technology technicians, nurses, medical assistants, and billing personnel in two states may be manageable with practice, negotiating differences among state and federal privacy laws is likely to be increasingly more difficult with each additional state, thereby deterring providers from expanding their services and possibly hindering patients’ access to specialized mental health care[95]. In addition, though more recent technological advances and using secure or closed networks have improved security of online data transmission[91,95] regulations are not clear regarding where or how the data should be stored (e.g., online, at the providers’ institution, or at the institution of the place of service). While providers should follow good safety procedures such as a personalized login, automatic time-out setting when not in use, encrypted data storage, and encrypted data transmission, they must also deliver informed consent and ensure a patient’s understanding of the potential and ever-evolving risks of transmitting health information over the internet or mobile networks[95,105,106]. These challenges require that a provider maintain understanding not only of the HIPAA and PHI laws but also of technological capabilities and challenges to technological safety, which is a daunting task for even the most informed provider.
CONCLUSION AND FUTURE DIRECTIONS IN THTaken together, research has shown the effectiveness and promise of TH in improving several primary (i.e., glycemic control, adherence) and secondary (i.e., social support, comorbidity, and knowledge) outcomes in T1D patients. Although a previous review suggested that phone interventions appear to have more promise than internet interventions in improving targeted T1D outcomes, the reviewed literature above suggests promise in both web based and mobile interventions. Broadly, TH interventions must strive to be theory driven, integrate multiple platforms, be secure, and be user-friendly[87,107,108]. In doing so, TH interventions will expand to a broader audience and have an improved chance at reaching those most in need for TH, the underserved individuals who have difficulty accessing traditional services.
In order to improve current TH interventions, resear-chers and providers should invest in portability. As mobile computing and mobile phones reach a larger and larger share of the United States[65] TH interventions must be optimized to provide an efficient, appealing, and
interactive environment on the smaller screen of mobile phones and tablets to reduce attrition and maintain participant engagement. This is especially true for adolescents and children, of whom a large portion (74%) access the internet from their phone and an increasing portion (25%) use their mobile phones as their primary device for navigating the web[109]. Researchers who do not adjust their TH interventions to take advantage of the increased computing power and accessibly of portable technology will continue to struggle with participant engagement and attrition as fewer and fewer young individuals use a traditional desktop computer.
Researchers and clinicians should also seek to integrate their TH interventions into existing technological infrastructure both to increase participant familiarity and ease of use. By intentionally creating interventions that integrate with diabetes technology (i.e., blood glucose monitors, insulin pumps), providers improve their ability to obtain objective health information and increase participant engagement through ease in integration. Integration, especially automated or hands free integration, with these technologies also has the benefit of providing a method for providers to easily view and provide feedback without relying on patients to physically produce their blood glucose meters at appointments. This improves the quality of information that providers have to deliver care, as well as decreases the burden of appointment preparation on the patient and potentially improving compliance.
In addition to technological integration, effective TH interventions should seek to involve family members, providers, and other supporting members of the patient’s T1D management team. An effective way to do this may be by building on the preexisting support networks targeting these individuals and developing ways to improve how individuals find, interoperate, and communicate the information they find online. Recent research has demonstrated that parents of children with T1D are especially likely to use these online sources and actively incorporate them into how they care for their child’s T1D[6,72]. Providers may benefit from this predisposition of parents to search for information online by designing and implementing procedures to inform users of appropriate sources of information and increase awareness of effective TH interventions that may provide similar information. Building atop of pre-existing resources may also reduce the infrastructure cost that contributes to the short term growth and long term discontinuation of existing TH interventions.
Finally, clinical care providers are encouraged to advocate for improved legislation regarding TH. Notwithstanding substantial improvement over the last decade regarding TH reimbursement and rules, there continues to be a lack of guidelines regarding TH interventions delivered by the mental health profession and the associated technological, privacy, and security issues created by these interventions. In fact, many of the privacy and security concerns related to TH interventions may only be feasibly addressed by policy
375 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Balkhi AM et al . Telehealth and management in diabetes

makers and technology manufacturers. The development and national recognition of TH guidelines in conjunction with improved licensure recognition across state lines may provide increased support to mental health pro-fessionals who wish to pursue TH interventions. As a part of these guidelines, providers and researchers are encouraged to pair with technology consultants whom are informed and educated both on technological advances and advances in privacy and security laws. With proper support and the development of structured guidelines, TH interventions can grow to fit within the evolving scope of health care policy, reimbursement, and technological advancement while reducing the number of individuals who are underserved.
REFERENCES1 Alwan A. Global status report on noncommunicable diseases 2010.
World Health Organization, 2011. Available form: URL: http: //www.who.int/nmh/publications/ncd_report_full_en.pdf
2 Chumbler NR, Kobb R, Harris L, Richardson LC, Darkins A, Sberna M, Dixit N, Ryan P, Donaldson M, Kreps GL. Healthcare utilization among veterans undergoing chemotherapy: the impact of a cancer care coordination/home-telehealth program. J Ambul Care Manage 2007; 30: 308-317 [PMID: 17873662 DOI: 10.1097/01.JAC.0000290399.43543.2e]
3 Dickinson R, Hall S, Sinclair JE, Bond C, Murchie P. Using technology to deliver cancer follow-up: a systematic review. BMC Cancer 2014; 14: 311 [PMID: 24885758 DOI: 10.1186/1471-2407-14-311]
4 Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 2010; 87: 4-14 [PMID: 19896746 DOI: 10.1016/j.diabres.2009.10.007]
5 Wootton R. Telemedicine. BMJ 2001; 323: 557-560 [DOI: 10.1136/bmj.323.7312.557]
6 Balkhi AM, Reid AM, McNamara JP, Geffken GR. The diabetes online community: the importance of forum use in parents of children with type 1 diabetes. Pediatr Diabetes 2014; 15: 408-415 [PMID: 24372986 DOI: 10.1111/pedi.12110]
7 Collie K, Kreshka MA, Ferrier S, Parsons R, Graddy K, Avram S, Mannell P, Chen XH, Perkins J, Koopman C. Videoconferencing for delivery of breast cancer support groups to women living in rural communities: a pilot study. Psychooncology 2007; 16: 778-782 [PMID: 17253594 DOI: 10.1002/pon.1145]
8 Head BA, Keeney C, Studts JL, Khayat M, Bumpous J, Pfeifer M. Feasibility and Acceptance of a Telehealth Intervention to Promote Symptom Management during Treatment for Head and Neck Cancer. J Support Oncol 2011; 9: e1-e11 [PMID: 21499540 DOI: 10.1016/j.suponc.2010.12.006]
9 Lounsberry JJ, Macrae H, Angen M, Hoeber M, Carlson LE. Feasibility study of a telehealth delivered, psychoeducational support group for allogeneic hematopoietic stem cell transplant patients. Psychooncology 2010; 19: 777-781 [PMID: 19653332 DOI: 10.1002/pon.1617]
10 Anker SD, Koehler F, Abraham WT. Telemedicine and remote management of patients with heart failure. Lancet 2011; 378: 731-739 [PMID: 21856487 DOI: 10.1016/S0140-6736(11)61229-4]
11 Meystre S. The current state of telemonitoring: a comment on the literature. Telemed J E Health 2005; 11: 63-69 [PMID: 15785222 DOI: 10.1089/tmj.2005.11.63]
12 Paré G, Poba-Nzaou P, Sicotte C. Home telemonitoring for chronic disease management: an economic assessment. Int J Technol Assess Health Care 2013; 29: 155-161 [PMID: 23514722 DOI: 10.1017/S0266462313000111]
13 Noel HC, Vogel DC, Erdos JJ, Cornwall D, Levin F. Home telehealth reduces healthcare costs. Telemed J E Health 2004; 10: 170-183 [PMID: 15319047 DOI: 10.1089/tmj.2004.10.170]
14 Jennett PA, Affleck Hall L, Hailey D, Ohinmaa A, Anderson C, Thomas R, Young B, Lorenzetti D, Scott RE. The socio-economic impact of telehealth: a systematic review. J Telemed Telecare 2003; 9: 311-320 [PMID: 14680514 DOI: 10.1258/135763303771005207]
15 Koch S. Home telehealth--current state and future trends. Int J Med Inform 2006; 75: 565-576 [PMID: 16298545 DOI: 10.1016/j.ijmedinf.2005.09.002]
16 Nickelson DW. Telehealth and the evolving health care system: Strategic opportunities for professional psychology. Professional Psychology: Research and Practice, 1998; 527-535 [DOI: 10.1037/0735-7028.29.6.527]
17 Kvedar J, Coye MJ, Everett W. Connected health: a review of technologies and strategies to improve patient care with telemedicine and telehealth. Health Aff (Millwood) 2014; 33: 194-199 [PMID: 24493760 DOI: 10.1377/hlthaff.2013.0992]
18 Perle JG, Langsam LC, Nierenberg B. Controversy clarified: an updated review of clinical psychology and tele-health. Clin Psychol Rev 2011; 31: 1247-1258 [PMID: 21963670 DOI: 10.1016/-j.cpr.2011.08.003]
19 Centers for Disease Control and Prevention. Diabetes report card 2012. Atlanta, GA: Centers for Disease Control and Prevention, US Department of Health and Human Services, 2012. Available from: URL: http: //www.cdc.gov/diabetes/pubs/pdf/diabetesreportcard.pdf
20 National Center for Chronic Disease Prevention and Health Promotion. National diabetes statistics report, 2014. 2014. Available from: URL: http: //www.cdc.gov/diabetes/pubs/statsreport14/national-diabetes-report-web.pdf
21 Daneman D. Type 1 diabetes. Lancet 2006; 367: 847-858 [PMID: 16530579 DOI: 10.1016/S0140-6736(06)68341-4]
22 Silverstein J, Klingensmith G, Copeland K, Plotnick L, Kaufman F, Laffel L, Deeb L, Grey M, Anderson B, Holzmeister LA, Clark N. Care of children and adolescents with type 1 diabetes: a statement of the American Diabetes Association. Diabetes Care 2005; 28: 186-212 [PMID: 15616254 DOI: 10.2337/diacare.28.1.186]
23 Harris MA, Hood KK, Mulvaney SA. Pumpers, skypers, surfers and texters: technology to improve the management of diabetes in teenagers. Diabetes Obes Metab 2012; 14: 967-972 [PMID: 22443170 DOI: 10.1111/j.1463-1326.2012.01599.x]
24 Kovacs M, Goldston D, Obrosky DS, Iyengar S. Prevalence and predictors of pervasive noncompliance with medical treatment among youths with insulin-dependent diabetes mellitus. J Am Acad Child Adolesc Psychiatry 1992; 31: 1112-1119 [PMID: 1429414 DOI: 10.1097/00004583-199211000-00020]
25 Rapoff M, Barnard M. Compliance with pediatric medical regimens. Cramer JA, Spilker B, editors. Patient compliance in medical practice and clinical trials. New York: Raven Press, 1991: 78-93
26 Anderson BJ, Holmbeck G, Iannotti RJ, McKay SV, Lochrie A, Volkening LK, Laffel L. Dyadic measures of the parent-child relationship during the transition to adolescence and glycemic control in children with type 1 diabetes. Fam Syst Health 2009; 27: 141-152 [PMID: 19630455 DOI: 10.1037/a0015759]
27 Ingerski LM, Anderson BJ, Dolan LM, Hood KK. Blood glucose monitoring and glycemic control in adolescence: contribution of diabetes-specific responsibility and family conflict. J Adolesc Health 2010; 47: 191-197 [PMID: 20638012 DOI: 10.1016/j.jadohealth.2010.01.012]
28 McNamara JPH, Reid AM, Freedland A, Righi S, Geffken GR. Contributing factors to poor adherence and glycemic control in pediatric type 1 diabetes: Facilitating a move toward telehealth. Liu C, editor. Type 1 Diabetes-Complications, Pathogenesis, and Alternative Treatments. Croatia: InTech Inc, 2011: 141-159 [DOI: 10.5772/24838]
29 McGrady ME, Laffel L, Drotar D, Repaske D, Hood KK. Depressive symptoms and glycemic control in adolescents with type 1 diabetes: mediational role of blood glucose monitoring. Diabetes Care 2009; 32: 804-806 [PMID: 19228870 DOI: 10.2337/dc08-2111]
30 Stewart SM, Rao U, Emslie GJ, Klein D, White PC. Depressive
376 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Balkhi AM et al . Telehealth and management in diabetes

symptoms predict hospitalization for adolescents with type 1 diabetes mellitus. Pediatrics 2005; 115: 1315-1319 [PMID: 15867041 DOI: 10.1542/peds.2004-1717]
31 Van Tilburg MA, McCaskill CC, Lane JD, Edwards CL, Bethel A, Feinglos MN, Surwit RS. Depressed mood is a factor in glycemic control in type 1 diabetes. Psychosom Med 2001; 63: 551-555 [PMID: 11485108 DOI: 10.1097/00006842-200107000-00005]
32 Delamater AM, Patiño-Fernández AM, Smith KE, Bubb J. Measurement of diabetes stress in older children and adolescents with type 1 diabetes mellitus. Pediatr Diabetes 2013; 14: 50-56 [PMID: 22913570 DOI: 10.1111/j.1399-5448.2012.00894.x]
33 Hassan K, Loar R, Anderson BJ, Heptulla RA. The role of socioeconomic status, depression, quality of life, and glycemic control in type 1 diabetes mellitus. J Pediatr 2006; 149: 526-531 [PMID: 17011326 DOI: 10.1016/j.jpeds.2006.05.039]
34 Herzer M, Hood KK. Anxiety symptoms in adolescents with type 1 diabetes: association with blood glucose monitoring and glycemic control. J Pediatr Psychol 2010; 35: 415-425 [PMID: 19684117 DOI: 10.1093/jpepsy/jsp063]
35 Cameron LD, Young MJ, Wiebe DJ. Maternal trait anxiety and diabetes control in adolescents with type 1 diabetes. J Pediatr Psychol 2007; 32: 733-744 [PMID: 17264087 DOI: 10.1093/jpepsy/jsl053]
36 Hilliard ME, Monaghan M, Cogen FR, Streisand R. Parent stress and child behaviour among young children with type 1 diabetes. Child Care Health Dev 2011; 37: 224-232 [PMID: 21083686 DOI: 10.1111/j.1365-2214.2010.01162.x]
37 Lewin AB, Geffken GR, Heidgerken AD, Duke DC, Novoa W, Williams LB, Storch EA. The diabetes family behavior checklist: A psychometric evaluation. J Clin Psychol Med Settings 2005; 12: 315-322 [DOI: 10.1007/s10880-005-7817-x]
38 Reid AM, Balkhi AM, St Amant J, McNamara JP, Silverstein JH, Navia L, Geffken G. Relations Between Quality of Life, Family Factors, Adherence, and Glycemic Control in Pediatric Patients With Type 1 Diabetes Mellitus. Children’s Health Care 2013; 42: 295-310 [DOI: 10.1080/02739615.2013.842455]
39 Maas-van Schaaijk NM, Roeleveld-Versteegh AB, Odink RR, van Baar AL. Behavioral Problems and Depressive Symptoms in Adolescents with Type 1 Diabetes Mellitus: Self and Parent Reports. Diabetes Mellitus-Insights And Perspectives, 2013: 47-58
40 Davis CL, Delamater AM, Shaw KH, La Greca AM, Eidson MS, Perez-Rodriguez JE, Nemery R. Parenting styles, regimen adherence, and glycemic control in 4- to 10-year-old children with diabetes. J Pediatr Psychol 2001; 26: 123-129 [PMID: 11181888 DOI: 10.1093/jpepsy/26.2.123]
41 Hauser ST, Jacobson AM, Lavori P, Wolfsdorf JI, Herskowitz RD, Milley JE, Bliss R, Wertlieb D, Stein J. Adherence among children and adolescents with insulin-dependent diabetes mellitus over a four-year longitudinal follow-up: II. Immediate and long-term linkages with the family milieu. J Pediatr Psychol 1990; 15: 527-542 [PMID: 2258799 DOI: 10.1093/jpepsy/15.4.527]
42 Mackey ER, Streisand R. Brief report: The relationship of parental support and conflict to physical activity in preadolescents with type 1 diabetes. J Pediatr Psychol 2008; 33: 1137-1141 [PMID: 18477630 DOI: 10.1093/jpepsy/jsn045]
43 Rosilio M, Cotton JB, Wieliczko MC, Gendrault B, Carel JC, Couvaras O, Ser N, Gillet P, Soskin S, Garandeau P, Stuckens C, Le Luyer B, Jos J, Bony-Trifunovic H, Bertrand AM, Leturcq F, Lafuma A, French Pediatric Diabetes Group PF. Factors associated with glycemic control. A cross-sectional nationwide study in 2,579 French children with type 1 diabetes. The French Pediatric Diabetes Group. Diabetes Care 1998; 21: 1146-1153 [PMID: 9653610 DOI: 10.2337/diacare.21.7.1146]
44 Rovner AJ, Mehta SN, Haynie DL, Robinson EM, Pound HJ, Butler DA, Laffel LM, Nansel TR. Perceived benefits, barriers, and strategies of family meals among children with type 1 diabetes mellitus and their parents: focus-group findings. J Am Diet Assoc 2010; 110: 1302-1306 [PMID: 20800121 DOI: 10.1016/j.jada.2010.06.010]
45 Heidgerken AD, Merlo L, Williams LB, Lewin AB, Gelfand K,
Malasanos T, Silverstein JH, Storch EA, Geffken G. Diabetes awareness and reasoning test: A preliminary analysis of development and psychometrics. Children’s Healthcare 2007; 36: 117-136 [DOI: 10.1080/02739610701334624]
46 Koontz MB, Cuttler L, Palmert MR, O’Riordan M, Borawski EA, McConnell J, Kern EO. Development and validation of a questionnaire to assess carbohydrate and insulin-dosing knowledge in youth with type 1 diabetes. Diabetes Care 2010; 33: 457-462 [PMID: 20007940 DOI: 10.2337/dc09-0390]
47 Murphy HR, Rayman G, Skinner TC. Psycho-educational interventions for children and young people with Type 1 diabetes. Diabet Med 2006; 23: 935-943 [PMID: 16922699 DOI: 10.1111/j.1464-5491.2006.01816.x]
48 Borus JS, Laffel L. Adherence challenges in the management of type 1 diabetes in adolescents: prevention and intervention. Curr Opin Pediatr 2010; 22: 405-411 [PMID: 20489639 DOI: 10.1097/MOP.0b013e32833a46a7]
49 Knight KM, Bundy C, Morris R, Higgs JF, Jameson RA, Unsworth P, Jayson D. The effects of group motivational interviewing and externalizing conversations for adolescents with Type-1 diabetes. Psychology, Health & Medicine 2003; 8: 149-157 [DOI: 10.1080/1354850031000087528]
50 Wysocki T, Harris MA, Buckloh LM, Mertlich D, Lochrie AS, Taylor A, Sadler M, Mauras N, White NH. Effects of behavioral family systems therapy for diabetes on adolescents’ family relationships, treatment adherence, and metabolic control. J Pediatr Psychol 2006; 31: 928-938 [PMID: 16401678 DOI: 10.1093/jpepsy/jsj098]
51 Wysocki T, Harris MA, Greco P, Bubb J, Danda CE, Harvey LM, McDonell K, Taylor A, White NH. Randomized, controlled trial of behavior therapy for families of adolescents with insulin-dependent diabetes mellitus. J Pediatr Psychol 2000; 25: 23-33 [PMID: 10826241 DOI: 10.1093/jpepsy/25.1.23]
52 Adolfsson P, Veijola R, Huot C, Hansen HD, Lademann JB, Phillip M. Safety and patient perception of an insulin pen with simple memory function for children and adolescents with type 1 diabetes--the REMIND study. Curr Med Res Opin 2012; 28: 1455-1463 [PMID: 22640459 DOI: 10.1185/03007995.2012.698258]
53 Clarke SF, Foster JR. A history of blood glucose meters and their role in self-monitoring of diabetes mellitus. Br J Biomed Sci 2012; 69: 83-93 [PMID: 22872934]
54 Korytkowski M, Bell D, Jacobsen C, Suwannasari R. A multicenter, randomized, open-label, comparative, two-period crossover trial of preference, efficacy, and safety profiles of a prefilled, disposable pen and conventional vial/syringe for insulin injection in patients with type 1 or 2 diabetes mellitus. Clin Ther 2003; 25: 2836-2848 [PMID: 14693308 DOI: 10.1016/S0149-2918(03)80337-5]
55 Griffin SJ. Lost to follow-up: the problem of defaulters from diabetes clinics. Diabet Med 1998; 15 Suppl 3: S14-S24 [PMID: 9829764 DOI: 10.1002/(SICI)1096-9136(1998110)15:3]
56 Shore JH, Brooks E, Savin DM, Manson SM, Libby AM. An economic evaluation of telehealth data collection with rural populations. Psychiatr Serv 2007; 58: 830-835 [PMID: 17535944 DOI: 10.1176/appi.ps.58.6.830]
57 Currie CJ, Peyrot M, Morgan CL, Poole CD, Jenkins-Jones S, Rubin RR, Burton CM, Evans M. The impact of treatment non-compliance on mortality in people with type 1 diabetes. J Diabetes Complications 2013; 27: 219-223 [PMID: 23157988 DOI: 10.1016/j.jdiacomp.2012.10.006]
58 Russell-Minda E, Jutai J, Speechley M, Bradley K, Chudyk A, Petrella R. Health technologies for monitoring and managing diabetes: a systematic review. J Diabetes Sci Technol 2009; 3: 1460-1471 [PMID: 20144402 DOI: 10.1177/193229680900300628]
59 Farmer A, Gibson OJ, Tarassenko L, Neil A. A systematic review of telemedicine interventions to support blood glucose self-monitoring in diabetes. Diabet Med 2005; 22: 1372-1378 [PMID: 16176199 DOI: 10.1111/j.1464-5491.2005.01627.x]
60 Howe CJ, Jawad AF, Tuttle AK, Moser JT, Preis C, Buzby M,
377 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Balkhi AM et al . Telehealth and management in diabetes

Murphy KM. Education and telephone case management for children with type 1 diabetes: a randomized controlled trial. J Pediatr Nurs 2005; 20: 83-95 [PMID: 15815568 DOI: 10.1016/j.pedn.2004.12.010]
61 Bin-Abbas B, Jabbari M, Al-Fares A, El-Dali A, Al-Orifi F. Effect of mobile phone short text messages on glycaemic control in children with type 1 diabetes. J Telemed Telecare 2014; 20: 153-156 [PMID: 24643953 DOI: 10.1177/1357633X14529244]
62 Franklin VL, Waller A, Pagliari C, Greene SA. A randomized controlled trial of Sweet Talk, a text-messaging system to support young people with diabetes. Diabet Med 2006; 23: 1332-1338 [PMID: 17116184 DOI: 10.1111/j.1464-5491.2006.01989.x]
63 Liang X, Wang Q, Yang X, Cao J, Chen J, Mo X, Huang J, Wang L, Gu D. Effect of mobile phone intervention for diabetes on glycaemic control: a meta-analysis. Diabet Med 2011; 28: 455-463 [PMID: 21392066 DOI: 10.1111/j.1464-5491.2010.03180.x]
64 Holtz B, Lauckner C. Diabetes management via mobile phones: a systematic review. Telemed J E Health 2012; 18: 175-184 [PMID: 22356525 DOI: 10.1089/tmj.2011.0119]
65 Duggan M. Cell Phone Activities 2013. Pew Research Internet Project. [Cited 2013-09-19]. Available from: URL: http: //pewinternet.org/Reports/2013/Cell-Activities.aspx
66 Lenhart A. Teens, smartphones & texting. Pew Research Internet Project. Cited 2012-03-19. Available from: URL: http: //www.pewinternet.org/2012/03/19/teens-smartphones-texting/
67 Cafazzo JA, Casselman M, Hamming N, Katzman DK, Palmert MR. Design of an mHealth app for the self-management of adolescent type 1 diabetes: a pilot study. J Med Internet Res 2012; 14: e70 [PMID: 22564332 DOI: 10.2196/jmir.2058]
68 Frøisland DH, Arsand E, Skårderud F. Improving diabetes care for young people with type 1 diabetes through visual learning on mobile phones: mixed-methods study. J Med Internet Res 2012; 14: e111 [PMID: 22868871 DOI: 10.2196/jmir.2155]
69 Louch G, Dalkin S, Bodansky J, Conner M. An exploratory randomised controlled trial using short messaging service to facilitate insulin administration in young adults with type 1 diabetes. Psychol Health Med 2013; 18: 166-174 [PMID: 22646659 DOI: 10.1080/13548506.2012.689841]
70 Fox S, Duggan M. The Diagnosis Difference. Pew Research Internet Project. Cited 2013-11-26. Available from: URL: http: //www.pewinternet.org/2013/11/26/the-diagnosis-difference/
71 Zickuhr K. Who’s Not Online and Why. Pew Research Internet Project. Cited 2013-9-25. Available from: URL: //www.pewinternet.org/2013/09/25/whos-not-online-and-why/
72 Balkhi AM, Olsen B, Lazaroe L, Silverstein J, Geffken GR. Managing Diabetes Online: A Clinic Based Prevalence Study of Internet Use in Parents of Children with Type 1 Diabetes. Washington, DC: American Psychological Association Annual Conference, 2014
73 Grey M, Whittemore R, Jeon S, Murphy K, Faulkner MS, Delamater A. Internet psycho-education programs improve outcomes in youth with type 1 diabetes. Diabetes Care 2013; 36: 2475-2482 [PMID: 23579179 DOI: 10.2337/dc12-2199]
74 Mulvaney SA, Rothman RL, Wallston KA, Lybarger C, Dietrich MS. An internet-based program to improve self-management in adolescents with type 1 diabetes. Diabetes Care 2010; 33: 602-604 [PMID: 20032275 DOI: 10.2337/dc09-1881]
75 Shalitin S, Ben-Ari T, Yackobovitch-Gavan M, Tenenbaum A, Lebenthal Y, de Vries L, Phillip M. Using the Internet-based upload blood glucose monitoring and therapy management system in patients with type 1 diabetes. Acta Diabetol 2014; 51: 247-256 [PMID: 23982170 DOI: 10.1007/s00592-013-0510-x]
76 Tildesley HD, Conway ME, Ross SA, Lee AM, Chan JH, Mazanderani AB, Tildesley HG, White AS. Review of the effect of internet therapeutic intervention in patients with type 1 and type 2 diabetes. Diabetes Care 2014; 37: e31-e32 [PMID: 24459161 DOI: 10.2337/dc13-1940]
77 Toma T, Athanasiou T, Harling L, Darzi A, Ashrafian H. Online social networking services in the management of patients with diabetes mellitus: systematic review and meta-analysis of
randomised controlled trials. Diabetes Res Clin Pract 2014; 106: 200-211 [PMID: 25043399 DOI: 10.1016/j.diabres.2014.06.008]
78 Azar M, Gabbay R. Web-based management of diabetes through glucose uploads: has the time come for telemedicine? Diabetes Res Clin Pract 2009; 83: 9-17 [PMID: 19056140 DOI: 10.1016/j.diabres.2008.09.055]
79 Glasgow RE, Boles SM, McKay HG, Feil EG, Barrera M. The D-Net diabetes self-management program: long-term implementation, outcomes, and generalization results. Prev Med 2003; 36: 410-419 [PMID: 12649049 DOI: 10.1016/S0091-7435(02)00056-7]
80 Ralston JD, Hirsch IB, Hoath J, Mullen M, Cheadle A, Goldberg HI. Web-based collaborative care for type 2 diabetes: a pilot randomized trial. Diabetes Care 2009; 32: 234-239 [PMID: 19017773 DOI: 10.2337/dc08-1220]
81 Izquierdo R, Morin PC, Bratt K, Moreau Z, Meyer S, Ploutz-Snyder R, Wade M, Weinstock RS. School-centered telemedicine for children with type 1 diabetes mellitus. J Pediatr 2009; 155: 374-379 [PMID: 19464030 DOI: 10.1016/j.jpeds.2009.03.014]
82 Hanauer DA, Wentzell K, Laffel N, Laffel LM. Computerized Automated Reminder Diabetes System (CARDS): e-mail and SMS cell phone text messaging reminders to support diabetes management. Diabetes Technol Ther 2009; 11: 99-106 [PMID: 19848576 DOI: 10.1089/dia.2008.0022]
83 Wangberg SC. An Internet-based diabetes self-care intervention tailored to self-efficacy. Health Educ Res 2008; 23: 170-179 [PMID: 17412717 DOI: 10.1093/her/cym014]
84 van Bastelaar KM, Pouwer F, Cuijpers P, Riper H, Snoek FJ. Web-based depression treatment for type 1 and type 2 diabetic patients: a randomized, controlled trial. Diabetes Care 2011; 34: 320-325 [PMID: 21216855 DOI: 10.2337/dc10-1248]
85 Suh S, Jean C, Koo M, Lee SY, Cho MJ, Sim KH, Jin SM, Bae JC, Kim JH. A randomized controlled trial of an internet-based mentoring program for type 1 diabetes patients with inadequate glycemic control. Diabetes Metab J 2014; 38: 134-142 [PMID: 24851207 DOI: 10.4093/dmj.2014.38.2.134]
86 Holtslander L, Kornder N, Letourneau N, Turner H, Paterson B. Finding straight answers: identifying the needs of parents and service providers of adolescents with type 1 diabetes to aid in the creation of an online support intervention. J Clin Nurs 2012; 21: 2419-2428 [PMID: 22889443 DOI: 10.1111/j.1365-2702.2012.04182.x]
87 Weymann N, Härter M, Dirmaier J. Quality of online information on type 2 diabetes: a cross-sectional study. Health Promot Int 2014; Epub ahead of print [PMID: 24688114 DOI: 10.1093/heapro/dau019]
88 Eysenbach G, Powell J, Kuss O, Sa ER. Empirical studies assessing the quality of health information for consumers on the world wide web: a systematic review. JAMA 2002; 287: 2691-2700 [PMID: 12020305 DOI: 10.1001/jama.287.20.2691]
89 Ravert RD, Hancock MD, Ingersoll GM. Online forum messages posted by adolescents with type 1 diabetes. Diabetes Educ 2003; 30: 827-834 [PMID: 15510534 DOI: 10.1177/014572170403000518]
90 Weitzman ER, Cole E, Kaci L, Mandl KD. Social but safe? Quality and safety of diabetes-related online social networks. J Am Med Inform Assoc 2011; 18: 292-297 [PMID: 21262920 DOI: 10.1136/jamia.2010.009712]
91 Soares NS, Langkamp DL. Telehealth in developmental-behavioral pediatrics. J Dev Behav Pediatr 2012; 33: 656-665 [PMID: 23027140 DOI: 10.1097/DBP.0b013e3182690741]
92 Antoniotti NM, Drude KP, Rowe N. Private payer telehealth reimbursement in the United States. Telemed J E Health 2014; 20: 539-543 [PMID: 24654748 DOI: 10.1089/tmj.2013.0256]
93 Whitten P, Buis L. Private payer reimbursement for telemedicine services in the United States. Telemed J E Health 2007; 13: 15-23 [PMID: 17309350 DOI: 10.1089/tmj.2006.0028]
94 Center for Teleheatlh and e-Health Law. Reimbursement Overview. 2011. Available from: URL: http: //www2.vgz.nl/sitecollectiondocuments/zakelijk/2011/d0456-201011_vgz_verg
378 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Balkhi AM et al . Telehealth and management in diabetes

collectief_eng.web.pdf95 Hyler SE, Gangure DP. Legal and ethical challenges in
telepsychiatry. J Psychiatr Pract 2004; 10: 272-276 [PMID: 15552552 DOI: 10.1097/00131746-200407000-00011]
96 Guljas R, Ahmed A, Chang K, Whitlock A. Impact of telemedicine in managing type 1 diabetes among school-age children and adolescents: an integrative review. J Pediatr Nurs 2014; 29: 198-204 [PMID: 24269308 DOI: 10.1016/j.pedn.2013.10.013]
97 Kaufman N. Internet and information technology use in treatment of diabetes. Int J Clin Pract Suppl 2010; (166): 41-46 [PMID: 20377663 DOI: 10.1111/j.1742-1241.2009.02277.x]
98 Franc S, Daoudi A, Mounier S, Boucherie B, Dardari D, Laroye H, Neraud B, Requeda E, Canipel L, Charpentier G. Telemedicine and diabetes: achievements and prospects. Diabetes Metab 2011; 37: 463-476 [PMID: 21889388 DOI: 10.1016/j.diabet.2011.06.006]
99 Japuntich SJ, Zehner ME, Smith SS, Jorenby DE, Valdez JA, Fiore MC, Baker TB, Gustafson DH. Smoking cessation via the internet: a randomized clinical trial of an internet intervention as adjuvant treatment in a smoking cessation intervention. Nicotine Tob Res 2006; 8 Suppl 1: S59-S67 [PMID: 17491172 DOI: 10.1080/14622200601047900]
100 Glasgow RE, Fisher L, Skaff M, Mullan J, Toobert DJ. Problem solving and diabetes self-management: investigation in a large, multiracial sample. Diabetes Care 2007; 30: 33-37 [PMID: 17192329 DOI: 10.2337/dc06-1390]
101 Steele R, Mummery WK, Dwyer T. Using the Internet to promote physical activity: a randomized trial of intervention delivery modes. J Phys Act Health 2007; 4: 245-260 [PMID: 17846455]
102 Wangberg SC, Bergmo TS, Johnsen JA. Adherence in Internet-based interventions. Patient Prefer Adherence 2008; 2: 57-65 [PMID: 19920945]
103 Armfield NR, Edirippulige SK, Bradford N, Smith AC. Telemedicine--is the cart being put before the horse? Med J Aust 2014; 200: 530-533 [PMID: 24835716 DOI: 10.5694/mja13.11101]
104 United States Department of Health and Human Services. Health Information Privacy. Available from: URL: http: //www.hhs.gov/ocr/privacy
105 Denton DR. Ethical and legal issues related to telepractice. Semin Speech Lang 2003; 24: 313-322 [PMID: 14722804 DOI: 10.1055/s-2004-815584]
106 Henriksen E, Burkow TM, Johnsen E, Vognild LK. Privacy and information security risks in a technology platform for home-based chronic disease rehabilitation and education. BMC Med Inform Decis Mak 2013; 13: 85 [PMID: 23937965 DOI: 10.1186/1472-6947-13-85]
107 El-Gayar O, Timsina P, Nawar N, Eid W. A systematic review of IT for diabetes self-management: are we there yet? Int J Med Inform 2013; 82: 637-652 [PMID: 23792137 DOI: 10.1016/j.ijmedinf.2013.05.006]
108 Nordfeldt S, Angarne-Lindberg T, Nordwall M, Ekberg J, Berterö C. As Facts and Chats Go Online, What Is Important for Adolescents with Type 1 Diabetes? PLoS One 2013; 8: e67659 [PMID: 23805322 DOI: 10.1371/journal.pone.0067659]
109 Madden M, Lenhart A, Duggan M, Cortesi A, Gasser U. Teens and Technology. Pew Research Internet Project, 2013. Available from: URL: http: //www.pewinternet.org/2013/03/13/teens-and-technology-2013/
P- Reviewer: Charoenphandhu N, Daltro C S- Editor: Tian YL L- Editor: A E- Editor: Liu SQ
379 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Balkhi AM et al . Telehealth and management in diabetes

Kimber M Simmons, Aaron W Michels
Kimber M Simmons, Aaron W Michels, Barbara Davis Center for Childhood Diabetes, University of Colorado Denver, Aurora, CO 80045, United StatesAuthor contributions: Simmons KS and Michels AW contributed to this paper.Supported by Grants from the National Institute of Diabetes and Digestive Kidney Diseases, No. R01 DK032083, K08 DK095995; Juvenile Diabetes Research Foundation, the Children’s Diabetes Foundation, and the Brehm Coalition.Conflict-of-interest: The authors have no conflicts-of-interest relevant to this manuscript.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Aaron W Michels, MD, Barbara Davis Center for Childhood Diabetes, University of Colorado Denver, Mail Stop A140, 1775 Aurora Court, Aurora, CO 80045, United States. [email protected]: +1-303-7241923 Fax: +1-303-7246784Received: September 11, 2014Peer-review started: September 11, 2014First decision: November 14, 2014Revised: November 26, 2014Accepted: January 9, 2015Article in press: January 12, 2015Published online: April 15, 2015
AbstractType 1 diabetes (T1D) is an autoimmune disease cha-racterized by loss of insulin producing beta cells and reliance on exogenous insulin for survival. T1D is one of the most common chronic diseases in childhood and the incidence is increasing, especially in children less than 5 years of age. In individuals with a genetic predisposition, an unidentified trigger initiates an abnormal immune response and the development of islet autoantibodies
directed against proteins in insulin producing beta cells. There are currently four biochemical islet autoantibodies measured in the serum directed against insulin, glutamic decarboxylase, islet antigen 2, and zinc transporter 8. Development of islet autoantibodies occurs before clinical diagnosis of T1D, making T1D a predictable disease in an individual with 2 or more autoantibodies. Screening for islet autoantibodies is still predominantly done through research studies, but efforts are underway to screen the general population. The benefits of screening for islet autoantibodies include decreasing the incidence of diabetic ketoacidosis that can be life threatening, initiating insulin therapy sooner in the disease process, and evaluating safe and specific therapies in large randomized clinical intervention trials to delay or prevent progression to diabetes onset.
Key words: Autoimmunity; Autoantibodies; Diabetes prevention; Screening; Type 1 diabetes
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Type 1 diabetes (T1D), the immune mediated form of diabetes, is now a predictable disease with the measurement of islet autoantibodies. The presence of two or more antibodies defines preclinical disease as nearly everyone with multiple antibodies progresses to clinical diabetes. With improved platforms to measure islet autoantibodies, screening the general population is now a goal. Early identification of preclinical diabetes allows for less diabetic ketoacidosis, early initiation of insulin therapy, and the potential to delay or prevent diabetes onset. Clinical trials using safe and specific therapies to block disease specific immune cells are underway in T1D.
Simmons KM, Michels AW. Type 1 diabetes: A predictable disease. World J Diabetes 2015; 6(3): 380-390 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/380.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.380
REVIEW
380 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Type 1 diabetes: A predictable disease
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.380
World J Diabetes 2015 April 15; 6(3): 380-390ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

INTRODUCTIONType 1 diabetes (T1D) is a chronic disease caused by immune-mediated destruction of insulin producing beta cells in the pancreas[1]. The destruction of beta cells results in insulin insufficiency, and patients develop life-threatening hyperglycemia that clinically manifests with weight loss, polyuria, and polydipsia. The majority of patients who develop T1D have high-risk human leukocyte antigen (HLA) genes. Islet autoantibodies can be measured in the serum of these high-risk individuals years before the onset of any clinical symptoms, making T1D a predictable disease. Multiple prevention trials in patients with high-risk HLA genes or in patients who have measureable autoantibodies have been completed. To date, no trial has prevented the onset of T1D, but data indicates that the disease process may be delayed by administering oral insulin to induce insulin specific regulatory T-cells in the gut, resulting in decreased inflammation in the pancreas. This review summarizes the epidemiology, risk factors and pathogenesis of T1D. The review also examines the goal of screening the general population for T1D risk and preventing disease onset in individuals with preclinical disease.
EPIDEMIOLOGYT1D is one of the most common chronic diseases in childhood and is diagnosed at an increasing rate in adults. The incidence rate varies significantly by geographical region. Sweden, Finland, Norway, United Kingdom, and Sardinia have the highest incidence of T1D at an age-adjusted rate of > 20/100000 patient years. For comparison, the United States has an incidence rate of 17.8/100000 patient years in a predominantly Caucasian population. China and South America have the lowest incidence of T1D, reported as < 1/100000 patient years[2-5]. The rate of T1D diagnosis is increasing in most countries, with rates dramatically increasing in children less than 5 years of age[6]. The annual incidence of T1D is increasing globally by 2.3% per year and is estimated to be increasing by 2.7%-2.8% in non-Hispanic white youth in the United States[7]. Large registries in both Europe and the United States show that the incidence of T1D peaks between 5 to 7 years of age and again when children enter puberty[8]. Unlike most autoimmune diseases, T1D is more common in males than females. The risk of T1D development in the general population is 1:300[9]. In children who have a genetically related sibling, the risk is increased to 1:7 and is greatest in children under 5 years of age[10,11]. Offspring of mothers with T1D carry approximately 3% risk and offspring of fathers with T1D carry approximately 5% risk[12]. Genetics confer risk for development of T1D, as does seasonal variation and birth month suggesting an environmental influence on disease pathogenesis. Children born in the spring tend to be at a greater risk for developing T1D, while diagnosis is increased during climatically cold
seasons[13-16]. This is an epidemiological association that requires further investigation.
RISK FACTORSGeneticT1D is a polygenic disorder with many genes contri-buting varying amounts of genetic risk for disease development. The genes conferring risk for diabetes are generally classified as HLA and non-HLA genes. Large genome wide association studies show that over 40 genes increase susceptibility to T1D[17,18]. The major determinant of genetic susceptibility to T1D, contributing greater than 50% of the genetic risk, is conferred by genes in the HLA complex located on chromosome 6[9]. The HLA complex is divided into 3 regions: classes Ⅰ, Ⅱ, and Ⅲ. Alleles of the class Ⅱ genes, DQ and DR (and to a lesser extent DP), are the most important determinants of T1D. These class Ⅱ molecules are expressed on antigen-presenting cells (macrophages, dendritic cells, and B cells) and present antigens to CD4 T lymphocytes. DQ and DR genes are in close linkage disequilibrium on chromosome 6 with specific DQ and DR genes inherited together. The presence of the DR4/DQ8 haplotype increases the odds ratio for T1D development to approximately 11, indicating an individual with this haplotype is 11 times more likely to develop T1D than those without. Approximately 90% of all individuals with T1D have either or both the DR4/DQ8 or DR3/DQ2 haplotypes. Interestingly, HLA genes also confer protection from T1D development. Individuals who have the specific DQ6 allele (DQB1*06:02) are dominantly protected from T1D, with an odds ratio of 0.03 for disease development[19].
Of the non-HLA genes, insulin and protein tyrosine phosphatase non-receptor type 22 (PTPN22) confer risk for T1D development but to lesser degrees than HLA genes[20]. Similar to HLA class Ⅱ genes, insulin gene polymorphisms can confer both susceptibility to and protection from T1D development. At the 5’ end of the insulin gene, there are variable numbers of tandem repeats. Having more repeats correlates to more insulin message being expressed in the thymus. The thymus responds by developing central tolerance to insulin. In individuals with fewer repeats, autoreactive T-cells can persist, and the risk for T1D development is increased[21]. PTPN22 helps regulate antigen receptor signaling and T cell activation, and a single nucleotide polymorphism (arginine to tryptophan at position 620) has been associated with a number of autoimmune disorders including T1D. A gain of function polymorphism decr-eases T cell receptor signaling which confers diabetes risk. It is unknown why decreased T cell activation leads to T1D risk, but it can be hypothesized that deficient negative selection of thymic cells may be involved[22,23].
EnvironmentGenetics alone does not lead to T1D; the environment also plays a pivotal role. This is evidenced by the fact
Simmons KM et al . Type 1 diabetes: A predictable disease
381 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

that not all individuals with high-risk genes develop T1D. In fact, the majority of individuals with high-risk HLA class Ⅱ genes (DR4/DQ8 and DR3/DQ2) do not develop T1D. There are likely one or more environmental factors that trigger and perpetuate the autoimmune disease process prior to hyperglycemia and a clinical diagnosis of hyperglycemia and T1D. Large natural history studies indicate that the development of islet autoantibodies (the first laboratory evidence of beta cell autoimmunity) in high-risk individuals often occurs between 9 mo and 2 years of age[24]. This suggests that an environmental trigger is present early in life, possibly in utero.
One of the most extensively evaluated environmental triggers is viral infection. Many viruses are implicated in the development of T1D including enteroviruses such as coxsackie B virus, cytomegalovirus, congenital rubella syndrome, and rotavirus[25-32]. Enterovirus is the leading candidate for contributing to T1D development. Epidemiologic studies in Finland show that the develop-ment of beta cell autoimmunity parallels the seasonal pattern of enterovirus infection and clinical symptoms of enteroviral infection[33,34]. Enterovirus infection was strongly associated with the development of autoantibodies in the Diabetes Autoimmunity Study in the Young (DAISY) cohort[35]. Laboratory evidence of enterovirus infection is reproducibly present in individuals with new onset T1D, pregnant women whose children develop T1D, and donor pancreata of individuals with T1D[36,37]. The exact mechanism of how viruses induce autoimmunity is not clear. The molecular mimicry hypothesis proposes that because the P2-C protein sequence of enterovirus is similar to glutamic decarboxylase (GAD), which is expressed in islet cells, the immune system erroneously targets destruction of beta cells[38]. The other leading hypothesis is that viral infection activates autoreactive T cells. As evidence, Cytomegalovirus B4 has tropism for pancreatic tissue and infection results in release of beta cell antigens that are phagocytized by macrophages and presented to autoreactive T cells[39].
Another potential environmental influence relates to the north-south division of diabetes development in the world, with a higher incidence of T1D in northern climates compared to southern. The north-south hypothesis implicates that a lack of vitamin A and/or D exposure early in life predisposes individuals to the development of autoimmune diseases including T1D. Offspring of mothers supplemented with vitamin D during pregnancy and young children supplemented with vitamin D have shown a reduced risk of T1D development that may be dose responsive[40,41]. However, an analysis from the DAISY Study found that vitamin D intake and 25(OH) vitamin D levels throughout childhood were not associated with the development of islet autoantibodies or T1D development[42].
Early introduction of cow’s milk and gluten have also been extensively studied. The introduction of gluten into an infant’s diet prior to three months and after 7 mo has been associated with increased auto-antibody development[24,43]. Some studies also indicate
that breastfeeding or using elemental formula may be protective against T1D. Other environmental factors that continue to be explored include nitrosamine compounds, maternal age, pre-eclampsia, and childhood obesity. There is no evidence to suggest that vaccines increase the risk of T1D development[44]. To date, there are no causal environmental factors that trigger the develop-ment of islet autoantibodies or increase the risk of progression to clinical T1D development. However, there is a large international prospective longitudinal study, The Environmental Determinants of Diabetes in the Young, currently underway to evaluate potential environmental factors in T1D[45].
NATURAL HISTORYThree decades ago, it was hypothesized that T1D is a chronic autoimmune disorder that develops in stages, and the model remains valid today (Figure 1). In genetically predisposed individuals (those with DR4/DQ8 and/or DR3/DQ2 haplotypes) there is an environmental trigger that leads to a break in immunologic tolerance and loss of beta cell mass. Over a period of time, usually years, there is autoimmune destruction of insulin producing beta cells that is marked by the presence of serum islet autoantibodies (Figure 2). As the process continues, very likely in a relapsing and remitting manner, there is a loss of glucose stimulated insulin release, and eventually insulin deficiency such that overt hyperglycemia results and clinical T1D is diagnosed[4].
How an inciting event leads to an aberrant immune response is not completely understood. Most hypotheses focus on immunologic abnormalities in antigen pre-sentation by HLA molecules to T cells in the thymus and peripheral lymph organs. T cells are educated in the thymus to self-antigens, such as insulin, and if there are dysregulated immune processes, self-reactive T cells can escape central tolerance and exist in the periphery[46,47]. Once these cells encounter their target antigen or peptide in peripheral lymph organs, they become activated to target beta cells. Other hypotheses focus on environmental triggers leading to immune activation and targeting of beta cells. The molecular mimicry theory proposes that a viral or bacterial protein shares amino acid sequence homology with beta cells and induces immune system activation through targeting beta cell antigen that is molecularly similar to a foreign antigen[38]. Finally an infectious triggering event may allow beta cells to become more sensitive to cytokine and free radical induced inflammation[48].
Recently the network for pancreatic organ donors has been established to study the pancreata of deceased donors with islet autoantibodies (preclinical disease) or established T1D[49]. The goal is to understand mechanisms of disease pathogenesis and interactions between beta cells and the immune system[50]. What we have gleaned from the initial efforts is that islet infiltrates (insulitis) are present in a lobular pattern in the pancreas, and there is a predominance of CD8 and CD4 T cells,
382 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Simmons KM et al . Type 1 diabetes: A predictable disease

Approximately 90% of individuals have two or more islet cell autoantibodies at diagnosis, and it is likely that the remaining 10% of individuals (islet autoantibody negative) have autoantibodies against antigens that have yet to be discovered. In children, IAA is usually the first antibody to develop, and the progression to T1D is 100% in children with a persistently high level of IAA[55,56]. This is in contrast to adults who tend to have higher levels of GAD at diagnosis. Islet autoantibodies can be easily measured in the serum, with the gold standard method for detecting antibodies being fluid phase radioimmunoassays (RIA)[57]. More recently, islet autoantibodies are now able to be measured from smaller volumes of serum and without the use of radioactivity using electrochemiluminescense as a detection method while maintaining similar sensitivity and specificity to RIA[58,59]. The rate at which individuals with positive islet autoantibodies progress to clinical T1D is dependent upon the age of appearance, insulin autoantibody level, and the number of autoantibodies present[55]. Hemoglobin A1c rises 1 to 1.5 years prior to diagnosis. Therefore, reduced insulin secretion and resultant hyperglycemia occur before T1D is clinically diagnosed[60,61]. Once T1D is clinically diagnosed, indivi-duals must commit to lifelong blood glucose monitoring and intensive insulin administration via multiple daily injections or an insulin pump to achieve good glycemic control. With improved diabetes management, the risk for long-term complications such as renal failure, myocardial infarctions, stroke, and lower extremity amputations has decreased over the last two decades[62]. However despite the decreasing prevalence of complications in diabetes, the need still exists to understand the underlying pathogenesis of complications such as diabetic cardiomyopathy and
B-lymphocytes, and macrophages[51,52]. Pancreata from established T1D patients also show an overall decrease in weight compared to age matched controls, potentially related to atrophy of the exocrine pancreas with the loss of beta cells[52]. The first serological evidence of an autoimmune response to beta cells is the appearance of autoantibodies to insulin (IAA), GAD, islet antigen 2, and zinc transporter 8[53]. Placental antibodies are no longer present after approximately 6 mo, so any antibodies in serum after that time reflect endogenous antibodies. If an individual develops two or more of these antibodies, they will eventually progress to clinical onset of T1D[54].
383 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
t
Primary prevention Secondary prevention Tertiary prevention
Genetic predisposition Preclinical diabetes T1D
Loss of insulin release
↓Increasing glucose
excursions
Minimal to no insulin release
↓Abnormal glucose
levels
Abnormal immune response↓
Dysregulated T cell responses and islet autoantibodies
detectable in serum
Beta
cel
l mas
s
UnknownPrecipitating
Eventmay occur in utero
or early in life
Figure 1 Stages in the development of type 1 diabetes adapted from the initial model proposed by George Eisenbarth. In genetically at risk individuals an unknown trigger, presumably environmental, initiates an autoimmune response that results in loss of beta cell mass. Before metabolic disturbances occur, islet autoantibodies (insulin, glutamic decarboxylase, islet antigen 2, zinc transporter 8) are measureable in serum. As beta cell mass decreases, potentially in a relapsing-remitting manner, there is loss of endogenous insulin release and ensuing hyperglycemia. Within this model, there are opportunities for type 1 diabetes (T1D) prevention in genotypically high risk individuals (primary prevention) and in autoantibody positive individuals (secondary prevention). Interventions to preserve remaining beta cell mass at diagnosis are also possible (tertiary prevention).
Figure 2 Insulin autoantibodies are often the first antibody to develop in young children. In contrast, adults most often are GAD65 and IA-2 autoantibody positive at diagnosis. The ZnT8 antibody is the most recently identified autoantibody with commercial testing now available. T1D: Type 1 diabetes; GAD65: Glutamic decarboxylase; IA-2: Islet antigen 2; ZnT8: Zinc transporter 8.
IA-2GAD65
Most common antibody seen in adults diagnosed with T1D
Islet autoantibodies
ZnT8Clinical antibody testing
available as of 2014
Insulin(mlAA)
Levels predict age of diabetes onsetOften first antibody to appear in
young children
Simmons KM et al . Type 1 diabetes: A predictable disease

novel approaches for treating complications such as neovascularization in diabetic foot disease[63,64].
SCREENING AND PREVENTIONThe American Diabetes Association recently adapted their guidelines to recommend screening for islet auto-antibodies in high-risk individuals[65]. Highly sensitive serological assays are not widely available, and all screening is recommended to be done in the setting of a clinical research study. To date, general population screening has been done through large clinical trial networks such as the National Institutes of Health sponsored TrialNet, which enroll and screen first or second degree family members of individuals with T1D. By identifying individuals with positive islet autoantibodies, the rate of diabetic ketoacidosis (DKA) at diagnosis is reduced[66]. Preventing DKA is important as altered mental status, coma, and even death can occur[67]. In fact, DKA is the most common cause of death in children with T1D[68]. Without screening, DKA at diagnosis is relatively common[69]. In the EURODIAB study, 42% of children presented in DKA (pH < 7.3) at the time of diagnosis with T1D[3]. By identifying individuals with positive autoantibodies, insulin therapy can be initiated early, and these children can enroll in studies aimed at preserving beta cell mass. In adults, maintaining endogenous insulin secretion reduces hemoglobin A1C, reduces the risk of severe hypoglycemia, decreases reliance on exogenous insulin, and decreases the rate of long-term complications[70-77]. In children, there has been very little data collected regarding residual beta cell mass beyond the first year after diagnosis[78-82]. A case-control study did show that children without severe hypoglycemia had increased residual beta cell mass compared to those children with severe hypoglycemia[83]. An effective method of preserving beta cell mass is not yet available, and the benefit of increased residual beta cell mass in children remains to be confirmed.
According to the World Health Organization’s principles of early disease detection, T1D is a condition that meets criteria for the establishment of a screening program. These principles include the condition is an important health problem, there is a recognizable latent stage of the disease, the natural history of the disease is understood, there is an adequate and accepted laboratory screening test, providers agree on who should receive treatment and there is a treatment available, there are adequate resources for diagnosis and treatment, and the cost of overall medical care would not increase[84]. Islet autoantibodies can be reliably measured in serum, with each antibody assay having a specificity of 99% when measured by radioimmunoassay in tertiary referral centers such as the Barbara Davis Center for Diabetes. The sensitivity for each autoantibody assay ranges from 70%-80%. We view these radioimmunoassays as a confirmatory test for T1D. A desired screening test needs to be reliable with high sensitivity, cost effective, and technically feasible, likely as a multiplex assay in
which all four autoantibodies are measured in a single well of an assay plate. Currently, to measure islet autoantibodies a blood draw is required with subsequent shipping of venous or capillary blood samples to a reference laboratory. This is not feasible for population wide screening due to technical requirements of sample collection and high cost. Screening large populations of infants for metabolic diseases and other congenital disorders has been successfully done using dried blood spots[85]. To establish an accepted screening program for T1D, the sensitivity and specificity of islet autoantibodies, specifically insulin autoantibody, needs to be established using a feasible collection method such as dried blood spots on filter paper, which would be a simplified collection method and more cost effective. Overall, T1D would not be over diagnosed with general population screening as diagnosis of the disorder requires both the presence of islet autoantibodies and metabolic abnormalities.
Ideally, individuals who screen positive for islet autoantibodies can be offered a treatment to prevent or delay the progression to T1D. Many secondary prevention trials have been completed with more currently underway[86]. As population based screening may be feasible in the near future, it is important to continue secondary prevention trials with the goal of delaying or preventing progression to T1D in islet autoantibody positive individuals. Patients enrolled in clinical inter-vention trials benefit from close follow up by medical professionals, early diagnosis of T1D, decreased incidence of DKA, and early initiation of insulin therapy (Figure 3).
Secondary prevention trialsAs T1D is a predictable disease with the measurement of islet autoantibodies, it logically follows that the disease should be preventable. To date, the majority of secondary prevention trials (enrolled individuals with preclinical disease) have administered different preparations of insulin to autoantibody positive individuals in an attempt to slow the progression to T1D onset[87]. The first such trial was the diabetes prevention trial-type 1 in which at risk patients were either administered subcutaneous insulin or oral insulin in randomized, doubleblinded, placebo controlled trials. Oral insulin has no metabolic effect; however, orally administered insulin does encounter mucosal gut-associated lymphoid tissue. The role of this lymphoid tissue is to provide protection from orally acquired pathogens and to keep individuals from developing reactions to ingested proteins. By administering low doses of oral insulin, insulin-specific T-regulatory cells are produced which may release cytokines that inhibit the inflammatory cascade that leads to β-cell destruction[88-90]. Relatives of patients with T1D who were 3 to 45 years of age and had high-risk HLA genes and one or more positive autoantibodies were evaluated for abnormal glucose metabolism. Those individuals who had abnormal glucose tolerance (n = 339) were administered 0.25 units/kg per day of Ultralente insulin twice daily and received an intravenous insulin infusion for four
384 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Simmons KM et al . Type 1 diabetes: A predictable disease

days at the beginning of the study and then annually. There was no effect of low-dose subcutaneous insulin on delaying the progression to T1D[91]. Participants with normal glucose tolerance received 7.5 mg/kg per day of oral insulin (n = 372). An oral glucose tolerance test was completed every 6 mo during a 6-year follow up, and there was not a delay in progression to T1D. Of interest, a post-hoc analysis showed in participants with persistently high levels of IAA (≥ 80 nU/mL) there was a delay in disease onset of approximately five years[92]. Also, the rate of progression to T1D onset after stopping insulin was more rapid[93]. A follow-up oral insulin trial through TrialNet is currently enrolling participants in order to determine if oral insulin can delay the progression to T1D in individuals with high IAA levels (ClinicalTrials.gov Identifier: NCT00419562).
Another insulin intervention trial from the Belgian T1D Registry identified study participants who were insulin autoantibody positive and did not have a HLA haplotype conferring protection (DQB*0602). Study participants were given two subcutaneous injections of insulin daily for 3 years (n = 25) or observed and prospectively followed (n = 25). The participants who were treated with insulin and those who refused treatment or agreed to observation developed T1D at the same rate[94].
Many preclinical studies have suggested that administration of intranasal insulin may delay T1D development through mucosal tolerance, in which mucosal antigens have been shown to impact regulatory T cell development[95]. To translate these findings to humans, individuals with high-risk HLA haplotypes and one or more islet autoantibodies were enrolled in the Intranasal Insulin Trial (INIT-Ⅰ) (n = 38). This randomized, doubleblinded, crossover pilot study suggested that intranasal insulin protects against the development of T1D by increasing antibody formation and decreasing T cell responsiveness[96]. The INIT-Ⅱ, a randomized, doubleblinded, placebo controlled trial, is now enrolling individuals to determine if intranasal insulin can delay or prevent the progression to T1D (ClinicalTrials.gov Identifier: NCT 00336674).
However, a large study in Finland, the T1D Prediction and Prevention Trial enrolled and followed siblings of children with T1D or infants of mothers with T1D who had high-risk HLA genes for islet autoantibody development. Once two or more autoantibodies were detected (n = 264), they were randomized to intranasal insulin (n = 137) or placebo (n = 127). Interim analyses showed no benefit of intranasal insulin in delaying the onset of T1D[97]. This indicates that intranasal insulin may not be effective at delaying diabetes onset at the administered dose and timing in the disease process. Potentially insulin antigen specific therapies may need to be administered earlier in the disease course to have an impact on delaying progression to T1D.
Several trials using non-antigen specific therapies including bacille calmette-guerin injections, Ketotifen (histamine antagonist), oral cyclosporine, and nicotina-mide (B6) have been completed. No study has prevented or delayed T1D development[98-104].
Clinical trials with drugs aimed at modulating the immune response and preserving endogenous insulin secretion in patients with new-onset T1D are termed tertiary prevention trials[51]. Only recently have these drugs expanded to prevention trials in islet auto-antibody positive individuals (Figure 3). The CTLA4-Ig antibody (Abatacept) for Prevention of Abnormal Glucose Tolerance and Diabetes in Relatives At-Risk (ClinicalTrials.gov Identifier: NCT 01773707) and Anti-CD3 monoclonal antibody (Teplizumab) for Prevention Of Diabetes In Relatives At Risk For T1D mellitus (ClinicalTrials.gov Identifier: NCT01030861) are both TrialNet studies currently enrolling participants. Abatacept is a fusion antibody that binds to antigen presenting cells and blocks co-stimulation to T cells. Anti-CD3 monoclonal antibodies bind the CD3 molecule which is present on CD8 and CD4 T cells, thereby inhibiting T cell activation[105]. Both of these drugs have shown some degree of success when used in new-onset trials[106-111]. DIAPREV-IT is an antigen-based treatment currently enrolling individuals who are positive for GAD and one or more additional autoantibodies (ClinicalTrials.gov Identifier: NCT 01122446). A GAD/alum vaccine
385 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 3 The measurement of serum islet autoantibodies has made type 1 diabetes a predictable disease. Early identification of islet autoantibody positive individuals leads to improved clinical outcomes by decreasing the risk for diabetic ketoacidosis and potentially preserving beta cell mass through clinical prevention trials. T1D: Type 1 diabetes; INIT-Ⅱ: Intranasal Insulin Trial.
Screen for isletautoantibodies
Enroll autoantibody positive individuals in secondary
prevention trials
Increased clinical monitoring in autoantibody positive individuals
Oral insulin for prevention of T1D
Anti-CD3 mAb (teplizumab) for prevention of diabetes in relatives
at risk for T1D mellitus
CTLA4-lg (abatacept) for prevention of T1D in relatives at-risk
INIT-Ⅱ
Decreased rate of diabetic ketoacidosisIncreased beta cell mass at diagnosis
Simmons KM et al . Type 1 diabetes: A predictable disease

is given at enrollment and 1 mo later. Although GAD vaccination is safe and easily administered, new-onset intervention trials have not shown long-term preservation of endogenous insulin secretion[108,112,113].
NOVEL APPROACHES TO PREVENT T1DCurrently, insulin is the only medication approved by the United States Food and Drug Administration for the treatment of T1D. Despite T1D being a predictable chronic autoimmune disorder, there are not any therapiesto preserve endogenous insulin production. As mentioned above, many large clinical intervention trials have not slowed the progression or prevented disease onset. We believe T1D will be preventable and that safe and specific therapies targeting the immune system are needed. One such approach is to target the trimolecular complex, which consists of a self-reactive CD4 T cell, insulin, and HLA molecule[114]. It is well established that specific HLA alleles, namely HLA DQ8 which is present in approximately 60% of all T1D patients, confer significant disease risk. DQ8 is a molecular target for diabetes intervention by using small “drug-like” molecules to block antigen presentation, thereby inhibiting specific T cell activation. Preclinical studies have shown this to be a potential pathway for diabetes intervention[115]. This concept has been advanced from bench to bedside as a clinical trial in which methyldopa (Aldomet), a clinically well-established antihypertensive drug, is being investigated to block DQ8 antigen presentation. The phase 1b dose escalation trial is using personalized medicine as methyldopa is being administered to recent onset adult T1D patients with the presence of the DQ8 gene (ClinicalTrials.gov Identifier: NCT01883804). Methyldopa is orally administered, safe as it has been used clinically for the last 50 years, and currently indicated for the treatment of pregnancy induced hypertension. Furthermore, all individuals have three class Ⅱ molecules (DQ, DR, and DP), and by blocking a single class Ⅱ molecule, there are two others to permit normal immune system function.
Other approaches have targeted components of the insulin trimolecular complex including antibodies that specifically bind to an insulin peptide in the HLA molecule. Preclinical studies in an animal model of spontaneous autoimmune diabetes indicate that this approach can delay diabetes onset[116]. Efforts are currently being made to make a human antibody, which again is a very specific immune therapy for diabetes intervention. Finally, insulin antigen specific therapy has the potential to evolve with recent advances in the field of immunology. A peptide from the insulin B chain amino acids 9-23 (B:9-23) has been extensively studied in animal models and human T1D[117,118]. It is now appreciated that insulin B:9-23 is a key autoantigen in the disease process of both mice and humans, sharing an identical amino acid sequence in both species[119,120]. A mutated insulin B:9-23 peptide, but not the native peptide sequence, induced protective immune responses (regulatory T cells) and prevented
diabetes onset in preclinical animal models[121]. With a deeper understanding of how the insulin peptide binds to HLA molecules and activates T cells, an insulin vaccine again holds promise for diabetes prevention.
In conclusion, T1D is now a predicable disease with the measurement of islet autoantibodies and prevention will naturally follow. To prevent T1D, general population screening for islet autoantibodies is needed along with a safe and specific therapy for disease intervention. The genes that confer diabetes risk are now molecular targets, and tailoring therapies to specific HLA genes is personalized medicine. The future holds promise for delaying the progression and ultimately preventing diabetes.
REFERENCES1 Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes.
Lancet 2014; 383: 69-82 [DOI: 10.1016/s0140-6736(13)60591-7]2 Rewers M, Bugawan TL, Norris JM, Blair A, Beaty B, Hoffman
M, McDuffie RS, Hamman RF, Klingensmith G, Eisenbarth GS, Erlich HA. Newborn screening for HLA markers associated with IDDM: diabetes autoimmunity study in the young (DAISY). Diabetologia 1996; 39: 807-812 [PMID: 8817105]
3 Lévy-Marchal C, Patterson CC, Green A; EURODIAB ACE Study Group. Europe and Diabetes. Geographical variation of presentation at diagnosis of type I diabetes in children: the EURODIAB study. European and Dibetes. Diabetologia 2001; 44 Suppl 3: B75-B80 [PMID: 11724421]
4 Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 1986; 314: 1360-1368 [PMID: 3517648 DOI: 10.1056/NEJM198605223142106]
5 DIAMOND Project Group. Incidence and trends of childhood Type 1 diabetes worldwide 1990-1999. Diabet Med 2006; 23: 857-866 [PMID: 16911623 DOI: 10.1111/j.1464-5491.2006.01925.x]
6 Patterson CC, Dahlquist GG, Gyürüs E, Green A, Soltész G. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet 2009; 373: 2027-2033 [PMID: 19481249 DOI: 10.1016/S0140-6736(09)60568-7]
7 Lawrence JM, Imperatore G, Dabelea D, Mayer-Davis EJ, Linder B, Saydah S, Klingensmith GJ, Dolan L, Standiford DA, Pihoker C, Pettitt DJ, Talton JW, Thomas J, Bell RA, D’Agostino RB. Trends in incidence of type 1 diabetes among non-Hispanic white youth in the U.S., 2002-2009. Diabetes 2014; 63: 3938-3945 [PMID: 24898146 DOI: 10.2337/db13-1891]
8 Harjutsalo V, Sjöberg L, Tuomilehto J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet 2008; 371: 1777-1782 [PMID: 18502302 DOI: 10.1016/S0140-6736(08)60765-5]
9 Redondo MJ, Eisenbarth GS. Genetic control of autoimmunity in Type I diabetes and associated disorders. Diabetologia 2002; 45: 605-622 [PMID: 12107741 DOI: 10.1007/s00125-002-0781-1]
10 Nokoff N, Rewers M. Pathogenesis of type 1 diabetes: lessons from natural history studies of high-risk individuals. Ann N Y Acad Sci 2013; 1281: 1-15 [PMID: 23360422 DOI: 10.1111/nyas.12021]
11 Gillespie KM, Gale EA, Bingley PJ. High familial risk and genetic susceptibility in early onset childhood diabetes. Diabetes 2002; 51: 210-214 [PMID: 11756343]
12 Hämäläinen AM, Knip M. Autoimmunity and familial risk of type 1 diabetes. Curr Diab Rep 2002; 2: 347-353 [PMID: 12643195]
13 Rothwell PM, Gutnikov SA, McKinney PA, Schober E, Ionescu-Tirgoviste C, Neu A. Seasonality of birth in children with diabetes in Europe: multicentre cohort study. European Diabetes Study Group. BMJ 1999; 319: 887-888 [PMID: 10506043]
14 Vaiserman AM, Carstensen B, Voitenko VP, Tronko MD,
386 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Simmons KM et al . Type 1 diabetes: A predictable disease

Kravchenko VI, Khalangot MD, Mechova LV. Seasonality of birth in children and young adults (0-29 years) with type 1 diabetes in Ukraine. Diabetologia 2007; 50: 32-35 [PMID: 17093948 DOI: 10.1007/s00125-006-0456-4]
15 Ostman J, Lönnberg G, Arnqvist HJ, Blohmé G, Bolinder J, Ekbom Schnell A, Eriksson JW, Gudbjörnsdottir S, Sundkvist G, Nyström L. Gender differences and temporal variation in the incidence of type 1 diabetes: results of 8012 cases in the nationwide Diabetes Incidence Study in Sweden 1983-2002. J Intern Med 2008; 263: 386-394 [PMID: 18205768 DOI: 10.1111/j.1365-2796.2007.01896.x]
16 Kordonouri O, Shuga N, Lewy H, Ashkenazi I, Laron Z. Seasonality of month of birth of children and adolescents with type 1 diabetes mellitus in Berlin differs from the general population. Eur J Pediatr 2002; 161: 291-292 [PMID: 12012228 DOI: 10.1007/s00431-002-0941-9]
17 Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Plagnol V, Pociot F, Schuilenburg H, Smyth DJ, Stevens H, Todd JA, Walker NM, Rich SS. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009; 41: 703-707 [PMID: 19430480 DOI: 10.1038/ng.381]
18 Noble JA, Valdes AM, Cook M, Klitz W, Thomson G, Erlich HA. The role of HLA class II genes in insulin-dependent diabetes mellitus: molecular analysis of 180 Caucasian, multiplex families. Am J Hum Genet 1996; 59: 1134-1148 [PMID: 8900244]
19 Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, Lavant E, Louey A, Moonsamy P. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 2008; 57: 1084-1092 [PMID: 18252895 DOI: 10.2337/db07-1331]
20 Polychronakos C, Li Q. Understanding type 1 diabetes through genetics: advances and prospects. Nat Rev Genet 2011; 12: 781-792 [PMID: 22005987 DOI: 10.1038/nrg3069]
21 Pugliese A, Zeller M, Fernandez A, Zalcberg LJ, Bartlett RJ, Ricordi C, Pietropaolo M, Eisenbarth GS, Bennett ST, Patel DD. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet 1997; 15: 293-297 [PMID: 9054945 DOI: 10.1038/ng0397-293]
22 Bradfield JP, Qu HQ, Wang K, Zhang H, Sleiman PM, Kim CE, Mentch FD, Qiu H, Glessner JT, Thomas KA, Frackelton EC, Chiavacci RM, Imielinski M, Monos DS, Pandey R, Bakay M, Grant SF, Polychronakos C, Hakonarson H. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet 2011; 7: e1002293 [PMID: 21980299 DOI: 10.1371/journal.pgen.1002293]
23 Steck AK, Baschal EE, Jasinski JM, Boehm BO, Bottini N, Concannon P, Julier C, Morahan G, Noble JA, Polychronakos C, She JX, Eisenbarth GS. rs2476601 T allele (R620W) defines high-risk PTPN22 type I diabetes-associated haplotypes with preliminary evidence for an additional protective haplotype. Genes Immun 2009; 10 Suppl 1: S21-S26 [PMID: 19956096 DOI: 10.1038/gene.2009.87]
24 Snell-Bergeon JK, Smith J, Dong F, Barón AE, Barriga K, Norris JM, Rewers M. Early childhood infections and the risk of islet autoimmunity: the Diabetes Autoimmunity Study in the Young (DAISY). Diabetes Care 2012; 35: 2553-2558 [PMID: 23043167 DOI: 10.2337/dc12-0423]
25 Hyöty H, Leinikki P, Reunanen A, Ilonen J, Surcel HM, Rilva A, Käär ML, Huupponen T, Hakulinen A, Mäkelä AL. Mumps infections in the etiology of type 1 (insulin-dependent) diabetes. Diabetes Res 1988; 9: 111-116 [PMID: 3243043]
26 Hyöty H, Taylor KW. The role of viruses in human diabetes. Diabetologia 2002; 45: 1353-1361 [PMID: 12378375 DOI: 10.1007/s00125-002-0852-3]
27 Honeyman MC, Coulson BS, Stone NL, Gellert SA, Goldwater PN, Steele CE, Couper JJ, Tait BD, Colman PG, Harrison LC. Association between rotavirus infection and pancreatic islet autoimmunity in
children at risk of developing type 1 diabetes. Diabetes 2000; 49: 1319-1324 [PMID: 10923632]
28 Honeyman MC, Stone NL, Harrison LC. T-cell epitopes in type 1 diabetes autoantigen tyrosine phosphatase IA-2: potential for mimicry with rotavirus and other environmental agents. Mol Med 1998; 4: 231-239 [PMID: 9606176]
29 Pak CY, Eun HM, McArthur RG, Yoon JW. Association of cytomegalovirus infection with autoimmune type 1 diabetes. Lancet 1988; 2: 1-4 [PMID: 2898620]
30 Forrest JM, Menser MA, Burgess JA. High frequency of diabetes mellitus in young adults with congenital rubella. Lancet 1971; 2: 332-334 [PMID: 4105044]
31 Menser MA, Forrest JM, Bransby RD. Rubella infection and diabetes mellitus. Lancet 1978; 1: 57-60 [PMID: 74564]
32 Devendra D, Liu E, Eisenbarth GS. Type 1 diabetes: recent developments. BMJ 2004; 328: 750-754 [PMID: 15044291 DOI: 10.1136/bmj.328.7442.750]
33 Kimpimäki T, Kupila A, Hämäläinen AM, Kukko M, Kulmala P, Savola K, Simell T, Keskinen P, Ilonen J, Simell O, Knip M. The first signs of beta-cell autoimmunity appear in infancy in genetically susceptible children from the general population: the Finnish Type 1 Diabetes Prediction and Prevention Study. J Clin Endocrinol Metab 2001; 86: 4782-4788 [PMID: 11600541 DOI: 10.1210/jcem.86.10.7907]
34 Lönnrot M, Korpela K, Knip M, Ilonen J, Simell O, Korhonen S, Savola K, Muona P, Simell T, Koskela P, Hyöty H. Enterovirus infection as a risk factor for beta-cell autoimmunity in a prospectively observed birth cohort: the Finnish Diabetes Prediction and Prevention Study. Diabetes 2000; 49: 1314-1318 [PMID: 10923631]
35 Stene LC, Oikarinen S, Hyöty H, Barriga KJ, Norris JM, Klingensmith G, Hutton JC, Erlich HA, Eisenbarth GS, Rewers M. Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY). Diabetes 2010; 59: 3174-3180 [PMID: 20858685 DOI: 10.2337/db10-0866]
36 Hyöty H, Hiltunen M, Knip M, Laakkonen M, Vähäsalo P, Karjalainen J, Koskela P, Roivainen M, Leinikki P, Hovi T. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes 1995; 44: 652-657 [PMID: 7789630]
37 Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009; 52: 1143-1151 [PMID: 19266182 DOI: 10.1007/s00125-009-1276-0]
38 Kaufman DL, Erlander MG, Clare-Salzler M, Atkinson MA, Maclaren NK, Tobin AJ. Autoimmunity to two forms of glutamate decarboxylase in insulin-dependent diabetes mellitus. J Clin Invest 1992; 89: 283-292 [PMID: 1370298 DOI: 10.1172/JCI115573]
39 Horwitz MS, Ilic A, Fine C, Balasa B, Sarvetnick N. Coxsackieviral-mediated diabetes: induction requires antigen-presenting cells and is accompanied by phagocytosis of beta cells. Clin Immunol 2004; 110: 134-144 [PMID: 15003810 DOI: 10.1016/j.clim.2003.09.014]
40 Zipitis CS, Akobeng AK. Vitamin D supplementation in early childhood and risk of type 1 diabetes: a systematic review and meta-analysis. Arch Dis Child 2008; 93: 512-517 [PMID: 18339654 DOI: 10.1136/adc.2007.128579]
41 Hyppönen E, Läärä E, Reunanen A, Järvelin MR, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet 2001; 358: 1500-1503 [PMID: 11705562 DOI: 10.1016/S0140-6736(01)06580-1]
42 Simpson M, Brady H, Yin X, Seifert J, Barriga K, Hoffman M, Bugawan T, Barón AE, Sokol RJ, Eisenbarth G, Erlich H, Rewers M, Norris JM. No association of vitamin D intake or 25-hydroxyvitamin D levels in childhood with risk of islet autoimmunity and type 1 diabetes: the Diabetes Autoimmunity Study in the Young (DAISY). Diabetologia 2011; 54: 2779-2788 [PMID: 21858504 DOI: 10.1007/s00125-011-2278-2]
43 Hummel S, Pflüger M, Hummel M, Bonifacio E, Ziegler AG. Primary dietary intervention study to reduce the risk of islet
387 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Simmons KM et al . Type 1 diabetes: A predictable disease

autoimmunity in children at increased risk for type 1 diabetes: the BABYDIET study. Diabetes Care 2011; 34: 1301-1305 [PMID: 21515839 DOI: 10.2337/dc10-2456]
44 Schattner A. Consequence or coincidence? The occurrence, pathogenesis and significance of autoimmune manifestations after viral vaccines. Vaccine 2005; 23: 3876-3886 [PMID: 15917108 DOI: 10.1016/j.vaccine.2005.03.005]
45 Lee HS, Burkhardt BR, McLeod W, Smith S, Eberhard C, Lynch K, Hadley D, Rewers M, Simell O, She JX, Hagopian B, Lernmark A, Akolkar B, Ziegler AG, Krischer JP. Biomarker discovery study design for type 1 diabetes in The Environmental Determinants of Diabetes in the Young (TEDDY) study. Diabetes Metab Res Rev 2014; 30: 424-434 [PMID: 24339168 DOI: 10.1002/dmrr.2510]
46 Michels AW, Gottlieb PA. Autoimmune polyglandular syndromes. Nat Rev Endocrinol 2010; 6: 270-277 [PMID: 20309000 DOI: 10.1038/nrendo.2010.40]
47 Michels AW, Nakayama M. The anti-insulin trimolecular complex in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17: 329-334 [PMID: 20489610 DOI: 10.1097/MED.0b01-3e32833aba41]
48 Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci 2013; 1281: 16-35 [PMID: 23323860 DOI: 10.1111/j.1749-6632.2012.06826.x]
49 Campbell-Thompson M, Wasserfall C, Kaddis J, Albanese-O’Neill A, Staeva T, Nierras C, Moraski J, Rowe P, Gianani R, Eisenbarth G, Crawford J, Schatz D, Pugliese A, Atkinson M. Network for Pancreatic Organ Donors with Diabetes (nPOD): developing a tissue biobank for type 1 diabetes. Diabetes Metab Res Rev 2012; 28: 608-617 [PMID: 22585677 DOI: 10.1002/dmrr.2316]
50 Pugliese A, Yang M, Kusmarteva I, Heiple T, Vendrame F, Wasserfall C, Rowe P, Moraski JM, Ball S, Jebson L, Schatz DA, Gianani R, Burke GW, Nierras C, Staeva T, Kaddis JS, Campbell-Thompson M, Atkinson MA. The Juvenile Diabetes Research Foundation Network for Pancreatic Organ Donors with Diabetes (nPOD) Program: goals, operational model and emerging findings. Pediatr Diabetes 2014; 15: 1-9 [PMID: 24325575 DOI: 10.1111/pedi.12097]
51 Michels AW, Eisenbarth GS. Immune intervention in type 1 diabetes. Semin Immunol 2011; 23: 214-219 [PMID: 21852151 DOI: 10.1016/j.smim.2011.07.003]
52 Spencer J, Peakman M. Post-mortem analysis of islet pathology in type 1 diabetes illuminates the life and death of the beta cell. Clin Exp Immunol 2009; 155: 125-127 [PMID: 19128357 DOI: 10.1111/j.1365-2249.2008.03864.x]
53 Watkins RA, Evans-Molina C, Blum JS, DiMeglio LA. Established and emerging biomarkers for the prediction of type 1 diabetes: a systematic review. Transl Res 2014; 164: 110-121 [PMID: 24662515 DOI: 10.1016/j.trsl.2014.02.004]
54 Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, Winkler C, Ilonen J, Veijola R, Knip M, Bonifacio E, Eisenbarth GS. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013; 309: 2473-2479 [PMID: 23780460 DOI: 10.1001/jama.2013.6285]
55 Steck AK, Johnson K, Barriga KJ, Miao D, Yu L, Hutton JC, Eisenbarth GS, Rewers MJ. Age of islet autoantibody appearance and mean levels of insulin, but not GAD or IA-2 autoantibodies, predict age of diagnosis of type 1 diabetes: diabetes autoimmunity study in the young. Diabetes Care 2011; 34: 1397-1399 [PMID: 21562325 DOI: 10.2337/dc10-2088]
56 Parikka V, Näntö-Salonen K, Saarinen M, Simell T, Ilonen J, Hyöty H, Veijola R, Knip M, Simell O. Early seroconversion and rapidly increasing autoantibody concentrations predict prepubertal manifestation of type 1 diabetes in children at genetic risk. Diabetologia 2012; 55: 1926-1936 [PMID: 22441569 DOI: 10.1007/s00125-012-2523-3]
57 Yu L, Rewers M, Gianani R, Kawasaki E, Zhang Y, Verge C, Chase P, Klingensmith G, Erlich H, Norris J, Eisenbarth GS. Antiislet autoantibodies usually develop sequentially rather than
simultaneously. J Clin Endocrinol Metab 1996; 81: 4264-4267 [PMID: 8954025 DOI: 10.1210/jcem.81.12.8954025]
58 Yu L, Miao D, Scrimgeour L, Johnson K, Rewers M, Eisenbarth GS. Distinguishing persistent insulin autoantibodies with differential risk: nonradioactive bivalent proinsulin/insulin autoantibody assay. Diabetes 2012; 61: 179-186 [PMID: 22124462 DOI: 10.2337/db11-0670]
59 Miao D, Guyer KM, Dong F, Jiang L, Steck AK, Rewers M, Eisenbarth GS, Yu L. GAD65 autoantibodies detected by electro-chemiluminescence assay identify high risk for type 1 diabetes. Diabetes 2013; 62: 4174-4178 [PMID: 23974918 DOI: 10.2337/db13-0534]
60 Sosenko JM, Skyler JS, Krischer JP, Greenbaum CJ, Mahon J, Rafkin LE, Cuthbertson D, Cowie C, Herold K, Eisenbarth G, Palmer JP; Diabetes Prevention Trial-Type 1 Study Group. Glucose excursions between states of glycemia with progression to type 1 diabetes in the diabetes prevention trial-type 1 (DPT-1). Diabetes 2010; 59: 2386-2389 [PMID: 20682683 DOI: 10.2337/db10-0534]
61 Ferrannini E, Mari A, Nofrate V, Sosenko JM, Skyler JS. Progression to diabetes in relatives of type 1 diabetic patients: mechanisms and mode of onset. Diabetes 2010; 59: 679-685 [PMID: 20028949 DOI: 10.2337/db09-1378]
62 Gregg EW, Li Y, Wang J, Burrows NR, Ali MK, Rolka D, Williams DE, Geiss L. Changes in diabetes-related complications in the United States, 1990-2010. N Engl J Med 2014; 370: 1514-1523 [PMID: 24738668 DOI: 10.1056/NEJMoa1310799]
63 Ciccone MM, Scicchitano P, Cameli M, Cecere A, Cortese F, Dentamaro I, Gentile F, Gesualdo M, Maiello M, Modesti PA, Muiesan ML, Novo S, Palmiero P, Saba PS, Zito A, Mattioli AV, Pedrinelli R. Endothelial Function in Pre-diabetes, Diabetes and Diabetic Cardiomyopathy: A Review. J Diabetes Metab 2014; 5: 364 [DOI: 10.4172/2155-6156.1000364]
64 Ciccone MM, Marchese A, Generali A, Loiodice C, Cortese F, Carbonara R, Scicchitano P, Laviola L, Giorgino F. Interventional therapy in diabetic foot: risk factors, clinical events and prognosis at one year follow-up (a study of 103 cases). Pak J Biol Sci 2012; 15: 789-794 [PMID: 24175420]
65 American Diabetes Association. Standards of medical care in diabetes--2014. Diabetes Care 2014; 37 Suppl 1: S14-S80 [PMID: 24357209 DOI: 10.2337/dc14-S014]
66 Elding Larsson H, Vehik K, Gesualdo P, Akolkar B, Hagopian W, Krischer J, Lernmark Å, Rewers M, Simell O, She JX, Ziegler A, Haller MJ. Children followed in the TEDDY study are diagnosed with type 1 diabetes at an early stage of disease. Pediatr Diabetes 2014; 15: 118-126 [PMID: 24034790 DOI: 10.1111/pedi.12066]
67 Rewers A. Current concepts and controversies in prevention and treatment of diabetic ketoacidosis in children. Curr Diab Rep 2012; 12: 524-532 [PMID: 22864672 DOI: 10.1007/s11892-012-0307-2]
68 Edge JA, Ford-Adams ME, Dunger DB. Causes of death in children with insulin dependent diabetes 1990-96. Arch Dis Child 1999; 81: 318-323 [PMID: 10490436]
69 Dabelea D, Rewers A, Stafford JM, Standiford DA, Lawrence JM, Saydah S, Imperatore G, D’Agostino RB, Mayer-Davis EJ, Pihoker C; SEARCH for Diabetes in Youth Study Group. Trends in the prevalence of ketoacidosis at diabetes diagnosis: the SEARCH for diabetes in youth study. Pediatrics 2014; 133: e938-e945 [PMID: 24685959 DOI: 10.1542/peds.2013-2795]
70 Effects of age, duration and treatment of insulin-dependent diabetes mellitus on residual beta-cell function: observations during eligibility testing for the Diabetes Control and Complications Trial (DCCT). The DCCT Research Group. J Clin Endocrinol Metab 1987; 65: 30-36 [PMID: 2884229 DOI: 10.1210/jcem-65-1-30]
71 Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care 2003; 26: 832-836 [PMID: 12610045]
72 Sjöberg S, Gjötterberg M, Berglund L, Möller E, Ostman J. Residual C-peptide excretion is associated with a better long-term glycemic control and slower progress of retinopathy in type I (insulin-dependent) diabetes mellitus. J Diabet Complications
388 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Simmons KM et al . Type 1 diabetes: A predictable disease

1991; 5: 18-22 [PMID: 1830314]73 Nakanishi K, Kobayashi T, Miyashita H, Ohkubo M, Sugimoto
T, Murase T, Kosaka K, Inouye K, Kono M. Relationships among islet cell antibodies, residual beta-cell function, and metabolic control in patients with insulin-dependent diabetes mellitus of long duration: use of a sensitive C-peptide radioimmunoassay. Metabolism 1990; 39: 925-930 [PMID: 2202883]
74 Nakanishi K, Watanabe C. Rate of beta-cell destruction in type 1 diabetes influences the development of diabetic retinopathy: protective effect of residual beta-cell function for more than 10 years. J Clin Endocrinol Metab 2008; 93: 4759-4766 [PMID: 18826998 DOI: 10.1210/jc.2008-1209]
75 Panero F, Novelli G, Zucco C, Fornengo P, Perotto M, Segre O, Grassi G, Cavallo-Perin P, Bruno G. Fasting plasma C-peptide and micro- and macrovascular complications in a large clinic-based cohort of type 1 diabetic patients. Diabetes Care 2009; 32: 301-305 [PMID: 19017769 DOI: 10.2337/dc08-1241]
76 Mühlhauser I, Overmann H, Bender R, Bott U, Berger M. Risk factors of severe hypoglycaemia in adult patients with Type I diabetes--a prospective population based study. Diabetologia 1998; 41: 1274-1282 [PMID: 9833933 DOI: 10.1007/s001250051065]
77 Effect of intensive therapy on residual beta-cell function in patients with type 1 diabetes in the diabetes control and complications trial. A randomized, controlled trial. The Diabetes Control and Complications Trial Research Group. Ann Intern Med 1998; 128: 517-523 [PMID: 9518395]
78 Mortensen HB, Swift PG, Holl RW, Hougaard P, Hansen L, Bjoerndalen H, de Beaufort CE, Knip M. Multinational study in children and adolescents with newly diagnosed type 1 diabetes: association of age, ketoacidosis, HLA status, and autoantibodies on residual beta-cell function and glycemic control 12 months after diagnosis. Pediatr Diabetes 2010; 11: 218-226 [PMID: 19708904 DOI: 10.1111/j.1399-5448.2009.00566.x]
79 Picardi A, Visalli N, Lauria A, Suraci C, Buzzetti R, Merola MK, Manfrini S, Guglielmi C, Gentilucci UV, Pitocco D, Crinò A, Bizzarri C, Cappa M, Pozzilli P. Metabolic factors affecting residual beta cell function assessed by C-peptide secretion in patients with newly diagnosed type 1 diabetes. Horm Metab Res 2006; 38: 668-672 [PMID: 17075776 DOI: 10.1055/s-2006-954586]
80 Böber E, Dündar B, Büyükgebiz A. Partial remission phase and metabolic control in type 1 diabetes mellitus in children and adolescents. J Pediatr Endocrinol Metab 2001; 14: 435-441 [PMID: 11327378]
81 Bonfanti R, Bognetti E, Meschi F, Brunelli A, Riva MC, Pastore MR, Calori G, Chiumello G. Residual beta-cell function and spontaneous clinical remission in type 1 diabetes mellitus: the role of puberty. Acta Diabetol 1998; 35: 91-95 [PMID: 9747961]
82 Komulainen J, Knip M, Lounamaa R, Vähäsalo P, Karjalainen J, Sabbah E, Akerblom HK. Poor beta-cell function after the clinical manifestation of type 1 diabetes in children initially positive for islet cell specific autoantibodies. The Childhood Diabetes in Finland Study Group. Diabet Med 1997; 14: 532-537 [PMID: 9223390 DOI: 10.1002/(SICI)1096-9136(199707)14:7<532::AID-DIA403>3.0.CO;2-6]
83 Egger M, Gschwend S, Smith GD, Zuppinger K. Increasing incidence of hypoglycemic coma in children with IDDM. Diabetes Care 1991; 14: 1001-1005 [PMID: 1797479]
84 Strong K, Wald N, Miller A, Alwan A. Current concepts in screening for noncommunicable disease: World Health Organization Consultation Group Report on methodology of noncommunicable disease screening. J Med Screen 2005; 12: 12-19 [PMID: 15825234]
85 Clague A, Thomas A. Neonatal biochemical screening for disease. Clin Chim Acta 2002; 315: 99-110 [PMID: 11728413]
86 Skyler JS. Primary and secondary prevention of Type 1 diabetes. Diabet Med 2013; 30: 161-169 [PMID: 23231526 DOI: 10.1111/dme.12100]
87 Michels AW, von Herrath M. 2011 Update: antigen-specific therapy in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes 2011; 18: 235-240 [PMID: 21844706 DOI: 10.1097/MED.0b013e32834803ae]
88 Mayer L, Shao L. Therapeutic potential of oral tolerance. Nat Rev Immunol 2004; 4: 407-419 [PMID: 15173830 DOI: 10.1038/nri1370]
89 Weiner HL. Oral tolerance for the treatment of autoimmune diseases. Annu Rev Med 1997; 48: 341-351 [PMID: 9046967 DOI: 10.1146/annurev.med.48.1.341]
90 Weiner HL, Mayer LF. Oral tolerance: mechanisms and applications. Introduction. Ann N Y Acad Sci 1996; 778: xiii-xviii [PMID: 8610961]
91 Diabetes Prevention Trial--Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med 2002; 346: 1685-1691 [PMID: 12037147 DOI: 10.1056/NEJMoa012350]
92 Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, Cuthbertson D, Rafkin-Mervis LE, Chase HP, Leschek E. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial--Type 1. Diabetes Care 2005; 28: 1068-1076 [PMID: 15855569]
93 Vehik K, Cuthbertson D, Ruhlig H, Schatz DA, Peakman M, Krischer JP; DPT-1 and TrialNet Study Groups. Long-term outcome of individuals treated with oral insulin: diabetes prevention trial-type 1 (DPT-1) oral insulin trial. Diabetes Care 2011; 34: 1585-1590 [PMID: 21610124 DOI: 10.2337/dc11-0523]
94 Vandemeulebroucke E, Gorus FK, Decochez K, Weets I, Keymeulen B, De Block C, Tits J, Pipeleers DG, Mathieu C; Belgian Diabetes Registry. Insulin treatment in IA-2A-positive relatives of type 1 diabetic patients. Diabetes Metab 2009; 35: 319-327 [PMID: 19647467 DOI: 10.1016/j.diabet.2009.02.005]
95 Martinez NR, Augstein P, Moustakas AK, Papadopoulos GK, Gregori S, Adorini L, Jackson DC, Harrison LC. Disabling an integral CTL epitope allows suppression of autoimmune diabetes by intranasal proinsulin peptide. J Clin Invest 2003; 111: 1365-1371 [PMID: 12727928 DOI: 10.1172/JCI17166]
96 Harrison LC, Honeyman MC, Steele CE, Stone NL, Sarugeri E, Bonifacio E, Couper JJ, Colman PG. Pancreatic beta-cell function and immune responses to insulin after administration of intranasal insulin to humans at risk for type 1 diabetes. Diabetes Care 2004; 27: 2348-2355 [PMID: 15451899]
97 Näntö-Salonen K, Kupila A, Simell S, Siljander H, Salonsaari T, Hekkala A, Korhonen S, Erkkola R, Sipilä JI, Haavisto L, Siltala M, Tuominen J, Hakalax J, Hyöty H, Ilonen J, Veijola R, Simell T, Knip M, Simell O. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet 2008; 372: 1746-1755 [PMID: 18814906 DOI: 10.1016/S0140-6736(08)61309-4]
98 Böhmer KP, Kolb H, Kuglin B, Zielasek J, Hübinger A, Lampeter EF, Weber B, Kolb-Bachofen V, Jastram HU, Bertrams J. Linear loss of insulin secretory capacity during the last six months preceding IDDM. No effect of antiedematous therapy with ketotifen. Diabetes Care 1994; 17: 138-141 [PMID: 8137684]
99 Carel JC, Boltard C, Eisenbarth G, Bach JF, Bougneres PF. Cyclosporine Delays but does not Prevent Clinical Onset in Glucose Intolerant Pre-type 1 Diabetic Children. J Autoimmun 1996; 9: 739-745
100 Huppmann M, Baumgarten A, Ziegler AG, Bonifacio E. Neonatal Bacille Calmette-Guerin vaccination and type 1 diabetes. Diabetes Care 2005; 28: 1204-1206 [PMID: 15855590]
101 Ziegler AG, Schmid S, Huber D, Hummel M, Bonifacio E. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA 2003; 290: 1721-1728 [PMID: 14519706 DOI: 10.1001/jama.290.13.1721]
102 Dulin WE, Wyse BM. Reversal of streptozotocin diabetes with nicotinamide. Proc Soc Exp Biol Med 1969; 130: 992-994 [PMID: 4304354]
103 Yamada K, Nonaka K, Hanafusa T, Miyazaki A, Toyoshima H, Tarui S. Preventive and therapeutic effects of large-dose nicotinamide injections on diabetes associated with insulitis. An observation in nonobese diabetic (NOD) mice. Diabetes 1982; 31: 749-753 [PMID: 6219022]
389 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Simmons KM et al . Type 1 diabetes: A predictable disease

104 Lazarow A. Protection against alloxan diabetes. Anat Rec 1947; 97: 353 [PMID: 20289367]
105 Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol 2007; 7: 622-632 [PMID: 17641665 DOI: 10.1038/nri2134]
106 Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002; 346: 1692-1698 [PMID: 12037148 DOI: 10.1056/NEJMoa012864]
107 Herold KC , Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 2005; 54: 1763-1769 [PMID: 15919798]
108 Wherrett DK, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Herold KC, Marks JB, Monzavi R, Moran A, Orban T, Palmer JP, Raskin P, Rodriguez H, Schatz D, Wilson DM, Krischer JP, Skyler JS. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet 2011; 378: 319-327 [PMID: 21714999 DOI: 10.1016/S0140-6736(11)60895-7]
109 Herold KC, Gitelman SE, Ehlers MR, Gottlieb PA, Greenbaum CJ, Hagopian W, Boyle KD, Keyes-Elstein L, Aggarwal S, Phippard D, Sayre PH, McNamara J, Bluestone JA. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013; 62: 3766-3774 [PMID: 23835333 DOI: 10.2337/db13-0345]
110 Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ, Bode B, Aronoff S, Holland C, Carlin D, King KL, Wilder RL, Pillemer S, Bonvini E, Johnson S, Stein KE, Koenig S, Herold KC, Daifotis AG. Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011; 378: 487-497 [PMID: 21719095 DOI: 10.1016/S0140-6736(11)60931-8]
111 Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Marks JB, Monzavi R, Moran A, Raskin P, Rodriguez H, Russell WE, Schatz D, Wherrett D, Wilson DM, Krischer JP, Skyler JS. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 2011; 378: 412-419 [PMID: 21719096 DOI: 10.1016/S0140-6736(11)60886-6]
112 Ludvigsson J, Faresjö M, Hjorth M, Axelsson S, Chéramy M, Pihl M, Vaarala O, Forsander G, Ivarsson S, Johansson C, Lindh A, Nilsson NO, Aman J, Ortqvist E, Zerhouni P, Casas R. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med 2008; 359: 1909-1920 [PMID: 18843118 DOI: 10.1056/NEJMoa0804328]
113 Ludvigsson J, Krisky D, Casas R, Battelino T, Castaño L, Greening J, Kordonouri O, Otonkoski T, Pozzilli P, Robert JJ, Veeze HJ, Palmer J, Samuelsson U, Elding Larsson H, Åman J, Kärdell G, Neiderud Helsingborg J, Lundström G, Albinsson E, Carlsson A, Nordvall M, Fors H, Arvidsson CG, Edvardson S, Hanås R, Larsson K, Rathsman B, Forsgren H, Desaix H, Forsander G, Nilsson NÖ, Åkesson CG, Keskinen P, Veijola R, Talvitie T, Raile K, Kapellen T, Burger W, Neu A, Engelsberger I, Heidtmann B, Bechtold S, Leslie D, Chiarelli F, Cicognani A, Chiumello G, Cerutti F, Zuccotti GV, Gomez Gila A, Rica I, Barrio R, Clemente M, López Garcia MJ, Rodriguez M, Gonzalez I, Lopez JP, Oyarzabal M, Reeser HM, Nuboer R, Stouthart P, Bratina N, Bratanic N, de Kerdanet M, Weill J, Ser N, Barat P, Bertrand AM, Carel JC, Reynaud R, Coutant R, Baron S. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med 2012; 366: 433-442 [PMID: 22296077 DOI: 10.1056/NEJMoa1107096]
114 Michels AW. Targeting the trimolecular complex. Clin Immunol 2013; 149: 339-344 [PMID: 23537861 DOI: 10.1016/j.clim.2013.02.020]
115 Michels AW, Ostrov DA, Zhang L, Nakayama M, Fuse M, McDaniel K, Roep BO, Gottlieb PA, Atkinson MA, Eisenbarth GS. Structure-based selection of small molecules to alter allele-specific MHC class II antigen presentation. J Immunol 2011; 187: 5921-5930 [PMID: 22043012 DOI: 10.4049/jimmunol.1100746]
116 Zhang L, Crawford F, Yu L, Michels A, Nakayama M, Davidson HW, Kappler JW, Eisenbarth GS. Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes. Proc Natl Acad Sci USA 2014; 111: 2656-2661 [PMID: 24550292 DOI: 10.1073/pnas.1323436111]
117 Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005; 435: 220-223 [PMID: 15889095 DOI: 10.1038/nature03523]
118 Alleva DG, Crowe PD, Jin L, Kwok WW, Ling N, Gottschalk M, Conlon PJ, Gottlieb PA, Putnam AL, Gaur A. A disease-associated cellular immune response in type 1 diabetics to an immunodominant epitope of insulin. J Clin Invest 2001; 107: 173-180 [PMID: 11160133 DOI: 10.1172/JCI8525]
119 Crawford F, Stadinski B, Jin N, Michels A, Nakayama M, Pratt P, Marrack P, Eisenbarth G, Kappler JW. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci USA 2011; 108: 16729-16734 [PMID: 21949373 DOI: 10.1073/pnas.1113954108]
120 Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci USA 2010; 107: 10978-10983 [PMID: 20534455 DOI: 10.1073/pnas.1006545107]
121 Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med 2011; 208: 1501-1510 [PMID: 21690251 DOI: 10.1084/jem.20110574]
P- Reviewer: Ciccone MM, Faienza MF, Haidara M, Ilangumaran S, Gray SG
S- Editor: Song XX L- Editor: A E- Editor: Liu SQ
390 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Simmons KM et al . Type 1 diabetes: A predictable disease

deep pressure pain perception threshold at musculus abductor hallucis was beyond 1400 kPa (equivalent to 14 kg; limit of measurement) only in every fifth case. These discrepancies of pain perception between forefoot and hindfoot, and between skin and muscle, demand further study. Measuring nociception at the feet in diabetes opens promising clinical perspectives. A critical nociception threshold may be quantified (probably corresponding to a critical number of intraepidermal nerve fibre endings), beyond which the individual risk of a diabetic foot rises appreciably. Staging of diabetic neuropathy according to nociception thresholds at the feet is highly desirable as guidance to an individualised injury prevention strategy.
Key words: Foot ulcer; Neuroarthropathy; Insensitivity to pain; Pain perception; Diabetes mellitus; Amputation; Diabetic neuropathy
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: The diabetic foot is characterised by painless ulcers and/or arthropathy. Although painless diabetic neuropathy is known as the underlying condition, little is known quantitatively about the pain evoked by noxious stimuli (nociception) at the diabetic foot. Preliminary evidence shows that pinprick pain perception threshold at plantar digital skinfolds is supranormal, beyond the upper limits of measurement. It is suggested that measuring nociception at the foot in diabetes could specify the individual risk of painless ulcers and/or arthropathy and, thereby, provide the basis of an individualised graded injury prevention strategy.
Chantelau EA. Nociception at the diabetic foot, an uncharted territory. World J Diabetes 2015; 6(3): 391-402 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/391.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.391
INTRODUCTIONThe diabetic foot (also called diabetic foot syndrome,
Ernst A Chantelau
Ernst A Chantelau, Diabetic Foot Clinic, Heinrich-Heine-University Düsseldorf, 40001 Düsseldorf, GermanyAuthor contributions: Chantelau EA solely contributed to this article.Conflict-of-interest: None.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Ernst A Chantelau, Professor, Diabetic Foot Clinic, Heinrich-Heine-University Düsseldorf, Moorenstraße 5, 40001 Düsseldorf, Germany. [email protected]: +49-421-637196 Received: July 23, 2014Peer-review started: July 25, 2014First decision: September 16, 2014Revised: September 18, 2014Accepted: January 18, 2015Article in press: January 20, 2015Published online: April 15, 2015
AbstractThe diabetic foot is characterised by painless foot ulceration and/or arthropathy; it is a typical complication of painless diabetic neuropathy. Neuropathy depletes the foot skin of intraepidermal nerve fibre endings of the afferent A-delta and C-fibres, which are mostly nociceptors and excitable by noxious stimuli only. However, some of them are cold or warm receptors whose functions in diabetic neuropathy have frequently been reported. Hence, it is well established by quantitative sensory testing that thermal detection thresholds at the foot skin increase during the course of painless diabetic neuropathy. Pain perception (nociception), by contrast, has rarely been studied. Recent pilot studies of pinprick pain at plantar digital skinfolds showed that the perception threshold was always above the upper limit of measurement of 512 mN (equivalent to 51.2 g) at the diabetic foot. However,
REVIEW
391 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Nociception at the diabetic foot, an uncharted territory
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.391
World J Diabetes 2015 April 15; 6(3): 391-402ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

or diabetic podopathy) is characterised by painless foot ulceration and/or arthropathy in diabetes. Foot injuries and inflammations do not hurt, as illustrated on Figure 1.
Painless diabetic neuropathy (PLDN) is the underlying condition. Skin ulcers extending to the subcutaneous tissues are completely anaesthetic, while arthropathy may display faint deep dull aching upon load bearing. Nociception, that is the perception of pain originating from neural processes of encoding and processing noxious stimuli, is failing at the diabetic foot. Although insensitivity to pain at the diabetic foot is common knowledge, details are largely unknown[1,2].
Research into painlessness in diabetes is scarce. Early studies date back to Pamela Margaret Le Quesne[3] and her group almost 30 years ago. They tried to measure the pain perception (nociception) at the diabetic foot by pinching the skin with a custom made “pinchometer”. The results were inconclusive at best[4]. Other authors designed calibrated tools for assessing hypersensitivity of so-called symptomatic, i.e., painful, diabetic neuropathy (SDN, see below). To this end, pinprick pain perception, axon-reflex reaction and temperature detection of the skin were studied. These modalities represent the functions of the afferent A-delta and C-fibres (so-called small fibres; see below) whose contributions to SDN, however, remain controversial[5,6]. SDN will not be discussed here.
To date, measuring pain perception (i.e., noci-ception) at the diabetic foot was deemed futile with the excuse that “The threshold of sensation that protects normal feet from injury is difficult to define…it is extremely difficult to define a ‘significant loss of sensation’, or at what level sensory loss becomes ‘critical’..”[7]. However, there is some dissent on this issue, as expressed by Dyck et al[8]: “Sadly, the clinical assessment of decreased sensation by physicians is generally inadequate because it is not performed or is performed badly.” Undeniably, many a clinical physician or expert was biased against measuring diminished nociception in diabetic neuropathy, and for several possible reasons: (1) quantitative sensory testing (QST, see below) was considered inferior to nerve conduction velocity studies in the detection of symptomatic (painful) diabetic neuropathy; (2) Experts of QST not always knew enough of the diabetic foot, this typical complication of symmetrical diabetic neuropathy, to understand what they read. For example those reviewers, who commented on a paper: “In a group of 20 patients (...) the authors compared sensation in ulcerated vs nonulcerated feet and were unable to find any difference. Had a difference been found, one might interpret the sensory dysfunction as a pathophysiological factor in ulcer development”[9]; (3) QST findings were assumed to be poorly reproducible, an argument which was often mixed up with the natural inter-individual variability (on average 5-fold[10]). Intra-individual coefficients of variation are acceptable, for example those of pain perception thresholds are well
below 25%[10-14]; and (4) A really important obstacle against routine QST of nociceptive functions was the lack of normative data. However, since 2005, extensive reference material using a particular QST protocol was published[12,14-17], see below. Although this protocol had primarily been devised for examining hyperalgesia in non-nociceptive pain syndromes like fibromyalgia, or neuropathic pains, and not for assessing hypoalgesia[18], the author (Chantelau EA) believes that parts of it may well be applied for studying the failing nociception at the foot in diabetes. Diabetic hyperglycaemia in general does not interfere with QST[19-21]; however, subclinical or clinical hypoglycaemia makes QST virtually impossible.
PLDN: THE PRINCIPAL RISK FACTOR FOR THE DIABETIC FOOTPeripheral diabetic (poly) neuropathy is the most frequent sequel of diabetes mellitus. The nature of diabetic neuropathy has been clearly established by Martin[22,23] in 1953 and 1954, and by Catterall et al[24] in 1956. While today many people still believe that the condition is always symptomatic with spontaneous pains and dysaesthesias in the legs and feet, these authors had already noted more than half a century ago: “non-myelinated nerve-fibre degeneration occurs in a high proportion of diabetic patients, who, on clinical examination, show no evidence of ‘clinical’ diabetic neuropathy…the presence in diabetics of such nerve-fibre disturbance explains the frequent finding of persistently cold feet and their proneness to traumatic lesions”[24]. Indeed, the basic feature of diabetic neuropathy is small fibre degeneration[25] with reduced or absent nociception at the feet as the key sign (PLDN). By contrast, SDN develops secondary to PLDN, superimposes on PLDN, and affects only a minority of 15%-30% of patients[25,26]. The symptoms of SDN like spontaneous neuropathic pains and tingling in the feet may be alleviated by drug treatment; they resolve gradually while nerve degeneration[27] and PLDN progress. However, diminished nociception in PLDN does
Chantelau EA. Nociception at the diabetic foot
392 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 1 Sketch reproduced from Brand P[2].
Pain makes a healthy man fall when he begins to twist his foot.The man who feels no pain walks on without realising the damage he is doing.

not resolve, nor does PLDN respond to drug treatment. Prevalence and incidence rates of PLDN are not known. However, the prevalence of the diabetic foot, indicative of severe, end-stage PLDN, in patients with SDN is about 10%, while the incidence is about 5%-7% per year[28,29].
Most of the patients with PLDN never have complaints until they are facing a foot injury that -unexpectedly - does not hurt. To once more quote Catterall et al[24]: “Cutaneous neuropathic changes commonly start as blisters about the tips of the toes or at the site of a corn or callosity in places constantly exposed to irritation by an ill-fitting shoe. Frequently the deceptive lack of normal sensation, and particular pain sensation, leads a patient to ignore the lesion and delay treatment until secondary infection with deep penetration of the tissues or severe inflammation is present.” PLDN is associated to the duration of diabetes and to the cumulative effects of increased blood glucose (or deficient insulin function). It is symmetrical, age- and length-dependent (it starts in advanced age by affecting the endings of the longest axons in the body, in the skin of the toes[30]). Thus, deficits in nociceptive function develop in the forefeet first, and subsequently ascend to the rearfeet, the ankles and the lower legs. The hands are affected only in most severe cases.
PLDN is a disease of the small primary afferent A-delta and C-fibres: they are thin and their conduction velocity is relatively slow. (A fraction of about 15% of small fibres extending to the skin are efferents; they belong to the sympathetic nervous system and are restricted to the dermis, innervating sweat glands, blood vessels, and the arrector pili muscles. Small fibre efferents will not be discussed here). Primary C-afferents (also named group Ⅳ fibres, axon diameter < 1.5 μm, conduction velocity 0.5-2 m/s) are unmyelinated nerve fibres. Primary A-delta afferents (also named group Ⅲ fibres, axon diameter 1-6 μm, conduction velocity 5-30 m/s) consist of thinly myelinated axons whose intracutaneous endings, however, are unmyelinated[31]. The endings of A-delta and C-afferents do not carry corpuscles but are “free”; they serve as receptors for noxious thermal,
mechanical and chemical stimuli (termed nociceptors), and as receptors for innocuous thermal stimuli. In the epidermis they are densely distributed, up to 1000 per 1 cm², which is much more than in muscles, ligaments, joints and bones (periosteum). Most nociceptors are specific of one single modality, while some are polymodal. C-fibre afferents are estimated to account for 70% of all nociceptors in the skin (the remainder are A-delta nociceptors). Cutaneous A-delta nociceptor stimulation results in a short sharp, stinging “first” pain, whereas C-fibre nociceptor stimulation results in prolonged dull, burning “second” pain. In muscles, A-delta and C-fibre nociceptors evoke the same dull pain character[32], see section 4.
In PLDN, the nociceptors degenerate progressively (“die back”) by unknown molecular mechanisms. It is not clear, whether this insidious process of central-peripheral distal axonopathy[33,34] begins already in a prediabetic stage[25]. However, PLDN superimposes on the physiological age-related decline in sensory functions after the age of 50[34-36], and is aggravated by other neuropathic conditions like vitamin B12 deficiency, alcohol toxicity, end-stage renal failure, or exposition to neurotoxic substances or conditions. Clinically, PLDN remains undetected until most nociceptors have died and painlessness of a foot trauma astounds the patient (and the doctor)[24]. The vanishing of intraepidermal free nerve endings can be quantified by histomor-phology[31,37,38]; it correlates to increasing severity of diabetic neuropathy[39-41] (Figure 2), and to rising thermal and pain perception thresholds[34,38,40].
A-delta and C- fibre nociceptors and thermal receptors “die back” not only in the skin, but presumably also inside the adjacent subcutaneous tissues (equivalent to group Ⅲ and Ⅳ fibre nociceptors in muscles, fascia, ligaments and joints) of the diabetic foot. However, this remains to be established by histopathology. A-delta (group Ⅲ) fibres seem to be particularly susceptible to the neuropathic processes, as their functions deteriorate somewhat earlier than C- (group Ⅳ) fibre functions[42].
Diabetes affects small fibres prior to large ones. When large myelinated A-beta afferents (axon diameter 6-12 μmol/L, conduction velocity 30-70 m/s) become affected, the axon and the myelin sheet get damaged, and conduction velocity decreases. A-beta fibres mediate touch, deep pressure and vibration sensation by means of their corpuscular endings: Meissner’s, Merkel’s, and Pacinian corpuscles. A-beta fibres are not involved in nociception, but have a role in allodynia[43] which, however, will not be discussed here.
Diabetic foot: End-stage complication of PLDN The diabetic foot is characterised by painless ulceration and/or arthropathy. Since 60 years or so[22,23,24,44] the condition is well known as an end-stage complication of diabetic neuropathy, of PLDN rather than SDN. The basic defect has frequently been termed “loss of protective sensation”[44], which is, in fact, “loss of
393 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
12
10
8
6
4
2
0
INEF density (number/mm)
HC DC Dmi Dmo Dsev
Figure 2 Intraepidermal nerve fibre ending density at the lower limb in relation to severity of diabetic neuropathy (adapted from Quattrini et al[40,41]). HC: Healthy controls; DC: Diabetic controls; Dmi: Mild SDN; Dmo: Moderate SDN; Dsev: Severe SDN; SDN: Symptomatic diabetic neuropathy; INEF: Intraepidermal nerve fibre ending.
Chantelau EA. Nociception at the diabetic foot

needs to be ascertained”[48]. However, measuring loss of nociception (LON) as yet has no place in assessing neuropathy in diabetes[47].
Current clinical practice of diagnosing a diabetic foot/diabetic podopathyLoss of touch or vibration sensation is not diagnostic of an active diabetic foot, while painlessness of a foot injury (soft tissue or skeletal lesion) or infection in a patient with diabetes mellitus is. Three different clinical pictures of active diabetic foot lesions may be discerned, according to the dominant clinical component: (1) septic; (2) ischaemic; and (3) arthropathic. After healing of such a lesion, the foot is an inactive diabetic foot (with scars, deformities, etc.) and remains a locus minoris resistentiae for the rest of the life. LON traditionally is not assessed systematically, but is taken as matter of fact when a patient with an injured foot does not limp. The patient history of traumatic events is often unproductive as far as normal symptomatology (pain!) is concerned. However, when concurrent symptoms of trauma onset other than pain are concerned, like swelling and erythema, patient history may be very well productive.
QST The QST protocol, published by the German Research Network on Neuropathic Pain and supported by pharmaceutical companies, was designed for non-invasively diagnosing pain syndromes like spontaneous neuropathic pains, fibromyalgia or chronic lower back pain[15,16,18]. It comprises 13 somatosensory sensory tests that measure the function of large (A-beta) and small (A-delta and C-) afferent nerve fibres, and the corresponding functions of the spinothalamic tract (cold detection threshold, warm detection threshold, thermal sensory limen, paradoxical heat sensation, cold pain threshold, heat pain threshold, mechanical detection threshold, mechanical pain threshold, mechanical pain sensitivity, dynamic mechanical allodynia, wind-up ratio, vibration detection threshold, and deep pressure pain threshold). Using this protocol in healthy subjects, a pool of normative reference data from face, hand and foot had been generated[16,17].
QST of pain perception in diabetesOf all sensory modalities that are impaired by diabetes mellitus, the nociceptive pain system is the most relevant. In the following sections, the focus will therefore be directed on pain perception thresholds: elevated thresholds indicate reduced perception of noxious stimuli, and vice versa. Pain tolerance or other sensory modalities (vibration, electrical current, chemicals, touch, thermal perception) will not be addressed. Some technical features of the threshold studies considered here are outlined below, concerning thermal (cutaneous heat and cold), and mechanical (cutaneous and deep pressure)
nociception” (see below). Ulcers and arthropathy start from single injuries (mechanical, thermal or chemical skin wound, or skeletal trauma) which are subjected to persisting repetitive stress (load bearing by walking, because they do not hurt - Figure 1), whereby they enlarge enormously until they become apparent to the patient. The initial injury may also be caused by uninterrupted repetitive mechanical wear and tear (fatigue from overuse, remaining unperceived because of lack of nociception!) The ulcers become infected, and the infection spreads to the adjacent subcutaneous tissues. Bone and joint damage aggravates due to ongoing unprotected walking. When properly treated immediately (like any other acute foot injury in subjects with preserved nociception!), the initial injuries will heal uneventfully. Human experiments showed that wound healing is not impaired by PLDN[45,46]. Perforating neuropathic foot ulcers heal if they are unloaded completely[24], and if arterial blood flow is sufficient and infection is cured. The ulcers frequently break down again when repetitive stress from walking is resumed, “due to faulty and often excessive weight-bearing by the ulcerated area”[23]. Hence, the overall amputation risk is increased: the diabetic foot is the most frequent cause of amputation in industrialised countries.
CURRENT CLINICAL PRACTICE OF DIAGNOSING DIABETIC NEUROPATHYAccording to the common misconception that neur-opathy in diabetes in general is SDN, current guidelines state that the condition be diagnosed and staged semi-quantitatively according to various symptom scores (e.g., Neuropathy Symptoms Score, Neuropathy Disability Score, Michigan Neuropathy Screening Instrument, Toronto Clinical Scoring System). However, testing sensory functions is also accepted: five simple nerve tests are considered diagnostic for symptomatic neuropathy, the 10-g monofilament plus 1 of the following 4: vibration using 128-Hz tuning fork, pinprick sensation, ankle reflexes, vibration perception threshold[47]. All except vibration perception threshold are qualitative tests, all but pinprick sensation are unsuitable for diagnosing PLDN.
Vibration and touch represent A-beta fibre functions, not nociceptive functions. Nevertheless, vibration and touch sensation is used as surrogate markers of the risk of sustaining painless foot injuries (ulceration/arthropathy). To identify loss of protective sensation (LOPS) - without assessing nociception- “any of the five tests listed could be used (…). One or more abnormal tests would suggest LOPS, while at least two normal tests (and no abnormal test) would rule out LOPS”[47].
Current guidelines claim that “estimating the severity of diabetic sensorimotor polyneuropathy has not received the attention it deserves. For a given patient with diabetes it is not sufficient to simply identify patients as having sensorimotor polyneuropathy - severity also
394 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Chantelau EA. Nociception at the diabetic foot

pain perception.
Cutaneous thermal pain perception: Thermal pain perception threshold is measured by applying warm or cold stimuli to the skin, generated by appropriate thermodes (Figure 3). The temperature of the thermode is gradually increased or decreased. The subject under study is asked to indicate whether a thermal sensation becomes painful. Heat pain represents C-fibre function, and cold pain represents A-delta fibre function. In healthy subjects, heat pain perception threshold may range from approximately 41 ℃ to approximately 47 ℃ (average approximately 44 ℃), and cold pain perception threshold ranges from approximately 1 ℃ to approximately 30 ℃ (average approximately 12 ℃).
Deep pressure pain perception: Deep pressure pain perception threshold (DPPPT) may be examined, for instance, by a hand held device (algometer, Figure 4) containing a mechanical or electronic force gauge, and a plunger with a flat blunt rubber tip. The diameter of the tip can be 11 mm (surface approximately 1 cm²), or lager, as appropriate. The plunger is pressed perpendicularly onto the skin and the underlying structures, with slowly increasing stimulus ramp (50 kPa/s, equivalent to 0.5 kg/s). The subject under study communicates when his pressure sensation turns to pain.
The deep pressure pain sensation evoked by an algometer at muscle or joint is as yet not fully under-stood. For instance, the afferents involved (high and low threshold mechanosensitive units[32]), the anatomical structures subjected to algometer pressure, and the role of spatial summation (pressure area 1 cm²)[49,50], still have to be elucidated. The contribution of fascia, which is more pain-sensitive than muscle, and of periosteum, which is more pain-sensitive than bone marrow, remains to be determined. The pain quality at reaching DPPPT is dull and burning and probably more of a pressure discomfort than of a stinging or pricking[49,50]. Inside deep tissues, stimulation of the few A-delta (group Ⅲ) nociceptors simultaneously probably does not elicit a separate pricking pain sensation like a single punctuate stimulation at the skin does[49,51]. DPPPT decreases at chronically ischaemic muscle[52,53]. DPPPT at muscle (blunt stimulation) decreases with age, at variance to cutaneous pressure pain perception threshold (CPPPT, punctuate stimulation)[42]. Using a pressure algometer with 1 cm² contact area to stimulate musculus abductor hallucis, DPPPT in healthy subjects may range from approximately 200 kPa to approximately 1000 kPa (average approximately 500 kPa, equivalent to 5 kg). Algometer stimulation of a joint evokes a DPPPT of the same range.
Cutaneous pressure pain perception: Cutaneous pressure pain perception threshold (CPPPT) is examined by the use of calibrated pinprick stimulators with a sharp edge, comparable to von Frey hairs. These punctuate stimulators are filaments of 50-350 μm diameter, with calibrated bending forces (or weight loads) over a range from 8 mN to 512 mN (equivalent to 0.8 g to 51.2 g) (Figure 5).
The investigator presses each single stimulator perpendicular to the skin (until bending of the filament) and the subject under study communicates, whether a painless touch is felt, or a painful (pricking or stinging) sensation, or nothing at all. The pain threshold is determined by the method of limits, or by the forced-choice-method. The stimulators excite single nociceptors representing A-delta (rather than C-fibre) functions; the
395 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 3 MSA Thermotest® (electronic device, expensive!). Figure 4 Algometer® (electronic device, expensive!).
Figure 5 MARSTOCKnervtest® PinPrick stimulator 512 mN (fibreglass, cheap!).
Chantelau EA. Nociception at the diabetic foot

pain quality evoked is that of a stinging, pricking “first” pain. CPPPT increases with age (at variance to DPPPT, see above). At the non-callous plantar skin of healthy subjects, CPPPT may range from approximately 4 mN to approximately 450 mN (average approximately 100 mN, equivalent to 10 g).
Limitations of pain threshold measurements: QST is a subjective psychophysical method, as it is based on the patient report. “A critical element in pain threshold determination is the particular sensory experience an individual considers painful. This factor will be influenced by, among other things, the subject’s pain experience history, and the instructions given by the experimenter. For instance, thresholds are likely to be different if a subject is instructed to indicate when he perceives ‘pain’ vs when he perceives ‘a sharp or burning sensation’ or ‘an uncomfortable situation’….”[54] In particular, pressure pain measurement is affected by the hardness of the skin at the site of measurement, and by the ramp rate of increasing the stimulus of hand-held algometers, amongst others.
QST of nociception at the diabetic foot Early studies of pain perception thresholds: Between 1988 and 2011, not a single study could be identified on cold pain perception threshold, or on DPPPT. However, there are at least 4 studies on CPPPT and/or heat pain perception threshold[29,55-57], all of which found significantly elevated thresholds at the active (?) diabetic foot and the contralateral foot, as compared to neuropathic patients without diabetic foot (Table 1). In neuropathic patients with and without diabetic foot, pain perception thresholds were significantly higher than in non-neuropathic (healthy or diabetic) control subjects. Lang et al[52] reported that perception thresholds for cold pain, heat pain and pinprick were elevated at legs with chronic severe foot ischaemia (rest pain with/without tissue loss); the 16 patients (8 of whom had diabetes mellitus) had polyneuropathy according to a vibration perception threshold < 4/8 (graduated tuning fork). DPPPT at ischaemic muscle, however, was lower than at healthy controls’ muscle (199 kPa vs 295 kPa[52]). In another study of non-neuropathic patients with chronic leg ischaemia, CPPPT, and DPPPT at muscle, were lower
than in control subjects but cutaneous cold and heat pain perception thresholds were not[53].
Recent pilot studies of pressure pain perception thresholds: Between 2012 and 2014, we conducted four studies on the matter[58-61] including explorative cross-sectional and follow-up studies. Our overall database comprised 123 subjects in whom pressure pain perception thresholds were measured: 43 control subjects, 23 of whom had an acute trauma of the foot skeleton (sprain, minor fracture), 59 diabetic patients with PLDN (46 of whom with a diabetic foot), and 21 diabetic patients without PLDN. Among the 46 patients with a diabetic foot, there were 33 with active painless foot ulcer, 13 with healed foot ulcer, 11 with healed Charcot foot (grade 1, inactive[62]), and 13 with acute skeletal trauma (elective surgery). CPPPT and DPPPT were studied, as well as vibration perception threshold (not reported here). According to our study protocol, CPPPT was measured only at the forefoot (plantar digital skinfold), while DPPPT was measured at the hindfoot (m. abductor hallucis) and at the forefoot (metatarsophalangeal joint) (Figure 6).
By cross-sectional comparison, CPPPT at a plantar digital skinfold (glabrous skin) was above the safety limit of measurement of 512 mN (approximately 51.2 g) in 98% of cases with a past or present painless foot ulcer or healed arthropathy (Charcot foot). This is in line with the older reports cited above. Simultaneously, DPPPT at abductor hallucis muscle was above the safety limit of measurement (1400 kPa, equivalent to 14 kg) only in about 20% of cases. The DPPPT at metatarsaophalangeal joint was above 1400 kPa in about 50% of cases (Table 2).
Serial pressure nociception studies to monitor pain thresholds after an acute foot trauma (ankle sprain, toe fracture) in otherwise healthy subjects revealed that CPPPT and DPPPT at the site of the trauma decreased slightly, indicating normal, inflammation-mediated posttraumatic hyperalgesia (Figure 7). This observation is consistent with previous reports (Martinez et al[63]:
396 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Pinprick stimulator CPPPT
Algometer DPPPT
Algometer DPPPT
Figure 6 Pressure pain perception threshold measurements done in own studies[58-61]. CPPPT: Cutaneous pressure pain perception threshold; DPPPT: Deep pressure pain perception threshold.
Table 1 Early studies of cutaneous pain perception thresholds (pressure, heat) at the diabetic foot, using various, non-standardised methods[29,55,56,57]
Test Diabetic subjects under study Healthy Ref.
Diabetic foot Neuropathy controlsCPPPT 1001 100 0 [29]CPPPT 1001 631 - [55]CPPPT 68 30 2 [56]Heat 1001 100 0 [29]Heat 1001 100 0 [57]
Percentages of supranormal results are shown. 1Above upper limit of measurement. CPPPT: Cutaneous pressure pain perception threshold.
Chantelau EA. Nociception at the diabetic foot

decrements in CPPPT on day 1-4 at an operated knee; Lang et al[53]: reduction in CPPPT and DPPPT in a chronic ischaemic leg up to 3 mo after successful endovascular revascularisation, Dominguez et al[64]: reduced DPPPT at the foot immediately after a sprain). This physiologic, inflammation-mediated posttraumatic hyperalgesia is the essential trigger of avoidance behaviour to preserve the injured limb from further noxious and innocuous impacts.
By contrast, at the diabetic foot CPPPT was extremely elevated (above the safety limit of measurement) prior to an acute foot trauma (elective surgery), and also immediately thereafter (Figure 7A); there was no posttraumatic decrement of CPPPT at the site of the trauma[61]. This suggests absence of A-delta “first” pain quality and inflammation-mediated hyperalgesia. DPPPT at the diabetic foot, being elevated prior to the surgery, showed a small posttraumatic decrement from this baseline level (Figure 7B and C) which, however, was not accompanied by clinical signs of hyperalgesia, or avoidance reaction, respectively.
These data, however, have to be interpreted with caution since our study groups were small (only 12 and 13 subjects, respectively) and heterogeneous. Furthermore, it cannot be ruled out that our study subjects may have behaved like those young healthy women, who reacted to consecutive daily measurements of DPPPT over 4 d by increased pain sensitivity[65] - a reaction that may be an exception rather than the rule[13].
CPPPT within the normal reference range may decrease slightly upon re-testing[14] but remains consistently above the safety limit of measurement in patients with diabetic foot[58-61].
Miscellaneous studies of A-delta and C-fibre sensory functions at the diabetic foot: At least 7 studies[66-72] could be ascertained, all of which reporting that the axon-reflex reaction is not much smaller in neuropathic patients with a diabetic foot than in those without. Axon-reflex reaction in the feet of neuropathic patients was consistently smaller than in the feet of non-neuropathic control subjects.
At least 11 studies[57,67,73-81] could be ascertained, all of which reporting that neuropathic patients with a diabetic foot have higher thermal detection thresholds than neuropathic patients without. Thresholds were higher in the feet of neuropathic patients than of control subjects, and were particularly high in patients with more severe, relapsing diabetic podopathy and/or Charcot foot[76-79]. One study[79] showed a lower cooling detection threshold in the active or inactive Charcot foot vs the contralateral foot, while the warming detection threshold was similar on both feet.
CONCLUSIONAt the diabetic foot, cutaneous thermal and mechanical (pinprick) nociception is reduced dramatically. CPPPT
397 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Pres
sure
(m
N)
600
500
400
300
200
100
0
t / d-1 0 1 2 3 4 5 6 7
Trauma
Healthy Diabetic foot Healthy average
Pres
sure
(kP
a)
800
700
600
500
400
300
200
100
0
t / d-1 0 1 2 3 4 5 6 7
Trauma
Healthy Diabetic foot Healthy average
Figure 7 Lowered pain perception thresholds (hyperalgesia) after acute skeletal trauma of the foot in 13 healthy control subjects (sprain, toe fracture) and in 12 diabetic foot patients (elective surgery) at the injured site. Data are combined from ref. [59,61]. A: Cutaneous pressure pain perception threshold (mN) (pinprick) at plantar digital skinfold (medians); B: Deep pressure pain perception threshold (kPa) over metatarsophalangeal joint (medians); C: Deep pressure pain perception threshold (kPa) over musculus abductor hallucis (medians).
A B C
Table 2 Simultaneous measurements of pressure pain perception thresholds at the foot in subjects with and without painless diabetic neuropathy, with and without diabetic foot
Subject populations Diabetic foot1 Healthy2 PLDN3 DM-controls4
No. of subjects 46 43 13 21% of subjects with CPPPT > 512 mN at toe skinfold 98% 4% 46% 5%% of subjects with DPPPT > 1400 kPa at m. abductor hallucis 22% 0% 0% 0%% of subjects with DPPPT > 1400 kPa at metatarsophalangeal joint 50% 2% 0% 5%
1With previous or active ulcer/arthropathy; 2Non-neuropathic, healthy; 3PLDN, no ulcer; 4Diabetes, no neuropathy, no ulcer. Percent subjects with thresholds above the safety limit of measurement are shown. Adapted from ref. [58-60]. CPPPT: Cutaneous pressure pain perception threshold (512 mN upper limit of measurement); DPPPT: Deep pressure pain perception threshold (1400 kPa upper limit of measurement).
Chantelau EA. Nociception at the diabetic foot
Pres
sure
(kP
a)
800
800700600500
400
300200100
0
t / d-1 0 1 2 3 4 5 6 7
Trauma
Healthy Diabetic foot Healthy average

was elevated beyond the safety limit of measurement in most of the small number of published studies. Patients with diabetic foot ulcers unanimously display a CPPPT at a digital plantar skinfold which is beyond the safety limit of 512 mN. However, details of the causal role of increased CPPPT (indicating lack of A-delta nociception) for the manifestation of diabetic podopathy remain to be established, as well as the anatomical distribution of PLDN at the foot. Concerning DPPPT at the diabetic foot, the data is less clear, as differences to control subjects often were surprisingly small. Compared to baseline DPPPT, inflammation-mediated posttraumatic DPPPT was reduced at the diabetic foot, albeit at a high level and far from indicating hyperalgesia. Pending confirmation, this may suggest that DPPPT probably does not need to be extremely elevated for a diabetic foot to develop.
There are many more open questions, for instance: what number of residual intraepidermal free nerve fibre endings corresponds to the elevation of CPPPT at the diabetic foot? What elevation of CPPPT is necessary to appreciably increase a patient’s risk of sustaining painless foot ulceration and/or arthropathy? How much pain threshold elevation, i.e., how much “LON”, is clinically meaningful? Is the nociception at healthy tissues relevant, or is it the pain perception (hyperalgesia, allodynia), that is typically caused by stimulation of inflamed tissue? Is superficial and deep tissue nociception similarly impaired by PLDN? How much are “silent” nociceptors[82] affected? Is the stinging “first” pain more reduced than the burning “second” pain? Is there a defect in posttraumatic ongoing (non-stimulated) pain? Is absolute or relative reduction of individual pain perception essential for the diabetic foot to develop? Is pain perception or pain tolerance more relevant? Do abnormalities of the central nervous system contribute to the painlessness of the diabetic foot? To what extent is sensitization, either peripheral or central, affected at the diabetic foot? Concerning the discrepancies between CPPPT (at the forefoot) and DPPPT (at muscle of hindfoot) in our own studies[58-61] - does the distal-to-proximal gradient of PLDN provide an explanation?
Measuring nociception at the diabetics' feet-what is it good for?LON is the principal pathogenic component of diabetic podopathy, as it is the principal functional component
of PLDN. Loss of pressure perception does not increase the risk of diabetic foot ulceration, whereas a history of foot ulceration (equivalent to cutaneous LON) does[83]. Measuring nociception provides the opportunity for a clinically meaningful staging of the severity of PLDN. A critical LON needs to be elaborated (probably corresponding to a critical number of intraepidermal nerve fibre endings), “to be able to inform the patient adequately about the risk of ulcer formation and to prescribe preventative measures… otherwise, sensation loss and resultant ulcers may lead to amputation”, as Assal et al[84] advocated 25 years ago. Perhaps every patient may have his/her own individual critical LON to be taken into account. Small reductions in intraepidermal nerve fibre density may be mirrored by respective increases in CPPPT, as recent work by Selim et al[85] shows. Hence, measuring CPPPT prospectively may be suitable for monitoring progression of PLDN.
Shifting the emphasis from amputation prevention by wound healing to amputation prevention by injury prevention: Once an active diabetic foot/podo-pathy has become manifest, a multitude of cumbersome, laborious, tedious, expensive and costly therapeutic interventions is necessary to save the foot and prevent an amputation. Injury prevention in high-risk PLDN - to prevent the diabetic foot- is certainly much more efficient. Preventing the first or recurrent ulcers[86] should be no question. Having had a first ulcer strongly predisposes to subsequent ones, most likely due to comorbidities from structural abnormalities that resulted from the (healed) first ulcer (scars, deformities, contractures, tissue loss). The same holds true for arthropathy. Hence, preventing the first ulcer/arthropathy must have absolute priority. To this end, intensive prophylactic measures are necessary in high-risk patients who should be identifiable by simple means. As a proposal, the following tentative risk-stratification is construed from various published data (assuming a “normal” average walking activity of 5000 strides per day [87]); (Table 3).
Subjects with critical LON at their feet require signals other than foot pain to control foot usage and avoid over-usage. These signals can only come from technical devices, like step counters or load monitors incorporated into footwear, that send acoustic or optical alarms when a critical load (or an equivalent number of steps) has accumulated. Then, the feet have to be rested - for an
398 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 3 Percentages of subjects with active diabetic podopathy (ulcer, arthropathy) according to loss of nociception
Foot morbidity Degree of loss of nociception Ref.
None Mild/moderate Severe(CPPPT1 approximately 100 mN) (CPPPT1 < 512 mN) (CPPPT1 > 512 mN) [59]
Past/present painless ulcer 0% 0% 100% [28,59,77,86-101]Foot ulcer, incidence per year Approximately 0%-1% Approximately 6%-15% Approximately 20%-100%2 [28,77,86-101]Charcot-foot, prevalence Approximately 0% Approximately 0.05% Approximately 2% [102]
1Cutaneous pressure pain perception threshold, pinprick stimulator, upper limit of measurement 512 mN, measured at a plantar digital skinfold; 2Depending on compliance with prophylactic podiatry and/or special footwear. CPPPT: Cutaneous pressure pain perception threshold.
Chantelau EA. Nociception at the diabetic foot

appropriate period of time as physiologically required to regenerate the stressed tissues - before their usage (walking) may be resumed. Moreover, feet with critical LON need special footwear to avoid rubbing of the skin while walking in order to prevent skin abrasion and blister formation.
Pain is a homeostatic emotion that drives behaviour, just as hunger and thirst are. Patients who cannot have it, either from acquired or inherited insensitivity to pain[103,104], either in the feet or elsewhere in the body, need our special protection, and much more specific than we, their caregivers, may have been willing to concede in the past. Research into nociception at the foot in diabetes must continue and expand. Chances to improve injury prevention will be missed, if this uncharted territory remains unexplored.
REFERENCES 1 Brand PW. Tenderizing the foot. Foot Ankle Int 2003; 24: 457-461
[PMID: 12854665]2 Brand P. Insensitive Feet: A Practical Handbook on Foot Problems
in Leprosy. London, UK: The Leprosy Mission, 19663 McDonald WI. Pamela Margaret Le Quesne. B 6 August 1931 d 2
August 1999. Br Med J 1999; 319: 12724 Le Quesne PM, Fowler CJ. A study of pain threshold in diabetics
with neuropathic foot lesions. J Neurol Neurosurg Psychiatry 1986; 49: 1191-1194 [PMID: 3783181 DOI: 10.1136/jnnp.49.10.1191]
5 Chao CC, Tseng MT, Lin YJ, Yang WS, Hsieh SC, Lin YH, Chiu MJ, Chang YC, Hsieh ST. Pathophysiology of neuropathic pain in type 2 diabetes: skin denervation and contact heat-evoked potentials. Diabetes Care 2010; 33: 2654-2659 [PMID: 20841612 DOI: 10.2337/dc10-1135]
6 Schley M, Bayram A, Rukwied R, Dusch M, Konrad C, Benrath J, Geber C, Birklein F, Hägglöf B, Sjögren N, Gee L, Albrecht PJ, Rice FL, Schmelz M. Skin innervation at different depths correlates with small fibre function but not with pain in neuropathic pain patients. Eur J Pain 2012; 16: 1414-1425 [PMID: 22556099 DOI: 10.1002/j.1532-2149.2012.00157.x]
7 Boulton AJ. The pathway to foot ulceration in diabetes. Med Clin North Am 2013; 97: 775-790 [PMID: 23992891 DOI: 10.1016/j.mcna.2013.03.007]
8 Dyck PJ, Herrmann DN, Staff NP, Dyck PJ. Assessing decreased sensation and increased sensory phenomena in diabetic polyneuro-pathies. Diabetes 2013; 62: 3677-3686 [PMID: 24158999 DOI: 10.2337/db13-0352]
9 Zaslansky R, Yarnitsky D. Clinical applications of quantitative sensory testing (QST). J Neurol Sci 1998; 153: 215-238 [PMID: 9511880 DOI: 10.1016/S0022-510X(97)00293-1]
10 Persson AL, Brogårdh C, Sjölund BH. Tender or not tender: test-retest repeatability of pressure pain thresholds in the trapezius and deltoid muscles of healthy women. J Rehabil Med 2004; 36: 17-27 [PMID: 15074434 DOI: 10.1080/16501970310015218]
11 Brennum J, Kjeldsen M, Jensen K, Jensen TS. Measurements of human pressure-pain thresholds on fingers and toes. Pain 1989; 38: 211-217 [PMID: 2780075 DOI: 10.1016/0304-3959(89)90240-6]
12 Rolke R, Andrews Campbell K, Magerl W, Treede RD. Deep pain thresholds in the distal limbs of healthy human subjects. Eur J Pain 2005; 9: 39-48 [PMID: 15629873 DOI: 10.1016/j.ejpain.2004.04.001]
13 Nussbaum EL, Downes L. Reliability of clinical pressure-pain algometric measurements obtained on consecutive days. Phys Ther 1998; 78: 160-169 [PMID: 9474108]
14 Geber C, Klein T, Azad S, Birklein F, Gierthmühlen J, Huge V, Lauchart M, Nitzsche D, Stengel M, Valet M, Baron R, Maier C, Tölle T, Treede RD. Test-retest and interobserver reliability
of quantitative sensory testing according to the protocol of the German Research Network on Neuropathic Pain (DFNS): a multi-centre study. Pain 2011; 152: 548-556 [PMID: 21237569 DOI: 10.1016/j.pain.2010.11.013]
15 Rolke R, Magerl W, Campbell KA, Schalber C, Caspari S, Birklein F, Treede RD. Quantitative sensory testing: a comprehensive protocol for clinical trials. Eur J Pain 2006; 10: 77-88 [PMID: 16291301 DOI: 10.1016/j.ejpain.2005.02.003]
16 Rolke R, Baron R, Maier C, Tölle TR, Treede RD, Beyer A, Binder A, Birbaumer N, Birklein F, Bötefür IC, Braune S, Flor H, Huge V, Klug R, Landwehrmeyer GB, Magerl W, Maihöfner C, Rolko C, Schaub C, Scherens A, Sprenger T, Valet M, Wasserka B. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): standardized protocol and reference values. Pain 2006; 123: 231-243 [PMID: 16697110 DOI: 10.1016/j.pain.2006.01.041]
17 Magerl W, Krumova EK, Baron R, Tölle T, Treede RD, Maier C. Reference data for quantitative sensory testing (QST): refined stratification for age and a novel method for statistical comparison of group data. Pain 2010; 151: 598-605 [PMID: 20965658 DOI: 10.1016/j.pain.2010.07.026]
18 Krumova EK, Geber C, Westermann A, Maier C. Neuropathic pain: is quantitative sensory testing helpful? Curr Diab Rep 2012; 12: 393-402 [PMID: 22623149 DOI: 10.1007/s11892-012-0282-7]
19 Chan AW, MacFarlane IA, Bowsher DR, Wells JC. Does acute hyperglycaemia influence heat pain thresholds? J Neurol Neurosurg Psychiatry 1988; 51: 688-690 [PMID: 3404165 DOI: 10.1136/jnnp.51.5.688]
20 Damci T, Oşar Z, Beyhan S, Ilkova H, Ozyazar M, Gorpe U, Bagriacik N. Does instantaneous blood glucose affect vibration perception threshold measurement using biothesiometer? Diabetes Res Clin Pract 1999; 46: 19-22 [PMID: 10580611 DOI: 10.1016/S0168-8227(99)00064-9]
21 Thye-Rønn P, Sindrup SH, Arendt-Nielsen L, Brennum J, Hother-Nielsen O, Beck-Nielsen H. Effect of short-term hyperglycemia per se on nociceptive and non-nociceptive thresholds. Pain 1994; 56: 43-49 [PMID: 8159440 DOI: 10.1016/0304-3959(94)90148-1]
22 Martin MM. Diabetic neuropathy; a clinical study of 150 cases. Brain 1953; 76: 594-624 [PMID: 13115576 DOI: 10.1093/brain/76.4.594]
23 Martin MM. Neuropathic lesions of the feet in diabetes mellitus. Proc R Soc Med 1954; 47: 139-140 [PMID: 13134195]
24 Catterall RC, MARTIN MM, OAKLEY W. Aetiology and management of lesions of the feet in diabetes. Br Med J 1956; 2: 953-957 [PMID: 13364361 DOI: 10.1136/bmj.2.5006.1432]
25 Malik RA. Which test for diagnosing early human diabetic neuropathy? Diabetes 2014; 63: 2206-2208 [PMID: 24962918 DOI: 10.2337/db14-0492]
26 Tesfaye S, Boulton AJ, Dickenson AH. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care 2013; 36: 2456-2465 [PMID: 23970715 DOI: 10.2337/dc12-1964]
27 Tesfaye S, Tandan R, Bastyr EJ, Kles KA, Skljarevski V, Price KL. Factors that impact symptomatic diabetic peripheral neuropathy in placebo-administered patients from two 1-year clinical trials. Diabetes Care 2007; 30: 2626-2632 [PMID: 17623822 DOI: 10.2337/dc07-0608]
28 Abbott CA, Vileikyte L, Williamson S, Carrington AL, Boulton AJ. Multicenter study of the incidence of and predictive risk factors for diabetic neuropathic foot ulceration. Diabetes Care 1998; 21: 1071-1075 [PMID: 9653597 DOI: 10.2337/diacare.21.7.1071]
29 Benbow SJ, Chan AW, Bowsher D, MacFarlane IA, Williams G. A prospective study of painful symptoms, small-fibre function and peripheral vascular disease in chronic painful diabetic neuropathy. Diabet Med 1994; 11: 17-21 [PMID: 8181246 DOI: 10.1111/j.1464-5491.1994.tb00223.x]
30 Pittenger GL, Ray M, Burcus NI, McNulty P, Basta B, Vinik AI. Intraepidermal nerve fibers are indicators of small-fiber neuropathy in both diabetic and nondiabetic patients. Diabetes Care 2004; 27: 1974-1979 [PMID: 15277426 DOI: 10.2337/diacare.27.8.1974]
399 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Chantelau EA. Nociception at the diabetic foot

31 Provitera V, Nolano M, Pagano A, Caporaso G, Stancanelli A, Santoro L. Myelinated nerve endings in human skin. Muscle Nerve 2007; 35: 767-775 [PMID: 17405136 DOI: 10.1002/mus.20771]
32 Mense S. Algesic agents exciting muscle nociceptors. Exp Brain Res 2009; 196: 89-100 [PMID: 19139871 DOI: 10.1007/s00221-008-1674-4]
33 Spencer PS, Schaumburg HH. Ultrastructural studies of the dying-back process. IV. Differential vulnerability of PNS and CNS fibers in experimental central-peripheral distal axonopathies. J Neuropathol Exp Neurol 1977; 36: 300-320 [PMID: 190358 DOI: 10.1097/00005072-197703000-00006]
34 Le Quesne PM. Persisting nutritional neuropathy in former war prisoners. Br Med J (Clin Res Ed) 1983; 286: 917-918 [PMID: 6299447 DOI: 10.1136/bmj.286.6369.917-a]
35 Bartlett G, Stewart JD, Tamblyn R, Abrahamowicz M. Normal distributions of thermal and vibration sensory thresholds. Muscle Nerve 1998; 21: 367-374 [PMID: 9486866 DOI: 10.1002/(SICI)1097-4598(199803)21:3<367::AID-MUS11>3.0.CO;2-X]
36 Lautenbacher S. Experimental approaches in the study of pain in the elderly. Pain Med 2012; 13 Suppl 2: S44-S50 [PMID: 22497747 DOI: 10.1111/j.1526-4637.2012.01326.x]
37 Beiswenger KK, Calcutt NA, Mizisin AP. Epidermal nerve fiber quantification in the assessment of diabetic neuropathy. Acta Histochem 2008; 110: 351-362 [PMID: 18384843 DOI: 10.1016/j.acthis.2007.12.004]
38 Ragé M, Van Acker N, Knaapen MW, Timmers M, Streffer J, Hermans MP, Sindic C, Meert T, Plaghki L. Asymptomatic small fiber neuropathy in diabetes mellitus: investigations with intraepidermal nerve fiber density, quantitative sensory testing and laser-evoked potentials. J Neurol 2011; 258: 1852-1864 [PMID: 21472496 DOI: 10.1007/s00415-011-6031-z]
39 Arimura A, Deguchi T, Takashima H, Nishio Y. Intraepidermal nerve fiber density decreases in advance of large fiber neuropathy of diabetic patient. Diabetes 2012; 61 Suppl 1: A152
40 Quattrini C, Tavakoli M, Jeziorska M, Kallinikos P, Tesfaye S, Finnigan J, Marshall A, Boulton AJ, Efron N, Malik RA. Surrogate markers of small fiber damage in human diabetic neuropathy. Diabetes 2007; 56: 2148-2154 [PMID: 17513704 DOI: 10.2337/db07-0285]
41 Quattrini C, Jeziorska M, Boulton AJ, Malik RA. Reduced vascular endothelial growth factor expression and intra-epidermal nerve fiber loss in human diabetic neuropathy. Diabetes Care 2008; 31: 140-145 [PMID: 17934147 DOI: 10.2337/dc07-1556]
42 Lautenbacher S, Kunz M, Strate P, Nielsen J, Arendt-Nielsen L. Age effects on pain thresholds, temporal summation and spatial summation of heat and pressure pain. Pain 2005; 115: 410-418 [PMID: 15876494 DOI: 10.1016/j.pain.2005.03.025]
43 Schaible HG, Richter F. Pathophysiology of pain. Langenbecks Arch Surg 2004; 389: 237-243 [PMID: 15034717 DOI: 10.1007/s00423-004-0468-9]
44 Mueller MJ. Etiology, evaluation, and treatment of the neuropathic foot. Critical Reviews in Physical and Rehabilitation Medicine 1992; 3: 289-309
45 Krishnan STM, Baker NR, Rayman G. Wound healing and microvascular responses in the foot skin of type 2 diabetic subjects with neuropathy. Diabetic Med 2005; 22 Suppl 2: 44
46 Krishnan STM, Quattrini C, Jeriyoska M, Malik R, Rayman G. Role of small fibre neuropathy in wound healing in type 2 diabetes. Diabetic Med 2006; 23 Suppl 2: 69
47 Boulton AJ, Armstrong DG, Albert SF, Frykberg RG, Hellman R, Kirkman MS, Lavery LA, Lemaster JW, Mills JL, Mueller MJ, Sheehan P, Wukich DK. Comprehensive foot examination and risk assessment: a report of the task force of the foot care interest group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care 2008; 31: 1679-1685 [PMID: 18663232 DOI: 10.2337/dc08-9021]
48 Tesfaye S, Boulton AJ, Dyck PJ, Freeman R, Horowitz M, Kempler P, Lauria G, Malik RA, Spallone V, Vinik A, Bernardi L, Valensi P. Diabetic neuropathies: update on definitions, diagnostic
criteria, estimation of severity, and treatments. Diabetes Care 2010; 33: 2285-2293 [PMID: 20876709 DOI: 10.2337/dc10-1303]
49 Defrin R, Ronat A, Ravid A, Peretz C. Spatial summation of pressure pain: effect of body region. Pain 2003; 106: 471-480 [PMID: 14659531 DOI: 10.1016/j.pain.2003.09.010]
50 Xiong S, Goonetilleke RS, Jiang Z. Pressure thresholds of the human foot: measurement reliability and effects of stimulus characteristics. Ergonomics 2011; 54: 282-293 [PMID: 21390958 DOI: 10.1080/00140139.2011.552736]
51 Mense SS. [Functional neuroanatomy for pain stimuli. Reception, transmission, and processing]. Schmerz 2004; 18: 225-237 [PMID: 15221425]
52 Lang PM, Schober GM, Rolke R, Wagner S, Hilge R, Offenbächer M, Treede RD, Hoffmann U, Irnich D. Sensory neuropathy and signs of central sensitization in patients with peripheral arterial disease. Pain 2006; 124: 190-200 [PMID: 16716518 DOI: 10.1016/j.pain.2006.04.011]
53 Lang PM, Vock G, Schober GM, Kramer S, Abahji T, Crispin A, Irnich D, Hoffmann U. Impact of endovascular intervention on pain and sensory thresholds in nondiabetic patients with intermittent claudication: a pilot study. J Pain 2009; 10: 264-273 [PMID: 19010739 DOI: 10.1016/j.jpain.2008.09.001]
54 Greenspan JD. Pain in Humans, Thresholds. Schmidt RF, Willis WD, editors. Encyclopedia of Pain. Berlin, Heidelberg, New York: Springer, 2007: 1694-1695 [DOI: 10.1007/978-3-540-29805-2_3100]
55 Tjon-A-Tsien AML, Ellis JA, Lemkes HHPJ. Specific patterns of sensory loss in diabetic neuroarthropathic patients. Diabetologia 1995: 38 Suppl 1: A273
56 Perkins BA, Olaleye D, Zinman B, Bril V. Simple screening tests for peripheral neuropathy in the diabetes clinic. Diabetes Care 2001; 24: 250-256 [PMID: 11213874 DOI: 10.2337/diacare.24.2.250]
57 Krishnan ST, Baker NR, Carrington AL, Rayman G. Comparative roles of microvascular and nerve function in foot ulceration in type 2 diabetes. Diabetes Care 2004; 27: 1343-1348 [PMID: 15161786 DOI: 10.2337/diacare.27.6.1343]
58 Chantelau E, Wienemann T, Richter A. Pressure pain thresholds at the diabetic Charcot-foot: an exploratory study. J Musculoskelet Neuronal Interact 2012; 12: 95-101 [PMID: 22647283]
59 Wienemann T, Chantelau EA. The diagnostic value of measuring pressure pain perception in patients with diabetes mellitus. Swiss Med Wkly 2012; 142: w13682 [PMID: 23037453]
60 Wienemann T, Chantelau EA, Richter A. Pressure pain perception at the injured foot: the impact of diabetic neuropathy. J Musculoskelet Neuronal Interact 2012; 12: 254-261 [PMID: 23196268]
61 Wienemann T, Chantelau EA, Koller A. Effect of painless diabetic neuropathy on pressure pain hypersensitivity (hyperalgesia) after acute foot trauma. Diabetic Foot & Ankle 2014; 5: 24926 [DOI: 10.3402/DFA.V5.24926]
62 Chantelau EA, Grützner G. Is the Eichenholtz classification still valid for the diabetic Charcot foot? Swiss Med Wkly 2014; 144: w13948 [PMID: 24764120 DOI: 10.4414/smw.2014.13948]
63 Martinez V, Fletcher D, Bouhassira D, Sessler DI, Chauvin M. The evolution of primary hyperalgesia in orthopedic surgery: quantitative sensory testing and clinical evaluation before and after total knee arthroplasty. Anesth Analg 2007; 105: 815-821 [PMID: 17717244 DOI: 10.1213/01.ane.0000278091.29062.63]
64 Dominguez S, Salom-Moreno J, Abian-Vicent J, Cleland JA, Fernández-de-Las-Peñas C. Efficacy of thrust and nonthrust manipulation and exercise with or without the addition of myofascial therapy for the management of acute inversion ankle sprain: a randomized clinical trial. J Orthop Sports Phys Ther 2013; 43: 300-309 [PMID: 23485845 DOI: 10.2519/jospt.2013.4467]
65 Jones DH, Kilgour RD, Comtois AS. Test-retest reliability of pressure pain threshold measurements of the upper limb and torso in young healthy women. J Pain 2007; 8: 650-656 [PMID: 17553750 DOI: 10.1016/j.jpain.2007.04.003]
66 Parkhouse N, Le Quesne PM. Impaired neurogenic vascular
400 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Chantelau EA. Nociception at the diabetic foot

response in patients with diabetes and neuropathic foot lesions. N Engl J Med 1988; 318: 1306-1309 [PMID: 3362188 DOI: 10.1056/NEJM198805193182005]
67 Le Quesne PM, Fowler CJ, Parkhouse N. Peripheral neuropathy profile in various groups of diabetics. J Neurol Neurosurg Psychiatry 1990; 53: 558-563 [PMID: 2391517 DOI: 10.1136/jnnp.53.7.558]
68 Walmsley D, Wiles PG. Early loss of neurogenic inflammation in the human diabetic foot. Clin Sci (Lond) 1991; 80: 605-610 [PMID: 1647924]
69 Veves A, Akbari CM, Donaghue VM, Zacharoulis D, Chrzan JS, Freeman R, LoGerfo FW. The effect of diabetes, neuropathy, Charcot arthropathy and arterial disease on the foot microcirculation. Diabetologia 1996; 39 Suppl 1: A3
70 Hamdy O, Abou-Elenin K, LoGerfo FW, Horton ES, Veves A. Contribution of nerve-axon reflex-related vasodilation to the total skin vasodilation in diabetic patients with and without neuropathy. Diabetes Care 2001; 24: 344-349 [PMID: 11213890 DOI: 10.2337/diacare.24.2.344]
71 Krishnan ST, Rayman G. The LDIflare: a novel test of C-fiber function demonstrates early neuropathy in type 2 diabetes. Diabetes Care 2004; 27: 2930-2935 [PMID: 15562209 DOI: 10.2337/diacare.27.12.2930]
72 Baker N, Green A, Krishnan S, Rayman G. Microvascular and C-fiber function in diabetic charcot neuroarthropathy and diabetic peripheral neuropathy. Diabetes Care 2007; 30: 3077-3079 [PMID: 17804679 DOI: 10.2337/dc07-1063]
73 Guy RJ, Clark CA, Malcolm PN, Watkins PJ. Evaluation of thermal and vibration sensation in diabetic neuropathy. Diabetologia 1985; 28: 131-137 [PMID: 3996794]
74 Sosenko JM, Kato M, Soto R, Bild DE. Comparison of quantitative sensory-threshold measures for their association with foot ulceration in diabetic patients. Diabetes Care 1990; 13: 1057-1061 [PMID: 2209302 DOI: 10.2337/diacare.13.10.1057]
75 Marshall A, Young MJ, Boulton AJM. The neuropathy of patients with diabetic Charcot feet: is there a specific deficit? Diabetic Med 1993; 10 Suppl 1: S39
76 Mantey L, Foster A, Spencer S, Edmonds M. Why do foot ulcers recur in diabetic patients? Diabetic Med 1996; 13 Suppl 7: S44
77 Carrington AL, Shaw JE, Van Schie CH, Abbott CA, Vileikyte L, Boulton AJ. Can motor nerve conduction velocity predict foot problems in diabetic subjects over a 6-year outcome period? Diabetes Care 2002; 25: 2010-2015 [PMID: 12401748 DOI: 10.2337/diacare.25.11.2010]
78 Stevens MJ, Edmonds ME, Foster AV, Watkins PJ. Selective neuropathy and preserved vascular responses in the diabetic Charcot foot. Diabetologia 1992; 35: 148-154 [PMID: 1547919 DOI: 10.1007/BF00402547]
79 van Schie CHM, Carrington AL, Abouaesha F, Boulton JMN, Jude EB. Neuropathic and vascular profiles in diabetic patients with Charcot neuroarthropathy. Diabetes 2002; 51 Suppl 2: A543
80 Ali Z, Carroll M, Robertson KP, Fowler CJ. The extent of small fibre sensory neuropathy in diabetics with plantar foot ulceration. J Neurol Neurosurg Psychiatry 1989; 52: 94-98 [PMID: 2540287 DOI: 10.1136/jnnp.52.1.94]
81 Purewal TS, Wilson S, Watkins PJ, Edmonds ME. Predictive features of the acute diabetic Charcot foot: a prospective study. Abstract. Diabetic Med 1995; 12 Suppl 1: S47
82 Schaible HG, Richter F, Ebersberger A, Boettger MK, Vanegas H, Natura G, Vazquez E, Segond von Banchet G. Joint pain. Exp Brain Res 2009; 196: 153-162 [PMID: 19363606 DOI: 10.1007/s00221-009-1782-9]
83 Lavery LA, Armstrong DG, Wunderlich RP, Tredwell J, Boulton AJ. Predictive value of foot pressure assessment as part of a population-based diabetes disease management program. Diabetes Care 2003; 26: 1069-1073 [PMID: 12663575 DOI: 10.2337/diacare.26.4.1069]
84 Assal JP, Lindblom U. Sensory thresholds. Diabetes Care 1989; 12: 43-44 [PMID: 2714169]
85 Selim MM, Wendelschafer-Crabb G, Hodges JS, Simone DA,
Foster SX, Vanhove GF, Kennedy WR. Variation in quantitative sensory testing and epidermal nerve fiber density in repeated measurements. Pain 2010; 151: 575-581 [PMID: 20851518 DOI: 10.1016/j.pain.2010.06.034]
86 Lavery LA, La Fontaine J, Kim PJ. Preventing the first or recurrent ulcers. Med Clin North Am 2013; 97: 807-820 [PMID: 23992893 DOI: 10.1016/j.mcna.2013.05.001]
87 Kanade RV, van Deursen RW, Harding K, Price P. Walking performance in people with diabetic neuropathy: benefits and threats. Diabetologia 2006; 49: 1747-1754 [PMID: 16758177 DOI: 10.1007/s00125-006-0309-1]
88 Abbott CA, Carrington AL, Ashe H, Bath S, Every LC, Griffiths J, Hann AW, Hussein A, Jackson N, Johnson KE, Ryder CH, Torkington R, Van Ross ER, Whalley AM, Widdows P, Williamson S, Boulton AJ. The North-West Diabetes Foot Care Study: incidence of, and risk factors for, new diabetic foot ulceration in a community-based patient cohort. Diabet Med 2002; 19: 377-384 [PMID: 12027925 DOI: 10.1046/j.1464-5491.2002.00698.x]
89 McGill M, Molyneaux L, Yue DK. Which diabetic patients should receive podiatry care? An objective analysis. Intern Med J 2005; 35: 451-456 [PMID: 16176466 DOI: 10.1111/j.1445-5994.2005.00880.x]
90 Pham H, Armstrong DG, Harvey C, Harkless LB, Giurini JM, Veves A. Screening techniques to identify people at high risk for diabetic foot ulceration: a prospective multicenter trial. Diabetes Care 2000; 23: 606-611 [PMID: 10834417 DOI: 10.2337/diacare.23.5.606]
91 Tanudjaja T, Chantelau E. Recurrent neuropathic foot ulcer disease in diabetes mellitus. Diabetologia 1996; 39 Suppl 1: A264
92 Peters EJ, Lavery LA. Effectiveness of the diabetic foot risk classification system of the International Working Group on the Diabetic Foot. Diabetes Care 2001; 24: 1442-1447 [PMID: 11473084 DOI: 10.2337/diacare.24.8.1442]
93 Benbow SJ, Chan AW, Bowsher DR, Williams G, Macfarlane IA. The prediction of diabetic neuropathic plantar foot ulceration by liquid-crystal contact thermography. Diabetes Care 1994; 17: 835-839 [PMID: 7956627 DOI: 10.2337/diacare.17.8.835]
94 Striesow F. [Special manufactured shoes for prevention of recurrent ulcer in diabetic foot syndrome]. Med Klin (Munich) 1998; 93: 695-700 [PMID: 10024836 DOI: 10.1007/BF03044805]
95 Uccioli L, Faglia E, Monticone G, Favales F, Durola L, Aldeghi A, Quarantiello A, Calia P, Menzinger G. Manufactured shoes in the prevention of diabetic foot ulcers. Diabetes Care 1995; 18: 1376-1378 [PMID: 8721941 DOI: 10.2337/diacare.18.10.1376]
96 Chantelau E, Haage P. An audit of cushioned diabetic footwear: relation to patient compliance. Diabet Med 1994; 11: 114-116 [PMID: 8181241 DOI: 10.1111/j.1464-5491.1994.tb00240.x]
97 Busch K, Chantelau E. Effectiveness of a new brand of stock ‘diabetic’ shoes to protect against diabetic foot ulcer relapse. A prospective cohort study. Diabet Med 2003; 20: 665-669 [PMID: 12873296 DOI: 10.1046/j.1464-5491.2003.01003.x]
98 Köhler G, Haas W, Rakovac I, Schintler M, Bock G, Korsatko S, Plank J, Pieber TR. Evaluation of the impact of chiropodist care in the secondary prevention of foot ulcerations in diabetic subjects over a period of 6 years in Austria. Diabetologia 2007; 50 Suppl.2: S459
99 Plank J, Haas W, Rakovac I, Görzer E, Sommer R, Siebenhofer A, Pieber TR. Evaluation of the impact of chiropodist care in the secondary prevention of foot ulcerations in diabetic subjects. Diabetes Care 2003; 26: 1691-1695 [PMID: 12766095 DOI: 10.2337/diacare.26.6.1691]
100 Rizzo L, Tedeschi A, Fallani E, Coppelli A, Vallini V, Iacopi E, Piaggesi A. Custom-made orthesis and shoes in a structured follow-up program reduces the incidence of neuropathic ulcers in high-risk diabetic foot patients. Int J Low Extrem Wounds 2012; 11: 59-64 [PMID: 22336901 DOI: 10.1177/1534734612438729]
101 Waaijman R, Keukenkamp R, de Haart M, Polomski WP, Nollet F, Bus SA. Adherence to wearing prescription custom-made footwear in patients with diabetes at high risk for plantar foot ulceration.
401 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Chantelau EA. Nociception at the diabetic foot

Diabetes Care 2013; 36: 1613-1618 [PMID: 23321218 DOI: 10.2337/dc12-1330]
102 Margolis D. Epidemiology of Charcot neuroarthropathy. NIDDK Workshop September 9-11, 2008. Bethesda, Maryland/USA. National Institute of Diabetes and Digestive and Kidney Diseases, 2008. Available from: URL: http//archives.niddk.nih.gov/
neuroarthropathy/SummaryReport.pdf103 Minde JK. Norrbottnian congenital insensitivity to pain. Acta
Orthop Suppl 2006; 77: 2-32 [PMID: 16768023]104 Nagasako EM, Oaklander AL, Dworkin RH. Congenital insensitivity
to pain: an update. Pain 2003; 101: 213-219 [PMID: 12583863 DOI: 10.1016/S0304-3959(02)00482-7]
P- Reviewer: Cui WP, Trohman RG S- Editor: Tian YL L- Editor: A E- Editor: Liu SQ
402 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Chantelau EA. Nociception at the diabetic foot

Francesco Fallucca, Lucia Fontana, Sara Fallucca, Mario Pianesi
Francesco Fallucca, “In Unam Sapientiam” Foundation, University La Sapienza, 00161 Rome, Italy Francesco Fallucca, Mario Pianesi, International Study Center for Environment, Agriculture, Food, Health and Economics, 00161 Rome, ItalyLucia Fontana, Unit of Dietology and Diabetology, Sandro Pertini Hospital, 00157 Rome, ItalySara Fallucca, Department of Endocrinology and Diabetes, University Campus Bio-Medico, 00128 Rome, ItalyAuthor contributions: Fallucca F and Pianesi M conceived and designed the manuscript; Fallucca F, Fontana L and Fallucca S searched the literature and selected relevant studies; Fallucca F and Fontana L drafted the manuscript; Fallucca S and Pianesi M critically reviewed the manuscript drafts; all authors read and approved the final manuscript.Conflict-of-interest: The authors declare that they have no conflicts of interest.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Francesco Fallucca, Professor, “In Unam Sapientiam” Foundation, University La Sapienza, Largo Ettore Marchiafava 1, 00161 Rome, Italy. [email protected]: +39-06-8605616Received: August 28, 2014 Peer-review started: August 30, 2014 First decision: December 17, 2014Revised: January 10, 2015Accepted: January 30, 2015Article in press: February 2, 2015Published online: April 15, 2015
AbstractIn the past 10 years the prevalence of type 2 diabetes mellitus (T2DM) has increased hugely worldwide, driven by
a rise in the numbers of overweight and obese individuals. A number of diets have been shown to be effective for the management of T2DM: the Mediterranean diet, the vegetarian diet and the low-calorie diet. Results of studies clearly indicate, however, that the efficacy of these diets is not solely related to the biochemical structure of the individual nutrients they contain. This review discusses this point with reference to the potential role of the intestinal microbiota in diabetes. The macrobiotic Ma-Pi 2 diet is rich in carbohydrates, whole grains and vegetables, with no animal fat or protein or added sugar. In short- and medium-term trials conducted in patients with T2DM, the Ma-Pi 2 diet has been found to significantly improve indicators of metabolic control, including fasting blood glucose, glycosylated hemoglobin, the serum lipid profile, body mass index, body weight and blood pressure. The diet may also alter the gut microbiota composition, which could additionally affect glycemic control. As a result, the Ma-Pi 2 diet could be considered a valid additional short- to medium-term treatment for T2DM.
Key words: Ma-Pi 2 macrobiotic diet; Type 2 diabetes; Low-grade inflammation; Gut microbiota; Metabolic control
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Imbalances in the intestinal microbiota (dysbiosis) have been linked to diseases, including diabetes. In short- and medium-term trials conducted in patients with type 2 diabetes mellitus (T2DM), the Ma-Pi 2 diet, which is rich in carbohydrates, whole grains and vegetables, with no animal fat or protein or added sugar, has been found to significantly improve indicators of metabolic control, including fasting blood glucose, glycosylated hemoglobin, the serum lipid profile, body mass index, body weight and blood pressure. The diet may also alter the gut microbiota composition. Hence, the Ma-Pi 2 diet could be considered a valid additional short- to medium-term treatment for T2DM.
REVIEW
403 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Gut microbiota and Ma-Pi 2 macrobiotic diet in the treatment of type 2 diabetes
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.403
World J Diabetes 2015 April 15; 6(3): 403-411ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

Fallucca F, Fontana L, Fallucca S, Pianesi M. Gut microbiota and Ma-Pi 2 macrobiotic diet in the treatment of type 2 diabetes. World J Diabetes 2015; 6(3): 403-411 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/403.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.403
INTRODUCTIONIn the past 10 years, the prevalence of type 2 diabetes mellitus (T2DM) in Italy has increased greatly; currently, approximately 3.3 million people are diagnosed with T2DM (5.5% of the total population) and another million people are thought to be affected but as yet undiagnosed. These numbers are expected to increase in the near future, and it has been estimated that 9.0% of the population will have T2DM by 2030[1]. Similar increases are anticipated in other European countries and, globally, the number of people with diabetes is predicted to reach 592 million by the year 2035 - an increase of 55% from 2013 data[2].
This increase in the prevalence of T2DM, which accounts for approximately 90% of all diabetes cases, is clearly being driven by an increase in the numbers of individuals who are overweight or obese[3]. Indeed, the risk of diabetes appears to literally soar as the weight piles on; a rise in body mass index (BMI) from 21 kg/m2 (healthy) to 35 kg/m2 (obese) can increase the likelihood of developing the disease by a factor of 80[4]. In line with the increase in prevalence of diabetes, the number of obese adults, together with the rates of obesity-related diseases (e.g., coronary heart disease and stroke, hypertension and arthritis) and associated healthcare costs, are all expected to increase dramatically worldwide over the next 20 years[4,5].
T2DM and obesity, and their associated com-plications and costs, are therefore among the most pressing healthcare problems that society is currently facing. Despite improvements in our understanding of T2DM and the development of new diagnostic and treatment methods, we are still failing to control this epidemic. There is a crucial need to find effective, simple, pragmatic, sustainable and cheap interventions (e.g., those aimed at changing lifestyle, such as exercise and diet).
Recent reviews highlight the benefits of physical exercise in T2DM[6]. In Italy, the Italian Diabetes and Exercise Study[7-10] has contributed to this evidence and has helped provide definitions and standards used by the American College of Sports and Medicine and the American Diabetes Association (ADA). Yet, despite the accepted benefits of physical exercise for patients with T2DM, the implementation of exercise recommendations has proved difficult. Reasons for this include a lack of patient compliance, insufficient knowledge/awareness of benefits among general practitioners, diabetologists or exercise professionals, and a lack of dedicated facilities[11]. In addition, in order to be successful, exercise interventions in individuals
with T2DM tend to require intensive counseling and supervision from medical or exercise professionals.
As a result, recent attention has focused less on exercise and more on diet; dietary therapy (as a life-style intervention), with or without additional drug treatment, represents an efficacious and safe alternative for T2DM management[12]. Various cohort studies have shown that selected healthy eating patterns, mainly characterized by a higher intake of fruit and vegetables, are associated with a lower risk of diabetes[13]. In this review, we look at some of the various diets that have been shown to be effective for the management of T2DM, in particular the Ma-Pi 2 macrobiotic diet, and explore the potential role of the intestinal microbiota in T2DM.
MEDITERRANEAN DIETA Mediterranean-type diet is one involving: a high consumption of cereals, grains, fruits, vegetables, legumes, nuts; olive oil as the principal source of fat; low-to-moderate consumption of fish and poultry; relatively low consumption of red meat; and moderate consumption of wine, normally with meals[14].
Based on this definition, numerous epidemiological studies have indicated the favorable effects of this diet[15-17]. Such diets have been reported in two pro-spective studies[18,19] and one intervention study[20] to be associated with a reduced risk of diabetes. However, the role of the Mediterranean diet in weight control remains controversial[21], which suggests that the protective effect of this diet against diabetes are not based on weight control but through several of its dietary characteristics. Currently it is unclear which component of the Mediterranean diet contributes most to its favorable effects.
LOW CALORIE DIETWeight loss following a reduction in energy intake (caloric restriction) and/or an increase in energy expenditure (through exercise) improves insulin sensitivity, hyper-glycemia and other cardio-metabolic risk factors[22]. Caloric restriction (CR) is defined as a reduction in caloric intake by around 20%-40%, with adequate intakes of protein and micronutrients to avoid malnutrition[23,24]. Caloric intake can be reduced by eliminating the consump-tion of energy-dense foods (e.g., refined carbohydrates, potatoes, white bread, white rice, sweets and sweetened drinks) and by increasing the intake of a nutrient-dense foods (a wide variety of vegetables, fruits, nuts, low-fat dairy products, egg whites, wheat and soy proteins, fish and meat). The energy intake of such a diet is around 1100-2000 kcal/d, with approximately 26% of calories obtained from protein, 28% from fat and 46% from complex carbohydrates[23]. CR may be administered as a short- (1 mo-1 year) or long-term diet. CR in healthy individuals as well as those who are obese or have T2DM has been reported to lower blood glucose
Fallucca F et al . Gut microbiota, Ma-Pi 2 diet and T2DM
404 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

levels, fasting insulin values and BMI[25]. Weight loss induced by caloric restriction mediates a reduction in inflammatory markers and improves insulin resistance and sensitivity[26]. With adequate nutrition, CR can improve cardio-metabolic health, prevent T2DM and may be a powerful tool against obesity and insulin resistance.
VEGETARIAN DIETVegetarian diets typically include large amount of fruits, vegetables and legumes, nuts and soya-protein and usually aim to eliminate all animal products. Studies have shown that, generally, vegetarians residing in Western countries have a lower BMI and a higher ratio of polyunsaturated to saturated fat intake than individuals consuming a non-vegetarian diet (people eating meat and/or fish)[27,28]. Vegetarians have also been shown to have lower concentrations of low-density lipoprotein (LDL)-cholesterol[29-31], lower blood pressure[32,33] and a lower risk of diabetes[34] than non-vegetarians. Factors associated with the higher fiber content in vegetarian diets promote increased insulin sensitivity[35]. Vegetarian diets are also associated with other health benefits. According to the largest study ever conducted in the United Kingdom from the University of Oxford, the risk of hospitalization or death from heart disease was 32% lower in vegetarians than in people who ate meat and fish, with most of the difference in risk probably caused by effects on cholesterol and blood pressure[36]. In addition to a lower risk of cardiovascular disease[37,38], vegetarian diets have been reported to reduce the incidence of cancer in a low-risk population[39,40].
MICROBIOTAThe results of some studies indicate that the efficacy of a diet is not solely related to the biochemical structure
of the individual nutrients it contains and the current standard definition of macronutrients fails to capture important information[41]. This has become more obvious as knowledge of the gut microbiota[42-46] has been gained and the “food as hormone” hypothesis[47] has emerged.
The human gut microbiota weighs about 1500 g and the number of intestinal microbial cells it contains is tenfold greater than the total number of human body cells[48]. The gut microbiome, which represents the collective genomes of all gut microbiota, is 150 times greater than the human gene complement[46]. The gut microflora plays several metabolic active roles: it produces vitamins, synthesizes amino-acids, transforms bile acid, and is able to ferment non-digestible substrates and endogenous mucus, stimulating bacterial growth and producing short-chain fatty-acids (SCFA)[49], which work in the gut and at distance after absorption[49]. Moreover, the gut microbiota also causes pathogen displacement, by competing for attachment sites and nutrients, and by secreting antimicrobials[50]. The microbiota plays a fundamental role in the development of the immune system[51], as SCFA have a strong immunomodulatory activity, leading to the production and release of cytokines, chemokines and phagocytes[52].
The gut microbiota is also involved in the development of cells and tissues[53], and is responsible for the integrity of the gut barrier[53] through its involvement in glucagon-like peptide (GLP)-2 production by enteroendocrine L-cells, and the circulation of endotoxins[54]. In addition, the microbiota triggers the production of peptide YY, which regulates gut motility and the hungry sensation[55].
Dysbiosis is a bacterial imbalance in the gut resulting in a modification of the normal local distribution of microbial communities that correlates with biochemical and clinical modifications in the host: hyperglycemia and T2DM have been observed in the presence of a low percentage of bacterial Firmicutes and Clostridia species[56]. Diet and increase in bodyweight are linked to gut microbial imbalance, in both the presence and absence of obesity[57].
Recent studies have shown a link between dysbiosis and several diseases, including diabetes (Figure 1).
Obesity and T2DM are associated with chronic low-grade inflammation and endotoxemia. Lipopolysaccharide (LPS) consumption and a diet rich in fat increase adiposity, impaired glucose tolerance, and insulin resistance those raises are reversible by the consumption of a pre-probiotic rich diet[44,58,59], or a meal based on fiber and fruit, as recommended by the American Heart Association[60]. Bacterial LPS derived from Gram-negative bacteria residing in the gastrointestinal microflora could act as a trigger for the development of diabetes and obesity; in a series of experiments in mice, it was observed that high-fat feeding favors an increase in the Gram-negative to Gram-positive colonizing bacteria ratio, which has been associated with an increased intestinal permeability that precedes the development of metabolic endotoxemia, inflammation, and associated disorders (obesity and diabetes)[44]. Different nutrients have different enhancing-
405 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Dysbiosis
Microbiota and human diseasesPubMed July 2014
Obesity (661)Diabetes (408)
Allergy (462)
Autoimmune diseases (179)
CVD (157)
Atherosclerosis (56)
Ageing (158)
ALS (2)Others???
Alzheimer (3)
Multiple sclerosis (23)
Hypertension (39)
Metabolic syndrome (182)
Liver diseases (518)
Cancer (671)
Hyperlipemia (10)
Figure 1 Articles indexed in PubMed concerning the microbiota and human diseases. Results of a research in PubMed looking for articles concerning gut microbiota that associated dysbiosis and human diseases. Number of articles for each kind of disease is indicated in brackets. CVD: Cardiovascular diseases; ALS: Amyotrophic lateral sclerosis.
Fallucca F et al . Gut microbiota, Ma-Pi 2 diet and T2DM

ability to work in a manner similar to that of hormones. Identifying these food- and food metabolite-receptor interactions will provide new opportunities for studying the relationship between the food we eat and diseases, including obesity.
MA-PI 2 MACROBIOTIC DIETMacrobiotic diets are originally derived from an ancient Eastern philosophy of life based on two ancient Asian theories (Yin/Yang and the Five Transformations), formulated for Western culture by the Japanese philo-sopher Georges Ohsawa[76] and further updated by Mario Pianesi who created the 5 Ma-Pi diets[77]. The Ma-Pi 2 diet was specifically designed for patients with metabolic disorders, and consists of 50%-55% whole-grain (rice, millet and barley), 35%-40% vegetables (carrots, savoy cabbage, chicory, red radish, onions, parsley, cabbage) and 8%-10% legumes (adzuki beans, chickpeas, lentils and black beans), plus gomashio (roasted ground sesame seeds with unrefined sea salt), and fermented products [miso, wandadou jiangyou (soy sauce) and yanzimei (pickled ume plums)], which could have probiotic effect; seaweeds [Kunbu (Laminaria japonica, Aresch), Qundaicai (Undaria pinnatifida, Harv.), Haitai (Porphyria tenera, Kjell.) and Hiziki (Sargassum fusiforme, Harv.)] and Beicha tea (caffeine-free green tea). The daily average energy intake is in the range of 1700-2200 kcal (12% from proteins, 18% from fats and 70% from carbohydrates, mainly complex). The Ma-Pi 2 diet contains, on average, 18% saturated, 46% monounsaturated and 36% polyunsaturated fat, with no trans-fatty acids and an n-6:n-3 polyunsaturated fatty acid ratio of 5:1. It provides nutrients and phytocompounds with antioxidant, hypoglycemic and hypolipidemic effects, such as vitamin C, β carotene, magnesium (average 700 mg/d), manganese (average 16 mg/d), zinc (average 15 mg/d), chromium, phytosterols (average 326 mg/d), dietary fiber (average 50-60 g/d), inulin (average 9 g/d), polyphenols, tocotrienols, folates (more than 500 µg/d), quercetin, and prebiotic and probiotic products. The Ma-Pi 2 diet excludes all animal products, (including egg and dairy), and has no added sugars[77].
All of the ingredients used to prepare the Ma-Pi 2 diets are grown, stored and processed without the use of synthetic chemicals. The crops are from old seed varieties, which were produced using natural methods. All of the products used are labeled, providing detailed information on the origin and characteristics of the product and its supply chain (the Pianesian Transparent Label)[78]. The nutritional content of one Ma-Pi 2 diet used in a clinical study in T2DM patients is presented in Table 1[79].
Since 2001, various clinical studies have been carried out to assess the effect of the Ma-Pi 2 diet in T2DM patients.
Single-arm studies of the Ma-Pi 2 diet in patients with T2DM conducted in several countries and continents(e.g., America, Asia, Africa and Europe) have consistently
effects on endotoxemia (LPS production by microflora or for higher gut permeability) and the consequent inflammatory response[61]. Other factors may also influence our microbiota, such as age[62-64], chemical products and antibiotics[65], and prebiotics and probiotics[58,66-68].
The internationally endorsed definition of probiotics is “live microorganisms which when administered in adequate amounts confer a health benefit on the host”[69],whereas, definition for prebiotics is “a selectively fermented ingredient that allows specific changes, both in the composition and/or activity in the gastrointestinal microflora, that confer benefits upon host well-being and health”[66]. Therefore, in order to be classified as a prebiotic, the ingredient should resist gastric acidity, hydrolysis and should be fermented by the intestinal microflora. The consumption of prebiotic foods enhances a reduction of triglyceride and postprandial glucose levels, gut permeability and inflammatory effect[66,70]. It has been observed that administering prebiotics to obese mice induces a microbiota modulation, lowering both diet-induced LPS endotoxemia, and systemic inflammation and liver inflammation[44]. The main positive effects of pro- and prebiotic administration are increases in GLP-1 and peptide YY secretion[71-73], a reduction in the complications of pregnancy[74], and improvements in metabolic diseases (obesity, T2DM, cardiovascular disease)[75].
New findings in microbiota knowledge are opening new fields of study about human health and disease. However, the research regarding the interaction between bacteria and human health is at the beginning and many aspects are still not properly understood; well-defined clinical studies are needed in order to develop the potential of this new therapeutic area.
Until now, scientists have focused primarily on identifying human genetic markers, but research has shown that other factors, such as the intestinal flora, play a role in the development of T2DM[45,46]. The results of metagenome-wide association studies in T2DM patients and non-diabetic controls[45,46] clearly indicate that T2DM is a significant factor in the observed variation in gut microbial samples, and suggest that the gut microbiota in T2DM patients features dysbiosis. These studies[45,46] indicate that a gut-microbiota-based index could be more effective and accurate in assessing and predicting the risk of T2DM than a human genome variation-based one.
Knowledge of the microbiota and the general concept of “food as hormone”, as proposed by Ryan et al[47], suggest that diet has an enormous impact on our health. In fact, circulating substrates derived from food have both direct and indirect actions and, ultimately, food may be considered a cocktail of hormones that exert their effects on target tissues, activating cell-surface or nuclear receptors. Food components also interact with the gut flora to induce indirect signals. Therefore, if we consider this new role for food, we can make dietary recommendations to promote health or treat specific diseases, taking into account that until now macronutrients have been classified according to their energy-yielding biochemical properties and not by their
406 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Fallucca F et al . Gut microbiota, Ma-Pi 2 diet and T2DM

shown that consumption of the diet for 3 or 6 mo resulted in statistically significant improvements from baseline in indicators of metabolic control, including fasting blood glucose (FBG), glycosylated hemoglobin (HbA1c), the serum lipid profile [including reductions in total cholesterol, LDL cholesterol and triglyceride values, and the LDL:high-density lipoprotein (HDL) cholesterol ratio], BMI, and insulin resistance, as well as body weight and blood pressure (P < 0.001) (Table 2)[79,80-84]. These results were apparent even in patients with poor glycemic control (HbA1c > 8.5%)[85]. In patients being treated with insulin, use of this agent fell during the intervention[79,85].
Similar results were obtained later in short-term (21-d) intervention studies[86,87], thus demonstrating the ability of the diet to achieve rapid metabolic control.
The first randomized, controlled trial comparing the Ma-Pi 2 diet (in 25 participants) with Italian dietary recommendation for T2DM (in 26 participants)-the MADIAB trial-was carried out in 2013[88]. After 21 d on the prescribed diet, which was administered under supervised conditions, the average daily energy intake was 1803 kcal (12% protein, 15% fat, and 73% complex carbohydrates, with 29 g/1000 kcal fiber) in the Ma-Pi 2 group and 1798 kcal (18% protein, 32% fat,
and 49% complex carbohydrates, with 20.5 g/1000 kcal fiber) in the control group (P = 0.860).
The multivariate analysis (adjusted for age, gender, BMI at baseline, and physical activity) showed that significantly higher percentage reductions in FBG, post-prandial blood glucose (PPBG), HbA1c, total cholesterol, LDL-cholesterol, LDL:HDL ratio, BMI, weight, waist and hip circumference, and insulin resistance were seen in the Ma-Pi 2 group than in the control group. Although the triglyceride levels were reduced in all subjects, the reduction detected in the controls was significantly higher than that in intervention subjects. Furthermore, all subjects in the Ma-Pi 2 group achieved FBG and PPBG target levels (< 110 mg/dL for FBG and < 140 mg/dL for PPBG) at the end of the 21-d dietary treatment[88].
Further results from this trial, presented at the 2014 ADA 74th Scientific Sessions, have also demonstrated that the Ma-Pi 2 diet is a safe strategy by which to reduce markers of insulin resistance and inflammation. In addition, a significant reduction in insulin-like growth factor-1 was observed in the Ma-Pi 2 subjects vs the controls (P < 0.001)[89].
The MADIAB trial is also currently assessing thehypothesis that the Ma-Pi 2 diet may affect the com-position of the gut microbiota to a greater extent than the control diet. The results of this analysis will be published later this year.
These results strengthen the idea that the Ma-Pi 2 diet could play an important role in the treatment of T2DM,
407 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 1 Example of the average daily energy and nutrient intake for the Ma-Pi 2 diet during in one dietary intervention study[79]
Nutrient Ma-Pi 2 diet
Energy (kcal) 2174Protein (g) 66Tryptophan1 13Threonine1 35Isoleucine1 41Leucine1 73Lysine1 42Met + cystine1 34Phen + tyrosine1 78Valine1 50Total fat (g) 38Cholesterol (mg) 0Carbohydrates (g) 392Fiber (g) 54Vitamin C (mg) 164Folic acid (g) 751Vitamin B1 (mg) 3.52Vitamin B2 (mg) 1.3Vitamin B6 (mg) 5.55Niacin (mg) 25Vitamin B12 (g) 0.45Vitamin E (mg) 10Vitamin A (g) 3266Potassium (mg) 3646Manganese (mg) 16Iron (mg) 24Calcium (mg) 982Phosphorus (mg) 1632Zinc (mg) 15.4Magnesium (mg) 754Sodium (mg) 1724
1mg of amino acid per gram of protein.
Table 2 Metabolic effects of the Ma-Pi 2 dietary intervention after variable periods of time
Metabolic parameters Duration of Ma-Pi 2 intervention Effect
Body mass index 3 wk-6 mo ↓Body weight (kg) 3 wk-6 mo ↓Body fat mass (%) 3 wk-6 mo ↓Body fat free mass (%) 3 wk-6 mo ↔Fasting glucose (mg/dL) 3 wk-6 mo ↓Fasting insulin (mIU/mL) 3 wk ↓HbA1c 3 wk-6 mo ↓HOMA-R index 3 wk-6 mo ↓Triglycerides (mg/dL) 3 wk-6 mo ↓Total cholesterol (mg/dL) 3 wk-6 mo ↓HDL-cholesterol (mg/dL) 3 wk-6 mo ↔LDL-cholesterol (mg/dL) 3 wk-6 mo ↓Systolic blood pressure (mmHg)
3 wk-6 mo ↓
Diastolic blood pressure (mmHg)
3 wk ↓
Urinary pH 3 wk ↑Serum anion gap (mEq/L) 3 wk ↓Serum bicarbonate (mEq/L) 3 wk ↑Tumor necrosis factor alpha (ng/mL)
3 wk ↓
Interleukin 6 (pg/mL) 3 wk ↔Insulin-like growth factor 1 (ng/mL)
↓
Taken from the results of studies investigating the Ma-Pi 2 diet in men and women with type 2 diabetes over variable periods. ↑: Increase; ↓: Decrease; ↔: No effect; HbA1c: Glycosylated hemoglobin; HDL: High-density lipoprotein; LDL: Low-density lipoprotein.
Fallucca F et al . Gut microbiota, Ma-Pi 2 diet and T2DM

as whole cereals, non-animal fats and protein, and the high fiber content (> 50 g/d) and probiotic presence could improve gut microbiota composition, favoring a reduction in inflammation and insulin resistance[90].
Eliminating all animal products from the diet may increase the risk of certain nutritional deficiencies in some micronutrients such as vitamin D, calcium, iron, zinc and long-chain n-3 (omega-3) fatty acids[40]. However, the Ma-Pi 2 diet content of those micronutrients seems to be adequate[77,79]. Special concern should be given to possible deficiencies of vitamin B-12[77], even though clinical evidences for deficiency in this vitamin are described only after several years of insufficient consumption[91]. The MA-PI 2 diet was especially conceived for the treatment of T2DM and studies in adults with T2DM consistently showed that a 3 or 6-mo consumption of the Ma-Pi 2 diet was nutritionally safe, at least for the evaluated time[79-83,85]. These evidences suggest that Ma-Pi 2 diet can be used as a short- and medium-term treatment, aimed to achieve a good metabolic control and that further research is needed to demonstrate the safety of this diet in the long-term.
However, currently available studies on the Ma-Pi 2 diet in patients with T2DM have a number of limitations, such as the use of relatively small sample sizes. This was due to the particular design of the studies, where subjects were required to dwell in isolated environment 24 h per day, in order to ensure a good compliance to the diets[80-85,87-89]. However the MADIAB randomized trial was adequately powered and corroborative analyses were performed to ensure consistency and robustness of the clinical trial results[88]. Other limitations in available studies on the Ma-Pi 2 diets include their short- or medium-term durations and the fact that, in some studies, participants (in both the intervention and control groups) were studied in a supervised environment[84,85,87-89]. Subject compliance to dietary recommendations for T2DM are required to obtain clinically significant changes in outcomes, and improvements obtained with dietary interventions in clinical trials are not easy to reproduce in daily life. Future studies should aim to address all of these issues.
CONCLUSIONA variety of diets have been shown to be effective for the management of T2DM, including the Mediterranean-style, vegetarian and low-calorie diets. However, the Ma-Pi 2 diet, in both uncontrolled and controlled short- and medium-term trials conducted in patients with T2DM in several countries and continents, has been found to achieve a speed of metabolic control that has not been reported in studies of other diets.
This finding could be attributable to the composition of the Ma-Pi 2 diet, which is high in whole grains, vegetables and legumes, and fermented products, with no animal products and no added sugars. These characteristics allow to improve glycemic control, reduce insulin requirements, improve insulin sensitivity, lower blood cholesterol and
triglycerides, reduce body weight control, and lower systemic blood pressure. The dietary habits may also modify the gastrointestinal microflora composition, which could influence glucose control.
Hence, the Ma-Pi 2 diet could be considered a valid additional short- to medium-term treatment for T2DM, particularly when glycemic control needs to be rapidly improved. Further research is needed to demonstrate the efficacy of this diet in the long-term management of T2DM.
REFERENCES1 Nicolucci A, Rossi C, Lucisano G. Facts and figures about
diabetes in Italy. Italian Diabetes Monitor: Fondazione Mario Negri Sud, 2014. Available from: URL: http: //www.ibdo.it/pdf/DiabetesMonitor-FactandFigure2014.pdf
2 International Diabetes Federation. IDF Diabetes Atlas, 6th ed. Brussels, Belgium: International Diabetes Federation, 2013
3 Haslam DW, James WP. Obesity. Lancet 2005; 366: 1197-1209 [PMID: 16198769 DOI: 10.1016/S0140-6736(05)67483-1]
4 Bilous R, Donnelly R. Handbook of Diabetes, 4th ed. Oxford: Wiley Blackwell, 2010: 46
5 Levi J, Segal LM, St Laurent R, Lang A, Rayburn J. F as in Fat: How Obesity Threatens America’s Future 2012. Project Report. Princeton, NJ: Trust for America’s Health/Robert Wood Johnson Foundation, 2012
6 Balducci S, Sacchetti M, Haxhi J, Orlando G, D’Errico V, Fallucca S, Menini S, Pugliese G. Physical exercise as therapy for type 2 diabetes mellitus. Diabetes Metab Res Rev 2014; 30 Suppl 1: 13-23 [PMID: 24353273 DOI: 10.1002/dmrr.2514]
7 Balducci S, Zanuso S, Massarini M, Corigliano G, Nicolucci A, Missori S, Cavallo S, Cardelli P, Alessi E, Pugliese G, Fallucca F. The Italian Diabetes and Exercise Study (IDES): design and methods for a prospective Italian multicentre trial of intensive lifestyle intervention in people with type 2 diabetes and the metabolic syndrome. Nutr Metab Cardiovasc Dis 2008; 18: 585-595 [PMID: 18061415 DOI: 10.1016/j.numecd.2007.07.006]
8 Balducci S, Zanuso S, Nicolucci A, De Feo P, Cavallo S, Cardelli P, Fallucca S, Alessi E, Fallucca F, Pugliese G. Effect of an intensive exercise intervention strategy on modifiable cardiovascular risk factors in subjects with type 2 diabetes mellitus: a randomized controlled trial: the Italian Diabetes and Exercise Study (IDES). Arch Intern Med 2010; 170: 1794-1803 [PMID: 21059972 DOI: 10.1001/archinternmed.2010.380]
9 Nicolucci A, Balducci S, Cardelli P, Zanuso S, Pugliese G. Improvement of quality of life with supervised exercise training in subjects with type 2 diabetes mellitus. Arch Intern Med 2011; 171: 1951-1953 [PMID: 22123809 DOI: 10.1001/archinternmed.2011.561]
10 Balducci S, Zanuso S, Cardelli P, Salvi L, Bazuro A, Pugliese L, Maccora C, Iacobini C, Conti FG, Nicolucci A, Pugliese G. Effect of high- versus low-intensity supervised aerobic and resistance training on modifiable cardiovascular risk factors in type 2 diabetes; the Italian Diabetes and Exercise Study (IDES). PLoS One 2012; 7: e49297 [PMID: 23185314 DOI: 10.1371/journal.pone.0049297]
11 American Diabetes Association. Standards of medical care in diabetes--2010. Diabetes Care 2010; 33 Suppl 1: S11-S61 [PMID: 20042772 DOI: 10.2337/dc10-S011]
12 Knowler WC, Fowler SE, Hamman RF, Christophi CA, Hoffman HJ, Brenneman AT, Brown-Friday JO, Goldberg R, Venditti E, Nathan DM. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009; 374: 1677-1686 [PMID: 19878986 DOI: 10.1016/S0140-6736(09)61457-4]
13 Montonen J, Knekt P, Härkänen T, Järvinen R, Heliövaara M, Aromaa A, Reunanen A. Dietary patterns and the incidence of type
408 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Fallucca F et al . Gut microbiota, Ma-Pi 2 diet and T2DM

2 diabetes. Am J Epidemiol 2005; 161: 219-227 [PMID: 15671254 DOI: 10.1093/aje/kwi039]
14 Willett WC, Sacks F, Trichopoulou A, Drescher G, Ferro-Luzzi A, Helsing E, Trichopoulos D. Mediterranean diet pyramid: a cultural model for healthy eating. Am J Clin Nutr 1995; 61: 1402S-1406S [PMID: 7754995]
15 Esposito K, Giugliano D. Mediterranean diet and type 2 diabetes. Diabetes Metab Res Rev 2014; 30 Suppl 1: 34-40 [PMID: 24357346 DOI: 10.1002/dmrr.2516]
16 Esposito K, Kastorini CM, Panagiotakos DB, Giugliano D. Prevention of type 2 diabetes by dietary patterns: a systematic review of prospective studies and meta-analysis. Metab Syndr Relat Disord 2010; 8: 471-476 [PMID: 20958207 DOI: 10.1089/met.2010.0009]
17 Esposito K, Maiorino MI, Ceriello A, Giugliano D. Prevention and control of type 2 diabetes by Mediterranean diet: a systematic review. Diabetes Res Clin Pract 2010; 89: 97-102 [PMID: 20546959 DOI: 10.1016/j.diabres.2010.04.019]
18 Martínez-González MA, de la Fuente-Arrillaga C, Nunez-Cordoba JM, Basterra-Gortari FJ, Beunza JJ, Vazquez Z, Benito S, Tortosa A, Bes-Rastrollo M. Adherence to Mediterranean diet and risk of developing diabetes: prospective cohort study. BMJ 2008; 336: 1348-1351 [PMID: 18511765 DOI: 10.1136/bmj.39561.501007]
19 Romaguera D, Guevara M, Norat T, Langenberg C, Forouhi NG, Sharp S, Slimani N, Schulze MB, Buijsse B, Buckland G, Molina-Montes E, Sánchez MJ, Moreno-Iribas MC, Bendinelli B, Grioni S, van der Schouw YT, Arriola L, Beulens JW, Boeing H, Clavel-Chapelon F, Cottet V, Crowe FL, de Lauzon-Guillan B, Franks PW, Gonzalez C, Hallmans G, Kaaks R, Key TJ, Khaw K, Nilsson P, Overvad K, Palla L, Palli D, Panico S, Quirós JR, Rolandsson O, Romieu I, Sacerdote C, Spijkerman AM, Teucher B, Tjonneland A, Tormo MJ, Tumino R, van der AD, Feskens EJ, Riboli E, Wareham NJ. Mediterranean diet and type 2 diabetes risk in the European Prospective Investigation into Cancer and Nutrition (EPIC) study: the InterAct project. Diabetes Care 2011; 34: 1913-1918 [PMID: 21788627 DOI: 10.2337/dc11-0891]
20 Salas-Salvadó J, Bulló M, Babio N, Martínez-González MÁ, Ibarrola-Jurado N, Basora J, Estruch R, Covas MI, Corella D, Arós F, Ruiz-Gutiérrez V, Ros E. Reduction in the incidence of type 2 diabetes with the Mediterranean diet: results of the PREDIMED-Reus nutrition intervention randomized trial. Diabetes Care 2011; 34: 14-19 [PMID: 20929998 DOI: 10.2337/dc10-1288]
21 Rossi M, Negri E, Bosetti C, Dal Maso L, Talamini R, Giacosa A, Montella M, Franceschi S, La Vecchia C. Mediterranean diet in relation to body mass index and waist-to-hip ratio. Public Health Nutr 2008; 11: 214-217 [PMID: 17686205 DOI: 10.1017/S1368980007000833]
22 Goldstein DJ. Beneficial health effects of modest weight loss. Int J Obes Relat Metab Disord 1992; 16: 397-415 [PMID: 1322866]
23 Fontana L, Klein S, Holloszy JO. Effects of long-term calorie restriction and endurance exercise on glucose tolerance, insulin action, and adipokine production. Age (Dordr) 2010; 32: 97-108 [PMID: 19904628 DOI: 10.1007/s11357-009-9118-z]
24 Fontana L, Klein S, Holloszy JO, Premachandra BN. Effect of long-term calorie restriction with adequate protein and micronutrients on thyroid hormones. J Clin Endocrinol Metab 2006; 91: 3232-3235 [PMID: 16720655 DOI: 10.1210/jc.2006-0328]
25 Weiss EP, Racette SB, Villareal DT, Fontana L, Steger-May K, Schechtman KB, Klein S, Holloszy JO. Improvements in glucose tolerance and insulin action induced by increasing energy expenditure or decreasing energy intake: a randomized controlled trial. Am J Clin Nutr 2006; 84: 1033-1042 [PMID: 17093155]
26 Fontana L. Neuroendocrine factors in the regulation of inflammation: excessive adiposity and calorie restriction. Exp Gerontol 2009; 44: 41-45 [PMID: 18502597 DOI: 10.1016/j.exger.2008.04.005]
27 Davey GK, Spencer EA, Appleby PN, Allen NE, Knox KH, Key TJ. EPIC-Oxford: lifestyle characteristics and nutrient intakes in a cohort of 33 883 meat-eaters and 31 546 non meat-eaters in the UK. Public Health Nutr 2003; 6: 259-269 [PMID: 12740075]
28 Tonstad S, Butler T, Yan R, Fraser GE. Type of vegetarian diet,
body weight, and prevalence of type 2 diabetes. Diabetes Care 2009; 32: 791-796 [PMID: 19351712 DOI: 10.2337/dc08-1886]
29 Burslem J, Schonfeld G, Howald MA, Weidman SW, Miller JP. Plasma apoprotein and lipoprotein lipid levels in vegetarians. Metabolism 1978; 27: 711-719 [PMID: 206801 DOI: 10.1016/0026-0495(78)90009-4]
30 Lock DR, Varhol A, Grimes S, Patsch W, Schonfeld G. ApoA-I/ApoA-II ratios in plasmas of vegetarians. Metabolism 1983; 32: 1142-1145 [PMID: 6417448 DOI: 10.1016/0026-0495(83)90061-6]
31 Thorogood M, Carter R, Benfield L, McPherson K, Mann JI. Plasma lipids and lipoprotein cholesterol concentrations in people with different diets in Britain. Br Med J (Clin Res Ed) 1987; 295: 351-353 [PMID: 3115444 DOI: 10.1136/bmj.295.6594.351]
32 Armstrong B, van Merwyk AJ, Coates H. Blood pressure in Seventh-day Adventist vegetarians. Am J Epidemiol 1977; 105: 444-449 [PMID: 871119]
33 Appleby PN, Davey GK, Key TJ. Hypertension and blood pressure among meat eaters, fish eaters, vegetarians and vegans in EPIC-Oxford. Public Health Nutr 2002; 5: 645-654 [PMID: 12372158 DOI: 10.1079/PHN2002332]
34 Tonstad S, Stewart K, Oda K, Batech M, Herring RP, Fraser GE. Vegetarian diets and incidence of diabetes in the Adventist Health Study-2. Nutr Metab Cardiovasc Dis 2013; 23: 292-299 [PMID: 21983060 DOI: 10.1016/j.numecd.2011.07.004]
35 Fukagawa NK, Anderson JW, Hageman G, Young VR, Minaker KL. High-carbohydrate, high-fiber diets increase peripheral insulin sensitivity in healthy young and old adults. Am J Clin Nutr 1990; 52: 524-528 [PMID: 2168124]
36 Crowe FL, Appleby PN, Travis RC, Key TJ. Risk of hospita-lization or death from ischemic heart disease among British vegetarians and nonvegetarians: results from the EPIC-Oxford cohort study. Am J Clin Nutr 2013; 97: 597-603 [PMID: 23364007 DOI: 10.3945/ajcn.112.044073]
37 Key TJ, Fraser GE, Thorogood M, Appleby PN, Beral V, Reeves G, Burr ML, Chang-Claude J, Frentzel-Beyme R, Kuzma JW, Mann J, McPherson K. Mortality in vegetarians and nonvegetarians: detailed findings from a collaborative analysis of 5 prospective studies. Am J Clin Nutr 1999; 70: 516S-524S [PMID: 10479225]
38 Key TJ, Appleby PN, Spencer EA, Travis RC, Roddam AW, Allen NE. Mortality in British vegetarians: results from the European Prospective Investigation into Cancer and Nutrition (EPIC-Oxford). Am J Clin Nutr 2009; 89: 1613S-1619S [PMID: 19297458 DOI: 10.3945/ajcn.2009.26736L]
39 Tantamango-Bartley Y, Jaceldo-Siegl K, Fan J, Fraser G. Vegetarian diets and the incidence of cancer in a low-risk population. Cancer Epidemiol Biomarkers Prev 2013; 22: 286-294 [PMID: 23169929 DOI: 10.1158/1055-9965.EPI-12-1060]
40 World Cancer Research Fund/American Institute for Cancer Research. Food, Nutrition, Physical Activity, and the Prevention of Cancer: a Global Perspective. Washington DC: AICR, 2007
41 Colditz GA, Manson JE, Stampfer MJ, Rosner B, Willett WC, Speizer FE. Diet and risk of clinical diabetes in women. Am J Clin Nutr 1992; 55: 1018-1023 [PMID: 1315120]
42 Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science 2005; 307: 1915-1920 [PMID: 15790844 DOI: 10.1126/science.1104816]
43 Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007; 56: 1761-1772 [PMID: 17456850 DOI: 10.2337/db06-149]
44 Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008; 57: 1470-1481 [PMID: 18305141 DOI: 10.2337/db07-1403]
45 Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S,
409 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Fallucca F et al . Gut microbiota, Ma-Pi 2 diet and T2DM

Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012; 490: 55-60 [PMID: 23023125 DOI: 10.1038/nature11450]
46 Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich SD, Wang J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464: 59-65 [PMID: 20203603 DOI: 10.1038/nature08821]
47 Ryan KK, Seeley RJ. Physiology. Food as a hormone. Science 2013; 339: 918-919 [PMID: 23430646 DOI: 10.1126/science.1234062]
48 Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol 1977; 31: 107-133 [PMID: 334036]
49 Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 2006; 40: 235-243 [PMID: 16633129]
50 Tlaskalová-Hogenová H, Stepánková R, Hudcovic T, Tucková L, Cukrowska B, Lodinová-Zádníková R, Kozáková H, Rossmann P, Bártová J, Sokol D, Funda DP, Borovská D, Reháková Z, Sinkora J, Hofman J, Drastich P, Kokesová A. Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunol Lett 2004; 93: 97-108 [PMID: 15158604 DOI: 10.1016/j.imlet.2004.02.005]
51 Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 2009; 9: 313-323 [PMID: 19343057 DOI: 10.1038/nri2515]
52 Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009; 461: 1282-1286 [PMID: 19865172 DOI: 10.1038/nature08530]
53 Salminen S, Bouley C, Boutron-Ruault MC, Cummings JH, Franck A, Gibson GR, Isolauri E, Moreau MC, Roberfroid M, Rowland I. Functional food science and gastrointestinal physiology and function. Br J Nutr 1998; 80 Suppl 1: S147-S171 [PMID: 9849357]
54 Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, Muccioli GG, Delzenne NM. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009; 58: 1091-1103 [PMID: 19240062 DOI: 10.1136/gut.2008.165886]
55 Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, Hammer RE, Williams SC, Crowley J, Yanagisawa M, Gordon JI. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci USA 2008; 105: 16767-16772 [PMID: 18931303 DOI: 10.1073/pnas.0808567105]
56 Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sørensen SJ, Hansen LH, Jakobsen M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One 2010; 5: e9085 [PMID: 20140211 DOI: 10.1371/journal.pone.0009085]
57 Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, Knight R, Ahima RS, Bushman F, Wu GD. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009; 137: 1716-24.e1-2 [PMID: 19706296 DOI: 10.1053/j.gas-tro.2009.08.042]
58 Geurts L, Neyrinck AM, Delzenne NM, Knauf C, Cani PD. Gut microbiota controls adipose tissue expansion, gut barrier and
glucose metabolism: novel insights into molecular targets and interventions using prebiotics. Benef Microbes 2014; 5: 3-17 [PMID: 23886976 DOI: 10.3920/BM2012.0065]
59 Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, Gibson GR, Delzenne NM. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007; 50: 2374-2383 [PMID: 17823788 DOI: 10.1007/s00125-007-0791-0]
60 Ghanim H, Abuaysheh S, Sia CL, Korzeniewski K, Chaudhuri A, Fernandez-Real JM, Dandona P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care 2009; 32: 2281-2287 [PMID: 19755625 DOI: 10.2337/dc09-0979]
61 Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylo-microns promote intestinal absorption of lipopolysaccharides. J Lipid Res 2009; 50: 90-97 [PMID: 18815435 DOI: 10.1194/jlr.M800156-JLR200]
62 Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol 2007; 5: e177 [PMID: 17594176 DOI: 10.1371/journal.pbio.0050177]
63 Zoetendal EG, Vaughan EE, de Vos WM. A microbial world within us. Mol Microbiol 2006; 59: 1639-1650 [PMID: 16553872 DOI: 10.1111/j.1365-2958.2006.05056.x]
64 Biagi E, Candela M, Fairweather-Tait S, Franceschi C, Brigidi P. Aging of the human metaorganism: the microbial counterpart. Age (Dordr) 2012; 34: 247-267 [PMID: 21347607 DOI: 10.1007/s11357]
65 Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 2008; 6: e280 [PMID: 19018661 DOI: 10.1371/journal.pbio.0060280]
66 Gibson GR, Probert HM, Loo JV, Rastall RA, Roberfroid MB. Dietary modulation of the human colonic microbiota: updating the concept of prebiotics. Nutr Res Rev 2004; 17: 259-275 [PMID: 19079930 DOI: 10.1079/NRR200479]
67 Roberfroid M, Gibson GR, Hoyles L, McCartney AL, Rastall R, Rowland I, Wolvers D, Watzl B, Szajewska H, Stahl B, Guarner F, Respondek F, Whelan K, Coxam V, Davicco MJ, Léotoing L, Wittrant Y, Delzenne NM, Cani PD, Neyrinck AM, Meheust A. Prebiotic effects: metabolic and health benefits. Br J Nutr 2010; 104 Suppl 2: S1-63 [PMID: 20920376 DOI: 10.1017/S0007114510003363]
68 Cani PD, Lecourt E, Dewulf EM, Sohet FM, Pachikian BD, Naslain D, De Backer F, Neyrinck AM, Delzenne NM. Gut microbiota fermentation of prebiotics increases satietogenic and incretin gut peptide production with consequences for appetite sensation and glucose response after a meal. Am J Clin Nutr 2009; 90: 1236-1243 [PMID: 19776140 DOI: 10.3945/ajcn.2009.28095]
69 Food and Agriculture Organization/World Health Organi-zation. Health and Nutritional properties of probiotics in food including powder milk with live lactic acid bacteria. Report of joint FAO/WHO expert consultation on evaluation of health and nutritional properties of probiotics in food including powder milk with live lactic acid bacteria. Switzerland, Basel: World Health Organization, 2001
70 Strowski MZ, Wiedenmann B. Probiotic carbohydrates reduce intestinal permeability and inflammation in metabolic diseases. Gut 2009; 58: 1044-1045 [PMID: 19592687 DOI: 10.1136/gut.2009.179325]
71 Nilsson AC, Ostman EM, Holst JJ, Björck IM. Including indigestible carbohydrates in the evening meal of healthy subjects improves glucose tolerance, lowers inflammatory markers, and increases satiety after a subsequent standardized breakfast. J Nutr 2008; 138: 732-739 [PMID: 18356328]
72 Solga SF, Buckley G, Clark JM, Horska A, Diehl AM. The effect of a probiotic on hepatic steatosis. J Clin Gastroenterol 2008; 42: 1117-1119 [PMID: 18936646 DOI: 10.1097/MCG.0b013e31816d9
410 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Fallucca F et al . Gut microbiota, Ma-Pi 2 diet and T2DM

20c]73 Parnell JA, Reimer RA. Weight loss during oligofructose
supplementation is associated with decreased ghrelin and increased peptide YY in overweight and obese adults. Am J Clin Nutr 2009; 89: 1751-1759 [PMID: 19386741 DOI: 10.3945/ajcn.2009.27465]
74 Luoto R, Kalliomäki M, Laitinen K, Isolauri E. The impact of perinatal probiotic intervention on the development of overweight and obesity: follow-up study from birth to 10 years. Int J Obes (Lond) 2010; 34: 1531-1537 [PMID: 20231842 DOI: 10.1038/ijo.2010.50]
75 Delzenne NM, Neyrinck AM, Cani PD. Modulation of the gut microbiota by nutrients with prebiotic properties: consequences for host health in the context of obesity and metabolic syndrome. Microb Cell Fact 2011; 10 Suppl 1: S10 [PMID: 21995448 DOI: 10.1186/1475-2859-10-S1-S10]
76 Ohsawa G. Le zen macrobiotique ou l’art du rajeunissement et de la longevitè. Paris: Librairie Philosophique J Vrin, 2004
77 Porrata C, Hernández M, Abuín A, Campa C, Pianesi M. Caracterización y evaluación nutricional de las dietas macrobióticas Ma-Pi. Rev Cubana Investig Bioméd 2008; 27: 1-36
78 Mancini L. Conventional, organic and polycultural farming practices: material intensity of italian crops and Foodstuffs. Resources 2013; 2: 628-650 [DOI: 10.3390/resources2040628]
79 Porrata-Maury C, Hernández-Triana M, Rodríguez-Sotero E, Vilá-Dacosta-Calheiros R, Hernández-Hernández H, Mirabal-Sosa M, Campa-Huergo C, Pianesi M. Medium- and short-term interventions with ma-pi 2 macrobiotic diet in type 2 diabetic adults of bauta, havana. J Nutr Metab 2012; 2012: 856342 [PMID: 23097695]
80 Bhumisawasdi J, Vanna O, Surinpang N. The self-reliant system for alternative care of diabetes mellitus patients--experience macrobiotic management in Trad Province. J Med Assoc Thai 2006; 89: 2104-2115 [PMID: 17214064]
81 Porrata C, Abuin Landin A, Zayas AM, Vilà Dacosta-Calheiros R, Hernandez Triana M, Menendez Triana J, Diaz Sanchez ME, Sosa MM, Huergo CC, Pianesi M. Efecto terapeutico de la dieta macrobiotica MAPI-2 en 25 adultos con diabetes mellitus tipo 2. Rev Cubana Invest Biomed 2007; 26: 71-87
82 Monsan Yapo M, Katchè Adueni V, Ehouman G, Porrata C, Pianesi M. MAPI-2 macrobiotic diet intervention during six momths in adults with type 2 diabetes mellitus, Cote D’Ivoire,
2010. Revue Soc Sci Nat de Tunisie 2010; 37: 62-7183 Kablan BJ, Kouassi D, N’guetta KF, Adoueni KV, Konè M,
Diafouka F, Boa YF, Lokrou A, Gbanè A, Ehouman G, Pianesi M. Curative effects of MAPI-2 macrobiotic diet in Ivoirian type 2 diabetic. Cahier de Santè Publique 2011; 10: 96-125
84 Fallucca F, Fallucca S, Pianesi M. The effects of the MA-PI 2 macrobiotic diet in the treatment of type 2 diabetes and diet-induced metabolic acidosis. Diabetes Metab Res Rev 2014; 30: 659-660 [PMID: 25400068 DOI: 10.1002/dmrr.2602]
85 Porrata C, Sánchez J, Correa V, Abuín A, Hernández-Triana M, Dacosta-Calheiros RV, Díaz ME, Mirabal M, Cabrera E, Campa C, Pianesi M. Ma-pi 2 macrobiotic diet intervention in adults with type 2 diabetes mellitus. MEDICC Rev 2009; 11: 29-35 [PMID: 21483296]
86 Porrata-Maury C, Hernández-Triana M, Ruiz-Álvarez V, Díaz-Sánchez ME, Fallucca F, Bin W, Baba-Abubakari B, Pianesi M. Ma-Pi 2 macrobiotic diet and type 2 diabetes mellitus: pooled analysis of short-term intervention studies. Diabetes Metab Res Rev 2014; 30 Suppl 1: 55-66 [PMID: 24532293 DOI: 10.1002/dmrr.2519]
87 Fallucca F, Porrata C, Monaco G, Bufacchi A, Pianesi M. MAPI macrobiotic diet intervention during 21 days in adults with type 2 diabetes mellitus, Rome. Minerva Endocrinol 2012; 37 Suppl 1: 116
88 Soare A, Khazrai YM, Del Toro R, Roncella E, Fontana L, Fallucca S, Angeletti S, Formisano V, Capata F, Ruiz V, Porrata C, Skrami E, Gesuita R, Manfrini S, Fallucca F, Pianesi M, Pozzilli P. The effect of the macrobiotic Ma-Pi 2 diet vs. the recommended diet in the management of type 2 diabetes: the randomized controlled MADIAB trial. Nutr Metab (Lond) 2014; 11: 39 [PMID: 25302069 DOI: 10.1186/1743-7075-11-39]
89 Behavioral medicine, clinical nutrition, education, and exercise. Diabetes 2014; 63 Suppl 1: A170-A212 [PMID: 24928966]
90 Fallucca F, Porrata C, Fallucca S, Pianesi M. Influence of diet on gut microbiota, inflammation and type 2 diabetes mellitus. First experience with macrobiotic Ma-Pi 2 diet. Diabetes Metab Res Rev 2014; 30 Suppl 1: 48-54 [PMID: 24532292 DOI: 10.1002/dmrr.2518]
91 Dong A, Scott SC. Serum vitamin B12 and blood cell values in vegetarians. Ann Nutr Metab 1982; 26: 209-216 [PMID: 6897159 DOI: 10.1159/000176565]
P- Reviewer: Bagyánszki M S- Editor: Gong XM L- Editor: A E- Editor: Liu SQ
411 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Fallucca F et al . Gut microbiota, Ma-Pi 2 diet and T2DM

of hippocampus structure and function. Although, the exact mechanism by which maternal diabetes affects the developing hippocampus remains to be defined. Multiple biological alterations, including hyperglycemia, hyperinsulinemia, oxidative stress, hypoxia, and iron deficiency occur in pregnancies with diabetes and affect the development of central nervous system (CNS) of the fetus. The conclusion from several studies is that disturbance in glucose and insulin homeostasis in mothers and infants are major teratogenic factor in the development of CNS. Insulin and Insulin-like growth factor-1 (IGF-1) are two key regulators of CNS function and development. Insulin and IGF-1 receptors (IR and IGF1R, respectively) are distributed in a highly specific pattern with the high density in some brain regions such as hippocampus. Recent researches have clearly established that maternal diabetes disrupts the regulation of both IR and IGF1R in the hippocampus of rat newborn. Dissecting out the mechanisms responsible for maternal diabetes-related changes in the development of hippocampus is helping to prevent from impaired cognitive and memory functions in offspring.
Key words: Maternal diabetes; Cognition complications; Teratogenic factor; Hippocampus
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Diabetes mellitus is the most seriously metabolic condition in pregnancy that affects the hippo-campal development and function of the offspring. Multiple biological alterations, including hyperglycemia and hyperinsulinemia are occurring in maternal diabetes and impair the neurodevelopment of the fetus. Insulin-like growth factor-1 (IGF-1) and insulin are important regulators of development of central nervous system. It has clearly showed that maternal diabetes disturb the regulation of both insulin receptors and IGF-1 receptors in the hippocampus of rat newborn. This article is a brief review of the literatures that suggests a probable
Javad Hami, Fatemeh Shojae, Saeed Vafaee-Nezhad, Nasim Lotfi, Hamed Kheradmand, Hossein Haghir
Javad Hami, Fatemeh Shojae, Saeed Vafaee-Nezhad, Nasim Lotfi, Department of Anatomy, School of Medicine, Birjand University of Medical Sciences, Birjand 97178, IranNasim Lotfi, Hossein Haghir, Department of Anatomy and Cell Biology, School of Medicine, Mashhad University of Medical Sciences, Mashhad 91857, IranHamed Kheradmand, Hazrat Rasoul Hospital, Tehran University of Medical Sciences, Tehran 19168, IranHossein Haghir, Medical Genetic Research Center, School of Medicine, Mashhad University of Medical Sciences, Mashhad 91857, IranAuthor contributions: All authors contributed to this manuscript.Conflict-of-interest: All authors claim no conflict of interest.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Hossein Haghir, MD, PhD, Department of Anatomy and Cell Biology, School of Medicine, Mashdad University of Medical Sciences, Azadi Square, Mashhad 91857, Iran. [email protected]: +98-513-8002486 Fax: +98-513-8002486Received: August 27, 2014 Peer-review started: August 31, 2014 First decision: October 14, 2014Revised: December 22, 2014 Accepted: January 9, 2015 Article in press: January 12, 2015Published online: April 15, 2015
AbstractDiabetes mellitus during pregnancy is associated with an increased risk of multiple congenital anomalies in progeny. There are sufficient evidence suggesting that the children of diabetic women exhibit intellectual and behavioral abnormalities accompanied by modification
REVIEW
412 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Some of the experimental and clinical aspects of the effects of the maternal diabetes on developing hippocampus
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.412
World J Diabetes 2015 April 15; 6(3): 412-422ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

mechanism of how diabetes during pregnancy affects the hippocampus development in the offspring.
Hami J, Shojae F, Vafaee-Nezhad S, Lotfi N, Kheradmand H, Haghir H. Some of the experimental and clinical aspects of the effects of the maternal diabetes on developing hippocampus. World J Diabetes 2015; 6(3): 412-422 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/412.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.412
INTRODUCTIONDiabetes mellitus (DM) is a chronic common metabolic illness characterized by hyperglycaemia associated with insulin deficiency or insulin resistance. There are two major types of DM: type 1 DM (T1DM) results from chronic and progressive destruction of the beta-cells of the islets of Langerhans in the pancreas. T2DM is primarily characterized by both insulin resistance and relative insulin deficiency[1,2].
DM is one of the most common and most important metabolic condition affecting up to 7% of pregnancies. Several population studies show that the diabetes during pregnancy affects the health of both mothers and their infants[3-6].
There are two main types of DM occurs in pregnancy period: pregestational DM (PGDM) and gestational DM (GDM)[7]. GDM accounts for approximately 90% of all cases of diabetes in pregnancy. Of the remaining cases, 60% have preexisting T2DM before pregnancy, while 40% have a diagnosis of pre-conceptual T1DM[8].
In a study in the United Kingdom, it was found that PGDM occurs in approximately 1 out of 250 preg-nancies, with two-thirds of them having T1DM and the remaining one-third having T2DM[9]. At present, type 2 diabetes is the most prevalent form of diabetes affecting women of reproductive age in the developed countries, with a mean prevalence of 27.6% in United Kingdom[10]. Last data from Australia also showed that T2DM affected about 55% of pregnancies[11]. Overall, GDM complicated from 2% to 12% of all pregnancies. So, however, the incidence of T1DM appears to be relatively constant and similar to the rate in young adults throughout the world; the incidence of T2DM is specially rising in developed world[9,12]. Furthermore, the overall numbers of children born to mothers with diabetes will significantly rise in the next decades.
COMMON CONGENITAL MALFORMATIONS IN DIABETIC MOTHER INFANTSAn increasing number of evidence clearly showed that infants born to diabetic mothers are in increased risk of fetal and neonatal anomalies which results in increased infant mortality and morbidity rates. Although, the
wide spectrum of the effects of diabetes in pregnancy on the fetus development are determined by the time of onset and the severity of DM[6,13,14].
Despite considerable progress in the clinical mana-gement of pregnant women who have diabetes, the prevalence of major congenital anomalies is approximately 3 times more in infants of diabetic women in comparison to the offspring of normal mothers[12,15,16].
According to previous works, diabetes in pregnancy has been accompanied by increased risk of a wide range of fetal complications and malformations, in-cluding respiratory distress syndrome, macrosomia, organomegaly, obstetric, metabolic and hematological complications[13,14,16,17].
The overall rate of major congenital anomalies in infants born to diabetic mothers is about 6% to 13%, which is approximately 2-3 times higher than that of the normal condition. However, the prevalence of major structural malformations in the children born to mothers with diabetes are to 10 times greater in comparison to the general population. The frequencies of perinatal mortality are also five-fold higher in women with diabetes compared with normal women[12,14,16,18].
The prevalence of Minor congenital anomalies in infants born to 802 mothers with GDM, 117 mothers with PGDM, and 380 normal mothers were assessed by Hod et al[19] (1992). They found a range between 19.4% and 20.5% in prevalence of minor congenital anomalies in their infant in all groups studied without any marked difference between groups. In their study, there was no correlation between the type and prevalence of minor congenital anomalies with the severity and the time of onset of the diabetes in mothers. Neither there was any relationship between the type or prevalence of minor congenital anomalies with the type or appearance of major congenital anomalies[19].
Overall, the defects in central nervous system, heart, kidney, and skeletal system are among the commonest congenital malformations in the offspring of diabetic pregnancies[3,14,16,18].
PATHOGENESIS OF COMMON COMPLICATIONS IN DIABETIC INFANTS As yet, the exact mechanisms by which maternal diabetes alters the development of growing fetuses are not completely understood, and no distinct teratogenic mechanism has been identified to clearly explain a reason for this range of congenital anomalies observed in the infants born to diabetic mothers[20,21].
Many of the developmental effects of diabetic pregnancies on the fetus can be attributed to maternal glucose (metabolic) control. In normal pregnancies, the blood glucose level in women remains within a tight range. Consequently, the blood glucose concentration in the fetus is fairly constant as the glucose in the mother’sblood crosses the placenta freely. Nevertheless, in pregnancies complicated by DM, there are great
Hami J et al . Effects of maternal diabetes on developing hippocampus
413 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

variations in the maternal blood glucose level[12,14,22]. Pedersen et al[23] (1954) emphasized the correlation between high blood glucose concentration in pregnant women with hyperglycemia during fetal period[23], which stimulated the pancreas of the fetuses, leads to beta-cell hyperplasia and hypertrophy with increased insulin secretion and content[23,24].
Several lines of studies also emphasized the direct correlation between high blood glucose concentration in mothers as revealed by increased level of glycosy-lated hemoglobin (HbA1c) with increased frequency of congenital malformations in offspring[22,25]. Towner et al[26] (1995) found a striking correlation between pregestational elevated HbA1c in early pregnancy with the increased risk of fetal congenital malformations[26]. There is enough evidence accumulates to support this hypothesis that the tight glycaemic control in early and before pregnancy may reduce the frequency of anomalies in the infants born to diabetic mothers[22]. Therefore, the prevalence of congenital abnormalities in children born to mothers with diabetes has decreased in the last three decades probably as a result of the overall progress in monitoring of blood glucose concentrations in diabetic pregnant women[27-29].
In addition, fetal hyperinsulinemia also has been shown in the offspring of diabetic mothers. On separation of the newborn from the mother, the glucose former no longer is supported by placental glucose transfer, may develop neonatal hypoglycemia[30,31].
In earlier works, chronic fetal hyperinsulinemia in the third trimester of pregnancy has been produced in monkeys. The researchers showed that in utero hyperinsulinemia results in an organomegaly and fetal macrosomia, except for kidney and brain[32,33]. Increased plasma erythropoietin levels and body fat content was also evident in infants of diabetic mothers[34].
Insulin is a hormone that primarily functions as an anabolic hormone of fetal development and growth resulting in macrosomia and visceromegaly[14]. On the basis of in vitro and animal studies on the pancreas, the simplified hyperinsulinemia - hyperglycemia hypothesis has been expanded in earlier studies[35,36].
Ketones in the mother’s blood readily cross the placenta, but they cannot be cause fetal hyperinsu-linemia; so, they might not influence fetal growth and development[14].
In experimental diabetic rodents, fetal hyperglycemia, neonatal hypoglycemia, and disturbances in pro-staglandin and arachidonic acid metabolism have been reported to result in congenital anomalies in their offspring[14,37]. In diabetic pregnancies in rats, it is clearly established that the elevated intracellular free oxygen radicals concentrations have been accompanied by the increased risk of embryopathies[38]. Nevertheless, hyperglycemia-induced fetal malformations in rodents have been demonstrated to associate with the number of genomic DNA mutations[39]. Therefore, there are animal
studies showing that the free radical scavengers and antioxidants may decrease the risk of teratogenicity of maternal diabetes[40,41].
DIABETIC PREGNANCY AND NEURODEVELOPMENTAL SEQUELAE IN HUMAN Both PGDM and GDM can affect fetal neurodeve-lopment[42-45]. Although the untoward effects of maternal diabetes on pregnancy outcomes with respect to congenital malformations have generally been appreciated, the effect of maternal diabetes on the development of central nervous system (CNS) in the fetuses and its behavioral sequelae remains to be completely defined. A limited number of long-term follow-up studies of neurodevelopmental outcomes in diabetic pregnancies have been reported[46-50]. Nevertheless, it is demonstrated that children born to diabetic mothers are more likely to have neurodevelopmental abnormalities including impairments in learning ability, activity level, attention span, and motor functioning. Interestingly, some of these deficiencies are well-known as risk factors in children who develop schizophrenia later, but the degree of risk is variable and may be related to the severity of metabolic derangement in the mothers with diabetes[42,46,50,51].
Examination of cognitive functioning and the behavior of the children born to diabetic mothers offers the opportunity to functionally assess the CNS development[52]. Hence the assessment of the behavior and cognition in the offspring of diabetic mothers may clarify the maternal diabetes effects on the development of CNS.
The results of earlier studies clearly demonstrated that the diabetes during pregnancies may results in intellectual and behavioral functioning disturbances in the infants[45,46,49,53]. These data suggest a teratogenic effect of diabetes in pregnancy on the function of CNS in fetuses and provides the earliest indicator of postnatal CNS deficiencies reflected in intellectual and behavioral problems observed in the children of mothers with diabetes[54].
Earlier reports on the neurologic development in infants of diabetic mothers revealed serious CNS deficits, even in the absence of structural malformations. These alterations were significantly less severe when maternal diabetes was medically controlled and treated, but some alterations in cognitive function may persist throughout childhood[6,42,45,48,51].
To elucidate the effects of diabetes during pregnancy period on cognitive functioning in the children, Yamashita et al[45] (1996) studied 33 pregnant women (24 with T2DM, 6 with T1DM, and 3 with GDM). Their long term follow-up research showed that maternal diabetes significantly affects the development of intellectual functioning in infants. Although, they report no differences in IQ score among offspring of three groups[45]. In another
414 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Hami J et al . Effects of maternal diabetes on developing hippocampus

DIABETES IN PREGNANCY AND NEURODEVELOPMENTAL ABERRATION IN ANIMALSNeurodevelopmental assessment of the offspring born to diabetic dams have been revealed a wide spectrum of behavioral, neurochemical, cellular, and molecular impairments[69-72]. Experimental models subjected to streptozotocin - induced type 1 diabetic pregnancies developed significant deficits in cognitive behaviors[68]. Kinney et al[73] (2003) found that the only female offspring born to diabetic dams showed deficits in long-term memory and learning. These results have suggested that the in utero diabetic condition has gender-specific effects on CNS development[73].
In a study by Plagemann et al[74] (1998), alterations in catecholamines levels in the hypothalamic nuclei of newborns born to diabetic animals were evaluated. They reported an increased hypothalamic dopamine (DA) and norepinephrine (NE) concentrations in the offspring born to diabetic rats at birth. Twenty one-day-old pups born to diabetic mothers, NE levels were strikingly increased in the ventromedial hypothalamic nucleus and the lateral hypothalamic area (LHA), while DA levels were significantly elevated in the paraventricular hypothalamic nucleus and the LHA. The authors concluded that there are strikingly differences in hypothalamic catecholaminergic systems during early development in the rat newborns born to diabetic animals[74].
It has been shown that PGDM is associated with an increases risk of neural tube defects (NTDs), also known as diabetic embryopathy. The prevalence of NTDs in the offspring of diabetic mothers is 3 to 10-fold higher than that of in general population[75-77]. In earlier studies, the mitochondrial morphological changes has been shown in developing neural tubes subjected to a diabetic environment during the same time period when the NTDs are induced in diabetic pregnancies[72,78]. Altered activity of cytochrome-c oxidase also has been manifested in the rat hippocampus that have iron deficiency in their brains due to fetal hyperglycemia[79]. It has also been hypothesized that dysfunction in energy metabolism of developing brain leads to oxidative stress elevation and abnormal regulation of intracellular calcium[80-82].
NEUROPATHOPHYSIOLOGY OF DIABETIC PREGNANCYSeveral well-known biological alterations occur in mothers with diabetes that affect fetal neurodevelopment. These biological changes lead to alterations in neurotransmitter systems, synaptic membranes, neuronal integrity, and growth factors expression that have also been implicated in the development of neurodevelopmental and neurocognitive sequelae in offspring born to diabetic mothers[14,42,69,72,74].
study, Churchill et al[55] (1969), found that diabetic mothers had offspring with significantly lower mean IQs than control infants. In a cohort study of fifty infants at 1, 3, and 5 years of age born to diabetic mothers, Stehbens et al[56] (1977) reported an adverse effect of diabetes in pregnancy on CNS structure and function in their children. They also found that three infants born to diabetic mothers had major neurologic anomalies and six of them had IQs less than 80.
Rizzo et al[57] (1991) reported a strikingly correlation between diabetes in mothers during pregnancy and lower IQ in their children. In another study, the same researcher found significant correlations between second- and third-trimester regulation of glycemia and results on the Brazelton Neonatal Behavioral Assessment Scale; they reported an association between increases in maternal glucose levels with poorer infant responses[58].
Other investigators have not found any differences in cognitive scores[59-61]. Children born to diabetic mothers may sustain minor neurological damage which does not necessarily affect their scores in IQ tests[49]. For example, Persson et al[60] reported more encouraging results in 73 infants born either to mothers with T1DM or to mothers with GDM; all subjects available for follow-up at 5 years of age had normal results on neurologic examinations, as well as normal IQs, and there was no relation between maternal diabetes and IQ[60].
In a retrospective study at considerable variance with most other reported studies, Yssing et al[62] (1975) reported a 36% incidence of cerebral dysfunction or related conditions in 740 infants of diabetic pregnancies; 18% had a major cerebral disability. In a study by Haworth et al[63] (1976) on infants of diabetic pregnancies, noted an about 30% increase in the incidence of neurological and intellectual development impairments[63]. No differences in behavioral adjustment and academic achievement between children born to T1DM mothers and children of normal mothers were reported in Hadden et al[64] study.
In addition, poorer habituation performance were also found in fetuses of diabetic pregnancies when compared to fetuses born to normal mothers[65]. Since habituation reflects the fetal CNS performance, the observed habituation abilities differences between the diabetic and normal groups suggests differences in their CNS function or maturation[65,66]. Recent investigations also have been demonstrated that there is a significant correlation between diabetes during pregnancy and increased risk of some psychologic disturbances including schizophrenia in their children[67,68].
Together, these studies suggest a wide-range of teratogenic effects of maternal diabetes on the development and function of fetal CNS[67]. However, no single molecular mechanism can fully explain the effects of maternal diabetes on fetal neurodevelopment because the CNS development is complex and regulated by a number of signaling molecules and transcription factors.
415 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Hami J et al . Effects of maternal diabetes on developing hippocampus

The hyperglycemia is the most distinct mechanism by which these predispositions might be mediated. The defining characteristics of diabetes during pregnancy, these are clearly showed to have effects on developing CNS and to induce fetal hyperinsulinemia, chronic tissue hypoxia, decreased in fetal iron levels and increased oxidative stress[20,83-85].
Hyperinsulinemia, hypoxia, polycythemia and iron deficiencyIt is revealed that fetal polycythemia induced by DM in pregnancy is likely triggered by the chronic in utero hyperglycemia and hyperinsulinemia and develops in response to higher concentration of blood glucose in mothers[20,42]. A study by Mimouni et al[86] (1986) showed an incidence of 29.4% fetal polycythemia in infants born to diabetic mothers in comparison to 5.9% in normal conditions. One of the reasonable explanations is that in utero hyperglycemia and hyperinsulinemia produces a hypermetabolic state in the developing fetuses, which results in relative tissue hypoxia, leading to polycythemia via excess production of erythropoietin. Elevated fetal plasma erythropoietin concentrations in diabetic pregnancies suggests an increased prevalence of chronic fetal hypoxia during development. Although there are no distinct mechanisms for fetal hypoxia in diabetic pregnancies[34].
Hypoxia affects the fetal CNS development in-cluding alterations in myelination, changes in cortical connectivity, excitotoxicity, and neuronal or glial cell death[87-89].
In the hypoxia state, excess erythropoietin and hemoglobin are produced[90]. So, the developing fetuses need for iron exceeds its supply that leads to Iron mobilization from vital tissues including the fetal brain[91,92]. It is showed that human infants born to diabetic women possess brain iron content 40% less than that in normal pregnancies[93].
Several authors have reviewed the role of iron on CNS development and function[94-96]. Iron is one of the key component of the many enzymes involve in essential reduction or oxidation reactions, neuronal replication, synthesis and catabolism of neurotransmitters and myelin production[97-101]. The uptake of Iron into the brain is maximal during the rapid brain development and growth, which coincides with the peak of myelinogenesis[99-101]. In experimental animals, iron deficiency demonstrated to affect the development of neurotransmitter systems in the brain results in behavioral alteration[98,102]. Moreover, iron deficiency in fetuses is also known to manifest as increased negative emotionality and higher levels of irritability in infants and is a predictive factor in the behavioral and developmental problems at 5 age old children[103]. There are studies suggesting that iron deficiency during brain development leads to alterations in the hippocampal structure and function[104].
Moreover, in animal models, hyperinsulinemia during in utero period reduced the fetal amino acid
concentrations, including levels of the nonprotein amino acid taurine, which is showed to be involved in the development of the brain[105].
Diabetes during pregnancy as a proinflammatory milieuDiabetes during pregnancy is associated with the disbalance of pro-inflammatory pathways supported by increased circulating concentrations of inflammatory molecules[106,107]. It is demonstrated that inflammatory cytokines affect the neuronal development and metabolism of neurotransmitters[108]. In experimental models, cytokines reduced the survival of dopaminergic and serotonergic cells[109]. Earlier investigations demonstrated that cytokines, including tumor necrosis factor alpha and interleukin-6, are increased in infants exposed to diabetes in utero and have been implicated in neuronal damage[110,111]. It is also clearly showed that imbalances in the regulation of cytokines have been increasingly associated by a number of prevalent and severe neurodevelopmental disorders[112,113].
Currently, a limited number of human studies demonstrating the effects of inflammatory prenatal environment on neurobehavioural development in children[114]. Evidence from a few studies suggests that immune alterations that occur in prenatal period may persist into postnatal life[115,116]. Therefore, increased inflammatory cytokines in diabetic pregnancies may be relevant to neurodevelopment alterations exhibited by the infants born to diabetic women.
Diabetes in pregnancy and oxidative stressEnhanced oxidative stress plays important roles in embryo development and implicated in the pathogenesis of some neurodevelopmental disorders observed in infants of diabetic mothers[83-85]. Free radicals can inactivate the biological functions of proteins and lipids and potentially leading to cell death[117,118]. It is showed that GDM is associated with an increased concentration of oxidative stress, due to both defect in the antioxidant defenses and/or overproduction of free radicals[83-85]. Although, there are several studies demonstrating the presence of elevated oxidative stress in PGDM and GDM states. Some works also suggested that this milieu can be shared with the developing fetus[83-85,119].
In experimental animals, it is emphasized that oxygen radicals play crucial roles in the progression and timing of the development and differentiation of neurons, and synaptic plasticity; so, imbalance in these signals can lead to changes in development of CNS[120,121]. On the other hands, the developing CNS is particularly susceptible to increase in oxidative stress, owing to its poor antioxidant defenses and/or high oxygen consumption[122].
Oxidative stress might contribute to the pathogenesis of some of neurodevelopmental disorders via an attenuation with gamma-aminobutyric acid receptor function in the brain, decreased synaptic efficiency in hippocampal cells, and inhibition of dopamine β-hydr-oxylase[123-125]. On the other hand, there are evidence
416 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Hami J et al . Effects of maternal diabetes on developing hippocampus

indicating that maternal diabetes-induced NTDs are associated with some metabolic difficulties such as increase in superoxide dismutase activity and decreased in arachidonic acid level[126]. There are evidence from human models indicating a potential role of elevated oxidative stress in neurodevelopmental diseases[127-131]. Moreover, the incidence of congenital anomalies in infants of diabetic mothers has been reported to be significantly reduced by antioxidants supplements therapy including Ascorbic Acid and vitamin E. These data suggest that elevated oxidative stress is associated with the pathophysiology of fetal dysmorphogenesis[132,133].
Role of insulin-like growth factor-1 and insulin in maternal diabetes - induced neuropathiesInsulin- like growth factor-1 (IGF-1) and insulin belong to the insulin superfamily and exert profound effects in the CNS development and function. Evidence of these peptides actions on CNS comes from a wide variety of in vitro and in vivo experimental data, the latter predominantly derived from rodent studies. In several studies, the authors demonstrated that the insulin and IGF-1 have a wide spectrum of biological actions in the developing CNS including neuronal/glial cell proliferation, differentiation, survival, synaptogenesis, longevity, and neuroregeneration. Most of these biological functions are mediated via two transmembrane receptors: the insulin receptor (IR) and the IGF-1 receptor (IGF1R)[69,70,134-142].
The IR and IGF1R, which are homologus in structure, are composed of two α-subunits and two β-subunits linked by disulphide bonds. The extracellular α- subunit binds to their cognate ligand. The β-subunit comprises an extracellular domain and a cytoplasmic domain that contains intrinsic protein tyrosine kinase activity. The binding of either IGF-1 or insulin to α-subunit of their cognate receptors stimulates phosphorylation of the β subunit on serine and tyrosine residues. The autophosphorylation of the β subunit then results in the phosphorylation of cellular substrates and signal transfer cascade for IGF-1 and insulin[141,143,144].
Using ligand binding autoradiography and in situ hybridization, several line of human and animal inves-tigations have found that IR and IGF1R are irregularly distributed in developing and mature brain with the highest densities in the hippocampus, cerebral cortex, olfactory bulb, cerebellum, and hypothalamus. Interestingly, some of brain regions show a marked difference in the density of InsR and IGF1R between embryonic vs mature brain, implying a key role for these ligands in brain development[145-147].
There are evidence from several researches impli-cating defects in IR and IGF1R signaling result in congenital developmental defects seen in offspring of diabetic mothers[145,148-150]. Ramsay et al[151] (1994) in their study was found that expression of IGF-1 in the brain were declined in swine newborns born to diabetic dams when compared to that of control animals.
The hippocampal formation subserves important physiological and behavioral functions including spatial
learning and memory and is a part of brain that parti-cularly vulnerable to changes in blood glucose level[152-154]. In an investigation by Tehranipour et al[155] (2008) the effects of maternal T1DM on density of hippocampal pyramidal cells immediately after birth were examined. Those study results were clearly indicated that maternal diabetes can decreased the numerical density of pyramidal cells in the hippocampus of rat newborns, especially in CA3[155].
Interestingly, the studies By Hami et al[156] (2012) showed that there are prominent gender-and laterality- differences in expression and distribution pattern of InsR and IGF1R in the developing rat hippocampus. The authors concluded that these differences may be a probable mechanism for the control of sex and laterality differences in development and function of the rat hippocampus[156].
In another study by Hami et al[69] (2013), the effects of diabetes in pregnancy on gene expression and protein concentration of IGF1R and IR in the developing rat hippocampus at postnatal days 0, 7, and 14 were evaluated. In that study, the authors found a markedly upregulation of both IR and IGF1R expression in the hippocampus of diabetic group newborns at first postnatal day. At the same time point, they showed only slight changes in their hippocampal protein transcripts. In 7-d old rats, there was a significant decreased in IGF-1R gene expression and protein levels in the newborns born to diabetic dams. Moreover, they found a down regulation in hippocampal IGF1R transcripts in 14-d old diabetic group offspring. Two weeks after birth, the IR gene expression was significantly declined in the hippocampus of diabetic newborns. The authors claimed that maternal diabetes strongly altered the regulation of both IR and IGF1R during development of rat hippocampus[67].
CONCLUSIONThe Incidence of congenital anomalies in infants born to diabetic women is more common in comparison to children of normal population. There are multiple lines of evidence that suggest the disturbances in intellectual and behavioral functioning observed in the children of diabetic women are accompanied by modification of hippocampus structure and function. The etiology and pathogenesis of these impairments induced by diabetes during pregnancy have spurred considerable efforts for clinically and basically researches. The final goal at these investigations was to find the teratogenic factors, which may enable preventive or protective measures to be taken in pregnancies with diabetes. Nevertheless, the exact mechanism by which diabetes during pregnancy affects the CNS development remains to be defined. Until now, several biological changes are defined to occur in diabetic mothers and to affect development of CNS (i.e., disturbance in glucose and insulin homeostasis, oxidative stress, hypoxia, and iron deficiency). Moreover, the new researches on genetic
417 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Hami J et al . Effects of maternal diabetes on developing hippocampus

predisposition involves in teratogenicity of diabetes in pregnancy starts to define new genes and their products involved in the etiology of CNS malfunctions and malformations observed in offspring born to diabetic mothers. Hyperglycaemia and hyperinsulinemia in the mothers and their fetuses are thought to be major factors in teratogenicity of maternal diabetes on the CNS development. There are sufficient evidences for this hypothesis as concise control of glycemia in mothers reduces the incidence of anomalies exhibited with the offspring born to diabetic mothers. Recent evidence clearly indicated that maternal diabetes markedly influences the regulation of both IR and IGF1R - as two important regulators of development and function of CNS - in the developing rat hippocampus. Dissecting out the mechanisms responsible for maternal diabetes-related changes in the development of hippocampus is helping to prevent from impaired cognitive and memory functions in offspring.
REFERENCES1 Khan MN, Khan FA, Sultana S, Dilawar M, Ijaz A, Khan MJ,
Mahmood T. Impact of new diagnostic criteria of diabetes mellitus. J Coll Physicians Surg Pak 2007; 17: 327-330 [PMID: 17623579]
2 Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 2003; 26 Suppl 1: S5-20 [PMID: 12502614]
3 Reece EA. The fetal and maternal consequences of gestational diabetes mellitus. J Matern Fetal Neonatal Med 2010; 23: 199-203 [PMID: 20121460 DOI: 10.3109/14767050903550659]
4 Fetita LS, Sobngwi E, Serradas P, Calvo F, Gautier JF. Con-sequences of fetal exposure to maternal diabetes in offspring. J Clin Endocrinol Metab 2006; 91: 3718-3724 [PMID: 16849402 DOI: 10.1210/jc.2006-0624]
5 Aerts L, Holemans K, Van Assche FA. Maternal diabetes during pregnancy: consequences for the offspring. Diabetes Metab Rev 1990; 6: 147-167 [PMID: 2091909]
6 Persaud OD. Maternal diabetes and the consequences for her offspring. J Develop Disab 2007; 1: 101-134
7 US Preventive Services Task Force (USPSTF). Screening for gestational diabetes mellitus: recommendation and rationale. Am Fam Physician 2003; 68: 331-335 [PMID: 12892353]
8 Reece EA, Homko CJ. Diabetes mellitus in pregnancy. What are the best treatment options? Drug Saf 1998; 18: 209-220 [PMID: 9530539]
9 Chaudry R, Gilby P, Carroll PV. Pre-existing (type 1 and type 2) diabetes in pregnancy. Obst Gyn Repro Med 2007; 17: 339-344 [DOI: 10.1016/j.ogrm.2007.09.001]
10 Health CEiMaC. Pregnancy in women with type 1 and type 2 diabetes in 2002-03, england, wales and northern ireland. London: CEMACH, 2005
11 McElduff A, Ross GP, Lagström JA, Champion B, Flack JR, Lau SM, Moses RG, Seneratne S, McLean M, Cheung NW. Pregestational diabetes and pregnancy: an Australian experience. Diabetes Care 2005; 28: 1260-1261 [PMID: 15855607]
12 Meur S , Mann NP. Infant outcomes following diabetic pregnancies. Paediatrics and Child Health 2007; 17: 217-222
13 ter Braak EW, Evers IM, Willem Erkelens D, Visser GH. Maternal hypoglycemia during pregnancy in type 1 diabetes: maternal and fetal consequences. Diabetes Metab Res Rev 2002; 18: 96-105 [PMID: 11994900 DOI: 10.1002/dmrr.271]
14 Schwartz R, Teramo KA. Effects of diabetic pregnancy on the fetus and newborn. Semin Perinatol 2000; 24: 120-135 [PMID: 10805168]
15 Gabbe SG. Congenital malformations in infants of diabetic mothers. Obstet Gynecol Surv 1977; 32: 125-132 [PMID: 857205]
16 Mills JL. Malformations in infants of diabetic mothers. Teratology 1982; 25: 385-394 [PMID: 7051398 DOI: 10.1002/tera.1420250316]
17 Suevo DM. The infant of the diabetic mother. Neonatal Netw 1997; 16: 25-33 [PMID: 9325870]
18 Bánhidy F, Acs N, Puhó EH, Czeizel AE. Congenital abnor-malities in the offspring of pregnant women with type 1, type 2 and gestational diabetes mellitus: a population-based case-control study. Congenit Anom (Kyoto) 2010; 50: 115-121 [PMID: 20184644]
19 Hod M, Merlob P, Friedman S, Litwin A, Mor N, Rusecki Y, Schoenfeld A, Ovadia J. Prevalence of minor congenital anomalies in newborns of diabetic mothers. Eur J Obstet Gynecol Reprod Biol 1992; 44: 111-116 [PMID: 1587375]
20 Eidelman AI, Samueloff A. The pathophysiology of the fetus of the diabetic mother. Semin Perinatol 2002; 26: 232-236 [PMID: 12099314]
21 Styrud J, Thunberg L, Nybacka O, Eriksson UJ. Correlations between maternal metabolism and deranged development in the offspring of normal and diabetic rats. Pediatr Res 1995; 37: 343-353 [PMID: 7784144]
22 Ray JG, O’Brien TE, Chan WS. Preconception care and the risk of congenital anomalies in the offspring of women with diabetes mellitus: a meta-analysis. QJM 2001; 94: 435-444 [PMID: 11493721]
23 Pedersen J. Weight and length at birth of infants of diabetic mothers. Acta Endocrinol (Copenh) 1954; 16: 330-342 [PMID: 13206643]
24 Pedersen J. The pregnant diabetic and her newborn. Baltimore: Williams & Wilkins, 1977
25 Taylor R, Lee C, Kyne-Grzebalski D, Marshall SM, Davison JM. Clinical outcomes of pregnancy in women with type 1 diabetes(1). Obstet Gynecol 2002; 99: 537-541 [PMID: 12039106]
26 Towner D, Kjos SL, Leung B, Montoro MM, Xiang A, Mestman JH, Buchanan TA. Congenital malformations in pregnancies complicated by NIDDM. Diabetes Care 1995; 18: 1446-1451 [PMID: 8722068]
27 Eriksson U, Dahlström E, Larsson KS, Hellerström C. Increased incidence of congenital malformations in the offspring of diabetic rats and their prevention by maternal insulin therapy. Diabetes 1982; 31: 1-6 [PMID: 6759206]
28 Eriksson UJ, Bone AJ, Turnbull DM, Baird JD. Timed interruption of insulin therapy in diabetic BB/E rat pregnancy: effect on maternal metabolism and fetal outcome. Acta Endocrinol (Copenh) 1989; 120: 800-810 [PMID: 2658457]
29 Lapolla A, Dalfrà MG, Fedele D. Insulin therapy in pregnancy complicated by diabetes: are insulin analogs a new tool? Diabetes Metab Res Rev 2005; 21: 241-252 [PMID: 15818714 DOI: 10.1002/dmrr.551]
30 Salvesen DR, Freeman J, Brudenell JM, Nicolaides KH. Prediction of fetal acidaemia in pregnancies complicated by maternal diabetes mellitus by biophysical profile scoring and fetal heart rate monitoring. Br J Obstet Gynaecol 1993; 100: 227-233 [PMID: 8476827]
31 Schwartz R, Gruppuso PA, Petzold K, Brambilla D, Hiilesmaa V, Teramo KA. Hyperinsulinemia and macrosomia in the fetus of the diabetic mother. Diabetes Care 1994; 17: 640-648 [PMID: 7924772]
32 Susa JB, Neave C, Sehgal P, Singer DB, Zeller WP, Schwartz R. Chronic hyperinsulinemia in the fetal rhesus monkey. Effects of physiologic hyperinsulinemia on fetal growth and composition. Diabetes 1984; 33: 656-660 [PMID: 6376221]
33 Susa JB, Widness JA, Hintz R, Liu F, Sehgal P, Schwartz R. Somatomedins and insulin in diabetic pregnancies: effects on fetal macrosomia in the human and rhesus monkey. J Clin Endocrinol Metab 1984; 58: 1099-1105 [PMID: 6373810]
34 Widness JA, Susa JB, Garcia JF, Singer DB, Sehgal P, Oh W, Schwartz R, Schwartz HC. Increased erythropoiesis and elevated erythropoietin in infants born to diabetic mothers and in hyperinsulinemic rhesus fetuses. J Clin Invest 1981; 67: 637-642
418 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Hami J et al . Effects of maternal diabetes on developing hippocampus

[PMID: 7009647 DOI: 10.1172/JCI110078]35 Freinkel N. Diabetic embryopathy and fuel-mediated organ
teratogenesis: lessons from animal models. Horm Metab Res 1988; 20: 463-475 [PMID: 3053387 DOI: 10.1055/s-2007-1010861]
36 Milner RD, Hill DJ. Interaction between endocrine and paracrine peptides in prenatal growth control. Eur J Pediatr 1987; 146: 113-122 [PMID: 3552691]
37 Cousins L. Etiology and prevention of congenital anomalies among infants of overt diabetic women. Clin Obstet Gynecol 1991; 34: 481-493 [PMID: 1934700]
38 Eriksson UJ, Borg LA. Diabetes and embryonic malformations. Role of substrate-induced free-oxygen radical production for dysmorphogenesis in cultured rat embryos. Diabetes 1993; 42: 411-419 [PMID: 8432412]
39 Lee AT, Reis D, Eriksson UJ. Hyperglycemia-induced embryonic dysmorphogenesis correlates with genomic DNA mutation frequency in vitro and in vivo. Diabetes 1999; 48: 371-376 [PMID: 10334316]
40 Hagay ZJ, Weiss Y, Zusman I, Peled-Kamar M, Reece EA, Eriksson UJ, Groner Y. Prevention of diabetes-associated embryopathy by overexpression of the free radical scavenger copper zinc superoxide dismutase in transgenic mouse embryos. Am J Obstet Gynecol 1995; 173: 1036-1041 [PMID: 7485290]
41 Wentzel P, Thunberg L, Eriksson UJ. Teratogenic effect of diabetic serum is prevented by supplementation of superoxide dismutase and N-acetylcysteine in rat embryo culture. Diabetologia 1997; 40: 7-14 [PMID: 9028712 DOI: 10.1007/s001250050636]
42 Georgieff MK. The effect of maternal diabetes during pregnancy on the neurodevelopment of offspring. Minn Med 2006; 89: 44-47 [PMID: 16669433]
43 Ornoy A. Growth and neurodevelopmental outcome of children born to mothers with pregestational and gestational diabetes. Pediatr Endocrinol Rev 2005; 3: 104-113 [PMID: 16361984]
44 Nelson CA, Wewerka S, Thomas KM, Tribby-Walbridge S, deRegnier R, Georgieff M. Neurocognitive sequelae of infants of diabetic mothers. Behav Neurosci 2000; 114: 950-956 [PMID: 11085609]
45 Yamashita Y, Kawano Y, Kuriya N, Murakami Y, Matsuishi T, Yoshimatsu K, Kato H. Intellectual development of offspring of diabetic mothers. Acta Paediatr 1996; 85: 1192-1196 [PMID: 8922082]
46 Sells CJ, Robinson NM, Brown Z, Knopp RH. Long-term developmental follow-up of infants of diabetic mothers. J Pediatr 1994; 125: S9-17 [PMID: 8021756]
47 Van Assche FA, Holemans K, Aerts L. Long-term consequences for offspring of diabetes during pregnancy. Br Med Bull 2001; 60: 173-182 [PMID: 11809625]
48 Ornoy A. The impact of intrauterine exposure versus postnatal environment in neurodevelopmental toxicity: long-term neuro-behavioral studies in children at risk for developmental disorders. Toxicol Lett 2003; 140-141: 171-181 [PMID: 12676464]
49 Ornoy A, Ratzon N, Greenbaum C, Peretz E, Soriano D, Dulitzky M. Neurobehaviour of school age children born to diabetic mothers. Arch Dis Child Fetal Neonatal Ed 1998; 79: F94-F99 [PMID: 9828733]
50 Ornoy A, Ratzon N, Greenbaum C, Wolf A, Dulitzky M. School-age children born to diabetic mothers and to mothers with gestational diabetes exhibit a high rate of inattention and fine and gross motor impairment. J Pediatr Endocrinol Metab 2001; 14 Suppl 1: 681-689 [PMID: 11393563]
51 Delascio Lopes C, Sinigaglia-Coimbra R, Mazzola J, Camano L, Mattar R. Neurofunctional evaluation of young male offspring of rat dams with diabetes induced by streptozotocin. ISRN Endocrinol 2011; 2011: 480656 [PMID: 22363880 DOI: 10.5402/2011/480656]
52 Hepper PG. The behaviour of the foetus as an indicator of neural functioning. In: Lecanuet JP, Fifer W, Krasnegor N, Smotherman W, eds. Fetal development. A psychobiological perspective. Lawrence Erlbaum: Hillsdale, NJ, 1995: 405-417
53 Rizzo TA, Silverman BL, Metzger BE, Cho NH. Behavioral adjustment in children of diabetic mothers. Acta Paediatr 1997;
86: 969-974 [PMID: 9343277]54 Freinkel N. Banting Lecture 1980. Of pregnancy and progeny.
Diabetes 1980; 29: 1023-1035 [PMID: 7002669]55 Churchill JA, Berendes HW, Nemore J. Neuropsychological
deficits in children of diabetic mothers. A report from the Collaborative Sdy of Cerebral Palsy. Am J Obstet Gynecol 1969; 105: 257-268 [PMID: 4980345]
56 Stehbens JA, Baker GL, Kitchell M. Outcome at ages 1, 3, and 5 years of children born to diabetic women. Am J Obstet Gynecol 1977; 127: 408-413 [PMID: 835641]
57 Rizzo T, Metzger BE, Burns WJ, Burns K. Correlations between antepartum maternal metabolism and child intelligence. N Engl J Med 1991; 325: 911-916 [PMID: 1881416 DOI: 10.1056/NEJM199109263251303]
58 Rizzo T, Freinkel N, Metzger BE, Hatcher R, Burns WJ, Barglow P. Correlations between antepartum maternal metabolism and newborn behavior. Am J Obstet Gynecol 1990; 163: 1458-1464 [PMID: 2240088]
59 Cummins M, Norrish M. Follow-up of children of diabetic mothers. Arch Dis Child 1980; 55: 259-264 [PMID: 7416774]
60 Persson B, Gentz J. Follow-up of children of insulin-dependent and gestational diabetic mothers. Neuropsychological outcome. Acta Paediatr Scand 1984; 73: 349-358 [PMID: 6741537]
61 Rizzo TA, Dooley SL, Metzger BE, Cho NH, Ogata ES, Silverman BL. Prenatal and perinatal influences on long-term psychomotor development in offspring of diabetic mothers. Am J Obstet Gynecol 1995; 173: 1753-1758 [PMID: 8610757]
62 Yssing M. Long-term prognosis of children born to mothers diabetic when pregnant. In: Camerini-Davalus RA, Cole HS, editors. Early diabetics in early life. New York: Academic Press, 1975: 575-586
63 Haworth JC, McRae KN, Dilling LA. Prognosis of infants of diabetic mothers in relation to neonatal hypoglycaemia. Dev Med Child Neurol 1976; 18: 471-479 [PMID: 955311]
64 Hadden DR, Byrne E, Trotter I, Harley JM, McClure G, McAuley RR. Physical and psychological health of children of Type 1 (insulin-dependent) diabetic mothers. Diabetologia 1984; 26: 250-254 [PMID: 6376231]
65 Doherty NN, Hepper PG. Habituation in fetuses of diabetic mothers. Early Hum Dev 2000; 59: 85-93 [PMID: 10996746]
66 Gonzalez-Gonzalez NL, Medina V, Padron E, Domenech E, Diaz Gomez NM, Armas H, Bartha JL. Fetal and neonatal habituation in infants of diabetic mothers. J Pediatr 2009; 154: 492-497 [PMID: 19054526 DOI: 10.1016/j.jpeds.2008.10.020]
67 Van Lieshout RJ, Voruganti LP. Diabetes mellitus during pregnancy and increased risk of schizophrenia in offspring: a review of the evidence and putative mechanisms. J Psychiatry Neurosci 2008; 33: 395-404 [PMID: 18787655]
68 Ramanathan M, Jaiswal AK, Bhattacharya SK. Hyperglycaemia in pregnancy: effects on the offspring behaviour with special reference to anxiety paradigms. Indian J Exp Biol 2000; 38: 231-236 [PMID: 10927864]
69 Hami J, Sadr-Nabavi A, Sankian M, Balali-Mood M, Haghir H. The effects of maternal diabetes on expression of insulin-like growth factor-1 and insulin receptors in male developing rat hippocampus. Brain Struct Funct 2013; 218: 73-84 [PMID: 22241286 DOI: 10.1007/s00429-011-0377-y]
70 Haghir H, Rezaee AA, Sankian M, Kheradmand H, Hami J. The effects of induced type-I diabetes on developmental regulation of insulin & amp; insulin like growth factor-1 (IGF-1) receptors in the cerebellum of rat neonates. Metab Brain Dis 2013; 28: 397-410 [PMID: 23397157 DOI: 10.1007/s11011-013-9386-2]
71 Yamano T, Shimada M, Fujizeki Y, Kawasaki H, Onaga A. Quantitative synaptic changes on Purkinje cell dendritic spines of rats born from streptozotocin-induced diabetic mothers. Brain Dev 1986; 8: 269-273 [PMID: 2945494]
72 Yang X, Borg LA, Eriksson UJ. Altered mitochondrial morphology of rat embryos in diabetic pregnancy. Anat Rec 1995; 241: 255-267 [PMID: 7710141 DOI: 10.1002/ar.1092410212]
73 Kinney BA, Rabe MB, Jensen RA, Steger RW. Maternal
419 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Hami J et al . Effects of maternal diabetes on developing hippocampus

hyperglycemia leads to gender-dependent deficits in learning and memory in offspring. Exp Biol Med (Maywood) 2003; 228: 152-159 [PMID: 12563021]
74 Plagemann A, Harder T, Lindner R, Melchior K, Rake A, Rittel F, Rohde W, Dörner G. Alterations of hypothalamic catecholamines in the newborn offspring of gestational diabetic mother rats. Brain Res Dev Brain Res 1998; 109: 201-209 [PMID: 9729385]
75 Becerra JE, Khoury MJ, Cordero JF, Erickson JD. Diabetes mellitus during pregnancy and the risks for specific birth defects: a population-based case-control study. Pediatrics 1990; 85: 1-9 [PMID: 2404255]
76 Correa A, Gilboa SM, Besser LM, Botto LD, Moore CA, Hobbs CA, Cleves MA, Riehle-Colarusso TJ, Waller DK, Reece EA. Diabetes mellitus and birth defects. Am J Obstet Gynecol 2008; 199: 237.e1-237.e9 [PMID: 18674752 DOI: 10.1016/j.ajog.2008.06.028]
77 Ramos-Arroyo MA, Rodriguez-Pinilla E, Cordero JF. Maternal diabetes: the risk for specific birth defects. Eur J Epidemiol 1992; 8: 503-508 [PMID: 1397216]
78 Xu C, Li X, Wang F, Weng H, Yang P. Trehalose prevents neural tube defects by correcting maternal diabetes-suppressed autophagy and neurogenesis. Am J Physiol Endocrinol Metab 2013; 305: E667-E678 [PMID: 23880312]
79 Reece EA, Pinter E, Leranth CZ, Garcia-Segura M, Sanyal MK, Hobbins JC, Mahoney MJ, Naftolin F. Ultrastructural analysis of malformations of the embryonic neural axis induced by in vitro hyperglycemic conditions. Teratology 1985; 32: 363-373 [PMID: 4082067 DOI: 10.1002/tera.1420320306]
80 Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci 2012; 322: 254-262 [PMID: 22669122 DOI: 10.1016/j.jns.2012.05.030]
81 Faizi M, Salimi A, Rasoulzadeh M, Naserzadeh P, Pourahmad J. Schizophrenia induces oxidative stress and cytochrome C release in isolated rat brain mitochondria: a possible pathway for induction of apoptosis and neurodegeneration. Iran J Pharm Res 2014; 13: 93-100 [PMID: 24711834]
82 Foster KA, Galeffi F, Gerich FJ, Turner DA, Müller M. Optical and pharmacological tools to investigate the role of mitochondria during oxidative stress and neurodegeneration. Prog Neurobiol 2006; 79: 136-171 [PMID: 16920246 DOI: 10.1016/j.pneurobio.2006.07.001]
83 Zein S, Rachidi S, Hininger-Favier I. Is oxidative stress induced by iron status associated with gestational diabetes mellitus? J Trace Elem Med Biol 2014; 28: 65-69 [PMID: 24238846 DOI: 10.1016/j.jtemb.2013.09.009]
84 Lappas M, Hiden U, Desoye G, Froehlich J, Hauguel-de Mouzon S, Jawerbaum A. The role of oxidative stress in the pathophysiology of gestational diabetes mellitus. Antioxid Redox Signal 2011; 15: 3061-3100 [PMID: 21675877 DOI: 10.1089/ars.2010.3765]
85 Chen X, Scholl TO. Oxidative stress: changes in pregnancy and with gestational diabetes mellitus. Curr Diab Rep 2005; 5: 282-288 [PMID: 16033680]
86 Mimouni F, Miodovnik M, Siddiqi TA, Butler JB, Holroyde J, Tsang RC. Neonatal polycythemia in infants of insulin-dependent diabetic mothers. Obstet Gynecol 1986; 68: 370-372 [PMID: 3737061]
87 Curristin SM, Cao A, Stewart WB, Zhang H, Madri JA, Morrow JS, Ment LR. Disrupted synaptic development in the hypoxic newborn brain. Proc Natl Acad Sci USA 2002; 99: 15729-15734 [PMID: 12438650 DOI: 10.1073/pnas.232568799]
88 McQuillen PS, Sheldon RA, Shatz CJ, Ferriero DM. Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J Neurosci 2003; 23: 3308-3315 [PMID: 12716938]
89 Johnston MV, Trescher WH, Ishida A, Nakajima W. Neurobiology of hypoxic-ischemic injury in the developing brain. Pediatr Res 2001; 49: 735-741 [PMID: 11385130 DOI: 10.1203/00006450-200106000-00003]
90 Carrapato MR, Marcelino F. The infant of the diabetic mother: The critical developmental windows. Early Pregnancy 2001; 5: 57-58 [PMID: 11753515]
91 Yehuda S, Youdim MB. Brain iron: a lesson from animal models.
Am J Clin Nutr 1989; 50: 618-625; discussion 625-629 [PMID: 2570524]
92 Yehuda S. Neurochemical basis of behavioral effects of brain iron deficiency in anemia. In: Dobbing J, editor. Brain, behavior and iron in the infant diet. London: Springer-Verlag, 1990: 63-81
93 Petry CD, Eaton MA, Wobken JD, Mills MM, Johnson DE, Georgieff MK. Iron deficiency of liver, heart, and brain in newborn infants of diabetic mothers. J Pediatr 1992; 121: 109-114 [PMID: 1625067]
94 Lozoff B, Georgieff MK. Iron deficiency and brain development. Semin Pediatr Neurol 2006; 13: 158-165 [PMID: 17101454 DOI: 10.1016/j.spen.2006.08.004]
95 Beard J. Iron deficiency alters brain development and functioning. J Nutr 2003; 133: 1468S-1472S [PMID: 12730445]
96 Georgieff MK. The role of iron in neurodevelopment: fetal iron deficiency and the developing hippocampus. Biochem Soc Trans 2008; 36: 1267-1271 [PMID: 19021538 DOI: 10.1042/BST0361267]
97 Larkin EC, Rao GA. Mportance of fetal and neonatal iron: Adequacy for normal development of central nervous system. In: Dobbing J, editor. Brain, behavior and iron in the infant diet. London: Springer-Verlag, 1990: 43-62
98 Felt BT, Beard JL, Schallert T, Shao J, Aldridge JW, Connor JR, Georgieff MK, Lozoff B. Persistent neurochemical and behavioral abnormalities in adulthood despite early iron supplementation for perinatal iron deficiency anemia in rats. Behav Brain Res 2006; 171: 261-270 [PMID: 16713640 DOI: 10.1016/j.bbr.2006.04.001]
99 Rytych JL, Elmore MR, Burton MD, Conrad MS, Donovan SM, Dilger RN, Johnson RW. Early life iron deficiency impairs spatial cognition in neonatal piglets. J Nutr 2012; 142: 2050-2056 [PMID: 23014488 DOI: 10.3945/jn.112.165522]
100 Eden AN. Iron deficiency and impaired cognition in toddlers: an underestimated and undertreated problem. Paediatr Drugs 2005; 7: 347-352 [PMID: 16356022]
101 Walter T. Impact of iron deficiency on cognition in infancy and childhood. Eur J Clin Nutr 1993; 47: 307-316 [PMID: 7686486]
102 Felt BT, Lozoff B. Brain iron and behavior of rats are not normalized by treatment of iron deficiency anemia during early development. J Nutr 1996; 126: 693-701 [PMID: 8598555]
103 Wachs TD, Pollitt E, Cueto S, Jacoby E, Creed-Kanashiro H. Relation of neonatal iron status to individual variability in neonatal temperament. Dev Psychobiol 2005; 46: 141-153 [PMID: 15732057 DOI: 10.1002/dev.20049]
104 Siddappa AM, Georgieff MK, Wewerka S, Worwa C, Nelson CA, Deregnier RA. Iron deficiency alters auditory recognition memory in newborn infants of diabetic mothers. Pediatr Res 2004; 55: 1034-1041 [PMID: 15155871 DOI: 10.1203/01.pdr.0000127021.38207.62]
105 Aerts L, Van Assche FA. Animal evidence for the transgenerational development of diabetes mellitus. Int J Biochem Cell Biol 2006; 38: 894-903 [PMID: 16118061]
106 Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M, Quagliaro L, Ceriello A, Giugliano D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation 2002; 106: 2067-2072 [PMID: 12379575]
107 Loukovaara M, Leinonen P, Teramo K, Alfthan H, Stenman UH, Andersson S. Fetal hypoxia is associated with elevated cord serum C-reactive protein levels in diabetic pregnancies. Biol Neonate 2004; 85: 237-242 [PMID: 14718755 DOI: 10.1159/000076132]
108 Mehler MF, Kessler JA. Hematolymphopoietic and inflammatory cytokines in neural development. Trends Neurosci 1997; 20: 357-365 [PMID: 9246730]
109 Jarskog LF, Xiao H, Wilkie MB, Lauder JM, Gilmore JH. Cytokine regulation of embryonic rat dopamine and serotonin neuronal survival in vitro. Int J Dev Neurosci 1997; 15: 711-716 [PMID: 9402221]
110 Kinalski M, Telejko B, Kuźmicki M, Kretowski A, Kinalska I. Tumor necrosis factor alpha system and plasma adiponectin concentration in women with gestational diabetes. Horm Metab Res 2005; 37: 450-454 [PMID: 16034719 DOI: 10.1055/s-2005-870238]
420 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Hami J et al . Effects of maternal diabetes on developing hippocampus

111 Bo S, Signorile A, Menato G, Gambino R, Bardelli C, Gallo ML, Cassader M, Massobrio M, Pagano GF. C-reactive protein and tumor necrosis factor-alpha in gestational hyperglycemia. J Endocrinol Invest 2005; 28: 779-786 [PMID: 16370555]
112 Deverman BE, Patterson PH. Cytokines and CNS development. Neuron 2009; 64: 61-78 [PMID: 19840550 DOI: 10.1016/j.neu-ron.2009.09.002]
113 Zhao B, Schwartz JP. Involvement of cytokines in normal CNS development and neurological diseases: recent progress and perspectives. J Neurosci Res 1998; 52: 7-16 [PMID: 9556025]
114 Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V, Perrin M, Gorman JM, Susser ES. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry 2004; 161: 889-895 [PMID: 15121655]
115 Kaan A, Dimich-Ward H, Manfreda J, Becker A, Watson W, Ferguson A, Chan H, Chan-Yeung M. Cord blood IgE: its determinants and prediction of development of asthma and other allergic disorders at 12 months. Ann Allergy Asthma Immunol 2000; 84: 37-42 [PMID: 10674563 DOI: 10.1016/S1081-1206(10)62738-X]
116 Spencer SJ, Hyland NP, Sharkey KA, Pittman QJ. Neonatal immune challenge exacerbates experimental colitis in adult rats: potential role for TNF-alpha. Am J Physiol Regul Integr Comp Physiol 2007; 292: R308-R315 [PMID: 16973935 DOI: 10.1152/ajpregu.00398.2006]
117 Stadtman ER. Protein oxidation and aging. Science 1992; 257: 1220-1224 [PMID: 1355616]
118 Ames BA, Shingenaga MK, Park EM. Oxyradicals and DNA damage. In: Davies KJ, editor. Oxidation damage and repair: Chemical, biological and medical aspects. Elmsford: Pergamon Press, 1991: 181-187
119 Bis-Głuchowska M, Marciniak B, Szpringer-Boguń E, Rola R, Leszczyńska-Gorzelak B, Oleszczuk J. [Determination of antioxidative-peroxidative balance in the cord blood of newborns delivered to mothers with diabetes type G1]. Ginekol Pol 2001; 72: 1255-1258 [PMID: 11883260]
120 Allen RG, Venkatraj VS. Oxidants and antioxidants in deve-lopment and differentiation. J Nutr 1992; 122: 631-635 [PMID: 1542023]
121 Rafalowska U, Liu GJ, Floyd RA. Peroxidation induced changes in synaptosomal transport of dopamine and gamma-aminobutyric acid. Free Radic Biol Med 1989; 6: 485-492 [PMID: 2744581]
122 Mahadik SP, Mukherjee S. Free radical pathology and antioxidant defense in schizophrenia: a review. Schizophr Res 1996; 19: 1-17 [PMID: 9147491]
123 Schwartz RD, Skolnick P, Paul SM. Regulation of gamma-aminobutyric acid/barbiturate receptor-gated chloride ion flux in brain vesicles by phospholipase A2: possible role of oxygen radicals. J Neurochem 1988; 50: 565-571 [PMID: 2447244]
124 Pellmar T. Electrophysiological correlates of peroxide damage in guinea pig hippocampus in vitro. Brain Res 1986; 364: 377-381 [PMID: 3947975]
125 LEVIN EY, KAUFMAN S. Studies on the enzyme catalyzing the conversion of 3,4-dihydroxyphenylethylamine to norepinephrine. J Biol Chem 1961; 236: 2043-2049 [PMID: 13761407]
126 Dheen ST, Tay SS, Boran J, Ting LW, Kumar SD, Fu J, Ling EA. Recent studies on neural tube defects in embryos of diabetic pregnancy: an overview. Curr Med Chem 2009; 16: 2345-2354 [PMID: 19519395]
127 De Felice C, Signorini C, Leoncini S, Pecorelli A, Durand T, Valacchi G, Ciccoli L, Hayek J. The role of oxidative stress in Rett syndrome: an overview. Ann N Y Acad Sci 2012; 1259: 121-135 [PMID: 22758644 DOI: 10.1111/j.1749-6632.2012.06611.x]
128 Rossignol DA, Frye RE. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front Physiol 2014; 5: 150 [PMID: 24795645 DOI: 10.3389/fphys.2014.00150]
129 Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysiology 2006; 13: 171-181 [PMID: 16766163 DOI: 10.1016/j.pathoph-ys.2006.05.007]
130 Flatow J, Buckley P, Miller BJ. Meta-analysis of oxidative stress in schizophrenia. Biol Psychiatry 2013; 74: 400-409 [PMID: 23683390 DOI: 10.1016/j.biopsych.2013.03.018]
131 Tylec A, Jarzab A, Stryjecka-Zimmer M, Wójcicka A. [Stress oxidative in schizophrenia]. Pol Merkur Lekarski 2007; 23: 74-77 [PMID: 18051835]
132 Cederberg J, Simán CM, Eriksson UJ. Combined treatment with vitamin E and vitamin C decreases oxidative stress and improves fetal outcome in experimental diabetic pregnancy. Pediatr Res 2001; 49: 755-762 [PMID: 11385134 DOI: 10.1203/00006450-200106000-00007]
133 Cederberg J, Eriksson UJ. Antioxidative treatment of pregnant diabetic rats diminishes embryonic dysmorphogenesis. Birth Defects Res A Clin Mol Teratol 2005; 73: 498-505 [PMID: 15959875 DOI: 10.1002/bdra.20144]
134 Anlar B, Sullivan KA, Feldman EL. Insulin-like growth factor-I and central nervous system development. Horm Metab Res 1999; 31: 120-125 [PMID: 10226791 DOI: 10.1055/s-2007-978708]
135 Baron-Van Evercooren A, Olichon-Berthe C, Kowalski A, Visciano G, Van Obberghen E. Expression of IGF-I and insulin receptor genes in the rat central nervous system: a developmental, regional, and cellular analysis. J Neurosci Res 1991; 28: 244-253 [PMID: 1851850 DOI: 10.1002/jnr.490280212]
136 Dou JT, Chen M, Dufour F, Alkon DL, Zhao WQ. Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn Mem 2005; 12: 646-655 [PMID: 16287721]
137 Haghir H, Rezaee AA, Nomani H, Sankian M, Kheradmand H, Hami J. Sexual dimorphism in expression of insulin and insulin-like growth factor-I receptors in developing rat cerebellum. Cell Mol Neurobiol 2013; 33: 369-377 [PMID: 23322319 DOI: 10.1007/s10571-012-9903-6]
138 Hami J, Kheradmand H, Haghir H. Gender differences and lateralization in the distribution pattern of insulin-like growth factor-1 receptor in developing rat hippocampus: an immunohistochemical study. Cell Mol Neurobiol 2014; 34: 215-226 [PMID: 24287499 DOI: 10.1007/s10571-013-0005-x]
139 Popken GJ, Hodge RD, Ye P, Zhang J, Ng W, O’Kusky JR, D’Ercole AJ. In vivo effects of insulin-like growth factor-I (IGF-I) on prenatal and early postnatal development of the central nervous system. Eur J Neurosci 2004; 19: 2056-2068 [PMID: 15090033 DOI: 10.1111/j.0953-816X.2004.03320.xEJN3320]
140 Rother KI, Accili D. Role of insulin receptors and IGF receptors in growth and development. Pediatr Nephrol 2000; 14: 558-561 [PMID: 10912518]
141 Zhao WQ, Chen H, Quon MJ, Alkon DL. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol 2004; 490: 71-81 [PMID: 15094074 DOI: 10.1016/j.ejphar.2004.02.045S001429990400202X]
142 Nelson TJ, Sun MK, Hongpaisan J, Alkon DL. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur J Pharmacol 2008; 585: 76-87 [PMID: 18402935]
143 Zhao WQ, Alkon DL. Role of insulin and insulin receptor in learning and memory. Mol Cell Endocrinol 2001; 177: 125-134 [PMID: 11377828]
144 Navarro I, Leibush B, Moon TW, Plisetskaya EM, Baños N, Méndez E, Planas JV, Gutiérrez J. Insulin, insulin-like growth factor-I (IGF-I) and glucagon: the evolution of their receptors. Comp Biochem Physiol B Biochem Mol Biol 1999; 122: 137-153 [PMID: 10327604]
145 Chiu SL, Cline HT. Insulin receptor signaling in the development of neuronal structure and function. Neural Dev 2010; 5: 7 [PMID: 20230616]
146 Zemva J, Schubert M. Central insulin and insulin-like growth factor-1 signaling: implications for diabetes associated dementia. Curr Diabetes Rev 2011; 7: 356-366 [PMID: 21916834]
147 Zhang J, Moats-Staats BM, Ye P, D’Ercole AJ. Expression of insulin-like growth factor system genes during the early postnatal neurogenesis in the mouse hippocampus. J Neurosci Res 2007; 85: 1618-1627 [PMID: 17455296 DOI: 10.1002/jnr.21289]
421 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Hami J et al . Effects of maternal diabetes on developing hippocampus

148 Lauszus FF. The clinical significance of IGF-I in maternal serum during pregnancy in type 1 diabetes. Curr Diabetes Rev 2007; 3: 194-197 [PMID: 18220671]
149 Brussee V, Cunningham FA, Zochodne DW. Direct insulin signaling of neurons reverses diabetic neuropathy. Diabetes 2004; 53: 1824-1830 [PMID: 15220207]
150 Russo VC, Gluckman PD, Feldman EL, Werther GA. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr Rev 2005; 26: 916-943 [PMID: 16131630]
151 Ramsay TG, Wolverton CK, Steele NC. Alteration in IGF-I mRNA content of fetal swine tissues in response to maternal diabetes. Am J Physiol 1994; 267: R1391-R1396 [PMID: 7977870]
152 Thompson CL, Pathak SD, Jeromin A, Ng LL, MacPherson CR, Mortrud MT, Cusick A, Riley ZL, Sunkin SM, Bernard A, Puchalski RB, Gage FH, Jones AR, Bajic VB, Hawrylycz MJ, Lein ES. Genomic anatomy of the hippocampus. Neuron 2008; 60:
1010-1021 [PMID: 19109908]153 Förster E, Zhao S, Frotscher M. Laminating the hippocampus. Nat
Rev Neurosci 2006; 7: 259-267 [PMID: 16543914]154 McNay EC, Fries TM, Gold PE. Decreases in rat extracellular
hippocampal glucose concentration associated with cognitive demand during a spatial task. Proc Natl Acad Sci USA 2000; 97: 2881-2885 [PMID: 10706633 DOI: 10.1073/pnas.050583697050583697]
155 Tehranipour M, Khakzad MR. Effect of maternal diabetes on hippocampus neuronal density in neonatal rats. J Biol Sci 2008; 6: 1027-1032 [DOI: 10.3923/jbs.2008.1027.1032]
156 Hami J, Sadr-Nabavi A, Sankian M, Haghir H. Sex differences and left-right asymmetries in expression of insulin and insulin-like growth factor-1 receptors in developing rat hippocampus. Brain Struct Funct 2012; 217: 293-302 [PMID: 22042446 DOI: 10.1007/s00429-011-0358-1]
P- Reviewer: Budziszewska B, Hernandez SRZ S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
422 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Hami J et al . Effects of maternal diabetes on developing hippocampus

which carries a risk of vascular impairment. DM type 2 (DM2) can be characterized by the dysfunction of haemostasis manifesting by stimulated coagulation process, disorder of platelet function and decreased fibrinolytic activity. These all are the reasons why DM2 is the most common acquired thrombophilia. Endothelial dysfunction along with platelet hyperactivity are unquestionably involved in the hyperactivation of platelets and clotting factors in DM. As a natural consequence of continuous investigation, many markers of endothelial dysfunction and diabetic thrombocytopathy have been identified and considered for implementation in clinical practice. Endothelial function can be assessed by the evaluation of endothelial markers, circulating molecules synthesised in various amounts by the endothelium. These markers precede the signs of evident microangiopathy. Platelets have an ethiopathogenic relation to the microangiopathy in DM. Their increased activity was confirmed in both types of DM. Predictors of endothelial and platelet disorder could improve the screening of individuals at increased risk, thus leading to the early diagnosis, appropriate treatment, as well as to the effective prevention of the complications of DM2. In the article we deal with the mechanisms involved in the pathogenesis of endothelial and platelet functional abnormalities, endothelial and platelet markers of DM2 considered for implementation in clinical practice and possibilities of their detection.
Key words: Diabetic thrombocytopathy; Endothelial markers; Platelet markers; Endothelial dysfunction
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Number of diabetics increases, what leads to worldwide increasing diabetes-associated vascular events. Moreover, diabetes mellitus type 2 is the most common acquired thrombophilia. Therefore, to prevent life-threatening vascular complications in subjects with diabetes, mechanisms and markers of endothelial and platelet dysfunction have been investigated. In order to contribute to better management of discussed patients and to increase knowledge about their origin, in this article we tried to summarize the pathogenesis of
Peter Kubisz, Lucia Stančiaková, Ján Staško, Peter Galajda, Marián Mokáň
Peter Kubisz, Lucia Stančiaková, Ján Staško, National Centre of Haemostasis and Thrombosis, Clinic of Haematology and Transfusiology, Comenius University in Bratislava, Jessenius Faculty of Medicine and University Hospital in Martin, 03659 Martin, Slovak RepublicPeter Galajda, Marián Mokáň, Department of Internal Medicine I, Comenius University in Bratislava, Jessenius Faculty of Medicine and University Hospital in Martin, 03659 Martin, Slovak RepublicAuthor contributions: Kubisz P, Staško J and Stančiaková L wrote the paper; Kubisz P, Staško J, Galajda P and Mokáň M designed and performed the research with obtaining the results, on the basis of which the review was partially written; all authors had read and approved the final version of the manuscript.Supported by Projects CEPV I (ITMS 26220120016), CEVYPET (ITMS 26220120053), APVV 0222-11 and Vega 1/0016/12.Conflict-of-interest: The authors declare no conflict of interest.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Peter Kubisz, MD, DSc, Professor, National Centre of Haemostasis and Thrombosis, Clinic of Haematology and Transfusiology, Comenius University in Bratislava, Jessenius Faculty of Medicine and University Hospital in Martin, Kollarova Str. N. 2, 03659 Martin, Slovak Republic. [email protected] Telephone: +421-43-4203232Fax: +421-43-4132061Received: September 1, 2014 Peer-review started: September 1, 2014 First decision: November 27, 2014Revised: January 12, 2015 Accepted: January 30, 2015Article in press: February 2, 2015Published online: April 15, 2015
AbstractDiabetes mellitus (DM) is an extremely common disorder
REVIEW
423 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Endothelial and platelet markers in diabetes mellitus type 2
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.423
World J Diabetes 2015 April 15; 6(3): 423-431ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

endothelial and platelet dysfunction and to characterize possible predictors of abnormalities of endothelium and platelets, as well as methods of their detection.
Kubisz P, Stančiaková L, Staško J, Galajda P, Mokáň M. Endothelial and platelet markers in diabetes mellitus type 2. World J Diabetes 2015; 6(3): 423-431 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/423.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.423
INTRODUCTIONDiabetes mellitus (DM) is an extremely common disorder which carries a risk of vascular impairment. In 1970s the attention has shifted towards haemostatic mechanisms with particular emphasis on the relationship between platelets and the vessel wall[1].
The term “endothelial dysfunction” is usually defined as a disorder of the endothelial capacity to maintain vascular homeostasis[2]. Endothelial dysfunction along with platelet hyperactivity are unquestionably involved in the hyperactivation of platelets and clotting factors in DM[3]. It is an early sign of vascular damage in DM type 2 (DM2) but late in DM1. Endothelial dysfunction occurs even in normoalbuminuric (NAU) patients with DM2[4]. Endothelial function can be assessed by the evaluation of endothelial “markers”, circulating molecules synthesised in various amounts by the endothelium[5]. In fact, markers precede the signs of evident microangiopathy[4].
Microalbuminuria conventionally represents albumin excretion rate 30-300 mg in a 24 h urine collection. It is associated with endothelial dysfunction and serves as a predictor of DM at levels out of the characterized reference range[4,6].
DM2 can be characterized by the dysfunction of haemostasis manifesting by stimulated coagulation process, disorder of platelet function and decreased fibrinolytic activity. These all are the reasons why DM2 is the most common acquired thrombophilia[7].
Markers of activated haemostasis, for instance prothrombin activation fragment 1 + 2 (F1 + 2) and thrombin-anti-thrombin complexes, plasma levels of fibrinogen, factor Ⅶ, factor Ⅷ, factor XI, factor XII, kallikrein, and von Willebrand factor (vWF) are elevated in DM[3]. Impairment of fibrinolytic system because of imbalance between regulators of plasminogen represents the typical sign of thrombophilia present in diabetes[7]. Increased amount of platelet aggregates in circulation, increased aggregation of platelets after addition of platelet agonists, increased platelet contractility, and the presence of elevated plasma levels of their contents, for example beta-thromboglobulin (β-TG), platelet factor 4 (PF4), and thromboxane B2 (TXB2), show platelet hyperreactivity in DM[3].
Eighty percent of individuals with DM are dying because of thrombosis[3]. The most effective tool to preserve dysfunction of the endothelium and vascular
impairment in DM is the management of hyperglycae-mia[4].
This review will characterize mechanisms responsible for the development of endothelial and platelet functional abnormalities, endothelial and platelet markers of DM2 considered for implementation in clinical practice and possibilities of their detection.
MECHANISMS OF ENDOTHELIAL DYSFUNCTION IN DM2 The pathogenesis of endothelial dysfunction in both DM1 and DM2 is multifactorial[4].
Altered cell signalization The alteration of the insulin-mediated activation of nitric oxid synthase derived from endothelium is important factor of endothelial damage in the setting of DM and insulin resistance (IR). On the other hand, the activation of insulin-signalling cascade is associated with the expression of endothelin-1 (ET-1), a vasoconstrictor and mitogenic substance, and proinflammatory molecules as intercellular adhesion molecule 1 (ICAM-1)[2].
Increased oxidative stressReactive oxygen species and circulating markers of oxidative stress are elevated in DM, IR and obesity[2].
Pro-inflammatory activation of the endotheliumSystemic inflammation present in DM can impair the function of the endothelium and lead to atherosclerosis. Individuals with diabetes or obesity have elevated circulating levels of markers of inflammation, e.g., C-reactive protein, tumor necrosis factor α, interleukin 6 (IL-6), and ICAM-1. Moreover, elevated levels of pro-inflammatory substances predict vascular complications in diabetics[2].
Activation of protein kinase CActivation of protein kinase C beta can be the cause of the association between inflammation, endothelial damage, and IR in DM[2].
Mitochondrial dysfunctionRecent works relationship between endothelial dysfunction and impaired mitochondrial biogenesis in subjects with DM[2].
ENDOTHELIAL MARKERS OF DM2vWFvWF represents a marker of the activation of the endothelium[8]. It is synthesised, stored and secreted both by megakaryocytes/platelets and by endothelial cells[9]. vWF takes part in the processes of platelet adhesion, platelet aggregation and acts as a plasma carrier for factor VIII, thus enabling its stability in the circulation. Its role in platelet adhesion is particularly
Kubisz P et al . Endothelial and platelet markers in diabetes
424 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

important under conditions of high shear stress[10] (Figure 1).
According to the results of the Framingham Off-spring Study there was strong evidence that vWF is an independent risk factor for DM (relative risk = 1.37, 95%CI: 1.07-1.75; P = 0.01)[12]. vWF level is already increased in NAU individuals, while in a few studies vWF level was increased firstly in microalbuminuric (MAU) individuals[8]. For instance, Galajda et al[13] have found significant increase of vWF in DM2 subjects without vascular impairment comparing to healthy individuals (1.33 ± 0.39 IU/mL vs 1.01 ± 0.27 IU/mL, P < 0.01)[13].
On the contrary, in the study of Kubisz et al[8] circulating vWF levels did not significantly increase in diabetic subgroups when comparing with the controls. Since then, many studies have confirmed no or no independent link between vWF and DM[14,15]. This can be explained by the fact that the association between vWF and DM relates to the concentrations of the proinflammatory cytokine IL-6. Thus, vWF as a product of the acute phase response is associated with the risk of DM complications, but the relation between vWF and DM is probably indirect[14,16].
Thrombomodulin Plasma (soluble) thrombomodulin (TM) is a marker of endothelial damage[8]. TM represents major substance of the protein C anticoagulant system[17].
It was suggested that the elevation of plasma TM concentration in patients with DM2 could be the consequence of widespread vascular damage in diabetic patients with incipient nephropathy[18]. Actually, vWF and TM levels are increased in group of subjects with DM2 and endothelial dysfunction[19].
Widespread endothelial dysfunction is present in diabetic nephropathy. Hence, plasma vWF and TM represent valuable markers of endothelial dysfunction potentially useful in early confirmation of diabetic
microvascular complications[20]. Surprisingly, our recent study found soluble TM more sensitive marker than vWF in individuals diagnosed DM2 with both, the normo- and microalbuminuria (TM P < 0.0001 in NAU, P < 0.005 in MAU)[8]. Galajda et al[13] reported on the contrary to the previous studies, when comparing the DM2 patients without vasculopathy and control group, the concentrations of TM as calcium-independent marker of endothelial injury to be similar. This is the reason to assume that only elevated concentrations of intracellular calcium (Cai)-depending endothelial and platelet indicators can predict the dysfunction of the endothelium[13].
Thrombin-activatable fibrinolysis inhibitorThrombin-activatable fibrinolysis inhibitor (TAFI) removes C-terminal lysine part from fibrin and inhibits plasminogen activation. Taking all the facts into the consideration, TAFI represents a substantial link between coagulation and fibrinolytic processes[7]. Positive correlation between TAFI and F1 + 2 in MAU group (r = 0.427, P = 0.001) as well as TAFI and fibrinogen in the NAU group confirmed activation of TAFI in the course of stimulated coagulation[6].
TAFI antigen levels and activity are significantly (P < 0.05) increased in diabetics when comparing with healthy subjects. Inverse and significant correlation of TAFI antigen and D-dimers was found in diabetic subjects supporting the function of TAFI in diabetes-induced inhibition of fibrinolytic activity[6,7,21]. Elevated levels of TAFI may also be important in endothelial injury in MAU individuals[6,8].
A significantly elevated plasma level of TAFI was present in diabetics with microalbuminuria when compared with diabetics with normoalbuminuria (P ≤ 0.001)[8]. Progression of DM2 therefore contributes to marked TAFI mediated downregulation of fibrinolytic activity. It is also suggested that fibrinolysis inhibition can be mediated by TAFI in early stages of diabetes
425 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Platelet
GPⅡb/Ⅲa GPⅡb/Ⅲa
vWFAggregation
Adhesion
Gplb bindinginduces CpⅡb/ⅢaexpressionGpIb GpIb
Endothelium Endothelium
Subendothelialcolagen receptorsexposed at injury site
Endothelium
Platelet
GPⅡb/Ⅲa GPⅡb/Ⅲa
vWF
vWF vWF
Figure 1 The role of von Willebrand factor in the platelet adhesion to the damaged vessel wall and the subsequent platelet aggregation. Adapted from: Trombose[11]. vWF: Von Willebrand factor; GP: Glycoprotein.
Kubisz P et al . Endothelial and platelet markers in diabetes

and body mass index (r = 0.43, P < 0.05) in the MAU patients and between PAI-1 and triglycerides (TAG) (r = 0.67, P = 0.01) in NAU subjects, proving that obesity, dyslipidaemia and decreased fibrinolytic activity are related to each other[6,7]. Subjects with DM2 and hyperinsulinaemia non-diabetics had significantly increased PAI-1 levels which correlated with the C-peptide (r = 0.519, P < 0.001), TAG (r = 0.685, P < 0.001), body mass index (r = 0.607, P < 0.001) and with levels vWF and soluble TM[25].
Moreover, increased PAI-1 levels are an independent risk factor for DM2. It was proposed that a a decrease of PAI-1 level may be linked with a decrease in trans-formation to DM2. On the other hand, the finding of elevation of PAI-1 preceding DM2 thus being, in the preexistence of IR, independent of glycaemia gives unquestionable evidence that abnormality of fibrinolytic system characterized with increased PAI-1 and tPA antigen occurs nearly at the beginning of the development of metabolic disorders and is considered a risk factor for future DM and metabolic complications[7].
Tissue factor pathway inhibitor Tissue factor pathway inhibitor (TFPI) acts as an inhibitor of tissue factor (TF)-initiated coagulation. TF binds to the activated factor X, subsequently TFPI-Xa complex binds to the TF/factor Ⅶa (FⅦa) complex and regulates its action[26].
Enhanced TFPI activity was confirmed in individuals with DM and mainly in those with microalbuminuria. Increases in TFPI reflect endothelial dysfunction or impaired binding of TFPI to endothelial cells by glucosa-minoglycans because TFPI is predominantly synthesized in endothelial cells[26]. It may be the first consequence of increased glycosylation, and thus the deterioration of the antithrombotic potential of endothelial cells. Mentioned increase in TFPI activity does not compensate the procoagulant state associated with increased thrombinogenesis (excess of F1 + 2), and inversely correlates with the internal thrombin potential[27,28]. Plasma levels of TF, TFPI and FⅦa are significantly increased in DM2[26].
Total TFPI (t-TFPI) has a poor anticoagulant effect. On the contrary, free form of TFPI (f-TFPI) has an increased anticoagulant capacity. Moreover, the study of Morange et al[29] showed that f-TFPI has a strong correlation with endothelial markers and t-TFPI has increased relation to parameters indicating cardiovascular risk. Normal values of f-TFPI varying according to gender, as well as informative normal values of t-TFPI can be seen in Table 1.
tPA tPA is a serine protease which converts plasminogen to plasmin, the form active in the process of fibrinolysis, thus being the most valuable initiator of fibrinolytic process. tPA is synthesised particularly in the endothelial cells, smooth muscle cells, monocytes and megakar-yocytes[6,7]. Quick tPA release to the circulation occurs
defined by microalbuminuria in DM2 subjects without macrovascular complications. It seems that discussed process is independent from the level of plasminogen activator inhibitor-1 (PAI-1)[6,8].
Elevated circulating levels of TAFI were present not only in individuals diagnosed DM2, but also in patients with obesity, IR and arterial hypertension[8]. On the contrary, increased total cholesterol level probably downregulates TAFI[6].
PAI-1 PAI-1 represents one of the most significant and quick natural inhibitors of tissue plasminogen activator (tPA) and urokinase-type plasminogen activator[7].
PAI-1 is produced in vascular and metabolic tissues[7]. Therefore, up-regulation of PAI-1 occurs predominantly in obese patients and presence of endothelial dysfunction[6].
PAI-1 is released to plasma predominantly from the endothelial cells[7]. This is the cause of the elevation of PAI-1 concentrations in individuals with DM2 and hypertension with endothelial dysfunction[6,22]. The inflammatory cytokines, as well as metabolic components are activators of PAI-1 production in the endothelial tissue[7,23]. Insulin has inhibiting influence on the PAI-1 synthesis in endothelial cells. Significant decrease of PAI-1 level in DM2 individuals with endothelial impair-ment on long-term insulin therapy was confirmed[7,24]. However, the mechanism of insulins action depend upon the metabolism in hepatic cells and endothelium[7].
PAI-1 overproduction belongs to the most typical signs of thrombophilia in DM2 leading to the dysfunction of fibrinolytic system[7]. Plasma levels of PAI-1 are significantly increased in both cohorts (MAU and NAU) compared with the controls (P < 0.0001 in both subgroups)[8].
When comparing with other parameters, PAI-1 is the most complex one associated with IR. Level of PAI-1 highly correlates with parameters of the IR syndrome in healthy individuals, patients with IR, DM2, or subjects with coronary artery disease. Positive correlation was found between PAI-1 concentrations
426 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 1 The most important endothelial markers of diabetes mellitus type 2 with the method used for their detection
Endothelial markers Method of detection Normal values
PAI-1 antigen ELISA 5-40 ng/mLTAFI antigen ELISA 5.8-10.0 μg/mL
(100-172 nmol/L)t-TFPI antigen EIA 70 (40-110) ng/mLf-TFPI (men) EIA 16 ± 4 ng/mLf-TFPI (women) EIA 16 ± 3 ng/mLTM antigen EIA 10-50 ng/mLtPA antigen ELISA 1-20 ng/mLvWF antigen EIA Less than 1.4 IU/mL (140%)
EIA: Enzyme immunoassay; ELISA: Enzyme-linked immunosorbent assay; PAI-1: Plasminogen activator inhibitor 1; TAFI: Thrombin-activatable fibrinolysis inhibitor; f-TFPI: Free tissue factor pathway inhibitor; t-TFPI: Total tissue factor pathway inhibitor; TM: Thrombomodulin; tPA: Tissue plasminogen activator; vWF: Von Willebrand factor.
Kubisz P et al . Endothelial and platelet markers in diabetes

following the Cai - dependent stimulus. Minority of tPA is present in plasma in the form of complex binding PAI-1[7].
tPA is present mainly on the endothelial surface and may be released to circulation following its injury[7]. tPA levels are elevated in subjects with DM and metabolic syndrome. Increased tPA concentrations were proposed as a risk factor of DM in healthy subjects, because total tPA antigen levels were elevated in DM, but the level of free tPA and the tPA activity not. Therefore, the elevated total tPA is a facade masking the dysfunctional fibrinolysis in DM2[6]. Moreover, an elevated tPA antigen concentration is a substantial feature of the IR syndrome and also relates to the inflammation. tPA antigen levels have strong correlations with IR, and can serve as helpful markers of diabetes[14]. The increased tPA levels correlate with vWF and soluble TM in individuals with various locations of atherosclerotic lesions. This supports the position of tPA as an indicator of endothelial injury[7].
The tPA levels were described to be increased in DM2 in an early phase of disease while tPA levels in DM1 are increased later in presence of vascular complications. Dependence between tPA levels and PAI-1 and IR is higher in the phase of complications than dependence between tPA and presence of complications in DM2. There is a correlation between tPA and PAI-1 found in patients with DM2 and the principle cause of the tPA increase is formation of the tPA/PAI-1 complexes[7].
As it was mentioned earlier, insulin can inhibit the endothelial production of PAI-1 with the following decrease of tPA/PAI-1 complexes levels but it does not influence the tPA release from endothelium and tPA plasma levels. This can be explanation of the absent correlation between tPA and PAI-1 found in DM2 patients treated with insulin[7,25].
E-selectin E-selectin (CD62E), expressed by endothelium, is rapidly enhanced by proinflammatory molecules[30].
At first the adhesion molecules were considered to be endothelial markers because of their general elevation in various vasculopathies[25]. It was confirmed that individuals with increased E-selectin had a significantly higher probability of the development of DM2[4]. Moreover, in the study of Thorand et al[15] soluble E-selectin and ICAM-1 significantly preceded DM2. In particular, soluble E-selectin remained an independent marker of diabetes after exclusion of inflammation, levels of insulin and hemoglobin A1c in both men and women[15,30].
ICAM-1 and vascular cell adhesion molecule 1 ICAM-1 and vascular cell adhesion molecule 1 (VCAM-1) are synthesised in the endothelium and leukocytes. Increased concentrations of these cell adhesion molecules (CAMs) are early markers of endothelial dysfunction, cardiovascular disease and DM2[30].
Increased concentrations of E-selectin, ICAM-1,
and VCAM-1 raise the relative risk of DM by 1.5- to 7.5-fold[30]. Taking everything into account, elevated concentrations of CAMs representing an increased probability of the development of diabetes highlight the link between dysfunction of the endothelium and IR[15].
Vascular endothelial growth factor Vascular endothelial growth factor (VEGF) is a growth factor derived from endothelial cells, stimulating their proliferation, differentiation and survival, mediating endothelium-dependent vasodilatation, inducing increased permeability in the microcirculation and parti-cipating in other supporting functions. VEGF enhances the glomerular permeability to macromolecules and leads to profound proteinuria including albuminuria. Described action has probably a role in the development of endothelial dysfunction in diabetes[8]. The beneficial effects of VEGF-A (principal member of VEGF family) inhibition in the early phases of diabetic glomerulopathy was also confirmed[31].
Highly increased plasma VEGF levels were measured in individuals with retinopathy and nephropathy when compared with the control group. Widespread endothelial dysfunction occurs at the beginning of the development of diabetic nephropathy and precedes biochemical finding of the disorder of renal functions since the significantly increased VEGF level was present already in NAU diabetics without manifestations of microvascular impairment. Because VEGF levels were significantly elevated in the NAU cohort and had a tendency to be significant in the MAU patients, VEGF can be concluded as an indicator of diabetic endotheliopathy. VEGF ought to be evaluated only along with further markers representing the dysfunction of the endothelium, particularly soluble TM[8]. It is assumed that VEGF may be a more sensitive indicator of renal changes than microalbuminuria[8,32]. On the other hand, since there were no marked differences in VEGF concentrations in individuals with DM with physiological renal functions and at the beginning of the development of diabetic nephropathy, VEGF was not confirmed as a reliable marker of the progression in DM2 patients[8].
ET-1ET-1 is considered to be the most potent vasoconstrictor form of endothelin and is produced by the endothelial cells. During prolonged periods of stress caused by hyperglycaemia and IR in the course of DM2, there is a shift in the balance with more vasoconstrictors being produced compared to vasodilating agents. This leads to endothelial dysfunction and affects the permeability of the glomerular filtration barrier leading to microalbuminuria[33].
Even experimental studies suggested that ET-1 drives development of glomerulosclerosis and podocyte loss[34]. ET-1 is really significantly associated with early development of the endothelial dysfunction in the
427 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Kubisz P et al . Endothelial and platelet markers in diabetes

glomerulus[33].
MECHANISMS OF DIABETIC THROMBOCYTOPATHY Primary dysfunction of megakaryocyte-platelet system One of the manifestations of DM is the production of subpopulation of large and hyperreactive platelets in the bone marrow. Thrombocytopoesis is increased[35,36]. IR in plateletsIn obesity and DM2 increased aggregability of platelets, as well as decreased sensitivity to antiplatelet effect of insulin was confirmed[36].
Influence of endothelial dysfunctionSubstances produced by the endothelium, represented by nitric oxide, prostacyclin and adenosine normally inhibit platelets. In diabetic patients due to the hyperglycaemia and oxidative stress the capacity of endothelium to produce prostacyclin and nitric oxide is decreased. In the cases of microangiopathy consumption of platelets in the sites of damaged endothelium causes paradoxically the decrease in the subpopulation of large activated platelets[36].
Changes of the activity of membrane pumps and increase of Cai in platelets[36] Platelet markers of DM2: Platelets have an ethiopathogenic relation to the microangiopathy in DM. Their increased activity was confirmed in both types of
DM[36,37]. The structure of the platelet with components involved in its physiological function and markers of platelet activity are illustrated in Figure 2.
Mean platelet volumeMean platelet volume (MPV) is the parameter informing about the platelet size and activation Bigger platelets indicate more activity[38]. Patients with DM have significantly higher MPV due to the elevated alpha-granule content in cytoplasm as the sign of dysfunction of megakaryocyte-platelet system[36,38]. Thus, platelets can be a useful tool to evaluate complications in DM[38]. Further studies concluded that MPV is strongly and independently associated with the presence and severity of diabetes and higher MPV is the manifestation of early diabetic thrombocytopathy in both types of DM without vascular complications[39,40].
Soluble P-selectin P-selectin (CD62P) is a highly glycosylated membrane glycoprotein constitutively expressed in endothelial cells and megakaryocytes and present in the membrane of Weibel-Palade bodies of endothelium and in α granules of platelets[36,41]. It is a marker of platelets’ activation. Its increased levels in patients with arterial hypertension, lower extremity peripheral artery disease and coronary artery disease correlate with levels of β-TG, and they do not have a relation to vWF and TM[42-44]. Increased levels of P-selectin occured already in early stages of DM1 and DM2 without vascular complications. Elevated levels of P-selectin were also found in subjects diagnosed IR and impaired glucose tolerance[45-47].
Elevated synthesis of P-selectin and ICAM-1 in diabetic human retina and choroid was confirmed. This is the reason why circulating levels of soluble adhesion molecules have been proposed as biomarkers playing a significant role in the pathogenesis of macrovascular and microvascular dysfunction and neuropathy of DM[48]. Galajda et al[45] found in patients with DM2 significantly elevated P-selectin that paradoxically did not correlate with levels of PF4, but they correlated with vWF. This can propose also its function in the endothelial dysfunction in patients with DM2[45].
P-selectin was significantly increased in patients with DM suffering myocardial infarction than in control group or those without myocardial infarction. It indi-cates contribution of elevated levels of P-selectin to the atherosclerosis leading to coronary heart disease in diabetics[49]. Thus, detection of P-selectin level could be useful marker of possible cardiovascular event in individuals with DM[50].
PF4PF4 is a basic protein found exclusively in α granules of platelets. After release it is able to modulate haemostasis. PF4 has both procoagulant and anticoagulant properties. It has also angiostatic effect - it inhibits proliferation and migration of endothelial cells[36].
428 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Alpha granule
Dense granule
Canalicularsystem
vWFfibrinogenTSPPF4
PDGFβ-TGfactor V
Fibrinogen
GP Ⅱ
b/Ⅲ
a
GPⅡb/ⅢaADP ATPcalcium ++serotonin
Exposedcolagen
GPIb/Ⅸ/Ⅴ
vWF
Area of denuded endotheliumBasement menbrane Vascular endothelial cell
Figure 2 The structure of the platelet and its most important functions, including the secretion of stored products, as well as the binding of specific surface glycoproteins to the endothelium (bottom), and other platelet (left side). Adapted from: Trombose[11]. ADP: Adenosine diphosphate; ATP: Adenosine triphosphate; β-TG: Beta-thromboglobulin; GP: Glycoprotein; PDGF: Platelet-derived growth factor; PF4: Platelet factor 4; TSP: Thrombospondin; vWF: Von Willebrand factor.
Kubisz P et al . Endothelial and platelet markers in diabetes

In patients with both types of DM the increased levels of PF4 and β-TG as the manifestation of degranulation of activated platelets without influence on their lifespan were confirmed. In DM2 these levels are increased already in the early stage of the disease without the presence of vascular complications[36].
In the study of Galajda et al[13] the correlation between vWF concentrations and calcium dependent substances as C-peptide (r = 0.680, P < 0.001) and PF4 (r = 0.613, P < 0.01) and no correlation with calcium-independent markers of endothelial damage, as TM (r = 0.287, P = 0.196) or TFPI (r = 0.296, P = 0.181) in diabetics was confirmed[13]. In advanced cases of DM associated with retinopathy and nephropathy increased levels of PF4, β-TG, P-selectin, Cai in platelets, as well as their increased aggregability were confirmed[36]. Moreover, patients with diabetes type 2 treated with sulphonylurea preparates and individuals treated 2-3 mo with insulin did not differ by endothelial and platelet parameters (vWF, TM and PF4)[7].
β-TG β-TG is a protein with 50% structural homology with PF4 acting as a leukocyte chemoattractant. Measurement of β-TG is considered to be a golden standard in the detection of activation of platelets in patients with normal renal function. Its levels are increased in renal insufficiency and correlate with levels of PF4[39].
Glycoprotein V (CD42d)Glycoprotein V (GPV) is exclusively expressed in platelets and megakaryocytes, where it is non-covalently bound to the complex GPIb/GPIX as the part of the receptor for vWF and thrombin[51].
According to the fact that platelets are the only source of GPV it can be considered as a marker of specific activation of platelets by thrombin[51].
TXB2Metabolism of arachidonic acid with incorporation of its metabolites to membrane phospholipids of platelets in DM is upregulated. Production of TXA2 in platelets is increased. Plasma levels of its stable catabolite TXB2 produced in liver, kidneys, platelets and lungs are also
increased and correlate with raised levels of β-TG and PF4[52].
Increased synthesis of TXA2 indicates that “resting” platelets of diabetics in general tend to be activated. TXA2 is a vasoconstrictor and prothrombotic factor with the risk of atherothrombogenesis present in DM[53].
Moreover, synthesis of TXB2 has inverse correlation with the minimal level of arachidonic acid inevitable to 50% platelet aggregation. As the consequence, platelet aggregates in the circulation of patients with DM2 than in the healthy subjects. Platelets of diabetics therefore exhibit the signs of hyperactivity. This fact may also be very important for the arise of vascular disease in DM[52].
Thrombospondin-1This molecule represents a multifunctional glycoprotein synthesised in a wide range of cell types, to be more exact in platelets (increased expression), vascular smooth muscle cells and various kinds of renal cells[53]. Unlike PF4 and β-TG, after release of thrombospondin (TSP) from activated platelets it binds to platelet surface, so levels of TSP-1, PF4 and β-TG usually do not correlate with each other. Last but not at least, its levels are influenced by hepatopathy. These are the reasons why analysis of TSP is not considered to be the golden standard in detection of platelet activity. Therefore, TSP is evaluated as the sign of activation of the whole vascular system[36].
TSP-1 with antiangiogenic and proatherogenic properties is involved in diabetic vasculopathy. The endothelial dysfunction in DM was confirmed in many studies, and TSP-1 surely may contribute to this impaired function because it shows antiproliferative and apoptotic effect on the endothelium. It was confirmed that TSP-1 level has a relation with renal damage and vascular impairment[53].
DETECTION OF MARKERS OF DM2Informative normal values of the most important endothelial and platelet markers with the method used for their detection are documented in Tables 1[36,54-56] and 2[36].
Size of the platelet up to 7 μm and its volume more than 10 fl contribute to increased MPV[36].
Levels of soluble P-selectin are measured using enzyme immunoassay method and their normal values vary between laboratories and are dependent on the commercial kit used for the detection[36].
Values of β-TG 50 ng/mL and more are considered to be pathological[36].
Plasma levels of VEGF, F1 + 2 and TXB2 can also be detected using enzyme-linked immunosorbent assay[6,8,57].
CONCLUSION Number of patients with DM2 is increasing incredibly and is expected to rise even in the future. Researchers
429 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 2 The most important platelet markers of diabetes mellitus type 2 with the method used for their detection
Platelet markers Method of detection Normal values
βTG EIA/RIA Less than 40 ng/mLGPV EIA 80 (40-160) ng/mLMPV Electric impedance or
laser diffraction6.5 (4.5-8.5) fl
PF4 EIA/RIA Less than 10 ng/mLTSP-1 EIA 40 (10-90) ng/mL,
respectively 20-300 ng/mL
βTG: Beta-thromboglobulin; EIA: Enzyme immunoassay; GPV: Glycoprotein V; MPV: Mean platelet volume; PF4: Platelet factor 4; RIA: Radioimmunoassay; TSP-1: Thrombospondin 1.
Kubisz P et al . Endothelial and platelet markers in diabetes

all over the world have made strong efforts to increase knowledge and improve understanding of mechanisms of abnormalities of endothelium and platelets unquestionably involved in the pathogenesis of this challenging health problem. As a natural consequence of continuous investigation, many markers of diabetic thrombocytopathy and endothelial dysfunction have been identified and are considered for implementation in clinical practice. These substances may improve the screening of high-risk individuals and lead to the early diagnosis, appropriate treatment, as well as to the effective prevention of the complications of DM2. However, these conclusions have to be confirmed by further research.
REFERENCES1 Preston FE. Disorders of haemostasis in diabetes mellitus. Ric
Clin Lab 1982; 12: 425-438 [PMID: 7134747]2 Tabit CE, Chung WB, Hamburg NM, Vita JA. Endothelial
dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord 2010; 11: 61-74 [PMID: 20186491 DOI: 10.1007/s11154-010-9134-4]
3 Carr ME. Diabetes mellitus: a hypercoagulable state. J Diabetes Complications 2001; 15: 44-54 [PMID: 11259926]
4 Hadi HA, Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag 2007; 3: 853-876 [PMID: 18200806]
5 Storey AM, Perry CJ, Petrie JR. Endothelial dysfunction in type 2 diabetes. Br J Diabetes Vasc Dis 2001; 1: 22-27
6 Chudý P, Kotuličová D, Staško J, Kubisz P. The relationship among TAFI, t-PA, PAI-1 and F1 + 2 in type 2 diabetic patients with normoalbuminuria and microalbuminuria. Blood Coagul Fibrinolysis 2011; 22: 493-498 [PMID: 21519232 DOI: 10.1097/MBC.0b013e328346f8ca]
7 Staško J, Chudý P, Kotuličová D, Galajda P, Mokáň M, Kubisz P. Type 2 Diabetes and Fibrinolysis. In: Zimering M. Recent Advances in the Pathogenesis, Prevention and Management of Type 2 Diabetes and its Complications. InTech Rijeka: InTech, 2011: 67-90
8 Kubisz P, Chudý P, Stasko J, Galajda P, Hollý P, Vysehradský R, Mokán M. Circulating vascular endothelial growth factor in the normo- and/or microalbuminuric patients with type 2 diabetes mellitus. Acta Diabetol 2010; 47: 119-124 [PMID: 19436948 DOI: 10.1007/s00592-009-0127-2]
9 Vischer UM, Emeis JJ, Bilo HJ, Stehouwer CD, Thomsen C, Rasmussen O, Hermansen K, Wollheim CB, Ingerslev J. von Willebrand factor (vWf) as a plasma marker of endothelial activation in diabetes: improved reliability with parallel determination of the vWf propeptide (vWf: AgII). Thromb Haemost 1998; 80: 1002-1007 [PMID: 9869174]
10 Vischer UM. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J Thromb Haemost 2006; 4: 1186-1193 [PMID: 16706957]
11 Trombose. [Cited August 5, 2014]. Available from: URL: http://www.prd-online.com/booklets/boekje-trombose/
12 Meigs JB, O’donnell CJ, Tofler GH, Benjamin EJ, Fox CS, Lipinska I, Nathan DM, Sullivan LM, D’Agostino RB, Wilson PW. Hemostatic markers of endothelial dysfunction and risk of incident type 2 diabetes: the Framingham Offspring Study. Diabetes 2006; 55: 530-537 [PMID: 16443791]
13 Galajda P, Martinka E, Mokán M, Kubisz P. Endothelial markers in diabetes mellitus. Thromb Res 1997; 85: 63-65 [PMID: 8983126]
14 Wannamethee SG, Sattar N, Rumley A, Whincup PH, Lennon L, Lowe GD. Tissue plasminogen activator, von Willebrand factor, and risk of type 2 diabetes in older men. Diabetes Care 2008; 31: 995-1000 [PMID: 18235054 DOI: 10.2337/dc07-1569]
15 Thorand B, Baumert J, Chambless L, Meisinger C, Kolb H,
Döring A, Löwel H, Koenig W. Elevated markers of endothelial dysfunction predict type 2 diabetes mellitus in middle-aged men and women from the general population. Arterioscler Thromb Vasc Biol 2006; 26: 398-405 [PMID: 16322530]
16 Tousoulis D, Papageorgiou N, Androulakis E, Siasos G, Latsios G, Tentolouris K, Stefanadis C. Diabetes mellitus-associated vascular impairment: novel circulating biomarkers and therapeutic approaches. J Am Coll Cardiol 2013; 62: 667-676 [PMID: 23948511 DOI: 10.1016/j.jacc.2013.03.089]
17 Khan S, Dickerman JD. Hereditary thrombophilia. Thromb J 2006; 4: 15 [PMID: 16968541 DOI: 10.1186/1477-9560-4-15]
18 Mormile A, Veglio M, Gruden G, Girotto M, Rossetto P, D’Este P, Cavallo-Perin P. Physiological inhibitors of blood coagulation and prothrombin fragment F 1 + 2 in type 2 diabetic patients with normoalbuminuria and incipient nephropathy. Acta Diabetol 1996; 33: 241-245 [PMID: 8904933]
19 Galajda P, Baláž D, Martinka E, Mokáň M, Kubisz P. Insulin treatment inhibits PAI-1 production in NIDDM patients with endothelial dysfunction. Clin Appl Thromb Hemost 1998; 4: 250-252
20 Samy N, Afifya M, Maksouda NAE, Sayeda M, Imamb A. Circulating markers of endothelial dysfunction in type 2 diabetic patients with microalbuminuria. Asian Biomed 2012; 6: 175-183 [DOI: 10.5372/10465]
21 Hori Y, Gabazza EC, Yano Y, Katsuki A, Suzuki K, Adachi Y, Sumida Y. Insulin resistance is associated with increased circulating level of thrombin-activatable fibrinolysis inhibitor in type 2 diabetic patients. J Clin Endocrinol Metab 2002; 87: 660-665 [PMID: 11836301]
22 Galajda P, Kubisz P, Mokán M. [A multicomparmental and multifactorial model of production of plasminogen activator inhibitor (PAI-1). I. Experimental studies]. Vnitr Lek 1998; 44: 718-721 [PMID: 10422516]
23 Galajda P, Sutarík L, Vladár L, Stasko J, Kubisz P, Mokán M. [Plasminogen activator inhibitor (PAI-1) and diabetes mellitus. I. Regulation of PAI-1 levels]. Vnitr Lek 1997; 43: 804-808 [PMID: 9601894]
24 Galajda P, Martinka M, Staško J, Mokáň M, Kubisz P. Plas-minogen Activator Inhibitor Type-1 (PAI-1) Levels Are Decreased in NIDDM Patients treated with Insulin. Clin Appl Thromb Hemost 1998; 4: 138-139 [DOI: 10.1177/107602969800400212]
25 Galajda P, Martinka E, Baláz D, Stasko J, Kerný J, Planková E, Mokán M, Kubisz P. [Plasminogen activator inhibitor (PAI-1) and markers of endothelial dysfunction in patients with diabetes mellitus]. Vnitr Lek 1997; 43: 744-747 [PMID: 9650506]
26 El-Hagracy RS, Kamal GM, Sabry IM, Saad AA, Abou El Ezz NF, Nasr HA. Tissue Factor, Tissue Factor Pathway Inhibitor and Factor VII Activity in Cardiovascular Complicated Type 2 Diabetes Mellitus. Oman Med J 2010; 25: 173-178 [PMID: 22043333 DOI: 10.5001/omj.2010.52]
27 Galajda P, Martinka E, Kubisz P, Ivanková J, Ochodnický M, Polko J, Mokán M. [Hemostasis in patients with diabetes mellitus. II. Tissue factor and the extrinsic blood coagulation inhibitor]. Vnitr Lek 1996; 42: 776-778 [PMID: 9012122]
28 Leurs PB, van Oerle R, Wolffenbuttel BH, Hamulyak K. Increased tissue factor pathway inhibitor (TFPI) and coagulation in patients with insulin-dependent diabetes mellitus. Thromb Haemost 1997; 77: 472-476 [PMID: 9065996]
29 Morange PE, Renucci JF, Charles MA, Aillaud MF, Giraud F, Grimaux M, Juhan-Vague I. Plasma levels of free and total TFPI, relationship with cardiovascular risk factors and endothelial cell markers. Thromb Haemost 2001; 85: 999-1003 [PMID: 11434709]
30 Meigs JB, Hu FB, Rifai N, Manson JE. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA 2004; 291: 1978-1986 [PMID: 15113816]
31 Karalliedde J, Gnudi L. Endothelial factors and diabetic nephropathy. Diabetes Care 2011; 34 Suppl 2: S291-S296 [PMID: 21525471 DOI: 10.2337/dc11-s241]
32 Kubisz P, Chudý P, Staško J, Hollý P, Galajda P. Controversies about VEGF in type 2 diabetics with normo- and microalbuminuria. Diabetes & Metabolic Syndrome: Clinical Research & Reviews
430 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Kubisz P et al . Endothelial and platelet markers in diabetes

2009; 3: 176–177 [DOI:10.1016/j.dsx.2009.04.010]33 Use of Endothelin-1 (ET-1) and von Willenbrand Factor (vWF) as
biological markers in patients with normo and microalbuminuria. [Cited August 1, 2014]. Available from: URL: http: //www.scribd.com/doc/221523488/Use-of-Endothelin-1-ET-1-and-von-Willenbrand-Factor-vWF-as-biological-markers-in-patients-with-normo-and-micro-albuminuria
34 Lenoir O, Milon M, Virsolvy A, Hénique C, Schmitt A, Massé JM, Kotelevtsev Y, Yanagisawa M, Webb DJ, Richard S, Tharaux PL. Direct action of endothelin-1 on podocytes promotes diabetic glomerulosclerosis. J Am Soc Nephrol 2014; 25: 1050-1062 [PMID: 24722437 DOI: 10.1681/ASN.2013020195]
35 Brown AS, Hong Y, de Belder A, Beacon H, Beeso J, Sherwood R, Edmonds M, Martin JF, Erusalimsky JD. Megakaryocyte ploidy and platelet changes in human diabetes and atherosclerosis. Arterioscler Thromb Vasc Biol 1997; 17: 802-807 [PMID: 9108797]
36 Galajda P, Baláž D, Vladár Ľ, Ivanková J, Mokáň M. Dysfun-ction of platelets in patients with diabetes mellitus (diabetic thrombocytopathy). Slov Lek 1998; 8: 18-21
37 Patrono C, Davi G. Antiplatelet agents in the prevention of diabetic vascular complication. Diabet Metabol Rew 1993; 9: 177-188
38 Kodiatte TA, Manikyam UK, Rao SB, Jagadish TM, Reddy M, Lingaiah HKM, Lakshmaiah V. Mean Platelet Volume in Type 2 Diabetes Mellitus. J Lab Physicians 2012; 4: 5-9 [DOI: 10.4103/0974-2727.98662]
39 Shah B, Sha D, Xie D, Mohler ER, Berger JS. The relationship between diabetes, metabolic syndrome, and platelet activity as measured by mean platelet volume: the National Health And Nutrition Examination Survey, 1999-2004. Diabetes Care 2012; 35: 1074-1078 [PMID: 22410814 DOI: 10.2337/dc11-1724]
40 Galajda P , Baláž D, Országh A, Šutarík Ľ, Mokáň M. Hyperfibirnogenemia in patients with diabetes mellitus. Slov Lek 1998; 8: 22-25
41 Kansas GS. Selectins and their ligands: current concepts and controversies. Blood 1996; 88: 3259-3287 [PMID: 8896391]
42 Blann AD, Dobrotova M, Kubisz P, McCollum CN. von Willebrand factor, soluble P-selectin, tissue plasminogen activator and plasminogen activator inhibitor in atherosclerosis. Thromb Haemost 1995; 74: 626-630 [PMID: 8584997]
43 Blann AD, Lip GY, Beevers DG, McCollum CN. Soluble P-selectin in atherosclerosis: a comparison with endothelial cell and platelet markers. Thromb Haemost 1997; 77: 1077-1080 [PMID: 9241735]
44 Lip GY, Blann AD, Zarifis J, Beevers M, Lip PL, Beevers DG. Soluble adhesion molecule P-selectin and endothelial dysfunction in essential hypertension: implications for atherogenesis? A
preliminary report. J Hypertens 1995; 13: 1674-1678 [PMID: 8903631]
45 Galajda P, Baláž D, Šutarík Ľ, Mokáň M, Kubisz P. P-selectin in patients with diabetes mellitus. Hematol Transfuziol 1998; 8: 21-24
46 Jilma B, Fasching P, Ruthner C, Rumplmayr A, Ruzicka S, Kapiotis S, Wagner OF, Eichler HG. Elevated circulating P-selectin in insulin dependent diabetes mellitus. Thromb Haemost 1996; 76: 328-332 [PMID: 8883265]
47 Kopp HP, Hopmeier P, Schernthaner G. Concentrations of circulating P-selectin are increased in patients with newly diagnosed insulin-dependent diabetes mellitus. Exp Clin Endocrinol Diabetes 1998; 106: 41-44 [PMID: 9516058]
48 Pieper GM. Hyperglycemia and diabetes-induced vascular dysfunction: role of oxidative stress. In: Keaney JF, editor. Oxidative Stress and Vascular Disease. Massachusetts: Springer Science and Business Media, 1999: 305-322
49 El-Mesallamy H, Hamdy N, Suwailem S, Mostafa S. Oxidative stress and platelet activation: markers of myocardial infarction in type 2 diabetes mellitus. Angiology 2010; 61: 14-18 [PMID: 19759031 DOI: 10.1177/0003319709340891]
50 Aref S, Sakrana M, Hafez AA, Hamdy M. Soluble P-selectin levels in diabetes mellitus patients with coronary artery disease. Hematology 2005; 10: 183-187 [PMID: 16019467]
51 Blann AD, Lanza F, Galajda P, Gurney D, Moog S, Cazenave JP, Lip GY. Increased platelet glycoprotein V levels in patients with coronary and peripheral atherosclerosis--the influence of aspirin and cigarette smoking. Thromb Haemost 2001; 86: 777-783 [PMID: 11583307]
52 Kubisz P, Arabi A, Holan J, Cronberg S. Investigations on platelet function in diabetes mellitus. Haemostasis 1984; 14: 347-353 [PMID: 6238882]
53 Abdelwhab S, Fooda O, Abdelmaksoud S. Thrombospondin-1 in Patients with Diabetic Nephropathy. Kidney 2010; 19: 229-235
54 Plasminogen Activator Inhibitor Type 1 [PAI-1] Assays. [Cited August 9, 2014]. Available from: URL: http: //practical-haemostasis.com/Fibrinolysis/pai_1.html
55 Thrombin Activatable Fibrinolysis Inhibitor [TAFI]. [Cited August 9, 2014]. Available from: URL: http: //practical-haemostasis.com/Fibrinolysis/tafi.html
56 Tissue-type Plasminogen Activity [T-PA] Assays. [Cited August 10, 2014]. Available from: URL: http: //practical-haemostasis.com/Fibrinolysis/t_pa.html
57 Véricel E, Januel C, Carreras M, Moulin P, Lagarde M. Diabetic patients without vascular complications display enhanced basal platelet activation and decreased antioxidant status. Diabetes 2004; 53: 1046-1051 [PMID: 15047620 DOI: 10.2337/diabetes.53.4.1046]
P- Reviewer: Pistrosch F, Rezzani R, Taguchi I S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
431 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Kubisz P et al . Endothelial and platelet markers in diabetes

neuropathy, improving glycemic control as a prophylactic therapy and using medications to alleviate pain. First line drugs for pain relief include anticonvulsants, such as pregabalin and gabapentin and antidepressants, especially those that act to inhibit the reuptake of serotonin and noradrenaline. In addition, there is experimental and clinical evidence that opioids can be helpful in pain control, mainly if associated with first line drugs. Other agents, including for topical application, such as capsaicin cream and lidocaine patches, have also been proposed to be useful as adjuvants in the control of diabetic neuropathic pain, but the clinical evidence is insufficient to support their use. In conclusion, a better understanding of the mechanisms underlying diabetic neuropathic pain will contribute to the search of new therapies, but also to the improvement of the guidelines to optimize pain control with the drugs currently available. Key words: Diabetes; Neuropathic pain; Hyperglycemia; Anticonvulsants; Antidepressants
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Diabetic neuropathic pain is a common complication of diabetes and the most common form of neuropathic pain. In this review, we will discuss the various factors that may contribute to the pathogenesis of diabetic neuropathic pain, including metabolic, vascular, autoimmune and oxidative stress-related mechanisms. In addition, we will review the possibilities of pain treatment, taken into consideration the first line drugs clinically used, the antidepressants and anticonvulsants, but also other options such as opioids, tapentadol and drugs for topical use, such as lidocaine and capsaicin cream.
Schreiber AK, Nones CFM, Reis RC, Chichorro JG, Cunha JM. Diabetic neuropathic pain: Physiopathology and treatment. World J Diabetes 2015; 6(3): 432-444 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/432.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.432
Anne K Schreiber, Carina FM Nones, Renata C Reis, Juliana G Chichorro, Joice M Cunha
Anne K Schreiber, Carina FM Nones, Renata C Reis, Juliana G Chichorro, Joice M Cunha, Department of Pharmacology, Biological Sciences Sector, Federal University of Parana, Curitiba 81540-970, BrazilAuthor contributions: All authors contributed equally to this work.Conflict-of-interest: None.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Dr. Joice M Cunha, Professor, Department of Pharmacology, Biological Sciences Sector, Federal University of Parana, Rua XV de Novembro, 1299 - Centro, Curitiba 81540-970, Brazil. [email protected]: +55-41-33611720Fax: +55-41-32662042Received: August 29, 2014Peer-review started: August 30, 2014First decision: November 14, 2014Revised: November 26, 2014Accepted: February 4, 2015Article in press: February 9, 2015Published online: April 15, 2015
AbstractDiabetic neuropathy is a common complication of both type 1 and type 2 diabetes, which affects over 90% of the diabetic patients. Although pain is one of the main symptoms of diabetic neuropathy, its pathophysiolo-gical mechanisms are not yet fully known. It is widely accepted that the toxic effects of hyperglycemia play an important role in the development of this complication, but several other hypotheses have been postulated. The management of diabetic neuropathic pain consists basically in excluding other causes of painful peripheral
REVIEW
432 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Diabetic neuropathic pain: Physiopathology and treatment
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.432
World J Diabetes 2015 April 15; 6(3): 432-444ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

INTRODUCTIONAccording to the International Diabetes Federation, 382 million people worldwide are currently affected by diabetes[1], one of the leading causes of neuropathy[2]. The distal symmetrical polyneuropathy (DSPN) is the commonest clinical form of diabetic neuropathy, affecting more than 90% of the patients[3]. Generally, DSPN affects the toes and distal foot, but slowly progresses proximally to involve the feet and legs in a stocking distribution. It is also characterized by a progressive loss of nerve fibers affecting both the autonomic and somatic divisions, thereby diabetic retinopathy and nephropathy can occur[3]. Foot ulceration and painful neuropathy are the main clinical consequences of DSPN, linked with higher morbidity and mortality[4]. Frequently, patients look for medical help only when pain appears[5], a symptom that affects 10% to 26% of this population[6,7].
Diabetic neuropathic pain (DNP) is characterized by tingling, burning, sharp, shooting, and lancinating or even as electric shock sensations[3,8]. It is usually considered moderate to severe and often worse at night, causing sleeping disturbs. The pain can be constant and accompanied of cutaneous allodynia, which can substantially affect the quality of life of patients, impacting the ability to perform daily activities and having a negative influence on mood. The pain may also be a reason of withdrawal of recreational and social activities and may be associated with depression[3,9,10].
The pathogenesis of DNP is not fully understood. Several theories have been proposed to explain the pain related to the diabetic neuropathy, such as changes in the blood vessels that supply the peripheral nerves; metabolic and autoimmune disorders accompanied by glial cell activation, changes in sodium and calcium channels expression and more recently, central pain mechanisms, such as increased thalamic vascularity and imbalance of the facilitatory/inhibitory descending pathways[3]. Additionally, several risk factors are associated with DNP including worsening glucose tolerance, older age, longer diabetes duration, drinking alcohol and cigarette smoking[10].
Currently, only three agents are approved in the United States for the treatment of DNP: duloxetine, a selective serotonin and norepinephrine reuptake inhibitor, pregabalin, an anticonvulsant, and the dualeffect drug tapentadol, an opioid receptor agonist and norepinephrine reuptake inhibitor[11]. However, as pain relief is unsatisfactory for most patients, several pharmacological interventions have been used based on preclinical and/or clinical evidence, as well as an inference of mechanism of action.
PHYSIOPATHOLOGY OF NEUROPATHIC PAIN IN DIABETESAlthough there is a great advance in understanding
the pathophysiological mechanisms leading to the development of diabetic complications, there is not yet a plausible hypothesis to explain why some patients develop the painful form of disease while others do not. In general, researchers seek to elucidate neuropathy underlying mechanisms as a bigger event, and include pain and other sensorial manifestations as direct consequences of neuropathy. However, interestingly, pain intensity normally is not associated with neuropathy severity, and can occur even in the absence of nerve injuries[12,13]. In this review, it will be addressed the pathophysiological mechanisms currently believed to promote the DNP.
In this sense, the mechanisms that lead to DNP are not fully understood, although there is a consensus that toxic effects of hyperglycemia represent an important factor for the development of this complication[14,15]. Nonetheless, other factors besides hyperglycemia should not be discarded[16], and will be discussed as follows.
Polyol pathway hyperactivityMetabolic disorders are the primary cause of diabetic neuropathy. Hyperglycemia, induced through decreased of insulin secretion or insulin resistance, is responsible for the enhanced of the polyol pathway activity. The ratelimiting first enzyme of this pathway aldose reductase catalyzes the formation of sorbitol from glucose, with the oxidation of nicotinamide adenine dinucleotide phosphate (NADPH) to NADP+. Sorbitol is further oxidized to fructose by sorbitol dehydrogenase, which is coupled with the reduction of nicotinamide adenine dinucleotide (NAD+) to NADH. It is described that during hyperglycemic states, the affinity of aldose reductase for glucose is higher, generating intracellular osmotic stress due to accumulation of sorbitol, since sorbitol does not cross cell membranes. Interesting, the nerve damage following the diabetic state seems not to be due to this osmotic stress since it has been reported insignificant sorbitol concentrations in the nerves of diabetic patients[1719]. However, the current accepted hypothesis states that polyol pathway hyperactivity is pathogenic primarily by increasing the turnover of cofactors such as NADPH and NAD+, which leads to a decrease in the reduction and regeneration of glutathione, as well as to an increase of advanced glycation end products (AGEs) production and activation of diacylglycerol and protein kinase C (PKC) isoforms. Depletion of glutathione could be the primary cause of oxidative stress and be related to the accumulation of toxic species[19]. In fact, aldose reductase inhibitors are effective in preventing the development of diabetic neuropathy in animal models, but they have demonstrated disappointing results and doselimiting toxicity in human trials[20].
Oxidative and nitrosative stressAs already mentioned, the polyol pathway activation could be the primary cause of oxidative stress associated
Schreiber AK et al . Diabetic neuropathic pain
433 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

with diabetes. However, oxidative stress could be also initiated by autoxidation of glucose and their metabolites, increased intracellular formation of AGEs, increased expression of the receptor for AGEs and its activating ligands, altered mitochondrial function, activation of PKC isoforms and overactivity of the hexosamine pathway[2123]. Moreover, there is mounting evidence that oxidative stress is caused by enhanced free radical formation due to glucose metabolism itself and/or deficits in antioxidant defense and it may play a major role among the putative pathogenic mechanisms of diabetic neuropathy. It seems that, in addition to oxidative stress, reactive nitrogen species, especially the peroxynitrite also play an important role in the development of diabetes and its complications[2426]. Although it has been clearly demonstrated significant changes in oxidative status in animal models of diabetes[27], tissue concentrations of known carbonyl compounds are nearly negligible and plasma ET1, nitric oxide, catalase and glutathione levels did not differ in neuropathic diabetic patients when compared to nonneuropathic diabetic ones[28]. In line with this observation, clinical results have been contradictory for antioxidants as alpha lipoic acid, ranging from little benefit[29,30] to interesting advantages[31,32].
Microvascular changesDNP is frequently associated with microvascular impairment[33,34]. In clinical and preclinical studies, it was found that peripheral perfusion is reduced, not only in the nervous tissue[35,36], but also in the skin[37], being an important physiological evidence of microvasculature alteration. As a result, nerve ischemia occurs, caused by raise in wall thickness and hyalinization of the basal lamina of vessels that nurse peripheral nerves[38,39], together with luminal reduction[38]. These alterations are caused by plasma protein scape of capillary membrane to endoneurium, promoting swelling and augmented interstitial pressure in the nerves, accompanied by higher capillary pressure, deposition of fibrin and thrombus development[40]. Hyperglycemia per se can evoke nerve hypoxia, especially in sensory nerves, altering their electrical stability[41]. Apparently controversial data from clinical studies described that diabetic patients suffering from the DNP presented higher levels of intravascular oxygen and augmented blood flow in the lower limbs than painless patients. Nevertheless, authors still consider a hypoxic state inside the endoneurium[42]. Alternatively, a potential sympathetic dysfunction can be the cause of higher blood flow[43].
As a result of nerve ischemia, both diabetic patients and animals have shown a progressive nerve loss in proximal and distal segments[44,45], resulting in reduction of intraepidermal nerve fiber density[12]. Consequently, axonal degeneration and regeneration also occurs, but more frequently in patients that do not experience pain. Besides axonal retraction and regeneration, another structural modification related to hyperglycemia is myelin sheath alteration. The observed demyelinization
can be related to Schwann cells altered capacity to support normal myelin sheath[46].
It is also important to point out that endothelial function in patients with DNP is also altered. The vaso dilatation induced by acetylcholine (i.e., the endotheliumdependent response) in dermal vessels of diabetic patients was reduced in comparison with healthy volunteers. In addition, vasoconstriction mediate by the sympathetic system (i.e., endotheliumindependent response) was also defective, what can also be implicated in the pathophysiology of diabetic neuropathy and then, in the DNP[47].
It is believed that one potential cause of the microvascular changes described above may be the oxidative stress, since the treatment with antioxidant agents can maintain regular perfusion, restoring sensory transmission in type 1 diabetes model[48].
Channels sprouting Damaged nerve endings are believed to contribute to pain in DNP[49,50]. The most accepted hypothesis states that disturbed action potentials can be produced by damaged nerve endings, being interpreted by central nervous system (CNS) as pain or dysesthesias[51]. Changes in ion channel expression in peripheral fibers are direct consequences of nerve injury, leading to hyperexcitability[52], that is far linked with neuropathic pain[53].
In this regard, upregulation of voltagegated sodium channels (Nav) has been widely demonstrated in neuropathic pain models[54,55]. These channels are involved in generation and transmission of action potential, and can be classified into sensitive (TTXS) or resistant (TTXR) to tetrodotoxin[56]. There are several reports that the TTXS channels Nav1.3, which primary function relays during the embrionary development[57], and Nav1.7, that is constitutively expressed in peripheral sensory neurons[58], are both upregulated in the dorsal root ganglion (DRG) of diabetic animals[5961]. Nav1.3 expression was also found increased in small and large diameter DRG neurons of diabetic rats presenting allodynia[62]. Considering TTXR sodium channels, Nav1.8 and Nav1.9 are normally expressed in peripheral nociceptive neurons[63], playing an important role in the generation of electrical activity in DRG[64]. Intriguingly, DRGs of allodynic diabetic rats showed a reduction of Nav1.8 expression in the following days after diabetes induction[62], and this reduction lasted 6 mo post diabetes induction[61], indicating that other sodium channels may play an important role in DNP. The same reduction was detected for Nav1.6, another TTXS channel, normally present at Ranvier nodes[60,62].
In addition, an increase of Nav1.6, Nav1.7 and Nav1.8 phosphorylation is another feature observed in the diabetic state, which leads to augmentation in their activity. Thus, both abnormal expression and function of TTXR and TTXS sodium channels is linked to abnormal activity of nociceptive fibers[60]. In this way, Sun et al[65] showed that TTXS and TTXR sodium
434 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

and selectively reverses hyperalgesia in a preclinical model of type 2 diabetes[80].
Resting membrane potential can also be altered by K+ voltage dependent channels (Kv), also participating in the electrical properties of neurons[81]. Regarding the currents generated by activation of these Kv channels in primary afferents there are two main types: rapidly inactivating Atype currents (IA), and slowly inactivating currents (IK)[82,83]. It was verified that the total density of Kv currents as well as the mRNA of IA subunits of KV 1.4, 3.4, 4.2 and 4.3 were reduced in large and medium size DRG neurons of diabetic rats[84]. So, this down regulation can increase neuronal excitability and peptide release[83], which might also participate in hyperexcitability of peripheral nerves of diabetic subjects.
Microglial activationIt is becoming increasingly recognized that glial cells play an important role in the pathogenesis of many diseases of the nervous system, including chronic pain states[85]. Glia comprises both macroglia (including astrocytes, radial cells and oligodendrocytes) and microglia cells, which are mainly responsible for maintain homeostasis, form myelin, and provide support and protection for neurons from both central and peripheral nervous system[85]. Normally, microglial cells comprise less than 20% of spinal glial cells but in response to dorsal root ganglia and spinal cord after nerve injury there is a robust proliferation at spinal level[86]. Activation of microglia occurs right after peripheral nerve injury, lasting for less than 3 mo, and is responsible for a production of several inflammatory mediators as cytokines, chemokines, and cytotoxic substances such as nitric oxide and free radicals, prompting to a pro inflammatory milieu[83]. Diabetes has impact on all glial cells of the spinal cord since persistent microglial activation was observed in streptozotocininduced diabetic rats lasting from 4 wk[87,88] to 6 or 8 mo[61,89]. This microglial activation has been associated with sensorial changes and upregulation of Nav1.3 sodium channels in the DRG[61], possible through p38 mitogen activated protein kinase dependent mechanism[90,91]. Conversely, diabetes is associated with a reduction in glial fibrillary acidic protein (i.e., glial fibrillary acidic protein) immunoreactive astrocytes in the spinal cord, which may affect the functional support and role of astrocytic cells in the nervous tissue, such as the clearance of neurotransmitters within the synaptic cleft[92]. Considering the potential of microglial activation in driving spinal sensitization, in the near future, drugs that target these cells may become an important therapeutic alternative in chronic pain control.
Central sensitization As already demonstrated in different neuropathic pain states, DNP may be a consequence of both peripheral and CNS changes[93,94]. It was well described that during DNP, primary afferents are sensitized, inducing dorsal horn hyperactivity and neuroplastic changes in
currents are increased in small neurons in the DRG of diabetic animals, being this related not only with sensory disturbances, but also with the rise of efficiency of conductance in polymodal C fibers, which in turn, facilitates nociceptive transmission.
In a recent in vitro study, it has been described that hyperglycemia evokes higher TTXR Na+ currents in a time and concentrationdependent manner, demonstrating a straight relationship between glucose levels and biophysical changes[66]. In DNP patients, it was reported an increase in nodal Na+ currents when compared to painless diabetic patients, what can also contribute to hyperexcitability in peripheral nerves[67].
A new concept proposed by Hoeijmakers et al[68], links the beginning of pancreatic beta cells failure and DNP with genetic disruptions on Nav1.7 channels. Sinceboth pancreatic beta cells and peripheral neurons express Nav1.7 channels, a susceptible genetic background could facilitate generation of Nav1.7 mutations, leading to gainoffunction that evokes beta cell lesions,and thereafter, diabetes and hyperexcitability in neurons[69]. According to these authors, this theory could explain why some patients have neuropathy before diabetes onset[68].
Another interesting finding related to sodium channels modulation is the increased levels of methylglyoxal in type 2 diabetes DNP patients, when compared with those painless[70,71], and in complicationfree type 1 diabetic patients[72]. This glycolytic metabolite can activate nerve endings through transient receptor potential cation channel subfamily A member 1 activation in the DRG[73], and also change the Nav1.7 and Nav1.8 function through posttranslational changes[70]. In line with this clinical observation, in preclinical models methylglyoxal was found to reduce nerve conduction velocity, to elevate calcitonin generelated peptide release from sensory nerves and to induce thermal and mechanical sensibility[70]. In addition, in diabetic states methylglyoxal is also involved in the formation of AGEs[74].
Calcium channels can also be misregulated in a diabetic condition, leading to an enhanced calcium influx in sensory neurons[75], what can deflagrate both substance P and glutamate release[76]. It was verified in two different animal models of insulin dependent diabetes that high voltage activated Ca2+ current amplitudes were increased in small diameter neurons[75,77] and the activity of Ttype channels (Cav3.2) is also augmented in small diameter fibers[78], what could be normalized by molecular knockdown of this calcium channel[79]. However, there was no translation of these results to patients in clinical trials[80]. A possible future target for pharmacological intervention over calcium channels has been proposed by Orestes and colleagues (2013), which observed that glycosilation of Cav3.2 augments the current density, accelerates kinetics, and also increases the number of channels on neuron membrane, which can be directly involved in DNP. Interestingly, the deglycosylation treatment with neuraminidase inhibits native calcium currents in nociceptors and completely
435 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

central sensory neurons[93]. The common occurrence of allodynia in DNP patients supports the idea that CNS pain processing is altered[95].
Among the factors that can lead to the hyperactivity of spinal neurons in diabetic neuropathy is the increased glutamate release from primary afferents in the spinal cord[96,97]. Moreover, spinal NMethylDaspartate (NMDA) receptor expression is augmented in this condition[98], generating increased and more frequent excitatory post synaptic currents in the lamina Ⅱ[97]. Additionally, it has been described that cAMP response elementbinding protein signaling, which directly regulates NMDA receptors activity[99] is enhanced in DNP[98,100]. Thus, it is plausible that augmented NMDA expression and glutamate release might contribute to spinal cord hyperactivity. On the other hand, GABAB receptors seem to be downregulated in the spinal cord in diabetic neuropathy[98]. Activation of GABAB receptors results in inhibition of NMDA receptor activity through a direct inhibition of voltagesensitive Ca2+ channels[101] and opening of inwardly rectifying K+ channels[102]. Furthermore, GABAB receptor activation also causes downregulation of NMDA receptors at the spinal level in diabetic rats[98]. Considering the importance of central sensitization in the hypersensitivity associated with DNP, strategies that aim to control spinal neurons hyperexcitability are very useful in pain control in this condition, as will be discussed bellow.
Brain plasticityFunctional changes in pain processing areas in the CNS, besides the spinal cord, have been ultimately linked with DNP[103], in a tight relation to increased peripheral input[93,104]. Among these areas, marked changes in the thalamus, cortex and rostroventromedial medulla (RVM) have been reported in DNP patients and or experimental models.
The ventral posterolateral nucleus (VPL) of the thalamus is the main receiving area of nociceptive stimuli that is processed in the spinal cord[105]. Projection neurons reach the thalamus trough the spinothalamic tract (STT), which represents a major ascending nociceptive pathway. It has been demonstrated that in diabetic rats, these neurons present increased spontaneous activity, enlargement of the receptive field and augmented responses to mechanical noxious and innocuous stimuli. The hyperexcitability of STT neurons probably accounts to hypersensitivity to external stimuli and spontaneous pain[93], increased in primary afferents activity[93,104] and to plastic changes in spinal neurons[93].
In addition, in studies that assessed brain imaging in diabetic rats it was reported increased activity not only at VPL, but also in different thalamic nuclei that control sensorymotor aspects[106]. In diabetic patients, a recent study showed increased activation of diverse brain areas, including medial thalamus after application of noxious thermal stimuli in feet[107]. Moreover, it has been described that DNP patients has a marked reduction in the levels of Nacetylaspartate (NAA) levels in the
thalamus compared to painless diabetic individuals[108]. It is important to point out that patients with brain disorders in which neuronal loss or dysfunction are involved have consistently decreases in brain NAA concentrations[109]. Other clinical finding related to thalamus alterations in diabetic patients is that subjects with painful type 1 diabetic neuropathy presented increased thalamus blood flow, when compared with those without pain, which was considered to reflect higher neuronal activity[110]. Taking account the thalamus relevance in the nociceptive pathway, it is plausible to suggest that the alterations reported in this area might contribute to the development and/or maintance of DNP[108,110].
Likewise, in a model of type 1 diabetes, increased glutamate transmission was reported in the anterior cingulate cortex a brain area involved in the processing of the affectivemotivational dimension of pain[111]. The consequence of higher stimulation of this area by glutamate is suggested to be a sustained negative perception of affective component of pain[111].
Changes in the endogenous pain control system have also been described in preclinical and clinical studies of DNP. The RVM is a structure that receives direct influences of periaqueductal gray matter, which is, in turn, affected by other structures, such as amygdala and hypothalamus[112]. Three different populations of cells have been describe within the RVM: activation of ON cells act in a pronociceptive way, while activation of OFF cells has the opposite effect[113] and neural cells which activation is still contradictory and remains to be better clarified[114,115]. In diabetic animals, there is evidence of a reduction on the OFF cells and increase on the ON cells population. In addition, basal activity is augmented in ON cells, and reduced in OFF cells, in a resultant misbalance between pain facilitatory and inhibitory descending modulation in diabetic animals[103]. After noxious mechanical stimulation in the periphery, there was no difference between diabetic and control ON cells activity. Thus, the mechanical hyperalgesia detected in diabetic rats could be associated with OFF cells impairment and consequently reduction on descending inhibitory tone[103].
Some studies have also addressed the levels of the main neurotransmitters of the endogenous pain control system in different areas of the CNS in diabetic rats, but they have shown discrepant results. While some researchers found reduced release of norepinephrine in the spinal cord in diabetic rats[116], others have described opposite findings[117]. There is also evidence of diminished norepinephrine levels in supra spinal areas, such as brainstem and thalamus, but higher concentration in the cortex of diabetic animals[118]. Additionally, impaired spinal opioidinduced release of serotonin (5HT) has been demonstrated in diabetic rats, and this finding may be related to opioid hyporesponsiveness in experimental DNP[119]. Increased norepinephrine and 5HT levels in the spinal cord, as well as, augmented expression of norepinephrine and 5HT in RVM neurons
436 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

was also demonstrated in diabetic rats[117]. Considering the facilitatory role of serotoninergic and noradrenergic descending modulation during chronic pain, these changes may probably account for enhanced pain during diabetic neuropathy[117]. There is also clinical evidence for misbalance between excitatory and inhibitory neurotransmitters in the CNS of diabetic patients with positive symptoms of neuropathy. In this regard, it has been found reduced levels of GABA and higher levels of glutamate in the posterior insula of diabetic patients, as well as a higher glutamate/GABA ratio within the thalamus[120]. These changes may contribute to pain development in DNP, but further studies are necessary to determine their clinical significance.
TREATMENT OF DNPDNP continues to represent a therapeutic challenge as its pathophysiology is not yet fully understood and pain relief is still unsatisfactory. The pharmacological treatments, with exception to those targeted to the glycemic control, are symptomatic, not focused on the pathophysiological mechanisms, limited by side effects[3,121] and by the development of tolerance[121].
A wide variety of drugs, used alone or in combination, has been shown to significantly reduce neuropathic pain compared with placebo in randomized controlled trials, but pain relief remains inadequate for most patients[122]. Generally, in clinical trials, treatment is considered successful if patients would obtain 50% of reduction in the pain level[123125] associated with some additional beneficial effects on sleep, fatigue, depression and quality of life[125]. Thus, the management of this condition basically consists of excluding other causes of painful peripheral neuropathy, improving glycemic control as a prophylactic therapy and using medications to alleviate pain[126].
Despite of multimodal and multidisciplinary approaches to the treatment, the primary pathway is pharmacologically based[127]. Three different agents have regulatory approval in the United States for the treatment of DNP: pregabalin, duloxetine and tapentadol[11,128]. However, as pain relief is still suboptimal and challenging for clinicians[95], drugs from various pharmacological classes have been used and some of them are included in this review.
AnticonvulsantsPregabalin was the first anticonvulsant to receive approval from the Food and Drug Administration (FDA) for the treatment of postherpetic neuralgia, DNP[129,130] and neuropathic pain after spinal cord injury[131]. Pregabalin is a GABA analogue that selectively binds to presynaptic voltagegated calcium channels containing the α2δ subunit in the brain and spinal cord, causing inhibition of the release of excitatory neurotransmitters[128,132]. Moreover, α2δ1 subunits are responsible for increasing the functional expression of these channels, as a consequence of increased trafficking. Thus, the analgesic
action of pregabalin is also proposed to be the result of impaired trafficking of α2δ1 subunit with a consequent diminished expression of functional calcium channels[133].
Several clinical trials evaluating pregabalin in DNP showed efficacy in the management of this condition[3,
134,135] with a number needed to treat (NNT) of 6.3[125]. In addition to its analgesic effects, pregabalin presents anxiolytic activity[132,135] and it has a beneficial effect on sleep and quality of life[132], contributing, therefore, to improve the general condition of the patients. The side effects include dizziness, somnolence, peripheral edema, headache and weight gain[3].
Some guidelines have also recommended gabapentin to treat DNP[136]. Besides pregabalin, gabapentin is the only other anticonvulsant drug that demonstrated efficacy in the treatment of this condition[128] with an NNT of 5.8[137]. Gabapentin and pregabalin have a similar mechanism of action and the first is licensed for neuropathic pain in the United Kingdom, but not in the United States[128]. Some clinical trials have suggested that gabapentin and pregabalin present better analgesic efficacy than tricyclic antidepressants or opioids[138] and other important aspects of these drugs include their tolerability and lack of serious toxicity[139].
AntidepressantsAntidepressants represent the first line drugs in DNP management. Duloxetine, a serotonin and norepinephrine reuptake inhibitor, is rated level A for efficacy and is approved in the United States for the treatment of this condition. Additionally, some clinical trials have pointed out the effectiveness of duloxetine in other chronic pain conditions, such as fibromyalgia and chronic musculoskeletal pain[140,141].
Results from a metaanalysis that included randomized, double blind, placebo controlled studies in patients with DNP showed the superiority of duloxetine over placebo in reduction of pain severity and in Patient Global Impression of Improvement/Change, as well as efficacy similar to gabapentin and pregabalin[142]. Moreover, in a 2wk openlabel randomized trial in diabetic patients poorly responsive to gabapentin, duloxetine was able to reduce the pain score to levels similar to those achieved with pregabalin[143,144]. Furthermore, analgesic efficacy of duloxetine in the treatment of DNP is maintained over a 6mo period[145], reinforcing its importance as a treatment option for this condition. The NNT for duloxetine varies from 1.3 to 5.1 in DNP patients[146,147], which experience more frequently nausea, somnolence and dizziness as side effects[146].
Venlafaxine is also a selective serotonin and noradrenaline reuptake inhibitor, that predominantly inhibits serotonin reuptake at low doses and noradrenaline at higher doses[148]. Venlafaxine was also shown to be effective in reducing pain intensity in diabetic neuropathic patients[149], with an NNT between 2.2 and 5.1 and a number needed to harm (NNH) of 9.6, to minor adverse effects, and of 16.2, for major adverse effects[150].
Tricyclic antidepressants can also be an alternative to
437 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

treat DNP[151]. Amitriptyline was shown to be as effective as gabapentin in a direct metaanalysis study[152] and as duloxetine in a randomized, doubleblind, crossover trial[153]. Likewise, nortriptyline was reported as being as effective as gabapentin in attenuating neuropathic pain in a doubleblind crossover trial enrolling diabetic patients[154].
Tricyclic antidepressants are estimated to have an NNT of 1.3[151,155], and an NNH from 4.2 to 10.7[151] in DNP. The most common side effects related to the use of these drugs are dry mouth, postural hypotension, arrhythmias, cognitive impairment, constipation and urinary retention, which are more frequently observed after amitriptyline than nortriptyline treatment[155].
OpioidsOpioids are recommended to be used as second or third line treatment for DNP[3,151]. One multicenter, randomized, placebocontrolled study reported the tramadol effectiveness to significantly improve scores on physical and social functioning ratings in patients with DNP, but beneath some side effects such as nausea, constipation, headache and somnolence[156]. Morphine was also shown to be effective in reducing mean daily pain scores related to diabetic neuropathy and postherpetic neuralgia[128,157]. Moreover, results of clinical trials indicated that diabetic neuropathic patients experienced a significant reduction in pain intensity and an improvement on quality of life during oxycodone treatment, compared to placeboexposed group[158,159]. Besides, oxycodone improved gabapentin but not the pregabalin effectiveness in promoting DNP relief[160,161].
The clinical evidence for the effectiveness of opioids in the control of DNP is corroborated by some preclinical data, which have reported the effectiveness of morphine[162,163] and buprenorphine[164] in reducing thermal or mechanical hypersensitivity in DNP animal models.
There is also evidence that the antihyperalgesic effect of opioids is improved by the association with some drugs, such as the antidepressants amitriptyline, moclobemide and reboxetine[165]. In line with this idea, new molecules that integrate additional mechanisms to the opioid receptor agonism have been shown efficacy in reducing nociceptive behavior in animal models of DNP, such as the cebranopadol, a nociceptin/orphanin FQ peptide and opioid receptor agonist[166], and the muopioid receptor agonist and norepinephrine reuptake inhibitor, tapentadol[162,167]. The latter was approved by FDA for DNP treatment since 2012[11]. Tapentadol has been shown to be effective in the management of different types of chronic pain, including osteoarthritis knee pain, low back pain and DNP, with a tolerable safety profile[168,169]. Specifically concerning DNP, a randomizedwithdrawal, placebocontrolled trial reported reduction of at least 30% in pain intensity in about 50% of the patients that received tapentadol[170]. Similar data were obtained in a recent clinical trial in diabetic
neuropathic patients with moderate to severe pain, which experienced nausea (21.1%) and vomiting (12.7%) as side effects[171].
Others agents The drugs discussed below are currently associated to the pharmacological treatments already described according to the patients’ symptoms and needs in order to achieve a better relief of pain in DNP conditions. However, further studies are necessary, specially controlled clinical trials, to determine the more efficacious, safe and successful combinations to be applied in the management of DNP[128].
Capsaicin topical cream: Topical agents may be associated with fewer and less clinically significant adverse events than systemic agents[172]. In addition, the possibilities of drug interactions are markedly reduced with the use of local treatments, which represent good options for patients with multiple medical problems[128]. The capsaicin cream has been shown to be effective in the treatment of neuropathic conditions[150] and is approved for topical relief of neuropathic pain[128]. Capsaicin is the pungent component of hot chilli peppers[11] and an agonist of the transient receptor potential vanilloid 1. This receptor is a ligandgated, nonselective cation channel, predominantly expressed on unmyelinated C nerve fibers[173], which, after repeated exposure to topical capsaicin, are depleted of their content of substance P and other neurotransmitters[173,174]. The C fibers depletion and desensitization reduce painful stimuli transmission from peripheral nerves to the central nervous system[173]. Some clinical trials have demonstrated the effectiveness of lowconcentration (from 0.025% to 0.075%) capsaicin cream in DNP[11,174,175]. Higher concentrations are not indicated because of desensitization of nociceptive sensory nerve endings, which may increase the risk of skin injuries in DNP patients[173,174]. Some adverse effects include itching, stinging, erythema, transient burning sensation and initial pain at the application site, that diminishes with repeated use[132,175], leading many patients to withdraw from the treatment[128,173].
Lidocaine patch: Lidocaine patches act as peripheral analgesics with minimal systemic absorption and are used in combination with other analgesic drugs[132,172]. Lidocaine blocks sodium channels and counteracts the hyperexcitability of peripheral nociceptors that contributes to neuropathic pain[132,176]. The blockade reduces ectopic discharges and raises the peripheral sensory neuron discharge threshold[176]. The few DNP clinical trials that compared topical lidocaine with other relevant interventions suggested that the effects in pain reduction are comparable to other drugs, such as capsaicin, gabapentin, amitriptyline[172] and pregabalin[172,174]. Adverse events include local irritation[172], contact dermatitis and itching[132].
Alpha lipoic acid: The benefit provided by alpha
438 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

lipoic acid (ALA) in the treatment of DNP possibly is due to its direct effects on the neuropathy, by reducing the oxidative stress, which has been defined as an important factor in the physiopatology of the diabetic neuropathy[122]. Its antioxidant and antiinflammatory actions may contribute to an allround improvement of diabetic neuropathy symptoms[135]. In some clinical trials that evaluated ALA effect in diabetic patients, pain was not a primary end point. However, they have shown a moderate benefit in terms of pain reduction[132]. In a randomized doubleblinded trial, ALAtreated patients reported a greater reduction in neuropathic pain when compared to placebotreated subjects[122]. Compared to several drugs currently in use for DNP treatment, ALA has fewer side effects[30], being nausea and vomiting the most common[132].
Isosorbide dinitrate spray: Isosorbide dinitrate is a nitric oxidedependent vasodilator with effects on both arteries and veins[177]. The improvement of pain and burning sensation could be associated with the increased generation of nitric oxide, improving microvascular blood flow[178]. In a clinical trial with diabetic patients, the isosorbide dinitrate spray reduced overall neuropathic pain and burning sensation in about 50% of the patients, which also reported improvement in their quality of life, with improvements in sleep, mobility and mood[178,179].
Final considerations about DNP treatment: Besides the fact of many diabetic complications can be reduced with improved blood glucose control and other lifestyle interventions[132,150], such as quit smoking and reducing alcohol consumption[150], the efficacy of these measures, as well as the pharmacological treatments on DNP are not predictable. The medications rated as level A based on their efficacy are able to reduce pain and improve some aspects of patients’ quality of life, but are not able to fully eliminate pain or prevent/revert the neuropathy. Even their combination does not result in satisfactory pain control, being the best improvement in pain, restricted to 50% of relief for the majority of the patients. Considering the available pharmacological options, DNP treatment has to be based mainly on patients’ symptoms, pain level and tolerance of side effects.
REFERENCES 1 International Diabetes Federation. IDF Diabetes Atlas. 6th ed.
Brussels, Belgium: International Diabetes Federation, 20132 Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman
R, Malik RA, Maser RE, Sosenko JM, Ziegler D. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 2005; 28: 956-962 [PMID: 15793206 DOI: 10.2337/diacare.28.4.956]
3 Tesfaye S, Boulton AJ, Dickenson AH. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care 2013; 36: 2456-2465 [PMID: 23970715 DOI: 10.2337/dc12-1964]
4 Boulton AJ, Kirsner RS, Vileikyte L. Clinical practice. Neuropathic diabetic foot ulcers. N Engl J Med 2004; 351: 48-55 [PMID:
15229307 DOI: 10.1056/NEJMcp032966]5 Tesfaye S, Chaturvedi N, Eaton SE, Ward JD, Manes C, Ionescu-
Tirgoviste C, Witte DR, Fuller JH. Vascular risk factors and diabetic neuropathy. N Engl J Med 2005; 352: 341-350 [PMID: 15673800 DOI: 10.1056/NEJMoa032782]
6 Low PA, Dotson RM. Symptomatic treatment of painful neuropathy. JAMA 1998; 280: 1863-1864 [PMID: 9846782 DOI: 10.1001/jama.280.21.1863]
7 Harris M, Eastman R, Cowie C. Symptoms of sensory neuropathy in adults with NIDDM in the U.S. population. Diabetes Care 1993; 16: 1446-1452 [PMID: 8299433 DOI: 10.2337/diacare.16.11.1446]
8 Bansal V, Kalita J, Misra UK. Diabetic neuropathy. Postgrad Med J 2006; 82: 95-100 [PMID: 16461471]
9 Quattrini C, Tesfaye S. Understanding the impact of painful diabetic neuropathy. Diabetes Metab Res Rev 2003; 19 Suppl 1: S2-S8 [PMID: 12577252 DOI: 10.1002/dmrr.360]
10 Gore M, Brandenburg NA, Dukes E, Hoffman DL, Tai KS, Stacey B. Pain severity in diabetic peripheral neuropathy is associated with patient functioning, symptom levels of anxiety and depression, and sleep. J Pain Symptom Manage 2005; 30: 374-385 [PMID: 16256902 DOI: 10.1016/j.jpainsymman.2005.04.009]
11 Freeman R. New and developing drugs for the treatment of neuropathic pain in diabetes. Curr Diab Rep 2013; 13: 500-508 [PMID: 23771401 DOI: 10.1007/s11892-013-0396-6]
12 Sorensen L, Molyneaux L, Yue DK. The relationship among pain, sensory loss, and small nerve fibers in diabetes. Diabetes Care 2006; 29: 883-887 [PMID: 16567832 DOI: 10.2337/diacare.29.04.06.dc05-2180]
13 Young RJ, Zhou YQ, Rodriguez E, Prescott RJ, Ewing DJ, Clarke BF. Variable relationship between peripheral somatic and autonomic neuropathy in patients with different syndromes of diabetic polyneuropathy. Diabetes 1986; 35: 192-197 [PMID: 3002887 DOI: 10.2337/diab.35.2.192]
14 Dobretsov M, Hastings SL, Romanovsky D, Stimers JR, Zhang JM. Mechanical hyperalgesia in rat models of systemic and local hyperglycemia. Brain Res 2003; 960: 174-183 [PMID: 12505670 DOI: 10.1016/S0006-8993(02)03828-3]
15 Oyibo SO, Prasad YD, Jackson NJ, Jude EB, Boulton AJ. The relationship between blood glucose excursions and painful diabetic peripheral neuropathy: a pilot study. Diabet Med 2002; 19: 870-873 [PMID: 12358878 DOI: 10.1046/j.1464-5491.2002.00801.x]
16 Romanovsky D, Wang J, Al-Chaer ED, Stimers JR, Dobretsov M. Comparison of metabolic and neuropathy profiles of rats with streptozotocin-induced overt and moderate insulinopenia. Neuroscience 2010; 170: 337-347 [PMID: 20600635 DOI: 10.1016/j.neuroscience.2010.06.059]
17 Sheetz MJ, King GL. Molecular understanding of hypergly-cemia’s adverse effects for diabetic complications. JAMA 2002; 288: 2579-2588 [PMID: 12444865 DOI: 10.1001/jama.288.20.2579]
18 Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 1999; 48: 1-9 [PMID: 9892215 DOI: 10.2337/diabetes.48.1.1]
19 Oates PJ. Polyol pathway and diabetic peripheral neuropathy. Int Rev Neurobiol 2002; 50: 325-392 [PMID: 12198816]
20 Chalk C, Benstead TJ, Moore F. Aldose reductase inhibitors for the treatment of diabetic polyneuropathy. Cochrane Database Syst Rev 2007; (4): CD004572 [PMID: 17943821 DOI: 10.1002/14651858.CD004572.pub2]
21 Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001; 414: 813-820 [PMID: 11742414 DOI: 10.1038/414813a]
22 Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 2010; 107: 1058-1070 [PMID: 21030723 DOI: 10.1161/CIRCRESAHA.110.223545]
23 Baynes JW. Role of oxidative stress in development of com-plications in diabetes. Diabetes 1991; 40: 405-412 [PMID: 2010041 DOI: 10.2337/diab.40.4.405]
24 Obrosova IG, Mabley JG, Zsengellér Z, Charniauskaya T, Abatan OI, Groves JT, Szabó C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite
439 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

decomposition catalyst. FASEB J 2005; 19: 401-403 [PMID: 15611153]
25 Drel VR, Pacher P, Vareniuk I, Pavlov I, Ilnytska O, Lyzogubov VV, Tibrewala J, Groves JT, Obrosova IG. A peroxynitrite decomposition catalyst counteracts sensory neuropathy in streptozotocin-diabetic mice. Eur J Pharmacol 2007; 569: 48-58 [PMID: 17644085 DOI: 10.1016/j.ejphar.2007.05.055]
26 Vareniuk I, Pavlov IA, Drel VR, Lyzogubov VV, Ilnytska O, Bell SR, Tibrewala J, Groves JT, Obrosova IG. Nitrosative stress and peripheral diabetic neuropathy in leptin-deficient (ob/ob) mice. Exp Neurol 2007; 205: 425-436 [PMID: 17475250 DOI: 10.1016/j.expneurol.2007.03.019]
27 Cunha JM, Jolivalt CG, Ramos KM, Gregory JA, Calcutt NA, Mizisin AP. Elevated lipid peroxidation and DNA oxidation in nerve from diabetic rats: effects of aldose reductase inhibition, insulin, and neurotrophic factors. Metabolism 2008; 57: 873-881 [PMID: 18555826 DOI: 10.1016/j.metabol.2008.01.021]
28 Ozkul A, Ayhan M, Yenisey C, Akyol A, Guney E, Ergin FA. The role of oxidative stress and endothelial injury in diabetic neuropathy and neuropathic pain. Neuro Endocrinol Lett 2010; 31: 261-264 [PMID: 20424576]
29 Ziegler D, Low PA, Litchy WJ, Boulton AJ, Vinik AI, Freeman R, Samigullin R, Tritschler H, Munzel U, Maus J, Schütte K, Dyck PJ. Efficacy and safety of antioxidant treatment with α-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes Care 2011; 34: 2054-2060 [PMID: 21775755 DOI: 10.2337/dc11-0503]
30 Mijnhout GS, Alkhalaf A, Kleefstra N, Bilo HJ. Alpha lipoic acid: a new treatment for neuropathic pain in patients with diabetes? Neth J Med 2010; 68: 158-162 [PMID: 20421656]
31 Ruessmann HJ. Switching from pathogenetic treatment with alpha-lipoic acid to gabapentin and other analgesics in painful diabetic neuropathy: a real-world study in outpatients. J Diabetes Complications 2009; 23: 174-177 [PMID: 18403218 DOI: 10.1016/j.jdiacomp.2008.02.002]
32 Tang J, Wingerchuk DM, Crum BA, Rubin DI, Demaerschalk BM. Alpha-lipoic acid may improve symptomatic diabetic polyneuropathy. Neurologist 2007; 13: 164-167 [PMID: 17495764 DOI: 10.1097/01.nrl.0000263703.78318.2b]
33 Arora S, Pomposelli F, LoGerfo FW, Veves A. Cutaneous microcirculation in the neuropathic diabetic foot improves significantly but not completely after successful lower extremity revascularization. J Vasc Surg 2002; 35: 501-505 [PMID: 11877698 DOI: 10.1067/mva.2002.121126]
34 Doupis J, Lyons TE, Wu S, Gnardellis C, Dinh T, Veves A. Microvascular reactivity and inflammatory cytokines in painful and painless peripheral diabetic neuropathy. J Clin Endocrinol Metab 2009; 94: 2157-2163 [PMID: 19276232 DOI: 10.1210/jc.2008-2385]
35 Cameron NE, Eaton SE, Cotter MA, Tesfaye S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia 2001; 44: 1973-1988 [PMID: 11719828 DOI: 10.1007/s001250100001]
36 Tuck RR, Schmelzer JD, Low PA. Endoneurial blood flow and oxygen tension in the sciatic nerves of rats with experimental diabetic neuropathy. Brain 1984; 107 (Pt 3): 935-950 [PMID: 6478183 DOI: 10.1093/brain/107.3.935]
37 Jin HY, Lee KA, Song SK, Liu WJ, Choi JH, Song CH, Baek HS, Park TS. Sulodexide prevents peripheral nerve damage in streptozotocin induced diabetic rats. Eur J Pharmacol 2012; 674: 217-226 [PMID: 21641343 DOI: 10.1016/j.ejphar.2011.05.059]
38 Malik RA, Tesfaye S, Thompson SD, Veves A, Sharma AK, Boulton AJ, Ward JD. Endoneurial localisation of microvascular damage in human diabetic neuropathy. Diabetologia 1993; 36: 454-459 [PMID: 8314451 DOI: 10.1007/BF00402283]
39 Pavy-Le Traon A, Fontaine S, Tap G, Guidolin B, Senard JM, Hanaire H. Cardiovascular autonomic neuropathy and other complications in type 1 diabetes. Clin Auton Res 2010; 20: 153-160 [PMID: 20354891 DOI: 10.1007/s10286-010-0062-X]
40 Czyzyk A. [Epidemiology of vascular disease in diabetes mellitus].
Pol Arch Med Wewn 1986; 76: 129-136 [PMID: 3575142]41 Fuchs D, Birklein F, Reeh PW, Sauer SK. Sensitized peripheral
nociception in experimental diabetes of the rat. Pain 2010; 151: 496-505 [PMID: 20832942 DOI: 10.1016/j.pain.2010.08.010]
42 Eaton SE, Harris ND, Ibrahim S, Patel KA, Selmi F, Radatz M, Ward JD, Tesfaye S. Increased sural nerve epineurial blood flow in human subjects with painful diabetic neuropathy. Diabetologia 2003; 46: 934-939 [PMID: 12819899 DOI: 10.1007/s00125-003-1127-3]
43 Archer AG, Roberts VC, Watkins PJ. Blood flow patterns in painful diabetic neuropathy. Diabetologia 1984; 27: 563-567 [PMID: 6530051 DOI: 10.1007/BF00276968]
44 Jelicic Kadic A, Boric M, Vidak M, Ferhatovic L, Puljak L. Changes in epidermal thickness and cutaneous innervation during maturation in long-term diabetes. J Tissue Viability 2014; 23: 7-12 [PMID: 24361118 DOI: 10.1016/j.jtv.2013.11.002]
45 Shun CT, Chang YC, Wu HP, Hsieh SC, Lin WM, Lin YH, Tai TY, Hsieh ST. Skin denervation in type 2 diabetes: correlations with diabetic duration and functional impairments. Brain 2004; 127: 1593-1605 [PMID: 15128619 DOI: 10.1093/brain/awh180]
46 Said G, Baudoin D, Toyooka K. Sensory loss, pains, motor deficit and axonal regeneration in length-dependent diabetic polyneuropathy. J Neurol 2008; 255: 1693-1702 [PMID: 18825430 DOI: 10.1007/s00415-008-0999-z]
47 Quattrini C, Harris ND, Malik RA, Tesfaye S. Impaired skin microvascular reactivity in painful diabetic neuropathy. Diabetes Care 2007; 30: 655-659 [PMID: 17327336 DOI: 10.2337/dc06-2154]
48 Inkster ME, Cotter MA, Cameron NE. Treatment with the xanthine oxidase inhibitor, allopurinol, improves nerve and vascular function in diabetic rats. Eur J Pharmacol 2007; 561: 63-71 [PMID: 17291486 DOI: 10.1016/j.ejphar.2006.12.029]
49 Brown MJ, Asbury AK. Diabetic neuropathy. Ann Neurol 1984; 15: 2-12 [PMID: 6370098 DOI: 10.1002/ana.410150103]
50 Said G, Slama G, Selva J. Progressive centripetal degeneration of axons in small fibre diabetic polyneuropathy. Brain 1983; 106 (Pt 4): 791-807 [PMID: 6652463 DOI: 10.1093/brain/106.4.791]
51 Stewart JD, Low PA, Fealey RD. Distal small fiber neuropathy: results of tests of sweating and autonomic cardiovascular reflexes. Muscle Nerve 1992; 15: 661-665 [PMID: 1324425 DOI: 10.1002/mus.880150605]
52 Dickenson AH, Matthews EA, Suzuki R. Neurobiology of neuropathic pain: mode of action of anticonvulsants. Eur J Pain 2002; 6 Suppl A: 51-60 [PMID: 11888242]
53 Ochoa JL, Torebjörk HE. Paraesthesiae from ectopic impulse generation in human sensory nerves. Brain 1980; 103: 835-853 [PMID: 6254609 DOI: 10.1093/brain/103.4.835]
54 Black JA, Cummins TR, Plumpton C, Chen YH, Hormuzdiar W, Clare JJ, Waxman SG. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J Neurophysiol 1999; 82: 2776-2785 [PMID: 10561444]
55 He XH, Zang Y, Chen X, Pang RP, Xu JT, Zhou X, Wei XH, Li YY, Xin WJ, Qin ZH, Liu XG. TNF-α contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain 2010; 151: 266-279 [PMID: 20638792 DOI: 10.1016/j.pain.2010.06.005]
56 Roy ML, Narahashi T. Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci 1992; 12: 2104-2111 [PMID: 1318956]
57 Felts PA, Yokoyama S, Dib-Hajj S, Black JA, Waxman SG. Sodium channel alpha-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res Mol Brain Res 1997; 45: 71-82 [PMID: 9105672 DOI: 10.1016/S0169-328X(96)00241-0]
58 Ogata N, Ohishi Y. Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn J Pharmacol 2002; 88: 365-377 [PMID: 12046980 DOI: 10.1254/jjp.88.365]
59 Galloway C, Chattopadhyay M. Increases in inflammatory mediators in DRG implicate in the pathogenesis of painful neuropathy in Type 2 diabetes. Cytokine 2013; 63: 1-5 [PMID:
440 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

23664770 DOI: 10.1016/j.cyto.2013.04.009]60 Hong S, Morrow TJ, Paulson PE, Isom LL, Wiley JW. Early painful
diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J Biol Chem 2004; 279: 29341-29350 [PMID: 15123645 DOI: 10.1074/jbc.M404167200]
61 Cheng KI, Wang HC, Chuang YT, Chou CW, Tu HP, Yu YC, Chang LL, Lai CS. Persistent mechanical allodynia positively correlates with an increase in activated microglia and increased P-p38 mitogen-activated protein kinase activation in streptozotocin-induced diabetic rats. Eur J Pain 2014; 18: 162-173 [PMID: 23868758 DOI: 10.1002/j.1532-2149.2013.00356.x]
62 Craner MJ, Klein JP, Renganathan M, Black JA, Waxman SG. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann Neurol 2002; 52: 786-792 [PMID: 12447933 DOI: 10.1002/ana.10364]
63 Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 1996; 379: 257-262 [PMID: 8538791 DOI: 10.1038/379257a0]
64 Herzog RI, Cummins TR, Waxman SG. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J Neurophysiol 2001; 86: 1351-1364 [PMID: 11535682]
65 Sun W, Miao B, Wang XC, Duan JH, Wang WT, Kuang F, Xie RG, Xing JL, Xu H, Song XJ, Luo C, Hu SJ. Reduced conduction failure of the main axon of polymodal nociceptive C-fibres contributes to painful diabetic neuropathy in rats. Brain 2012; 135: 359-375 [PMID: 22271663 DOI: 10.1093/brain/awr345]
66 Singh JN, Jain G, Sharma SS. In vitro hyperglycemia enhances sodium currents in dorsal root ganglion neurons: an effect attenuated by carbamazepine. Neuroscience 2013; 232: 64-73 [PMID: 23262239 DOI: 10.1016/j.neuroscience.2012.12.011]
67 Misawa S, Sakurai K, Shibuya K, Isose S, Kanai K, Ogino J, Ishikawa K, Kuwabara S. Neuropathic pain is associated with increased nodal persistent Na(+) currents in human diabetic neuropathy. J Peripher Nerv Syst 2009; 14: 279-284 [PMID: 20021569 DOI: 10.1111/j.1529-8027.2009.00239.x]
68 Hoeijmakers JG, Faber CG, Merkies IS, Waxman SG. Channe-lopathies, painful neuropathy, and diabetes: which way does the causal arrow point? Trends Mol Med 2014; 20: 544-550 [PMID: 25008557 DOI: 10.1016/j.molmed.2014.06.003]
69 Han C, Hoeijmakers JG, Ahn HS, Zhao P, Shah P, Lauria G, Gerrits MM, te Morsche RH, Dib-Hajj SD, Drenth JP, Faber CG, Merkies IS, Waxman SG. Nav1.7-related small fiber neuropathy: impaired slow-inactivation and DRG neuron hyperexcitability. Neurology 2012; 78: 1635-1643 [PMID: 22539570 DOI: 10.1212/WNL.0b013e3182574f12]
70 Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C, Sauer SK, Eberhardt M, Schnölzer M, Lasitschka F, Neuhuber WL, Kichko TI, Konrade I, Elvert R, Mier W, Pirags V, Lukic IK, Morcos M, Dehmer T, Rabbani N, Thornalley PJ, Edelstein D, Nau C, Forbes J, Humpert PM, Schwaninger M, Ziegler D, Stern DM, Cooper ME, Haberkorn U, Brownlee M, Reeh PW, Nawroth PP. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med 2012; 18: 926-933 [PMID: 22581285 DOI: 10.1038/nm.2750]
71 Wood H. Diabetes: Methylglyoxal mediates hyperalgesia in patients with painful diabetic neuropathy. Nat Rev Neurol 2012; 8: 354 [PMID: 22688780 DOI: 10.1038/nrneurol.2012.103]
72 Han Y, Randell E, Vasdev S, Gill V, Gadag V, Newhook LA, Grant M, Hagerty D. Plasma methylglyoxal and glyoxal are elevated and related to early membrane alteration in young, complication-free patients with Type 1 diabetes. Mol Cell Biochem 2007; 305: 123-131 [PMID: 17594057 DOI: 10.1007/s11010-007-9535-1]
73 Andersson DA, Gentry C, Light E, Vastani N, Vallortigara J, Bierhaus A, Fleming T, Bevan S. Methylglyoxal evokes pain by stimulating TRPA1. PLoS One 2013; 8: e77986 [PMID: 24167592 DOI: 10.1371/journal.pone.0077986]
74 Thornalley PJ. Glycation in diabetic neuropathy: characteristics, consequences, causes, and therapeutic options. Int Rev Neurobiol
2002; 50: 37-57 [PMID: 12198817 DOI: 10.1016/S0074-7742(02)50072-6]
75 Hall KE, Liu J, Sima AA, Wiley JW. Impaired inhibitory G-protein function contributes to increased calcium currents in rats with diabetic neuropathy. J Neurophysiol 2001; 86: 760-770 [PMID: 11495948]
76 White DM, Zimmermann M. The bradykinin-induced release of substance P from nerve fibre endings in the rat saphenous nerve neuroma is not related to electrophysiological excitation. Neurosci Lett 1988; 92: 108-113 [PMID: 2460804 DOI: 10.1016/0304-3940(88)90751-3]
77 Hall KE, Sima AA, Wiley JW. Opiate-mediated inhibition of calcium signaling is decreased in dorsal root ganglion neurons from the diabetic BB/W rat. J Clin Invest 1996; 97: 1165-1172 [PMID: 8636427 DOI: 10.1172/JCI118530]
78 Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, Bayliss DA, Jevtovic-Todorovic V, Todorovic SM. Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci 2007; 27: 3305-3316 [PMID: 17376991 DOI: 10.1523/JNEUROSCI.4866-06.2007]
79 Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ, Orestes P, Latham JR, Todorovic SM, Jevtovic-Todorovic V. In vivo silencing of the Ca(V)3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain 2009; 145: 184-195 [PMID: 19577366 DOI: 10.1016/j.pain.2009.06.012]
80 Mendis S, Kumarasunderam R. The effect of daily consumption of coconut fat and soya-bean fat on plasma lipids and lipoproteins of young normolipidaemic men. Br J Nutr 1990; 63: 547-552 [PMID: 2383532 DOI: 10.2337/db13-0813]
81 Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol 2005; 569: 41-57 [PMID: 16141270 DOI: 10.1113/jphysiol.2005.095042]
82 Everill B, Rizzo MA, Kocsis JD. Morphologically identified cutaneous afferent DRG neurons express three different potassium currents in varying proportions. J Neurophysiol 1998; 79: 1814-1824 [PMID: 9535950]
83 Vydyanathan A, Wu ZZ, Chen SR, Pan HL. A-type voltage-gated K+ currents influence firing properties of isolectin B4-positive but not isolectin B4-negative primary sensory neurons. J Neurophysiol 2005; 93: 3401-3409 [PMID: 15647393 DOI: 10.1152/jn.01267.2004]
84 Cao XH, Byun HS, Chen SR, Cai YQ, Pan HL. Reduction in voltage-gated K+ channel activity in primary sensory neurons in painful diabetic neuropathy: role of brain-derived neurotrophic factor. J Neurochem 2010; 114: 1460-1475 [PMID: 20557422 DOI: 10.1111/j.1471-4159.2010.06863.x]
85 Mika J, Zychowska M, Popiolek-Barczyk K, Rojewska E, Przewlocka B. Importance of glial activation in neuropathic pain. Eur J Pharmacol 2013; 716: 106-119 [PMID: 23500198 DOI: 10.1016/j.ejphar.2013.01.072]
86 Kettenmann H, Verkhratsky A. Neuroglia: the 150 years after. Trends Neurosci 2008; 31: 653-659 [PMID: 18945498 DOI: 10.1016/j.tins.2008.09.003]
87 Tsuda M, Ueno H, Kataoka A, Tozaki-Saitoh H, Inoue K. Activation of dorsal horn microglia contributes to diabetes-induced tactile allodynia via extracellular signal-regulated protein kinase signaling. Glia 2008; 56: 378-386 [PMID: 18186080 DOI: 10.1002/glia.20623]
88 Wodarski R, Clark AK, Grist J, Marchand F, Malcangio M. Gabapentin reverses microglial activation in the spinal cord of streptozotocin-induced diabetic rats. Eur J Pain 2009; 13: 807-811 [PMID: 18977160 DOI: 10.1016/j.ejpain.2008.09.010]
89 Toth CC, Jedrzejewski NM, Ellis CL, Frey WH. Cannabinoid-mediated modulation of neuropathic pain and microglial accumulation in a model of murine type I diabetic peripheral neuropathic pain. Mol Pain 2010; 6: 16 [PMID: 20236533 DOI: 10.1186/1744-8069-6-16]
441 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

90 Suzuki N, Hasegawa-Moriyama M, Takahashi Y, Kamikubo Y, Sakurai T, Inada E. Lidocaine attenuates the development of diabetic-induced tactile allodynia by inhibiting microglial activation. Anesth Analg 2011; 113: 941-946 [PMID: 21788310 DOI: 10.1213/ANE.0b013e31822827a2]
91 Crown ED. The role of mitogen activated protein kinase signaling in microglia and neurons in the initiation and maintenance of chronic pain. Exp Neurol 2012; 234: 330-339 [PMID: 22062045 DOI: 10.1016/j.expneurol.2011.10.019]
92 Afsari ZH, Renno WM, Abd-El-Basset E. Alteration of glial fibrillary acidic proteins immunoreactivity in astrocytes of the spinal cord diabetic rats. Anat Rec (Hoboken) 2008; 291: 390-399 [PMID: 18360886 DOI: 10.1002/ar.20678]
93 Chen SR, Pan HL. Hypersensitivity of spinothalamic tract neurons associated with diabetic neuropathic pain in rats. J Neurophysiol 2002; 87: 2726-2733 [PMID: 12037174]
94 Maier C, Baron R, Tölle TR, Binder A, Birbaumer N, Birklein F, Gierthmühlen J, Flor H, Geber C, Huge V, Krumova EK, Landwehrmeyer GB, Magerl W, Maihöfner C, Richter H, Rolke R, Scherens A, Schwarz A, Sommer C, Tronnier V, Uçeyler N, Valet M, Wasner G, Treede RD. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): somatosensory abnormalities in 1236 patients with different neuropathic pain syndromes. Pain 2010; 150: 439-450 [PMID: 20627413 DOI: 10.1016/j.pain.2010.05.002]
95 Aslam A, Singh J, Rajbhandari S. Pathogenesis of painful diabetic neuropathy. Pain Res Treat 2014; 2014: 412041 [PMID: 24891949 DOI: 10.1155/2014/412041]
96 Li JQ, Chen SR, Chen H, Cai YQ, Pan HL. Regulation of increased glutamatergic input to spinal dorsal horn neurons by mGluR5 in diabetic neuropathic pain. J Neurochem 2010; 112: 162-172 [PMID: 19840219 DOI: 10.1111/j.1471-4159.2009.06437.x]
97 Wang XL, Zhang HM, Chen SR, Pan HL. Altered synaptic input and GABAB receptor function in spinal superficial dorsal horn neurons in rats with diabetic neuropathy. J Physiol 2007; 579: 849-861 [PMID: 17218355 DOI: 10.1113/jphysiol.2006.126102]
98 Bai HP, Liu P, Wu YM, Guo WY, Guo YX, Wang XL. Activation of spinal GABAB receptors normalizes N-methyl-D-aspartate receptor in diabetic neuropathy. J Neurol Sci 2014; 341: 68-72 [PMID: 24787504 DOI: 10.1016/j.jns.2014.04.002]
99 Kim JH, Roberts DS, Hu Y, Lau GC, Brooks-Kayal AR, Farb DH, Russek SJ. Brain-derived neurotrophic factor uses CREB and Egr3 to regulate NMDA receptor levels in cortical neurons. J Neurochem 2012; 120: 210-219 [PMID: 22035109 DOI: 10.1111/j.1471-4159.2011.07555.x]
100 Sanchez AP, Sharma K. Transcription factors in the pathogenesis of diabetic nephropathy. Expert Rev Mol Med 2009; 11: e13 [PMID: 19397838 DOI: 10.1017/S1462399409001057]
101 Pérez-Garci E, Gassmann M, Bettler B, Larkum ME. The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron 2006; 50: 603-616 [PMID: 16701210 DOI: 10.1016/j.neuron.2006.04.019]
102 Sodickson DL, Bean BP. GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J Neurosci 1996; 16: 6374-6385 [PMID: 8815916]
103 Silva M, Amorim D, Almeida A, Tavares I, Pinto-Ribeiro F, Morgado C. Pronociceptive changes in the activity of rostroventromedial medulla (RVM) pain modulatory cells in the streptozotocin-diabetic rat. Brain Res Bull 2013; 96: 39-44 [PMID: 23644033 DOI: 10.1016/j.brainresbull.2013.04.008]
104 Chen X, Levine JD. Hyper-responsivity in a subset of C-fiber nociceptors in a model of painful diabetic neuropathy in the rat. Neuroscience 2001; 102: 185-192 [PMID: 11226682 DOI: 10.1016/S0306-4522(00)00454-1]
105 Naderi A, Asgari AR, Zahed R, Ghanbari A, Samandari R, Jorjani M. Estradiol attenuates spinal cord injury-related central pain by decreasing glutamate levels in thalamic VPL nucleus in male rats. Metab Brain Dis 2014; 29: 763-770 [PMID: 24879046 DOI: 10.1007/s11011-014-9570-z]
106 Paulson PE, Wiley JW, Morrow TJ. Concurrent activation of
the somatosensory forebrain and deactivation of periaqueductal gray associated with diabetes-induced neuropathic pain. Exp Neurol 2007; 208: 305-313 [PMID: 17936273 DOI: 10.1016/j.expneurol.2007.09.001]
107 Tseng MT, Chiang MC, Chao CC, Tseng WY, Hsieh ST. fMRI evidence of degeneration-induced neuropathic pain in diabetes: enhanced limbic and striatal activations. Hum Brain Mapp 2013; 34: 2733-2746 [PMID: 22522975 DOI: 10.1002/hbm.22105]
108 Sorensen L, Siddall PJ, Trenell MI, Yue DK. Differences in metabolites in pain-processing brain regions in patients with diabetes and painful neuropathy. Diabetes Care 2008; 31: 980-981 [PMID: 18299445 DOI: 10.2337/dc07-2088]
109 Auer DP, Wilke M, Grabner A, Heidenreich JO, Bronisch T, Wetter TC. Reduced NAA in the thalamus and altered membrane and glial metabolism in schizophrenic patients detected by 1H-MRS and tissue segmentation. Schizophr Res 2001; 52: 87-99 [PMID: 11595395 DOI: 10.1016/S0920-9964(01)00155-4]
110 Selvarajah D, Wilkinson ID, Gandhi R, Griffiths PD, Tesfaye S. Microvascular perfusion abnormalities of the Thalamus in painful but not painless diabetic polyneuropathy: a clue to the pathogenesis of pain in type 1 diabetes. Diabetes Care 2011; 34: 718-720 [PMID: 21282344 DOI: 10.2337/dc10-1550]
111 Li W, Wang P, Li H. Upregulation of glutamatergic transmission in anterior cingulate cortex in the diabetic rats with neuropathic pain. Neurosci Lett 2014; 568: 29-34 [PMID: 24686190 DOI: 10.1016/j.neulet.2014.03.038]
112 Millan MJ. Descending control of pain. Prog Neurobiol 2002; 66: 355-474 [PMID: 12034378 DOI: 10.1016/S0301-0082(02)00009-6]
113 Morgan MM, Fields HL. Pronounced changes in the activity of nociceptive modulatory neurons in the rostral ventromedial medulla in response to prolonged thermal noxious stimuli. J Neurophysiol 1994; 72: 1161-1170 [PMID: 7807201]
114 Carlson JD, Maire JJ, Martenson ME, Heinricher MM. Sen-sitization of pain-modulating neurons in the rostral ventromedial medulla after peripheral nerve injury. J Neurosci 2007; 27: 13222-13231 [PMID: 18045916 DOI: 10.1523/JNEUROSCI.3715-07.2007]
115 Sugino S, Namiki A, Yamakage M. Genetic differences in response properties of rostral ventromedial medulla neurons to the μ-opioid receptor agonist DAMGO in mouse inbred strains. Neurosci Lett 2012; 517: 107-112 [PMID: 22546603 DOI: 10.1016/j.neulet.2012.04.038]
116 Bitar MS, Bajic KT, Farook T, Thomas MI, Pilcher CW. Spinal cord noradrenergic dynamics in diabetic and hypercortisolaemic states. Brain Res 1999; 830: 1-9 [PMID: 10350553 DOI: 10.1016/S0006-8993(99)01284-6]
117 Morgado C, Silva L, Pereira-Terra P, Tavares I. Changes in serotoninergic and noradrenergic descending pain pathways during painful diabetic neuropathy: the preventive action of IGF1. Neurobiol Dis 2011; 43: 275-284 [PMID: 21515376 DOI: 10.1016/j.nbd.2011.04.001]
118 Ezzeldin E, Souror WA, El-Nahhas T, Soudi AN, Shahat AA. Biochemical and neurotransmitters changes associated with tramadol in streptozotocin-induced diabetes in rats. Biomed Res Int 2014; 2014: 238780 [PMID: 24971322 DOI: 10.1155/2014/238780]
119 Suh HW, Song DK, Wie MB, Jung JS, Hong HE, Choi SR, Kim YH. The reduction of antinociceptive effect of morphine administered intraventricularly is correlated with the decrease of serotonin release from the spinal cord in streptozotocin-induced diabetic rats. Gen Pharmacol 1996; 27: 445-450 [PMID: 8723523 DOI: 10.1016/0306-3623(95)02059-4]
120 Petrou M, Pop-Busui R, Foerster BR, Edden RA, Callaghan BC, Harte SE, Harris RE, Clauw DJ, Feldman EL. Altered excitation-inhibition balance in the brain of patients with diabetic neuropathy. Acad Radiol 2012; 19: 607-612 [PMID: 22463961 DOI: 10.1016/j.acra.2012.02.004]
121 Boyle J, Eriksson ME, Gribble L, Gouni R, Johnsen S, Coppini DV, Kerr D. Randomized, placebo-controlled comparison of amitriptyline, duloxetine, and pregabalin in patients with chronic diabetic peripheral neuropathic pain: impact on pain,
442 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

polysomnographic sleep, daytime functioning, and quality of life. Diabetes Care 2012; 35: 2451-2458 [PMID: 22991449 DOI: 10.2337/dc12-0656]
122 Singleton JR, Smith AG. The diabetic neuropathies: practical and rational therapy. Semin Neurol 2012; 32: 196-203 [PMID: 23117944 DOI: 10.1055/s-0032-1329195]
123 Huizinga MM, Peltier A. Painful Diabetic Neuropathy: A Management-Centered Review. Clinical Diabetes 2007; 25: 6-15 [DOI: 10.2337/diaclin.25.1.6]
124 Tanenberg RJ. Diabetic Peripheral Neuropathy: Painful or Painless. Hospital Physician 2009; 45: 1-8
125 Moore RA, Wiffen PJ, Derry S, Toelle T, Rice AS. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev 2014; 4: CD007938 [PMID: 24771480 DOI: 10.1002/14651858.CD007938]
126 Snedecor SJ, Sudharshan L, Cappelleri JC, Sadosky A, Mehta S, Botteman M. Systematic review and meta-analysis of pharmacological therapies for painful diabetic peripheral neuropathy. Pain Pract 2014; 14: 167-184 [PMID: 23534696 DOI: 10.1111/papr.12054]
127 Hurley RW, Lesley MR, Adams MC, Brummett CM, Wu CL. Pregabalin as a treatment for painful diabetic peripheral neuropathy: a meta-analysis. Reg Anesth Pain Med 2008; 33: 389-394 [PMID: 18774507 DOI: 10.1016/j.rapm.2008.02.012]
128 Peltier A, Goutman SA, Callaghan BC. Painful diabetic neuropathy. BMJ 2014; 348: g1799 [PMID: 24803311 DOI: 10.1136/bmj.g1799]
129 Verma V, Singh N, Singh Jaggi A. Pregabalin in neuropathic pain: evidences and possible mechanisms. Curr Neuropharmacol 2014; 12: 44-56 [PMID: 24533015 DOI: 10.2174/1570159X1201140117162802]
130 Blommel ML, Blommel AL. Pregabalin: an antiepileptic agent useful for neuropathic pain. Am J Health Syst Pharm 2007; 64: 1475-1482 [PMID: 17617497 DOI: 10.2146/ajhp060371]
131 Guy S, Mehta S, Leff L, Teasell R, Loh E. Anticonvulsant medication use for the management of pain following spinal cord injury: systematic review and effectiveness analysis. Spinal Cord 2014; 52: 89-96 [PMID: 24296804 DOI: 10.1038/sc.2013.146]
132 Zilliox L, Russell JW. Treatment of diabetic sensory poly-neuropathy. Curr Treat Options Neurol 2011; 13: 143-159 [PMID: 21274758 DOI: 10.1007/s11940-011-0113-1]
133 Stahl SM, Porreca F, Taylor CP, Cheung R, Thorpe AJ, Clair A. The diverse therapeutic actions of pregabalin: is a single mechanism responsible for several pharmacological activities? Trends Pharmacol Sci 2013; 34: 332-339 [PMID: 23642658 DOI: 10.1016/j.tips.2013.04.001]
134 Zychowska M, Rojewska E, Przewlocka B, Mika J. Mechanisms and pharmacology of diabetic neuropathy - experimental and clinical studies. Pharmacol Rep 2013; 65: 1601-1610 [PMID: 24553008]
135 Patel N, Mishra V, Patel P, Dikshit RK. A study of the use of carbamazepine, pregabalin and alpha lipoic acid in patients of diabetic neuropathy. J Diabetes Metab Disord 2014; 13: 62 [PMID: 24926454 DOI: 10.1186/2251-6581-13-62]
136 Ziegler D, Schneider E, Boess FG, Berggren L, Birklein F. Impact of comorbidities on pharmacotherapy of painful diabetic neuropathy in clinical practice. J Diabetes Complications 2014; 28: 698-704 [PMID: 24862108 DOI: 10.1016/j.jdiacomp.2014.04.004]
137 Moore A, Wiffen P, Kalso E. Antiepileptic drugs for neuropathic pain and fibromyalgia. JAMA 2014; 312: 182-183 [PMID: 25005656 DOI: 10.1001/jama.2014.6336]
138 Gilron I, Flatters SJL. Gabapentin and pregabalin for the treatment or neuropathic pain: A review of laboratory and clinical evidence. Pain Res Manage 2006; 11 Suppl A: S16A-29A
139 Freeman R, Durso-Decruz E, Emir B. Efficacy, safety, and tolerability of pregabalin treatment for painful diabetic peripheral neuropathy: findings from seven randomized, controlled trials across a range of doses. Diabetes Care 2008; 31: 1448-1454 [PMID: 18356405 DOI: 10.2337/dc07-2105]
140 Attal N, Cruccu G, Baron R, Haanpää M, Hansson P, Jensen TS,
Nurmikko T; European Federation of Neurological Societies. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol 2010; 17: 1113-1e88 [PMID: 20402746 DOI: 10.1111/j.1468-1331.2010.02999.x]
141 Pergolizzi JV, Raffa RB, Taylor R, Rodriguez G, Nalamachu S, Langley P. A review of duloxetine 60 mg once-daily dosing for the management of diabetic peripheral neuropathic pain, fibromyalgia, and chronic musculoskeletal pain due to chronic osteoarthritis pain and low back pain. Pain Pract 2013; 13: 239-252 [PMID: 22716295 DOI: 10.1111/j.1533-2500.2012.00578.x]
142 Quilici S, Chancellor J, Löthgren M, Simon D, Said G, Le TK, Garcia-Cebrian A, Monz B. Meta-analysis of duloxetine vs. pregabalin and gabapentin in the treatment of diabetic peripheral neuropathic pain. BMC Neurol 2009; 9: 6 [PMID: 19208243 DOI: 10.1186/1471-2377-9-6]
143 Tanenberg RJ, Irving GA, Risser RC, Ahl J, Robinson MJ, Skljarevski V, Malcolm SK. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open-label, randomized, noninferiority comparison. Mayo Clin Proc 2011; 86: 615-626 [PMID: 21719618 DOI: 10.4065/mcp.2010.0681]
144 Tesfaye S, Wilhelm S, Lledo A, Schacht A, Tölle T, Bouhassira D, Cruccu G, Skljarevski V, Freynhagen R. Duloxetine and pregabalin: high-dose monotherapy or their combination? The “COMBO-DN study”--a multinational, randomized, double-blind, parallel-group study in patients with diabetic peripheral neuropathic pain. Pain 2013; 154: 2616-2625 [PMID: 23732189 DOI: 10.1016/j.pain.2013.05.043]
145 Skljarevski V, Desaiah D, Zhang Q, Chappell AS, Detke MJ, Gross JL, Ziegler D. Evaluating the maintenance of effect of duloxetine in patients with diabetic peripheral neuropathic pain. Diabetes Metab Res Rev 2009; 25: 623-631 [PMID: 19637208 DOI: 10.1002/dmrr.1000]
146 Kalso E, Aldington DJ, Moore RA. Drugs for neuropathic pain. BMJ 2013; 347: f7339 [PMID: 24355386 DOI: 10.1136/bmj.f7339]
147 Mercier A, Auger-Aubin I, Lebeau JP, Schuers M, Boulet P, Hermil JL, Van Royen P, Peremans L. Evidence of prescription of antidepressants for non-psychiatric conditions in primary care: an analysis of guidelines and systematic reviews. BMC Fam Pract 2013; 14: 55 [PMID: 23641784 DOI: 10.1186/1471-2296-14-55]
148 Cegielska-Perun K, Bujalska-Zadrożny M, Tatarkiewicz J, Gąsińska E, Makulska-Nowak HE. Venlafaxine and neuropathic pain. Pharmacology 2013; 91: 69-76 [PMID: 23183148 DOI: 10.1159/000345035]
149 Rowbotham MC, Goli V, Kunz NR, Lei D. Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo-controlled study. Pain 2004; 110: 697-706 [PMID: 15288411 DOI: 10.1016/j.pain.2004.05.010]
150 Deli G, Bosnyak E, Pusch G, Komoly S, Feher G. Diabetic neuropathies: diagnosis and management. Neuroendocrinology 2013; 98: 267-280 [PMID: 24458095 DOI: 10.1159/000358728]
151 Page N, Deluca J, Crowell K. Clinical inquiry: what medications are best for diabetic neuropathic pain? J Fam Pract 2012; 61: 691-693 [PMID: 23256101]
152 Chou R, Carson S, Chan BK. Gabapentin versus tricyclic antidepressants for diabetic neuropathy and post-herpetic neuralgia: discrepancies between direct and indirect meta-analyses of randomized controlled trials. J Gen Intern Med 2009; 24: 178-188 [PMID: 19089502 DOI: 10.1007/s11606-008-0877-5]
153 Kaur H, Hota D, Bhansali A, Dutta P, Bansal D, Chakrabarti A. A comparative evaluation of amitriptyline and duloxetine in painful diabetic neuropathy: a randomized, double-blind, cross-over clinical trial. Diabetes Care 2011; 34: 818-822 [PMID: 21355098 DOI: 10.2337/dc10-1793]
154 Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet 2009; 374: 1252-1261 [PMID: 19796802 DOI:
443 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

10.1016/S0140-6736(09)61081-3]155 Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a
Cochrane review. J Neurol Neurosurg Psychiatry 2010; 81: 1372-1373 [PMID: 20543189 DOI: 10.1136/jnnp.2008.144964]
156 Harati Y, Gooch C, Swenson M, Edelman S, Greene D, Raskin P, Donofrio P, Cornblath D, Sachdeo R, Siu CO, Kamin M. Double-blind randomized trial of tramadol for the treatment of the pain of diabetic neuropathy. Neurology 1998; 50: 1842-1846 [PMID: 9633738]
157 Gilron I, Bailey JM, Tu D, Holden RR, Weaver DF, Houlden RL. Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med 2005; 352: 1324-1334 [PMID: 15800228 DOI: 10.1056/NEJMoa042580]
158 Watson CP, Moulin D, Watt-Watson J, Gordon A, Eisenhoffer J. Controlled-release oxycodone relieves neuropathic pain: a randomized controlled trial in painful diabetic neuropathy. Pain 2003; 105: 71-78 [PMID: 14499422 DOI: 10.1016/S0304-3959(03)00160-x]
159 Gimbel JS, Richards P, Portenoy RK. Controlled-release oxycodone for pain in diabetic neuropathy: a randomized controlled trial. Neurology 2003; 60: 927-934 [PMID: 12654955]
160 Hanna M, O’Brien C, Wilson MC. Prolonged-release oxycodone enhances the effects of existing gabapentin therapy in painful diabetic neuropathy patients. Eur J Pain 2008; 12: 804-813 [PMID: 18262450 DOI: 10.1016/j.ejpain.2007.12.010]
161 Zin CS, Nissen LM, O’Callaghan JP, Duffull SB, Smith MT, Moore BJ. A randomized, controlled trial of oxycodone versus placebo in patients with postherpetic neuralgia and painful diabetic neuropathy treated with pregabalin. J Pain 2010; 11: 462-471 [PMID: 19962354 DOI: 10.1016/j.jpain.2009.09.003]
162 Christoph T, De Vry J, Tzschentke TM. Tapentadol, but not morphine, selectively inhibits disease-related thermal hyperalgesia in a mouse model of diabetic neuropathic pain. Neurosci Lett 2010; 470: 91-94 [PMID: 20026182 DOI: 10.1016/j.neulet.2009.12.020]
163 Nones CF, Reis RC, Jesus CH, Veronez DA, Cunha JM, Chichorro JG. Orofacial sensory changes after streptozotocin-induced diabetes in rats. Brain Res 2013; 1501: 56-67 [PMID: 23313875 DOI: 10.1016/j.brainres.2013.01.002]
164 Canta A, Chiorazzi A, Meregalli C, Carozzi V, Oggioni N, Lauria G, Lombardi R, Bianchi R, Porretta-Serapiglia C, Cavaletti G. Continuous buprenorphine delivery effect in streptozotocine-induced painful diabetic neuropathy in rats. J Pain 2009; 10: 961-968 [PMID: 19595641 DOI: 10.1016/j.jpain.2009.04.003]
165 Cegielska-Perun K, Bujalska-Zadrożny M, Gąsińska E, Makulska-Nowak HE. Enhancement of antinociceptive effect of morphine by antidepressants in diabetic neuropathic pain model. Pharmacol Rep 2014; 66: 228-234 [PMID: 24911074 DOI: 10.1016/j.pharep.2013.09.003]
166 Linz K, Christoph T, Tzschentke TM, Koch T, Schiene K, Gautrois M, Schröder W, Kögel BY, Beier H, Englberger W, Schunk S, De Vry J, Jahnel U, Frosch S. Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J Pharmacol Exp Ther 2014; 349: 535-548 [PMID: 24713140 DOI: 10.1124/jpet.114.213694]
167 Christoph T, Schröder W, Tallarida RJ, De Vry J, Tzschentke
TM. Spinal-supraspinal and intrinsic μ-opioid receptor agonist-norepinephrine reuptake inhibitor (MOR-NRI) synergy of tapentadol in diabetic heat hyperalgesia in mice. J Pharmacol Exp Ther 2013; 347: 794-801 [PMID: 24051022 DOI: 10.1124/jpet.113.207704]
168 Afilalo M, Morlion B. Efficacy of tapentadol ER for managing moderate to severe chronic pain. Pain Physician 2013; 16: 27-40 [PMID: 23340531]
169 Taylor R, Pergolizzi JV, Raffa RB. Tapentadol extended release for chronic pain patients. Adv Ther 2013; 30: 14-27 [PMID: 23328938 DOI: 10.1007/s12325-013-0002-y]
170 Schwartz S, Etropolski M, Shapiro DY, Okamoto A, Lange R, Haeussler J, Rauschkolb C. Safety and efficacy of tapentadol ER in patients with painful diabetic peripheral neuropathy: results of a randomized-withdrawal, placebo-controlled trial. Curr Med Res Opin 2011; 27: 151-162 [PMID: 21162697 DOI: 10.1185/03007995.2010.537589]
171 Vinik AI, Shapiro DY, Rauschkolb C, Lange B, Karcher K, Pennett D, Etropolski MS. A randomized withdrawal, placebo-controlled study evaluating the efficacy and tolerability of tapentadol extended release in patients with chronic painful diabetic peripheral neuropathy. Diabetes Care 2014; 37: 2302-2309 [PMID: 24848284 DOI: 10.2337/dc13-2291]
172 Wolff RF, Bala MM, Westwood M, Kessels AG, Kleijnen J. 5% lidocaine medicated plaster in painful diabetic peripheral neuropathy (DPN): a systematic review. Swiss Med Wkly 2010; 140: 297-306 [PMID: 20458651]
173 Kulkantrakorn K, Lorsuwansiri C, Meesawatsom P. 0.025% capsaicin gel for the treatment of painful diabetic neuropathy: a randomized, double-blind, crossover, placebo-controlled trial. Pain Pract 2013; 13: 497-503 [PMID: 23228119 DOI: 10.1111/papr.12013]
174 Vinik AI, Nevoret ML, Casellini C, Parson H. Diabetic neuropathy. Endocrinol Metab Clin North Am 2013; 42: 747-787 [PMID: 24286949 DOI: 10.1016/j.ecl.2013.06.001]
175 Groninger H, Schisler RE. Topical capsaicin for neuropathic pain #255. J Palliat Med 2012; 15: 946-947 [PMID: 22849599 DOI: 10.1089/jpm.2012.9571]
176 Casale R, Mattia C. Building a diagnostic algorithm on localized neuropathic pain (LNP) and targeted topical treatment: focus on 5% lidocaine-medicated plaster. Ther Clin Risk Manag 2014; 10: 259-268 [PMID: 24790451 DOI: 10.2147/TCRM.S58844]
177 Sánchez-Vázquez R, Briseño-Rodríguez G, Cardona-Muñoz EG, Gálvez-Gastélum FJ, Totsuka-Sutto SE, Garcia-Benavides L. Isosorbide dinitrate spray as therapeutic strategy for treatment of chronic venous ulcers. Angiology 2008; 59: 64-71 [PMID: 18319224 DOI: 10.1177/0003319707303700]
178 Yuen KC, Baker NR, Rayman G. Treatment of chronic painful diabetic neuropathy with isosorbide dinitrate spray: a double-blind placebo-controlled cross-over study. Diabetes Care 2002; 25: 1699-1703 [PMID: 12351464 DOI: 10.2337/diacare.25.10.1699]
179 Rayman G, Baker NR, Krishnan ST. Glyceryl trinitrate patches as an alternative to isosorbide dinitrate spray in the treatment of chronic painful diabetic neuropathy. Diabetes Care 2003; 26: 2697-2698 [PMID: 12941745 DOI: 10.2337/diacare.26.9.2697-a]
P- Reviewer: Georgescu A, Li CJ, Panchu P S- Editor: Tian YL L- Editor: A E- Editor: Liu SQ
444 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Schreiber AK et al . Diabetic neuropathic pain

family history for diabetes mellitus, polycystic kidney disease, and previously diagnosed glucose intolerance. Modifiable risk factors for NODAT include obesity and the metabolic syndrome, hepatitis C virus and cytomegalovirus infection, corticosteroids, calcineurin inhibitor drugs (especially tacrolimus), and sirolimus. NODAT affects graft and patient survival, and increases the incidence of post-transplant cardiovascular disease.The incidence and impact of NODAT can be minimized through pre- and post-transplant screening to identify patients at higher risk, including by oral glucose tole-rance tests, as well as multi-disciplinary care, lifestyle modification, and the use of modified immunosuppressive regimens coupled with glucose-lowering therapies including oral hypoglycemic agents and insulin. Since NODAT is a major cause of post-transplant morbidity and mortality, measures to reduce its incidence and impact have the potential to greatly improve overall transplant success.
Key words: Cyclosporine; Graft; Kidney; New-onset diabetes; Tacrolimus; Transplantation
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: New-onset diabetes after kidney trans-plantation (NODAT) is detrimental to patient and graft survival. Early diagnosis through the identification of modifiable and non-modifiable risk factors for NODAT and appropriate screening, accompanied by good glycemic control that involves a multidisciplinary care approach will help result in good short- and long-term kidney transplant outcomes.
Palepu S, Prasad GVR. New-onset diabetes mellitus after kidney transplantation: Current status and future directions. World J Diabetes 2015; 6(3): 445-455 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/445.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.445
Sneha Palepu, G V Ramesh Prasad
Sneha Palepu, G V Ramesh Prasad, Division of Nephrology, University of Toronto, St. Michael’s Hospital, Toronto ON M5C 2T2, CanadaAuthor contributions: Palepu S and Prasad GVR contributed to this paper. Conflict-of-interest: Both authors (Sneha Palepu and G V Ramesh Prasad) have no conflict of interest to declare.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: G V Ramesh Prasad, MBBS, MSc, MA, FRCPC, FACP, FASN, Associate Professor of Medicine, Division of Nephrology, University of Toronto, St. Michael’s Hospital, 61 Queen Street East, 9th Floor, Toronto ON M5C 2T2, Canada. [email protected]: +1-416-8673722 Fax: +1-416-8673709 Received: August 25, 2014Peer-review started: August 25, 2014First decision: October 14, 2014Revised: November 14, 2014Accepted: January 9, 2015Article in press: January 9, 2015Published online: April 15, 2015
AbstractA diagnosis of new-onset diabetes after transplantation (NODAT) carries with it a threat to the renal allograft, as well as the same short- and long-term implications of type 2 diabetes seen in the general population. NODAT usually occurs early after transplantation, and is usually diagnosed according to general population guidelines. Non-modifiable risk factors for NODAT include advancing age, African American, Hispanic, or South Asian ethnicity, genetic background, a positive
REVIEW
445 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
New-onset diabetes mellitus after kidney transplantation: Current status and future directions
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.445
World J Diabetes 2015 April 15; 6(3): 445-455ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

INTRODUCTIONKidney transplantation is widely considered to be the therapy of choice for patients with end-stage renal disease (ESRD) because of its ability to provide the maximum amount of replacement of renal function and a consequent improvement in patient morbidity and mortality. Among the various complications experienced by kidney transplant recipients (KTR), new-onset diabetes after transplantation (NODAT) is a well-known condition by which an organ transplant recipient such as a KTR develops diabetes mellitus (DM) at some point after the transplant procedure. The purpose of this review is to discuss the epidemiology and management of this common and important post-transplant disorder. A special emphasis will be placed on studies performed during the past ten years, with the intent of providing management recommendations for both diabetes and transplant professionals.
A diagnosis of NODAT carries with it a threat to the renal allograft, as well as the same short- and long-term implications of type 2 DM, but these occur at an accelerated pace[1]. DM can both cause and expedite the course of cardiovascular disease (CVD) as well as the failure of multiple organ systems[2]. According to the World Health Organization (WHO), the number of patients worldwide with DM has doubled from 170 million to 340 million over the past ten years. In addition, the WHO predicts that DM will be the seventh leading cause of death by 2030. This increase in incidence of DM has extended across many population groups, including perhaps transplant recipients.
Over the past decade, concomitantly with DM, there has also been a corresponding increase in the number of patients seeking kidney transplants. According to the 2010 United States Renal Data System Annual Data Report, the annual number of kidney transplants in that country increased from 13425 to 17350 over the decade between 1998 and 2008[3]. Thus, there is also a larger KTR population at increased risk for NODAT. Although type 2 DM is an important cause of ESRD and consequently an important contributor to clinical consequences in incident KTR cohorts, we focus here on NODAT, as an entity distinct from pre-existing DM in a patient receiving a kidney transplant.
INCIDENCE AND PREVALENCE OF NODATThe prevalence of NODAT after kidney transplantation has increased[4], and varies between 2% and 53% based on several estimates[5]. One reason for the wide variation in reported prevalence may be due to the challenges faced in making the diagnosis. Until about a decade ago, there was no consensus definition for NODAT. Previously, it was defined by fasting or random blood glucose levels of varying thresholds, or perhaps quite naively, by the need for insulin or oral
hypoglycemic agents in the post-transplant period[6]. This issue was addressed by the development of the 2003 Consensus Guidelines, developed by the American Diabetes Association (ADA) and the WHO[7]. According to these guidelines, NODAT is present if the patient has symptoms of DM, casual plasma glucose ≥ 11.1 mmol/L, or 8-h fasting plasma glucose ≥ 7.0 mmol/L. In 2010, the definition for DM was revised by the ADA to include the 2-h oral glucose tolerance test (OGTT), with a 2-h value of ≥ 11.1 mmol/L being indicative of DM. The general principle behind standardized NODAT incidence reporting is that the diagnostic criteria should reflect what is used in the general population. Hopefully, these developments will allow for more consistent reporting of NODAT in the future, leading in turn to more precise estimates of incidence rates.
It is well established that NODAT usually occurs early after kidney transplantation. One long-term study[8] conducted between 1976 and 2004 in 1580 Egyptian KTR demonstrated a diagnosis of NODAT in 18.2% patients overall, in whom 52.4% were diagnosed by 6 mo and 11.5% between 6 and 12 mo. A French study of 527 KTR indicated a median time-to-onset of 1.6 mo, with an incidence of 7% over 2 years[9]. Incidence estimates in the United States are 9.1% at 3 mo, 16% at 12 mo, and 24% at 36 mo[10]. The incidence may be increasing even in children[11]. Most studies reporting NODAT prevalence do not include OGTT data, as a result of which significant underestimation of incidence may occur. Variation in studies with regards to length of follow-up, intensity of routine screening, and degree of use of standardized definitions will all have an impact on incidence estimates. Furthermore, glycemic control improves in some patients with increasing time post-transplant, as a result of immunosuppressive medication dose reduction and other interventions. This in turn affects prevalence estimates and interpretation of incidence rates. Addition of HbA1c testing may also modify reported incidence rates. However, HbA1c testing has not been routinely employed at most trans-plant centres.
RISK FACTORS FOR NODATThe risk factors specific to NODAT have been well-delineated. These can be broadly classified into non-modifiable and modifiable risk factors. These are summarized in Table 1.
NON-MODIFIABLE RISK FACTORS FOR NODATNon-modifiable risk factors include advancing age, black race, genetic background, a positive family history for DM, and previously diagnosed glucose intolerance[12]. Polycystic kidney disease is also believed to be a risk factor. All of these risk factors can usually be identified
Palepu S et al . New-onset diabetes after kidney transplantation
446 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

before transplantation. Since the ESRD population continues to age, it is
possible that the incidence of NODAT will increase as a result of more elderly patients being transplanted for their ESRD. Age is considered to be the strongest risk factor for NODAT[13]. It was long ago shown that patients above the age of 45 may be at higher risk for NODAT[14]. A later study estimated that risk at about 2.9-fold[15]. The incidence of NODAT may increase by as much as 50% for every 10-year increase in age[16]. Age over 60 has been associated with a risk of 2.6-fold[10]. However, age per se is not a precluding factor to kidney transplantation[17] and the risk for NODAT does not typically dissuade transplant centres from performing transplants in older patients.
Besides African-American or Hispanic descent[12], KTR of South Asian origin may also be at higher risk for NODAT[18]. The relative risk for NODAT has been estimated at 1.68 for blacks and 1.35 for Hispanics compared to Caucasians[10]. Using the 2003 consensus criteria, blacks are considered to be at a two-fold risk for NODAT[19]. The increased risk for NODAT, at least in blacks, is believed to be at least partly due to variation in the pharmacokinetics of various immunosuppressive agents[1]. Variability in dosing requirements based on ethnicity has been demonstrated recently for tacrolimus, with increased amounts of this diabetogenic agent needed in East Asians[20]. It is unclear at this time, however, if the diabetogenic effects of immunosuppressive medication are influenced by ethnicity, although this has been speculated[7].
Genetic predisposition to NODAT includes traditional associations with the alleles human leukocyte antigen A28, A30, B27, and Bw42. A number of genetic associations with NODAT have been identified in the last decade. For example, there has been a reported association with the R325W polymorphism in the SLC30A8 zinc transporter gene of pancreatic islets, with the R/r inheritance being associated with higher risk[21]. Another study from the same group suggested the association of TCF7L2 gene variants with NODAT[22]. This
association with TCF7L2 has also been demonstrated by another group[23]. Other associations of specific gene variants with NODAT include KCNQ1[24], NFATc4[25], adiponectin 276G/T[26], and mitochondrial haplotype H[27]. While all of these associations are interesting, it remains to be seen if such findings can be replicated in different populations, and whether they can be demonstrated to be independent risk factors for NODAT in prospectively conducted studies. Furthermore, the expense and inconvenience of such assessments greatly limits the ability to study these and other gene candidates further.
In the past decade, NODAT has also been associated with autosomal dominant polycystic kidney disease (ADPKD)[28,29], as well as autosomal recessive polycystic kidney disease[30]. ADPKD in particular is a common disease and cause of ESRD, particularly in Caucasians, and so it remains to be definitively proven as a risk factor for NODAT since both ADPKD and NODAT are common in KTR. A plausible mechanism for the association also needs to be developed.
Finally, previously diagnosed glucose intolerance is a risk factor for NODAT[13]. Glucose intolerance may have been diagnosed at the time of pregnancy, or when corticosteroids were used as part of the therapy for the primary renal disease or a related or unrelated systemic disease. In such cases, even though DM may have resolved with cessation of steroid therapy, the risk for DM may persist lifelong. To our knowledge, these risks have not been evaluated prospectively. Also, donor factors for NODAT considered “non-modifiable” include male gender and deceased, as opposed to living donor kidneys. However, these risk factors are unlikely to ever be evaluated prospectively, and so will not be discussed further.
MODIFIABLE RISK FACTORS FOR NODATThere are many more risk factors for NODAT that may be considered modifiable, at least on a theoretical basis. The most significant risk factors are as follows.
Obesity has detrimental effects to transplantation for a variety of reasons. Obesity has been estimated to increase the risk for NODAT, with a relative risk of 1.73[13]. The risk for NODAT increases linearly for every 1 kg above 45 kg[13]. While obesity in the context of transplantation is traditionally understood as body mass index > 30 kg/m2, body fat percentage may also be a useful marker[31]. It remains undetermined, however, whether it is pre-transplant weight that increases the risk for NODAT, or whether it is the weight gain that occurs soon after transplantation that is the cause. At least one study indicates that is the former[32]. Nonetheless, longer-term studies may be needed to demonstrate the association of weight gain with NODAT[13]. Adiponectin is a hormone that is reduced with increasing adiposity. Increased adipose tissue is also associated with inflammation. NODAT has been associated with reduced
447 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 1 Risk factors for new-onset diabetes after transplantation
Non-modifiable Modifiable
Advanced age ObesityAfrican American, Hispanic, or South Asian descent
Sedentary lifestyle
Genetic, e.g., HLA B27 Metabolic syndromeAdult polycystic kidney disease Viral infections, e.g., HCV,
cytomegalovirus Previous glucose intolerance, e.g., during pregnancy, steroid therapy for renal or non-renal disease
Corticosteroids
Male donor Calcineurin-inhibitors (tacrolimus > cyclosporine)
Deceased donor SirolimusAcute rejection
HCV: Hepatitis C virus; HLA: Human leukocyte antigen.
Palepu S et al . New-onset diabetes after kidney transplantation

due to its demonstrated superior efficacy and safety[44], despite the risk for diabetes. However, it is possible to use much lower doses presently[44]. Deficiency of calcineurin leads to decreased insulin production. CNI inhibit the uptake of glucose molecules by cells due to a reduction in the number of glucose transporter type 4 (GLUT-4) receptor molecules on the cell membrane surface of adipocytes[45]. GLUT-4 is an insulin-regulated protein present primarily in adipose tissue and striated muscle, enabling the translocation of glucose into the cell cytoplasm[46]. Thus, reduction in GLUT-4 leads to hyperglycemia. Tacrolimus also reduces glucokinase activity in pancreatic islets, thereby suppressing glucose-induced insulin release[47]. Although both impaired insulin release and increased insulin resistance are both believed to be mechanisms for CNI-induced NODAT, the former may be more important. Islet cell damage in the form of cytoplasmic swelling, vacuolization, and altered insulin staining can be demonstrated as a result of CNI therapy[48]. CNI also cause reduced insulin gene expression[49].
Sirolimus is another immunosuppressive agent used in transplantation that has been contextualized to NODAT. The association of sirolimus with NODAT has been believed to be due to its combination with CNI, and so conversion from a CNI to sirolimus as the main immunosuppression has been perceived as beneficial[50]. However, some studies seem to indicate that sirolimus is a risk factor for NODAT. These include both large database studies[51] and smaller single-centre reviews[52]. The effect may be mediated by hypertriglyceridemia[53]. Sirolimus may also inhibit pancreatic beta cell proliferation. At best, sirolimus is neutral with respect to NODAT risk[13].
The potential pathogenic mechanisms for drug-induced NODAT are summarized in Table 2.
CLINICAL SIGNIFICANCE OF NODATRTR who develop NODAT are most likely to be at the same risk for developing the short- and long-term complications of diabetes as people with type 2 diabetes. However, there have been few prospective, long-term, or interventional studies with adequate statistical power to support this statement. It is clear, however, from smaller studies that NODAT has an adverse effect on important transplant- and patient-related outcomes.
An older study has demonstrated that after 12 years of post-transplant follow-up, graft survival in patients with NODAT was only 48% compared to 70% in those who did not develop NODAT, with NODAT predicting a relative risk of graft loss of 3.72[54]. In the shorter term, graft survival was shown to be reduced by 17% after 3 years and 34% after 4 years in those with DM compared to those without DM[55]. In one prospective study, more than 1400 KTR underwent an OGTT at 10 wk post-transplant and were followed for a median of 6.7 years. Impaired glucose tolerance was found to be associated
adiponectin and increased C-reactive protein levels[33], indicating that these are also possible mechanisms for the association seen.
The metabolic syndrome is often associated with obesity. When a definition for metabolic syndrome such as the National Cholesterol Education Program Adult Treatment Panel Ⅲ is used, which does not contain diabetes as a mandatory component, the presence of metabolic syndrome has been associated with an increased risk for NODAT. In the Patient Outcomes in Renal Transplantation study[34], metabolic syndrome was independently associated with NODAT (HR = 3.46, 95%CI: 2.40-4.98, P < 0.0001).
Hepatitis C virus (HCV) infection has been associated with DM in the general population, particularly over the age of 40[35]. Just like DM, HCV also causes ESRD by causing glomerular disease, and HCV can be acquired through blood contamination in hemodialysis units. As a result, it is not uncommon for HCV-infected patients to be considered for kidney transplantation when there is no evidence for hepatic dysfunction and viral titres are sufficiently low or undetectable. The one-year incidence of NODAT was found to be 25.6% in those who were HCV-positive, compared to 14.4% in those who were negative[10]. Another study found an adjusted Odds ratio for NODAT approaching 4.0 in those with HCV[36]. The risk for NODAT with HCV may be exacerbated by the use of tacrolimus[37]. Cytomegalovirus (CMV) infection has also been considered as a risk factor for NODAT, with the mechanism being impaired insulin release[38]. Unlike HCV however, CMV is much more easily treated in the post-transplant setting and so receives considerably less attention as a risk factor.
Corticosteroids remain a mainstay of post-transplant immunosuppression and are a part of most medication regimens. The risk for NODAT with steroids relates both to the dose used and the duration of therapy[13]. Steroids induce gluconeogenesis and glycolysis, increa-sing both fasting and post-prandial glucose levels. Glycogenesis is decreased. Insulin resistance, to which KTR may be predisposed, is an important effect of steroid therapy. They also impair pancreatic beta-cell function[39]. While a maintenance dose of 5 mg/d of prednisone is typically used for KTR, higher doses (such as 1 mg/kg per day) are used in the early post-transplant phase. Higher doses are also used as bolus therapy when acute rejection of the transplant occurs. There is a dose dependent relationship between steroid dose and glucose levels[40].
Tacrolimus and cyclosporine are widely used calcineurin-inhibitor (CNI) drugs in KTR. Most patientsreceive one or the other drug as part of their immu-nosuppressive drug regimen. Between these two, tacrolimus is considered to be more diabetogenic by about 50%[10,41]. Unlike in the case of steroids, this effect is not believed to be dose-dependent[16,42], although this is controversial[43]. Tacrolimus is being increasingly preferred as the CNI of choice at most transplant centres
448 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Palepu S et al . New-onset diabetes after kidney transplantation

with a 40% greater mortality risk[56]. This increased risk was not seen with impaired fasting glucose (IFG). In a prospective, single-centre study of 201 consecutive KTR in Norway, the 8-year major adverse cardiac event rate was 7% in those without DM, 21% in those with DM before transplantation, and 20% in those with NODAT[57]. Perhaps due to low statistical power, NODAT was not associated with mortality in this study. In another single centre cohort of over 1800 KTR, NODAT was associated with a hazard ratio of 1.80 for mortality[58]. The main cause of mortality is CVD[58].
An important distinction to be made is if NODAT leads to patient mortality independent of transplant graft function. In one study[59], an association was detected between NODAT and death with a functioning graft, but there was no impact on graft survival when censored for death. It is easier to demonstrate associations when NODAT is combined with pre-existing DM[60]. In order to identify the unique contribution of NODAT to mortality, independent of graft function, longer follow-up is required.
NODAT also imposes a significant cost to health care systems. In the United States, NODAT was estimated to cost more than $12000 in the first post-transplant year, and over $19000 in the year after this[41]. This cost is most likely related to the treatment for, and morbidity associated with DM.
Regardless of the implications of NODAT for cardio-vascular risk, graft function, or mortality, a diagnosis of NODAT carries great significance for the individual patient. Transplant patients typically require three immunosuppressive medications, to which prophylactic antibiotics, antihypertensive agents, and others are often added. Patients with NODAT are prescribed oral hypoglycemic agents and sometimes insulin in addition to all of these. They are also subject to new dietary restrictions, which will often be superimposed on those mandated by a chronic kidney disease state (such as potassium restriction). Hospitalization may occur for both hyperglycemia and hypoglycemia[61]. All of these have an important impact on quality of life. Studies assessing this aspect of NODAT implications are few. NODAT has been associated in older studies with
peripheral neuropathy and diabetic nephropathy[54], as well as more infections[62], including severe infections[63].
PREVENTION AND MANAGEMENT OF NODATPrevention of NODATSome common preventative strategies for NODAT are summarized in Table 3. Preventing NODAT has the potential to prevent many of the short- and long-term complications of NODAT. Screening is an important part of any preventative strategy. Screening for NODAT risk prior to transplantation, as well as the risk for cardiac events, will allow for better informed consent and also help to guide post-transplant management. One recommendation is to obtain a fasting glucose level in all transplant candidates, with a subsequent OGTT performed if IFG is detected[64]. A pre-transplant OGTT may be justified if the fasting blood glucose (FBG) is as low as 5.1 mmol/L[65]. Even if the OGTT is normal, a pre-transplant random BG > 6.0 mmol/L is associated with a NODAT risk of over 25%, and > 7.2 mmol/L with a risk exceeding 50%[66]. Use of OGTT may be justifiable in all transplant candidates if the population is at particularly high risk, such as a multi-ethnic population. More sophisticated testing such as HOMA-IR assessment is not feasible routinely. HbA1c testing has not been evaluated as a pre-transplant screening strategy. If a transplant candidate is determined to be at high risk for NODAT, this should be discussed with the candidate before the transplant, and if appropriate, their proposed post-transplant immunosuppressive strategy discussed as well.
Screening for NODAT has also been employed in the post-transplant setting. Self-testing of blood glucose in the afternoon during the early post-transplant phase has been associated with an increased rate of detection of NODAT[67]. An OGTT performed at 10 wk post-transplant may help to predict longer-term hyperglycemia[68]. One difficulty with OGTT in large post-transplant clinics is that it may interfere with CNI blood level measurements, which are strictly timed. It is reasonable to measure fasting glucose at least monthly, and random blood glucose at least twice weekly for the first few months after transplantation. The HbA1c can also be checked. Since the published incidence and prevalence rates published in the literature are too variable to be helpful to individual transplant centres, it behooves every transplant centre to properly estimate its own incidence and prevalence rates, particularly in the early post-transplant period. Making an early diagnosis of NODAT is important because preventative measures can enhance the KTR’s chances for a better quality of life and also prolong their graft survival[12].
As with those at-risk for DM in other populations, lifestyle modification is likely to have a favorable impact on NODAT incidence. Recommendations for weight loss in obese patients prior to transplantation may be beneficial for preventing NODAT, but remains difficult
449 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 2 Potential pathogenic mechanisms for drug-induced new-onset diabetes mellitus after transplantation
Immunosuppressive drug Mechanism for new-onset diabetes after transplantation
Corticosteroids Increased gluconeogenesisIncreased insulin resistanceReduced glycogenesisDecreased insulin releaseImpaired pancreatic beta cell function
Calcineurin-inhibitors(cyclosporine, tacrolimus)
Reduced glucose uptakeDecreased insulin releaseReduced insulin gene expressionDirect pancreatic beta cell toxicity
Sirolimus Hypertriglyceridemia? Decreased pancreatic beta cell proliferation
?: Requires further investigation.
Palepu S et al . New-onset diabetes after kidney transplantation

to enforce due to the predisposition for malnutrition in patients on dialysis. Nonetheless, safe and closely supervised dietary and exercise recommendations for dialysis patients should be encouraged, particularly in those identified to be at higher risk for NODAT. Prompt attention from allied health professionals such as nurse practitioners and dieticians may help prevent NODAT from being established when hyperglycemia is detected.
The role of pharmacotherapy in the prevention of NODAT has also been investigated. Since early post-transplant hypomagnesemia is common, magnesium oxide supplementation has been investigated and has been associated with a reduction in FBG[69]. The use of statins in the post-transplant period has been associated with a reduced incidence of NODAT[70]. However, rosuvastatin has not been associated with improved insulin sensitivity in non-diabetic KTR[71], and so this pleiotropic effect of statins remains to be established[72]. Although angiotensin-converting enzyme inhibitors have also been associated with reduced NODAT incidence[70], their beneficial effect has not been demonstrated prospectively. Similarly, metformin has not been tested as a preventative strategy.
Since immunosuppressive medication remains the most readily available tool at the disposal of transplant clinicians to reduce the incidence of NODAT, much attention has been given to the reduction in exposure to existing immunosuppressive drugs such as corticosteroids and tacrolimus. Such reduction is often facilitated through the testing of newer pharmacological therapies. In a large, randomized controlled trial of corticosteroid withdrawal at 7 d post-transplant vs no withdrawal, a trend was noted towards better glycemic control in the withdrawal arm[73]. However, there was no difference in NODAT rates[73]. Despite this, fewer patients in the corticosteroid withdrawal arm required insulin for NODAT at 5 years[73]. On the other hand, a large retrospective study involving more than 25000 KTR reported significant benefits of steroid avoidance at initial hospital discharge when compared to a steroid-containing regimen with respect to NODAT at three years[74]. Corticosteroid withdrawal has also been shown to be beneficial when combined
with tacrolimus reduction[75]. Other studies have shown that complete corticosteroid withdrawal has no additional benefit beyond only dose reduction[40].
Although tacrolimus has a 50% greater association with NODAT than cyclosporine[10], it is the preferred CNI in many transplant programs for other reasons such as graft function in large clinical trials[44]. Reduced exposure to tacrolimus has also been associated with a similar incidence of NODAT to cyclosporine monitored using the 2-h instead of trough level[76]. In the randomized DIRECT study[77], which also used 2-h cyclosporine monitoring, a marginal increase in the composite safety endpoint of NODAT and IFG (33.6% vs 26.0%, P = 0.046) was noted with tacrolimus. Sometimes cyclosporine is switched to tacrolimus late after transplantation for reasons such as the development of other side effects, or rejection. In such instances, the risk of impaired glucose metabolism does not appear to be increased[78]. In addition, either tacrolimus or cyclosporine may be switched to sirolimus in order to reduce the burden of CNI nephrotoxicity. However, conversion to sirolimus has been associated with worsening insulin sensitivity[79]. The use of alemtuzumab, which is an anti-lymphocyte induction agent, has been associated with a reduced risk for NODAT[80]. Belatacept, which is a recently introduced selective costimulation blocker in kidney transplantation, has been associated with a reduced incidence of NODAT compared to cyclosporine when studied as a prespecified endpoint in two Phase Ⅲ studies[81]. Both alemtuzumab and belatacept are induction therapies and so cannot be used for this purpose if a higher risk for NODAT is detected post-transplant.
Management of NODATManagement strategies for NODAT are summarized in Table 3. Even if attempts to prevent NODAT are meticulous, it is likely that NODAT will occur in at least a proportion of KTR. The goals for management at this juncture include adequate glycemic control, perhaps with complete resolution of hyperglycemia without pharmacotherapy, and minimizing the short- and long-term complications of hyperglycemia. At our centre, non-fasting blood tests including glucose measurements
450 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Prevention strategies Management strategies
Identification of risk factors (Table 1) with pre-transplant counseling Regular blood glucose monitoring with appropriate follow-upPre- and post-transplant screening: random blood glucose, fasting blood glucose, 2-h oral glucose tolerance test with appropriate follow-up
Multi-disciplinary care
Lifestyle modification: weight control, diet, exercise (subject to dialysis-imposed restrictions)
Lifestyle modification: weight control, diet, exercise
Rapid corticosteroid reduction or avoidance Rapid corticosteroid reductionSelective calcineurin-inhibitor use (e.g., cyclosporine instead of tacrolimus) Conversion of cyclosporine to tacrolimus? Newer immunosuppressive agents (e.g., alemtuzumab, belatacept) Oral hypoglycemic agents: metformin, sulfonylureas, meglitinides,
dipeptidyl peptidase-4 antagonists (alone and/or in combination)? Magnesium oxide Insulin? Statins Monitoring for complications
Table 3 Management strategies for new-onset diabetes after transplantation
?: Requires further investigation.
Palepu S et al . New-onset diabetes after kidney transplantation

are obtained twice weekly for the first three months and weekly for the next three months. Fasting blood tests including glucose are obtained at least monthly. Our centre also employs an intensive multidisciplinary approach in the post-transplant clinic setting that includes a nurse practitioner with specialized expertise in diabetes prevention and management. This health care professional selectively reviews all clinic patients identified as having NODAT or being at high risk for NODAT. In addition, KTR have access to a dietician and pharmacist at all times in the clinic. Reading material about NODAT is also readily available. While such resource-intensive measures are probably helpful to individual patients, their overall effectiveness at a population level remains to be demonstrated. At other centres, adoption of a healthy diet and exercise program coupled with weight control strategies has demonstrated improvement in glycemic control over 6 mo[82]. One challenge in this regard will be multi-ethnicity of KTR[83] and possible language barriers. Collaboration between nephrologists and endocrinologists will help in the delivery of optimal care.
When antihypertensive medication is necessary for blood pressure control, it is reasonable to avoid or minimize the use of beta-blockers and thiazide diuretics in the absence of a compelling indication in those deemedat risk for NODAT. While clinical trials of antihypertensive agents in KTR are especially rare, there have also been no prospective studies of antihypertensive medications in KTR with NODAT as a pre-specified endpoint.
It is tempting for clinicians to alter transplant-related immunosuppression once NODAT has developed, with the view of optimizing glycemic control and perhaps avoiding the use of diabetes-specific medication. Reducing corticosteroid exposure carries an enhanced risk of transplant rejection[84]. Switching from tacrolimus to cyclosporine once hyperglycemia or NODAT has developed is controversial. Cyclosporine possesses a side effect profile somewhat different from tacrolimus that includes hypertrichosis and gingival hyperplasia. Nonetheless, this conversion has been attempted as rescue therapy from NODAT in small studies, with demonstration that glycemic control can be improved[85-87]. However, acute rejection remains a risk[88]. Specific features related to NODAT in patients that can predict a successful conversion have not yet been identified.
If it is clear that lifestyle modification alone will beinsufficient to control hyperglycemia, then pharmaco-therapy targeting glucose metabolism should be initiated. In the very early post-transplant phase, whencorticosteroids are being rapidly tapered, additional pharmacotherapy may not be required if the hyper-glycemia is mild. The choice between insulin and oral hypoglycemic agents depends on the severity, timing, and expected duration of hyperglycemia[49]. Insulin therapy is safe[50] particularly when graft function is not yet established or is unstable. Since corticosteroids are typically administered in the morning in KTR, a combination of intermediate and short-acting insulin administered several
times during the day and corresponding to mealtimes may be required. In less urgent instances, oral hypoglycemic agents can be commenced without resorting to insulin firsthand.
The choice of oral hypoglycemic agent is dictated by the level of renal function. No agent is specifically contraindicated in KTR, and there are no significant drug interactions with CNI or other immunosuppressive drugs. Pharmaceutical approaches generally mirror those utilized in the general population, with no studies specific to KTR available. In addition, the target HbA1c for KTR has not been defined[49]. Metformin improves insulin sensitivity, which is often affected in NODAT. The safety of metformin in KTR with sufficient renal function has been formally demonstrated[88]. Since the estimated GFR in KTR with well-functioning grafts often exceeds 50 mL/min per 1.73 m2, metformin can be safely used for most patients. A theoretical disadvantage to using metformin in KTR would be gastrointestinal upset, to which KTR are already prone by virtue of immunosuppressive medications such as mycophenolate mofetil. However, metformin may counter the post-transplant weight gain associated with corticosteroid administration, but this has not been demonstrated. Keeping these in mind, metformin is often employed as a first-line agent in KTR with NODAT.
Sulfonylureas, which enhance insulin secretion, are also widely used in KTR. Their use is again dictated by level of renal function, and unlike metformin, they may exacerbate post-transplant weight gain. Newer sulfonylureas like glipizide and gliclazide may be used in KTR, with appropriate monitoring for hypoglycemia. Amongst the thiazolidinediones, which are selective agonists of the peroxisomal proliferator-activated receptor gamma, rosiglitazone has been shown to improve glucose tolerance, insulin sensitivity, and even endothelial function in a small group of KTR[89]. Its short-term efficacy has also been shown by other groups[90]. Thiazolidinediones should again be used with extreme caution if graft function is severely impaired. Congestive heart failure is a significant concern, and many KTR already have impaired cardiac function. Furthermore, cyclosporine may promote a sodium-retentive state. As a result, thiazolidinediones are usually not used.
Amongst the meglitinides, which are short-acting drugs that induce insulin secretion, repaglinide has been shown to be safe in KTR[91]. Repaglinide can also be used in severe renal graft dysfunction. Dipeptidyl peptidase-4 (DPP-4) antagonists have been employed in KTR. These drugs inhibit DPP-4, which is responsible for the rapid degradation of numerous substrates including the glucagon-like peptide 1, whose role is to increase pancreatic insulin secretion. Among the DPP-4 antagonists, sitagliptin has been shown to be both safe and efficacious in KTR[92,93]. A theoretical risk in KTR is an increased risk of infections, to which they may already be prone by virtue of being in an immunosuppressed state.
It is reasonable to employ rationally selected
451 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Palepu S et al . New-onset diabetes after kidney transplantation

combinations of two or three of the above drugs before proceeding to insulin therapy. However, the initiation of insulin should not be delayed unnecessarily, since prolonged hyperglycemia may result in hospitalization from intravascular volume depletion and serious comorbidities such as infections. Opportunistic infections of many kinds typically occur in the first 6 mo post-transplant, which corresponds to the phase when NODAT usually occurs.
FUTURE DIRECTIONSRenal transplant survival has significantly improved over the last 50 years. Death with graft function, often as a result of CVD, has become a major cause of graft loss. As a result, increasing attention is being given to cardiovascular risk factors such as DM, within which NODAT is a significant component. Intensive screening for NODAT should be the norm in all transplant centres. Efforts to combat NODAT have to be balanced against the risks for graft rejection. Large clinical trials of newer immunosuppressive agents in kidney transplantation are few, but the inclusion of NODAT as a prespecified endpoint will help to better understand not only the most important contributing risks for NODAT, but also identify those interventions that are most likely to result in a lower NODAT incidence. Correspondingly, clinical trials that address treatment strategies for diabetes post-transplant have received very little attention. This is indeed unfortunate given the magnitude of NODAT prevalence and its financial implications for society, not to mention the profound impact of NODAT on transplant recipients. Until such trials are performed, multidis-ciplinary care focused on intensive glucose control in keeping with general population guidelines, along with management of other cardiovascular risk factors should remain the standard.
REFERENCES1 First MR, Dhadda S, Croy R, Holman J, Fitzsimmons WE. New-
onset diabetes after transplantation (NODAT): an evaluation of definitions in clinical trials. Transplantation 2013; 96: 58-64 [PMID: 23619735 DOI: 10.1097/TP.0b013e318293fcf8]
2 Lv C, Chen M, Xu M, Xu G, Zhang Y, He S, Xue M, Gao J, Yu M, Gao X, Zhu T. Influencing factors of new-onset diabetes after a renal transplant and their effects on complications and survival rate. PLoS One 2014; 9: e99406 [PMID: 24911157 DOI: 10.1371/journal.pone.0099406]
3 Chakkera HA, Weil EJ, Pham PT, Pomeroy J, Knowler WC. Can new-onset diabetes after kidney transplant be prevented? Diabetes Care 2013; 36: 1406-1412 [PMID: 23613600 DOI: 10.2337/dc12-2067]
4 Sarno G, Muscogiuri G, De Rosa P. New-onset diabetes after kidney transplantation: prevalence, risk factors, and management. Transplantation 2012; 93: 1189-1195 [PMID: 22475764]
5 Balla A, Chobanian M. New-onset diabetes after transplantation: a review of recent literature. Curr Opin Organ Transplant 2009; 14: 375-379 [PMID: 19542891 DOI: 10.1097/MOT.0b013e32832dbb98]
6 Pham PT, Pham PM, Pham SV, Pham PA, Pham PC. New onset diabetes after transplantation (NODAT): an overview. Diabetes
Metab Syndr Obes 2011; 4: 175-186 [PMID: 21760734 DOI: 10.2147/DMSO.S19027]
7 Davidson J, Wilkinson A, Dantal J, Dotta F, Haller H, Hernández D, Kasiske BL, Kiberd B, Krentz A, Legendre C, Marchetti P, Markell M, van der Woude FJ, Wheeler DC. New-onset diabetes after transplantation: 2003 International consensus guidelines. Proceedings of an international expert panel meeting. Barcelona, Spain, 19 February 2003. Transplantation 2003; 75: SS3-S24 [PMID: 12775942]
8 Elmagd MM, Bakr MA, Metwally AH, Wahab AM. Clini-coepidemiologic study of posttransplant diabetes after living-donor renal transplant. Exp Clin Transplant 2008; 6: 42-47 [PMID: 18405244]
9 Kamar N, Mariat C, Delahousse M, Dantal J, Al Najjar A, Cassuto E, Lefrançois N, Cointault O, Touchard G, Villemain F, Di Giambattista F, Benhamou PY. Diabetes mellitus after kidney transplantation: a French multicentre observational study. Nephrol Dial Transplant 2007; 22: 1986-1993 [PMID: 17400559]
10 Kasiske BL, Snyder JJ, Gilbertson D, Matas AJ. Diabetes mellitus after kidney transplantation in the United States. Am J Transplant 2003; 3: 178-185 [PMID: 12603213]
11 Greenspan LC, Gitelman SE, Leung MA, Glidden DV, Mathias RS. Increased incidence in post-transplant diabetes mellitus in children: a case-control analysis. Pediatr Nephrol 2002; 17: 1-5 [PMID: 11793126]
12 Chakkera HA, Weil EJ, Swanson CM, Dueck AC, Heilman RL, Reddy KS, Hamawi K, Khamash H, Moss AA, Mulligan DC, Katariya N, Knowler WC. Pretransplant risk score for new-onset diabetes after kidney transplantation. Diabetes Care 2011; 34: 2141-2145 [PMID: 21949218]
13 Rodrigo E, Fernández-Fresnedo G, Valero R, Ruiz JC, Piñera C, Palomar R, González-Cotorruelo J, Gómez-Alamillo C, Arias M. New-onset diabetes after kidney transplantation: risk factors. J Am Soc Nephrol 2006; 17: S291-S295 [PMID: 17130277 DOI: 10.1681/ASN.2006080929]
14 Boudreaux JP, McHugh L, Canafax DM, Ascher N, Sutherland DE, Payne W, Simmons RL, Najarian JS, Fryd DS. The impact of cyclosporine and combination immunosuppression on the incidence of posttransplant diabetes in renal allograft recipients. Transplantation 1987; 44: 376-381 [PMID: 3307061]
15 Cosio FG, Pesavento TE, Osei K, Henry ML, Ferguson RM. Post-transplant diabetes mellitus: increasing incidence in renal allograft recipients transplanted in recent years. Kidney Int 2001; 59: 732-737 [PMID: 11168956]
16 Gourishankar S, Jhangri GS, Tonelli M, Wales LH, Cockfield SM. Development of diabetes mellitus following kidney transplantation: a Canadian experience. Am J Transplant 2004; 4: 1876-1882 [PMID: 15476489 DOI: 10.1111/j.1600-6143,2004.00591.x]
17 Impedovo SV, Ditonno P, Ricapito V, Bettocchi C, Gesualdo L, Grandaliano G, Selvaggi FP, Battaglia M. Advanced age is not an exclusion criterion for kidney transplantation. Transplant Proc 2013; 45: 2650-2653 [PMID: 24034014 DOI: 10.1016/j.transproceed.2013.08.003]
18 Moore R, Boucher A, Carter J, Kim SJ, Kiberd B, Loertscher R, Mongeau JG, Prasad GV, Vautour L. Diabetes mellitus in transplantation: 2002 consensus guidelines. Transplant Proc 2003; 35: 1265-1270 [PMID: 12826134]
19 Sulanc E, Lane JT, Puumala SE, Groggel GC, Wrenshall LE, Stevens RB. New-onset diabetes after kidney transplantation: an application of 2003 International Guidelines. Transplantation 2005; 80: 945-952 [PMID: 16249743]
20 Glick L, Shamy F, Nash M, Sokwala A, Malavade T, Prasad GR, Zaltzman JS. A prospective cohort conversion study of twice-daily to once-daily extended-release tacrolimus: role of ethnicity. Transplant Res 2014; 3: 7 [PMID: 24606676 DOI: 10.1186/2047-1440-3-7]
21 Kang ES, Kim MS, Kim YS, Kim CH, Han SJ, Chun SW, Hur KY, Nam CM, Ahn CW, Cha BS, Kim SI, Lee HC. A polymorphism in the zinc transporter gene SLC30A8 confers resistance against posttransplantation diabetes mellitus in renal allograft recipients.
452 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Palepu S et al . New-onset diabetes after kidney transplantation

Diabetes 2008; 57: 1043-1047 [PMID: 18162509]22 Kang ES, Kim MS, Kim YS, Hur KY, Han SJ, Nam CM, Ahn
CW, Cha BS, Kim SI, Lee HC. A variant of the transcription factor 7-like 2 (TCF7L2) gene and the risk of posttransplantation diabetes mellitus in renal allograft recipients. Diabetes Care 2008; 31: 63-68 [PMID: 17934151]
23 Ghisdal L, Baron C, Le Meur Y, Lionet A, Halimi JM, Rerolle JP, Glowacki F, Lebranchu Y, Drouet M, Noël C, El Housni H, Cochaux P, Wissing KM, Abramowicz D, Abramowicz M. TCF7L2 polymorphism associates with new-onset diabetes after transplantation. J Am Soc Nephrol 2009; 20: 2459-2467 [PMID: 19713311 DOI: 10.1681/ASN.2008121314]
24 Tavira B, Coto E, Díaz-Corte C, Ortega F, Arias M, Torres A, Díaz JM, Selgas R, López-Larrea C, Campistol JM, Ruiz-Ortega M, Alvarez V; Pharmacogenetics of Tacrolimus REDINREN Study Group. KCNQ1 gene variants and risk of new-onset diabetes in tacrolimus-treated renal-transplanted patients. Clin Transplant 2011; 25: E284-E291 [PMID: 21355884 DOI: 10.1111/j.1399-0012.2011.01417.x]
25 Chen Y, Sampaio MS, Yang JW, Min D, Hutchinson IV. Genetic polymorphisms of the transcription factor NFATc4 and development of new-onset diabetes after transplantation in Hispanic kidney transplant recipients. Transplantation 2012; 93: 325-330 [PMID: 22234350 DOI: 10.1097/TP.0b013e31823f7f26]
26 Nicoletto BB, Souza GC, Fonseca NK, Centenaro A, Manfro RC, Canani LH, Gonçalves LF. Association between 276G/T adiponectin gene polymorphism and new-onset diabetes after kidney transplantation. Transplantation 2013; 96: 1059-1064 [PMID: 23985723 DOI: 10.1097/TP.0b013e3182a45283]
27 Tavira B, Gómez J, Díaz-Corte C, Llobet L, Ruiz-Pesini E, Ortega F, Coto E. Mitochondrial DNA haplogroups and risk of new-onset diabetes among tacrolimus-treated renal transplanted patients. Gene 2014; 538: 195-198 [PMID: 24445060 DOI: 10.1016/j.gene.2014.01.036]
28 de Mattos AM, Olyaei AJ, Prather JC, Golconda MS, Barry JM, Norman DJ. Autosomal-dominant polycystic kidney disease as a risk factor for diabetes mellitus following renal transplantation. Kidney Int 2005; 67: 714-720 [PMID: 15673321]
29 Hamer RA, Chow CL, Ong AC, McKane WS. Polycystic kidney disease is a risk factor for new-onset diabetes after transplantation. Transplantation 2007; 83: 36-40 [PMID: 17220788]
30 Carter SA, Kitching AR, Johnstone LM. Four pediatric patients with autosomal recessive polycystic kidney disease developed new-onset diabetes after renal transplantation. Pediatr Transplant 2014; 18: 698-705 [PMID: 25118046 DOI: 10.1111/petr.12332]
31 Tokodai K, Amada N, Kikuchi H, Haga I, Takayama T, Nakamura A. Body fat percentage as a marker of new-onset diabetes mellitus after kidney transplantation. Transplant Proc 2013; 45: 1544-1547 [PMID: 23726616 DOI: 10.106/j.transporiceed.2012.12.009]
32 Marrero D, Hernandez D, Tamajón LP, Rivero M, Lampreabe I, Checa MD, Gonzalez-Posada JM; For the Spanish Late Allograft Dysfunction Study Group. Pre-transplant weight but not weight gain is associated with new-onset diabetes after transplantation: a multi-centre cohort Spanish study. NDT Plus 2010; 3: ii15-ii20 [PMID: 20508859]
33 Bayés B, Granada ML, Pastor MC, Lauzurica R, Salinas I, Sanmartí A, Espinal A, Serra A, Navarro M, Bonal J, Romero R. Obesity, adiponectin and inflammation as predictors of new-onset diabetes mellitus after kidney transplantation. Am J Transplant 2007; 7: 416-422 [PMID: 17229078]
34 Israni AK, Snyder JJ, Skeans MA, Kasiske BL. Clinical diagnosis of metabolic syndrome: predicting new-onset diabetes, coronary heart disease, and allograft failure late after kidney transplant. Transpl Int 2012; 25: 748-757 [PMID: 22548293 DOI: 10.1111/j.1432-2277.2012.01488.x]
35 Mehta SH, Brancati FL, Sulkowski MS, Strathdee SA, Szklo M, Thomas DL. Prevalence of type 2 diabetes mellitus among persons with hepatitis C virus infection in the United States. Ann Intern Med 2000; 133: 592-599 [PMID: 11033586]
36 Fabrizi F, Martin P, Dixit V, Bunnapradist S, Kanwal F, Dulai
G. Post-transplant diabetes mellitus and HCV seropositive status after renal transplantation: meta-analysis of clinical studies. Am J Transplant 2005; 5: 2433-2440 [PMID: 16162192]
37 Bloom RD, Rao V, Weng F, Grossman RA, Cohen D, Mange KC. Association of hepatitis C with posttransplant diabetes in renal transplant patients on tacrolimus. J Am Soc Nephrol 2002; 13: 1374-1380 [PMID: 11961026]
38 Hjelmesaeth J, Sagedal S, Hartmann A, Rollag H, Egeland T, Hagen M, Nordal KP, Jenssen T. Asymptomatic cytomegalovirus infection is associated with increased risk of new-onset diabetes mellitus and impaired insulin release after renal transplantation. Diabetologia 2004; 47: 1550-1556 [PMID: 15338129]
39 van Raalte DH, Nofrate V, Bunck MC, van Iersel T, Elassaiss Schaap J, Nässander UK, Heine RJ, Mari A, Dokter WH, Diamant M. Acute and 2-week exposure to prednisolone impair different aspects of beta-cell function in healthy men. Eur J Endocrinol 2010; 162: 729-735 [PMID: 20124412 DOI: 10.1530/EJE-09-1034]
40 Midtvedt K, Hjelmesaeth J, Hartmann A, Lund K, Paulsen D, Egeland T, Jenssen T. Insulin resistance after renal transplantation: the effect of steroid dose reduction and withdrawal. J Am Soc Nephrol 2004; 15: 3233-3239 [PMID: 15579527 DOI: 10.1097/01.ASN.0000145435.80]
41 Woodward RS, Schnitzler MA, Baty J, Lowell JA, Lopez-Rocafort L, Haider S, Woodworth TG, Brennan DC. Incidence and cost of new onset diabetes mellitus among U.S. wait-listed and transplanted renal allograft recipients. Am J Transplant 2003; 3: 590-598 [PMID: 12752315]
42 Romagnoli J, Citterio F, Violi P, Cadeddu F, Nanni G, Castagneto M. Post-transplant diabetes mellitus: a case-control analysis of the risk factors. Transpl Int 2005; 18: 309-312 [PMID: 15730491]
43 Maes BD, Kuypers D, Messiaen T, Evenepoel P, Mathieu C, Coosemans W, Pirenne J, Vanrenterghem YF. Posttransplantation diabetes mellitus in FK-506-treated renal transplant recipients: analysis of incidence and risk factors. Transplantation 2001; 72: 1655-1661 [PMID: 11726827]
44 Ekberg H, Tedesco-Silva H, Demirbas A, Vítko S, Nashan B, Gürkan A, Margreiter R, Hugo C, Grinyó JM, Frei U, Vanrenterghem Y, Daloze P, Halloran PF. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med 2007; 357: 2562-2575 [PMID: 18094377]
45 Pereira MJ, Palming J, Rizell M, Aureliano M, Carvalho E, Svensson MK, Eriksson JW. Cyclosporine A and tacrolimus reduce the amount of GLUT4 at the cell surface in human adipocytes: increased endocytosis as a potential mechanism for the diabetogenic effects of immunosuppressive agents. J Clin Endocrinol Metab 2014; 99: E1885-E1894 [PMID: 25004245 DOI: 10.1210/jc.2014-1266]
46 Al-Hasani H, Kunamneni RK, Dawson K, Hinck CS, Müller-Wieland D, Cushman SW. Roles of the N- and C-termini of GLUT4 in endocytosis. J Cell Sci 2002; 115: 131-140 [PMID: 11801731]
47 Radu RG, Fujimoto S, Mukai E, Takehiro M, Shimono D, Nabe K, Shimodahira M, Kominato R, Aramaki Y, Nishi Y, Funakoshi S, Yamada Y, Seino Y. Tacrolimus suppresses glucose-induced insulin release from pancreatic islets by reducing glucokinase activity. Am J Physiol Endocrinol Metab 2005; 288: E365-E371 [PMID: 15479952]
48 Drachenberg CB, Klassen DK, Weir MR, Wiland A, Fink JC, Bartlett ST, Cangro CB, Blahut S, Papadimitriou JC. Islet cell damage associated with tacrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation 1999; 68: 396-402 [PMID: 10459544]
49 Yates CJ, Fourlanos S, Hjelmesaeth J, Colman PG, Cohney SJ. New-onset diabetes after kidney transplantation-changes and challenges. Am J Transplant 2012; 12: 820-828 [PMID: 22123607 DOI: 10.1111/j.1600-6143.2011.03855.x]
50 Veroux M, Tallarita T, Corona D, Sinagra N, Giaquinta A, Zerbo D, Guerrieri C, D’Assoro A, Cimino S, Veroux P. Conversion to sirolimus therapy in kidney transplant recipients with new onset
453 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Palepu S et al . New-onset diabetes after kidney transplantation

diabetes mellitus after transplantation. Clin Dev Immunol 2013; 2013: 496974 [PMID: 23762090 DOI: 10.1155/2013/496974]
51 Johnston O, Rose CL, Webster AC, Gill JS. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J Am Soc Nephrol 2008; 19: 1411-1418 [PMID: 18385422 DOI: 10.1681/ASN.2007111202]
52 Gyurus E, Kaposztas Z, Kahan BD. Sirolimus therapy predisposes to new-onset diabetes mellitus after renal transplantation: a long-term analysis of various treatment regimens. Transplant Proc 2011; 43: 1583-1592 [PMID: 21693238 DOI: 10.1016/j.transproceed.2011.05.001]
53 Roland M, Gatault P, Doute C, Büchler M, Al-Najjar A, Barbet C, Chatelet V, Marlière JF, Nivet H, Lebranchu Y, Halimi JM. Immunosuppressive medications, clinical and metabolic parameters in new-onset diabetes mellitus after kidney transplantation. Transpl Int 2008; 21: 523-530 [PMID: 18266773 DOI: 10.1111/j.1432-2277.2008.00640.x]
54 Miles AM, Sumrani N, Horowitz R, Homel P, Maursky V, Markell MS, Distant DA, Hong JH, Sommer BG, Friedman EA. Diabetes mellitus after renal transplantation: as deleterious as non-transplant-associated diabetes? Transplantation 1998; 65: 380-384 [PMID: 9484755]
55 Roth D, Milgrom M, Esquenazi V, Fuller L, Burke G, Miller J. Posttransplant hyperglycemia. Increased incidence in cyclosporine-treated renal allograft recipients. Transplantation 1989; 47: 278-281 [PMID: 2645712]
56 Valderhaug TG, Hjelmesæth J, Hartmann A, Røislien J, Bergrem HA, Leivestad T, Line PD, Jenssen T. The association of early post-transplant glucose levels with long-term mortality. Diabetologia 2011; 54: 1341-1349 [PMID: 21409415 DOI: 10.1007/s00125-011-2105-9]
57 Hjelmesaeth J, Hartmann A, Leivestad T, Holdaas H, Sagedal S, Olstad M, Jenssen T. The impact of early-diagnosed new-onset post-transplantation diabetes mellitus on survival and major cardiac events. Kidney Int 2006; 69: 588-595 [PMID: 16395250]
58 Cosio FG, Pesavento TE, Kim S, Osei K, Henry M, Ferguson RM. Patient survival after renal transplantation: IV. Impact of post-transplant diabetes. Kidney Int 2002; 62: 1440-1446 [PMID: 12234317]
59 Cole EH, Johnston O, Rose CL, Gill JS. Impact of acute rejection and new-onset diabetes on long-term transplant graft and patient survival. Clin J Am Soc Nephrol 2008; 3: 814-821 [PMID: 18322046 DOI: 10.2215/CJN.04681107]
60 Kuo HT, Sampaio MS, Vincenti F, Bunnapradist S. Associations of pretransplant diabetes mellitus, new-onset diabetes after transplant, and acute rejection with transplant outcomes: an analysis of the Organ Procurement and Transplant Network/United Network for Organ Sharing (OPTN/UNOS) database. Am J Kidney Dis 2010; 56: 1127-1139 [PMID: 20934793 DOI: 10.1053/j.ajkd.2010.06.027]
61 Burroughs TE, Swindle J, Takemoto S, Lentine KL, Machnicki G, Irish WD, Brennan DC, Schnitzler MA. Diabetic complications associated with new-onset diabetes mellitus in renal transplant recipients. Transplantation 2007; 83: 1027-1034 [PMID: 17452891]
62 Siraj ES, Abacan C, Chinnappa P, Wojtowicz J, Braun W. Risk factors and outcomes associated with posttransplant diabetes mellitus in kidney transplant recipients. Transplant Proc 2010; 42: 1685-1689 [PMID: 20620501 DOI: 10.1016/j.transproceed.2009.12.062]
63 von Kiparski A, Frei D, Uhlschmid G, Largiadèr F, Binswanger U. Post-transplant diabetes mellitus in renal allograft recipients: a matched-pair control study. Nephrol Dial Transplant 1990; 5: 220-225 [PMID: 2113651]
64 Wilkinson A, Davidson J, Dotta F, Home PD, Keown P, Kiberd B, Jardine A, Levitt N, Marchetti P, Markell M, Naicker S, O’Connell P, Schnitzler M, Standl E, Torregosa JV, Uchida K, Valantine H, Villamil F, Vincenti F, Wissing M. Guidelines for the treatment and management of new-onset diabetes after transplantation. Clin Transplant 2005; 19: 291-298 [PMID: 15877787]
65 Caillard S, Eprinchard L, Perrin P, Braun L, Heibel F, Moreau F, Kessler L, Moulin B. Incidence and risk factors of glucose metabolism disorders in kidney transplant recipients: role of systematic screening by oral glucose tolerance test. Transplantation 2011; 91: 757-764 [PMID: 21336240 DOI: 10.1097/TP.0b013e3820f0877]
66 Ramesh Prasad GV, Huang M, Bandukwala F, Nash MM, Rapi L, Montada-Atin T, Meliton G, Zaltzman JS. Pre-transplantation glucose testing for predicting new-onset diabetes mellitus after renal transplantation. Clin Nephrol 2009; 71: 140-146 [PMID: 19203506]
67 Clore JN, Thurby-Hay L. Glucocorticoid-induced hyperglycemia. Endocr Pract 2009; 15: 469-474 [PMID: 19454391 DOI: 10.4158/EP08331.RAR]
68 Hjelmesaeth J, Hagen M, Hartmann A, Midtvedt K, Egeland T, Jenssen T. The impact of impaired insulin release and insulin resistance on glucose intolerance after renal transplantation. Clin Transplant 2002; 16: 389-396 [PMID: 12437616]
69 Van Laecke S, Nagler EV, Taes Y, Van Biesen W, Peeters P, Vanholder R. The effect of magnesium supplements on early post-transplantation glucose metabolism: a randomized controlled trial. Transpl Int 2014; 27: 895-902 [PMID: 24909487 DOI: 10.1111/tri.12287]
70 Prasad GV, Kim SJ, Huang M, Nash MM, Zaltzman JS, Fenton SS, Cattran DC, Cole EH, Cardella CJ. Reduced incidence of new-onset diabetes mellitus after renal transplantation with 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors (statins). Am J Transplant 2004; 4: 1897-1903 [PMID: 15476492]
71 Sharif A, Ravindran V, Moore R, Dunseath G, Luzio S, Owens D, Baboolal K. The effect of rosuvastatin on insulin sensitivity and pancreatic beta-cell function in nondiabetic renal transplant recipients. Am J Transplant 2009; 9: 1439-1445 [PMID: 19459810 DOI: 10.1111/j.1600-6143.2009.02644.x]
72 Sasaki J, Iwashita M, Kono S. Statins: beneficial or adverse for glucose metabolism. J Atheroscler Thromb 2006; 13: 123-129 [PMID: 16835466]
73 Woodle ES, First MR, Pirsch J, Shihab F, Gaber AO, Van Veldhuisen P; Astellas Corticosteroid Withdrawal Study Group. A prospective, randomized, double-blind, placebo-controlled multicenter trial comparing early (7 day) corticosteroid cessation versus long-term, low-dose corticosteroid therapy. Ann Surg 2008; 248: 564-577 [PMID: 18936569 DOI: 10.1097/SLA.0b013e318187d1da]
74 Luan FL, Steffick DE, Ojo AO. New-onset diabetes mellitus in kidney transplant recipients discharged on steroid-free immunosuppression. Transplantation 2011; 91: 334-341 [PMID: 21242885 DOI: 10.1097/TP.0b013e318203c25f]
75 Boots JM, van Duijnhoven EM, Christiaans MH, Wolffenbuttel BH, van Hooff JP. Glucose metabolism in renal transplant recipients on tacrolimus: the effect of steroid withdrawal and tacrolimus trough level reduction. J Am Soc Nephrol 2002; 13: 221-227 [PMID: 11752041]
76 Kim SJ, Prasad GV, Huang M, Nash MM, Famure O, Park J, Thenganatt MA, Chowdhury N, Cole EH, Fenton SS, Cattran DC, Zaltzman JS, Cardella CJ. A comparison of the effects of C2-cyclosporine and C0-tacrolimus on renal function and cardiovascular risk factors in kidney transplant recipients. Transplantation 2006; 82: 924-930 [PMID: 17038908]
77 Vincenti F, Friman S, Scheuermann E, Rostaing L, Jenssen T, Campistol JM, Uchida K, Pescovitz MD, Marchetti P, Tuncer M, Citterio F, Wiecek A, Chadban S, El-Shahawy M, Budde K, Goto N. Results of an international, randomized trial comparing glucose metabolism disorders and outcome with cyclosporine versus tacrolimus. Am J Transplant 2007; 7: 1506-1514 [PMID: 17359512]
78 Luan FL, Zhang H, Schaubel DE, Miles CD, Cibrik D, Norman S, Ojo AO. Comparative risk of impaired glucose metabolism associated with cyclosporine versus tacrolimus in the late posttransplant period. Am J Transplant 2008; 8: 1871-1877 [PMID: 18786231 DOI: 10.1111/j.1600-6143.2008.02328.x]
454 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Palepu S et al . New-onset diabetes after kidney transplantation

79 Teutonico A, Schena PF, Di Paolo S. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol 2005; 16: 3128-3135 [PMID: 16107580]
80 Shah T, Kasravi A, Huang E, Hayashi R, Young B, Cho YW, Bunnapradist S. Risk factors for development of new-onset diabetes mellitus after kidney transplantation. Transplantation 2006; 82: 1673-1676 [PMID: 17198258]
81 Vanrenterghem Y, Bresnahan B, Campistol J, Durrbach A, Grinyó J, Neumayer HH, Lang P, Larsen CP, Mancilla-Urrea E, Pestana JM, Block A, Duan T, Glicklich A, Gujrathi S, Vincenti F. Belatacept-based regimens are associated with improved cardiovascular and metabolic risk factors compared with cyclosporine in kidney transplant recipients (BENEFIT and BENEFIT-EXT studies). Transplantation 2011; 91: 976-983 [PMID: 21372756 DOI: 10.1097/TP.0b013e31820c10eb]
82 Sharif A, Moore R, Baboolal K. Influence of lifestyle modification in renal transplant recipients with postprandial hyperglycemia. Transplantation 2008; 85: 353-358 [PMID: 18301331 DOI: 10.1097/TP.0b013e3181605ebf]
83 Prasad GV, Vangala SK, Silver SA, Wong SC, Huang M, Rapi L, Nash MM, Zaltzman JS. South Asian ethnicity as a risk factor for major adverse cardiovascular events after renal transplantation. Clin J Am Soc Nephrol 2011; 6: 204-211 [PMID: 20884776 DOI: 10.2215/CJN.03100410]
84 Pascual J, Zamora J, Galeano C, Royuela A, Quereda C. Steroid avoidance or withdrawal for kidney transplant recipients. Cochrane Database Syst Rev 2009; 21: CD005632 [DOI: 10.1002/14651858.CD005632.pub2]
85 Wyzgal J, Oldakowska-Jedynak U, Paczek L, Michalska M, Ziolkowski J, Soluch L, Zygier D, Sanko-Resmer J, Gradowska L, Niewczas M, Galazka Z, Pacholczyk M, Durlik M. Posttransplantation diabetus mellitus under calcineurin inhibitor.
Transplant Proc 2003; 35: 2216-2218 [PMID: 14529893]86 Oberholzer J, Thielke J, Hatipoglu B, Testa G, Sankary HN,
Benedetti E. Immediate conversion from tacrolimus to cyclosporine in the treatment of posttransplantation diabetes mellitus. Transplant Proc 2005; 37: 999-1000 [PMID: 15848603]
87 Abouljoud MS, Kumar MS, Brayman KL, Emre S, Bynon JS; OLN-452 Study Group. Neoral rescue therapy in transplant patients with intolerance to tacrolimus. Clin Transplant 2002; 16: 168-172 [PMID: 12010138]
88 Pilmore HL. Review: metformin: potential benefits and use in chronic kidney disease. Nephrology (Carlton) 2010; 15: 412-418 [PMID: 20609092 DOI: 10.1111/j.1440-1797.2010.01328.x]
89 Voytovich MH, Simonsen C, Jenssen T, Hjelmesaeth J, Asberg A, Hartmann A. Short-term treatment with rosiglitazone improves glucose tolerance, insulin sensitivity and endothelial function in renal transplant recipients. Nephrol Dial Transplant 2005; 20: 413-418 [PMID: 15615809]
90 Villanueva G, Baldwin D. Rosiglitazone therapy of posttransplant diabetes mellitus. Transplantation 2005; 80: 1402-1405 [PMID: 16340782]
91 Türk T, Pietruck F, Dolff S, Kribben A, Janssen OE, Mann K, Philipp T, Heemann U, Witzke O. Repaglinide in the management of new-onset diabetes mellitus after renal transplantation. Am J Transplant 2006; 6: 842-846 [PMID: 16539642]
92 Boerner BP, Miles CD, Shivaswamy V. Efficacy and safety of sitagliptin for the treatment of new-onset diabetes after renal transplantation. Int J Endocrinol 2014; 2014: 617638 [PMID: 24817885 DOI: 10.1155/2014/617638]
93 Strøm Halden TA, Åsberg A, Vik K, Hartmann A, Jenssen T. Short-term efficacy and safety of sitagliptin treatment in long-term stable renal recipients with new-onset diabetes after transplantation. Nephrol Dial Transplant 2014; 29: 926-933 [PMID: 24452849 DOI: 10.1093/ndt/gft536]
P- Reviewer: Cardoso CRL, Ray S S- Editor: Song XX L- Editor: A E- Editor: Liu SQ
455 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Palepu S et al . New-onset diabetes after kidney transplantation

insulin resistance, dyslipidemia, β-cell dysfunction, impaired glucose tolerance and ultimately leading to T2DM. Chronic oxidative stress, hyperglycemia and dyslipidemia are particularly dangerous for β-cells from lowest levels of antioxidant, have high oxidative energy requirements, decrease the gene expression of key β-cell genes and induce cell death. If β-cell functioning is impaired, it results in an under production of insulin, impairs glucose stimulated insulin secretion, fasting hyperglycemia and eventually the development of T2DM.
Key words: Insulin resistance; Dyslipidemia; Type 2 diabetes mellitus; Oxidative stress
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Oxidative stress is underling in the development of cardiovascular disease, type 2 diabetes mellitus (T2DM) and diabetic complications. Increased oxidative stress appears to be a deleterious factor leading to insulin resistance, dyslipidemia, β-cell dysfunction, impaired glucose tolerance and ultimately leading to T2DM.
Tangvarasittichai S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J Diabetes 2015; 6(3): 456-480 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/456.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.456
INTRODUCTIONAerobic life uses oxygen to oxidize (metabolism) food substrates (carbon- and hydrogen-rich) to obtain the heat energy and chemical essential for life. When we oxidize molecules with oxygen, the oxygen molecule itself becomes reduced and forms intermediates.
Surapon Tangvarasittichai
Surapon Tangvarasittichai, Chronic Disease Research Unit, Department of Medical Technology, Faculty of Allied Health Sciences, Naresuan University, Phitsanulok 65000, ThailandAuthor contributions: Tangvarasittichai S and his assistances performed the literatures review search and wrote the manuscript; Tangvarasittichai S designed essential ideas, drawing figures and sequencing of this review manuscript, also provided references and edited the manuscript, addressed the responses to reviewers’ concerns and contributed to the edition of the manuscript.Conflict-of-interest: The author declares that there are no conflicts of interest.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Dr. Surapon Tangvarasittichai, Associate Professor, Chronic Disease Research Unit, Department of Medical Technology, Faculty of Allied Health Sciences, Naresuan University, 99 Moo 9 Tambon Tha Pho, Muang, Phitsanulok 65000, Thailand. [email protected]: +66-08-96388382 Fax: +66-08-55966300Received: September 3, 2014 Peer-review started: September 4, 2014 First decision: November 14, 2014Revised: December 25, 2014 Accepted: January 9, 2015Article in press: January 12, 2015Published online: April 15, 2015
AbstractOxidative stress is increased in metabolic syndrome and type 2 diabetes mellitus (T2DM) and this appears to underlie the development of cardiovascular disease, T2DM and diabetic complications. Increased oxidative stress appears to be a deleterious factor leading to
REVIEW
456 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.456
World J Diabetes 2015 April 15; 6(3): 456-480ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

In eukaryotic cells, reactive oxygen species (ROS) are always produced as the consequence of regular physiological metabolism[1]. These ROS (pro-oxidants) productions are counter-balanced by cellular antioxidant defense mechanisms in the normal physiological conditions. ROS define as diverse chemical that have reactive properties are capable to accommodate or donate electrons (e-) to the broad range of biological molecules. Normally, the production and neutralization of ROS are balance with antioxidants in a living system and does not cause any oxidative damage, determines as physiological state[2]. The imbalance between these prooxidants and antioxidants in the living organism system to determine as oxidative stress state, brings to cellular disruption and damage[3]. The free-radical can attack polyunsaturated fatty acids oxidation in physiological systems known as lipid peroxidation. Lipid peroxidation is an autocatalytic free radical mediated destructive process whereby poly-unsaturated fatty acids in cell membranes undergo degradation to form lipid hydroperoxides[4,5]. By-products of lipid peroxidation such as conjugated dienes and malondialdehyde (MDA) are increased in the patients with obesity, metabolic syndrome and type 2 diabetes mellitus (T2DM). Car-bohydrates, lipids, proteins and DNA are the targets of oxidative stress modification biomolecules generally as the principal of ROS induced cellular damage. Therefore, these ROS modified biomolecules are used as oxidative stress markers both in vivo and in vitro measurement. Recent study suggests that ROS may act as the mechanical link of salt sensitive hypertension, over nutrition and high fat diet, metabolic syndrome and T2DM animal models[6]. ROS levels are increased in obesity, especially in abdominal obesity which is the major component of metabolic syndrome and it can be reduced by weight loss[7]. Many studies demonstrated that increased oxidative stress is associated with insulin resistance pathogenesis by insulin signals inhibition and adipokines dysregulation[8,9]. In animal studies, oxidative stress enhances insulin resistance. The evidence suggested that angiotensin Ⅱ (Ang Ⅱ) infused rats required the increased glucose load to maintain normal glucose levels during hyperinsulinemic clamp to stimulate ROS production[10]. Thus, ROS may also contribute and accelerate the insulin resistance development in insulin-targeted organs of the over nutrition and the excess salt individuals.
In the large general population studies demonstrated that insulin resistance is multifactorial[11,12] and the genetic component[11,13,14]. Insulin resistance most often precedes in many years before the onset of T2DM. Insulin resistance and the consequence of declined of insulin secretion are the principle of the T2DM patho-genesis[11,12,15,16]. The late complications of diabetes have been associated and implicated in their etiology with oxidative stress[17-19]. The influence of oxidative stress on insulin resistance, dyslipidemia, abnormal lipoprotein production and the pathophysiology of T2DM by using in vivo, in vitro and animal models data on these effects
were also included in this review.
ROSOxygen exists in air known as oxygen molecule (O2) or dioxygen. Oxygen on the surface of earth appeared in significant amounts approximately 2.5 × 109 years ago. It was created by the photosynthetic activity of plants and microorganisms (blue green algae). Increased atmospheric oxygen concentration was followed by the ozone layer formation in the stratosphere. Both oxygen and ozone layer were filters against the solar ultraviolet radiation reaching surface of the Earth. In eukaryotic cells, ROS is produced as the consequence of the normal aerobic physiological metabolism[1]. These ROS levels are counter-balanced with the cellular antioxidants in the normal physiological conditions. ROS define as diverse chemical that have reactive properties are capable to accommodate or donate electrons (e-) to the broad range of biological molecules. These species includeinstability radicals arise from an unpaired e-. Existence of the presence of oxygen and the aerobic organisms on the earth is possible[20].
O2 + e + H+ → HO2• (hydroperoxyl radical)
HO2• → H+ + O2•
- (superoxide radical)O2•
- + 2H+ + e → H2O2 (hydrogen peroxide)H2O2 + e → OH- + OH• (hydroxyl radical)
However, these molecules are also played an adverse role in the biological systems as oxidative stress. At the steady state of the living systems, oxygen metabolism always produce oxygen-derived free radicals such as superoxide O2
•-, hydroxyl OH•, alkoxyl RO•, peroxyl RO2
•, peroxynitrite ONOO- and oxygen-derived non-radicals such as hydrogen peroxide H2O2, hypochlorous acid HOCl and hypobromous acid HOBr. Both free radicals and non-radicals groups are the important factors of the oxidative stress mediated cellular damages[21]. Normally, the neutralization of ROS productions by cellular antioxidant defense mechanisms are determine as the physiological state and do not cause any oxidative damage[2]. The imbalance of the ROS production and antioxidants defense system in the living systems caused oxidative stress brings to cellular function disruption and damage[3].
This imbalance occurs due to over production of ROS and reduction of the antioxidant defense mechanisms. The electron transport chain in mitochondrial, peroxisomes and cytochrome P450 system are the most important sources of ROS production (involves in O2
•- production)[22]. Moreover, various enzymes can be accelerated ROS production such as cyclooxygenases[23,24], xanthine oxidase[25], uncoupled nitric oxide synthases (NOS)[26-28] and NADPH oxidases[29]. Drugs such as doxorubicin[30,31], cisplatin, acetaminophen[32-34] and nimesulide[35]. Heavy metals (Fe, Cd, Pb, Hg) as the toxic substances [36-39], acrolein, chloroform, carbon tetrachloride[40], tertiary butyl hydroperoxide[41-44], environmental pollutants (oxides of
Tangvarasittichai S. Oxidative stress and T2DM
457 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

nitrogen, SO2, CO2), xenobiotics, UV irradiation and the other factors induce ROS overproduction.
In metabolic disorders assist the increased ROS production in the physiological system such as obesity, insulin resistance and diabetes mellitus[45-48]. In Figure 1 summarized of obesity and metabolic syndrome elevate in oxidative stress. Superoxide radical (O2
•-), hydroxyl radical (OH•) and hydrogen peroxide (H2O2) are the three major ROS in physiological organisms[49]. Superoxide radical (O2
•-) acts as the parent ROS molecules caused from the one electron reduction of oxygen molecule by electron transport chain enzymes in mitochondrial such as enzymes in cytochrome P450, cyclooxygenase and NADPH oxidase. Various reactions of enzymes and non-enzymes system further convert these ROS molecules to hydroxyl radical (OH•), peroxynitrite ion (ONOO-) and hyperchlorous acid (HOCl). For example superoxide dismutase converts O2
•- to H2O2 by the dismutase reaction[50,51].
Elevated ROS molecules caused the cellular ma-cromolecules damage such as lipids[52], proteins[53] and nucleic acids[54]. In the anti-oxidants system of the living system, possess own antioxidant defense mechanisms[55] includes enzymes and non-enzyme molecules such as SOD, catalase (CAT) and glutathione peroxidases (GPx). Enzyme SOD catalyzes O2
•- conversion to H2O2, while CAT converts H2O2 to H2O and O2. For reduction of two peroxide molecules use non-enzymatic glutathione (GSH; reduced and oxidized forms), reduced glutathione (GSH) and GPx catalyze to produce oxidized glutathione (GSSG) and water[56]. Various enzymes play the important combination roles in the series of antioxidant defense systems such as glutathione reductase, glutathione S-transferase, and glutathione disulfide (GSSG).
ROS production is identified as endogenous and exogenous source. UV exposure and xenobiotic agents has been shown to generate these ROS[57]. In fact, dietary is the major source of these oxidant compounds, especially in animal fat as the source of high lipid peroxides[58]. ROS may also be derived from the general biochemical reactions in living organism to generate ROS as by-products or end products. In the transition heavy metals such as iron (Fe2+) and copper (Cu+) are pose the oxidative stress production, especially in
Fe2+ may cause autooxidation to cause O2•- generation
and/or interaction with H2O2 can generate OH• via the Fenton and Harber Weiss reactions[59]. Fenton chemical reaction may also causes lipid peroxides generation and propagation[60].Auto oxidation of Fe2+:
Fe2+ + O2 → Fe3+ + O2•-
Fenton reaction:H2O2 + Fe2+ → Fe3+ + OH- + OH•
Haber-Weiss reaction:
The major cellular oxidative stress is come from mitochondrial respiration. Heart, brain, kidney, liver and skeletal muscle are the effective oxygen consumption organs is converted oxygen to O2
•-, approximate releasing 0.1%-0.2% while the liver is converted oxygen to O2
•-, approximately releasing 2%[61]. Electron transport chain complex of the mitochondrial has been sourced to O2
•- generation and have been estimated upto 107 ROS molecules per mitochondria per day[62].
In the enzymatic systems of xanthine oxidase generated via xanthine dehydrogenase, which utilize oxygen molecule as e- acceptor during catabolism of xanthine. Xanthine oxidase is the generator of O2
•-, H2O2
[63] and OH• producer[64], highly expressed in epithelial, injured and diseased tissues as shown in Figure 2. Xanthine oxidase has been involved to peroxynitrite (OONO-) and nitric oxide (NO) productions through nitrite reduction[65,66]. Intracellular nitric oxide synthases (NOS) catalyze L-arginine to form citrulline and NO. Endothelial NOS and neuronal NOS are activated by calcium-induced calmodulin binding to produce NO levels[67]. Inducible NOS (iNOS) has also calmodulin bound molecule. It may rapid and chronic expression in many cell types such as smooth muscle cells, hepatocytes and macrophages. INOS is induced by the many inflammatory cytokines
458 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
FeH2O2 + O2•- O2 + OH- + OH•
Obesity/MetS
Oxidative stress ↑
Inflammation
Dyslipidemia
T2DM
Hyperinsulinemia insulin resistance
Atherosclerosis
Figure 1 Summarized of obesity and metabolic syndrome elevate in oxidative stress. T2DM: Type 2 diabetes mellitus; MetS: Metabolic syndrome.
NAD(P)H
NAD(P)+GRed
NAD(P)H oxidase
GSSG
GSH GPxH2O
O2
SOD
Xanthine
Oxidase
Hypoxanthinexanthine
Xanthineuric acid
Mitochondria
Fe2+
Fe3+
O2•-
Lipid peroxidation
DNAdamage
•OH
H2O2
O2 O2•-
Figure 2 Increased oxidative stress by xanthine oxidase. NADH: Nicotinamide adenine dinucleotide.
Tangvarasittichai S. Oxidative stress and T2DM

the chain-breaking antioxidant (vitamin E) agent is added to terminate the chain reaction. The three stages of lipid peroxidation are initiation, propagation and termination. Hydroxyl radical (•OH), alkoxyl radical (RO•), peroxyl radical (ROO•), and HO2
• species can abstract the first hydrogen atom of polyunsaturated fatty acid but not H2O2 or O2
•-[70]. Variety of lipid hydroperoxides and cyclic peroxides are the end products of the chain reaction. Lipid peroxides are stable molecules in the physiological temperatures. Lipid peroxides decomposition is catalyzed by transition heavy metals. For example, iron ion-active complexes present in circulating can participate in the Fenton reaction to promote lipid peroxide decomposition. Hemoglobin and the cytochromes molecules can also facilitate peroxide decomposition, although they do not directly catalyze Fenton chemistry. However, hemeproteins can release chelatable iron that can participate in Fenton chemistry[71]. Ferritin and hemosiderin are effective at stimulating lipid peroxidation and catalase is weakly effective, caused problems to use catalase as a probe for H2O2 in lipid peroxidation systems[72].
Reduced heavy metal [Fe+2, Cu+] react with lipid peroxides (LOOH) to alkoxyl radical or Cu+ react with LOOH to alkoxyl radical.
LOOH + Mn+ → LO• + M(n+1)+ + OH-
In the reaction oxidized-heavy metals [Fe+3, Cu+2] slowly react with LOOH to produce alkoxyl and peroxyl radicals. Both peroxyl and alkoxyl radicals initiate the chain reaction by reducing hydrogen atoms (Figure 3). The fixed oxidation metals ions can affect the rate of lipid peroxidation (Ca2+, Pb2+ and Al3+ ions). Lipid peroxidation accelerates by the iron salts stimulation result in the membrane structure changes and important implications for environmental toxicology[73].
Rawls et al[74] demonstrated that singlet O2• is
formed during the lipid peroxidation degradation and might contribute to cause more initiation in the chain
[tumor necrosis factor-α (TNF-α), interleukin-6 and growth factors] regulation at the transcriptional level, results in micromolar NO production[67]. INOS can poduce O2
•- and OONO- when lower in L-arginine substrate[68].
LIPID PEROXIDATIONFats and oils oxidized with characteristic changes in texture, color, taste and odor. This process, known as rancidity, was chemically defined in the 1940s as an autoxidative free-radical chain reaction[69]. The most powerful oxidant formed in biological systems is hydroxyl radical. It can attack any biological molecule. The initiation step of lipid peroxidation occurred when hydroxyl radicals attack to polyunsaturated fatty acids, to cause the free-radical polyunsaturated fatty acids oxidation in biological systems. Lipid peroxidation is autocatalytic lipid hydroperoxides radical production mediated poly-unsaturated fatty acids in cell membranes destruction and degradation process[4,5]. Conjugated dienes and MDA, by-products of lipid peroxidation are increased in the circulation of obesity, metabolic syndrome and T2DM patients.
First-peroxidation chain initiation, results from the attack by any species to reduce a hydrogen atom from methylene (-CH2-) group of polyunsaturated fatty acid or membrane. Because one hydrogen atom contains one electron, reduction leaves an unpaired electron on the carbon of -CH-, double bond in the fatty acid weakens the C-H bonds on the carbon atom adjacent to the other double bond and facilitates it removal. Then, the polyunsaturated fattyacid chains in lipids membrane are sensitive to cause lipid peroxidation. The carbon-centered radical forms a conjugated diene by the molecular rearrangement (Figure 3), which combines with oxygen to form a peroxyl radical that able to reduce a hydrogen atom from another fatty acid to start a chain reaction. Peroxidation continues to use up the polyunsaturated fatty acid substrate unless
459 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Fatty acid with 3 double bond
RH
R•
R•
RO2•
ROOH
Fragmentation to aldehyde(including malondialdehyde and polymerization products)
H•
Molecularrearrangement
O2 oxygenuptakes
O
O H•
O
O
H
Hydrogen abstraction
Conjugated diene with UV absorbance at 234 nm
Peroxylradical: abstracted H+ from another fatty acid causing an autocatalytic chain reaction
Lipid hydroperoxide
Cyclic peroxide
Cyclic endoperoxide
Figure 3 The chain reaction of lipid peroxidation.
Tangvarasittichai S. Oxidative stress and T2DM

reaction. Initiation in the first-chain initiation should be used as lipid peroxide decomposition reactions to start the new chain reaction. Iron ions and ferrous ions are free radicals[55], can act in electron transfer reactions with oxygen molecule. Then, the presence of iron ions can promote the hydroxyl radicals formation by Fenton reaction. Bielski et al[75] demonstrated that the •OH radical production in any source can initiate lipid peroxidation reaction.
LH + OH• → L• + H2O
Superoxide-dependent Fenton reaction (superoxide resulting H2O2 and reducing Fe3+ to Fe2+) did not demonstrate any substantial involvement of the hydroxyl radical in liposomal peroxidation systems as detected by the scavengers action[76]. Hydroxyl radicals in the systems can be measured by spin trapping[77] or deoxyribose degradation measurements[76] but do not contribute to the lipid peroxidation rate[76]. The addition of iron ion in any preparations can stimulate peroxidation reaction by lipid hydroperoxide degradation to generate peroxyl (LO2
•) and alkoxyl (LO•) radicals.
The rate constant of the reaction when ferrous ions are reacted as 1.5 × 103 /mol/L per second[78], which is higher than the rate reaction constant of ferrous ions with H2O2 reaction (76 /mol/L per second)[79]. The iron ions stimulate lipid peroxidation by the lipid degradation reactions from the present of abundant hydroperoxide.
Iron or copper in a biological system attach to biological molecules at the specific location of OH radicals formation to cause lipid, protein and DNA damage. On lipid membrane, the propagation step of lipid peroxidation reactions does not proceedes further until the reaction reach the protein portion. Thus, lipid peroxidation in vivo causes proteins membrane damage[80,81]. This damage has more biologically important than those lipids membrane damage. Cells also contain mechanisms for recognizing and removing oxidative modified proteins[80,81].
OXIDATIVE STRESSOxidative stress occurs at the molecular level as the cellular event when increased ROS overwhelm the antioxidant defense capabilities systems. Oxidative stress was defines as the increasing ROS production, vary in intensities, the different cellular locations and may be occurred either acutely or chronically[82]. Oxidative damage to macromolecules including carbo-hydrates, proteins, lipids and DNA typically viewed as increased ROS induced cellular damage to cause the irreversible macromolecules modifications. Therefore, the by-products of these oxidative modified biomo-lecules are used as oxidative stress biomarkers in vivo and in vitro. Many research studies demonstrated the association of oxidative stress and the pathogenesis
of insulin resistance via insulin signals inhibition and adipocytokines dysregulation[8,9]. Oxidative stress biomarkers included MDA[83], 4-hydroxy-2-nonenal and isoprostanes species[84], protein carbonyls, 3-nitrotyrosine, hydroperoxides, protein oxidation products[85], glycation end products, carbohydrate modifications[86] and 8-hydroxy-2′-deoxyguanosine (8-OH-dG), an oxidized DNA product[84].
Assaying lipid peroxidationThe lipid peroxidation contributes to the pathogenesis of atherosclerosis. It is occurred in the blood vessel walls and does not occur from low density lipoproteins (LDL) in circulation[87,88]. LDL can enter to the blood vessel walls. The modified LDL (oxidized LDL) may escape from the scavenger recognition receptors and back to the circulation. Therefore, this circulating LDL peroxidation is a potentially useful biomarker of lipid peroxidation in circulation. Indeed, this assay is used for the demonstration of in vivo antioxidants inhibit the effects of lipid peroxidation[89,90].
Thiobarbituric acid-reactive substanceMDA from the oxidative polyunsaturated fatty acids (PUFA) degradation is determined by the reaction of thiobarbituric acid (TBA) with MDA to generate the stable end product of MDA-TBA adduct[91-95]. This MDA free radical has been demonstrated as a causative of the atherosclerosis pathogenesis[96,97], aging[98], cancer[99] and Alzheimer’s disease[100,101]. Serum MDA levels have been used as the lipid peroxidation biomarker and indicator of free radical damage[37,83,102]. MDA, the three-carbon dialdehyde, can exist in many forms in the aqueous circulation. This method was used the reaction of MDA with TBA and heated under acidic conditions but the TBA can react with many chemical species such as proteins, phospholipids, aldehydes, amino acid and nucleic acids[103,104]. One MDA molecule reacts with TBA two molecules to form a stable pink to red chromophore that absorbs maximally at 532 nm[105] or fluorescence detection. This chromophore is termed thiobarbituric acid reacting substances. Elevated MDA levels in T2DM patients are associated with cardiovascular disease risk[83].
IsoprostanesThe most valuable of lipid peroxidation biomarker in the biological system is the isoprostanes, elevated from the PUFA peroxidation[106-113]. Isoprostanes identified as free form and the most are esterified to lipids in circulation. Isoprostanes can be analyzed by mass spectrometry techniques, so that can easily be detected in human body fluids[108,109,112,113]. Isoprostanes appear to turn over rapidly in metabolized and excreted[108,109]. Isoprostanes and their metabolites detection in urine may be the useful biomarker for lipid peroxidation[113]. Isoprostanes assay have focused on the F2-isoprostanes measurement, which elevate from the arachidonic acid peroxidation[109]. Elevation of F2-isoprostanes levels have
460 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Fe2LOOH LO• + LO2
• + H2Ocomplexes
Tangvarasittichai S. Oxidative stress and T2DM

been shown in conditions of the cardiovascular disease, diabetes development[114,115], cigarette smoking[111,116,
117], hyperhomocysteinaemia[118] and hypercholestero-laemia[110,119]. F2-isoprostane levels have also been shown to decrease by antioxidants supplementation both in animal models and humans subjects[120-124].
Oxidative stress in metabolic syndromeThe components of metabolic syndrome consist with abdominal obesity, dyslipidemia, hypertension and diabetes[125,126]. It is the major modern lifestyle com-plication cause from physical inactivity and overeating and associated with the increased risk of cardiovascular diseases, hypertension and T2DM that summarized in Figure 4.
Over nutrition and oxidative stress: In metabolism of glucose through glycolysis and tricarboxylic acid (TCA) cycle to generate nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) as the electron donors. In over nutrition, the excessive glucose occur and a large amount of glucose is oxidized in the glycolysis and TCA cycle to increase NADH and FADH2 generation in electron transport chain of mitochondrial and increased superoxide generation[127]. The excessive of free fatty acids (FFAs) leads to increase FFA-oxidation and acetyl coenzyme A (CoA) oxidation in TCA cycle generate the NADH and FADH2 electron donors as glucose oxidation results in mitochondrial ROS overproduction[127]. Furthermore, NADPH oxidase in the plasma membrane can convert oxygen molecule to superoxide radical and involve in ROS nutrient-based generation. In adipocytes, ROS is generated by in fused
with FFAs, treatment with NADPH oxidase inhibitor can block this ROS generation. This indicates that NADPH oxidase involves in fatty acids ROS generation[8]. Palmitate can activate diacylglycerol synthesis and protein kinase C (PKC) leading to activate NADPH oxidase[128]. Thus, over accumulated fat result in the increased fatty acids oxidation and lead to activate NADPH oxidase (in local or remotely cells) to cause ROS over production in over nutrition or obesity (Figure 4). Conversely, calorie restriction may be associated with normal physiological system[129] and may involve in normal cellular redox state[130]. In aged animals models treated with antioxidant agents or hypocaloric diets led to ameliorate in oxidative stress status and tissue function[131,132]. Treatment with resveratrol, a polyphenol reduced atherosclerosis and diabetes development[133]. These studies demonstrate that nutrition is associated with increased or decreased redox status and over nutrition result to increase oxidative stress to contribute pathogenesis of atherosclerosis, cancer and other diseases.
Oxidative stress in adipose tissue: Increased fat accumulation in human has been associated with oxidative stress biomarkers[134]. Similarly, obese mice were significantly higher oxidative stress levels in circulation[8]. Moreover, lipid peroxidation and H2O2 levels were increased in adipose tissue[8]. These mean that adipose tissue may the major source of ROS production and can be released to the circulation potentially affecting various distance organs functions and damage (Figure 4).
Increased NADPH oxidase expression in adipose
461 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 4 Summarized the increasing reactive oxygen species in obesity, metabolic syndrome and salt sensitive hypertension. FFA: Free fatty acid; MetS: Metabolic syndrome; HT: Hypertension; IGT: Impaired glucose tolerance.
Tangvarasittichai S. Oxidative stress and T2DM
Oxidative stress
MetS-salt-sensitive HT
CVD MetS
Diabetes (chronic hyperglycemia)
IGT(post prandial hyperglycemia)
Over nutritionDecreased physical activity
GlucoseFFAOrgan-celluar overload
AdipocyteMuscleLiver ↓Insulin resistance
Sodium balance ↓Renin-angiotensin system
Endothelial cells↓
Endothelial cells dysfunction
Insulin secretion ↑(β-cells)HyperinsulinemiaInsulin activty ↓

tissue associated with increased oxidative stress levels. Increased mRNA expression was found in adipose tissue of obese mice[8]. Increased ROS generation in lipid accumulation and further elevating ROS generation with FFA treatment were found in 3T3-L1 adipocytes cultured[8]. These ROS generation processes can be blocked by NADPH oxidase inhibitors, apocynin or diphenyleneiodonium. Many studies suggest that NADPH oxidase induces adipocytes ROS production[8]. Moreover, obese mice ameliorated hyperinsulinemia, hypertriglyceridemia, hyperglycemia and hepatic steatosis by supplementation with apocynin[8]. These data demonstrate that NADPH oxidase increase ROS production in obesity and metabolic syndrome may play the important roles in the atherosclerosis, T2DM and cancer pathogenesis. Adipose tissue tries to increase antioxidant enzymes levels to against ROS over production. However, these antioxidant enzymes activity and expression are decreased in adipose tissue[8,135-137]. Then, increased ROS-production enzymes and decreased antioxidant enzymes may cause oxidative stress in obese and metabolic syndrome.
Oxidative stress and salt-sensitive hypertension: As in mention above, ROS levels are increased in obesity and can be ameliorated by weight loss[7]. Obese rats induced by refined sugar or high fat diet leading to ROS overproduction and increase oxidative stress[6,138]. Many research evidences suggest that metabolic syndrome was associated with the salt-sensitive hypertension. ROS play the roles as mechanical link of metabolic syndrome and salt-sensitive hypertension[125,126], which itself leads to ROS overproduction[139-142]. Salt restriction in hypertensive obesity was more effective reduction in blood pressure than in hypertensive non-obesity patients, and weight loss in obesity and salt sensitive hypertensive patients caused the successful of blood pressure reduction[143]. Salt-sensitive hyper-tensive patients were significantly more prevalent in metabolic syndrome patients than without metabolic syndrome[144]. Oxidative stress in abdominal adipocytes due to increase adipocytokines secretion such as TNF-α, angiotensinogen, non-esterified fatty acids[126]. Interestingly, infused Ang Ⅱ-rats disturbed sodium balance to cause ROS overproduction in salt-sensitive rats[139-141]. Moreover, in salt-sensitive hypertensive patients are also increased 8-isoprostane levels[142]. Thus, ROS may the underling pathogenesis of diseases in metabolic syndrome, obese and non-obese intake excessive salt as the salt-sensitive hypertensive patients.
In high-renin patients (non-modulating salt sensitive hypertension) had elevated the homeostasis model assessment of insulin resistance (HOMA-IR) levels[145]. Insalt-sensitive hypertensive non-obesity patients had significantly lower insulin sensitivity than in non-salt-sensitive hypertensive patients[146]. Insulin resistance caused salt-sensitive hypertensive obesity and/or metabolic syndrome patients[125]. Increased renal ROS overproduction may increase the salt sensitive
hypertension[147]. Then, increased renal oxidative stress may contribute to cause salt-sensitive hypertension development. Moreover, ROS overproduction in vascular endothelial cells suppresses the NO-dependent vaso-dilation[148] and may play the role in the salt-sensitive hypertension development.
Oxidative stress in type 2 diabetesMany research studies demonstrated that T2DM patients have increased ROS production-induced higher oxidative damage in the circulation and also have reduced antioxidant defenses mechanisms[149-152]. Increased ROS production in T2DM patients is thought to activate many detrimental pathways including hexosamine pathways, advanced glycation end-products (AGEs) formation, and PKCβ1/2[127]. Hyperglycemia condition can induce oxidative stress by several mechanisms such as glucose autoxidation, polyol pathway, AGE formation and PKCβ1/2 kinase. Elevated free fatty acids, leptin and other circulating factors in T2DM patients may also contribute to cause ROS overproduction. Figure 5 demonstrates the association of increased ROS production with atherosclerosis and sources of ROS generations in T2DM patients.
Glucose autoxidationHyperglycemia due to cause increased glucose metabolism leading to increase NADH and FADH2 overproduction, which are used by the electron transport chain of mitochondria to generate ATP[153]. NADH overproduction can cause the higher proton gradient production in mitochondria. These electrons are transferred to oxygen to produce higher superoxide[154]. The NADH dehydrogenase of the complex Ⅰ ubiquinone oxidoreductase and complex Ⅲ cytochrome c reductase are the two main site of superoxide production via the electron transport chain[155].
The polyol pathwayOxidative stress increased in circulation of T2DM patients from the polyol pathway. ROS was generated by two enzymes: (1) Aldose reductase in the reaction use NADPH to change glucose to sorbitol. Sorbitol production is a minor reaction in normal physiological conditions. However, 30%-35% of glucose in T2DM conditions is metabolized by polyol pathway[156]. In the condition of sorbitol overproduction, the availability of NADPH is reduced this reflect to reduce glutathione regeneration and NOS synthase activity to cause increased oxidativestress[153]; and (2) Sorbitol dehydrogenase in the second step oxidizes sorbitol to fructose concomitant with NADH overproduction. Increased NADH may be used by NADH oxidases to increase superoxide production[157] include in mitochondrial over superoxide production.
PKCβ1/2Many structures and biochemical components changed in the circulation of T2DM patients were caused from PKCβ1/2 activation via diacylglycerol leading to cause dysfunction in endothelial contractility and permeability,
462 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

hemodynamics (retinal blood flow) changes, extra-cellular matrix protein synthesis, VEGF production and intracellular signaling in the vascular[128,158,159].
Non-enzymatic glycationGlycation end-product is the binding of ketone or aldehyde groups of glucose with the free amino groups of proteins leading Schiff bases formation without enzymes, then to form the Amadori product and rearrangements of the structure to the irreversible AGEs in the final[160,161]. AGEs has been demonstrated in atherosclerotic lesions and their tissue of T2DM patients and increased AGEs levels associated with severity of the diseases[162]. Moreover, binding of AGEs to specific cell surface receptor for AGE can activate intracellular redox signaling and subsequent to activate the expression of redox-sensitive transcription factors and inflammatory mediator[163-165].
InflammationOxidative stress is the major factor underlying in the CVD, insulin resistance and T2DM pathogenesis. These may explain by the presence of the inflammation conditions. Now, inflammation recognized as the onemanifestation of oxidative stress[166] and can be gene-rate the inflammatory mediators including adhesion molecules and interleukins to induce oxidative stress[166]. The concept of atherosclerosis is an inflammatory disease now well established. This chronic inflammation may be involved in the insulin resistance and T2DM pathogenesis[167]. Recent clinical research indicates that sub-clinical inflammation may impact in the development and progression of diabetic complications[165,168]. Moreover, excessive FFA and glucose induce inflammation effect through oxidative stress and reduced antioxidants[169]. Interestingly, the subclinical pro-inflammatory state
observed in many pathogenesis conditions such as atherosclerosis, aging, T2DM and cancer, is caused from mitochondrial ROS overgeneration[170].
Other sources of oxidative stress in diabetesNon-esterified FFAs are elevated in T2DM patients[171]. These excessive FFAs enter the citric acid cycle to generate acetyl-CoA to receive NADH overproduction to cause mitochondrial superoxide over production. In humans, infused FFA has been shown increased lipid peroxidation by elevated isoprostanes marker levels[172,173]. Adipocytokine, leptin is secreted from the adipocytes to act on the central nervous system to decrease food intake. It reflects all effects on the vascular smooth muscle cells, endothelial cells, macrophages and monocytes[174]. Leptin levels are increased and associated with cardiovascular disease in T2DM patients[175-177]. In culture of endothelial cells incubated with leptin to cause ROS production[178,179].
AntioxidantsRegulation of the cellular redox status is depends on the rate of ROS counterbalance and elimination from the enzymatic and/or non-enzymatic antioxidants. Superoxide is converted by SOD to H2O2 and O2 molecule. There are 3 isoforms of SOD such as cytosolic Cu/Zn SOD (SOD1), mitochondrial Mn-SOD (SOD2) and extracellular SOD (SOD3). Catalase, the heme metalloenzyme is expressed in peroxisomes, mitochondria, cytoplasm and nucleus. H2O2 is catalyzed by catalase to oxygen and water[180]. While glutathione peroxidase the selenoprotein, was found in both intracellular and extracellular. Gluta-thione peroxidase has a highly sensitive function for lipid peroxides degradation, converses H2O2 to water by using the thiol group of glutathione[181]. Their H2O2
463 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Atherosclerosis
Diabetes
Glucose ↑ FFA ↑
Auto oxidation Polyol pathway AGEs
Endothelial celldysfunction
oxLDL ↑
Cytokinesadipokinesprostanoids
Insulin resistance
β-cell dysfunction
Oxidative stressROS
Inflammation Monocyte activation Cytokines VSMC migration/proliferation
Figure 5 Summarizes the reactive oxygen species associations with atherosclerosis and sources of reactive oxygen species production in type 2 diabetes. oxLDL: Oxidized low density lipoprotein; FFA: Free fatty acids; AGEs: Advanced glycation end-products; VSMC: Vascular smooth muscle cells; ROS: Reactive oxygen species.
Tangvarasittichai S. Oxidative stress and T2DM

detoxification plays the important roles to prevent lipid peroxidation production and regulation of the cellular redox status[182]. The glutathione system, thioredoxin peroxidase is key enzyme to regulate the cellular levels of thiol/disulfide while the production of antioxidant enzymes is regulated by the redox-cellular transcription factors[183]. For example, the expressed transcription factor NF-E2 related factor in the cytosolic is interrupted binding with Keap-1 as the responsible to increase oxidative stress and translocate to the nucleus for initiation of the transcription of the various antioxidant enzymes[184] as the strategy to develop many class of antioxidant, anti-inflammatory, and anticancer agents. Reduction in non-enzymatic antioxidants, thiol glutathione and thioredoxin are the major dysregulation of the cellular redox status[185]. The cellular redox status is reflected by the reduction of glutathione (GSH), oxidized glutathione (GSSG) ratio (or GSH:GSSG ratio), ascorbic acid, tocopherols and methionine and cysteine amino acids. Exogenous herbal antioxidants compounds in dietary foods include flavanoids, anthocyanins and polyphenolics act as ROS scavenging[186,187]. The direct interaction of ROS with non-enzymatic antioxidants is based on chemical structure properties. In free radicals participate in 1e- oxidation while non-radical species was 2e- oxidation. For example, O2
•- and OH• radicals react with the ascorbic acid and thiols. While the OH• more activity and instability react with methionine and tocopherols. H2O2 and the non-radical may react with thiols and methionine, and the OONO- discriminate to react with thiols, ascorbic acid, tocopherols and methionine[188].
Oxidative stress induces insulin resistanceOxidative stress plays the major role in the association with the insulin resistance pathogenesis by insulin signals disruption and adipocytokines dysregulation[8,9]. In rat models, oxidative stress enhances insulin resistance. The evidence suggested that Ang Ⅱ infused rats required the increased glucose infusion to maintain euglycemia during hyperinsulinemic clamp to stimulate ROS production[10]. For this example, Ang Ⅱ-infused rats were caused insulin resistance from the suppression on insulin-induced glucose uptake in skeletal muscle and increased in oxidative stress biomarkers in this animal experiment. In experimental model, superoxide dismutase and tempol can reduce the insulin resistance. Many evidences indicated that ROS overproduction may induce insulin resistance and confirmed by the supplementation of antioxidant tempol to cause insulin resistance amelioration in Ren-2 transgenic rats[189]. Insulin-target organs of the obese and diabetic KKAy mice were stimulated and caused ROS over production (skeletal muscle, liver and adipose tissue)[8] and to cause insulin resistance in these organs. High fat-fed mice found ROS overproduction in liver and adipose tissue of these obese mices to induce insulin resistance[190]. Many research studies suggested that antioxidant agents decreased plasma insulin, glucose, triglycerides levels and ameliorate insulin resistance in KKAy mice
with no weight loss[8]. Antioxidant coenzyme Q10 supplementation can ameliorate the increased insulin levels in circulation of SHR/cp rats[191]. As mention above, in over nutrition, the excessive glucose occur and a large amount of glucose is metabolized in the glycolysis and TCA cycle leading to increased NADH and FADH2 production in electron transport chain of mitochondrial and increased superoxide production[127]. In aged animals models treated with antioxidant agents or hypocaloric diets led to ameliorate in oxidative stress status and tissue function[131,132].
Insulin resistanceIn general population, insulin resistance precede in many years before onset of T2DM and it is also multi-factorial[11,12] such as genetic component[11,13]. Insulin resistance and reduction in insulin production are the major characteristics of the T2DM pathogenesis[11,12,14-16]. Modern lifestyle, physical inactivity, abdominal obesity and excessive of adipokines can cause insulin resistance[11,15]. In early stage, normal glucose tolerance is preserved by compensation hyperinsulinemia. About 25% of non-diabetic subject cause insulin resistance in the same ranges that found in T2DM patients[12]. Insulin resistance continuous increases and/or decreases in insulin secretory compensation responses, the deterioration into impaired glucose tolerance occurred. Increased glucose, FFA and insulin levels lead to ROS overproduction, increased oxidative stress and activate stress transduction factor pathways. This can cause insulin activity inhibition and secretion to accelerate the onset of T2DM as shown in Figure 6.
Oxidative stress has been demonstrated the implication and association in the late complications of diabetes mellitus[17,18] as in the schematic of Figure 5. Many studies have demonstrated ROS overproduction and increased oxidative stress to insulin resistance[192-194]. Both in vitro studies and in animal models demonstrated that α-lipoic acid (LA), antioxidant agent increase insulin sensitivity[194-196]. In clinical trials, supplementation with vitamin C, vitamin E, glutathione increases insulin sensitivity in both insulin-resistance and T2DM pa-tients[197,198]. LA act as insulin sensitizer agent, it increased insulin sensitivity about approximately 25% and approximately 20% higher than metformin and rosiglitazone, respectively[199,200]. Oral supplementation with LA formulation for 6 wk decreased circulating fructosamine levels[201] and increase insulin sensitivity[202] in T2DM patients and the other studies have confirmed 2.5 mmol/L of LA to cause GLUT4 activation and translocation[203-205].
Because insulin resistance occurred before chronic hyperglycemia development[12], that difference from insulin resistance in the pre-diabetic state result from oxidative stress activation by increased glucose levels. However, obesity demonstrated the strong association with insulin resistance. In this regard, the mediator of oxidative stress-induced insulin resistance of the pre-diabetic state might be from the adipocyte-derived
464 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

factor such as TNF-α[206], leptin[207], FFAs[208-210] and resistin[211]. However, the FFAs elevations are associated with insulin resistance and obesity[208,209,212]. Many studies found that increased FFA levels decrease insulin sensitivity, as in the Randle hypothesis[210] and insulin-signaling inhibition[212]. The increased fasting FFA levels are significantly correlated with decreased reduced/oxidized glutathione ratio in T2DM patients[190]. Elevated FFA concentrations cause mitochondrial dysfunction such as uncouplers of oxidative phosphorylation in mitochondria[213] and increased superoxide produc-tion[214]. These caused the exacerbated situation from FFAs induce oxidative stress and reduce intracellular glutathione caused impaired endogenous antioxidant defenses[190,215,216]. Supplementation with glutathione improves insulin sensitivity and β-cell function by the restoration of redox status in T2DM patients and healthy subjects[217].
FFA mediated the nuclear factor-κB (NF-κB) activation, as the consequence of FFAs increased ROS overproduction and glutathione reduction[216,218-220] and also linked to FFA-activated PKC-θ [221] to caused NF-κB activation[222]. Vitamin E supplementation inhibits the FFA-induced NF-κB activation[216] indicated that FFAs act as pro-inflammatory agent effects the alteration of the cellular redox status.
The HOMA-IR was proposed by Matthews et al[223] that can be used to estimate insulin resistance and insulin sensitivity in individuals. HOMA-IR is easy to calculate and no more laborious technique. HOMA-IR method derives from the mathematic calculation from fasting plasma insulin and glucose concentrations.
Oxidative stress and β-cells dysfunctionIncreased circulating glucose levels stimulate the β-Cells
function by sensing and secreting of insulin in appropriate amount[224] and as the target of oxidative stress. The processes are complex and depend on many factors[16]. The critical glucose metabolism in mitochondrial is the importance linking stimulus the insulin secretion[224-226]. Therefore, mitochondria damage and markedly blunt insulin secretion is also occur by the ability of oxidative stress (H2O2)[226]. Many studies in T2DM patients have suggested that chronic exposure to high glucose and/or high FFA levels impaired β-cells function and β-cells dysfunction[16,227]. Because β-Cells are lower in antioxidant enzymes levels (superoxide dismutase, catalase and glutathione peroxidase) and higher sensitive to oxidative stress[228]. Oxidative stress exposure to β-cells activated the increased p21 cyclin-dependent kinase inhibitor production, decreased insulin mRNA, ATP and calcium flux reductions in mitochondria and cytosol to cause apoptosis[226]. Glucose or methyl succinate can stimulate insulin secretion and inhibit by response to K+ within 30 min[226]. The results indicate that mitochondria in β-cells involved in the processes of glucose induced insulin secretion are affected by increased oxidative stress. Lipid peroxidation, oxidative stress products exposed to islets, inhibited insulin secretion and also caused glucose oxidation[229]. Conversely, antioxidants can protect β-cell against the toxicity of oxidative stress, AGEs production and inhibit NF-κB activation[230-234]. These antioxidants are N-acetyl cysteine (NAC), α-phenyl-tert butylnitrone, aminoguanidine and zinc. Recent research study evaluated β-cells function after over expression of glutamine. Hexosamine over production resulted from the deterioration of insulin signaling of glucose-stimulated insulin secretion. Fructose-6-phosphate amidotransferase is the rate-limiting enzyme increase in hexosamine pathway[235], coincident with increased
465 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Hepatic glucose, FFA production ↑
Organ glucose, FFA uptake ↓
Glucose transport (uptake) ↓
Hyperglycemia
Hyperinsulinemia
Insulin activity ↓
Insulin resistance
Organ response to insulin ↓
Insulin receptor deficit
Glucose-inducedinsulin secretion ↓
Impaired β-cell function
Insulin deficiency
Figure 6 Insulin resistancedevelopment and consequence of β-cell dysfunction. FFA: Free fatty acid.
Tangvarasittichai S. Oxidative stress and T2DM

H2O2 production[235] that can ameliorate by NAC supplementation.
β-cells glucose-induced toxicityWest[19] demonstrated that insulin secretion in T2DM patients improved by the reduction of hyperglycemia with diet, insulin or sulfonylureas. On the other hand, in healthy normal, high glucose infused as a clamp reduces insulin secretion[236]. In the study of long term culture of HIT-T15 and/or βTC-6 cells demonstrated that increased glucose levels cause decreased insulin secretion, insulin mRNA and decreased binding of transcription factors[237,238]. Thus, glucose toxicity, the concept of the condition of hyperglycaemia itself can decrease insulin secretion which implies the irreversible damage to cellular components of β-cells[239]. Generally in β-cells, excessive glucose oxidation and metabolism will always cause to ROS over production. Superoxide dismutase and catalase are normally as the detoxified antioxidant enzymes. β-Cells are low amount of these antioxidant enzymes and also low in glutathione peroxidase, a redox-regulating enzyme[240]. Then, hyperglycaemia condition leads to increase ROS production and accumulation in β-cells and subsequent of cellular components damage. Pancreas duodenum homeobox-1 is an insulin promoter activity regulator was loss leading to β-cell dysfunction[240]. Supplementation with NAC and/or aminoguanidine can ameliorate the glucotoxic effects on insulin gene activity[230], reduced insulin levels and increased insulin mRNA and insulin sensitivity[230].
β-cells lipid-induced toxicityLipotoxicity to β-cells concept, elevation of non-esterified fatty acids concentrations in diabetic and non-diabetic obese patients, result of the enhanced adipocyte lipolysis. In the presence of the excessive fatty acid oxidation in β-cells is caused increased long-chain acyl CoA accumulation leading to inhibite β-cells function[241]. This process is as an integral part of the normal insulin secretory function. This long-chain acyl CoA can inhibit the insulin secretory function by opening β-cell K+-sensitive ATP channels. In the second mechanism, in long-term culture of β-cells formulas with FFAs can effect the potential reduction on mitochondrial membrane and uncoupler proteins-2 over expression to cause the K+-sensitive ATP channels opening which lead to decreased ATP production and insulin secretion[242,243]. Third mechanism, β-cells apoptosis might possess from triglyceride or fatty acid induced ceramide synthesis and/or nitric oxide production. Thus, impaired insulin secretion and β-cell dysfunction strongly associated with the FFA-stimulated ROS overproduction[244].
β-cells combined glucose/lipid toxicityElevation of glucose and FFA levels are the major characteristic of T2DM patients. This combination is the major β-cells toxicity and require the maximize protection. In culture cells of islets or HIT cells were
exposed to high concentrations of glucose and FFA levels. There was decrease in insulin-gene activity and insulin mRNA[245]. In the study of islets co-culture with high glucose and palmitate levels caused impaired insulin signaling of the glucose-stimulated insulin secretion[244]. Recent studies have confirmed that β-cells lipotoxicity is the concurrent status as the amplifying effect mediated by glucose toxicity in hyperglycemia condition[246,247].
DyslipidemiaInsulin resistance and T2DM are characterized by dyslipidemia one major risk factor for cardiovascular disease. Lipid triad is the complex metabolic milieu associated with dyslipidaemia[248] comprise with hyper-triglyceridemia, low levels of high-density lipoprotein cholesterol (HDL-C) and the appearance of small, dense, LDL (sdLDL) - and caused excessive post prandial lipemia[249,250]. Diabetic dyslipidemia caused from the disturbance of lipid metabolism, an early event cardiovascular complications development and was preceded in T2DM patients by several years[249-253]. Indeed, insulin resistance status in both with and without T2DM patients was display qualitatively similar lipid abnormalities[250]. The different components of diabetic dyslipidemia are closely linked to each other metabolically[249-253] and are initiated by the elevation of triglyceriderich very LDL (VLDL) from hepatic over production[249,251]. It is the key importance mechanisms to elucidate the over production of VLDL involved in diabetic dyslipidemia[249].
In insulin resistance state, decrease insulin function and lack of insulin inhibits lipolysis leads to increase FFAs generation of and lower lipoprotein lipase activity. This occurs after meal consumption, generates a chylomicron remnant rich in TG[254], caused elevated hepatic FFAs and VLDL TG-rich particles secretion. These processes affects HDL-C metabolism through the interchange with TG-rich lipoproteins via cholesteryl ester transfer protein to produce HDL particles containing high TG concentrations. These HDL-TG particles were hydrolyzed with hepatic lipase to TG and HDL. This HDL becomes smaller and less antiatherogenic activity, easily to remove from the circulation by the kidneys. Moreover, insulin resistance in T2DM patients associated with endothelial dysfunction led to increase risk of CVD[255]. The most atherogenic subfractions of sdLDL are elevated in circulation of obesity individuals, as a key feature in association with elevated triglyceride and low HDL cholesterol. Elevated sdLDL concentrations are also founded in abdominal obesity subjects and demonstrated greater myocardial risk.The mechanisms are related to excess accumulation of abdominal adipose tissues, elevated total cholesterol and LDL-C and related to high saturated-fat consumption, weight gain and obesity.
Dyslipidemia is commonly occurred in T2DM patients and might play the major role in accelerated
466 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

HDL-C can be used as markers of insulin levels, insulin resistance and CVD risk factor[258,263]. The highest % sensitivity and % specificity cut-off points corresponding to the TC/HDL-C, TG/HDL-C ratios and non-HDL-C are 3.58, 2.48 and 130.4, respectively[258]. Because of TC/HDL-C, TG/HDL-C ratios and non-HDL-C are easily calculated and ordered with every lipid profiles available to the clinician and no costs addition. The cut-off value of these ratios in Tangvarasittichai et al[258] study was lower than the results from Western populations[266-268]. Then, insulin resistance was significantly predicted by these markers. For atherosclerotic risk assessment in obesity, metabolic syndrome and T2DM patients requires more attention to lipid screening.
Development of T2DM from insulin resistanceInsulin resistance often occurs with T2DM but is insufficient for the T2DM development. β-cells dysfunction are important event for the T2DM development and progression. In early stage of insulin resistance, β-cells increase the secretory function try to compensate and control hyperglycemia. In Pima Indian population study caused acute insulin response dysfunction or decreased β-cell responses was found during the normal glucose tolerance state in individuals who eventually progressed from normal glucose tolerance to impaired glucose tolerance or T2DM when compared with individuals who persisted in the state of normal glucose tolerance[269]. There was evidence of early defects in glucose disposal by decreased insulin sensitivity before the develop-ment of glucose intolerance state, although output of circulating glucose did not increase until the progression from impaired glucose tolerance to T2DM revealed. Interestingly, individuals who demonstrated transient glucose intolerance but were able to recover and to reach normal glucose tolerance and did not show the early secretory defect observed in progressed individuals[269]. β-cells failure or dysfunction occurred as the results of the combination of increased oxidative stress, glucose and lipids accumulation to cause glucotoxicity and lipotoxicity to β-cells to progress increased apoptosis and loss of the insulin granule secretory components expression[270].
T2DMThe World Health Organization updated the prevalence of T2DM estimated by the year 2025 those 30.3 million people in the United States and total of 380 million people worldwide will be diagnosed as DM[271]. By the year 2050, those 45.6 million Americans will be diagnosed as DM[272]. T2DM is associated with obesity, sedentary lifestyle and lack of exercise in the aging population. There are a number of gene abnormalities related to T2DM, that showed significant differences exist in the abnormalities gene associated with T2DM among the various ethnic populations, such as African Americans, Asians and Europids[273,274]. The contribution of any one of these genes to T2DM is small and total
macrovascular atherosclerotic disease and increased CVD risk in T2DM patients[256]. Dyslipidemia in T2DM patients as lipids triad is characterized by increased insulin levels, hypertriglyceridemia, low HDL-C levels and increased sdLDL-particles (independent of LDL-cholesterol) and increased TG-rich remnant lipoprotein (TGRLs) concentrations[257,258]. In this manner, low HDL-C levels associated with hyperinsulinemia or insulin resistance and insulin signaling for insulin-mediated glucose disposal[259] characterized by higher fasting plasma glucose and insulin levels. Then, these major changes associated with the insulin resistance syndrome are increased TGRLs and decreased HDL-C levels. Thus, in dyslipidemia, using the lipoprotein concentration ratios are associated with insulin resistance and increased CVD risk conditions. Lipoprotein ratios might be useful to identify insulin resistance individuals even different in fasting glucose or insulin levels. Obesity, metabolic syndrome, and T2DM may also show the same dysli-pidemia characteristic[12,257,259] and measuring TG, HDL-C, TC/HDL-C and TG/HDL-C ratio in circulation may also use as insulin resistance estimation. For example, these TG, HDL-C, TC/HDL-C and TG/HDL-C ratio are independently associated with insulin levels, insulin resistance and CVD risk[258,260,261].
Lipoprotein ratios: In description above, the major change is increased TGRLs and decreased HDL-C levels are associated with insulin resistance syndrome. Insulin plays the important role in TG metabolism, in normal condition TGRLs particles reduces synthesis by the distinct pathways when compared with VLDL particles synthesis[249,258]. Insulin fails to suppress VLDL particles synthesis[262]. Insulin resistance is significantly associated with increased lipid synthesis in the liver, increased FFAs flow to the liver and decreased VLDL particles clearance resulting in increased VLDL levels in the circulation[251]. Thus, dyslipidemia (as lipoprotein ratios) may associate with insulin resistance and increased CVD risk. On this basis, waist circumference, LDL-C, TG levels, insulin resistance and the CVD risk are estimated[263]. The major features of dyslipidemia are determined by hypertriglyceridemia, low HDL-C levels and slightly high or normal LDL-C levels with altered composition. Hypertriglyceridemia is indicate as elevated atherogenic chylomicron and VLDL remnant and associated with increased CVD risk[264,265]. These phenomenons demonstrated the problems of VLDL and HDL levels but not the LDL levels and concurrent with increased insulin levels. Low HDL-C level is associated with the hyperinsulinemia and/or insulin resistance and insulin signaling for insulin-mediated glucose disposal[259]. All of these features are associated with coronary heart disease risk in obesity, metabolic syndrome and T2DM patients. The TC/HDL-C, TG/HDL-C ratios and non-HDL-C (as TC - HDL-C) were used as surrogate markers for insulin levels and insulin resistance estimation. In Tangvarasittichai et al[258] study suggests that TC/HDL-C, TG/HDL-C ratios and non-
467 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

aggregate of all described genes accounts for < 15% of the predisposition[273,275]. It is typically diagnosed in patients older than 30 years with overweight or obesity and positive in family history of T2DM. However, insulin resistance may occur and develop in many years before diagnosed as T2DM[276]. Figure 7 summarized the etiology of the T2DM pathogenesis.
Patients are diagnosed as T2DM when plasma glucose levels reach at the diagnostic criteria (Table 1). These T2DM patients are at high risk for microvascular complications (e.g., nephropathy, retinopathy and neuropathy) and macrovascular complications (e.g., peripheral vascular disease, cerebrovascular disease and cardiovascular disease). T2DM patients with good controlled plasma glucose levels demonstrated to delay the progression of microvascular and macrovascular complications[271,277].
MANAGEMENT OF DYSLIPIDEMIA AND HYPERGLYCEMIA IN T2DM PATIENTSFasting serum lipids profile should be determined annually in T2DM patients as in the recommendation by the American Diabetes Association (ADA)[278]. ADA recommended for the satisfied lipids profile level as low-risk by LDL-C < 100 mg/dL (2.6 mmol/L), triglycerides < 150 mg/dL (1.7 mmol/L) and HDL-C > 50 mg/dL (1.3 mmol/L)[276].
TreatmentLifestyle interventions: The American Diabetes Asso-ciation and the American Heart Association recommend that increased physical activity and lifestyle modifications should be advised for all T2DM patients[278,279]. Combination with such interventions included nutrition therapy or supplementation, weight loss and non-smoking. These have been help T2DM patients to receive better controlled their lipid concentrations. Nutrition interventions and supplementations should be designed according to the condition of T2DM individuals such as diabetes status,
age, other comorbidities and avoidance to intake transfat, saturated fat, cholesterol and should increase intake of fiber (fiber in oats, legumes, citrus), omega-3 fatty acids and plant stanols/sterols[278]. Glycemic control can also modify circulating triglycerides levels, especially in T2DM patients with hypertriglyceridemia and poor glycemic control[278].
Pharmacological interventions of dyslipidemiaThere are many pharmacological classes available for dyslipidemia treatment.
Statins: Statins inhibit enzyme 3-hydroxy-3-methyl-glutaryl CoA reductase suppress cholesterol synthesis and increase number and activity of LDL-receptor. Statins are effective drug for lowering LDL-cholesterol, raising HDL-C and reducing TG levels. There are seven pharmaceutical forms of statins including lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin and pitavastatin available in the market. Statins also have the other pharmacodynamic actions such as vascular inflammation reduction, immune suppression, improved endothelial function, platelet aggregability, enhanced fibrinolysis, antithrombotic action, increase neovascularization in ischemic tissue and stabilization of atherosclerotic plaques[280].
FibratesFibrates control the lipid metabolism by mediated through peroxisome proliferator-activated receptors-α activation, stimulation of β-oxidation of fatty acids in peroxisomes and mitochondria to cause lowering fatty acid and triglycerides levels in circulation. The first drug of this class is Clofibrate. Eventually, the revolution in lipid-lowering drugs research discover of many other fibrate drugs such as fenofibrate, bezafibrate, gemfibrozil and ciprofibrate. These drugs demonstrated the adverse effect to cause hepatomegaly and tumor formation in the liver of rodents. Then, they had restricted for the widely
468 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
TNF-α ↑NF-κB ↑Adiponectin ↓
FFA ↑
Liver Muscle, organs
Obesity and abdominal obesityOxidative stress ↑
Insulin resistance
Impaired glucose tolerance
Diabetes mellitusGenes Candidate/susceptibility
Environmental factors Overnutrition High energy intake High fat, carbohydrate, etc . Physical inactivity
Genes
Figure 7 Summarized the etiology of the type 2 diabetes mellitus patho-genesis. FFA: Free fatty acid; NF-κB: Nuclear factor-κB; TNF-α: Tumor necrosis factor α.
Table 1 Type 2 diabetes mellitus and glucose levels for diagnostic criteria1
Glucose management test Range Diagnosis
Fasting plasma glucose (mg/dL) (at least 8 h fast)
≥ 126 Diabetes mellitus
100-125 Impaired fasting glucose ≤ 99 Normal
2-h oral glucose tolerance test of 75 g glucose load (mg/dL) WITH
≥ 200 Diabetes
Random screening with common symptoms of diabetes (polyuria,
140-199 Impaired glucose tolerance
polydipsia, weight loss, etc.) ≤ 139 NormalHemoglobin A1c (%) ≥ 6.5 Diabetes
5.7-6.4 Prediabetes/high risk ≤ 5.7 Normal
1All tests in diabetes range must be repeated after 24 h, to be confirmed diagnosis.
Tangvarasittichai S. Oxidative stress and T2DM

use in humans. Gemfibrozil and fenofibrate are Food and Drug Administration (FDA)-approved for lipid lowering drugs due to milder effect on peroxisome proliferation.
Nicotinic acidLong term study of the coronary drug project demon-strated that niacin is the effective drug to increase HDL-C levels and reduced CVD events[281] in a non-diabetic subjects. Niacin cause adverse effects on the glycemic control levels in T2DM patients. In high doses treatment with niacin may increase blood glucose levels. The modest doses of 750-2000 mg/d of niacin are significantly increased HDL-C levels and decreased LDL-C, triglyceride levels and accompanied with modest changes in glucose levels for diabetes therapy[282,283]. However, there is no evidence for the CVD outcomes reduction with niacin supplementation in T2DM patients.
Antihyperglycemic drugs: The standard care for T2DM patients is mainly in controlled blood glucose levels by using glycemic lowering drugs and concomitant with controlled diet and increased physical activity. With proper controlled and managed these contributors such as circulating glucose levels, hemoglobin A1c, lifestyle modifications, these can be effectively controlled and reduced the progression and complications disease. In general, only approximately 50% to 60% of T2DM patients have achieved their glycemic goals[284]. There are many reasons for poor control of T2DM includingmedication efficacy, adverse effects, access to medi-cations and health care education, poor adherence, lack of lifestyle changes and no physical activity. Now a day, more pharmacologicals for T2DM treatment have been approved for use. There are 12 classes of antihy-perglycemic drugs FDA-approved in the United States[285]
such as sulfonylureas, meglitinides, thiazolidinediones, dipeptidyl peptidase-4 (DPP-4) inhibitors, biguanides, sodium glucose transporter 2 inhibitors, α-gluco-sidase inhibitors, amylin analogues and glucagon-like peptide-1 (GLP-1) receptor agonists. These are insulin analogues. Metformin is one of the most commonly prescribed medications for T2DM management. Met-formin treatment ameliorate the insulin resistance especially in liver and skeletal muscle but less effect in adipose tissue[286,287], decreased inflammatory response, improved glycemic control[288,289] and enhance β-cell function in T2DM patients by increased insulin sensitivity and glucotoxicity reduction[290]. Metformin reduces fatty acid oxidation in adipose tissue[291], increased GLUT4 translocation in muscle and adipose tissues by activated enzyme adenosine monophosphate kinase and reduced gluconeogenesis in liver[292-295]. There are many developed non-conventional drugs to improve glycemic control such as Cycloset is used together with diet and exercise to treat type 2 diabetes. Cycloset is not for treating type 1 diabetes. Welchol is a non-absorbed, polymeric form, lipid-lowering and glucose-lowering agent for oral administration. Welchol is a high-capacity bile acid-binding molecule. Afrezza Inhalation Powder is
the FDA approved the inhalation form of insulin. The new drug is not a substitute for long-acting insulin and use as the combination with conventional long-acting insulin drug for both types of diabetes and many drugs are in the late clinical trials state.
There are new medications and treatments were identified from the FDA, they are in the clinical trials or waiting for approval treatment in dyslipidemia, obesity and T2DM[296]. Recent research study reports that metformin treatment cause metabolic effects to increase GLP-1 concentration in the circulation[297,298]. GLP-1 is an incretin generated from the transcription product of the proglucagon gene. Incretin is a signaling polypeptide contained with 30-amino acid. GLP-1 secretion by ileal L-cells is not depend on the presence of nutrients in the small intestine and responsible for stimulated insulin secretion to limit glucose elevations with the higher efficacy at high glucose levels[276,299]. Elevated GLP-1 secretion might possibly cause increased glucose absorption in the distal segments of small intestine.
Incretins are the gastrointestinal hormone secreted from the intestine and stomach responsible for oral food intake and stimulated the secretion of insulin during meals in healthy peoples[276]. Two major incretin molecules are (1) GLP-1; and (2) Glucose-dependent insulinotopic peptide knows as gastric inhibitory polypeptide (GIP) and to neutralize stomach acid to protect the small intestine and no therapeutic efficacy in T2DM. GLP-1 has lower glucose levels by stimulated insulinproduction and increased glucose metabolism in adipose tissue and muscle. GLP-1 promote the pancreatic β-cells proliferation, reduce apoptosis, increase cardiac chronotropic, inotropic activity, decreases glucagon secretion, reduces glucose production, increase appetite suppression for food intake reduction and slow gastric emptying[271,276,299]. GLP-1 is degraded by enzyme DPP-4 and this enzyme does not inhibit by metformin[298]. The prevention of GLP-1degradation by DPP-4 is one method to increase the effects of GLP-1. DPP-4 inhibitor drugs inhibit the glucagon secretion which in turn increases secretion of insulin to decrease blood glucose levels and decreases gastric emptying. The FDA-approved the DPP-4 inhibitor drugs including sitagliptin (Januvia), alogliptin (Nesina), saxagliptin (Onglyza), linagliptin (Tradjenta), anagliptin, vildagliptin, teneligliptin, gemigliptin and dutogliptin. The adverse effects are dose-dependent to cause headache, vomiting, nausea, nasopharyngitis, hypersensitivity and other conditions. Other side effects of exenatide (GLP-1 agonist) note for abdominal pain, acid stomach, diarrhea, altered renal function, weight loss, dysgeusia, belching and cause pruritus, urticaria and rash reactions at the injection site.
CONCLUSIONIn this present review has described the detrimental effects from chemicals and biochemicals reaction, metals, medications, over nutrition, obesity and diseases in oxidative stress, insulin resistance development and
469 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

the progression of T2DM and the progression of diabetic complications and organ dysfunctions. Oxidative stress played underling associated with the pathogenesis of diseases, leading to increases risk of insulin resistance, dyslipidemia, elevated blood pressure, metabolic syndrome, inflammation and endothelial dysfunction. This reviewed support the oxidative stress contribution of the multifactorial etiology of oxidative stress and insulin resistance in the whole body. ROS act as the signal transduction factor and plays the important role in oxidative stress-mediated downstream signaling pathways and enhances the cell death. Furthermore, risk for several chronic diseases development associated with oxidative stress and metabolic syndrome including T2DM, hypertension, arthritis, congestive heart failure, chronic renal failure, cancer and Alzheimer’s. These diseases may be substantially reduced by dietary modifications, increased physical activity and antioxidant drugs ameliorated oxidative stress. The therapeutic approaches target on oxidative stress may delay or prevent the progression and onset of diseases. Then, antioxidants supplementation may curtail the progression and onset of the metabolic disease complications. Antioxidant interventions, an importance goal of future clinical investigations should be implementation and to improve oral bioavailability targeted to the oxidant overproduction site. Lifestyle change remains the best prevention and therapeutic approach to oppose the increasing epidemic of cardiovascular diseases, obesity, hypertension, dyslipidemia and T2DM. Finally, the connection between oxidative stress, insulin resistance, dyslipidemia, inflammation, life style, atherosclerosis and diabetes as demonstrated in the schematic in Figure 8.
REFERENCES1 Cossarizza A, Ferraresi R, Troiano L, Roat E, Gibellini L,
Bertoncelli L, Nasi M, Pinti M. Simultaneous analysis of reactive oxygen species and reduced glutathione content in living cells by polychromatic flow cytometry. Nat Protoc 2009; 4: 1790-1797 [PMID: 20010930 DOI: 10.1038/nprot.2009.189]
2 Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 1980; 191: 421-427 [PMID: 6263247]
3 Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol 1997; 82: 291-295 [PMID: 9129943]
4 Freeman BA, Crapo JD. Biology of disease: free radicals and tissue injury. Lab Invest 1982; 47: 412-426 [PMID: 6290784]
5 Slater TF. Free-radical mechanisms in tissue injury. Biochem J 1984; 222: 1-15 [PMID: 6383353]
6 Dobrian AD, Davies MJ, Schriver SD, Lauterio TJ, Prewitt RL. Oxidative stress in a rat model of obesity-induced hypertension. Hypertension 2001; 37: 554-560 [PMID: 11230334 DOI: 10.1161/01.HYP.37.2.554]
7 Vincent HK, Taylor AG. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int J Obes (Lond) 2006; 30: 400-418 [PMID: 16302012 DOI: 10.1038/sj.ijo.0803177]
8 Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004; 114: 1752-1761 [PMID: 15599400]
9 Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006; 440: 944-948 [PMID: 16612386 DOI: 10.1038/nature04634]
10 Ogihara T, Asano T, Ando K, Chiba Y, Sakoda H, Anai M, Shojima N, Ono H, Onishi Y, Fujishiro M, Katagiri H, Fukushima Y, Kikuchi M, Noguchi N, Aburatani H, Komuro I, Fujita T. Angiotensin II-induced insulin resistance is associated with enhanced insulin signaling. Hypertension 2002; 40: 872-879 [PMID: 12468572 DOI: 10.1161/01.HYP.0000040262.48405.A8]
11 Dedoussis GV, Kaliora AC, Panagiotakos DB. Genes, diet and type 2 diabetes mellitus: a review. Rev Diabet Stud 2007; 4: 13-24 [PMID: 17565412 DOI: 10.1900/RDS.2007.4.13]
12 Reaven GM. Insulin resistance: the link between obesity and cardiovascular disease. Med Clin North Am 2011; 95: 875-892 [PMID: 21855697 DOI: 10.1016/j.mcna.2011.06.002]
13 Kahn CR, Vicent D, Doria A. Genetics of non-insulin-dependent (type-II) diabetes mellitus. Annu Rev Med 1996; 47: 509-531 [PMID: 8712800 DOI: 10.1146/annurev.med.47.1.509]
14 Unger RH. Reinventing type 2 diabetes: pathogenesis, treatment, and prevention. JAMA 2008; 299: 1185-1187 [PMID: 18334695 DOI: 10.1001/jama.299.10.1185]
15 Kahn CR. Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes 1994; 43: 1066-1084 [PMID: 8039601]
16 Grodsky GM. The importance of rapid insulin secretion: revisited. Diabetes Technol Ther 1999; 1: 259-260 [PMID: 11475271]
17 Rösen P, Nawroth PP, King G, Möller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored
470 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM
Figure 8 Connection between life style, oxidative stress, insulin resistance, inflammation and atherosclerosis. FFA: Free fatty acid; NO: Nitric oxide; MNC: Mononuclear cells; PMN: Polymorpho nuclear cells; CRP: C-reactive protein; MIF: Migration inhibitory factor.
Oxidativestress
Lipolysis ↑ Obesity
FFA ↑
Endothelial NO
AtherosclerosisVasodilatory reserve
Excess calories physical activity
Hyperglycemia(diabetes)
Inflammation inMNC/PMN cells↑ CRP, ↑ MIF
Glucose transport ↓ Hyperinsulinemia
Insulin resistant

by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab Res Rev 2001; 17: 189-212 [PMID: 11424232 DOI: 10.1002/dmrr.196]
18 Nishikawa T, Edelstein D, Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney Int Suppl 2000; 77: S26-S30 [PMID: 10997687]
19 West IC. Radicals and oxidative stress in diabetes. Diabet Med 2000; 17: 171-180 [PMID: 10784220 DOI: 10.1046/j.1464-5491.2000.00259.x]
20 Farrugia G, Balzan R. Oxidative stress and programmed cell death in yeast. Front Oncol 2012; 2: 64 [PMID: 22737670 DOI: 10.3389/fonc.2012.00064]
21 Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem 2004; 266: 37-56 [PMID: 15646026]
22 Narayanan D, Xi Q, Pfeffer LM, Jaggar JH. Mitochondria control functional CaV1.2 expression in smooth muscle cells of cerebral arteries. Circ Res 2010; 107: 631-641 [PMID: 20616314 DOI: 10.1161/CIRCRESAHA.110.224345]
23 Didion SP, Hathaway CA, Faraci FM. Superoxide levels and function of cerebral blood vessels after inhibition of CuZn-SOD. Am J Physiol Heart Circ Physiol 2001; 281: H1697-H1703 [PMID: 11557560]
24 Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res 2001; 88: 600-608 [PMID: 11282894 DOI: 10.1161/01.RES.88.6.600]
25 Kinugawa S, Huang H, Wang Z, Kaminski PM, Wolin MS, Hintze TH. A defect of neuronal nitric oxide synthase increases xanthine oxidase-derived superoxide anion and attenuates the control of myocardial oxygen consumption by nitric oxide derived from endothelial nitric oxide synthase. Circ Res 2005; 96: 355-362 [PMID: 15637297 DOI: 10.1161/01.RES.0000155331.09458.A7]
26 Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 2003; 111: 1201-1209 [PMID: 12697739 DOI: 10.1172/JCI14172]
27 Dikalova AE, Góngora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am J Physiol Heart Circ Physiol 2010; 299: H673-H679 [PMID: 20639222 DOI: 10.1152/ajpheart.00242.2010]
28 Santhanam AV, d’Uscio LV, Smith LA, Katusic ZS. Uncoupling of eNOS causes superoxide anion production and impairs NO signaling in the cerebral microvessels of hph-1 mice. J Neurochem 2012; 122: 1211-1218 [PMID: 22784235 DOI: 10.1111/j.1471-4159.2012.07872.x]
29 Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 2011; 10: 453-471 [PMID: 21629295 DOI: 10.1038/nrd3403]
30 Das J, Roy A, Sil PC. Mechanism of the protective action of taurine in toxin and drug induced organ pathophysiology and diabetic complications: a review. Food Funct 2012; 3: 1251-1264 [PMID: 22930035 DOI: 10.1039/c2fo30117b]
31 Das J, Ghosh J, Manna P, Sil PC. Taurine suppresses doxorubicin-triggered oxidative stress and cardiac apoptosis in rat via up-regulation of PI3-K/Akt and inhibition of p53, p38-JNK. Biochem Pharmacol 2011; 81: 891-909 [PMID: 21295553 DOI: 10.1016/j.bcp.2011.01.008]
32 Ghosh J, Das J, Manna P, Sil PC. Acetaminophen induced renal injury via oxidative stress and TNF-alpha production: therapeutic potential of arjunolic acid. Toxicology 2010; 268: 8-18 [PMID: 19922764 DOI: 10.1016/j.tox.2009.11.011]
33 Sarkar K, Sil PC. Attenuation of Acetaminophen-Induced Hepatotoxicity In Vivo and In Vitro by a 43-kD Protein Isolated from the Herb Cajanus indicus L. Toxicol Mech Methods 2007; 17: 305-315 [PMID: 20020954 DOI: 10.1080/15376510601031919]
34 Sarkar K, Sil PC. Cajanus indicus leaf protein: Beneficial role in
experimental organ pathophysiology. A review. Pathophysiology 2011; 18: 295-303 [PMID: 21628093 DOI: 10.1016/j.pathophys.2011.05.001]
35 Chatterjee M, Sil PC. Protective role ofPhyllanthus niruri against nimesulide induced hepatic damage. Indian J Clin Biochem 2007; 22: 109-116 [PMID: 23105663 DOI: 10.1007/BF02912892]
36 Bhattacharyya S, Ghosh J, Sil PC. Iron induces hepatocytes death via MAPK activation and mitochondria-dependent apoptotic pathway: beneficial role of glycine. Free Radic Res 2012; 46: 1296-1307 [PMID: 22817335 DOI: 10.3109/10715762.2012.712690]
37 Kayankarnna W, Thessomboon D, Niyomtam S, Pingmuangkaew P, Nunthawarasilp P, Tangvarasittichai S. Elevated cadmium exposure associated with oxidative stress and oxidative DNA damage in population of cadmium-contaminated area. IJTPR 2013; 5: 102-108
38 Pal PB, Pal S, Das J, Sil PC. Modulation of mercury-induced mitochondria-dependent apoptosis by glycine in hepatocytes. Amino Acids 2012; 42: 1669-1683 [PMID: 21373768 DOI: 10.1007/s00726-011-0869-3]
39 Pal PB, Sinha K, Sil PC. Mangiferin, a natural xanthone, protects murine liver in Pb(II) induced hepatic damage and cell death via MAP kinase, NF-κB and mitochondria dependent pathways. PLoS One 2013; 8: e56894 [PMID: 23451106 DOI: 10.1371/journal.pone.0056894]
40 Jia L, Liu Z, Sun L, Miller SS, Ames BN, Cotman CW, Liu J. Acrolein, a toxicant in cigarette smoke, causes oxidative damage and mitochondrial dysfunction in RPE cells: protection by (R)-alpha-lipoic acid. Invest Ophthalmol Vis Sci 2007; 48: 339-348 [PMID: 17197552 DOI: 10.1167/iovs.06-0248]
41 Roy A, Sil PC. Tertiary butyl hydroperoxide induced oxidative damage in mice erythrocytes: Protection by taurine. Pathophysiology 2012; 19: 137-148 [PMID: 22626456 DOI: 10.1016/j.patho-phys.2012.05.001]
42 Bhattacharya S, Chatterjee S, Manna P, Das J, Ghosh J, Gachhui R, Sil PC. Prophylactic role of D-Saccharic acid-1,4-lactone in tertiary butyl hydroperoxide induced cytotoxicity and cell death of murine hepatocytes via mitochondria-dependent pathways. J Biochem Mol Toxicol 2011; 25: 341-354 [PMID: 21538728 DOI: 10.1002/jbt.20393]
43 Ghosh A, Mandal AK, Sarkar S, Das N. Hepatoprotective and neuroprotective activity of liposomal quercetin in combating chronic arsenic induced oxidative damage in liver and brain of rats. Drug Deliv 2011; 18: 451-459 [PMID: 21554158 DOI: 10.3109/10717544.2011.577110]
44 Sarkar MK, Sil PC. Prevention of tertiary butyl hydroperoxide induced oxidative impairment and cell death by a novel antioxidant protein molecule isolated from the herb, Phyllanthus niruri. Toxicol In Vitro 2010; 24: 1711-1719 [PMID: 20510348 DOI: 10.1016/j.tiv.2010.05.014]
45 Bhattacharya S, Manna P, Gachhui R, Sil PC. D-saccharic acid 1,4-lactone protects diabetic rat kidney by ameliorating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via NF-κB and PKC signaling. Toxicol Appl Pharmacol 2013; 267: 16-29 [PMID: 23261973 DOI: 10.1016/j.taap.2012.12.005]
46 Rashid K, Bhattacharya S, Sil PC. Protective role of D-saccharic acid-1,4-lactone in alloxan induced oxidative stress in the spleen tissue of diabetic rats is mediated by suppressing mitochondria dependent apoptotic pathway. Free Radic Res 2012; 46: 240-252 [PMID: 22239106 DOI: 10.3109/10715762.2011.650694]
47 Manna P, Ghosh J, Das J, Sil PC. Streptozotocin induced activation of oxidative stress responsive splenic cell signaling pathways: protective role of arjunolic acid. Toxicol Appl Pharmacol 2010; 244: 114-129 [PMID: 20053369 DOI: 10.1016/j.taap.2009.12.024]
48 Das J, Vasan V, Sil PC. Taurine exerts hypoglycemic effect in alloxan-induced diabetic rats, improves insulin-mediated glucose transport signaling pathway in heart and ameliorates cardiac oxidative stress and apoptosis. Toxicol Appl Pharmacol 2012; 258: 296-308 [PMID: 22138235 DOI: 10.1016/j.taap.2011.11.009]
49 Matés JM, Segura JA, Alonso FJ, Márquez J. Oxidative stress in apoptosis and cancer: an update. Arch Toxicol 2012; 86: 1649-1665
471 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

[PMID: 22811024 DOI: 10.1007/s00204-012-0906-3]50 Chrissobolis S, Faraci FM. The role of oxidative stress and NADPH
oxidase in cerebrovascular disease. Trends Mol Med 2008; 14: 495-502 [PMID: 18929509 DOI: 10.1016/j.molmed.2008.09.003]
51 Miller AA, Budzyn K, Sobey CG. Vascular dysfunction in cerebrovascular disease: mechanisms and therapeutic intervention. Clin Sci (Lond) 2010; 119: 1-17 [PMID: 20370718 DOI: 10.1042/CS20090649]
52 Biliński T, Litwińska J, Błaszczyński M, Bajus A. Superoxide dismutase deficiency and the toxicity of the products of autooxidation of polyunsaturated fatty acids in yeast. Biochim Biophys Acta 1989; 1001: 102-106 [PMID: 2563227]
53 Cabiscol E, Piulats E, Echave P, Herrero E, Ros J. Oxidative stress promotes specific protein damage in Saccharomyces cerevisiae. J Biol Chem 2000; 275: 27393-27398 [PMID: 10852912 DOI: 10.1074/jbc.M003140200]
54 Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA 1997; 94: 514-519 [PMID: 9012815]
55 Gutteridge JM, Halliwell B. Comments on review of Free Radicals in Biology and Medicine, second edition, by Barry Halliwell and John M. C. Gutteridge. Free Radic Biol Med 1992; 12: 93-95 [PMID: 1537574]
56 Savaskan NE, Ufer C, Kühn H, Borchert A. Molecular biology of glutathione peroxidase 4: from genomic structure to developmental expression and neural function. Biol Chem 2007; 388: 1007-1017 [PMID: 17937614 DOI: 10.1515/BC.2007.126]
57 Djordjević VB. Free radicals in cell biology. Int Rev Cytol 2004; 237: 57-89 [PMID: 15380666 DOI: 10.1016/S0074-7696(04)37002-6]
58 Addis PB. Occurrence of lipid oxidation products in foods. Food Chem Toxicol 1986; 24: 1021-1030 [PMID: 3542756 DOI: 10.1016/0278-6915(86)90283-8]
59 Rada B, Hably C, Meczner A, Timár C, Lakatos G, Enyedi P, Ligeti E. Role of Nox2 in elimination of microorganisms. Semin Immunopathol 2008; 30: 237-253 [PMID: 18574584 DOI: 10.1007/s00281-008-0126-3]
60 Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem 1995; 41: 1819-1828 [PMID: 7497639]
61 Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med 2009; 46: 1283-1297 [PMID: 19245829 DOI: 10.1016/j.freeradbiomed.2009.02.008]
62 Richter C. Do mitochondrial DNA fragments promote cancer and aging? FEBS Lett 1988; 241: 1-5 [PMID: 3197826 DOI: 10.1016/0014-5793(88)81018-4]
63 Kelley EE, Khoo NK, Hundley NJ, Malik UZ, Freeman BA, Tarpey MM. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic Biol Med 2010; 48: 493-498 [PMID: 19941951 DOI: 10.1016/j.freeradbiomed.2009.11.012]
64 Granger DN, Rutili G, McCord JM. Superoxide radicals in feline intestinal ischemia. Gastroenterology 1981; 81: 22-29 [PMID: 6263743]
65 Godber BL, Doel JJ, Durgan J, Eisenthal R, Harrison R. A new route to peroxynitrite: a role for xanthine oxidoreductase. FEBS Lett 2000; 475: 93-96 [PMID: 10858495 DOI: 10.1016/S0014-5793(00)01639-2]
66 Vorbach C, Harrison R, Capecchi MR. Xanthine oxidoreductase is central to the evolution and function of the innate immune system. Trends Immunol 2003; 24: 512-517 [PMID: 12967676 DOI: 10.1016/S1471-4906(03)00237-0]
67 Luiking YC, Engelen MP, Deutz NE. Regulation of nitric oxide production in health and disease. Curr Opin Clin Nutr Metab Care 2010; 13: 97-104 [PMID: 19841582 DOI: 10.1097/MCO.0b013e328332f99d]
68 Xia Y, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Natl Acad Sci USA 1997; 94: 6954-6958 [PMID: 9192673]
69 Farmer EH, Sutton DA. The course of autoxidation reactions in polyisoprenes and allied compounds: V. Observations on fish-oil acids. J Chem Soc 1943; 24: 122-125 [DOI: 10.1039/JR9430000122]
70 Halliwell B, Gutteridge JM. Lipid peroxidation in brain homogenates: the role of iron and hydroxyl radicals. J Neurochem 1997; 69: 1330-1331 [PMID: 9282962 DOI: 10.1046/j.1471-4159.1997.69031330.x]
71 Gutteridge JM. Iron promoters of the Fenton reaction and lipid peroxidation can be released from haemoglobin by peroxides. FEBS Lett 1986; 201: 291-295 [PMID: 2423372 DOI: 10.1016/0014-5793(86)80626-3]
72 Gutteridge JM, Beard AP, Quinlan GJ. Superoxide-dependent lipid peroxidation. Problems with the use of catalase as a specific probe for fenton-derived hydroxyl radicals. Biochem Biophys Res Commun 1983; 117: 901-907 [PMID: 6320819 DOI: 10.1016/0006-291X(83)91681-9]
73 Gutteridge JM. Free radicals in disease processes: a compilation of cause and consequence. Free Radic Res Commun 1993; 19: 141-158 [PMID: 8244084]
74 Rawls HR, Van Santen PJ. Singlet oxygen: a possible source of the original hydroperoxides in fatty acids. Ann NY Acad Sci 1970; 171: 135-137 [DOI: 10.1111/j.1749-6632.1970.tb39316.x]
75 Bielski BH, Arudi RL, Sutherland MW. A study of the reactivity of HO2/O2- with unsaturated fatty acids. J Biol Chem 1983; 258: 4759-4761 [PMID: 6833274]
76 Gutteridge JM. The role of superoxide and hydroxyl radicals in phospholipid peroxidation catalysed by iron salts. FEBS Lett 1982; 150: 454-458 [PMID: 6297981 DOI: 10.1016/0014-5793(82)80788-6]
77 Schaich KM, Borg DC. Solvent effects in the spin trapping of lipid oxyl radicals. Free Radic Res Commun 1990; 9: 267-278 [PMID: 2167265]
78 Garnier-Suillerot A, Tose L, Paniago E. Kinetic and mechanism of vesicle lipoperoxide decomposition by Fe(II). Biochim Biophys Acta 1984; 794: 307-312 [DOI: 10.1016/0005-2760(84)90160-7]
79 Hardwick TJ. The rate constant of the reaction between ferrous ions and hydrogen peroxide in acid solution. Can J Chem 1957; 35: 428-436 [DOI: 10.1139/v57-062]
80 Davies KJ. Protein damage and degradation by oxygen radicals. I. general aspects. J Biol Chem 1987; 262: 9895-9901 [PMID: 3036875]
81 Wolff SP, Dean RT. Fragmentation of proteins by free radicals and its effect on their susceptibility to enzymic hydrolysis. Biochem J 1986; 234: 399-403 [PMID: 3718475]
82 Dröge W. Free radicals in the physiological control of cell function. Physiol Rev 2002; 82: 47-95 [PMID: 11773609 DOI: 10.1152/physrev.00018.2001]
83 Tangvarasittichai S, Poonsub P, Tangvarasittichai O, Sirigulsatien V. Serum levels of malondialdehyde in type 2 diabetes mellitus Thai subjects. Siriraj Med J 2009; 61: 20-23
84 Moreira PI, Sayre LM, Zhu X, Nunomura A, Smith MA, Perry G. Detection and localization of markers of oxidative stress by in situ methods: application in the study of Alzheimer disease. Methods Mol Biol 2010; 610: 419-434 [PMID: 20013193 DOI: 10.1007/978-1-60327-029-8_25]
85 Rahmanto AS, Morgan PE, Hawkins CL, Davies MJ. Cellular effects of peptide and protein hydroperoxides. Free Radic Biol Med 2010; 48: 1071-1078 [PMID: 20109544 DOI: 10.1016/j.freeradbiomed.2010.01.025]
86 Hunt JV, Dean RT, Wolff SP. Hydroxyl radical production and autoxidative glycosylation. Glucose autoxidation as the cause of protein damage in the experimental glycation model of diabetes mellitus and ageing. Biochem J 1988; 256: 205-212 [PMID: 2851978]
87 Rosenfeld ME. Inflammation, lipids, and free radicals: lessons learned from the atherogenic process. Semin Reprod Endocrinol 1998; 16: 249-261 [PMID: 10101807]
88 Steinberg D. Lewis A. Conner Memorial Lecture. Oxidative modification of LDL and atherogenesis. Circulation 1997; 95:
472 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

1062-1071 [PMID: 9054771 DOI: 10.1161/01.CIR.95.4.1062]89 Esterbauer H, Puhl H, Dieber-Rotheneder M, Waeg G, Rabl H.
Effect of antioxidants on oxidative modification of LDL. Ann Med 1991; 23: 573-581 [PMID: 1756027]
90 Porkkala-Sarataho EK, Nyyssönen MK, Kaikkonen JE, Poulsen HE, Hayn EM, Salonen RM, Salonen JT. A randomized, single-blind, placebo-controlled trial of the effects of 200 mg alpha-tocopherol on the oxidation resistance of atherogenic lipoproteins. Am J Clin Nutr 1998; 68: 1034-1041 [PMID: 9808219]
91 Horton AA, Fairhurst S. Lipid peroxidation and mechanisms of toxicity. Crit Rev Toxicol 1987; 18: 27-79 [PMID: 3311640 DOI: 10.3109/10408448709089856]
92 Mihara M, Uchiyama M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem 1978; 86: 271-278 [PMID: 655387 DOI: 10.1016/0003-2697(78)90342-1]
93 Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979; 95: 351-358 [PMID: 36810]
94 Rankinen T, Hietanen E, Väisänen S, Lehtiö M, Penttilä I, Bouchard C, Rauramaa R. Relationship between lipid peroxidation and plasma fibrinogen in middle-aged men. Thromb Res 2000; 99: 453-459 [PMID: 10973673 DOI: 10.1016/S0049-3848(00)00271-1]
95 Pasaoglu H, Sancak B, Bukan N. Lipid peroxidation and resistance to oxidation in patients with type 2 diabetes mellitus. Tohoku J Exp Med 2004; 203: 211-218 [PMID: 15240931 DOI: 10.1620/tjem.203.211]
96 Cavalca V, Cighetti G, Bamonti F, Loaldi A, Bortone L, Novembrino C, De Franceschi M, Belardinelli R, Guazzi MD. Oxidative stress and homocysteine in coronary artery disease. Clin Chem 2001; 47: 887-892 [PMID: 11325893]
97 Witztum JL. The oxidation hypothesis of atherosclerosis. Lancet 1994; 344: 793-795 [PMID: 7916078 DOI: 10.1016/S0140-6736(94)92346-9]
98 Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis 2000; 21: 361-370 [PMID: 10688856 DOI: 10.1093/carcin/21.3.361]
99 Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem 2003; 278: 31426-31433 [PMID: 12775726 DOI: 10.1074/jbc.M212549200]
100 Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med 1989; 320: 915-924 [PMID: 2648148 DOI: 10.1056/NEJM198904063201407]
101 Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging 1998; 19: 33-36 [PMID: 9562500 DOI: 10.1016/S0197-4580(98)00009-8]
102 Staruchova M, Collins AR, Volkovova K, Mislanová C, Kova-cikova Z, Tulinska J, Kocan A, Staruch L, Wsolova L, Dusinska M. Occupational exposure to mineral fibres. Biomarkers of oxidative damage and antioxidant defence and associations with DNA damage and repair. Mutagenesis 2008; 23: 249-260 [PMID: 18281292 DOI: 10.1093/mutage/gen004]
103 Nair V, Cooper CS, Vietti DE, Turner GA. The chemistry of lipid peroxidation metabolites: crosslinking reactions of malondialdehyde. Lipids 1986; 21: 6-10 [PMID: 3959768]
104 Requena JR, Fu MX, Ahmed MU, Jenkins AJ, Lyons TJ, Thorpe SR. Lipoxidation products as biomarkers of oxidative damage to proteins during lipid peroxidation reactions. Nephrol Dial Transplant 1996; 11 Suppl 5: 48-53 [PMID: 9044307]
105 Knight JA, Pieper RK, McClellan L. Specificity of the thiobarbituric acid reaction: its use in studies of lipid peroxidation. Clin Chem 1988; 34: 2433-2438 [PMID: 3197281]
106 Praticò D, Iuliano L, Mauriello A, Spagnoli L, Lawson JA, Rokach J, Maclouf J, Violi F, FitzGerald GA. Localization of distinct F2-isoprostanes in human atherosclerotic lesions. J Clin Invest 1997; 100: 2028-2034 [PMID: 9329967 DOI: 10.1172/jci119735]
107 Mallat Z, Philip I, Lebret M, Chatel D, Maclouf J, Tedgui A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo
oxidant stress in ventricular dilatation and progression to heart failure. Circulation 1998; 97: 1536-1539 [PMID: 9593557 DOI: 10.1161/01.CIR.97.16.1536]
108 Lawson JA, Rokach J, FitzGerald GA. Isoprostanes: formation, analysis and use as indices of lipid peroxidation in vivo. J Biol Chem 1999; 274: 24441-24444 [PMID: 10455102 DOI: 10.1074/jbc.274.35.24441]
109 Roberts LJ, Morrow JD. The generation and actions of isoprostanes. Biochim Biophys Acta 1997; 1345: 121-135 [PMID: 9106492]
110 Reilly MP, Praticò D, Delanty N, DiMinno G, Tremoli E, Rader D, Kapoor S, Rokach J, Lawson J, FitzGerald GA. Increased formation of distinct F2 isoprostanes in hypercholesterolemia. Circulation 1998; 98: 2822-2828 [PMID: 9860782 DOI: 10.1161/01.CIR.98.25.2822]
111 Mori TA, Croft KD, Puddey IB, Beilin LJ. An improved method for the measurement of urinary and plasma F2-isoprostanes using gas chromatography-mass spectrometry. Anal Biochem 1999; 268: 117-125 [PMID: 10036170 DOI: 10.1006/abio.1998.3037]
112 Praticò D. F(2)-isoprostanes: sensitive and specific non-invasive indices of lipid peroxidation in vivo. Atherosclerosis 1999; 147: 1-10 [PMID: 10525118 DOI: 10.1016/S0021-9150(99)00257-9]
113 Li H, Lawson JA, Reilly M, Adiyaman M, Hwang SW, Rokach J, FitzGerald GA. Quantitative high performance liquid chro-matography/tandem mass spectrometric analysis of the four classes of F(2)-isoprostanes in human urine. Proc Natl Acad Sci USA 1999; 96: 13381-13386 [PMID: 10557329]
114 Gopaul NK, Anggård EE, Mallet AI, Betteridge DJ, Wolff SP, Nourooz-Zadeh J. Plasma 8-epi-PGF2 alpha levels are elevated in individuals with non-insulin dependent diabetes mellitus. FEBS Lett 1995; 368: 225-229 [PMID: 7628610 DOI: 10.1016/0014-5793(95)00649-T]
115 Davì G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, Pennese E, Vitacolonna E, Bucciarelli T, Costantini F, Capani F, Patrono C. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation 1999; 99: 224-229 [PMID: 9892587 DOI: 10.1161/01.CIR.99.2.224]
116 Morrow JD, Frei B, Longmire AW, Gaziano JM, Lynch SM, Shyr Y, Strauss WE, Oates JA, Roberts LJ. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N Engl J Med 1995; 332: 1198-1203 [PMID: 7700313 DOI: 10.1056/NEJM199505043321804]
117 Bachi A, Zuccato E, Baraldi M, Fanelli R, Chiabrando C. Measurement of urinary 8-Epi-prostaglandin F2alpha, a novel index of lipid peroxidation in vivo, by immunoaffinity extraction/gas chromatography-mass spectrometry. Basal levels in smokers and nonsmokers. Free Radic Biol Med 1996; 20: 619-624 [PMID: 8904305 DOI: 10.1016/0891-5849(95)02087-X]
118 Voutilainen S, Morrow JD, Roberts LJ, Alfthan G, Alho H, Nyyssönen K, Salonen JT. Enhanced in vivo lipid peroxidation at elevated plasma total homocysteine levels. Arterioscler Thromb Vasc Biol 1999; 19: 1263-1266 [PMID: 10323778 DOI: 10.1161/01.ATV.19.5.1263]
119 Davi G, Alessandrini P, Mezzetti A, Minotti G, Bucciarelli T, Costantini F, Cipollone F, Bon GB, Ciabattoni G, Patrono C. In vivo formation of 8-Epi-prostaglandin F2 alpha is increased in hypercholesterolemia. Arterioscler Thromb Vasc Biol 1997; 17: 3230-3235 [PMID: 9409316 DOI: 10.1161/01.ATV.17.11.3230]
120 Praticò D, Tangirala RK, Rader DJ, Rokach J, FitzGerald GA. Vitamin E suppresses isoprostane generation in vivo and reduces atherosclerosis in ApoE-deficient mice. Nat Med 1998; 4: 1189-1192 [PMID: 9771755 DOI: 10.1038/2685]
121 Schnackenberg CG, Wilcox CS. Two-week administration of tempol attenuates both hypertension and renal excretion of 8-Iso prostaglandin f2alpha. Hypertension 1999; 33: 424-428 [PMID: 9931141 DOI: 10.1161/01.HYP.33.1.424]
122 Palmer AM, Thomas CR, Gopaul N, Dhir S, Anggård EE, Poston
473 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

L, Tribe RM. Dietary antioxidant supplementation reduces lipid peroxidation but impairs vascular function in small mesenteric arteries of the streptozotocin-diabetic rat. Diabetologia 1998; 41: 148-156 [PMID: 9498647 DOI: 10.1007/s001250050883]
123 Laight DW, Kengatharan KM, Gopaul NK, Anggård EE, Carrier MJ. Investigation of oxidant stress and vasodepression to glyceryl trinitrate in the obese Zucker rat in vivo. Br J Pharmacol 1998; 125: 895-901 [PMID: 9831930 DOI: 10.1038/sj.bjp.0702132]
124 England T, Beatty E, Rehman A, Nourooz-Zadeh J, Pereira P, O’Reilly J, Wiseman H, Geissler C, Halliwell B. The steady-state levels of oxidative DNA damage and of lipid peroxidation (F2-isoprostanes) are not correlated in healthy human subjects. Free Radic Res 2000; 32: 355-362 [PMID: 10741856 DOI: 10.1080/10715760000300351]
125 Fujita T. Insulin resistance and salt-sensitive hypertension in metabolic syndrome. Nephrol Dial Transplant 2007; 22: 3102-3107 [PMID: 17602196 DOI: 10.1093/ndt/gfm409]
126 Fujita T. Aldosterone in salt-sensitive hypertension and metabolic syndrome. J Mol Med (Berl) 2008; 86: 729-734 [PMID: 18437332 DOI: 10.1007/s00109-008-0343-1]
127 Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes 2005; 54: 1615-1625 [PMID: 15919781 DOI: 10.2337/diabetes.54.6.1626]
128 Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000; 49: 1939-1945 [PMID: 11078463 DOI: 10.2337/diabetes.49.11.1939]
129 Anderson RM, Weindruch R. The caloric restriction paradigm: implications for healthy human aging. Am J Hum Biol 2012; 24: 101-106 [PMID: 22290875 DOI: 10.1002/ajhb.22243]
130 Rebrin I, Kamzalov S, Sohal RS. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic Biol Med 2003; 35: 626-635 [PMID: 12957655 DOI: 10.1016/S0891-5849(03)00388-5]
131 Grattagliano I, Portincasa P, Cocco T, Moschetta A, Di Paola M, Palmieri VO, Palasciano G. Effect of dietary restriction and N-acetylcysteine supplementation on intestinal mucosa and liver mitochondrial redox status and function in aged rats. Exp Gerontol 2004; 39: 1323-1332 [PMID: 15489055 DOI: 10.1016/j.exger.2004.06.001]
132 Cocco T, Sgobbo P, Clemente M, Lopriore B, Grattagliano I, Di Paola M, Villani G. Tissue-specific changes of mitochondrial functions in aged rats: effect of a long-term dietary treatment with N-acetylcysteine. Free Radic Biol Med 2005; 38: 796-805 [PMID: 15721990 DOI: 10.1016/j.freeradbiomed.2004.11.034]
133 Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006; 444: 337-342 [PMID: 17086191 DOI: 10.1038/nature05354]
134 Keaney JF, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol 2003; 23: 434-439 [PMID: 12615693 DOI: 10.1161/01.ATV.0000058402.34138.11]
135 Okuno Y, Matsuda M, Kobayashi H, Morita K, Suzuki E, Fukuhara A, Komuro R, Shimabukuro M, Shimomura I. Adipose expression of catalase is regulated via a novel remote PPARgamma-responsive region. Biochem Biophys Res Commun 2008; 366: 698-704 [PMID: 18073138 DOI: 10.1016/j.bbrc.2007.12.001]
136 Okuno Y, Matsuda M, Miyata Y, Fukuhara A, Komuro R, Shimabukuro M, Shimomura I. Human catalase gene is regulated by peroxisome proliferator activated receptor-gamma through a
response element distinct from that of mouse. Endocr J 2010; 57: 303-309 [PMID: 20075562 DOI: 10.1507/endocrj.K09E-113]
137 Kobayashi H, Matsuda M, Fukuhara A, Komuro R, Shimomura I. Dysregulated glutathione metabolism links to impaired insulin action in adipocytes. Am J Physiol Endocrinol Metab 2009; 296: E1326-E1334 [PMID: 19366877 DOI: 10.1152/ajpendo.90921.2008]
138 Roberts CK, Barnard RJ, Sindhu RK, Jurczak M, Ehdaie A, Vaziri ND. Oxidative stress and dysregulation of NAD(P)H oxidase and antioxidant enzymes in diet-induced metabolic syndrome. Metabolism 2006; 55: 928-934 [PMID: 16784966 DOI: 10.1016/j.metabol.2006.02.022]
139 Meng S, Roberts LJ, Cason GW, Curry TS, Manning RD. Superoxide dismutase and oxidative stress in Dahl salt-sensitive and -resistant rats. Am J Physiol Regul Integr Comp Physiol 2002; 283: R732-R738 [PMID: 12185008 DOI: 10.1152/ajpregu.00346.2001]
140 Kido M, Ando K, Oba S, Fujita T. Renoprotective effect of prav-astatin in salt-loaded Dahl salt-sensitive rats. Hypertens Res 2005; 28: 1009-1015 [PMID: 16671341 DOI: 10.1291/hypres.28.1009]
141 Wang H, Shimosawa T, Matsui H, Kaneko T, Ogura S, Uetake Y, Takenaka K, Yatomi Y, Fujita T. Paradoxical mineralocorticoid receptor activation and left ventricular diastolic dysfunction under high oxidative stress conditions. J Hypertens 2008; 26: 1453-1462 [PMID: 18551023 DOI: 10.1097/HJH.0b013e328300a232]
142 Laffer CL, Bolterman RJ, Romero JC, Elijovich F. Effect of salt on isoprostanes in salt-sensitive essential hypertension. Hypertension 2006; 47: 434-440 [PMID: 16432053 DOI: 10.1161/01.HYP.0000202480.06735.82]
143 Rocchini AP, Key J, Bondie D, Chico R, Moorehead C, Katch V, Martin M. The effect of weight loss on the sensitivity of blood pressure to sodium in obese adolescents. N Engl J Med 1989; 321: 580-585 [PMID: 2668763 DOI: 10.1056/NEJM198908313210905]
144 Uzu T, Kimura G, Yamauchi A, Kanasaki M, Isshiki K, Araki S, Sugiomoto T, Nishio Y, Maegawa H, Koya D, Haneda M, Kashiwagi A. Enhanced sodium sensitivity and disturbed circadian rhythm of blood pressure in essential hypertension. J Hypertens 2006; 24: 1627-1632 [PMID: 16877966 DOI: 10.1097/01.hjh.0000239299.71001.77]
145 Sanchez RA, Masnatta LD, Pesiney C, Fischer P, Ramirez AJ. Telmisartan improves insulin resistance in high renin nonmodulating salt-sensitive hypertensives. J Hypertens 2008; 26: 2393-2398 [PMID: 19008718 DOI: 10.1097/HJH.0b013e328312677e]
146 Giner V, Coca A, de la Sierra A. Increased insulin resistance in salt sensitive essential hypertension. J Hum Hypertens 2001; 15: 481-485 [PMID: 11464258]
147 Dobrian AD, Schriver SD, Lynch T, Prewitt RL. Effect of salt on hypertension and oxidative stress in a rat model of diet-induced obesity. Am J Physiol Renal Physiol 2003; 285: F619-F628 [PMID: 12799306 DOI: 10.1152/ajprenal.00388.2002]
148 Annuk M, Zilmer M, Fellström B. Endothelium-dependent vasodilation and oxidative stress in chronic renal failure: impact on cardiovascular disease. Kidney Int Suppl 2003; (84): S50-S53 [PMID: 12694308 DOI: 10.1046/j.1523-1755.63.s84.2.x]
149 Martín-Gallán P, Carrascosa A, Gussinyé M, Domínguez C. Biomarkers of diabetes-associated oxidative stress and antioxidant status in young diabetic patients with or without subclinical complications. Free Radic Biol Med 2003; 34: 1563-1574 [PMID: 12788476 DOI: 10.1016/S0891-5849(03)00185-0]
150 Varvarovská J, Racek J, Stozický F, Soucek J, Trefil L, Pomahacová R. Parameters of oxidative stress in children with Type 1 diabetes mellitus and their relatives. J Diabetes Complications 2003; 17: 7-10 [PMID: 12505749 DOI: 10.1016/S1056-8727(01)00228-8]
151 Seghrouchni I, Drai J, Bannier E, Rivière J, Calmard P, Garcia I, Orgiazzi J, Revol A. Oxidative stress parameters in type I, type II and insulin-treated type 2 diabetes mellitus; insulin treatment efficiency. Clin Chim Acta 2002; 321: 89-96 [PMID: 12031597 DOI: 10.1016/S0009-8981(02)00099-2]
152 VanderJagt DJ, Harrison JM, Ratliff DM, Hunsaker LA, Vander Jagt DL. Oxidative stress indices in IDDM subjects with and
474 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

without long-term diabetic complications. Clin Biochem 2001; 34: 265-270 [PMID: 11440725 DOI: 10.1016/S0009-9120(01)00204-1]
153 Bonnefont-Rousselot D. Glucose and reactive oxygen species. Curr Opin Clin Nutr Metab Care 2002; 5: 561-568 [PMID: 12172481]
154 Ceriello A, Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol 2004; 24: 816-823 [PMID: 14976002 DOI: 10.1161/01.ATV.0000122852.22604.78]
155 Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404: 787-790 [PMID: 10783895 DOI: 10.1038/35008121]
156 Ramana KV, Chandra D, Srivastava S, Bhatnagar A, Srivastava SK. Nitric oxide regulates the polyol pathway of glucose metabolism in vascular smooth muscle cells. FASEB J 2003; 17: 417-425 [PMID: 12631581 DOI: 10.1096/fj.02-0722com]
157 Morré DM, Lenaz G, Morré DJ. Surface oxidase and oxidative stress propagation in aging. J Exp Biol 2000; 203: 1513-1521 [PMID: 10769214]
158 Shams N, Ianchulev T. Role of vascular endothelial growth factor in ocular angiogenesis. Ophthalmol Clin North Am 2006; 19: 335-344 [PMID: 16935208 DOI: 10.1016/j.ohc.2006.05.005]
159 Bhisitkul RB. Vascular endothelial growth factor biology: clinical implications for ocular treatments. Br J Ophthalmol 2006; 90: 1542-1547 [PMID: 17114590 DOI: 10.1136/bjo.2006.098426]
160 Stitt AW. The role of advanced glycation in the pathogenesis of diabetic retinopathy. Exp Mol Pathol 2003; 75: 95-108 [PMID: 12834631 DOI: 10.1016/S0014-4800(03)00035-2]
161 Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res 2004; 63: 582-592 [PMID: 15306213 DOI: 10.1016/j.cardiores.2004.05.00]
162 Nakamura Y, Horii Y, Nishino T, Shiiki H, Sakaguchi Y, Kagoshima T, Dohi K, Makita Z, Vlassara H, Bucala R. Immunohistochemical localization of advanced glycosylation end products in coronary atheroma and cardiac tissue in diabetes mellitus. Am J Pathol 1993; 143: 1649-1656 [PMID: 8256853]
163 Yamagishi S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp Gerontol 2011; 46: 217-224 [PMID: 21111800 DOI: 10.1016/j.exger.2010.11.007]
164 Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, Pinsky D, Stern D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 1994; 269: 9889-9897 [PMID: 8144582]
165 Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J, Cao R, Yan SD, Brett J, Stern D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest 1995; 96: 1395-1403 [PMID: 7544803 DOI: 10.1172/JCI118175]
166 Roebuck KA. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: differential activation and binding of the transcription factors AP-1 and NF-kappaB (Review). Int J Mol Med 1999; 4: 223-230 [PMID: 10425270 DOI: 10.3892/ijmm.4.3.223]
167 Hu FB, Stampfer MJ. Is type 2 diabetes mellitus a vascular condition? Arterioscler Thromb Vasc Biol 2003; 23: 1715-1716 [PMID: 14555640 DOI: 10.1161/01.ATV.0000094360.38911.71]
168 Kaul K, Hodgkinson A, Tarr JM, Kohner EM, Chibber R. Is inflammation a common retinal-renal-nerve pathogenic link in diabetes? Curr Diabetes Rev 2010; 6: 294-303 [PMID: 20594163 DOI: 10.2174/157339910793360851]
169 Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M, Quagliaro L, Ceriello A, Giugliano D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in
humans: role of oxidative stress. Circulation 2002; 106: 2067-2072 [PMID: 12379575 DOI: 10.1161/01.CIR.0000034509.14906.AE]
170 Lane N. A unifying view of ageing and disease: the double-agent theory. J Theor Biol 2003; 225: 531-540 [PMID: 14615212 DOI: 10.1016/S0022-5193(03)00304-7]
171 Steinberg HO, Baron AD. Vascular function, insulin resistance and fatty acids. Diabetologia 2002; 45: 623-634 [PMID: 12107742 DOI: 10.1007/s00125-002-0800-2]
172 Lopes HF, Morrow JD, Stojiljkovic MP, Goodfriend TL, Egan BM. Acute hyperlipidemia increases oxidative stress more in African Americans than in white Americans. Am J Hypertens 2003; 16: 331-336 [PMID: 12745192 DOI: 10.1016/S0895-7061(03)00041-4]
173 Stojiljkovic MP, Lopes HF, Zhang D, Morrow JD, Goodfriend TL, Egan BM. Increasing plasma fatty acids elevates F2-isoprostanes in humans: implications for the cardiovascular risk factor cluster. J Hypertens 2002; 20: 1215-1221 [PMID: 12023694]
174 Peelman F, Waelput W, Iserentant H, Lavens D, Eyckerman S, Zabeau L, Tavernier J. Leptin: linking adipocyte metabolism with cardiovascular and autoimmune diseases. Prog Lipid Res 2004; 43: 283-301 [PMID: 15234549 DOI: 10.1016/j.plipres.2004.03.001]
175 Chan WB, Ma RC, Chan NN, Ng MC, Lee ZS, Lai CW, Tong PC, So WY, Chan JC. Increased leptin concentrations and lack of gender difference in Type 2 diabetic patients with nephropathy. Diabetes Res Clin Pract 2004; 64: 93-98 [PMID: 15063601 DOI: 10.1016/j.diabres.2003.10.023]
176 Wauters M, Considine RV, Yudkin JS, Peiffer F, De Leeuw I, Van Gaal LF. Leptin levels in type 2 diabetes: associations with measures of insulin resistance and insulin secretion. Horm Metab Res 2003; 35: 92-96 [PMID: 12734788 DOI: 10.1055/s-2003-39054]
177 Reilly MP, Iqbal N, Schutta M, Wolfe ML, Scally M, Localio AR, Rader DJ, Kimmel SE. Plasma leptin levels are associated with coronary atherosclerosis in type 2 diabetes. J Clin Endocrinol Metab 2004; 89: 3872-3878 [PMID: 15292320 DOI: 10.1210/jc.2003-031676]
178 Yamagishi SI, Edelstein D, Du XL, Kaneda Y, Guzmán M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem 2001; 276: 25096-25100 [PMID: 11342529 DOI: 10.1074/jbc.M007383200]
179 Bouloumie A, Marumo T, Lafontan M, Busse R. Leptin induces oxidative stress in human endothelial cells. FASEB J 1999; 13: 1231-1238 [PMID: 10385613]
180 Yamamoto K, Völkl A, Hashimoto T, Fahimi HD. Catalase in guinea pig hepatocytes is localized in cytoplasm, nuclear matrix and peroxisomes. Eur J Cell Biol 1988; 46: 129-135 [PMID: 3396586]
181 Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J 1999; 13: 1169-1183 [PMID: 10385608]
182 Zhang P, Liu B, Kang SW, Seo MS, Rhee SG, Obeid LM. Thioredoxin peroxidase is a novel inhibitor of apoptosis with a mechanism distinct from that of Bcl-2. J Biol Chem 1997; 272: 30615-30618 [PMID: 9388194 DOI: 10.1074/jbc.272.49.30615]
183 Galter D, Mihm S, Dröge W. Distinct effects of glutathione disulphide on the nuclear transcription factor kappa B and the activator protein-1. Eur J Biochem 1994; 221: 639-648 [PMID: 8174544 DOI: 10.1111/j.1432-1033.1994.tb18776.x]
184 Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol 2003; 43: 233-260 [PMID: 12359864 DOI: 10.1146/annurev.pharmtox.43.100901.140229]
185 Thomas JA, Poland B, Honzatko R. Protein sulfhydryls and their role in the antioxidant function of protein S-thiolation. Arch Biochem Biophys 1995; 319: 1-9 [PMID: 7771771 DOI: 10.1006/abbi.1995.1261]
186 Shirwaikar A, Shirwaikar A, Rajendran K, Punitha IS. In vitro antioxidant studies on the benzyl tetra isoquinoline alkaloid
475 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

berberine. Biol Pharm Bull 2006; 29: 1906-1910 [PMID: 16946507 DOI: 10.1248/bpb.29.1906]
187 Ulrich-Merzenich G, Zeitler H, Vetter H, Kraft K. Synergy research: vitamins and secondary plant components in the maintenance of the redox-homeostasis and in cell signaling. Phytomedicine 2009; 16: 2-16 [PMID: 19118991 DOI: 10.1016/j.phymed.2008.11.007]
188 Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med 2008; 45: 549-561 [PMID: 18544350 DOI: 10.1016/j.freeradbiomed.2008.05.004]
189 Blendea MC, Jacobs D, Stump CS, McFarlane SI, Ogrin C, Bahtyiar G, Stas S, Kumar P, Sha Q, Ferrario CM, Sowers JR. Abrogation of oxidative stress improves insulin sensitivity in the Ren-2 rat model of tissue angiotensin II overexpression. Am J Physiol Endocrinol Metab 2005; 288: E353-E359 [PMID: 15494608 DOI: 10.1152/ajpendo.00402.2004]
190 Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H, Ota T, Yokoyama M, Honda M, Miyamoto K, Kaneko S. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism 2008; 57: 1071-1077 [PMID: 18640384 DOI: 10.1016/j.metabol.2008.03.010]
191 Kunitomo M, Yamaguchi Y, Kagota S, Otsubo K. Beneficial effect of coenzyme Q10 on increased oxidative and nitrative stress and inflammation and individual metabolic components developing in a rat model of metabolic syndrome. J Pharmacol Sci 2008; 107: 128-137 [PMID: 18544898]
192 Paolisso G, Giugliano D. Oxidative stress and insulin action: is there a relationship? Diabetologia 1996; 39: 357-363 [PMID: 8721784]
193 Rudich A, Kozlovsky N, Potashnik R, Bashan N. Oxidant stress reduces insulin responsiveness in 3T3-L1 adipocytes. Am J Physiol 1997; 272: E935-E940 [PMID: 9176196]
194 Maddux BA, See W, Lawrence JC, Goldfine AL, Goldfine ID, Evans JL. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of alpha-lipoic acid. Diabetes 2001; 50: 404-410 [PMID: 11272154 DOI: 10.2337/diabetes.50.2.404]
195 Rudich A, Tirosh A, Potashnik R, Khamaisi M, Bashan N. Lipoic acid protects against oxidative stress induced impairment in insulin stimulation of protein kinase B and glucose transport in 3T3-L1 adipocytes. Diabetologia 1999; 42: 949-957 [PMID: 10491755 DOI: 10.1007/s001250051253]
196 Packer L, Kraemer K, Rimbach G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition 2001; 17: 888-895 [PMID: 11684397]
197 Teachey MK, Taylor ZC, Maier T, Saengsirisuwan V, Sloniger JA, Jacob S, Klatt MJ, Ptock A, Kraemer K, Hasselwander O, Henriksen EJ. Interactions of conjugated linoleic acid and lipoic acid on insulin action in the obese Zucker rat. Metabolism 2003; 52: 1167-1174 [PMID: 14506623 DOI: 10.1016/S0026-0495(03)00145-8]
198 Evans JL, Goldfine ID. Alpha-lipoic acid: a multifunctional antioxidant that improves insulin sensitivity in patients with type 2 diabetes. Diabetes Technol Ther 2000; 2: 401-413 [PMID: 11467343]
199 Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, Walton V, Shulman GI. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med 1998; 338: 867-872 [PMID: 9516221 DOI: 10.1056/NEJM199803263381303]
200 Mayerson AB, Hundal RS, Dufour S, Lebon V, Befroy D, Cline GW, Enocksson S, Inzucchi SE, Shulman GI, Petersen KF. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 2002; 51: 797-802 [PMID: 11872682]
201 Evans JL, Heymann CJ, Goldfine ID, Gavin LA. Pharma-cokinetics, tolerability, and fructosamine-lowering effect of a novel, controlled-release formulation of alpha-lipoic acid. Endocr Pract 2002; 8: 29-35 [PMID: 11951812 DOI: 10.4158/EP.8.1.29]
202 Ansar H, Mazloom Z, Kazemi F, Hejazi N. Effect of alpha-lipoic acid on blood glucose, insulin resistance and glutathione peroxidase of type 2 diabetic patients. Saudi Med J 2011; 32:
584-588 [PMID: 21666939]203 Estrada DE, Ewart HS, Tsakiridis T, Volchuk A, Ramlal T,
Tritschler H, Klip A. Stimulation of glucose uptake by the natural coenzyme alpha-lipoic acid/thioctic acid: participation of elements of the insulin signaling pathway. Diabetes 1996; 45: 1798-1804 [PMID: 8922368 DOI: 10.2337/diab.45.12.1798]
204 Konrad D, Somwar R, Sweeney G, Yaworsky K, Hayashi M, Ramlal T, Klip A. The antihyperglycemic drug alpha-lipoic acid stimulates glucose uptake via both GLUT4 translocation and GLUT4 activation: potential role of p38 mitogen-activated protein kinase in GLUT4 activation. Diabetes 2001; 50: 1464-1471 [PMID: 11375349 DOI: 10.2337/diabetes.50.6.1464]
205 Ramrath S, Tritschler HJ, Eckel J. Stimulation of cardiac glucose transport by thioctic acid and insulin. Horm Metab Res 1999; 31: 632-635 [PMID: 10668913 DOI: 10.1055/s-2007-978811]
206 Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes 1994; 43: 1271-1278 [PMID: 7926300 DOI: 10.2337/diab.43.11.1271]
207 Cohen B, Novick D, Rubinstein M. Modulation of insulin activities by leptin. Science 1996; 274: 1185-1188 [PMID: 8895466 DOI: 10.1126/science.274.5290.1185]
208 McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002; 51: 7-18 [PMID: 11756317 DOI: 10.2337/diabetes.51.1.7]
209 Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 1997; 46: 3-10 [PMID: 8971073 DOI: 10.2337/diab.46.1.3]
210 Randle PJ, Kerbey AL, Espinal J. Mechanisms decreasing glucose oxidation in diabetes and starvation: role of lipid fuels and hormones. Diabetes Metab Rev 1988; 4: 623-638 [PMID: 3069395]
211 Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature 2001; 409: 307-312 [PMID: 11201732 DOI: 10.1038/35053000]
212 Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest 2000; 106: 171-176 [PMID: 10903330 DOI: 10.1172/JCI10583]
213 Wojtczak L, Schönfeld P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim Biophys Acta 1993; 1183: 41-57 [PMID: 8399375 DOI: 10.1016/0005-2728(93)90004-Y]
214 Bakker SJ, IJzerman RG, Teerlink T, Westerhoff HV, Gans RO, Heine RJ. Cytosolic triglycerides and oxidative stress in central obesity: the missing link between excessive atherosclerosis, endothelial dysfunction, and beta-cell failure? Atherosclerosis 2000; 148: 17-21 [PMID: 10580166 DOI: 10.1016/S0021-9150(99)00329-9]
215 Toborek M, Hennig B. Fatty acid-mediated effects on the glutathione redox cycle in cultured endothelial cells. Am J Clin Nutr 1994; 59: 60-65 [PMID: 8279404]
216 Hennig B, Meerarani P, Ramadass P, Watkins BA, Toborek M. Fatty acid-mediated activation of vascular endothelial cells. Metabolism 2000; 49: 1006-1013 [PMID: 10954018 DOI: 10.1053/meta.2000.7736]
217 Paolisso G, Di Maro G, Pizza G, D’Amore A, Sgambato S, Tesauro P, Varricchio M, D’Onofrio F. Plasma GSH/GSSG affects glucose homeostasis in healthy subjects and non-insulin-dependent diabetics. Am J Physiol 1992; 263: E435-E440 [PMID: 1415522]
218 Dichtl W, Nilsson L, Goncalves I, Ares MP, Banfi C, Calara F, Hamsten A, Eriksson P, Nilsson J. Very low-density lipoprotein activates nuclear factor-kappaB in endothelial cells. Circ Res 1999; 84: 1085-1094 [PMID: 10325246 DOI: 10.1161/01.RES.84.9.1085]
219 Hennig B, Meerarani P, Toborek M, McClain CJ. Antioxidant-like properties of zinc in activated endothelial cells. J Am Coll Nutr 1999; 18: 152-158 [PMID: 10204831]
220 Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem 2001; 276: 16683-16689 [PMID: 11278967 DOI: 10.1074/jbc.M011695200]
221 Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee
476 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999; 48: 1270-1274 [PMID: 10342815 DOI: 10.2337/diabetes.48.6.1270]
222 Coudronniere N, Villalba M, Englund N, Altman A. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc Natl Acad Sci USA 2000; 97: 3394-3399 [PMID: 10716728 DOI: 10.1073/pnas.060028097]
223 Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28: 412-419 [PMID: 3899825]
224 Meglasson MD, Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev 1986; 2: 163-214 [PMID: 2943567]
225 Malaisse WJ. Physiology, pathology and pharmacology of insulin secretion: recent acquisitions. Diabetes Metab 1997; 23 Suppl 3: 6-15 [PMID: 9342537]
226 Maechler P, Jornot L, Wollheim CB. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem 1999; 274: 27905-27913 [PMID: 10488138 DOI: 10.1074/jbc.274.39.27905]
227 Robertson RP, Harmon JS. Diabetes, glucose toxicity, and oxidative stress: A case of double jeopardy for the pancreatic islet beta cell. Free Radic Biol Med 2006; 41: 177-184 [PMID: 16814095 DOI: 10.1016/j.freeradbiomed.2005.04.030]
228 Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997; 46: 1733-1742 [PMID: 9356019 DOI: 10.2337/diab.46.11.173]
229 Miwa I, Ichimura N, Sugiura M, Hamada Y, Taniguchi S. Inhibition of glucose-induced insulin secretion by 4-hydroxy-2-nonenal and other lipid peroxidation products. Endocrinology 2000; 141: 2767-2772 [PMID: 10919261 DOI: 10.1210/endo.141.8.7614]
230 Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci USA 1999; 96: 10857-10862 [PMID: 10485916]
231 Ho E, Bray TM. Antioxidants, NFkappaB activation, and diabetogenesis. Proc Soc Exp Biol Med 1999; 222: 205-213 [PMID: 10601879]
232 Tajiri Y, Möller C, Grill V. Long-term effects of aminoguanidine on insulin release and biosynthesis: evidence that the formation of advanced glycosylation end products inhibits B cell function. Endocrinology 1997; 138: 273-280 [PMID: 8977414 DOI: 10.1210/endo.138.1.4851]
233 Ho E, Chen G, Bray TM. Supplementation of N-acetylcysteine inhibits NFkappaB activation and protects against alloxan-induced diabetes in CD-1 mice. FASEB J 1999; 13: 1845-1854 [PMID: 10506589]
234 Ho E, Chen G, Bray TM. Alpha-phenyl-tert-butylnitrone (PBN) inhibits NFkappaB activation offering protection against chemically induced diabetes. Free Radic Biol Med 2000; 28: 604-614 [PMID: 10719242 DOI: 10.1016/S0891-5849(99)00271-3]
235 Kaneto H, Xu G, Song KH, Suzuma K, Bonner-Weir S, Sharma A, Weir GC. Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function through the induction of oxidative stress. J Biol Chem 2001; 276: 31099-31104 [PMID: 11390407 DOI: 10.1074/jbc.M104115200]
236 Boden G, Ruiz J, Kim CJ, Chen X. Effects of prolonged glucose infusion on insulin secretion, clearance, and action in normal subjects. Am J Physiol 1996; 270: E251-E258 [PMID: 8779946]
237 Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest 1992; 90: 320-325 [PMID: 1644911 DOI: 10.1172/JCI115865]
238 Poitout V, Olson LK, Robertson RP. Chronic exposure of
betaTC-6 cells to supraphysiologic concentrations of glucose decreases binding of the RIPE3b1 insulin gene transcription activator. J Clin Invest 1996; 97: 1041-1046 [PMID: 8613527 DOI: 10.1172/JCI118496]
239 Yki-Järvinen H. Glucose toxicity. Endocr Rev 1992; 13: 415-431 [PMID: 1425483 DOI: 10.1210/edrv-13-3-415]
240 Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H. Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003; 52: 581-587 [PMID: 12606496 DOI: 10.2337/diabetes.52.3.581]
241 Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004; 53 Suppl 1: S119-S124 [PMID: 14749276 DOI: 10.2337/diabetes.53.2007.S119]
242 Lameloise N, Muzzin P, Prentki M, Assimacopoulos-Jeannet F. Uncoupling protein 2: a possible link between fatty acid excess and impaired glucose-induced insulin secretion? Diabetes 2001; 50: 803-809 [PMID: 11289045 DOI: 10.2337/diabetes.50.4.803]
243 Segall L, Lameloise N, Assimacopoulos-Jeannet F, Roche E, Corkey P, Thumelin S, Corkey BE, Prentki M. Lipid rather than glucose metabolism is implicated in altered insulin secretion caused by oleate in INS-1 cells. Am J Physiol 1999; 277: E521-E528 [PMID: 10484365]
244 Carlsson C, Borg LA, Welsh N. Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology 1999; 140: 3422-3428 [PMID: 10433196 DOI: 10.1210/endo.140.8.6908]
245 Jacqueminet S, Briaud I, Rouault C, Reach G, Poitout V. Inhibition of insulin gene expression by long-term exposure of pancreatic beta cells to palmitate is dependent on the presence of a stimulatory glucose concentration. Metabolism 2000; 49: 532-536 [PMID: 10778881 DOI: 10.1016/S0026-0495(00)80021-9]
246 Poitout V, Robertson RP. Minireview: Secondary beta-cell failure in type 2 diabetes--a convergence of glucotoxicity and lipotoxicity. Endocrinology 2002; 143: 339-342 [PMID: 11796484 DOI: 10.1210/endo.143.2.8623]
247 Harmon JS, Gleason CE, Tanaka Y, Poitout V, Robertson RP. Antecedent hyperglycemia, not hyperlipidemia, is associated with increased islet triacylglycerol content and decreased insulin gene mRNA level in Zucker diabetic fatty rats. Diabetes 2001; 50: 2481-2486 [PMID: 11679425 DOI: 10.2337/diabetes.50.11.2481]
248 Grundy SM. Hypertriglyceridemia, atherogenic dyslipidemia, and the metabolic syndrome. Am J Cardiol 1998; 81: 18B-25B [PMID: 9526809]
249 Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 2003; 46: 733-749 [PMID: 12774165 DOI: 10.1007/s00125-003-1111-y]
250 Ginsberg HN, Zhang YL, Hernandez-Ono A. Metabolic syndrome: focus on dyslipidemia. Obesity (Silver Spring) 2006; 14 Suppl 1: 41S-49S [PMID: 16642962 DOI: 10.1038/oby.2006.281]
251 Adiels M, Olofsson SO, Taskinen MR, Borén J. Diabetic dyslipidaemia. Curr Opin Lipidol 2006; 17: 238-246 [PMID: 16680028 DOI: 10.1097/01.mol.0000226115.97436.c0]
252 Vergès B. New insight into the pathophysiology of lipid abnormalities in type 2 diabetes. Diabetes Metab 2005; 31: 429-439 [PMID: 16357786]
253 Packard CJ. Triacylglycerol-rich lipoproteins and the generation of small, dense low-density lipoprotein. Biochem Soc Trans 2003; 31: 1066-1069 [PMID: 14505481]
254 Coppack SW, Evans RD, Fisher RM, Frayn KN, Gibbons GF, Humphreys SM, Kirk ML, Potts JL, Hockaday TD. Adipose tissue metabolism in obesity: lipase action in vivo before and after a mixed meal. Metabolism 1992; 41: 264-272 [PMID: 1542265]
255 Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 2006; 113: 1888-1904 [PMID: 16618833 DOI: 10.1161/CIRCULATIONAHA.105.563213]
256 Garg A, Grundy SM. Management of dyslipidemia in NIDDM. Diabetes Care 1990; 13: 153-169 [PMID: 2190770 DOI: 10.2337/diacare.13.2.153]
477 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

257 Sheu WH, Shieh SM, Fuh MM, Shen DD, Jeng CY, Chen YD, Reaven GM. Insulin resistance, glucose intolerance, and hyperinsulinemia. Hypertriglyceridemia versus hypercholesterolemia. Arterioscler Thromb 1993; 13: 367-370 [PMID: 8443140 DOI: 10.1161/01.ATV.13.3.367]
258 Tangvarasittichai S, Poonsub P, Tangvarasittichai O. Association of serum lipoprotein ratios with insulin resistance in type 2 diabetes mellitus. Indian J Med Res 2010; 131: 641-648 [PMID: 20516535]
259 Laws A, Reaven GM. Evidence for an independent relationship between insulin resistance and fasting plasma HDL-cholesterol, triglyceride and insulin concentrations. J Intern Med 1992; 231: 25-30 [PMID: 1732395]
260 Miller GJ , Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet 1975; 1: 16-19 [PMID: 46338 DOI: 10.1016/S0140-6736(75)92376-4]
261 Hokanson JE, Austin MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk 1996; 3: 213-219 [PMID: 8836866 DOI: 10.1177/174182679600300214]
262 Malmström R, Packard CJ, Caslake M, Bedford D, Stewart P, Yki-Järvinen H, Shepherd J, Taskinen MR. Defective regulation of triglyceride metabolism by insulin in the liver in NIDDM. Diabetologia 1997; 40: 454-462 [PMID: 9112023 DOI: 10.1007/s001250050700]
263 Stampfer MJ, Sacks FM, Salvini S, Willett WC, Hennekens CH. A prospective study of cholesterol, apolipoproteins, and the risk of myocardial infarction. N Engl J Med 1991; 325: 373-381 [PMID: 2062328 DOI: 10.1056/NEJM199108083250601]
264 Sacks FM, Alaupovic P, Moye LA, Cole TG, Sussex B, Stampfer MJ, Pfeffer MA, Braunwald E. VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the Cholesterol and Recurrent Events (CARE) trial. Circulation 2000; 102: 1886-1892 [PMID: 11034934 DOI: 10.1161/01.CIR.102.16.1886]
265 Wilson PW, Kannel WB, Anderson KM. Lipids, glucose intolerance and vascular disease: the Framingham Study. Monogr Atheroscler 1985; 13: 1-11 [PMID: 4088270]
266 Jeppesen J, Facchini FS, Reaven GM. Individuals with high total cholesterol/HDL cholesterol ratios are insulin resistant. J Intern Med 1998; 243: 293-298 [PMID: 9627143 DOI: 10.1046/j.1365-2796.1998.00301.x]
267 Li C, Ford ES, Meng YX, Mokdad AH, Reaven GM. Does the association of the triglyceride to high-density lipoprotein cholesterol ratio with fasting serum insulin differ by race/ethnicity? Cardiovasc Diabetol 2008; 7: 4 [PMID: 18307789 DOI: 10.1186/1475-2840-7-4]
268 McLaughlin T, Reaven G, Abbasi F, Lamendola C, Saad M, Waters D, Simon J, Krauss RM. Is there a simple way to identify insulin-resistant individuals at increased risk of cardiovascular disease? Am J Cardiol 2005; 96: 399-404 [PMID: 16054467 DOI: 10.1016/j.amjcard.2005.03.085]
269 Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 1999; 104: 787-794 [PMID: 10491414 DOI: 10.1172/JCI7231]
270 Muoio DM, Newgard CB. Mechanisms of disease: Molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 2008; 9: 193-205 [PMID: 18200017 DOI: 10.1038/nrm2327]
271 Nicholson G, Hall GM. Diabetes mellitus: new drugs for a new epidemic. Br J Anaesth 2011; 107: 65-73 [PMID: 21610015 DOI: 10.1093/bja/aer120]
272 Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr 2010; 8: 29 [PMID: 20969750 DOI: 10.1186/1478-7954-8-29]
273 Morris AP, Voight BF, Teslovich TM, Ferreira T, Segrè AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan
A, Prokopenko I, Kang HM, Dina C, Esko T, Fraser RM, Kanoni S, Kumar A, Lagou V, Langenberg C, Luan J, Lindgren CM, Müller-Nurasyid M, Pechlivanis S, Rayner NW, Scott LJ, Wiltshire S, Yengo L, Kinnunen L, Rossin EJ, Raychaudhuri S, Johnson AD, Dimas AS, Loos RJ, Vedantam S, Chen H, Florez JC, Fox C, Liu CT, Rybin D, Couper DJ, Kao WH, Li M, Cornelis MC, Kraft P, Sun Q, van Dam RM, Stringham HM, Chines PS, Fischer K, Fontanillas P, Holmen OL, Hunt SE, Jackson AU, Kong A, Lawrence R, Meyer J, Perry JR, Platou CG, Potter S, Rehnberg E, Robertson N, Sivapalaratnam S, Stančáková A, Stirrups K, Thorleifsson G, Tikkanen E, Wood AR, Almgren P, Atalay M, Benediktsson R, Bonnycastle LL, Burtt N, Carey J, Charpentier G, Crenshaw AT, Doney AS, Dorkhan M, Edkins S, Emilsson V, Eury E, Forsen T, Gertow K, Gigante B, Grant GB, Groves CJ, Guiducci C, Herder C, Hreidarsson AB, Hui J, James A, Jonsson A, Rathmann W, Klopp N, Kravic J, Krjutškov K, Langford C, Leander K, Lindholm E, Lobbens S, Männistö S, Mirza G, Mühleisen TW, Musk B, Parkin M, Rallidis L, Saramies J, Sennblad B, Shah S, Sigurðsson G, Silveira A, Steinbach G, Thorand B, Trakalo J, Veglia F, Wennauer R, Winckler W, Zabaneh D, Campbell H, van Duijn C, Uitterlinden AG, Hofman A, Sijbrands E, Abecasis GR, Owen KR, Zeggini E, Trip MD, Forouhi NG, Syvänen AC, Eriksson JG, Peltonen L, Nöthen MM, Balkau B, Palmer CN, Lyssenko V, Tuomi T, Isomaa B, Hunter DJ, Qi L, Shuldiner AR, Roden M, Barroso I, Wilsgaard T, Beilby J, Hovingh K, Price JF, Wilson JF, Rauramaa R, Lakka TA, Lind L, Dedoussis G, Njølstad I, Pedersen NL, Khaw KT, Wareham NJ, Keinanen-Kiukaanniemi SM, Saaristo TE, Korpi-Hyövälti E, Saltevo J, Laakso M, Kuusisto J, Metspalu A, Collins FS, Mohlke KL, Bergman RN, Tuomilehto J, Boehm BO, Gieger C, Hveem K, Cauchi S, Froguel P, Baldassarre D, Tremoli E, Humphries SE, Saleheen D, Danesh J, Ingelsson E, Ripatti S, Salomaa V, Erbel R, Jöckel KH, Moebus S, Peters A, Illig T, de Faire U, Hamsten A, Morris AD, Donnelly PJ, Frayling TM, Hattersley AT, Boerwinkle E, Melander O, Kathiresan S, Nilsson PM, Deloukas P, Thorsteinsdottir U, Groop LC, Stefansson K, Hu F, Pankow JS, Dupuis J, Meigs JB, Altshuler D, Boehnke M, McCarthy MI. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012; 44: 981-990 [PMID: 22885922 DOI: 10.1038/ng.2383]
274 Palmer ND, McDonough CW, Hicks PJ, Roh BH, Wing MR, An SS, Hester JM, Cooke JN, Bostrom MA, Rudock ME, Talbert ME, Lewis JP, Ferrara A, Lu L, Ziegler JT, Sale MM, Divers J, Shriner D, Adeyemo A, Rotimi CN, Ng MC, Langefeld CD, Freedman BI, Bowden DW, Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, McCulloch LJ, Ferreira T, Grallert H, Amin N, Wu G, Willer CJ, Raychaudhuri S, McCarroll SA, Langenberg C, Hofmann OM, Dupuis J, Qi L, Segrè AV, van Hoek M, Navarro P, Ardlie K, Balkau B, Benediktsson R, Bennett AJ, Blagieva R, Boerwinkle E, Bonnycastle LL, Boström KB, Bravenboer B, Bumpstead S, Burtt NP, Charpentier G, Chines PS, Cornelis M, Couper DJ, Crawford G, Doney AS, Elliott KS, Elliott AL, Erdos MR, Fox CS, Franklin CS, Ganser M, Gieger C, Grarup N, Green T, Griffin S, Groves CJ, Guiducci C, Hadjadj S, Hassanali N, Herder C, Isomaa B, Jackson AU, Johnson PR, Jørgensen T, Kao WH, Klopp N, Kong A, Kraft P, Kuusisto J, Lauritzen T, Li M, Lieverse A, Lindgren CM, Lyssenko V, Marre M, Meitinger T, Midthjell K, Morken MA, Narisu N, Nilsson P, Owen KR, Payne F, Perry JR, Petersen AK, Platou C, Proença C, Prokopenko I, Rathmann W, Rayner NW, Robertson NR, Rocheleau G, Roden M, Sampson MJ, Saxena R, Shields BM, Shrader P, Sigurdsson G, Sparsø T, Strassburger K, Stringham HM, Sun Q, Swift AJ, Thorand B, Tichet J, Tuomi T, van Dam RM, van Haeften TW, van Herpt T, van Vliet-Ostaptchouk JV, Walters GB, Weedon MN, Wijmenga C, Witteman J, Bergman RN, Cauchi S, Collins FS, Gloyn AL, Gyllensten U, Hansen T, Hide WA, Hitman GA, Hofman A, Hunter DJ, Hveem K, Laakso M, Mohlke KL, Morris AD, Palmer CN, Pramstaller PP, Rudan I, Sijbrands E, Stein LD, Tuomilehto J, Uitterlinden A, Walker M, Wareham NJ, Watanabe RM, Abecasis GR, Boehm BO, Campbell H, Daly
478 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

MJ, Hattersley AT, Hu FB, Meigs JB, Pankow JS, Pedersen O, Wichmann HE, Barroso I, Florez JC, Frayling TM, Groop L, Sladek R, Thorsteinsdottir U, Wilson JF, Illig T, Froguel P, van Duijn CM, Stefansson K, Altshuler D, Boehnke M, McCarthy MI, Soranzo N, Wheeler E, Glazer NL, Bouatia-Naji N, Mägi R, Randall J, Johnson T, Elliott P, Rybin D, Henneman P, Dehghan A, Hottenga JJ, Song K, Goel A, Egan JM, Lajunen T, Doney A, Kanoni S, Cavalcanti-Proença C, Kumari M, Timpson NJ, Zabena C, Ingelsson E, An P, O’Connell J, Luan J, Elliott A, McCarroll SA, Roccasecca RM, Pattou F, Sethupathy P, Ariyurek Y, Barter P, Beilby JP, Ben-Shlomo Y, Bergmann S, Bochud M, Bonnefond A, Borch-Johnsen K, Böttcher Y, Brunner E, Bumpstead SJ, Chen YD, Chines P, Clarke R, Coin LJ, Cooper MN, Crisponi L, Day IN, de Geus EJ, Delplanque J, Fedson AC, Fischer-Rosinsky A, Forouhi NG, Frants R, Franzosi MG, Galan P, Goodarzi MO, Graessler J, Grundy S, Gwilliam R, Hallmans G, Hammond N, Han X, Hartikainen AL, Hayward C, Heath SC, Hercberg S, Hicks AA, Hillman DR, Hingorani AD, Hui J, Hung J, Jula A, Kaakinen M, Kaprio J, Kesaniemi YA, Kivimaki M, Knight B, Koskinen S, Kovacs P, Kyvik KO, Lathrop GM, Lawlor DA, Le Bacquer O, Lecoeur C, Li Y, Mahley R, Mangino M, Manning AK, Martínez-Larrad MT, McAteer JB, McPherson R, Meisinger C, Melzer D, Meyre D, Mitchell BD, Mukherjee S, Naitza S, Neville MJ, Oostra BA, Orrù M, Pakyz R, Paolisso G, Pattaro C, Pearson D, Peden JF, Pedersen NL, Perola M, Pfeiffer AF, Pichler I, Polasek O, Posthuma D, Potter SC, Pouta A, Province MA, Psaty BM, Rayner NW, Rice K, Ripatti S, Rivadeneira F, Rolandsson O, Sandbaek A, Sandhu M, Sanna S, Sayer AA, Scheet P, Seedorf U, Sharp SJ, Shields B, Sijbrands EJ, Silveira A, Simpson L, Singleton A, Smith NL, Sovio U, Swift A, Syddall H, Syvänen AC, Tanaka T, Tönjes A, Uitterlinden AG, van Dijk KW, Varma D, Visvikis-Siest S, Vitart V, Vogelzangs N, Waeber G, Wagner PJ, Walley A, Ward KL, Watkins H, Wild SH, Willemsen G, Witteman JC, Yarnell JW, Zelenika D, Zethelius B, Zhai G, Zhao JH, Zillikens MC, Borecki IB, Loos RJ, Meneton P, Magnusson PK, Nathan DM, Williams GH, Silander K, Salomaa V, Smith GD, Bornstein SR, Schwarz P, Spranger J, Karpe F, Shuldiner AR, Cooper C, Dedoussis GV, Serrano-Ríos M, Lind L, Palmer LJ, Franks PW, Ebrahim S, Marmot M, Kao WH, Pramstaller PP, Wright AF, Stumvoll M, Hamsten A, Buchanan TA, Valle TT, Rotter JI, Siscovick DS, Penninx BW, Boomsma DI, Deloukas P, Spector TD, Ferrucci L, Cao A, Scuteri A, Schlessinger D, Uda M, Ruokonen A, Jarvelin MR, Waterworth DM, Vollenweider P, Peltonen L, Mooser V, Sladek R. A genome-wide association search for type 2 diabetes genes in African Americans. PLoS One 2012; 7: e29202 [PMID: 22238593 DOI: 10.1371/journal.pone.0029202]
275 Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends Mol Med 2010; 16: 407-416 [PMID: 20728409 DOI: 10.1016/j.molmed.2010.06.004]
276 Tahrani AA, Bailey CJ, Del Prato S, Barnett AH. Management of type 2 diabetes: new and future developments in treatment. Lancet 2011; 378: 182-197 [PMID: 21705062 DOI: 10.1016/S0140-6736(11)60207-9]
277 Handelsman Y, Mechanick JI, Blonde L, Grunberger G, Bloomgarden ZT, Bray GA, Dagogo-Jack S, Davidson JA, Einhorn D, Ganda O, Garber AJ, Hirsch IB, Horton ES, Ismail-Beigi F, Jellinger PS, Jones KL, Jovanovič L, Lebovitz H, Levy P, Moghissi ES, Orzeck EA, Vinik AI, Wyne KL. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for developing a diabetes mellitus comprehensive care plan. Endocr Pract 2011; 17 Suppl 2: 1-53 [PMID: 21474420]
278 Haffner SM. Management of dyslipidemia in adults with diabetes. Diabetes Care 2003; 26 Suppl 1: S83-S86 [PMID: 12502625 DOI: 10.2337/diacare.26.2007.S83]
279 Tangvarasittichai S, Lertsinthai P, Taechasubamorn P, Veerapun O, Tangvarasittichai O. Effect of Moderate-Intensity Exercise Training on Body Weight, Serum Uric Acid, Serum hs-CRP, and Insulin Sensitivity in Type 2Diabetic Patients. Siriraj Med J 2009; 61: 310-313
280 Mills EJ, Wu P, Chong G, Ghement I, Singh S, Akl EA, Eyawo
O, Guyatt G, Berwanger O, Briel M. Efficacy and safety of statin treatment for cardiovascular disease: a network meta-analysis of 170,255 patients from 76 randomized trials. QJM 2011; 104: 109-124 [PMID: 20934984 DOI: 10.1093/qjmed/hcq165]
281 Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, Friedewald W. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol 1986; 8: 1245-1255 [PMID: 3782631 DOI: 10.1016/S0735-1097(86)80293-5]
282 Elam MB, Hunninghake DB, Davis KB, Garg R, Johnson C, Egan D, Kostis JB, Sheps DS, Brinton EA. Effect of niacin on lipid and lipoprotein levels and glycemic control in patients with diabetes and peripheral arterial disease: the ADMIT study: A randomized trial. Arterial Disease Multiple Intervention Trial. JAMA 2000; 284: 1263-1270 [PMID: 10979113 DOI: 10.1001/jama.284.10.1263]
283 Grundy SM, Vega GL, McGovern ME, Tulloch BR, Kendall DM, Fitz-Patrick D, Ganda OP, Rosenson RS, Buse JB, Robertson DD, Sheehan JP. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the assessment of diabetes control and evaluation of the efficacy of niaspan trial. Arch Intern Med 2002; 162: 1568-1576 [PMID: 12123399 DOI: 10.1001/archinte.162.14.1568]
284 Centers for Disease Control and Prevention. [Accessed 2014 June 15]. Available from: URL: http: www.cdc.gov
285 Qaseem A, Humphrey LL, Sweet DE, Starkey M, Shekelle P. Oral pharmacologic treatment of type 2 diabetes mellitus: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2012; 156: 218-231 [PMID: 22312141 DOI: 10.7326/0003-4819-156-3-201202070-00011]
286 Giannarelli R, Aragona M, Coppelli A, Del Prato S. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab 2003; 29: 6S28-6S35 [PMID: 14502098]
287 Staels B. Metformin and pioglitazone: Effectively treating insulin resistance. Curr Med Res Opin 2006; 22 Suppl 2: S27-S37 [PMID: 16914073 DOI: 10.1185/030079906X112732]
288 Bulcão C, Ribeiro-Filho FF, Sañudo A, Roberta Ferreira SG. Effects of simvastatin and metformin on inflammation and insulin resistance in individuals with mild metabolic syndrome. Am J Cardiovasc Drugs 2007; 7: 219-224 [PMID: 17610348]
289 Fidan E, Onder Ersoz H, Yilmaz M, Yilmaz H, Kocak M, Karahan C, Erem C. The effects of rosiglitazone and metformin on inflammation and endothelial dysfunction in patients with type 2 diabetes mellitus. Acta Diabetol 2011; 48: 297-302 [PMID: 21424914 DOI: 10.1007/s00592-011-0276-y]
290 Ferner RE, Rawlins MD, Alberti KG. Impaired beta-cell responses improve when fasting blood glucose concentration is reduced in non-insulin-dependent diabetes. Q J Med 1988; 66: 137-146 [PMID: 3051084]
291 Perriello G, Misericordia P, Volpi E, Santucci A, Santucci C, Ferrannini E, Ventura MM, Santeusanio F, Brunetti P, Bolli GB. Acute antihyperglycemic mechanisms of metformin in NIDDM. Evidence for suppression of lipid oxidation and hepatic glucose production. Diabetes 1994; 43: 920-928 [PMID: 8013758 DOI: 10.2337/diab.43.7.920]
292 Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001; 108: 1167-1174 [PMID: 11602624 DOI: 10.1172/JCI13505]
293 Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG, Brett CM, Giacomini KM. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest 2007; 117: 1422-1431 [PMID: 17476361 DOI: 10.1172/JCI30558]
294 Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest 2010; 120: 2355-2369 [PMID: 20577053 DOI: 10.1172/JCI40671]
479 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

295 Kim YD, Park KG, Lee YS, Park YY, Kim DK, Nedumaran B, Jang WG, Cho WJ, Ha J, Lee IK, Lee CH, Choi HS. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 2008; 57: 306-314 [PMID: 17909097 DOI: 10.2337/db07-0381]
296 Food and Drug Administration. [Accessed 2014 June 9]. Available from: URL: http: //www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm
297 Lindsay JR, Duffy NA, McKillop AM, Ardill J, O’Harte FP, Flatt
PR, Bell PM. Inhibition of dipeptidyl peptidase IV activity by oral metformin in Type 2 diabetes. Diabet Med 2005; 22: 654-657 [PMID: 15842525 DOI: 10.1111/j.1464-5491.2005.01461.x]
298 Sinha Roy R, Bergeron R, Zhu L, He H, Jiang G, Liu F, Lyons K, Pryor K, Yao J, Zhang BB, Thornberry N. Metformin is a GLP-1 secretagogue, not a dipeptidyl peptidase-4 inhibitor. Diabetologia 2007; 50 (suppl 1): S284 [DOI: 10.1007/s00125-007-0809-7]
299 Sisson EM. Liraglutide: clinical pharmacology and considerations for therapy. Pharmacotherapy 2011; 31: 896-911 [PMID: 21923591 DOI: 10.1592/phco.31.9.896]
P- Reviewer: Sicari R, Soare A S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
480 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tangvarasittichai S. Oxidative stress and T2DM

Rinat Gabbay-Benziv, E Albert Reece, Fang Wang, Peixin Yang
Rinat Gabbay-Benziv, E Albert Reece, Fang Wang, Peixin Yang, Department of Obstetrics, Gynaecology and Reproductive Sciences, University of Maryland School of Medicine, Baltimore, MD 21201, United StatesE Albert Reece, Peixin Yang, Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD 21201, United StatesAuthor contributions: Gabbay-Benziv R and Wang F collected the data and wrote the manuscript; Reece EA contributed to the writing of the manuscript; Yang P designed the aim of the editorial and wrote the manuscript; all authors reviewed the manuscript, made their revision and finally approved it for publication. Conflict-of-interest: None of the authors have a conflict of interest.Supported by NIH R01DK083243, R01DK101972 and R56 DK095380 (to Yang P), R01DK103024 (to Yang P and Reece EA) and Basic Science Award (1-13-BS-220), American Diabetes Association (to Yang P).Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Peixin Yang, PhD, Department of Obstetrics, Gynaecology and Reproductive Sciences, University of Maryland School of Medicine, BRB11-039, 655 W. Baltimore Street, Baltimore, MD 21201, United States. [email protected] Telephone: +1-410-7068402 Fax: +1-410-7065747Received: August 29, 2014 Peer-review started: August 30, 2014 First decision: November 27, 2014Revised: December 9, 2014 Accepted: January 9, 2015 Article in press: January 12, 2015Published online: April 15, 2015
Abstract Currently, 60 million women of reproductive age (18-44 years old) worldwide, and approximately 3 million American women have diabetes mellitus, and it has been estimated that this number will double by 2030. Pregestational diabetes mellitus (PGD) is a significant public health problem that increases the risk for structural birth defects affecting both maternal and neonatal pregnancy outcome. The most common types of human structural birth defects associated with PGD are congenital heart defects and central nervous system defects. However, diabetes can induce birth defects in any other fetal organ. In general, the rate of birth defects increases linearly with the degree of maternal hyperglycemia, which is the major factor that mediates teratogenicity of PGD. Stringent prenatal care and glycemic control are effective means to reduce birth defects in PGD pregnancies, but cannot reduce the incidence of birth defects to the rate of that is seen in the nondiabetic population. Studies in animal models have revealed that PGD induces oxidative stress, which activates cellular stress signalling leading to dysregulation of gene expression and excess apoptosis in the target organs, including the neural tube and embryonic heart. Activation of the apoptosis signal-regulating kinase 1 (ASK1)-forkhead transcription factor 3a (FoxO3a)-caspase 8 pathway causes apoptosis in the developing neural tube leading to neural tube defects (NTDs). ASK1 activates the c-Jun-N-Terminal kinase 1/2 (JNK1/2), which leads to activation of the unfolded protein response and endoplasmic reticulum (ER) stress. Deletion of the ASK1 gene, the JNK1 gene, or the JNK2 gene, or inhibition of ER stress by 4-Phenylbutyric acid abrogates diabetes-induced apoptosis and reduces the formation of NTDs. Antioxidants, such as thioredoxin, which inhibits the ASK1-FoxO3a-caspase 8 pathway or ER stress inhibitors, may prevent PGD-induced birth defects.
MINIREVIEWS
481 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Birth defects in pregestational diabetes: Defect range, glycemic threshold and pathogenesis
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.481
World J Diabetes 2015 April 15; 6(3): 481-488ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

Key words: Pregestational diabetes; Birth defects; Glycemic threshold; Diabetic embryopathy; Range of defects
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Pregestational diabetes is a rising problem with gravid impact on adverse pregnancy outcomes. This review concentrates on diabetes-induced birth defects and the underlying mechanism of diabetic embryopathy derived from animal studies. The main defects associated with pregestational diabetes are in the cardiovascular and central nervous systems, and are linearly related to maternal glycemic control. Animal studies reveal oxidative stress and stress kinase signalling-induced apoptosis as key factors in pathogenesis. However, many questions remain unanswered, and the rate of congenital defects in human diabetic pregnancies is still high. The cause of diabetic embryopathy warrants further investigations.
Gabbay-Benziv R, Reece EA, Wang F, Yang P. Birth defects in pregestational diabetes: Defect range, glycemic threshold and pathogenesis. World J Diabetes 2015; 6(3): 481-488 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/481.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.481
INTRODUCTIONMajor structural or genetic birth defects affect approximately 3% of births[1], and are the leading cause of infant mortality in the United States[2]. The pathogenesis of most birth defects is unknown[3,4]. Pregestational diabetes (PGD) is one of the leading known causes with up to a nine-fold increase in birth defects, compared with the rate seen in nondiabetic pregnancies[5]. Although the risk for birth defects in PGD pregnancies can be significantly reduced by appropriate pregestational counselling and meticulous control of maternal glycemic levels[6], it cannot be equalized to the background risk partially because at least 40% of diabetic pregnancies are usually unplanned[6,7].
The pathophysiology of maternal diabetes induced birth defects is complex, however, clearly relates to maternal glucose levels. The mechanism is not entirely understood, but animal studies have shown it to be associated with decreased cell proliferation and increased cell apoptosis due to high oxidative stress. The second major change seen in animal models of PGD is altered gene expression causing deviation from the normal developmental process.
PGD can affect almost any organ. However, most congenital defects associated with diabetes occur in the cardiovascular, central nervous and musculoskeletal systems[8-10].
In this review we will discuss how PGD increases the risk of birth defects, the range of defects seen in diabetic
pregnancies and the relationship of these defects to maternal glucose control. As the risk of birth defects is well established in type 1 and type 2 diabetes mellitus, while it is still controversial in gestational diabetes[11], we will focus this review on PGD only.
RANGE OF DEFECTSPGD is associated with a wide range of anomalies in almost any fetal organ. Based on available literature, women with PGD have 2- to 9-fold higher risk for having babies with birth defects, with a prevalence of birth defects of 2.7%-18.6%, compared with the healthy population[5,12-17], having a prevalence of birth defects of 2%-3%.
Although any birth defect can be associated with PGD, more anomalies are seen in the cardiovascular, central nervous and skeletal systems[9,10,18]. Rare abnormalities almost exclusively associated with diabetes include caudal regression syndrome with femoral shortening and sacral agenesis. Caudal regression syndrome consists of a spectrum of congenital anomalies of the lower spine and hips, is associated with genitourinary and lower limb defects, and is one of the few syndromes in which the presence of maternal diabetes has always to be considered as the cause, if not confirmed[19]. Although very rare, some studies found this syndrome to be 200-600 times more prevalent in infants of diabetic mothers compared with nondiabetic mothers[16]. Be that as it may, it can still be seen also in non-diabetic mothers as described in a case report by Versiani et al[20] who reported 2 cases of caudal dysplasia sequence - one woman had PGD while the other had gestational diabetes.
Previous studies assessed the prevalence of con-genital anomalies in the diabetic population. Eidem et al[21] Studied the risk of congenital anomalies related to type 1 diabetes. They compared major congenital anomalies (excluding minor anomalies as defined by the EUROCAT system) between women with pregestational type 1 diabetes and controls. All women registered at the Norway national delivery registry from 1999 to 2004 were included. Anomalies were registered in 5.7% (91/1583) offspring of women with type 1 diabetes, compared with 2.9% in the background population. The risk for cardiovascular anomalies was 3 fold higher in the diabetic group (3.2% vs 0.94%, respectively) with ventricular septal defect (VSD) (12/1583) and patent ductus arteriosus (10/1583) being the most prevalent birth defects in the cardiovascular system and the most common compared with all other birth defects.
Garne et al[22] used the same EUROCAT classification to describe birth defects in 18 population-based EUROCAT registries of congenital anomalies, which included births between 1990 to 2005, and compared 669 diabetes cases with 92976 non-diabetes cases. The authors showed significantly increased odds ratios for neural tube defects (anencephaly and encephalocele, but not spina bifida) and several subgroups of congenital heart defects. Other birth defects found in increased odds ratios in
Gabbay-Benziv R et al . Birth defects in pregestational diabetes
482 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

infants of pregnancies complicated by pregestational diabetes were anotia, omphalocele and bilateral renal agenesis. Multiple congenital anomalies were present in 13.6% of the diabetes cases and 6.1% of nondiabetes cases. The odds ratio for caudal regression sequence was very high (26.4, 95%CI: 8.98-77.64), but only 17% of all caudal regression cases occurred in pregnancies complicated by PGD.
Most recently, Correa et al[23] reviewed the association of type 1 and type 2 PGD with 39 birth defects using the National Birth Defects Prevention Study. The authors examined databases from 10 birth defects surveillance systems in the United States looking for isolated (one or more major defects in same organ system or with known sequence) or multiple birth defects (2 or more major, unrelated defects) from births between 1997 to 2003. They found an association between PGD and 11 cardiac defects and 7 non cardiac defects.
Galindo et al[12] prospectively followed 126 women with pregestational diabetes in a single tertiary centre in Spain. They reported 17 offspring with congenital anomalies (13.5%). Although chromosomal abnormalities are not generally associated with PGD, one fetus had trisomy 21 and atriventricular septal defects. Eight fetuses were listed with major birth defects (6.3%), 35.3% of fetuses had cardiovascular defects, and geni-tourinary defects accounted for another 23.5%. They noted positive correlation between high levels of haemoglobin A1c (HgA1c) > 7% at early pregnancy and major congenital anomalies. Despite this study including only small number of women, the prospective nature of it allows strength in addressing its results.
Wender-Ozegowska et al[16] studied a group of 198 diabetic women and found 17 pregnancies that resulted in infants with birth defects (8.6%). The most common defects were in the cardiovascular system (5.5%), with 7/198 (3.5%) of cases being atrioventricular septal defects (3 cases of VSD and 3 cases of atrial septal defect). Wren et al[17] focused their research on cardiovascular birth defects. They followed 609 diabetic pregnancies and found that 3.6% of the women delivered infants with cardiovascular defects, the most common being transposition of great arteries, truncus arteriosus and tricuspid atresia. Janssen et al[18] studied 1511 PGD cases, with congenital birth defects prevalence of 7.2%. The most common defects occurred in the cardiovascular system, NTDs, cleft lip/palate and skeletal defects.
Although many studies discuss diabetes associated congenital anomalies, the leading type of birth defects and the exact rate of diabetes associated birth defects are difficult to determine. The literature varies widely in what is considered the “leading” type of anomaly, as well as the rate of anomalies, because of suboptimal coding of the diabetic pregnancies according to White’s classification, differences in anomaly coding and reporting bias. Moreover, the recognition of a birth defect may not always occur in the immediate neonatal period, thus, may not be entered into a registry. On the other
hand, over documentation of diabetes related birth defects, compared with birth defects in the nondiabetic population, may occur because of the known association of defects to diabetes, or the relatively high transfer of infants of diabetic pregnancies to neonatal intensive care units, both leading to a more thorough neonatal examination.
Based upon the available data, it seems that the major organ systems affected by PGD are the cardio-vascular and the central nervous systems, which could be related to their embryonic origin in the neural crest. However, PGD can increase the rate of any other birth defects described above. Therefore, all pregnant women with PGD are advised to undergo a detailed anatomical scan and full fetal echocardiography to screen for possible birth defects, regardless of their having type 1 or type 2 diabetes mellitus or glycemic control at the time of pregnancy.
GLYCEMIC THRESHOLD FOR BIRTH DEFECTSPericonceptional HgA1c is used as a surrogate marker for glycemic control, and is almost linearly related to PGD-induced birth defects. Previous studies have shown that stringent glucose control prior to or at early pregnancy, during organogenesis, can significantly reduce the incidence of birth defects[12,24-29]. However, there are still questions to be answered. Hanson et al[25] studied 532 type 1 PGD women and compared their malformation rate to 222 nondiabetic women. The rate of malformations did not differ significantly between the diabetic and the control groups (4.3% vs 2.4%) although significant different was find in levels of first trimester HgA1c. The median value of HgA1c was 7.7% in the diabetic and 5.3% in the control group (P < 0.001). However, when higher levels of HgA1c were evaluated (HgA1c greater than 10.1% - equal to 8 SD above the normal mean control value), there was statistically significant higher occurrence rate of congenital malformation (P < 0.01).
Wender-Ozegowska et al[16] tried to determine the cut-off for first trimester glycemic levels for prediction of congenital anomalies. They used whole day glycemic profile as well as HgA1c to assess glycemic control. Their cohort included 198 diabetic pregnancies and 4700 nondiabetic pregnancies. The rates of malformation were 8.6% and 3.6%, respectively. They determined the cut-off of value of HgA1c that could prediction congenital malformations as 9.3% (measured up to 16 wk).
Other studies demonstrated the connection between poor glycemic control before or early at pregnancy: Ylinen et al[29] found that the mean HgA1c for women whose infants had congenital malformations was higher than that measured for women with healthy infants prior to 15 gestational weeks (9.5% vs 8.0%, res-pectively). Mironiuk et al[27] compared an occurrence rate of congenital malformations in newborn to mothers with
483 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Gabbay-Benziv R et al . Birth defects in pregestational diabetes

it difficult to differentiate PGD-induced anomalies from others encountered in nondiabetic pregnancies. Third, most studies use HgA1c to reflect the level of glycemic control, but HgA1c only measures a woman’s average glycemic control over a 3-mo period. Therefore, it does not necessarily reflect a woman’s level of glycemic control during organogenesis and embryogenesis. Moreover, because it is an average measurement, the same level of HgA1c may reflect completely different glycemic patterns, one being constant around the average, and the other with larger fluctuations lower and above from the mean HgA1c value[35]. The effects of such fluctuations (i.e., episodes of acute hyperglycemia and hypoglycemia with “normal” HgA1c) are yet to be determined. Fourth, given the complicated metabolic nature of diabetes, coupled with metabolic changes that are normal during pregnancy, other factors may influence the developing embryo. Finally, lower prenatal detection rates of fetal anomalies (due to obesity, lack of prenatal care, etc.) in diabetic women may lead to statistic skewing when studying infants with congenital anomalies born to PGD mothers[36,37].
DOES DIABETES TYPE AFFECT THE RATES OF CONGENITAL ANOMALIES? Diabetes can be classified by the mechanism (type 1, type 2 or gestational diabetes) or alternatively, by White’sclassification[38]. Rates of diabetes, both type 1 and types 2 are increasing all over the world[39,40] causing an increase in the incidence of maternal diabetes in pregnancy[41]. Although hyperglycemia is a common mechanism for teratogenicity, differences in disease characteristics, such as age of onset, ethnicity, obesity and duration of disease, may affect the disease impact on the perinatal outcome and the rate of congenital anomalies. Current literature does not specify the risk for anomalies for any type of diabetes separately, as some studies include only type 1 diabetes[25,42], others only type 2 diabetes[32] and only a few compare the outcomes between them[43,44].
Gonzalez-Gonzalez et al[44] retrospectively compared the outcomes of type 1 diabetes (n = 904), type 2 diabetes (n = 516), gestational diabetes (n = 3188) and control (n = 115996) deliveries in Ontario, Canada. Congenital anomalies were most frequently observed in women with type 1 diabetes and gestational diabetes, with risk approaching 1.5- to 2-times that of controls. They found a total of 44 anomalies (6.1%) among all women with diabetes, most commonly in the cardiovascular system. Adjusted OR for anomalies was 3.5, 1.7, 2.5 and 1.9 for type 1, type 2, gestational diabetes and control deliveries, respectively. Peticca et al[45] found similar results in a multicentre study in Italy. They prospectively compared 504 type 1 PGD to 164 type 2 PGD pregnancies. The rate of birth defects was significantly higher in women with type 1 diabetes (5.9%), compared with in women with type 2 diabetes
type 1 diabetes (n = 170), newborns of healthy mothers (n = 26368) and mothers with GDM (n = 56). They demonstrated that type 1 diabetes was associated with congenital malformations (11.2%, 1.8% and 2.2%, respectively) and that the risk of major birth defects was directly proportional to the level of maternal blood glucose control during the first trimester. These studies all support the idea that lack of glycemic control leads to congenital malformations, but do not indicate what HgA1c value should be maintained to reduce the risk for infant anomalies. Moreover, Shields et al[30] tried to find cut-off value associated with congenital anomalies but failed to do so, and Lucas et al[31] proved the association of PGD with congenital anomalies only when HgA1c was combined with other factors such as diabetic classification (White’s classification) and maternal vascular complications. The Atlantic-Diabetes in Pregnancy (DIP) trial compared PGD pregnancy outcomes in the same population after a change in pre-pregnancy care policy and improved glycemic control throughout pregnancy. In the Atlantic DIP study, the first trial conducted from 2005-2007, included 104 diabetic pregnancies (87% type 1 PGD). The authors reported two pregnancies with birth defects that had HgA1c values of 6.6% and 5.4% at early pregnancy. The second trial, conducted from 2008-2010, included 168 pregnancies, with more women affected by type 2 PGD (81/168, 48%) and lower HgA1c values (7.3% compared to 6.9%); however, despite of presumed better prenatal care and the lower HgA1c - rate of malformation reported was not changed between the trials. The authors did not report the type of birth defects seen in either of the two studies[32,33].
Other studies have demonstrated a linear relation-ship between HgA1c and major congenital defects. Greene et al[34] showed an increase in PGD-induced birth defects correlating to the level HgA1c. Their risk for major malformation was 3.0% when HgA1c taken at first trimester was less than or equal to 9.3% and 40% with HgA1c was greater than 14.4% (RR = 13.2; 95%CI: 4.3-40.4). Todorova et al[35] studied 124 pregnancies complicated by pregestational diabetes. The mean values of HgA1c were significantly higher in pregnancies complicated by fetal malformations (n = 19/15.3%) than those values measured in pregnancies without fetal malformations [9.01% (SD ± 1.53) vs 8.06% (SD ± 1.64) P = 0.022, respectively].
In conclusion, it is clear that glycemic control is associated with a reduced risk of congenital anomalies. However, the recommended threshold of HgA1c for pregestational diabetic women planning pregnancy is still not known. Reaching a consensus as to what HgA1c level a diabetic pregnant woman should strive for has remained elusive for many reasons. First, differing definitions for major and minor anomalies, and taking into account the various forms of diabetes, limit the ability to derive conclusions from the published data. Second, PGD pregnancies can have the same spectrum of anomalies as nondiabetic pregnancies which makes
484 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Gabbay-Benziv R et al . Birth defects in pregestational diabetes

(2.0%). Difficulties in glycemic control, increased disease severity and frequent episodes of ketoacidosis and hypoglycaemia were postulated as explanations for this difference. Other studies have found the opposite results[46,47]. Clausen et al[47] compared 389 type 1 and 146 type 2 diabetics delivering in the United Kingdom. Pregnancies affected by type 2 diabetes had worse perinatal outcomes with congenital abnormalities (12.3% in type 2 vs 4.4% in type 1; P = 0.002) accounting for most of this difference. In 2005, Clausen et al[47] showed similar results with almost doubled number of congenital anomalies among pregnant women with type 2 diabetes (6.6%) compared to type 1 diabetes (2.9%)[46]. Both studies cited a delay in antenatal care, suboptimal glycemic control prior to conception, inadequate folate supplementation and maternal obesity, as possible reasons to explain the worse outcomes in pregnancies complicated by type 2 diabetes. Another explanation could be the tendency toward oral glycemic agents in type 2 diabetes, which may lead to less than optimal glycemic control compared to the use of insulin only in type 1 diabetes. Conversely, Roland et al[48], showed a high rate of congenital anomalies in women with type 1 diabetes and type 2 diabetes, compared with the nondiabetic population, with no significant difference between them. Jensen et al[43] found similar results.
Over the past years, there has been a great improvement in perinatal outcomes for women with type 1 diabetes. However, with rising incidence of obesity and type 2 diabetes, it seems that type 2 diabetes has become a more prominent concern. Women with type 2 diabetes tend to be under less stringent control of their glucose values, either because they use oral glycemic agents, have lower compliance with management strategies or have a lower prevalence of pre-pregnancy counselling, as sometimes type 2 diabetes is regarded as a “lesser” problem compared with type 1 diabetes. Moreover, lack of folate supplementation, obesity with lower ultrasound detection rates and other demographic differences between the women with type 1 or type 2 diabetes may account for the differences showed by different studies comparing the types of diabetes. There is no doubt that pre-pregnancy care and good glycemic control is equally important in type 2 and type 1 diabetes.
PATHOGENESIS OF DIABETES INDUCED BIRTH DEFECTSMost data regarding the pathophysiology of diabetes associated birth defects originates from animal studies. Our research group, as well as others, have shown that maternal diabetes triggers multiple cellular stress responses and subsequent aberrant signal transduction pathways[49-51]. All these may contribute to gene dysregulation and apoptosis in the affected organs of the developing embryo leading to structural birth defects. Studies from animal models have shown that maternal
diabetes increases the production of cellular reactive oxygen species and simultaneously impairs endogenous cellular antioxidant capacity, leading to an overall oxidative stress in the embryo[50,52-55]. Maternal diabetes also appears to increase the expression of inducible nitric oxide synthase (iNOS)[56], whose enzymatic activity catalyzes the reaction of superoxide with nitric oxide to produce reactive nitrogen species. Work in animal models indicates that reactive nitrogen species create a severe form of oxidative stress, nitrosative stress, which is responsible for the activation of cellular stress signalling. Our laboratory has shown that over expression of an antioxidant enzyme, superoxide dismutase 1 (SOD1), in SOD1 transgenic mice, mitigates maternal diabetes-induced oxidative stress and reduces embryonic malformations in diabetic pregnancies[57-59]. Likewise, studies indicate that eliminating the iNOS gene in iNOS knockout mice reduces the incidence of embryonic malformations caused by maternal diabetes[60]. Therefore, our work and that of others has shown that oxidative and nitrosative stress mediates the teratogenicity of maternal diabetes in the developing embryo.
In our studies, maternal diabetes-induced oxidative stress activates the c-Jun-N-terminal kinase 1/2 (JNK1/2)[49,50,61-63]. Deletion of either JNK1 or JNK2 gene ameliorates maternal diabetes-induced NTDs formation[49,50], supporting the hypothesis that activation of the cellular stress kinases, JNK1/2, mediates the adverse effect of maternal diabetes on neural tube closure. In the absence of JNK1 or JNK2, maternal diabetes-induced apoptosis in the neuroepithelial cells are blunted[49,50]. Thus, JNK1/2 activation induced by maternal diabetes transmits the pro-apoptotic signal emanating from oxidative stress under diabetic conditions[49,50].
JNK1/2 belongs to the mitogen-activated protein (MAP) kinase family, whose activation follows a three-tier cascade: MAP three kinases activate MAP kinase kinases, which in turn trigger JNK1/2 phosphorylation. Subsequent studies have revealed the upstream kinases in diabetic embryopathy, including apoptosis signal-regulating kinase 1 (ASK1)[64]. ASK1 is a three kinase that leads to JNK1/2 activation. We have observed that ASK1 gene deletion abolishes maternal diabetes-induced JNK1/2 activation, as well as activation of four major transcription factors downstream of JNK1/2. Similar to our findings in the JNK1 and JNK2 gene deletion studies, ASK1 gene deletion blocks maternal diabetes-induced apoptosis in the developing neural tube, and consequently reduces the number of embryos with NTDs[64]. Thus, the ASK1-JNK1/2 pathway, which is activated by oxidative stress, appears to play a causal role in the induction of diabetic embryopathy.
Recently, we have worked to understand how cellular ASK1-JNK1/2 kinase signalling relays its pro-apoptotic signals to the nucleus[64]. We have observed that ASK1 activation increases the activity of Forkhead transcription factor 3a (FoxO3a), and that FoxO3a
485 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Gabbay-Benziv R et al . Birth defects in pregestational diabetes

induces TNFR1 associated death domain (TRADD)[64]. TRADD up-regulation triggers caspase 8 activation, which, in turn, activates the caspase cascade and leads to apoptosis[64]. Both germline deletion and conditional deletion of the FoxO3a gene significantly reduce maternal diabetes-induced apoptosis and NTDs formation, underscoring the potential importance of FoxO3a in the induction of diabetic embryopathy[64]. We have successfully inhibited the whole stress pathway, ASK1-JNK1/2-FoxO3a-TRADD-caspase 8, using an endogenous ASK1 inhibitor, thioredoxin. Thioredoxin treatment ameliorates NTDs formation in embryos of diabetic dams and cultured embryos[64]. These studies reveal the ASK1 initiated stress signalling with the activity of a transcription factor and pro-apoptotic gene expression.
We and others have shown that JNK1/2 activation leads to endoplasmic reticulum (ER) stress[50]. Under ER stress, the unfolded protein response (UPR) is activated, and prolonged UPR activation induces apoptosis[65,66]. Maternal diabetes in vivo and high glucose in vitro induce ER stress and activate the UPR pathway[50]. Treatment with an ER chemical inhibitor, 4-Phenylbutyric acid, reduces high glucose-induced JNK1/2 activation, neuroepithelial cell apoptosis and NTD formation[50]. Deletion of either the JNK1 or the JNK2 gene abrogates maternal diabetes-induced UPR and ER stress[50]. These findings support a reciprocal causation between JNK1/2 and ER stress in diabetic embryopathy. Other studies have found that activation of the inositol-requiring enzyme 1 alpha, one of the major UPR arms, activates ASK1 and subsequently leads to JNK1/2 activation and apoptosis[67,68], which support our hypothesis that ASK1 plays a major role in diabetic embryopathy.
CONCLUSIONWhile the risk for major congenital defects has been well established in women with pregestational diabetes, many questions remained unanswered. For example, it is still unknown why some organs display vulnerability to glucose teratological effects more than others, or why meticulous control of maternal blood glucose does not reduce the rate of birth defects to the background risk of non-diabetic population, or why some women with pregestational diabetes will have normal pregnancies even when their levels of HgA1c are way above the accepted threshold for birth defects.
The key to understanding the true relationship between PGD and birth defects lies in a basic under-standing of the pathogenesis of congenital defects. Based on our research, the oxidative stress-triggered ASK1 pathway mediates the pro-apoptotic effect of maternal diabetes and high glucose in vitro by inducing pro-apoptotic gene expression and causing UPR and ER stress. The endogenous ASK1 inhibitor, thioredoxin, and the ER stress inhibitor, a Food and Drug Administration approved drug, may be potential therapeutics against maternal diabetes-induced structural birth defects.
We believe further research to evaluate glucose teratological effects on different embryonic organs, via animal models that can encompass the diversity seen in human diabetes, is needed. Research should utilize type 1 and type 2 models, as well as models of chronic and acute glycemia and how these two conditions may affect organogenesis. Our hope is that, by combining all known clinical data with current and future basic and translational science studies, we will be able to establish better ways to care for the diabetic mother and prevent the mal-effects of maternal diabetes on the developing embryo.
ACKNOWLEDGMENTSWe would like to thank Dr. Julie A Wu from the University of Maryland, School of Medicine, for editing assistance in preparing the manuscript.
REFERENCES1 Centers for Disease Control and Prevention (CDC). Update
on overall prevalence of major birth defects--Atlanta, Georgia, 1978-2005. MMWR Morb Mortal Wkly Rep 2008; 57: 1-5 [PMID: 18185492]
2 Heron M, Tejada-Vera B. Deaths: leading causes for 2005. Natl Vital Stat Rep 2009; 58: 1-97 [PMID: 20361522]
3 Brent RL. Environmental causes of human congenital malfor-mations: the pediatrician’s role in dealing with these complex clinical problems caused by a multiplicity of environmental and genetic factors. Pediatrics 2004; 113: 957-968 [PMID: 15060188]
4 Nelson K , Holmes LB. Malformations due to presumed spontaneous mutations in newborn infants. N Engl J Med 1989; 320: 19-23 [PMID: 2909875]
5 Temple R, Aldridge V, Greenwood R, Heyburn P, Sampson M, Stanley K. Association between outcome of pregnancy and glycaemic control in early pregnancy in type 1 diabetes: population based study. BMJ 2002; 325: 1275-1276 [PMID: 12458245]
6 Cemac H. Pregnancy in women with type 1 and type 2 diabetes in 2002-2003. Confidential Enquiry into Maternal and Child Health, 2005
7 Wender-Ozegowska E, Gutaj P, Szczepanek U, Ozegowska K, Zawiejska A, Brazert J. [Influence of pregnancy planning on obstetrical results in women with pregestational diabetes mellitus]. Ginekol Pol 2010; 81: 762-767 [PMID: 21117305]
8 Aberg A, Westbom L, Källén B. Congenital malformations among infants whose mothers had gestational diabetes or preexisting diabetes. Early Hum Dev 2001; 61: 85-95 [PMID: 11223271]
9 Schaefer-Graf UM, Buchanan TA, Xiang A, Songster G, Montoro M, Kjos SL. Patterns of congenital anomalies and relationship to initial maternal fasting glucose levels in pregnancies complicated by type 2 and gestational diabetes. Am J Obstet Gynecol 2000; 182: 313-320 [PMID: 10694330]
10 Yang J, Cummings EA, O’connell C, Jangaard K. Fetal and neonatal outcomes of diabetic pregnancies. Obstet Gynecol 2006; 108: 644-650 [PMID: 16946226 DOI: 10.1097/01.AOG.0000231688.08263.47]
11 Balsells M, García-Patterson A, Gich I, Corcoy R. Major congenital malformations in women with gestational diabetes mellitus: a systematic review and meta-analysis. Diabetes Metab Res Rev 2012; 28: 252-257 [PMID: 22052679 DOI: 10.1002/dmrr.1304]
12 Galindo A, Burguillo AG, Azriel S, Fuente Pde L. Outcome of fetuses in women with pregestational diabetes mellitus. J Perinat Med 2006; 34: 323-331 [PMID: 16856824 DOI: 10.1515/JPM.2006.062]
13 Lemons JA, Vargas P, Delaney JJ. Infant of the diabetic mother: review of 225 cases. Obstet Gynecol 1981; 57: 187-192 [PMID: 7193318]
486 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Gabbay-Benziv R et al . Birth defects in pregestational diabetes

14 Schaefer UM, Songster G, Xiang A, Berkowitz K, Buchanan TA, Kjos SL. Congenital malformations in offspring of women with hyperglycemia first detected during pregnancy. Am J Obstet Gynecol 1997; 177: 1165-1171 [PMID: 9396914]
15 Moore TR. Diabetes in pregnancy. In: Maternal-fetal medicine, 4th edition. 1999: 964-965
16 Wender-Ozegowska E, Wróblewska K, Zawiejska A, Pietryga M, Szczapa J, Biczysko R. Threshold values of maternal blood glucose in early diabetic pregnancy--prediction of fetal malformations. Acta Obstet Gynecol Scand 2005; 84: 17-25 [PMID: 15603562 DOI: 10.1111/j.0001-6349.2005.00606.x]
17 Wren C, Birrell G, Hawthorne G. Cardiovascular malformations in infants of diabetic mothers. Heart 2003; 89: 1217-1220 [PMID: 12975424]
18 Janssen PA, Rothman I, Schwartz SM. Congenital malformations in newborns of women with established and gestational diabetes in Washington State, 1984-91. Paediatr Perinat Epidemiol 1996; 10: 52-63 [PMID: 8746431]
19 Jones KL. Smith’s recognizable patterns of human malformation. 5th edn. Philadelphia: WB Saunders, 1997: 635
20 Versiani BR, Gilbert-Barness E, Giuliani LR, Peres LC, Pina-Neto JM. Caudal dysplasia sequence: severe phenotype presenting in offspring of patients with gestational and pregestational diabetes. Clin Dysmorphol 2004; 13: 1-5 [PMID: 15127755]
21 Eidem I, Stene LC, Henriksen T, Hanssen KF, Vangen S, Vollset SE, Joner G. Congenital anomalies in newborns of women with type 1 diabetes: nationwide population-based study in Norway, 1999-2004. Acta Obstet Gynecol Scand 2010; 89: 1403-1411 [PMID: 20929418 DOI: 10.3109/00016349.2010.518594]
22 Garne E, Loane M, Dolk H, Barisic I, Addor MC, Arriola L, Bakker M, Calzolari E, Matias Dias C, Doray B, Gatt M, Melve KK, Nelen V, O’Mahony M, Pierini A, Randrianaivo-Ranjatoelina H, Rankin J, Rissmann A, Tucker D, Verellun-Dumoulin C, Wiesel A. Spectrum of congenital anomalies in pregnancies with pregestational diabetes. Birth Defects Res A Clin Mol Teratol 2012; 94: 134-140 [PMID: 22371321 DOI: 10.1002/bdra.22886]
23 Correa A, Gilboa SM, Besser LM, Botto LD, Moore CA, Hobbs CA, Cleves MA, Riehle-Colarusso TJ, Waller DK, Reece EA. Diabetes mellitus and birth defects. Am J Obstet Gynecol 2008; 199: 237.e1-237.e9 [PMID: 18674752 DOI: 10.1016/j.ajog.2008.06.028]
24 Fuhrmann K, Reiher H, Semmler K, Glöckner E. The effect of intensified conventional insulin therapy before and during pregnancy on the malformation rate in offspring of diabetic mothers. Exp Clin Endocrinol 1984; 83: 173-177 [PMID: 6373320 DOI: 10.1055/s-0029-1210327]
25 Hanson U, Persson B, Thunell S. Relationship between hae-moglobin A1C in early type 1 (insulin-dependent) diabetic pregnancy and the occurrence of spontaneous abortion and fetal malformation in Sweden. Diabetologia 1990; 33: 100-104 [PMID: 2328844]
26 Kitzmiller JL, Gavin LA, Gin GD, Jovanovic-Peterson L, Main EK, Zigrang WD. Preconception care of diabetes. Glycemic control prevents congenital anomalies. JAMA 1991; 265: 731-736 [PMID: 1990188]
27 Mironiuk M, Kietlińska Z, Jezierska-Kasprzyk K, Piekosz-Orzechowska B. A class of diabetes in mother, glycemic control in early pregnancy and occurrence of congenital malformations in newborn infants. Clin Exp Obstet Gynecol 1997; 24: 193-197 [PMID: 9478316]
28 Reece EA, Gabrielli S, Abdalla M. The prevention of diabetes-associated birth defects. Semin Perinatol 1988; 12: 292-301 [PMID: 3065942]
29 Ylinen K, Aula P, Stenman UH, Kesäniemi-Kuokkanen T, Teramo K. Risk of minor and major fetal malformations in diabetics with high haemoglobin A1c values in early pregnancy. Br Med J (Clin Res Ed) 1984; 289: 345-346 [PMID: 6432090]
30 Shields LE, Gan EA, Murphy HF, Sahn DJ, Moore TR. The prognostic value of hemoglobin A1c in predicting fetal heart disease in diabetic pregnancies. Obstet Gynecol 1993; 81: 954-957 [PMID: 8497362]
31 Lucas MJ, Leveno KJ, Williams ML, Raskin P, Whalley PJ. Early pregnancy glycosylated hemoglobin, severity of diabetes, and fetal malformations. Am J Obstet Gynecol 1989; 161: 426-431 [PMID: 2669494]
32 Dunne FP, Avalos G, Durkan M, Mitchell Y, Gallacher T, Keenan M, Hogan M, Carmody LA, Gaffney G. ATLANTIC DIP: pregnancy outcome for women with pregestational diabetes along the Irish Atlantic seaboard. Diabetes Care 2009; 32: 1205-1206 [PMID: 19564472 DOI: 10.2337/dc09-1118]
33 Owens LA, Avalos G, Kirwan B, Carmody L, Dunne F. ATLANTIC DIP: closing the loop: a change in clinical practice can improve outcomes for women with pregestational diabetes. Diabetes Care 2012; 35: 1669-1671 [PMID: 22826448 DOI: 10.2337/dc12-0120]
34 Greene MF, Hare JW, Cloherty JP, Benacerraf BR, Soeldner JS. First-trimester hemoglobin A1 and risk for major malformation and spontaneous abortion in diabetic pregnancy. Teratology 1989; 39: 225-231 [PMID: 2727930 DOI: 10.1002/tera.1420390303]
35 Todorova K, Mazneĭkova V, Ivanov S, Genova M. [The frequency of mild and severe fetal malformations in diabetic women with high values of glycosilated hemoglobin in early pregnancy]. Akush Ginekol (Sofiia) 2005; 44: 3-10 [PMID: 16028383]
36 Kerssen A, Evers IM, de Valk HW, Visser GH. Poor glucose control in women with type 1 diabetes mellitus and ‘safe’ hemoglobin A1c values in the first trimester of pregnancy. J Matern Fetal Neonatal Med 2003; 13: 309-313 [PMID: 12916680 DOI: 10.1080/jmf.13.5.309.313]
37 Dashe JS, McIntire DD, Twickler DM. Effect of maternal obesity on the ultrasound detection of anomalous fetuses. Obstet Gynecol 2009; 113: 1001-1007 [PMID: 19384114 DOI: 10.1097/AOG.0b013e3181a1d2f5]
38 Wong SF, Chan FY, Cincotta RB, Oats JJ, McIntyre HD. Routine ultrasound screening in diabetic pregnancies. Ultrasound Obstet Gynecol 2002; 19: 171-176 [PMID: 11876810 DOI: 10.1046/j.0960-7692.2001.00560.x]
39 White P. Pregnancy complicating diabetes. Am J Med 1949; 7: 609-616 [PMID: 15396063]
40 Centers for Disease Control and Prevention. National diabetes fact sheet: National estimates and general information on diabetes and prediabetes in the united states, 2011. Altanta, GA: US Department of Health and Human Services, 2011
41 Imkampe AK, Gulliford MC. Trends in Type 1 diabetes incidence in the UK in 0- to 14-year-olds and in 15- to 34-year-olds, 1991-2008. Diabet Med 2011; 28: 811-814 [PMID: 21395679 DOI: 10.1111/j.1464-5491.2011.03288.x]
42 Lapolla A, Dalfrà MG, Fedele D. Pregnancy complicated by type 2 diabetes: an emerging problem. Diabetes Res Clin Pract 2008; 80: 2-7 [PMID: 18201793 DOI: 10.1016/j.diabres.2007.11.009]
43 Jensen DM, Damm P, Moelsted-Pedersen L, Ovesen P, Wes-tergaard JG, Moeller M, Beck-Nielsen H. Outcomes in type 1 diabetic pregnancies: a nationwide, population-based study. Diabetes Care 2004; 27: 2819-2823 [PMID: 15562191]
44 Gonzalez-Gonzalez NL, Ramirez O, Mozas J, Melchor J, Armas H, Garcia-Hernandez JA, Caballero A, Hernandez M, Diaz-Gomez MN, Jimenez A, Parache J, Bartha JL. Factors influencing pregnancy outcome in women with type 2 versus type 1 diabetes mellitus. Acta Obstet Gynecol Scand 2008; 87: 43-49 [PMID: 18158626 DOI: 10.1080/00016340701778732]
45 Peticca P, Keely EJ, Walker MC, Yang Q, Bottomley J. Pregnancy outcomes in diabetes subtypes: how do they compare? A province-based study of Ontario, 2005-2006. J Obstet Gynaecol Can 2009; 31: 487-496 [PMID: 19646313]
46 Lapolla A, Dalfrà MG, Di Cianni G, Bonomo M, Parretti E, Mello G. A multicenter Italian study on pregnancy outcome in women with diabetes. Nutr Metab Cardiovasc Dis 2008; 18: 291-297 [PMID: 17433638 DOI: 10.1016/j.numecd.2006.12.001]
47 Clausen TD, Mathiesen E, Ekbom P, Hellmuth E, Mandrup-Poulsen T, Damm P. Poor pregnancy outcome in women with type 2 diabetes. Diabetes Care 2005; 28: 323-328 [PMID: 15677787]
48 Roland JM, Murphy HR, Ball V, Northcote-Wright J, Temple RC. The pregnancies of women with Type 2 diabetes: poor outcomes
487 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Gabbay-Benziv R et al . Birth defects in pregestational diabetes

but opportunities for improvement. Diabet Med 2005; 22: 1774-1777 [PMID: 16401329 DOI: 10.1111/j.1464-5491.2005.01784.x]
49 Macintosh MC, Fleming KM, Bailey JA, Doyle P, Modder J, Acolet D, Golightly S, Miller A. Perinatal mortality and congenital anomalies in babies of women with type 1 or type 2 diabetes in England, Wales, and Northern Ireland: population based study. BMJ 2006; 333: 177 [PMID: 16782722 DOI: 10.1136/bmj.38856.692986.AE]
50 Li X, Weng H, Xu C, Reece EA, Yang P. Oxidative stress-induced JNK1/2 activation triggers proapoptotic signaling and apoptosis that leads to diabetic embryopathy. Diabetes 2012; 61: 2084-2092 [PMID: 22688338 DOI: 10.2337/db11-1624]
51 Xu C, Li X, Wang F, Weng H, Yang P. Trehalose prevents neural tube defects by correcting maternal diabetes-suppressed autophagy and neurogenesis. Am J Physiol Endocrinol Metab 2013; 305: E667-E678 [PMID: 23880312 DOI: 10.1152/ajpendo.00185.2013]
52 Reece EA , Ma XD, Zhao Z, Wu YK, Dhanasekaran D. Aberrant patterns of cellular communication in diabetes-induced embryopathy in rats: II, apoptotic pathways. Am J Obstet Gynecol 2005; 192: 967-972 [PMID: 15746699 DOI: 10.1016/j.ajog.2004.10.592]
53 Yang X, Borg LA, Eriksson UJ. Altered metabolism and superoxide generation in neural tissue of rat embryos exposed to high glucose. Am J Physiol 1997; 272: E173-E180 [PMID: 9038867]
54 Sivan E, Lee YC, Wu YK, Reece EA. Free radical scavenging enzymes in fetal dysmorphogenesis among offspring of diabetic rats. Teratology 1997; 56: 343-349 [PMID: 9485543 DOI: 10.1002/(SICI)1096-9926(199712)56:6<343::AID-TERA1>3.0.CO;2-X]
55 Sakamaki H, Akazawa S, Ishibashi M, Izumino K, Takino H, Yamasaki H, Yamaguchi Y, Goto S, Urata Y, Kondo T, Nagataki S. Significance of glutathione-dependent antioxidant system in diabetes-induced embryonic malformations. Diabetes 1999; 48: 1138-1144 [PMID: 10331421]
56 Yang P, Li H. Epigallocatechin-3-gallate ameliorates hyper-glycemia-induced embryonic vasculopathy and malformation by inhibition of Foxo3a activation. Am J Obstet Gynecol 2010; 203: 75.e1-75.e6 [PMID: 20417490 DOI: 10.1016/j.ajog.2010.02.008]
57 Yang P, Cao Y, Li H. Hyperglycemia induces inducible nitric oxide synthase gene expression and consequent nitrosative stress via c-Jun N-terminal kinase activation. Am J Obstet Gynecol 2010; 203: 185.e5-185.11 [PMID: 20541731 DOI: 10.1016/j.ajog.2010.05.003]
58 Li X, Weng H, Reece EA, Yang P. SOD1 overexpression in vivo blocks hyperglycemia-induced specific PKC isoforms:
substrate activation and consequent lipid peroxidation in diabetic embryopathy. Am J Obstet Gynecol 2011; 205: 84.e1-84.e6 [PMID: 21529760 DOI: 10.1016/j.ajog.2011.02.071]
59 Wang F, Reece EA, Yang P. Superoxide dismutase 1 overexpression in mice abolishes maternal diabetes-induced endoplasmic reticulum stress in diabetic embryopathy. Am J Obstet Gynecol 2013; 209: 345.e1-345.e7 [PMID: 23791840 DOI: 10.1016/j.ajog.2013.06.037]
60 Weng H, Li X, Reece EA, Yang P. SOD1 suppresses maternal hyperglycemia-increased iNOS expression and consequent nitrosative stress in diabetic embryopathy. Am J Obstet Gynecol 2012; 206: 448.e1-448.e7 [PMID: 22425406 DOI: 10.1016/j.ajog.2012.02.011]
61 Sugimura Y, Murase T, Oyama K, Uchida A, Sato N, Hayasaka S, Kano Y, Takagishi Y, Hayashi Y, Oiso Y, Murata Y. Prevention of neural tube defects by loss of function of inducible nitric oxide synthase in fetuses of a mouse model of streptozotocin-induced diabetes. Diabetologia 2009; 52: 962-971 [PMID: 19283362 DOI: 10.1007/s00125-009-1312-0]
62 Yang P, Zhao Z, Reece EA. Blockade of c-Jun N-terminal kinase activation abrogates hyperglycemia-induced yolk sac vasculopathy in vitro. Am J Obstet Gynecol 2008; 198: 321.e1-321.e7 [PMID: 18177823 DOI: 10.1016/j.ajog.2007.09.010]
63 Yang P, Zhao Z, Reece EA. Activation of oxidative stress signaling that is implicated in apoptosis with a mouse model of diabetic embryopathy. Am J Obstet Gynecol 2008; 198: 130.e1-130.e7 [PMID: 18166327 DOI: 10.1016/j.ajog.2007.06.070]
64 Yang P, Zhao Z, Reece EA. Involvement of c-Jun N-terminal kinases activation in diabetic embryopathy. Biochem Biophys Res Commun 2007; 357: 749-754 [PMID: 17449011 DOI: 10.1016/j.bbrc.2007.04.023]
65 Yang P, Li X, Xu C, Eckert RL, Reece EA, Zielke HR, Wang F. Maternal hyperglycemia activates an ASK1-FoxO3a-caspase 8 pathway that leads to embryonic neural tube defects. Sci Signal 2013; 6: ra74 [PMID: 23982205 DOI: 10.1126/scisignal.2004020]
66 Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8: 519-529 [PMID: 17565364 DOI: 10.1038/nrm2199]
67 Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol 2011; 23: 143-149 [PMID: 21146390 DOI: 10.1016/j.ceb.2010.11.003]
68 Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 2002; 16: 1345-1355 [PMID: 12050113 DOI: 10.1101/gad.992302]
P- Reviewer: Ren AG S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
488 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Gabbay-Benziv R et al . Birth defects in pregestational diabetes

nations by 20% and in developing countries by 69% until the year 2030. Due to the expected rise in diabetic patients, the need for ophthalmic care of patients (i.e. , exams and treatments) will also increase and represents a challenge for eye-care providers. Development of optimized screening programs, which respect available resources of the ophthalmic infrastructure, will become even more important. Main reasons for loss of vision in patients with diabetes mellitus are diabetic macular edema and proliferative diabetic retinopathy. Incidence or progression of these potentially blinding complications can be greatly reduced by adequate control of blood glucose and blood pressure levels. Additionally, regular ophthalmic exams are mandatory for detecting ocular complications and initiating treatments such as laser photocoagulation in case of clinical significant diabetic macular edema or early proliferative diabetic retinopathy. In this way, the risk of blindness can considerably be reduced. In advanced stages of diabetic retinopathy, pars-plana vitrectomy is performed to treat vitreous hemorrhage and tractional retinal detachment. In recent years, the advent of intravitreal medication has improved therapeutic options for patients with advanced diabetic macular edema.
Key words: Laser photocoagulation; Diabetic macular edema; Diabetic retinopathy; Intravitreal injection; Prevention
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Diabetic retinopathy is a potentially blinding complication of diabetes mellitus. In patients with diabetes, regular retinal exams are essential. While laser photocoagulation is effective, if performed in time, advanced stages of diabetic retinopathy need to be treated by vitreo-retinal surgery and have limited visual prognosis. Even though new therapeutic options such as intravitreal medical therapy and sutureless pars-plana vitrectomy have improved ophthalmic care of patients
Martin M Nentwich, Michael W Ulbig
Martin M Nentwich, Michael W Ulbig, Department of Ophthalmology, Ludwig-Maximilians University, 80336 Munich, GermanyAuthor contributions: Nentwich MM and Ulbig MW contributed to this paper.Conflict-of-interest: None.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Dr. Martin M Nentwich, Department of Ophthalmology, Ludwig-Maximilians University, Mathildenstr 8, 80336 Munich, Germany. [email protected]: +49-89-440053811Fax: +49-89-440054569Received: July 17, 2014Peer-review started: July 17, 2014First decision: November 27, 2014Revised: January 3, 2015Accepted: January 18, 2015Article in press: January 20, 2015Published online: April 15, 2015
AbstractIn industrialized nations diabetic retinopathy is the most frequent microvascular complication of diabetes mellitus and the most common cause of blindness in the working-age population. In the next 15 years, the number of patients suffering from diabetes mellitus is expected to increase significantly. By the year 2030, about 440 million people in the age-group 20-79 years are estimated to be suffering from diabetes mellitus worldwide (prevalence 7.7%), while in 2010 there were 285 million people with diabetes mellitus (prevalence 6.4%). This accounts for an increase in patients with diabetes in industrialized
MINIREVIEWS
489 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Diabetic retinopathy - ocular complications of diabetes mellitus
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.489
World J Diabetes 2015 April 15; 6(3): 489-499ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

with diabetes, interdisciplinary care of these patients is essential. Good metabolic and blood pressure control is indispensable for reducing the risk of ophthalmic complications.
Nentwich MM, Ulbig MW. Diabetic retinopathy - ocular complications of diabetes mellitus. World J Diabetes 2015; 6(3): 489-499 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/489.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.489
INTRODUCTIONDiabetic retinopathy is a potentially blinding complication of diabetes mellitus. Reasons for loss of vision are diabetic maculopathy and complications of proliferative diabetic retinopathy (PDR) such as vitreous hemorrhage, tractional retinal detachment, and neovascular glaucoma. By 2030 developing countries will face an increase by 69% and industrialized countries by 20% of the number of patients with diabetes compared to 2010[1]. For Africa more than 18 million, according to some estimations even 24 million, diabetic patients are predicted for the year 2030[1,2].
Probability of retinal complications increases with increasing duration of disease. In up to 50% of patients with type 1 diabetes and 30% of those with type 2 diabetes potentially visionthreatening retinal changes develop over time, while early retinal changes are not noticed by the patients[3].
Diabetic retinopathy is the most common microvascular complication of diabetes mellitus and affects between 3%4% of people in Europe, while the relative risk for developing diabetic retinopathy is higher in type 1 diabetes compared to type 2[46]. Diabetes mellitus is responsible for about 15% of all cases of legal blindness (best corrected visual acuity less than 0.02) in Germany[7]. It is the main cause of blindness within the workingage population in industrialized nations[4].
While retinal changes are rarely seen in patients with type 1 diabetes before adolescence, about one third of patients have signs of diabetic retinopathy at time of initial diagnosis of diabetes mellitus. The risk of PDR is higher in type 1 diabetes than in type 2, while diabetic macular edema is more commonly found in type 2 diabetes (prevalence after 15 years of disease: type 1 vs type 2 = 15% vs 25%)[8].
Kramer et al[9] reported in a recent study, that in patients with type 1 diabetes progression of diabetic retinopathy and development of nephropathy each increase the risk for incidence of the other. This association was independent of established risk factors for microvascular complications and the authors suggested a shared etiologic basis of these two complications of diabetes mellitus[9]. Another group also found proliferative diabetic retinopathy to be an independent marker of longterm nephropathy in patients with type 1 diabetes[10].
Other studies indicated association of the presence of diabetic retinopathy and increased overallmortality and cardiovascular events both in type 1 and type 2 diabetes[11].
Therefore an interdisciplinary approach of physicians, endocrinologists, and ophthalmologists is needed for optimal care.
PATHOGENESIS OF DIABETIC RETINOPATHYMicroangiopathy due to hyperglycemia in patients with diabetes mellitus results in vascular leakage, which causes diabetic macular edema on one hand, and capillary occlusion on the other hand. Capillary occlusion then again causes retinal ischemia and increased levels of vascular endothelial growth factor (VEGF) which are responsible for the development of neovascularization and the proliferative stage of diabetic retinopathy.
More recently new pathways which may be involved in the pathogenesis of diabetic retinopathy have been identified, such as inflammation, nerve growth factor autophagy and epigenetics. A detailed discussion of all these pathways would go beyond the scope of this minireview about clinical aspects of diabetic retinopathy, however some aspects should be addressed.
Biochemical alterations such as oxidative stress, activation of protein kinase C and formation of advanced glycation end products have been detected as a response of the retina to hyperglycemia[12]. Also kinin B1 and B2 are thought to increase vascular permeability, infiltration of leukocytes and inflammation. Especially kinin B1, which is almost nonexistent in normal tissue, is upregulated in the retina of diabetic patients. These findings may be important for developing new therapeutic strategies aiming at antagonizing kinin receptors or at inhibiting kallikreins[13].
Recent investigations showed that the whole retinal neurovascular system is impaired by diabetes mellitus resulting in loss of neurovascular coupling, neurodegeneration and neuroinflammation, which can be detected even before the advent of vascular damage[14]. Clinically, reduced dark adaption, impaired colour and/or contrast vision and visual field defects are found during functional examinations of diabetic patients[15].
Diabetic retinopathy tends to deteriorate during hormonal changes such as adolescence and pregnancy[16].
CLASSIFICATION AND PATHOPYSIOLOGY OF DIABETIC RETINOPATHYDuring fundoscopy, which should be performed after dilation of the pupil (mydriasis) to allow visualization of the entire retina, presence and grade of diabetic retinopathy can be assessed clinically. Typical changes seen in early diabetic retinopathy (nonPDR) are micro
Nentwich MM et al . Diabetes mellitus: Ocular complications
490 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

aneurysms, retinal hemorrhages, and exudates (Figure 1). In the beginning, these alterations are often found slightly temporal to the central area of the macula (Figure 2). These are caused by deranged vascular integrity and loss of pericytes. In the later course of disease, intraretinal microvascular anomalies may develop, which represent dilated and therefore during fundoscopy visible retinal capillaries, and indicate possible risk of neovascularization and proliferative diabetic retinopathy.
Proliferative diabetic retinopathy is caused by increased intraocular VEGFlevels due to retinal ischemia because of capillary occlusion of retinal vessels. Proliferation may
grow at the optic disc (neovascularization at the disk) or elsewhere in the retina (neovascularization elsewhere ) into the vitreous (Figure 3). These newly formed vessels leak on fluorescein angiography and may cause vitreous hemorrhage and finally tractional retinal detachment (Figures 4 and 5). Tractional retinal detachment results in separation of the neurosensory retina from the retinal pigment epithelium. Detached sections of the retina cause relative visual-field defects (scotoma) and loss of visual acuity in cases with macular involvement.
Further, neovascularization on the iris in the anterior segment of the eye may evolve as anterior segment sequel of ischemia (Figure 6). These new blood vessels grow towards the anterior chamber angle and may obstruct the trabecular meshwork, thus increasing outflow resistance of aqueous humor. This results in increased intraocular pressure followed by optic atrophy (neovascular glaucoma).
CLASSIFICATION AND PATHOPHYSIOLOGY OF DIABETIC MACULOPATHYDiabetic maculopathy may develop in the nonproliferative and in the proliferative stage of diabetic retinopathy. While complications of untreated proliferative diabetic
491 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 3 Proliferative diabetic retinopathy with neovascularization at disk. Figure 4 Advanced proliferative diabetic retinopathy with neovascularization and limited vitreous hemorrhage.
Figure 5 Advanced proliferative diabetic retinopathy with tractional retinal detachment.
Figure 1 Non-proliferative diabetic retinopathy. Wide-field fundus photo of a 65-year-old female patient (right eye) showing several retinal hemorrhages.
Figure 2 Non-proliferative diabetic retinopathy. Color fundus photo of a 51-year-old male patient with micro-aneurysms and lipid exudates.
Nentwich MM et al . Diabetes mellitus: Ocular complications

domain OCT). Vitrectomy is needed in patients with symptomatic tractional macular edema in order to release traction by surgical removal of the vitreous.
Apart from edema, diabetic maculopathy may also present as occlusion of perifoveal capillaries resulting in distinct visual impairment in the absence of edema on fundoscopy. In these cases, fluorescein angiography is needed to confirm the diagnosis by visualizing capillary occlusion in around the fovea.
Table 1 summarizes the different types of diabetic maculopathy (adapted from Nentwich et al[19]).
CLINICAL DIAGNOSICSDilated fundoscopy is the most important clinical investigation in patients with diabetes during screening examinations by an ophthalmologist. Dilation of the pupil is necessary in order to enable a proper stereoscopic view, which is needed for evaluation of macular edema, and to allow visualization of the peripheral retina.
Fluorescein angiography was introduced by Novotny et al[20] in the 1960s into the clinical ophthalmic practice and allows evaluation of the retinal vascular status. Fluorescein, a fluorescent dye, is injected intravenously and distributes throughout the body. In the eye, fluorescence is activated by blue light of 490 nm. During fluorescein angiography vascular leakage, capillary occlusion, ischemic areas of the retina and neovascularization can be seen (Figure 9A and C). It provides information on the area of leakage as well as of the location of nonperfused parts of the retina. In patients with suspected diabetic maculopathy, fluorescein angiography is mandatory for excluding ischemic maculopathy and guiding possible focal laser photocoagulation.
Wide-field fundus photography (2-laser wavelength nonmydriatic 200° ultrawidefield scanning laser ophthalmoscopy) has been introduced recently. Widefield images and traditional color fundus photography
retinopathy, such as vitreous hemorrhage and tractional retinal detachment involving the macula may cause the most severe loss of vision in diabetic retinopathy, diabetic maculopathy is the main cause of visual impairment in patients with type 2 diabetes[17].
Diabetic macular edema is caused by a disruption of the inner bloodretinal barrier, formed by the retinal vascular endothelium, due to hyperglycemia, increased levels of growthfactors, inflammation and cytokines. This also causes impairment of pericytes and leads to consecutive exudation of fluid, proteins and lipids by para cellular and trans cellular transport mechanisms[18]. Clinically, areas of thickened retina are often demarcated by surrounding yellowish lipid exudates (Figure 7). Diabetic macular edema may be limited or affect a large retinal area and it may also involve the macula (centerinvolving) or spare the central area (noncenterinvolving) (Figure 8).
Traction caused by vitreous attachment to the fovea may result in macular thickening, as well. Vitreous traction can excellently be documented by highresolution optical coherence tomography (OCT, spectral
492 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 6 Neovascularization of the iris. These neovascular vessels may block the trabecular meshwork and cause neovascular glaucoma.
Table 1 Types of diabetic maculopathy[19]
Focal Localized edemaLipid exudatesIntraretinal hemorrhagesFocal hyperfluorescence in late fluorescein angiography
Clinically significant without foveal thickening (non-center-involving) (sight-threatening)
Edema within 500 μm around the foveolaExudates within 500 μm around the foveola accompanied by edemaEdema ≥ 1 optic-disk diameter within one optic-disk diameter around the foveola
Clinically significant with foveal thickening (center-involving) Ill-defined edema, which may be cystoidExudatesIntraretinal hemorrhagesOrigin of leakage often not clearly identifiable by fluorescein angiography
Tractional Due to vitreous traction to the foveaThickened posterior hyaloid membraneOCT visualizes vitreal traction
Ischemic maculopathy (occlusion of the perifoveal capillaries) Loss of vision without any clearly visible cause on fundoscopyFluorescein angiography needed for diagnosisDifficult to diagnose by fundoscopy onlyEdema may be present or absent
OCT: Optical coherence tomography.
Nentwich MM et al . Diabetes mellitus: Ocular complications

in mydriasis seem to correlate with regard to the classification of diabetic retinopathy and visualization of diabetic macular edema according to recent studies (Figure 10A)[2123]. This widefield imaging technique can be combined with fluorescein angiography and provides information about peripheral retinal ischemia or peripheral neovascularization (Figure 10B)[24].
Probably the most important new imaging technique, which was introduced in ophthalmologic practice, is OCT. Recently, resolution and recording speed have been greatly improved by spectraldomain OCT technique (SDOCT). OCT provides data on retinal volume and configuration of the macular region. In diabetic macular edema SDOCT is a very valuable examination technique comparing followup visits with baseline data, as modern OCT-devices use eye-tracking for finding the same imaging position during followup examinations (Figure 11)[25].
IMPORTANCE OF PREVENTIONIn early diabetic retinopathy, laser photocoagulation can be used effectively for preventing loss of vision[26]. However, early diabetic retinopathy is not noticed by patients because of the absence of visual loss in early diabetic retinopathy. Thus, regular retinal exams with dilation of the pupil are necessary in patients with
diabetes mellitus in order to detect sightthreatening changes timely and enable ophthalmologists to perform treatment.
Patients with type 1 diabetes should undergo retinal exams starting at the age 11 and/or after 5 years of diagnosis on a yearly basis. In case of retinal changes, shorter followup intervals are recommended.
In type 2 diabetes, the first retinal exam should be performed immediately after first diagnosis of diabetes, because the previous duration of the disease is unknown. Annual followup examinations are recommended in the absence of retinal changes, otherwise shorter intervals are recommended.
Pregnancy bears an increased risk of worsening of diabetic retinopathy due to hormonal changes. During pregnancy diabetic retinopathy may start in about 10% of cases and may worsen in an even higher percentage in case of preexisting diabetic retinopathy at time of conception[27]. In case of proliferative diabetic retinopathy before or shortly after conception, panretinal laser photocoagulation should be performed as retinal changes may worsen in one out of two women in this population. This risk can be reduced by half by thorough retinal laser photocoagulation. Therefore all females with diabetes, who are planning to get pregnant, should undergo fundoscopy prior to conception and then every three months during pregnancy to enable early intervention, if needed. Additionally good metabolic control should be achieved before conception[28].
On the other hand, the presence of diabetic retinopathy is no indication for cesarean section per se, as no study has indicated so far, that Valsalvamaneuvers during vaginal delivery bear an increased risk of vitreous hemorrhages[29].
The German Diabetes Society recommends scheduling of followup examinations depending on the grade of diabetic retinopathy as summarized in Table 2 (Adapted from[30]).
These recommended examination intervals will result in an increased workload for ophthalmologists due to the rising number of diabetic patients. Studies examined the effect of an extension of followup intervals and found no increased risk of progression to sight
493 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 7 Clinically significant diabetic macular edema without involvement of the fovea. A: Fundus photo; B: Fluorescein angiogram depicting leakage of perifoveal retinal blood vessels.
A B
Figure 8 Center-involving diabetic macular edema with subfoveal edema and numerous lipid exudates.
Nentwich MM et al . Diabetes mellitus: Ocular complications

threatening diabetic retinopathy in patients with good metabolic control (HbA1c < 8%) and no known diabetic retinopathy[3135]. By adequate selection of patients, the number of necessary screening examinations could be reduced by 40% according to one study and even by 59% in case a special mathematical algorism is used[31,33]. However, feasibility of these strategies remains to be proven in routine clinical practice and until then
ophthalmic screening intervals as indicated above are recommended.
GENERAL MEDICAL THERAPY AND INTERVENTIONSBlood glucose and blood pressure controlGood metabolic and bloodpressure control are essential for successful ophthalmic care of patients with diabetes.
In type 2 diabetes, a reduction of HbA1c from 7.9% to 7.0% resulted in a decline in the frequency of laser treatments needed[36]. In type 1 patients, improved blood glucose control with a reduction of HbA1c values from 9.1% to 7.1% reduced the risk of developing diabetic retinopathy within 6.5 years by 76%, the risk of progression of diabetic retinopathy by 54%, and the risk of developing proliferative diabetic retinopathy by 47%[37]. Therefore an HbA1c level of about 7% should be aimed for from the ophthalmologist’s point of view[19], for the individual patient a bespoke treatment regime may be needed.
Rapid improvement of metabolic control may result in temporary worsening of diabetic retinopathy (“earlyworsening”) in patients with longlasting disease and
494 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
A B
Figure 10 Wide-field picture of the right eye of a 65-year-old female patient. A: On scanning-laser-ophthalmoscope-Imaging some micro-aneurysms and lipid exudates can be seen; B: Fluorescein angiogram shows leakage from micro-aneurysms and extensive areas of retinal non-perfusion.
Figure 11 Spectral-domain optical coherence tomography of a female patient with center-involving diabetic macular edema. On the left side of the picture, an infra-red image shows the exact location of the OCT-scan on the right. The OCT-scan visualizes intraretinal edema with thickening of the fovea. OCT: Optical coherence tomography.
Nentwich MM et al . Diabetes mellitus: Ocular complications
Figure 9 Fluorescein angiogram of a 49-year-old female patient. A: Fluorescein angiogram of the right eye 50 s after intravenous injection of fluorescein dye. Here, leaking micro-aneurysms in the macula can be seen; B: Fluorescein angiogram of the left eye 25 s after intravenous injection of fluorescein dye. Leakage from neovascular blood vessels causes spots of increased fluorescence at the optic disk and temporal to the fovea; C: Fluorescein angiogram of the temporal part of the left eye 30 s after intravenous injection of fluorescein dye. Areas of retinal non-perfusion can be seen as reason for neovascularization.
A B C
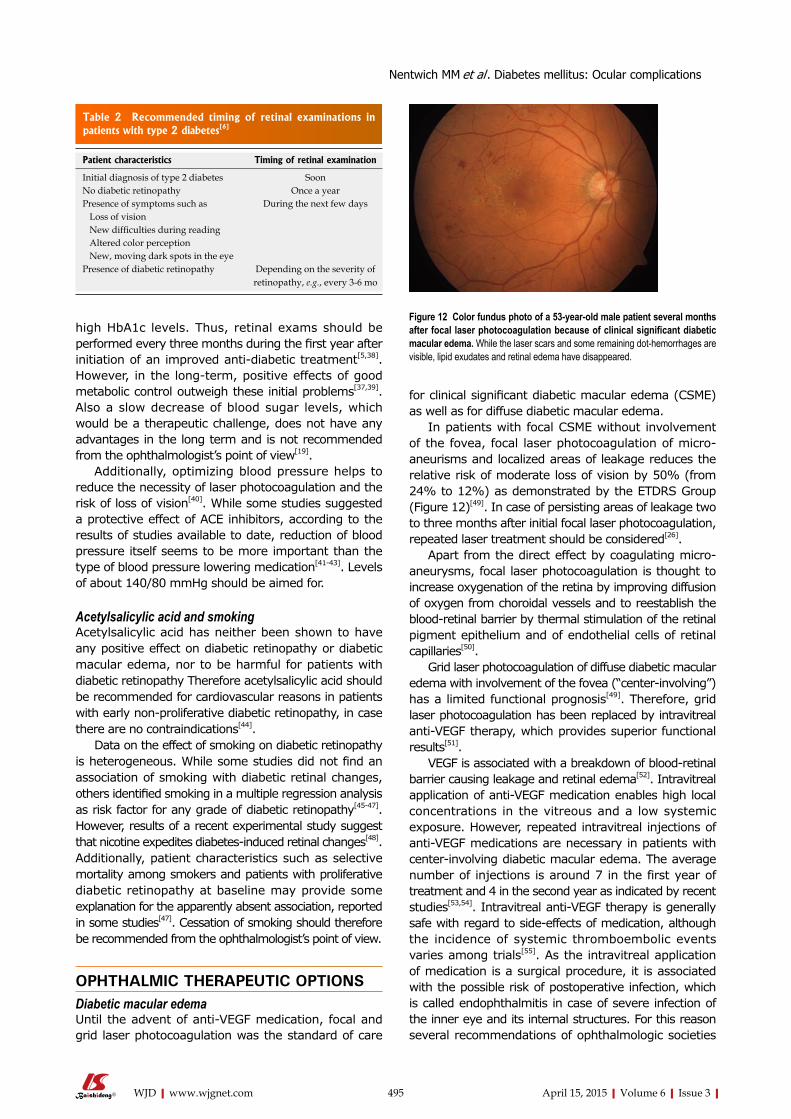
high HbA1c levels. Thus, retinal exams should be performed every three months during the first year after initiation of an improved antidiabetic treatment[5,38]. However, in the longterm, positive effects of good metabolic control outweigh these initial problems[37,39]. Also a slow decrease of blood sugar levels, which would be a therapeutic challenge, does not have any advantages in the long term and is not recommended from the ophthalmologist’s point of view[19].
Additionally, optimizing blood pressure helps to reduce the necessity of laser photocoagulation and the risk of loss of vision[40]. While some studies suggested a protective effect of ACE inhibitors, according to the results of studies available to date, reduction of blood pressure itself seems to be more important than the type of blood pressure lowering medication[4143]. Levels of about 140/80 mmHg should be aimed for.
Acetylsalicylic acid and smokingAcetylsalicylic acid has neither been shown to have any positive effect on diabetic retinopathy or diabetic macular edema, nor to be harmful for patients with diabetic retinopathy Therefore acetylsalicylic acid should be recommended for cardiovascular reasons in patients with early nonproliferative diabetic retinopathy, in case there are no contraindications[44].
Data on the effect of smoking on diabetic retinopathy is heterogeneous. While some studies did not find an association of smoking with diabetic retinal changes, others identified smoking in a multiple regression analysis as risk factor for any grade of diabetic retinopathy[4547]. However, results of a recent experimental study suggest that nicotine expedites diabetesinduced retinal changes[48]. Additionally, patient characteristics such as selective mortality among smokers and patients with proliferative diabetic retinopathy at baseline may provide some explanation for the apparently absent association, reported in some studies[47]. Cessation of smoking should therefore be recommended from the ophthalmologist’s point of view.
OPHTHALMIC THERAPEUTIC OPTIONSDiabetic macular edemaUntil the advent of antiVEGF medication, focal and grid laser photocoagulation was the standard of care
for clinical significant diabetic macular edema (CSME) as well as for diffuse diabetic macular edema.
In patients with focal CSME without involvement of the fovea, focal laser photocoagulation of microaneurisms and localized areas of leakage reduces the relative risk of moderate loss of vision by 50% (from 24% to 12%) as demonstrated by the ETDRS Group (Figure 12)[49]. In case of persisting areas of leakage two to three months after initial focal laser photocoagulation, repeated laser treatment should be considered[26].
Apart from the direct effect by coagulating microaneurysms, focal laser photocoagulation is thought to increase oxygenation of the retina by improving diffusion of oxygen from choroidal vessels and to reestablish the bloodretinal barrier by thermal stimulation of the retinal pigment epithelium and of endothelial cells of retinal capillaries[50].
Grid laser photocoagulation of diffuse diabetic macular edema with involvement of the fovea (“centerinvolving”) has a limited functional prognosis[49]. Therefore, grid laser photocoagulation has been replaced by intravitreal antiVEGF therapy, which provides superior functional results[51].
VEGF is associated with a breakdown of bloodretinal barrier causing leakage and retinal edema[52]. Intravitreal application of antiVEGF medication enables high local concentrations in the vitreous and a low systemic exposure. However, repeated intravitreal injections of antiVEGF medications are necessary in patients with centerinvolving diabetic macular edema. The average number of injections is around 7 in the first year of treatment and 4 in the second year as indicated by recent studies[53,54]. Intravitreal antiVEGF therapy is generally safe with regard to sideeffects of medication, although the incidence of systemic thromboembolic events varies among trials[55]. As the intravitreal application of medication is a surgical procedure, it is associated with the possible risk of postoperative infection, which is called endophthalmitis in case of severe infection of the inner eye and its internal structures. For this reason several recommendations of ophthalmologic societies
495 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 2 Recommended timing of retinal examinations in patients with type 2 diabetes[6]
Patient characteristics Timing of retinal examination
Initial diagnosis of type 2 diabetes SoonNo diabetic retinopathy Once a yearPresence of symptoms such as During the next few days Loss of vision New difficulties during reading Altered color perception New, moving dark spots in the eyePresence of diabetic retinopathy Depending on the severity of
retinopathy, e.g., every 3-6 mo
Figure 12 Color fundus photo of a 53-year-old male patient several months after focal laser photocoagulation because of clinical significant diabetic macular edema. While the laser scars and some remaining dot-hemorrhages are visible, lipid exudates and retinal edema have disappeared.
Nentwich MM et al . Diabetes mellitus: Ocular complications

have been published concerning prevention of postinjection endophthalmitis. Recent studies reported very low rates of post-injection endophthalmitis of less than 1 in 8000 injections performed in an operating theater setting after diligent disinfection of the conjunctiva with povidoneiodine, using sterile gloves and wearing facemasks[56,57]. Other studies report somewhat higher rates of infectious endophthalmitis after intravitreal injection, both after procedures performed in an office-setting and in a theatersetting[58]. The effect of meticulous povidoneiodine prophylaxis on the conjunctival bacterial load and contamination of needles used for injection prophylaxis has been shown in several studies[5962].
Apart from antiVEGF medication intravitreal steroids have been evaluated for the treatment of diabetic macular edema. Compared to antiVEGF medication, steroids have additional antiinflammatory effects and sustainedrelease devices can lengthen the intervals between retreatments[63]. In Europe, a nonabsorbable implant containing 190 μg fluocinolone acetonide has been approved as secondline treatment of chronic diabetic macular edema not responding to other therapeutic options. Also approval of an absorbable dexamethasone implant for the treatment of diabetic macular edema is expected by the end of 2014. While
intravitreal steroids are effective in reducing diabetic macular edema, patients need to be informed about possible progression of cataract and increased intraocular pressure[6467].
Proliferative diabetic retinopathyIn early proliferative diabetic retinopathy, panretinal laser photocoagulation is effective in reducing the risk of visual loss, while surgery needs to be performed in very advanced proliferative diabetic retinopathy[68]. The suggested screening intervals of patients with diabetes are intended to enable the treating ophthalmologist to detect proliferation amendable for laser treatment early. Panretinal laser photocoagulation should be performed when retinal neovascularization is detected during fundoscopy or via fluorescein angiography in order to prevent complications of proliferative diabetic retinopathy such as vitreous hemorrhage or tractional retinal detachment (Figure 13)[68]. Panretinal laser photocoagulation aims at eliminating nonperfused parts of the retina, thus reducing ischemia, intravitreal VEGFlevels, and the stimulus for proliferation. The efficacy of panretinal laser photocoagulation in reducing the risk of loss of vision has already been shown more than 30 years ago[68]. However, patients need to be informed about sideeffects of panretinal laser treatments such as constriction of visual fields and reduced dark-adaption due to loss of rod function. These sideeffects may also interfere with the patients’ driving ability.
Parsplana vitrectomy is performed in advanced cases of proliferative diabetic retinopathy, the presence of extensive tractional membranes, vitreous hemorrhage, and tractional retinal detachment (Figure 14). About ten years ago trans conjunctival sutureless 23 gauge vitrectomy started to replace conventional 20 gauge technique and offers comparable safety and efficacy as well as reduced surgerytimes and faster rehabilitation of patients[69].
Combination of diabetic macular edema and proliferative diabetic retinopathyIn patients with diabetic macular edema and proliferation, treatment of macular edema should be performed before panretinal laser photocoagulation in order to prevent worsening of macular edema due to peripheral panretinal laser photocoagulation.
CONCLUSIONIn patients with diabetes, regular retinal exams are essential. While laser photocoagulation is effective, if performed in time, advanced stages of diabetic retinopathy need to be treated by vitreoretinal surgery and have limited visual prognosis. Even though new therapeutic options such as intravitreal medical therapy and sutureless parsplana vitrectomy have improved ophthalmic care of patients with diabetes, interdisciplinary care remains essential. Good metabolic and blood pressure control is indispensable for reducing the risk of
496 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 13 Wide-field picture of the right eye of a 48-year-old male patient after pan-retinal laser photocoagulation because of proliferative diabetic retinopathy. The peripheral laser scars can be seen in the picture, while neovascularization have regressed.
Figure 14 Proliferative diabetic retinopathy with extensive fibro-vascular membranes.
Nentwich MM et al . Diabetes mellitus: Ocular complications

ophthalmic complications.
REFERENCES1 Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence
of diabetes for 2010 and 2030. Diabetes Res Clin Pract 2010; 87: 4-14 [PMID: 19896746 DOI: 10.1016/j.diabres.2009.10.007]
2 World Health Organization. Diabetes Action Now: An Initiative of the World Health Organization and the International Diabetes Federation. Switzerland: World Health Organization, 2004
3 Stefánsson E, Bek T, Porta M, Larsen N, Kristinsson JK, Agardh E. Screening and prevention of diabetic blindness. Acta Ophthalmol Scand 2000; 78: 374-385 [PMID: 10990036 DOI: 10.1034/j.1600-0420.2000.078004374.x]
4 Prokofyeva E, Zrenner E. Epidemiology of major eye diseases leading to blindness in Europe: a literature review. Ophthalmic Res 2012; 47: 171-188 [PMID: 22123077 DOI: 10.1159/000329603]
5 Hammes HP. Optimal treatment of diabetic retinopathy. Ther Adv Endocrinol Metab 2013; 4: 61-71 [PMID: 23626903 DOI: 10.1177/2042018813477886]
6 Deutsche DG. Nationale VersorgungsLeitlinie Typ-2-Diabetes Prävention und Therapie von Netzhautkomplikationen. Berlin: Bundesärztekammer (BÄK), 2010: 1-42
7 Giani G, Janka HU, Hauner H, Standl E, Schiel R, Neu, A. Epidemiologie und Verlauf des Diabetes mellitus in Deutschland. Evidenzbasierte Leitlinie DDG-Aktualisierung 2004; 5: 1-12
8 Nentwich MM, Ulbig M. Diabetische Retinopathie. Der Diabetologe 2010; 6: 491-502 [DOI: 10.1007/s11428-010-0605-8]
9 Kramer CK, Retnakaran R. Concordance of retinopathy and nephropathy over time in Type 1 diabetes: an analysis of data from the Diabetes Control and Complications Trial. Diabet Med 2013; 30: 1333-1341 [PMID: 23909911 DOI: 10.1111/dme.12296]
10 Karlberg C, Falk C, Green A, Sjølie AK, Grauslund J. Pro-liferative retinopathy predicts nephropathy: a 25-year follow-up study of type 1 diabetic patients. Acta Diabetol 2012; 49: 263-268 [PMID: 21688016 DOI: 10.1007/s00592-011-0304-y]
11 Kramer CK, Rodrigues TC, Canani LH, Gross JL, Azevedo MJ. Diabetic retinopathy predicts all-cause mortality and cardiovascular events in both type 1 and 2 diabetes: meta-analysis of observational studies. Diabetes Care 2011; 34: 1238-1244 [PMID: 21525504 DOI: 10.2337/dc11-0079]
12 Ahsan H. Diabetic retinopathy--biomolecules and multiple pathophysiology. Diabetes Metab Syndr 2015; 9: 51-54 [PMID: 25450817 DOI: 10.1016/j.dsx.2014.09.011]
13 Bhat M, Pouliot M, Couture R, Vaucher E. The kallikrein-kinin system in diabetic retinopathy. Prog Drug Res 2014; 69: 111-143 [PMID: 25130041 DOI: 10.1007/978-3-319-06683-7_5]
14 Abcouwer SF, Gardner TW. Diabetic retinopathy: loss of neuroretinal adaptation to the diabetic metabolic environment. Ann N Y Acad Sci 2014; 1311: 174-190 [PMID: 24673341 DOI: 10.1111/nyas.12412]
15 Verbraak FD. Neuroretinal degeneration in relation to vasculopathy in diabetes. Diabetes 2014; 63: 3590-3592 [PMID: 25342732 DOI: 10.2337/db14-0888]
16 Hammes HP, Bertram B, Bornfeld N, Danne T, Kroll P, Lemmen KD. Diagnostik, Therapie und Verlaufskontrolle der diabetischen Retinopathie und Makulopathie. Germany: Evidenzbasierte Leitlinie DDG Aktualisierung, 2004
17 Garweg JG, Wenzel A. [Diabetic maculopathy and retinopathy. Functional and sociomedical significance]. Ophthalmologe 2010; 107: 628-635 [PMID: 20533047 DOI: 10.1007/s00347-010-2176-x]
18 Klaassen I, Van Noorden CJ, Schlingemann RO. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res 2013; 34: 19-48 [PMID: 23416119 DOI: 10.1016/j.preteyeres.2013.02.001]
19 Nentwich MM, Ulbig M. Diabetes und Auge. Diabetologe 2014; 10: 69-82 [DOI: 10.1007/s11428-013-1180-6]
20 Novotny HR, Alvis DL. A method of photographing fluorescence
in circulating blood in the human retina. Circulation 1961; 24: 82-86 [PMID: 13729802 DOI: 10.1161/01.CIR.24.1.82]
21 Kernt M, Hadi I, Pinter F, Seidensticker F, Hirneiss C, Haritoglou C, Kampik A, Ulbig MW, Neubauer AS. Assessment of diabetic retinopathy using nonmydriatic ultra-widefield scanning laser ophthalmoscopy (Optomap) compared with ETDRS 7-field stereo photography. Diabetes Care 2012; 35: 2459-2463 [PMID: 22912430 DOI: 10.2337/dc12-0346]
22 Silva PS, Cavallerano JD, Sun JK, Noble J, Aiello LM, Aiello LP. Nonmydriatic ultrawide field retinal imaging compared with dilated standard 7-field 35-mm photography and retinal specialist examination for evaluation of diabetic retinopathy. Am J Ophthalmol 2012; 154: 549-559.e2 [PMID: 22626617 DOI: 10.1016/j.ajo.2012.03.019]
23 Liegl R, Liegl K, Ceklic L, Haritoglou C, Kampik A, Ulbig MW, Kernt M, Neubauer AS. Nonmydriatic ultra-wide-field scanning laser ophthalmoscopy (Optomap) versus two-field fundus photography in diabetic retinopathy. Ophthalmologica 2014; 231: 31-36 [PMID: 24247157 DOI: 10.1159/000355092]
24 Wessel MM, Aaker GD, Parlitsis G, Cho M, D’Amico DJ, Kiss S. Ultra-wide-field angiography improves the detection and classification of diabetic retinopathy. Retina 2012; 32: 785-791 [PMID: 22080911 DOI: 10.1097/IAE.0b013e3182278b64]
25 Chin EK, Sedeek RW, Li Y, Beckett L, Redenbo E, Chandra K, Park SS. Reproducibility of macular thickness measurement among five OCT instruments: effects of image resolution, image registration, and eye tracking. Ophthalmic Surg Lasers Imaging 2012; 43: 97-108 [PMID: 22201525 DOI: 10.3928/15428877-20111222-02]
26 Nentwich MM, Lemmen K-D, Ulbig MW. Stadieneinteilung und Therapie der diabetischen Retinopathie und Makulopathie. Z prakt Augenheilk 2010; 31: 491-499
27 Pescosolido N, Campagna O, Barbato A. Diabetic retinopathy and pregnancy. Int Ophthalmol 2014; 34: 989-997 [PMID: 24482250 DOI: 10.1007/s10792-014-9906-z]
28 Nentwich MM, Ulbig M. Diabetische Retinopathie. CME Springer 2010; 7: 47-56
29 Kleinwechter H, Schäfer-Graf U, Bührer C, Hösli I, Kainer F, Kautzky-Willer A, Pawlowski B, Schunck K, Somville T, Sorger M. Diabetes und Schwangerschaft. Diabetologie und Stoffwechsel 2012; 7: S185-S191 [DOI: 10.1055/s-0032-1325334]
30 Deutsche DG. Nationale VersorgungsLeitlinie Typ-2-Diabetes – Prävention und Therapie von Netzhautkomplikationen (Klinisch relevante Auszüge aus der Leitlinie). Dtsch Arztebl International 2007; 104: 211-214
31 Aspelund T, Thornórisdóttir O, Olafsdottir E, Gudmundsdottir A, Einarsdóttir AB, Mehlsen J, Einarsson S, Pálsson O, Einarsson G, Bek T, Stefánsson E. Individual risk assessment and information technology to optimise screening frequency for diabetic retinopathy. Diabetologia 2011; 54: 2525-2532 [PMID: 21792613 DOI: 10.1007/s00125-011-2257-7]
32 Echouffo-Tcheugui JB, Ali MK, Roglic G, Hayward RA, Narayan KM. Screening intervals for diabetic retinopathy and incidence of visual loss: a systematic review. Diabet Med 2013; 30: 1272-1292 [PMID: 23819487 DOI: 10.1111/dme.12274]
33 Looker HC, Nyangoma SO, Cromie DT, Olson JA, Leese GP, Philip S, Black MW, Doig J, Lee N, Briggs A, Hothersall EJ, Morris AD, Lindsay RS, McKnight JA, Pearson DW, Sattar NA, Wild SH, McKeigue P, Colhoun HM. Predicted impact of extending the screening interval for diabetic retinopathy: the Scottish Diabetic Retinopathy Screening programme. Diabetologia 2013; 56: 1716-1725 [PMID: 23689796 DOI: 10.1007/s00125-013-2928-7]
34 Olafsdóttir E, Stefánsson E. Biennial eye screening in patients with diabetes without retinopathy: 10-year experience. Br J Ophthalmol 2007; 91: 1599-1601 [PMID: 17627978 DOI: 10.1136/bjo.2007.123810]
35 Stratton IM, Aldington SJ. Risk stratification for diabetic eye screening. Diabetologia 2014; 57: 259 [PMID: 24057137 DOI: 10.1007/s00125-013-3060-4]
497 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Nentwich MM et al . Diabetes mellitus: Ocular complications

36 Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352: 837-853 [PMID: 9742976 DOI: 10.1016/S0140-6736(98)07019-6]
37 The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329: 977-986 [PMID: 8366922 DOI: 10.1056/NEJM199309303291401]
38 Rodriguez-Fontal M, Kerrison JB, Alfaro DV, Jablon EP. Metabolic control and diabetic retinopathy. Curr Diabetes Rev 2009; 5: 3-7 [PMID: 19199891 DOI: 10.2174/157339909787314176]
39 The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes 1995; 44: 968-983 [PMID: 7622004 DOI: 10.2337/diab.44.8.968]
40 Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. UK Prospective Diabetes Study Group. BMJ 1998; 317: 713-720 [PMID: 9732338 DOI: 10.1136/bmj.317.7160.713]
41 Chaturvedi N, Porta M, Klein R, Orchard T, Fuller J, Parving HH, Bilous R, Sjølie AK. Effect of candesartan on prevention (DIRECT-Prevent 1) and progression (DIRECT-Protect 1) of retinopathy in type 1 diabetes: randomised, placebo-controlled trials. Lancet 2008; 372: 1394-1402 [PMID: 18823656 DOI: 10.1016/S0140-6736(08)61412-9]
42 Chaturvedi N, Sjolie AK, Stephenson JM, Abrahamian H, Keipes M, Castellarin A, Rogulja-Pepeonik Z, Fuller JH. Effect of lisinopril on progression of retinopathy in normotensive people with type 1 diabetes. The EUCLID Study Group. EURODIAB Controlled Trial of Lisinopril in Insulin-Dependent Diabetes Mellitus. Lancet 1998; 351: 28-31 [PMID: 9433426 DOI: 10.1016/S0140-6736(97)06209-0]
43 Sjølie AK, Klein R, Porta M, Orchard T, Fuller J, Parving HH, Bilous R, Chaturvedi N. Effect of candesartan on progression and regression of retinopathy in type 2 diabetes (DIRECT-Protect 2): a randomised placebo-controlled trial. Lancet 2008; 372: 1385-1393 [PMID: 18823658 DOI: 10.1016/S0140-6736(08)61411-7]
44 Effects of aspirin treatment on diabetic retinopathy. ETDRS report number 8. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology 1991; 98: 757-765 [PMID: 2062511 DOI: 10.1016/s0161-6420(13)38010-5]
45 Nittala MG, Keane PA, Zhang K, Sadda SR. Risk factors for proliferative diabetic retinopathy in a Latino American population. Retina 2014; 34: 1594-1599 [PMID: 24662751 DOI: 10.1097/IAE.0000000000000117]
46 Yang JY, Kim NK, Lee YJ, Noh JH, Kim DJ, Ko KS, Rhee BD, Kim DJ. Prevalence and factors associated with diabetic retinopathy in a Korean adult population: the 2008-2009 Korea National Health and Nutrition Examination Survey. Diabetes Res Clin Pract 2013; 102: 218-224 [PMID: 24268633 DOI: 10.1016/j.diabres.2013.10.016]
47 Gaedt Thorlund M, Borg Madsen M, Green A, Sjølie AK, Grauslund J. Is smoking a risk factor for proliferative diabetic retinopathy in type 1 diabetes? Ophthalmologica 2013; 230: 50-54 [PMID: 23751972 DOI: 10.1159/000350813]
48 Boretsky A, Gupta P, Tirgan N, Liu R, Godley BF, Zhang W, Tilton RG, Motamedi M. Nicotine accelerates diabetes-induced retinal changes. Curr Eye Res 2015; 40: 368-377 [PMID: 24911405 DOI: 10.3109/02713683.2014.924147]
49 Early photocoagulation for diabetic retinopathy. ETDRS report number 9. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology 1991; 98: 766-785 [PMID: 2062512 DOI: 10.1016/jsurvophthal.2008.10.001]
50 Bhagat N, Grigorian RA, Tutela A, Zarbin MA. Diabetic macular edema: pathogenesis and treatment. Surv Ophthalmol 2009; 54: 1-32 [PMID: 19171208 DOI: 10.1016/j.survophthal.2008.10.001]
51 Nentwich MM, Ulbig MW. Diabetisches Makulaödem: Alternative Indikation der Anti-VEGF-Therapie. Z prakt Augenheilk 2011; 32:
162-16652 Qaum T, Xu Q, Joussen AM, Clemens MW, Qin W, Miyamoto K,
Hassessian H, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP. VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci 2001; 42: 2408-2413 [PMID: 11527957]
53 Mitchell P, Bandello F, Schmidt-Erfurth U, Lang GE, Massin P, Schlingemann RO, Sutter F, Simader C, Burian G, Gerstner O, Weichselberger A; RESTORE study group. The RESTORE study: ranibizumab monotherapy or combined with laser versus laser monotherapy for diabetic macular edema. Ophthalmology 2011; 118: 615-625 [DOI: 10.1016/j.ophtha.2011.01.031]
54 Stefanini FR, Badaró E, Falabella P, Koss M, Farah ME, Maia M. Anti-VEGF for the management of diabetic macular edema. J Immunol Res 2014; 2014: 632307 [PMID: 24741610 DOI: 10.1155/2014/632307]
55 Stewart MW. Anti-VEGF therapy for diabetic macular edema. Curr Diab Rep 2014; 14: 510 [PMID: 24919750 DOI: 10.1007/s11892-014-0510-4]
56 Nentwich MM, Yactayo-Miranda Y, Schwarzbach F, Wolf A, Kampik A, Mino de Kaspar H. Endophthalmitis after intravitreal injection: decreasing incidence and clinical outcome-8-year results from a tertiary ophthalmic referral center. Retina 2014; 34: 943-950 [PMID: 24136408 DOI: 10.1097/IAE.0000000000000011]
57 Brynskov T, Kemp H, Sørensen TL. No cases of endophthalmitis after 20,293 intravitreal injections in an operating room setting. Retina 2014; 34: 951-957 [PMID: 24317292 DOI: 10.1097/IAE.0000000000000071]
58 Tabandeh H, Boscia F, Sborgia A, Ciracì L, Dayani P, Mariotti C, Furino C, Flynn HW. Endophthalmitis associated with intravitreal injections: office-based setting and operating room setting. Retina 2014; 34: 18-23 [PMID: 24362413 DOI: 10.1097/IAE.0000000000000008]
59 Nentwich MM, Rajab M, Ta CN, He L, Grueterich M, Haritoglou C, Gandorfer A, Kampik A, Mino De Kaspar H. Application of 10% povidone iodine reduces conjunctival bacterial contamination rate in patients undergoing cataract surgery. Eur J Ophthalmol 2012; 22: 541-546 [PMID: 22180155 DOI: 10.5301/ejo.5000093]
60 Gines JC, Nentwich MM, Peggy Bedoya AH, Cibils P, Esteche A, Laspina F, Samudio M, Fariña N, de Kaspar HM. [Bacterial contamination of needles after intravitreal injection in Paraguay]. Ophthalmologe 2012; 109: 782-787 [PMID: 22733287 DOI: 10.1007/s00347-012-2591-2]
61 Nentwich M, Yactayo-Miranda Y, Weimann S, Froehlich S, Wolf A, Kampik A, Mino De Kaspar H. Bacterial contamination of needle points after intravitreal injection. Eur J Ophthalmol 2009; 19: 268-272 [PMID: 19253245]
62 Li B, Nentwich MM, Hoffmann LE, Haritoglou C, Kook D, Kampik A, Sheng M, Miño de Kaspar H. Comparison of the efficacy of povidone-iodine 1.0%, 5.0%, and 10.0% irrigation combined with topical levofloxacin 0.3% as preoperative prophylaxis in cataract surgery. J Cataract Refract Surg 2013; 39: 994-1001 [PMID: 23680628 DOI: 10.1016/j.jcrs.2013.02.039]
63 Nentwich MM, Ulbig MW. The therapeutic potential of intraocular depot steroid systems: developments aimed at prolonging duration of efficacy. Dtsch Arztebl Int 2012; 109: 584-590 [PMID: 23093988 DOI: 10.3238/arztebl.2012.0584]
64 Boyer DS, Yoon YH, Belfort R, Bandello F, Maturi RK, Augustin AJ, Li XY, Cui H, Hashad Y, Whitcup SM. Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology 2014; 121: 1904-1914 [PMID: 24907062 DOI: 10.1016/j.ophtha.2014.04.024]
65 Dutra Medeiros M, Postorino M, Navarro R, Garcia-Arumí J, Mateo C, Corcóstegui B. Dexamethasone intravitreal implant for treatment of patients with persistent diabetic macular edema. Ophthalmologica 2014; 231: 141-146 [PMID: 24356099 DOI: 10.1159/000356413]
66 Ciulla TA, Harris A, McIntyre N, Jonescu-Cuypers C. Treatment of diabetic macular edema with sustained-release glucocorticoids: intravitreal triamcinolone acetonide, dexamethasone implant, and fluocinolone acetonide implant. Expert Opin Pharmacother 2014;
498 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Nentwich MM et al . Diabetes mellitus: Ocular complications

15: 953-959 [PMID: 24661081 DOI: 10.1517/14656566.2014.896899]
67 Campochiaro PA, Brown DM, Pearson A, Chen S, Boyer D, Ruiz-Moreno J, Garretson B, Gupta A, Hariprasad SM, Bailey C, Reichel E, Soubrane G, Kapik B, Billman K, Kane FE, Green K. Sustained delivery fluocinolone acetonide vitreous inserts provide benefit for at least 3 years in patients with diabetic macular edema. Ophthalmology 2012; 119: 2125-2132 [PMID: 22727177 DOI: 10.1016/j.ophtha.2012.04.030]
68 Photocoagulation treatment of proliferative diabetic retinopathy. Clinical application of Diabetic Retinopathy Study (DRS) findings, DRS Report Number 8. The Diabetic Retinopathy Study Research Group. Ophthalmology 1981; 88: 583-600 [PMID: 7196564 DOI: 10.1016/s0161-6420(81)34978-1]
69 Park DH, Shin JP, Kim SY. Comparison of clinical outcomes between 23-gauge and 20-gauge vitrectomy in patients with proliferative diabetic retinopathy. Retina 2010; 30: 1662-1670 [PMID: 20661174 DOI: 10.1097/IAE.0b013e3181d95261]
P- Reviewer: Mitra A, Sui RF S- Editor: Tian YL L- Editor: A E- Editor: Liu SQ
499 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Nentwich MM et al . Diabetes mellitus: Ocular complications

Gergő A Molnár, Esztella Zsóka Mikolás, István András Szijártó, Szilárd Kun, Eszter Sélley, István Wittmann
Gergő A Molnár, Esztella Zsóka Mikolás, István András Szijártó, Szilárd Kun, Eszter Sélley, István Wittmann, 2nd Department of Medicine and Nephrological Center, University of Pécs, H-7624 Pécs, HungaryAuthor contributions: Molnár GA and Wittmann I designed the research; Mikolás EZ, Szijártó IA, Kun S and Sélley E performed the research; Molnár GA and Wittmann I wrote the paper.Supported by The European Union and the State of Hungary and co-financed by the European Social Fund in the framework of TÁMOP 4.2.4. A/2-11-1-2012-0001 National Excellence Program.Conflict-of-interest: All authors have nothing to declare.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: István Wittmann, MD, PhD, Professor of Medicine, Head of the Department, 2nd Department of Medicine and Nephrological Center, University of Pécs, Pacsirta Str. 1, H-7624 Pécs, Hungary. [email protected]: +36-72-536050 Fax: +36-72-536051Received: September 3, 2014 Peer-review started: September 4, 2014 First decision: November 19, 2014Revised: December 5, 2014 Accepted: January 9, 2015Article in press: January 12, 2015Published online: April 15, 2015
AbstractOxidative stress processes play a major role in the development of the complications associated with diabetes and other diseases via non-enzymatic glycation, the hexosamine pathway, the polyol pathway and diacylglycerol-protein kinase C. Oxidative stress may lead to the production of hydroxyl free radicals, which
can attack macromolecules, such as lipids, nucleic acids or amino acids. Phenylalanine (Phe) can be enzymatically converted to the physiological para-tyrosine (p-Tyr); however, a hydroxyl free radical attack on Phe may yield meta- and ortho-tyrosine (m- and o-Tyr, respectively) in addition to p-Tyr. Hence, m- and o-Tyr may be regarded as markers of hydroxyl free radical-induced damage. Their accumulation has been described; e.g. , this accumulation has been found in the urine of patients with diabetes mellitus (DM) and/or chronic kidney disease, in cataract lenses, in vessel walls, in irradiated food and in amniotic fluid, and it may serve as an indicator of oxidative stress. The use of resveratrol to treat patients with type 2 DM led to a decrease in the urinary excretion of o-Tyr and concomitantly led to an improvement in insulin signaling and insulin sensitivity. Literature data also suggest that m- and o-Tyr may interfere with intracellular signaling. Our group has shown that erythropoietin (EPO) has insulin-like metabolic effects on fat cells in addition to its ability to promote the proliferation of erythroid precursor cells. We have shown that the supplementation of cell culture medium with m- and o-Tyr inhibits erythroblast cell proliferation, which could be ameliorated by p-Tyr. Additionally, in vivo , the o-Tyr/p-Tyr ratio is higher in patients with renal replacement therapy and a greater need for EPO. However, the o-Tyr/p-Tyr ratio was an independent determinant of EPO-resistance indices in our human study. The o-Tyr content of blood vessel walls inversely correlates with insulin- and acetylcholine-induced vasodilation, which could be further impaired by artificial oxidative stress and improved by the use of antioxidants. In rats that receive o-Tyr supplements, decreased vasorelaxation is detected in response to insulin. Additionally, o-Tyr supplementation led to the incorporation of the unnatural amino acid into cellular proteins and caused a decrease in the insulin-induced phosphorylation of endothelial nitric oxide synthase. Our data suggest that m- and o-Tyr may not only be markers of oxidative stress; instead, they may also be incorporated into cellular proteins, leading to resistance to insulin, EPO and acetylcholine.
MINIREVIEWS
500 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Tyrosine isomers and hormonal signaling: A possible role for the hydroxyl free radical in insulin resistance
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.500
World J Diabetes 2015 April 15; 6(3): 500-507ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

Key words: Acetylcholine; Insulin resistance; Hormone resistance; Oxidative stress; Para-tyrosine; Ortho-tyrosine; Meta-tyrosine; Erythropoietin
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Hydroxyl-free radical-derived amino acids, such as meta- and ortho-tyrosine (m- and o-Tyr, respectively) are regarded as free radical markers, but they may also be taken up and incorporated into blood vessel walls, erythroid precursors and endothelial cells. These pathological amino acids can induce vascular insulin and acetylcholine, as well as cellular erythropoietin resistance.
Molnár GA, Mikolás EZ, Szijártó IA, Kun S, Sélley E, Wittmann I. Tyrosine isomers and hormonal signaling: A possible role for the hydroxyl free radical in insulin resistance. World J Diabetes 2015; 6(3): 500-507 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/500.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.500
OXIDATIVE STRESS IN THE OLD PERSPECTIVESOxidative stress plays a role in the pathogenesis of many diseases, such as diabetes mellitus (DM), chronic kidney disease (CKD) and inflammatory diseases, as well as in the development of the complications associated with these diseases. Oxidative stress, free radicals and reactive oxygen species (ROS) are tightly connected to DM in several ways. Hyperglycemia may lead to a shift in cellular metabolism toward the polyol pathway, which leads to an oxidative shift in the NADPH/NADP ratio. NADPH is in turn required for antioxidant defense, e.g., for the reduction of oxidized glutathione by glutathione reductase. Furthermore, hyperglycemia activates the diacylglycerol-protein kinase C intracellular signaling pathway, which can activate NADPH oxidases; this oxidation leads to the translocation of nuclear factor κB into the nucleus and then to the transcription of proinflammatory cytokines, which results in increased oxidative stress. Additionally, hyperglycemia increases the rate of non-enzymatic glycation, which produces advanced glycation end-products (AGEs) that can bind to their receptors of AGE (RAGE), and the AGE-RAGE interaction also leads to inflammation and oxidative stress. DM involves the enhancement of not only non-enzymatic glycation but also enzymatic glycation via the hexosamine pathway; this enhanced glycation may also result in a proinflammatory response and oxidative stress. However, this interplay is complex; the ROS arising from oxidative stress may also contribute to the activation of the abovementioned pathways and reactions and can thus generate a vicious circle[1,2].
DETECTION OF OXIDATIVE STRESSThe study of oxidative stress processes is therefore important, albeit somewhat cumbersome. Per their definition, free radicals are highly reactive molecules with a very short lifetime; therefore, detecting these molecules requires spin traps and an electron spin resonance device[3,4], but the sensitivity of this method is usually too low for many diseases. Because of the high reactivity of free radicals, they can react with macromolecules and yield oxidation products, some of which are more chemically stable molecules. These products may include lipid peroxidation products (such as malonyldialdehyde derivatives), nucleobase products (such as 8-hydroxydeoxyguanosine), so-called advanced oxidation products or amino acid derivatives[5,6].
DETECTION OF HYDROXYL FREE RADICALS BY STABILE L-PHENYLALANINE DERIVATIVESL-Phenylalanine (Phe) is a highly abundant essential amino acid in proteins of the human body, and Phe plays a role in forming peptides and proteins; Phe also gives rise to the semi-essential amino acid L-para-tyrosine (p-Tyr) and its derivatives, such as 3,4 dihydroxy-phenylalanine (DOPA), and derivatives thereof[7]. p-Tyr is mainly produced via the enzymatic reactions catalyzed by the Phe hydroxylase enzyme, especially in the kidney and liver[8]. p-Tyr synthesis becomes impaired in patients with renal failure (e.g., CKD); therefore, the serum levels of p-Tyr in these patients are lower than those in patients/controls with normal renal function [CKD: 28 (26-34), DM + CKD: 32 (29-39) μmol/L vs controls: 56 (36-57) μmol/L][9]. However, other isomers of tyrosine, namely meta- and ortho-tyrosine (m-Tyr and o-Tyr, respectively) also exist in humans. These amino acids cannot be formed enzymatically in humans; instead, they are stable products of the reaction between the hydroxyl free radical and Phe. Additionally, p-Tyr may be formed non-enzymatically, via the hydroxyl radical, but the enzymatically produced p-Tyr is much more abundant than the free radical-derived p-Tyr. Therefore, p-Tyr is regarded as the physiologic isoform, whereas m- and o-Tyr are considered to be free radical markers[10-14].
All four aromatic amino acids (p-Tyr, m-Tyr, o-Tyr and Phe) can be readily detected in the nanomolar range via their autofluorescence and reverse-phase high performance liquid chromatography[9,15-22], as well as by gas chromatography combined with mass spectrometric detection[23] or by ultra-performance liquid chromatography combined with mass spectrometry[24].
ABUNDANCE OF THE HYDROXYL FREE RADICAL MARKERS M- AND O-TYRIn vitro measurements[25] and in silico calculations[26] have shown that in free radical reactions, the three isoforms (p-,
Molnár GA et al . Tyrosine isomers and hormone resistance
501 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
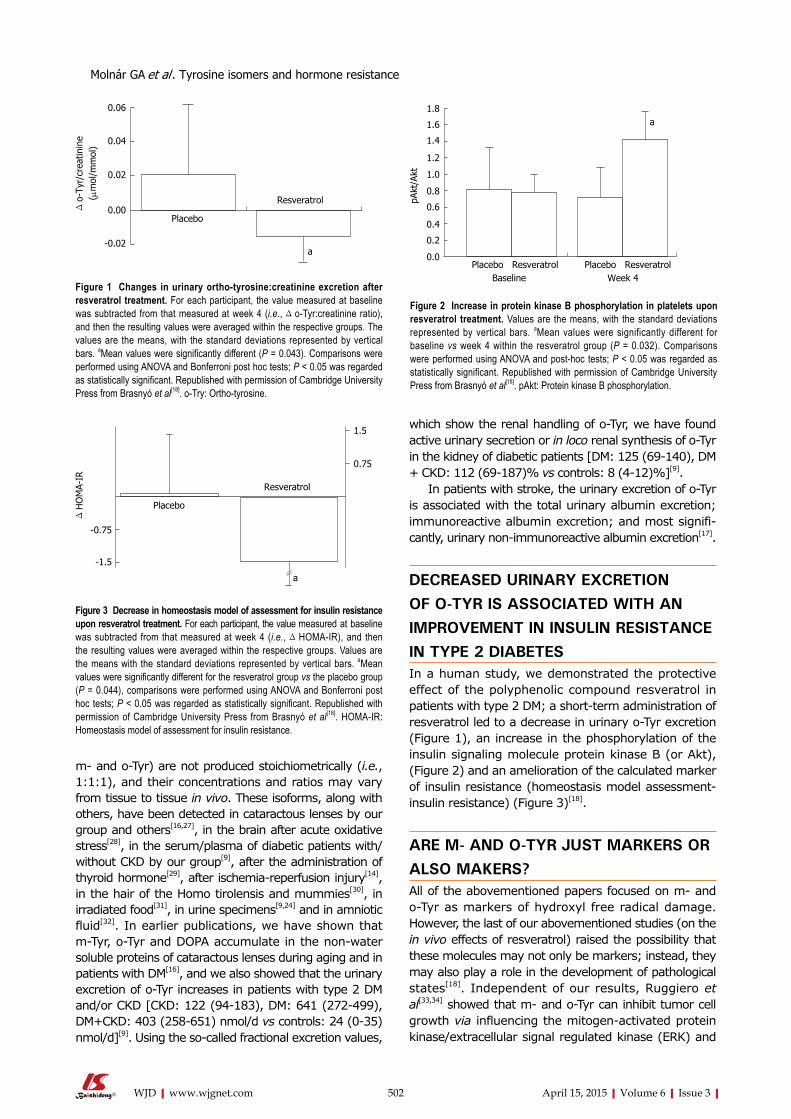
m- and o-Tyr) are not produced stoichiometrically (i.e., 1:1:1), and their concentrations and ratios may vary from tissue to tissue in vivo. These isoforms, along with others, have been detected in cataractous lenses by our group and others[16,27], in the brain after acute oxidative stress[28], in the serum/plasma of diabetic patients with/without CKD by our group[9], after the administration of thyroid hormone[29], after ischemia-reperfusion injury[14], in the hair of the Homo tirolensis and mummies[30], in irradiated food[31], in urine specimens[9,24] and in amniotic fluid[32]. In earlier publications, we have shown that m-Tyr, o-Tyr and DOPA accumulate in the non-water soluble proteins of cataractous lenses during aging and in patients with DM[16], and we also showed that the urinary excretion of o-Tyr increases in patients with type 2 DM and/or CKD [CKD: 122 (94-183), DM: 641 (272-499), DM+CKD: 403 (258-651) nmol/d vs controls: 24 (0-35) nmol/d][9]. Using the so-called fractional excretion values,
which show the renal handling of o-Tyr, we have found active urinary secretion or in loco renal synthesis of o-Tyr in the kidney of diabetic patients [DM: 125 (69-140), DM + CKD: 112 (69-187)% vs controls: 8 (4-12)%][9].
In patients with stroke, the urinary excretion of o-Tyr is associated with the total urinary albumin excretion; immunoreactive albumin excretion; and most signifi-cantly, urinary non-immunoreactive albumin excretion[17].
DECREASED URINARY EXCRETION OF O-TYR IS ASSOCIATED WITH AN IMPROVEMENT IN INSULIN RESISTANCE IN TYPE 2 DIABETESIn a human study, we demonstrated the protective effect of the polyphenolic compound resveratrol in patients with type 2 DM; a short-term administration of resveratrol led to a decrease in urinary o-Tyr excretion (Figure 1), an increase in the phosphorylation of the insulin signaling molecule protein kinase B (or Akt), (Figure 2) and an amelioration of the calculated marker of insulin resistance (homeostasis model assessment-insulin resistance) (Figure 3)[18].
ARE M- AND O-TYR JUST MARKERS OR ALSO MAKERS?All of the abovementioned papers focused on m- and o-Tyr as markers of hydroxyl free radical damage. However, the last of our abovementioned studies (on the in vivo effects of resveratrol) raised the possibility that these molecules may not only be markers; instead, they may also play a role in the development of pathological states[18]. Independent of our results, Ruggiero et al[33,34] showed that m- and o-Tyr can inhibit tumor cell growth via influencing the mitogen-activated protein kinase/extracellular signal regulated kinase (ERK) and
502 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 1 Changes in urinary ortho-tyrosine:creatinine excretion after resveratrol treatment. For each participant, the value measured at baseline was subtracted from that measured at week 4 (i.e., Δ o-Tyr:creatinine ratio), and then the resulting values were averaged within the respective groups. The values are the means, with the standard deviations represented by vertical bars. aMean values were significantly different (P = 0.043). Comparisons were performed using ANOVA and Bonferroni post hoc tests; P < 0.05 was regarded as statistically significant. Republished with permission of Cambridge University Press from Brasnyó et al[18]. o-Try: Ortho-tyrosine.
Δ o-
Tyr/
crea
tinin
e( μ
mol
/mm
ol)
0.06
0.04
0.02
0.00
-0.02
Resveratrol
Placebo
a
pAkt
/Akt
1.8
1.6
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0Placebo Resveratrol Placebo Resveratrol Baseline Week 4
a
Figure 2 Increase in protein kinase B phosphorylation in platelets upon resveratrol treatment. Values are the means, with the standard deviations represented by vertical bars. aMean values were significantly different for baseline vs week 4 within the resveratrol group (P = 0.032). Comparisons were performed using ANOVA and post-hoc tests; P < 0.05 was regarded as statistically significant. Republished with permission of Cambridge University Press from Brasnyó et al[18]. pAkt: Protein kinase B phosphorylation.
Δ H
OM
A-IR
1.5
0.75
-0.75
-1.5
a
Placebo
Resveratrol
Figure 3 Decrease in homeostasis model of assessment for insulin resistance upon resveratrol treatment. For each participant, the value measured at baseline was subtracted from that measured at week 4 (i.e., Δ HOMA-IR), and then the resulting values were averaged within the respective groups. Values are the means with the standard deviations represented by vertical bars. aMean values were significantly different for the resveratrol group vs the placebo group (P = 0.044), comparisons were performed using ANOVA and Bonferroni post hoc tests; P < 0.05 was regarded as statistically significant. Republished with permission of Cambridge University Press from Brasnyó et al[18]. HOMA-IR: Homeostasis model of assessment for insulin resistance.
Molnár GA et al . Tyrosine isomers and hormone resistance

phosphorylation of STAT5 and ERK1/2 (Figure 8). This finding indicates that the unnatural isoforms, o- and m-Tyr, can be incorporated into cellular proteins and interfere with the hormonal signaling of EPO[21].
This finding is consistent with a clinical observation of our group that the ratio of the pathological o-Tyr to the physiological p-Tyr (o-Tyr/p-Tyr ratio) is higher in patients who receive renal replacement therapy compared with controls. Additionally, the o-Tyr/p-Tyr ratio in the blood was higher in the individuals receiving hemodialysis and EPO replacement than in those patients receiving hemodialysis who did not require EPO replacement or in patients receiving peritoneal dialysis and requiring markedly lower EPO doses. Furthermore, the plasma o-Tyr/p-Tyr ratio was an independent predictor of the EPO dose and EPO-resistance indices in these patients. This finding is another indirect demonstration that o-Tyr may interfere with EPO signaling and lead to EPO resistance[22].
VASCULAR INSULIN RESISTANCE ACCORDING TO THE O-TYR CONTENT OF THE VESSEL WALLIn a further set of experiments, we analyzed the o-Tyr levels in the blood vessel walls of rodents and found that the o-Tyr content of blood vessels decreases toward the peripheral vasculature (i.e., thoracic aorta > abdominal aorta > femoral artery) (Figure 9A). The o-Tyr content could be increased by treatment with hydrogen peroxide and aminotriazole or by aortic banding, and it could be inhibited using superoxide dismutase and catalase (Figure 9B).
We have shown that vascular segments with higher o-Tyr content show lower vasorelaxation in response to insulin; i.e., the insulin-induced vasorelaxation is lowest in the thoracic aorta, higher in the abdominal aorta and the highest in the femoral artery (Figure 10).
Furthermore, the insulin-induced relaxation could be increased by an antioxidant (superoxide dismutase and catalase) treatment in the thoracic aorta. By contrast, pro-oxidant therapy further diminished the vasorelaxation in an ERK1/2-dependent manner[19].
VASCULAR ACETYLCHOLINE RESISTANCE ACCORDING TO THE O-TYR CONTENT OF THE VESSEL WALLIn the same set of experiments, the vasorelaxation in response to acetylcholine was also tested, and we also found an inverse relationship between the vessel wall o-Tyr content and the vasorelaxation in response to acetylcholine; i.e., the blood vessels with high o-Tyr content (see Figure 9A) show less vasorelaxation in response to Ach (Figure 11, previously unpublished data).
Based on these experiments, we subjected rats
the signal transducer and activator of transcription (STAT) pathway. In plants, m-Tyr inhibits cell growth and plant root growth and may be considered a natural herbicide[35,36]. This finding raises the possibility that the unnatural isoforms, m- and o-Tyr, might affect cellular function in mammals and plants and may interfere with hormonal signaling.
INSULIN-LIKE EFFECT OF ERYTHROPOIETIN ON GLUCOSE METABOLISMIn a subsequent study, we have shown that under hyperglycemic circumstances, erythropoietin (EPO) exerts insulin-like effects on the uptake of isotope-labeled glucose by 3T3-L1-type fat cells (Figure 4) and can lead to the translocation of glucose transporters transporters (GLUTs) from their intracellular pools to the membrane (Figure 5). EPO also improves glucose metabolism in streptozotocin-induced diabetic rats[37].
M- AND O-TYR ARE INCORPORATED INTO CELLULAR PROTEINS AND LEAD TO ERYTHROPOIETIN RESISTANCEIn further studies, we showed that the administration of m- and o-Tyr to erythroblasts inhibited erythroblast proliferation in a time- and concentration-dependent manner. Increasing doses of p-Tyr could overcome the inhibition by m- and o-Tyr, suggesting potential competition among the structural isoforms (Figure 6).
Erythroblasts grown in cell culture medium supple-mented with m- or o-Tyr incorporated the Tyr isoforms into their cellular proteins (Figure 7).
Supplementing erythroblast cells with o- or m-Tyr inhibited the EPO-dependent increase in the rate of
503 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
[3H
] de
oxy-
D-g
luco
se u
ptak
e(%
of
cont
rol)
300
250
200
150
100
50
0C 0.15 0.3 0.625 1.25 2.5
EPO (ng/mL)
a,c
a,c a,c
5 mmol/L glucose
25 mmol/L glucose
Figure 4 Effects of treatment with r-mo-erythropoietin on the rate of [3H]-deoxy-D-glucose uptake in 3T3-L1 adipocytes cultured in either normal (5 mmol/L) or high (25 mmol/L) glucose medium (insulin: n = 8 and n = 6; r-mo-erythropoietin: n = 6 and n = 5, respectively). aP < 0.05 vs control in the same medium; cP < 0.05 between the two media; Comparisons with control were performed using a one-sample t-test; pairwise comparisons between 5 and 25 mmol/L glucose-cultured cells were performed using a paired samples t-test. Republished with permission of Georg Thieme Verlag KG Stuttgart, New York from Mikolás et al[37]. EPO: Erythropoietin.
Molnár GA et al . Tyrosine isomers and hormone resistance

504 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 5 GLUT4 translocation (red fluorescence) after a 30 min treatment with insulin (400 nmol/L) or r-mo-erythropoietin (40 ng/mL) in 3T3-L1 adipocytes cultured in high glucose (25 mmol/L) or normal glucose (5 mmol/L) medium compared with that in untreated cells (Control). Nuclei are shown with blue fluorescence. The GLUT4 translocation was apparent after both the erythropoietin (EPO) and insulin treatments, whereas it was not present in untreated cells. Representative images are shown from n = 3 independent experiments. Republished with permission of Georg Thieme Verlag KG Stuttgart, New York from Mikolás et al[37].
A
Cell
coun
t EP
O/c
ontr
ol (
%)
Cell
coun
t EP
O/c
ontr
ol (
%)
180
160
140
120
100
80
60
40
20
0
b
bb
Para Ortho Ortho + 20 Ortho + 40 Ortho + 80 Meta Meta + 20 Meta + 40 Meta + 80 mg/L para mg/L para mg/L para mg/L para mg/L para mg/L para
B
250
200
150
100
50
01st day 2nd day 3rd day
Pare
Ortho
Meta
a a aa
Figure 6 Effect of para-, ortho-, or meta-tyrosine supplementation on the proliferation of cells cultured in medium with or without erythropoietin. A: The time-dependent increase in cell count in medium supplemented with p-Tyr (open bars), o-Tyr (black bars) or m-Tyr (grey bars) (aP < 0.05 vs p-Tyr cultured cells on the same day; n = 10); B: The alterations in the cell counts after incubation for 3 d in medium containing o- or m-Tyr and the indicated additional amount of para-tyrosine (bP < 0.001 vs p-Tyr-containing medium; n = 5). The results are expressed as the ratio of the protein content of erythropoietin (EPO) and non-EPO (control) cells (aP < 0.05 vs p-Tyr cultured cells; n = 10). Comparisons were performed using ANOVA and Bonferroni’s post-hoc test. Republished with permission of S Karger AG, Basel from Mikolás et al[21]. p-Tyr: Para-tyrosine; o-Tyr: Ortho-tyrosine; m-Tyr: Meta-tyrosine.
Control EPO
Control Insulin
5 mmol/L
25 mmol/L
Molnár GA et al . Tyrosine isomers and hormone resistance

to vehicle or o-Tyr supplementation for 4 wk. By the end of the 4 wk, we showed decreased vasorelaxation in response to insulin in the o-Tyr-supplemented rats
compared with the controls. Additionally, the endothelial cells that received o-Tyr supplementation incorporated o-Tyr into their cellular proteins. In these cells, the
505 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
A B
Eryt
hrob
last
o-T
yr/
tota
l-tyr
osin
e ra
tio (
%)
b
b,d
Pare
Ortho
Meta
0.14
0.12
0.10
0.08
0.06
0.04
0.02
0.00Control EPO
Eryt
hrob
last
m-T
yr/
tota
l-tyr
osin
e ra
tio (
%)
f
f
Pare
Ortho
Meta
0.25
0.20
0.15
0.10
0.05
0.00Control EPO
Figure 7 Relative meta- and ortho-tyrosine content (i.e., ratios of meta- and ortho-tyrosine/total tyrosine) of the cellular proteins of TF-1 cells (A and B, respectively). bP < 0.001 vs p- and m-Tyr cultured cells; dP < 0.001 vs o-Tyr cultured control cells; fP < 0.001 vs p- and o-Tyr cultured erythroblasts; n = 10. Comparisons were performed using ANOVA and Bonferroni’s post-hoc test. Republished with permission of S Karger AG, basel from Mikolás et al[21]. p-Tyr: Para-tyrosine; o-Tyr: Ortho-tyrosine; m-Tyr: Meta-tyrosine.
pSTA
T5/S
TAT5
(%
) Pare
Ortho
Meta
Control EPO
1600140012001000800600400200
0
a
c c
pERK
2/ER
K2 (
%)
Pare
Ortho
Meta
Control EPO
300
250
200
150
100
50
0
a
c c
A
pERK
1/ER
K1 (
%) Pare
Ortho
Meta
Control EPO
300
250
200
150
100
50
0
a
c c
B
C
Figure 8 Densitometric analyses of the phosphorylation of signal transducer and activator of transcription 5 (A: n = 3); densitometric analysis of extracellular signal regulated kinase 1 (B: n = 4) and extracellular signal regulated kinase 2 (C: n = 4). The data are expressed as the percent of untreated (control) cells. aP < 0.05; erythropoietin (EPO) vs control (one-sample t-test); cP < 0.05 vs para EPO (one-way ANOVA). Republished with permission of S Karger AG, basel from Mikolás et al[21].
o-Ty
r/Ph
e (n
mol
/ μm
ol)
a
a a
Thoracic aorta Abdominal aorta Femoral artery
0.15
0.10
0.05
0.00 o-Ty
r/Ph
e (%
of
untr
eate
d ve
ssel
) c
Thoracic aorta Abdominal aorta Femoral artery
200
150
100
50
0
NSNSNS NSNS
cc
c
Control H2O2 + AT Aortic banding SOD + CATA B
Figure 9 The ortho-tyrosine levels of consecutive arterial segments isolated from rats (A). There were significant differences (aP < 0.05) in the o-Tyr levels among the thoracic aorta (n = 7), abdominal aorta (n = 7), and the femoral artery (n = 9) of rats. The changes in the o-Tyr levels in the consecutive arterial segments of rats after H2O2 + AT treatment (i.e., high oxidative state), SOD + CAT treatment (i.e., low oxidative state), and aortic banding were compared with those of the control vessels (B). Control: untreated, control vessels (thoracic: n = 7; abdominal: n = 7; femoral: = 9); H2O2 + AT: hydrogen peroxide- and aminotriazole-incubated vessels (aminotriazole is an inhibitor of catalase) (thoracic: n = 5; abdominal: n = 5; femoral: n = 4); SOD + CAT: superoxide dismutase- and catalase-incubated vessels (thoracic: n = 5; abdominal: n = 5; femoral: n = 5); aortic banding (thoracic: n = 5; abdominal: n = 4; femoral: n = 4). Data are the mean ± SEM. The o-Tyr levels are relative to the phenylalanine (Phe) levels (Panels A and B) and are expressed as the percentage of the control vessels (Pane B). cP < 0.05; NS, P > 0.05 (ANOVA). Republished with permission of Informa Healthcare from Szijártó et al[19]. o-Tyr: Ortho-tyrosine; NS: Non-significant; SEM: Standard error of the mean.
Molnár GA et al . Tyrosine isomers and hormone resistance

insulin-induced increase in endothelial nitric oxide synthase phosphorylation was diminished compared with that in the control cells[20] (data not shown).
CONCLUSIONOur results, which are consistent with previous findings, indicate that m- and o-Tyr are valuable markers of oxidative stress and other types of stress in patients or experimental animals with DM. Furthermore, the results suggest that these unnatural amino acids may also perform a pathogenic role, i.e., interfere with the signaling of three distinct hormones: insulin (vascular and perhaps metabolic signaling), acetylcholine and erythropoietin (also has metabolic effects). This inhibition may be even more pronounced in patients who have high levels of the pathological amino acids m- or o-Tyr (e.g., in DM) and simultaneously have lower levels of physiological p-Tyr (e.g., patients with impaired kidney function). This finding, together with the effect in which p-Tyr overcomes the inhibitory effect of m- and o-Tyr, raises the possibility that the physiological amino acid p-Tyr could be a therapeutic tool in hormone resistance in states with increased oxidative stress (e.g.,
in DM).
REFERENCES1 Brownlee M. The pathobiology of diabetic complications: a
unifying mechanism. Diabetes 2005; 54: 1615-1625 [PMID: 15919781]
2 Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 2010; 107: 1058-1070 [PMID: 21030723]
3 Rashba-Step J, Turro NJ, Cederbaum AI. ESR studies on the production of reactive oxygen intermediates by rat liver microsomes in the presence of NADPH or NADH. Arch Biochem Biophys 1993; 300: 391-400 [PMID: 8380968]
4 Berliner LJ, Khramtsov V, Fujii H, Clanton TL. Unique in vivo applications of spin traps. Free Radic Biol Med 2001; 30: 489-499 [PMID: 11182519]
5 Maritim AC, Sanders RA, Watkins JB. Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol 2003; 17: 24-38 [PMID: 12616644]
6 Ghiselli A, Serafini M, Natella F, Scaccini C. Total antioxidant capacity as a tool to assess redox status: critical view and experimental data. Free Radic Biol Med 2000; 29: 1106-1114 [PMID: 11121717]
7 Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003; 25: 207-218 [PMID: 14661084]
8 Bergeron M, Scriver CR. Pathophysiology of renal hyperamin-oacidurias and glucosuria. In Seldin DW, Giebisch G, editors. The kidney. Physiology and pathophysiology, New York: Raven Press,
506 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Rela
xatio
n (%
)
Insulin [log(mol/L)]
-8 -7 -6 -5 -40
20
40
60
80
100
Thoracic aorta
Abdominal aorta
Femoral artery
a
a
a
Figure 10 The dose–response curve of the insulin-induced relaxation of consecutive arterial segments isolated from rats. The relaxation in response to insulin was the greatest in the femoral artery (n = 7) and was less pronounced in the abdominal aorta (n = 7) and even less in the thoracic aorta (n = 7). The relaxation is depicted as a function of the logarithm of the insulin dose. Data are the mean ± SEM. aP < 0.05; NS, P > 0.05 (Extra sum-of-squares F test). Republished with permission of Informa Healthcare from Szijártó et al[19]. NS: Non-significant; SEM: Standard error of the mean.
Rela
xatio
n (%
)
Acetylcholine [log(mol/L)]
-10 -8 -6 -40
20
40
60
80
100
Thoracic aorta
Abdominal aorta
Femoral artery
a
a
a
Figure 11 Acetylcholine-induced vasorelaxation in the abdominal aorta and femoral artery. Data are the means ± SEM. aP < 0.05; NS, P > 0.05 (Extra sum-of-squares F test). NS: Non-significant.
Molnár GA et al . Tyrosine isomers and hormone resistance

1985: 1725-17459 Molnár GA, Wagner Z, Markó L, Kó Szegi T, Mohás M, Kocsis
B, Matus Z, Wagner L, Tamaskó M, Mazák I, Laczy B, Nagy J, Wittmann I. Urinary ortho-tyrosine excretion in diabetes mellitus and renal failure: evidence for hydroxyl radical production. Kidney Int 2005; 68: 2281-2287 [PMID: 16221230]
10 Themann C, Teismann P, Kuschinsky K, Ferger B. Comparison of two independent aromatic hydroxylation assays in combination with intracerebral microdialysis to determine hydroxyl free radicals. J Neurosci Methods 2001; 108: 57-64 [PMID: 11459618]
11 Stadtman ER, Berlett BS. Fenton chemistry. Amino acid oxidation. J Biol Chem 1991; 266: 17201-17211 [PMID: 1894614]
12 Davies MJ, Fu S, Wang H, Dean RT. Stable markers of oxidant damage to proteins and their application in the study of human disease. Free Radic Biol Med 1999; 27: 1151-1163 [PMID: 10641706]
13 Brennan ML, Hazen SL. Amino acid and protein oxidation in cardiovascular disease. Amino Acids 2003; 25: 365-374 [PMID: 14661097]
14 Sun JZ, Kaur H, Halliwell B, Li XY, Bolli R. Use of aromatic hydroxylation of phenylalanine to measure production of hydroxyl radicals after myocardial ischemia in vivo. Direct evidence for a pathogenetic role of the hydroxyl radical in myocardial stunning. Circ Res 1993; 73: 534-549 [PMID: 8394226]
15 Ishimitsu S, Fujimoto S, Ohara A. Determination of m-tyrosine and o-tyrosine in human serum by high-performance liquid chromatography with fluorimetric detection. J Chromatogr 1986; 378: 222-225 [PMID: 3733974]
16 Molnár GA, Nemes V, Biró Z, Ludány A, Wagner Z, Wittmann I. Accumulation of the hydroxyl free radical markers meta-, ortho-tyrosine and DOPA in cataractous lenses is accompanied by a lower protein and phenylalanine content of the water-soluble phase. Free Radic Res 2005; 39: 1359-1366 [PMID: 16298866]
17 Toth P, Koller A, Pusch G, Bosnyak E, Szapary L, Komoly S, Marko L, Nagy J, Wittmann I. Microalbuminuria, indicated by total versus immunoreactive urinary albumins, in acute ischemic stroke patients. J Stroke Cerebrovasc Dis 2011; 20: 510-516 [PMID: 20813547]
18 Brasnyó P, Molnár GA, Mohás M, Markó L, Laczy B, Cseh J, Mikolás E, Szijártó IA, Mérei A, Halmai R, Mészáros LG, Sümegi B, Wittmann I. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br J Nutr 2011; 106: 383-389 [PMID: 21385509]
19 Szijártó IA, Molnár GA, Mikolás E, Fisi V, Laczy B, Gollasch M, Koller A, Wittmann I. Increase in insulin-induced relaxation of consecutive arterial segments toward the periphery: Role of vascular oxidative state. Free Radic Res 2014; 48: 749-757 [PMID: 24628420]
20 Szijártó IA, Molnár GA, Mikolás E, Fisi V, Cseh J, Laczy B, Kovács T, Böddi K, Takátsy A, Gollasch M, Koller A, Wittmann I. Elevated vascular level of ortho-tyrosine contributes to the impairment of insulin-induced arterial relaxation. Horm Metab Res 2014; 46: 749-752 [PMID: 25208272]
21 Mikolás E, Kun S, Laczy B, Molnár GA, Sélley E, Kőszegi T, Wittmann I. Incorporation of ortho- and meta-tyrosine into cellular proteins leads to erythropoietin-resistance in an erythroid cell line. Kidney Blood Press Res 2013; 38: 217-225 [PMID: 24751667]
22 Kun S, Mikolás E, Molnár GA, Sélley E, Laczy B, Csiky B, Kovács T, Wittmann I. Association of plasma ortho-tyrosine/para-tyrosine ratio with responsiveness of erythropoiesis-stimulating agent in dialyzed patients. Redox Rep 2014; 19: 190-198 [PMID: 24693974]
23 Leeuwenburgh C, Rasmussen JE, Hsu FF, Mueller DM, Pennathur
S, Heinecke JW. Mass spectrometric quantification of markers for protein oxidation by tyrosyl radical, copper, and hydroxyl radical in low density lipoprotein isolated from human atherosclerotic plaques. J Biol Chem 1997; 272: 3520-3526 [PMID: 9013599]
24 Kuligowski J, Torres-Cuevas I, Quintás G, Rook D, van Goudoever JB, Cubells E, Asensi M, Lliso I, Nuñez A, Vento M, Escobar J. Assessment of oxidative damage to proteins and DNA in urine of newborn infants by a validated UPLC-MS/MS approach. PLoS One 2014; 9: e93703 [PMID: 24695409]
25 Molnár GA. Pathogenesis of the chronic complications of type 2 diabetes mellitus, chronic kidney disease and ageing. The role of oxidative stress, endothelial dysfunction and the renin-angiotensin system (PhD Theses). University of Pécs, 2007. Available from: URL: http: //aok.pte.hu/en/egyseg/phd_dolgozatok/1670/docs/phd/file/dolgozatok/2007/Molnar_Gergo_Attila_PhD_dolgozat.pdf
26 Galano A, Cruz-Torres A. OH radical reactions with phenylalanine in free and peptide forms. Org Biomol Chem 2008; 6: 732-738 [PMID: 18264574]
27 Fu S, Dean R, Southan M, Truscott R. The hydroxyl radical in lens nuclear cataractogenesis. J Biol Chem 1998; 273: 28603-28609 [PMID: 9786852]
28 Lubec B, Hayn M, Denk W, Bauer G. Brain lipid peroxidation and hydroxy radical attack following the intravenous infusion of hydrogen peroxide in an infant. Free Radic Biol Med 1996; 21: 219-223 [PMID: 8818637]
29 Magsino CH, Hamouda W, Ghanim H, Browne R, Aljada A, Dandona P. Effect of triiodothyronine on reactive oxygen species generation by leukocytes, indices of oxidative damage, and antioxidant reserve. Metabolism 2000; 49: 799-803 [PMID: 10877210]
30 Lubec G, Weninger M, Anderson SR. Racemization and oxidation studies of hair protein in the Homo tirolensis. FASEB J 1994; 8: 1166-1169 [PMID: 7958623]
31 Hein WG, Simat TJ, Steinhart H: Detection of irradiated food. Determination of non-protein bound o-tyrosine as a marker for the detection of irradiated shrimps. Eur Food Res Technol 2000; 210: 299-304
32 Escobar J, Teramo K, Stefanovic V, Andersson S, Asensi MA, Arduini A, Cubells E, Sastre J, Vento M. Amniotic fluid oxidative and nitrosative stress biomarkers correlate with fetal chronic hypoxia in diabetic pregnancies. Neonatology 2013; 103: 193-198 [PMID: 23295371]
33 Ruggiero RA, Bruzzo J, Chiarella P, di Gianni P, Isturiz MA, Linskens S, Speziale N, Meiss RP, Bustuoabad OD, Pasqualini CD. Tyrosine isomers mediate the classical phenomenon of concomitant tumor resistance. Cancer Res 2011; 71: 7113-7124 [PMID: 22084446]
34 Ruggiero RA, Bruzzo J, Chiarella P, Bustuoabad OD, Meiss RP, Pasqualini CD. Concomitant tumor resistance: the role of tyrosine isomers in the mechanisms of metastases control. Cancer Res 2012; 72: 1043-1050 [PMID: 22315349]
35 Bertin C, Weston LA, Huang T, Jander G, Owens T, Meinwald J, Schroeder FC. Grass roots chemistry: meta-tyrosine, an herbicidal nonprotein amino acid. Proc Natl Acad Sci USA 2007; 104: 16964-16969 [PMID: 17940026]
36 Bertin C, Harmon R, Akaogi M, Weidenhamer JD, Weston LA. Assessment of the phytotoxic potential of m-tyrosine in laboratory soil bioassays. J Chem Ecol 2009; 35: 1288-1294 [PMID: 19924485]
37 Mikolás E, Cseh J, Pap M, Szijárto IA, Balogh A, Laczy B, Bekő V, Fisi V, Molnár GA, Mérei A, Szeberényi J, Wittmann I. Effects of erythropoietin on glucose metabolism. Horm Metab Res 2012; 44: 279-285 [PMID: 22351476]
P- Reviewer: Tsuruya K S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
507 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Molnár GA et al . Tyrosine isomers and hormone resistance

Mami Yamaoka, Toshimasa Ishizaki, Toshihide Kimura
Mami Yamaoka, Toshimasa Ishizaki, Toshihide Kimura, Department of Pharmacology, Oita University Faculty of Medicine, Oita 879-5593, JapanAuthor contributions: Yamaoka M, Ishizaki T and Kimura T developed the concept and wrote the paper.Supported by JSPS KAKENHI Grant, Nos. 19790636, 21790876, 23791039, and 26461342; the joint research program of the Institute for Molecular and Cellular Regulation, Gunma University, Takeda Science Foundation, Novo Nordisk Pharma, Oita Broadcasting System Cultural Foundation, and Research Fund at the Discretion of the President, Oita University.Conflict-of-interest: The authors declare no competing financial interests.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Toshihide Kimura, Associate Professor, Department of Pharmacology, Oita University Faculty of Medicine, 1-1 Idaigaoka, Hasama, Yufu, Oita 879-5593, Japan. [email protected]: +81-97-5865722Fax: +81-97-5865729Received: August 25, 2014Peer-review started: August 25, 2014First decision: December 17, 2014Revised: December 24, 2014Accepted: February 4, 2015 Article in press: February 9, 2015Published online: April 15, 2015
AbstractThe small GTPase Rab27a is a member of the Rab family that is involved in membrane trafficking in various kinds of cells. Rab27a has GTP- and GDP-bound forms, and their interconversion regulates intracellular signaling pathways. Typically, only a GTP-bound GTPase binds its specific effectors with the resulting downstream signals controlling specific cellular functions. We previously
identified novel Rab27a-interacting proteins. Surprisingly, some of these proteins interacted with GDP-bound Rab27a. The present study reviews recent progress in our understanding of the roles of Rab27a and its effectors in the secretory process. In pancreatic β-cells, GTP-bound Rab27a regulates insulin secretion at the pre-exocytotic stages via its GTP-specific effectors such as Exophilin8/Slac2-c/MyRIP and Slp4/Granuphilin. Glucose stimulation causes insulin exocytosis. Glucose stimulation also converts Rab27a from its GTP- to its GDP-bound form. GDP-bound Rab27a interacts with GDP-specific effectors and controls endocytosis of the secretory membrane. Thus, Rab27a cycling between GTP- and GDP-bound forms synchronizes with the recycling of secretory membrane to re-use the membrane and keep the β-cell volume constant.
Key words: Insulin; Exocytosis; Endocytosis; Rab27a; IQGAP1; Coronin 3; Glucose; Small GTPase
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: The small GTPase Rab27a is a member of the Rab family that is involved in membrane trafficking in pancreatic β-cells. GTP-bound Rab27a regulates insulin secretion at pre-exocytotic stages via its GTP-specific effectors such as Exophilin8/Slac2-c/MyRIP and Slp4/Granuphilin. Glucose stimulation causes insulin exocytosis. Glucose stimulation also converts Rab27a from its GTP- to its GDP-bound form. GDP-bound Rab27a interacts with GDP-specific effectors and controls endocytosis of the secretory membrane. Thus, Rab27a cycling between GTP- and GDP-bound forms synchronizes with the recycling of secretory membrane to re-use the membrane and keep the β-cell volume constant.
Yamaoka M, Ishizaki T, Kimura T. Interplay between Rab27a effectors in pancreatic β-cells. World J Diabetes 2015; 6(3): 508-516 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/508.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.508
MINIREVIEWS
508 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Interplay between Rab27a effectors in pancreatic β-cells
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.508
World J Diabetes 2015 April 15; 6(3): 508-516ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

INTRODUCTIONDiabetes mellitus is defined as chronic hyperglycemia due to relative insulin deficiency. Impairment of secretory activity in pancreatic β-cells plays an important role in the pathogenesis of this disease. In particular, decreased output in the early phase of glucose-induced insulin release precedes the onset of type 2 diabetes mellitus[1,2]. Insulin secretion from pancreatic β-cells is finely tuned for glucose homeostasis by multiple physiological factors such as nutrients, hormones and neurotransmitters. Some of these factors induce or enhance insulin release whereas others decrease it, thereby enabling sophisticated glucose regulation. One characteristic of this regulation in pancreatic β-cells is the control of secretion by several nutrients. Glucose, the most important insulin secretagogue, stimulates insulin secretion mainly by the generation of ATP via glucose metabolism, although there appear to be other signaling pathways involved that have not been fully identified.
The insulin secretory process comprises insulin synthesis and its packaging into secretory granules, granule transport in the cytoplasm, interaction with the cell membrane, exocytosis as a result of an increase in cytoplasmic Ca2+, endocytosis and retrograde transport. The present study reviews recent progress in our understanding of the roles of the small GTPase Rab27a and its effectors in the secretory process.
SMALL GTPASE CYCLESmall GTPases are GTP-binding proteins with molecular masses ranging from 20 to 30 kDa. These proteins, comprising Ras, Rho, Rab, Ran and Sar/Arf, are expressed in almost all eukaryotic cells and participate in a wide variety of cellular functions including proliferation, cytoskeletal rearrangement and intracellular transport[3]. Small GTPases have GTP- and GDP-bound forms, and their interconversion regulates intracellular signaling pathways (Figure 1). Typically, only the GTP-bound small GTPase binds its specific effectors and the resulting downstream signals control specific cellular functions[4]. Therefore, GTP- and GDP-bound small GTPases are considered as active and inactive forms, respectively. The activation of small GTPases is modulated by guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and GDP-dissociation inhibitors (GDIs). Small GTPases localize in the cytosol as a GDP-bound form under unstimulated conditions. Cell stimulation recruits GDP-bound small GTPases to the vicinity of the plasma membrane and converts them to the GTP-bound form through the action of GEFs[4,5]. These GTP-bound forms interact with their specific effectors and transduce signals. GAPs promote the intrinsic GTPase activity of small GTPases and induce the conversion of the GTP- to the GDP-bound form[4,6]. GDIs form a complex with GDP-bound small GTPases and induce their intracellular redistribution from the
plasma membrane to the cytosol[7-9].
RAB27AThe Rab family, which consists of more than 60 members, regulates membrane trafficking[10-12]. Rab27 is a Rab-family member that controls vesicle transport in various kinds of cells[13]. There are two isoforms of Rab27; Rab27a and Rab27b. Mutations in Rab27a have been reported to cause Griscelli syndrome[14]. This disorder results in pigment dilution in the skin and the hair. The same symptoms are observed in ashen mice with a natural mutation in Rab27a[15]. To date, three forms of Griscelli syndrome have been reported. Mutations in Myosin Va[14,16-18], Rab27a[14,15], and its effector Slac2-a/melanophilin[19,20] cause type 1 (GS1), type 2 (GS2), and type 3 (GS3) Griscelli syndrome, respectively. These proteins play an important role in melanosome transport in melanocytes. Peripheral melanosome distribution is regulated by the molecular motor protein myosin Va. GTP-bound Rab27a localizes on the surface of melanosomes where it interacts with Slac2-a/melanophilin. Since Slac2-a/melanophilin binds myosin Va, this complex functions as a linker protein between the melanosome and the motor protein[10,21,22]. GTP-bound Rab27 also interacts with another effector protein Slp2-a, thereby regulating docking of the melanosome to the plasma membrane[23].
Griscelli syndrome also results in immunodefici-ency[14,17]. Exocytosis of granzyme-A-containing granules is decreased in ashen mice cytotoxic T lymphocytes[24,25]. Although both Rab7 and Rab11 are also present on the surface of these granzyme-A-containing granules, Rab27a is the main regulator of granule exocytosis[26]. Indeed, GTP-bound Rab27a interacts with phosphatidylinositol 4,5-bisphosphate (PIP2) via its effector protein Munc13-4 and regulates the docking of the granules to the synapse. These results suggest that Rab27a regulates the secretion of lytic granules in cytotoxic T lymphocytes.
Rab27b, a closely related isoform of Rab27a, also participates in intracellular transport and secretion. Rab27b and its effector proteins including Slac2-c and Slp4-a are expressed in parotid acinar cells[27]. In these cells, GTP-bound Rab27a interacts with both Slac2-c and Slp4-a. Amylase release was inhibited when streptolysin O-permeabilized cells were treated with anti-Slac2-c antibody[28]. Amylase release was also inhibited when the Rab27b/Slp4-a complex was dissociated with a GST-Slp4-a-linker[29]. Isoproterenol (IPR) stimulation promoted intracellular redistribution of Slac2-c from the cytosol to a luminal site[30]. Since Slac2-c potentially interacts with Myosin Va, it is possible that the Rab27b/Slac2-c complex regulates F-actin-dependent intracellular transport of secretory vesicles. In contrast, the intracellular distribution of Slp4-a was not changed in the presence or absence of IPR stimulation[30]. The Rab27b/Slp4-a complex may regulate docking of the secretory vesicles to the membrane[31].
Amylase is also released from pancreatic acinar
Yamaoka M et al . Rab27a effectors in pancreatic β-cells
509 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

cells in which Rab27b is localized on the surface of zymogen granules[32]. Rab27b-Q78L, a constitutively active Rab27b mutant, enhances amylase release. In contrast, Rab27b-N133I, a dominant negative mutant, inhibits amylase release. These results suggest that Rab27b regulates amylase release in pancreatic acinar cells. Exophilin7/Slp1/JFC1 is a GTP-bound Rab27a interacting protein. The number of zymogen granules was increased in the pancreatic acinar cells of fasted Exophilin7/Slp1/JFC1-KO mice[33]. Moreover, amylase release was promoted in pancreatic acinar cells from Exophilin7/Slp1/JFC1-KO mice that were treated with carbamylcholine chloride or cholecystokinin-8, which mimic fed conditions. Thus, Rab27b may be involved in amylase release via Exophilin7/Slp1/JFC1 in pancreatic acinar cells.
Rab3a, which has the highest homology to Rab27, is highly expressed in synaptic vesicles[27]. In PC12 cells, Rab27a and Rab3a are co-localized on the surface of dense-core granules[34]. Silencing of both Rabs caused a significantly greater decrease in the number of these granules docked to the plasma membrane compared to silencing of either Rab alone. Since, Rab27a and Rab3a interact with the same effector proteins, these Rabs must cooperatively regulate the docking step of dense-core granules. In contrast, each of these Rabs has a specific function in pancreatic β-cells[35]. Rab3a-GAP inhibited the dot-like distribution of Rab3a in the insulin-secreting β-cell line MIN6. In contrast, the distribution of Rab27a was not changed in Rab27a-GAP expressing
cells. Furthermore, whereas Rab3a is localized on insulin granules as a GTP-bound form, the Rab27a on these granules is present as both GTP- and GDP-bound forms. Moreover, these Rabs direct unique kinetic and functional properties of the exocytic pathway.
RAB27A IN PANCREATIC β-CELLSRab27a is highly expressed in pancreatic β-cells and is involved in insulin secretion[36]. In ashen mice, blood glucose levels after a glucose load were higher, and insulin secretion in response to high glucose was lower than that in control mice. Interestingly, the decrease in insulin secretion was specific to glucose stimulation. These data suggest that Rab27a signaling in pancreatic β-cells is glucose-specific.
Insulin granules that are synthesized in pancreatic β-cells are eventually released by exocytosis via a series of stages (Figure 2). Granules that are transported in the cytoplasm attach to the inner surface of the cell membrane (docking). The contents of both docked and undocked granules are eventually released following elevation of intracellular Ca2+ levels. The granules are categorized into three types[37,38]. Residents are granules that are pre-docked to the plasma membrane before fusion. Passengers are granules that fused without stably docking. Visitors are granules that remain near the plasma membrane for some time before fusion. GTP-bound Rab27a regulates insulin secretion by modulating the transport and docking steps of insulin granules via its effector proteins (Table 1)[39-41]. To date, Exophilin8/Slac2-c/MyRIP, Slp4/Granuphilin, Exophilin7/Slp1/JFC1 and Exophilin1/Rabphilin3A are known to act as GTP-dependent Rab27a effectors in pancreatic β-cells. Exophilin8/Slac2-c/MyRIP possesses a potential
510 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Exophilin8/Slac2-c/MyRIP
Rab27aGTP
Noc2Slp4/Granuphilin
Insulin
Plasma membranePassengersVisitors, Residents
Pancreatic beta-cell
Translocation
Docking
Fusion
?
Figure 2 Schematic model of GTP-bound Rab27a function in pancreatic β-cells. Insulin-containing granules transported in the cytoplasm attach to the inner surface of the cell membrane (docking). The contents of both docked and undocked granules are eventually released following elevation of intracellular Ca2+ levels. GTP-bound Rab27a regulates insulin secretion by modulating the granule transport and docking steps via its effector proteins. Residents are granules that are pre-docked to the plasma membrane before fusion. Passengers are granules that fused without stably docking. Visitors are granules that remain near the plasma membrane for some time before fusion.
GDP
GTPase
GDIs
GAPs
PiGTP
GDP
GEFs
GTP
GTPase
Effectors
Cellular function
ON
OFF
Figure 1 Small GTPase GTP/GDP cycle. Small GTPases localize in the cytosol as a GDP-bound form under unstimulated conditions. Cell stimulation recruits GDP-bound small GTPases to the vicinity of the plasma membrane and converts these proteins to the GTP-bound form through GEF activity. The GTP-bound form interacts with its specific effectors and thereby transduces signals. GAPs promote the intrinsic GTPase activity of small GTPases and induce conversion of the GTP- to the GDP-bound form. GDIs form a complex with GDP-bound small GTPases and induce their intracellular redistribution from the plasma membrane to the cytosol. GEFs: Guanine nucleotide exchange factors; GAPs: GTPase-activating proteins; GDIs: GDP-dissociation inhibitors.
Yamaoka M et al . Rab27a effectors in pancreatic β-cells

pancreatic β-cells.
GDP-DEPENDENT EFFECTORS OF RAB27AWe have identified novel Rab27a-interacting proteins. Surprisingly, some of these proteins interacted with GDP-bound Rab27a (Table 2)[52,53]. Since GDP-bound GTPases have been considered to be an inactive form, these proteins might be suspected to be regulators of Rab27a GTP/GDP cycles. Indeed, protrudin, which was identified as a GDP-bound small GTPase interacting protein, forms a complex with GDP-bound Rab11 via its GDI consensus sequence and regulates neurite formation[54]. Therefore, protrudin is thought to be a GDI that regulates GTPase cycles. In contrast, the GDP-bound Rab27a interacting proteins that we identified do not contain any GDI consensus sequences. Moreover, these proteins interact with GDP-bound Rab27a and transduce downstream signals in a similar manner to classical GTP-dependent effectors of small GTPases. We consider that these proteins are GDP-dependent effectors of Rab27a[40,41].
Coronin 3We identified coronin 3 as a GDP-bound Rab27a interacting protein[52]. Coronins have been purified from precipitated actin of Dictyostelium discoideum cells[55]. To date, more than ten coronin subfamilies have been reported. Coronin 3 is a ubiquitously expressed coronin that shares a central domain with other coronin family members that contains five WD40 repeats, which are known to form β-propeller structures and to mediate protein-protein interactions (Figure 3). Initially, these repeats were thought to form a five-bladed β-propeller structure. However, recent studies of coronin structure demonstrated that the N-terminal region of coronin forms two additional blades (not identified from the sequence). Therefore, coronin 3 is now considered to possess a seven-bladed β-propeller structure[56,57]. It was noted that this propeller structure of coronin 3 is a GDP-bound Rab27a binding site[52]. Oligomerization of coronins and their interaction with F-actin are mediated by the C-terminal 30-40 amino acids, which form a coiled-coil structure. These interactions modulate actin assembly and participate in various cellular functions. Human coronin 3 also modulates F-actin through binding to Arp2/3[57], a key protein in the formation of a branched actin filament network[57-59]. These direct and indirect modulations of F-actin must play a crucial role in the cellular function of coronins, because they are conserved in eukaryote cells[60]. Both F-actin-binding and -bundling activities of coronin 3 were promoted by its interaction with GDP-bound Rab27a[61]. In contrast, GDP-bound Rab27a did not affect the interaction between coronin 3 and Arp2/3. GDP-bound Rab27a regulates F-actin bundling by modulating a direct effect of coronin 3 on F-actin (Figure 3).
myosin binding site. Although Exophilin8/Slac2-c/MyRIP functions as a linker protein between GTP-bound Rab27a and Myosin Va in melanocytes[42,43], this effector may form a different complex in pancreatic β-cells. Indeed, some reports suggest that there are several conditions under which Exophilin8/Slac2-c/MyRIP does not interact with Myosin Va[44]. Slp4/Granuphilin and Exophilin1/Rabphilin3A interact with Myosin Va in pancreatic β-cells[45]. Moreover, they are linked to a different subset of insulin granules. Further studies are required to investigate the regulation of the transport of insulin granules to the plasma membrane.
Slp4/Granuphilin was identified as a molecule that associates with insulin granules in pancreatic β-cells[46]. This molecule forms a complex with Syntaxin1a and Munc18-1 and regulates the docking step of insulin granules in the insulin secretion pathway[47]. The number of insulin granules docked to the plasma membrane was decreased in pancreatic β-cells from Slp4/Granuphilin-KO mice[48]. Interestingly, insulin secretion was promoted in these mice. These results suggest that Slp4/Granuphilin regulates the docking state and inhibits the fusion of the insulin granule membrane and the plasma membrane in unstimulated pancreatic β-cells (Figure 2). Exophilin7/Slp1/JFC1 also tethers insulin granules to the plasma membrane[49]. There seem to be multiple docking states of insulin granules in pancreatic β-cells.
Noc2 is a potential Rab27a effector. Noc2 displays 78% similarity to the N-terminus of Exophilin1/Rabphilin3A[50]. Ca2+ triggered insulin secretion from pancreatic islets of Noc2-KO mice was impaired, but was restored by treatment with the G-protein inhibitor pertussis toxin[51]. Although Noc2 is involved in insulin secretion through G-protein Gi/o signaling, its role in pancreatic β cells has not been identified. A yeast two-hybrid experiment identified zyxin as a Noc2-interacting protein. Because zyxin has been reported to bind the actin-binding protein α-actinin in fibroblasts[50], the interaction between Noc2 and zyxin may regulate insulin secretion by modulating actin dynamics in
511 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 1 GTP-bound Rab27a effectors in pancreatic β-cells
GTP-bound Rab27a effectors Interacting proteins Function
Exophillin8/Slac2-c/MyRIP Myosin Va, Actin, PKA TranslocationSlp4/Granuphilin Munc18-1,
Syntaxin1a,Myosin VaDocking
Noc2 Munc13-1 -Exophilin7/Slp1/JFC1 - DockingExophilin1/Rabphilin3A Myosin Va Translocation
Table 2 GDP-bound Rab27a effectors in pancreatic β-cells
GDP-bound Rab27a effectors Interacting proteins Function
Coronin 3/coronin 1c F-actin Actin bundlingEndocytosis
IQGAP1 GTP-bound Cdc42 ScaffoldGTP-bound Rac1
Yamaoka M et al . Rab27a effectors in pancreatic β-cells

Coronins regulate membrane internalization in some types of cells through F-actin remodeling. In pancreatic β-cells, silencing of coronin 3 inhibited the internalization of both FM4-64 and phogrin[52]. Internalization of these molecules was also inhibited when Rab27a-coronin 3 binding was inhibited by a dominant negative mutant of coronin 3. Moreover, the inhibition of F-actin binding to coronin 3 had the same effect[61]. Thus, coronin 3 regulates the endocytosis of secretory membranes via the modulation of actin assembly in pancreatic β-cells.
Endocytosis is a complex process that involves cargo sorting, membrane invagination, vesicle scission and vesicle targeting (Figure 4). The uptake of an antibody against the extracellular domain of phogrin was inhibited and phogrin was located near the plasma membrane in MIN6 cells expressing a dominant negative mutant of coronin 3[62]. Interestingly, phogrin staining near the plasma membrane remained when the cells were treated with acid wash, suggesting that the anti-phogrin antibody near the plasma membrane is separated from the plasma membrane. Thus the formation of GDP-bound Rab27a-coronin 3 complexes regulates the retro-grade transport of internalized secretory membrane, at a stage after scission from the plasma membrane.
Insulin secretagogue glucose stimulation induced the intracellular redistribution of both Rab27a and coronin 3 from the cytosol to the plasma membrane[62]. These redistributions were inhibited in Rab27a-silenced and Rab27a-Q78L-expressing MIN6 cells. Glucose-induced translocation of coronin 3 is due to its interaction with GDP-bound Rab27a. It has been reported that coronin 3 forms a closed conformation through an intramolecular interaction between its C- and N-termini[59]. Therefore, the glucose-dependent binding of its N-terminus to GDP-Rab27a may shift coronin 3 to the open conformation, which enables this molecule to interact with F-actin[41].
IQGAP1We identified IQGAP1 as another GDP-bound Rab27a interacting protein[53]. IQGAP1, an effector for Cdc42 and Rac1, is a member of the IQGAP family[63]. IQGAP1 regulates cell-cell contacts and cell migration[64-70]. In pancreatic β-cells, IQGAP1 interacts with vesicle-tethering exocysts under basal conditions. Moreover, this IQGAP-exocyst complex is dissociated by GTP-bound Cdc42[71]. GTP-bound Cdc42 regulates insulin secretion by modulating F-actin bundling[72,73]. Active Cdc42 also interacts with SNARE proteins such as VAMP2 and Syntaxin1a, and promotes the fusion step in insulin secretion[74]. Since glucose converts Cdc42 from the GDP- to the GTP-bound form[75], the IQGAP1-vesicle tethering exocyst complex is dissociated by glucose. It has been reported that vesicle-tethering to the plasma membrane is not a prerequisite for, but instead temporarily inhibits glucose-induced membrane fusion[37]. This finding raises the possibility that the IQGAP1-exocyst complex may inhibit subsequent fusion events. IQGAP1 also binds A kinase anchoring protein 79 and functions as a scaffold protein[76].
IQGAP1 binds GDP-bound Rab27a through its RasGAP related domain (Figure 3)[53]. This domain lacks GAP activity and forms part of the GTP-bound Cdc42 and Rac1 interacting site[63,70,77]. Moreover, IQGAP1 interacts with GDP-bound Rab27a when it forms a complex with GTP-bound Cdc42[53]. IQGAP1 is distributed in the vicinity of the plasma membrane in both glucose-stimulated and - unstimulated cells. In contrast, glucose-induced redistribution of Rab27a and its binding protein coronin 3 was inhibited in both IQGAP1-silenced cells and in cells expressing the dominant negative mutant Cdc42-T17N. Since endocytosis of secretory membrane was also inhibited in these cells, these data indicate that activated Cdc42-bound IQGAP1, to which GDP-bound Rab27a binds,
512 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Figure 4 Schematic model of GDP-bound Rab27a function in pancreatic β-cells. Endocytosis is a complex process that involves cargo sorting, membrane invagination, vesicle scission and vesicle targeting. GDP-bound Rab27a modulates F-actin assembly and regulates the retrograde transport of the internalized secretory membrane, at the stage after scission from the plasma membrane. Scission is indicated by the dynamin step.
Pancreatic beta-cell
F-actinGDP
Rab27a Coronin 3IQGAP1
Clathrin
Dynamin 2 Plasma membrane
Insulin
Pancreatic beta-cellF-actin
Coronin 3
N WD C
β-propeller structure GDP
Rab27a GTP
Cdc42IQGAP1
CHD NTR IQR GRD CT
Plasma membrane
Figure 3 Interplay between Rab27a, coronin 3 and IQGAP1. GDP-bound Rab27a simultaneously binds coronin 3 and IQGAP1, resulting in the formation of a trimeric complex. WD: Five WD40 repeats; CHD: Calponin homology domain; NTR: N-terminal repeats; IQR: IQ repeats; GRD: RasGAP related domain; CT: Carboxy terminus of IQGAP1.
Yamaoka M et al . Rab27a effectors in pancreatic β-cells

recruits endocytic machinery including coronin 3 and regulates endocytosis of secretory membrane.
In summary, IQGAP1 functions at pre-exocytotic stages via interaction with the exocyst complex[71] and this complex is dissociated by GTP-bound Cdc42. Our results indicate that IQGAP1 also plays a crucial role in the control of endocytosis via interaction with GTP-bound Cdc42 and GDP-bound Rab27a[53]. Based on the combined data, we propose a model where IQGAP1 functions as a scaffold protein and is a key molecule for membrane recycling.
IQGAP1 also interacts with GTP-bound Rac1[63,77]. Interestingly, IQGAP1 interacts with GDP-bound Rab27a when it forms a complex with GTP-bound Rac1[53]. Moreover, endocytosis of secretory membrane was inhibited in MIN6 cells expressing the dominant negative Rac1-T17N mutant. These results suggest that Rac1 also recruits endocytic machinery and regulates endocytosis of secretory membrane. Cdc42 and Rac1 display some different characteristics in pancreatic β-cells. The most important difference is the timing of the glucose-induced conversion from the GDP- to the GTP-bound form. Glucose stimulation causes a shift from GDP-bound Cdc42 to GTP-bound Cdc42 within 3 min. In contrast, the glucose induced conversion of GDP- to GTP-bound Rac1 requires 20 min[73,75]. These findings raises the possibility that Cdc42 regulates rapid endocytosis whereas Rac1 regulates subsequent, prolonged endocytosis, a pattern that may be associated with the biphasic release of insulin in response to glucose.
CONCLUSIONIn the basal state, GTP-bound Rab27a controls insulin secretion at pre-exocytotic stages via its GTP-specific effectors (Figure 5). Glucose stimulation causes insulin exocytosis. Glucose stimulation also converts Rab27a
from its GTP- to its GDP-bound form, which interacts with IQGAP1, recruits coronin 3, and controls endocytosis of the secretory granule membrane. A long-term overexpression of a dominant negative coronin 3 mutant caused β-cell death (unpublished data), suggesting that the membrane recycling system controlled by Rab27a may be necessary for β-cell survival. Thus, we consider that Rab27a GTP/GDP cycling synchronizes with the recycling of secretory membrane to re-use the membrane and to keep the β-cell volume constant. It raises the possibility that a pharmacological agent that modulates the recycling system may become a new therapeutic option for the treatment of β-cell dysfunction in diabetes. Further studies are required to investigate whether Rab27a is involved in the pathogeneses of diabetes mellitus.
Typically, small GTPases are predominantly present in the GDP-bound form under unstimulated conditions and are converted to the GTP-bound form by stimulation. In contrast, glucose stimulation causes a shift of Rab27a in pancreatic β-cells from its GTP- to its GDP-bound form[52]. The same conversion also occurs in thrombin stimulated platelets[78]. These findings suggest that specific Rab27a-GAPs are activated by these stimulations. Two candidate Rab27a-GAPs, EPI64A and EPI64B, have been reported[79]. In melanocytes, EPI64A has the higher GAP activity and functions as the main Rab27a-GAP. In pancreatic acinar cells, EPI64B regulates amylase secretion by modulating Rab27a GTP/GDP cycles[80]. Further studies are needed to identify and characterize Rab27a-GAPs in pancreatic β-cells.
ACKNOWLEDGMENTSWe thank all members of our laboratory for helpful suggestions.
REFERENCES1 Ashcroft FM, Rorsman P. Molecular defects in insulin secretion
in type-2 diabetes. Rev Endocr Metab Disord 2004; 5: 135-142 [PMID: 15041789 DOI: 10.1023/B:REMD.0000021435.87776.a7]
2 Kahn SE. Clinical review 135: The importance of beta-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab 2001; 86: 4047-4058 [PMID: 11549624 DOI: 10.1210/jcem.86.9.7713]
3 Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci 2005; 118: 843-846 [PMID: 15731001 DOI: 10.1242/jcs.01660]
4 Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev 2001; 81: 153-208 [PMID: 11152757]
5 Novick P, Zerial M. The diversity of Rab proteins in vesicle transport. Curr Opin Cell Biol 1997; 9: 496-504 [PMID: 9261061 DOI: 10.1016/S0955-0674(97)80025-7]
6 Fukui K, Sasaki T, Imazumi K, Matsuura Y, Nakanishi H, Takai Y. Isolation and characterization of a GTPase activating protein specific for the Rab3 subfamily of small G proteins. J Biol Chem 1997; 272: 4655-4658 [PMID: 9030515 DOI: 10.1074/jbc.272.8.4655]
7 Dirac-Svejstrup AB, Soldati T, Shapiro AD, Pfeffer SR. Rab-GDI presents functional Rab9 to the intracellular transport machinery and contributes selectivity to Rab9 membrane recruitment. J Biol
513 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
GTP GDP
Rab27aGAP
Rab27a
Effectors Glucose Coronin 3 IQGAP1
Pre-exocytosis Fusion Endocytosis
Pancreatic beta-cell
Insulin
?
Figure 5 Rab27a GTP/GDP cycling synchronizes with the recycling of secretory membrane. In the basal state, GTP-bound Rab27a controls insulin secretion at pre-exocytotic stages via its GTP-specific effectors. Glucose stimulation causes insulin exocytosis. Glucose stimulation also converts Rab27a from its GTP- to its GDP-bound form. GDP-bound Rab27a interacts with IQGAP1, recruits coronin 3, and controls endocytosis of the secretory granule membrane. GAPs: GTPase-activating proteins.
Yamaoka M et al . Rab27a effectors in pancreatic β-cells

Chem 1994; 269: 15427-15430 [PMID: 8195183]8 Garrett MD, Zahner JE, Cheney CM, Novick PJ. GDI1 encodes
a GDP dissociation inhibitor that plays an essential role in the yeast secretory pathway. EMBO J 1994; 13: 1718-1728 [PMID: 8157010]
9 Ullrich O, Horiuchi H, Bucci C, Zerial M. Membrane association of Rab5 mediated by GDP-dissociation inhibitor and accompanied by GDP/GTP exchange. Nature 1994; 368: 157-160 [PMID: 8139660 DOI: 10.1038/368157a0]
10 Fukuda M. Versatile role of Rab27 in membrane trafficking: focus on the Rab27 effector families. J Biochem 2005; 137: 9-16 [PMID: 15713878 DOI: 10.1093/jb/mvi002]
11 Grosshans BL, Ortiz D, Novick P. Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA 2006; 103: 11821-11827 [PMID: 16882731 DOI: 10.1073/pnas.0601617103]
12 Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2001; 2: 107-117 [PMID: 11252952 DOI: 10.1038/35052055]
13 Chen D, Guo J, Miki T, Tachibana M, Gahl WA. Molecular cloning and characterization of rab27a and rab27b, novel human rab proteins shared by melanocytes and platelets. Biochem Mol Med 1997; 60: 27-37 [PMID: 9066979 DOI: 10.1006/bmme.1996.2559]
14 Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, Wulffraat N, Bianchi D, Fischer A, Le Deist F, de Saint Basile G. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 2000; 25: 173-176 [PMID: 10835631]
15 Wilson SM, Yip R, Swing DA, O’Sullivan TN, Zhang Y, Novak EK, Swank RT, Russell LB, Copeland NG, Jenkins NA. A mutation in Rab27a causes the vesicle transport defects observed in ashen mice. Proc Natl Acad Sci USA 2000; 97: 7933-7938 [PMID: 10859366 DOI: 10.1073/pnas.140212797]
16 Pastural E, Barrat FJ, Dufourcq-Lagelouse R, Certain S, Sanal O, Jabado N, Seger R, Griscelli C, Fischer A, de Saint Basile G. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet 1997; 16: 289-292 [PMID: 9207796 DOI: 10.1038/ng0797-289]
17 Pastural E, Ersoy F, Yalman N, Wulffraat N, Grillo E, Ozkinay F, Tezcan I, Gediköglu G, Philippe N, Fischer A, de Saint Basile G. Two genes are responsible for Griscelli syndrome at the same 15q21 locus. Genomics 2000; 63: 299-306 [PMID: 10704277 DOI: 10.1006/geno.1999.6081]
18 Takagishi Y, Murata Y. Myosin Va mutation in rats is an animal model for the human hereditary neurological disease, Griscelli syndrome type 1. Ann N Y Acad Sci 2006; 1086: 66-80 [PMID: 17185506 DOI: 10.1196/annals.1377.006]
19 Ménasché G, Ho CH, Sanal O, Feldmann J, Tezcan I, Ersoy F, Houdusse A, Fischer A, de Saint Basile G. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest 2003; 112: 450-456 [PMID: 12897212 DOI: 10.1172/JCI18264]
20 Westbroek W, Lambert J, Naeyaert JM. The dilute locus and Griscelli syndrome: gateways towards a better understanding of melanosome transport. Pigment Cell Res 2001; 14: 320-327 [PMID: 11601653 DOI: 10.1034/j.1600-0749.2001.140503.x]
21 Bahadoran P, Aberdam E, Mantoux F, Buscà R, Bille K, Yalman N, de Saint-Basile G, Casaroli-Marano R, Ortonne JP, Ballotti R. Rab27a: A key to melanosome transport in human melanocytes. J Cell Biol 2001; 152: 843-850 [PMID: 11266474 DOI: 10.1083/jcb.152.4.843]
22 Hume AN, Collinson LM, Rapak A, Gomes AQ, Hopkins CR, Seabra MC. Rab27a regulates the peripheral distribution of melanosomes in melanocytes. J Cell Biol 2001; 152: 795-808 [PMID: 11266470 DOI: 10.1083/jcb.152.4.795]
23 Kuroda TS, Fukuda M. Rab27A-binding protein Slp2-a is required for peripheral melanosome distribution and elongated cell shape in melanocytes. Nat Cell Biol 2004; 6: 1195-1203 [PMID: 15543135 DOI: 10.1038/ncb1197]
24 Haddad EK, Wu X, Hammer JA, Henkart PA. Defective granule
exocytosis in Rab27a-deficient lymphocytes from Ashen mice. J Cell Biol 2001; 152: 835-842 [PMID: 11266473 DOI: 10.1083/jcb.152.4.835]
25 Stinchcombe JC, Barral DC, Mules EH, Booth S, Hume AN, Machesky LM, Seabra MC, Griffiths GM. Rab27a is required for regulated secretion in cytotoxic T lymphocytes. J Cell Biol 2001; 152: 825-834 [PMID: 11266472 DOI: 10.1083/jcb.152.4.825]
26 van der Sluijs P, Zibouche M, van Kerkhof P. Late steps in secretory lysosome exocytosis in cytotoxic lymphocytes. Front Immunol 2013; 4: 359 [PMID: 24302923 DOI: 10.3389/fimmu.2013.00359]
27 Fukuda M. Regulation of secretory vesicle traffic by Rab small GTPases. Cell Mol Life Sci 2008; 65: 2801-2813 [PMID: 18726178 DOI: 10.1007/s00018-008-8351-4]
28 Imai A, Yoshie S, Nashida T, Shimomura H, Fukuda M. The small GTPase Rab27B regulates amylase release from rat parotid acinar cells. J Cell Sci 2004; 117: 1945-1953 [PMID: 15039459 DOI: 10.1242/jcs.01048]
29 Fukuda M , Imai A, Nashida T, Shimomura H. Slp4-a/granuphilin-a interacts with syntaxin-2/3 in a Munc18-2-dependent manner. J Biol Chem 2005; 280: 39175-39184 [PMID: 16186111 DOI: 10.1074/jbc.M505759200]
30 Imai A, Fukuda M, Yoshie S, Nashida T, Shimomura H. Redistribution of Rab27-specific effector Slac2-c, but not Slp4-a, after isoproterenol-stimulation in rat parotid acinar cells. Arch Oral Biol 2009; 54: 361-368 [PMID: 19185850 DOI: 10.1016/j.archoralbio.2008.12.007]
31 Imai A, Nashida T, Shimomura H. Roles of Munc18-3 in amylase release from rat parotid acinar cells. Arch Biochem Biophys 2004; 422: 175-182 [PMID: 14759605 DOI: 10.1016/j.abb.2003.12.021]
32 Chen X, Li C, Izumi T, Ernst SA, Andrews PC, Williams JA. Rab27b localizes to zymogen granules and regulates pancreatic acinar exocytosis. Biochem Biophys Res Commun 2004; 323: 1157-1162 [PMID: 15451418 DOI: 10.1016/j.bbrc.2004.08.212]
33 Saegusa C, Kanno E, Itohara S, Fukuda M. Expression of Rab27B-binding protein Slp1 in pancreatic acinar cells and its involvement in amylase secretion. Arch Biochem Biophys 2008; 475: 87-92 [PMID: 18477466 DOI: 10.1016/j.abb.2008.04.031]
34 Tsuboi T, Fukuda M. Rab3A and Rab27A cooperatively regulate the docking step of dense-core vesicle exocytosis in PC12 cells. J Cell Sci 2006; 119: 2196-2203 [PMID: 16684812 DOI: 10.1242/jcs.02962]
35 Cazares VA, Subramani A, Saldate JJ, Hoerauf W, Stuenkel EL. Distinct actions of Rab3 and Rab27 GTPases on late stages of exocytosis of insulin. Traffic 2014; 15: 997-1015 [PMID: 24909540 DOI: 10.1111/tra.12182]
36 Kasai K, Ohara-Imaizumi M, Takahashi N, Mizutani S, Zhao S, Kikuta T, Kasai H, Nagamatsu S, Gomi H, Izumi T. Rab27a mediates the tight docking of insulin granules onto the plasma membrane during glucose stimulation. J Clin Invest 2005; 115: 388-396 [PMID: 15690086 DOI: 10.1172/JCI22955]
37 Kasai K, Fujita T, Gomi H, Izumi T. Docking is not a prerequisite but a temporal constraint for fusion of secretory granules. Traffic 2008; 9: 1191-1203 [PMID: 18397364 DOI: 10.1111/j.1600-0854.2008.00744.x]
38 Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, Seino S. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci USA 2007; 104: 19333-19338 [PMID: 18040047 DOI: 10.1073/pnas.0707054104]
39 Izumi T. Physiological roles of Rab27 effectors in regulated exocytosis. Endocr J 2007; 54: 649-657 [PMID: 17664848 DOI: 10.1507/endocrj.KR-78]
40 Kimura T, Niki I. Rab27a in pancreatic beta-cells, a busy protein in membrane trafficking. Prog Biophys Mol Biol 2011; 107: 219-223 [PMID: 21762718 DOI: 10.1016/j.pbiomolbio.2011.06.016]
41 Kimura T, Niki I. Rab27a, actin and beta-cell endocytosis. Endocr J 2011; 58: 1-6 [PMID: 21187662 DOI: 10.1507/endocrj.K10E-391]
42 Brozzi F, Lajus S, Diraison F, Rajatileka S, Hayward K, Regazzi R,
514 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Yamaoka M et al . Rab27a effectors in pancreatic β-cells

Molnár E, Váradi A. MyRIP interaction with MyoVa on secretory granules is controlled by the cAMP-PKA pathway. Mol Biol Cell 2012; 23: 4444-4455 [PMID: 22993210 DOI: 10.1091/mbc.E12-05-0369]
43 Mizuno K, Ramalho JS, Izumi T. Exophilin8 transiently clusters insulin granules at the actin-rich cell cortex prior to exocytosis. Mol Biol Cell 2011; 22: 1716-1726 [PMID: 21441305 DOI: 10.1091/mbc.E10-05-0404]
44 Waselle L, Coppola T, Fukuda M, Iezzi M, El-Amraoui A, Petit C, Regazzi R. Involvement of the Rab27 binding protein Slac2c/MyRIP in insulin exocytosis. Mol Biol Cell 2003; 14: 4103-4113 [PMID: 14517322 DOI: 10.1091/mbc.E03-01-0022]
45 Brozzi F, Diraison F, Lajus S, Rajatileka S, Philips T, Regazzi R, Fukuda M, Verkade P, Molnár E, Váradi A. Molecular mechanism of myosin Va recruitment to dense core secretory granules. Traffic 2012; 13: 54-69 [PMID: 21985333 DOI: 10.1111/j.1600-0854.2011.01301.x]
46 Wang J, Takeuchi T, Yokota H, Izumi T. Novel rabphilin-3-like protein associates with insulin-containing granules in pancreatic beta cells. J Biol Chem 1999; 274: 28542-28548 [PMID: 10497219 DOI: 10.1074/jbc.274.40.28542]
47 Torii S, Zhao S, Yi Z, Takeuchi T, Izumi T. Granuphilin modulates the exocytosis of secretory granules through interaction with syntaxin 1a. Mol Cell Biol 2002; 22: 5518-5526 [PMID: 12101244 DOI: 10.1128/MCB.22.15.5518-5526.2002]
48 Gomi H, Mizutani S, Kasai K, Itohara S, Izumi T. Granuphilin molecularly docks insulin granules to the fusion machinery. J Cell Biol 2005; 171: 99-109 [PMID: 16216924 DOI: 10.1083/jcb.200505179]
49 Wang H, Ishizaki R, Xu J, Kasai K, Kobayashi E, Gomi H, Izumi T. The Rab27a effector exophilin7 promotes fusion of secretory granules that have not been docked to the plasma membrane. Mol Biol Cell 2013; 24: 319-330 [PMID: 23223571 DOI: 10.1091/mbc.E12-04-0265]
50 Kotake K, Ozaki N, Mizuta M, Sekiya S, Inagaki N, Seino S. Noc2, a putative zinc finger protein involved in exocytosis in endocrine cells. J Biol Chem 1997; 272: 29407-29410 [PMID: 9367993 DOI: 10.1074/jbc.272.47.29407]
51 Matsumoto M, Miki T, Shibasaki T, Kawaguchi M, Shinozaki H, Nio J, Saraya A, Koseki H, Miyazaki M, Iwanaga T, Seino S. Noc2 is essential in normal regulation of exocytosis in endocrine and exocrine cells. Proc Natl Acad Sci USA 2004; 101: 8313-8318 [PMID: 15159548 DOI: 10.1073/pnas.0306709101]
52 Kimura T, Kaneko Y, Yamada S, Ishihara H, Senda T, Iwamatsu A, Niki I. The GDP-dependent Rab27a effector coronin 3 controls endocytosis of secretory membrane in insulin-secreting cell lines. J Cell Sci 2008; 121: 3092-3098 [PMID: 18768935 DOI: 10.1242/jcs.030544]
53 Kimura T, Yamaoka M, Taniguchi S, Okamoto M, Takei M, Ando T, Iwamatsu A, Watanabe T, Kaibuchi K, Ishizaki T, Niki I. Activated Cdc42-bound IQGAP1 determines the cellular endocytic site. Mol Cell Biol 2013; 33: 4834-4843 [PMID: 24100016 DOI: 10.1128/MCB.00895-13]
54 Shirane M, Nakayama KI. Protrudin induces neurite formation by directional membrane trafficking. Science 2006; 314: 818-821 [PMID: 17082457 DOI: 10.1126/science.1134027]
55 de Hostos EL, Bradtke B, Lottspeich F, Guggenheim R, Gerisch G. Coronin, an actin binding protein of Dictyostelium discoideum localized to cell surface projections, has sequence similarities to G protein beta subunits. EMBO J 1991; 10: 4097-4104 [PMID: 1661669]
56 Appleton BA, Wu P, Wiesmann C. The crystal structure of murine coronin-1: a regulator of actin cytoskeletal dynamics in lymphocytes. Structure 2006; 14: 87-96 [PMID: 16407068 DOI: 10.1016/j.str.2005.09.013]
57 Rosentreter A, Hofmann A, Xavier CP, Stumpf M, Noegel AA, Clemen CS. Coronin 3 involvement in F-actin-dependent processes at the cell cortex. Exp Cell Res 2007; 313: 878-895 [PMID: 17274980 DOI: 10.1016/j.yexcr.2006.12.015]
58 Hasse A, Rosentreter A, Spoerl Z, Stumpf M, Noegel AA, Clemen
CS. Coronin 3 and its role in murine brain morphogenesis. Eur J Neurosci 2005; 21: 1155-1168 [PMID: 15813925 DOI: 10.1111/j.1460-9568.2005.03917.x]
59 Spoerl Z, Stumpf M, Noegel AA, Hasse A. Oligomerization, F-actin interaction, and membrane association of the ubiquitous mammalian coronin 3 are mediated by its carboxyl terminus. J Biol Chem 2002; 277: 48858-48867 [PMID: 12377779 DOI: 10.1074/jbc.M205136200]
60 Galletta BJ, Chuang DY, Cooper JA. Distinct roles for Arp2/3 regulators in actin assembly and endocytosis. PLoS Biol 2008; 6: e1 [PMID: 18177206 DOI: 10.1371/journal.pbio.0060001]
61 Kimura T, Taniguchi S, Niki I. Actin assembly controlled by GDP-Rab27a is essential for endocytosis of the insulin secretory membrane. Arch Biochem Biophys 2010; 496: 33-37 [PMID: 20138020 DOI: 10.1016/j.abb.2010.01.017]
62 Kimura T, Taniguchi S, Toya K, Niki I. Glucose-induced translocation of coronin 3 regulates the retrograde transport of the secretory membrane in the pancreatic beta-cells. Biochem Biophys Res Commun 2010; 395: 318-323 [PMID: 20362548 DOI: 10.1016/j.bbrc.2010.03.173]
63 Kuroda S, Fukata M, Kobayashi K, Nakafuku M, Nomura N, Iwamatsu A, Kaibuchi K. Identification of IQGAP as a putative target for the small GTPases, Cdc42 and Rac1. J Biol Chem 1996; 271: 23363-23367 [PMID: 8798539 DOI: 10.1074/jbc.271.38.23363]
64 Briggs MW, Sacks DB. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep 2003; 4: 571-574 [PMID: 12776176 DOI: 10.1038/sj.embor.embor867]
65 Fukata M, Nakagawa M, Itoh N, Kawajiri A, Yamaga M, Kuroda S, Kaibuchi K. Involvement of IQGAP1, an effector of Rac1 and Cdc42 GTPases, in cell-cell dissociation during cell scattering. Mol Cell Biol 2001; 21: 2165-2183 [PMID: 11238950 DOI: 10.1128/MCB.21.6.2165-2183.2001]
66 Fukata M, Nakagawa M, Kaibuchi K. Roles of Rho-family GTPases in cell polarisation and directional migration. Curr Opin Cell Biol 2003; 15: 590-597 [PMID: 14519394 DOI: 10.1016/S0955-0674(03)00097-8]
67 Fukata M, Watanabe T, Noritake J, Nakagawa M, Yamaga M, Kuroda S, Matsuura Y, Iwamatsu A, Perez F, Kaibuchi K. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 2002; 109: 873-885 [PMID: 12110184 DOI: 10.1016/S0092-8674(02)00800-0]
68 Kuroda S, Fukata M, Nakagawa M, Fujii K, Nakamura T, Ookubo T, Izawa I, Nagase T, Nomura N, Tani H, Shoji I, Matsuura Y, Yonehara S, Kaibuchi K. Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin- mediated cell-cell adhesion. Science 1998; 281: 832-835 [PMID: 9694656 DOI: 10.1126/science.281.5378.832]
69 Kuroda S, Fukata M, Nakagawa M, Kaibuchi K. Cdc42, Rac1, and their effector IQGAP1 as molecular switches for cadherin-mediated cell-cell adhesion. Biochem Biophys Res Commun 1999; 262: 1-6 [PMID: 10448058 DOI: 10.1006/bbrc.1999.1122]
70 Noritake J, Fukata M, Sato K, Nakagawa M, Watanabe T, Izumi N, Wang S, Fukata Y, Kaibuchi K. Positive role of IQGAP1, an effector of Rac1, in actin-meshwork formation at sites of cell-cell contact. Mol Biol Cell 2004; 15: 1065-1076 [PMID: 14699063 DOI: 10.1091/mbc.E03-08-0582]
71 Rittmeyer EN, Daniel S, Hsu SC, Osman MA. A dual role for IQGAP1 in regulating exocytosis. J Cell Sci 2008; 121: 391-403 [PMID: 18216334 DOI: 10.1242/jcs.016881]
72 Kowluru A, Seavey SE, Li G, Sorenson RL, Weinhaus AJ, Nesher R, Rabaglia ME, Vadakekalam J, Metz SA. Glucose- and GTP-dependent stimulation of the carboxyl methylation of CDC42 in rodent and human pancreatic islets and pure beta cells. Evidence for an essential role of GTP-binding proteins in nutrient-induced insulin secretion. J Clin Invest 1996; 98: 540-555 [PMID: 8755667 DOI: 10.1172/JCI118822]
73 Nevins AK, Thurmond DC. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am J Physiol Cell Physiol 2003; 285: C698-C710 [PMID:
515 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Yamaoka M et al . Rab27a effectors in pancreatic β-cells

12760905 DOI: 10.1152/ajpcell.00093.2003]74 Nevins AK, Thurmond DC. A direct interaction between Cdc42
and vesicle-associated membrane protein 2 regulates SNARE-dependent insulin exocytosis. J Biol Chem 2005; 280: 1944-1952 [PMID: 15537656 DOI: 10.1074/jbc.M409528200]
75 Wang Z, Oh E, Thurmond DC. Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J Biol Chem 2007; 282: 9536-9546 [PMID: 17289663 DOI: 10.1074/jbc.M610553200]
76 Nauert JB, Rigas JD, Lester LB. Identification of an IQGAP1/AKAP79 complex in beta-cells. J Cell Biochem 2003; 90: 97-108 [PMID: 12938160 DOI: 10.1002/jcb.10604]
77 Hart MJ, Callow MG, Souza B, Polakis P. IQGAP1, a calmodulin-binding protein with a rasGAP-related domain, is a potential
effector for cdc42Hs. EMBO J 1996; 15: 2997-3005 [PMID: 8670801]
78 Kondo H, Shirakawa R, Higashi T, Kawato M, Fukuda M, Kita T, Horiuchi H. Constitutive GDP/GTP exchange and secretion-dependent GTP hydrolysis activity for Rab27 in platelets. J Biol Chem 2006; 281: 28657-28665 [PMID: 16880209 DOI: 10.1074/jbc.M603227200]
79 Itoh T, Fukuda M. Identification of EPI64 as a GTPase-activating protein specific for Rab27A. J Biol Chem 2006; 281: 31823-31831 [PMID: 16923811 DOI: 10.1074/jbc.M603808200]
80 Hou Y, Chen X, Tolmachova T, Ernst SA, Williams JA. EPI64B acts as a GTPase-activating protein for Rab27B in pancreatic acinar cells. J Biol Chem 2013; 288: 19548-19557 [PMID: 23671284 DOI: 10.1074/jbc.M113.472134]
P- Reviewer: Elia E S- Editor: Tian YL L- Editor: A E- Editor: Liu SQ
516 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Yamaoka M et al . Rab27a effectors in pancreatic β-cells

Orit Pinhas-Hamiel, Uri Hamiel, Yael Levy-Shraga
Orit Pinhas-Hamiel, Uri Hamiel, Yael Levy-Shraga, Pediatric Endocrinology and Diabetes Unit, Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, Tel Hashomer, Ramat-Gan 52621, IsraelOrit Pinhas-Hamiel, Uri Hamiel, Yael Levy-Shraga, Sackler School of Medicine, Tel-Aviv University, Juvenile Diabetes Center, Maccabi Health Care Services, Ramat-Gan 52621, IsraelAuthor contributions: Pinhas-Hamiel O, Hamiel U and Levy-Shraga Y performed literature review, wrote the paper, and conceived the three level models and created the graphs.Conflict-of-interest: None.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Dr. Orit Pinhas-Hamiel, Pediatric Endocrine and Diabetes Unit, Edmond and Lily Safra Children’sHospital, Sheba Medical Center, Tel Hashomer, Ramat-Gan 52621, Israel. [email protected] Telephone: +972-3-5305015Fax: +972-3-5305055Received: August 27, 2014Peer-review started: August 28, 2014First decision: November 27, 2014Revised: December 9, 2014Accepted: January 15, 2015Article in press: January 19, 2015Published online: April 15, 2015
AbstractEating disorders (ED) are characterized by a persistent disturbance of eating that impairs health or psychosocial functioning. They are associated with increased rates of medical complications and mortality. Insulin omission is a unique purging behavior available to individuals with type 1 diabetes mellitus (T1DM). The standard treatment regimen for T1DM requires a major focus on food and
eating patterns. Moreover, intensive insulin therapy is associated with increasing body weight. These factors, combined with the psychological burden of chronic disease management and depression, may contribute to ED. The comorbidity of ED in T1DM patients is associated with poorer glycemic control and consequently higher rates of diabetes complications. Early recognition and adequate treatment of ED in T1DM is essential.
Key words: Type 1 diabetes; Eating disorders; Insulin omission
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Intentional insulin omission for the purpose of preventing weight gain is a unique behavior available to individuals with type 1 diabetes mellitus (T1DM). It is classified as either an inappropriate compensatory feature of bulimia nervosa or as a purging disorder component of other specified feeding or eating disorder (ED). Its prevalence increases with age, affecting up to 40% of young adult females with T1DM. The comorbid of ED in T1DM patients is associated with higher rates of short and long-term diabetes complications. A high index of suspicion is needed since ED behaviors are often well hidden and denied. Treatment involves a complex interplay of psychosocial, dietary and medical aspects and requires a multidisciplinary team.
Pinhas-Hamiel O, Hamiel U, Levy-Shraga Y. Eating disorders in adolescents with type 1 diabetes: Challenges in diagnosis and treatment. World J Diabetes 2015; 6(3): 517-526 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/517.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.517
DEFINITIONS OF EATING DISORDERSEating disorders (ED) are characterized by a persistent
MINIREVIEWS
517 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Eating disorders in adolescents with type 1 diabetes: Challenges in diagnosis and treatment
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.517
World J Diabetes 2015 April 15; 6(3): 517-526ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

disturbance of eating that impairs health or psychosocial functioning[1,2]. Based upon the fifth edition of the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders (DSM-Ⅴ), which was released in May 2013, ED are divided into eight mutually exclusive categories[3]. These categories are based upon observed symptoms and include anorexia nervosa (AN), bulimia nervosa (BN), binge eating disorder, avoidant/restrictive food intake disorder, pica, rumination disorder, other specified feeding or eating disorder (OSFED) and unspecified feeding or eating disorder. Some diagnoses include a dimensional component that enables clinicians to specify the severity of illness[4-6].
In patients with type 1 diabetes mellitus (T1DM), intentional insulin omission or reduction for the purpose of preventing weight gain is recognized as either an inappropriate compensatory feature of bulimia nervosa or as a purging disorder, a component of OSFED. The latter is relevant when the recurrent purging behavior to influence weight or shape (e.g., insulin omission) occurs in the absence of binge eating.
The two categories: “eating disorders not otherwise specified (EDNOS)” and “disturbed eating behavior (DEB)”, compromised a wide spectrum of eating disorder pathologies, at a frequency or severity that does not merit a formal ED diagnosis. Though these categories are not included in DSM-Ⅴ, they appear in the current review, due to their use by several studies involving individuals with diabetes.
ARE ADOLESCENTS WITH T1DM AT INCREASED RISK TO DEVELOP ED?This question should be answered for the specific ED studied, according to the tools used for assessment and the prevalence rates in the general population.
Diagnostic tools of ED in persons with T1DMThe diagnosis of ED in individuals with T1DM is difficult, since eating behaviors are often well hidden and denied. The use of different questionnaires for assessing prevalence rates of ED makes comparisons across studies difficult. Furthermore, as Markowitz et al[7] pointed out, that general diagnostic questionnaires for detecting ED are not appropriate for individuals with T1DM for two main reasons. Firstly, these questionnaires do not identify eating disorder behaviors that are unique to T1DM, such as insulin omission[7]. Secondly, such questionnaires may inflate the prevalence of eating problems in those with T1DM, because behaviors that are considered disturbed, such as particular concern about diet, reduced intake of certain food groups, and eating when not hungry, are integral to diabetes care[8].
Prevalence rates of ED among persons with T1DM Prevalence rates of ED among persons with TIDM vary according to the different ED categories and the populations studied. Some studies examined prevalence
rates of AN and BN only, while others examined the prevalence of insulin omission only, and some reported the prevalence of a combination of all ED together. Some studies reported the prevalence of full-threshold diagnoses of ED, whereas others also included subclinical ED. Reported prevalence rates differ also according to characteristics of the study populations, such as age range, and whether males were included or only females.
Prevalence of AN: A meta-analysis of controlled studies that defined ED in females according to DSM Ⅲ-R or DSM Ⅳ criteria reported the prevalence of AN in T1DM subjects was not significantly different from that of controls[9].
Prevalence of BN: This same meta-analysis showed a significantly higher prevalence of BN among the females with T1DM than those without diabetes[9].
Overall prevalence of AN, BN and EDNOS: In a recently published meta-analysis[10] of six studies of adolescents[11-16], 7.0% of the 825 individuals with T1DM had ED, compared with 2.8% of the 2282 individuals without T1DM.
Prevalence of DEB: In a meta-analysis[10] of five studies[11,15-18], a higher proportion of adolescents with T1DM than without T1DM were classified as having DEB (39.3% vs 32.5%). The prevalence rate of DEB increased significantly with weight and age, from 7.2% in the underweight group to 32.7% in the obese group, and from 8.1% in the youngest age-group (11-13 years) to 38.1% in the oldest age-group (17-19 years)[19].
Prevalence of insulin omission in T1DM: Insulin omission is a unique purging behavior available to individuals with T1DM. As presented in Table 1, insulin omission for weight loss is identified mainly in females; its prevalence increases with age, affecting up to 40% of young adult females with T1DM.
PREDISPOSING FACTORS FOR DEVELOPING EDWe suggest a three level model to describe the development of disturbed eating in adolescents with T1DM. This model comprises a hierarchically arranged continuum of potential effects on health problems (Figure 1). The first circle in the model, the premorbid status, includes a tendency to overweight, and factors relating to personality and family characteristics and dynamics. The second circle comprises factors arising at diagnosis of diabetes, such as the age of diabetes onset and satisfaction from weight loss. The third circle comprises factors associated with the chronic management of diabetes, such as recurrent hypoglycemic episodes,
Pinhas-Hamiel O et al . Eating disorders in type 1 diabetes
518 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

strict insulin treatment and carbohydrate counting.
FIRST CIRCLE - PREMORBID FACTORSIncreased body weight and a tendency to gain weight It has been suggested that the single question “Have you ever been overweight?” may be sufficient to screen for those with T1DM who are at a high risk for disordered eating attitudes/behaviors[20]. Indeed, adolescent girls with T1DM who reported ever being overweight endorsed more disordered eating attitudes and behaviors[21]. In addition, a higher body mass index percentile among teenagers and adults with T1DM, particularly among females, one to two years prior to the onset of ED, was found to be associated with disordered eating[22]. Furthermore, youth at risk for disordered eating have been shown to have poorer
diet quality, including a higher intake of total fat and saturated fat[23].
Low self-esteem and body dissatisfaction Higher levels of weight dissatisfaction tended to be associated with unhealthy weight control/disordered eating among adolescent females[24]. Low self-esteem predicts eating disturbances in the general population. Lower self-esteem was associated with disturbed eating behavior in girls with T1DM[25]. Of note, low self-esteem and body dissatisfaction could also arise along the course of the disease. In a longitudinal study the development of DEB in girls with T1DM, was predicted by lower self-esteem related to physical appearance, and lower global self-esteem[26].
Personality characteristicsPerfectionism has been associated with attitudinal aspects of eating disorders such as weight preoccupation. Borderline personality characteristics were related to insulin omission[27]. Adolescent girls with T1DM and an ED showed deficits in self-regulation and narcissistic gain from illness[28], and tended to blame themselves for the situation[29].
Comparing adolescents with ED and adolescents with T1DM and ED, those with T1DM were less pathologically compromised, scored lower on several affective areas and had a lower prevalence of depression and anxiety[30].
Family characteristics and dynamics Several factors involving family characteristics and dynamics are associated with an increased risk for ED. Firstly, in many families of adolescent girls with T1DM,
519 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 1 Prevalence rates of insulin omission according to age range and gender
Prevalence of insulin omission (%)
Age range (yr)
Number and sex
Ref.
2 9-13 101 females Colton et al[12]
26.24.5
11-19 390 females380 males
Wisting et al[19]
11 12-19 361 females Jones et al[11]
14 12–19 356 females Rodin et al[61]
101
12-21 70 females73 males
Neumark-Sztainer et al[24]
34 16-22 91 females Rydall et al[46]
40 18-30 59 females Stancin et al[74]
110
12-56 141 females58 males
Philippi et al[75]
Pinhas-Hamiel O et al . Eating disorders in type 1 diabetes
Increased BMI
Personality factors
Age of T1DM onset
Family factors
Weight loss at diagnosis
Intensive insulin treatment
Recurrent hypoglycemia
Insulinomission
Weight gain
Carb/calories counting
Strict eating
Bingeeating
Depression
Ⅲ
Ⅱ
Ⅰ
Eatingdisorders
Figure 1 Three level models describing the development of eating disorders in adolescents with type 1 diabetes mellitus. The outer circle delineates the premorbid status. The middle circle delineates weight changes at diagnosis and with initiation of treatment. The inner circle includes factors associated with the management of T1DM that predispose to eating disorders. Dashed arrows imply that the specific factor extends on all levels. T1DM: Type 1 diabetes mellitus; BMI: Body mass index.
Weight gain atinitiation of insulin

proposed by Goebel-Fabbri et al[38,39] suggests that the increased weight gain causes negative feelings. Fear of further weight gain can lead to insulin restriction for caloric purging or weight regulation[40].
Dietary restraintDiabetes management imposes dietary restraint. This may lead to the yearning and craving of “forbidden foods”, and result in binging, without administration of the appropriate insulin dose[41].
Hypoglycemic episodesThe delicate balance between dietary regimen, insulin and exercise that is integral to T1DM management can result in recurrent hypoglycemic episodes. Hypoglycemia is accompanied by intense hunger and by eating sweetened foods and drinks that are normally forbidden. Patients may subsequently feel guilty about consuming these foods and restrict eating, which may result in yet another hypoglycemic episode. This vicious cycle of dietary restriction, over-eating and guilt is similar to that experienced by individuals with bulimia[42].
Chronic disease and depression The risk of significant depressive symptoms in individuals with T1DM has been assessed as about double that of the general population[43]. Depression may increase the susceptibility for developing disturbed eating behavior. Indeed, among girls with T1DM, those diagnosed with depression scored higher on the Eating Disorder Examination than did those without depression; 75% and 45%, respectively[44]. Furthermore, anxiety disorders and eating disorders co-occur in the general population, and 21% of children and adolescents with T1DM were reported to screen positively for anxiety[45].
CLINICAL SIGNS OF ED AMONG INDIVIDUALS WITH T1DMThe diagnosis of subclinical or clinical eating disorders in individuals with T1DM is not easily established. Due to the frequent concealment and denial of eating disorder behaviors, a high index of suspicion is needed. In addition to the predisposing characteristics described above, a number of clinical signs should alert health care providers to the possibility of ED.
Poor glycemic control Among females with T1DM, mean HbA1c levels were shown to be significantly higher in those with disturbed eating than among those with non-disturbed eating[46]. HbA1c level of patients who confessed intentional under-dosing of insulin was 9.0% ± 1.6% compared with 7.8% ± 1.2% of compliant patien[47]. Similarly, in a nationwide population-based study, conducted in Norway, a significantly higher HbA1c was documented in patients reporting insulin restriction (9.0% ± 1.7%) than in nonrestrictors (8.3% ± 1.2%)[19] In a meta-
interactions within the family center around food and weight. The prevalence of ED was found to be higher in families in which parents tended to make negative comments about eating or weight[31]. Secondly, mothers who engaged in dieting and binge-eating themselves were more likely to have daughters with disturbed eating behavior. Maternal weight and shape concerns and impaired mother-daughter relationships significantly predicted eating disturbances in girls with T1DM, accounting for 57% of the variance[32]. Finally, family dysfunctioning has been identified as an important risk factor for eating disorders among the general population. Specifically, among girls with T1DM, less support from, poorer communication with, and less trust in their relationships with their parents, were reported among those with eating disturbances than among those without[33]. Families of girls with ED more often reported having infrequent family meals than did families without ED[34].
SECOND CIRCLE - FACTORS ASSOCIATED WITH DISEASE ONSETAge of diabetes onsetThe age of onset of T1DM that is most closely related to the subsequent development of a severe eating disorder such as AN and BN was studied in 53 women with ED compared with 49 with T1DM with T1DM who had no eating disorder - related problem[35]. It was demonstrated that for diabetes onset age between 7 to 18 years, the density of the “eating disorder” group was higher, but for the younger and older onset ages the densities were lower. Thus the development of T1DM in preadolescence or adolescence seems to place girls at risk for the subsequent development of AN or BN. Similarly, frequent insulin reduction or omission was reported in type 1 diabetic patients with later disease onset (mean age at onset between 8 and about 17 years)[36]. It was suggested that later age at diabetes onset, in particular during pubertal age with hormonal changes and gain in weight and fat mass may be associated with a greater risk for ED especially in female patients.
Weight issuesThe cycle of weight loss at disease onset and subsequent weight gain with the initiation of insulin treatment is a risk factor for the development of ED in susceptible patients[11].
THIRD CIRCLE - FACTORS ASSOCIATED WITH THE CHRONIC MANAGEMENT OF DIABETESIntensive insulin treatmentIntensive insulin treatment conveys an increased risk of weight gain[37]. A model of disordered eating and T1DM
520 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Pinhas-Hamiel O et al . Eating disorders in type 1 diabetes

analysis, eating problems, including both DEB and ED, were found to be associated with poorer glycemic control[10]. Using a computer model of data mining, we recently showed that adolescent females with intentional insulin omission were discriminated by HbA1c > 9.2% and by more than 20% of HbA1c measurements above the 90th percentile[48].
Recurrent episodes of diabetic ketoacidosis Intentional insulin omission has long been recognized as a cause of recurrent episodes of recurrent episodes of diabetic ketoacidosis (DKA) in adolescents with T1DM[49]. Thus, any recurrent episode of DKA in established diabetes should arouse suspicion[50].
Recurrent hypoglycemic episodes Recurrent hypoglycemic episodes may occur among individuals with T1DM and ED, mainly among those participating in binge eating and self-induced vomiting. Deliberately inducing hypoglycemia to justify eating sweets and high carbohydrate meals were described in individuals with T1DM. Eighteen percent of a cohort of males and females with T1DM, aged 10-22 years, reported intentional overdosing of insulin[47]. The most common reason (49%) stated was the desire for uncontrolled binge eating, followed by self-destructive behavior in stressful situations.
Other characteristics and signs of ED Other characteristic and clinical signs of ED are: frequently missed medical appointments, refusal to be weighed, preoccupation with appearance, a tendency to vegetarianism, and the calculating of caloric values and weighing of foods. Apparently, due to the availability of insulin omission as a weight loss strategy, females with diabetes are less likely to use other unhealthy weight control behaviors like vomiting, laxatives or diuretics,
and skipping meals or fasting, compared to their peers without diabetes[51].
DIAGNOSISSeveral questionnaires were developed specifically for adolescents with T1DM. The revised Diabetes Eating Problem Survey (DEPS-R) is a 16-item diabetes-specific self-report measure of disordered eating that was designed specifically to identify patients with T1DM who are at risk for early intervention for disordered eating behaviors[7]. The mSCOFF is a simple 5 item screening tool, with demonstrated reliability and validity, which can easily be used during a follow-up visit[52]. Using data mining methods, a clinical prediction model that provides a decision support system for the detection of intentional insulin omission for weight loss in adolescent females with T1DM was developed[48]. The model is based on identifying a pattern of HbA1c levels indicative of intentional insulin omission. After a period of apparently stable HbA1c levels, the onset of insulin omission is characterized by both high HbA1c levels and wide fluctuations between clinical visits. A typical example of the course of HbA1c levels in a patient who developed ED is depicted in Figure 2.
MORBIDITY AND MORTALITYED result in poor metabolic control and cause short and long-term complications.
Short term complicationsInsulin omission is associated with recurrent events of DKA, and disturbed eating behavior is associated with recurrent episodes of severe hypoglycemia[53]. Data from the diabetes patienten verlaufsdokumentation study, including 52215 T1DM males and females aged 8 to 30 years, revealed significantly higher rates of severe hypoglycemic episodes in adolescents with AN, BN or EDNOS, than with no ED: 12.1, 18.0, 12.9 and 5.7, respectively[54]. Moreover, rates of hypoglycemia with coma, and DKA with hospitalization were higher, and the duration of hospital stay longer, among those with eating disorders[54].
Long term complications Long-term complications have been shown to be markedly increased among individuals with both T1DM and ED[54]. Data from the DPV study revealed that both hypertension and dyslipidemia were more common in persons with T1DM and either EDNOS or BN than in those with T1DM without ED[54].
Of T1DM patients who developed serious micro-vascular complications, 21% had a probable clinical ED, 47% had a history of DEB, and 48% had a history of insulin omission. The development of two or more serious complications was associated with the presence of a probable clinical eating disorder[55].
Different ED are associated with varying prevalence
521 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
HbA
1c (
%)
Age (yr)2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
10.5
10.0
9.5
9.0
8.5
8.0
7.5
7.0
6.5
6.0
5.5
5.0
90
75
50
25
10
Figure 2 Typical example of the course of HbA1c levels in a patient, with intentional insulin omission for weight loss, plotted on the HbA1c percentile reference curves. HbA1c levels were around the 10th percentile from age 10 to 14 years. From age 18 the pattern changed and was characterized by sharp increases to levels above the 90th percentile, sharp decreases and multiple measurements above the 90th percentile.
Pinhas-Hamiel O et al . Eating disorders in type 1 diabetes

rates of morbidities. For example, data from the DPV study revealed a 2.5-fold higher risk for retinopathy in T1DM patients with BN than in those without ED. Patients with EDNOS also had a higher risk but without statistical significance, whereas those with AN had no increased risk for retinopathy[54].
The duration of ED may also affect diabetes complications. The duration of severe insulin omission was the factor most closely associated with the development of retinopathy and nephropathy in T1DM females[56].
As seen in Table 2, higher rates of retinopathy[46] and nephropathy[57] were documented in young TIDM patients with ED than in those without ED.
MortalitySeveral studies documented increased relative risk of death among patients with ED. Mortality rates per 1000 person years were reported as 2.2 in girls with T1DM, 7.3 in girls with ED and 34.6 in girls with both T1DM and an ED[58]. During an 11-year period, self-reported insulin restriction at baseline increased the relative risk of death by 3.2 times in women, mean age 45 years, mean diabetes duration 28 years; those with ED were younger when they died (aged 44 vs 58 years, P < 0.01)[57]. In a 12-year follow-up of a cohort of 14 women with T1DM and ED (12 with AN), 5 died (36%)[59]. The median age of the cohort was 37 years (range 25-46), with a median duration of diabetes of 26 years (14-33). The age of death of the 5 patients was 30 years (25-42).
PREVENTIONPromoting the understanding and awareness among healthcare providers of factors involved in the deve-lopment of ED in young vulnerable individuals with T1DM may help prevent these disorders[60]. These factors include the weight gain with the initiation of insulin treatment[11], the dietary restraint necessitated by diabetes management, and recurrent hypoglycemic episodes. Diabetes treatment should therefore afford flexibility where possible, and non-depriving approaches to eating[61]. As current clinical practice includes a focus on measuring body weight, health care professionals should be sensitive to preoccupation with body weight. Interventions aimed at increasing self-esteem and body acceptance, as well as family-based interventions aimed
to improve family management of obesity and diabetes, may also help to diminish the risk of developing ED in individuals with diabetes[62].
Girls aged between 10 and 12 with a diagnosis of T1DM for at least 1 year, attended two group sessions, each of 4-h duration[63]. The content of the sessions targeted perfectionism, media literacy, self-esteem. The program was found to have impact on self-efficacy relating to diabetes management. As lower T1DM regimen adherence is associated with lower self-efficacy in adolescents, it was concluded that a brief interactive program had a favorable impact on protective factors for disordered eating.
Among college-age women at highest risk for an eating disorder an internet-based intervention, has successfully reduced weight/shape concerns and prevented eating disorders[64]. Specifically guided discussion group resulted in reduction in weight/shape concern. We did not find similar reports among adolescent girls with T1DM, however it is possible that online interventions may be successfully used to reduce eating disorder risk factors and preventing eating disorders in this group.
TREATMENTTreatment of ED in patients with T1DM is challenging, especially since the clinical improvement achieved with insulin treatment is associated with weight gain. Compliance to the therapeutic regimen may therefore be poor.
Treatment should mirror the three circle model presented above, and address the factors predisposing to the development of ED (Figure 3).
Treating the first circle - premorbid variablesIncreased body weight and a tendency to gain weight should be addressed by prudent dietary management: Since a tendency to gain weight was found to be associated with ED[22], particular attention should be given to the constellation of excess weight and preoccupation with food.
Low self-esteem, body dissatisfaction and personality variables should be addressed by individual psychological treatment or group therapy: The concurrence of eating behavior disorder and mood disorder in patients with T1DM supports the importance of treating depression as a means of
522 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 2 Prevalence rates of retinopathy and nephropathy among type 1 diabetes mellitus patients with eating disorders
Complication Average T1DM duration (yr)
T1DM patients with severe ED (%)
T1DM patients with moderate ED (%)
T1DM patients without ED (%)
Retinopathy[46] 11 ± 4 86 42 24Nephropathy[46] 11 ± 4 43 20 18Nephropathy[57] 28 25 10Foot problems[57] 28 25 12
ED: Eating disorders; T1DM: Type 1 diabetes mellitus.
Pinhas-Hamiel O et al . Eating disorders in type 1 diabetes

treating ED. If depression and anxiety are suspected, a psychiatric consultation is needed. Psychiatrists have a distinct role in the diabetes care of children under their care, specifically in working through issues associated with the burden of the disease[65]. The impact of anti-depressant and anti-anxiety medications on recovery from eating disorders in adolescents with T1DM has not been studied systematically.
The effect of psychoeducation on adolescents with T1DM and ED is controversial. A psychoeducation approach with six weekly group sessions was associated with reductions in dieting, in body dissatisfaction and with preoccupation with thinness and eating; the reduction in disturbed eating behaviour was maintained for at least 6 mo[66]. In another study, a six week intervention was as effective as a wait list control group[67].
Group cognitive-behavioral therapy intervention has been shown to improve glycemic control, well-being and diabetes related stress among poorly controlled adult T1DM patients[68], and among patients with T1DM and depression[69]; however, it was not studied in patients with insulin omission.
As family dynamics may influence the development of ED[34], family education is an important component of the treatment. However, we could not find data on the impact of family intervention as a treatment modality for adolescents with T1DM and ED.
Treating the second circle - factors associated with disease onsetCurrent T1DM management involves carbohydrate counting, which may lead to excessive preoccupation with food and diets. The cycle of weight loss at disease
onset, and subsequent weight gain with the initiation of insulin treatment, is a risk factor for the development of ED in susceptible patients[11]. Dietary treatment should focus on healthy choices and low calorie alternatives with a high satiety index.
Treating the third circle - factors associated with the chronic management of diabetesIntensive insulin treatment, dietary restraint, and hypoglycemic episodes should be addressed by the diabetes management team: As good metabolic control is associated with weight gain, changes in target blood glucose levels should be gradual. Setting higher than standard target blood glucose ranges (preprandial 120 to 150 mg/dL and postprandial < 200 mg/dL) may yield more benefit in the long run. In contrast, achieving excellent control may result in marked weight gain. Moreover, since low glucose target levels are associated with an increased risk of recurrent hypoglycemic episodes, which may result in additional increased calorie intake, setting higher target levels may be a better initial objective. In girls who were skipping insulin dosing at almost every meal, starting with adequate insulin injection at a single meal is sometimes helpful as a first step. Caregivers should be aware that patients often switch from one disturbed behavior to another, ie from insulin omission to restrictive eating or binge eating; thus, a prescribed diet should be flexible.
Treatment with an insulin pump was postulated as a means of enabling better physiological insulin delivery to adolescents with T1DM than achieved with multiple daily injections. Patients with insulin pumps require less insulin and thus gain less weight. Furthermore, the occurrence of hypoglycemic episodes is decreased, and
523 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Individual psychological treatment
Family treatment
Group treatment
Dietary guidance for healthy eating
Realistic blood glucose targets
Flexible dietary regimen
Prevention of hypoglycemia
Treating depression and anxiety
ⅢⅡ
Ⅰ
Hospitalization
Figure 3 Three level models describing treatment modalities of eating disorders in adolescents with type 1 diabetes mellitus. The outer circle addresses treatment for premorbid variables. The middle circle delineates. The inner circle includes factors associated with the management of T1DM that predispose to eating disorders. T1DM: Type 1 diabetes mellitus.
Pinhas-Hamiel O et al . Eating disorders in type 1 diabetes

thus the need to consume extra calories. A recently published multicenter study demonstrated a decrease in disturbed eating behaviors, as assessed by the DEPS-R, in youth with T1DM, 6 mo after pump initiation[70]. Moreover, among adolescent girls with T1DM and ED, the mean HbA1c level was significantly lower in those who were treated by insulin pumps than in those who were treated with multiple daily injections (9.07% ± 1.33% vs 10.40% ± 2.01%; P = 0.04)[71].
Failure of outpatient treatment, the presence of a severe psychopathological state and poor glycemiccontrol are key elements in the decision for hospi-talization. Information is limited regarding the long-term outcomes of hospitalized adolescents with ED. Significantly lower levels of HbA1c and lower psychological test scores related to eating disorder psychopathology, depressiveness and anxiety were reported for 9 patients who were hospitalized, compared to 10 who were not[72]. At 3-year follow-up, 78% of the hospitalized patients in that study no longer fulfilled any criteria for clinical or subclinical ED.
Thus, treatment of ED in individuals with T1DM involves a complex interplay of psychosocial, dietary and medical aspects and requires a multidisciplinary team. It is important to realize that adolescents with T1DM and ED tend to have lower motivation to change their eating habits[73]. Furthermore, they are characterized by giving up easily when confronted with frustration and with low levels of accomplishment. These personality characteristics may explain the high dropout rate from therapy (79%) among patients with T1DM and ED[73].
CONCLUSIONDeliberate insulin omission as a weight loss strategy is recognized as either an inappropriate compensatory feature of bulimia nervosa or as a purging disorder, a component of OSFED. About one-third of individuals with T1DM intentionally omit insulin. Diagnosis of eating disorders in individuals with T1DM is difficult, since eating disorder behaviors are often well hidden. Weight loss that is related to deteriorated glycemic control and recurrent DKA should raise suspicion. Early diagnosis is essential, as the combination of eating disorders and diabetes is associated with increased morbidity and mortality.
ACKNOWLEDGMENTSWe thank Ms. Cindy Cohen for her editorial assistance.
REFERENCES1 Treasure J, Claudino AM, Zucker N. Eating disorders. Lancet
2010; 375: 583-593 [PMID: 19931176 DOI: 10.1016/S0140-6736(09)61748-7]
2 Zipfel S, Löwe B, Reas DL, Deter HC, Herzog W. Long-term prognosis in anorexia nervosa: lessons from a 21-year follow-up study. Lancet 2000; 355: 721-722 [PMID: 10703806 DOI: 10.1016/S0140-6736(99)05363-5]
3 American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. 5th ed. Arlington, VA: American Psychiatric Association, 2013
4 Wildes JE, Marcus MD. Incorporating dimensions into the classification of eating disorders: three models and their implications for research and clinical practice. Int J Eat Disord 2013; 46: 396-403 [PMID: 23658078 DOI: 10.1002/eat.22091]
5 Lavender JM, Crosby RD, Wonderlich SA. Dimensions in the eating disorders: past, present, and future. Commentary on Wildes and Marcus: Incorporating dimensions into the classification of eating disorders. Int J Eat Disord 2013; 46: 404-407 [PMID: 23658079 DOI: 10.1002/eat.22121]
6 Pike KM. Classification, culture, and complexity: a global look at the diagnosis of eating disorders: Commentary on Wildes and Marcus: Incorporating dimensions into the classification of eating disorders. Int J Eat Disord 2013; 46: 408-411 [PMID: 23658080 DOI: 10.1002/eat.22122]
7 Markowitz JT, Butler DA, Volkening LK, Antisdel JE, Anderson BJ, Laffel LM. Brief screening tool for disordered eating in diabetes: internal consistency and external validity in a contemporary sample of pediatric patients with type 1 diabetes. Diabetes Care 2010; 33: 495-500 [PMID: 20032278 DOI: 10.2337/dc09-1890]
8 Young-Hyman DL, Davis CL. Disordered eating behavior in individuals with diabetes: importance of context, evaluation, and classification. Diabetes Care 2010; 33: 683-689 [PMID: 20190297 DOI: 10.2337/dc08-1077]
9 Mannucci E, Rotella F, Ricca V, Moretti S, Placidi GF, Rotella CM. Eating disorders in patients with type 1 diabetes: a meta-analysis. J Endocrinol Invest 2005; 28: 417-419 [PMID: 16075924 DOI: 10.1007/BF03347221]
10 Young V, Eiser C, Johnson B, Brierley S, Epton T, Elliott J, Heller S. Eating problems in adolescents with Type 1 diabetes: a systematic review with meta-analysis. Diabet Med 2013; 30: 189-198 [PMID: 22913589 DOI: 10.1111/j.1464-5491.2012.03771.x]
11 Jones JM, Lawson ML, Daneman D, Olmsted MP, Rodin G. Eating disorders in adolescent females with and without type 1 diabetes: cross sectional study. BMJ 2000; 320: 1563-1566 [PMID: 10845962 DOI: 10.1136/bmj.320.7249.1563]
12 Colton P, Olmsted M, Daneman D, Rydall A, Rodin G. Disturbed eating behavior and eating disorders in preteen and early teenage girls with type 1 diabetes: a case-controlled study. Diabetes Care 2004; 27: 1654-1659 [PMID: 15220242 DOI: 10.2337/diacare.27.7.1654]
13 Smith FM, Latchford GJ, Hall RM, Dickson RA. Do chronic medical conditions increase the risk of eating disorder? A cross-sectional investigation of eating pathology in adolescent females with scoliosis and diabetes. J Adolesc Health 2008; 42: 58-63 [PMID: 18155031 DOI: 10.1016/j.jadohealth.2007.08.008]
14 Svensson M, Engström I, Aman J. Higher drive for thinness in adolescent males with insulin-dependent diabetes mellitus compared with healthy controls. Acta Paediatr 2003; 92: 114-117 [PMID: 12650311 DOI: 10.1111/j.1651-2227.2003.tb00480.x]
15 Engström I, Kroon M, Arvidsson CG, Segnestam K, Snellman K, Aman J. Eating disorders in adolescent girls with insulin-dependent diabetes mellitus: a population-based case-control study. Acta Paediatr 1999; 88: 175-180 [PMID: 10102151 DOI: 10.1111/j.1651-2227.1999.tb01078.x]
16 García-Reyna NI, Gussinyer S, Raich RM, Gussinyer M, Tomàs J, Carrascosa A. [Eating disorders in young adolescents with type 1 diabetes]. Med Clin (Barc) 2004; 122: 690-692 [PMID: 15171828 DOI: 10.1016/S0025-7753(04)74357-2]
17 Pinar R. Disordered eating behaviors among Turkish adolescents with and without Type 1 diabetes. J Pediatr Nurs 2005; 20: 383-388 [PMID: 16182098 DOI: 10.1016/j.pedn.2005.07.001]
18 Alice Hsu YY, Chen BH, Huang MC, Lin SJ, Lin MF. Disturbed eating behaviors in Taiwanese adolescents with type 1 diabetes mellitus: a comparative study. Pediatr Diabetes 2009; 10: 74-81 [PMID: 18680544 DOI: 10.1111/j.1399-5448.2008.00422.x]
19 Wisting L, Frøisland DH, Skrivarhaug T, Dahl-Jørgensen K, Rø O. Disturbed eating behavior and omission of insulin in adolescents
524 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Pinhas-Hamiel O et al . Eating disorders in type 1 diabetes

receiving intensified insulin treatment: a nationwide population-based study. Diabetes Care 2013; 36: 3382-3387 [PMID: 23963896 DOI: 10.2337/dc13-0431]
20 Markowitz JT, Lowe MR, Volkening LK, Laffel LM. Self-reported history of overweight and its relationship to disordered eating in adolescent girls with Type 1 diabetes. Diabet Med 2009; 26: 1165-1171 [PMID: 19929996 DOI: 10.1111/j.1464-5491.2009.02844.x]
21 Domargård A, Särnblad S, Kroon M, Karlsson I, Skeppner G, Aman J. Increased prevalence of overweight in adolescent girls with type 1 diabetes mellitus. Acta Paediatr 1999; 88: 1223-1228 [PMID: 10591423 DOI: 10.1111/j.1651-2227.1999.tb01021.x]
22 Hyrich KL, Watson KD, Lunt M, Symmons DP. Changes in disease characteristics and response rates among patients in the United Kingdom starting anti-tumour necrosis factor therapy for rheumatoid arthritis between 2001 and 2008. Rheumatology (Oxford) 2011; 50: 117-123 [PMID: 20671021 DOI: 10.1093/rheumatology/keq209]
23 Tse J, Nansel TR, Haynie DL, Mehta SN, Laffel LM. Disordered eating behaviors are associated with poorer diet quality in adolescents with type 1 diabetes. J Acad Nutr Diet 2012; 112: 1810-1814 [PMID: 23102180 DOI: 10.1016/j.jand.2012.06.359]
24 Neumark-Sztainer D, Patterson J, Mellin A, Ackard DM, Utter J, Story M, Sockalosky J. Weight control practices and disordered eating behaviors among adolescent females and males with type 1 diabetes: associations with sociodemographics, weight concerns, familial factors, and metabolic outcomes. Diabetes Care 2002; 25: 1289-1296 [PMID: 12145223]
25 Colton PA, Olmsted MP, Daneman D, Rydall AC, Rodin GM. Natural history and predictors of disturbed eating behaviour in girls with Type 1 diabetes. Diabet Med 2007; 24: 424-429 [PMID: 17298588 DOI: 10.1111/j.1464-5491.2007.02099.x]
26 Olmsted MP, Colton PA, Daneman D, Rydall AC, Rodin GM. Prediction of the onset of disturbed eating behavior in adolescent girls with type 1 diabetes. Diabetes Care 2008; 31: 1978-1982 [PMID: 18628570 DOI: 10.2337/dc08-0333]
27 Pollock-BarZiv SM, Davis C. Personality factors and disordered eating in young women with type 1 diabetes mellitus. Psychosomatics 2005; 46: 11-18 [PMID: 15765816 DOI: 10.1176/appi.psy.46.1.11]
28 Grylli V, Wagner G, Berger G, Sinnreich U, Schober E, Karwautz A. Characteristics of self-regulation in adolescent girls with type 1 diabetes with and without eating disorders: a cross-sectional study. Psychol Psychother 2010; 83: 289-301 [PMID: 20188019 DOI: 10.1348/147608309X481180]
29 Grylli V, Wagner G, Hafferl-Gattermayer A, Schober E, Karwautz A. Disturbed eating attitudes, coping styles, and subjective quality of life in adolescents with Type 1 diabetes. J Psychosom Res 2005; 59: 65-72 [PMID: 16186000 DOI: 10.1016/j.jpsychores.2005.02.010]
30 Powers MA, Richter S, Ackard D, Gerken S, Meier M, Criego A. Characteristics of persons with an eating disorder and type 1 diabetes and psychological comparisons with persons with an eating disorder and no diabetes. Int J Eat Disord 2012; 45: 252-256 [PMID: 21495056 DOI: 10.1002/eat.20928]
31 Maharaj S, Rodin G, Connolly J, Olmsted M, Daneman D. Eating problems and the observed quality of mother-daughter interactions among girls with type 1 diabetes. J Consult Clin Psychol 2001; 69: 950-958 [PMID: 11777122]
32 Maharaj SI, Rodin GM, Olmsted MP, Connolly JA, Daneman D. Eating disturbances in girls with diabetes: the contribution of adolescent self-concept, maternal weight and shape concerns and mother-daughter relationships. Psychol Med 2003; 33: 525-539 [PMID: 12701673]
33 Maharaj SI, Rodin GM, Olmsted MP, Daneman D. Eating disturbances, diabetes and the family: an empirical study. J Psychosom Res 1998; 44: 479-490 [PMID: 9587890]
34 Mellin AE, Neumark-Sztainer D, Patterson J, Sockalosky J. Unhealthy weight management behavior among adolescent girls with type 1 diabetes mellitus: the role of familial eating patterns and weight-related concerns. J Adolesc Health 2004; 35: 278-289
[PMID: 15450541 DOI: 10.1016/j.jadohealth.2003.10.006]35 Takii M, Uchigata Y, Kishimoto J, Morita C, Hata T, Nozaki
T, Kawai K, Iwamoto Y, Sudo N, Kubo C. The relationship between the age of onset of type 1 diabetes and the subsequent development of a severe eating disorder by female patients. Pediatr Diabetes 2011; 12: 396-401 [PMID: 20723101 DOI: 10.1111/j.1399-5448.2010.00708.x]
36 Baechle C, Castillo K, Straßburger K, Stahl-Pehe A, Meissner T, Holl RW, Giani G, Rosenbauer J. Is disordered eating behavior more prevalent in adolescents with early-onset type 1 diabetes than in their representative peers? Int J Eat Disord 2014; 47: 342-352 [PMID: 24375553 DOI: 10.1002/eat.22238]
37 McErlane F, Foster HE, Davies R, Lunt M, Watson KD, Symmons DP, Hyrich KL. Biologic treatment response among adults with juvenile idiopathic arthritis: results from the British Society for Rheumatology Biologics Register. Rheumatology (Oxford) 2013; 52: 1905-1913 [PMID: 23873820 DOI: 10.1093/rheumatology/ket248]
38 Goebel-Fabbri AE, Fikkan J, Connell A, Vangsness L, Anderson BJ. Identification and treatment of eating disorders in women with type 1 diabetes mellitus. Treat Endocrinol 2002; 1: 155-162 [PMID: 15799208 DOI: 10.2165/00024677-200201030-00003]
39 Goebel-Fabbri AE. Disturbed eating behaviors and eating disorders in type 1 diabetes: clinical significance and treatment recommendations. Curr Diab Rep 2009; 9: 133-139 [PMID: 19323958 DOI: 10.1007/s11892-009-0023-8]
40 Thompson CJ, Cummings JF, Chalmers J, Gould C, Newton RW. How have patients reacted to the implications of the DCCT? Diabetes Care 1996; 19: 876-879 [PMID: 8842607 DOI: 10.2337/diacare.19.8.876]
41 Rodin GM, Daneman D. Eating disorders and IDDM. A problematic association. Diabetes Care 1992; 15: 1402-1412 [PMID: 1425109 DOI: 10.2337/diacare.15.10.1402]
42 Verrotti A, Catino M, De Luca FA, Morgese G, Chiarelli F. Eating disorders in adolescents with type 1 diabetes mellitus. Acta Diabetol 1999; 36: 21-25 [PMID: 10436248 DOI: 10.1007/s005920050140]
43 Dantzer C, Swendsen J, Maurice-Tison S, Salamon R. Anxiety and depression in juvenile diabetes: a critical review. Clin Psychol Rev 2003; 23: 787-800 [PMID: 14529698 DOI: 10.1016/S0272-7358(03)00069-2]
44 Colton PA, Olmsted MP, Daneman D, Rodin GM. Depression, disturbed eating behavior, and metabolic control in teenage girls with type 1 diabetes. Pediatr Diabetes 2013; 14: 372-376 [PMID: 23418901 DOI: 10.1111/pedi.12016]
45 Bernstein CM, Stockwell MS, Gallagher MP, Rosenthal SL, Soren K. Mental health issues in adolescents and young adults with type 1 diabetes: prevalence and impact on glycemic control. Clin Pediatr (Phila) 2013; 52: 10-15 [PMID: 22988007 DOI: 10.1177/0009922812459950]
46 Rydall AC, Rodin GM, Olmsted MP, Devenyi RG, Daneman D. Disordered eating behavior and microvascular complications in young women with insulin-dependent diabetes mellitus. N Engl J Med 1997; 336: 1849-1854 [PMID: 9197212 DOI: 10.1056/NEJM199706263362601]
47 Schober E , Wagner G, Berger G, Gerber D, Mengl M, Sonnenstatter S, Barrientos I, Rami B, Karwautz A, Fritsch M. Prevalence of intentional under- and overdosing of insulin in children and adolescents with type 1 diabetes. Pediatr Diabetes 2011; 12: 627-631 [PMID: 21435136 DOI: 10.1111/j.1399-5448.2011.00759.x]
48 Pinhas-Hamiel O, Hamiel U, Greenfield Y, Boyko V, Graph-Barel C, Rachmiel M, Lerner-Geva L, Reichman B. Detecting intentional insulin omission for weight loss in girls with type 1 diabetes mellitus. Int J Eat Disord 2013; 46: 819-825 [PMID: 23674378 DOI: 10.1002/eat.22138]
49 Golden MP, Herrold AJ, Orr DP. An approach to prevention of recurrent diabetic ketoacidosis in the pediatric population. J Pediatr 1985; 107: 195-200 [PMID: 3926977 DOI: 10.1016/S0022-3476(85)80124-4]
50 Goebel-Fabbri AE. Diabetes and eating disorders. J Diabetes Sci
525 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Pinhas-Hamiel O et al . Eating disorders in type 1 diabetes

Technol 2008; 2: 530-532 [PMID: 19885221 DOI: 10.1177/193229680800200326]
51 Ackard DM, Vik N, Neumark-Sztainer D, Schmitz KH, Hannan P, Jacobs DR. Disordered eating and body dissatisfaction in adolescents with type 1 diabetes and a population-based comparison sample: comparative prevalence and clinical implications. Pediatr Diabetes 2008; 9: 312-319 [PMID: 18466215 DOI: 10.1111/j.1399-5448.2008.00392.x]
52 Zuijdwijk CS, Pardy SA, Dowden JJ, Dominic AM, Bridger T, Newhook LA. The mSCOFF for screening disordered eating in pediatric type 1 diabetes. Diabetes Care 2014; 37: e26-e27 [PMID: 24459158 DOI: 10.2337/dc13-1637]
53 Weinert LS, Scheffel RS, Severo MD, Cioffi AP, Teló GH, Boschi A, Schaan BD. Precipitating factors of diabetic ketoacidosis at a public hospital in a middle-income country. Diabetes Res Clin Pract 2012; 96: 29-34 [PMID: 22153415 DOI: 10.1016/j.diabres.2011.11.006]
54 Scheuing N, Bartus B, Berger G, Haberland H, Icks A, Knauth B, Nellen-Hellmuth N, Rosenbauer J, Teufel M, Holl RW. Clinical characteristics and outcome of 467 patients with a clinically recognized eating disorder identified among 52,215 patients with type 1 diabetes: a multicenter german/austrian study. Diabetes Care 2014; 37: 1581-1589 [PMID: 24623022 DOI: 10.2337/dc13-2156]
55 McErlane F, Beresford MW, Baildam EM, Thomson W, Hyrich KL. Recent developments in disease activity indices and outcome measures for juvenile idiopathic arthritis. Rheumatology (Oxford) 2013; 52: 1941-1951 [PMID: 23630368 DOI: 10.1093/rheumatology/ket150]
56 Takii M, Uchigata Y, Tokunaga S, Amemiya N, Kinukawa N, Nozaki T, Iwamoto Y, Kubo C. The duration of severe insulin omission is the factor most closely associated with the microvascular complications of Type 1 diabetic females with clinical eating disorders. Int J Eat Disord 2008; 41: 259-264 [PMID: 18095311 DOI: 10.1002/eat.20498]
57 Goebel-Fabbri AE, Fikkan J, Franko DL, Pearson K, Anderson BJ, Weinger K. Insulin restriction and associated morbidity and mortality in women with type 1 diabetes. Diabetes Care 2008; 31: 415-419 [PMID: 18070998 DOI: 10.2337/dc07-2026]
58 Nielsen S, Emborg C, Mølbak AG. Mortality in concurrent type 1 diabetes and anorexia nervosa. Diabetes Care 2002; 25: 309-312 [PMID: 11815501 DOI: 10.2337/diacare.25.2.309]
59 Walker JD, Young RJ, Little J, Steel JM. Mortality in concurrent type 1 diabetes and anorexia nervosa. Diabetes Care 2002; 25: 1664-1665 [PMID: 12196451 DOI: 10.2337/diacare.25.9.1664-a]
60 Daneman D, Olmsted M, Rydall A, Maharaj S, Rodin G. Eating disorders in young women with type 1 diabetes. Prevalence, problems and prevention. Horm Res 1998; 50 Suppl 1: 79-86 [PMID: 9677005]
61 Rodin G, Olmsted MP, Rydall AC, Maharaj SI, Colton PA, Jones JM, Biancucci LA, Daneman D. Eating disorders in young women with type 1 diabetes mellitus. J Psychosom Res 2002; 53: 943-949 [PMID: 12377307]
62 Nansel TR, Anderson BJ, Laffel LM, Simons-Morton BG, Weissberg-Benchell J, Wysocki T, Iannotti RJ, Holmbeck GN, Hood KK, Lochrie AS. A multisite trial of a clinic-integrated intervention for promoting family management of pediatric type 1 diabetes: feasibility and design. Pediatr Diabetes 2009; 10: 105-115 [PMID: 18721167 DOI: 10.1111/j.1399-5448.2008.00448.
x]63 Wilksch SM, Starkey K, Gannoni A, Kelly T, Wade TD.
Interactive programme to enhance protective factors for eating disorders in girls with type 1 diabetes. Early Interv Psychiatry 2013; 7: 315-321 [PMID: 23347796 DOI: 10.1111/eip.12012]
64 Kass AE, Trockel M, Safer DL, Sinton MM, Cunning D, Rizk MT, Genkin BH, Weisman HL, Bailey JO, Jacobi C, Wilfley DE, Taylor CB. Internet-based preventive intervention for reducing eating disorder risk: A randomized controlled trial comparing guided with unguided self-help. Behav Res Ther 2014; 63C: 90-98 [PMID: 25461783 DOI: 10.1016/j.brat.2014.09.010]
65 Dahan A, McAfee SG. A proposed role for the psychiatrist in the treatment of adolescents with type I diabetes. Psychiatr Q 2009; 80: 75-85 [PMID: 19408118 DOI: 10.1007/s11126-009-9099-1]
66 Olmsted MP, Daneman D, Rydall AC, Lawson ML, Rodin G. The effects of psychoeducation on disturbed eating attitudes and behavior in young women with type 1 diabetes mellitus. Int J Eat Disord 2002; 32: 230-239 [PMID: 12210667 DOI: 10.1002/eat.10068]
67 Alloway SC, Toth EL, McCargar LJ. Effectiveness of a group psychoeducation program for the treatment of subclinical disordered eating in women with type 1 diabetes. Can J Diet Pract Res 2001; 62: 188-192 [PMID: 11742560]
68 Amsberg S, Anderbro T, Wredling R, Lisspers J, Lins PE, Adamson U, Johansson UB. A cognitive behavior therapy-based intervention among poorly controlled adult type 1 diabetes patients--a randomized controlled trial. Patient Educ Couns 2009; 77: 72-80 [PMID: 19297117 DOI: 10.1016/j.pec.2009.01.015]
69 Markowitz SM, Carper MM, Gonzalez JS, Delahanty LM, Safren SA. Cognitive-behavioral therapy for the treatment of depression and adherence in patients with type 1 diabetes: pilot data and feasibility. Prim Care Companion CNS Disord 2012; 14: pii: PCC.11m01220 [PMID: 22943030 DOI: 10.4088/PCC.11m01220]
70 Battaglia MR, Alemzadeh R, Katte H, Hall PL, Perlmuter LC. Brief report: disordered eating and psychosocial factors in adolescent females with type 1 diabetes mellitus. J Pediatr Psychol 2006; 31: 552-556 [PMID: 16014821 DOI: 10.1093/jpepsy/jsj047]
71 Pinhas-Hamiel O, Graph-Barel C, Boyko V, Tzadok M, Lerner-Geva L, Reichman B. Long-term insulin pump treatment in girls with type 1 diabetes and eating disorders--is it feasible? Diabetes Technol Ther 2010; 12: 873-878 [PMID: 20879965 DOI: 10.1089/dia.2010.0049]
72 Takii M, Uchigata Y, Komaki G, Nozaki T, Kawai H, Iwamoto Y, Kubo C. An integrated inpatient therapy for type 1 diabetic females with bulimia nervosa: a 3-year follow-up study. J Psychosom Res 2003; 55: 349-356 [PMID: 14507546]
73 Custal N, Arcelus J, Agüera Z, Bove FI, Wales J, Granero R, Jiménez-Murcia S, Sánchez I, Riesco N, Alonso P, Crespo JM, Virgili N, Menchón JM, Fernandez-Aranda F. Treatment outcome of patients with comorbid type 1 diabetes and eating disorders. BMC Psychiatry 2014; 14: 140 [PMID: 24885411 DOI: 10.1186/1471-244X-14-140]
74 Stancin T, Link DL, Reuter JM. Binge eating and purging in young women with IDDM. Diabetes Care 1989; 12: 601-603 [PMID: 2791824]
75 Philippi ST, Cardoso MG, Koritar P, Alvarenga M. Risk behaviors for eating disorder in adolescents and adults with type 1 diabetes. Rev Bras Psiquiatr 2013; 35: 150-156 [PMID: 23904020]
P- Reviewer: Codoner-Franch P, Marchesini G S- Editor: Tian YL L- Editor: A E- Editor: Liu SQ
526 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Pinhas-Hamiel O et al . Eating disorders in type 1 diabetes

Tiffany M Potts, Jacqueline L Nguyen, Kanika Ghai, Kathy Li, Lawrence Perlmuter
Tiffany M Potts, Jacqueline L Nguyen, Kathy Li, Lawrence Perlmuter, Department of Psychology, Rosalind Franklin University of Medicine and Science - College of Health Professions, North Chicago, IL 60064, United StatesKanika Ghai, Pediatric Endocrinology, Advocate Children’s Hospital, Park Ridge, IL 60068, United StatesAuthor contributions: Potts TM and Perlmuter L designed the research project; Ghai K provided the patient population and the testing space; Potts TM performed the research; Potts TM, Nguyen JL, Li K, and Perlmuter L analyzed the data; Potts TM, Nguyen JL, Li K and Perlmuter L wrote the paper. Ethics approval: The study was reviewed and approved by the Rosalind Franklin University of Medicine and Science Institutional Review Board and the Advocate Health Care Institutional Review Board.Clinical trial registration: Not available.Informed consent: All study participants, or their legal guardian, provided informed written consent prior to study enrollment.Conflict-of-interest: There are no conflicts of interest related to this study.Data sharing: No additional data are available. Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: Lawrence Perlmuter, PhD, Professor, Department of Psychology, Rosalind Franklin University of Medicine and Science - College of Health Professions, 3333 Green Bay Road, North Chicago, IL 60064, United States. [email protected] Telephone: +1-847-5788754Fax: +1-847-5788765Received: August 6, 2014Peer-review started: August 6, 2014First decision: August 28, 2014Revised: October 7, 2014Accepted: February 4, 2015 Article in press: February 9, 2015
Published online: April 15, 2015
AbstractAIM: To investigate whether perceptions of task difficulty on neuropsychological tests predicted academic achievement after controlling for glucose levels and depression.
METHODS: Participants were type 1 diabetic adolescents, with a mean age = 12.5 years (23 females and 16 males), seen at a northwest suburban Chicago hospital. The sample population was free of co-morbid clinical health conditions. Subjects completed a three-part neuropsychological battery including the Digit Symbol Task, Trail Making Test, and Controlled Oral Word Association test. Following each task, individuals rated task difficulty and then completed a depression inventory. Performance on these three tests is reflective of neuropsychological status in relation to glucose control. Blood glucose levels were measured immediately prior to and after completing the neuropsychological battery using a glucose meter. HbA1c levels were obtained from medical records. Academic performance was based on self-reported grades in Math, Science, and English. Data was analyzed using multiple regression models to evaluate the associations between academic performance, perception of task difficulty, and glucose control.
RESULTS: Perceptions of difficulty on a neuro-psychological battery significantly predicted academic performance after accounting for glucose control and depression. Perceptions of difficulty on the neuro-psychological tests were inversely correlated with academic performance (r = -0.48), while acute (blood glucose) and long-term glucose levels increased along with perceptions of task difficulty (r = 0.47). Additionally, higher depression scores were associated
527 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.527
World J Diabetes 2015 April 15; 6(3): 527-533ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.
Perception of difficulty and glucose control: Effects on academic performance in youth with type Ⅰ diabetes
Prospective Study
ORIGINAL ARTICLE

with poorer academic performance (r = -0.43). With the first regression analysis, perception of difficulty on the neuropsychological tasks contributed to 8% of the variance in academic performance after controlling for peripheral blood glucose and depression. In the second regression analysis, perception of difficulty accounted for 11% of the variance after accounting for academic performance and depression. The final regression analysis indicated that perception of difficulty increased with peripheral blood glucose, contributing to 22% of the variance. Most importantly, after controlling for perceptions of task difficulty, academic performance no longer predicted glucose levels. Finally, subjects who found the cognitive battery difficult were likely to have poor academic grades.
CONCLUSION: Perceptions of difficulty on neurological tests exhibited a significant association with academic achievement, indicating that deficits in this skill may lead to academic disadvantage in diabetic patients.
Key words: Type 1 diabetes; Adolescents; Perception of difficulty; Academic performance; Glucose control
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: The objective of the current study was to investigate the association between perceptions of difficulty and academic performance in adolescents with type 1 diabetes. Perceptions of difficulty are reflected in executing cognitive activities as well as in the task of glucose regulation. Glucose control needs to be understood, not so much from a biological perspective but as an effortful process that is also reflected in academic challenges. The problem of anergia observed generally in patients with diabetes seems to broadly affect a variety of tasks and challenges. Thus, the novelty of the study is in showing that the regulation of glucose in diabetic patients is another example of the broad challenges confronted in the life of patients with diabetes.
Potts TM, Nguyen JL, Ghai K, Li K, Perlmuter L. Perception of difficulty and glucose control: Effects on academic performance in youth with type Ⅰ diabetes. World J Diabetes 2015; 6(3): 527-533 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/527.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.527
INTRODUCTIONType 1 diabetes is one of the most common chronic diseases of childhood. In the United States, type 1 diabetes affects approximately 1 in 300 children and is usually diagnosed between the ages of 5 and 11 years[1,2]. The course of this autoimmune version of diabetes is characterized by ongoing beta cell destruction
and the need for exogenous insulin[3]. Since the central nervous system (CNS) is dependent on a supply of glucose for normal neural functioning, reduced glucose concentrations may result in temporary and/or enduring impairments in CNS function[4]. Dysregulation of the CNS may have negative effects on neuropsychological functioning, such as impaired memory, attention, motor skills, and executive functioning[5].
Adolescents with type 1 diabetes face many challenges that can impact metabolic control along with physiological and psychological complications. The need for adherence to metabolic control and compliance with regimen is effortful. Beyond the demands for medical compliance, adolescents are often burdened by depression[6] as well as anergia[7] and thus may be at academic disadvantage[8].
Along with academic adversity, individuals with type 1 diabetes may be challenged by self-evaluative factors such as the perception of control, which affects behavioral and social functioning[9,10]. The literature has described three prominent types of control: perceived control, perceived confidence (self efficacy), and perceived difficulty[11]. Perceived difficulty, a subjective experience, can be defined as the extent to which individuals believe that performance on a task or behavior would range from easy to difficult[11]. If adolescents believe they are substandard at a task because of its difficulty, they are likely to devalue the activity and decrease effort expenditure[12].
Perceived difficulty has also been shown to relate to an individual’s beliefs regarding how much effort is needed to succeed at a task and whether success is possible[13]. The effort expended in performing a task is predicted to increase proportionally with the level of perceived task difficulty[14]. Indeed, effort has been associated with academic performance. Studies[15] have found that self-reported grades are positively correlated with effort. Moreover, adherence to regimen[16] and metabolic control[17] also reflect the expenditure of effort. Therefore, poor glucose control may be positively correlated with perceptions of task difficulty and inversely associated with academic performance[18]. Indeed, self-control and willpower are both functions of available glucose levels[19]. Thus, glucose control and effort are two highly related processes that need to be differentiated.
Perceptions of difficulty rely on executive functioning skills, such as problem solving and decision-making. These skills require an individual to make a decision regarding the degree of difficulty associated with the task and whether he or she will be successful in carrying out the task. Since problem solving and decision-making skills contribute to academic performance, these executive functioning skills may be impaired in diabetic individuals[20]. A meta-analytic finding indicated that children with type 1 diabetes performed slightly, yet significantly, lower on attention/executive functioning tasks than non-diabetic individuals[21]. This finding, in turn, was associated with poor glucose control and
Potts TM et al . Perception of difficulty, glucose, and academics
528 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

hypoglycemic episodes as well[21]. To open the question of effort, three brief but well
validated neuropsychological tests were administered[22] and participants rated the difficulty of each test. Secondly, academic performance and depression levels were evaluated and analyzed. Since long-term glucose control (HbA1c) is associated with cognition[18], HbA1c levels from within the past three months were collected from medical records. Acute glucose levels immediately prior to and following neuropsychological testing were also collected and evaluated. These procedures enabled the distinction between the assessment of effort-associated processes supporting academic and neuropsychological processes vs those putatively supporting glucose control.
MATERIALS AND METHODSParticipantsThis study was designed as a cross-sectional, pro-spective study. Participants were type 1 diabetic patients who attended a diabetes clinic in a northwest suburban Chicago hospital. Adolescents were approached to participate in the study during their regularly scheduled doctor’s appointment. Adolescents were excluded from participation if they had co-morbid clinical health conditions, such as asthma, anemia, vascular disease,kidney disease, chronic tiredness, depression, psychiatric history, or obesity. Parents of the patients filled out consent forms that outlined the procedure and risks of participation. The subjects also signed an assent form. The study measures were carried out in a quiet, separate room for each participant individually. Pre-doctoral psychology students administered the assessments. Participants were provided modest monetary compensation for participation in the study. There were a total of 39 participants, consisting of 23 females and 16 males. The population was comprised of 71.8% Caucasian, 20.5% Hispanic, 5.1% Asian, and 2.6% African American. The mean age of the participants was 12.5 years (SD = 2.73). This study was approved by the Institutional Review Board at Rosalind Franklin University as well as the hospital’s review board. There is no conflict of interest related to this study.
MaterialsGlucose control: Blood glucose measurements were performed immediately prior to administering and again after completing the neuropsychological battery with glucose meters (One Touch, Blood Glucose Monitoring System). This procedure was used to obtain an average peripheral blood glucose measurement. A single HbA1c level was recorded from the medical records retrieved from the previous three months, prior to testing. Neuropsychological battery: The neuropsychological battery included the Digit Symbol task (Wechsler Intelligence Scale for Children-Forth Edition), Trail Making test Parts A and B, and Controlled Oral Word
Association (COWA) in that order. This three-part neuropsychological battery was selected because the tests are brief, well researched, psychometrically sound, and are commonly used[23]. Performance on these tests is reflective of neuropsychological status in diabetic patients in relation to glucose control[22].
The Digit Symbol task of the Wechsler Intelligence Scale for Children-Fourth Edition (DS; WISC-Ⅳ)[24] was used to assess psychomotor performance, attention, memory, and perceptual organization. It consists of Coding and Incidental Learning. In Coding, the individual was presented with several rows of a series of numbers (1 through 9). Each number was paired with a corresponding unique symbol. The individual was asked to fill in as many symbols as possible in boxes directly under the corresponding number and was allowed 120 s to complete this task. In Incidental Learning, individuals were asked to carry out two tasks: to recall as many digit-symbol pairs as possible (Pairing) and to recall as many symbols as possible without the corresponding numbers (Free Recall). Internal consistency of the Digit Symbol Coding task is 0.85.
The Trail Making Test[23] was used to assess attention, visual-motor tracking, and speed of information processing. It consists of two parts: in Part A, individuals were asked to draw a line that accurately connected the correct sequence of numbers as quickly as possible. In Part B, individuals were asked to draw a line that accurately connects the correct sequence of numbers and letters in an alternating pattern as quickly as possible. Reliability for Part A is 0.78 and for Part B 0.67.
The COWA test[25] assessed verbal functions. Individuals were asked to generate as many words as possible that began with the letter S in 60 s and then the letter F in 60 s as the examiner recorded these. Test-retest reliability is 0.88 and inter-scorer reliability was almost perfect.
Rating of task difficulty: After completing three tasks (the Digit Symbol task of the Wechsler Intelligence Scale for Children-Fourth Edition, Trail Making test Part A and B, and Controlled Oral Word Association test), participants were asked to rate the level of perceived difficulty of each task on a scale from 1-5, where 1 = very easy and 5 = very difficult. These evaluations were summed and averaged to develop a task difficulty measure.
Academic performance: Participants self reported academic grades in several courses during the current school year: Math, Science, and English. For the purposes of tabulation, grades were converted into a scale score from 1-5, where 1 = F, 2 = D, 3 = C, 4 = B, and 5 = A. Grades were then averaged for each participant.
Depression: To avoid possible reactive effects[26], participants were lastly administered the Children’sDepression Inventory (CDI)[27] which is a 27 item self report inventory used to measure depressive symp-
529 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Potts TM et al . Perception of difficulty, glucose, and academics

that higher levels of difficulty perceived on the neuropsychological test was inversely associated with academic performance after taking into account peripheral blood glucose and depression. The result of this analysis indicated that the perception of difficulty on the neuropsychological tasks contributed to 8% of the variance in academic performance after controlling for peripheral blood glucose and depression. Thus, stronger ratings of laboratory task difficulty were associated with poorer academic performance.
After controlling for academic performance and depression, a regression analysis was conducted with peripheral blood glucose as the dependent variable to test if perceptions of task difficulty were associated with glucose levels (Table 4). The predictors in the regression analysis were entered in the following order: (1) academic performance; (2) CDI; and (3) perceptions of difficulty on the neurological tests with peripheral blood glucose as the dependent variable. Results indicated that the first predictor (higher academic performance) was inversely related to peripheral blood glucose, contributing to 14% of the variance. The second predictor (CDI) showed that the levels of depression had insignificant effects on peripheral blood glucose. The third predictor (perception of difficulty) showed that the higher level of difficulty perceived on the test accounted for the higher peripheral blood glucose level. Perception of difficulty accounted for 11% of the variance after accounting for academic performance and depression. This indicated that higher perceptions of task difficulty were associated with higher blood glucose levels while depression seemed to play no significant role in this model. Individuals, who perceived the neuropsychological battery as challenging, may also experience difficulty controlling glucose levels.
The purpose of the final analysis (Table 5) was to determine whether academic performance would predict glucose levels after controlling for perceptions of task difficulty. The order of predictors was: (1) perception of difficulty; (2) academic performance; and (3) CDI with peripheral glucose levels as the dependent variable. The first predictor (perception of difficulty) showed that perception of difficulty increased with peripheral blood glucose, contributing to 22% of the variance. Most importantly, after controlling for perceptions of task difficulty, academic performance no longer predicted glucose levels. Additionally, it was found that the level of depression was not associated with peripheral glucose levels. Results from Table 5 indicated that peripheral blood glucose was positively correlated with the perception of task difficulty.
DISCUSSIONThe general purpose underlying this research was to critically evaluate the various explanations for the problems that diabetic adolescents experience in the performance of cognitive tasks. In addition to elevations
tomatology in youth aged 7-17. Respondents indicated which of three options best represented their mood in the context of everyday life: absence of symptom = 0; mild symptom = 1; and definite symptom = 2. The CDI provides a total score on five domains of depression. The reliability coefficient for the total inventory was 0.86[27].
Data was collected and entered into Statistic Package for Social Science (SPSS) Version 13. Zero order correlation tables were examined to assess the degree of relationship between the variables. P-values of less than 0.05 were considered statistically significant. Multiple regression analyses were used to assess outcome variables.
RESULTSBasic description of the sample population is presented in Table 1. The primary results (Table 2) indicated that perceptions of difficulty on the neuropsychological tests were inversely correlated with academic per-formance (r = -0.48), while acute (blood glucose) and long-term glucose levels (HbA1c) increased along with perceptions of task difficulty (r = 0.47). Finally, higher depression scores were associated with poorer academic performance (r = -0.43).
To evaluate the relationship of difficulty on neuro-psychological tests and academic performance after controlling for peripheral glucose levels and depression, a multiple regression analysis was conducted (Table 3). The predictors in the regression analysis were entered in the following order: (1) peripheral blood glucose; (2) CDI; and (3) perception of difficulty with academic performance as the dependent variable. Results from the first predictor indicated that as peripheral blood glucose increased, academic performance decreased. The second predictor (CDI) indicated that increasing depression was associated with poorer academic performance after controlling for peripheral blood glucose. Lastly, the third predictor (perception of difficulty) showed
530 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 1 Characteristics of participants
Mean SD
Disease duration (mo) 48.05 41.08BMI (kg/m2) 21.18 3.85Peripheral blood glucose (mm/dL) 190.99 89.27HbA1c (%) 8.39 1.63Digit symbol coding 55.38 14.7Digit symbol paring 11.44 5.16Digit symbol free recall 7.38 1.41Trails A (s) 39.67 19.02Trails B (s) 92.74 64.84COWA 20.26 6.62CDI 7.23 5.54Perception of difficulty1 10.15 2.95Grades2 4.1 0.77
1Range 1-20; 2Range low to high; 1-5. BMI: Body mass index; HbA1c: Hemoglobin A1c; COWA: Controlled Oral Word Association; CDI: Child depression inventory.
Potts TM et al . Perception of difficulty, glucose, and academics

in blood glucose, there are two alternatives, namely anergia[7] and depression[6]. Since these processes each exacerbate or contribute to the diminution of effort, it is possible that the actual mediator of compromised cognition can be found in reduced effort. Thus, the primary purpose of this introductory study was to assess the contribution of effort[14] to both metabolic control and reduced cognition in these patients.
Through a series of multiple regression analyses, the results showed that after controlling for levels of effort, the role of metabolic control was compromised with respect to cognition. A key assumption in this process is that metabolic control is per se an effortful process. If effort is viewed as the primary process, metabolic control remains as an adjunctive player in cognition.
To examine this model, we turn to the results of the multiple regression analyses presented earlier. That is, adolescents who perceived the neuropsychological battery as more difficult were more likely to have lower grades in Math, Science, and English. Thus, perceiving the battery as difficult may indicate a broad weakness in executive functioning skills, such as problem solving and decision-making. Furthermore, increased levels of depression also negatively contributed to academic performance. As a result, academic performance was
poorer in adolescents with elevated peripheral blood glucose, which in itself may be a reflection of the difficulty or effort necessary for exercising metabolic control.
This study provided insight into how academic performance may be associated with self-ratings of difficulty on neuropsychological tasks after controlling for blood glucose levels and depression. Further, the study showed how peripheral blood glucose levels can be derived from perceptions of difficulty based on neuropsychological testing after controlling for academic performance and depression. Each of these observations will be discussed serially.
Previous findings[28,29] have suggested a relationship between glucose control and academic performance. Greater difficulty in maintaining strong glucose control was associated with poorer academic performance. The list of predictors of academic performance included a neuropsychological battery that assessed psychomotor performance, attention, memory, speed of information processing, and verbal functioning.
Participants who perceived the neuropsychological battery as difficult were also more likely to exhibit poor grades (Table 4). However, this finding may not be surprising, since performance on the neuropsychological battery and academic performance share some conceptual space. Thus, a basic question in this study is whether the commonly observed relationship between glucose levels and cognitive functioning represents a basic physiological relationship between glucose and neuropsychological functioning? Alternatively, is the index of glucose control merely a reflection of the basic challenge between compliance with regimen, an effort-demanding task, and the challenge intrinsic to the cognitive task?
After controlling for appraisals of task difficulty, blood glucose levels seem to no longer account for cognitive
531 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Table 2 Correlations among the variables
Academic performance HbA1c Peripheral blood glucose CDI Perceptions of difficulty
Academic performance1 - -0.26 -0.37a -0.43b -0.48b
HbA1c - 0.42b 0.03 0.34a
Peripheral blood glucose2 - 0.22 0.47b
CDI3 - 0.27
1Low scores indicate poor grades; 2Low score-lower glucose levels; 3Higher scores more depression. aP < 0.05; bP < 0.01. HbA1c: Hemoglobin A1c; CDI: Child depression inventory.
Table 3 Linear multiple regression analysis for variables predicting academic performance1
β R2 change
Peripheral blood glucose2 -0.37 0.14a
Child depression inventory3 -0.36 0.12a
Perceptions of difficulty on neurological tests -0.33 0.08a
1Low scores indicate poor grades; 2Low score-lower glucose levels; 3Higher score more depression. aP < 0.05. β: Standardized coefficient beta.
β R2 change
Academic performance2 -0.37 0.14b
Child depression inventory3 0.08 0.01Perceptions of difficulty on neurological test 0.38 0.11b
Table 4 Linear multiple regression analysis for variables predicting peripheral blood glucose1
1Low score-lower glucose levels; 2Low scores indicate poor grades; 3Higher scores more depression. bP < 0.01. β: Standardized coefficient beta.
Table 5 Linear multiple regression analysis for variables predicting peripheral blood glucose1
β R2 change
Perceptions of difficulty on neurological test 0.47 0.22b
Academic performance2 -0.18 0.03Child depression inventory3 0.05 0.00
1Low score-lower glucose levels; 2Low scores indicate poor grades; 3Higher scores more depression. bP < 0.01. β: Standardized coefficient beta.
Potts TM et al . Perception of difficulty, glucose, and academics

performance once task difficulty is factored into the equation (Table 5). Finally, by eliminating the influence of perceptions of difficulty in maintaining blood glucose control and complying with medical regimen, peripheral blood glucose and academic performance were no longer significantly related.
This may be the first study to illustrate how perceptions of task difficulty on neuropsychological tests are associated with poor academic performance in diabetic patients. While several studies have reported that individuals with diabetes exhibit relatively poor academic performance, the present study identifies how perceptual evaluative factors diminish academic performance.
Thus, careful disease management is needed to prevent or effectively treat potentially life-threatening complications such as hypoglycemia, diabetic ketoa-cidosis, and hyperglycemia as well as effort.
Limitations to the study include a relatively small sample size as well as the use of a single item to assess perceptions of task difficulty. However, this method of assessing perceptions of difficulty with one item was also utilized in a previous study[11]. In their study, Rogers and colleagues assessed self-efficacy, perceived control and perceived difficulty with the use of a single item Likert scale question for each construct[11]. Therefore, single item indicators may be regarded as similarly robust as multiple item indicators[30,31]. Lastly, academic performance was assessed by participant self-report. Systematic biases may influence the validity of self-reported grades, but self-reported grades generally predict outcomes as effectively as actual or objectively culled grades[32].
In conclusion, this study demonstrated that per-ceptions of task difficulty can predict academic difficulty in a sample of diabetic adolescents. Task difficulty was also associated with the relationship between glucose levels and academic performance. Hence, perceptions of difficulty can be viewed as a generalized executive function. When individuals perceive a task as more difficult, this may promote academic disadvantage along with increased challenges in controlling glucose. Overall, such information may be useful in training and counseling students in enhancing academic success and performance as well as in assistance with disease control.
COMMENTSBackgroundType 1 diabetes is one of the most common chronic diseases of childhood. In the United States, type 1 diabetes affects approximately 1 in 300 children and is usually diagnosed between the ages of 5 and 11 years. Research frontiersAdolescents with type 1 diabetes face many challenges that can impact metabolic control along with physiological and psychological complications. Cognitive functioning is an important aspect of life and can be impaired in individuals with diabetes. Academic performance in diabetic adolescents may also be compromised due to fluctuations in glucose.Innovations and breakthroughsAlong with academic adversity, individuals with type 1 diabetes may be
challenged by self-evaluative factors such as the perception of control, which affects behavioral and social functioning. Perceived difficulty has also been shown to relate to an individual’s beliefs regarding how much effort is needed to succeed at a task and whether success is possible. Perceptions of difficulty rely on executive functioning skills, such as problem solving and decision-making. Since problem solving and decision-making skills contribute to academic performance, these executive functioning skills may be impaired in diabetic individuals. The current study sought to examine the role of effort on glucose and academic performance. ApplicationsThis may be the first study to illustrate how perceptions of task difficulty on neuropsychological tests are associated with poor academic performance in diabetic patients. While several studies have reported that individuals with diabetes exhibit relatively poor academic performance, the present study identifies how perceptual evaluative factors diminish academic performance.TerminologyDiabetes mellitus is a major health problem in the United States affecting approximately 29.1 million people. There are two major classifications of diabetes: insulin dependent diabetes (type 1) and non-insulin dependent diabetes (type 2). Diabetes is a group of metabolic diseases that either cause defects in insulin action, insulin secretion, or a combination of both, resulting in hyperglycemia. Perceived difficulty, a subjective experience, can be defined as the extent to which individuals believe that performance on a task or behavior would range from easy to difficult. Peer-reviewThe study addresses a relevant scientific question and is well-conducted. The results are significant indicating that deficits in perception of difficulty may lead to academic disadvantage in diabetic patients. The manuscript is interesting and may provide important information for the reader of the journal.
REFERENCES1 Lamb MM, Norris JM. Epidemiology of type 1 diabetes.
Eisenbarth GS, editor. Immunoendocrinology: Scientific and Clinical Aspects, Contemporary Endocrinology. New York: Springer Science, 2011: 267-278 [DOI: 10.1007/978-1-60327-478-4_16]
2 Peterson L, Reach K, Grabe S. Health-related disorders. Mash EJ, Barkley RA, editors. Child Psychopathology. New York: The Guilford Press, 2003: 716-749
3 Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. Epidemiology of type 1 diabetes. Endocrinol Metab Clin North Am 2010; 39: 481-497 [PMID: 20723815 DOI: 10.1016/j.ecl.2010.05.011]
4 Lin A, Northam EA, Rankins D, Werther GA, Cameron FJ. Neuropsychological profiles of young people with type 1 diabetes 12 yr after disease onset. Pediatr Diabetes 2010; 11: 235-243 [PMID: 20070555 DOI: 10.1111/j.1399-5448.2009.00588.x]
5 Kucera M, Sullivan AL. The educational implications of type 1 diabetes mellitus: A review of research and recommendations for school psychological practice. Psychology in the Schools 2011; 48: 587-603 [DOI: 10.1002/pits.20573]
6 Lernmark B, Persson B, Fisher L, Rydelius PA. Symptoms of depression are important to psychological adaptation and metabolic control in children with diabetes mellitus. Diabet Med 1999; 16: 14-22 [PMID: 10229288]
7 Perlmuter LC, Flanagan BP, Shah PH, Singh SP. Glycemic control and hypoglycemia: is the loser the winner? Diabetes Care 2008; 31: 2072-2076 [PMID: 18820231 DOI: 10.2337/dc08-1441]
8 Taras H, Potts-Datema W. Chronic health conditions and student performance at school. J Sch Health 2005; 75: 255-266 [PMID: 16102088 DOI: 10.1111/j.1746-1561.2005.00034.x]
9 Perlmuter LC. Choice enhances performance in non-insulin dependent diabetics and controls. J Gerontol 1991; 46: P218-P223 [PMID: 1890288 DOI: 10.1093/geronj/46.5.P218]
10 Skinner EA. A guide to constructs of control. J Pers Soc Psychol 1996; 71: 549-570 [PMID: 8831161 DOI: 10.1037/0022-3514.71.3.549]
11 Rodgers WM, Conner M, Murray TC. Distinguishing among perceived control, perceived difficulty, and self-efficacy as determinants of intentions and behaviours. Br J Soc Psychol 2008;
532 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
COMMENTS
Potts TM et al . Perception of difficulty, glucose, and academics

47: 607-630 [PMID: 17945040 DOI: 10.1348/014466607X248903]12 Brehm JW, Self EA. The intensity of motivation. Annu Rev
Psychol 1989; 40: 109-131 [PMID: 2648973 DOI: 10.1146/annurev.ps.40.020189.000545]
13 Horvath M, Herleman HA, McKie RL. Goal orientation, task difficulty, and task interest: A multilevel analysis. Motivation and Emotions 2006; 30: 171-178 [DOI: 10.1007/s11031-006-9029-6]
14 Capa RL, Audiffren M, Ragot S. The effects of achievement motivation, task difficulty, and goal difficulty on physiological, behavioral, and subjective effort. Psychophysiology 2008; 45: 859-868 [PMID: 18627535 DOI: 10.1111/j.1469-8986.2008.00675.x]
15 Ramaswamy R, Mirochna M, Perlmuter LC. The negative association of BMI with classroom effort in elementary school children. J Child Health Care 2010; 14: 161-169 [PMID: 20388720 DOI: 10.1177/1367493509359222]
16 Malik FS, Taplin CE. Insulin therapy in children and adolescents with type 1 diabetes. Paediatr Drugs 2014; 16: 141-150 [PMID: 24458650 DOI: 10.1007/s40272-014-0064-6]
17 LeRoith D, Smith DO. Monitoring glycemic control: the cornerstone of diabetes care. Clin Ther 2005; 27: 1489-1499 [PMID: 16330287 DOI: 10.1016/j.clinthera.2005.10.010]
18 McCarthy AM, Lindgren S, Mengeling MA, Tsalikian E, Engvall J. Factors associated with academic achievement in children with type 1 diabetes. Diabetes Care 2003; 26: 112-117 [PMID: 12502666 DOI: 10.2337/diacare.26.1.112]
19 Gailliot MT, Baumeister RF. The physiology of willpower: linking blood glucose to self-control. Pers Soc Psychol Rev 2007; 11: 303-327 [PMID: 18453466 DOI: 10.1177/1088868307303030]
20 Cato MA, Mauras N, Ambrosino J, Bondurant A, Conrad AL, Kollman C, Cheng P, Beck RW, Ruedy KJ, Aye T, Reiss AL, White NH, Hershey T. Cognitive functioning in young children with type 1 diabetes. J Int Neuropsychol Soc 2014; 20: 238-247 [PMID: 24512675 DOI: 10.1017/S1355617713001434]
21 Gaudieri PA, Chen R, Greer TF, Holmes CS. Cognitive function in children with type 1 diabetes: a meta-analysis. Diabetes Care 2008; 31: 1892-1897 [PMID: 18753668 DOI: 10.2337/dc07-2132]
22 Galanina N, Surampudi V, Ciltea D, Singh SP, Perlmuter LC. Blood glucose levels before and after cognitive testing in diabetes mellitus. Exp Aging Res 2008; 34: 152-161 [PMID: 18351501 DOI: 10.1080/03610730701876979]
23 Groth-Marnat G. Handbook of Psychological Assessment. 4th ed. Hoboken, New Jersey: John Wiley & Sons Inc., 2003: 517-577
24 Wechsler D. Wechsler Intelligence Scale for Children: Administration and scoring manual. 4th ed. San Antonio, TX: Psychological Corporation, 2003
25 Spreen O, Strauss E. A compendium of neuropsychological tests. New York: Oxford University Press, 1991
26 Perlmuter LC, Noblin CD, Hakami M. Reactive effects of tests for depression: Theoretical and methodological considerations. Journal of Social and Clinical Psychology 1983; 1: 128-139 [DOI: 10.1521/jscp.1983.1.2.128]
27 Kovacs M. Child Depression Inventory Technical Manual. North Tonawanda, New York: Multi-Health Systems Inc., 1992
28 Dahlquist G, Källén B. School performance in children with type 1 diabetes--a population-based register study. Diabetologia 2007; 50: 957-964 [PMID: 17333107 DOI: 10.1007/s00125-007-0615-2]
29 McCarthy AM, Lindgren S, Mengeling MA, Tsalikian E, Engvall JC. Effects of diabetes on learning in children. Pediatrics 2002; 109: E9 [PMID: 11773577 DOI: 10.1542/peds.109.1.e9]
30 Herman AO, Fairchild DG, Hefner JE. Single-Question Screening Can Help Identify Teens with Substance Misuse. New England Journal of Medicine Physician’s First Watch, 2014. Cited 2014-08-04. Available from: URL: http: //www.jwatch.org/fw109105/2014/07/29/single-question-screening-can-help-identify-teens-with
31 Gardner DG, Cummings LL, Dunham RB, Pierce JL. Single item versus multiple-item measurement scales: An empirical example. Educ Psychol Meas 1998; 58: 898-906 [DOI: 10.1177/0013164498058006003]
32 Kuncel NR, Crede M, Thomas LL. The validity of self-reported grade point averages, class ranks, and test scores: Meta-analysis and review of the literature. Review of Educational Research 2005; 75: 63-82 [DOI: 10.3102/00346543075001063]
P- Reviewer: Akbulut S, Cerwenka HR, Das UN, Hassan M, Hortobagyi T
S- Editor: Tian YL L- Editor: A E- Editor: Liu SQ
533 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Potts TM et al . Perception of difficulty, glucose, and academics

diabetes. Only peer-reviewed articles in English were included published in the last 15 years. It was selected from 1999 to 2014. Glycemic control was measured with HbA1c. Studies with an intervention lasting at least 12 wk were included if the HbA1c was measured before and after the intervention.
RESULTS: A total of nine articles were found, and they were published between the years of 2002-2011. The sample size was 401 diabetic youths (166 males and 235 females) with an age range of 10-19 years except one study, in which the age range was 13-30 years. Study participants were from Australia, Tunisia, Lithuania, Taiwan, Turkey, Brazilia, Belgium, Egypt and France. Four studies were aerobic-based, four were combined aerobic and anaerobic programs, and one compared aerobic exercise to anaerobic one. Available studies had insufficient evidence that any type of exercise or combined training would clearly improve the glycemic control in type 1 diabetic youth. Only three (two aerobic-based and one combined) studies could provide a significant positive change in glycemic control.
CONCLUSION: The regular physical exercise has several other valuable physiological and health benefits that justify the inclusion of exercise in pediatric diabetes treatment and care.
Key words: Type 1 diabetes mellitus; Glycemic control; Exercise; Aerobic; Anaerobic
© The Author(s) 2015. Published by Baishideng Publishing Group Inc. All rights reserved.
Core tip: Diabetic patients should be aware that exercise interferes with the glucose homeostasis. Anaerobic exercise can increase glycemia, whereas the aerobic exercise may cause a decrease during the exercise and post-exercise. By evaluating the long-term effect of exercise on glycemic control in type 1 diabetic youths
Andrea Lukács, László Barkai
Andrea Lukács, László Barkai, Faculty of Health Care, University of Miskolc, 3515 Miskolc-Egyetemváros, HungaryLászló Barkai, Velkey László Center for Child Health, 3501 Miskolc, HungaryAuthor contributions: Both authors contributed to this manuscript.Conflict-of-interest: No conflict of interest stated by any authors.Data sharing: This manuscript does not describe basic or clinical research, therefore data sharing statement is not relevant.Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/Correspondence to: László Barkai, MD, PhD, DSc, Faculty of Health Care, University of Miskolc, 3515 Miskolc-Egyetemváros, Hungary. [email protected]: +36-46-565111Fax: +36-46-366961Received: July 29, 2014 Peer-review started: July 30, 2014 First decision: October 14, 2014Revised: January 2, 2015 Accepted: January 15, 2015Article in press: January 19, 2015Published online: April 15, 2015
AbstractAIM: To evaluate the long-term effect of aerobic and/or anaerobic exercise on glycemic control in youths with type 1 diabetes.
METHODS: Literature review was performed in spring and summer 2014 using PubMed/MEDLINE, Google Scholar, Scopus, and ScienceDirect with the following terms: aerobic, anaerobic, high-intensity, resistance, exercise/training, combined with glycemic/metabolic control, glycated haemoglobin A1c (HbA1c) and type 1
SYSTEMATIC REVIEWS
534 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Effect of aerobic and anaerobic exercises on glycemic control in type 1 diabetic youths
Submit a Manuscript: http://www.wjgnet.com/esps/Help Desk: http://www.wjgnet.com/esps/helpdesk.aspxDOI: 10.4239/wjd.v6.i3.534
World J Diabetes 2015 April 15; 6(3): 534-542ISSN 1948-9358 (online)
© 2015 Baishideng Publishing Group Inc. All rights reserved.

according to the major metabolic pathway involved in energy utilization (aerobic or anaerobic), we found insufficient evidence in the latest literature that any type of exercise or combined aerobic and anaerobic training would clearly improve the glycemic control. The regular physical exercise has several other valuable benefits that justify the inclusion of exercise in pediatric diabetes treatment and care.
Lukács A, Barkai L. Effect of aerobic and anaerobic exercises on glycemic control in type 1 diabetic youths. World J Diabetes 2015; 6(3): 534-542 Available from: URL: http://www.wjgnet.com/1948-9358/full/v6/i3/534.htm DOI: http://dx.doi.org/10.4239/wjd.v6.i3.534
INTRODUCTIONPhysiological, social and emotional benefits of regular exercise and physically active lifestyle are well documented mostly in a healthy population[1-4], but they are also important for diabetic patients regardless of the type of diabetes[5-8]. For type 1, successful diabetes management is based on individualized insulin therapy, adjusted diet and regular exercise[9].
Exercise used as prevention and therapy in diabetes is not a new concept in the literature. Even the ancient Ayurvedic physician, Susruta Shamita (born around 600 B.C.), noted the reduction in the sweetness of urine from diabetic patients with excecise, and included moderate exercise within his treatment regimens[10]. In 1919, Allen proved that exercise reduced blood glucose level and improved acutely the tolerance to a carbohydrate load[11]. After the discovery of insulin in 1921[12], Lawrence demonstrated that exercise has an effect on insulin requirements and glucose uptake[13]. Joslin believed in “troika” (group of three) in the treatment of diabetes, symbolizing insulin, diet and exercise correlation[14]. Regular exercise helps to improve overall health and fitness, and reduces risk factors for vascular complications. Diabetic youths with regular exercise have improved blood lipid profile, and increased insulin sensitivity, primarily in the skeletal muscles, which leads to a reduced need for insulin[15-17]. The American Diabetes Association recommends all levels of physical exercise including leisure activities, recreational sport and competitive sport for youths with type 1 diabetes mellitus (T1DM) if they are in good blood glucose control and have no long-term complications[18]. Until the sympathetic nervous and endocrine systems control the blood glucose level at the physiological levels during and after physical exercise in healthy subjects[19], the regulation is external in diabetic patients considering many internal influences. Youths with T1DM should be aware that exercise interferes with the glucose homeostasis, although there are individual differences in blood glucose response due to type, duration and intensity of the exercise, the pre-exercise level of counterregulatory hormones, and blood glucose
concentrations[20,21]. Several studies examined the role of physical activity and exercise in the treatment of T1DM and considered it an important component. The evidence for improvement in glycemic control is equivocal. Some studies suggest a positive effect[22-27], whereas others fail to show this effect[28-34]. Scientific questions remain concerning what is the exact effect of different types of exercise on glycemic control in youths with T1DM.
In this systematic literature review, we evaluated the latest studies examining the long-term effect of aerobic and/or anaerobic exercise on glycemic control in youths with T1DM. We also made distinguishing between concepts of physical activity, physical fitness, and aerobic-anaerobic exercise. Finally, we considered clinical applications and future directions.
ConceptualisationPhysical activity: Physical activity encompasses body movement produced by skeletal muscles which requires energy consumption[35]. Physical activity can range from sports to any other lifestyle activities including school and out-of-school activities, weekend activities where youths play, are active and expend energy. There is evidence that behavioural patterns of physical activity in childhood are maintained throughout adulthood[36,37]. This term is often used interchangeably with regular exercise and physical fitness in studies, although they are different concepts[38].
Exercise: Exercise has been defined as any form of body movement that results in an increase in metabolic demand with the intention of developing one or more components of physical fitness. It is generally planned, structured and systematic[35]. Regular exercise improves physical fitness. Exercise is characterised by five com-ponents: type [dynamic (isotonic) or static (isometric), and both of them can be performed aerobically or anaerobically on the basis of energy utilization][39], intensity (degree of effort that individual puts into exercise), duration (the length of each training session), repetition (number of times individual performs a complete movement of a given exercise) and frequency (number of exercise sessions per week)[40].
Fitness: Fitness refers to the “possession of adequate levels of strength, endurance, and mobility to provide for successful participation in occupational effort, recreational pursuits, familial obligation, and that is consistent with a functional phenotyp expression of the human genotype”[41]. Physical fitness determines cardiovascular, respiratory, and musculosceletal systems in order to perform physically demanding activities such as exercise, sport, or work. Fit youth has normal body fat, good immune system and, in general, is in a physiologic state of well-being.
Aerobic-anaerobic exercise: Whether an activity is aerobic or anaerobic depends primarily on its intensity and duration. Most physical exercises are characterised
Lukács A et al . Glycemic control and type of exercise
535 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com

by both static and dynamic contractions as well as aerobic and anaerobic metabolism. Thus, exercise is classified by their dominant features.
Aerobic exercise includes any type of exercise, typically those performed at moderate levels of intensity for extended periods of time that maintains an increased heart rate. Activities such as cycling, swimming, jogging, rowing, cross-country skiing, and aerobic dancing require oxygen to produce ATP. Regular aerobic exercise improves maximal oxygen consumption and overall endurance performances. Anaerobic exercise is used to promote strength, power, and speed. Generally, anaerobic exercise has a short duration and high intensity activity. Unlike aerobic exercise, it does not depend on exogenous oxygen. Activities such as heavy weightlifting, all types of sprint (running, cycling, or swimming) or any hard exercises require anaerobic metabolism. During exercise various forms of energy sources are utilized markedly depending on the intensity and duration of exercise, but the activity is classified typically based on the predominantly used system. The anaerobic energy system is used for resistance training and increasing speed[42]. During high intensity (anaerobic) exercise almost the entire metabolic fuel source is glucose, whereas during low intensity (aerobic) exercise fat utilization increases and glucose oxidation decrease[43]. Both types of exercise increase the mechanical efficiency of the heart (cardiac adaptation), changes in morphology and functionality of the left ventricle[44].
Measuring the intensity of exercise using maximal heart rate: Maximal heart rate (HRmax) is one of the most commonly used values in clinical settings and physiology. HR increases nearly linearly with the increase in the intensity of exercise; and this is the simplest physiological response to measure. As maximal exercise intensity is approached, HR begins to plateau even as the exercise workload continues to increase. This HRmax is a reliable value and it remains constant for a longer period[45]. To estimate HRmax for children, formula 208 - 0.7 × age in years is applied[46,47]. There are several more and less accurate types of estimation existing for determining the exercise intensity[48]. Aerobic training zone is around between 60%-70% of the HRmax. Between 70%-85% of HRmax, the youths are in the mixed zone, and above 85% of HRmax the anaerobic metabolism will come to the fore.
Effects of different types of exercise on blood glucose level in type 1 diabetes patientsDifferent types of exercise produce different effects on blood glucose level. If the carbohydrate intake and the insulin dosage are not in line with the exercise the patient does, it will result in metabolic disturbances[49]. The most common problems are the evaluation of pre-exercise level of blood glucose, evaluation of the intensity and duration of the expected exercise, and to
take into consideration the time of day when the patient exercises, because of the body’s different physiological insulin need[49]. Exercise functions like insulin, so the balance between the insulin therapy and diet could be facilitated if the daily schedule for exercise and the exercise parameters are consistent[50], although this goal is almost impossible to obtain in the real life for youths. Findings from exercise training studies support the concept that moderate intensity aerobic workload increases the risk of hypoglycemia during the exercise and several hours after the exercise. High-intensity training with anaerobic utilization may increase the blood glucose level due to the release of adrenaline and noradrenalin in the blood, which then stimulates the liver to release glucose faster than normal. The exercise-induced rise in glucose level is followed by hypoglycemia after hours of finishing the exercise as counterregulatory hormone levels decrease[21,51-53]. Both aerobic and anaerobic exercise training regimens improve glucose uptake and insulin sensitivity[21,54]. School-aged children engage a combination of moderate- and high-intensity sessions in their everyday sport activities. Their exercise is often spontaneous and unplanned. Thus, a different plan is needed for each type of exercise to optimize blood glucose level, but problems with the management are well recognised. Patients differ in tolerance to exercise and insulin requirements and it is impossible to give precise guidelines suitable for everyone with T1DM, therefore experiences with regular blood glucose testing are inevitable. Continuous glucose monitoring may assist the active diabetic participants to follow the changes in blood glucose level and give information about the individual response to the exercise[21,54]. There are several guidelines to discuss safe sport participation in children and adolescents with T1DM[5,55-59]. These guidelines can be adjusted to own measurements and awareness of the usual responses to a particular type of exercise.
MATERIALS AND METHODSStudy eligibilityA systematic review was performed in spring and summer 2014 using PubMed/Medline, Google Scholar, Scopus, and ScienceDirect with the following terms: aerobic, anaerobic, high-intensity, resistance, exercise/training, combined with glycemic/metabolic control and type 1 diabetes. Only peer-reviewed articles in English were included published in the last 15 years with at least five subjects per group. Studies were selected from 1999 to 2014 that measured the long-term effect of aerobic and/or anaerobic exercise on glycemic control in type 1 diabetic children, adolescents and young adults without diabetes complications. Glycemic control was measured with glycated haemoglobin A1c (HbA1c). As the HbA1c provides an average of blood glucose control over a 12-wk period, we included studies with an intervention lasting at least 12 wk if the HbA1c was
536 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Lukács A et al . Glycemic control and type of exercise

glycemic control, exercise three times a week (one hour) for three months had no significant impact on glycemic control, but exercise four times a week (one hour) for 6 mo resulted in a significant improvement[60]. Neither guided or self-directed home-based aerobic exercise program nor supervised Pilates exercise had a significant effect on glycosylated haemoglobin after a 12-wk session[62,63].
Study investigating anaerobic exercise: We found only one study that measured the efficacy of resistance (anaerobic) and aerobic exercise in parallel. Both groups were trained three times a week (40 min) for 12 wk under supervision by a physical trainer and an endocrinologist. Interestingly, the HbA1c increased in the aerobic group significantly after 12 wk, whereas a non-significant reduction was observed in the resistance training group[64].
Studies investigating combined exercise program: Four randomised controlled trials examined the effect of combined (aerobic and anaerobic) exercise on glycemic control[30,65-67]. Supervised combined aerobic and resis-tance training twice a week (70 min) for 20 wk resulted in no significant change in glycemic control[65]. In a 12-wk supervised training followed by a 12-wk unsupervised training, HbA1c level remained unchanged regardless of the baseline HbA1c level[30]. There were two trials that explored the effect of supervised combined exercise on glycemic control for a longer duration (6 mo)[66,67]. The Egyptian study found significant improvements in HbA1c level in both training groups exercising three times a week and once a week. However, patients exercising three times a week produced significantly greater improvements[66]. In the Heyman’s study, the participants completed a two-hour supervised training and a one-hour unsupervised training a week including aerobic and strength exercises. The author could not prove a significant effect on glycated hemoglobin after a 6-mo session[67].
DISCUSSIONThe primary aim of diabetes treatment and care is to achieve as stable glycemic control as possible in order to prevent or delay the long-term complications[69]. Regular exercise is recommended for diabetic youths for several psychological and health benefits[7,70]. It is evidence-based that regular exercise has a preventive and curative role in type 2 diabetes, but its physiological role in type 1 diabetes is not fully explored. Exercise physiology in diabetes is described thoroughly by Robertson et al[7] and Riddell et al[53,59]. Another issue that remains unresolved is related to the type of exercise: aerobic or anaerobic workout is most beneficial for glycemic control. We evaluated the studies from the previous 15 years that investigated the long-term effect of exercise on glycemic control in youths with T1DM according
measured before and after the intervention. The flow diagram of study selection is shown in Figure 1.
Statistical analysisThis manuscript does not describe basic or clinical research using biostatistics for raw data, therefore biostatistics statement is not relevant.
RESULTSA total of nine articles was evaluated from nine different countries including Australia[30], Tunisia[60], Lithuania[61], Taiwan[62], Turkey[63], Brazilia[64], Belgium[65], Egypt[66] and France[67], and they were published between the years of 2002-2011. The list of articles is presented in Table 1. Across included studies the total sample size was 401 (166 males and 235 females) with an age range of 10-19 years except one study, in which the age range was 13-30 years[64]. Four studies were aerobic-based[60-63], four were combined aerobic and anaerobic programs[30,65-67], and one compared aerobic exercise to anaerobic one[64]. The Turkish study[63] conducted a Pilates program that is considered low-impact aerobic workout[68]. All studies had diabetic control group except one[61].
Effects of different types of exercise on glycemic control in youths with T1DMStudies investigating aerobic exercise: Studies measuring the effect of aerobic exercise on glycemic control presented variable results[60-63]. Only one study recognised a significant improvement in HbA1c after 12-wk exercise (girls swimming twice a week for 45 min a week)[61]. In a study investigating the impact of the frequency of the supervised aerobic training on
537 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Articles identified from electronic search n = 685
Excluded at first screening (title) n = 456
Abstracts screened n = 229
Addition articles from reference lists n = 2
Excluded at second screening: Based on abstract n = 128
Duplicates n = 56
Total articles included n = 9 with total sample size of 401
Excluded at final screening n = 38
Total articles evaluated n = 47
Figure 1 Flow diagram illustrating selection of the included studies.
Lukács A et al . Glycemic control and type of exercise

to t
he m
ajor
met
abol
ic p
athw
ay in
volv
ed in
ene
rgy
utiliza
tion
(pre
dom
inan
tly a
erob
ic o
r pr
edom
inan
tly a
naer
obic).
Res
ults
of
inve
stig
ated
stu
dies
are
ope
n to
deb
ate.
M
ost
stud
ies
foun
d no
sig
nific
ant
impr
ovem
ent
in g
lyce
mic c
ontr
ol r
egar
ding
the
aer
obic
[62,
63] a
nd c
ombi
ned
exer
cise
inte
rven
tion[3
0,65
,67]. Th
e on
ly s
tudy
inve
stig
atin
g th
e an
aero
bic
effe
ct o
n gl
ycem
ic c
ontr
ol p
rese
nted
a p
ositi
ve t
ende
ncy,
alth
ough
the
re w
as n
o st
atistic
ally
sig
nific
ant
effe
ct[6
4].
One
of
the
poss
ible
rea
sons
for
the
se v
arie
d re
sults
cou
ld b
e th
e sm
all s
ampl
e size
s. T
he g
luco
se p
rofil
e va
ries
grea
tly in
pat
ient
s w
ith T
1DM
bef
ore,
dur
ing
and
afte
r ex
ercise
, an
d ca
n be
ver
y di
ffere
nt in
pat
ient
s w
ith s
imila
r HbA
1c. A
ll ex
cept
one
stu
dy[6
6] h
ad le
ss tha
n 20
par
ticip
ants
in a
gro
up in
our
stu
dies
. The
oth
er r
easo
n co
uld
be the
sho
rt p
erio
d. I
t se
ems
that
at le
ast 6
mo
of e
xerc
ise
inte
rven
tion
mig
ht le
ad to
sig
nific
ant r
esul
ts[6
0,66
] . Ken
nedy
et a
l[71] a
lso
notic
ed in
thei
r sy
stem
atic rev
iew
and
met
a-an
alys
is th
at lo
nger
dur
atio
n of
inte
rven
tion
show
s a
tren
d fo
r HbA
1c r
educ
tion.
We
had
a to
tal o
f 401
sam
ples
in o
ur s
tudi
es a
nd t
he E
gypt
ian
stud
y[66] a
lone
offe
red
187
part
icip
ants
. W
eigh
ting
its r
esul
ts, it
mig
ht
be s
uppo
sed
that
com
bine
d lo
ng-t
erm
exe
rcise
inte
rven
tion
is m
ore
bene
ficia
l for
the
gly
cem
ic c
ontr
ol, th
an a
erob
ic o
r an
aero
bic
alon
e. I
rvin
e et
al[7
2] in
the
ir sy
stem
atic
revi
ew w
ith 3
72 p
atie
nts
with
T2D
M h
ave
repo
rted
that
(an
aero
bic)
pro
gres
sive
resist
ance
exe
rcise
is n
ot s
igni
fican
tly b
ette
r tha
n ae
robi
c ex
ercise
in im
prov
ing
glyc
osyl
ated
ha
emog
lobi
n. T
here
was
a la
ck o
f evi
denc
e to
sug
gest
tha
t on
e ty
pe o
f exe
rcise
was
bet
ter
than
ano
ther
[72]. Y
ang
et a
l[73] in
the
ir sy
stem
atic r
evie
w w
ith 5
95 p
atie
nts
with
538 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Sour
ceRC
TO
bjec
tive
Sam
ple
size
/gen
der
Age
(y/
o) g
ende
rIn
terv
ention
Dur
atio
n, f
requ
ency
Sign
ifica
nt p
ositiv
e ch
ange
in G
C
Aou
adi e
t al[6
0], (
2011
) Tu
nisi
anN
oEf
fect
of a
erob
ic tr
aini
ng o
n gl
ycem
ic
cont
rol a
nd li
pid
profi
leEG
1: 1
1 (tw
ice)
EG2:
11
(4 ti
mes
) D
C: 1
1 no
exe
rc
12-1
433
MSu
perv
ised
aer
obic
exe
rcis
e3
and
6 m
o, tw
ice
vs fo
ur ti
mes
a
wee
k (6
0 m
in)
Yes,
but
onl
y 6
mo
with
four
tim
es a
wee
k du
ratio
n
Side
ravi
ciūt
e et
al[6
1], (
2006
)Li
thua
nian
No
Effe
ct o
f lon
g-te
rm p
hysi
cal a
ctiv
ity in
w
ater
on
gluc
ose
cont
rol
EG: 1
9(H
C: 2
1)14
-19
19 F
Supe
rvis
ed a
erob
ic e
xerc
ise
14 w
k, tw
ice
a w
eek
(45
min
)Ye
s
Won
g et
al[6
2], (
2010
)Ta
iwan
No
Effe
ct o
f hom
e-ba
sed
exer
cise
pr
ogra
mm
e on
HbA
1c a
nd p
eak
oxyg
en u
ptak
e
EG: 1
2 vi
deo-
, 5 s
elf
dire
cted
DC
: 11
7-17
8 M
, 20
FU
nsup
ervi
sed
aero
bic
exer
cise
12
wk,
thre
e tim
es a
wee
k (3
0 m
in)
No
Tuna
r et a
l[63], (
2012
)Tu
rkis
hYe
sEf
fect
of p
ilate
s tr
aini
ng o
n m
etab
olic
co
ntro
l and
phy
sica
l per
form
ance
EG
: 17
DC
: 14
12-1
715
M, 1
6 F
Supe
rvis
ed a
erob
ic e
xerc
ise
12 w
k, th
ree
times
a w
eek
(40
min
)N
o
Ram
alho
et a
l[64], (
2006
)Br
azili
anYe
sEf
fect
of a
erob
ic v
s res
ista
nce
trai
ning
on
met
abol
ic c
ontr
ol
EG: 7
DC
: 613
-30
3 M
, 10
FBo
th a
erob
ic a
nd a
naer
obic
ex
erci
se w
ere
supe
rvis
ed12
wk,
thre
e tim
es a
wee
k (4
0 m
in)
Sig.
incr
ease
in a
erob
ic g
roup
, not
si
g. d
ecre
ase
in a
naer
obic
gro
upD
'hoo
ge et
al[6
5], (
2011
)Be
lgia
nYe
sEf
fect
of c
ombi
ned
exer
cise
trai
ning
on
met
abol
ic c
ontr
ol, p
hysi
cal fi
tnes
s an
d qu
ality
of l
ife
EG: 8
DC
: 810
-17
7 M
, 9 F
Supe
rvis
edco
mbi
ned
exer
cise
20 w
k, tw
ice
a w
eek
(70
min
)N
o
Robe
rts
et a
l[30], (
2002
)A
ustr
alia
nYe
sW
heth
er th
e ch
ange
of t
he g
lyce
mic
co
ntro
l afte
r int
erve
ntio
n is
dep
ende
nt
on th
e in
itiat
ion
of q
ualit
y of
gly
cem
ic
cont
rol
EG1:
12
HbA
1c >
9%
EG2:
12
HbA
1c <
9%
DC
1: 1
2 H
bA1c
> 9
%
DC
2: 1
2 H
bA1c
< 9
%
Cir
ca 1
1-17
24 M
, 24
F12
wk
supe
rvis
ed 1
2 w
k un
supe
r-vi
sed
com
bine
d ex
erci
se
24 w
k, th
ree
times
a w
eek
(45
min
)N
o
Sale
m et
al[6
6], (
2010
)Eg
yptia
nYe
sEf
fect
of e
xerc
ise
on g
lyce
mic
con
trol
, pl
asm
a lip
ids,
blo
od p
ress
ure,
sev
erity
an
d fr
eque
ncy
of h
ypog
lyce
mia
, an
thro
pom
etri
c m
easu
rem
ents
and
in
sulin
dos
e
EG1:
75
(onc
e)EG
2: 7
3 (3
tim
es)
DC
: 48
12-1
876
M, 1
21 F
Supe
rvis
ed c
ombi
ned
exer
cise
6 m
o, o
nce
and
thre
e tim
es a
w
eek
(70
min
)Ye
s, in
bot
h ex
erci
se g
roup
s
Hey
man
et a
l[67], (
2007
)Fr
ench
Yes
Effe
ct o
f exe
rcis
e on
qua
lity
of li
fe,
phys
ical
fitn
ess,
bod
y co
mpo
sitio
n,
lipid
and
apo
lipop
rote
in p
rofil
es, a
nd
adip
onec
tin a
nd le
ptin
leve
ls
EG: 9
DC
: 713
-18
16 F
Supe
rvis
ed c
ombi
ned
exer
cise
6 m
o, 2
h s
uper
vise
d, a
nd 1
h
unsu
perv
ised
No
Tabl
e 1 St
udy
char
acte
rist
ics,
obj
ective
and
res
ult
rega
rdin
g gl
ycem
ic c
ontr
ol
HbA
1c: H
aem
oglo
bin
A1c
; RC
T: R
ando
miz
ed c
ontr
olle
d tr
ial;
EG: E
xerc
ise
grou
p; D
C: D
iabe
tic c
ontr
ol; H
C: H
ealth
y co
ntro
l; M
: Mal
es; F
: Fem
ales
; y/o
: Yea
rs o
ld; G
C: G
lyce
mic
con
trol
; sig
.: Si
gnifi
cant
.
Lukács A et al . Glycemic control and type of exercise

T2DM also enunciated that either form of exercise appears to have comparable effects on glycemic control. A meta-analysis by Tonoli et al[74] with type 1 diabetic participants explored a tendency for improvement in glycaemic control due to aerobic or combined training, but they could not confirm this statistically. Individual studies on aerobic training had no significant evidence, but “in total” demonstrated a reduction in HbA1c, although they carefully interpret their results because of insufficient data on these topics[74]. Neither Kennedy’s meta-analysis in children and adults[71] nor Kavookjian’s systematic review in the adult population[75] revealed evidence for statistically significant glycemic benefits of exercise. Kennedy et al[71] suggest that HbA1c may not be a sensitive indicator of glycaemic control, and that improvement in glycaemic variability may not be reflected in this measure. Tight metabolic control is very important for diabetic individuals, and the regular physical exercise is part of the diabetes management. Available studies provide insufficient evidence that any type of exercise or combined training would clearly improve the glycemic control in type 1 diabetic young patients. Long-term exercise-induced glycemic benefits are based on the continuous effect of each successive bout of exercise. Glycemic variability can be detected by continuous glucose monitoring that could help individuals to learn their own response to different type of physical exercise; and it might be a more appropriate marker to explore the exercise-induced beneficial effect than HbA1c[71]. Youths with T1DM are recommended to be engaged in a sport they like, and do it regularly as much as possible at the same period of the day. Having the appropriate exercise physiology knowledge, they can accumulate own experiences regarding exercise in addition to insulin requirement and dietary program that may result in stable glucose levels. The insulin-diet-exercise adjustment must be personalized and discussed with the patient’s endocrinologist. The regular exercise is associated with numerous health benefits and chronically ill youths enjoy the same benefits as the healthy counterparts[1,70].
There are miscellaneous results regarding the long-term effect of various forms of exercise on glycemic control. Meta-analyses also suffered from the insufficient randomised controlled trials with greater sample sizes to examine the short- and long-term effect of glycemic control on different exercise modalities in youths with T1DM. Patients and health professionals may want more information they currently received, but there are concerns that the results would be overestimated. Different forms of exercise generate different skills in youths. They need a comprehensive form of exercise to help their healthy many-sided physical development. Diabetic patients should be aware that glycemic control is influenced by multiple factors (initial blood glucose level, insulin absorption, time of the day), and shows individual responses to exercise. It is advisable that young patients choose some kind of sports or physical exercise they like and be engaged with it for a longer
time. Clinical treatment and management should be adjusted to the exercise form and the diabetic patients should acquire how to change nutrition and insulin dose according to their daily physical exercise. Patients who can monitor themselves intensively around periods of activity can learn how to keep glucose levels in an acceptable range.
Although current studies have insufficient evidence of the beneficial effect of any type of exercise on glycemic control, any type of regular physical exercise has several other valuable physiological and health benefits that justify the inclusion of exercise in pediatric diabetes treatment and care.
ACKNOWLEDGMENTSThis research was partially carried out with the con-tribution of the Workgroup for Health Sciences, Center of Excellence of Applied Materials Science and Nano-Technology at the University of Miskolc.
COMMENTSBackgroundRegular exercise is recommended for diabetic youth for several psychological and health benefits, but its physiological role in type 1 diabetes is not fully explored. So far, there are only few studies evaluating the long-term effect of different types of exercise on glycemic control.Research frontiersIn this systematic literature review, the authors evaluated the latest studies examining the long-term effect of aerobic and/or anaerobic exercise on glycemic control in youths with type 1 diabetes. They also made distinguishing between concepts of physical activity, physical fitness, and aerobic-anaerobic exercise. Innovations and breakthroughsMost studies found no significant improvement in glycemic control regarding the aerobic and combined exercise intervention. The only one study evaluating anaerobic exercise observed a non-significant reduction in glycemic control. It might be supposed that longer duration (at least 6 mo) and more frequent (more than twice a week) exercise has a positive effect on glycemic control, but it is not proved yet. Applications Although current studies have insufficient evidence of the beneficial effect of any type of exercise on glycemic control, any type of regular physical exercise has several other valuable physiological and health benefits that justify the inclusion of exercise in pediatric diabetes treatment and care.TerminologyAerobic exercise includes any type of exercise, typically those performed at moderate levels of intensity for extended periods of time that maintains an increased heart rate. Anaerobic exercise is used to promote strength, power, and speed. Generally, it has a short duration and high intensity activity. Unlike aerobic exercise, it does not depend on exogenous oxygen. Peer-reviewThe manuscript covers an important topic in type 1 diabetes management. The contribution that the manuscript provides is an overview of the dearth of scientific evidence to support exercise intervention in this population.
REFERENCES1 Janssen I, Leblanc AG. Systematic review of the health benefits
of physical activity and fitness in school-aged children and youth. Int J Behav Nutr Phys Act 2010; 7: 40 [PMID: 20459784 DOI: 10.1186/1479-5868-7-40]
539 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
COMMENTS
Lukács A et al . Glycemic control and type of exercise

2 Tremblay MS, LeBlanc AG, Kho ME, Saunders TJ, Larouche R, Colley RC, Goldfield G, Connor Gorber S. Systematic review of sedentary behaviour and health indicators in school-aged children and youth. Int J Behav Nutr Phys Act 2011; 8: 98 [PMID: 21936895 DOI: 10.1186/1479-5868-8-98]
3 Downward P, Rasciute S. Does sport make you happy? An analysis of the well being derived from sports participation. Int Rev Appl Econ 2011; 25: 331-348 [DOI: 10.1080/02692171.2010.511168]
4 Eime RM, Young JA, Harvey JT, Charity MJ, Payne WR. A systematic review of the psychological and social benefits of participation in sport for children and adolescents: informing development of a conceptual model of health through sport. Int J Behav Nutr Phys Act 2013; 10: 98 [PMID: 23945179 DOI: 10.1186/1479-5868-10-98]
5 Zinman B, Ruderman N, Campaigne BN, Devlin JT, Schneider SH. Physical activity/exercise and diabetes mellitus. Diabetes Care 2003; 26 Suppl 1: S73-S77 [PMID: 12502622 DOI: 10.2337/diacare.27.2007.S58]
6 Giannini C, de Giorgis T, Mohn A, Chiarelli F. Role of physical exercise in children and adolescents with diabetes mellitus. J Pediatr Endocrinol Metab 2007; 20: 173-184 [PMID: 17396433 DOI: 10.1515/JPEM.2007.20.2.173]
7 Robertson K, Adolfsson P, Scheiner G, Hanas R, Riddell MC. Exercise in children and adolescents with diabetes. Pediatr Diabetes 2009; 10 Suppl 12: 154-168 [PMID: 19754626 DOI: 10.1111/j.1399-5448.2009.00567.x]
8 Leclair E, de Kerdanet M, Riddell M, Heyman E. Type 1 Diabetes and Physical Activity in Children and Adolescents. J Diabetes Metab 2013; S10: 004 [DOI: 10.4172/2155-6156.S10-004]
9 Silverstein J, Klingensmith G, Copeland K, Plotnick L, Kaufman F, Laffel L, Deeb L, Grey M, Anderson B, Holzmeister LA, Clark N. Care of children and adolescents with type 1 diabetes: a statement of the American Diabetes Association. Diabetes Care 2005; 28: 186-212 [PMID: 15616254 DOI: 10.2337/diacare.28.1.186]
10 Tipton CM. Susruta of India, an unrecognized contributor to the history of exercise physiology. J Appl Physiol (1985) 2008; 104: 1553-1556 [PMID: 18356481 DOI: 10.1152/japplphysiol.00925.2007]
11 Allen FM. Note concerning exercise in the treatment of severe diabetes. Boston Med Surg J 1915; 173: 743-744 [DOI: 10.1056/NEJM191511111732005]
12 Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. Pancreatic extracts in the treatment of diabetes mellitus. 1922. Indian J Med Res 2007; 125: 141-146 [PMID: 17580419]
13 Lawrence RD. The Effect of Exercise on Insulin Action in Diabetes. Br Med J 1926; 1: 648-650 [PMID: 20772477 DOI: 10.1136/bmj.1.3406.648]
14 Joslin EP. The Prevention of Diabetes Mellitus. JAMA 1922; 76: 79-84 [DOI: 10.1001/jama.1921.02630020001001]
15 Herbst A, Bachran R, Kapellen T, Holl RW. Effects of regular physical activity on control of glycemia in pediatric patients with type 1 diabetes mellitus. Arch Pediatr Adolesc Med 2006; 160: 573-577 [PMID: 16754817]
16 Miculis CP, Mascarenhas LP, Boguszewski MC, Campos Wd. Physical activity in children with type 1 diabetes. J Pediatr (Rio J) 2010; 86: 271-278 [PMID: 20711549 DOI: 10.2223/JPED.2003]
17 Schmitz KH, Jacobs DR, Hong CP, Steinberger J, Moran A, Sinaiko AR. Association of physical activity with insulin sensitivity in children. Int J Obes Relat Metab Disord 2002; 26: 1310-1316 [PMID: 12355326 DOI: 10.1038/sj.ijo.0802137]
18 American Diabetes Association. Physical activity/exercise and diabetes mellitus. Diabetes Care 2004; 27 (Suppl 1): S58-S62
19 Suh SH, Paik IY, Jacobs K. Regulation of blood glucose homeostasis during prolonged exercise. Mol Cells 2007; 23: 272-279 [PMID: 17646701]
20 Giannini C, Mohn A, Chiarelli F. Physical exercise and diabetes during childhood. Acta Biomed 2006; 77 Suppl 1: 18-25 [PMID: 16918068]
21 Riddell M, Perkins BA. Exercise and glucose metabolism in persons with diabetes mellitus: perspectives on the role for continuous glucose monitoring. J Diabetes Sci Technol 2009; 3: 914-923 [PMID: 20144341 DOI: 10.1177/193229680900300439]
22 Zoppini G, Carlini M, Muggeo M. Self-reported exercise and quality of life in young type 1 diabetic subjects. Diabetes Nutr Metab 2003; 16: 77-80 [PMID: 12848309]
23 Massin MM, Lebrethon MC, Rocour D, Gérard P, Bourguignon JP. Patterns of physical activity determined by heart rate monitoring among diabetic children. Arch Dis Child 2005; 90: 1223-1226 [PMID: 15941770 DOI: 10.1136/adc.2005.075283]
24 Valerio G, Spagnuolo MI, Lombardi F, Spadaro R, Siano M, Franzese A. Physical activity and sports participation in children and adolescents with type 1 diabetes mellitus. Nutr Metab Cardiovasc Dis 2007; 17: 376-382 [PMID: 17562573 DOI: 10.1016/j.numecd.2005.10.012]
25 Schweiger B, Klingensmith G, Snell-Bergeon JK. Physical activity in adolescent females with type 1 diabetes. Int J Pediatr 2010; 2010: 328318 [PMID: 20652080]
26 Lukács A, Mayer K, Juhász E, Varga B, Fodor B, Barkai L. Reduced physical fitness in children and adolescents with type 1 diabetes. Pediatr Diabetes 2012; 13: 432-437 [PMID: 22353226 DOI: 10.1111/j.1399-5448.2012.00848]
27 Lukács A, Mayer K, Török A, Kiss-Tóth E, Barkai L. Better cardiorespiratory fitness associated with favourable metabolic control and health-related quality of life in youths with type 1 diabetes mellitus. Acta Physiol Hung 2013; 100: 77-83 [PMID: 23471043 DOI: 10.1556/APhysiol.100.2013.1.7]
28 Zinman B, Zuniga-Guajardo S, Kelly D. Comparison of the acute and long-term effects of exercise on glucose control in type I diabetes. Diabetes Care 1984; 7: 515-519 [PMID: 6439529 DOI: 10.2337/diacare.7.6.515]
29 Raile K, Kapellen T, Schweiger A, Hunkert F, Nietzschmann U, Dost A, Kiess W. Physical activity and competitive sports in children and adolescents with type 1 diabetes. Diabetes Care 1999; 22: 1904-1905 [PMID: 10546027 DOI: 10.2337/diacare.22.11.1904]
30 Roberts L, Jones TW, Fournier PA. Exercise training and glycemic control in adolescents with poorly controlled type 1 diabetes mellitus. J Pediatr Endocrinol Metab 2002; 15: 621-627 [PMID: 12014521 DOI: 10.1515/JPEM.2002.15.5.621]
31 Särnblad S, Ekelund U, Aman J. Physical activity and energy intake in adolescent girls with Type 1 diabetes. Diabet Med 2005; 22: 893-899 [PMID: 15975105 DOI: 10.1111/j.1464-5491.2005.01544.x]
32 Aman J, Skinner TC, de Beaufort CE, Swift PG, Aanstoot HJ, Cameron F. Associations between physical activity, sedentary behavior, and glycemic control in a large cohort of adolescents with type 1 diabetes: the Hvidoere Study Group on Childhood Diabetes. Pediatr Diabetes 2009; 10: 234-239 [PMID: 19140898 DOI: 10.1111/j.1399-5448.2008.00495.x]
33 Edmunds S, Roche D, Stratton G. Levels and patterns of physical activity in children and adolescents with type 1 diabetes and associated metabolic and physiologic health outcomes. J Phys Act Health 2010; 7: 68-77 [PMID: 20231757]
34 Galler A, Lindau M, Ernert A, Thalemann R, Raile K. Asso-ciations between media consumption habits, physical activity, socioeconomic status, and glycemic control in children, adolescents, and young adults with type 1 diabetes. Diabetes Care 2011; 34: 2356-2359 [PMID: 21926289 DOI: 10.2337/dc11-0838]
35 Caspersen CJ, Powell KE, Christenson GM. Physical activity, exercise, and physical fitness: definitions and distinctions for health-related research. Public Health Rep 1985; 100: 126-131 [PMID: 3920711]
36 Twisk JW, Kemper HC, van Mechelen W. Tracking of activity and fitness and the relationship with cardiovascular disease risk factors. Med Sci Sports Exerc 2000; 32: 1455-1461 [PMID: 10949012 DOI: 10.1097/00005768-200008000-00014]
37 Mikkelsson L, Kaprio J, Kautiainen H, Kujala U, Mikkelsson M, Nupponen H. School fitness tests as predictors of adult
540 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Lukács A et al . Glycemic control and type of exercise

health-related fitness. Am J Hum Biol 2006; 18: 342-349 [PMID: 16634020 DOI: 10.1002/ajhb.20498]
38 Vanhees L, Lefevre J, Philippaerts R, Martens M, Huygens W, Troosters T, Beunen G. How to assess physical activity? How to assess physical fitness? Eur J Cardiovasc Prev Rehabil 2005; 12: 102-114 [PMID: 15785295 DOI: 10.1097/01.hjr.0000161551.73095.9c]
39 Mitchell JH, Haskell W, Snell P, Van Camp SP. Task Force 8: classification of sports. J Am Coll Cardiol 2005; 45: 1364-1367 [PMID: 15837288 DOI: 10.1016/j.jacc.2005.02.015]
40 Rodriguez NR, Di Marco NM, Langley S. American College of Sports Medicine position stand. Nutrition and athletic performance. Med Sci Sports Exerc 2009; 41: 709-731 [PMID: 19225360 DOI: 10.1249/MSS.0b013e31890eb86]
41 Kilgore L, Rippetoe M. Redefining fitness for health and fitness professionals. J Exerc Physiol 2007; 10: 34-39
42 Plowman S, Smith D. Exercise Physiology for Health, Fitness, and Performance. 4th ed. Lippincott Williams & Wilkins, 2004
43 Romijn JA, Coyle EF, Sidossis LS, Rosenblatt J, Wolfe RR. Substrate metabolism during different exercise intensities in endurance-trained women. J Appl Physiol (1985) 2000; 88: 1707-1714 [PMID: 10797133]
44 Dickhuth HH, Scharhag J, Röcker K, König D. Cardiovascular adaptation and exercise. Int SportMed J 2012; 13: 1-7
45 Kenney WL, Wilmore JH, Costill DL. Physiology of Sport and Exercise. 5th ed. Human Kinetics: Champaign, IL, 2011
46 Mahon AD, Marjerrison AD, Lee JD, Woodruff ME, Hanna LE. Evaluating the prediction of maximal heart rate in children and adolescents. Res Q Exerc Sport 2010; 81: 466-471 [PMID: 21268470 DOI: 10.1080/02701367.2010.10599707]
47 Machado FA, Denadai BS. Validity of maximum heart rate prediction equations for children and adolescents. Arq Bras Cardiol 2011; 97: 136-140 [PMID: 21739069 DOI: 10.1590/S0066-782X2011005000078]
48 American College of Sports Medicine. ACSM’s Guidelines for Exercise Testing and Prescription. 9th ed. Baltimore (MD): Lippincott Williams & Wilkins, 2013: 162-174
49 Yardley JE, Kenny GP, Perkins BA, Riddell MC, Malcolm J, Boulay P, Khandwala F, Sigal RJ. Effects of performing resistance exercise before versus after aerobic exercise on glycemia in type 1 diabetes. Diabetes Care 2012; 35: 669-675 [PMID: 22374639 DOI: 10.2337/dc11-1844]
50 Apor P. [Physical activity in prevention and treatment of diabetes]. Orv Hetil 2009; 150: 579-587 [PMID: 19293059 DOI: 10.1556/OH.2009.28550]
51 Francescato MP, Geat M, Fusi S, Stupar G, Noacco C, Cattin L. Carbohydrate requirement and insulin concentration during moderate exercise in type 1 diabetic patients. Metabolism 2004; 53: 1126-1130 [PMID: 15334372 DOI: 10.1016/j.metabol.2004.03.015]
52 Guelfi KJ, Jones TW, Fournier PA. The decline in blood glucose levels is less with intermittent high-intensity compared with moderate exercise in individuals with type 1 diabetes. Diabetes Care 2005; 28: 1289-1294 [PMID: 15920041 DOI: 10.2337/diacare.28.6.1289]
53 Riddell M, Perkins B. Type 1 diabetes and vigorous exercise: Applications of exercise physiology to patient management. Can J Diab 2006; 30: 63-71 [DOI: 10.1016/S1499-2671(06)01010-0]
54 Iscoe KE, Campbell JE, Jamnik V, Perkins BA, Riddell MC. Efficacy of continuous real-time blood glucose monitoring during and after prolonged high-intensity cycling exercise: spinning with a continuous glucose monitoring system. Diabetes Technol Ther 2006; 8: 627-635 [PMID: 17109594]
55 Mullooly CA, Kemmis KL. Diabetes Educators and the Exercise prescription. Diabetes Spectr 2005; 18: 108-113 [DOI: 10.2337/diaspect.18.2.108]
56 Hornsby WG, Chetlin RD. Management of competitive athletes with diabetes. Diabetes Spectr 2005; 18: 102-107 [DOI: 10.2337/diaspect.18.2.102]
57 Draznin MB. Managing the adolescent athlete with type 1 diabetes mellitus. Pediatr Clin North Am 2010; 57: 829-837 [PMID:
20538159 DOI: 10.1016/j.pcl.2010.02.003]58 Gallen I. Type 1 Diabetes. Clinical management of the Athlete.
Springer-Verlag London Limited, 2012: 73-99 [DOI: 10.1007/978-0-85729-754-9]
59 Riddell MC, Iscoe KE. Physical activity, sport, and pediatric diabetes. Pediatr Diabetes 2006; 7: 60-70 [PMID: 16489976 DOI: 10.1111/j.1399-543X.2006.00146.x]
60 Aouadi R, Khalifa R, Aouidet A, Ben Mansour A, Ben Rayana M, Mdini F, Bahri S, Stratton G. Aerobic training programs and glycemic control in diabetic children in relation to exercise frequency. J Sports Med Phys Fitness 2011; 51: 393-400 [PMID: 21904277]
61 Sideraviciūte S, Gailiūniene A, Visagurskiene K, Vizbaraite D. The effect of long-term swimming program on glycemia control in 14-19-year aged healthy girls and girls with type 1 diabetes mellitus. Medicina (Kaunas) 2006; 42: 513-518 [PMID: 16816547]
62 Wong CH, Chiang YC, Wai JP, Lo FS, Yeh CH, Chung SC, Chang CW. Effects of a home-based aerobic exercise programme in children with type 1 diabetes mellitus. J Clin Nurs 2011; 20: 681-691 [PMID: 21320197 DOI: 10.1111/j.1365-2702.2010.03533.x]
63 Tunar M, Ozen S, Goksen D, Asar G, Bediz CS, Darcan S. The effects of Pilates on metabolic control and physical performance in adolescents with type 1 diabetes mellitus. J Diabetes Complications 2012; 26: 348-351 [PMID: 22609217 DOI: 10.1016/j.jdiacomp.2012.04.006]
64 Ramalho AC, de Lourdes Lima M, Nunes F, Cambuí Z, Barbosa C, Andrade A, Viana A, Martins M, Abrantes V, Aragão C, Temístocles M. The effect of resistance versus aerobic training on metabolic control in patients with type-1 diabetes mellitus. Diabetes Res Clin Pract 2006; 72: 271-276 [PMID: 16406128 DOI: 10.1016/j.diabres.2005.11.011]
65 D’hooge R, Hellinckx T, Van Laethem C, Stegen S, De Schepper J, Van Aken S, Dewolf D, Calders P. Influence of combined aerobic and resistance training on metabolic control, cardiovascular fitness and quality of life in adolescents with type 1 diabetes: a randomized controlled trial. Clin Rehabil 2011; 25: 349-359 [PMID: 21112904 DOI: 10.1177/0269215510386254]
66 Salem MA, Aboelasrar MA, Elbarbary NS, Elhilaly RA, Refaat YM. Is exercise a therapeutic tool for improvement of cardiovascular risk factors in adolescents with type 1 diabetes mellitus? A randomised controlled trial. Diabetol Metab Syndr 2010; 2: 47 [PMID: 20618996 DOI: 10.1186/1758-5996-2-47]
67 Heyman E, Toutain C, Delamarche P, Berthon P, Briard D, Youssef H, Dekerdanet M, Gratas-Delamarche A. Exercise training and cardiovascular risk factors in type 1 diabetic adolescent girls. Pediatr Exerc Sci 2007; 19: 408-419 [PMID: 18089908]
68 Adamany K, Loigerot D. The Pilates Edge: An Athlete’s Guide to Strength and Performance. New York, USA: Penguin Group Inc, 2004: 17
69 American Diabetes Association. Standards of Medical Care in Diabetes - 2013. Diabetes Care 2013; 36 (Suppl 1): S11-S66 [DOI: 10.2337/dc13-S011]
70 Chimen M, Kennedy A, Nirantharakumar K, Pang TT, Andrews R, Narendran P. What are the health benefits of physical activity in type 1 diabetes mellitus? A literature review. Diabetologia 2012; 55: 542-551 [PMID: 22189486 DOI: 10.1007/s00125-011-2403-2]
71 Kennedy A, Nirantharakumar K, Chimen M, Pang TT, Hemming K, Andrews RC, Narendran P. Does exercise improve glycaemic control in type 1 diabetes? A systematic review and meta-analysis. PLoS One 2013; 8: e58861 [PMID: 23554942 DOI: 10.1371/journal.pone.0058861]
72 Irvine C, Taylor NF. Progressive resistance exercise improves glycaemic control in people with type 2 diabetes mellitus: a systematic review. Aust J Physiother 2009; 55: 237-246 [PMID: 19929766 DOI: 10.1016/S0004-9514(09)70003-0]
73 Yang Z, Scott CA, Mao C, Tang J, Farmer AJ. Resistance exercise versus aerobic exercise for type 2 diabetes: a systematic review and meta-analysis. Sports Med 2014; 44: 487-499 [PMID: 24297743 DOI: 10.1007/s40279-013-0128-8]
74 Tonoli C, Heyman E, Roelands B, Buyse L, Cheung SS, Berthoin S,
541 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Lukács A et al . Glycemic control and type of exercise

Meeusen R. Effects of different types of acute and chronic (training) exercise on glycaemic control in type 1 diabetes mellitus: a meta-analysis. Sports Med 2012; 42: 1059-1080 [PMID: 23134339 DOI: 10.2165/11635380-000000000-00000]
75 Kavookjian J, Elswick BM, Whetsel T. Interventions for being active among individuals with diabetes: a systematic review of the literature. Diabetes Educ 2007; 33: 962-988; discussion 989-990 [PMID: 18057265 DOI: 10.1177/0145721707308411]
P- Reviewer: Faulkner MS, Sanlioglu AD S- Editor: Ji FF L- Editor: Wang TQ E- Editor: Liu SQ
542 April 15, 2015|Volume 6|Issue 3|WJD|www.wjgnet.com
Lukács A et al . Glycemic control and type of exercise

© 2015 Baishideng Publishing Group Inc. All rights reserved.
Published by Baishideng Publishing Group Inc8226 Regency Drive, Pleasanton, CA 94588, USA
Telephone: +1-925-223-8242Fax: +1-925-223-8243
E-mail: [email protected] Desk: http://www.wjgnet.com/esps/helpdesk.aspx
http://www.wjgnet.com




















