
Determination of Molecular Weight Distribution and ... - Nature

REVIEW
www.nature.com/natureimmunology • march 2003 • volume 4 no 3 • nature immunology 217
Considerable progress has been made in charac-terizing four key sets of interactions controllingantigen responsiveness in T cells, involving the fol-lowing: the T cell antigen receptor, its corecep-tors CD4 and CD8, the costimulatory receptorsCD28 and CTLA-4, and the accessory moleculeCD2. Complementary work has defined the gen-eral biophysical properties of interactionsbetween cell surface molecules.Among the majorconclusions are that these interactions are struc-turally heterogeneous, often reflecting clear-cutfunctional constraints, and that, although they allinteract relatively weakly, hierarchical differencesin the stabilities of the signaling complexesformed by these molecules may influence thesequence of steps leading to T cell activation.Here we review these developments and high-light the major challenges remaining as the fieldmoves toward formulating quantitative models ofT cell recognition.
1Nuffield Department of Clinical Medicine and 2MRC Human Immunology Unit,Weatherall Institute of Molecular Medicine,The University of Oxford, John Radcliffe Hospital,Headington, Oxford, OX3 9DU, UK. 3Division of Structural Biology, Graduate School of Pharmaceutical Sciences, Kumamoto University, 5-1 Oe-honmachi, Kumamoto 862-0973, Japan. 4Sir William Dunn School of Pathology,The University of Oxford, South Parks Road, Oxford, OX1 3RE, UK. Correspondence should be addressed to S.J.D. and
P.A.v.d.M. ([email protected] and [email protected]).
The nature of molecular recognition by T cells
Simon J. Davis1, Shinji Ikemizu3, Edward J. Evans1, Lars Fugger2,Talitha R. Bakker4 and P. Anton van der Merwe4
The analysis of leukocyte cell surface molecules has been a key factordriving our understanding of immunological phenomena. The identifi-cation of these molecules began with the characterization of allotypicdifferences between inbred mouse strains, and was transformed by theadvent of monoclonal antibody technology1. The antibody approachhas been so successful that by the time of the next Leukocyte TypingWorkshop, to be held in 2004, it is possible that more than 350 clusterof differentiation (CD) antigens will have been identified on leuko-cytes. Protein sequencing and conventional gene cloning provided thefirst insights into the structures of these molecules, emphasizing in par-ticular the ubiquity of the immunoglobulin superfamily (IgSF)2.Expression cloning3 greatly accelerated the isolation of the genesencoding these molecules during the early 1990s, although the rate ofgene identification using these methods has since declined4 in favor ofgenomics-based approaches.
Soluble forms of B cell antigen receptors (that is, Bence-Jones pro-teins and antibody Fab fragments) were the first leukocyte recognition
structures determined, preceding the crystal structure of the iconichuman leukocyte antigen (HLA)-A2 peptide complex and major histo-compatibility complex (MHC) class II, CD4 and CD8 structures. 1H-Nuclear magnetic resonance (NMR) spectroscopy5 and crystallograph-ic6 analyses of rat CD2 yielded the earliest structures of a cell surfaceprotein not directly involved in antigen recognition. Subsequently,binding studies7 of rat CD2 using surface plasmon resonance (SPR)technology, which proved particularly well suited to the analysis ofweak protein-protein interactions, provided the first opportunity to con-sider structural data in the context of rigorous affinity and kinetic dataobtained with the same protein.
Considerable progress has since been made in characterizing thegeneral biophysical properties of interactions between cell surface mol-ecules. In particular, alongside work on CD2, the structures and inter-actions of three other sets of T cell surface molecules that initiate ormodulate responses to antigen have been characterized in detail: the Tcell antigen receptor (TCR) itself; the ‘coreceptor’ molecules CD4 andCD8; and the costimulatory proteins CD28 and cytotoxic T lympho-cyte-associated antigen (CTLA)-4. Here we summarize the general fea-tures of cell-cell recognition and then present an overview of recentwork on the four sets of interactions involving these T cell surface mol-ecules. Because an exhaustive review is not possible here, we focus onthe studies that best illustrate the way in which the binding and struc-tural data have together enhanced our understanding of T cell recogni-tion and activation.
Binding properties—an overviewFrom the wide range of interactions studied by SPR (Fig. 1), it emergesthat cell surface molecules interact with generally low affinities, with dis-sociation constants (Kd) between 1 and 100 µM, mainly as a result of fastdissociation rate constants (koff > 1 s–1 at 37 °C; reviewed in refs. 8, 9).However, there are intriguing exceptions, such as CTLA-4 and CD80,which interact with a relatively high affinity (Kd = 0.2 µM), and the core-ceptors CD4 and CD8, which bind with exceptionally low affinities (Kd
≥ 200 µM). As will be discussed below, in each case there are plausiblereasons why these interactions have atypical affinities.
Binding properties measured by techniques such as SPR, in which atleast one molecule is soluble, depend on the molar concentration, a three-dimensional (3D) parameter. However, the movement of membrane-teth-ered molecules is largely restricted to the two-dimensional (2D) plane ofthe membrane, so the surface density (a 2D parameter), rather than themolar concentration, is important. Measuring 2D Kd values is technicallydifficult, but Dustin and colleagues have achieved this for the T cell sur-face molecules CD210,11 and CD2812 using glass-supported planar lipid
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

nature immunology • volume 4 no 3 • march 2003 • www.nature.com/natureimmunology
REVIEW
218
bilayers. In this system, fluorescence microscopy is used to determine theamount of binding of fluorescently labeled, bilayer-tethered counter-receptors to receptors expressed on T cells interacting with the bilayers.Two-dimensional Kd values, expressed in molecules/µm2, can be thoughtof as the density of counter-receptors at which 50% of the receptor isengaged. Values obtained for the CD2-CD5810 and CD28-CD8012 inter-actions are very similar at 1.1 and 0.7 molecules/µm2, respectively, andthese interactions have similar 3D Kd values (10–20 µM for both at37 ºC), suggesting a good correlation between 2D and 3D Kd values.However, it is notable that for the rat CD2-CD48 interaction11, the 3D Kd
is one-third to one-sixth that of human CD2 or CD28 interactions and the2D Kd is approximately one-fortieth of that for human CD2 and CD28,suggesting that there is a nonlinear relationship between 3D and 2D Kd.
Consequences of membrane tetheringThe fact that cell-cell recognition molecules are tethered to membranesand interact at membrane interfaces has several important consequences.First, it limits the ability of molecules to encounter each other by requir-ing the membranes to come into proximity and the receptors to diffuselaterally in the membrane. It follows that binding will be affected byother receptor-ligand interactions that help bring the membranes intoproximity and by changes in the lateral mobility of receptors and/or theirligands. The importance of these effects has been demonstrated for theCD28-CD80 interaction in the glass-supported lipid bilayer system12.The CD28-CD80 interaction is enhanced by CD2-CD48 binding and,although it has higher 3D and 2D affinities than the CD2-CD48 interac-tion, CD28-CD80 is far less effective at mediating adhesion of T cells tothe bilayers—due in part to limitations in CD28 mobility12.
A second, related issue is the importance of the size of cell surfacemolecules or, more precisely, the distance of the membrane tether tothe binding site. In addition to flexibility and mobility considerations,for any interaction to occur the molecules must be able to span the dis-tance between apposing membranes. This is of particular significancein T cell antigen recognition because many of the molecules involvedare small compared with the most abundant cell surface moleculessuch as CD45 and CD43 and other molecules such as integrins, whichare thought to initiate cell-cell adhesion13,14. The importance of size hasbeen directly demonstrated by experimentally manipulating thedimensions of the CD2-CD48 interaction15. Whereas the wild-typeCD2-CD48 interaction (spanning ∼ 14 nm) substantially enhances T cell antigen recognition, elongated CD2-CD48 complexes (spanning∼ 21 nm) inhibit it. The inhibition of TCR engagement by an increasein intermembrane distance from ∼ 14 to ∼ 21 nm supports the hypothe-sis13,16 that short molecules will be segregated from larger molecules atthe cell-cell interface. Segregation according to size has now been
observed in several studies by confocal or deconvolution fluorescencemicroscopy14. It should be stressed, however, that such segregation isnot always detectable and is usually only seen after several minutes ofcell contact, by which time TCR triggering has been initiated14. This isprobably a consequence of the fact that segregation on a smaller scale(<100 nm) is below the resolution of the techniques used.
A third important consequence of membrane tethering is that cell-cell recognition molecule interactions will be subjected to mechanicalstress or unbinding forces. The origins of this force include the activemigration of cells, the extension or withdrawal of cellular processes andthermally driven fluctuations in intermembrane distance. Determina-tion of the functional significance of mechanical properties requiresdirect measurements using, for example, the surface force apparatus(SFA), which relies on measurements of force as a function of the sep-aration distance between materials bound to two crossed cylinders,yielding their interaction energies17. Only the CD2-CD48 interactionhas been analyzed thus far18; it is exceptionally weak, with a bindingenergy of ∼ 1 kT. Given the difficulties of performing force measure-ments, another approach is to measure the solution binding propertythat correlates best with unbinding force. Theoretical considerationsand empirical studies of the avidin-biotin system indicate that the acti-vation enthalpy of dissociation (∆‡H), measured from the temperaturedependence of the koff (Fig. 2), correlates best with unbinding force17.However, an analysis of some antibody-antigen interactions observed abetter correlation with koff
19, suggesting a contribution from entropicfactors (Fig. 2). Thus, the solution binding property that correlates bestwith unbinding force may vary between interactions17.
Regulation of affinity and avidityIt is often observed that the activation of T cells increases their ability tobind or adhere to a particular ligand without an increase in the amount ofreceptor at the cell surface. This has been demonstrated for integrins suchas LFA-120, as well as CD221, CD822 and even the TCR23. Such changeslikely result from increases in the valency and hence the avidity of bind-ing (due to redistribution or clustering of molecules), or enhanced asso-ciation with the cytoskeleton. Only for integrins is there compelling evi-dence for an increase in the intrinsic binding affinity of a protein inducedby changes inside the cell24. Structural studies have revealed that theectodomain of integrins, which consists of α- and β-subunits, undergoesa ‘switchblade’-like change from a closed to an open conformation, withthe latter revealing the ligand-binding site. The coupling of perturbationsof the cytoplasmic domains of the α- and β-chains to ectodomain con-formational changes represents a plausible mechanism by whichectodomain affinity could be regulated from within the cell. Althoughthere are several examples of other types of receptors that undergo
Figure 1.The wide variation in the 2D and 3D affinitiesof leukocyte cell-cell recognition molecules. (Top scale)The 3D (solution) affinities measured using SPR methods9 areshown on a logarithmic scale.Actual Kd values (in µM) are indi-cated after the name of each molecule. A typical antibody-pro-tein antigen affinity is included for comparison. (Bottom scale)The 2D affinities for the human (h) CD2-CD5810, rat (r)CD2–rat CD4811 and human CD28–mouse (m) B7-112 interac-tions, measured as molecules (mols) per µm2, are shown direct-ly below the values for their respective 3D affinities.The scale iscolored according to whether T cells bound strongly10 (red) orvery weakly11 (red/blue) via the human and rat CD2 ligands,respectively, to the model bilayer system10. The 3D affinities ofcoreceptors for MHC ligands are much lower than that of therat CD2–rat CD48 interaction, suggesting that the 2D affinitiesof coreceptor ectodomains alone will be insufficient to sustainspontaneous recognition of their MHC ligands at the cell surface.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

REVIEW
www.nature.com/natureimmunology • march 2003 • volume 4 no 3 • nature immunology 219
conformational changes after ligand binding (that is, the aspartate recep-tor, bacterial photoreceptors and the erythropoietin receptor), the absenceof convincing evidence for analogous processes in cell adhesion mole-cules other than the integrins suggests that, overall, transducing confor-mational changes across membranes is structurally difficult and unusual.Consequently, when a receptor evolves this ability, it tends to be reusedby expansion of its family through gene duplication and sequence diver-gence. This is illustrated by the G-protein coupled receptors, whose abil-ity to couple ligand binding by its extracellular region to a conforma-tional change in its intracellular region is likely to be one of the reasonsthat it is by far the largest family of receptors in mammals. It is remark-able that, of all the possible structural homologs, the head domain of theαvβ3 integrin exhibits striking similarity to the intracellular ligands of G-protein coupled receptors (that is, G-proteins), which are also capable ofsubstantial conformational changes25.
GlycosylationAlmost all cell surface proteins are glycosylated, suggesting that gly-cosylation has important general roles that may include protein folding,transport and the prevention of nonspecific interactions26. The negativecharge on sialic-acid–rich, mucin-like portions of cell surface proteinsmay direct these proteins away from the membranes as a result ofrepulsion from the negatively charged cell surface, just as conserved N-glycans in the membrane-proximal portions of molecules such as CD28
and ICAM-227 are proposed to help orient these molecules at the cellsurface. Another role of glycans is to serve as ligands for lectin-like cellsurface molecules26.
There is also a considerable body of circumstantial evidence that dif-ferential glycosylation of specific cell surface proteins has an importantrole in T cell function28. However, because many proteins are affected bythese manipulations and glycosylation can affect proteins in severalways, it is often difficult to establish the precise role of glycosylation andto obtain definitive evidence that glycosylation of a particular protein isimportant. For example, CD45 dimerization is influenced by changes inthe glycosylation of its ectodomains, but the functional importance of thiseffect has yet to be demonstrated29. Similarly, alteration in the glycosyla-tion of the CD8β chain correlates with changes in binding to MHC-pep-tide class I tetramers22,30, but the mechanism of this effect (that is, con-formational change or increase in valency) is undefined.
T cell receptor interactionsMore than 17 structures of intact TCRs, TCR-MHC-peptide complex-es and TCR fragments or isolated chains have confirmed that the TCRis structurally similar to the antigen-binding fragment of an antibody
molecule, with the peptide-MHC binding site formed primarily fromthree complementarity-determining regions (CDRs) or loops con-tributed by the variable domains31,32. Unexpectedly, for the αβ-TCR atleast, TCR-peptide-MHC interactions are topologically constrainedinsofar as the Vα and Vβ domains are restricted to binding the N- andC-terminal halves of the peptide, respectively. There is considerablevariation in the twist around the long axis of the TCR-peptide-MHCcomplex, but, for most complexes, this is limited to within ∼ 35°.
An important property of the TCR that sets it apart from other leuko-cyte recognition molecules is its unusually slow binding kinetics: boththe on- and off-rates for TCR-MHC-peptide interactions are generallyless than one-tenth those for the CD2-CD58 interaction, for example,and far more sensitive to temperature (Fig. 2a)33,34. These properties aresuggestive of conformational flexibility at the binding interface. NMR35
and thermodynamic33,34 studies suggest that the conformational flexibil-ity of the TCR and/or peptide-MHC binding surfaces decreases uponbinding. Crystallographic analyses of TCR structures in bound andunbound states36,37, or bound to different peptide-MHC ligands38, indi-cate that one or more of the CDR loops adopts different conformations,with the CDR3 loops showing the largest changes (Fig. 2b). The find-ing that mutations of individual MHC residues have smaller effects onTCR-peptide-MHC binding than mutations of TCR-contacting residuesin the peptide has been interpreted as evidence for a two-step bindingmechanism39 in which peptide contacts, formed after initial, weakMHC contact and structural rearrangements of the CDR3 loops, arelargely responsible for stabilizing the complex. However, because moreMHC residues than peptide residues contribute to the interface, thesedata do not rule out the possibility that TCR-MHC interactions con-tribute the bulk of the binding energy.
Conformational flexibility is likely to be responsible for the highdegree of promiscuity or cross-reactivity evident in TCR recognition ofpeptide-MHC. Theoretical considerations and experimental data suggestthat each TCR can recognize more than 105 peptide-MHC complexes40,enabling the available repertoire of TCR specificities present in a singleindividual to recognize the much larger repertoire of peptide-MHC com-plexes that can be presented in that individual. In this way, ‘holes’ in theTCR repertoire are eliminated, which would otherwise allow organismsto escape detection by the adaptive immune response. Slow dissociationis also significant because it indicates that, once formed, TCR-peptide-MHC complexes are more stable than those formed by other cell-cellrecognition molecules with similar affinities, such as CD2, by as muchas two orders of magnitude. Long-lived complexes might help compen-sate for the relative paucity of TCR ligands, and increase the dynamicrange over which discriminatory signaling is effective.
Figure 2. Structural rearrangements accompany TCR-peptide-MHC inter-actions. (a) Temperature-dependence of TCR binding kinetics.A soluble form of theJM22 TCR (sTCR) was injected (7.5 µM, filled bar) at the indicated temperaturesover a surface to which an HLA-A2-influenza peptide complex had been immobilized(data from ref. 33).According to activation complex theory, the variation in kon andkoff with temperature can be used to determine the activation enthalpy (∆‡H) of asso-ciation and dissociation, respectively.The ∆‡H of dissociation, which is very high forthis interaction (∼ 31 kcal mol–1), represents the number of bonds that need to bebroken for the interacting proteins to dissociate.The koff, which can be expressed interms of the free energy (∆‡G) of dissociation, depends on this as well as entropiccontributions (∆‡S) such as solvent reorganization and changes in protein conforma-tion and rotational and translational degrees of freedom.The ∆‡H of dissociation wasshown to correlate with mechanical strength in the avidin-biotin interaction17, where-as the koff (∆‡G) correlated best in an antibody-antigen interaction19. (b)Conformational differences between the CDR regions of the KB5-C20 TCR before(magenta) and after (cyan) binding of the TCR (blue) to an allogeneic H-2Kb MHCclass I molecule (red/yellow)37. CDR3β (asterisk) undergoes the largest change.
a b
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y
nature immunology • volume 4 no 3 • march 2003 • www.nature.com/natureimmunology
REVIEW
220
Several studies examining the correlation between the solution bindingproperties and functional consequences of TCR–peptide-MHC interac-tions suggest that a broad correlation exists between affinity or half-lifeand functional effect41, although there have been some exceptions42. Thebroad correlation supports models of TCR triggering in which the dura-tion of binding, rather than a specific conformational change, determinesthe outcome of TCR–peptide-MHC interactions. Regarding the excep-tions, one possibility is that TCR–peptide-MHC interactions are subjectto mechanical stress43. Although this will generally enhance dissociation,the degree of enhancement will vary. Thus, two TCR–peptide-MHCinteractions having identical half-lives in solution can have different half-lives when subjected to mechanical stress. Although direct measurementsare yet to be performed, there is indirect evidence that TCR interactionsare mechanically strong. TCR–peptide-MHC interactions have relativelyslow koff and high ∆H and ∆‡H values33,34 (Fig. 2). Moreover, T cells leaveTCRs behind when migrating over surfaces presenting peptide-MHC lig-and44, and can extract and internalize peptide-MHC from target cells45.
Coreceptor interactionsCD4 and CD8 have evolved very different solutions to the problem ofligand binding-site presentation. In contrast to CD4, which ismonomeric in solution46 if not in the crystal lattice47 and has four extra-cellular IgSF domains, CD8 consists of two chains with single IgSFdomains supported on O-glycosylated stalks. CD4 only binds MHCclass II, and CD8αβ is the primary coreceptor for conventional MHCclass I–restricted T cells. CD8αα , on the other hand, is the principalligand of the nonclassical MHC class I molecule thymus leukemia anti-gen48. Structures of human49 and mouse50 CD8αα–MHC class I com-plexes, and of a human CD4 fragment complexed with mouse MHCclass II51, show that CD4 and CD8 bind to nonpolymorphic surfaces ofthe membrane-proximal domains of MHC molecules.
Of all the molecules studied so far, the binding properties of the core-ceptors are the most enigmatic. SPR-based analyses indicate that CD8αα
and CD8αβ bind MHC class I with similar affinities that, surprisingly,are less than one-tenth that of most other interactions involving cell sur-face molecules (>200 µM at 37 ºC; Fig. 1)48,52,53. The differences in thedissociation rate constants are even more profound. The CD8-MHCinteraction has a koff at least two orders of magnitude faster thanTCR–MHC-peptide interactions, and the CD4–MHC class II interactionseems to be even less stable. Mouse MHC class II is proposed to bindhuman CD4 with a Kd of ∼ 200 µM at 25 ºC54, but the exceptionally lowbinding response relative to background (∼ 1%) and low apparent CD4activity (<2%) suggest that this result should be treated with caution. Inour SPR experiments, we have been unable to detect any bindingbetween human CD4 and MHC class II, at concentrations of human sol-uble CD4 as high as 2 mM (G.F. Gao, J. Wyer, L.F., P.A.v.d.M. & S.J.D.,unpublished data). In addition to highlighting the unusual properties ofCD4, this implies that some biologically meaningful interactions may beundetectable using monomeric proteins and SPR methods.
The remarkable differences in the binding properties of coreceptorsand more conventional cell surface molecules are likely to be of con-siderable significance. The observation that the 2D affinity of theCD2–CD48 interaction (solution Kd = 60 µM) barely sustains adhesiveinteractions11 implies that an affinity threshold exists, below whichbinding will not occur between monovalent cell surface moleculesexpressed at physiological levels (Fig. 1). If the threshold is close to100 µM, as implied by the CD2–CD48 data, this suggests that the inter-actions of coreceptors with their MHC ligands are unlikely to be sus-tained by the extracellular domains of these molecules alone.Supporting this concept, adhesive coreceptor-MHC interactions areonly seen when the molecules are overexpressed55,56.
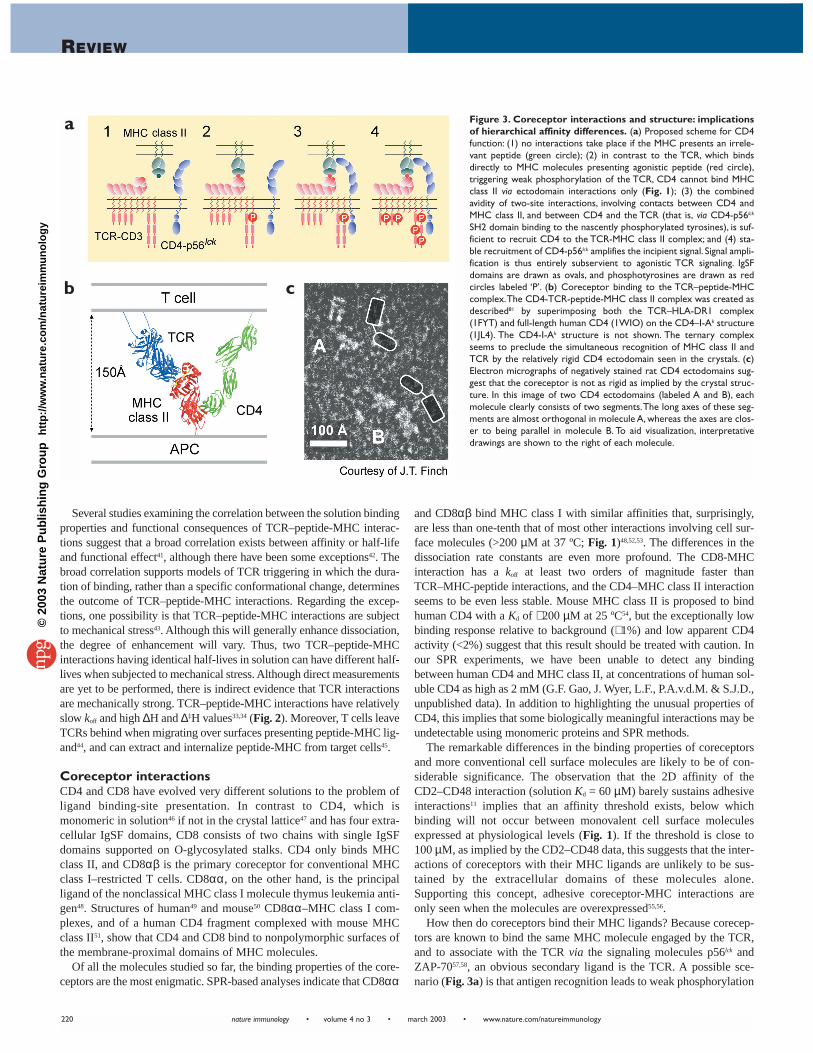
How then do coreceptors bind their MHC ligands? Because corecep-tors are known to bind the same MHC molecule engaged by the TCR,and to associate with the TCR via the signaling molecules p56lck andZAP-7057,58, an obvious secondary ligand is the TCR. A possible sce-nario (Fig. 3a) is that antigen recognition leads to weak phosphorylation
Figure 3. Coreceptor interactions and structure: implicationsof hierarchical affinity differences. (a) Proposed scheme for CD4function: (1) no interactions take place if the MHC presents an irrele-vant peptide (green circle); (2) in contrast to the TCR, which bindsdirectly to MHC molecules presenting agonistic peptide (red circle),triggering weak phosphorylation of the TCR, CD4 cannot bind MHCclass II via ectodomain interactions only (Fig. 1); (3) the combinedavidity of two-site interactions, involving contacts between CD4 andMHC class II, and between CD4 and the TCR (that is, via CD4-p56lck
SH2 domain binding to the nascently phosphorylated tyrosines), is suf-ficient to recruit CD4 to the TCR-MHC class II complex; and (4) sta-ble recruitment of CD4-p56lck amplifies the incipient signal. Signal ampli-fication is thus entirely subservient to agonistic TCR signaling. IgSFdomains are drawn as ovals, and phosphotyrosines are drawn as redcircles labeled ‘P’. (b) Coreceptor binding to the TCR–peptide-MHCcomplex.The CD4-TCR-peptide-MHC class II complex was created asdescribed81 by superimposing both the TCR–HLA-DR1 complex(1FYT) and full-length human CD4 (1WIO) on the CD4–I-Ak structure(1JL4). The CD4-I-Ak structure is not shown. The ternary complexseems to preclude the simultaneous recognition of MHC class II andTCR by the relatively rigid CD4 ectodomain seen in the crystals. (c)Electron micrographs of negatively stained rat CD4 ectodomains sug-gest that the coreceptor is not as rigid as implied by the crystal struc-ture. In this image of two CD4 ectodomains (labeled A and B), eachmolecule clearly consists of two segments.The long axes of these seg-ments are almost orthogonal in molecule A, whereas the axes are clos-er to being parallel in molecule B. To aid visualization, interpretativedrawings are shown to the right of each molecule.
a
b c
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

REVIEW
www.nature.com/natureimmunology • march 2003 • volume 4 no 3 • nature immunology 221
of the TCR and that the combined avidity of two-site interactions involv-ing coreceptor binding to the ectodomain of the MHC molecule and tointracellular components of the TCR (for example, via p56lck SH2domain interactions with phosphorylated CD3 immunoreceptor tyrosineactivation motifs or TCR-associated Zap70) is sufficient to recruit core-ceptor and p56lck to the triggered TCR–MHC-peptide complex. If thiswas to occur, the stably recruited p56lck could then be expected to ampli-fy the initial phosphorylation signal. According to this scheme, hierar-chical differences in TCR–MHC-peptide and coreceptor-MHC affinitieswould ensure that these events occur sequentially, and that signal ampli-fication remains entirely subservient to agonistic TCR signaling.
A potential problem for this idea is that when coreceptor-MHC com-plex structures49,51 are superimposed on structures of TCR–peptide-MHC complexes, the angle at which CD4 and CD8 engage peptide-MHC seems to preclude direct association with a TCR binding thesame peptide-MHC complexes (Fig. 3b). How can this be reconciledwith evidence that CD4 and CD8 physically associate with TCR-CD3complexes? It is possible that the coreceptors and the TCR associateindirectly via other proteins, such as CD3. A second possibility is thatthe coreceptor bridges TCRs interacting with agonist- and self-peptide-MHC complexes, creating a TCR ‘pseudodimer’ capable of intracellu-lar signaling59. However, this proposal fails to account for the signalfeature of coreceptor interactions, that is, their very low affinity. A thirdpossibility is that the coreceptor extracellular domains are more flexi-ble than is implied by the crystal structures. This is not an issue for theO-glycosylated stalk of CD8, given its similarity to flexible, mucin-likeproteins such as CD43. In the case of CD4, electron micrographs sug-gest a greater degree of segmental flexibility in the protein than may begenerally thought (Fig. 3c).
Costimulatory interactionsCD28 and CTLA-4 are closely related type I membrane proteins con-sisting of single, moderately to highly glycosylated, V-set IgSF domainsand highly conserved cytoplasmic domains, and are expressed at the cellsurface in the form of disulphide-linked homodimers. CTLA-4 dimer-izes via residues at the beginning and ends of the domains60,61, and,although there is no equivalent CD28 data, the amino acids in theseregions are not conserved in CD28, suggesting that the dimerizationmodes for CD28 and CTLA-4 are different62. B7-1 is chimeric insofar asthe ligand-binding, V-set domain is similar to that of conventional recog-nition molecules such as CD2 and CD4, whereas the membrane-proxi-mal domain of B7-1 has the topology of the C1-set domains present inantigen receptors and MHC antigens63. In this sense, B7-related proteinsconstitute the ‘missing link’ between conventional cell-cell recognitionmolecules and antigen receptors63. In crystal lattices61,63 and in solution63,B7-1 forms bivalent homodimers capable of generating periodic, linear
arrays with bivalent CTLA-4 homodimers. As anticipated by mutation-al studies, binding to B7-1 and B7-2 is dominated by the hydrophobicMYPPPY sequence conserved in CTLA-4 and CD28. Surface-shapecomplementarity at the interface with B7-1 (Fig. 4a) is as high as thatseen in any protein complex61 and much higher than is observed forcomplexes of cell surface proteins such as CD2–CD58 (Fig. 4b) orTCR–peptide-MHC.
The activating and inhibitory functions of the CD28 and CTLA-4receptors are now well established, but the reason why two sequential-ly expressed B7 proteins exist has been a mystery. Part of the reason forthis uncertainty is that early binding studies tended to emphasize theuniformity of costimulatory interactions. In fact, costimulatory recep-tors and ligands have distinct binding properties and form signalingcomplexes of unanticipated structural diversity62. The key findings,summarized in Fig. 5a, are that B7-1 binds CTLA-4 13-fold morestrongly than B7-2; that, relative to its CTLA-4 binding affinity, B7-2binds CD28 2- to 3-fold more effectively than B7-1 (that is, theCD28/CTLA-4 Kd ratios are ∼ 8 and 20 for B7-2 and B7-1, respective-ly); and that, in contrast to CTLA-4 homodimers, which are bivalent,CD28 homodimers are monovalent—presumably because CD28homodimers assemble differently. Because the CD28 homodimer ismonovalent, it cannot participate in avidity-enhanced interactions sim-ilar to those proposed for CTLA-4. As a result, the organization and sta-bility of the signaling complexes CD28 forms will be very differentfrom those formed by CTLA-4. Similarly, in contrast to soluble B7-1,B7-2 is monomeric in solution62,64, implying that it is monovalent andthus incapable of forming periodic arrays with CTLA-4 in vivo. Theseobservations indicate that B7-1 favors CTLA-4 over CD28 engage-ment, whereas B7-2 exhibits much less bias. In this context, the delayedexpression of B7-1 on APCs seems to be timed to enhance inhibitorysignaling by CTLA-4, explaining why sequentially expressed costimu-latory ligands may have evolved.
Overall, the ligand binding affinities of costimulatory molecules varymore than 100-fold (CD28–B7-2, Kd = 20 µM; CTLA-4–B7-1, Kd = 0.2µM; Fig. 5a). Taking avidity effects into account, the stabilities of thesignaling complexes that these molecules form probably vary by morethan four orders of magnitude62. Why is such extreme variation neces-sary? The affinity of the CD28–B7-2 interaction is comparable to thoseof TCR-peptide-MHC and CD2–CD58 interactions (Kd = 10–20 µM),but there are generally many more CD28 counter-receptors than TCRligands on an APC. Because CD28 amplifies TCR-dependent signals65
and in some circumstances can even circumvent the requirement forTCR signaling66, it may be important that mechanisms exist that preventCD28 from accentuating inappropriate TCR-dependent signals undernormal circumstances. This seems to be achieved by coupling relativelylow binding affinity with low CD28 expression and restricted mobility,
Figure 4. Contrasting modes of low-affinity recognition at theleukocyte surface. Structures of the interacting surfaces of complex-es of CTLA-4 (cyan) and B7-1 (magenta)61 (a) and of CD2 (blue) andCD58 (red)72 (b) after ‘undocking’ the proteins by 5–10 Å, revealing theprofound differences in the degree of shape complementarity of thebinding sites (shown semi-transparently).The algorithm of Lawrence andColman82, which measures the degree of geometric fit between two pro-tein surfaces, gives scores of 0.77 for the CTLA-4–B7-1 interface. Thisvalue is similar to those for constitutive oligomeric proteins (0.7–0.76)and much higher than scores for protein antigen-antibody interfaces(0.64–0.68) or interacting cell surface molecules, such as CD2 and theTCR (0.45–0.58).
a b
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

nature immunology • volume 4 no 3 • march 2003 • www.nature.com/natureimmunology
REVIEW
222
to the extent that CD28 ligation cannot occur until adequate agonisticTCR signaling creates a more favorable microenvironment for binding,in the form of an immunological synapse12. Subsequent CD28-depen-dent transport of cell surface molecules to the synapse67,68 might con-tribute to its maturation. Extremely stable CTLA-4 interactions with B7-1, on the other hand, may be required to overturn ongoing activationevents within the synapse, into which CTLA-4 is inserted from secreto-ry granules. These observations are compatible with the emerging viewof the synapse as a post-TCR signaling structure whose principal func-tion is to enhance secondary activation or differentiation events69.
The binding and structural studies also have important implicationsfor the mechanism of signaling by the costimulatory molecules. CTLA-4 does not undergo significant structural rearrangements upon complexformation60, so signaling cannot be driven by conformational changes.For CD28, the interchain disulphide bond in the homodimer is likely toprevent such changes from being transmitted across the membrane, ifthey occur at all. The formation of periodic arrays similar to those seenin crystals of soluble CTLA-4 and soluble B7-1 is conjectured to be part
of the signaling mechanism60,64, but this is unlikely to be generally truebecause murine B7-1 seems to be prevented from self-associating byglycosylation of the putative dimerization interface (A. Iaboni andS.J.D., unpublished data). Dimerization- and aggregation-based signal-ing models are likely to be even less relevant to signaling by CD28–B7-2 complexes, because CD28 is monovalent and B7-2 is monomeric62. Inthis regard, the TCR resembles CD28 insofar as it also responds tomonovalent ligands59 (Fig. 5b). Other parallels are that both signalingproteins lack intrinsic enzymatic activity and that signal initiation ineach case apparently requires the phosphorylation, by p56lck, of tyrosinespresent in their cytoplasmic domains.
CD2 interactionsThe CD2 subset of the IgSF consists of monomeric type I membrane gly-coproteins, most of which have two extracellular IgSF domains13. InitialSPR-based analyses revealed that the affinity of rat CD2-CD48 binding(Kd = 60 µM) is almost two orders of magnitude weaker than expected andthat the kinetics are extremely fast (kon ≥ 105 M–1s–1, koff ≥ 6 s–1), empha-sizing the importance of transient interactions at the leukocyte surface70.Similar findings were obtained for human CD2–CD58 binding, althoughthe affinity of this interaction is somewhat higher (Kd = 10 µM)71.
Why are such weak interactions also specific? Mutational and struc-tural studies have confirmed that CD2-ligand contacts involve relative-ly large binding surfaces (∼ 700 Å2), so binding is not weak and specif-ic because these surfaces are small and exhibit a high degree of com-plementarity, as seen for the CD28–B7-1 complex (Fig. 4a). The bind-ing surfaces of CD2 and CD58 are characterized by a high degree ofcharged residue clustering (Fig. 6a,b), however, which is unusual forsites of protein-protein interaction. The structure of the humanCD2–CD58 complex72 showed that nine mostly basic CD2 residuesform an inter-digitating network of ten salt bridges and five H-bondswith 11 predominantly acidic CD58 residues. Unexpectedly, substitu-tion of nearly half the CD2 residues mediating these contacts with ala-nine resulted in modest increases or decreases in affinity73. Similarly,the binding of rat CD2 to CD48 was unaffected by increases in ionicstrength or the removal of more than half the charged or polar sidechains in the binding site of CD274. The explanation for these effects isthat the favorable electrostatic complementarity merely compensatesfor the unfavorable removal, upon binding, of water bound to chargedresidues, with the net result that interactions between oppositelycharged residues at the binding interface make little favorable contri-bution to binding. Such contacts are therefore ideally suited to cellularinteractions because they allow increases in specificity to be uncoupledfrom increases in affinity74. The idea that electrostatic contacts might bethe major source of binding specificity in the absence of a high degreeof shape complementarity (Fig. 4b), which is the hallmark of mosthigh-affinity interactions, was directly tested via an attempt to predictthe structure of the CD2–CD58 complex before it was solved75. Thisprediction was based on maximizing electrostatic complementarity atthe interface. The agreement between the predicted75 and actual72 struc-tures (Fig. 6c,d) lends strong support to the concept. Overall, however,CD2 may prove to be a rather extreme example of weak interactions inwhich electrostatic contacts are dominant.
The very low 2D affinity of the CD2-CD48 interaction, which is bare-ly sufficient to sustain interactions with model bilayers11, implies thatCD2 is not a conventional cell adhesion molecule. What other possiblefunction is compatible with its binding and structural properties?Mutational76, structural6 and SFA18 experiments all indicate that thelongest dimension of the extracellular region of CD2-ligand complexesis similar to that of TCR-peptide-MHC complexes (130–150 Å). This
Figure 5.Diverse stabilities and organization of signaling structures formedby costimulatory molecules. (a) Signaling complexes formed sequentially (blackarrows) during the course of an immune response are shown. Colored arrows referto the nature of the signals generated by the complexes.Affinities (Kd values, µM) areshown for each interaction. Bivalent binding may stabilize a given complex ∼ 100-fold62.(b) Are CD28–B7-2 and TCR–peptide-MHC signaling complexes representatives of adistinct class of signaling structures? Both complexes are monovalent, lack enzymaticactivity and are dependent on extrinsic kinases, such as p56lck, for the initiation of sig-naling. CD4 or CD8 are not shown as part of the basic TCR signaling structure, as Tcells can respond to antigen in the absence of coreceptor83.
a
b
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

REVIEW
www.nature.com/natureimmunology • march 2003 • volume 4 no 3 • nature immunology 223
implies that the spontaneous interaction of CD2 with its ligands duringthe earliest stages of T cell–APC contact will facilitate the scanning ofMHC-peptide complexes by the TCR by bringing the membranes intoclose proximity76. This is consistent with the observation that, in murineCD2–/–, TCR-transgenic models, CD2 lowers thresholds for TCR trig-gering and activation77.
ConclusionsThe well-characterized interactions discussed here involve compact,structurally related cell surface molecules directly or indirectly associat-ed with antigen recognition. In spite of their similar structures and func-tional properties, their recognition mechanisms exhibit considerablestructural diversity. The relatively poor surface-shape complementarityseen in CD2- and TCR-ligand complexes is likely to be a common fea-ture of such interactions, whereas the intense charged-residue clusteringseen in CD2, and the extreme surface complementarity of the B7-1–CTLA-4 interface, more likely reflects the distinct IgSF subgroup ori-gins of these proteins. Some of the structural properties clearly reflectfunctional constraints, such as the repertoire-extending effects of con-formational flexibility in the MHC-peptide binding surface of the TCR.For others, for example CD2, it is probably more important that bindingis weak and specific than how this is achieved.
Among the most significant and least expected features of theseinteractions is that, although the binding affinities are all relatively low,signaling complex stability varies enormously. Interactions between
leukocyte surface proteins were expected to be weak because their cel-lular contacts are generally transient. This view is confirmed by studiesof adhesion molecules that form stable cellular interactions, such as C-cadherin78, but does not explain the unprecedented variation in thestrength of binding. We suggest that the regulated expression of theseproteins, coupled with hierarchical differences in their binding affini-ties, represents a simple mechanism for leading T cells sequentiallythrough the signaling steps leading to full activation, as is best illus-trated by coreceptor and costimulatory interactions.
What does the future hold? It will be important to generalize theseconcepts in studies of other proteins with distinct structural propertiesand functions not directly involved in antigen recognition, such as inte-grins. The relationship between 2D and 3D affinity also needs to beexplored more fully. Ongoing efforts will also be important to determinewhether or not these interactions can be targeted therapeutically. Theexisting structural data, which show that recognition by these moleculesgenerally involves relatively flat surfaces lacking deep hydrophobicpockets suited to binding small-molecule inhibitors, suggest that thiswill generally be difficult. Nevertheless, high-throughput screening hassucceeded twice in identifying compounds that bind with sub-micromo-lar affinity to B7-179 (P. Sørensen, personal communication).
The ultimate goal is to generate quantitative models of T cell recog-nition and activation. Foremost among the theoretical constraintsremaining is the mechanism of initial signal transduction, or ‘receptortriggering’, for proteins such as the TCR. Classical models of receptortriggering rely on receptor dimerization or aggregation, or on ligand-induced conformational changes in the receptors14. Although it has beenreported80 that antibody binding to the CD3ε ectodomain can modulate,through a conformational change, the association of a signaling mole-cule with the CD3ε cytoplasmic domain, there is no evidence thatnative TCR ligands have the same effects on CD3ε, and most crystal-lographic data argue against the possibility of such changes occurringfor the αβ-heterodimer. The finding59 that T cells can ‘sense’ the pres-ence of single MHC agonists present on APCs, which rules out con-ventional dimerization models of TCR triggering, is a major step for-ward that will have to be incorporated into all TCR triggering models.It now also seems that, like the TCR, CD28 is a monovalent signalingmolecule62, and that conformational change- and dimerization-basedsignaling models are as unlikely to apply to CD28 as to the TCR. Thissuggests that, rather than the TCR being a special case, a general mech-anism for signaling by monovalent cell surface proteins lacking intrin-sic enzymatic activity must exist. Molecular redistribution-based recep-tor triggering models13,14,16, which are not dependent on aggregation ordimerization, or on the transmission of conformational changes, arecompatible with the shared structural and signaling properties of CD28and the TCR. A recent analysis of the transcriptome of a resting T cellsuggests that most, if not all, of the key cell surface molecules consti-tuting the basic triggering apparatus of this cell may be known (E.J.E.et al., manuscript submitted). Once the triggering puzzle is solved,therefore, we should be close to having the necessary framework forconstructing quantitative models of T cell recognition.
AcknowledgmentsWe thank E.Y. Jones, D.I. Stuart,A.N. Barclay and P. Sørensen for valuable discussion, andJ.T. Finch for permission to use the electron micrographs of rat CD4 ectodomains. S.J.D.and P.A.v.d.M. are supported by the Wellcome Trust and the UK Medical ResearchCouncil, respectively. S.I. is supported by Grants-in-Aid from the Ministry of Education,Culture, Sports, Science and Technology of Japan. L.F. is supported by the Karen EliseJensen Foundation,The Danish MS Society and the UK Medical Research Council.
1. Williams A.F., Galfre, G. & Milstein, C.Analysis of cell surfaces by xenogeneic myeloma-hybrid anti-bodies: differentiation antigens of rat lymphocytes. Cell 12, 663–673 (1977).
2. Williams,A.F.The immunoglobulin superfamily takes shape. Nature 308, 12–13 (1984).
Figure 6.Electrostatic contacts determine the specificity of CD2 binding toCD58. Native electrostatic potential is shown projected onto the structures ofdomain 1 of CD2 (a) and CD58 (b), determined as a complex72. Blue represents pos-itive potential, white represents neutral potential and red represents negative poten-tial.The line of view in each case is approximately perpendicular to the ligand-bindingsurface.The complex formed by CD2 (blue) and CD58 (red) (c), predicted by maxi-mizing electrostatic potential at the interface75, is compared with the native complex(d) determined subsequently72.
a b
c d
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y

nature immunology • volume 4 no 3 • march 2003 • www.nature.com/natureimmunology
REVIEW
224
3. Seed, B. & Aruffo,A. Molecular cloning of the CD2 antigen, the T-cell erythrocyte receptor, by a rapidimmunoselection procedure. Proc. Natl. Acad. Sci. USA 84, 3365–3369 (1987).
4. Barclay,A.N. et al. The Leukocyte Antigen Factsbook (Academic Press, London, 1997).5. Driscoll, P.C., Cyster, J.G., Campbell, I.D. & Williams,A.F. Structure of domain 1 of rat T lymphocyte
CD2 antigen. Nature 353, 762–765 (1991).6. Jones, E.Y. et al. Crystal structure at 2.8 Å resolution of a soluble form of the cell adhesion molecule
CD2. Nature 360, 232–239 (1992).7. van der Merwe, P.A., Brown, M.H., Davis, S.J. & Barclay,A.N.Affinity and kinetic analysis of the inter-
action of the cell-adhesion molecules rat CD2 and CD48. EMBO J. 12, 4945–4954 (1993).8. Davis, S.J., Ikemizu, S.,Wild, M.K. & van der Merwe, P.A. CD2 and the nature of protein interactions
mediating cell-cell recognition. Immunol. Rev. 163, 217–236 (1998).9. van der Merwe, P.A. & Davis, S.J. Molecular interactions mediating T cell antigen recognition. Annu.
Rev. Immunol. (in the press).10. Dustin, M.L. et al.Visualization of the CD2 interaction with LFA-3 and determination of the two-dimen-
sional dissociation constant for adhesion receptors in a contact area. J. Cell Biol. 132, 465–474 (1996).11. Dustin, M.L. et al. Low affinity interaction of human or rat T cell adhesion molecule CD2 with its
ligand aligns adhering membranes to achieve high physiological affinity. J. Biol. Chem. 272,30889–30898 (1997).
12. Bromley, S.K. et al.The immunological synapse and CD28-CD80 interactions. Nat. Immunol. 2,1159–1166 (2001).
13. Davis, S.J. & van der Merwe, P.A.The structure and ligand interactions of CD2: implications for T-cellfunction. Immunol.Today 17, 177–187 (1996).
14. van der Merwe, P.A., Davis, S.J., Shaw,A.S. & Dustin, M.L. Cytoskeletal polarization and redistributionof cell surface molecules during T cell antigen recognition. Semin. Immunol. 12, 5–21 (2000).
15. Wild, M.K. et al. Dependence of T cell antigen recognition on the dimensions of an accessory recep-tor-ligand complex. J. Exp. Med. 190, 31–41 (1999).
16. Shaw,A.S. & Dustin, M.L. Making the T cell receptor go the distance: a topological view of T cell acti-vation. Immunity 6, 361–369 (1997).
17. Leckband, D. Measuring the forces that control protein interactions. Annu. Rev. Biophys. Biomol. Struct.29, 1–26 (2000).
18. Zhu, B. et al. Direct measurements of heterotypic adhesion between the cell surface proteins CD2and CD48. Biochemistry 41, 12163–12170 (2002).
19. Schwesinger, F. et al. Unbinding forces of single antibody-antigen complexes correlate with their ther-mal dissociation rates. Proc. Natl. Acad. Sci. USA 97, 9972–9977 (2000).
20. Dustin, M.L. & Springer,T.A.T-cell receptor cross-linking transiently stimulates adhesiveness throughLFA-1. Nature 341, 619–624 (1989).
21. Hahn,W.C. et al. A distinct cytoplasmic domain of CD2 regulates ligand avidity and T-cell responsive-ness to antigen. Proc. Natl. Acad. Sci. USA 89, 7179–7183 (1992).
22. Moody,A.M. et al. Developmentally regulated glycosylation of the CD8αβ coreceptor stalk modu-lates ligand binding. Cell 107, 501–512 (2001).
23. Fahmy,T.M., Bieler, J.G., Edidin, M. & Schneck, J.P. Increased TCR avidity after T cell activation: a mech-anism for sensing low-density antigen. Immunity 14, 135–143 (2001).
24. Hynes, R. Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 (2002).25. Xiong, J.P. et al. Crystal structure of the extracellular segment of integrin α Vβ3. Science 294,
339–345 (2001).26. Rudd, P.M. et al. Glycosylation and the immune system. Science 291, 2370–2376 (2001).27. Wang, J. & Springer,T.A. Structural specializations of immunoglobulin superfamily members for adhe-
sion to integrins and viruses. Immunol. Rev. 163, 197–215 (1998).28. Daniels, M.A., Hogquist, K.A. & Jameson, S.C. Sweet ‘n’ sour: the impact of differential glycosylation
on T cell responses. Nat. Immunol. 3, 903–910 (2002).29. Xu, Z. & Weiss,A. Negative regulation of CD45 by differential homodimerization of the alternatively
spliced isoforms. Nat. Immunol. 3, 764–771 (2002).30. Daniels, M.A. et al. CD8 binding to MHC class I molecules is influenced by T cell maturation and gly-
cosylation. Immunity 15, 1051–1061 (2001).31. Rudolph, M.G., Luz, J.G. & Wilson, I.A. Structural and thermodynamic correlates of T cell signaling.
Annu. Rev. Biophys. Biomol. Struct. 31, 121–149 (2002).32. Hennecke, J. & Wiley, D.C.T cell receptor-MHC interactions up close. Cell 104, 1–4 (2001).33. Willcox, B.E. et al.TCR binding to peptide-MHC stabilises a flexible recognition interface. Immunity
10, 357–365 (1999).34. Boniface, J.J., Reich, Z., Lyons, D.S. & Davis, M.M.Thermodynamics of T cell receptor binding to pep-
tide-MHC: evidence for a general mechanism of molecular scanning. Proc. Natl. Acad. Sci. USA 96,11446–11451 (1999).
35. Hare, B.J. et al. Structure, specificity and CDR mobility of a class II restricted single- chain T-cellreceptor. Nat. Struct. Biol. 6, 574–581 (1999).
36. Garcia, K.C. et al. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHCantigen. Science 279, 1166–1172 (1998).
37. Reiser, J.B. et al.A T cell receptor CDR3β loop undergoes conformational changes of unprecedentedmagnitude upon binding to a peptide/MHC class I complex. Immunity 16, 345–354 (2002).
38. Ding,Y.-H. et al. Four A6-TCR/peptide/HLA-A2 structures that generate very different T cell signalsare nearly identical. Immunity 11, 45–56 (1999).
39. Wu, L.C.,Tuot, D.S., Lyons, D.S., Garcia, K.C. & Davis, M.M.Two-step binding mechanism for T-cellreceptor recognition of peptide MHC. Nature 418, 552–556 (2002).
40. Mason, D.A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol.Today 19, 395–404 (1998).
41. Davis, M.M. et al. Ligand recognition by α β T cell receptors. Annu. Rev. Immunol. 16, 523–544 (1998).42. al-Ramadi, B.K. et al. Lack of strict correlation of functional sensitization with the apparent affinity of
MHC/peptide complexes for the TCR. J. Immunol. 155, 662–673 (1995).43. van der Merwe, P.A. Leukocyte adhesion: high-speed cells with ABS. Curr. Biol. 9, R419–422 (1999).44. Dustin, M.L. et al.TCR-mediated adhesion of T cell hybridomas to planar bilayers containing purified MHC
class II/peptide complexes and receptor shedding during detachment. J. Immunol. 157, 2014–2021 (1996).
45. Huang, J.-F. et al.TCR-mediated internalization of peptide-MHC complexes acquired by T cells.Science 286, 952–954 (1999).
46. Davis, S.J. et al. High level expression in Chinese hamster ovary cells of soluble forms of CD4 T lym-phocyte glycoprotein including glycosylation variants. J. Biol. Chem. 265, 10410–10418 (1990).
47. Wu, H., Kwong, P.D. & Hendrickson,W.A. Dimeric association and segmental variability in the struc-ture of human CD4. Nature 387, 527–530 (1997).
48. Leishman,A.J. et al.T cell responses modulated through interaction between CD8αα and the non-classical MHC class I molecule,TL. Science 294, 1936–1939 (2001).
49. Gao, G.F. et al. Crystal structure of the complex between human CD8αα and HLA-A2. Nature 387,630–634 (1997).
50. Kern, P.S. et al. Structural basis of CD8 coreceptor function revealed by crystallographic analysis of amurine CD8αα ectodomain fragment in complex with H-2Kb. Immunity 9, 519–530 (1998).
51. Wang, J.H. et al. Crystal structure of the human CD4 N-terminal two-domain fragment complexedto a class II MHC molecule. Proc. Natl. Acad. Sci. USA 98, 10799–10804 (2001).
52. Wyer, J.R. et al.T cell receptor and co-receptor CD8αα bind peptide-MHC independently and withdistinct kinetics. Immunity 10, 219–225 (1999).
53. Kern, P. et al. Expression, purification, and functional analysis of murine ectodomain fragments ofCD8αα and CD8αβ dimers. J. Biol. Chem. 274, 27237–27243 (1999).
54. Xiong,Y., Kern, P., Chang, H. & Reinherz, E.T Cell receptor binding to a pMHCII ligand is kineticallydistinct from and independent of CD4. J. Biol. Chem. 276, 5659–5667 (2001).
55. Doyle, C. & Strominger, J.L. Interaction between CD4 and class II MHC molecules mediates celladhesion. Nature 330, 256–259 (1987).
56. Norment,A.M. et al. Cell-cell adhesion mediated by CD8 and MHC class I molecules. Nature 336,79–81 (1988).
57. Janeway, C.A.The T cell receptor as a signalling machine: CD4/CD8 coreceptors and CD45 in T cellactivation. Annu. Rev. Immunol. 10, 645–674 (1992).
58. Thome, M., Duplay, P., Guttinger, M. & Acuto, O. Syk and ZAP-70 mediate recruitment of p56lck/CD4to the activated T cell receptor/CD3/ζ complex. J. Exp. Med. 181, 1997–2006 (1995).
59. Irvine, D.J., Purbhoo, M.A., Krogsgaard, M. & Davis, M.M. Direct observation of single ligand recogni-tion by T cells. Nature 419, 845–849 (2002).
60. Schwartz, J.C. et al. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature410, 604–608 (2001).
61. Stamper, C.C. et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immuneresponses. Nature 410, 608–611 (2001).
62. Collins,A.V. et al.The interaction properties of costimulatory molecules revisited. Immunity 17,201–210 (2002).
63. Ikemizu, S. et al. Structure and dimerization of a soluble form of B7-1. Immunity 12, 51–60 (2000).64. Schwartz, J.C., Zhang, X., Nathenson, S.G. & Almo SC. Structural mechanisms of costimulation. Nat.
Immunol. 3, 427–434 (2002).65. Diehn, M. et al. Genomic expression programs and the integration of the CD28 costimulatory signal
in T cell activation. Proc. Natl. Acad. Sci. USA 99, 11796–11801 (2002).66. Tacke, M., Hanke, G., Hanke,T. & Hunig,T. CD28-mediated induction of proliferation in resting T cells
in vitro and in vivo without engagement of the T cell receptor: evidence for functionally distinctforms of CD28. Eur. J. Immunol. 27, 239–247 (1997).
67. Wulfing, C. & Davis, M.M.A receptor/cytoskeletal movement triggered by costimulation during T cellactivation. Science 282, 2266–2269 (1998).
68. Viola,A., Schroeder, S., Sakakibara,Y. & Lanzavecchia,A.T lymphocyte costimulation mediated byreorganization of membrane microdomains. Science 283, 680–682 (1999).
69. van der Merwe, P.A. & Davis, S.J. Immunology.The immunological synapse—a multitasking system.Science 295, 1479–1480 (2002).
70. van der Merwe, P.A. & Barclay,A.N.Transient intercellular adhesion: the importance of weak protein-protein interactions. Trends Biochem. Sci. 19, 354–358 (1994).
71. van der Merwe, P.A. et al.The human cell-adhesion molecule CD2 binds CD58 with a very low affin-ity and an extremely fast dissociation rate but does not bind CD48 or CD59. Biochemistry 33,10149–10160 (1994).
72. Wang, J.H. et al. Structure of a heterophilic adhesion complex between the human CD2 and CD58(LFA-3) counterreceptors. Cell 97, 791–803 (1999).
73. Kim, M. et al. Molecular dissection of the CD2-CD58 counter-receptor interface identifies CD2Tyr86 and CD58 Lys34 residues as the functional “hot spot”. J. Mol. Biol. 312, 711–720 (2001).
74. Davis, S.J. et al.The role of charged residues mediating low affinity protein-protein recognition at thecell surface by CD2. Proc. Natl. Acad. Sci. USA 95, 5490–5494 (1998).
75. Ikemizu, S. et al. Crystal structure of the CD2-binding domain of CD58 (lymphocyte function-associ-ated antigen 3) at 1.8-Å resolution. Proc. Natl. Acad. Sci. USA 96, 4289–4294 (1999).
76. van der Merwe, P.A. et al.Topology of the CD2-CD48 cell-adhesion molecule complex: implicationsfor antigen recognition by T cells. Curr. Biol. 5, 74–84 (1995).
77. Bachmann, M.F., Barner, M. & Kopf, M. CD2 sets quantitative thresholds in T cell activation. J. Exp.Med. 190, 1383–1392 (1999).
78. Sivasankar, S., Brieher,W., Lavrik, N., Gumbiner, B. & Leckband, D. Direct molecular force measure-ments of multiple adhesive interactions between cadherin ectodomains. Proc. Natl. Acad. Sci. USA 96,11820–11824 (1999).
79. Erbe, D.V.,Wang, S., Xing,Y. & Tobin J.F. Small molecule ligands define a binding site on the immuneregulatory protein B7.1. J. Biol. Chem. 277, 7363–7368 (2002).
80. Gil, D. et al. Recruitment of Nck by CD3ε reveals a ligand-lnduced conformational change essentialfor T cell receptor signaling and synapse formation. Cell 109, 901–912 (2002).
81. Gao, G.F., Rao, Z. & Bell, J.I. Molecular coordination of αβ T-cell receptors and coreceptors CD8 andCD4 in their recognition of peptide-MHC ligands. Trends Immunol. 23, 408–413 (2002).
82. Lawrence, M.C. & Colman, P.M. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234,946–950 (1993).
83. Locksley, R.M., Reiner, S.L., Hatam, F., Littman, D.R. & Killeen, N. Helper T cells without CD4: controlof leishmaniasis in CD4-deficient mice. Science 261, 1448–1451 (1993).
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
reim
mu
no
log
y
Copyright © 2022 FDOKUMEN




















