
Ion channel hypothesis for Alzheimer amyloid peptide neurotoxicity
14
Cellular and Molecular Neurobiology, Vol. 15, No. 5. 1995 Ion Channel Hypothesis for Alzheimer Amyloid Peptide Neurotoxicity Harvey B. Pollard, 1'" Nelson Arispe, ~ and Eduardo Rojas ~ Received January 5, 1995; accepted February 20, 1995 KEY WORDS: Alzheimer's Disease: calcium channels: amyloid. SUMMARY 1. Alzheimer's disease (AD) is a chronic dementia and neurodegenerative disorder affecting the oldest portions of the population. Brains of AD patients accumulate large amount of the A/3P peptide in amyloid plaques. 2. The A/3P[1-40] peptide is derived by proteolytic processing from a much larger amyloid precursor protein (APP), and has been circumstantially identified as the toxic principle causing cell damage in the disease. 4. The A/3P[1-40] peptide is able to form quite characteristic calcium channels in planar lipid bilayers. These channels have conductances in the nS range, and can dissipate ion gradients quickly. The peptide can also cause equivalent cation conductances in cells. 5. We suggest that amyloid channel blocking agents might be therapeutically useful in Alzheimer's Disease, and have constructed molecular models of the channels to aid in the design of such compounds. INTRODUCTION Alzheimer's disease (AD) is a chronic dementia and neurodegenerative disorder affecting the oldest portions of the population. It is characterized pathologically by extracellular amyloid plaques, intraneuronal neurofibrillary tangles, and vascular and neuronal damage (Masters et al., 1985; Neve et al., 1990: Selkoe, 1991; Hardy and Higgins, 1992). The major component of brain amyloid is a ~Laboratory of Cell Biology and Genetics, NIDDSK, National Institutes of Health, Bethesda. MD 20892. 2 To whom correspondence should be addressed at Laboratorw of Cell Biology and Genetics, Building 8, Room 402, National Institutes of Health, Bethesda, MD -50892. 513 0272.4340/95tlOOO.0513507.50to ~ 1995PlenumPubhshmg Corporation
Transcript of Ion channel hypothesis for Alzheimer amyloid peptide neurotoxicity

Cellular and Molecular Neurobiology, Vol. 15, No. 5. 1995
Ion Channel Hypothesis for Alzheimer Amyloid Peptide Neurotoxicity
Harvey B. Pollard, 1'" Nelson Arispe, ~ and Eduardo Rojas ~
Received January 5, 1995; accepted February 20, 1995
KEY WORDS: Alzheimer's Disease: calcium channels: amyloid.
SUMMARY
1. Alzheimer's disease (AD) is a chronic dementia and neurodegenerative disorder affecting the oldest portions of the population. Brains of AD patients accumulate large amount of the A/3P peptide in amyloid plaques.
2. The A/3P[1-40] peptide is derived by proteolytic processing from a much larger amyloid precursor protein (APP), and has been circumstantially identified as the toxic principle causing cell damage in the disease.
4. The A/3P[1-40] peptide is able to form quite characteristic calcium channels in planar lipid bilayers. These channels have conductances in the nS range, and can dissipate ion gradients quickly. The peptide can also cause equivalent cation conductances in cells.
5. We suggest that amyloid channel blocking agents might be therapeutically useful in Alzheimer's Disease, and have constructed molecular models of the channels to aid in the design of such compounds.
INTRODUCTION
Alzheimer's disease (AD) is a chronic dementia and neurodegenerative disorder affecting the oldest portions of the population. It is characterized pathologically by extracellular amyloid plaques, intraneuronal neurofibrillary tangles, and vascular and neuronal damage (Masters et al., 1985; Neve et al., 1990: Selkoe, 1991; Hardy and Higgins, 1992). The major component of brain amyloid is a
~ Laboratory of Cell Biology and Genetics, NIDDSK, National Institutes of Health, Bethesda. MD 20892.
2 To whom correspondence should be addressed at Laboratorw of Cell Biology and Genetics, Building 8, Room 402, National Institutes of Health, Bethesda, MD -50892.
513
0272.4340/95tlOOO.0513507.50to ~ 1995 Plenum Pubhshmg Corporation

514 Pollard, Arispe, and Rojas
38-42 residue peptide, termed amyloid /3 protein (Al3P or/3A/4) (Glenner and Wong, 1984; Masters et al., 1985; Kang et al., 1987), which is a proteolytic product of the widely distributed amyloid precursor protein (APP7s~) defined by a locus on chromosome 21 (Goldgaber et al., 1987; Tanzi et al., 1987; Goate et al., 1991). A/3P has been circumstantially identified as the toxic principle causing cell damage in the disease, although the mechanism of this toxicity has been a matter of contention (e.g., Price et al., 1992).
Amyloid peptides do not appear to interact with target cells via receptors, since no binding sites have been detected. Indeed, direct interaction of the peptide with the target cell membrane has been most often observed (Mattson et al., 1993). In addition, increases in intracellular calcium have been noted to follow addition of amyloid peptide to the target cells (Mattson et al., 1992), indicating that toxicity might involve disordering of intracellular calcium homeostasis. The toxicity of amyloid peptide has not been ameliorated by u-type calcium channel blockers (Whitson and Appel, 1995), further indicating some specificity in the actions of amyloid peptides on calcium metabolism. Finally, amyloid peptide toxicity has been shown to be associated with the formation of free radicals in target cells (Behl et al., 1994), and the characteristic antiparallel t3-sheet conformation of the amyloid peptide has been implicated in the toxic process (Schubert et al., 1995). The centrality of calcium, even in the latter processes, is manifest by the fact that elevation of intracellular calcium in neuronal cells is closely associated with the generation of free radicals (Olanow and Arendash, 1994).
It was for these reasons that we have given particular emphasis to our original observation that the amyloid peptide (ACP[1-40]) was able to form quite characteristic calcium channels in planar lipid bilayers (Arispe et al., 1993a,b). This observation led us to propose that the toxic action of amyloid peptide on target cells in the brain might be due to these channels. We further suggested that amyloid channel blocking agents might be therapeutically useful, and have constructed molecular models of the amyloid channels to aid in the design of such compounds (Durrell et al., 1994). In the remainder of this article we will summarize the data leading to this conclusion, and mention studies supporting the concept that similar channels may indeed be formed in intact cells.
AMYLOID PRECURSOR PROTEIN (APP) AND THE Af~P[1-40] PEPTIDE
The A/3P[1-40] peptide is derived by proteolytic processing from a much larger amyloid precursor protein (APP). The mRNA for APP is said to represent as much as 2.5% of the total mRNA in the brain, and its synthesis is regulated by physical and chemical damage (Siman et al., 1989; Kawarabayashi et al., 1991; Roberts et aL, 1991; Heurteaux et al., 1993). As shown in Fig. 1, this precursor is a glycosylated integral membrane protein, which is synthesized from a single copy gene in three alternatively spliced forms: APP 695, 751, and 770 (Ponte et aL, 1988; Tanzi et aI., 1988; Kitaguchi et at., 1988). The larger 751 and 770 forms of

Amyloid Peptide Calcium Channels 515
A. extracellular
membrane
Lysine-16 "') Amyloid Beta Protein[I-40]
. cytoplasmic
I '",','~ C I
B.
H 2N'D I"A2"E3-F4"R5"Hs'D7"S8-Gg"Ylo'E1 l"V12"H13"H 14"Q 15"Kls'L17-
V t 8-F19-F20-A21-E22- D23-V24-G25-S 26-N27-K2s-G 29-A30-131-132-G 33 -
L34-M 35-V36-G37-G38-V39.-V40-OH.
Fig. 1. Structure of the amyloid precursor protein (APP). (A) The alterna- tively spliced forms are shown numbered. The Kunitz sequence is in the alternative spliced forms. (B) The sequence of the A/3P[l-40] domain is shown. The internally numbered sequence correlates with the amyloid domain in part A. K~ is lysine-16 in part A.
APP contain an exon-7-defined Kunitz type serine proteinase inhibitor ( "KPI ' ) sequence (Ponte et aL, 1988; Tanzi et al., 1988; Kitaguchi et al., 1988), and a large soluble fragment of APP, containing the Kunitz sequence, is eventually released from cells. This fragment is the previously described protease inhibitor "protease nexin II" (Hardy and Allsop, 1991). The remaining short segment of trans- membrane and cytosolic sequence is subsequently processed into the toxic amyloid /3 peptide (A/3P, /3A/4). The 40 residue peptide [1-40] is the m o s t common, but [1-41] and [1-42] peptides also occur with specific anatomical distributions.
Prior to the major cleavage step, APP is also processed within the cell by sequential glycosylation and other post-translational modifications reactions. Consequently, using antibodies to the distal N-terminal domain, three bands of "APP" are routinely seen by Western analysis (e.g., Sambamurti et aL, 1992a,b). In PC12 cells, for example, a core glycosylated form of endogenous APP appears in the endoplasmic reticulum as a 100 kDa species. This protein is then further glycosylated and sulfated to a mature 140kDa form in the trans-golgi network (TGN). Finally, after reaching the TGN, the mature form is proteolyzed to release the above-mentioned 120kDa soluble fragment. However, only the mature 140kDa and immature 110kDa forms can be detected by antibodies against the proximal C-terminal or transmembrane domains (Setkoe et aL, t988; Sambamurti et al., 1992a,b).
Similar results have been reported for endogenous APP in secretory vesicles

516 Pollard, Arispe, and Rojas
such as alpha granules in platelets (Van Nostrand et al., 1990) and chromaffin granules (Vassilacopoulou et al., 1995), and for recombinant AAP in CHO cells (Caporaso et al., 1994). Cleavage to form the soluble APP species is not a lysosomal process (Sambamurti et al., 1992a,b), and actually has been shown to occur within the chromaffin granule (Vassilacopoulou et al., 1995). In rat hippocampal slices, electrical stimulation causes the release of soluble forms of the APP lacking the APP carboxy terminus (Nitsch et al., 1993).
Depending on where the major proteotytic secretase step occurs on te APP, the remainder of the protein left in the membrane is further processed to form either a non-toxic 3 kDa species, or a longer, toxic 4kDa species (A/3P, or "/3A/4") (Haas and Selkoe, 1993). This processing step may occur in lysosomes (Cole et al., 1989; Caporaso et al., 1992; Golde et al., 1992), although there is evidence that the 4 kDa A/3P may not occur by normal processing (Sisodia er al., 1990). The sequence of the toxic At3P[1-40] product, numbered internally as in Fig. 1, is as follows:
H2N-D1-A2-E3-F4-Rs-H6"DT-Ss-G9-Ylo-E1 l"V12-H13-H~4-Q15"K~6-L17-V18-Flg- F20-A21-E22-D23-V24-G25-S26"N27"K28-G29-A30-I31-I32-G333"L34M35-V36-G37" G38-V39-V40-OH.
The most frequent cleavage occurs between lysine16 (KI6) and leucine 17 (L17), leaving the sequence [1-16] attached to the secreted soluble APP. The protease has been termed the "a-secretase," although the hydrolysis position on the APP is not sequence specific (Maruyama et al., 1991). Following further processing of the cytosolic domain, the resulting 3 kDa sequence [17-40] is either retained in the membrane or released, but is not toxic.
Historically, it was presumed that patients with Alzheimer's Disease suffered because of aberrant proteolysis by a "13-secretase" just before the N-terminal aspartic acid residue (viz. D1). This generates the toxic AI3P[1-40] peptide, which is either retained in the membrane or released extracellularly. However, it is now known that the toxic A/3P[1-40] peptide also occurs naturally in the CSF of "non-affected controls," as well as Alzheimer's disease patients (Gold et al., 1992; Shoji et al., 1992; Haas et aL, 1992; Seubert et al., 1993; Busciglio et aL, 1993). The interpretation of this result is still evolving. Perhaps there are no real "controls" in the case of Alzheimer's disease, since the incidence increases with age. Or, perhaps target cells may express some prodromal property that renders them uniquely susceptible to attack by the toxic A/3P[1-40] peptide (Pollard et al., 1994a). It is well known that the potency of the A/3P[1-40] is cell specific, and from this perspective the A/3P[1-40] may act by delivering a " c o u p de g r a c e " to a susceptible cell, leaving otherwise robust cells intact.
CATION CHANNEL ACTIVITY OF AI3P[1-40]
We noted that the first 16 residues of the toxic A~P[1-40] peptide had the remarkable property of alternating charged or neutral residues with hydrophobic residues. This was consistent with the amphipathic beta sheet structure occurring
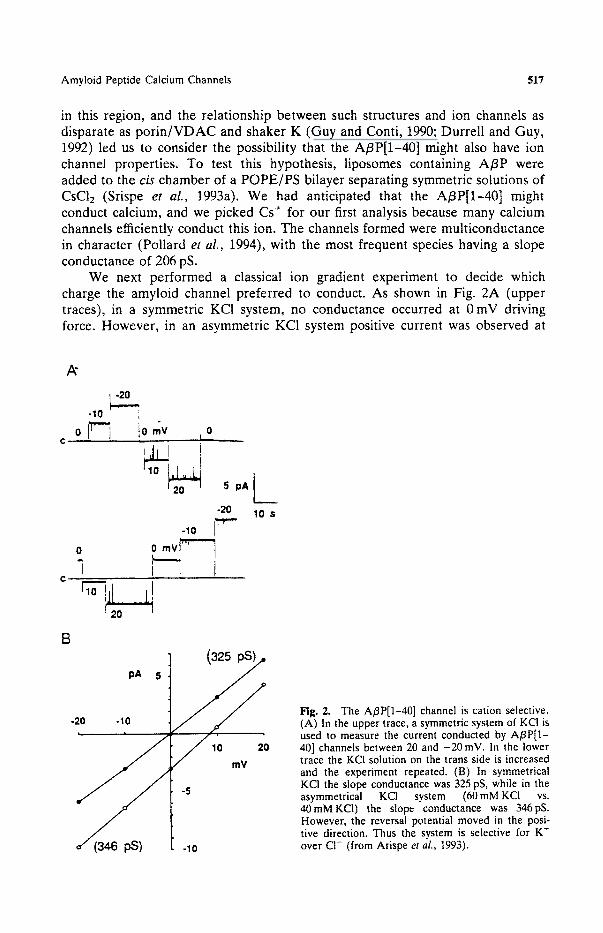
Amyloid Peptide Calcium Channels 517
in this region, and the relationship between such structures and ion channels as disparate as porin/VDAC and shaker K (Guy and Conti, 1990; Durrell and Guy, 1992) led us to consider the possibility that the A/3P[1-40] might also have ion channel properties. To test this hypothesis, liposomes containing A/3P were added to the cis chamber of a POPE/PS bilayer separating symmetric solutions of CsC12 (Srispe et al., 1993a). We had anticipated that the A/3P[1-40] might conduct calcium, and we picked Cs+ for our first analysis because many calcium channels efficiently conduct this ion. The channels formed were multiconductance in character (Pollard et al., 1994), with the most frequent species having a slope conductance of 206 pS.
We next performed a classical ion gradient experiment to decide which charge the amyloid channel preferred to conduct. As shown in Fig. 2A (upper traces), in a symmetric KC1 system, no conductance occurred at 0 mV driving force. However, in an asymmetric KC1 system positive current was observed at
A"
-20 . Io "!
o,,F7 o 'J l i ! ++++ F10
5 pa
-20 -10
o o mvF- 7 ~,, "7 j I l C .... I10 i ~
I,,, 10 s
B
pA 5
J ' ° mV -5
-10
Fig. 2. The A/3P[1-40] channel is cation selective. (A) In the upper trace, a symmetric system of KCI is used to measure the current conducted by A/3P[1- 40] channels between 20 and -20 mV. In the lower trace the KC1 solution on the trans side is increased and the experiment repeated. (B) In symmetrical KCI the slope conductance was 325 pS, while in the asymmetrical KCI system (60 mM KCI vs. 40mMKC1) the slope conductance was 346pS. However, the reversal potential moved in the posi- tive direction. Thus the system is selective for K ~ over C1- (from Arispe et aL, 1993).

518 Pollard. Arispe, and Rojas
0mV. From the appropriate application of the Nernst equation (Fig. 2B) we found that the A/3P[l-40] channels indeed preferred to conduct cations. Furthermore, from the new reversal potential we were able to calculate a permeability ratio corresponding to PK/Pcl of ca. 11.
Since the channel conducted both Cs + and K ÷, it appeared that the A/3P[l-40] might be a traditional calcium channel. We tested this hypothesis further by studying the ability of different cations, including Ca 2÷, to be conducted. We also tested the ability of low concentrations of Ca 2* to block conduction of high concentrations of monovalent cations (discussed in Tsien et
al., 1987). As summarized by Arispe et al. (1993a), the selectivities for different cations could be computed from the ratios of the permeability coefficients derived from bicationic reversal potentials. These data indicated a permeability sequence as follows:
Pcs > PLi > Pca ~ PK > PNa
This is typical of calcium channels. In addition, as predicted, Ca"* was able to block the conduction of Cs+ by the A13P[1-40] channels (see Fig. 3).
Studies with planar lipid bilayers and well defined exogenous channel forming proteins have some significant advantages over studies with cells, since the cells possess many additional endogenous channels. However, it would be important to know if the amyloid channels formed in the bilayers also were formed in cells. To this end, evidence has recently been reported for the occurrence of cation channels in mouse N1E-115 neuroblastoma cells treated with A/3P[1-40] (Davidson et al . , 1994). Whole cell recordings were performed on
A
- 6 0 m V
i F
• 4 0
- 2 0
2O
l o s
• 6 0
60
Fig. 3. Effects of C a 2÷ on A/3P[I-40] channel activity. (A) A/3P[1-40] channels conduct symmetrical Cs- (200raM) with electrical driving forces between -60 and 60inV. The time scale is very compressed, and several conductance levels are being sampled. (B) Ca 2- (10 raM) is added to the cis compartment only, and records were collected 10 minutes later over the voltage range of -60 to +60 inV. Only the ±60 mV records are shown (Arispe et aL. 1993a).

Amyloid Peptide Calcium Channels 519
cells treated with A/3P[1-40] for 24 hours. During this period ca. 50% of the cells were killed. Among the surviving cells, the median peak inward current was significantly increased, and the voltage at peak current was shifted to more positive potentials. At the single channel level, addition of A/3P[1-40] increased the open probability of cation conducting ion channels. The authors cautiously ascribed the changes to either direct formation of channels in the membranes, or to alteration of endogenous cation channels. The problems intrinsic to interpret- ing such studies is exemplified by studies showing that addition of A~gP[l-40] to human fibroblasts can actually abolish certain potassium channels (Etcheber- rigaray et al . , 1994).
GIANT, MULTILEVEL A[~P[1-40] CHANNELS
While the initial data on A~IP[1-40] channels were collected using peptide pre-complexed with phosphatidylserine liposomes, it was soon learned that the peptide could also form channels directly from solution (Arispe et al . , 1993b). This was an important finding because A/3P[l-40] peptides occur free in solution in the CSF, and it was a legitimate concern whether the peptides might also form channels from this state. In addition to the multiconductance channels observed in the large size region of <400 pS, channels in the giant range (>400 pS) were also observed. The example in Fig. 4 shows that channels in the large range can spontaneously move into the giant range, and still exhibit multi-conductance states. Channel conductances as high as 5 nS were seen routinely. A biological perspective on this value can be achieved by considering that such a conductance is in the range of that for cytotoxic complement channels.
With knowledge of the physical properties of single amyloid channels, it is possible to estimate whether the function of this channel could be potentially deleterious to a cell. This question can be approached by computing the ion flux across a membrane from the equation,
¢bj = ( p i I i ) / F
where ~bi is the ion flux, Pi is the fractional open time, I~ is the ion current through the open channel, and F is the Faraday constant (96,480 C/mole). Therefore, if a single 5 nS channel became active in a nerve cell of 25 microns diameter, the calcium influx could be calculated as 0.3 fmol/sec, and the intracellular calcium concentration change would be 5/.LM/sec. Such an increase in internal calcium is known to be associated with profound cytotoxicity (Choi, 1988). Furthermore, since the amyloid channel is permeable to other ions as well, the loss of general homeostatic ionic balance across the membrane would be catastrophic.
BLOCKADE OF AI3P[1-401 CHANNELS
If A/3P[1-40] ion channel activity were indeed the basis of amyloid peptide neurotoxicity, then agents capable of blocking the channels would be useful candidate therapeutics. The first compound to be found capable of blocking the

5 2 0 Pollard, Arispe, and Rojas
(3506)
o If '% .0 , (3°9) Ii I (2257) r',
I [I (2097) ,~ I (2145) ] ~1 ' i c,696) u ,fFI I , / , , = l , !,!!111 1(669)11 , , I=.iW i(1os6)
¢ J ~ ) V ~ J " ~ , - . t l l ' , , ~ J . , , L ~ I I (4so) I I ~1L4[(674) I (4500)
-30 1' - 2 0 / m V "I' ..... 10 1' 0 1' -1 ....... 20 s "-
j a i I i I M I ' "
Fig. 4. Fluctuations of A/3P[1-40] channels between large and giant levels. The upper record shows an experiment in which the channel is seen primarily in the large conductance region with a driving force of -30 inV. Conductances for individual transitions are given in parentheses. Occasional transitions to much higher conductances are seen. Suddenly. a stable transition to the giant nS conductance occurs. The lower record shows a portion of the upper record on a 20-fold expanded time base. Figure is from Arispe et aL, 1994.
A/3P[1-40] channel was the weak base tromethamine, also known as the buffer TRIS (Arispe et al., 1993a,b). The large channels (<400pS) were blocked completely by 1-2 mM tromethamine. However, the giant channels (>400pS) were much less affected. Tromethamine is of potential therapeutic interest because it has been used chronically in humans at much higher concentrations. Cardiac surgery patients are preequilibrated with tromethamine to a concentra- tion of 16 mM to control tissue acidosis during surgery. Tromethamine also has a very low intrinsic toxicity in cell cultures (Swim, 1961) and in humans (Nahas, 1963; Nahas, 1964). From these data on tromethamine, one can also conclude that selective blockade of the A/3P[1-40] channel is possible.
Aluminum, given as AIC13, was also discovered to block the A/3P[l-40] channel (Arispe et aL, 1993a). Even at concentrations as low as 10/xM, A13+ w a s
found to block conductance to driving forces as large as 100 mV. Aluminum has an interesting history insofar as Alzheimer's disease is concerned, because of early reports that amyloid plaques contained large amounts of this metal. Aluminium is thought to be toxic to the brain (Klatzo et al., 1965; Terry and Pefia, 1965), but the selective action of low concentrations of A13+ o n the A/3P[l-40] channel again supports the concept that selective blockade is

Amyloid Peptide Calcium Channels 521
possible. Interestingly, normal brain levels of aluminum are 1-2/.tg/gram wet weight, or ca. 15/zM (Crapper et aL, 1978), which compares favorably with the levels needed to achieve complete blockade of the A/3P[1-40] channels.
On the other hand, the L-type calcium channel blocking drug nitrendipine was found to be inactive as a blocker on A/3P[1-40] channels (Arispe et al., 1993b). Concentrations as high as 100/xm failed to affect the conductivity, added either to the cis, or trans, or both compartments of the bilayer. Since dihydropyridine drugs act at much lower concentrations on true L-type calcium channels, these data indicate that the structure of the A/3P[1-40] channel is unique from at least one other calcium channel. We also tested the compound Cognex ® (tetrahydroacridine), an inhibitor of acetylcholine esterase, which is the only drug cleared by the FDA for treatment of Alzheimer's Disease. Although reportedly effective on potassium channels but not calcium channels, this compound, at a concentration of 100/xM, was totally inactive as a blocking agent of Cs 2+ permeation of A/3P[1-40] channels.
MOLECULAR MODELS OF THE Ai~P[1-40] CHANNEL
Extensive molecular modeling studies have been performed with other peptides which form ion channels, including cecropin, magainin, 6-hemolysin, and pardaxin (Durrell et aL, 1992; Cruciani et aL, 1992; Raghunathan et al., 1990: Lazarovici et al., 1992). The techniques developed with these peptides have also been used to create molecular models for the A/3P[1-40] channel (Durrell et al., 1994). Figure 5A shows the A/3P[1-40] sequence, with specific residues de- lineated as to their likely conformation in the lipid bilayer. The charged residues are circled, and shown to alternate with hydrophobic or neutral residues. The boxes DSG domain is predicted to form a beta turn, and is the location of the turn between the two rectangular beta sheets in Fig. 5B. These beta sheets form the walls of the pore for the type I model. A similar boxed GSN domain forms a predicted beta turn which breaks the C-terminal alpha helix to form a second middle helix (see Fig. 5B).
The type I model, shown in Fig. 6, is preferred for energetic and conformational reasons (Durrell et al., 1994), and is a hexamer of the monomeric structure shown in Fig. 5B. The C-terminal helix partially spans the membrane, and the turn structure formed by the GSN motif leads to a middle helix which "floats" on the membrane surface. Following the lysine 16 (K16) residue, the sequence QHH breaks the middle helix to form the hairpin walls of the conducting pore. This leaves lysine 16 on all the monomers exposed to the aqueous phase, where it can form a charge pair with the D or E on the C-terminal portion of the succeeding middle helix of the next monomer.
Other models can be considered. We have illustrated a Type II model in which the lysine and four glycines on one face of the C-terminal alpha helix ([28-40]) form the walls of the pore. Such a structure is reminiscent of the gramacidin channel, in that glycines also predicted to form the walls of the pore. The predicted structure is not very stable, and so we do not favor it over the type

522
A. 1 5
Pollard, Arispe, and Rojas
I0 15 20
A
25 30
I I G.L M V G G V V-CO~ 35 40
m Fig. 5. Molecular models of A/3P[1-40] channel. (A) Amino acid sequence of A/3P[1-40]. The circles indicate the charged residues. The underscores indicate the polar-neutral residues. The remaining residues are considered hydrophobic. The rectangles indicate predicted turn regions (from Durrell et aL, 1994). (B) Secondary structure prediction for membrane-bound A/3P[1-40] molecule. Rectangles indicate the /3 strands of the hairpin, The cylinders indicate the middle and C-terminal alpha helices, The walls of the pore can be formed from the charged surfaces of the hairpin (Type I), the glycine residues of the C-terminal helix (Type II), or the charged residues on the middle helix (Type II1), We favor the Type I model for reasons mentioned in the text (from Durrell et al., 1994).

Amyloid Peptide Calcium Channels 523
Fig. 6. Schematic view of the Type I A/3P[I-40] channel. Six peptides insert into the membrane and form a conducting pathway from the charged surfaces of the hairpin structure. These six hairpins form a 12-stranded. antiparallel /3-barrel. The middle helices greatly stabilize the structure through interactions between the K-16 on the end of one helix and the D and E residues on the next middle helix (from Durrell et al.. 1994).
I model. However, an eleven residue fragment of A/3P[1-40], composed of [25-35], is also toxic (Joseph and Han, 1992), and is reported to cause a nonspecific ionic conductance in acutely dissociated rat cortical neurons (Furu- kawa et al., 1994). Weakly selective channels in bilayers have also been reported for the [25-35] sequence (Mirzabekov et al., 1994). Perhaps this alternative structure, present only in the [25-35] fragment, might explain these data. One caveat leads one to beware of giving physiological significance to this sequence, since it is actually not a naturally occurring breakdown product of A/3P[1-40]. The other model, termed Type III, is based on charged residues in the middle helix providing the walls of the pore. This also is a relatively unstable structure, but was included in the original set of models for completeness.
CONCLUSION
At present the cause of Alzheimer's disease is not known. It could be due to endogenous degenerative changes in neurons, or to assaults on the neurons from exogenous causes. However, in either case the common denominator appears to be the beta amyloid peptide. Thus, by focusing on the mechanism of toxicity of this material, it may eventually be possible to treat Alzheimer's disease without complete knowledge of its origins. We have discussed here the data underlying the hypothesis that the intrinsic calcium channel properties of the A/3P[1-40] peptide could be the basis of the neurotoxicity. Certainly the availability of molecular models for the peptide channels sets the stage for using rational drug

524 Pollard, Arispe, and Rojas
design to discover blockers. The models also supply hypothetical structures for experimentally testing, and refining the models presently available.
REFERENCES
Arispe, N., Rojas, E., and Pollard, H. B. Alzheimer's Disease amyloid /3 protein forms calcium channels in bilayer membranes (1993a). Blockade by tromethamine and aluminum. Proc. Nat. Acad. Sci (USA) 9t):567-571.
Arispe, N., Pollard, H. B., and Rojas, E. (1993b). Giant, multi-level cation channels formed by Alzheimer's Disease Amyloid Protein (A/3P[1-40]) in bilayer membranes. Proc. Nat. Acad Sei. ( USA ) 90:10573-10577.
Arispe, N., Pollard, H. B., and Rojas, E. (1994). /3-amyloid Ca2*-channel hypothesis for neuronal death in Alzheimer's Disease. Mol. Cell. Biochem. 140:119-125.
Beal, M. F. (1992). Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illness? Ann. Neurol. 31:119-130.
Behl, C., Davis, J. B., Lesley, R., and Schubert, D. (I994). Hydrogen peroxide mediates amyloid beta protein toxicity. Celt 77:817-827.
Busciglio, J., Gabuzda, D., Matsudiera, P., and Yankner, B. (1993). Generation of beta-amyloid in the secretory pathway in neuronal and non-neuronal cells. Proc. Nat. Acad. Sci. (USA) 90:2092-2096.
Caporaso, G. L., Gandy, S., Buxbaum, J. D., and Greengard, P. (1992). Chloroquine inhibits intracellulat degradation but not secretion of the Alzheimer /3/A4 amyloid precursor protein. Proc. Natl. Acad. Sci. (USA)89:2252-2256.
Caporaso. G. L., Takei, K., Gandy, S. E., Matteoli, M., Mundigl. O., Greengard, P., and de Camilli, P. (1994). Morphological and biochemical analysis of the intracellular trafficking of the alzheimer /3/A4 amyloid precursor protein. J. Neuroscience 14:3122-3138.
Choi, D. W. (1988). Calcium-mediated neurotoxicity: Relationship to specific channel types and role in ischemic damage. Trends Neurosci. 11:465-469.
Cote, G. M., Huynh, T. V., and Saitoh, T. (1989). Evidence for lysosomaI processing of amyloid /3-protein precursor in cultured cells. Neurochem. Res. 14:933-939.
Crapper, D. R., Karlik, S., and De Boni, U. (1978). Aluminum and other metals in senile (Alzheimer) dementia. In Katzman, R., Terry, R. D.. and Bicki, K. L.. Alzheimer's Disease: Senile Dementia and Related Disorders (Aging, Vol. 7), Raven Press, New York, pp. 471-485.
Cruciani, R. A., Barker, J. L., Durrell, S. R., Raghunathan, G., Guy, H. R., Zasloff, M., and Stanley, E. F. (1992). Magainin 2, a natural antibiotic from frog skin, forms ion channels in lipid bilayer membranes. Eur. J. Pharm. 226:287-296.
Davidson, R. M., Shajenko, L., and Donta. T. S. (1994). Amyloid beta peptide (A/3P) potentiates a nimodipine-sensitive L-type barium conductance in N1E-115 neuroblastoma cells. Brahz Res. 643:324-327.
Durrell, S. R., and Guy, H. R. (1992). Atomic scale structure and functional models of voltage-gated potassium channels. Biophysical J. 62:238-250.
Durrell, S. R., Guy, H. R., Arispe, N., Rojas. E., and Pollard, H. B. (1994). Theoretical models of the ion channel structure of amyloid-/3-protein. Biophysical J. 67:2137-2145.
Dyrkes, T., Weidemann, A., Multhaup, G., Salbaum, J. M., Lemaire, H-G., Kang, J., Muller-Hill, B., Masters, C. L., and Beyreuther, K. (1988). Identification, transmembrane orientation and biogenesis of the amyloid A4 precursor of Alzheimer's disease. EMBO J. 7:949-957.
Etcheberrigaray, R., ito, E., Kim, C. S., and Alkon, D. L. (1994). Soluble beta amyloid induction of Alzheimer's phenotype for human fibroblast K+ channels. Science 264:276-279.
Furukawa, K., Abe, Y., and Akaike, N. (1994). Amyloid /3 protein-induced current in rat corucal neurones. NeuroReport 5:2016-2018.
Glenner~ G. G., and Wong, C. W. (1984). Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun 120:.885-890.
Goate, A., Chartier-Harlan, M-C., Mullan, M., Brown, J., Crawford, F., Fidani, L., Giuffra, L., Haynes, A., Irving, N., James, L., Mant, R., Newton, P., Rooke, K., Roques, P., Talbot, C., Pericak-Vance, M., Roses, A., Williamson, R., Rossor, M., Owen, M., and Hardy, J. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349:.704-706.

Amyloid Peptide Calcium Channels 525
Golde, T., Estus, S., Yountkin, L., Selkoe, D., and Yountkin, S. (1992). Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science 255:728-730.
Goldgaber, D., Lerman, M. I., McBride, O. W., Saffioti. U., and Gajdusek, C. (1987). Characteriza- tion and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science 235:877-880.
Guy, H. R., and Conti, F. (1990). Pursuing the structure and function of voltage-gated channels. Trends Neurosci. 13:201-206.
Haas, C., and Selkoe, D. J. (1993). Cellular processing of/3-amyloid precursor protein and the genesis of the amyloid/3-peptide. Cell 75:1039-1042.
Haas, C., Schlossmacher, M., Hung, A., Vigo-Pelfrey, C., Mellon, A., Ostaszewski, B., Lieberberg, I., Koo, E., Schenk, D., Teplow, D., and Selkoe, D. J. (1992). Amyloid beta peptide is produced by cultured cells during normal metabolism. Nature 359:.322-327.
Hardy, J., and Altsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer's disease. TIPS 12:383-388.
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: The amyloid cascade hypothesis. Science 256:780-783.
Heurteaux, C., Bertaina, V., Widmann, C., and Lazdunski, M. (1993). K+ channel openers prevent global iachemia-induced expression of c-los, c-jun, heat shock protein, and amyloid beta-protein precursor genes and neuronal death in rat hippocampus. Proc. Natl. Acad. Sci. (USA) 90:9431-9435.
Joseph, R., and Han, E. (1992). Amyloid /3-protein fragment 25-35 causes activation of cytoplasmic calcium in neurons. Biochern. Biophys. Res. Commun. 184:1441-1447.
Kang, J., Lemaire, H-G., Unterbeck, A., Salbaum, J. M., Masters, C. L.. Grszeschik, K-H., Multhaup, G., Beyreuther, K., and MUller-Hill, B. (1987). The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 325:733-736.
Kawarabayashi, T., Shoji, M., Harigaya, Y., Yamaguchi, H., and Hirai, S. (1991). Expression of APP in early stages of brain damage. Brain Res. 563:334-338.
Kitaguchi. N., Takahashi, Y., Tokoshima, S, Shiojiri, S., and ho, H. (1988). Novel precursor of Alzheimer's disease amyloid protein shows protease inhibitory activity. Nature 331:530-532.
Klatzo, I., Wisniewski, H., and Streicher, E. (1965). Experimental production of neurofibrillary degeneration. J. Neuropath. ExptL NeuroL 24:187-t99.
Lazarovici, P., Edwards, C., Raghunathan, G., and Guy, H. R. (1992). Secondary structure, permeability, and molecular modeling of pardaxin pores. J. Natural Toxins. 1:1-15.
Maruyama, K., Kametani, F., Usami, M., Yamao-Harigaya, W., and Tanaka, K. (1991). "Secretase" Alzheimer amyloid protein precursor secreting enzyme is not sequence-specific. Biochem. Biophys. Res. Commun. 179:1670-1676.
Masters, C. L., Simms, G., Weinman, N. A., Malthaup, G., McDonald, B. I., and Beyreuther, K. (1985). Amyloid plaque core protein Alzheimer disease and Down syndrome. Proc. NatL Acad. Sci. (USA) 82:4245-4249.
Matson, M. P., Cheng, B., Davis, D., Bryant, K., Lieberberg, I., and Rydel, R. E. (1992). /3-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable t o excitotoxicity. J. Neurosci. 12:376-389.
Mattson, M. P., Barger, S. W., Cheng, B., Lieberberg, I., Smith-Swintosky, V. L., and Rydel, R. E. (1993). /3-amyloid precursor protein metabolites and loss of neuronal Ca 2- homeostasis in Alzheimer's disease. Trends Neurosci. 16:409-414.
Mirzebekov, T., Lin, M. C., Marshall, P. J., Carman, M., Tomaselli, K., Lieberberg, I., and Kagan, B. L. (1994). Biochem. Biophys. Res. Comrnun. 202:1142-1148.
Nahas, G. G. (1962). The pharmacology of tris (hydroxymethyl)aminomethane (THAM). Pharm. Rev. 14:447-472.
Nahas, G. G. (1963). The clinical pharmacology of THAM (tris(hydroxymethyl)aminomethane. Clin. Pharm. Ther. 4:784-803.
Neve, R, L., Dawes, L. R., Yankner, B. A., Benewitz, L. L., Rodriguez, W., and Higgins, G. A. (1990). Genetics and biology of the Alzheimer's disease precursor. Prog. Brain Res. 86',257-267.
Nitsch, R. M., Farber, S. A., Growdon. J. H., and Wurtman, R. J. (1993). Release of amyloid beta-protein precursor derivatives by electrical depolarization of rat hippocampal slices. Proc. Natl. Acad. Sci. (USA) 90:5191-5195.
Olanow, C. W., and Arendash, G. W. (1994). Metals and free radicals in neurodegeneration. Curr. Opin. Neurol. 7:548-558.
Pollard, H. B., Rojas, E., and Arispe, N. (1994a). /3-Amyloid in Alzheimer's Disease. Therapeutic implications. CNS Drugs 2:1-6.
Pollard, J. R., Arispe, N., Rojas, E., and Pollard, H. B. (1994b). A geometric sequence that

526 Pollard, Arispe, and Rojas
accurately describes allowed multiple conductance levels of ion channels: The "'Three- Halves(3/2) Rule. Biophysical J. 67:647-655.
Ponte, P., Gonzalez-DeWitt, P., Schilling. J., Miller. J., Hsu, D., Greenberg, B., Davis, K.0 Wallace, W., Lieberberg, I., Fuller, F., and Cordell. B. (1988). A new A4 amyloid mRNA contains a domain homologous to serine proteinase inhibitors. Nature 331:525-527.
Price, D., Borchelt, D., Walker, L., and Sisodia, S. (1992). Toxicity of synthetic A-beta peptides and modelling of Atzheimer's disease. NeurobioL Aging 13:623-625.
Raghunathan, G., Seetharamulu, P., Brooks, B. R.. and Guy, H. R. (1990). Models of 6-hemolysin membrane channels and crystal strucutre. Prot. Struct. Funct. Genet. 8:213-225.
Roberts, G. W., Gentleman, S. M., Lynch, A.. and Graham, D. I. (1991). /3-amyloid protein deposition in brain after head trauma. Lancet 338:1421-1423.
Sambamurti, K., Refolo, M., Shioi, J., Pappolla, M. A., and Robak~s. N. K. (1992a). The Alzheimer's Amyloid precursor is cleaved intracellularly in the trans-golgi network or in a post-golgi compartment. Ann. N.Y. Acad. Sci. 674:118-128.
Sambamurti, K., Shioi, J., Anderson. J. P.. Pappolla, M. A.. and Robakis. N. K. (1992b). Evidence for intracellular cleavage of the Alzheimer's amyloid precursor in PC 12 cells. J. Neurosci. Res 33:319-329.
Schubert, D., Behl, C., Lesley. R., Brack, A., Dargusch, R., Sagara, Y., and Kimura, H. (1995). Amyloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. (USA) 92:1989-1993.
Selkoe, D. J. (1991). The molecular pathology of Alzheimer's disease. Neuron 6:487-498. Selkoe, D. J., Podlisny, M. B., Joachim, C. L., Vickers, E. A.. and Lee, G. (1988). Beta-amyloid
precursor protein of Alzheimer's disease occurs as l10-135-kilodalton membrane associated proteins in neural and nonneural tissue. Proc. Natl. Acad. Sci. (USA) 85:7341-7345.
Seubert, P., Oltersdorf. T., Lee, M. G., Barbour, R., Blomquist, C., Davis. D. L., Bryant, K.. Fritz, L. C., Galasko, D., Thai, L. J., Lieberberg. I., and Schenk, D. B. (1993). Secretion of beta amvloid precursor protein cleaved at the amino terminus of the beta-amyloid peptide. Nature 361:260- 263.
Shoji, M., Golde, T., Ghiso, J., Cheung, T., Estus, S., Shaffer, L., Cai, X., McKay, D., Tintner. R., Frangione, B., and Younkin, S. (1992). Production of the Alzheimer's amvloid beta protein by normal proteotytic processing. Science 258:126-129.
Siman, R., Card, P., Nelson, R. B., and Davis, L. G. (1989). Expression of /3 amyloid precursor protein in reactive astrocytes following neuronal damage. Neuron 3:275-285.
Sisodia, S. S., Koo, E. H., Beyreuther, K., Unterbeck. A., and Price, D. L. (1990). Evidence that beta-amyloid protein in Alzheimer's Disease is not derived by normal processing. Science 248:492-495.
Swim, H. E. (1961). Amine and other noncarbonate buffers in cell culture media. Ann. N.Y. Acad. Sci. 92:440-445.
Tanzi, R. E., Gusella, J. F., Watkins, P. C., Bruns, G. A. P., St. George-Hyslop, P., van Keuren. M. L., Patterson, D., Pagan, S., Kurnit, D. M., and Neve, R. L. (1987). Amytoid /3 protein gene: cDNA, mRNA distribution and genetic linkage near the Alzheimer locus. Science 235:880-882.
Tanzi, R. E., McClatchey, A. I., Lamperti, E. D., Villa-Kamaroff, L.. Gusella, J. F., and Neve, R. L. (1988). Protease inhibitor domain encoded by an amyloid precursor mRNA associated with Alzheimer's disease. Nature 331:528-530.
Terry, R. D., and Pefia, C. (1965). Experimental production of neurofibrillary degeneration. Electron microscopy, phosphatase histochemistry, and electron probe analysis. J. Neuropath. Exptt. NeuroL 2,4:200-2t0.
Tsien, R. W., Hess, P., McClesky, E. W., and Rosenberg, R. L. (1987). Calcium channels: mechanisms of selectivity, permeation and block. Ann. Rev. Biophys. Chem, 16:265-290.
Van Nostrand, W. E., Schmaier, A. H., Farrow, J. S., and Cunningham, D. D. (1990). Protease nexin II (amyloid/3-protein precursor): A platlet ~ granule protein. Science 248:745-748.
Vassilacopoulou, D., Ripellino, J. A., Tezapsidis, N., Hook, V. Y. H., and Robakis, N. K. (1995). Full-length and truncated Alzheimer amyloid precursors in chromaffin granules: Solubilization of membrane amyloid precursor is mediated by an enzymatic mechanism. J. Neurochern. 64:2140- 2146.
Whitson, J. S., and Appel, S. H. (1995). Neurotoxicity of A beta amyloid protein in vitro is not altered by calcium channel blockade. Neurobiol. Aging 16:5-10.




















