
The refined structure of ribonuclease-A at 1.45 � resolution
Upload
independentCategory
view
0download
0
proteinsSTRUCTURE O FUNCTION O BIOINFORMATICS
Computational studies of the structure,dynamics and native content of amyloid-likefibrils of ribonuclease AGiorgio Colombo,1* Massimiliano Meli,1 and Alfonso De Simone2
1 Istituto di Chimica del Riconoscimento Molecolare, CNR, Via Mario Bianco, 9, 20131 Milano, Italy
2Dipartimento delle Scienze Biologiche, Sezione Biostrutture, Universit�a di Napoli Federico II, Via Mezzocannone 16,
I-80134 Naples, Italy
INTRODUCTION
The self-assembly of proteins and peptides into ordered amyloid
fibrils is associated with the evolution of more than 20 syndromes
in humans and animals.1–6 The conversion to amyloid aggregates
is however not restricted to disease-related sequences but appears
to be a general property of polypeptides under appropriate condi-
tions.7–11 Amyloid fibrils share a number of similarities that
include a common cross-b diffraction pattern typical of an elon-
gated stack of b-strands perpendicular to the fibril axis and b-sheets parallel to the axis.
Despite important progress,12–17 the fibrous nature of the
aggregates has hampered the experimental characterization of
many fundamental biophysical properties of the amyloid
state,17,18 and important issues regarding the structural and dy-
namical properties of polypeptides in fibrils are still open. More-
over, there is little consensus on whether native-like domains are
conserved or refolded in the final amyloid state and whether these
domains may retain their catalytic activity.
In this context, Sambashivan et al.19 showed that a designed
amyloid-like fibril of ribonuclease A (RNase A) could contain
native-like molecules capable of enzymatic activity. The amyloid
fibril was engineered starting from the structure of the C-terminal
domain swapped RNase A dimer, expanding with an amyloido-
genic segment (GQ10G) the hinge loop (residues 112–115) con-
necting the core domain (residues 1–111) with the swapped do-
main (residues 116–124). In this model, the C-terminal strand of
each RNase A monomer breaks its noncovalent connections to the
core domain, exposing the Q10 segment, and the C-terminal
strand of another monomer swaps in to take its place. The Q10
stretches from two domain-swapped monomers form an antiparal-
lel b-sheet, which is then complemented by an analogous antipar-
allel b-sheet formed by two more domain-swapped monomers.19
Self-recognition of proteins is completed by precise complementation
The Supplementary Material referred to in this article can be found online at http://www.
interscience.wiley.com/jpages/0887-3585/suppmat/
*Correspondence to: Giorgio Colombo, Istituto di Chimica del Riconoscimento Molecolare,
CNR, Via Mario Bianco, 9, 20131 Milano, Italy. E-mail: [email protected]
Received 20 March 2007; Revised 11 May 2007; Accepted 23 May 2007
Published online 5 September 2007 in Wiley InterScience (www.interscience.wiley.com).
DOI: 10.1002/prot.21648
ABSTRACT
The characterization at atomic resolution of amyloid-
like protein aggregates is one of the fundamental
problems of modern biology. In particular, the ques-
tion whether native-like domains are retained or com-
pletely refolded in the amyloid state and the identifi-
cation of possible mechanisms for macromolecular or-
dered aggregation represent major unresolved puzzles.
To address these issues, in this article we examine the
stability, dynamics, and conservation of native-like
properties of several models of a previously designed
amyloid-like fibril of RNase A (Sambashivan et al.,
Nature 2005; 437:266–269). Through the use of molec-
ular dynamics (MD) simulations, we have provided
molecular-level insights into the role of different parts
of the sequence on the stability of fibrils, the collective
properties of supramolecular complexes, and the pres-
ence of native-like conformations and dynamics in
supramolecular aggregates. We have been able to
show that within the fibrils the three-dimensional
globular domain-swapped units preserve the confor-
mational, dynamical, and hydration properties typical
of the monomeric state, providing a rationalization
for the experimentally observed catalytic activity of
fibrils. The nativeness of the globular domains is not
affected by the amyloidogenic stretches, which deter-
mine the molecular recognition process underlying
aggregation through the formation of a stable steric
zipper motif. Moreover, through the study of the
hydration features of a single sheet model, we have
been able to show that polyglutamine stretches of the
domain-swapped ribonuclease tend to minimize the
interaction with water in favor of sidechain–sidechain
interactions, shedding light on the factors leading to
the supramolecular assembly of b-sheet layers into dry
steric zippers.
Proteins 2008; 70:863–872.VVC 2007 Wiley-Liss, Inc.
Key words: amyloid formation; fibrils; folding mis-
folding; molecular recognition; molecular dynamics;
aggregation.
VVC 2007 WILEY-LISS, INC. PROTEINS 863

of the Q10 side chains stacking in the so-called zipper
spine motif.17
This model was used to explain the conservation of
RNase A enzymatic activity in the fibrillar state through
the formation of complemented active sites via domain-
swapping, implying in turn that fibrils can contain
native-like functional units. However, an experimental
atomic resolution structure is not yet available so that
one has to turn to computational-theoretical methods to
investigate the stability, dynamics, and conservation of
native-like properties of the globular domains in the
fibril model.
To obtain atomic-level resolution insights into these
factors, we have used atomistic molecular dynamics
(MD) simulations in explicit water of several models of
different sizes of the designed RNase A fibrils. We focus
on the role of different parts of the sequence on the sta-
bility of aggregates, the collective properties of supramo-
lecular complexes, and the presence of catalytically active
conformations and motions. Moreover, we analyze the
(de)hydration properties of the cross-b-spine motif and
of the globular domains to gain information on the fac-
tors leading to the supramolecular assembly of b-sheetlayers into dry steric zippers, observed in X-ray studies of
related amyloidogenic sequences.
MATERIALS AND METHODS
The starting structures for the all-atom MD simula-
tions of Q10 RNase A were taken from the Protein Data
Bank with accession numbers 2APQ and 2APU.19 The
former, consisting of a domain-swapped dimer was used
as a starting structure for simulation f2, the latter, con-
taining a model of a whole fibril, was used to construct
the starting structures for simulations f4, f8, and f16.
In all simulations, to mimic proper solution condi-
tions, Lysine amino groups were considered protonated,
while the carboxyl groups were considered to bear a neg-
ative charge. The systems were solvated in a parallel-
piped-shaped box large enough to contain 0.6 nm of sol-
vent around each aggregate. For the single sheet octamer
simulation, the distance between the protein and the box
edge was extended to 1.2 nm. The simple point charge
(SPC) water model was used20 to solvate each protein in
the simulation box. The SPCE flexible water model was
used for the single sheet octamer. Each system was subse-
quently energy minimized with a steepest descent
method for 5000 steps. The calculation of electrostatic
forces utilized the PME implementation of the Ewald
summation method. The LINCS21 algorithm was used to
constrain all bond lengths. For the water molecules, the
SETTLE algorithm22 was used. A dielectric permittivity,
E 5 1, and a time step of 2 fs were used. All atoms were
given an initial velocity obtained from a Maxwellian dis-
tribution at the desired initial temperature of 300 K. The
density of the system was adjusted to perform the first
equilibration runs at NPT (constant Number of Particles,
Pressure, Temperature) condition by weak coupling to a
bath of constant pressure (P0 5 1 bar, coupling time sP5 0.5 ps).23 In all simulations, the temperature was
maintained close to the intended values by weak coupling
to an external temperature bath23 with a coupling con-
stant of 0.1 ps. The proteins and the rest of the system
were coupled separately to the temperature bath. Table I
summarizes the simulation conditions and the number of
atoms for each simulation.
In all cases, the proteins were simulated at 300 K for
50 ns, and all simulations were run at NPT conditions.
All simulations and analysis were carried out using the
GROMACS package (version 3.3),24,25 using the GRO-
MOS96 43A1 force field.26,27 All calculations were per-
formed on clusters of PCs, with Linux operating system.
Graphical display of structures was done using the
PyMOL software.
Water density function calculation
Our hydration analysis is largely based on the solvent
density map whose maxima are assumed to be the mo-
lecular dynamics hydration sites (MDHS).28 The space
surrounding the protein is divided in two shells: the first
shell describes the water within a distance of 0.6 nm
from the protein surface. The second shell extends from
0.6 to 0.8 nm from the protein surface and represents the
bulk solvent shell. The solvent density calculation is grid
based (step-size 0.05 nm). To eliminate protein transla-
tion and rotation, the coordinates of each frame are
transformed by superimposing the current model onto a
reference one. To prevent sweeping effects due to back-
bone flexibility, a selection of frames based on Ca-RMSD
is adopted; therefore, only structures with a Ca-RMSD,
from the reference set, lower than the cutoff of 0.09 nm
are considered.
Time autocorrelation functionand residence time
The time autocorrelation function P(s) is adopted to
provide the residence time of a given interaction. The
function is calculated for each residue to evaluate interac-
Table ISummary of Simulation Conditions and Number of Atoms
SimulationNumberof chains
Numberof atoms
Solventmolecules
Length(ns)
f2 2 36,590 1280 50f4 4 61,885 8795 50f8 8 97,991 28,997 50f16 16 151,572 42,525 50
G. Colombo et al.
864 PROTEINS DOI 10.1002/prot

tions with the remaining residues or with water. The for-
mula is
PðsÞ ¼X
t
d W ðtÞ;W ðt þ sÞð Þ
where the delta function d(W(t), W(t 1 s)) assigns 0 or
1, respectively, whether the indexes of the partners at
times t and t 1 s differ or not. The resulting time auto-
correlation function is then fitted by an exponential
model.
RESULTS
The different fibril models analyzed herein consist of
2, 4, 8, and 16 chains. Studying constructs of increasing
dimensions may yield information on the properties of
growing aggregates. In the models with 2 and 4 chains, a
single b-sheet is formed by the alignment of Q10 amyloi-
dogenic segments from domain swapped RNase A mole-
cules. In the models with 8 and 16 chains, a full cross-b-spine formed by the juxtaposition of two b-sheets is con-sidered (Figure 1, Table I). All the MD runs used for
analysis are 50 ns long and are labeled according to the
number of protein chains in the fibril model. Simulations
f2, f4, f8, and f16 are thus run on models consisting of
two, four, eight, and sixteen chains, respectively. One
more simulation of a single sheet of eight monomers was
run to analyze the possible role of hydration in the
aggregation mechanism.
Structural and dynamical properties
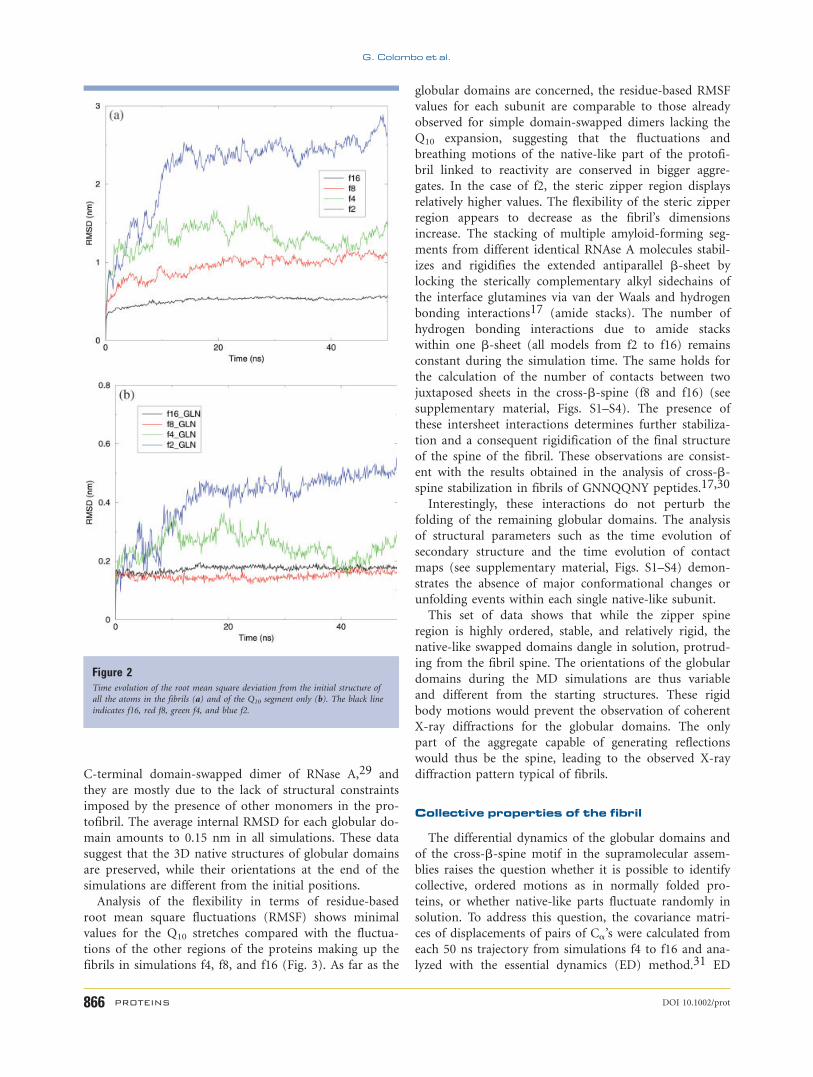
Figure 2(a) shows the time evolution of the Ca atom
root mean square deviation (RMSD) of the simulated
structures with respect to the initial X-ray based model.
The RMSD calculated over all the Ca’s of the proteins in
each construct increases in time with an inverse propor-
tionality to the number of chains in the system: simula-
tion f2 reaches the highest value, followed by f4, f8, and
f16.
Restricting the RMSD calculation only to the Q10 seg-
ments of the spine [Fig. 2(b)] shows that these regions
remain close to the starting geometry for simulations f4
to f16. In f4, the RMSD initially increases to around 0.3
nm followed by a decrease to 0.17 nm. In the cases of f8
and f16, the steric zipper RMSD value is consistently sta-
ble around 0.17 and 0.15 nm for the whole simulation
time. Only in the case of f2 does the RMSD of Q10
diverge to values as high as 0.5 nm due to the high flexi-
bility characterizing the dimer. Large quaternary structure
fluctuations were already noted in the case of the original
Figure 1Structures of the protofibril simulated models. f2 and f4 are a single b-sheet formed by the alignment of Q10 amyloidogenic segments from domain swapped RNase A
molecules. In f4 the Q10 segment is displayed using the representation of van der Waals radii of the atoms. In the models with 8 and 16 chains, f8 and f16, a full cross-b-spine is considered. In f16 the Q10 segment is displayed using the representation of van der Waals radii of the atoms. [Color figure can be viewed in the online issue,
which is available at www.interscience.wiley.com.]
MD Simulations of RNase A Fibrils
DOI 10.1002/prot PROTEINS 865
C-terminal domain-swapped dimer of RNase A,29 and
they are mostly due to the lack of structural constraints
imposed by the presence of other monomers in the pro-
tofibril. The average internal RMSD for each globular do-
main amounts to 0.15 nm in all simulations. These data
suggest that the 3D native structures of globular domains
are preserved, while their orientations at the end of the
simulations are different from the initial positions.
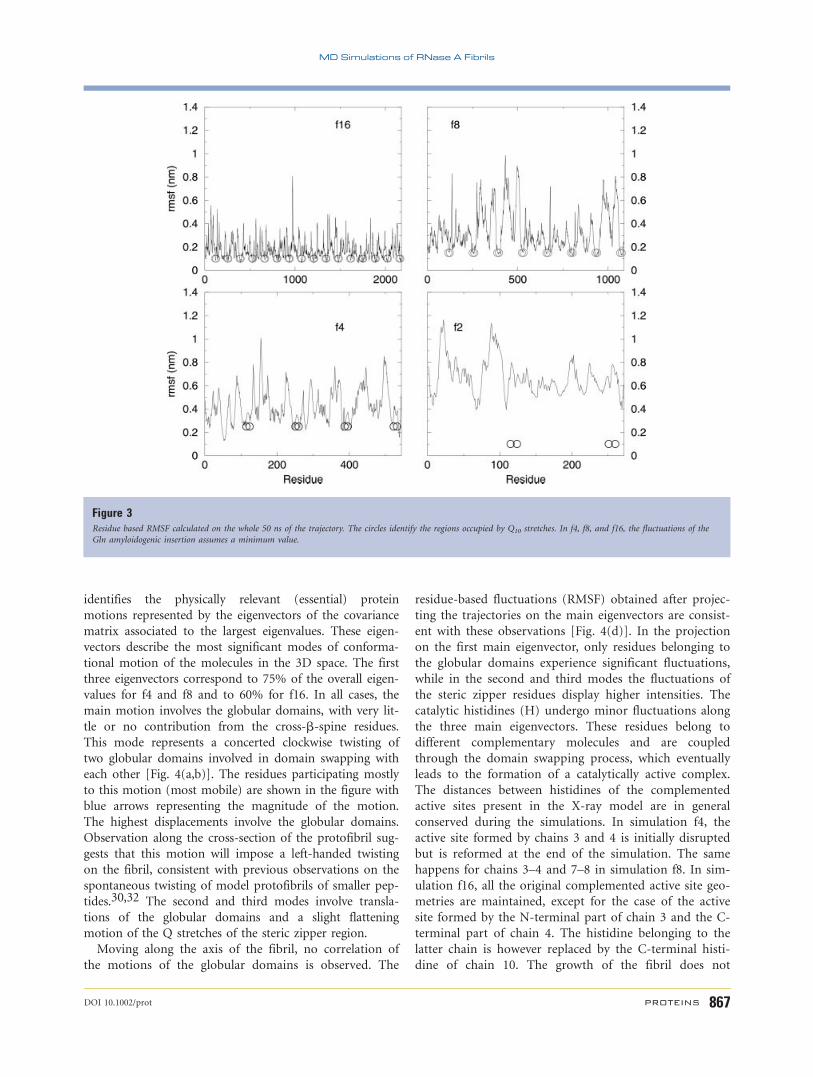
Analysis of the flexibility in terms of residue-based
root mean square fluctuations (RMSF) shows minimal
values for the Q10 stretches compared with the fluctua-
tions of the other regions of the proteins making up the
fibrils in simulations f4, f8, and f16 (Fig. 3). As far as the
globular domains are concerned, the residue-based RMSF
values for each subunit are comparable to those already
observed for simple domain-swapped dimers lacking the
Q10 expansion, suggesting that the fluctuations and
breathing motions of the native-like part of the protofi-
bril linked to reactivity are conserved in bigger aggre-
gates. In the case of f2, the steric zipper region displays
relatively higher values. The flexibility of the steric zipper
region appears to decrease as the fibril’s dimensions
increase. The stacking of multiple amyloid-forming seg-
ments from different identical RNAse A molecules stabil-
izes and rigidifies the extended antiparallel b-sheet by
locking the sterically complementary alkyl sidechains of
the interface glutamines via van der Waals and hydrogen
bonding interactions17 (amide stacks). The number of
hydrogen bonding interactions due to amide stacks
within one b-sheet (all models from f2 to f16) remains
constant during the simulation time. The same holds for
the calculation of the number of contacts between two
juxtaposed sheets in the cross-b-spine (f8 and f16) (see
supplementary material, Figs. S1–S4). The presence of
these intersheet interactions determines further stabiliza-
tion and a consequent rigidification of the final structure
of the spine of the fibril. These observations are consist-
ent with the results obtained in the analysis of cross-b-spine stabilization in fibrils of GNNQQNY peptides.17,30
Interestingly, these interactions do not perturb the
folding of the remaining globular domains. The analysis
of structural parameters such as the time evolution of
secondary structure and the time evolution of contact
maps (see supplementary material, Figs. S1–S4) demon-
strates the absence of major conformational changes or
unfolding events within each single native-like subunit.
This set of data shows that while the zipper spine
region is highly ordered, stable, and relatively rigid, the
native-like swapped domains dangle in solution, protrud-
ing from the fibril spine. The orientations of the globular
domains during the MD simulations are thus variable
and different from the starting structures. These rigid
body motions would prevent the observation of coherent
X-ray diffractions for the globular domains. The only
part of the aggregate capable of generating reflections
would thus be the spine, leading to the observed X-ray
diffraction pattern typical of fibrils.
Collective properties of the fibril
The differential dynamics of the globular domains and
of the cross-b-spine motif in the supramolecular assem-
blies raises the question whether it is possible to identify
collective, ordered motions as in normally folded pro-
teins, or whether native-like parts fluctuate randomly in
solution. To address this question, the covariance matri-
ces of displacements of pairs of Ca’s were calculated from
each 50 ns trajectory from simulations f4 to f16 and ana-
lyzed with the essential dynamics (ED) method.31 ED
Figure 2Time evolution of the root mean square deviation from the initial structure of
all the atoms in the fibrils (a) and of the Q10 segment only (b). The black line
indicates f16, red f8, green f4, and blue f2.
G. Colombo et al.
866 PROTEINS DOI 10.1002/prot
identifies the physically relevant (essential) protein
motions represented by the eigenvectors of the covariance
matrix associated to the largest eigenvalues. These eigen-
vectors describe the most significant modes of conforma-
tional motion of the molecules in the 3D space. The first
three eigenvectors correspond to 75% of the overall eigen-
values for f4 and f8 and to 60% for f16. In all cases, the
main motion involves the globular domains, with very lit-
tle or no contribution from the cross-b-spine residues.
This mode represents a concerted clockwise twisting of
two globular domains involved in domain swapping with
each other [Fig. 4(a,b)]. The residues participating mostly
to this motion (most mobile) are shown in the figure with
blue arrows representing the magnitude of the motion.
The highest displacements involve the globular domains.
Observation along the cross-section of the protofibril sug-
gests that this motion will impose a left-handed twisting
on the fibril, consistent with previous observations on the
spontaneous twisting of model protofibrils of smaller pep-
tides.30,32 The second and third modes involve transla-
tions of the globular domains and a slight flattening
motion of the Q stretches of the steric zipper region.
Moving along the axis of the fibril, no correlation of
the motions of the globular domains is observed. The
residue-based fluctuations (RMSF) obtained after projec-
ting the trajectories on the main eigenvectors are consist-
ent with these observations [Fig. 4(d)]. In the projection
on the first main eigenvector, only residues belonging to
the globular domains experience significant fluctuations,
while in the second and third modes the fluctuations of
the steric zipper residues display higher intensities. The
catalytic histidines (H) undergo minor fluctuations along
the three main eigenvectors. These residues belong to
different complementary molecules and are coupled
through the domain swapping process, which eventually
leads to the formation of a catalytically active complex.
The distances between histidines of the complemented
active sites present in the X-ray model are in general
conserved during the simulations. In simulation f4, the
active site formed by chains 3 and 4 is initially disrupted
but is reformed at the end of the simulation. The same
happens for chains 3–4 and 7–8 in simulation f8. In sim-
ulation f16, all the original complemented active site geo-
metries are maintained, except for the case of the active
site formed by the N-terminal part of chain 3 and the C-
terminal part of chain 4. The histidine belonging to the
latter chain is however replaced by the C-terminal histi-
dine of chain 10. The growth of the fibril does not
Figure 3Residue based RMSF calculated on the whole 50 ns of the trajectory. The circles identify the regions occupied by Q10 stretches. In f4, f8, and f16, the fluctuations of the
Gln amyloidogenic insertion assumes a minimum value.
MD Simulations of RNase A Fibrils
DOI 10.1002/prot PROTEINS 867

appear to negatively influence the determinants of enzy-
matic activity.
Interestingly, the principal component for the single
globular units [Fig. 4(c)] show the breathing motions of
the b-sheet regions, which have been identified as funda-
mental for the catalytic activity of the protein in mono-
meric RNase A, C-terminal, and N-terminal domain-
swapped dimers.29,33
Fibril hydration
The analysis of the hydration properties of a single
sheet octamer (representing half of the protofibrillar
unit) was used to investigate the role of water in the for-
mation of a full protofibril. The water density map was
calculated from the atomic coordinates of the protein
and solvent (see methods) collected during a 10-ns simu-
lation; peaks of the density function represent MDHS.28
Remarkable features immediately appear. A uniform dis-
tribution of MDHS is detected on the RNase A globular
domains, whereas the cross-b spine forming region
appears asymmetrically solvated. It has to be noted that
the single sheet presents a uniform plate of Gln side-
chains (polyQ plate) [Fig. 5(a–c)]. In particular, the Q10
surface not involved in the formation of the fibril dis-
plays stable MDHS and appears as a normally hydrated
hydrophilic surface [Fig. 5(a)]. We will refer to this sur-
face as ‘‘internal’’ since it faces the region where the glob-
ular moieties are placed. In contrast, the opposite surface
(‘‘external surface’’), which is supposed to engage the dry
interface in the double layer formation, does not display
any stable MDHS [Fig. 5(b,c)]. Here, in spite of the
hydrogen bonding properties of glutamines, water is
unable to find fixed and stable anchoring points and
undergoes very fast exchange with the bulk phase (see
residence time Table II); as a result this surface appears
on the average to be ‘‘dewetted.’’
Such peculiar behavior is due to sequence and struc-
tural aspects. On average, each Gln of the plate is sur-
rounded, in a 2D network, by four very accessible Gln
neighbors: two stacking on the flanking strands and the
two contiguous Gln on the same strand. This structural
arrangement determines a high crowding of Gln side-
chains, which are favored to interact with one another
through van der Waals packing of the alkyl part of the
side chain and through a complex dynamic network of
Figure 4The directions of the main displacements along the first eigenvector of the covariance matrix are shown as arrows of equal length. For the sake of visualization only a slice
of the whole fibril is represented in two different orientations (a,b). The Q10 appears not to participate in this motion, while the globular parts translate and rotate as
rigid bodies. Subpanel (c) reports the essential breathing motions of the globular part of the structure. These motions are very similar to what was previously observed for
simple monomers or dimers of RNase A. Subpanel (d) shows the residue based RMSF after projecting the trajectories along the first main eigenvector of the covariance
matrix. Small triangles indicate catalytic histidines. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
G. Colombo et al.
868 PROTEINS DOI 10.1002/prot

sidechain–sidechain H-bonds (amide stacks). This has
the effect of subtracting H-bonding donors and acceptors
for protein–water interaction. This crowding and the
consequent intramolecular H-bonding of Gln residues
prevent water molecules to stably localize on the polyQ
surface. Overall the sidechain–sidechain H-bonds have
the effect of disrupting sidechain–water H-bonds and
marginally subtracting H-bonding donors and acceptors
for protein–water interaction (see Table II).
In contrast, the opposite face (internal face) of the
polyQ sheet, although presenting the same 2D network
of Gln, is regularly hydrated because of the surrounding
globular domains. These domains slow down the
exchange between bulk solvent and protein hydration
layer (see residence time Table II). In addition, exposed
residues from the globular domains provide anchoring
points to water molecules, creating an H-bonding net-
work that propagates within the region enclosed by the
polyQ sheet and the globular domains.
Active site hydration
The analysis was extended to the hydration properties
of the globular ribonuclease domains in the fibril. Several
studies have shown that water molecules are important
to preserve the right active site conformation in the
ligand-free form. Brunger et al.34 showed the impor-
tance of Lys7-Lys41 interaction, which is mediated by a
water cluster. Moreover, a comparison between CMP-com-
plexed and ligand-free forms showed that water replaces
the substrate by keeping the active site in the active con-
formation. Our hydration analysis showed that in the
protofibrillar structures analyzed in this work, both the
water cluster that mediates Lys7–Lys41 interaction [Fig.
6(a)] and the pattern of hydration sites that preserves theFigure 5(a) RNase A single sheet octamer solvation map. MD water high-density sites
contoured at 2.5 times the bulk solvent density. The globular domains and the
internal surface appear regularly hydrated, while the external polyQ surface
appears to be dewetted. (b,c) Residence times of sidechain–water Hbonds. The
residence time is evaluated from the time autocorrelation function. The residence
times of Gln’s of the fourth and fifth strand (from the 8-mer single layer) are
reported in panels (b) and (c) respectively. Waters in contact with the external
face are very mobile and exchange rapidly with the bulk solution, whereas
waters in contact with the internal face are slowed down from the globular
moieties. As a result, the residence times present alternate profiles. Even numbers
indicate Gln residues on the external surface, whereas the odd numbers indicate
those on the internal surface. [Color figure can be viewed in the online issue,
which is available at www.interscience.wiley.com.]
Table IIResidence Times and Average Occurrences of Gln-Water and
Gln-Gln Hydrogen Bonds
Internal External
Residence time(ps) H-bonds Gln-H2O 286.04 49.47Average occurrence H-bond Gln-H2O 0.66 1.22
Residence time(ps) H-bonds Gln-Gln 323.73 279.32Average occurrence H-bond Gln-Gln 0.36 0.37
Figure 6Stable hydration sites in the complemented active site of one of the globular
domains of the whole fibril. Hydration is consistent with previous analogous
observations on the monomer.34 [Color figure can be viewed in the online issue,
which is available at www.interscience.wiley.com.]
MD Simulations of RNase A Fibrils
DOI 10.1002/prot PROTEINS 869

right active site conformation of His12 and His119 in the
domain-swapped arrangement are conserved [Fig. 6(b)].
This observation helps rationalize the conservation of en-
zymatic activity in full fibrils from RNase A.
DISCUSSION
The variety of dynamic, conformational, and hydration
features observed in our simulations of RNase A rever-
berates the complexity of the free energy surface that
may characterize the aggregated state of fibril forming
proteins.
The residues of the b-sheet region, formed by the Q10
amyloidogenic loop expansion, display low fluctuations
and high conformational rigidity. Highly dense packing
of Gln sidechains, through intrasheet hydrogen bonding
interactions of the amide stacks and intersheet van der
Waals interactions of their alkylic parts in the steric zip-
per, is the main determinant of the stabilization of the
cross-b-spine motif and of the fibrillar structure. These
conformational properties and the presence of strong
hydrophobic interactions in b-strands cores of amyloids
have already been largely recognized as important factors
for the stability of b-amyloid fibrils.35 Interestingly, the
high number of attractive interactions in the limited
space of the steric zipper is also useful to explain the
high resistance of fibrils to mechanical fracture, as
recently observed with force spectroscopy studies of insu-
lin fibrils.18
In contrast, the analysis of the domain-swapped globu-
lar regions reveals that they preserve all the properties
defining a ‘‘functional unit’’ even in the protofibrillar
models. In particular, the three-dimensional fold, the
breathing functional motions necessary for catalytic activ-
ity, the reactive geometry of catalytic sites and the hydra-
tion of critical residues appear to be essentially the same
as those observed for both the monomeric form and the
nonamyloidogenic C-terminal and N-terminal domain-
swapped dimeric form of RNase A.29,33 In terms of col-
lective properties, the globular domains move as rigid
bodies dangling from the central b-sheet, and the
motions of whole supramolecular aggregates appear to
impose a left-handed twisting on the fibril, a phenom-
enon observed previously on model small peptide proto-
fibrils30,32 and in several experimental low-resolution
structures. The rigid-body dangling motions of the glob-
ular domains around the central rigid spine help explain
the difficulties in obtaining coherent diffractions for these
regions of the aggregates. On the other hand, the rigidity
and order in the cross-b-spine motif explain the obser-
vation of typical fibril-like reflections only at 0.48 and
1 nm.
Overall our observations based on long timescale,
explicit solvent MD simulations validate the suggestion
by Eisenberg and coworkers that functional, globular
units can be considered ‘‘native-like’’ in aggregated fibril-
lar states and support the idea that domain swapping
may be considered a mechanism for amyloid formation.
In this context, Liu et al.36 already pointed out a high
degree of parallelism between 3D domain swapping and
amyloid fibril formation. The two phenomena display
the same type of specificity (being formed by only a sin-
gle type of protein), the presence of two (or more) stable
conformational states separated by high-energy barriers
and the possibility to form linear complexes. The pres-
ence of native-like folded states in higher order aggre-
gates was also confirmed recently by Gotte and co-
workers, who identified and partly characterized larger
domain-swapped oligomers of RNase A.37 The nativeness
of the globular domains and of their conformational dy-
namics is dictated uniquely by their sequence, which is
not changed in the aggregate. The aggregation process
and the stabilization of the fibril are driven principally by
the interactions among the Q10 expansions with no per-
turbation of the globular units. In this picture, there is
no need to invoke a complete unfolding and refolding to
a different structure of the whole sequence.
Our simulations and hydration analysis provide molec-
ular level insights into the role of water in fibril forma-
tion and suggest a possible picture of fibrillogenesis
mechanisms. The single layer polyQ sheet appears to be
dually solvated with both a hydrated and a dry surface.
The regularly hydrated face is on the site where globular
ribonucleases moieties are located (internal surface),
whereas the dewetted surface is fully exposed to the sol-
vent (external surface) and is on the side of the double
layer assembly. The same interactions among Gln side-
chains that are responsible for the stabilization of the
fibril are also responsible for favoring intramolecular
Gln–Gln H-bonding interactions, over intermolecular
Gln–water interactions. The net result of our hydration
analysis is that the face of the polyQ single layer that is
eventually engaged in the formation of the steric zipper
is intrinsically dewetted.
This finding is significant in terms of solution proper-
ties of the system. Indeed, tightly bound waters on the
protein surface are connected to high desolvation energy.
Accordingly, regions poorly surrounded by MDHS are
preferentially selected in processes requiring large desol-
vation energies. As a result, dewetted surfaces might
promote hydrophobic collapses38,39 and extended protein–
protein interactions. Finding a dewetted surface, com-
posed of partly polar sidechains, may be considered un-
usual. Noteworthy, this result is in large agreement with
the recent finding that monomeric polyQ forms collapsed
structures.40 Overall these data claim that polyQs have a
hydrophobic behavior, a singular property for a polymer
of an aminoacid considered hydrophilic in the hydropho-
bicity scales41–43; indeed polyQ stretches acquire a pecu-
liar hydrophobilicy that appears to be due to the intra-
molecular crowding of sidechain H-bonds donors and
G. Colombo et al.
870 PROTEINS DOI 10.1002/prot

acceptors. It is worth noting that we do find a local pro-
tein–water interaction (see Table II); however, the above-
described factors prevent the formation of stable and
long-lived sidechain–water H-bonds. This constitutes the
first rationalization of how the peculiar hydration of
polyQ surfaces makes them good candidates for the dry
steric zipper association.
The analysis of the properties of polyQ stretches may
contribute to the understanding of the role of hydrogen
bonding versus hydrophobic effects in the stabilization of
proteins and protein complexes. Gln residues (as well as
Asn residues) can form triple hydrogen bonds (two
involving main chain groups, one involving the amidic
sidechain moiety), which fit optimally into the antiparal-
lel b-sheet geometry of the polar zipper. In conditions of
thermodynamic control, this would lead to the net stabi-
lization of the state displaying the highest possible num-
ber of these interactions (the aggregate in this case).
Hydrophobic groups (such as the alkyl moieties of Gln,
Asn, or other aliphatic–aromatic side chains), in contrast,
may play a relevant role under kinetic control, imposing
the stereochemical constraints necessary for molecular
recognition in the formation of ordered protein struc-
tures and complexes. In this context, the height of the
barrier for complex formation is determined by the type
of residues present in the sequences and their sequence
and spatial relationships, while the final stability of the
complexes is determined by the net stabilization provided
by protein–protein hydrogen bonds.
To our knowledge, our study represents the first case
in which the properties of a complex, supramolecular ag-
gregate from a whole protein are analyzed with atomistic
methods. We could obtain a first rationalization of the
dynamic, flexibility and hydration properties of the fibril,
showing that the presence of the fibrillogenic region does
not affect the properties of the globular regions, which
preserve their fold and functional motions behaving as
native-like functional units. The extension of this
approach to other constructs of different proteins may
provide important and general information on the deter-
minants of self-organization of biological systems and
suggest ideas for their modification and use in biotech-
nological and medical applications.
ACKNOWLEDGEMENTS
This work was supported by funding under the Sixth
Research Framework Programme of the European
Union (ref. LSHB-CT-2006-037325) under the FIRB
program Folding e aggregazione di proteine: metalli e
biomolecole nelle malattie conformazionali, and under
support of the Ministero degli Esteri exchange program
Understanding the Molecular Determinants of Amyloid
Fibril Formation in Human Neurodegenerative Diseases.
The authors gratefully acknowledge Prof. David Eisen-
berg and his coworkers for helpful discussion and com-
ments. The authors also acknowledge Dr. Giacomo Car-
rea for support.
REFERENCES
1. Sunde M, Blake CCF. From the globular to the fibrous state: pro-
tein structure and structural conversion in amyloid formation.
Quart Rev Biophys 1998;31:1–39.
2. Rochet JC, Jr. PTL. Amyloid fibrillogenesis: themes and variations.
Curr Opin Struct Biol 2000;10:60–68.
3. Serpell LC. Alzheimer’s amyloid fibrils: structure and assembly. Bio-
chim Biophys Acta 2000;1502:16–30.
4. Chiti F, Dobson CM. Protein misfolding, functional amyloid, and
human disease. Annu Rev Biochem 2006;75:333–366.
5. Dobson CM. Protein misfolding diseases: getting out of shape. Na-
ture 2002;418:729–730.
6. Sacchettini JC, Kelly JW. Therapeutic strategies for human amyloid
diseases. Nat Rev Drug Discov 2002;1:267–275.
7. Gazit E. Mechanistic studies of the process of amyloid fibrils forma-
tion by the use of peptide fragments and analogues: implications
for the design of fibrillization inhibitors. Curr Med Chem 2002;9:
1725–1735.
8. Porat Y, Stepensky A, Ding FX, Naider F, Gazit E. Completely dif-
ferent amyloidogenic potential of nearly identical peptide frag-
ments. Bioplymers 2003;69:161–164.
9. Zanuy D, Porat Y, Gazit E, Nussinov R. Peptide sequence and amy-
loid formation: molecular simulations and experimental study of a
human islet amyloid polypeptide fragment and its analogs. Struc-
ture 2004;12:439–455.
10. Zurdo J, Guijarro JI, Jimenez JL, Saibil HR, Dobson CM. Depend-
ence on solution conditions of aggregation and amyloid formation
by an SH3 domain. J Mol Biol 2001;311:325–340.
11. Lopez de la Paz M, Serrano L. Sequence determinants of amyloid
fibril formation. Proc Natl Acad Sci USA 2004;101:87–92.
12. Lazar KL, Kurutz JW, Tycko R, Meredith SC. Encapsulation and
NMR on an aggregating peptide before fibrillogenesis. J Am Chem
Soc 2006;128:16460–16461.
13. Burkoth TS, Benzinger TLS, Urban V, Morgan DM, Gregory DM,
Thiyagarajan P, Botto RE, Meredith SC, Lynn DG. Structure of the
b-amyloid(10-35) fibril. J Am Chem Soc 2000;122:7883–7889.
14. Silva RAGD, Barber-Armstrong W, Decatur SM. The organization
and assembly of a b-sheet formed by a prion peptide in solution: An
isotope-edited FTIR study. J Am Chem Soc 2003;125:13674–13675.
15. Petty SA, Decatur SM. Experimental evidence for the reorganization
of b-strands within aggregates of the ab-(16-22) peptide. J Am
Chem Soc 2005;127:13488–13489.
16. Zanuy D, Gunasekaran K, Ma BY, Tsai HH, Tsai CJ, Nussinov R.
Insights into amyloid structural formation and assembly through
computational approaches. Amyloid 2004;11:143–161.
17. Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe
R, Eisenberg D. Structure of the cross-b spine of amyloid-like
fibrils. Nature 2005;435:773–778.
18. Smith JF, Knowles TP, Dobson CM, Macphee CE, Welland ME.
Characterization of the nanoscale properties of individual amyloid
fibrils. Proc Natl Acad Sci USA 2006;103:15806–15811.
19. Sambashivan S, Liu Y, Sawaya MR, Gingery M, Eisenberg D. Amy-
loid-like fibrils of ribonuclease A with three-dimensional domain-
swapped and native like structure. Nature 2005;437:266–269.
20. Berendsen HJC, Grigera JR, Straatsma PR. The missing term in
effective pair potentials. J Phys Chem 1987;91:6269–6271.
21. Hess B, Bekker H, Fraaije JGEM, Berendsen HJC. A linear con-
straint solver for molecular simulations. J Comput Chem 1997;
18:1463–1472.
22. Miyamoto S, Kollman PA. SETTLE: an analytical version of the
SHAKE and RATTLE algorithms for rigid water models. J Comput
Chem 1992;13:952–962.
MD Simulations of RNase A Fibrils
DOI 10.1002/prot PROTEINS 871

23. Berendsen HJC, Postma JPM, Gunsteren WFV, Nola AD, Haak JR.
Molecular dynamics with coupling to an external bath. J Chem
Phys 1984;81:3684.
24. Lindahl E, Hess B, van der Spoel D. Gromacs 3.0: a package for
molecular simulation and trajectory analysis. J Mol Mod 2001;
7:306–317.
25. van der Spoel D, Lindahl E, Hess B, van Buuren AR, Apol E, Meu-
lenhoff PJ, Tieleman DP, Sijbers ALTM, Feenstra KA, van Drunen
R, Berendsen HJC. Gromacs user manual, version 3.2, 2004. Avail-
able at: www.gromacs.org.
26. van Gunsteren WF, Daura X, Mark AE. GROMOS force field. Ency-
clopedia Comput Chem 1998;2:1211–1216.
27. van Gunsteren WF, Billeter SR, Eising AA, Hunenberger PH, Kruger
P, Mark AE, Scott WRP, Tironi IG. Biomolecular simulation: the
GROMOS96 manual and user guide. Switzerland: vdf Hochschul-
verlag; 1996.
28. De Simone A, Dodson GG, Verma CS, Zagari A, Fraternali F. Prion
and water: tight and dynamical hydration sites have a key role in
structural stability. Proc Natl Acad Sci USA 2005;102:7535–7540.
29. Merlino A, Ceruso MA, Vitagliano L, Mazzarella L. Open interface
and large quaternary structure movements in 3D domain swapped
proteins: insights from molecular dynamics simulations of the C-
terminal swapped dimer of ribonuclease A. Biophys J 2005;88:2003–
2012.
30. Esposito L, Pedone C, Vitagliano L. Molecular dynamics analyses of
cross-b-spine zipper models: b-sheet twisting and aggregation. Proc
Natl Acad Sci USA 2006;103:11533–11538.
31. Amadei A, Linssen ABM, Berendsen HJC. Essential dynamics of
proteins. Protein Struct Funct Genet 1993;17:412–425.
32. Soto P, Cladera J, Mark AE, Daura X. Stability of SIV gp32 fusion-
peptide single-layer protofibrils as monitored by molecular-dynam-
ics simulations. Angew Chem Int Ed Engl 2005;44:1065–1067.
33. Merlino A, Vitagliano L, Ceruso MA, Mazzarella L. Dynamic prop-
erties of the N-terminal swapped dimer of ribonuclease A. Biophys
J 2004;85:2383–2391.
34. Brunger AT, Brooks CL, III, Karplus M. Active site dynamics of ri-
bonuclease. Proc Natl Acad Sci USA 1985;82:8458–8462.
35. Buchete NV, Tycko R, Hummer G. Molecular dynamics simulations
of Alzheimer’s b-amyloid protofilaments. J Mol Biol 2005;353:804–
821.
36. Liu Y, Gotte G, Libonati M, Eisenberg D. A domain-swapped RNase
A dimer with implications for amyloid formation. Nat Struct Biol
2001;8:211–214.
37. Gotte G,Laurents, DV, Libonati M. Three-dimensional domain-
swapped oligomers of ribonuclease A: identification of a fifth tet-
ramer, pentamers and hexamers, and detection of trace heptameric,
octameric and nonameric species. Biochim Biophys Acta 2006;
1764:44–54.
38. Ball P. Chemical physics: how to keep dry in water. Nature
2003;423:25–26.
39. Liu P, Huang X, Zhou R, Berne BJ. Observation of a dewetting
transition in the collapse of the melittin tetramer. Nature
2005;437:159–162.
40. Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV. Fluores-
cence correlation spectroscopy shows that monomeric polyglut-
amine molecules form collapsed structures in aqueous solutions.
Proc Natl Acad Sci USA 2006;103:1674–1679.
41. Eisenberg D, Schwarz E, Komaromy M, Wall R. Analysis of mem-
brane and surface protein sequences with the hydrophobic moment
plot. J Mol Biol 1984;179:125–142.
42. Janin J. Surface and inside volumes in globular proteins. Nature
1979;157:491–492.
43. Kyte J, Doolittle RF. A simple method for displaying the hydro-
pathic character of a protein. J Mol Biol 1982;157:105–132.
G. Colombo et al.
872 PROTEINS DOI 10.1002/prot
Copyright © 2022 FDOKUMEN




















