
Molecular Recognition of Human Eosinophil-derived Neurotoxin (RNase 2) by Placental Ribonuclease...
19
Molecular Recognition of Human Eosinophil-derived Neurotoxin (RNase 2) by Placental Ribonuclease Inhibitor Shalini Iyer 1 , Daniel E. Holloway 1 , Kapil Kumar 2 , Robert Shapiro 2 and K. Ravi Acharya 1 * 1 Department of Biology and Biochemistry, 4 South University of Bath, Claverton Down, Bath BA2 7AY, UK 2 Center for Biochemical and Biophysical Sciences and Medicine, Department of Pathology, Harvard Medical School, Boston, MA 02115 USA Placental ribonuclease inhibitor (RI) binds diverse mammalian RNases with dissociation constants that are in the femtomolar range. Previous studies on the complexes of RI with RNase A and angiogenin revealed that RI utilises largely distinctive interactions to achieve high affinity for these two ligands. Here we report a 2.0 A ˚ resolution crystal structure of RI in complex with a third ligand, eosinophil-derived neurotoxin (EDN), and a mutational analysis based on this structure. The RI–EDN interface is more extensive than those of the other two complexes and contains a considerably larger set of interactions. Few of the contacts present in the RI–angiogenin complex are replicated; the correspondence to the RI–RNase A complex is somewhat greater, but still modest. The energetic contributions of various interface regions differ strikingly from those in the earlier complexes. These findings provide insight into the structural basis for the unusual combination of high avidity and relaxed stringency that RI displays. q 2005 Elsevier Ltd. All rights reserved. Keywords: ribonuclease inhibitor; eosinophil-derived neurotoxin; leucine- rich repeats; X-ray crystallography; molecular recognition *Corresponding author Introduction Accurate and highly selective recognition of proteins by other proteins drives most biological processes. However, there are numerous cases where an individual protein displays promiscuity in its ability to recognise multiple structurally related partners. Placental ribonuclease inhibitor (RI) is a remarkable example of such a promiscuous protein: it binds all of the different w14 kDa members of the mammalian pancreatic RNase superfamily with extraordinarily high avidity. 1–3 The affinity of RI for its strongest-binding ligand, angiogenin (Ang; K i Z0.7 fM), 4 is among the tightest on record for any protein-protein interaction; K i values for other ligands also lie in the femtomolar range. These affinities are comparable to, or higher than, those measured for the complexes of TIMP with matrix metalloproteinases, 5,6 barnase with barstar, 7 serpins with serine proteases, 8 and colicins with their immunity proteins. 9 Previous structural studies have shown that RI adopts a non-globular horseshoe shape 10 and that the ligands RNase A and Ang occupy the central cavity of the horseshoe and contact one face of the inhibitor in the C-terminal region. 11,12 In both cases, the interface is large, encompassing 26–28 residues on the inhibitor and 24 on the ligand. Nonetheless, mutational analyses 13–15 have revealed that in both complexes a single small region, containing the active site of the ligand and the 433–440 loop and C-terminal residue Ser460 of RI, contributes a substantial portion of the binding energy. In both cases, Asp435 of RI (human RI numbering) forms energetically important hydrogen bonds with the catalytic lysine of the ligand (Lys40 of Ang, Lys41 of RNase A). However, most of the other contacts in the two energetic “hot spots’ 16 do not correspond. Moreover, interactions within the hot spots exhibit opposite types of cooperativity in their functioning; 0022-2836/$ - see front matter q 2005 Elsevier Ltd. All rights reserved. Present address: K. Kumar, Biohelix Corporation, Beverly, MA 01915, USA. Abbreviations used: hRI, human ribonuclease inhibitor; pRI, porcine ribonuclease inhibitor; EDN, eosinophil- derived neurotoxin; RNase A, ribonuclease A; Ang, angiogenin; LRR, leucine rich repeat; r.m.s, root-mean- square. E-mail address of the corresponding author: [email protected] doi:10.1016/j.jmb.2005.01.035 J. Mol. Biol. (2005) 347, 637–655
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of Molecular Recognition of Human Eosinophil-derived Neurotoxin (RNase 2) by Placental Ribonuclease...

doi:10.1016/j.jmb.2005.01.035 J. Mol. Biol. (2005) 347, 637–655
Molecular Recognition of Human Eosinophil-derivedNeurotoxin (RNase 2) by Placental RibonucleaseInhibitor
Shalini Iyer1, Daniel E. Holloway1, Kapil Kumar2, Robert Shapiro2 andK. Ravi Acharya1*
1Department of Biology andBiochemistry, 4 SouthUniversity of Bath, ClavertonDown, Bath BA2 7AY, UK
2Center for Biochemical andBiophysical Sciences andMedicine, Department ofPathology, Harvard MedicalSchool, Boston, MA 02115USA
0022-2836/$ - see front matter q 2005 E
Present address: K. Kumar, BioheBeverly, MA 01915, USA.Abbreviations used: hRI, human r
pRI, porcine ribonuclease inhibitor;derived neurotoxin; RNase A, ribonangiogenin; LRR, leucine rich repeasquare.E-mail address of the correspond
Placental ribonuclease inhibitor (RI) binds diverse mammalian RNaseswith dissociation constants that are in the femtomolar range. Previousstudies on the complexes of RI with RNase A and angiogenin revealed thatRI utilises largely distinctive interactions to achieve high affinity for thesetwo ligands. Here we report a 2.0 A resolution crystal structure of RI incomplex with a third ligand, eosinophil-derived neurotoxin (EDN), and amutational analysis based on this structure. The RI–EDN interface is moreextensive than those of the other two complexes and contains aconsiderably larger set of interactions. Few of the contacts present in theRI–angiogenin complex are replicated; the correspondence to the RI–RNaseA complex is somewhat greater, but still modest. The energeticcontributions of various interface regions differ strikingly from those inthe earlier complexes. These findings provide insight into the structuralbasis for the unusual combination of high avidity and relaxed stringencythat RI displays.
q 2005 Elsevier Ltd. All rights reserved.
Keywords: ribonuclease inhibitor; eosinophil-derived neurotoxin; leucine-rich repeats; X-ray crystallography; molecular recognition
*Corresponding authorIntroduction
Accurate and highly selective recognition ofproteins by other proteins drives most biologicalprocesses. However, there are numerous caseswhere an individual protein displays promiscuityin its ability to recognise multiple structurallyrelated partners. Placental ribonuclease inhibitor(RI) is a remarkable example of such a promiscuousprotein: it binds all of the different w14 kDamembers of the mammalian pancreatic RNasesuperfamily with extraordinarily high avidity.1–3
The affinity of RI for its strongest-binding ligand,angiogenin (Ang; KiZ0.7 fM),4 is among the tighteston record for any protein-protein interaction; Ki
lsevier Ltd. All rights reserve
lix Corporation,
ibonuclease inhibitor;EDN, eosinophil-uclease A; Ang,t; r.m.s, root-mean-
ing author:
values for other ligands also lie in the femtomolarrange. These affinities are comparable to, or higherthan, those measured for the complexes of TIMPwith matrix metalloproteinases,5,6 barnase withbarstar,7 serpins with serine proteases,8 and colicinswith their immunity proteins.9
Previous structural studies have shown that RIadopts a non-globular horseshoe shape10 and thatthe ligands RNase A and Ang occupy the centralcavity of the horseshoe and contact one face of theinhibitor in the C-terminal region.11,12 In both cases,the interface is large, encompassing 26–28 residueson the inhibitor and 24 on the ligand. Nonetheless,mutational analyses13–15 have revealed that in bothcomplexes a single small region, containing theactive site of the ligand and the 433–440 loop andC-terminal residue Ser460 of RI, contributes asubstantial portion of the binding energy. In bothcases, Asp435 of RI (human RI numbering) formsenergetically important hydrogen bonds with thecatalytic lysine of the ligand (Lys40 of Ang, Lys41 ofRNase A). However, most of the other contacts inthe two energetic “hot spots’16 do not correspond.Moreover, interactions within the hot spots exhibitopposite types of cooperativity in their functioning;
d.

Figure 1 (legend next page)
638 EDN in Complex with Placental Ribonuclease Inhibitor
thus, effects of multiple mutations in the Angcomplex are largely superadditive, whereas in theRNase A complex they are subadditive.14,15
A recent mutational study of the complex ofhuman RI (hRI) with human eosinophil-derivedneurotoxin (EDN; also known as RNase 2), suggeststhat this complex does not contain an analogous hotspot.17 Single-residue mutations of residues in thehRI C-terminal segment that had been shown to befunctionally important in the Ang and RNase A
complexes had much smaller effects, if any, onaffinity for EDN. Individual replacements of RIresidues in other regions, selected by examining acomputational model of the hRI–EDN complex, didnot implicate any other residues as playing keyroles. Although multi-site mutagenesis revealedmore sizable combined contributions by theC-terminal segment and a Trp-rich region of RI,most of the binding energy still could not beaccounted for.

Figure 1. (a) Ribbon representation of the hRI–EDN complex, drawn with the program MOLSCRIPT.58 The hRImolecule has been coloured purple and the EDNmolecule is shown in olive green. (b) Stereo view of the superposed Ca
traces of the hRI–EDN complex (purple), the pRI–RNase A complex18 (olive green) and the hRI–Ang complex12 (cyan).Superpositioning was performed by aligning the RI components of the respective complexes. (c) Stereo view of thesuperposition of Ca traces of the two hRI molecules in the asymmetric unit. hRI from Complex 1 is coloured purple andthe hRI from Complex 2 is shown in blue. (d) Molecular surfaces of the inhibitor (left panel) and the enzymes (rightpanel) from the different RI complexes. The horseshoe was rotated byw208 from the standard orientation (b) in order tobetter show the interface. The enzymes were rotated by about 1808, along the x-axis, from the standard orientation (b) toshow their binding interface. The surfaces have been colour coded according to the type of contact atoms at the interface.Non-polar atoms are coloured green, uncharged polar atoms are shown in blue and charged atoms in red. The programGRASP was used to draw the molecular surfaces.59
EDN in Complex with Placental Ribonuclease Inhibitor 639
The three-dimensional structure of the hRI–EDNcomplex reported here provides a platform for amore complete understanding of the physical basisfor high-affinity binding of EDN by RI. Thestructure reveals that EDN interacts with RI over amore extensive area than do RNase A and Ang andconsequently forms a significantly larger number ofintermolecular contacts. Mutational analysis basedon this structure has now identified interactionsunique to the hRI–EDN complex that makeimportant contributions to the interfaceenergetics. Our findings help explain how RI can
bind such a diverse range of ligands with similarlyhigh affinity.
Results
Overall structure of the hRI–EDN complex
The hRI–EDN complex (Figure 1(a)) crystallisesin the trigonal space group, P3121. The structurewas determined at 2.0 A resolution with twocomplexes (Complex 1 and Complex 2) in the

Table 1. Crystallographic, data processing and refinement statistics
Space group Trigonal, P3121
Unit cell dimensions (A) aZbZ91.98, cZ257.65Resolution range (A) 40–2.0Total reflections measured 2,473,140Unique reflections measured 89,545Rsym (%)a 7.1I/s(I) (outermost shell)b 13.1 (3.6)Completeness (outermost shell) (%) 99.4 (99.7)Rcryst(%)c 20.6Rfree (%)d 24.3Contents of the asymmetric unit
Protein atoms 8882
Solvent molecules 616
Ligands 2 (one malonate ion and one glycerol molecule)
r.m.s. deviation from ideality
Bond lengths (A) 0.005
Bond angles (deg.) 1.2
Average B-factor (A2)
Complex 1 24.3
Complex 2 27.1
hRI (all atoms: chains A and B) 21.2 (A); 22.5 (B)
Main-chain atoms 20.3 (A); 21.6 (B)
Side-chain atoms 22.3 (A); 23.5 (B)
EDN (all atoms: chains C and D) 27.5 (C); 31.8 (D)
Main-chain atoms 27.2 (C); 31.8 (D)
Side-chain atoms 27.8 (C); 31.9 (D)
Solvent molecules 29.9Ligands 31.4 (malonate); 20.6 (glycerol)
Overall B-factor (A2/Da) from Wilson plot) 21.7
a RsymZShklSijIiðhklK hIðhklÞij=ShklSiIiðhklÞ, where hIi is the averaged intensity of the i observations of reflection hkl.b Outermost shell: the resolution range of the outermost shell is 2.06–1.99 A.c RcrystZSjjFojK jFcjj=SjFoj, where Fo and Fc are observed and calculated structure factors, respectively.d Rfree is equal to Rcryst for a random subset of reflections (3.7%) not used in refinement.54
640 EDN in Complex with Placental Ribonuclease Inhibitor
asymmetric unit (see Table 1 for crystallographicstatistics). The overall docking of EDN to hRI issimilar to that of RNase A18 and Ang12 in theirrespective complexes with porcine RI (pRI) and hRI(Figure 1(b)). The characteristic modular architec-ture of hRI comprises 16 tandem alternating 28- and29-residue leucine-rich repeat (LRR) elementsarranged symmetrically in the shape of a horseshoe.Each LRR element consists of a short b-strand, a b–aloop, an a-helix and an a–b loop. The b-strandsand the a-helices form the inner and outercircumferences of the horseshoe, respectively. Onelobe of EDN fills the central cavity of the horseshoeand the other sits on top of one face of hRI in theC-terminal region.
The dimer in the asymmetric unit is stabilised byseven hydrogen bonds (three between the two hRImolecules and four between hRI of one complexand EDN of the other) and 33 van der Waalsinteractions. A total of 46 water molecules lie at theinterface between the two complexes and four ofthese mediate hydrogen bonds. Complex 1 andComplex 2 superpose onto each other with an r.m.s.deviation of 1.25 A (Ca atoms). A closer look at thealignment of the hRIs from the two complexes(r.m.s. deviation of 1.54 A for 456 Ca atoms) showsthat the greatest differences lie in the N-terminal
segment of w150 residues (Figure 1(c)), where amaximum Ca deviation of about 5.2 A occurs at thebeginning of the polypeptide chain, and thedeviation gradually decreases to 0.53 A by residue150. Superposition of residues 150–460 from the twohRIs gives an r.m.s. deviation of only 0.53 A andresidues 5–149 can be overlaid with an r.m.s.deviation of 0.44 A. The relatively large deviationbetween the N-terminal segments in the twocomplexes may reflect contacts that stabilise thenon-crystallographic dimer: Ala4, Ser6, Glu21, andGln30 of hRI in Complex 2 form hydrogen bondswith both hRI and EDN in Complex 1, whereas theN-terminal segment of hRI in Complex 1 makes nocontacts with the other complex. Therefore, it seemslikely that the conformation in Complex 1 moreaccurately reflects the structure in solution, and alldescriptions below refer to Complex 1 unlessindicated otherwise. Least-squares superpositionof hRI from Complex 1 with hRI from the hRI–Angcomplex (molecule 1) yields an r.m.s. deviationof 0.69 A for Ca atoms. Similar comparison withthe RNase A-bound and free forms of pRI resultsin r.m.s. deviations of 1.83 A and 1.50 A,respectively. These alignments indicate that hRI inthe EDN complex is structurally more similar to thefree form of pRI than to the RNase A-bound pRI, as

Table 2. van der Waals contacts in the hRI–EDN complex structure
hRI residue EDN residue(s) No. of contacts
Gln10 Asn32(3), Ile93 4Cys11 Gln28, Val29, Asn32 3Arg33 Ser94 2Asp35 Gln28 7Asp36 Val29 1Arg63 Gln28, Ser94(2) 3Asp121 Asn25 1Tyr150 Gln22 7Trp261 Thr86(3), Thr87(2), Pro88 6Trp263 Thr86(8), Ala99(2) 10Trp318 Leu85, Thr86, Pro88(8) 10Lys320 Asn39(4), Asn84(2), Leu85(2) 8Gln346 Asn39 1Asn349 Gln40 1Trp375 Arg36(10), Leu85(2) 12Ala377 Asn39 1Glu401 Arg36 3Asp403 Arg36 3Asn406 Gln40 4Cys408 Asn65 1Gln430 Arg36 1Val432 Arg36 4Tyr434 Arg36(2), Lys38(11) 13Asp435 Lys38(4), His129(2) 6Ile436 His129 4Tyr437 Arg68(11), Asn70(2), Val128, His129(4)
Asp112(4)22
Trp438 Trp7 10Glu443 Trp7 7Leu446 Met0 4Ala447 Met0 2Val458 Met0 5Ile459 Gln34 6Ser460 Met0(7), Lys1(2), Gln34(4) 13
The maximum allowed values of contact distances are C–C, 4.1 A; C–N, 3.8 A; C–O, 3.7 A; O–O, 3.3 A; O–N, 3.4 A; N–N, 3.4 A; C–S,4.1 A; O–S, 3.7 A; N–S, 3.8 A. Superscript numbers in parentheses represent the number of contacts made by the indicated EDNresidue. The contact distances were calculated using the program CONTACT.51
Table 3. Potential hydrogen bonds between hRI and EDN in the complex structure
hRI atom B-factor (A2) EDN atom B-factor (A2) Distance D/A (A) Angle D–H/A (8)
Gln10 NE2 33.1 Asn32 OD1 23.0 2.6 158.2Cys11 SG 27.0 Gln28 NE2 29.6 3.4 167.6Asp35 OD2 23.7 Gln28 NE2 29.6 2.9 127.9Arg63 NH1 28.9 Asn25 OD1 25.0 3.2 144.2Arg63 NH2 29.1 Ser94 O 29.0 2.9 145.7Asp121 OD2 20.7 Asn25 ND2 25.3 2.9 158.1Trp261 NE1 14.1 Thr86 O 19.7 2.8 42.9Lys320 NZ 18.0 Asn39 OD1 16.2 2.8 148.5Lys320 NZ 18.0 Leu85 O 18.0 2.8 171.7Gln346 NE2 12.9 Asn39 OD1 16.2 3.1 124.6Asn349 ND2 13.7 Gln40 OE1 19.8 2.9 157.3Asn349 OD1 10.2 Asn39 ND2 14.3 2.9 158.3Glu401 OE2 25.3 Arg36 NE 22.5 2.7 135.3Glu401 OE2 25.3 Arg36 NH2 23.2 2.6 139.8Asp403 OD2 14.9 Arg36 NH1 22.1 2.9 140.1Cys408 SG 34.9 Ser64 O 39.8 3.6 177.8Tyr434 OH 18.2 Arg36 O 18.4 2.7 140.9Asp435 OD1 21.3 Lys38 NZ 15.2 2.8 158.0Asp435 O 17.5 His129 NE2 20.4 2.9 169.4Tyr437 OH 25.2 Asp112 OD2 28.7 2.7 166.6Arg457 NH2 32.3 Gln34 O 20.0 2.9 138.4Arg457 NH2 32.3 Arg35 O 18.8 3.2 142.9Val458 O 22.6 Met0 N 22.3 2.8 –Ser460 N 26.4 Gln34 OE1 16.8 2.4 168.9Ser460 OG 26.5 Trp10 NE1 15.2 3.0 178.3Ser460 O 28.9 Lys1 N 21.8 3.0 145.9
Hydrogen bond interactions were identified with the program HBPLUS.62 The upper limit for the donor–acceptor distance was 3.3 A,except for contacts involving a sulphur atom (limit 3.6 A); the lower limit for the donor–hydrogen–acceptor angle is 1208. Bond anglesare not given where the hydrogen position is ambiguous.
EDN in Complex with Placental Ribonuclease Inhibitor 641

Figure 2. Stereo views of Ca traces of hRI (a) and EDN (b), with stick representations of the side-chains of contactresidues. (c) The contact residues at the hRI–EDN interface in a stereo view. hRI residues are coloured purple and EDNresidues in olive green. The Figure was generated using MOLSCRIPT.58
642 EDN in Complex with Placental Ribonuclease Inhibitor
was also the case for hRI from the hRI–Angcomplex.
The overall topology of EDN in the complex issimilar to that of the free EDN molecule (PDB code1GQV.19) The r.m.s. difference between the Ca atomsof EDN from Complex 1 and free EDN is 0.89 A (thecorresponding value for EDN from Complex 2 is0.82 A). The two EDN molecules in the asymmetricunit can be superposed with an r.m.s. deviation of0.53 A. The residues at the active site are oriented
similarly in both the free and bound EDN struc-tures. The backbones in the two structures corre-spond well except for the regions containingresidues 32–37, 61–67 and 88–95, where the averager.m.s. deviation between Ca atoms is about 1.7 A(the maximum distance between correspondingatoms is w2.5 A). Residues that adopt differentside-chain conformations in the bound and freestructures are Met0, Phe5, Gln21, Gln22, Gln28,Asn32, Gln34, Arg35, Arg36, Lys38, Gln40, Arg68,

Figure 3.Amino acid sequence alignments for hRI and pRI (top) and for EDN, Ang, and RNase A (bottom), drawn byusing the program ALSCRIPT.60 The pRI sequence shows contact residues for RNase A shaded in black. The hRIsequence shows contact residues unique for Ang and EDN boxed in black and grey, respectively; those common to bothhave been boxed in white. Contact residues on EDN, Ang, and RNase A are boxed in black. The beginning of each repeatunit is marked for hRI.
EDN in Complex with Placental Ribonuclease Inhibitor 643

644 EDN in Complex with Placental Ribonuclease Inhibitor
His73, Asn84, Ser94 and Arg97. Most of theseresidues contact hRI in the complex. In somecases, the conformational rearrangements preventsteric clashes: the position of Gln34 in the freestructure severely conflicts with that of the side-chain of hRI Tyr434; Arg36 clashes with the side-chain of hRI Val432; and Gln40 bumps against theside-chain of hRI Asn349. Another notable differ-ence between the free and bound EDN structuresconcerns the loop between residues 89–95, which isdisordered in free EDN, but has well definedelectron density in both EDN molecules from thepresent study (with the exception of Gln91 of EDNfrom Complex 1). This could be because binding ofhRI constrains the loop and reduces its flexibility.
The enzyme–inhibitor interface: generalcharacteristics
Twelve repeat units (R1, R2, R3, R5, R6, R8, R10,R12, R13, R14, R15 and R16) and the terminalb-strand (b17, comprising residues 457–460) areinvolved in recognising the EDN molecule(Figure 3). Sixty-four residues (34 from hRI and 30from EDN) form direct contacts at the bindinginterface (Tables 2 and 3; Figure 2), and another fiveresidues (two from hRI and three from EDN) areinvolved in water-mediated interactions (Table 4).An additional 12 residues undergo some degree ofburial upon complex formation although they arenot involved in any well-defined interactions at theinterface. These are Ser64, Gln92, Asn93, Gln147,Glu149, Lys204, Glu344 and Asp378 from hRI andSer20, Thr24, Pro90 and Tyr98 from EDN. Thecontact residues on hRI are distributed throughoutthe sequence. The vast majority of the hRIresidues that form direct contacts with EDN lie onb-strands or b–a loops (15 residues on nine differentb-strands; 16 residues on 11 b–a loops); the
Table 4. Potential water-mediated hydrogen bonds between
Water number hRI atom Distance (A) EDN atom
11 Tyr434 OH 3.0 Gln34 NGln34 NE
12 Asp403 OD1 2.7 Asn39 N44 Asp435 N 2.8 Gln14 OE1
Ser460 OG 2.9123 Arg457 NE 2.9 Gln34 O
Arg457 NH2 3.2147 Asn406 OD1 2.7 Gln40 O167 Asp435 OD1 2.6 His15 NE2219 Gln10 O 2.9 Asn32 ND225 Glu264 OE2 2.9 Asn84 ND347 Glu264 OE2 2.7 Asn84 ND387 Tyr150 OH 3.1 Gln100 N425 Lys320 NZ 2.7 Asn84 OD
Asn349 OD1 3.0579 Gln430 NE2 2.8 Arg35 O587 Glu206 OE1 3.0 Thr87 OG
Solvent accessibility was calculated using the program XPLOR63 widentified with the program HBPLUS.62 The upper limit for the donsulphur atom (limit 3.6 A); the lower limit for the donor–hydrogen–a
remaining residues (Glu443, Leu446 and Ala447)are on a-helix 16. The contacts on EDN aredistributed over three of the protein’s four helices,four of its five b-strands and four of its seven loops.There are a total of 209 direct interactions at theinterface between 108 hRI atoms (27main-chain and81 side-chain atoms) and 84 EDN atoms (24 main-chain and 60 side-chain atoms). A fairly largenumber of the contacts involve at least onemain-chain atom: 72 van der Waals interactionsand 11 H-bonds (as compared to two in the Angcomplex and six in the RNase A complex).
Formation of the hRI–EDN complex buries3677 A2 of the total solvent-accessible surface areaof the two proteins. This area represents 16% of thecombined surface areas of the free proteins whereasonly w13% is buried in the Ang and RNase Acomplexes (w2900 A2). The hRI–EDN surface is72% non-polar, 16% uncharged polar and 12%charged. This distribution differs from those in theAng and RNase A complexes (Table 5), mostnotably in that the charged character is muchlower. The individual binding surfaces on theligands and their inhibitors are characteristicallyquite different (Figure 1(d)).
The degree of shape complementarity betweenhRI and EDN is equivalent to that observed in thehRI–Ang complex (ScZ0.69, where Sc is the shapecorrelation statistic),20 and indicates a moderatedegree of correlation between the topographies ofthe interacting protein surfaces.
The enzyme–inhibitor interface: details of theinteractions
EDN and hRI form a total of 184 van der Waalscontacts (Table 2), 26 potential hydrogen bonds(Table 3) and 17 water-mediated interactions(Table 4); detailed views of selected interactions
hRI and EDN in the complex structure
Distance (A)Solvent
accessibility B-factor (A2)
3.0 0.0 16.42 3.2 0.0 16.4
2.7 0.0 12.42.7 0.0 14.4
2.8 13.7 19.9
3.2 0.0 23.72.8 2.1 32.0
2 3.1 18.7 37.92 2.9 0.44 20.42 2.8 11.8 36.9
3.0 13.9 34.41 2.8 6.1 30.6
2.8 4.7 33.21 2.9 0.8 33.7
ith a probe radius of 1.4 A. Hydrogen bond interactions wereor–acceptor distance was 3.3 A, except for contacts involving acceptor angle is 1208.

Table 5. Comparison of the general features of the enzyme–inhibitor interface
General features EDN Angiogenin RNase A
Dissociation constanta (fM) 2.7 0.7 67Total buried surface areab (A2) 3677 2908 2878Shape correlation indexc 0.69 0.69 0.58No. of residues in contact 64 (34 from hRI and 30 from
EDN)50 (26 from hRI and 24 fromangiogenin)
52 (28 from pRI and 24 fromRNase A)
Chemical characterd of participating residuesNon-polar (%) 72 65 68Uncharged polar (%) 16 13 13Charged (%) 12 22 19No. of hydrogen bondse 26 11 18No. of van der Waals contactsf 189 124 117
a The dissociation constants for the hRI–EDN, hRI–Ang, and pRI–RNase A complexes are from the work done by Lee et al.,4 Teufel etal.,17 and Vincentini,64 respectively.
b Calculated with the program DSSP.65c Calculated with the program SHAPE.20 Sc determines the shape complementarity of two interacting molecular surfaces. Interfaces
with ScZ1 denotes perfect complementarity and those with ScZ0 means the two interfaces are effectively uncorrelated in theirtopography.
d Values listed for the Ang and RNase A complexes differ somewhat from those reported.12,18e Hydrogen bonds for the EDN and Ang complexes were determined with the program HBPLUS;62 those for the RNase A complex
are from the work done by Kobe & Deisenhofer.18f van der Waals contacts for the EDN and Ang complexes were determined with the program CONTACT;51 those for the RNase A
complex are from the work done by Kobe & Deisenhofer.18
EDN in Complex with Placental Ribonuclease Inhibitor 645
are shown in Figure 4. The numbers of van derWaals contacts and direct hydrogen bonds are bothmuch higher than those observed for the hRI–Ang(PDB code 1A4Y) and pRI–RNase A (PDB code1DFJ) complexes (Table 5).12,18 The inhibitor makesextensive contacts with the ribonucleolytic activesite of EDN. Catalytically important residues fromthe P1 subsite (Lys38, His129) and residues involvedonly in substrate binding (Arg68, Asn70 andAsp112 in the B2 nucleobase-binding subsite;Arg36, Asn39 and Gln40 in the putative PK1
phosphate-binding subsite) form a total of tendirect hydrogen bonds and 76 van der Waalscontacts (the composition of the various subsitesof EDN is discussed in the work done bySwaminathan et al.,19 Mosimann et al.,21 andLeonidas et al.22). Other residues with less well-defined roles in the active site region (Met0, Lys1,Trp7, and Trp10) make an additional threehydrogen bonds and 37 van der Waals contactswith hRI. Met0 is not present in natural EDN, andits interactions (one hydrogen bond and 18 van derWaals contacts) will not be considered further in ouranalysis.
The side-chains of the catalytic residues Lys38and His129 form direct hydrogen bonds with thecarboxylate group and the carbonyl oxygen of hRIAsp435; the third member of the catalytic triad ofEDN, His15, makes a water-mediated contact withthe carboxylate group of the same hRI residue.Lys38 and His129 also form 25 van der Waalscontacts with Tyr434, Asp435, Ile436 and Tyr437 ofhRI. The three putative PK1 subsite residues makeseven hydrogen bonds. Four of these involve Arg36of EDN: three between the guanidino group and thecarboxylate groups of hRI Glu401 and Asp403, andone between the carbonyl oxygen and the phenolicOH of Tyr434. Asn39 of EDN lies within hydrogen
bonding distance of the 3-amino group of hRILys320 and the carboxylate of hRI Asn349; the latterhRI residue makes a hydrogen bond with Gln40 ofEDN as well. Arg36 of the enzyme also forms 23 vander Waals interactions with hRI (11 of them withTrp375 and the others with Glu401, Asp403, Gln430,and Val432), whereas Asn39 and Gln40 form 11such contacts (with Lys320, Asn349, Ala377, andAsn406). The only B2 subsite residue to make ahydrogen bond with hRI is Asp112, which interactswith the hydroxyl group of Tyr437. The other two B2
subsite residues, Arg68 and Asn70, are involved in13 van der Waals contacts with this same inhibitorresidue. Also in the active site region, the indolegroup of EDN Trp7 stacks against that of hRI Trp438and forms seven van der Waals contacts with hRIGlu443, and EDN Lys1 and Trp10 make hydrogenbonds with Ser460 of hRI.Regions outside the active site of EDN are also a
part of the interface with hRI. Residues 84–88 fromstrand b2 make 32 contacts with the region of hRIthat contains Trp261, Trp263, Trp318 and Trp375,including two hydrogen bonds (both of whichinvolve main-chain oxygen atoms on the enzyme).This EDN segment also participates in five water-mediated hydrogen bonds, all but one of whichinvolve the side-chain of Asn84. EDN residuesGln22, Asn25, Gln28, Val29, Asn32, Gln34 andArg35, located on or adjacent to helix a2, interactwith hRI residues Gln10, Cys11, Asp35, Asp36,Arg63, Asp121, Tyr150, Arg457, Ile459 and Ser460,making 8 hydrogen bonds and 33 van der Waalscontacts.Complex 2 of the asymmetric unit contains nearly
all of the contacts described above for Complex 1,except for those involving the N-terminal hRIresidues 1–150. However, one of the two hydrogenbonds of hRI Arg457 in Complex 1 (the interaction
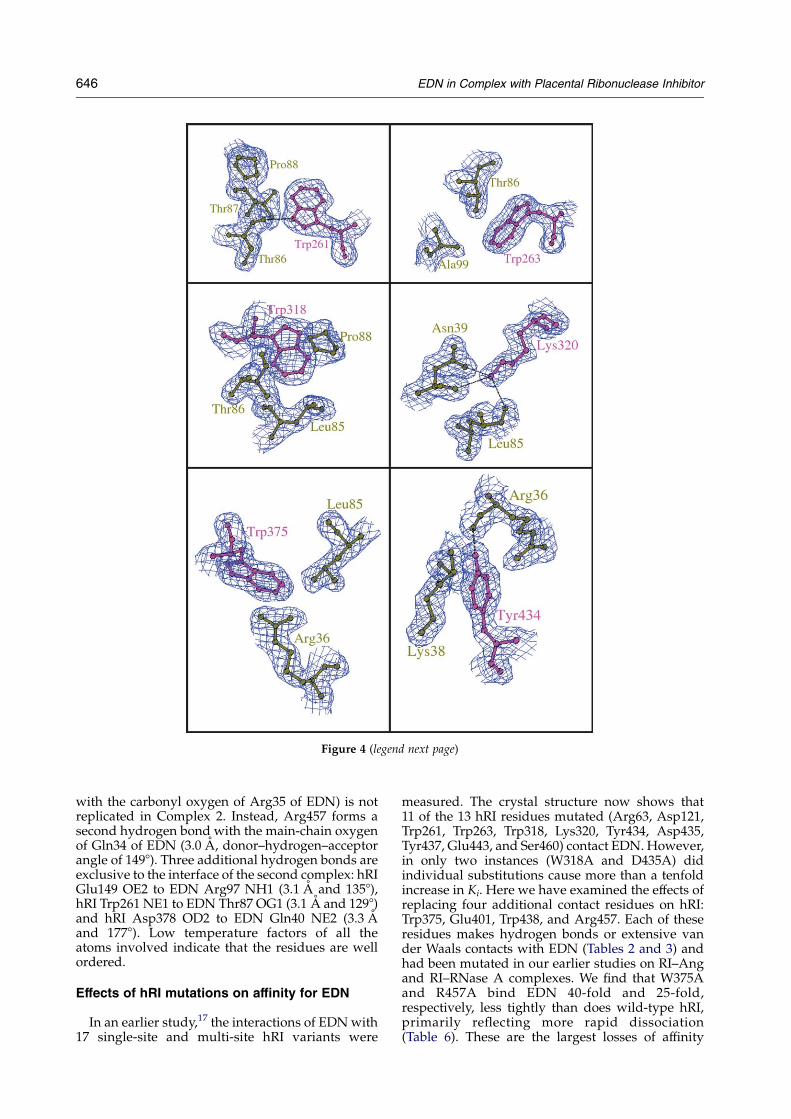
Figure 4 (legend next page)
646 EDN in Complex with Placental Ribonuclease Inhibitor
with the carbonyl oxygen of Arg35 of EDN) is notreplicated in Complex 2. Instead, Arg457 forms asecond hydrogen bond with the main-chain oxygenof Gln34 of EDN (3.0 A, donor–hydrogen–acceptorangle of 1498). Three additional hydrogen bonds areexclusive to the interface of the second complex: hRIGlu149 OE2 to EDN Arg97 NH1 (3.1 A and 1358),hRI Trp261 NE1 to EDN Thr87 OG1 (3.1 A and 1298)and hRI Asp378 OD2 to EDN Gln40 NE2 (3.3 Aand 1778). Low temperature factors of all theatoms involved indicate that the residues are wellordered.
Effects of hRI mutations on affinity for EDN
In an earlier study,17 the interactions of EDNwith17 single-site and multi-site hRI variants were
measured. The crystal structure now shows that11 of the 13 hRI residues mutated (Arg63, Asp121,Trp261, Trp263, Trp318, Lys320, Tyr434, Asp435,Tyr437, Glu443, and Ser460) contact EDN. However,in only two instances (W318A and D435A) didindividual substitutions cause more than a tenfoldincrease in Ki. Here we have examined the effects ofreplacing four additional contact residues on hRI:Trp375, Glu401, Trp438, and Arg457. Each of theseresidues makes hydrogen bonds or extensive vander Waals contacts with EDN (Tables 2 and 3) andhad been mutated in our earlier studies on RI–Angand RI–RNase A complexes. We find that W375Aand R457A bind EDN 40-fold and 25-fold,respectively, less tightly than does wild-type hRI,primarily reflecting more rapid dissociation(Table 6). These are the largest losses of affinity

Figure 4.Detailed interactions at the hRI–EDN interface. The electron-density maps (2FoKFc) around the residues arecontoured at 1s level. Potential hydrogen bonds are shown by dotted lines. The residues and labels for the hRI moleculeare coloured purple and those for the EDN molecule are shown in olive green. The Figure was generated usingBOBSCRIPT.61
EDN in Complex with Placental Ribonuclease Inhibitor 647
observed thus far for any single-residue variants. Incontrast, only minor changes were measured forE401R and W438A/S439A/E440A (the triplevariant was used in the latter case because it was
Table 6. Kinetic constants for the complexes of EDN with hR
hRI kd (sK1!10K7) t1⁄2a (days) ka (M
wild-typed 7.5G0.5 10.7 1W375A 160G8 0.5 0E401R 9.2G0.3 8.8 1W438A/S439A/E440A 9.4G0.3 8.5 0R457A 112G3 0.7 0
Kinetic constants were measured as described in Materials and Metha Half-life for dissociation of the complex.b Ki for variant hRI divided by that for wild-type hRI.c DDG values are the difference in binding free energies for the
DDGZKRT ln(Ki,var/Ki,wt). The standard errors for DDG are all !0.1d The Ki value for the wild-type complex reported here is somewha
may reflect minor variations in the EDN or hRI preparations, or in th
available from an earlier study,15 whereas a single-residue Trp438 variant had not been made; Ser439and Glu440 do not contact EDN in the crystalstructure).
I variants
K1 sK1!108) Ki (fM) Ki,var/Ki,wtb DDGc (kcal/mol)
.12G0.03 6.7G0.5
.59G0.02 271G16 40 2.2
.10G0.03 8.4G0.4 1.3 0.1
.59G0.01 16G1 2.4 0.5
.68G0.01 165G5 25 1.9
ods.
wild-type and variant complexes, calculated from the equationkcal/mol.t higher than that measured previously (2.7 fM).17 This differencee incubation conditions used.

ces
ion.
648 EDN in Complex with Placental Ribonuclease Inhibitor
Discussion
Table
7.Equivalen
tinteractionsin
variousRIcomplexes
Equivalen
tin
theEDN,Ang,an
dRNaseA
complexes
Equivalen
tonly
forEDN
andAng
Equivalen
tonly
forEDN
andRNaseA
Equivalen
tonly
forAngan
dRNaseA
RIresidue
EDN
residue
Ang
residue
RNaseA
residue
RI
residue
EDN
residue
Ang
residue
RI
residue
EDN
residue
RNaseA
residue
RI
residue
Ang
residue
RNaseA
residue
Hydrogen
bonds
D43
5K38
K40
K41
aD35
Q28
R31
D12
1bN25
N24
S46
0bW
10Q12
Q11
E40
1R36
(2)
R39
(2)
C40
8cS64
K66
Y43
7dD112
E111
S46
0bK1
K7
van
der
Waalscontacts
C11
V29
R32
S32
D35
Q28
R31
D12
1bN25
N24
Y437
A106
A109
Y43
4K38
K40
K41
K320
N39
D41
E40
1R36
R39
S/T43
9E10
8E111
Y43
7H12
9H114
H119
V43
2R36
R39
E44
0E10
8E111
Y43
7eN70
N71
Interactionsthat
areeq
uivalen
tin
thecrystalstructuresoftw
oormore
RI–ligan
dcomplexes.T
hecriterionforeq
uivalen
ceisstructuralanalogyrather
than
corresponden
cein
theam
inoacid
sequen
ofthedifferentligan
ds.Aninteractionis
listed
asavan
der
Waals
contact
ifitis
ahydrogen
bondin
oneofthecomplexes
indicated
,butis
avan
der
Waa
lscontact
intheother.
aThehydrogen
bondlisted
isnotseen
intheRNaseA
complexdueto
thepresence
ofasu
lphateionfrom
thecrystalliza
tionmed
ium,butmutagen
esis
resu
ltsindicateitis
presentin
solut
bLigan
dresidues
shownarenotat
correspondingpositionsin
theam
inoacid
sequen
ce,butoccupysimilar
positionsin
thethree-dim
ensional
structure.
cInteractioninvolves
main-chainatom(s)oftheligan
ds.
dE10
8ofAngalso
contactsRIresidueY43
7,butthedistance
isbey
ondthecu
toffforahydrogen
bond.
eThis
interactionis
ahydrogen
bondin
theRNaseA
complex.18
Structural basis for the recognition of differentligands by RI
RI was first characterized as an inhibitor ofpancreatic RNase23 and was subsequently shownto bind extremely tightly to angiogenin,4,24 EDN,25
and RNase 42,26 as well. The range of Ki valuesmeasured for non-orthologous ligands,w1–200 fM,is remarkably small in view of the modest level ofoverall sequence identity between these proteins(w25–40%)27 and the poor conservation of activesite residues in particular (only the P1 catalytic siteis universally maintained). In principle, RI mighthave achieved its non-stringent selectivity byrelying heavily on interactions with main-chainelements on the ligands or with the relatively fewside-chains that are well conserved. However, theactual mechanism that has evolved for the RIsystem is in fact rather different. This was alreadyapparent from the crystal structure of the pRI–RNase A complex,11 which revealed that most of theligand residues at the interface are not maintainedin Ang and EDN.28 The hRI–Ang crystal structure12
then showed that even the conserved interfaceresidues on RNase A and Ang form largely differentinteractions with RI.
The hRI–EDN structure reveals still greatervariability in the molecular basis for inhibitor-ligand recognition. The most striking generaldifference with respect to the earlier complexes isthe considerably larger size of the interface, whichburies 27%more solvent-accessible surface area andinvolves a 23–28% greater number of residues(Table 5; Figure 1(d)). (Although EDN is largerthan Ang and RNase A (134 residues versus 123 and124, respectively), the loop (114–122) that contrib-utes most of the additional residues is not part ofthe interface.) A majority of the specific inter-molecular contacts formed are again unique(Table 7). Only one hydrogen bond is equivalentin the crystal structures of all three complexes: thatof RI Ser460 OG with the side-chains of Gln12/11 ofAng/RNase A and Trp10 of EDN. (Although theresidue in the EDN sequence that corresponds toGln12/11 of Ang/RNase A is Gln14, it is Trp10 thatoccupies the analogous position in the three-dimensional structure.) It is highly likely that asecond hydrogen bond, between the carboxylate ofRI Asp435 and the 3-amino group of the catalyticlysine of the ligand (Lys38, Lys40, and Lys41 inEDN, Ang, and RNase A, respectively), is alsopresent in all three complexes, but in the pRI–RNase A structure a sulphate anion from thecrystallization medium binds to Lys41 and preventsany such interaction. Mutagenesis results stronglysupport the existence of a hydrogen bond betweenLys41 of RNase A and Asp435 of hRI in solution.13
Beyond these hydrogen bonds, the three complexesshare a small number of van der Waals contacts,most of which involve Tyr434 and Tyr437 of RI and

EDN in Complex with Placental Ribonuclease Inhibitor 649
Lys38/40/41 and His129/114/119 (EDN/Ang/RNase A) of the ligand.
The hRI–EDN interface also contains interactionsthat correspond to those seen in only one of theother two complex structures (Table 7; Figure 1(d)).Asp35 of hRI forms structurally equivalent hydro-gen bonding and van der Waals interactions withGln28 of EDN and Arg31 of Ang, and Lys320 of hRImakes corresponding van der Waals contacts withAsn39 of EDN and Asp41 of Ang. The correspon-dence between the EDN and RNase A complexstructures is somewhat more extensive, andincludes six hydrogen bonds. The carboxylategroup of Glu401 of RI forms fully analogoushydrogen bonds with the guanidino groups ofEDN Arg36 and RNase A Arg39, as does the SHgroup of RI Cys408 with the main-chain oxygen ofSer64/Lys66 of EDN/RNase A and the OH groupof RI Tyr437 with the side-chains of Asp112/Glu111of EDN/RNase A. Two other hydrogen bonds,made by RI Asp121 and Ser460, involve ligandresidues that are equivalent in the tertiary struc-tures but not in the sequence (Asn25/24 and Lys1/Lys7 of EDN/RNase A, respectively). The twocomplexes also share several van derWaals contactsthat are not present in the Ang complex. Overall, theEDN and RNase A complexes contain eightequivalent hydrogen bonds (assuming the existenceof a bond between Asp435 of RI and Lys41 of RNaseA), representing 32% and 42% of the totals,respectively; 15–20% of the van der Waals contactsare similar. This degree of correspondence, whilestill modest, is much greater than that between theEDN and Ang or RNase A and Ang complexes,where, for example, only three and two of thehydrogen bonds, respectively, are analogous(Table 7).
Energetics of the RI–ligand interfaces
A mutational study13 performed shortly after thedetermination of the RI–RNase A crystal structuresuggested that a small interface region, encompass-ing RI residues Tyr434, Asp435, Tyr437 and Ser460and the P1/B2/P2 part of the RNase A active site,provides a substantial fraction of the bindingenergy and thus constitutes a hot spot. Individualmutations of these four RI residues decreasedaffinity by factors ranging from 84 to 23,000 andthe sum of the losses in binding energy for themutations was 15.6 kcal/mol, compared with thetotal DG of K18.4 kcal/mol for complex formation.These same RI mutations also had marked, albeitmostly smaller, effects on the affinity for Ang,indicating that the same interface region in the RI–Ang complex might also be a hot spot. Moredetailed examination of the RNase A and Angcomplexes later presented a less straightforwardpicture of the interface energetics.14,15 In the RNaseA complex, mutational effects within the putativehot spot were shown to be strongly sub-additive,indicating that this region contributes less than theresults of single-site mutagenesis had suggested.
Moreover, replacements of RI residues from manyother areas of the interface were shown to producesignificant effects on affinity (DDGz1–2 kcal/mol).Thus, the binding energy is widely distributed,although interactions in the original hot spot stillseem to account for the majority. In contrast, multi-site mutagenesis in the Ang complex revealed thatlosses in binding energy for replacements in the hotspot are super-additive, and further exploration ofthe interface showed that only one other area(containing Trp261, Trp263, Trp318, and Trp375 ofRI and the 84–89 loop of Ang) provides anyappreciable binding energy. Thus, interactions inthe hot spot contribute even more to the stability ofthe RI–Ang complex than first appeared to be thecase.From both evolutionary and structural points of
view, it seemed reasonable that the existence of thishot spot would be a universal feature of RI–ligandcomplexes. If the driving force for RI evolution is todevelop and maintain highly efficient inhibition ofthe enzymatic activity of RNase targets, then stronginteractions with the active site might be expectedto play a key role. Moreover, hydrogen bonds of RIAsp435 with the strictly conserved catalytic lysineof the ligands would seem likely to provide a usefulanchoring point. However, when mutationalanalysis was extended to a third complex, that ofhRI with EDN,17 the results strongly challenged thisview. Thus, replacement of Asp435 in RI producedonly a moderate decrease in affinity for EDN, 14-fold as compared to 360 and 470-fold for Ang andRNase A, respectively. Mutations of RI Tyr434,Tyr437, and Ser460 caused even smaller effects onEDN binding: Ki values were increased by less thantwofold in all cases. Simultaneous replacements ofTyr434 and Asp435 or Tyr437 produced moredramatic increases in Ki (540-fold and 290-fold,respectively), signifying the same type of super-additivity seen in the Ang complex. However, thesechanges were still 2–3 orders of magnitude smallerthan those measured for the same multi-sitereplacements in the RNase A and Ang complexes.Thus, it appeared that the RI residues 434–437 and460 do not constitute a hot spot in the EDNcomplex. However, the possibility remained thatneighbouring residues in the C-terminal segment,not yet identified, play more substantial roles andconfer this property on the region.The crystal structure of the complex of hRI with
recombinant EDN determined here reveals that RIresidues 434, 435, 437, and 460 are all within theinterface. Ten additional residues in the RI segment430–460 also form contacts with EDN. The inter-actions of three of these residues (Leu446, Ala447,and Val458) are exclusively with Met0 of EDN andcannot contribute to stabilization of the complexwith natural EDN, which lacks this residue. Five ofthe RI residues (Gln430, Val432, Ile436, Glu443, andI459) make only van der Waals interactions that donot involve any substantial burying of hydrophobicgroups, suggesting that they contribute little bind-ing energy. This has been directly demonstrated for

650 EDN in Complex with Placental Ribonuclease Inhibitor
Glu443, where Ala substitution had no effect onaffinity.17 The remaining two RI residues thatcontact EDN, Trp438 and Arg457, seem to formstronger interactions in the crystal structure (stack-ing and hydrogen bonding, respectively), and theirroles were examined here by mutagenesis (Table 6).Replacement of Trp438 caused only a minordecrease in affinity whereas replacement of RIArg457 weakened affinity by a factor of 25. Overall,the combined structural and mutational findingsidentify the C-terminal region of RI as important forthe stability of the RI–EDN complex, but indicatethat its role is not as dominant as in the complexeswith the other two ligands. The present analysissuggests that the interactions of this RI segmentaccount for w30–40% of the total binding energy,although it remains possible that this estimate issomewhat low because additional negativecooperativities (as reflected in super-additive muta-tional effects) have not been identified. It shouldalso be noted that the contribution of the hydrogenbond formed by the main-chain O of RI residueAsp435 has not been assessed, and may besignificant.
The earlier mutational study on the RI–EDNcomplex17 had shown that the interactions of a Trp-rich region of RI, containing Trp261, Trp263, andTrp318, make a strong contribution, the magnitudeof which again became apparent only throughmulti-site mutagenesis. In the crystal structure, allthree tryptophan residues form numerous contactswith EDN. The neighbouring residue Trp375 alsomakes extensive interactions with the ligand. Wefind here that replacement of Trp375 by Alaincreases Ki by a factor of 40, the largest changemeasured for any single-site mutation. The sum ofthe losses in binding free energy for this substi-tution and the triple 261/263/318 replacement is7.2 kcal/mol, which equals 35% of the total bindingenergy for complex formation. The actual contri-bution of these residues depends on the nature andextent of any additional cooperativities within thisinterface region or between this region and others.15
The effects of several single-residue replacementsin RI outside the C-terminal and Trp-rich regions(Arg63, Asp121, Lys320, Ser405, and Asp411 to Alaand Glu401 to Arg) have also been determined.None of these substitutions diminished affinity forEDN. The first five of these residues had beenselected for examination on the basis of a modeledRI–EDN complex,17 and the crystal structure nowshows that two of them do not contact EDN. Inaddition, replacement of the RI segment Cys408-Leu409-Gly410 by Trp-Trp, which eliminates thehydrogen bond between Cys408 of RI and Ser64 ofEDN, increases Ki only by a factor 2.29
Further single- and multi-site mutational studieswill be required to determine the sources of one-third or so of the binding energy in the RI–EDNcomplex that is not yet accounted for. One or moreof the interface regions not yet investigated mayplay a significant role; the unexplored regionsinclude those containing the RI segments 10–36,
346–349, and 403–406. As noted above, it is alsopossible that additional negative cooperativitiesinvolving the C-terminal and Trp-rich regionsprovide some or all of the “missing” energy.
Reconciliation of structural and mutagenesisfindings
Views of the molecular basis for protein-proteinrecognition gleaned from crystal structures andsite-specific mutagenesis are often discordant.Replacements of residues that appear to formstrong interactions in crystal structures frequentlyhave little impact on binding affinity and, con-versely, mutations of some residues producereductions in avidity that seem to be out ofproportion to the strength of the interactionseliminated. The most widely appreciated reasonfor such discrepancies is that theoretical gaps inunderstanding thermodynamic and quantum-mechanical aspects of the interactions compromiseany quantitative assessments of the free energiesinvolved; indeed, these difficulties have not beenresolved even for the simpler case where the proteinligand is a small molecule.30 A second reason is thatinteractions often operate in a cooperative manner,as indicated empirically whenmutational effects arenon-additive (see the work done by Chen &Shapiro14 and references therein). Thus, eliminationof the interactions of one residue can interfere withthe function of another residue, producing arti-ficially large losses in affinity. In this case, thecombined result of mutating both residues will besubadditive. Replacement of an amino acid can alsohave the opposite effect, i.e. it can allow anotherresidue to optimize its interactions or form newones, resulting in an underestimation of thecontribution of the mutated residue. In this case,the energy changes measured for mutating bothresidues will be superadditive. Apparentinconsistencies between structural and mutationalobservations can also derive from a more trivialsource: lack of sufficient resolution or quality in thestructure to warrant any serious attempts to assessinteraction strength. In addition, we have notedpreviously that putative hydrogen bonds (includ-ing salt links) in crystal structures tend to contributelittle binding energy when the temperature factorsfor the participating atoms are relatively high.12
The effects of replacing 16 of the 34 hRI residuesthat contact EDN have now been measured, and inmany instances single-residue mutations werefound to produce losses in affinity that are muchsmaller than might be expected from the crystalstructures or from previous studies on the Ang andRNase A complexes (it should be noted that none ofthe temperature factors for the interacting RI andEDN residues are high). This discrepancy is moststriking for hRI residues Tyr434, Tyr437, and Ser460,which contribute to the hot spots seen in the earliercomplexes. These residues form sets of interactionswith EDN that are generally similar to those withRNase A, and even more extensive than those with

EDN in Complex with Placental Ribonuclease Inhibitor 651
Ang. Nonetheless, Y434A, Y437A, and des460 hRIvariants show only negligible decreases in affinityfor EDN. To some extent, this seems to reflectnegative cooperativity: e.g. the energetic effects ofreplacing Tyr434 and Tyr437 are markedly super-additive. Structural analysis suggests a possibleadditional factor for the Tyr434 mutation: theposition of Gln34 in free EDN conflicts with thatof Tyr434 in the complex, necessitating a confor-mational change (see above) that may have anenergetic cost. For another RI residue in thisinterface region, Asp435, the effect of Ala replace-ment on the Ki value with EDN is again consider-ably smaller than with Ang and RNase A (14-foldversus w400-fold), but here there are subtlestructural differences that may provide a partialexplanation. Lys40 of Ang forms two hydrogenbonds with the Asp435 carboxylate, whereas thecorresponding Lys of EDN makes only one hydro-gen bond with the 435 carboxylate (it is not clearhow many hydrogen bonds Asp435 forms with theanalogous Lys in RNase A because, as discussedabove, this site in the RI–RNase A crystal structureis distorted by a bound sulfate ion). The super-additivity of the effects of Asp435 and Tyr434mutations suggests that negative cooperativitymay be a factor as well.
Additional cases where mutational effects arequite small despite the existence of “good” inter-actions for the targeted residues in the crystalstructure are the hRI variants R63A, D121A,W261A, K320A, E401R, and W438A. Arg63,Asp121, Trp261, Lys320, and Glu401 each formhydrogen bonds and van der Waals contacts withEDN, whereas Trp438 stacks against a Trp residuein EDN. The hydrogen bonds of Arg63 and Asp121are seen only in Complex 1 of the asymmetric unit,suggesting that they are relatively weak. However,the interactions of Lys320 and Glu401 (includingtwo hydrogen bonds for each), Trp261 (onehydrogen bond), and Trp438 are present in bothcomplexes. The interaction partner for Trp438 onEDN, Trp7, is mannosylated at the C-2 position(atom CD1) in the natural enzyme used for kineticmeasurements, but not in the recombinant proteinused for structure determination; it is possible thatthe glycosylated residue does not form the samecontacts.
Individual Ala replacements of RI residuesTrp263 and Trp318 produced moderate losses inbinding energy (0.9–1.5 kcal/mol); each residueforms ten van der Waals contacts and has50–60 A2 of solvent-accessible surface area buriedupon complex formation with EDN. A largerchange (DDGZ2.2 kcal/mol) is observed for Alasubstitution of Trp375, which makes a similar set ofcontacts and buries a comparable amount ofhydrophobic surface area. Results with the triplevariant W261A/W263A/W318A-hRI suggest thatthe effects of individual mutations of Trp261,Trp263, and/or Trp318 underestimate the actualcontributions of these residues: the experimentalDDG value for the triple variant, 5.0 kcal/mol, is
twice the sum of the DDG values for the single-residue variants.17 Thus, the region of the interfacethat contains these residues may be somewhatplastic, so that when a single Trp is lost, one or bothof the others can form compensatory interactions.Interestingly, the effects of the 261, 263, and 318single-residue and multi-residue replacements onaffinity for EDN and Ang are nearly indistinguish-able, despite the complete lack of correspondencebetween the interactions of these tryptophanresidues in the two complex structures.The effects of the remaining mutations are readily
understood from the crystal structure. Losses ofCys408 and Glu443, which appear to form onlyweak interactions with EDN (a long SH-to-Ohydrogen bond and several van der Waals contactswith no significant hydrophobic burial), have littleimpact. Replacement of Arg457, which hydrogenbonds with two main-chain oxygen molecules ofEDN, causes the second largest decrease in bindingenergy measured for any single-residue mutation,1.9 kcal/mol.
Implications for design of ligand-specific RIderivatives
Pancreatic RNase and its homologues wereoriginally considered to be housekeeping ordigestive enzymes.31,32 However, many of themare now known to exert potent biological effects. Forexample, angiogenin induces the formation of newblood vessels33 and the eosinophil-associatedRNases EDN and ECP (eosinophil cationic protein)exhibit antiviral,34 antibacterial (ECP only) andantiparasitic activities35–37 and activate dendriticcells (EDN only),38 suggesting a possible role in hostdefense. In addition to their normal physiologicalroles, these RNases have been implicated in severalpathological processes. For instance, angiogenin isinvolved in the establishment and metastasis ofhuman tumours in athymic mice,39,40 probablythrough its angiogenic activity. EDN causes acerebellar dysfunction known as the Gordonphenomenon, when injected into rabbits.41,42
Although no direct counterpart to this phenomenonis known in humans, some diseases characterisedby elevated eosinophil levels (eosinophil-myalgiasyndrome and hypereosinophilic syndrome) pro-duce neurological manifestations ranging frommuscle ataxia to dementia to axonal neuropathy;43,44
these symptoms may reflect the toxic effects of highconcentrations of EDN or ECP. Increased levels ofEDN have also been associated with bronchialasthma and parasitic infections.45
The biological activities of Ang, EDN, and ECPthat are involved in human pathologies appear tobe strictly dependent on their enzymatic activi-ties.34,46–48 Therefore, the enzymatic active sites ofthese enzymes are attractive targets for the develop-ment of new drugs for treatment of cancer anddiseases caused by excess eosinophils.22,48,49 RI is ahighly effective inhibitor of Ang, EDN, and ECP, butits broad specificity for proteins in the pancreatic

652 EDN in Complex with Placental Ribonuclease Inhibitor
RNase superfamily undermines its potential thera-peutic utility; this lack of selectivity also limits theuse of RI as a tool to investigate the biologicalsignificance of individual RNase ligands. Althoughthe overall docking of the ligands in the crystalstructures of the three RI complexes determined sofar is similar, the specific interactions at the interfaceare quite distinct and mutational studies (discussedabove) have indicated that the versatility displayedby the inhibitor is a consequence of its ability torecognise features unique to each of its ligands.
This variability in the molecular basis for ligandrecognition was recently exploited to engineer an RIvariant that retains essentially native affinity forAng but does not bind RNase A detectably.29
Selectivity was achieved by remodelling the loopsegment 408–410 on RI, which contacts RNase A,but not Ang, so as to sterically hinder RNase Abinding. The remodelled RI, however, still had highavidity for EDN. It is possible that additionalengineering of the 408–410 region of RI, whichforms two contacts with EDN in the crystal, canfurther reduce affinity for EDN without impairingrecognition of Ang. Selectivity between Ang andEDN might also be achieved by introducing one ormore of the single residue mutations that have beenshown to produce differential effects on affinity. Forexample, Ala replacements of Trp375 and Arg457weaken binding of EDN more than that of Ang (bysevenfold and w30-fold, respectively), whereassubstituting Ala for Tyr434 diminishes affinity forAng 100-fold more than for EDN. Further structuraland mutational analyses of the Ang and EDNcomplexes should facilitate a better understandingof the functional roles of the individual contactresidues and may point to additional ways toremodel RI to achieve even greater selectivity.
Materials and Methods
Protein preparation, crystallisation and X-ray datacollection
Natural hRI was obtained from Promega. RecombinantMet0 EDN was a kind gift from Dr R. J. Youle (NIH,Bethesda, MD). The hRI–EDN complex was prepared bya modification of the method used for the preparation ofthe pRI–RNase A complex.50 All buffers were degassedthoroughly before use. Briefly, hRI (0.9 mg) was mixedwith EDN (0.4 mg) in 7.5 ml of 10 mM bis-Tris–HCl(pH 6.5), 20 mM DTT, and incubated on ice overnight.The mixture was injected onto a Mono-Q HR 5/5 anion-exchange column (Amersham Biosciences) equilibratedin the same buffer, and the complex was eluted with anascending NaCl gradient. Using an Ultrafree-4 concen-trator (10 kDa MWCO; Millipore), the complex wastransferred into 10 mM Hepes/NaOH (pH 7.5), 20 mMDTT, and concentrated to 4 mg mlK1.The complex was crystallised by the hanging drop-
vapour diffusion technique, using as reservoir a solutionof 1 M sodiummalonate (pH 7). Drops composed of equalvolumes of protein and reservoir solutions were incu-bated at 22 8C and crystals grew to their full size withintwo days. A cryoprotectant solution prepared by
supplementing the reservoir solution with 25% (v/v)glycerol enabled crystals to be flash-frozen in liquidnitrogen.Flash-cooled hRI–EDN crystals were used to collect
diffraction data to a minimum Bragg spacing of 2.0 A onPX 14.2 at the Synchrotron Radiation Source, Daresbury(UK). The crystals have the symmetry of the space group,P3121, with cell dimensions of aZbZ91.98 A and cZ257.65 A. There are two complexes in the asymmetric unitwith solvent content of about 48%. Data reduction usingthe program TRUNCATE51 estimated an overall B-factorof 21.7 A2/Da from the Wilson plot. Details of dataprocessing statistics are presented in Table 1.
Phase determination
The solution for the hRI–EDN complex structure wasfound by the molecular replacement method using theprogram AMoRe.52 hRI from a previously solved struc-ture of the hRI–Ang complex (PDB code 1A4Y)12 wasused as the search model. This resulted in distinctsolutions in both the rotation and translation searchesfor the two hRI molecules in the asymmetric unit. Rigidbody refinement of these two solutions in AMoRe, fordata between 10 A and 3.0 A, gave an R-factor of 46.6%and a correlation coefficient of 54.8%. The search for thetwo EDNmolecules, based on the native EDN structure at0.98 A as the search model (PDB code 1GQV),19 did notyield any clear solutions in AMoRe. Therefore, an electrondensity map was calculated using phase informationfrom the two hRI molecules. Examination of the mapsrevealed the location of the two EDN molecules in theasymmetric unit. A poly-alanine model was built into thedensity based on the interaction between Asp435 of hRIand Lys38 of EDN, as presumed from the hRI–Angstructure.
Refinement
Crystallographic refinement was carried out at 2.0 Aresolution against 91.2% of the measured data using theprogram CNS.53 A random set of reflections of 3.8% wasexcluded from the full data set for cross-validationpurposes by calculating the free R-factor (Rfree) to monitorthe refinement trend.54 The initial round of refinementwith the two hRI molecules and poly-alanine chains forthe two EDN molecules resulted in an Rcryst of 41.6% andan Rfree of 44.7%. Once most of the alanine residues werereplaced with the original EDN residues in the poly-alanine model and the model subjected to simulatedannealing, the Rcryst and the Rfree dropped to 31% and34.5%, respectively. Further refinement involving con-secutive cycles of energy minimisation, individual tem-perature factor (B-factor) refinement and simulatedannealing as implemented in CNS,53 alternated withmanual model building with the program O55 byreference to 2FoKFc and FoKFc maps progressivelyimproved the structure. In the final stages of refinement,water molecules with peaks greater than 3s in the FoKFcmaps were incorporated into the structure if theywere within hydrogen bond-forming distances fromappropriate atoms.The final refined structure at 2.0 A resolution has an
Rcryst of 20.6% and an Rfree of 24.3%. Chain A (hRI) andchain C (EDN) form one complex (Complex 1) and theother complex in the asymmetric unit (Complex 2) isformed by chain B (hRI) and chain D (EDN). The first fourresidues in chain A and the first three in chain B aremissing from the structure because of the lack of visible

EDN in Complex with Placental Ribonuclease Inhibitor 653
density. Residues 5, 13, 440, 447, 451 and 452 of both hRImolecules, residues 340, 419, 422 and 450 of chain A andresidues 4, 10, 45, 55, 85, 140, 237, 270, 351, 358 and 441 ofchain B have been modelled as alanine residues becauseof poor electron density. Similarly, residues 66, 69, 91 and117 of chain C (EDN) and residues 35, 53, 65, 66, 67, 69,116, 117 and 134 of chain D (EDN) have also beenrepresented as alanine residues. Analysis of theRamachandran plot using the program PROCHECK56
indicated that 83.7% of the residues lie in the mostfavourable region of the f–j plot and about 16% lie in theadditional allowed regions. Glu353 from both hRImolecules lies in the disallowed region. Inspection ofthese residues with the program O55 showed no obviousdistortion of geometry and they fit perfectly within theobserved density. Difference density for one molecule ofglycerol and one molecule of malonate ion were observedduring model building. The glycerol molecule makeshydrogen-bonding interactions with Ser105 and Arg109of hRI in Complex 1. The malonate binds at the interfaceof Complex 2, where it forms two direct hydrogen bondswith Glu264 of hRI and water-mediated interactions withthis same residue and with Asn84 and Thr101 of EDN.Details of the refinement statistics are given in Table 1.
Kinetics
The hRI variants W375A, E401R, W438A/S439A/E440A, and R457A were available from studies.13,15,29
The EDN preparation used for kinetic studies waspurified from human urine as described.57 Values of Ki
for EDN–inhibitor complexes were calculated from theindividual rate constants for association and dissociation(ka and kd, respectively; KiZkd/ka). The rate constantswere measured as reported.17 Briefly, kd values weredetermined by forming the complex, then adding excessAng as a scavenger for free inhibitor and following theappearance of free EDN by assaying for enzymaticactivity (Ang has no significant activity in the assayused). Values of ka were determined by monitoring theonset of inhibition of EDN activity in a highly sensitivefluorescence-based assay.
Protein Data Bank accession codes
The atomic coordinates and the structure factors of thehRI–EDN complex have been deposited with the RCSBProtein Data Bank (accession codes 2BEX and R2BEXSF,respectively).
Acknowledgements
We thank the scientists at station PX 14.2,Synchrotron Radiation Source, Daresbury (UK)and the EMBL X11 beamline at the DORIS storagering, DESY, Hamburg (Germany) for their supportduring X-ray data collection. This work wassupported by the Wellcome Trust (UK) programmegrant (067288) to K.R.A. and National Institutes ofHealth grant (CA-88738) to R.S. We would also liketo acknowledge the support of the EuropeanCommunity, Research Infrastructure Action underthe FP6 “Structuring the European Research AreaSpecific Programme” to the EMBL, Hamburg Out-station, contract number RII3-CT-2004–506008. The
authors declare that they do not have conflictingfinancial interests.
References
1. Lee, F. S. & Vallee, B. L. (1993). Structure and action ofmammalian ribonuclease (angiogenin) inhibitor. Prog.Nucl. Acid. Res. Mol. Biol. 44, 1–30.
2. Hofsteenge, J. (1997). Ribonuclease inhibitor. InRibonuclease Structures and Functions (D’Alessio, G. &Riordan, J. F., eds), pp. 621–658, Academic Press, NewYork.
3. Shapiro, R. (2001). Cytoplasmic ribonucleaseinhibitor. Methods Enzymol. 341, 611–628.
4. Lee, F. S., Shapiro, R. & Vallee, B. L. (1989). Tight-binding inhibition of Ang and ribonuclease A byplacental ribonuclease inhibitor. Biochemistry, 28,225–230.
5. Hutton, M., Willenbrock, F., Brocklehurst, K. &Murphy, G. (1998). Kinetic analysis of the mechanismof interaction of full-length TIMP-2 and gelatinase A:evidence for the existence of a low-affinity inter-mediate. Biochemistry, 37, 10094–10098.
6. Brew, K., Dinakarpandian, D. & Nagase, H. (2000).Tissue inhibitors of metalloproteinases: evolution,structure and function. Biochim. Biophys. Acta, 1477,277–283.
7. Schreiber, G. & Fersht, A. R. (1993). Interaction ofbarnase with its polypeptide inhibitor barstar studiedby protein engineering. Biochemistry, 32, 5145–5150.
8. Vincent, J. P. & Lazdunski, M. (1972). Trypsin–pancreatic trypsin inhibitor association. Dynamics ofthe interaction and the role of disulfide bridges.Biochemistry, 11, 2967–2977.
9. Wallis, R., Moore, G. R., James, R. & Kleanthous, C.(1995). Protein–protein interactions in colicin E9DNase-immunity protein complexes 1. Diffusion-controlled association and femtomolar binding forthe cognate complex. Biochemistry, 34, 13743–13750.
10. Kobe, B. & Deisenhofer, J. (1993). Crystal structure ofporcine ribonuclease inhibitor, a protein with leucine-rich repeats. Nature, 366, 751–756.
11. Kobe, B. & Deisenhofer, J. (1995). A structural basis ofthe interactions between leucine-rich repeats andprotein ligands. Nature, 374, 183–186.
12. Papageorgiou, A. C., Shapiro, R. & Acharya, K. R.(1997). Molecular recognition of human Ang byplacental ribonuclease inhibitor—an X-ray crystal-lographic study at 2.0 A resolution. EMBO. J. 16,5162–5177.
13. Chen, C. Z. & Shapiro, R. (1997). Site-specificmutagenesis reveals differences in the structuralbases for tight binding of RNase inhibitor to Angand RNase A. Proc. Natl Acad. Sci. USA, 94, 1761–1766.
14. Chen, C. Z. & Shapiro, R. (1999). Superadditive andsubadditive effects of hot spot mutations within theinterfaces of placental ribonuclease inhibitor withAng and ribonuclease A. Biochemistry, 38, 9273–9285.
15. Shapiro, R., Ruiz-Gutierrez, M. & Chen, C. Z. (2000).Analysis of the interactions of human ribonucleaseinhibitor with Ang and ribonuclease A by muta-genesis: importance of inhibitor residues insideversus outside the C-terminal hot spot. J. Mol. Biol.302, 497–519.
16. Clackson, T. & Wells, J. A. (1995). A hot spot ofbinding energy in a hormone–receptor interface.Science, 267, 383–386.
17. Teufel, D. P., Kao, R. Y., Acharya, K. R. & Shapiro, R.

654 EDN in Complex with Placental Ribonuclease Inhibitor
(2003). Mutational analysis of the complex of humanRNase inhibitor and human eosinophil-derivedneurotoxin (RNase 2). Biochemistry, 42, 1451–1459.
18. Kobe, B. & Deisenhofer, J. (1996). Mechanism ofribonuclease inhibition by ribonuclease inhibitorprotein based on the crystal structure of its complexwith ribonuclease A. J. Mol. Biol. 264, 1028–1043.
19. Swaminathan, G. J., Holloway, D. E., Veluraja, K. &Acharya, K. R. (2002). Atomic resolution (0.98 A)structure of eosinophil-derived neurotoxin.Biochemistry, 41, 3341–3352.
20. Lawrence, M. C. & Colman, P. M. (1993). Shapecomplementarity at protein/protein interfaces. J. Mol.Biol. 234, 946–950.
21. Mosimann, S. C., Newton, D. L., Youle, R. J. & James,M. N. (1996). X-ray crystallographic structure ofrecombinant eosinophil-derived neurotoxin at 1.83 Aresolution. J. Mol. Biol. 260, 540–552.
22. Leonidas, D. D., Boix, E., Prill, R., Suzuki, M., Turton,R., Minson, K. et al. (2001). Mapping the ribonucleo-lytic active site of eosinophil-derived neurotoxin(EDN). High resolution crystal structures of EDNcomplexes with adenylic nucleotide inhibitors. J. Biol.Chem. 276, 15009–15017.
23. Blackburn, P., Wilson, G. & Moore, S. (1977). Ribo-nuclease inhibitor from human placenta. Purificationand properties. J. Biol. Chem. 252, 5904–5910.
24. Shapiro, R. & Vallee, B. L. (1987). Human placentalribonuclease inhibitor abolishes both angiogenic andribonucleolytic activities of angiogenin. Proc. NatlAcad. Sci. USA, 84, 2238–2241.
25. Shapiro, R. & Vallee, B. L. (1991). Interaction of humanplacental ribonuclease with placental ribonucleaseinhibitor. Biochemistry, 30, 2246–2255.
26. Shapiro, R., Fett, J. W., Strydom, D. J. & Vallee, B. L.(1986). Isolation and characterization of a humancolon carcinoma-secreted enzyme with pancreaticribonuclease-like activity. Biochemistry, 25, 7255–7264.
27. Beintema, J. J., Breukelman, H. J., Carsana, A. & Furia,A. (1997). Evolution of vertebrate ribonucleases:ribonuclease A superfamily. In Ribonucleases:Structures and Functions (D’Alessio, G. & Riordan,J. F., eds), pp. 245–269, Academic Press, New York.
28. Shapiro, R., Riordan, J. F. & Vallee, B. L. (1995).LRRning the RIte of springs. Nature Struct. Biol. 2,350–354.
29. Kumar, K., Brady, M. & Shapiro, R. (2004). Selectiveabolition of pancreatic RNase binding to its inhibitorprotein. Proc. Natl Acad. Sci. USA, 101, 53–58.
30. Klon, A. E., Glick, M., Thoma, M., Acklin, P. & Davies,J. W. (2004). Finding more needles in the haystack: asimple and efficient method for improving high-throughput docking results. J. Med. Chem. 47,2743–2749.
31. Richards, F. M. & Wyckoff, H. W. (1971). Bovinepancreatic ribonuclease. In The Enzymes, (Boyer, P. D.,ed), 3rd Edit., vol. 4, pp. 647–806, Academic Press,New York.
32. Blackburn, P. & Moore, S. (1962). Pancreatic ribo-nuclease. In The Enzymes, 3rd edit., vol. 15,pp. 317–433, Academic Press, Orlando.
33. Fett, J. W., Strydom, D. J., Lobb, R. R., Alderman,E. M., Bethune, J. L., Riordan, J. F. & Vallee, B. L.(1985). Isolation and characterization of angiogenin,an angiogenic protein from human carcinoma cells.Biochemistry, 24, 5480–5486.
34. Domachowske, J. B., Dyer, K. D., Bonville, C. A. &Rosenberg, H. F. (1998). Recombinant human
eosinophil-derived neurotoxin/RNase 2 functions asan effective antiviral agent against respiratory syncy-tial virus. J. Infect. Dis. 177, 1458–1464.
35. Lehrer, R. A., Szklarek, D., Barton, A., Ganz, T.,Hamann, K. J. & Gleich, G. J. (1989). Antibacterialproperties of eosinophil major basic protein andeosinophil cationic protein. J. Immunol. 142,4428–4434.
36. Ackerman, S. J., Gleich, G. J., Loegering, D. A.,Richardson, B. A. & Butterworth, A. E. (1985).Comparative toxicity of purified human eosinophilgranule cationic proteins for schistosomula ofSchistosoma mansoni. Am. J. Trop. Med. Hyg. 34,735–745.
37. Hamann, K. J., Barker, R. L., Loegering, D. A. &Gleich, G. J. (1987). Comparative toxicity of purifiedhuman eosinophil granule proteins for newbornlarvae of Trichinella spiralis. J. Parasitol. 73, 523–529.
38. Yang, D., Chen, Q., Rosenberg, H. F., Rybak, S. M.,Newton, D. L., Wang, Z. Y. et al. (2004). Humanribonuclease A superfamily members, eosinophil-derived neurotoxin and pancreatic ribonuclease,induce dendritic cell maturation and activation.J. Immunol. 173, 6134–6142.
39. Olson, K. A., Fett, J. W., French, T. C., Key, M. E. &Vallee, B. L. (1995). Angiogenin antagonists preventtumour growth in vivo. Proc. Natl Acad. Sci. USA, 92,442–446.
40. Olson, K. A., Byers, H. R., Key, M. E. & Fett, J. W.(2001). Prevention of human prostate tumour meta-stasis in athymic mice by antisense targeting ofhuman angiogenin. Clin. Cancer Res. 7, 3598–3605.
41. Durack, D. T., Ackerman, S. J., Loegering, D. A. &Gleich, G. J. (1981). Purification of human eosinophil-derived neurotoxin. Proc. Natl Acad. Sci. USA, 78,5165–5169.
42. Fredens, K., Dahl, R. & Venge, P. (1982). The Gordonphenomenon induced by the eosinophil cationicprotein and eosinophil protein X. J. Allergy. Clin.Immunol. 70, 361–366.
43. Moore, P. M., Harley, J. B. & Fauci, A. S. (1985).Neurologic dysfunction in the idiopathic hyper-eosinophilic syndrome. Ann. Intern. Med. 102,109–114.
44. Prick, J. J. W., Gabreels-Festen, A. A. W. M., Korten,J. J. & van der Wiel, T. W. M. (1988). Neurologicmanifestations of the hypereosinophilic syndrome(HES). Neurol. Neurosurg. 90, 269–273.
45. Gleich, G. J., Kita, H. & Adolfson, C. R. (1995).Eosinophils. In Samter’s Immunologic Diseases (Frank,M. M., Austen, K. F., Claman, H. N. & Unanue, E. R.,eds), 5th edit., vol. 1, pp. 205–245, Brown and Co.,Boston.
46. Sorrentino, S., Glitz, D. G., Hamann, K. J., Loegering,D. A., Checkel, J. L. & Gleich, G. J. (1992). Eosinophil-derived neurotoxin and human liver ribonuclease.Identity of structure and linkage of neurotoxicity tonuclease activity. J. Biol. Chem. 267, 14859–14865.
47. Shapiro, R., Fox, E. A. & Riordan, J. F. (1989). Role oflysines in human angiogenin: chemical modificationand site-directed mutagenesis. Biochemistry, 28,1726–1732.
48. Kao, R. Y., Jenkins, J. L., Olson, K. A., Key, M. E., Fett,J. W. & Shapiro, R. (2002). A small molecule inhibitorof the ribonucleolytic activity of human angiogeninthat possesses antitumor activity. Proc. Natl Acad. Sci.USA, 99, 10066–10071.
49. Jenkins, J. L. & Shapiro, R. (2003). Identification ofsmall-molecule inhibitors of human angiogenin and

EDN in Complex with Placental Ribonuclease Inhibitor 655
characterization of their binding interactions guidedby computational docking. Biochemistry, 42,6674–6687.
50. Kobe, B., Ma, Z. & Deisenhofer, J. (1994). Complexbetween bovine ribonuclease A and porcine ribo-nuclease inhibitor crystallizes in a similar unit cell asfree ribonuclease inhibitor. J. Mol. Biol. 241, 288–291.
51. CCP4. (1994). The CCP4 suite: programs for proteincrystallography. Acta Crystallog. sect. D, 50, 760–763.
52. Navaza, J. (1994). AMoRe: an automated package formolecular replacement. Acta Crystallog. sect. A, 50,157–163.
53. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano,W. L., Gros, P., GrosseKunstleve, R. W. et al. (1998).Crystallography and NMR system: a new softwaresuite for macromolecular structure determination.Acta Crystallog. sect. D, 54, 905–921.
54. Brunger, A. T. (1992). The free R-value: a novelstatistical quantity for assessing the accuracy ofcrystal structures. Nature, 355, 472–474.
55. Jones, T. A., Zhou, J. Y., Cowman, S. W. & Kjeldgaard,M. (1991). Improved methods for building models inelectron-density maps and the location of errors inthese models. Acta Crystallog. sect. A, 47, 110–119.
56. Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK—a program tocheck the stereochemical quality of protein structures.J. Appl. Crystallog. 26, 283–291.
57. Russo, N. & Shapiro, R. (1999). Potent inhibition of
mammalian ribonucleases by 3 0, 5 0-pyrophosphate-linked nucleotides. J. Biol. Chem. 274, 14902–14908.
58. Kraulis, P. J. (1991). MOLSCRIPT—a program toproduce both detailed and schematic plots of proteinstructures. J. Appl. Crystallog. 24, 946–950.
59. Nicholls, A. & Honig, B. (1991). A rapid finite-difference algorithm, utilising successive over-relaxation to solve the Poisson–Boltzmann equation.J. Comput. Chem. 12, 435–445.
60. Barton, G. J. (1993). ALSCRIPT: a tool to formatmultiple sequence alignments. Protein Eng. 6, 37–40.
61. Esnouf, R. M. (1997). An extensively modified versionof Molscript that includes greatly enhanced colouringcapabilities. J. Mol. Graph. Model. 15, 132–134.
62. McDonald, I. K. & Thornton, J. M. (1994). Satisfyinghydrogen bonding potential in proteins. J. Mol. Biol.238, 777–793.
63. Brunger, A. T. (1992). XPLOR Manual Version 3.1, YaleUniversity, New Haven, CT.
64. Vincentini, A. M., Kieffer, B., Matthies, R., Meyhack,B., Hemmings, B. A., Stone, S. R. & Hofsteenge, J.(1990). Protein chemical and kinetic characterizationof recombinant porcine ribonuclease inhibitorexpressed in Saccharomyces cerevisiae. Biochemistry, 29,8827–8834.
65. Kabsch, W. & Sander, C. (1983). Dictionary of proteinsecondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22,2577–2637.
Edited by M. Guss
(Received 8 December 2004; received in revised form 8 January 2005; accepted 13 January 2005)




















