
MOLECULAR INTERACTIONS MEDIATING T CELL ANTIGEN RECOGNITION
32
Annu. Rev. Immunol. 2003. 21:659–84 doi: 10.1146/annurev.immunol.21.120601.141036 Copyright c 2003 by Annual Reviews. All rights reserved MOLECULAR INTERACTIONS MEDIATING TCELL ANTIGEN RECOGNITION P. Anton van der Merwe 1 and Simon J. Davis 2 1 Sir William Dunn School of Pathology, University of Oxford, Oxford OX1 3RE, UK; email: [email protected] 2 Nuffield Department of Clinical Medicine, University of Oxford, Oxford Radcliffe Hospital, Oxford OX3 9DU; email: [email protected] Key Words T cell receptor, coreceptor, costimulation, accessory molecules ■ Abstract Over the past decade, key protein interactions contributing to T cell antigen recognition have been characterized in molecular detail. These have included interactions involving the T cell antigen receptor (TCR) itself, its coreceptors CD4 and CD8, the accessory molecule CD2, and the costimulatory receptors CD28 and CTLA-4. A clear view is emerging of how these molecules interact with their ligands at the cell- cell interface. Structural and binding studies have confirmed that the proteins span small but comparable distances and that, overall, they interact very weakly. However, there have been important surprises as well: that TCR interactions with peptide-MHC are topologically constrained and characterized by considerable conformational flexibility at the binding interface; that coreceptors engage peptide-MHC with extraordinarily fast kinetics and at angles apparently precluding direct interactions with the TCR bound to the same peptide-MHC; that the structural mechanisms allowing recognition by costimulatory and accessory molecules to be weak and yet specific are very hetero- geneous; and that because of differences in both binding affinity and stoichiometry, there is enormous variation in the stability of the various costimulatory receptor/ligand complexes. These studies provide the necessary framework for exploring how these molecular interactions initiate T cell activation. INTRODUCTION Antigen recognition by T cells is the key event controlling the adaptive immune response. Its importance has made it the focus of intense study, making it possibly the best-understood cell-cell recognition process. Following an initial period when attention was directed at identifying the various molecules that contribute to T cell antigen recognition, in the past decade attention has shifted to understanding the mechanisms underlying antigen recognition. A key requirement for such under- standing is a detailed characterization of the structure and binding properties of each molecular interaction. In recent years considerable progress has been made in characterizing four key sets of interactions, and we review this progress here. 0732-0582/03/0407-0659$14.00 659 Annu. Rev. Immunol. 2003.21:659-684. Downloaded from arjournals.annualreviews.org by University of Oxford - Nuffield College on 07/02/09. For personal use only.
Transcript of MOLECULAR INTERACTIONS MEDIATING T CELL ANTIGEN RECOGNITION

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE10.1146/annurev.immunol.21.120601.141036
Annu. Rev. Immunol. 2003. 21:659–84doi: 10.1146/annurev.immunol.21.120601.141036
Copyright c© 2003 by Annual Reviews. All rights reserved
MOLECULAR INTERACTIONS MEDIATING T CELL
ANTIGEN RECOGNITION
P. Anton van der Merwe1 and Simon J. Davis21Sir William Dunn School of Pathology, University of Oxford, Oxford OX1 3RE, UK;email: [email protected] Department of Clinical Medicine, University of Oxford, Oxford RadcliffeHospital, Oxford OX3 9DU; email: [email protected]
Key Words T cell receptor, coreceptor, costimulation, accessory molecules
■ Abstract Over the past decade, key protein interactions contributing to T cellantigen recognition have been characterized in molecular detail. These have includedinteractions involving the T cell antigen receptor (TCR) itself, its coreceptors CD4 andCD8, the accessory molecule CD2, and the costimulatory receptors CD28 and CTLA-4.A clear view is emerging of how these molecules interact with their ligands at the cell-cell interface. Structural and binding studies have confirmed that the proteins span smallbut comparable distances and that, overall, they interact very weakly. However, therehave been important surprises as well: that TCR interactions with peptide-MHC aretopologically constrained and characterized by considerable conformational flexibilityat the binding interface; that coreceptors engage peptide-MHC with extraordinarily fastkinetics and at angles apparently precluding direct interactions with the TCR boundto the same peptide-MHC; that the structural mechanisms allowing recognition bycostimulatory and accessory molecules to be weak and yet specific are very hetero-geneous; and that because of differences in both binding affinity and stoichiometry,there is enormous variation in the stability of the various costimulatory receptor/ligandcomplexes. These studies provide the necessary framework for exploring how thesemolecular interactions initiate T cell activation.
INTRODUCTION
Antigen recognition by T cells is the key event controlling the adaptive immuneresponse. Its importance has made it the focus of intense study, making it possiblythe best-understood cell-cell recognition process. Following an initial period whenattention was directed at identifying the various molecules that contribute to T cellantigen recognition, in the past decade attention has shifted to understanding themechanisms underlying antigen recognition. A key requirement for such under-standing is a detailed characterization of the structure and binding properties ofeach molecular interaction. In recent years considerable progress has been madein characterizing four key sets of interactions, and we review this progress here.
0732-0582/03/0407-0659$14.00 659
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

19 Feb 2003 16:51 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
660 VAN DER MERWE ¥ DAVIS
The most important interaction is that of the T cell antigen receptor (TCR).Conventional T cells expressαβ TCRs, which engage processed antigen presentedon MHC molecules. T cells can also expressγ δ T cell receptors. These are lesswell characterized and appear to recognize unprocessed ligands directly, withouta requirement for presentation on MHC. This review focuses onαβ TCRs. T cellsalso express either CD8 or CD4 coreceptor molecules, which bind to MHC class Iand MHC class II molecules, respectively, enhancing TCR recognition of peptide-MHC. The third set of interactions we review are those between the related Tcell molecules CD28 and CTLA-4 and their ligands B7-1 and B7-2 expressed onantigen-presenting cells. Because interactions between CD28 and its ligands areessential for normal T cell responses, CD28 is termed a costimulatory receptor, andits ligands costimulatory ligands. Finally, we review interactions between CD2 andits related ligands. These interactions typically enhance T cell antigen recognition,but their contribution is more subtle and less well understood than is the case forcoreceptor and costimulatory molecules.
T CELL RECEPTOR INTERACTIONS
Structure
Theαβ TCR comprises disulphide-linkedα andβ chains, each of which has amembrane-distal variable (Vα or Vβ) and membrane proximal constant (Cα andCβ) immunoglobulin superfamily (IgSF) domain, transmembrane regions, andshort cytoplasmic segments. Based on primary sequence analysis it was correctlypredicted that the extracellular portion of the TCR would be structurally similar tothe antigen-binding fragment (Fab’) of an antibody molecule. The peptide-MHCbinding site is formed primarily from three complementarity-determining regions(CDRs) or loops contributed by each Vα and Vβ domain. MHC molecules havea membrane-distal binding platform comprised of aβ-sheet upon which the pre-sented peptide antigen is bound between twoα helices. Over the past 6 years thecrystal structures of several TCR:peptide-MHC complexes have been solved. Adetailed description of these structures is beyond the scope of this review, whichwill instead focus on some key features of these structures and the insights theyprovide.
THERE IS SIGNIFICANT VARIATION IN THE OVERALL STRUCTURE OF TCR:PEPTIDE-
MHC COMPLEXES In the complexes visualized thus far, TCRs dock onto thepeptide-MHC in a topologically constrained manner, i.e., with the Vα domainof the TCR positioned over the N-terminal half of the peptide and the Vβ domainover the C-terminus (Figure 1a) (1, 2). However, there is significant variation be-tween complexes. The greatest degree of variation is in the twist around the longaxis of the TCR:peptide-MHC complex, which varies by up to 35◦ (Figure 1a). Thisvariation will likely increase as more structures are solved, but it is neverthelessclear that there must be constraints on the binding orientation.
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 661
What might these constraints be? One possibility is the presence of conservedcontacts between conserved portions of the TCR and MHC. However, detailedcomparisons between available TCR:peptide-MHC structures show that there areno contacts that are conserved in all structures (2). A second possibility is thatthe shape of the TCR and/or peptide-MHC–binding surfaces limits the number ofdocking orientations. This possibility is supported by the fact that the peptide-MHCsurface is not completely flat, particularly in the case of MHC class I molecules.The N-terminal halves of theα1 andα2 helices are kinked, forming bumps onthe peptide-MHC surface (Figure 1b), which generates a shallow diagonal grooveacross the peptide. A diagonal orientation seems to be imposed on binding in orderto maximize the number of contacts between the binding surface of the TCR, whichhas an irregular oval shape, and this diagonal groove in the MHC-peptide surface.The variation in the fine structure of the antigen-binding surface of TCRs accountsfor the observed variation in binding orientation. It is notable that theα1 helix inMHC class II molecules lacks this kink (Figure 1b), making the groove somewhatshallower. This may explain why TCRs appear to engage peptide MHC class IIwith a slightly different orientation than MHC class I molecules, although there isoverlap between the two (Figure 1a) (3).
These MHCα-helix shape considerations do not entirely explain the bindingorientation because they would be compatible with TCR engaging at∼180◦ twistrelative to the observed orientation. One possibility is that the binding orientationis imposed by positive selection and that there are conserved contacts betweenpositively selecting self-peptide-MHC allele complexes and the TCRs they se-lect that are not identifiable in the limited number of structures solved to date.Comparison of two TCRs (A6 and B7), both of which were positively selected onHLA-A2, provides some support for this in that the CDR1α and CDR2α loops bindto the same portion of HLA-A2 (4). Conversely, because the V segments (whichdetermine the CDR1 and CDR2 sequences) are encoded in the germ line, evolu-tionary selection of Vα segments that maximize the size of the positively selectedrepertoire as a result of being selectable by multiple MHC alleles may explainwhy this orientation is conserved between MHC alleles. This mechanism is alsoconsistent with observations that T cells bearing TCRs with certain Vα segments(and therefore having identical CDR1α and CDR2α sequences) are preferentiallyselected into the CD4 or CD8 lineage (5). A second possibility that is not mutu-ally exclusive is that this orientation allows the coreceptors CD4 and CD8 to bindsimultaneously to the peptide-MHC in a conserved orientation with respect to theco-engaged TCR:CD3 complex. In any event, the significant variation in the ori-entation in which TCRs engage peptide-MHC rules out models of TCR triggeringproposing that TCR engagement with peptide-MHC produces a new compositebinding surface for other molecules.
BINDING IS ACCOMPANIED BY CONFORMATIONAL CHANGES AT THE INTERFACE Themost striking feature of the fine structure of the TCR:peptide-MHC binding inter-face is its variability [reviewed recently by Wilson and colleagues (2, 6)]. Indeed
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
662 VAN DER MERWE ¥ DAVIS
there are very few features common to all the interfaces visualized thus far. Oneconsistent feature is that the peptide contributes a smaller proportion of the buriedsurface area and a smaller number of contacts than the MHC surface. The potentialsignificance of this is discussed further below. A second consistent feature is that,where the same TCR structure has been solved in the bound and the unbound state(7, 8) or bound to different peptide-MHC ligands (9), one or more of the CDRloops adopt different conformations. In all these cases the CDR3 loops showed thegreatest conformational change. This indicates that the antigen-binding surface ofthe TCR exhibits conformational flexibility. Two other types of evidence supportthe existence of conformational flexibility.
First, NMR (nuclear magnetic resonance) structural analysis of a TCR showedincreased mobility of the CDR3 loops (10). Second, the unusual kinetic and ther-modynamic properties of TCR:peptide-MHC interactions are consistent with con-formational flexibility of the TCR and/or peptide-MHC binding surfaces, whichdecreases upon binding (11–13). It has been proposed that the TCR may be pre-disposed to having a flexible peptide-MHC binding site because it is formed fromloops generated (in the case of CDR3 loops) and assembled in a quasi-randommanner. This flexibility may contribute to the high degree of promiscuity or cross-reactivity evident in TCR recognition of peptide-MHC. Theoretical considerationsand experimental data suggest that a typical TCR can recognize in excess of 105
peptide-MHC complexes (14). This cross-reactivity is very likely a crucial fea-ture of TCRs because it may enable the available repertoire of TCR specificitiesin a single individual to recognize the much larger repertoire of peptide-MHCcomplexes that can be presented in that individual. This eliminates “holes” inthe TCR repertoire that would allow organisms easily to escape detection bythe adaptive immune response. It is important to stress, however, that the con-formational changes evident on TCR binding to peptide-MHC are restricted tothe binding interface, ruling out binding-induced conformational changes of theTCR itself as a mechanism of TCR triggering. Although it seems unlikely tous, these data nevertheless allow for the possibility that TCR engagement al-ters its position with respect to another TCR or to associated CD3 signalingmolecules.
DO PEPTIDE AND MHC MAKE DISTINCT CONTRIBUTIONS TO TCR BINDING? One un-resolved issue is the extent to which TCR:peptide and TCR:MHC contacts con-tribute to the affinity or binding energy of the TCR:peptide-MHC interaction. DoTCR:MHC contacts produce the bulk of the binding energy, with TCR:peptide con-tacts providing an incremental amount, sufficient to exceed an affinity threshold?Or do TCR:peptide contacts provide a major portion of the binding energy, withTCR:MHC contacts playing a primarily permissive role? One consistent featureof TCR:peptide-MHC complexes is that the peptide contributes a smaller portionof the binding interface (21–34%) and a smaller proportion of contacts (26–47%)than the MHC (2). However, because contacts do not necessarily contribute to thebinding energy, it does not necessarily follow that TCR:peptide contacts contribute
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 663
less to the binding energy than TCR:MHC contacts. The contribution of a contactto the binding energy needs to be directly measured, and this is usually done byanalyzing the effect of its elimination.
Recent studies have used this approach on different TCR:peptide-MHC interac-tions, mutating either the TCR (15, 16) or the peptide-MHC (17, 18). In studies ofthe 2C TCR binding to two different peptide-MHC complexes (15, 16), mutationsof TCR residues predicted to contact mainly the MHC (CDR1 and 2) had a greatereffect on binding than mutations of CDR3 residues, which are predicted to contactmainly peptide. Unfortunately not all mutations were informative (several werenot expressed), and it was not possible to assess the contribution of the glycine-rich CDR3β loop. In two studies in which all MHC residues either known (17) orpredicted (18) to make contact with the TCR were mutated to alanine (or glycine ifalready alanine), mutations of individual MHC residues did not have as profoundan effect on TCR:peptide-MHC binding as mutations of TCR contacting residuesin the peptide itself. However, because there are many more MHC residues thanpeptide residues contributing to the interface, these data do not rule out the pos-sibility that TCR:MHC interactions contribute the bulk of the binding energy. Itwas noteworthy that portions of the MHC that contributed the most binding energydiffered in these studies, consistent with the failure of structural analyses to iden-tify conserved TCR:MHC contacts that might account for the relatively conservedorientation of TCR binding.
STRUCTURAL STUDIES OF CD3 SUBUNITS Each TCR is constitutively associatedwith at least six CD3 polypeptides. These include CD3εγ and CD3εδ heterodimersand a CD3ζ homodimer. The solution structure of a refolded, chimeric ectodomainof a CD3εγ heterodimer has recently been determined, revealing two C2-set IgSFdomains, which associate via an unusual side-to-side interface involving paired Gstrands that continue into the conserved stalk region (19). It remains to be confirmedthat CD3ε andγ chains associate in this way, if at all, in vivo, and it is unclearhow such dimers might associate with each other and with the TCR. Two recentstudies have suggested that CD3 molecules may undergo structural changes thatinfluence signaling. In the first study the CD3ζ cytoplasmic domain underwenta conformational change and became resistant to phorphorylation by Src familykinases upon binding to acidic phospholipid-containing lipid vesicles (20). In asecond study the binding of an antibody Fab’ fragment to the CD3ε ectodomainmodulated association of the adaptor molecule Nck to its cytoplasmic tail (21),leading the authors to propose that the same phenomenon might be induced byphysiological engagement of the TCR by peptide-MHC.
Binding Properties
BINDING KINETICS TEND TO BE SLOW The main technique used for studying in-teractions of cell-cell recognition molecules, including the TCR, is to producesoluble forms of one or both binding partner(s) and to study the solution-binding
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
664 VAN DER MERWE ¥ DAVIS
TABLE 1 Binding properties of TCR:agonist peptide-MHC interactions1
Interaction Kd ( µM) k on (M−1 . s−1) koff (s−1) Reference
TCR:peptide-MHC class I2C/p2Ca/H2-Ld (allogeneic) 0.1–3 8–200 x 103 ∼0.03 (115–117)2
42.12(OT-1)/OVA/H2-Kb 7 3,000 0.02 (118)A6/tax/HLA-A2 1 100,000 0.1 (9)F5/flu/H2-Db 7 30,000 0.1 (11)JM22/tax/HLA-A2 6 40,000 0.2 (11)T1/PbCS(ABA)/H2-Kd 4 n.d. n.d. (56)
TCR:peptide-MHC class II2B4/MCC/I-Ek (early studies) 50–90 ∼1,000 ∼0.05 reviewed in (22)2
2B4/MCC/I-Ek(recent studies) 6 4,000 0.02 (12, 18)3
14.3.d/Flu/H2-Ed ∼10 n.d. n.d. (119)4
3.L2/Hb/I-Ek 10, 50 6,000 0.06 (120)5
172.10/MBP/I-Au 6 40,000 0.2 (13)1934.4/MBP/I-Au 30 5,000 0.2 (13)D10/CA/I-Ak 7 6,000 ∼0.05 (59)
1All measurements performed at 25◦C using surface plasmon resonance (SPR) except where indicated. All TCRs included,with the exception of the 2C TCR, were from syngeneic T cells, and the peptides used are agonist peptides. n.d., not done.2Includes measurements performed by techniques other than SPR.3These recent SPR measurements of the 2B4 TCR are likely to be more accurate because MCC/I-Ek was immobilized in amanner (via a biotinylated C-terminus) unlikely to affect TCR binding.4Performed by inhibition of T cell activation using soluble TCR at 37◦C.5The lower Kd value was calculated from kon and koff, whereas the higher value was determined by equilibrium binding.
properties. Most binding studies have used surface plasmon resonance as imple-mented in the BIAcore instrument, which is particularly well suited to measuringweak protein-protein interactions. One unanticipated finding is that TCR:peptide-MHC interactions have affinities that are at the high end of the range measured forcell-cell recognition molecule interactions (Tables 1 and 2). A second unexpectedfinding was that TCR:peptide-MHC interactions typically have slower kineticsthan interactions between cell-cell recognition molecules with comparable affini-ties, by as much as two orders of magnitude (Table 2). The slower association rateconstant is evidence for the existence of conformational flexibility at the bindinginterface. Conformational flexibility slows down binding because only a smallfraction of encounters will find the binding surface in a conformation compatiblewith binding. The slow dissociation rate constant is also highly significant becauseit indicates that, once formed, the TCR:peptide-MHC complex is more stable thanother cell-cell recognition molecule interactions. Thermodynamic analysis of theTCR peptide-MHC interactions supports this interpretation because it reveals thatbinding is characterized by unusually favorable enthalpic changes and highly un-favorable entropic changes (11–13). Large favorable enthalpic changes suggestthat a substantial number of favorable bonds form upon binding. Unfavorable
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 665
TABLE 2 Binding properties of some lymphocyte cell-cell recognition molecules
Interaction Temp (◦C) Kd (µM) k on (M−1 . s−1) koff (s−1) References
TCR:peptide-MHC 25 1–10 103–105 0.01–0.2 (See Table 1)1
CD8/MHC class I 25 50–200 — >20 (33, 53–56)2
CD4/MHC II 25 >200 — — (58, 59)
CD2/CD58 37 ∼10 4 x 105 4 (121)
CD2/CD48 (rodent) 37 60 >105 6 (122)
2B4/CD48 37 10 3 x 105 3 (123)
CD28/CD86 37 20 106 >20 (94)
CTLA-4/CD80 37 0.2 2 x 106 0.4 (100)
KIR/MHC I 25 10 2 x 105 ∼2 (124, 125)
OX2/OX2R 37 2 4 x 105 0.8 (126)
Selectin/ligand 37 0.3–100 105–106 1.4–10 reviewed in (127)
LFA-1/ICAM-1 25 0.13 2 x 105 0.03 (128)
1TCR:peptide-MHC Kd values at 37◦C are typically 1.5–2.5 fold higher than at 25◦C (see references cited in Table 1).2There is some variation between alleles but no difference between CD8αα and CD8αβ.
entropic changes are consistent with a decrease in conformational flexibility at thebinding interface upon binding.
RELATIONSHIP BETWEEN BINDING PROPERTIES AND FUNCTIONAL OUTCOME Se-veral studies have examined the correlation between the solution-binding prop-erties and functional consequences of TCR:peptide-MHC interactions. A broadcorrelation between affinity/half-life and functional effect [reviewed in (22, 23)]was observed, although there were some exceptions (24–28). This broad correla-tion supports models of TCR triggering in which the duration of binding, rather thana specific conformational change, determines the outcome of TCR:peptide-MHCinteractions.
What of the aforementioned exceptions to the correlation between affinity/half-life and functional outcome (24–28)? One possible explanation lies in the fact thatTCR:peptide-MHC interactions, like all interactions between membrane-anchoredmolecules, are subject to mechanical stress (29, 30). Whereas mechanical stresswill generally enhance dissociation, interactions can vary considerably in the de-gree of this enhancement (30). Thus, two TCR:peptide-MHC interactions thathave identical half-lives in solution can have different half-lives when subjectedto mechanical stress. Future studies that measure mechanical strength directly ormeasure solution-binding properties (such as the activation enthalpy of dissoci-ation) that are thought to correlate better than half-life/affinity with mechanicalstrength are needed to address this issue (31).
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

19 Feb 2003 16:52 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
666 VAN DER MERWE ¥ DAVIS
CORECEPTOR INTERACTIONS
Given their functional similarity, the CD4 and CD8 molecules have surprisinglydifferent structures (Figure 2). Whereas CD4 is a monomeric polypeptide withfour IgSF domains in its ectodomain, CD8 is a disulphide-linked dimer in whicheach chain has a single IgSF domain supported on a stalk. In most T cells CD8appears to exist exclusively as a heterodimer of anα andβ chain (CD8αβ), butsome T cells, notably intraepithelial lymphocytes, express the CD8αα homodimer(32). This pattern of expression, and the finding that CD8αα is substantially lesseffective than CD8αβ as a coreceptor (32), strongly suggests that CD8αβ is theprimary coreceptor for conventional MHC class I–restricted T cells. What then isthe function of CD8αα? Recently it was shown that CD8αα binds with a higheraffinity than CD8αβ to a nonclassical MHC class I molecule, thymus leukemiaantigen, and that this interaction modifies recognition of conventional peptide-MHC class I by these T cells (33).
Structural Studies
Both CD4 and CD8 bind to nonpolymorphic regions at the base of MHC molecules.Crystal structures have now been solved of human (34) and mouse (35) CD8αα incomplex with MHC class I, as well as, at low resolution, a mouse CD4 fragment incomplex with human MHC class II (36). When these structures are superimposedon structures of TCR:peptide-MHC complexes it is evident that the angle at whichCD4 and CD8 engage peptide-MHC precludes direct association with a TCR thatbinds the same peptide MHC complexes (Figure 2). How can this be reconciledwith evidence that CD4 and CD8 physically associate with the TCR:CD3 complex(37, 38)? One possibility is that the coreceptors and the TCR are linked indirectlythrough CD3 chains and associated molecules. This is supported by the observationthat CD4 and CD8 associate with TCRs through ZAP-70 and Lck (39, 40). Ac-cording to this model, nascent TCR triggering leads to recruitment of coreceptorsvia ZAP-70/lck, and the coreceptor thereby stabilizes further the TCR:peptide-MHC interaction (41). A second possibility, proposed more recently (42), is thatthe coreceptor and the TCR that it associates with bind different peptide-MHCcomplexes. This was proposed as part of a new “pseudodimerization” model ofTCR triggering, which was itself proposed to explain the observation that a singleagonist peptide-MHC complex could lead to TCR triggering (42). It was suggestedthat, when a TCR with an associated coreceptor binds to an agonist-peptide-MHC,the coreceptor binds a distinct self-peptide-MHC complex. A TCR pseudodimeris formed when a second TCR binds to this self-peptide-MHC complex.
Whereas it is believed that CD8αβ is likely to bind to MHC class I in much thesame way as CD8αα, there is disagreement as to which CD8α chain the CD8βchain will “replace.” Based on the analysis of electrostatic surface potential, Gaoet al. proposed that CD8 would bind in the position of the CD8α chain adjacent tothe peptide-binding platform (34). A subsequent mutagenesis experiment provides
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 667
some support for this (43). In contrast, Reinherz and colleagues have proposed that,because the CD8β stalk region is several amino acid residues longer than the CD8α
stalk, CD8β is more likely to bind in the more “distal” position (35).The presence of O-linked carbohydrates suggests that the CD8 stalk region
forms an extended structure, a possibility supported by low-resolution structuralstudies (44, 45). It was observed several years ago that glycosylation of the CD8β
stalk changes during thymocyte maturation and upon activation of mature CD8T cells (46). It was also shown several years ago that the binding propertiesof CD8 appear to be regulated by, for example, TCR engagement (47). Recentfindings (48, 49) suggest that these phenomena are linked because changes in CD8β
glycosylation correlate with changes in the binding of peptide-MHC tetramers, andmanipulations decreasing sialylation of T-cell surface molecules increase the bind-ing of peptide-MHC tetramers. These data have been interpreted to suggest thatsialylation induces structural changes in the CD8β stalk that decrease the affin-ity of CD8αβ for MHC class I (48). However, much earlier data indicate thatthe structural effects of O-glycans depend only on steric interactions between thepeptide-linked GalNAc and the adjacent amino acids of the polypeptide (50–52).The structural properties of the stalk region are therefore not expected to be af-fected by chain-branching or by changes in terminal sialylation. In light of this, andbecause binding was assessed using MHC multimers binding to cells, it is possiblethat changes in the valency of binding, rather than the CD8αβ:MHC affinity perse, were responsible for the observed changes in peptide-MHC tetramer binding.
Binding Properties
A number of groups have studied the binding properties of the CD8αα:MHCclass I and CD8αβ:MHC class I interactions using soluble, monomeric formsof these molecules (33, 53–56). The consensus is that CD8αα and CD8αβ bindwith a similar affinity, that this affinity is very low (Kd= 50–200µM at 25◦Cand>200µM at 37◦C), and that the binding of CD8 to peptide-MHC does notaffect binding of soluble TCR to the same peptide-MHC complex. Analysis of theCD4:MHC class II interactions has proved to be more difficult. Whereas an earlystudy measured a high affinity (57), attempts to reproduce this failed or could onlymeasure very weak binding (58). Recently, Xiong et al. (59) reported binding ofmouse MHC class II to human CD4 on the BIAcore with a Kd of∼200µM at25C, but the exceptionally low binding response relative to background response(∼1%) and the low level of apparent CD4 activity (<2%) suggest that this resultshould be treated with caution. Despite these problems it is reasonable to concludethat the affinity of the CD4:MHC class II interaction is at least as low and possiblymuch lower than that of the CD8:MHC class I interaction.
CD8αβ is better than CD8αα at enhancing TCR recognition of peptide-MHCclass I [reviewed in (32)], and several studies have investigated the basis for thisdifference. Studies using soluble (53, 54, 56) or cell surface forms of CD8αα and-αβ have failed to detect significant differences in binding to MHC class I. Whereas
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
668 VAN DER MERWE ¥ DAVIS
a recent study showed that MHC class I tetramers bind better to CD8αβ thanCD8αα expressed on cells (60), it is possible that CD8β induces, and the MHCclass I tetramers detect, an increase in the valency of CD8 rather than an increasein the affinity of the CD8:MHC class I interaction. A number of studies havedemonstrated that the transmembrane/cytoplasmic portion of the CD8β chain isimportant for enhancing CD8αβ function (56, 60–62) and that the cytoplasmicportion of CD8β enhances the association of CD8 with Lck and the TCR:CD3complex (62, 63). Although this might seem paradoxical given the fact that Lckbinds directly to the CD8α cytoplasmic domain, recent data suggest that the CD8β
cytoplasmic domain mediates association with lipid rafts, an effect dependent onits palmitoylation (56).
Coreceptor:MHC interactions have a much lower affinity than TCR:peptide-MHC interactions, but the differences in the dissociation rate constants are evenmore profound. The CD8:MHC interaction has a koff at least two orders of magni-tude faster than TCR:peptide-MHC interactions (Table 2). One important impli-cation of the exceptionally low affinity of coreceptor binding to MHC is that it isunlikely that the coreceptor:MHC interaction will occur at the cell-cell interface in-dependently of TCR:peptide-MHC interactions. These considerations suggest thatto have an effect, a coreceptor must physically associate with the TCR complexso that it can simultaneously bind to the TCR and the peptide-MHC. Coreceptorspreferentially interact with triggered TCR:CD3 complexes via signaling moleculesLck and ZAP-70 (39, 40), and there is evidence that mediating this interaction isan important function of CD4-associated Lck (41). Taken together these obser-vations support a recruitment model for coreceptor function, whereby low leveltriggering of a TCR:CD3 complex following initial/weak peptide-MHC engage-ment leads to recruitment of a coreceptor to that complex, which enhances TCRtriggering by stabilizing the TCR:peptide-MHC interaction and/or amplifying sig-naling. Because the TCR:peptide-MHC interaction has a much longer half-lifethan coreceptor:MHC interactions (Table 2), the latter will enhance the stabilityof the TCR:peptide-MHC:coreceptor complex but not dominate it.
CD2 INTERACTIONS
Binding Properties
Owing largely to genome sequencing, the CD2 subset of the IgSF has recentlyenlarged considerably and consists of at least 11 members [CD2, CD48, CD58(LFA-3), CD84, CD150 (SLAM), CD229, CD244, CS1, BLAME, CD2F-10,and Ly108)] (E.J. Evans, J.A. Fennelly & S.J. Davis, in preparation). System-atic analyses are yet to be done, but the known receptor-ligand interactions alloccur within the family, implying that the proteins evolved from a common, ho-mophilic precursor and suggesting that other interacting pairs of molecules willbe identified within the subset. CD2 binds LFA-3 in humans and CD48 in the rat,with low affinities (Kd 10µM and 60µM, respectively) and very rapid kinetics
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 669
(kon ≥ 105 M−1s−1, koff ≥ 4 s−1) [reviewed in (64, 65)]. The self-associationof the T-cell activation marker, SLAM, is the only other interaction character-ized in detail (66). It was initially thought that SLAM self-associates with sub-nanomolar affinity (67), whereas in fact it interacts even more weakly than ratCD2 and CD48.
Studies of T cells interacting with model bilayers indicate that, in contrast tothe CD2:LFA-3 interaction, which has a very favorable two-dimensional affin-ity, the murine counterpart barely sustains adhesive interactions (68, 69). Thisfinding highlights the fact that an affinity threshold must exist below which, atphysiological expression levels, significant levels of binding will not take placespontaneously between monovalent cell-surface molecules. This is likely to be rel-evant to the functions of coreceptors, whose affinity for MHC appears to be lowerthan Kd∼ 200µM at physiological temperatures. In support of this, adhesion me-diated by coreceptor:MHC interactions is only seen when one or another of thesemolecules are overexpressed on cells (70, 71). These considerations notwithstand-ing, the very low two-dimensional affinity of the murine CD2-CD48 interactionstrongly implies that CD2 is not a conventional cell adhesion molecule. This issupported by a recent study (72) using the surface force apparatus that measured amuch lower adhesion energy (∼1 kT) for this interaction than had been measuredpreviously for the homotypic adhesion molecule C-cadherin (∼8 kT). Studiesof TCR transgenic, CD2−/− mice nevertheless show that CD2 lowers thresholdsfor TCR triggering in vitro and T cell activation in vivo (73). Other members ofthe family containing the immunoreceptor tyrosine-based inhibitory motif, suchas 2B4, have been implicated in signaling events controlling lymphocyte and, inparticular, NK cell activation (74, 75).
Structures
With one exception, the CD2 subset of the IgSF consists of monomeric type Imembrane glycoproteins with two extracellular IgSF domains [Ly-9 has four IgSFdomains; reviewed in (76)]. The extracellular region of CD2 remains the only suchstructure from this subset to be solved in its entirety. The clustering of chargedresidues in the ligand-binding face revealed by the structural work provided the firstclue to the novel features of CD2-ligand recognition. The structure of CD2 has beenreviewed (65, 76), and we focus here on recent studies of ligand and CD2:ligandedcomplex structures and their implications for the nature of recognition by CD2.
As expected, the ligand-binding domain of LFA-3 closely resembles that ofCD2 (77, 78). The CD2-binding, AGFCC′C′′ β-sheet of LFA-3 has two key fea-tures. Unlike the ligand-binding face of CD2, which is very flat, the equivalentface of LFA-3 has two depressions separated by a “ridge” formed by residuesin the C and Fβ-strands, preventing any interaction between LFA-3 and CD2from having a high degree of surface-shape complementarity (77, 78). In addi-tion, like CD2, this surface is heavily populated with charged, predominantlyacidic residues and therefore exhibits considerable electrostatic complementarity
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

19 Feb 2003 16:56 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
670 VAN DER MERWE ¥ DAVIS
with the much more basic ligand binding site of CD2 (Figure 3) (77,78).
Studies of charge-switch mutants of CD2 and CD48 (79) confirmed that CD2forms ligand complexes similar to homophilic, orthogonal “head-to-head” latticecontacts seen in CD2 crystals and eliminated an alternative arrangement (80). Thisindicates that the CD2/ligand interactions would span∼134 A, which has nowbeen demonstrated directly for the mouse CD2/CD48 interaction using the surfaceforce apparatus (72). It was noted that the longest dimension of the “head-to-head”complex is very similar to that of TCR:peptide-MHC complexes (∼135A), sug-gesting that the spontaneous interaction of CD2 with its ligands in the T cellantigen–presenting cell contact zone would facilitate the scanning of MHC-peptidecomplexes by the TCR (79). This concept accounts for the observation that CD2significantly enhances TCR engagement in vitro and in vivo under conditions oflow antigenic load (73). The structure of the complex of the ligand-binding do-mains of CD2 and LFA-3 confirmed that the proteins interact orthogonally and thatbinding is dominated by electrostatic contacts between binding surfaces exhibitingpoor shape complementarity (Figure 4A) (81). The complementarity between thebinding surfaces is nevertheless better than that observed in TCR:peptide-MHCcomplexes (81, 82).
The Role of Electrostatics
In the CD2:LFA-3 complex 9 largely basic CD2 residues interact with 11 predom-inantly acidic residues from LFA-3, forming an interdigitating network of 10 saltbridges and 5 hydrogen bonds; only 3 hydrophobic residues are present at the inter-face (81). Considerable attention has focused on the role of these charged residuesin binding. Unexpectedly, substitution of over half the charged or polar residues inthe binding site of rat CD2 with alanine resulted in only modest changes in CD48binding affinity (83). Moreover, binding was unaffected by large increases in ionicstrength (83), confirming that the net electrostatic contribution to the binding en-ergy is zero. Similarly, almost half the residues involved in electrostatic contactsat the human CD2/LFA-3 interface could be substituted without profound lossesin binding (84). In contrast to the lack of effects on the energetics of binding,the specificity of CD48 recognition by rat CD2 was severely compromised whenthe charged residues were mutated to alanine (83). Electrostatic complementar-ity is required to compensate for the removal, upon binding, of water bound tothe charged residues. In this way electrostatic interactions uncouple increases inspecificity from increases in affinity and are thus ideally suited to mediating weak,specific protein recognition (83).
The Paradigmatic Value of CD2
Ligand recognition by CD2, based primarily as it is on electrostatic contacts be-tween binding sites and exhibiting little, if any, surface complementarity, repre-sents a departure from the paradigm that emerged from the analysis of high-affinity
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 671
interactions, such as that between antibodies and protein antigens. For these inter-actions, binding specificity and affinity are each dependent on substantially morehydrophobic binding surfaces sharing a much higher degree of surface-shape com-plementarity (85, 86). How representative is the CD2-ligand interaction? The de-gree of charged-residue clustering seen in the binding faces of CD2 and LFA-3is highly unusual although not unprecedented. The interaction of B7-1 and B7-2with CTLA-4, however, has an entirely different structural basis and is much more“antibody-like” (87, 88) (see below). Moreover, from a thermodynamic viewpoint,even the human and rat CD2-binding mechanism has not been conserved. Whereasligand binding by rat CD2 has equal, weakly favorable enthalpic and entropic com-ponents, a substantial entropic barrier limits the affinity of CD2:LFA-3 interac-tions (S.J. Davis, P.A. van der Merwe, M. Castro, A.M. Carmo, R. O’Brien & J.E.Ladbury, unpublished data). Thus, a variety of structural mechanisms underlieweak, specific recognition at the cell surface. Although important for highlightingthe issues involved, CD2 interactions may prove to be somewhat extreme examplesof these weak interactions in which electrostatic contacts are dominant.
COSTIMULATORY INTERACTIONS
Whereas the activating and inhibitory functions of the CD28 and CTLA-4 receptorsare well established, the reason why two, sequentially expressed ligands, B7-1 andB7-2, are necessary has been a mystery. Preservation of both genes in all mammalsexamined strongly suggests that they have each been subjected to distinct selectionpressures, but there has been little consensus regarding whether or not B7-1 andB7-2 have distinct regulatory functions and less agreement on what these functionsmight be. Part of the reason for this is that no obvious molecular basis for largefunctional differences emerged from early structural and binding studies of thesemolecules. The prevailing view has been that B7-1 and B7-2 have similar structuresand affinities for CD28 versus CTLA-4, and that CD28 and CTLA-4 are both biva-lent [see e.g., (89, 90)]. Recent work indicates instead that costimulatory receptorsand their ligands form signaling complexes of unexpected structural diversity andprovides an explanation for the existence of two costimulatory ligands.
Structures
CD28 AND CTLA-4 CD28 and CTLA-4 are type I membrane proteins consisting ofsingle, moderately to highly glycosylated V-set IgSF domains and highly conservedcytoplasmic domains, expressed at the cell surface in the form of disulphide-linkedhomodimers. In murine sCTLA-4 crystals the monomer forms dimeric contactsvia residues in theβ-sheet containing B, E, and Dβ strands (91). Because such adimer would impose a near orthogonal arrangement of CTLA-4:B7 axes incompat-ible with known membrane topologies for such molecules, this arrangement mustbe unphysiological (92). In B7:CTLA-4 complex crystals, CTLA-4 monomersdimerize via a much more limited interaction centered on the A and Gβ-strands
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
672 VAN DER MERWE ¥ DAVIS
(87, 88). This allows coligation of B7 molecules around an axis orthogonal to themembrane, maintaining the∼150-A intermembrane distance thought to be a crit-ical feature of the immunological synapse (93). There is currently no structuraldata available for CD28, but the monomer structure and ligand binding-site con-figuration are likely to be very similar to those of CTLA-4, given the degree ofprimary sequence conservation. Amino acid side chains mediating CTLA-4 dimer-ization are not conserved in CD28, however, suggesting a distinct arrangement ofmonomers in the CD28 homodimer (94).
B7-1 AND B7-2 The structure of the extracellular region of B7-1 (sB7-1) is chimericinsofar as the ligand-binding V-set domain is remarkably similar to that of acces-sory molecules, such as CD2 and CD4, whereas the membrane-proximal domainhas the GFC:DEBAβ-strand topology typical of the C1-set domains of antigenreceptors and MHC antigens (most IgSF proteins have C2-set domains) (95).B7-related proteins may thus constitute a “missing link” between conventionalcell-cell recognition molecules and the antigen receptors and MHC antigens me-diating adaptive immune responses that appeared relatively late in the evolutionof the IgSF (95).
Soluble B7-1 (sB7-1), whether crystallized in a deglycosylated state (95) orfully glycosylated in complexes with CTLA-4, formed side-by-side molecularcontacts, mediated exclusively by residues in the BED face of domain 1 thatformed a contiguous surface of∼600 A2, generating a potentially bivalent ho-modimer (95). The affinity of self-association of sB7-1 (Kd= 20–50µM) sug-gests that the native protein will spontaneously dimerize at the cell surface (95),although this has yet to be demonstrated. Because CTLA-4 is a constitutivebivalent homodimer, the two proteins were predicted to form periodic arraysat the cell surface wherein CTLA-4 homodimers are bridged by B7-1 homod-imers (95), similar to those seen in crystals of the sB7-1:sCTLA-4 complex(88). B7-2 domain 1 formed superficially similar dimeric contacts (87). Thesewere structurally asymmetric, however, contrary to the expectation that organized,self-assembled structures form via identical subunit interactions (96). In addi-tion, neither fully glycosylated sB7-2 (94) nor the unglycosylated bacterially ex-pressed B7-2 (97) dimerize in solution. Physiological B7-2 dimerization seemsunlikely.
B7:CTLA-4 COMPLEXES Like other IgSF proteins, such as CD2 and LFA-3 (81),and the coxsackievirus and adenovirus receptor (98), CTLA-4 interacts orthog-onally with B7-1 and B7-2, i.e., with an∼90◦ angle between the interactingβ-sheets. The best explanation for this is that this is how primordial IgSF proteinsinteracted (88). The sizes of the interacting surfaces are at the low end of therange for protein-protein binding sites (86, 88). As expected, binding is dominatedby the hydrophobic97MYPPPYY103 sequence conserved in CTLA-4 and CD28and implicated in binding by earlier mutational analyses (99). The very high de-gree of shape complementarity of the B7-1:CTLA-4 interface, which is as high
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 673
as has ever been seen in any other protein complex and much higher than ob-served for interacting cell surface proteins such as CD2:LFA-3 and TCR:peptide-MHC complexes, was not anticipated (Figure 4B) (88). Together, the small inter-acting surfaces and somewhat unfavorable entropy of binding (S.J. Davis, A.V.Collins, D.W. Brodie, J.E. Ladbury & P.A. van der Merwe, unpublished data)explain the relatively low affinity of these interactions (100). In the B7-2 crys-tals each CTLA-4 homodimer forms two different interfaces with B7-2, pos-sibly reflecting distinct lattice constraints on the association of each B7-2monomer (87).
IMPLICATIONS FOR SIGNALING CTLA-4 and B7-1 or B7-2 do not appear to un-dergo significant structural rearrangements upon complex formation (87, 88), rul-ing out the possibility that signaling by these molecules is driven by conformationalchanges. The dimensions of the periodic arrays formed by the sCTLA-4:sB7-1complexes in the crystal lattice (130–150A along the long axis of B7-1) suggestthat the interaction will be favored by the∼150-A intermembrane spacing likely toform within the immunological synapse (93). It was conjectured that the formationof these arrays is an essential feature of the CTLA-4/CD28 signaling mechanism(87, 92), and such an important property would be expected to be conserved. How-ever, murine B7-1 appears incapable of forming equivalent dimers owing to thelikely glycosylation of the putative dimerization surface (A. Iaboni & S.J. Davis,unpublished observations). Signaling by CD28 in this manner can also be ruled out,at least when interacting with B7-2, because B7-2 is monomeric and CD28 is mono-valent (94). Assuming that they occur at all at the T-cell-APC interface, periodicB7-1:CTLA-4 arrays are more likely simply to stabilize the inhibitory complexes.
Binding Properties
The initial studies of the interactions of these molecules, based on inconclusiveassays and incompletely characterized proteins, emphasized the uniformity oftheir binding properties, i.e., that B7-1 and B7-2 have comparable structures andaffinities for CD28 and CTLA-4, both of which in turn are bivalent (101, 102).More recent analyses, using exclusively monovalent forms of sB7-1 (100) andsB7-2 (94), and surface plasmon resonance–based technology, suggest an entirelydifferent view. The key findings are (a) that B7-1 binds CD28 and CTLA-4 at least10-fold more weakly than first thought, and with much faster kinetics; (b) that B7-2binds 13-fold more weakly to CTLA-4 than B7-1; (c) that relative to its CTLA-4binding affinity, B7-2 binds CD28 two- to threefold more effectively than B7-1 (i.e.,the CD28/CTLA-4 Kd ratios are∼8 and 20 for B7-2 and B7-1, respectively); in themouse B7-2 is an even poorer CTLA-4 ligand (A. Iaboni & S.J.Davis, unpublisheddata); and (d) that, in contrast to CTLA-4, which is bivalent, CD28 is monovalentand thus unable to participate in avidity-enhanced interactions similar to thoseproposed for CTLA-4. Together with the observation that B7-2 does not dimerize,these findings indicate that, relative to B7-1, the binding of B7-2 is biased against
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
674 VAN DER MERWE ¥ DAVIS
CTLA-4, reducing the likelihood that B7-2:CD28 interactions will be attenuatedby coincident CTLA-4 ligation, thereby enhancing the costimulatory effects ofB7-2 when CD28 and CTLA-4 are coexpressed. The reverse applies to B7-1; i.e.,its inhibitory activity is less likely to be affected by the presence of CD28. Inthis context the delayed expression of B7-1 on antigen-presenting cells appearsto be timed to specifically enhance the inhibitory function of CTLA-4, explainingthe existence of two sequentially expressed costimulatory ligands. In vivo studiesshowing that B7-1 and B7-2 antibody blockade enhances and attenuates immuneresponses, respectively [e.g., (103–106)], support the view that B7-2 is largelyactivating and B7-1 inhibitory. Studies that seemingly contradict this view [e.g.,(107)] may reflect the differential effects on TH1 versus TH2 T cell responses ofcostimulatory signaling.
Taking into account avidity effects, the earliest costimulatory signaling com-plexes to form during immune responses, i.e., between CD28 and B7-2, are likelyto be at least 10,000-fold less stable than inhibitory complexes formed later byCTLA-4 and B7-1 (Figure 5) (94). Why might such enormous stability differ-ences be necessary? It is possible that very stable interactions of B7-1 with CTLA-4 may confine CTLA-4 to the synapse where it can overturn ongoing activationsignals. In contrast, weak interactions involving B7-2 and CD28 may ensure thatcostimulatory signals are subservient to those generated by the TCR.
Figure 5 Costimulatory complexes. Quaternary structures of the costimulatory andinhibitory signaling complexes formed by human CD28, CTLA-4, B7-1, and B7-2 areshown in diagrammatic form. The solution dissociation constants of the monovalentinteractions, determined using surface plasmon resonance–based methods (94, 100),are shown above each complex.
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 675
Why do costimulatory proteins cross-react functionally? One possibility is thatthe unusual structural properties of the MYPPPY motif dominating the interac-tions of CD28 and CTLA-4 with B7-1 and B7-2 have made it resistant to struc-tural change, constraining the evolution of B7-2:CD28 and B7-1:CTLA-4 intoindependent binding and signaling units. This seems unlikely, however, given thatthe evolutionarily related, FDPPPF binding motif of the CD28-like protein ICOSfails to support B7-1 and B7-2 interactions [even though the ICOS ligand, LICOS,also binds CD28 and CTLA-4 (108)]. A second possibility, therefore, is that thecross-reaction is advantageous. Compared with CD28:B7-2 and CTLA-4:B7-1complexes, CD28:B7-1 and CTLA-4:B7-2 complexes will be intermediate instrength because CD28 is monovalent and B7-2 does not self-associate (Figure 5).The formation of these complexes may allow the intensity of costimulatory orinhibitory signaling to be varied with the stage of T cell or APC differentiation.
Costimulatory Interactions and ImmunologicalSynapse Formation
The affinity of the CD28:B7-2 interaction is similar to the affinities of TCR:peptide-MHC and CD2:LFA-3 interactions (Table 2), which occur spontaneously in twodimensions in model bilayers (68, 109). Unexpectedly, however, naive T cells failto interact with model bilayers containing B7 protein unless close membrane con-tact is induced by synapse formation or the introduction of other, topologicallysimilar molecules such as CD48 (110). As a consequence, the rate of synapseformation and the extent of TCR accumulation within the central zone of thesynapse are both unaffected by CD28 interactions (110). Rather than synergizingwith coincident signals from the TCR, costimulatory signaling now appears to bea secondary consequence of synapse formation. These findings support the ideathat, rather than enhancing or sustaining TCR signaling, the synapse generates amicroenvironment favoring secondary events such as costimulatory and cytokinesignaling and plays a central role in the delivery of full effector function via di-rected secretion (111, 112). TCR signaling, synapse formation, and costimulatorysignaling can be viewed as checkpoints coordinating the progression to full T cellactivation and commitment.
CONCLUDING REMARKS
Considerable progress has been made in understanding the nature of the key pro-tein interactions contributing to T cell antigen recognition. The well-characterizedinteractions reviewed here involve a select group of compact cell surface moleculesthat are probably highly specialized for cell-cell scanning functions in the contextof antigen presentation and reactivity. The broader significance of these findingsfor other cell-cell recognition processes, such as conventional cell adhesion, bothwithin and outside the immune system, remains to be established. With this caveat,several generalizations can be made concerning the nature of these interactions.
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
676 VAN DER MERWE ¥ DAVIS
1. The interactions are structurally heterogeneous.Some structural featuresare likely to be commonplace, such as relatively poor surface-shape complementar-ity as seen in CD2- and TCR-ligand complexes. The intense clustering of chargedresidues in the binding faces of CD2-related proteins and the extraordinarily highdegree of surface complementarity observed in the B7-1:CTLA-4 complex, how-ever, are more likely to be characteristic features of the CD2 and B7 subsets of theIgSF, perhaps reflecting the primordial properties of their progenitors.
2. The functional significance of given binding mechanisms varies case bycase.In some instances the structural features of the interactions appear to reflectclear-cut functional constraints. The best current example of this is the repertoire-extending effect of the conformational flexibility of the ligand-binding site of theTCR. For other interactions, as exemplified by ligand binding by human versusrat CD2, it probably matters far more that binding is both weak and specific thanhow this is achieved.
3. Binding mostly involves relatively flat, austere binding surfaces.Theabsence of hydrophobic pockets suited to binding small molecule inhibitors repre-sents a key obstacle limiting the potential of these otherwise excellent immunother-apeutic targets. In this context, it is a promising development that high throughputscreening has succeeded in identifying compounds that bind with submicromolaraffinity to B7-1 (113), and it will be of great interest to characterize the natureof the protein:compound interactions. Encouragement can also be taken from theobservation that gene dosage has clear-cut effects in animal models of human dis-ease. The distinct course of diabetes in wild-type versus B7−/+ NOD mice (114),for example, suggests that as little as 50% inhibition of these multivalent proteininteractions may yield real therapeutic benefit. Overall, however, it seems likelythat cell surface proteins with well-placed drug-binding pockets will at best berare. Identifying these targets, assuming they exist at all, could be among the mostimportant benefits of structural genomics initiatives.
4. Affinities are generally low, but signal complex stabilities vary enor-mously. The variation in the binding strengths of the complexes discussed herewas the least-expected feature of these interactions and perhaps the most significantfrom an immunological point of view. Even when disregarding CD4:MHC class IIinteractions, which remain truly enigmatic, the affinities of well-characterized in-teractions still vary by more than three orders of magnitude. Taking avidity effectsinto account, as is necessary, for example, in the case of costimulatiory molecules,extends the range of stabilities by perhaps two additional orders of magnitude.To our knowledge, this variation in stability is unprecedented among nonobligate,heterologous protein complexes. We favor the view that these binding differencesare highly significant and suggest that the regulated expression of these proteins,coupled with the hierarchical stabilities of the complexes they form, provides asimple mechanism for taking T cells sequentially through each of the checkpointsleading to full activation and commitment. The costimulatory system representsthe most compelling example of this process. Understanding the real significanceof these differences and using it to construct systematic, quantitative models of
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 677
immune function will be among the most important goals of the next phase ofwork.
ACKNOWLEDGMENTS
We thank S. Ikemizu and E.J. Evans for help with the figures and E.Y. Jones andD.I. Stuart for much valuable discussion. P.A.V. and S.J.D are supported by theUK Medical Research Council and the Wellcome Trust, respectively.
TheAnnual Review of Immunologyis online at http://immunol.annualreviews.org
LITERATURE CITED
1. Hennecke J, Wiley DC. 2001. T cellreceptor-MHC interactions up close.Cell104:1–4
2. Rudolph MG, Wilson IA. 2002. Thespecificity of TCR:pMHC interaction.Curr. Opin. Immunol.14:52–65
3. Hennecke J, Carfi A, Wiley DC. 2000.Structure of a covalently stabilized com-plex of a human alpha beta T-cell receptor,influenza HA peptide and MHC class IImolecule, HLA-DR1.EMBO J.19:5611–24
4. Ding YH, Smith KJ, Garboczi DN, UtzU, Biddison WE, Wiley DC. 1998. Twohuman T cell receptors bind in a similardiagonal mode to the HLA- A2/Tax pep-tide complex using different TCR aminoacids.Immunity8:403–11
5. Sim BC, Zerva L, Greene MI, GascoigneNR. 1996. Control of MHC restriction byTCR Valpha CDR1 and CDR2.Science273:963–66
6. Rudolph MG, Luz JG, Wilson IA. 2002.Structural and thermodynamic correlatesof T cell signaling.Annu. Rev. Biophys.Biomol. Struct.31:121–49
7. Garcia KC, Degano M, Pease LR, HuangM, Peterson PA, et al. 1998. Structural ba-sis of plasticity in T cell receptor recogni-tion of a self peptide-MHC antigen.Sci-ence279:1166–72
8. Reiser JB, Gregoire C, Darnault C,Mosser T, Guimezanes A, et al. 2002. A Tcell receptor CDR3 beta loop undergoes
conformational changes of unprecedentedmagnitude upon binding to a peptide/MHC class I complex.Immunity16:345–54
9. Ding Y-H, Baker BM, Garboczi DN,Biddison WE, Wiley DC. 1999. FourA6-TCR:peptide/HLA-A2 structures thatgenerate very different T cell signals arenearly identical.Immunity11:45–56
10. Hare BJ, Wyss DF, Osburne MS, Kern PS,Reinherz EL, Wagner G. 1999. Structure,specificity and CDR mobility of a classII restricted single-chain T-cell receptor.Nat. Struct. Biol.6:574–81
11. Willcox BE, Gao GF, Wyer JR, LadburyJE, Bell JI, et al. 1999. TCR binding topeptide-MHC stabilizes a flexible recog-nition interface.Immunity10:357–65
12. Boniface JJ, Reich Z, Lyons DS, DavisMM. 1999. Thermodynamics of T cellreceptor binding to peptide-MHC: evi-dence for a general mechanism of molec-ular scanning.Proc. Natl. Acad. Sci. USA96:11446–51
13. Garcia KC, Radu CG, Ho J, Ober RJ, WardES. 2001. Kinetics and thermodynamicsof T cell receptor-autoantigen interactionsin murine experimental autoimmune en-cephalomyelitis.Proc. Natl. Acad. Sci.USA98:6818–23
14. Mason D. 1998. A very high level of cross-reactivity is an essential feature of the T-cell receptor.Immunol. Today19:395–404
15. Manning TC, Schlueter CJ, Brodnicki TC,
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
678 VAN DER MERWE ¥ DAVIS
Parke EA, Speir JA, et al. 1998. Alaninescanning mutagenesis of an alpha beta Tcell receptor: mapping the energy of anti-gen recognition.Immunity8:413–25
16. Lee PU, Churchill HR, Daniels M, Jame-son SC, Kranz DM. 2000. Role of 2CT cellreceptor residues in the binding of self-and allo-major histocompatibility com-plexes.J. Exp. Med.191:1355–64
17. Baker BM, Turner RV, Gagnon SJ, Wi-ley DC, Biddison WE. 2001. Identifica-tion of a crucial energetic footprint on thealpha1 helix of human histocompatibil-ity leukocyte antigen (HLA)-A2 that pro-vides functional interactions for recogni-tion by Tax peptide/HLA-A2-specific Tcell receptors.J. Exp. Med.193:551–62
18. Wu LC, Tuot DS, Lyons DS, Garcia KC,Davis MM. 2002. A two-step bindingmechanism for T cell receptor recognitionof peptide-MHC.Nature418:552–56
19. Sun ZJ, Kim KS, Wagner G, Reinherz EL.2001. Mechanisms contributing to T cellreceptor signaling and assembly revealedby the solution structure of an ectodomainfragment of the CD3 epsilon gamma het-erodimer.Cell 105:913–23
20. Aivazian D, Stern LJ. 2000. Phosphoryla-tion of T cell receptor zeta is regulated bya lipid dependent folding transition.Nat.Struct. Biol.7:1023–26
21. Gil D, Schamel WW, Montoya M,Sanchez-Madrid F, Alarcon B. 2002. Re-cruitment of Nck by CD3epsilon revealsa ligand-induced conformational changeessential for T cell receptor signaling andsynapse formation.Cell 109:901–12
22. Davis MM, Boniface JJ, Reich Z, LyonsD, Hampl J, et al. 1998. Ligand recogni-tion by alpha beta T cell receptors.Annu.Rev. Immunol.16:523–44
23. Manning TC, Kranz DM. 1999. Bind-ing energetics of T-cell receptors: corre-lation with immunological consequences.Immunol. Today20:417–22
24. al-Ramadi BK, Jelonek MT, Boyd LF,Margulies DH, Bothwell AL. 1995.Lack of strict correlation of functional
sensitization with the apparent affinity ofMHC/peptide complexes for the TCR.J.Immunol.155:662–73
25. Sykulev Y, Vugmeyster Y, Brunmark A,Ploegh HL, Eisen HN. 1998. Peptide an-tagonism and T cell receptor interactionswith peptide-MHC complexes.Immunity9:475–83
26. Degano M, Garcia KC, ApostolopoulosV, Rudolph MG, Teyton L, Wilson IA.2000. A functional hot spot for antigenrecognition in a superagonist TCR:MHCcomplex.Immunity12:251–61
27. Baker MB, Gagnon JS, Biddison EW, Wi-ley CD. 2000. Conversion of a T cell an-tagonist into an agonist by repairing a de-fect in the TCR:peptide/MHC interface:implications for TCR signaling.Immunity13:475–84
28. Kalergis AM, Boucheron N, Doucey MA,Palmieri E, Goyarts EC, et al. 2001. Ef-ficient T cell activation requires an opti-mal dwell-time of interaction between theTCR and the pMHC complex.Nat. Im-munol.2:229–34
29. van der Merwe PA. 2001. The TCR trig-gering puzzle.Immunity14:665–68
30. van der Merwe PA. 1999. Leukocyte ad-hesion: high-speed cells with ABS.Curr.Biol. 9:R419–22
31. Leckband D. 2000. Measuring the forcesthat control protein interactions.Annu.Rev. Biophys. Biomol. Struct.29:1–26
32. Zamoyska R. 1994. The CD8 coreceptorrevisited: one chain good, two chains bet-ter. Immunity1:243–46
33. Leishman AJ, Naidenko OV, Attinger A,Koning F, Lena CJ, et al. 2001. T cellresponses modulated through interactionbetween CD8 alpha alpha and the nonclas-sical MHC class I molecule, TL.Science294:1936–39
34. Gao GF, Tormo J, Gerth UC, WyerJR, McMichael AJ, et al. 1997. Crystalstructure of the complex between humanCD8aa and HLA-A2.Nature387:630–34
35. Kern PS, Teng MK, Smolyar A, LiuJH, Liu J, et al. 1998. Structural basis
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 679
of CD8 coreceptor function revealed bycrystallographic analysis of a murineCD8 alpha alpha ectodomain fragment incomplex with H-2Kb.Immunity 9:519–30
36. Wang JH, Meijers R, Xiong Y, Liu JH,Sakihama T, et al. 2001. Crystal structureof the human CD4 N-terminal two-domain fragment complexed to a classII MHC molecule.Proc. Natl. Acad. Sci.USA98:10799–804
37. Janeway CA Jr. 1992. The T cell receptoras a multicomponent signalling machine:CD4/CD8 coreceptors and CD45 in T cellactivation.Annu. Rev. Immunol.10:645–74
38. Beyers AD, Spruyt LL, Williams AF.1992. Molecular associations between theT lymphocyte antigen receptor complexand the surface antigens CD2, CD4 orCD8 and CD5.Proc. Natl. Acad. Sci. USA89:2945–49
39. Thome M, Duplay P, Guttinger M, AcutoO. 1995. Syk and ZAP-70 mediate recruit-ment of p56lck/CD4 to the activated T cellreceptor/CD3/zeta complex.J. Exp. Med.181:1997–2006
40. Thome M, Germain V, DiSanto JP, AcutoO. 1996. The p56lck SH2 domain medi-ates recruitment of CD8/p56lck to the ac-tivated T cell receptor/CD3/zeta complex.Eur. J. Immunol.26:2093–100
41. Xu H, Littman DR. 1993. A kinase-independent function of Lck in potentiat-ing antigen-specific T cell activation.Cell74:633–43
42. Irvine DJ, Purbhoo MA, Krogsgaard M,Davis MM. 2002. Direct observation ofsingle ligand recognition by T cells.Na-ture.419:845–49
43. Devine L, Sun J, Barr MR, Kavathas PB.1999. Orientation of the Ig domains ofCD8 alpha beta relative to MHC class I.J. Immunol.162:846–51
44. Classon BJ, Brown MH, Garnett D, So-moza C, Barclay AN, et al. 1992. Thehinge region of the CD8 alpha chain:structure, antigenicity, and utility in ex-
pression of immunoglobulin superfamilydomains.Int. Immunol.4:215–25
45. Boursier JP, Alcover A, Herve F, Lais-ney I, Acuto O. 1993. Evidence for an ex-tended structure of the T-cell co-receptorCD8 alpha as deduced from the hydro-dynamic properties of soluble forms ofthe extracellular region.J. Biol. Chem.268:2013–20
46. Casabo LG, Mamalaki C, Kioussis D,Zamoyska R. 1994. T cell activationresults in physical modification of themouse CD8 beta chain.J. Immunol.152:397–404
47. O’Rourke AM, Rogers J, Mescher MF.1990. Activated CD8 binding to class Iprotein mediated by the T-cell receptor re-sults in signalling.Nature346:187–89
48. Moody AM, Chui D, Reche PA, PriatelJJ, Marth JD, Reinherz EL. 2001. Devel-opmentally regulated glycosylation of theCD8 alpha beta coreceptor stalk modu-lates ligand binding.Cell 107:501–12
49. Daniels MA, Devine L, Miller JD, MoserJM, Lukacher AE, et al. 2001. CD8 bind-ing to MHC class I molecules is influencedby T cell maturation and glycosylation.Immunity15:1051–61
50. Butenhof KJ, Gerken TA. 1993. Struc-ture and dynamics of mucin-like gly-copeptides. Examination of peptide chainexpansion and peptide-carbohydrate in-teractions by stochastic dynamics simu-lations.Biochemistry32:2650–63
51. Rose MC, Voter WA, Sage H, Brown CF,Kaufman B. 1984. Effects of deglycosyla-tion on the architecture of ovine submax-illary mucin glycoprotein.J. Biol. Chem.259:3167–72
52. Shogren RL, Jamieson AM, Blackwell J,Jentoft N. 1986. Conformation of mu-cous glycoproteins in aqueous solvents.Biopolymers25:1505–17
53. Wyer JR, Willcox BE, Gao GF, Gerth UC,Davis SJ, et al. 1999. T cell receptor andco-receptor CD8αα bind peptide-MHCindependently and with distinct kinetics.Immunity10:219–25
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
680 VAN DER MERWE ¥ DAVIS
54. Kern P, Hussey RE, Spoerl R, ReinherzEL, Chang HC. 1999. Expression, purifi-cation, and functional analysis of murineectodomain fragments of CD8 alpha al-pha and CD8 alpha beta dimers.J. Biol.Chem.274:27237–43
55. Gao GF, Willcox BE, Wyer JR, BoulterJM, O’Callaghan CA, et al. 2000. Classi-cal and nonclassical class I major histo-compatibility complex molecules exhibitsubtle conformational differences that af-fect binding to CD8 alpha alpha.J. Biol.Chem.275:15232–38
56. Arcaro A, Gregoire C, Bakker TR, BaldiL, Jordan M, et al. 2001. CD8β en-dows CD8 with efficient coreceptor func-tion by coupling T cell receptor/CD3 toraft-associated CD8/p56lck complexes.J.Exp. Med.194:1485–95
57. Cammarota G, Scheirle A, Takacs B, Do-ran DM, Knorr R, et al. 1992. Identifica-tion of the CD4 binding site on theβ2domain of HLA-DR molecules.Nature356:799–801
58. Weber S, Karjalainen K. 1993. MouseCD4 binds MHC class II with extremelylow affinity. Int. Immunol.5:695–98
59. Xiong Y, Kern P, Chang H, ReinherzE. 2001. T cell receptor binding to apMHCII ligand is kinetically distinct fromand independent of CD4.J. Biol. Chem.276:5659–67
60. Bosselut R, Kubo S, Guinter T, KopaczJL, Altman JD, et al. 2000. Role of CD8beta domains in CD8 coreceptor function:importance for MHC I binding, signaling,and positive selection of CD8+ T cells inthe thymus.Immunity12:409–18
61. Itano A, Cado D, Chan FK, Robey E.1994. A role for the cytoplasmic tail ofthe beta chain of CD8 in thymic selection.Immunity1:287–90
62. Irie HY, Mong MS, Itano A, Crooks ME,Littman DR, et al. 1998. The cytoplasmicdomain of CD8 beta regulates Lck kinaseactivation and CD8 T cell development.J.Immunol.161:183–91
63. Irie HY, Ravichandran KS, Burakoff SJ.
1995. CD8 beta chain influences CD8 al-pha chain-associated Lck kinase activity.J. Exp. Med.181:1267–73
64. van der Merwe PA, Barclay AN. 1994.Transient intercellular adhesion: the im-portance of weak protein-protein interac-tions.Trends Biochem. Sci.19:354–58
65. Davis SJ, Ikemizu S, Wild MK, van derMerwe PA. 1998. CD2 and the natureof protein interactions mediating cell-cellrecognition.Immunol. Rev.163:217–36
66. Mavaddat N, Mason DW, AtkinsonPD, Evans EJ, Gilbert RJ, et al.2000. Signaling lymphocytic activationmolecule (CDw150) is homophilic butself-associates with very low affinity.J.Biol. Chem.275:28100–9
67. Punnonen J, Cocks BG, Carballido JM,Bennett B, Peterson D, et al. 1997. Solubleand membrane-bound forms of signalinglymphocytic activation molecule (SLAM)induce proliferation and Ig synthesis byactivated human B lymphocytes.J. Exp.Med.185:993–1004
68. Dustin ML, Ferguson LM, Chan P,Springer TA, Golan DE. 1996. Visualiza-tion of the CD2 interaction with LFA-3and determination of the two-dimensionaldissociation constant for adhesion re-ceptors in a contact area.J. Cell Biol.132:465–74
69. Dustin ML, Golan DE, Zhu DM, MillerJM, Meier W, et al. 1997. Low affinity in-teraction of human or rat T cell adhesionmolecule CD2 with its ligand aligns ad-hering membranes to achieve high physio-logical affinity.J. Biol. Chem.272:30889–98
70. Doyle C, Strominger JL. 1987. Interactionbetween CD4 and class II MHC moleculesmediates cell adhesion.Nature330:256–59
71. Norment AM, Salter RD, Parham P, En-gelhard VH, Littman DR. 1988. Cell-celladhesion mediated by CD8 and MHCclass I molecules.Nature336:79–81
72. Zhu B, Davies EA, van der Merwe P,Calvert T, Leckband DE. 2002. Direct
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 681
measurements of heterotypic adhesion be-tween the cell surface proteins CD2 andCD48.Biochemistry.41:12163–70
73. Bachmann MF, Barner M, Kopf M. 1999.CD2 sets quantitative thresholds in T cellactivation.J. Exp. Med.190:1383–92
74. Moretta A, Bottino C, Vitale M, PendeD, Cantoni C, et al. 2001. Activating re-ceptors and coreceptors involved in hu-man natural killer cell-mediated cytolysis.Annu. Rev. Immunol.19:197–223
75. Boles KS, Stepp SE, Bennett M, Kumar V,Mathew PA. 2001. 2B4 (CD244) and CS1:novel members of the CD2 subset of theimmunoglobulin superfamily moleculesexpressed on natural killer cells andother leukocytes.Immunol. Rev.181:234–49
76. Davis SJ, van der Merwe PA. 1996. Thestructure and ligand interactions of CD2:implications for T-cell function.Immunol.Today17:177–87
77. Ikemizu S, Sparks LM, van der MerwePA, Harlos K, Stuart DI, et al. 1999. Crys-tal structure of the CD2-binding domainof CD58 (lymphocyte function-associatedantigen 3) at 1.8-A resolution.Proc. Natl.Acad. Sci. USA96:4289–94
78. Sun ZY, Dotsch V, Kim M, Li J, ReinherzEL, Wagner G. 1999. Functional glycan-free adhesion domain of human cell sur-face receptor CD58: design, productionand NMR studies.EMBO J.18:2941–49
79. van der Merwe PA, McNamee PN, DaviesEA, Barclay AN, Davis SJ. 1995. Topol-ogy of the CD2-CD48 cell-adhesionmolecule complex: implications for anti-gen recognition by T cells.Curr. Biol.5:74–84
80. Arulanandam ARN, Kister A, McGregorMJ, Wyss DF, Wagner G, Reinherz EL.1994. Interaction between human CD2and CD58 involves the majorβ sheet sur-face of their respective adhesion domains.J. Exp. Med.180:1861–71
81. Wang JH, Smolyar A, Tan K, Liu JH,Kim M, et al. 1999. Structure of a het-erophilic adhesion complex between the
human CD2 and CD58 (LFA-3) counter-receptors.Cell 97:791–803
82. Garcia KC, Teyton L, Wilson IA. 1999.Structural basis of T cell recognition.Annu. Rev. Immunol.17:369–97
83. Davis SJ, Davies EA, Tucknott MG, JonesEY, van der Merwe PA. 1998. The role ofcharged residues mediating low affinityprotein-protein recognition at the cell sur-face by CD2.Proc. Natl. Acad. Sci. USA95:5490–94
84. Kim M, Sun ZY, Byron O, Campbell G,Wagner G, et al. 2001. Molecular dissec-tion of the CD2-CD58 counter-receptorinterface identifies CD2 Tyr86 and CD58Lys34 residues as the functional “hotspot”.J. Mol. Biol.312:711–20
85. Chothia C, Janin J. 1975. Principlesof protein-protein recognition.Nature256:705–8
86. Lo Conte L, Chothia C, Janin J. 1999. Theatomic structure of protein-protein recog-nition sites.J. Mol. Biol.285:2177–98
87. Schwartz JC, Zhang X, Fedorov AA, Na-thenson SG, Almo SC. 2001. Structuralbasis for co-stimulation by the humanCTLA-4/B7-2 complex.Nature410:604–8
88. Stamper CC, Zhang Y, Tobin JF, ErbeDV, Ikemizu S, et al. 2001. Crystal struc-ture of the B7-1/CTLA-4 complex that in-hibits human immune responses.Nature410:608–11
89. Sharpe AH, Freeman GJ. 2002. The B7-CD28 superfamily.Nat. Rev. Immunol.2:116–26
90. Carreno BM, Collins M. 2002. The B7family of ligands and its receptors: newpathways for costimulation and inhibi-tion of immune responses.Annu. Rev. Im-munol.20:29–53
91. Ostrov DA, Shi W, Schwartz JC, AlmoSC, Nathenson SG. 2000. Structure ofmurine CTLA-4 and its role in modulatingT cell responsiveness.Science290:816–19
92. Schwartz JC, Zhang X, Nathenson SG,Almo SC. 2002. Structural mechanisms
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
682 VAN DER MERWE ¥ DAVIS
of costimulation.Nat. Immunol.3:427–34
93. van der Merwe PA, Davis SJ, Shaw AS,Dustin ML. 2000. Cytoskeletal polariza-tion and redistribution of cell surfacemolecules during T cell antigen recogni-tion. Semin. Immunol.12:5–21
94. Collins AV, Brodie DW, Gilbert RJC,Iaboni A, Manso-Sancho R, et al. 2002.The interaction properties of costim-ulatory molecules revisited.Immunity17:201–10
95. Ikemizu S, Gilbert RJ, Fennelly JA,Collins AV, Harlos K, et al. 2000. Struc-ture and dimerization of a soluble form ofB7-1. Immunity12:51–60
96. Klug A. 1969. Point groups and the de-sign of aggregates. InNobel Syposium 11:Symmetry and function of biological sys-tems at the macromolecular level. ed. AEngstrom, B Strandberg, pp. 425–436.Stockholm: Almqvist & Wiksell
97. Zhang X, Schwartz JC, Almo SC, Na-thenson SG. 2002. Expression, refolding,purification, molecular characterization,crystallization, and preliminary X-rayanalysis of the receptor binding domain ofhuman B7-2.Protein Expr. Purif.25:105–13
98. van Raaij MJ, Chouin E, van der Zandt H,Bergelson JM, Cusack S. 2000. Dimericstructure of the coxsackievirus and aden-ovirus receptor D1 domain at 1.7 A reso-lution. Struct. Fold. Des.8:1147–55
99. Metzler WJ, Bajorath J, Fenderson W,Shaw SY, Constantine KL, et al. 1997.Solution structure of human CTLA-4 anddelineation of a CD80/CD86 binding siteconserved in CD28.Nat. Struct. Biol.4:527–31
100. van der Merwe PA, Bodian DL, DaenkeS, Linsley P, Davis SJ. 1997. CD80 (B7-1) binds both CD28 and CTLA-4 with alow affinity and very fast kinetics.J. Exp.Med.185:393–403
101. Linsley PS, Greene JL, Brady W, Bajo-rath J, Ledbetter JA, Peach R. 1994. Hu-man B7-1 (CD80) and B7-2 (CD86) bind
with similar avidities but distinct kineticsto CD28 and CTLA-4 receptors.Immunity1:793–801
102. Greene JL, Leytze GM, Emswiler J, PeachR, Bajorath J, et al. 1996. Covalent dimer-ization of CD28/CTLA-4 and oligomer-ization of CD80/CD86 regulate T cellcostimulatory interactions.J. Biol. Chem.271:26762–71
103. Lenschow DJ, Ho SC, Sattar H, Rhee L,Gray G, et al. 1995. Differential effects ofanti-B7-1 and anti-B7-2 monoclonal an-tibody treatment on the development ofdiabetes in the nonobese diabetic mouse.J. Exp. Med.181:1145–55
104. Kearney ER, Walunas TL, Karr RW, Mor-ton PA, Loh DY, et al. 1995. Antigen-dependent clonal expansion of a tracepopulation of antigen-specific CD4+ Tcells in vivo is dependent on CD28 cos-timulation and inhibited by CTLA-4.J.Immunol.155:1032–36
105. Liang B, Gee RJ, Kashgarian MJ, SharpeAH, Mamula MJ. 1999. B7 costimula-tion in the development of lupus: au-toimmunity arises either in the absenceof B7.1/B7.2 or in the presence of anti-b7.1/B7.2 blocking antibodies.J. Im-munol.163:2322–29
106. Judge TA, Wu Z, Zheng XG, Sharpe AH,Sayegh MH, Turka LA. 1999. The role ofCD80, CD86, and CTLA4 in alloimmuneresponses and the induction of long-termallograft survival.J. Immunol.162:1947–51
107. Kuchroo VK, Das MP, Brown JA, RangerAM, Zamvil SS, et al. 1995. B7-1 and B7-2 costimulatory molecules activate differ-entially the Th1/Th2 developmental path-ways: application to autoimmune diseasetherapy.Cell 80:707–18
108. Brodie D, Collins AV, Iaboni A, FennellyJA, Sparks LM, et al. 2000. LICOS, a pri-mordial costimulatory ligand?Curr. Biol.10:333–36
109. Grakoui A, Bromley SK, Sumen C, DavisMM, Shaw AS, et al. 1999. The immuno-logical synapse: a molecular machine
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
T CELL ANTIGEN RECOGNITION 683
controlling T cell activation.Science285:221–27
110. Bromley SK, Iaboni A, Davis SJ, WhittyA, Green JM, et al. 2001. The immuno-logical synapse and CD28-CD80 interac-tions.Nat. Immunol.2:1159–66
111. Davis SJ, van der Merwe PA. 2001. Theimmunological synapse: required for Tcell receptor signaling or directing T celleffector function?Curr. Biol.11:R289–90
112. van der Merwe PA, Davis SJ. 2002. Im-munology. The immunological synapse—a multitasking system.Science295:1479–80
113. Erbe DV, Wang S, Xing Y, Tobin JF. 2002.Small molecule ligands define a bind-ing site on the immune regulatory proteinB7.1.J. Biol. Chem.277:7363–68
114. Salomon B, Lenschow DJ, Rhee L,Ashourian N, Singh B, et al. 2000.B7/CD28 costimulation is essential forthe homeostasis of the CD4+CD25+ im-munoregulatory T cells that control au-toimmune diabetes.Immunity12:431–40
115. Corr M, Slanetz AE, Boyd LF, JelonekMT, Khilko S, et al. 1994. T cellreceptor-MHC class I peptide interac-tions: affinity, kinetics, and specificity.Science265:946–49
116. Sykulev Y, Brunmark A, Jackson M, Co-hen RJ, Peterson PA, Eisen HN. 1994.Kinetics and affinity of reactions be-tween an antigen-specific T cell receptorand peptide-MHC complexes.Immunity1:15–22
117. Garcia KC, Tallquist MD, Pease LR,Brunmark A, Scott CA, et al. 1997. Al-pha beta T cell receptor interactions withsyngeneic and allogeneic ligands: affinitymeasurements and crystallization.Proc.Natl. Acad. Sci. USA94:13838–43
118. Alam SM, Travers PJ, Wung JL, NasholdsW, Redpath S, et al. 1996. T-cell-receptoraffinity and thymocyte positive selection.Nature381:616–20
119. Weber S, Traunecker A, Oliveri F, Ger-had W, Karjalainen K. 1992. Specific low-affinity recognition of major histocompat-
ibiliy complex plus peptide by soluble T-cell receptor.Nature356:793–96
120. Kersh GJ, Kersh EN, Fremont DH, AllenPM. 1998. High- and low-potency ligandswith similar affinities for the TCR: the im-portance of kinetics in TCR signaling.Im-munity9:817–26
121. van der Merwe PA, Barclay AN, MasonDW, Davies EA, Morgan BP, et al. 1994.The human cell-adhesion molecule CD2binds CD58 with a very low affinity and anextremely fast dissociation rate but doesnot bind CD48 or CD59.Biochemistry33:10149–60
122. van der Merwe PA, Brown MH, Davis SJ,Barclay AN. 1993. Affinity and kineticanalysis of the interaction of the cell-adhesion molecules rat CD2 and CD48.EMBO J.12:4945–54
123. Brown MH, Boles K, van der MerwePA, Kumar V, Mathew PA, Barclay AN.1998. 2B4, the natural killer and T cellimmunoglobulin superfamily surface pro-tein, is a ligand for CD48.J. Exp. Med.188:2083–90
124. Vales-Gomez M, Reyburn HT, Mandel-boim M, Strominger JL. 1998. Kinetics ofinteraction of HLA-C ligands with naturalkiller cell inhibitory receptors.Immunity9:892
125. Maenaka K, Juji T, Nakayama T, Wyer JR,Gao GF, et al. 1999. Killer cell immunog-lobulin receptors and T cell receptors bindpeptide- major histocompatibility com-plex class I with distinct thermodynamicand kinetic properties.J. Biol. Chem.274:28329–34
126. Wright GJ, Puklavec MJ, Willis AC, HoekRM, Sedgwick JD, et al. 2000. Lym-phoid/neuronal cell surface OX2 glyco-protein recognizes a novel receptor onmacrophages implicated in the control oftheir function.Immunity13:233–42
127. Wild MK, Huang MC, Schulze-Horsel U,van der Merwe PA, Vestweber D. 2001.Affinity, kinetics, and thermodynamics ofE-selectin binding to E-selectin ligand-1.J. Biol. Chem.276:31602–12
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

6 Feb 2003 11:33 AR AR180-IY21-20.tex AR180-IY21-20.sgm LaTeX2e(2002/01/18)P1: GCE
684 VAN DER MERWE ¥ DAVIS
128. Labadia ME, Jeanfavre DD, Caviness GO,Morelock MM. 1998. Molecular regula-tion of the interaction between leukocytefunction-associated antigen-1 and solubleICAM-1 by divalent metal cations.J. Im-munol.161:836–42
129. Garboczi DN, Ghosh P, Utz U, Fan QR,Biddison WE, Wiley DC. 1996. Structureof the complex between human T-cell re-ceptor, viral peptide and HLA-A2.Nature384:134–41
130. Gao GF, Rao Z, Bell JI. 2002. Molecu-lar coordination of alpha beta T-cell re-ceptors and coreceptors CD8 and CD4 intheir recognition of peptide-MHC ligands.Trends Immunol.23:408–13
131. Esnouf RM. 1997. An extensively mod-ified version of MolScript that includesgreatly enhanced coloring capabilities.J.Mol. Graph. Model.15:112–13, 132–34
132. Merritt EA, Murphy MEP. 1994.Raster3D version-2.0: a program forphotorealistic molecular graphics.ActaCrystallogr. D50:869–73
133. Nicholls A, Sharp KA, Honig B. 1991.Protein folding and association: in-sights from the interfacial and thermody-namic properties of hydrocarbons.Pro-teins11:281–96
134. Lawrence MC, Colman PM. 1993. Shapecomplementarity at protein/protein inter-faces.J. Mol. Biol.234:946–50
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

19 Feb 2003 21:7 AR AR180-20-COLOR.tex AR180-20-COLOR.SGM LaTeX2e(2002/01/18)P1: GDL
Figure 1 T cell antigen receptor (TCR) binding to peptide-MHC. (a) A view fromthe T cell of a peptide (red) presented on an MHC class I molecule (HLA-A2) with theapproximate position shown of the TCR “footprint” for the B7 TCR binding to the Tax/HLA-A2 complex (129). The approximate positions within the footprint of the TCRα
andβ chain complementarity-determining region loops are shown on the right. Alsoshown are the range of binding orientations observed for TCR binding to peptide-MHCclass I and class II in the structures solved to date [reviewed in (2)]. (b) A side viewof peptide/MHC class I and class II complexes to illustrate the positions of the bumpsproduced by kinks (black arrows) in theα-helices. The position of these kinks is alsodepicted ina. The complexes shown are HLA-A2 with an HIV peptide (PDB accessioncode 1HHG) and HLA-DR1 with an influenza virus peptide (1DLH). These figureswere generated using WebLab ViewerLite.
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

19 Feb 2003 21:7 AR AR180-20-COLOR.tex AR180-20-COLOR.SGM LaTeX2e(2002/01/18)P1: GDL
Fig
ure
2C
orec
epto
rbin
ding
toth
eT
cell
antig
enre
cept
or(T
CR
)/pe
ptid
e-M
HC
com
plex
.Mod
els
ofth
ehu
man
CD
8/T
CR
:pep
tide-
MH
Ccl
ass
Iand
CD
4/T
CR
:pep
tide-
MH
Ccl
ass
IIco
mpl
exes
wer
ecr
eate
das
desc
ribed
inR
efer
ence
130.
Brie
fly,t
heC
D8
com
plex
was
prod
uced
bysu
perim
posi
tion
ofH
LA-A
2fr
omth
eT
CR
:HLA
-A2
(acc
essi
onco
de1B
D2)
and
CD
8αα
/HLA
-A2
(1A
KJ)
stru
ctur
es,w
ithon
lyth
ela
tter
HLA
-A2
show
n.T
heC
D4
com
plex
was
prod
uced
bysu
perp
ositi
onof
both
the
TC
R:H
LA-D
R1
com
plex
(1F
YT
)an
dfu
ll-le
ngth
hum
anC
D4
(1W
IO)o
nth
eC
D4/
I-Ak
stru
ctur
e(1
JL4)
,with
the
latte
rstr
uctu
reno
tsho
wn.
The
figur
ew
asge
nera
ted
usin
gB
OB
SC
RIP
T(1
31)a
ndR
aste
r3D
(132
).
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.
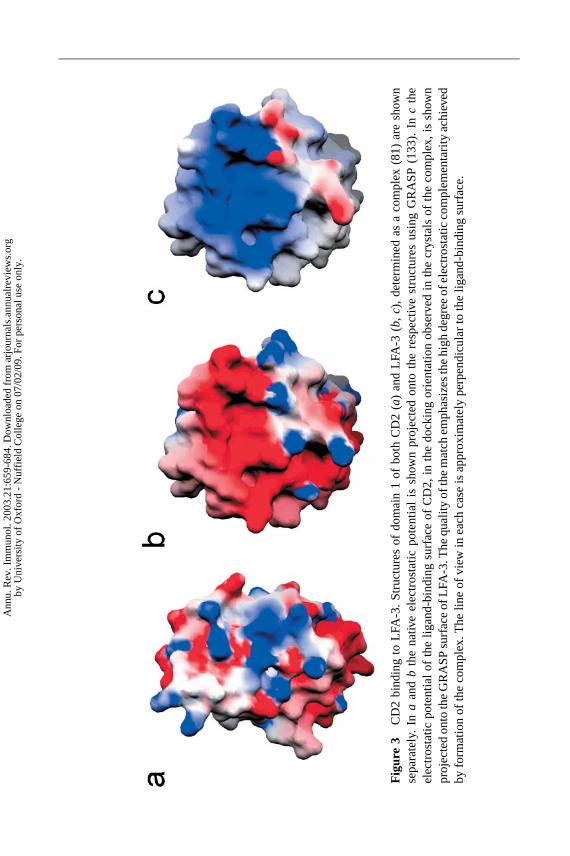
19 Feb 2003 21:7 AR AR180-20-COLOR.tex AR180-20-COLOR.SGM LaTeX2e(2002/01/18)P1: GDL
Fig
ure
3C
D2
bind
ing
toLF
A-3
.S
truc
ture
sof
dom
ain
1of
both
CD
2(
a)an
dLF
A-3
(b,
c),
dete
rmin
edas
aco
mpl
ex(8
1)ar
esh
own
sepa
rate
ly.
Ina
and
bth
ena
tive
elec
tros
tatic
pote
ntia
lis
show
npr
ojec
ted
onto
the
resp
ectiv
est
ruct
ures
usin
gG
RA
SP
(133
).In
cth
eel
ectr
osta
ticpo
tent
ialo
fth
elig
and-
bind
ing
surf
ace
ofC
D2,
inth
edo
ckin
gor
ient
atio
nob
serv
edin
the
crys
tals
ofth
eco
mpl
ex,
issh
own
proj
ecte
don
toth
eG
RA
SP
surf
ace
ofLF
A-3
.The
qual
ityof
the
mat
chem
phas
izes
the
high
degr
eeof
elec
tros
tatic
com
plem
enta
rity
achi
eved
byfo
rmat
ion
ofth
eco
mpl
ex.T
helin
eof
view
inea
chca
seis
appr
oxim
atel
ype
rpen
dicu
lar
toth
elig
and-
bind
ing
surf
ace.
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

19 Feb 2003 21:7 AR AR180-20-COLOR.tex AR180-20-COLOR.SGM LaTeX2e(2002/01/18)P1: GDL
Fig
ure
4S
urfa
ceco
mpl
emen
tarit
yof
CD
2:LF
A-3
and
CT
LA-4
:B7-
1co
mpl
exin
terf
aces
.(a)
Lack
ofsh
ape
com
plem
enta
rity
seen
atth
ein
terf
ace
ofth
eco
mpl
exof
CD
2(b
lue)
and
LFA
-3(r
ed),
(see
Ref
eren
ce81
).T
heso
lven
t-ac
cess
ible
surf
aces
ofce
ntra
lse
ctio
nsof
the
inte
ract
ingβ
-she
ets
are
show
nse
mitr
ansp
aren
tly.
(b)
The
very
good
fitbe
twee
nth
eM
YP
PP
Ym
otif
(dra
wn
inba
llan
dst
ick
form
)of
CT
LA-4
(cya
n),a
ndth
elig
and-
bind
ing
surf
ace
ofB
7-1
(mag
enta
;sho
wn
sem
itran
spar
ently
),re
veal
edby
the
stru
ctur
eof
the
B7-
1:C
TLA
-4co
mpl
ex(8
8).
The
algo
rithm
ofR
efer
ence
134,
whi
chm
easu
res
the
geom
etric
fitof
two
prot
ein
surf
aces
,w
ould
give
asc
ore
of1.
0to
ahy
poth
etic
alpe
rfec
tmat
ch.T
heC
TLA
-4:B
7-1
com
plex
scor
es0.
74–0
.77,
whi
chis
high
erth
anth
atfo
ran
tibod
y-pr
otei
nan
tigen
inte
rfac
es(0
.64–
0.68
),an
dm
uch
high
erth
anfo
rth
eC
D2:
LFA
-3in
tera
ctio
n(0
.58)
and
TC
R:M
HC
-pep
tide
com
plex
es(0
.45–
0.47
).T
heim
ages
wer
epr
epar
edus
ing
BO
BS
CR
IPT
(131
).
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

P1: FRK
January 24, 2003 20:2 Annual Reviews AR180-FM
Annual Review of ImmunologyVolume 21, 2003
CONTENTS
FRONTISPIECE, Thomas A. Waldmann x
THE MEANDERING 45-YEAR ODYSSEY OF A CLINICAL IMMUNOLOGIST,Thomas A. Waldmann 1
CD8 T CELL RESPONSES TO INFECTIOUS PATHOGENS, Phillip Wongand Eric G. Pamer 29
CONTROL OF APOPTOSIS IN THE IMMUNE SYSTEM: BCL-2,BH3-ONLY PROTEINS AND MORE, Vanessa S. Marsdenand Andreas Strasser 71
CD45: A CRITICAL REGULATOR OF SIGNALING THRESHOLDS INIMMUNE CELLS, Michelle L. Hermiston, Zheng Xu, and Arthur Weiss 107
POSITIVE AND NEGATIVE SELECTION OF T CELLS, Timothy K. Starr,Stephen C. Jameson, and Kristin A. Hogquist 139
IGA FC RECEPTORS, Renato C. Monteiro and Jan G.J. van de Winkel 177
REGULATORY MECHANISMS THAT DETERMINE THE DEVELOPMENTAND FUNCTION OF PLASMA CELLS, Kathryn L. Calame, Kuo-I Lin,and Chainarong Tunyaplin 205
BAFF AND APRIL: A TUTORIAL ON B CELL SURVIVAL,Fabienne Mackay, Pascal Schneider, Paul Rennert, and Jeffrey Browning 231
T CELL DYNAMICS IN HIV-1 INFECTION, Daniel C. Douek,Louis J. Picker, and Richard A. Koup 265
T CELL ANERGY, Ronald H. Schwartz 305
TOLL-LIKE RECEPTORS, Kiyoshi Takeda, Tsuneyasu Kaisho,and Shizuo Akira 335
POXVIRUSES AND IMMUNE EVASION, Bruce T. Seet, J.B. Johnston,Craig R. Brunetti, John W. Barrett, Helen Everett, Cheryl Cameron,Joanna Sypula, Steven H. Nazarian, Alexandra Lucas,and Grant McFadden 377
IL-13 EFFECTOR FUNCTIONS, Thomas A. Wynn 425
LOCATION IS EVERYTHING: LIPID RAFTS AND IMMUNE CELLSIGNALING, Michelle Dykstra, Anu Cherukuri, Hae Won Sohn,Shiang-Jong Tzeng, and Susan K. Pierce 457
v
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.

P1: FRK
January 24, 2003 20:2 Annual Reviews AR180-FM
vi CONTENTS
THE REGULATORY ROLE OF Vα14 NKT CELLS IN INNATE ANDACQUIRED IMMUNE RESPONSE, Masaru Taniguchi, Michishige Harada,Satoshi Kojo, Toshinori Nakayama, and Hiroshi Wakao 483
ON NATURAL AND ARTIFICIAL VACCINATIONS, Rolf M. Zinkernagel 515
COLLECTINS AND FICOLINS: HUMORAL LECTINS OF THE INNATEIMMUNE DEFENSE, Uffe Holmskov, Steffen Thiel, and Jens C. Jensenius 547
THE BIOLOGY OF IGE AND THE BASIS OF ALLERGIC DISEASE,Hannah J. Gould, Brian J. Sutton, Andrew J. Beavil, Rebecca L. Beavil,Natalie McCloskey, Heather A. Coker, David Fear, and Lyn Smurthwaite 579
GENOMIC ORGANIZATION OF THE MAMMALIAN MHC,Attila Kumanovics, Toyoyuki Takada, and Kirsten Fischer Lindahl 629
MOLECULAR INTERACTIONS MEDIATING T CELL ANTIGENRECOGNITION, P. Anton van der Merwe and Simon J. Davis 659
TOLEROGENIC DENDRITIC CELLS, Ralph M. Steinman, Daniel Hawiger,and Michel C. Nussenzweig 685
MOLECULAR MECHANISMS REGULATING TH1 IMMUNE RESPONSES,Susanne J. Szabo, Brandon M. Sullivan, Stanford L. Peng, andLaurie H. Glimcher 713
BIOLOGY OF HEMATOPOIETIC STEM CELLS AND PROGENITORS:IMPLICATIONS FOR CLINICAL APPLICATION, Motonari Kondo,Amy J. Wagers, Markus G. Manz, Susan S. Prohaska, David C. Scherer,Georg F. Beilhack, Judith A. Shizuru, and Irving L. Weissman 759
DOES THE IMMUNE SYSTEM SEE TUMORS AS FOREIGN OR SELF?Drew Pardoll 807
B CELL CHRONIC LYMPHOCYTIC LEUKEMIA: LESSONS LEARNEDFROM STUDIES OF THE B CELL ANTIGEN RECEPTOR,Nicholas Chiorazzi and Manlio Ferrarini 841
INDEXESSubject Index 895Cumulative Index of Contributing Authors, Volumes 11–21 925Cumulative Index of Chapter Titles, Volumes 11–21 932
ERRATAAn online log of corrections to Annual Review of Immunology chaptersmay be found at http://immunol.annualreviews.org/errata.shtml
Ann
u. R
ev. I
mm
unol
. 200
3.21
:659
-684
. Dow
nloa
ded
from
arj
ourn
als.
annu
alre
view
s.or
gby
Uni
vers
ity o
f O
xfor
d -
Nuf
fiel
d C
olle
ge o
n 07
/02/
09. F
or p
erso
nal u
se o
nly.




















