
Amyloid beta oligomers induce impairment of neuronal insulin receptors

Available online at www.sciencedirect.com
Biomaterials 29 (2008) 1553e1562www.elsevier.com/locate/biomaterials
Functionalised amyloid fibrils for roles in cell adhesion
Sally L. Gras a,b,c,*, Anna K. Tickler c, Adam M. Squires d, Glyn L. Devlin e,Michael A. Horton f, Christopher M. Dobson e, Cait E. MacPhee g
a Department of Chemical and Biomolecular Engineering, The University of Melbourne, Parkville VIC 3010, Australiab The Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville VIC 3010, Australia
c Cavendish Laboratory, University of Cambridge, JJ Thomson Avenue, Cambridge CB3 OHE, UKd Department of Chemistry, University of Reading, Whiteknights, Reading RG6 6AD, UK
e Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UKf The London Centre for Nanotechnology and Department of Medicine, University College London, London WC1H 0AH, UK
g School of Physics, University of Edinburgh, Mayfield Road, Edinburgh EH9 3JZ, UK
Received 23 August 2007; accepted 24 November 2007
Available online 31 December 2007
Abstract
We describe experiments designed to explore the possibility of using amyloid fibrils as new nanoscale biomaterials for promoting andexploiting cell adhesion, migration and differentiation in vitro. We created peptides that add the biological cell adhesion sequence (RGD) ora control sequence (RAD) to the C-terminus of an 11-residue peptide corresponding to residues 105e115 of the amyloidogenic protein trans-thyretin. These peptides readily self-assemble in aqueous solution to form amyloid fibrils, and X-ray fibre diffraction shows that they possess thesame strand and sheet spacing in the characteristic cross-b structure as do fibrils formed by the parent peptide. We report that the fibrils con-taining the RGD sequence are bioactive and that these fibrils interact specifically with cells via the RGD group displayed on the fibril surface. Asthe design of such functionalized fibrils can be systematically altered, these findings suggest that it will be possible to generate nanomaterialsbased on amyloid fibrils that are tailored to promote interactions with a wide variety of cell types.� 2007 Elsevier Ltd. All rights reserved.
Keywords: Self-assembly; Nanotopography; XRD (X-ray diffraction); Fibrils; Bioactivity; RGD peptide
1. Introduction
Self-assembling polypeptides offer great potential as scaf-folds for building bio-inspired materials featuring nanotopogra-phy. Polypeptides comprising repeating [1] or amphiphilic [2]sequences of amino acids have successfully been used to createa variety of fibre-like nanostructures with the ability to supportcells and promote cell differentiation. The self-assembly ofmisfolded proteins offers a further, less explored route for theproduction of protein-based nanofibres. Protein misfoldingpathways can produce non-native fibrillar aggregates that are
* Corresponding author. Department of Chemical and Biomolecular Engi-
neering, The University of Melbourne, Parkville VIC 3010, Australia. Tel.:
þ61 3 8344 6281; fax: þ61 3 8344 4153.
E-mail address: [email protected] (S.L. Gras).
0142-9612/$ - see front matter � 2007 Elsevier Ltd. All rights reserved.
doi:10.1016/j.biomaterials.2007.11.028
rich in b-sheet and contain a highly ordered ‘‘cross-b’’ corestructure [3,4] where b-strands are stacked in b-sheets alongthe fibril axis and adjacent b-sheets are stacked perpendicularto the fibril axis. This arrangement of proteins or peptides con-fers great strength [5] as well as stability under a variety ofenvironmental conditions; properties that have been exploitedin the construction of conductive nanowires [6,7].
Protein fibrils were initially observed in association withdisease, in which context they are known as ‘‘amyloid’’ fibrils.In many cases these fibrils appear to be benign and currentopinion suggests that it is the oligomeric species or pre-fibrillarintermediates which are toxic rather than mature fibrils [8,9].The occurrence of protein fibrils in nature is now thought tobe more widespread, with fibrillar structures identified withinfunctional bacterial biofilms [10] and fibrils found in asso-ciation with melanin machinery in mammalian cells [11].

1554 S.L. Gras et al. / Biomaterials 29 (2008) 1553e1562
Moreover, fibril formation has been demonstrated to be an in-herent property of the polypeptide chain [12e15] and a rangeof peptides and proteins unrelated to disease can be inducedto form amyloid-like fibrils in vitro [13,14,16] allowing thede novo design of fibrils[17e19] with tailored physico-chemi-cal and mechanical properties.
The properties of fibrils may be further extended by func-tionalization, resulting in ligands displayed on the fibril surfaceon a nano-length scale. Proposed applications for functional-ised fibrils include: scaffolds with enzyme activity, nanowiresfor the electronics industry, and diagnostic devices and tem-plates for assembly of inorganic materials [15,20e22]. Thebroad potential of this approach has been illustrated by studieswhich have successfully functionalised fibrils with fluorphores[15,20], biotin [21], cytochrome [22] and functional enzymes[23]. One particularly interesting application is the use of fibrilsto support cells and influence cell morphology and behaviour.A recent report [24] has raised the possibility that fibrils maybe designed for biological applications. The extensive scopefor fibrillar design and similarity between protein fibrils andother natural b-sheet rich assemblies, such as silk [25,26],has also stimulated interest in the possibility of developing pro-tein fibrils as biomaterials.
We have generated a series of bioactive and control fibrilsfrom short synthetic peptides. These peptides were designedto: (1) self-assemble and form model ‘amyloid-like’ fibrillarstructures identical to those formed by misfolded proteins,(2) incorporate ligands which are displayed on the fibril sur-face accessible for cell binding and (3) co-aggregate allowingthe modification of ligand density when mixed or doped fibrilsare formed. The transthyrethrin-(105e115)-peptide (hereafterreferred to as TTR1) from the human transthyretin sequence[27] was selected as the base self-assembling sequence.TTR1 was chosen as its molecular conformation within a fibrilhas been resolved [28], chemically modified forms of TTR1also form fibrils and the peptide has been shown to co-aggre-gate with other proteins[15]. Furthermore, patterned surfacearrays of TTR1 fibrils [29] may be useful for protein-based de-vices or for promoting cell adhesion.
We chose the arginine-glycine-aspartic acid (RGD) se-quence from the extra-cellular matrix protein fibronectin asa model bioactive ligand. This sequence interacts with cell sur-face integrin receptors, plays a critical role in cell adhesion,behaviour and signalling and is the minimum motif requiredfor cell adhesion on a surface [30]. The sequence arginine-alanine-aspartic acid (RAD) is an ideal negative control as ithas similar hydrophilicity to RGD but does not interact withcell integrins [31]. Together these three peptides form an inter-esting case study, with which to study the potential of fibrillarbiomaterials.
2. Materials and methods
2.1. Peptide synthesis
Peptides were synthesised using standard Fmoc-Solid-Phase Peptide
Synthesis chemistry with HBTU/DIEA activation and purified using High
Performance Liquid Chromatography where peptides were eluted in buffer
A (0.1% aqueous trichloroacetic acid) with a gradient of 10e40% buffer B
(0.1% trichloroacetic acid and 10% acetonitrile) over 30 min or on a Sephadex
column eluted with 20% acetic acid and freeze dried twice from a solution of
0.1% aqueous trifluoroacetic acid and 10% acetonitrile. TTR1-dansyl (TTR1-
Dan), TTR1-GGRGDS-dansyl (TTR1-RGD-Dan) and TTR1-GGRADS-
dansyl (TTR1-RAD-Dan) were synthesised using a Dansyl NovaTag resin
(Merck Biosciences Ltd., Nottingham, UK) and purified as described above.
All peptides contained a single dominant peak by mass spectrometry using
a MALDI-TOF/TOF 4700 system (Applied Biosystems, Warrington, UK)
and were >95% pure.
2.2. Fibril formation
The method for TTR1-based fibril formation was based on published pro-
tocols [15]. Peptides were dissolved at 10 mg/mL in purified H20 (dH20,
ELGA, Buckinghamshire, UK) with 10% (v/v) CH3CN, incubated at 37 �Cfor 24 h and then left at room temperature. The percentage of peptide con-
verted into fibrils was assessed by centrifuging samples at 313,000g at 4 �Cfor 50 min to separate fibrils from free peptide. The amino acid concentration
in the supernatant was determined by amino acid analysis (AAA) using a nin-
hydrin-based detection technique [32].
2.3. Transmission electron microscopy
TTR1-based fibrils were examined by transmission electron microscopy
(TEM, Technai 20 FEI Company, Eindhoven, The Netherlands). A 2-ml aliquot
of sample was placed on a formvar and carbon coated nickel electron micros-
copy grid (Agar Scientific, Essex, UK). The grid was rinsed three times with
10 ml of dH2O and stained with 10 ml of uranyl acetate (2 % (w/v), Agar Sci-
entific). Excess water and dye were removed by blotting and the grid air dried.
Images were collected using a LaB6 filament running at 120 kV, using a SIS
Megaview II CCD camera. Fibril dimensions were measured using Scion Im-
age (Scion Corporation, Maryland, USA).
2.4. X-ray diffraction
The structure of TTR1-based fibrils was examined by wide angle X-ray
scattering (WAXS) using reported protocols [33]. Samples were prepared by
air-drying an aliquot (10e20 ml) of TTR1-based fibrils between two wax-filled
capillary ends. X-ray scattering patterns were also obtained for a solution of hy-
drated TTR1 fibrils [34]. X-ray diffraction images were collected at user station
14.1 with an ADSC Quantum 4R CCD detector at Daresbury laboratory at War-
rington, UK (wavelength 1.488 A). The sample-to-detector distance, calibrated
using a wax standard was 153 mm, with exposure times of 10e30 s. Diffraction
patterns were converted to tiff files using the program fit-2d (Hammersley/
ESRF) and subsequently analysed using Matlab codes developed in-house.
The diffraction pattern was divided into 36 sections, each with a 10-degree
range in azimuthal angle. For reflections occurring in either the equatorial or
meridional directions two 10-degree sectors in this direction, on opposite sides
of the reflection, were chosen for analysis. The average intensity profile for
these two sections was used to assign reflection position d, where d was defined
using the position of maximum intensity. For reflections occurring in both the
equatorial and meridional directions four 10-degree sectors were used, two in
each direction. The difference between the positions (d ) of maximum intensity
for the two or four 10-degree sectors was used to determine the error in the po-
sition of each reflection. The structure of TTR1-based fibrils was also examined
by small angle X-ray scattering (SAXS) using the same stalks as for WAXS.
Images were collected at user station 2.1 (Daresbury) with a multi-wire 2D de-
tector (fixed wavelength 1.54 A) calibrated with silver behenate (AgC22H43O2),
with a sample-to-detector distance of 1 m and exposure times of 600 s. Patterns
were analysed as described for WAXS.
2.5. Proteolysis
The susceptibility of TTR1-based peptides and fibrils (10 mg) to trypsin di-
gestion (trypsin gold 200 ng, 50:1 peptide:proteinase weight ratio, Promega,
Southampton, UK) was examined overnight within a 10 ml reaction volume

1555S.L. Gras et al. / Biomaterials 29 (2008) 1553e1562
buffered by 25 mM ammonium bicarbonate at pH 8.0. Before digestion, fibril
samples were separated from monomeric peptide by ultracentrifugation
(313,000g in a TLA-100 rotor at 4 �C for 50 min, Beckman Coulter, Bucking-
hamshire, UK) and sample concentration determined by amino acid analysis.
Samples were then purified and concentrated using a C18 zip tip (Millipore,
Watford, UK) and eluted in 0.1% trifluoroacetic acid and 50% acetonitrile.
Digested and undigested control samples were analysed by mass spectrometry
and by MS/MS analysis (Applied Biosystems, Warrington, UK) using a 1:1
ratio of sample to alpha-cyano-4-hydroxy cinnamic acid matrix solution and
examined by TEM.
2.6. Immunogold labelling
Fibrils containing dansyl labelled peptides were immunogold labelled after
at least 9 days incubation at room temperature. Labelling was performed ac-
cording to published methods [15] using a primary anti-dansyl rabbit IgG
antibody (Invitrogen, Paisley, UK) and a secondary anti-rabbit IgG goat anti-
body (SigmaeAldrich, Dorset, UK). Negative controls were performed on fi-
brils assembled from 100% unlabelled peptide and on fibrils from samples of
TTR1 doped with 1% TTR1-RGD-Dan or 1% TTR1-RAD-Dan and fibrils
from 100% TTR1-Dan. Primary antibodies were not applied to these latter
controls but all other steps remained the same. Immunogold labelled samples
were examined by TEM and the spacing between successive gold particles on
the same side of each fibril was measured using Technai software (Technai).
2.7. Spectroscopy
The spectroscopic properties of C-terminally labelled TTR1-based pep-
tides and fibrils were examined using a protocol similar to a previous study
[15]. Fluorescent properties were measured on a spectrophotometer (Varian,
Oxford, UK) with a 1-cm path length quartz cuvette at room temperature.
Samples (6 mg/mL, labelled peptide or fibrils in water) were excited at
332 nm and emission spectra collected between 400 and 700 nm. Excitation
spectra were collected between 300 and 400 nm. Slit widths were 2.5 and
10 nm.
2.8. Cell culture
Mouse fibroblast cells (3T3) were a gift from Dr R. Brooks (Cambridge,
UK). Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) sup-
plemented with 10% fetal calf serum, 100 U/mL penicillin, 100 mg/mL strep-
tomycin and 0.25 mg/mL fungizone anti-mycotic liquid. Cells were maintained
at 37 �C and 5% CO2 in a humidified environment and were split (1:2) every
2e3 days. Cells were released for sub-cultivation using trypsineEDTA (1�liquid; 2 mL per T175 flask (Nunc, Roskilde, Denmark)). All reagents and me-
dia were from Invitrogen. Immuno-phenotyping of mouse fibroblast 3T3 cells
employing a fluorescence conjugated antibody (anti-CD49e, BD Pharmingen
California, USA) directed against the a5 integrin subunit and an isotype-
matched antibody control (IgG2a k isotype control), confirmed the presence
of a5b1 integrin fibronectin receptor on the cell surface under conditions di-
rectly relevant to bioactivity experiments (see Supplementary Fig. 1).
2.9. Peptide bioactivity assay
A solution-phase competitive adhesion assay was developed to measure the
bioactivity of soluble TTR1-based peptides based on the method used previ-
ously to test RGD bioactivity [30]. Fibronectin (100 ml of 10 mg/mL in
dH20, SigmaeAldrich) was added to black Fluoronunc 96-well Maxisorb
plates (Nunc) and incubated at 4 �C overnight. Wells were then blocked
with 100 ml of 1% bovine serum albumin (BSA; SigmaeAldrich) diluted in
dH20 for at least 1 h at room temperature. Trypsinised 3T3 cells (95% viable
by trypan blue exclusion) were rinsed twice in phosphate buffered saline (PBS)
containing 0.1% BSA, re-suspended at a concentration of 1� 106 cells/mL, in-
cubated with 2.5 mM 20,70-bis(carboxyethyl)-5(60)-carboxyfluorescein aceto-
methyl ester (BCECF) at 37 �C for 30 min and then rinsed twice in PBS
containing 0.1% BSA before being re-suspended in DMEM (serum free) at
1� 106 cells/mL. Peptides were re-suspended in DMEM (serum free). Equal
volumes of cells were then gently mixed with either peptides or additional se-
rum free media (five concentrations, six replicates) and incubated on either fi-
bronectin or BSA control layers for 30 min at 37 �C and 5% CO2 in
a humidified environment. Following incubation non-adherent cells were
removed by gently inverting the plate. Plates were then excited at 503 nm
and fluorescent intensity measured at 528 nm using a SpectraMAX Gemini
4.24 plate reader (Molecular Devices, Berkshire, UK) with SoftmaxPro
3.1.1. software. For calibration cells were fixed with glutaraldehye (1%,
100 ml in dH20; SigmaeAldrich) for 10 min following washing, and stained
with 100 ml of 0.1% crystal violet (SigmaeAldrich) in dH2O. Phase contrast
micrographs were taken using a Axiotech reflected light microscope (Zeiss,
Welwyn Garden City, UK) and cells counted in 10 random fields (at �40 mag-
nification) for each replicate using a calibrated graticule with a Millar Square
(1.96� 10�4 cm2 at �40 magnification).
2.10. Cell spreading assay
A cell adhesion assay was developed based on a protocol for cell spreading
on RGD coated surfaces [35]. TTR1-based fibrils (1.2� 10�10 mol, 100 ml)
were dried in duplicate onto the central wells of a 24-well plate. Fibronectin
was incubated, but not dried, in duplicate on a 24-well plate for 30 min. All
wells except surface controls were treated with BSA (1% (w/v) in water, at
room temperature 30 min). 3T3 cells were dissociated from tissue culture
flasks using trypsin, rinsed twice with Opti-MEM I Reduced Serum Medium
(Invitrogen) supplemented with 100 mg/L CaCl2 and 0.1% (w/v) BSA. Cells
were then seeded at a density of 0.5� 105 cells/well and incubated at 37 �Cand 5% CO2 in a humidified environment for 90 min. Cells were fixed, stained
and counted as outlined above.
2.11. Cell dissociation assay
Live cell imaging was used to examine the interaction between 3T3 mouse
fibroblast cells and TTR1-based fibrils based on the method used to examine
cell interactions with fibronectin fragments [36]. 3T3 cells were dissociated
from tissue culture flasks using enzyme free cell dissociation buffer (5 mL
per T175 flask, Invitrogen) to ensure that extra-cellular proteins contacting
the surface were not disrupted. Cells were seeded into the central 8 wells
of a 24-well plate (Corning, Surrey, UK) at a density of 0.5� 105 cells/
well. This density is less than peptide bioactivity tests to ensure that celle
surface and not cellecell interactions were measured. Cells were incubated
in complete DMEM media with serum overnight. The media were changed
to Opti-MEM I Reduced Serum Medium (Invitrogen) supplemented with
100 mg/L CaCl2 and 0.1% (w/v) BSA and cells equilibrated in these media
for 30 min. TTR1-based peptides or fibrils or RGDS peptide were added to
each well at a final concentration of 300 mM and final volume of 400 ml
and cell behaviour observed using a Nikon TE 2000 inverted microscope
with Simple PCI software in an environmentally controlled chamber set at
37 �C (Solent Scientific, Segensworth, UK). To determine whether TTR1-
based fibrils dissociate during bioassays, TTR1-based fibrils were incubated
with serum free media (300 mM fibrils, 50 ml, 37 �C, 30 min or 1 week).
The solution was centrifuged, and the contents of the pellet examined by
TEM.
3. Results and discussion
3.1. Peptide design
The designed bioactive peptide TTR1-RGD and the controlpeptide TTR1-RAD contain the base self-assembling TTR1peptide (YTIAALLSPYS) and six additional amino acidsat the C-terminal end consisting of: two glycine linker residues,the sequence RGD or RAD and a terminal serine residue. Theseamino acids increase ligand flexibility (glycine linker), enhancebioactivity (glycine and serine amino acids flanking RGD) and
1556 S.L. Gras et al. / Biomaterials 29 (2008) 1553e1562
promote ligand display on the fibril surface. The three peptidesTTR1, TTR1-RGD and TTR1-RAD are collectively referred toas TTR1-based peptides.
3.2. Amyloid fibril formation
The base TTR1 sequence drives assembly and TTR1-RGDand TTR1-RAD peptides formed fibrils under the same condi-tions as for TTR1. Fibril formation was confirmed by testingtraditional criteria [37] including: the ability of samples tobind Congo Red and Thioflavin T dyes, and the presence ofb-sheet rich secondary structure.
3.3. Fibril morphology
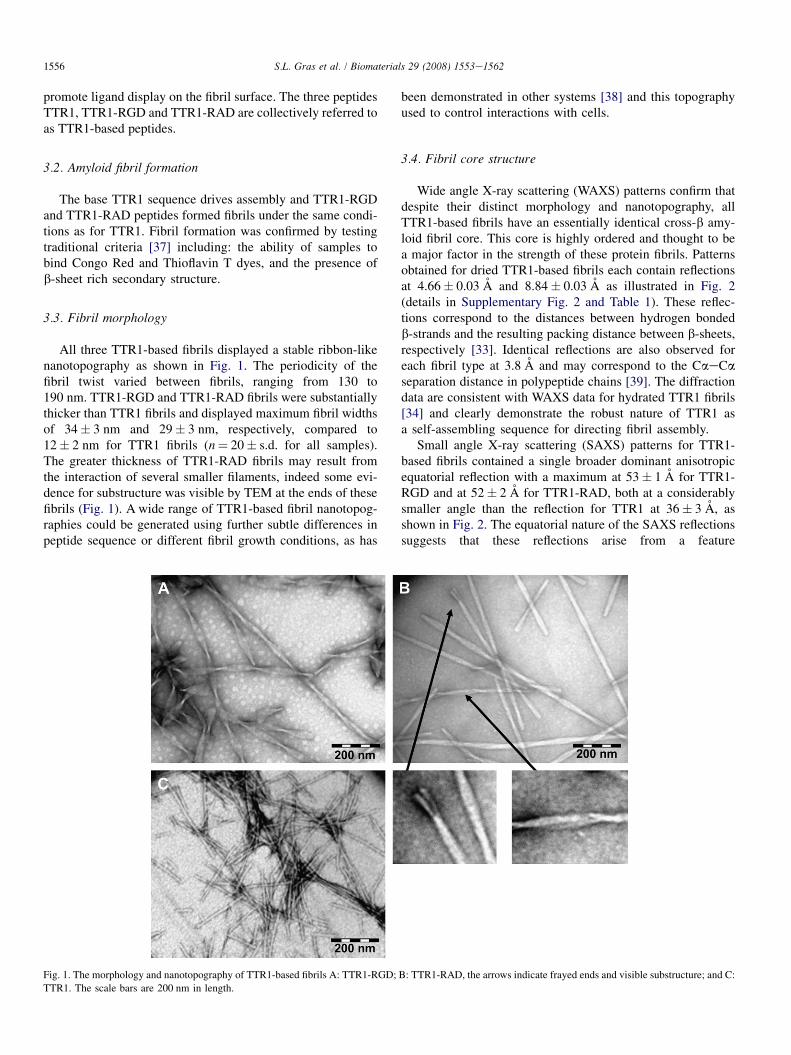
All three TTR1-based fibrils displayed a stable ribbon-likenanotopography as shown in Fig. 1. The periodicity of thefibril twist varied between fibrils, ranging from 130 to190 nm. TTR1-RGD and TTR1-RAD fibrils were substantiallythicker than TTR1 fibrils and displayed maximum fibril widthsof 34� 3 nm and 29� 3 nm, respectively, compared to12� 2 nm for TTR1 fibrils (n¼ 20� s.d. for all samples).The greater thickness of TTR1-RAD fibrils may result fromthe interaction of several smaller filaments, indeed some evi-dence for substructure was visible by TEM at the ends of thesefibrils (Fig. 1). A wide range of TTR1-based fibril nanotopog-raphies could be generated using further subtle differences inpeptide sequence or different fibril growth conditions, as has
Fig. 1. The morphology and nanotopography of TTR1-based fibrils A: TTR1-RGD;
TTR1. The scale bars are 200 nm in length.
been demonstrated in other systems [38] and this topographyused to control interactions with cells.
3.4. Fibril core structure
Wide angle X-ray scattering (WAXS) patterns confirm thatdespite their distinct morphology and nanotopography, allTTR1-based fibrils have an essentially identical cross-b amy-loid fibril core. This core is highly ordered and thought to bea major factor in the strength of these protein fibrils. Patternsobtained for dried TTR1-based fibrils each contain reflectionsat 4.66� 0.03 A and 8.84� 0.03 A as illustrated in Fig. 2(details in Supplementary Fig. 2 and Table 1). These reflec-tions correspond to the distances between hydrogen bondedb-strands and the resulting packing distance between b-sheets,respectively [33]. Identical reflections are also observed foreach fibril type at 3.8 A and may correspond to the CaeCaseparation distance in polypeptide chains [39]. The diffractiondata are consistent with WAXS data for hydrated TTR1 fibrils[34] and clearly demonstrate the robust nature of TTR1 asa self-assembling sequence for directing fibril assembly.
Small angle X-ray scattering (SAXS) patterns for TTR1-based fibrils contained a single broader dominant anisotropicequatorial reflection with a maximum at 53� 1 A for TTR1-RGD and at 52� 2 A for TTR1-RAD, both at a considerablysmaller angle than the reflection for TTR1 at 36� 3 A, asshown in Fig. 2. The equatorial nature of the SAXS reflectionssuggests that these reflections arise from a feature
B: TTR1-RAD, the arrows indicate frayed ends and visible substructure; and C:

Radius-1
(Å-1
)
0.00 0.01 0.02 0.03 0.04 0.05 0.06 0.07
Re
sc
ale
d in
te
ns
ity
(a
rb
itra
ry
u
nits
)
0.0
0.2
0.4
0.6
0.8
1.0
1.2BA C
Fig. 2. Wide and small angle X-ray scattering determined for TTR1-based fibrils. A: Wide angle X-ray fibre diffraction pattern of a stalk of dried TTR1-RGD
fibrils. Major reflections occur at 4.7 A (7) and at 8.8 A (;) parallel and perpendicular to the fibril axis, respectively, which is indicated by an arrow. B: Small
angle X-ray fibre diffraction pattern determined for a stalk of dried TTR1-RGD fibrils. C: Radial intensity profile for the X-ray diffraction patterns in the equatorial
direction for: TTR1-RGD fibrils (solid line), TTR1-RAD fibrils (dashed line) and TTR1 fibrils (dotted line). The peak in intensity for TTR1 fibrils is also marked
by an arrow. The intensities have been scaled to allow comparison.
1557S.L. Gras et al. / Biomaterials 29 (2008) 1553e1562
perpendicular to the fibril axis. We propose that this spacingrepresents a repeat in the direction of the b-strand arisingfrom two or more strands and reflects the length of one b-sheet. Fibrils from short peptides such as those studied hereare likely to consist of arrays of extended chains [28] andthe dimensions of the b-sheet are expected to be dependenton the length of the constituent peptides. The reflection ob-served for TTR1 at 36� 3 A is consistent with the peptidelength estimated for TTR1 when all residues are in an ex-tended b-strand conformation (w37� 2 A, assuming a lengthper residue of 3.4� 0.2 A [28]); suggesting that all TTR1 res-idues are included in the b-strand of these fibrils, consistentwith previous solid-state NMR results [28]. Not all residueswithin TTR1-RGD and TTR1-RAD peptides may be incorpo-rated within the b-strand and some residues may remain flex-ible and disordered. Indeed, the incorporation of two glycineresidues acting as a linker sequence was designed to promoteflexibility in this region. The positions of the SAXS reflectionsfor TTR1-RGD and TTR1-RAD fibrils are consistent withboth peptides adopting an extended b structure significantlylonger (w52 A) than that adopted by TTR1 (w36 A). How-ever, the observed spacings of 52.0e53.0 A are slightlyshorter than the calculated maximum peptide length ofw58� 3 A estimated for TTR1-RGD and TTR1-RAD whenall residues are included in a fully extended b-strand indicat-ing the presence of disordered structure. We suggest that thisdisordered structure is located at the C-terminus where the ad-ditional functional and control residues have been added,rather than the N-terminus, which is identical to the TTR1 se-quence. It is therefore possible that all six amino acids addedto the basic TTR1 framework may be disordered and accessi-ble for cell binding.
The display of RGD or RAD ligands on the surface of TTR1-RGD and TTR1-RAD fibrils was probed using the proteinasetrypsin, as this enzyme specifically cleaves the C-terminalside of arginine residues which occur within the ligand sequenceof both TTR1-RGD and TTR1-RAD but not the TTR1 peptide.
As a control, the digestion of soluble, non-fibrillar TTR1-RGDand TTR1-RAD peptides was examined. This resulted inidentical 1468.7 Da fragments (>99% complete digestion)and MS/MS analysis identified the sequence YTIAALLSPYSGGR, indicating that efficient cleavage of the ReG andReA bond had in fact occurred. Trypsin digestion of the fibrillarforms of TTR1-RGD and TTR1-RAD was consistent with ex-posure of the RGD and RAD ligands on the fibril surface. Fibrilsassembled from the TTR1-based peptides were purified by ul-tracentrifugation, subjected to trypsin digestion and examinedby MALDI-TOF MS, which is known to disrupt large non-covalent protein complexes [40]. As expected, the peptidewithin TTR1 fibrils was not affected by trypsin digestion.Within the TTR1-RGD or TTR1-RAD fibrils, full length pep-tides were also observed (1727.81 Da or 1741.89 Da, respec-tively); however, a significant proportion of the constituentpeptides was cleaved by trypsin, generating a 1468.7-Dafragment (see Supplementary Fig. 3). Digested fibrils werealso intact when examined by TEM.
3.5. Ligand display on fibrils
Cells are known to be highly responsive both to local topog-raphy [41,42] and to the density of ligands presented on a sur-face [43]. The ability to control the density and the spatialarrangement of RGD ligands on the fibril surface on the nano-scale, would therefore be highly advantageous and increase theutility of TTR1-RGD fibrils as biomaterials. We examinedwhether ligand density can be modulated when mixed fibrilsare formed from the co-aggregation of TTR1-RGD or TTR1-RAD peptides with the base TTR1 peptide sequence. Weemployed immunogold labelling to confirm the exposure ofthe C-terminus and to determine ligand density. The C-terminiof the TTR1-based peptides were covalently labelled with thedansyl fluorophore to provide an epitope for immunogoldlabelling, creating the peptides TTR1-Dan, TTR1-RGD-Danand TTR1-RAD-Dan. The chemical modification involved in

1558 S.L. Gras et al. / Biomaterials 29 (2008) 1553e1562
dansyl labelling did not detectably disrupt fibril assembly andfibrils formed from the labelled peptides largely resembled fi-brils from unlabelled TTR1. The dansyl fluorophore is also en-vironmentally sensitive and can provide further informationconcerning the environment of the fluorophore [15].
The C-terminus of dansyl labelled peptides was found to beexposed on the surface of fibrils and was successfully immu-nogold labelled with antibodies raised against the fluorophoreas illustrated in Fig. 3. Gold particles were observed to bebound specifically to dansyl groups exposed on the fibril sur-face. Very few gold particles resulted from non-specific attach-ment in samples of unlabelled fibrils where the dansyl epitopewas absent, or in controls with no primary antibody. Althoughsome gold particles did bind non-specifically to the grid sur-face (Fig. 3), it is likely that these particles were bound toa low level of labelled non-aggregated peptide molecules, asfibrils were not purified by ultracentrifugation. The successfulimmunogold labelling of these fibrils is consistent with trypsindigestion experiments. The successful immunogold labellingof fibrils assembled from 100% TTR1-Dan further suggeststhat chemical moieties or amino acid residues added afterthe C-terminus of the TTR1 peptide are exposed on the fibrilsurface, indicating the entire RGD ligand and linking glycineresidues will be accessible for cell binding.
Blends of both unlabelled TTR1 and dansyl labelled TTR1-based peptides were found to form mixed heterogenous fibrilsin which the ligand density could be modulated on the nano-scale. Labelled peptides were readily incorporated into mixedfibrils as no undecorated fibrils were found by immunogold la-belling (Fig. 3), indicating that peptides had not segregatedand formed homogeneous fibrils. A significant blue shift inthe emission spectrum was also observed when labelled pep-tides were mixed with unlabelled TTR1 (see SupplementaryTable 2). This shift indicates that the environment of at leastsome of the dansyl fluorophores has changed from an aqueousexposure, where fluorescence is largely quenched, to a morehydrophobic environment such as the interior of a mixed fibril.Unlabelled peptides are also readily incorporated into mixedfibrils and result in an increase in the measured spacing be-tween gold particles on immunogold labelled fibrils as the per-centage of labelled peptide is decreased (see SupplementaryTable 2). Ligand density was not directly proportional to theconcentration of labelled peptide used to form fibrils. Thiscould be due to the different rates at which labelled and unla-belled peptides are incorporated into the growing fibril or dueto subtle differences in the level of ligand exposure on the fi-bril surface for different mixed fibrils. Sections of high andlow ligand density were also observed within the immunogoldlabelled fibrils suggesting that the labelled and unlabelledTTR1-based peptides do not always distribute evenly withinthe mixed fibrils.
The successful gold labelling of fibrils containing dansylTTR1-RGD peptides indicates that integrin receptors willbind to the surface of these fibrils without steric hindrance,as the integrin receptor [44] is a similar size to the 10 nmgold particle used for labelling. The range of predictedRGD densities is also biologically relevant; 2000 RGD/mm
(100% doping) to 12.8 RGD/mm (1% doping), equating toa distance of 0.5e85 nm between ligands based on a simplemodel of a single b-sheet with parallel b-strands (see Supple-mentary Table 2). Integrin receptors are typically spacedw50 nm apart [45], suggesting RGD groups will be in excessof cell receptors, even if not all ligands are exposed. The den-sity of ligands can also be modulated to respond to cellularrequirements.
3.6. Peptide and fibril bioactivity
We next explored the bioactivity of the TTR1-based peptidesand fibrils. Fibrils were considered suitable for testing oncesamples had matured (>1 month) and at least 80% of peptidehad converted to fibrils, as assessed by amino acid analysis ofmonomeric peptide in the supernatant of ultracentrifugedsamples. A high proportion of TTR1-based peptide could besuccessfully converted to fibrils (w94% of TTR1-RGD orTTR1-RAD and w99% of TTR1 peptide) and TEM confirmedthat TTR1-based fibrils are the dominant species with no signif-icant quantity of amorphous aggregates evident within thesesamples. The bioactivity of TTR1-based peptides as well asthe positive control peptides RGDS and GRGDS was tested insolution using a typical competitive adhesion assay where analiquot of suspended cells, preincubated with soluble (i.e.non-fibrillar) peptide, was placed on a fibronectin coated sur-face [30]. Peptide bioactivity prevents the cells binding to thefibronectin surface in a competitive manner. Assays were per-formed in the absence of serum to prevent extra-cellular matrixproteins present in serum from blocking integrin receptors,ensuring optimal binding to extrinsic RGD ligands.
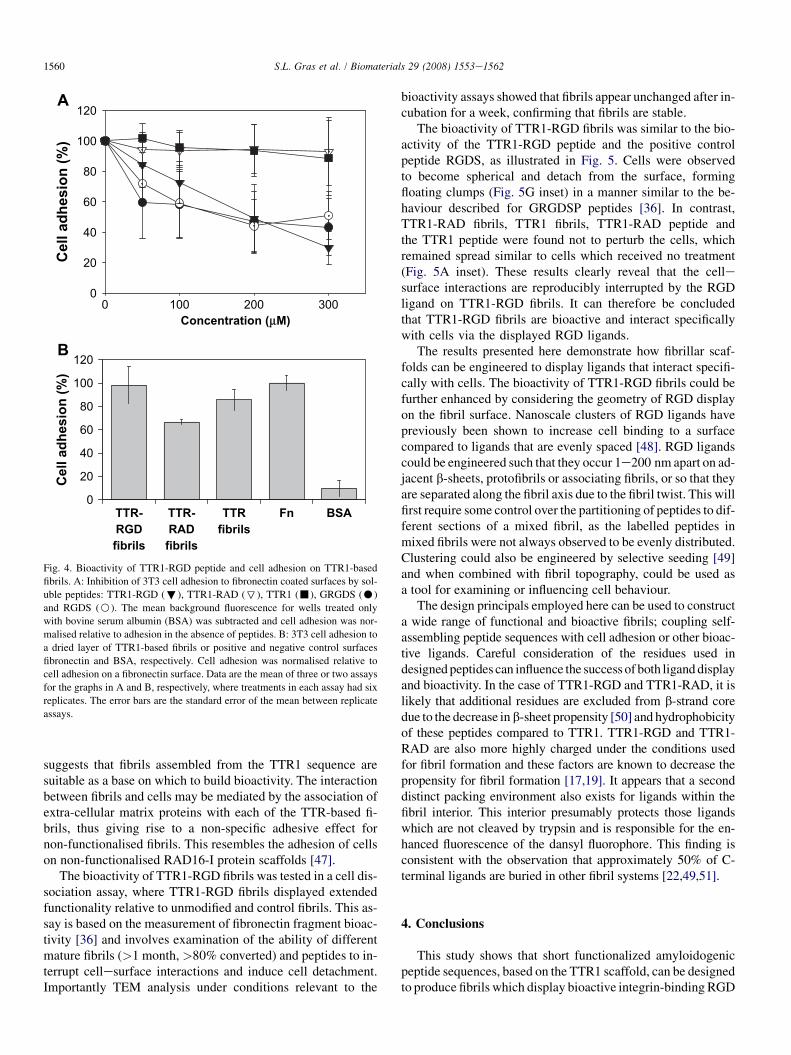
As expected, soluble TTR1-RGD peptide was found to havestrong cell binding properties and dramatically reduced celladhesion to fibronectin coated surfaces in a concentration-dependent manner. At a peptide concentration of 200 mM, forexample, TTR1-RGD reduced cell adhesion to fibronectin by50� 20% (n¼ 3� s.e.m.) and displayed similar behaviour topositive control peptides GRGDS (50� 20%) and RGDS(60� 20%) recorded here and in other studies [30,45,46]. Incontrast, TTR1-RAD and TTR1 peptides displayed limitedadhesive properties at the same concentration, reducing cellbinding by only 7� 17% and 5� 16%, respectively, as shownin Fig. 4. Although relatively high variability was observed be-tween replicate wells, such variability is typical of solution-phase adhesion assays.
All three TTR1-based fibrils also supported cell attachmentin a cell adhesion assay, where the ability of cells to adhere toa mature fibril (>1 month, >80% converted) coated surfacewas assessed relative to a bovine serum albumin (BSA) coatedcontrol surface. There was no substantial difference in the num-ber of attached cells to fibronectin (100� 6%; n¼ 2� s.e.m.)or TTR1-RGD (98� 16%) and TTR1 fibrils (85� 10%); how-ever, there were fewer cells on surfaces coated with TTR1-RADfibrils (67� 2%) as shown in Fig. 4. This assay does not neces-sarily measure the effect of fibril nanotopography but does sug-gest that a continuous layer of non-functionalised fibrils cansupport cell attachment for short periods of time. It also
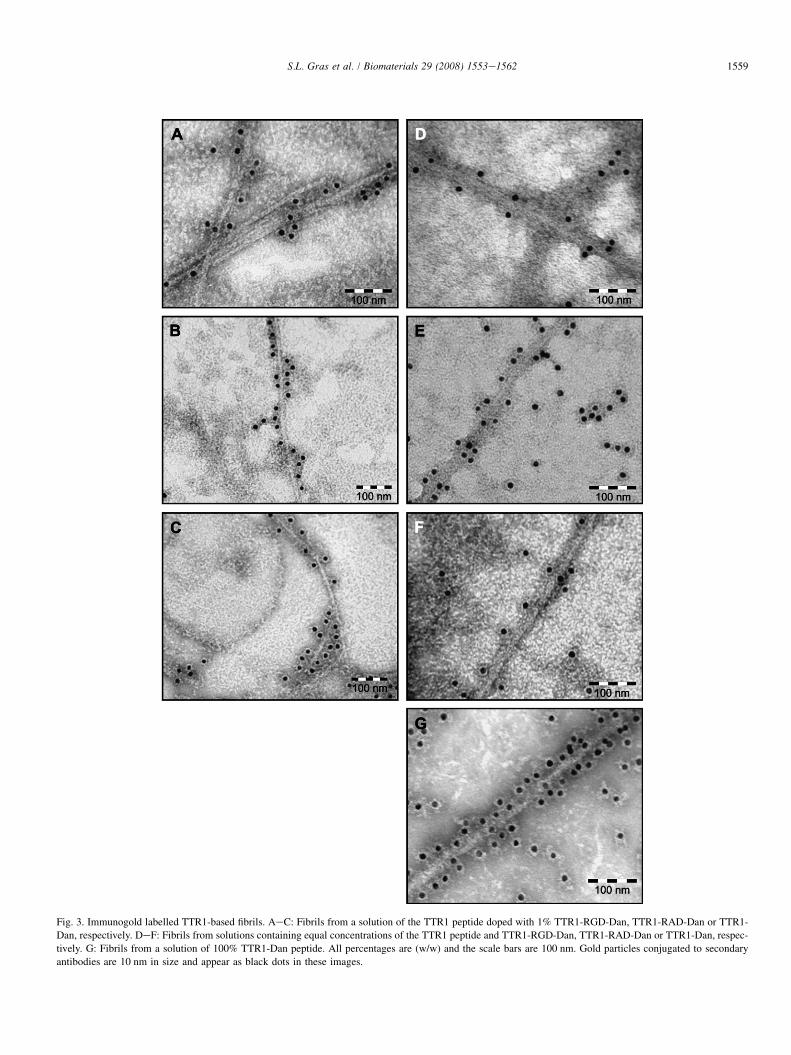
Fig. 3. Immunogold labelled TTR1-based fibrils. AeC: Fibrils from a solution of the TTR1 peptide doped with 1% TTR1-RGD-Dan, TTR1-RAD-Dan or TTR1-
Dan, respectively. DeF: Fibrils from solutions containing equal concentrations of the TTR1 peptide and TTR1-RGD-Dan, TTR1-RAD-Dan or TTR1-Dan, respec-
tively. G: Fibrils from a solution of 100% TTR1-Dan peptide. All percentages are (w/w) and the scale bars are 100 nm. Gold particles conjugated to secondary
antibodies are 10 nm in size and appear as black dots in these images.
1559S.L. Gras et al. / Biomaterials 29 (2008) 1553e1562
Concentration (µM)
0 100 200 300
Cell ad
hesio
n (%
)
0
20
40
60
80
100
120A
B
0
20
40
60
80
100
120
TTR-
RGD
fibrils
TTR-
RAD
fibrils
TTR
fibrils
Fn BSA
Ce
ll a
dh
es
io
n (%
)
Fig. 4. Bioactivity of TTR1-RGD peptide and cell adhesion on TTR1-based
fibrils. A: Inhibition of 3T3 cell adhesion to fibronectin coated surfaces by sol-
uble peptides: TTR1-RGD (;), TTR1-RAD (7), TTR1 (-), GRGDS (C)
and RGDS (B). The mean background fluorescence for wells treated only
with bovine serum albumin (BSA) was subtracted and cell adhesion was nor-
malised relative to adhesion in the absence of peptides. B: 3T3 cell adhesion to
a dried layer of TTR1-based fibrils or positive and negative control surfaces
fibronectin and BSA, respectively. Cell adhesion was normalised relative to
cell adhesion on a fibronectin surface. Data are the mean of three or two assays
for the graphs in A and B, respectively, where treatments in each assay had six
replicates. The error bars are the standard error of the mean between replicate
assays.
1560 S.L. Gras et al. / Biomaterials 29 (2008) 1553e1562
suggests that fibrils assembled from the TTR1 sequence aresuitable as a base on which to build bioactivity. The interactionbetween fibrils and cells may be mediated by the association ofextra-cellular matrix proteins with each of the TTR-based fi-brils, thus giving rise to a non-specific adhesive effect fornon-functionalised fibrils. This resembles the adhesion of cellson non-functionalised RAD16-I protein scaffolds [47].
The bioactivity of TTR1-RGD fibrils was tested in a cell dis-sociation assay, where TTR1-RGD fibrils displayed extendedfunctionality relative to unmodified and control fibrils. This as-say is based on the measurement of fibronectin fragment bioac-tivity [36] and involves examination of the ability of differentmature fibrils (>1 month, >80% converted) and peptides to in-terrupt cellesurface interactions and induce cell detachment.Importantly TEM analysis under conditions relevant to the
bioactivity assays showed that fibrils appear unchanged after in-cubation for a week, confirming that fibrils are stable.
The bioactivity of TTR1-RGD fibrils was similar to the bio-activity of the TTR1-RGD peptide and the positive controlpeptide RGDS, as illustrated in Fig. 5. Cells were observedto become spherical and detach from the surface, formingfloating clumps (Fig. 5G inset) in a manner similar to the be-haviour described for GRGDSP peptides [36]. In contrast,TTR1-RAD fibrils, TTR1 fibrils, TTR1-RAD peptide andthe TTR1 peptide were found not to perturb the cells, whichremained spread similar to cells which received no treatment(Fig. 5A inset). These results clearly reveal that the cellesurface interactions are reproducibly interrupted by the RGDligand on TTR1-RGD fibrils. It can therefore be concludedthat TTR1-RGD fibrils are bioactive and interact specificallywith cells via the displayed RGD ligands.
The results presented here demonstrate how fibrillar scaf-folds can be engineered to display ligands that interact specifi-cally with cells. The bioactivity of TTR1-RGD fibrils could befurther enhanced by considering the geometry of RGD displayon the fibril surface. Nanoscale clusters of RGD ligands havepreviously been shown to increase cell binding to a surfacecompared to ligands that are evenly spaced [48]. RGD ligandscould be engineered such that they occur 1e200 nm apart on ad-jacent b-sheets, protofibrils or associating fibrils, or so that theyare separated along the fibril axis due to the fibril twist. This willfirst require some control over the partitioning of peptides to dif-ferent sections of a mixed fibril, as the labelled peptides inmixed fibrils were not always observed to be evenly distributed.Clustering could also be engineered by selective seeding [49]and when combined with fibril topography, could be used asa tool for examining or influencing cell behaviour.
The design principals employed here can be used to constructa wide range of functional and bioactive fibrils; coupling self-assembling peptide sequences with cell adhesion or other bioac-tive ligands. Careful consideration of the residues used indesigned peptides can influence the success of both ligand displayand bioactivity. In the case of TTR1-RGD and TTR1-RAD, it islikely that additional residues are excluded from b-strand coredue to the decrease in b-sheet propensity [50] and hydrophobicityof these peptides compared to TTR1. TTR1-RGD and TTR1-RAD are also more highly charged under the conditions usedfor fibril formation and these factors are known to decrease thepropensity for fibril formation [17,19]. It appears that a seconddistinct packing environment also exists for ligands within thefibril interior. This interior presumably protects those ligandswhich are not cleaved by trypsin and is responsible for the en-hanced fluorescence of the dansyl fluorophore. This finding isconsistent with the observation that approximately 50% of C-terminal ligands are buried in other fibril systems [22,49,51].
4. Conclusions
This study shows that short functionalized amyloidogenicpeptide sequences, based on the TTR1 scaffold, can be designedto produce fibrils which display bioactive integrin-binding RGD

Fig. 5. Dissociation of 3T3 fibroblast cells from tissue culture surface after 4 h co-incubation with TTR1-based fibrils and peptides. A: Typical cell layer prior to
treatment. B: After incubation with the RGDS positive control peptide. C: Typical layer of cells not exposed to any treatment. D: After incubation with the TTR1-
RGD peptide. E: After incubation with the TTR1-RAD peptide. F: After incubation with the TTR1 peptide. G: After incubation with TTR1-RGD fibrils. H: After
incubation with TTR1-RAD fibrils. I: After incubation with TTR1 fibrils. Arrows indicate well-spread cells (A only) or clumps of spherical dissociated cells. These
images were representative of three different assays. Magnification �10.
1561S.L. Gras et al. / Biomaterials 29 (2008) 1553e1562
ligands on the fibril surface. These fibrils interact specificallywith cells, demonstrating the generation of fundamentallynew properties compared with non-functionalised fibrils. The fi-brils show some variation in nanotopography, yet the cross-b fi-bril core, including inter-strand and inter-sheet spacing, iscompletely preserved. A significant proportion of the ligandsincorporated into the fibril is solvent-exposed despite the choiceof a short (two residue) linker region and it was found to be sim-ple to generate mixed fibrils and adjust the density of ligands tosuit cellular requirements, although ligands were not alwaysevenly spaced. The results of these experiments strongly sug-gest that functionalized fibrils have real potential as componentsfor novel bio-mimetic materials or as tools to probe and exploitfundamental biological processes and cell behaviour.
Acknowledgement
The authors acknowledge Peter Sharrat, Ben Luisi andDarerca Owen for technical assistance and the Gates Cam-bridge Trust, the Wellcome Trust, the Leverhulme Centre forBiological Complexity, the Australian National Health andMedical Research Council, the IRC for Nanotechnology andthe Royal Society for financial support.
Appendix. Supplementary material
Supplementary material can be found, in the online version,at doi: 10.1016/j.biomaterials.2007.11.028.
References
[1] Genove E, Shen C, Zhang S, Semino CE. The effect of functionalized
self-assembling peptide scaffolds on human aortic endothelial cell func-
tion. Biomaterials 2005;16:3341e51.
[2] Hartgerink JD, Beniash E, Stupp SI. Self-assembly and mineralization of
peptide-amphiphile nanofibers. Science 2001;294:1684e8.
[3] Serpell LC, Sunde M, Benson MD, Tennent GA, Pepys MB, Fraser PE.
The protofilament substructure of amyloid fibrils. J Mol Biol 2000;300:
1033e9.
[4] Kelly JW. Mechanisms of amyloidogenesis. Nat Struct Mol Biol
2000;7:824e6.
[5] Smith JF, Knowles TPJ, Dobson CM, MacPhee CE, Welland ME.
Characterization of the nanoscale properties of individual amyloid fibrils.
Proc Natl Acad Sci U S A 2006;103:15806e11.
[6] Scheibel T, Parthasarathy R, Sawicki G, Lin X-M, Jaeger H,
Lindquist SL. Conducting nanowires built by controlled self-assembly
of amyloid fibres and selective metal deposition. Proc Natl Acad Sci
U S A 2003;100:4527e32.
[7] Reches M, Gazit E. Casting metal nanowires within discrete self-
assembled peptide nanotubes. Science 2003;300:625e7.

1562 S.L. Gras et al. / Biomaterials 29 (2008) 1553e1562
[8] Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, et al.
Inherent toxicity of aggregates implies a common mechanism for protein
misfolding diseases. Nature 2002;416:507e11.
[9] Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G,
et al. High-level neuronal expression of Abeta 1-42 in wild-type human
amyloid protein precursor transgenic mice: synaptotoxicity without
plaque formation. J Neurosci 2000;20:4050e8.
[10] Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M,
et al. Role of Escherichia coli curli operons in directing amyloid fibril
formation. Science 2002;295:851e5.
[11] Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW.
Functional amyloid formation within mammalian tissue. PLoS 2006;4:e6.
[12] Dobson CM. Protein misfolding, evolution and disease. Trends Biochem
Sci 1999;24:329e32.
[13] Fandrich M, Fletcher M, Dobson CM. Amyloid fibrils from muscle
myoglobin. Nature 2001;410:165e6.
[14] Guijarro JI, Sunde M, Jones JA, Campbell ID, Dobson CM. Amyloid
fibril formation by an SH3 domain. Proc Natl Acad Sci U S A
1998;95:4224e8.
[15] MacPhee CE, Dobson CM. Formation of mixed fibrils demonstrates the
generic nature and potential utility of amyloid nanostructures. J Am
Chem Soc 2000;122:12707e13.
[16] Litvinovich SV, Brew SA, Aota S, Akiyama SK, Haudenschild C,
Ingham KC. Formation of amyloid-like fibrils by self-association of a par-
tially unfolded fibronectin type III module. J Mol Biol 1998;280:245e58.
[17] Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization
of the effects of mutations on peptide and protein aggregation rates.
Nature 2003;424:805e8.
[18] Pawar AP, Dubay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM.
Prediction of ‘‘aggregation-prone’’ and ‘‘aggregation-susceptible’’ regions
in proteins associated with neurodegenerative diseases. J Mol Biol 2005;
350:379e92.
[19] Lopez de la Paz M, Goldie K, Zurdo J, Lacroix E, Dobson CM,
Hoenger A, et al. De novo designed peptide-based amyloid fibrils.
Proc Natl Acad Sci U S A 2002;99:16052e7.
[20] Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein
function in yeast. Mol Cell 2000;5:163e72.
[21] Kodama HM, Yamashita S, Mihara T. Construction of a protein array of
amyloid-like fibrils using co-assembly of designed peptides. Chem
Commun (Camb) 2004;24:2876e7.
[22] Baldwin AJ, Bader R, Christodoulou J, MacPhee CE, Dobson CM,
Barker PD. Cytochrome display on amyloid fibrils. J Am Chem Soc
2006;128:2162e3.
[23] Baxa U, Speransky V, Steven AC, Wickner RB. Mechanism of inactiva-
tion on prion conversion of the Saccharomyces cerevisiae Ure2 protein.
Proc Natl Acad Sci U S A 2002;99:5253e60.
[24] Kasai S, Ohga Y, Mochizuki M, Nishi N, Kadoya YK, Nomizu M.
Multifunctional peptide fibrils for biomedical materials. Biopolymers
2004;76:27e33.
[25] Slotta U, Hess S, Spiess K, Stromer T, Serpell L, Scheibel T. Spider silk and
amyloid fibrils: a structural comparison. Macromol Biosci 2007;7:183e8.
[26] Bini E, Foo CW, Huang J, Karageorgiou V, Kitchel B, Kaplan DL.
RGD-functionalized bioengineered spider dragline silk biomaterial.
Biomacromolecules 2006;7:3139e45.
[27] Gustavsson A, Engstrom U, Westermark P. Normal transthyretin and
synthetic transthyretin fragments from amyloid-like fibrils in vitro.
Biochem Biophys Res Commun 1991;175:1159e64.
[28] Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM,
Griffin RG. High-resolution molecular structure of a peptide in an
amyloid fibril determined by magic angle spinning NMR spectroscopy.
Proc Natl Acad Sci U S A 2004;101:711e6.
[29] Mesquida P, Ammann DL, MacPhee CE, McKendry RA. Microarrays of
peptide fibrils created by electrostatically controlled deposition. Advan
Mater 2005;17:893e7.
[30] Pierschbacher M, Ruoslahti E. Variants of the cell recognition site of
fibronectin that retain attachment-promoting activity. Proc Natl Acad
Sci U S A 1984;81:5985e8.
[31] Hautanen A, Gailit J, Mann D, Ruoslahti E. Effects of modifications of
the RGD sequence and its context on recognition by the fibronectin
receptor. J Biol Chem 1989;264:1437e42.
[32] Bader R, Bamford R, Zurdo JS, Luisi BF, Dobson CM. Probing the
mechanism of amyloidogenesis through a tandem repeat of the
PI3-SH3 domain suggests a generic model for protein aggregation and
fibril formation. J Mol Biol 2006;356:189e208.
[33] Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CCF.
Common core structure of amyloid fibrils by synchrotron X-ray
diffraction. J Mol Biol 1997;273:729e39.
[34] Squires AM, Devlin GD, Gras SL, Tickler AK, MacPhee CE,
Dobson CM. X-ray scattering study of the effect of hydration on the
cross-beta spine of amyloid fibrils. J Am Chem Soc 2006;128:11738e9.
[35] Reinhart-King CA, Dembo M, Hammer DA. The dynamics and
mechanics of endothelial cell spreading. Biophys J 2005;89:676e89.
[36] Hayman E, Pierschbacher M, Ruoslahti E. Synthetic peptide with cell
attachment activity of fibronectin. J Cell Biol 1985;100:1948e54.
[37] Nilsson MR. Techniques to study amyloid fibril formation in vitro.
Methods 2004;34:151e60.
[38] Petkova AT, Leapman RD, Guo Z, Yau W-M, Mattson MP, Tycko R.
Self-propagating, molecular-level polymorphism in Alzheimer’s {beta}-
amyloid fibrils. Science 2005;307:262e5.
[39] Blake CCF, Serpell LC. Synchrotron X-ray studies suggest that the core
of the transthyretin amyloid fibril is a continuous b-sheet helix. Structure
1996;4:989e98.
[40] Distler A, Alilison J, Hiser C, Qin L, Hilmi Y, Ferguson-Miller S. Mass
spectrometric detection of protein, lipid and heme components of
cytochrome c oxidase from R. sphaeroides and the stabilization of
non-covalent complexes from the enzyme. Eur J Mass Spectrom
(Chichester, Eng) 2004;10:295e308.
[41] Dalby MJ, Riehle MO, Sutherland DS, Agheli H, Curtis ASG. Changes
in fibroblast morphology in response to nano-columns produced by
colloidal lithography. Biomaterials 2004;25:5415e22.
[42] Teixeira AI, Abrams GA, Bertics PJ, Murphy CJ, Nealey PF. Epithelial
contact guidance on well-defined micro- and nanostructured substrates.
J Cell Sci 2003;116:1881e92.
[43] Maheshwari G, Brown G, Lauffenburger D, Wells A, Griffith L. Cell
adhesion and motility depend on nanoscale RGD clustering. J Cell Sci
2000;113:1677e86.
[44] Xiong J-P, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL,
et al. Crystal structure of the extracellular segment of integrin alphaVbeta
3 in complex with an Arg-Gly-Asp ligand. Science 2002;296:151e5.
[45] Puleo DA, Bizios R. RGDS tetrapeptide binds to osteoblasts and inhibits
fibronectin-mediated adhesion. Bone 1991;12:271e6.
[46] Akiyama S, Yamada KJ. Synthetic peptides competitively inhibit both
direct binding to fibroblasts and functional biological assays for the
purified cell-binding domain of fibronectin. Biol Chem 1985;260:
10402e5.
[47] Semino CE, Kasahara J, Hayashi Y, Zhang S. Entrapment of migrating
hippocampal neural cells in three-dimensional peptide nanofiber scaffold.
Tissue Eng 2004;10:643e55.
[48] Koo LY, Irvine DJ, Mayes AM, Lauffenburger DA, Griffith LG. Co-
regulation of cell adhesion by nanoscale RGD organization and mechan-
ical stimulus. J Cell Sci 2002;115:1423e33.
[49] Scheibel T, Kowall AS, Bloom JD. Bidirectional amyloid fibre growth
for a yeast prion determinant. Curr Biol 2001;11:366e9.
[50] Street AG, Mayo SL. Intrinsic beta-sheet propensities result from van der
Waals interactions between side chains and the local backbone. Proc Natl
Acad Sci U S A 1999;96:9074e6.
[51] Nilsson MR, Dobson CM. Chemical modification of insulin in amyloid
fibrils. Protein Sci 2003;12:2637e41.
Copyright © 2022 FDOKUMEN




















