
Function in Escherichia coli of the non-catalytic part of RNase E: role in the degradation of...
13
Molecular Microbiology (2002) 45 (5), 1231–1243 © 2002 Blackwell Science Ltd Blackwell Science, LtdOxford, UKMMIMolecular Microbiology0950-382XBlackwell Science, 200245Original Article The non-catalytic part of RNase E in vivoA. Leroy et al. Accepted 7 June, 2002. *For correspondence. E-mail [email protected]; Tel. ( + 33) 5 6133 5894; Fax ( + 33) 5 6133 5886. Present addresses: † Developmental Biology Programme, Euro- pean Molecular Biology Laboratory, Heidelberg, Germany. ‡ Unité des Interactions Bactéries-Cellules, Institut Pasteur, 28 rue du Dr Roux, 75724 Paris Cedex 15, France. Function in Escherichia coli of the non-catalytic part of RNase E: role in the degradation of ribosome-free mRNA Anne Leroy, 1 Nathalie F. Vanzo, 1† Sandra Sousa, 2‡ Marc Dreyfus 2 and Agamemnon J. Carpousis 1 * 1 Laboratoire de Microbiologie et Génétique Moléculaire (CNRS, UMR 5100), Paul Sabatier Université, 118 rue de Narbonne, 31062 Toulouse, France. 2 Laboratoire de Génétique Moléculaire (CNRS, UMR 8541), Ecole Normale Supérieure, 46 rue d'Ulm, 75230 Paris, France. Summary RNase E contains a large non-catalytic region that binds RNA and the protein components of the Escher- ichia coli RNA degradosome. The rne gene was replaced with alleles encoding deletions in the non- catalytic part of RNase E. All the proteins are stable in vivo . RNase E activity was tested using a P T7 – lacZ reporter gene, the message of which is particularly sensitive to degradation because translation is uncoupled from transcription. The non-catalytic region has positive and negative effectors of mRNA degradation. Disrupting RhlB and enolase binding resulted in hypoactivity, whereas disrupting PNPase binding resulted in hyperactivity. Expression of the mutant proteins in vivo anticorrelates with activity showing that autoregulation compensates for defec- tive function. There is no simple correlation between RNA binding and activity in vivo . An allele ( rne131 ), expressing the catalytic domain alone, was put under P lac control. In contrast to rne + , low expression of rne131 severely affects growth. Even with autoregu- lation, all the mutants are less fit when grown in com- petition with wild type. Although the catalytic domain of RNase E is sufficient for viability, our work demon- strates that elements in the non-catalytic part are nec- essary for normal activity in vivo . Introduction The RNA degradosome of Escherichia coli is a multien- zyme complex that was discovered during efforts to purify and characterize RNase E (Carpousis et al ., 1994; 1999; Py et al ., 1994; 1996; Miczak et al ., 1996). The other integral components of the degradosome are enolase, an RNA helicase (RhlB) and polynucleotide phosphorylase (PNPase). RhlB is a member of the DEAD-box family of RNA helicases (Schmid and Linder, 1992). PNPase, a single-strand-specific exonuclease, is a member of the RNase PH family of 3 ′ → 5 ′ RNA-degrading enzymes (Deutscher and Li, 2001; Symmons et al ., 2002). Members of both families are found in a wide range of prokaryotic and eukaryotic organisms. Experiments in vitro demonstrated that RhlB in the degradosome facili- tates the degradation of structured RNA by PNPase (Py et al ., 1996; Coburn et al ., 1999). Other ribonucleolytic complexes, e.g. the yeast exosome and mtEXO complex, also have associated factors that are putative RNA heli- cases (Margossian et al ., 1996; de la Cruz et al ., 1998; Jacobs et al ., 1998; Dziembowski and Stepien, 2001). The gene encoding RNase E was identified because of its role in the maturation of E. coli 5S ribosomal RNA (Ghora and Apirion, 1978; Misra and Apirion, 1979). RNase E is a single-strand-specific endonuclease (Cormack and Mackie, 1992; Ehretsmann et al ., 1992; Lin-Chao et al ., 1994). Subsequent work showed that RNase E has a more general role in RNA metabolism, and it is now believed to be the principal endonuclease in E. coli messenger RNA decay (Coburn and Mackie, 1999; Grunberg-Manago, 1999; Regnier and Arraiano, 2000). RNase E is a large, 1061 residue protein (Casaregola et al ., 1992). Its nucleolytic activity resides in the N- terminal half. The C-terminal half (CTH) of the protein contains a proline-rich linker, an arginine-rich RNA- binding domain (RBD) and a region that is the scaffold for protein–protein interactions with the other components of the degradosome (Taraseviciene et al ., 1995; McDowall and Cohen, 1996; Kaberdin et al ., 1998; Vanzo et al ., 1998). Proteins related to RNase E are found throughout the eubacterial kingdom and in some plants (Condon et al ., 2001). The plant homologues are presumably in the chloroplast, which is an organelle of eubacterial origin. An RNase E-based degradosome was recently identified in Rhodobacter capsulatus (Jager et al ., 2001). The complex contains two DEAD proteins and the transcription termi- nation factor Rho, but not PNPase and enolase. E. coli encodes a paralogue of RNase E now known as RNase G (Li et al ., 1999; Jiang et al ., 2000; Tock et al ., 2000). It has significant homology to the N-terminal catalytic
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Function in Escherichia coli of the non-catalytic part of RNase E: role in the degradation of...

Molecular Microbiology (2002)
45
(5), 1231–1243
© 2002 Blackwell Science Ltd
Blackwell Science, LtdOxford, UKMMIMolecular Microbiology0950-382XBlackwell Science, 200245Original Article
The non-catalytic part of RNase E in vivoA. Leroy et al.
Accepted 7 June, 2002. *For correspondence. [email protected]; Tel. (
+
33) 5 6133 5894; Fax (
+
33) 5 61335886. Present addresses:
†
Developmental Biology Programme, Euro-pean Molecular Biology Laboratory, Heidelberg, Germany.
‡
Unité desInteractions Bactéries-Cellules, Institut Pasteur, 28 rue du Dr Roux,75724 Paris Cedex 15, France.
Function in
Escherichia coli
of the non-catalytic part of RNase E: role in the degradation of ribosome-free mRNA
Anne Leroy,
1
Nathalie F. Vanzo,
1†
Sandra Sousa,
2‡
Marc Dreyfus
2
and Agamemnon J. Carpousis
1
*
1
Laboratoire de Microbiologie et Génétique Moléculaire (CNRS, UMR 5100), Paul Sabatier Université, 118 rue de Narbonne, 31062 Toulouse, France.
2
Laboratoire de Génétique Moléculaire (CNRS, UMR 8541), Ecole Normale Supérieure, 46 rue d'Ulm, 75230 Paris, France.
Summary
RNase E contains a large non-catalytic region thatbinds RNA and the protein components of the
Escher-ichia coli
RNA degradosome. The
rne
gene wasreplaced with alleles encoding deletions in the non-catalytic part of RNase E. All the proteins are stable
in vivo
. RNase E activity was tested using a P
T7
–
lacZ
reporter gene, the message of which is particularlysensitive to degradation because translation isuncoupled from transcription. The non-catalyticregion has positive and negative effectors of mRNAdegradation. Disrupting RhlB and enolase bindingresulted in hypoactivity, whereas disrupting PNPasebinding resulted in hyperactivity. Expression of themutant proteins
in vivo
anticorrelates with activityshowing that autoregulation compensates for defec-tive function. There is no simple correlation betweenRNA binding and activity
in vivo
. An allele (
rne131
),expressing the catalytic domain alone, was put underP
lac
control. In contrast to
rne
+
, low expression of
rne131
severely affects growth. Even with autoregu-lation, all the mutants are less fit when grown in com-petition with wild type. Although the catalytic domainof RNase E is sufficient for viability, our work demon-strates that elements in the non-catalytic part are nec-essary for normal activity
in vivo
.
Introduction
The RNA degradosome of
Escherichia coli
is a multien-zyme complex that was discovered during efforts to purify
and characterize RNase E (Carpousis
et al
., 1994; 1999;Py
et al
., 1994; 1996; Miczak
et al
., 1996). The otherintegral components of the degradosome are enolase, anRNA helicase (RhlB) and polynucleotide phosphorylase(PNPase). RhlB is a member of the DEAD-box family ofRNA helicases (Schmid and Linder, 1992). PNPase, asingle-strand-specific exonuclease, is a member of theRNase PH family of 3
′
→
5
′
RNA-degrading enzymes(Deutscher and Li, 2001; Symmons
et al
., 2002).Members of both families are found in a wide range ofprokaryotic and eukaryotic organisms. Experiments
invitro
demonstrated that RhlB in the degradosome facili-tates the degradation of structured RNA by PNPase (Py
et al
., 1996; Coburn
et al
., 1999). Other ribonucleolyticcomplexes, e.g. the yeast exosome and mtEXO complex,also have associated factors that are putative RNA heli-cases (Margossian
et al
., 1996; de la Cruz
et al
., 1998;Jacobs
et al
., 1998; Dziembowski and Stepien, 2001).The gene encoding RNase E was identified because
of its role in the maturation of
E. coli
5S ribosomal RNA(Ghora and Apirion, 1978; Misra and Apirion, 1979).RNase E is a single-strand-specific endonuclease(Cormack and Mackie, 1992; Ehretsmann
et al
., 1992;Lin-Chao
et al
., 1994). Subsequent work showed thatRNase E has a more general role in RNA metabolism,and it is now believed to be the principal endonuclease in
E. coli
messenger RNA decay (Coburn and Mackie, 1999;Grunberg-Manago, 1999; Regnier and Arraiano, 2000).RNase E is a large, 1061 residue protein (Casaregola
et al
., 1992). Its nucleolytic activity resides in the N-terminal half. The C-terminal half (CTH) of the proteincontains a proline-rich linker, an arginine-rich RNA-binding domain (RBD) and a region that is the scaffold forprotein–protein interactions with the other components ofthe degradosome (Taraseviciene
et al
., 1995; McDowalland Cohen, 1996; Kaberdin
et al
., 1998; Vanzo
et al
.,1998). Proteins related to RNase E are found throughoutthe eubacterial kingdom and in some plants (Condon
et al
., 2001). The plant homologues are presumably in thechloroplast, which is an organelle of eubacterial origin. AnRNase E-based degradosome was recently identified in
Rhodobacter capsulatus
(Jager
et al
., 2001). The complexcontains two DEAD proteins and the transcription termi-nation factor Rho, but not PNPase and enolase.
E. coli
encodes a paralogue of RNase E now known as RNaseG (Li
et al
., 1999; Jiang
et al
., 2000; Tock
et al
., 2000). Ithas significant homology to the N-terminal catalytic

1232
A. Leroy
et al.
© 2002 Blackwell Science Ltd,
Molecular Microbiology
,
45
, 1231–1243
domain of RNase E but is smaller because it lacks a CTH.The ‘RNase E/G’ family of proteins can thus be dividedinto two groups: the large RNase E-like enzymes that canform degradosomes and the small RNase G-like enzymesthat apparently act alone.
In
E. coli
, the tight coupling between transcription andtranslation is important for mRNA stability. BacteriophageT7 RNA polymerase (RNAP) elongates significantly fasterthan the
E. coli
enzyme. When
lacZ
mRNA is transcribedby T7 RNAP, long stretches of ribosome-free messageaccrue. These untranslated T7
–lacZ
mRNAs are veryunstable in uninfected cells, and this effect correlates withthe rate of elongation (Makarova
et al
., 1995). With wild-type T7 RNAP (eightfold faster than the
E. coli
enzyme),the
β
-galactosidase synthesized per message is only afew per cent compared with the same transcript from
E.coli
RNAP. RNase E is responsible for this rapid functionalinactivation (Iost and Dreyfus, 1995). In strains containingthe
rne131
mutation, which produces a truncated RNaseE lacking the CTH, there is a substantial increase in thefunctional stability of the T7
–lacZ
transcript (Lopez
et al
.,1999). This is a general effect, as the yield of many pro-teins expressed by T7 RNAP can be improved in the
rne131
background. Thus, elements in the CTH of RNaseE have an important role in the degradation of the T7messages. As stability can easily be measured by
β
-galactosidase activity, the T7–
lacZ
mRNA is a usefulreporter to study the effect of RNase E mutations onmRNA degradation
in vivo
.Another transcript that is sensitive to mutations in the
CTH of RNase E is the
rne
message. The expression ofRNase E is autoregulated by a mechanism involvingthe stability of its own mRNA (Mudd and Higgins, 1993;Jain and Belasco, 1995). A 361 nucleotide 5
′
untranslatedregion (5
′
UTR) in the
rne
message is essential for auto-regulation (Diwa
et al
., 2000). Catalytic activity is neces-sary for autoregulation but not sufficient, as the CTHsignificantly enhances the capacity of RNase E to regulateits own synthesis (Jiang
et al
., 2000).We previously made deletions within the CTH of RNase
E to elucidate the protein–protein interactions in the RNAdegradosome (Vanzo
et al
., 1998). In these constructs,the chromosomal gene was inactivated by an ambermutation and complemented with plasmids containing var-ious mutant alleles of
rne
. Ow
et al
. (2000) exploited asimilar system using a deletion of the chromosomal
rne
gene. Although relatively simple to construct, these sys-tems have disadvantages, including the need to work in arecombination-deficient background and the concern thatthe dose of the complementing gene could vary as a resultof changes in plasmid copy number. Here, we report theconstruction and characterization of strains in whichmutant alleles of
rne
, encoding proteins with deletions inthe CTH, have been substituted directly into the chromo-
some, replacing the wild-type gene. The activity of RNaseE
in vivo
was measured using the T7–
lacZ
reporter,RNase E expression was quantified by Western blotting,and RNA binding was examined by North-western blot-ting. In order to disrupt autoregulation and control expres-sion
in vivo
, we have also constructed strains in whichcertain RNase E mutants were put under the control of a
lac
promoter. The role of the RBD and protein scaffold incontrolling RNase E activity is discussed.
Results
In previous work, we constructed plasmid-based alleles of
rne
encoding RNase E with deletions in the CTH thatdisrupted the linker, RBD and/or protein scaffold (Vanzo
et al
., 1998). With these mutants, we measured the sta-bility of RNA1, which is a repressor of ColE1 plasmidreplication (unpublished results). Several of the mutantsshowed significantly impaired RNA I decay, but Northernblotting revealed that these strains were also defective inthe maturation of 5S rRNA, suggesting an intrinsic defectin ribonuclease activity. As a common feature of thesemutants was the deletion of the proline-rich ‘hinge’ flank-ing the catalytic domain, we decided to make new mutantsconserving this linker region (Fig. 1A). Another observa-tion was that several mutant proteins appeared to bestrongly overexpressed. To determine whether this effectresulted from defective autoregulation and avoid potentialvariations in gene copy number, we also decided to con-struct the new mutants as single-copy replacements of
rne
+
in the chromosome.
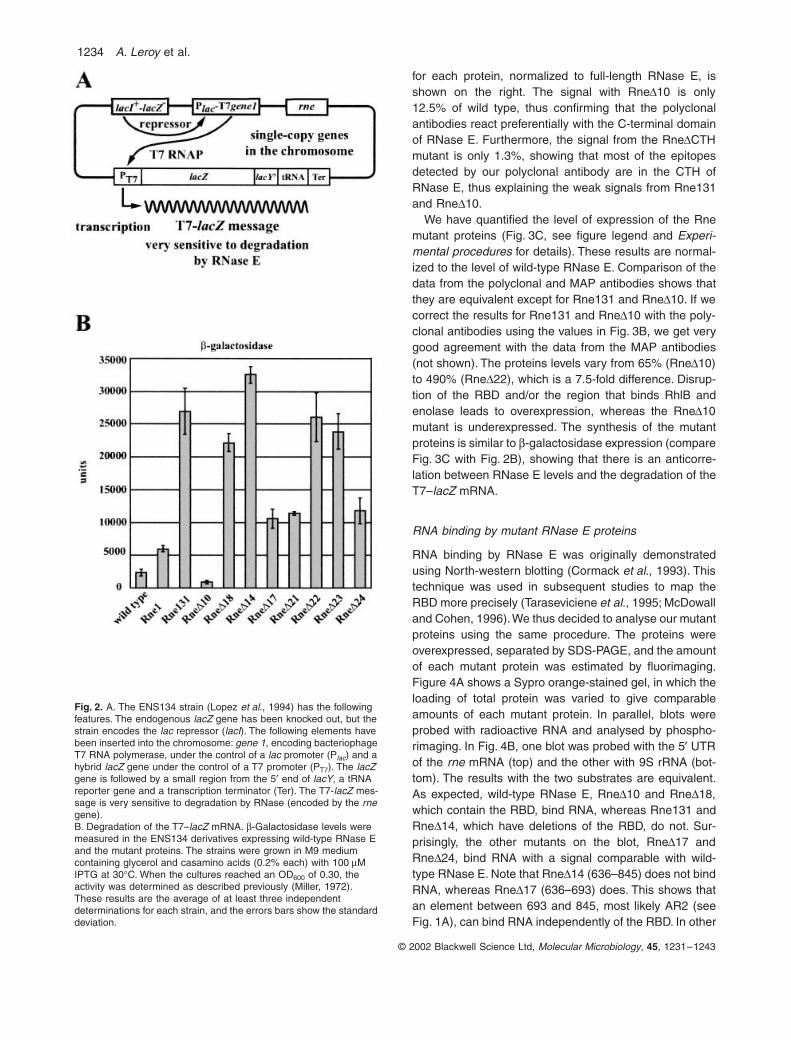
The ENS134 strain and degradation of the T7–lacZ message
Eight new strains were constructed. Figure 1B shows thestructure of the RNase E mutant proteins studied here andindicates the name of the ENS134 derivatives encodingthese proteins. Rne1 and Rne131 are controls that havebeen characterized previously (see figure legend). In allthe new strains, growth is normal in rich media at 30
°
C,37
°
C and 42
°
C, the maturation of 5S ribosomal RNA isnot perturbed, and the mutant proteins are stable, asjudged by experiments in which protein synthesis wasinhibited then RNase E levels were followed by Westernblotting (data not shown).
The ENS134 strain encodes a hybrid
lacZ
mRNA thatis expressed by the bacteriophage T7 RNAP (Fig. 2A).Although the 5
′
leader and more than 3000 nucleotides ofcoding sequence are identical to authentic
lacZ
mRNA,we will refer to this message as T7–
lacZ
to distinguish itfrom RNA transcribed by
E. coli
RNAP. Figure 2B showsthe level of expression of the T7–
lacZ
gene as measuredby
β
-galactosidase synthesis. As expression from a

The non-catalytic part of RNase E
in vivo 1233
© 2002 Blackwell Science Ltd,
Molecular Microbiology
,
45
, 1231–1243
single-copy T7–
lacZ
gene is not toxic, the strains werecultured continuously in the presence of IPTG, and activitywas measured during early logarithmic growth. Rne1 andRne131 have higher levels of
β
-galactosidase, in agree-ment with previous work showing that this results from thestabilization of the T7–
lacZ
message (Iost and Dreyfus,1995; Lopez
et al
., 1999). This experiment was performedat 30
°
C, which is the permissive temperature for growth
Fig. 1.
A. Primary structure of RNase E. The proline-rich (green), arginine-rich (orange) and glutamic acid–proline-rich (yellow) regions are colour coded. The N-terminal half, from residue 1 to 524, is the site of ribonucleolytic activity. This domain is followed by a proline-rich linker (green, 524–568). The central region of RNase E, which is highly charged, contains the arginine-rich RNA-binding domain (604–688) that has been shown to bind RNA by North-western blotting. The RNA-binding domain (RBD) is followed by another proline-rich stretch (743–796), a second arginine-rich region (796–818, 12 arginines out of 25 residues), which we call AR2, and a third proline-rich region (819–857). The C-terminus includes an acidic region rich in glutamic acid and proline (857–1036) followed by a region rich in proline. The ‘protein scaffold’ (688–1061) contains the binding sites for the major components of the RNA degradosome: RhlB, enolase and PNPase. Rh/En (red box) is the region where RhlB and enolase bind to RNase E. The site at which PNPase binds is shown by the yellow box.B. Deletions in the RBD and scaffold of RNase E. The construction of ENS134-1 and ENS134-2, encoding Rne1 and Rne131, was described previously (Iost and Dreyfus, 1995; Lopez
et al
., 1999). Rne1 is a temperature-sensitive enzyme with a glycine (G) to serine (S) substitution at residue 66. Rne131 is encoded by a gene with a
+
1 frameshift at codon 584. A short 32-amino-acid extension is encoded by the
+
1 reading frame (black box at the C-terminal end). In the strains with deletions, the region in RNase E that has been removed is indicated by a thin black line.
with the
rne1
allele. Even under these conditions, theRne1 protein is defective, as evidenced by theapproximately twofold increase in
β
-galactosidase activity.With the exception of Rne
∆
10, all the mutant proteinsexhibited increased
β
-galactosidase expression rangingfrom about fivefold (Rne
∆
17,
∆21 and ∆24) to 13-fold(Rne∆14). The β-galactosidase activity for several of theproteins (Rne∆18, ∆22 and ∆23) is comparable to thatof Rne131 lacking the entire CTH. The deletion of theRBD (Rne∆22 and ∆23) or the region that binds RhlB andenolase (Rne∆18) affects β-galactosidase levels equiva-lently (about 10-fold), whereas the deletion of both regions(Rne∆14) has the largest effect. Previous experimentsdemonstrated that the deletion in Rne∆18 disrupts theinteraction with RhlB and enolase, but not with PNPase(Vanzo et al., 1998). Thus, both the RBD and the regionof the scaffold interacting with RhlB and enolase are nec-essary for efficient degradation of the T7–lacZ mRNA.
The result with Rne∆10 was unexpected. We reproduc-ibly observed a reduction in β-galactosidase (40% of wildtype). This strain expresses the Rne∆10 protein, whichlacks the acidic C-terminal region of RNase E. Previousexperiments demonstrated that this deletion specificallydisrupts the interaction between RNase E and PNPase(Vanzo et al., 1998). Our results suggest that Rne∆10 ismore active in the degradation of the T7–lacZ mRNA thanwild-type RNase E.
Autoregulation of mutant RNase E expression
The expression of RNase E was quantified by Westernblotting using two antibodies. In Fig. 3A, a polyclonal rab-bit antibody raised against the entire Rne protein (top) ora MAP antibody (bottom) against the N-terminal 20 resi-dues of RNase E was used. The blots were developedusing a fluorescent reaction (ECF) and quantified with afluorimager (figure legend and Experimental procedures).Note that, with the polyclonal antibody, the signal fromRne∆10 and Rne131 is very weak compared with wild-type RNase E and the other mutants even though moreprotein was loaded and the fluorescent signal with theother proteins is saturated in this image. As Rne131 andRne∆10 are both missing the C-terminal region, whereasthe other mutants have this domain (Fig. 1B), we testedthe possibility that the polyclonal antibody reacts prefer-entially with the last 200 amino acids of RNase E. ThreeN-terminal His-tagged forms of RNase E were con-structed and purified (Fig. 3B). ∆10 is equivalent to theprotein discussed above except for the tag (black circle,left). ∆CTH is similar to Rne131 except that, like Rne∆10,it contains the C-terminal 16 amino acids of RNase E(black square, right). The reactivity of equivalent weightsof each protein was quantified by ECF Western blotting(data not shown). In Fig. 3B, the fluorescence per weight
1234 A. Leroy et al.
© 2002 Blackwell Science Ltd, Molecular Microbiology, 45, 1231–1243
for each protein, normalized to full-length RNase E, isshown on the right. The signal with Rne∆10 is only12.5% of wild type, thus confirming that the polyclonalantibodies react preferentially with the C-terminal domainof RNase E. Furthermore, the signal from the Rne∆CTHmutant is only 1.3%, showing that most of the epitopesdetected by our polyclonal antibody are in the CTH ofRNase E, thus explaining the weak signals from Rne131and Rne∆10.
We have quantified the level of expression of the Rnemutant proteins (Fig. 3C, see figure legend and Experi-mental procedures for details). These results are normal-ized to the level of wild-type RNase E. Comparison of thedata from the polyclonal and MAP antibodies shows thatthey are equivalent except for Rne131 and Rne∆10. If wecorrect the results for Rne131 and Rne∆10 with the poly-clonal antibodies using the values in Fig. 3B, we get verygood agreement with the data from the MAP antibodies(not shown). The proteins levels vary from 65% (Rne∆10)to 490% (Rne∆22), which is a 7.5-fold difference. Disrup-tion of the RBD and/or the region that binds RhlB andenolase leads to overexpression, whereas the Rne∆10mutant is underexpressed. The synthesis of the mutantproteins is similar to β-galactosidase expression (compareFig. 3C with Fig. 2B), showing that there is an anticorre-lation between RNase E levels and the degradation of theT7–lacZ mRNA.
RNA binding by mutant RNase E proteins
RNA binding by RNase E was originally demonstratedusing North-western blotting (Cormack et al., 1993). Thistechnique was used in subsequent studies to map theRBD more precisely (Taraseviciene et al., 1995; McDowalland Cohen, 1996). We thus decided to analyse our mutantproteins using the same procedure. The proteins wereoverexpressed, separated by SDS-PAGE, and the amountof each mutant protein was estimated by fluorimaging.Figure 4A shows a Sypro orange-stained gel, in which theloading of total protein was varied to give comparableamounts of each mutant protein. In parallel, blots wereprobed with radioactive RNA and analysed by phospho-rimaging. In Fig. 4B, one blot was probed with the 5′ UTRof the rne mRNA (top) and the other with 9S rRNA (bot-tom). The results with the two substrates are equivalent.As expected, wild-type RNase E, Rne∆10 and Rne∆18,which contain the RBD, bind RNA, whereas Rne131 andRne∆14, which have deletions of the RBD, do not. Sur-prisingly, the other mutants on the blot, Rne∆17 andRne∆24, bind RNA with a signal comparable with wild-type RNase E. Note that Rne∆14 (636–845) does not bindRNA, whereas Rne∆17 (636–693) does. This shows thatan element between 693 and 845, most likely AR2 (seeFig. 1A), can bind RNA independently of the RBD. In other
Fig. 2. A. The ENS134 strain (Lopez et al., 1994) has the following features. The endogenous lacZ gene has been knocked out, but the strain encodes the lac repressor (lacI). The following elements have been inserted into the chromosome: gene 1, encoding bacteriophage T7 RNA polymerase, under the control of a lac promoter (Plac) and a hybrid lacZ gene under the control of a T7 promoter (PT7). The lacZ gene is followed by a small region from the 5′ end of lacY, a tRNA reporter gene and a transcription terminator (Ter). The T7-lacZ mes-sage is very sensitive to degradation by RNase (encoded by the rne gene).B. Degradation of the T7–lacZ mRNA. β-Galactosidase levels were measured in the ENS134 derivatives expressing wild-type RNase E and the mutant proteins. The strains were grown in M9 medium containing glycerol and casamino acids (0.2% each) with 100 µM IPTG at 30°C. When the cultures reached an OD600 of 0.30, the activity was determined as described previously (Miller, 1972). These results are the average of at least three independent determinations for each strain, and the errors bars show the standard deviation.

The non-catalytic part of RNase E in vivo 1235
© 2002 Blackwell Science Ltd, Molecular Microbiology, 45, 1231–1243
Fig. 3. A. Western blot analysis of RNase E expression using poly-clonal rabbit antibody (top) or MAP antibody (bottom). The blots were developed using a fluorescence detection system and analysed by a fluorimager. RNase E (WT) and mutant proteins are indicated at the top of the blots. The asterisks indicate the position of each protein. All the lanes were loaded with equivalent amounts of total protein based on the OD of the cultures except where indicated, i.e. in the first lane, twofold more WT protein and fourfold more Rne∆10 and Rne131.B. His-tagged RNase E constructs. The black circle to the left signifies the N-terminal HIS tag; the black box to the right, the C-terminal last 16 amino acids, which is conserved in all these constructs. To the right is the fluorescence signal for each polypeptide (equivalent weights) normalized to the full-length construct.C. Quantification of the Western blots. The amount of protein relative to wild type is shown for each mutant, with the results from the polyclonal antibody in the left column and the MAP antibody at the right. These data represent at least six independent determinations for each mutant, and the error bars show the standard deviation.
experiments, we observed that Rne∆21 (603–627),Rne∆22 (603–693) and Rne∆23 (585–693) also bind RNA(data not shown).
In North-western blotting, Rne∆10 reproducibly has astronger signal (five- to 10-fold more radioactivity byweight) than wild type or the other mutants that bind RNA(Fig. 4B). This signal, which results from the retention ofsignificantly more RNA after washing the blot, suggeststhat the capacity and/or affinity of Rne∆10 for binding RNAis higher than wild type. This increased RNA bindingcorrelates with the higher activity of Rne∆10 in vivo, asjudged by T7–lacZ activity and the autoregulation ofRNase E expression. A model that could explain the influ-ence of the acidic C-terminal domain on RNA binding ispresented in the Discussion.
RNase E autoregulation compensates for defective degradation activity
The effect of the mutations on T7–lacZ mRNA stabilitymight be underestimated as a consequence of autoregu-lation. For instance, with the Rne131 mutant protein wesee 11-fold more β-galactosidase activity (Fig. 2B). How-ever, considering that Rne131 is threefold overexpressed(Fig. 3C), the defect appears to be more serious. Table 1shows an analysis of the activity of the mutant proteins invivo. Note that the activity of mutants such as rne∆18,rne∆14, rne∆22 and rne∆23 is at least as defective as thatof rne131 in both the autoregulation of RNase E synthesis(relative RNase E levels) and the degradation of the T7–lacZ mRNA (specific degradation efficiency). This analysisrests on the assumption that alterations in the concentra-
Table 1. Activity of the mutant proteins in the degradation of the T7–lacZ mRNA and the regulation of rne expressiona.
Allele
Relativedegradationefficiency
RelativeRNase Elevel
Specificdegradationefficiency
rne1 0.40 2.3 0.17rne131 0.090 2.7 0.034rne∆10 2.5 0.58 4.3rne∆18 0.11 3.2 0.034rne∆14 0.074 4.2 0.018rne∆17 0.23 1.7 0.13rne∆21 0.21 2.2 0.094rne∆22 0.092 4.8 0.019rne∆23 0.10 4.2 0.025rne∆24 0.20 2.7 0.075
a. The data from Figs 2B and 3C (MAP antibody) were analysed asfollows. Relative degradation efficiency of the T7–lacZ mRNA is theβ-galactosidase activity of wild type divided by the mutant, e.g.1.0 = wild type and 0.2 = fivefold more β-galactosidase. RelativeRNase E level is the amount of mutant RNase E divided by wild type,e.g. 1.0 = wild type and 5.0 = fivefold more RNase E. Specific degra-dation efficiency is obtained by dividing the value in the first columnby the value in the second column, e.g. 1.0 = wild type and0.2 = fivefold more β-galactosidase for the same amount of RNase E.

1236 A. Leroy et al.
© 2002 Blackwell Science Ltd, Molecular Microbiology, 45, 1231–1243
Fig. 4. A. Gel stained with Sypro orange showing the separation of extracts containing overexpressed RNase E or the mutant proteins (indicated by the asterisks). pET11a is a control extract with no overexpressed protein. The gel was loaded to give comparable amounts of RNase E and each mutant protein.B. Blots probed with 33P-labelled RNA: 5′ UTR of the rne mRNA (top) or 9S ribosomal RNA (bottom).
tion of the mutant proteins by autoregulation change thelevel of mRNA-degrading activity. To test this, we con-structed new strains in the ENS134 background in whichthe promoter and the 5′ UTR of rne were replaced withthe promoter and leader of the lacZ gene. The construc-tion and characterization of related strains with rne underPlac control has been described recently (Sousa et al.,2001). Table 2 shows a measurement of the degradationof the T7–lacZ mRNA when rne+, rne∆10 and rne131 areautoregulated (first column, Prne) or expressed equiva-lently (second column, Plac). By Western blotting, theexpression of RNase E and the mutant proteins from Plac
was about twofold higher than the wild-type protein withnormal autoregulation (data not shown). The lower β-galactosidase activity measured with wild-type rne underPlac control (770 versus 2370 units) could result fromdestabilization of the T7–lacZ mRNA by the twofold higherexpression of RNase E. Note that, under conditions ofequivalent expression (Plac), the ratio of β-galactosidaseactivity in wild type to mutant is 4.0 and 0.028 for rne∆10and rne131 respectively. This is in very good agreementwith the specific degradation efficiency of 4.3 and 0.034in Table 1, confirming the validity of the calculation. Thecomparison of the results in the first (Prne) and second(Plac) columns in Table 2 shows that autoregulation com-pensates, at least partially, for defective degradation of theT7–lacZ mRNA. Although it would have been interestingto perform this analysis over a range of IPTG concentra-tions, the growth defects with the Plac–rne131 straindescribed in the next section precluded further analysis.
Aberrant growth of the Plac–rne131 strain
The strains in which rne is under the control of Plac requireIPTG for growth. On solid media, there is no visible colonyformation in the absence of IPTG. Upon examining thegrowth of the Plac–rne131 strain at different IPTG concen-trations, we noticed significant filamentation at both lowand high inducer concentrations. Figure 5 shows repre-sentative images of microcolony formation of the Plac–rne131 strain over a range of IPTG concentrations. The
Table 2. Degradation of the T7–lacZ mRNA with rne∆10 and rne131under Plac control.
Prne Plac
rne+/rne∆10 2.5 4.0rne+/rne131 0.089 0.028
The strains contain rne+, rne∆10 and rne131 under the control of theirown promoter and 5′ UTR (Prne) or the lactose promoter and 5′ leader(Plac). Degradation of the T7–lacZ mRNA is expressed as the β-galactosidase activity of wild type divided by the mutant (compare withTable 1, first column). β-Galactosidase activity was measured duringgrowth at 30°C with 100 µM IPTG. The activity of rne+ was 2370 unitsunder Prne control and 770 units under Plac control (see text).

The non-catalytic part of RNase E in vivo 1237
© 2002 Blackwell Science Ltd, Molecular Microbiology, 45, 1231–1243
Plac–rne+ strain is shown for comparison. The morphologyat 16 µM IPTG is striking. The colony appears to be com-posed of a few very long filaments. At 25 and 50 µMinducer, long filaments predominate, although some nor-mal cells appear at the higher concentration. With thewild-type RNase E control, at 16 µM IPTG, we observedtwo different types of colonies: some filaments (a) versusnormal (b). Above this concentration, cell morphology isnormal in the Plac–rne+ strain. At 500 µM IPTG, the mor-phology of Plac–rne131 is normal, whereas at 1000 µM,we observed two different types of colonies: normal (a)versus long filaments (b). In control experiments, thePrne–rne131 strain, which has autoregulated expression,showed normal cell morphology throughout the range ofIPTG concentrations tested here (data not shown). Stain-ing of the filamented cells with DAPI showed that DNAreplication and partition of the nucleoid appears to benormal (data not shown). However, the cells fail to formsepta and divide. These results show that growth of thePlac–rne131 cells depends on the dose of IPTG, whereasthe Plac–rne+ strain is normal except at the lowest concen-tration of inducer. The Plac–rne131 strain has a clearlydiscernible defect in cell division, at levels of expressionat which wild type is normal, adding further support to theidea that autoregulation has an important role in compen-sating for defective RNase E activity.
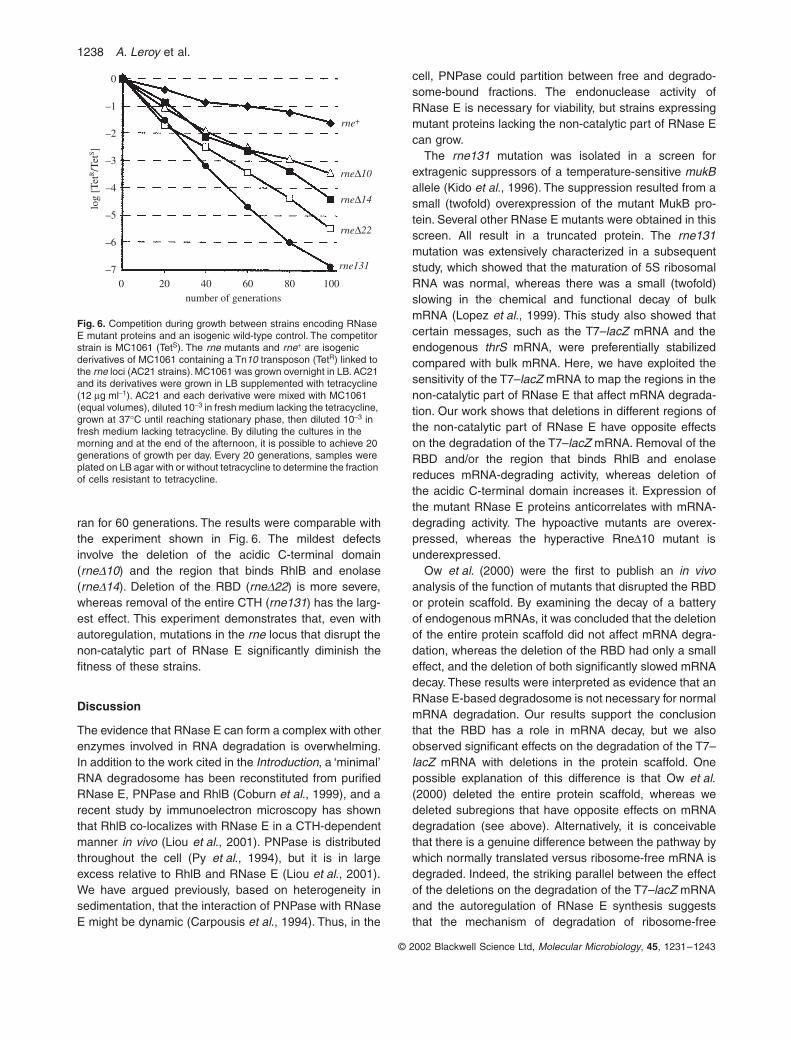
Fitness of strains with mutant RNase E proteins
The strains in which the expression of the mutant proteinsis autogenously controlled have a discernible phenotypewith respect to the stability of the T7–lacZ mRNA andRNase E expression. However, the rate of logarithmicgrowth in liquid media is unaffected, although small differ-ences might not be detected by this analysis. A sensitivetest of a mutant strain is its ‘fitness’ in competition with anisogenic ‘wild-type’ control. Figure 6 shows the results ofsuch an experiment. Wild type (rne+) is a derivative ofMC1061 containing a Tn10 transposon linked to the rnelocus. The mutants are genetically identical except for thealteration in the rne gene. The Tn10-containing strains canbe distinguished from the parent by their resistance totetracycline. MC1061 (TetS) and each of the Tn10 deriva-tives (TetR) were mixed together in equal proportions, thengrown for 100 generations. Every 20 generations, cellswere plated to measure the proportion that were tetracy-cline resistant. With the rne+ control, there is a small loss,<100-fold per 100 generations, as a result of the Tn10insertion. All the mutants are lost significantly faster. After100 generations, rne∆10 is ≈100-fold lower than rne+
rne ∆14, 1000-fold; rne∆22, 10 000-fold; and rne131,100 000-fold. Figure 6 shows the results of a single exper-iment. This work was repeated once in an experiment that
Fig. 5. Microcolonies of the Plac–rne131 strain, ENS134(Plac-rne)-2, grown in the presence of 16, 25, 50, 500 and 1000 µM IPTG. Wild-type rne under the control of Plac is shown for comparison. Micro-scope slides were covered with a thin layer (300 µl) of M9 agar containing glycerol and casamino acids (0.2% each). The strains were streaked on these slides and incubated at 37°C (7 h for rne131, 3 h for wild type). Microcolonies were visualized with a Leica DMRB 100× objective and a Coolsnap photometric camera. For the Plac–rne+ strain at 16 µM IPTG, we observed two types of colonies: (a) some of the cells were filamented; and (b) morphology was normal. For the Plac–rne131 strain at 1000 µM, we observed two types of colonies: (a) morphology was normal; and (b) many of the cells formed long filaments.
1238 A. Leroy et al.
© 2002 Blackwell Science Ltd, Molecular Microbiology, 45, 1231–1243
ran for 60 generations. The results were comparable withthe experiment shown in Fig. 6. The mildest defectsinvolve the deletion of the acidic C-terminal domain(rne∆10) and the region that binds RhlB and enolase(rne∆14). Deletion of the RBD (rne∆22) is more severe,whereas removal of the entire CTH (rne131) has the larg-est effect. This experiment demonstrates that, even withautoregulation, mutations in the rne locus that disrupt thenon-catalytic part of RNase E significantly diminish thefitness of these strains.
Discussion
The evidence that RNase E can form a complex with otherenzymes involved in RNA degradation is overwhelming.In addition to the work cited in the Introduction, a ‘minimal’RNA degradosome has been reconstituted from purifiedRNase E, PNPase and RhlB (Coburn et al., 1999), and arecent study by immunoelectron microscopy has shownthat RhlB co-localizes with RNase E in a CTH-dependentmanner in vivo (Liou et al., 2001). PNPase is distributedthroughout the cell (Py et al., 1994), but it is in largeexcess relative to RhlB and RNase E (Liou et al., 2001).We have argued previously, based on heterogeneity insedimentation, that the interaction of PNPase with RNaseE might be dynamic (Carpousis et al., 1994). Thus, in the
cell, PNPase could partition between free and degrado-some-bound fractions. The endonuclease activity ofRNase E is necessary for viability, but strains expressingmutant proteins lacking the non-catalytic part of RNase Ecan grow.
The rne131 mutation was isolated in a screen forextragenic suppressors of a temperature-sensitive mukBallele (Kido et al., 1996). The suppression resulted from asmall (twofold) overexpression of the mutant MukB pro-tein. Several other RNase E mutants were obtained in thisscreen. All result in a truncated protein. The rne131mutation was extensively characterized in a subsequentstudy, which showed that the maturation of 5S ribosomalRNA was normal, whereas there was a small (twofold)slowing in the chemical and functional decay of bulkmRNA (Lopez et al., 1999). This study also showed thatcertain messages, such as the T7–lacZ mRNA and theendogenous thrS mRNA, were preferentially stabilizedcompared with bulk mRNA. Here, we have exploited thesensitivity of the T7–lacZ mRNA to map the regions in thenon-catalytic part of RNase E that affect mRNA degrada-tion. Our work shows that deletions in different regions ofthe non-catalytic part of RNase E have opposite effectson the degradation of the T7–lacZ mRNA. Removal of theRBD and/or the region that binds RhlB and enolasereduces mRNA-degrading activity, whereas deletion ofthe acidic C-terminal domain increases it. Expression ofthe mutant RNase E proteins anticorrelates with mRNA-degrading activity. The hypoactive mutants are overex-pressed, whereas the hyperactive Rne∆10 mutant isunderexpressed.
Ow et al. (2000) were the first to publish an in vivoanalysis of the function of mutants that disrupted the RBDor protein scaffold. By examining the decay of a batteryof endogenous mRNAs, it was concluded that the deletionof the entire protein scaffold did not affect mRNA degra-dation, whereas the deletion of the RBD had only a smalleffect, and the deletion of both significantly slowed mRNAdecay. These results were interpreted as evidence that anRNase E-based degradosome is not necessary for normalmRNA degradation. Our results support the conclusionthat the RBD has a role in mRNA decay, but we alsoobserved significant effects on the degradation of the T7–lacZ mRNA with deletions in the protein scaffold. Onepossible explanation of this difference is that Ow et al.(2000) deleted the entire protein scaffold, whereas wedeleted subregions that have opposite effects on mRNAdegradation (see above). Alternatively, it is conceivablethat there is a genuine difference between the pathway bywhich normally translated versus ribosome-free mRNA isdegraded. Indeed, the striking parallel between the effectof the deletions on the degradation of the T7–lacZ mRNAand the autoregulation of RNase E synthesis suggeststhat the mechanism of degradation of ribosome-free
Fig. 6. Competition during growth between strains encoding RNase E mutant proteins and an isogenic wild-type control. The competitor strain is MC1061 (TetS). The rne mutants and rne+ are isogenic derivatives of MC1061 containing a Tn10 transposon (TetR) linked to the rne loci (AC21 strains). MC1061 was grown overnight in LB. AC21 and its derivatives were grown in LB supplemented with tetracycline (12 µg ml−1). AC21 and each derivative were mixed with MC1061 (equal volumes), diluted 10−3 in fresh medium lacking the tetracycline, grown at 37°C until reaching stationary phase, then diluted 10−3 in fresh medium lacking tetracycline. By diluting the cultures in the morning and at the end of the afternoon, it is possible to achieve 20 generations of growth per day. Every 20 generations, samples were plated on LB agar with or without tetracycline to determine the fraction of cells resistant to tetracycline.
0
–1
–2
–3
–4
–5
–6
–70 20 40 60 80 100
rne+
rne∆10
rne∆14
rne∆22
rne131
log
[Tet
R/T
etS ]
number of generations

The non-catalytic part of RNase E in vivo 1239
© 2002 Blackwell Science Ltd, Molecular Microbiology, 45, 1231–1243
mRNA and rne autoregulation are related. The T7–lacZmessage is ribosome free in the sense that the T7 RNAPlargely outpaces the ribosome. The 361 nucleotide rne 5′leader is an untranslated target. It is noteworthy that cer-tain other messages have been shown to be preferentiallystabilized by the rne131 allele, e.g. the thrS mRNA encod-ing threonyl-tRNA synthetase (Lopez et al., 1999). ThrSautogenously regulates its own expression by binding toan ‘operator’ in the 163 nucleotide 5′ leader of its mRNAand inhibiting translation. Recent work shows that, in rne+
cells, the repressed thrS mRNA is rapidly degraded,whereas with rne131, it accumulates significantly(Nogueira et al., 2001). Thus, the results with T7–lacZ, rneand thrS mRNA all suggest that elements in the non-catalytic part of RNase E facilitate the preferential degra-dation of ribosome-free mRNA. We speculate that thisprocess might represent a type of ‘mRNA surveillance’, inwhich the uncoupling of translation from transcription trig-gers decay.
All the RNase E mutants studied here bind RNA exceptRne131 and Rne∆14. This activity was initially localized tothe CTH (Cormack et al., 1993), then mapped more pre-cisely to the RBD (Taraseviciene et al., 1995; McDowalland Cohen, 1996). Our results suggest that a secondarginine-rich region (AR2, residues 796–819, 50% argin-ine) can also bind RNA. Analysis of the published work(Taraseviciene et al., 1995; McDowall and Cohen, 1996)shows that the constructs used in those studies would nothave permitted the detection of an independent RNA bind-ing site at AR2. An important conclusion of our work isthat there is no simple correlation between RNA bindingin vitro and mRNA-degrading activity in vivo. Mutants suchas Rne∆18, with an intact RBD and RNA-binding activity,exhibit significant defects in T7–lacZ mRNA degradationand rne autoregulation. Our results show that RNA bindingis more complex than anticipated: the RNA and proteinbinding sites overlap and cannot be separated by simpledeletions. Considering the effect of deletions locatedbetween residues 585 and 845 on the degradation of theT7–lacZ mRNA, this entire region appears to compriseeither a single functional domain or perhaps two domainsthat co-operate in the same reaction.
The observation that the deletion of the last 200 resi-dues of RNase E (Rne∆10) leads to destabilization of theT7–lacZ message and reduces expression of the mutantprotein is novel. These results suggest that Rne∆10 ismore active than wild type in vivo. Recent work (A. Leroy,unpublished) shows that the rne∆10 mutation also desta-bilizes the T7–lacZ mRNA in a pnp– strain. The pnp– allele,the same as that used by Lopez et al. (1999), abolishesproduction of PNPase, which is not detectable by Westernblotting. This shows that, even though the region deletedin rne∆10 is involved in an interaction with PNPase, thephenotype of rne∆10 does not involve PNPase per se. A
plausible explanation is that the acidic C-terminal domainis a negative effector of RNase E activity. As the removalof the acidic region stimulates RNA binding (Fig. 4), thisdomain appears to be an inhibitor of the basic RBD and/or AR2 regions. It should be interesting to explore thisinteraction in future work.
The growth defects in the Plac–rne131 strain at low andhigh IPTG concentrations is evidence that autoregulationis important in compensating for the defective activity(Fig. 5). As noted recently, using Plac control, wild-typeRNase E expression can be substantially reduced belownormal levels (Sousa et al., 2001; Jain et al., 2002). Thisis also observed with our Plac–rne+ construct, as growthis normal except at the lowest concentration (16 µM).However, the growth of the Plac–rne131 construct isalready defective at 50 µM IPTG and grossly aberrant atlower concentrations. These results demonstrate for thefirst time the role of autoregulation in compensating fordefective activity and the importance of the non-catalyticpart of RNase E for normal function in vivo. Even withautoregulation, the mutant strains are disadvantagedwhen grown in competition with wild type (Fig. 6). After100 generations of growth, there is only one cell harbour-ing the rne131 mutant for every 100 000 that are rne+.Even with the least impaired mutant (rne∆10), only about1% remains after 100 generations. In this protocol,because the cells reach stationary phase between dilu-tions, there is competition at each stage of growth, i.e. thetransition from stationary phase, logarithmic growth, thetransition to stationary phase and survival thereafter.Although we do not know the specific defect that causesthe growth phenotype, as already discussed, rne131slows bulk mRNA decay and preferentially stabilizescertain messages. Thus, it seems reasonable to believethat the disadvantage involves a problem with mRNAdegradation.
A feasible in vivo approach to characterize further theRNase E mutant proteins described here would be toanalyse mRNA levels globally by the hybridization ofcDNA to DNA arrays. This should permit the identificationof messages such as thrS, the decay of which is particu-larly sensitive to mutations disrupting the non-catalyticpart of RNase E. However, our work shows that the auto-regulation of RNase E expression partially compensatesfor defective degradation of the T7–lacZ mRNA. Thus, itwould also be interesting, in an analysis of the transcrip-tome, to examine strains such as those we have con-structed here, in which autoregulation is disrupted and theexpression of RNase E and the mutant proteins can becontrolled by the lactose promoter. This analysis shouldreveal what effect the mutations have on the degradationof normally translated messages. It should also clarifythe role of autoregulation in compensating for defectivemRNA-degrading activity.

1240 A. Leroy et al.
© 2002 Blackwell Science Ltd, Molecular Microbiology, 45, 1231–1243
Experimental procedures
Standard techniques, strains and plasmids
General techniques in genetics and molecular biology havebeen described previously (Miller, 1972; Sambrook andRussell, 2001). The strains and plasmids used here are listedin Tables 3 and 4.
The ENS134 derivatives were constructed as follows:rne∆10, rne∆14 and rne∆18 were generated in the pAM-rneplasmid by inverse polymerase chain reaction (PCR) asdescribed previously (Vanzo et al., 1998). The PstI fragmentscontaining these mutant rne alleles were cloned into the NsiIsite of pLN135.1 (Cornet et al., 1996) to generate pLN-rne∆10, pLN-rne∆14 and pLN-rne∆18. The others deletionswere generated by inverse PCR using the pLN-rne plasmid(Table 4). The pLN-∆rne plasmids were transformed intoAC23 (Vanzo et al., 1998). Using a previously described pro-tocol (Cornet et al., 1996), the endogenous temperature-sen-sitive rne1 allele was replaced by the mutant alleles. Briefly,the protocol relies on the following elements. The target strainis resistant to streptomycin and harbours a temperature-sensitive allele (rne1) that does not permit growth at 42°C.The plasmid contains a temperature-sensitive origin that doesnot permit replication at 42°C, a cat gene conferring resis-tance to chloramphenicol and an rpsL+ gene that renders theintegrants sensitive to streptomycin. After transformation andselection for chloramphenicol resistance at 30°C, coloniesare streaked onto plates with chloramphenicol at 42°C toselect integrants, which grow because the rne∆ alleles arenot temperature sensitive. Excision relies on the selection ofstreptomycin-resistant cells at 42°C, which were then testedfor sensitivity to chloramphenicol. The replacements were
verified by PCR and Western blotting. P1 transduction wasused to move the mutant rne alleles from the AC backgroundto ENS 134-1 (Lopez et al., 1999). The transductants wereselected at 42°C, and the replacement of the endogenoustemperature-sensitive rne1 allele was verified by PCR andWestern blotting. Throughout the experiments in this work,two independently derived strains were analysed for eachdeletion. In every case, both strains gave the same results.For simplicity, we only show the results for one set of strains.
The ENS134(Plac) derivatives were constructed as follows.Replacement of the promoter and 5′ UTR of the rne gene bythe corresponding region of the lac operon using a pKO3derivative has already been described (Sousa et al., 2001).To obtain ENS134(Plac-rne)-2 and ENS134(Plac-rne)-10, thepKO3 derivative was transformed into ENS134-2 andENS134-10, respectively, to replace the rne 5′ control region.The same method was used to obtain ENS134(Plac), exceptthat the rne 5′ control region was first replaced in a derivativeof MG1655 carrying a Tn10 transposon linked to the rne gene(zce-726::Tn10; Mudd et al., 1990). The Plac–rne+ gene wasthen transduced by P1 into ENS134 by selecting for tetracy-cline resistance and IPTG-dependent growth.
Antibodies and Western blotting
Rabbit polyclonal antibodies, raised against full-lengthRNase E, were described previously (Vanzo et al., 1998).MAP antibodies against RNase E, supplied by S. Kushner,were used in pilot experiments. We subsequently used anantibody that we produced ourselves. Briefly, a peptide cor-responding to the N-terminal 20 amino acids of RNase E wassynthesized using a MAP-8 matrix (Alta Bioscience). The
Table 3. Strains.
Strain Characteristic Reference
AC21 MC1061, zce-726::Tn10 Carpousis et al. (1994)AC23 MC1061, zce-726::Tn10, rne1(ams) Vanzo et al. (1998)AC24 AC23, rne∆10 (aa∆ 844–1045)a This workAC26 AC23, rne∆18 (aa∆ 728–845) This workAC27 AC23, rne131 (made by P1 transduction) This workAC28 AC23, rne∆14 (aa∆ 636–845) This workAC29 AC23, rne∆17 (aa∆ 636–693) This workAC31 AC23, rne∆21 (aa∆ 603–627) This workAC32 AC23, rne∆22 (aa∆ 603–693) This workAC33 AC23, rne∆23 (aa∆ 585–693) This workAC34 AC23, rne∆24 (aa∆ 585–627) This workENS134 BL21(DE3), Plac–T7 RNA polymerase, PT7–lacZ Lopez et al. (1994)ENS134-1 ENS134, zce-726::Tn10, rne1(ams) Iost and Dreyfus (1995)ENS134-2 ENS134, rne 131 Lopez et al. (1999)ENS134-10 ENS134, zce-726::Tn10, rne∆10 (aa∆ 844–1045) This workENS134-18 ENS134, zce-726::Tn10, rne∆18 (aa∆ 728–845) This workENS134-14 ENS134, zce-726::Tn10, rne∆14 (aa∆ 636–845) This workENS134-17 ENS134, zce-726::Tn10, rne∆17 (aa∆ 636–693) This workENS134-21 ENS134, zce-726::Tn10, rne∆21 (aa∆ 603–627) This workENS134-22 ENS134, zce-726::Tn10, rne∆22 (aa∆ 603–693) This workENS134-23 ENS134, zce-726::Tn10, rne∆23 (aa∆ 585–693) This workENS134-24 ENS134, zce-726::Tn10, rne∆24 (aa∆ 585–627) This workENS134(Plac–rne) ENS134, zce-726::Tn10, Plac–rne This workENS134(Plac–rne)-2 ENS134, Plac–rne131 This workENS134(Plac–rne)-10 ENS134, zce-726::Tn10, Plac–rne∆10 This work
a. This notation (aa∆ 844–1045) refers to the amino acids deleted in the protein expressed from the mutant rne allele. The alleles rne∆17, rne∆21,rne∆22, rne∆23 and rne∆24 were generated for the first time in this study (Experimental procedures). The alleles rne∆10, rne∆18, rne∆14 andrne131 were described previously (Kido et al., 1996; Lopez et al., 1999; Vanzo et al., 1998).

The non-catalytic part of RNase E in vivo 1241
© 2002 Blackwell Science Ltd, Molecular Microbiology, 45, 1231–1243
crude product was suspended in 2 ml of 10 mM Tris, pH 7.5,1 mM EDTA, 50 mM NaCl, 0.1% SDS, then sonicated anddialysed against three changes of the same buffer. A portionof this suspension was used without further purification toraise antisera in rabbits (Eurogentec).
SDS-PAGE (7.5%) was used to separate proteins fromcrude extracts, which were prepared by boiling cells in SDSsample buffer (Carpousis et al., 1994). The gel was blottedto a polyvinylidene difluoride (PVDF) membrane (Amersham)as described previously (Vanzo et al., 1998;). The blot wasblocked for 2 h in Tris-buffered saline (TBS) containing 5%non-fat milk, then incubated for either 1 h in TBS containing1% non-fat milk and the polyclonal antisera (1:10 000 dilu-tion) or 2 h in this buffer with the MAP antisera (1:20 dilution).The membrane was washed three times for 10 min with TBScontaining 1% non-fat milk, then incubated with an alkalinephosphatase-coupled secondary antibody (Sigma; 1:5000dilution) for 1 h. The membrane, washed twice for 30 min inTBS containing 1% non-fat milk and once for 30 min in TBS,was treated with ECF substrate (Amersham) and analysedwith a fluorimager (Molecular Dynamics). For quantification,each blot contained at least two different amounts ofprotein from the wild-type rne strain. During the ECF reaction,the blot was scanned several times to obtain a series ofimages with different intensities of fluorescence. To quantifythe levels of the wild-type and mutant proteins, images inwhich the signals from the mutant protein and at least one ofthe wild-type controls were not saturated were analysed dig-itally (IMAGEQUANT) in a procedure involving the subtractionof the background from regions just above and below theband.
North-western blotting
BL21(DE3) strains containing pET11a, pET11-rne or thepET11-rne∆ plasmids were plated on LA ampicillin(50 µg ml−1) and grown overnight. A single fresh colony wasinoculated in LB ampicillin (50 µg ml−1) and grown at 37°C for4 h (OD600 <0.3). The culture were then diluted in the samemedium to an OD600 of 0.1 and grown to an OD600 of 0.3.Expression was induced with 1 mM IPTG, and the cultureswere incubated for 2 h at 30°C. Total protein was preparedas described above for Western blotting. In a preliminarystep, the extracts were separated by SDS-PAGE, the gel wasstained with Sypro orange (Interchim; Steinberg et al., 1996),and the amount of RNase E or mutant protein was estimatedby fluorimaging. Based on this determination, the amount oftotal protein for North-western blotting was varied to givecomparable amounts of RNase E and the mutant proteins.
Two probes, 9S rRNA and the 5′ UTR of the rne mRNA,were synthesized as follows. Templates were generated byPCR as described previously (Ehretsmann et al., 1992;Carpousis et al., 1994) using the plasmids pLN-rne (5′ UTR-rne) or pKK238.8 (9S rRNA). The products, 415 bp rne-5′UTR template and 268 bp 9S rRNA template, were purifiedon 3% small-fragment agarose (Appligene) and extractedfrom the gel with a Qiaquick kit (Quiagen). The templateswere transcribed in vitro using a T7 RNA polymerase kit(Promega) and [α-33P]-UTP (Amersham), then desalted onSephadex G25. The 9S RNA was described previously(Carpousis et al., 1994). The rne-5′ UTR probe starts 4nucleotides before the normal transcription start of the rnemRNA (GGCCGUUUC, the underlined sequence is the nor-
Table 4. Plasmids.
Plasmid Characteristic Reference
pLN135.1 Low-copy-number, temperature-sensitive replicon, multiple cloning site, Cornet et al. (1996)cat gene and rpsL+ (SmS)
pLN-rne 6 kb PstI–PstI genomic fragment containing the entire rne gene cloned This workinto the NsiI site of pLN135.1
pLN-rne∆10 Derivative of pLN-rne (aa∆ 844–1045)a This workpLN-rne∆18 Derivative of pLN-rne (aa∆ 728–845) This workpLN-rne∆14 Derivative of pLN-rne (aa∆ 636–845) This workpLN-rne∆17 Derivative of pLN-rne (aa∆ 636–693) This workpLN-rne∆21 Derivative of pLN-rne (aa∆ 603–627) This workpLN-rne∆22 Derivative of pLN-rne (aa∆ 603–693) This workpLN-rne∆23 Derivative of pLN-rne (aa∆ 585–693) This workpLN-rne∆24 Derivative of pLN-rne (aa∆ 585–627) This workpET11a Vector for protein expression in E. coli Studier et al. (1990)pET11a-rne Contains complete rne coding sequence and transcription termination site Vanzo et al. (1998)pet11a-rne∆10 Derivative of pET11a-rne (aa∆ 844–1045) This workpet11a-rne∆18 Derivative of pET11a-rne (aa∆ 728–845) This workpet11a-rne∆14 Derivative of pET11a-rne (aa∆ 636–845) This workpet11a-rne∆17 Derivative of pET11a-rne (aa∆ 636–693) This workpet11a-rne∆22 Derivative of pET11a-rne (aa∆ 603–693) This workpet11a-rne∆23 Derivative of pET11a-rne (aa∆ 585–693) This workpet11a-rne∆24 Derivative of pET11a-rne (aa∆ 585–627) This workpet11a-rne∆26 Derivative of pET11a-rne (aa∆ ∆585–845) This workpet11a-rne∆CTH Derivative of pET11a-rne (aa∆ 585–1045) This workpET15b Derivative of pET11a with N-terminal histidine tag NovagenpET15b-rne rne transplanted from pET11a-rne to pET15b This workpET15b-rne∆10 Derivative of pET15b-rne (∆aa 844–1045) This workpET15b-rne∆CTH Derivative of pET15b-rne (∆aa 585–1045) This work
a. This notation (aa∆ 844–1045) refers to the amino acids deleted in the protein expressed from the mutant rne allele.

1242 A. Leroy et al.
© 2002 Blackwell Science Ltd, Molecular Microbiology, 45, 1231–1243
mal 5′ end) and ends 47 nucleotides after the AUG translationinitiation codon. Blotting and probing were performed asdescribed previously (Cormack et al., 1993), except that themembranes were incubated with the RNA probe at roomtemperature for 2 h and washed (30 min) in TEN buffer with0.02% Tween 20 and 50 mM NaCl, then washed in the samebuffer containing 200 mM NaCl and, finally, 500 mM NaCl.The radioactive RNA was visualized with a Phosphorimager(Fuji).
Acknowledgements
We thank S. Kushner (Georgia, USA) for the gift of a MAPantibody against RNase E that was used in pilot experiments,and L. Poljak for making the MAP antibody used in this work.We thank F. Cornet and C. Lesterlin for discussions regardingthe growth competition experiments and the microscopy ofmicrocolonies, V. Khemici and L. Poljak for discussions andcritical comments on the manuscript, and M. Lautier for helpwith the microcopy. We acknowledge aid from the technicalplatform of the Institut d'Exploration Fonctionnelle desGénomes (IFR 109). This research was supported by theCentre National de la Recherche Scientifique (CNRS), withadditional funding from the Cancer Research Association(ARC no. 5244 to A.J.C. and no. 5474 to M.D.) and the MidiPyrénées Région (A.J.C.). The groups of A.J.C. and M.D. arepart of a network funded by the Fundamental MicrobiologyProgram (MENRT). A.L. was supported by the MENRT andthe ARC.
References
Carpousis, A.J., Van Houwe, G., Ehretsmann, C., and Krisch,H.M. (1994) Copurification of E. coli RNAase E andPNPase: evidence for a specific association between twoenzymes important in RNA processing and degradation.Cell 76: 889–900.
Carpousis, A.J., Vanzo, N.F., and Raynal, L.C. (1999) mRNAdegradation. A tale of poly(A) and multiprotein machines.Trends Genet 15: 24–28.
Casaregola, S., Jacq, A., Laoudj, D., McGurk, G., Margarson,S., Tempete, M., et al. (1992) Cloning and analysis of theentire Escherichia coli ams gene. ams is identical to hmp1and encodes a 114 kDa protein that migrates as a 180 kDaprotein. J Mol Biol 228: 30–40.
Coburn, G.A., and Mackie, G.A. (1999) Degradation ofmRNA in Escherichia coli: an old problem with some newtwists. Prog Nucleic Acid Res Mol Biol 62: 55–108.
Coburn, G.A., Miao, X., Briant, D.J., and Mackie, G.A. (1999)Reconstitution of a minimal RNA degradosome demon-strates functional coordination between a-3′ exonucleaseand a DEAD-box RNA helicase. Genes Dev 13: 2594–2603.
Condon, C., Brechemier-Baey, D., Beltchev, B., Grunberg-Manago, M., and Putzer, H. (2001) Identification of thegene encoding the 5S ribosomal RNA maturase in Bacillussubtilis: mature 5S rRNA is dispensable for ribosome func-tion. RNA 7: 242–253.
Cormack, R.S., and Mackie, G.A. (1992) Structural require-
ments for the processing of Escherichia coli 5S ribosomalRNA by RNase E in vitro. J Mol Biol 228: 1078–1090.
Cormack, R.S., Genereaux, J.L., and Mackie, G.A. (1993)RNase E activity is conferred by a single polypeptide: over-expression, purification, and properties of the ams/rne/hmp1 gene product. Proc Natl Acad Sci USA 90: 9006–9010.
Cornet, F., Louarn, J., Patte, J., and Louarn, J.M. (1996)Restriction of the activity of the recombination site dif to asmall zone of the Escherichia coli chromosome. GenesDev 10: 1152–1161.
de la Cruz, J., Kressler, D., Tollervey, D., and Linder, P.(1998) Dob1p (Mtr4p) is a putative ATP-dependent RNAhelicase required for the 3′ end formation of 5.8S rRNA inSaccharomyces cerevisiae. EMBO J 17: 1128–1140.
Deutscher, M.P., and Li, Z. (2001) Exoribonucleases andtheir multiple roles in RNA metabolism. Prog Nucleic AcidRes Mol Biol 66: 67–105.
Diwa, A., Bricker, A.L., Jain, C., and Belasco, J.G. (2000) Anevolutionarily conserved RNA stem-loop functions as asensor that directs feedback regulation of RNase E geneexpression. Genes Dev 14: 1249–1260.
Dziembowski, A., and Stepien, P.P. (2001) Genetic andbiochemical approaches for analysis of mitochondrialdegradosome from Saccharomyces cerevisiae. MethodsEnzymol 342: 367–378.
Ehretsmann, C.P., Carpousis, A.J., and Krisch, H.M. (1992)Specificity of Escherichia coli endoribonuclease RNase E:in vivo and in vitro analysis of mutants in a bacteriophageT4 mRNA processing site. Genes Dev 6: 149–159.
Ghora, B.K., and Apirion, D. (1978) Structural analysis andin vitro processing to p5 rRNA of a 9S RNA moleculeisolated from an rne mutant of E. coli. Cell 15: 1055–1066.
Grunberg-Manago, M. (1999) Messenger RNA stability andits role in control of gene expression in bacteria andphages. Annu Rev Genet 33: 193–227.
Iost, I., and Dreyfus, M. (1995) The stability of Escherichiacoli lacZ mRNA depends upon the simultaneity of its syn-thesis and translation. EMBO J 14: 3252–3261.
Jacobs, J.S., Anderson, A.R., and Parker, R.P. (1998) The3′ to 5′ degradation of yeast mRNAs is a general mecha-nism for mRNA turnover that requires the SKI2 DEVH boxprotein and 3′ to 5′ exonucleases of the exosome complex.EMBO J 17: 1497–1506.
Jager, S., Fuhrmann, O., Heck, C., Hebermehl, M., Schiltz,E., Rauhut, R., and Klug, G. (2001) An mRNA degradingcomplex in Rhodobacter capsulatus. Nucleic Acids Res 29:4581–4588.
Jain, C., and Belasco, J.G. (1995) RNase E autoregulates itssynthesis by controlling the degradation rate of its ownmRNA in Escherichia coli: unusual sensitivity of the rnetranscript to RNase E activity. Genes Dev 9: 84–96.
Jain, C., Deana, A., and Belasco, J.G. (2002) Consequencesof RNase E scarcity in Escherichia coli. Mol Microbiol 43:1053–1064.
Jiang, X., Diwa, A., and Belasco, J.G. (2000) Regions ofRNase E important for 5′-end-dependent RNA cleavageand autoregulated synthesis. J Bacteriol 182: 2468–2475.
Kaberdin, V.R., Miczak, A., Jakobsen, J.S., Lin-Chao, S.,McDowall, K.J., and von Gabain, A. (1998) The endoribo-

The non-catalytic part of RNase E in vivo 1243
© 2002 Blackwell Science Ltd, Molecular Microbiology, 45, 1231–1243
nucleolytic N-terminal half of Escherichia coli RNase E isevolutionarily conserved in Synechocystis sp. and otherbacteria but not the C-terminal half, which is sufficient fordegradosome assembly. Proc Natl Acad Sci USA 95:11637–11642.
Kido, M., Yamanaka, K., Mitani, T., Niki, H., Ogura, T., andHiraga, S. (1996) RNase E polypeptides lacking acarboxyl-terminal half suppress a mukB mutation inEscherichia coli. J Bacteriol 178: 3917–3925.
Li, Z., Pandit, S., and Deutscher, M.P. (1999) RNase G (CafAprotein) and RNase E are both required for the 5′ matura-tion of 16S ribosomal RNA. EMBO J 18: 2878–2885.
Lin-Chao, S., Wong, T.T., McDowall, K.J., and Cohen, S.N.(1994) Effects of nucleotide sequence on the specificity ofrne-dependent and RNase E-mediated cleavages of RNAI encoded by the pBR322 plasmid. J Biol Chem 269:10797–10803.
Liou, G.G., Jane, W.N., Cohen, S.N., Lin, N.S., and Lin-Chao,S. (2001) RNA degradosomes exist in vivo in Escherichiacoli as multicomponent complexes associated with thecytoplasmic membrane via the N-terminal region of ribo-nuclease E. Proc Natl Acad Sci USA 98: 63–68.
Lopez, P.J., Iost, I., and Dreyfus, M. (1994) The use of atRNA as a transcriptional reporter: the T7 late promoter isextremely efficient in Escherichia coli but its transcripts arepoorly expressed. Nucleic Acids Res 22: 2434.
Lopez, P.J., Marchand, I., Joyce, S.A., and Dreyfus, M.(1999) The C-terminal half of RNase E, which organizesthe Escherichia coli degradosome, participates in mRNAdegradation but not rRNA processing in vivo. Mol Microbiol33: 188–199.
McDowall, K.J., and Cohen, S.N. (1996) The N-terminaldomain of the rne gene product has RNase E activity andis non-overlapping with the arginine-rich RNA-binding site.J Mol Biol 255: 349–355.
Makarova, O.V., Makarov, E.M., Sousa, R., and Dreyfus, M.(1995) Transcribing of Escherichia coli genes with mutantT7 RNA polymerases: stability of lacZ mRNA inverselycorrelates with polymerase speed. Proc Natl Acad Sci USA92: 12250–12254.
Margossian, S.P., Li, H., Zassenhaus, H.P., and Butow, R.A.(1996) The DExH box protein Suv3p is a component of ayeast mitochondrial 3′- to-5′ exoribonuclease that sup-presses group I intron toxicity. Cell 84: 199–209.
Miczak, A., Kaberdin, V.R., Wei, C.L., and Lin-Chao, S.(1996) Proteins associated with RNase E in a multicompo-nent ribonucleolytic complex. Proc Natl Acad Sci USA 93:3865–3869.
Miller, J.H. (1972) Experiments in Molecular Genetics. ColdSpring Harbor, NY: Cold Spring Harbor LaboratoryPress.
Misra, T.K., and Apirion, D. (1979) RNase E, an RNA pro-cessing enzyme from Escherichia coli. J Biol Chem 254:11154–11159.
Mudd, E.A., and Higgins, C.F. (1993) Escherichia coliendoribonuclease RNase E: autoregulation of expression
and site-specific cleavage of mRNA. Mol Microbiol 9: 557–568.
Mudd, E.A., Carpousis, A.J., and Krisch, H.M. (1990) Escher-ichia coli RNase E has a role in the decay of bacteriophageT4 mRNA. Genes Dev 4: 873–881.
Nogueira, T., de Smit, M., Graffe, M., and Springer, M. (2001)The relationship between translational control and mRNAdegradation for the Escherichia coli threonyl-tRNA syn-thetase gene. J Mol Biol 310: 709–722.
Ow, M.C., Liu, Q., and Kushner, S.R. (2000) Analysis ofmRNA decay and rRNA processing in Escherichia coli inthe absence of RNase E-based degradosome assembly.Mol Microbiol 38: 854–866.
Py, B., Causton, H., Mudd, E.A., and Higgins, C.F. (1994) Aprotein complex mediating mRNA degradation in Escheri-chia coli. Mol Microbiol 14: 717–729.
Py, B., Higgins, C.F., Krisch, H.M., and Carpousis, A.J.(1996) A DEAD-box RNA helicase in the Escherichia coliRNA degradosome. Nature 381: 169–172.
Regnier, P., and Arraiano, C.M. (2000) Degradation of mRNAin bacteria: emergence of ubiquitous features. Bioessays22: 235–244.
Sambrook, J., and Russell, D.W. (2001) Molecular Cloning.Cold Spring Harbor, NY: Cold Spring Harbor LaboratoryPress.
Schmid, S.R., and Linder, P. (1992) D-E-A-D protein familyof putative RNA helicases. Mol Microbiol 6: 283–291.
Sousa, S., Marchand, I., and Dreyfus, M. (2001) Autoregula-tion allows Escherichia coli RNase E to adjust continuouslyits synthesis to that of its substrates. Mol Microbiol 42:867–878.
Steinberg, T.H., Jones, L.J., Haugland, R.P., and Singer, V.L.(1996) SYPRO orange and SYPRO red protein gel stains:one-step fluorescent staining of denaturing gels for detec-tion of nanogram levels of protein. Anal Biochem 239: 223–237.
Studier, F.W., Rosenberg, A.H., Dunn, J.J., and Dubendorff,J.W. (1990) Use of T7 RNA polymerase to direct expres-sion of cloned genes. Methods Enzymol 185: 60–89.
Symmons, M., Williams, M., Luisi, B., Jones, G., andCarpousis, A.J. (2002) Running rings around RNA: asuperfamily of phosphate-dependent RNases. TrendsBiochem Sci 27: 11–18.
Taraseviciene, L., Bjork, G.R., and Uhlin, B.E. (1995) Evi-dence for an RNA binding region in the Escherichia coliprocessing endoribonuclease RNase E. J Biol Chem 270:26391–26398.
Tock, M.R., Walsh, A.P., Carroll, G., and McDowall, K.J.(2000) The CafA protein required for the 5′-maturation of16 S rRNA is a 5′-end-dependent ribonuclease that hascontext-dependent broad sequence specificity. J BiolChem 275: 8726–8732.
Vanzo, N.F., Li, Y.S., Py, B., Blum, E., Higgins, C.F., Raynal,L.C., et al. (1998) Ribonuclease E organizes the proteininteractions in the Escherichia coli RNA degradosome.Genes Dev 12: 2770–2781.




















