
REFARAT REVISI
70
BAB I PENDAHULUAN Beberapa masalah pediatri dapat ditangani dengan pembedahan diantaranya trauma, tumor, masalah-masalah gastrointestinal (misalnya perdarahan, anomali traktus gastrointestinal, peritonitis, ikterus obstruktif), distres pernapasan (misalnya yang disebabkan oleh obstruksi jalan napas atas, anomali diafragma), malformasi kongenital (misalnya defek dinding abdomen, malformasi anorektal), dan gangguan endokrin (misalnya hiperparatiroidisme primer, disorder of sex development). 1 Beberapa masalah di atas juga merupakan kelainan bawaan / kelainan kongenital, yaitu kelainan yang sudah ada sejak lahir yang dapat disebabkan oleh faktor genetik maupun non genetik; contohnya anensefalus, labiopalatoskisis, atresia esofagus, atresia bilier, omfalokel, penyakit Hirschprung, malformasi anorektal, disorder of sex development, tetralogy of Fallot, defek septum ventrikel, dan duktus arteriosus paten. 2 1
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of REFARAT REVISI

BAB I
PENDAHULUAN
Beberapa masalah pediatri dapat ditangani dengan
pembedahan diantaranya trauma, tumor, masalah-masalah
gastrointestinal (misalnya perdarahan, anomali traktus
gastrointestinal, peritonitis, ikterus obstruktif),
distres pernapasan (misalnya yang disebabkan oleh
obstruksi jalan napas atas, anomali diafragma),
malformasi kongenital (misalnya defek dinding abdomen,
malformasi anorektal), dan gangguan endokrin (misalnya
hiperparatiroidisme primer, disorder of sex development).1
Beberapa masalah di atas juga merupakan kelainan
bawaan / kelainan kongenital, yaitu kelainan yang sudah
ada sejak lahir yang dapat disebabkan oleh faktor
genetik maupun non genetik; contohnya anensefalus,
labiopalatoskisis, atresia esofagus, atresia bilier,
omfalokel, penyakit Hirschprung, malformasi anorektal,
disorder of sex development, tetralogy of Fallot, defek
septum ventrikel, dan duktus arteriosus paten.2
1

Pada refarat ini akan dibahas mengenai 2 dari
beberapa masalah pediatri yaitu disorder of sex development
(DSD) dan malformasi anorektal (atresia ani).
BAB II
TINJAUAN PUSTAKA
DISORDERS OF SEX DEVELOPMENT (DSD)
A. Definisi
Perkembangan normal sistem reproduksi terjadi
melalui dua fase yaitu fase determinasi dan fase
diferensiasi. Fase determinasi merupakan fase penentuan
jenis gonad yang dipengaruhi oleh faktor kromosom dan
faktor gonad; sedangkan fase diferensiasi dipengaruhi
oleh faktor hormonal. Jika terjadi gangguan pada salah2

satu dari kedua fase tersebut, maka sistem reproduksi
tidak akan berkembang sempurna. Hal ini kini dikenal
sebagai disorders of sex development (DSD). Istilah DSD
muncul dari pertemuan Lawson Wilkins Paediatric Endocrine Society
(LWPES) dan the European Society for Paediatric Endocrinology
(ESPE), untuk menggantikan terminologi lama yaitu
‘interseks’ atau ‘hermafrodit’.3,4
Terdapat beberapa terminologi lama yang sudah tidak
dipakai lagi, dan digantikan dengan istilah baru, yang
dapat dilihat pada tabel 1.
Tabel 1. Terminologi yang lama dan yang baru sehubungan dengan kasus DSD3,5
Terminologi lama Terminologi baruInterseks DSD
Male pseudohermaphrodite, undervirilized male,
atau undermasculinization of XY male46,XY DSD
Female pseudohermaphrodite, overvirilized of XX
female, atau masculinization of XX female46,XX DSD
True hermaphrodite DSD ovotestikulerXX male atau XX sex reversal 46,XX DSD testikuler
XY sex reversal atau XY female46,XY disgenesis gonad
komplit
DSD merupakan kelainan bawaan dimana terjadi
ketidakselarasan kromosom, perkembangan gonad dan
3

anatomi jenis kelamin, sehingga perkembangan sistem
reproduksi atipikal atau menyimpang.3-5
B. Perkembangan sistim reproduksi
Perkembangan genitalia terjadi pada masa gestasi 6-
14 minggu. Meskipun jenis kelamin embrio ditentukan
secara genetik pada waktu fertilisasi, tetapi gonad
tidak memperoleh karakteristik morfologi pria atau
wanita sampai usia gestasi 6 minggu. Jadi, sampai
dengan masa gestasi 6 minggu, gonad primordial bersifat
indiferen atau bipotensial (mampu untuk berkembang ke
dua arah yang mungkin yaitu menjadi testis atau menjadi
ovarium). Hingga usia 6 minggu masa gestasi, embrio
juga memiliki sepasang duktus Mulleri (duktus
mesonephros), sepasang duktus Wolffi (duktus
paramesonephros) dan bakal genitalia eksterna maupun
interna yang indiferen.3
Sekresi hormon androgen mulai terjadi pada masa
gestasi 7-8 minggu setelah testis terbentuk. Puncak
sekresi testosteron terjadi antara masa gestasi 14-164

minggu. Hormon androgen selanjutnya akan menyempurnakan
proses diferensiasi genitalia interna dan eksterna.3
a. Fase determinasi
Fase ini merupakan langkah awal perkembangan
sistim reproduksi. Setiap gangguan pada fase ini
sangat potensial untuk menyebabkan DSD disgenesis
gonad.3
Kromosom
Laki-laki memiliki kromosom 46,XY sedangkan
wanita 46,XX. Kromosom XY atau XX ditentukan
saat fertilisasi. Pada usia gestasi dini, gonad
yang terbentuk bersifat indiferen atau
bipotensial, baik pada embrio XY atau XX. Dalam
penelitian Jost dkk, disimpulkan bahwa
testislah yang berperan dalam diferensiasi
genitalia interna maupun eksterna; dan sejak
percobaan ini, upaya untuk mencari faktor
penentu testis (testis-determining factor / TDF)
berlangsung. Keberadaan faktor penentu testis
ini kemudian berhasil dilokalisir oleh Sinclair5

dkk tahun 1990, yang dikenal sebagai gen SRY
(sex-determining region on the Y chromosome), pada
lengan pendek kromosom Y (kromosom Yp11.31).
Pada ketiadaan gen SRY, maka gonad akan
berkembang menjadi ovarium; sebaliknya dengan
adanya gen SRY maka gonad bipotensial akan
berkembang menjadi testis.3,6
Gen SRY juga mengatur steroidogenesis factor 1 atau
SF1 (dalam hal ini upregulation) yang bekerja
melalui faktor transkripsi, SOX9, untuk
menginduksi diferensiasi dari sel-sel Leydig
dan Sertoli. SOX9 juga mempengaruhi gen yang
memproduksi MIS untuk regresi duktus Mulleri
(akan dijelaskan selanjutnya). Telah diketahui
pula pada diferensiasi seksual wanita (yang
akan dijelaskan selanjutnya), terdapat gen
spesifik yang menginduksi perkembangan ovarium,
yaitu DAX1, yang menghambat SOX9 dan berlokasi
pada lengan pendek kromosom X dan bekerja
dengan mengatur aktivitas SF1 (dalam hal ini6

downregulation), yang mencegah diferensiasi dari
sel Sertoli dan sel Leydig. Diketahui pula
bahwa faktor pertumbuhan yang disekresikan
yaitu WNT4 berkontribusi dalam diferensiasi
ovarium.7,8
Gonad
Cikal bakal dari gonad adalah tonjolan
urogenital (urogenital ridge). Tonjolan urogenital
ini berkembang dari mesoderm, dan terdiri dari
pronephros, mesonephros, dan metanephros, yang
akan berkembang menjadi gonad, ginjal dan
adrenal. Tonjolan gonad (gonadal / genital ridge)
terbentuk pada sisi ventromedial mesonephros
pada masa gestasi 10 hari. Gonad primordium ini
terdiri dari mesenkim mesonephros, sel epitel
serta sel-sel germinal. Sel-sel germinal tidak
muncul pada tonjolan gonad sampai usia
kehamilan 6 minggu. Sel-sel germinal primordial
pertama kali muncul pada tahap awal gestasi, di
antara sel endoderm di dinding yolk sac, dekat7

alantois (Gambar 1a). Sel-sel germinal
primordial ini bermigrasi dengan gerakan
ameboid sepanjang bagian dorsal mesenterium
hindgut dan tiba pada gonad primitif pada awal
minggu ke-5 dan menginvasi tonjolan gonad pada
minggu ke-6 masa gestasi (Gambar 1b). Jika sel-
sel germinal primordial gagal mencapai tonjolan
gonad, maka gonad tidak akan berkembang. Karena
itu, sel-sel germinal primordial memiliki
pengaruh induktif pada perkembangan gonad ke
arah ovarium atau testis. Pada stadium ini,
gonad bersifat indiferen atau bipotensial.3,8
Sebelum dan selama sel-sel germinal primordial
tiba, epitel tonjolan gonad berproliferasi, dan
sel-sel epitel memasuki mesenkim yang
mendasari. Disini, sel-sel epitel membentuk
sejumlah korda yang berbentuk ireguler, yaitu
korda seks primitif (primitive sex cords), yang
mampu berdiferensiasi antara gonad pria dan
wanita (Gambar 2).8
8

Gambar 1. (a) Embrio usia 3 minggu, menunjukkan sel-sel germinal
primordial pada dinding yolk sac dekat dengan alantois; (b)
Jalan migrasi sel-sel germinal primordial ke tonjolan
genital.8
Jika embrio secara genetik adalah laki-laki,
sel-sel germinal primordial membawa kompleks
kromosom seks XY. Di bawah pengaruh gen SRY
pada kromosom Y, maka primitive sex cords
melanjutkan proliferasi dan masuk lebih dalam
ke medula untuk membentuk testis atau medullary9
Gambar 2.Potongan melintang
melalui regio
lumbalis dari embrio
usia 6 minggu
menunjukkan gonad
indiferen dengan
korda seksual

cords (Gambar 3). Selama perkembangan
selanjutnya, sebuah lapisan padat jaringan ikat
fibrosa, yaitu tunika albuginea, memisahkan
korda testis dari epitel permukaan. Sel-sel
epitel permukaan ini kemudian berproliferasi
dan berdiferensiasi menjadi sel-sel Sertoli.
Ini terjadi pada usia gestasi 6 minggu. Sel-sel
interstitial Leydig berasal dari mesenkim
original dari gonadal ridge, yang mulai
berkembang secara singkat setelah diferensiasi
dari korda testis. Minggu ke-8 gestasi, sel-sel
Leydig mulai memproduksi testosteron dan testis
menjadi mampu untuk mempengaruhi diferensiasi
seksual dari genital interna dan eksterna.3,8
10
Gambar 3.Potongan
melintang melalui
testis pada minggu
ke-8 menunjukkan

Pada embrio wanita dengan kompleks kromosom sex
XX dan tidak ada kromosom Y, korda seksual
primitif berpisah menjadi kelompok-kelompok sel
ireguler. Kelompok-kelompok ini memuat kelompok
sel germinal primitif, yang menempati bagian
medula dari ovarium. Kemudian, mereka hilang
dan digantikan oleh stroma vaskuler yang
membentuk medula ovarium. Epitel permukaan
gonad wanita, tidak seperti gonad pria,
melanjutkan diri berproliferasi. Pada minggu
ke-7, epitel permukaan berkembang menjadi
cortical cords, yang memasuki mesenkim yang
mendasari (Gambar 4).8
11
Gambar 4.Potongan melintang ovarium pada minggu ke-7
menunjukkan degenerasi dari medullary cords dan
pembentukan cortical cords.8

Dengan demikian, dapat dinyatakan bahwa jenis
kelamin genetik dari embrio ditentukan pada waktu
fertilisasi, tergantung dari apakah sperma
membawa kromosom X atau kromosom Y. Pada embrio
dengan konfigurasi kromosom seks XX, medullary cords
dari gonad menjadi surut, dan generasi kedua dari
cortical cords berkembang (Gambar 4). Pada embrio
dengan kompleks kromosom seks XY, medullary cords
berkembang menjadi korda testis dan cortical cords
gagal berkembang (Gambar 3).8
b. Fase diferensiasi
Fase diferensiasi genital interna dan eksterna
bergantung pada faktor hormonal (Gambar 5). Hormon
androgen yang disekresikan oleh testis, pada awalnya
diatur oleh human chorionic gonadotropin (hCG) yang
berasal dari plasenta, yang mencapai kadar puncak
pada usia gestasi 8-12 minggu. Pada minggu ke-15
masa gestasi, pengaturan sekresi testosteron ini
mulai diambil alih oleh jaras hipotalamus-hipofisis12

janin dengan gonadotropinnya (dalam hal ini adalah
luteinizing hormone / LH), dan dipertahankan pada kadar
yang lebih rendah di usia kehamilan lanjut. Sekresi
gonadotropin ini akan berkurang hingga menjelang
akhir gestasi.3
Gambar 5. Skema peran hormon pada perkembangan normal
sistim reproduksi3
Genital interna
13

Perkembangan genital interna merupakan efek
parakrin dari gonad ipsilateral. Parakrin
merupakan tipe fungsi hormon dimana hormon
disintesis dan dilepaskan dari sel-sel endokrin
dan terikat pada reseptornya di sel-sel dekat
sel-sel endokrin tersebut dan mempengaruhi
fungsinya. Ketika tidak ada jaringan testis,
janin (fetus) secara morfologi, mulai dan
menyelesaikan perkembangan genital interna dan
perkembangan fenotip eksterna sebagai wanita.
Ketika ada jaringan testis, ada 2 substansi
yang diproduksi yang penting untuk perkembangan
genital interna pria dan fenotipe eksterna
pria, yaitu testosteron dan Mullerian-inhibiting
substance (MIS) atau Mullerian-inhibiting factor (MIF)
atau Anti-Mullerian hormone (AMH).3,9
Proses diferensiasi genital interna terjadi
sejak minggu ke-6 masa gestasi. Pada janin
pria, diferensiasi genital interna berlangsung
dengan terbentuknya sel Sertoli yang14
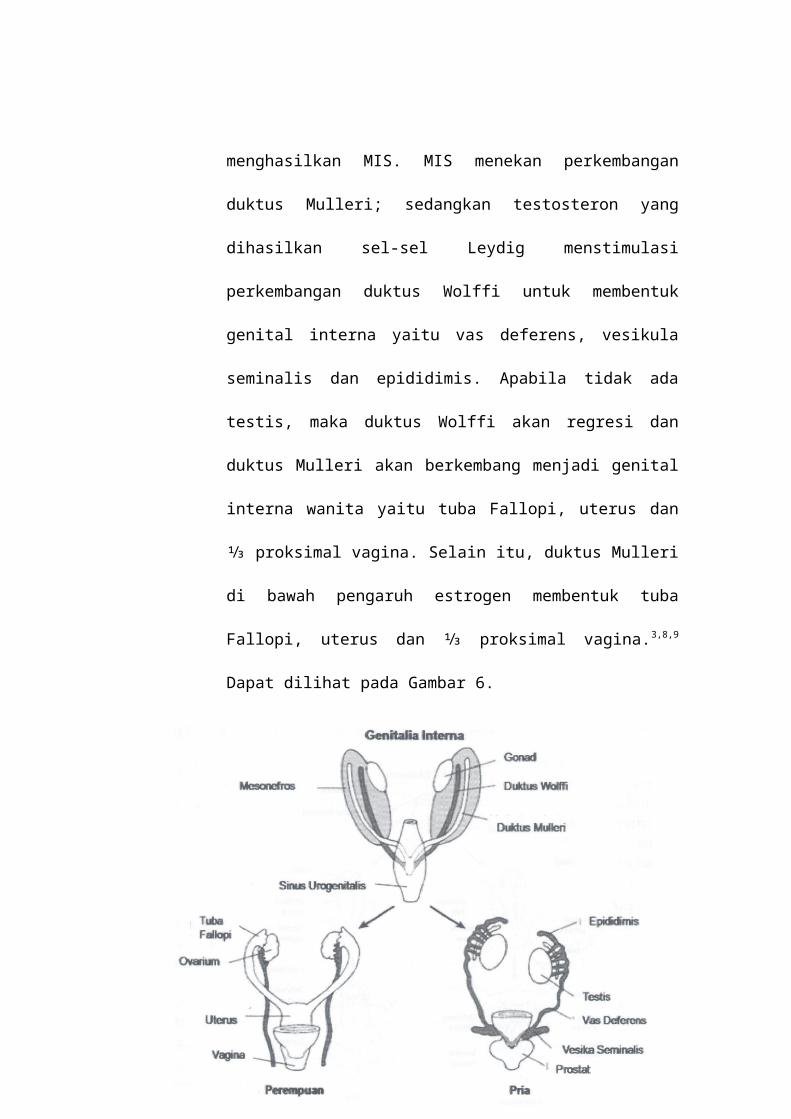
menghasilkan MIS. MIS menekan perkembangan
duktus Mulleri; sedangkan testosteron yang
dihasilkan sel-sel Leydig menstimulasi
perkembangan duktus Wolffi untuk membentuk
genital interna yaitu vas deferens, vesikula
seminalis dan epididimis. Apabila tidak ada
testis, maka duktus Wolffi akan regresi dan
duktus Mulleri akan berkembang menjadi genital
interna wanita yaitu tuba Fallopi, uterus dan
⅓ proksimal vagina. Selain itu, duktus Mulleri
di bawah pengaruh estrogen membentuk tuba
Fallopi, uterus dan ⅓ proksimal vagina.3,8,9
Dapat dilihat pada Gambar 6.
15

Genital eksterna
Sebelum terjadi diferensiasi genital eksterna,
maka baik janin laki-laki maupun perempuan
memiliki struktur embrional genital eksterna
yang indiferen / bipotensial yaitu sinus
urogenitalis, genital tubercle, genital fold, dan genital
swelling (Gambar 7). Ada tidaknya testosteron
yang dikonversi menjadi 5-DHT
(dihidrotestosteron) oleh enzim 5-α reduktase,
mempengaruhi berkembangnya struktur embrional
tersebut. Perkembangan struktur embrional
genital eksterna dapat dilihat pada tabel 2.3,9
Tabel 2. Perkembangan struktur embrional genital eksterna3
Struktur
embrionalPria Wanita
Sinus
urogenitalisProstat*
Vagina ⅔
inferior
16
Gambar 6. Diferensiasi
genital interna3
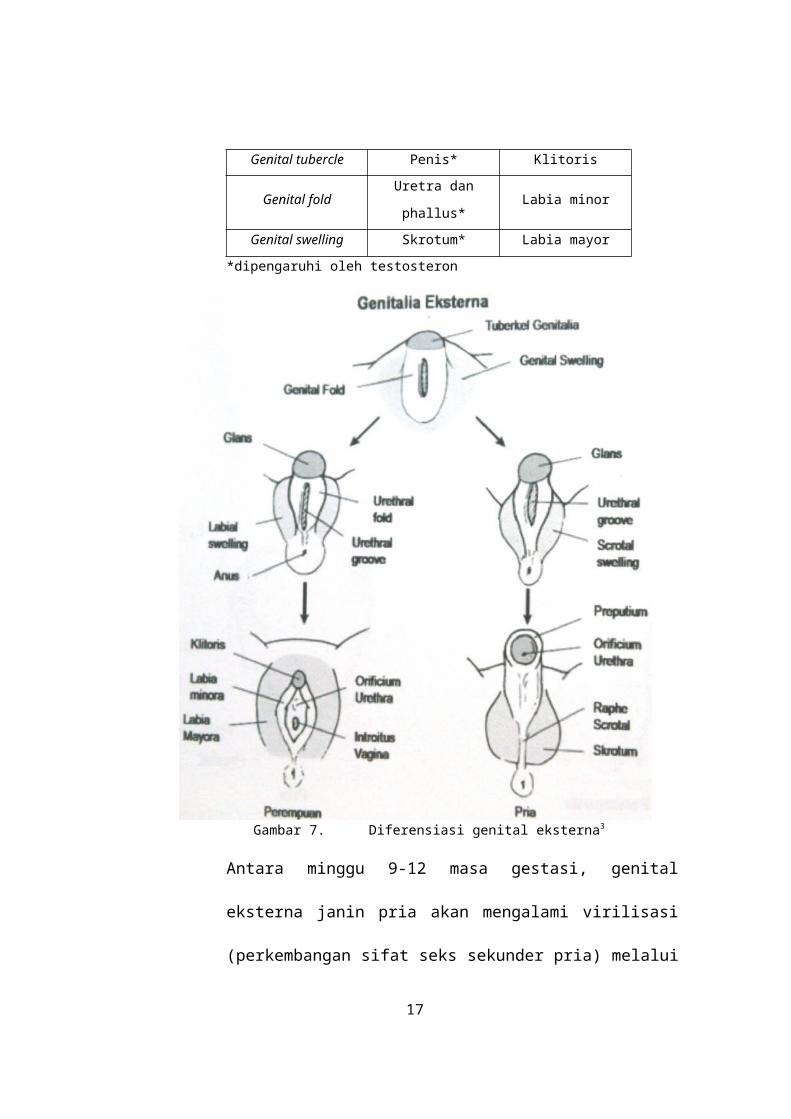
Genital tubercle Penis* Klitoris
Genital foldUretra dan
phallus*Labia minor
Genital swelling Skrotum* Labia mayor*dipengaruhi oleh testosteron
Gambar 7. Diferensiasi genital eksterna3
Antara minggu 9-12 masa gestasi, genital
eksterna janin pria akan mengalami virilisasi
(perkembangan sifat seks sekunder pria) melalui
17

DHT. DHT akan menyebabkan fusi lipatan
labioskrotal sehingga terbentuk skrotum. DHT
akan menyempurnakan bentuk anatomi genital
eksterna antara minggu 12-14 masa gestasi.
Apabila lipatan labioskrotal tidak mengalami
fusi pada akhir minggu ke-12, maka testosteron
akan tetap menyebabkan pertumbuhan phallus
tanpa menyempurnakan fusi yang gagal tersebut.
Pada trimester ketiga kehamilan, testis akan
turun (desensus) ke skrotum.3
Untuk perkembangan penis, terjadi dalam fase
intra- dan ekstrauterin, dan dipengaruhi oleh
testosteron. Pada fase intrauterin, terjadi
fase pembentukan (formative phase) dan fase
pertumbuhan (linear growth phase). Fase pembentukan
terjadi pada trimester pertama kehamilan
(antara 8-14 minggu). Fase pertumbuhan terjadi
pada trimester kedua dan ketiga kehamilan dan
berlanjut sampai usia pubertas. Pada keadaan
normal, panjangnya penis pada akhir trimester18

pertama adalah 3,5 mm, identik panjangnya
dengan klitoris. Gangguan hormonal pada fase
pembentukan akan mengakibatkan ukuran penis
yang kecil, dengan hipospadia. Dalam keadaan
normal, pada fase kedua intrauterin, panjang
penis bertambah panjang 10 kali lipat sehingga
pada saat lahir panjangnya adalah 35 mm.3
C. Epidemiologi
DSD merupakan keadaan yang relatif jarang ditemukan.
Insidensnya diperkirakan 1:4500 sampai 1:5500, atau
bervariasi sesuai dengan etiologi. Penyebab tersering
adalah hiperplasia adrenal kongenital (HAK) ditemukan
pada 50% bayi baru lahir dengan DSD; mixed gonadal disease
(MGD) diperkirakan merupakan penyebab tersering kedua
dengan insidens 1:10000. Hal ini sama pula dengan yang
terjadi di Amerika Serikat dimana HAK merupakan
penyebab tersering genital ambigu pada bayi baru lahir,
dan MGD merupakan penyebab kedua dari DSD. Analisis
dari skrining bayi di seluruh dunia bahwa dari 6,5 juta
19

bayi baru lahir ditemukan insidens HAK adalah 1:15.000
kelahiran hidup. Dengan teknologi terkini, hanya 50%
kasus 46,XY DSD yang dapat diketahui penyebabnya dan
hanya 20% kasus DSD secara keseluruhan yang dapat
didiagnosis secara molekuler.3-5,9,10
D. Etiologi
Etiologi DSD sangat luas, dapat dilihat pada tabel
3.
Tabel 3. Klasifikasi etiologi DSD3,5,6,10
Kromosom
seks DSD
46,XX DSD 46,XY DSD
- 45,XO
(sindrom
Turner dan
varian
mosaic)
- 47,XXY
(sindrom
Klinefelte
r dan
varian)
- 45,XO/
46,XY
(MDG,
ovotestiku
- Paparan androgen
berlebih yaitu dari
janin atau fetoplasenta
(misalnya karena
defisiensi P450 c21,
defisiensi P450 c11,
hiperplasia adrenal
kongenital / HAK,
defisiensi aromatase,
mutasi gen reseptor
glukokortikoid) dan
dari ibu (misalnya
karena obat-obat
androgenik, tumor
- Defek perkembangan testis
Disgenesis gonad komplit
(Swyer syndrome)
Disgenesis gonad parsial
(mutasi WT1 / Denys Drash
syndrome, SOX9, SF1)
DSD ovotestikuler
Sebab lain yang tidak
diketahui
- Defisiensi hormon
testikuler (misalnya karena
hipoplasia / aplasia sel-
sel Leydig, mutasi reseptor
LH, HAK, defisiensi enzim
20

ler DSD)
- 46,XX/
46,XY
(chimeric,
ovotestiku
ler DSD)
virilisasi)
- Gangguan perkembangan
ovarium (misalnya
disgenesis gonad XX,
DSD ovotestikuler)
- Sebab yang tidak dapat
ditentukan (berhubungan
dengan defek traktus
genitourinarius dan
gastrointestinal)
5-α reduktase, Smith-Lemli-
Opitz syndrome)
- Defek kerja androgen
(misalnya defek reseptor
androgen, sindrom
insensitivitas androgen
komplit dan parsial)
a. 46,XX DSD (bayi atau anak yang mengalami
virilisasi)
Sebagian besar kasus kategori ini ditandai dengan
adanya gonad berupa ovarium disertai genital interna
wanita. Genital eksterna mengalami maskulinisasi
karena pengaruh androgen. Sumber androgen
intrauterin seperti yang terdapat dalam tabel 3 di
atas yaitu dari janin, plasenta dan dari ibu.
Pengaruhnya bervariasi, dari klitoromegali ringan
sampai dengan fusi sempurna labia dengan bergesernya
sinus urogenitalis sebagai lubang uretra ke arah
21

ujung distal dari phallus yang membesar. Gonad pada
kelompok ini tidak akan teraba.3
Penyebab tersering adalah HAK. HAK merupakan
keadaan yang diturunkan secara autosomal resesif
dimana terdapat defek enzim pada salah satu dari
proses steroidogenesis adrenal sehingga terjadi
akumulasi steroid proksimal. Akumulasi steroid
proksimal pada akhirnya dikonversi menjadi androgen
yang mengakibatkan terjadinya virilisasi.3
Penyebab lain yang mungkin adalah pajanan
terhadap androgen eksogen, misalnya dari konsumsi
androgen atau progestin ibu, atau tumor ibu yang
menghasilkan androgen (tetapi hal ini jarang).
Selain itu, defisiensi enzim aromatase plasenta juga
dapat menyebabkan virilisasi pada janin (dan pada
ibu), karena enzim aromatase berfungsi untuk
mengubah testosteron menjadi estradiol pada unit
fetoplasenta, dan defisiensi enzim ini berakibat
meningkatnya kadar testosteron pada plasenta dan
janin.3
22

b. 46,XY DSD (bayi atau anak yang mengalami
undervirilisation)
Penyebab tersering kategori ini adalah sindrom
insensitivitas androgen (SIA), yang merupakan
keadaan yang diturunkan secara resesif X-linked
karena resistensi perifer (sel target) terhadap
kerja androgen akibat mutasi gen reseptor androgen.
SIA terbagi menjadi SIA komplit (SIAK) dan SIA
parsial (SIAP). Pada SIAK fenotip adalah perempuan
sempurna, sedangkan pada SIAP terjadi genital ambigu
yang bervariasi. Sebagian besar penderita SIAK akan
terdiagnosis pada masa pubertas atau setelahnya
karena keluhan amenore. Pada SIA, kadar testosteron
normal dengan genital interna tetap laki-laki.
Karena diturunkan secara X-linked, maka riwayat
keluarga sangat penting.3
Defisiensi enzim 5-α reduktase merupakan penyebab
lain kategori ini, yang diturunkan secara autosomal23

resesif, sehingga mengakibatkan gangguan konversi
testosteron menjadi DHT. Defisiensi DHT menyebabkan
virilisasi genital eksterna tidak sempurna.3
Bayi atau anak yang termasuk dalam kategori ini
memiliki phallus kecil, hipospadia posterior,
skrotum bifidum yang terbentuk tidak sempurna dengan
atau tanpa kriptorkismus.3
c. 46,XX DSD testikuler dan DSD ovotestikuler
Manifestasi klinis XX male dapat dikategorikan
sebagai DSD testikuler dengan fenotip lelaki normal,
DSD testikuler dengan genital ambigu dan DSD
ovotestikuler. Berdasarkan ada tidaknya unsur SRY
maka diklasifikasikan sebagai DSD testikuler Y (+)
dan DSD testikuler Y (-). Sebagian besar yang Y (+)
memiliki genital eksterna normal dan steril,
sedangkan pada Y (-) genital tampak ambigu dan
steril. Penelitian aspek molekuler pada kasus-kasus
DSD testikuler ini memperlihatkan bahwa pada 80%
kasus terjadi akibat translokasi Y-X, dan terjadi24

kecenderungan menginaktifkan kromosom X yang
mengandung Y. Sumber fenotip pria pada DSD
testikuler ini diperkirakan berasal dari: 1)
translokasi sekuens Y, termasuk gen SRY ke kromosom
X atau kromosom autosom; 2) mutasi yang belum
diketahui pada gen X-linked atau autosom yang
terlibat pada jalur pembentukan testis; 3) mosaicsm
kromosom Y yang kriptik.3
Gonad pada bayi atau anak dengan 46,XX DSD
testikuler adalah testis. Ciri utama dari tipe ini
adalah genital eksterna yang tidak berkembang
sempurna disertai testis yang kecil (mikrotestis).
Selain itu pada sebagian besar kasus, akan mengalami
kegagalan untuk menjalani fase pubertas dengan
rambut dada dan aksila yang jarang disertai
distribusi rambut pubis seperti perempuan.3
DSD ovotestikuler, yang dulu disebut true
hermaphrodite, merupakan keadaan ditemukannya
jaringan testis dan ovarium nomal, tanpa memandang
kariotipenya. Gonad biasanya berupa ovarium-testis25

atau ovarium-ovotestis. Genotipe tersering adalah
46,XX walaupun dapat pula ditemukan 46,XY atau
mosaik. Kariotipe yang paling sering dilaporkan pada
DSD ovotestikuler adalah 46XX, 46XY, 46XX/46XY,
45X/46XY. Gonad, genital interna dan eksterna
didapatkan asimetri. Sisi mana yang mengalami
virilisasi dan sisi mana yang mengalami feminisasi
bergantung gonad yang dominan pada sisi ipsilateral.
Fenotipenya sangat bervariasi dari perempuan hingga
laki-laki normal bergantung pada fungsi sel-sel
Leydig, namun sebagian besar kasus memperlihatkan
adanya virilisasi.3
d. DSD kromosom seks
Penyebab tersering kategori ini adalah MGD.
Seperti pada keadaan DSD ovotestikuler, pada
kategori ini ditemukan pula gambaran asimetris. Pada
individu 46,XY testis yang disgenetik walaupun masih
dapat mensekresikan testosteron, produksi MIS
biasanya rendah atau tidak ada sehingga organ-organ26

derivat duktus Mulleri seringkali ditemukan.
Sebagian besar kasus memiliki fenotip genital
interna testis atau ovotestis unilateral disertai
streak gonad (gonad pita) kontralateral, struktur
duktus Mulleri yang persisten pada sisi homolateral
dengan gonad yang disgenetik, dan berbagai tingkat
undervirilisation pada genital eksterna. Secara
histologis, pada streak gonad terdiri dari stroma
ovarium tanpa oosit.3
e. Disgenesis gonad
Disgenesis gonad dapat bersifat komplit / total
dan parsial / mixed. Dikatakan total apabila kedua
gonad adalah streak gonad, dikatakan parsial bila
ditemukan gonad (testis atau ovotestis) pada satu
sisi disertai gonad pita pada sisi kontralateral.
Secara klinis, individu dengan disgenesis gonad akan
memperlihatkan gejala hipogonadisme.3
f. DSD disgenesis gonad komplit
27

Disgenesis gonad total jarang ditemukan pada masa
neonatus karena fenotipenya adalah perempuan tanpa
genital ambigu. Pada kasus dengan kariotipe 46,XY
(sindrom Swyer) terjadi sex reversal sehingga
fenotipenya adalah perempuan. Klinis terlihat
sebagai perempuan dengan tinggi badan normal,
pubertas terlambat, amenore primer, sexual infantilism
(tidak ada perkembangan tanda-tanda seks sekunder,
hipoplasia uterus) dan streak gonad bilateral.3
E. Manifestasi klinis
Genital ambigu merupakan salah satu gejala klinis
yang memberikan indikasi adanya DSD, walaupun tidak
semua DSD akan bermanifestasi klinis genital ambigu,
sebagai contoh pada DSD kromosom seks yang disebabkan
oleh sindrom Turner (kariotipe 45,XO) dan sindrom
Klinefelter (kariotipe 47,XXY) yang memiliki gambaran
klinis anak perempuan. Genital ambigu adalah penampilan
atau fenotip genital eksterna yang tidak khas sehingga
ragu menggolongkan sebagai laki-laki atau perempuan.28

Manifestasi DSD sebagian besar terjadi pada masa
neonatus dan bayi dengan keluhan utama:3
Genital ambigu pada neonatus. Ditemukan pada
beberapa klasifikasi DSD seperti yang telah
dijelaskan sebelumnya.
Testis tidak teraba pada bayi ‘laki-laki’
Penonjolan di daerah inguinal pada bayi
‘perempuan’
Hipospadia berat
Klitoromegali
Gambar 8. Gambaran genital eksterna yang atipikal.
Gambaran ini ditemukan pada kasus (a) DSD29
F

ovotestikuler; (b)-(e) HAK; (f) Insensitivitas
androgen parsial.6
Manifestasi lambat dapat terjadi setelah masa
neonatus, baik pada anak maupun remaja dengan keluhan
utama:3,5
Virilisasi saat pubertas pada anak ‘perempuan’
Pubertas terlambat pada anak perempuan
Amenore primer
Hernia inguinalis pada anak perempuan
Ginekomastia pada anak laki-laki
Infertilitas pada laki-laki
F. Diagnosis
Penentuan jenis kelamin pada bayi yang lahir dengan
genital ambigu sebaiknya tidak segera dilakukan, hingga
pemeriksaan lengkap telah dilakukan. Diagnosis DSD
dilakukan berdasarkan anamnesis, pemeriksaan fisik dan
pemeriksaan penunjang.3,4
a. Anamnesis
30

Riwayat pranatal: ibu mengkonsumsi obat-obat
steroid, diagnosis antenatal adanya androgen
producing tumor, dan virilisasi ibu selama
kehamilan (defisiensi aromatase).3,4
Riwayat keluarga: riwayat serupa dalam keluarga
untuk beberapa kelainan DSD yang diturunkan
(misalnya sindrom insensitivitas androgen
secara X-linked, defisiensi 5-α reduktase
secara autosomal resesif, dan HAK secara
autosomal resesif), riwayat kematian perinatal
yang tidak jelas (misalnya akibat muntah-
muntah, dengan atau tanpa genital ambigu
merupakan petunjuk yang mengarah ke HAK),
riwayat infertilitas (dapat memberi petunjuk
adanya DSD pada SIAK).3,4
Riwayat penyakit: mulai timbulnya,
progresivitas, riwayat gagal tumbuh dan
pubertas, riwayat penyakit dahulu atau operasi
yang pernah dijalani.3
b. Pemeriksaan fisik31

Keadaan umum: Perlu diteliti keadaan umum
penderita seperti gagal tumbuh, retardasi
mental, mikrosefal. Penampilan fisik yang
dismorfik dengan genital ambigu cukup sering
ditemukan seperti pada sindrom Smith-Lemli-
Opitz. Sindrom Turner dengan perawakan pendek,
low posterior hairline, web neck, dan limfedema
merupakan tanda yang khas ditemui. Perlu
dicatat bahwa tampilan genital eksterna pada
DSD tidak selamanya adalah genital ambigu.3
Genital eksterna
Gonad. Pemeriksaannya merupakan langkah
strategis dalam diagnosis DSD. Gonad yang
teraba menandakan adanya gen SRY,
perkembangan testis dan regresi duktus
Mulleri ipsilateral. Tiga keadaan klinis yang
mungkin ditemukan yaitu:3
(1) Kedua gonad teraba dan simetris, artinya
bayi tersebut kemungkinan besar laki-laki
yang tidak mengalami virilisasi adekuat32

dan struktur duktus Mullerinya regresi.
Diagnosis bandingnya: produksi testosteron
inadekuat, defek reseptor androgen,
defisiensi 5-α reduktase. DSD
ovotestikuler merupakan pengecualian
dimana ditemukan ovotestis bilateral yang
simetris.3
(2) Asimetri gonad dengan hanya teraba 1
gonad, yang menandakan bahwa paling tidak,
ada 1 testis; yang satunya mungkin
ovarium, ovotestis, atau streak gonad. Bila
ditemukan perlu dipikirkan: DSD
ovotestikuler atau MGD.3
(3) Tidak teraba gonad. Pada kondisi ini
kondisi gonad dan duktus tidak diketahui.
Petunjuk tambahan mungkin dapat dilakukan
lebih teliti untuk mengetahui apakah
cincin inguinal terbuka atau tidak. Bila
terbuka, menandakan kemungkinan testis
yang tidak turun, sedangkan bila tertutup33

dapat dihubungkan dengan adanya ovarium
atau testis yang sangat displastik dengan
produksi testosteron yang sangat minimal.
Pemeriksaan rektal dengan menggunakan jari
kelingking akan mudah neraba serviks dan
mengkonfirmasi adanya uterus. Diagnosis
bandingnya: 46XX DSD, 46XY disgenesis
gonad, ovotestikuler DSD.3
Phallus. Pada bayi baru lahir, panjang penis
normal adalah 3,5 ± 0,7 cm. Panjang phallus <
2,0 cm dianggap sebagai mikropenis, dan pada
bayi perempuan bila panjang klitoris > 1 cm
dianggap sebagai klitoromegali. Mikropenis
merupakan ukuran penis < - 2,5 SD untuk
usianya, tanpa disertai kelainan struktural
penis lainnya (hipospadia), yang diukur
ketika penis telah diregang maksimal tanpa
ereksi.3
Orifisium uretra. Bila orifisium uretra dan
introitus vagina jelas terpisah, menandakan34

46,XX. Bila hanya terdapat 1 lubang genital
eksterna, selain kemungkinan 46,XY DSD dapat
juga merupakan sinus urogenital perempuan
yang mengalami virilisasi.3
Rasio anogenital, yang merupakan jarak antara
anus dengan posterior fourchette dibagi dengan
jarak antara anus dengan dasar phallus /
klitoris, Rasio > 0,5 menandakan adanya
virilisasi; dan pada laki-laki yang mengalami
virilisasi sempurna, rasionya adalah 1.3
Stadium Prader, yang menunjukkan berat
ringannya virilisasi, terbagi menjadi 6
stadium yaitu: Prader 0 (genital perempuan
normal); Prader 1 (phallus membesar /
hipertrofi klitoris saja sedangkan genital
eksterna lain normal fenotipe perempuan);
Prader 2 (phallus membesar / hipertrofi
klitoris dengan lubang uretra dan vagina yang
terpisah secara nyata); Prader 3 (phallus
membesar / hipertrofi klitoris dengan 135

lubang sinus urogenital); Prader 4 (phallus
membesar dengan hipospadia); Prader 5
(genital laki-laki normal).3,4
Gambar 9. Stadium Prader untuk genital ambigu.4
c. Pemeriksaan penunjang
Analisis kromosom. Merupakan pemeriksaan lini
pertama yang perlu dilakukan dengan kariotipe
dan flourescence in-situ hybridisation (FISH) dengan
probe DNA khusus kromosom X dan Y, dengan atau
tanpa pemeriksaan gen SRY. Lamanya hasil
pemeriksaan (3 minggu) merupakan hambatan utama
dalam percepatan langkah diagnosis. FISH dapat
mengatasi hal ini karena hanya membutuhkan 2-3
hari, namun harus dilakukan kariotyping untuk
36

mengetahui secara pasti ada tidaknya kelainan
struktural kromosom.3,4
Pencitraan. Dilakukan untuk visualiasi genital
interna, dapat merupakan genitogram dan/atau
ultrasonografi (USG), serta CT-scan atau MRI
bila diperlukan. USG juga dapat membantu
memvisualisasi ada tidaknya testis di regio
inguinal dan ada tidaknya hiperplasia
adrenal.3,4
Pemeriksaan hormonal. Merupakan langkah
strategis berikutnya ketika menghadapi pasien
dengan DSD. Pemeriksaan awal yang paling sering
dianjurkan adalah pemeriksaan kadar
testosteron, LH, FSH dan 17-OH progesteron.
Kadar LH dan FSH meningkat pada disgenesis
gonad atau defek reseptor LH. Pemeriksaan
hormonal ini dapat dipandu oleh teraba tidaknya
gonad. Pada gonad yang tidak teraba, dimana
sangat besar kemungkinan adalah suatu 46,XX
DSD, maka hormon yang diperiksa adalah kadar37

17-OH progesteron serum setelah usia 48 jam.
Bila normal, perlu diperiksa 11-deoksikortisol
dan 11-deoksikortikosteron untuk mendeteksi ada
tidaknya defisiensi 11-β-hidroksilase yang
merupakan kasus HAK tersering kedua. Pada gonad
yang teraba, maka kemungkinan besar adalah
suatu 46,XY DSD, dan yang perlu diperiksa
adalah kadar testosteron dan DHT. Bila rasio
testosteron : DHT tinggi, ini mengarah pada
defisiensi 5-α reduktase.3,4
Pemeriksaan molekuler genetik, dimana analisis
mutasi sangat membantu menegakkan diagnosis DSD
akibat HAK maupun DSD lainnya (misal pada
duplikasi DAX-1, WNT4).3
Pemeriksaan profil steroid pada urin, dengan
menggunakan gas chromatography assay untuk
menentukan secara tepat etiologi HAK.3
d. Diagnosis prenatal
USG dan MRI prenatal dapat pula mendiagnosis DSD,
namun hal ini diperumit dengan resolusi USG dan38

keahlian dari radiografer; juga memiliki
keterbatasan dalam diagnosis DSD misalnya pada janin
46,XX yang mengalami virilisasi berat dengan
tampilan genital eksterna berupa laki-laki (contoh
pada defisiensi enzim 21 hidroksilase) yang sulit
dibedakan dengan janin 46,XY normal; juga pada janin
46,XY yang mengalami virilisasi tidak lengkap akan
tampak genital eksterna perempuan (misal pada
sindrom insensitivitas androgen) dan sulit dibedakan
dengan janin 46,XX normal.11
Normalnya, pada usia kehamilan di atas 12 minggu,
jenis kelamin janin telah dapat diketahui; usia ini
merupakan usia dimana genital eksterna janin telah
berkembang sempurna. Abnormalitas yang dapat
ditemukan dari USG pada janin dengan kecurigaan
virilisasi tidak lengkap yaitu adanya struktur
phalus abnormal (tidak ada, pendek, atau bentuk
abnormal), skrotum (bifida atau tidak ada) dan
testis yang tidak turun pada kehamilan lanjut.
Abnormalitas yang dapat ditemukan dari USG pada39

janin dengan kecurigaan virilisasi berat genital
eksterna perempuan yaitu adanya struktur phalus yang
membesar dan labia yang abnormal, dengan uterus yang
dapat diidentifikasi.11
Selain USG dan MRI, dapat pula dilakukan prosedur
invasif untuk penentuan kromosom seks, misalnya
dengan chorionic villus sampling, amniosentesis dan
kordosentesis; namun prosedur ini membawa risiko
terjadinya keguguran atau persalinan prematur
(bergantung dari usia kehamilan sewaktu dilakukan
prosedur ini).11
G. Tatalaksana
Tujuan tatalaksana kasus DSD adalah: 1) untuk
menjamin semaksimal mungkin fertilitas / reproduksi; 2)
untuk menjamin semaksimal mungkin fungsi seksual; 3)
untuk menjamin kesesuaian hasil akhir fenotip dan
psikososial dengan jenis kelamin yang ditentukan.3
Untuk itu, setiap kasus DSD idealnya dievaluasi /
dirujuk ke dokter spesialis endokrin anak dan40

pendekatan dilakukan secara multidisipliner (yaitu
terdiri dari tim ahli di bidang endokrinologi anak,
bedah urologi / plastik anak, obstetri ginekologi,
radiologi, etik, psikiatri, patologi anatomi dan ahli
agama). Tim ahli bekerja sama dengan keluarga mengambil
keputusan terbaik untuk tatalaksana pasien. Perubahan
jenis kelamin dilakukan oleh pengadilan atas
rekomendasi tim medis. Jadi, perawatan optimal untuk
bayi / anak dengan DSD membutuhkan tim multidispliner
yang berpengalaman yang biasa ditemukan di pusat
pelayanan kesehatan tersier.3-5
a. Penentuan gender
Biasanya, untuk menentukan jenis kelamin seorang
anak diperlukan minimal 7 sifat, yaitu 5 sifat
organik dan 2 sifat psikologis. Ketujuh sifat itu
adalah:12
Susunan kromosom: XX pada perempuan dan XY pada
laki-laki.
Jenis gonad: ovarium pada perempuan dan testis
pada laki-laki.41

Morfologi genitalia eksterna
Morfologi genitalia interna
Hormon seks
Pengasuhan (the sex of rearing): cara anak
dibesarkan oleh orangtuanya akan menentukan
penampilan dalam kehidupan kelak. Ini merupakan
faktor psikologis. Bila seseorang sejak lahir
dibesarkan sebagai perempuan maka perilakunya
akan seperti perempuan. Inilah yang dilihat
oleh masyarakat.
Peranan dan orientasi (gender role and orientation):
yang dimaksudkan disini ialah apa yang
diperbuat atau dinyatakan oleh seseorang untuk
mewujudkan dirinya sebagai seorang perempuan
atau seorang lelaki. Yang perlu diperhatikan
ialah: kelakuan, pilihan permainan, minat,
khayalan, percakapan, impian, kebiasaan
erotisme, dan jawaban atas pertanyaan-
pertanyaan yang kadang-kadang menentukan.
42

Untuk pasien DSD, penentuan jenis kelamin
sebaiknya dilakukan sesegera mungkin setelah
evaluasi diagnostik secara menyeluruh. Beberapa
faktor yang mempengaruhi penentuan gender meliputi
kariotipe, diagnosis, fenotip genital, pilihan
operasi, kebutuhan terapi substitusi seumur hidup,
potensi fertilitas dan fungsi seksual, risiko
keganasan, pandangan keluarga, dan nilai-nilai
budaya masyarakat setempat.3
Untuk pasien 46,XX yang mengalami virilisasi
dengan HAK, tumbuh sebagai perempuan. Kira-kira 60%
pasien dengan defisiensi enzim 5-α reduktase yang
mana adalah perempuan sewaktu bayi dan mengalami
virilisasi saat pubertas, hidup sebagai laki-laki.
Untuk pasien DSD ovotestikuler, harus
mempertimbangkan potensi fertilitas berdasarkan
derajat diferensiasi gonad dan perkembangan genital.
Untuk pasien dengan MDG, faktor yang harus
dipertimbangkan adalah paparan androgen prenatal,
fungsi testis, struktur phalus dan lokasi gonad.10
43

b. Tatalaksana medis
Terapi sulih hormon, tujuannya bukan hanya
untuk memulai dan menjaga perkembangan tanda
seks sekunder, tetapi juga untuk perkembangan
psikososial. Pasien DSD yang perlu terapi
hormon adalah mereka dengan hipogonad atau
kegagalan gonad primer. Pada perempuan
digunakan estrogen, etinil estradiol; dan pada
laki-laki digunakan testosteron, untuk
menginduksi pubertas.3,4,10
Untuk HAK, diberikan hidrokortison 15-20
mg/m2/hari dalam dosis terbagi 2-3 kali/hari;
atau fludrokortison 25-50 µg/hari.4
c. Tatalaksana bedah
Tujuannya antara lain untuk diagnosis
(laparoskopi / laparotomi eksplorasi untuk melihat
struktur genital interna), juga untuk konstruksi
genital sesuai jenis kelamin (misalnya koreksi atau
pengangkatan testis bila ukuran phallus kecil, untuk
mengecilkan ukuran klitoris pada bayi yang akan44

dibesarkan sebagai perempuan), mencegah obstruksi
urin dan infeksi, serta memberikan fungsi seksual
dan reproduksi dewasa yang baik. Namun waktu dan
indikasi pembedahan pada kasus DSD ditentukan oleh
tim ahli multidisipliner, karena sangat tergantung
pada tiap kasus yang dihadapi.3,4,10
d. Tatalaksana psikososial
Melibatkan psikolog / psikiater, dan hal ini
sangat penting sebagai bagian dari evaluasi klinis
rutin dan manajemen. Hal ini ditunjukan untuk
skrining dan menolong keluarga yang berisiko
mengalami maladaptasi dengan kondisi medis pasien
dengan DSD.10
H. Prognosis
Prognosis bervariasi dari baik sampai buruk;
dipengaruhi oleh kemampuan dan komitmen orang tua untuk
mendukung anak dengan DSD, kepribadian pasien dan
kemampuan untuk menerima kondisi mereka, kualitas
pengobatan dan pembedahan, serta dukungan orang-orang
45

sekitar. Faktor-faktor ini dapat ditingkatkan dengan
teknik pembedahan terbaru dan lebih banyak dukungan
psikologis yang terlatih. Prinsip dasarnya adalah fakta
dalam kasus kompleks seperti ini, semua faktor tidak
dapat menjadi ideal, terutama yang berhubungan dengan
potensi fertilitas dan tanggung jawab seksual,
sedangkan dengan dukungan keluarga dan orang-orang yang
disayangi maka kualitas hidup dapat memuaskan dan
produktif.13
46

ATRESIA ANI / ANUS IMPERFORATA
A. Definisi
Atresia ani, disebut pula anus imperforata merupakan
keadaan tidak adanya pembukaan dimana anus seharusnya
berada. Anus imperforata merupakan bagian dari
malformasi anorektal, yang merupakan defek berspektrum
luas mengenai defek perkembangan bagian terbawah
traktus intestinal dan urogenital. Defek bervariasi
dari yang sangat kecil dan mudah ditangani dengan
prognosis fungsional yang baik, sampai defek yang
kompleks, sulit ditangani, biasanya berhubungan dengan47

anomali lainnya, dan memiliki prognosis fungsional yang
buruk.14,15
B. Epidemiologi
Malformasi anorektal merupakan anomali kongenital
yang terjadi pada kira-kira 1 dari 5000 kelahiran
hidup. Tidak ada laporan mengenai predileksi ras dan
jenis kelamin. Kebanyakan anak dengan malformasi
anorektal teridentifikasi selama pemeriksaan fisik
rutin bayi baru lahir. Malformasi yang hampir tidak
terlihat (subtle malformation) misalkan pada anak dengan
fistula perineal, dapat terlihat normal, sampai
beberapa bulan atau beberapa tahun anak tersebut datang
dengan keluhan konstipasi atau infeksi saluran
kemih.14,15
C. Patofisiologi
Normalnya, akibat perlipatan sefalokaudal dan
lateral dari embrio, maka bagian dari endoderm-lined
kavitas yolk sac bergabung ke embrio untuk membentuk
48

primitive gut. Perkembangan dari primitive gut dan derivatnya
yaitu: pharygeal gut yang penting untuk perkembangan
kepala dan leher; foregut yang akan berkembang menjadi
esofagus, gaster, duodenum, hepar dan kantung empedu,
pankreas; midgut dimulai dari kaudal hepar dan meluas
ke perhubungan 2/3 kanan dan 1/3 kiri kolon
transversum; hindgut membentuk 1/3 distal kolon
transversum, kolon descendens, kolon sigmoid, rektum,
dan bagian superior dari kanalis analis. Endoderm dari
hindgut juga membentuk lapisan dalam dari vesika
urinaria dan uretra.8
Untuk malformasi anorektal, embriogenesisnya masih
belum jelas. Gangguan pada perkembangan struktur
anorektal pada berbagai tingkatan menyebabkan
terjadinya beragam anomali, mulai dari stenosis anus,
ruptur membran anal inkomplit, atau agenesis anus,
sampai kegagalan proctodeum untuk invaginasi. Pada
atresia ani, defek terjadi karena kurangnya
rekanalisasi bagian bawah kanalis analis.14
49

D. Etiologi
Etiologi malformasi anorektal masih belum jelas dan
tampaknya multifaktorial. Namun dipercaya bahwa faktor
genetik yang berperan. Pada awal tahun 1950-an,
diketahui bahwa ada peningkatan risiko saudara kandung
pasien dengan malformasi anorektal untuk lahir pula
dengan malformasi, yaitu 1:100, dibandingkan dengan
1:5000 pada populasi umum. Selain itu, secara khusus,
mutasi pada gen spesifik yang mengkode faktor
transkripsi telah dilaporkan pada pasien yang memiliki
sindrom yang diturunkan secara autosomal dominan. Juga
telah ditemukan bahwa tidak hanya terjadi peningkatan
insidens malformasi anorektal pada pasien trisomi 21
(sindrom Down), tetapi 95% pasien dengan trisomi 21 dan
malformasi anorektal memiliki anus imperforata tanpa
fistula, dibandingkan dengan 5% dari seluruh pasien
malformasi anorektal. Berdasarkan bukti ini, terlihat
bahwa mutasi beragam gen yang berbeda dapat menyebabkan
malformasi anorektal, atau dapat dikatakan bahwa
50

etiologi dari malformasi anorektal bersifat
multigenik.15
E. Klasifikasi
Klasifikasi malformasi anorektal non-syndromic dapat
dilihat pada tabel 4.
Tabel 4. Klasifikasi malformasi anorektal non-syndromic15
Pria
Fistula recto-perineal
Fistula recto-uretra-bulbar
Fistula recto-uretra-
prostat
Anus imperforata tanpa
fistula
Defek yang kompleks dan
tidak lazim
Wanita
Fistula recto-perineal
Fistula recto-vestibuler
Kloaka dengan channel pendek
(< 3 cm)
Kloaka dengan channel
panjang (> 3 cm)
Anus imperforata tanpa
fistula
Defek kompleks dan tidak
lazim
Ekstrofi kloaka, menutup
ekstra kloaka
Kloaka posterior
Atresia rektum
Berhubungan dengan massa
presakrum51

Anus imperforata dapat dibagi pula menjadi lesi
letak rendah (low lesion) dimana rektum telah turun
melalui kompleks sfingter (sekelompok masa serat otot
yang mengelilingi anorektal dan merupakan kombinasi
dari muskulus puborektalis, levator ani, sfingter
interna dan eksterna); dan lesi letak tinggi (high lesion)
dimana rektum belum turun melalui kompleks sfingter.6
Dapat dilihat pada gambar 9.
Gambar 9. Anus imperforata pada laki-laki (a). Lesi letak rendah;
(b) Lesi letak tinggi.6
F. Manifestasi klinis
52

Kebanyakan bayi dengan anus imperforata dirujuk
karena tidak memiliki pembukaan anus yang dapat
diidentifikasi pada pemeriksaan bayi baru lahir, atau
karena gagal mengeluarkan mekonium. Dapat pula
ditemukan anomali lain seperti anomali genitourinaria,
anomali vertebra, anomali gastrointestinal, defek
jantung dan sindrom Down (yang biasa ditemukan pada
bayi laki-laki dengan anus imperforata tanpa fistula).
Berbagai anomali yang berhubungan dapat dilihat pada
tabel 5.1,6
Tabel 5. Berbagai anomali yang berhubungan dengan malformasi anorektal6
GenitourinariaVertebra Kardiovaskula
r
Gastrointestinal
Refluks
vesikoureter
Agenesis renal
Displasia renal
Duplikasi
ureter
Kriptorkidismus
Hipospadia
Massa
presakral:
Meningokel
Lipoma
Teratoma
Tetralogy of Fallot
Defek septum
ventrikel
Transposisi
arteri besar
Fistula
trakeoesofageal
Atresia duodenal
Malrotasi
Penyakit
Hirschsprung
53
Gambar 10. Anus
imperforata

Uterus bikonuat
Septum vagina
Pada bayi laki-laki dengan lesi letak rendah,
biasanya ada noda mekonium pada perineum sepanjang
medium raphe. Bayi perempuan dengan lesi letak rendah
memiliki berbagai tanda, dari anus yang hanya terletak
anterior dari perineum sampai ke fourchette fistula yang
membuka pada mukosa lembab dari introitus, distal
terhadap hymen. Lesi letak tinggi pada bayi laki-laki
tidak memiliki pembukaan kutaneus atau fistula yang
tampak, tetapi biasanya memiliki fistula ke traktus
urinarius, baik ke uretra maupun ke vesika urinaria.
Meskipun jarang terjadi fistula rektovaginalis, pada
bayi perempuan, lesi letak tinggi biasanya berupa
anomali kloaka (Gambar 11), yang mana rektum, vagina
dan uretra sama-sama membuka pada satu saluran dengan
panjang yang bervariasi.6
54

G. Diagnosis
1. Anamnesis
Riwayat keluarga dengan sindrom atau malformasi
juga penting. Riwayat USG prenatal biasanya normal,
namun USG prenatal dapat pula mendiagnosis anus
imperforata. Dalam suatu penelitian di Norwegia,
dari 69 kasus anus imperforata pada tahun 1987 -
2004, hanya 11 kasus yang murni anus imperforata
terdiagnosis, yaitu dengan adanya dilatasi rektum
atau bagian bawah dari anus, juga melalui temuan
berupa massa abdominopelvik atau klasifikasi
intraluminal dan dilatasi usus. Usia kehamilan
sewaktu terdiagnosis rata-rata 18 + 4 minggu.14,16
2. Pemeriksaan fisik55
Gambar 11. Anus
imperforata pada
bayi perempuan

Selama pemeriksaan fisik, harus difokuskan pada
daerah abdomen, genital, rektum dan vertebra bagian
bawah. Bayi baru lahir dengan anus imperforata
biasanya dapat diidentifikasi pada pemeriksaan fisik
pertama kali. Evaluasi lanjutan dilakukan bila tidak
ditemukan orifisium anal pada posisi yang tepat.
Posisi anus normal pada perineum kira-kira rasionya
0,5 antara koksigeus dan skrotum atau introitus
vagina. Malformasi pada bayi baru lahir yang
terlewati biasanya akan ditemukan dalam 24 jam
ketika bayi tersebut mengalami distensi abdomen dan
gagal mengeluarkan mekonium.6,14
Lesi letak rendah pada bayi laki-laki dapat
ditemukan fistula rektoperineal (kutaneus) dapat
berjalan anterior sepanjang median raphe melewati
skrotum dan kadang turun melalui penis. Traktus ini
biasanya tipis, dengan rektum normal hanya beberapa
milimeter tingginya dari kulit. Anomali
ekstraintestinal dapat terlihat pada < 10% pasien
dengan fistula rektoperineal. Pada bayi perempuan,56

lesi letak rendah memasuki vestibulum atau fourchette
(mukosa lembab di luar hymen tetap masih di dalam
introitus vagina). Bila ada fistula rektoperineal,
maka ketika pemeriksaan perineum didapatkan
pembukaan kecil, mekonium, atau mukus.6,14
Pada anus imperforata letak tinggi, perineum pada
bayi laki-laki terlihat datar. Dapat pula terlihat
mekonium yang melewati penis / uretra (fistula
rektouretra, merupakan yang paling sering pada bayi
laki-laki), dimana bila fistula letak tinggi akan
memasuki uretra pars prostatika atau bahkan vesika
urinaria. Pada fistula rektoprostat, skrotum bisa
bifida dan lengkung anus berada dekat skrotum. Pada
kasus seperti ini dimana tidak ditemukan adanya
pembukaan (lubang) pada perineum, bayi harus
diobservasi selama 24 jam. Pada bayi perempuan
dengan anus imperforata letak tinggi, terdapat
fistula rektovaginal, namun jarang, kebanyakan
fistula ke vestibulum di luar orifisium hymen. Labia
harus dipisahkan untuk mencari fistula vestibuler.57

Bila tidak ada fistula yang terlihat dan bayi hanya
memiliki 1 pembukaan di antara labia, maka bayi
dikatakan memiliki kloaka persisten. Kloaka
persisten didefinisikan sebagai defek dimana rektum,
vagina dan uretra semuanya bertemu dan bersatu
membentuk satu saluran. Lesi letak tinggi juga dapat
terjadi pada atresia rektum, yang merupakan defek
yang jarang, terjadi hanya pada 1% anomali
anorektal. Fitur unik dari atresia rektum bahwa
defek ini mengenai pasien dengan kanalis analis dan
anus yang normal. Defek ini biasanya ditemukan
ketika hendak memeriksa temperatur rektal. Obstruksi
ada pada sekitar 2 cm di atas kulit.6
3. Pemeriksaan penunjang
Urinalisis
Jika pasien memiliki fistula perineal atau
rektourinarius, mekonium mungkin tidak terlihat
dalam urin sebelum 16-24 jam setelah kelahiran.
Hal ini dikarenakan otot sfingter volunter yang58

mengelilingi bagian distal dari usus pada kasus
fistula perineal atau rektourinarius harus
‘diatasi’ dengan tekanan intrausus yang tinggi
sebelum mekonium terlihat dalam urin. Adanya
mekonium dalam urin merupakan indikasi untuk
dilakukannya kolostomi protektif.6
Radiologi
Pada kelahiran, usus belum distensi, sehingga
evaluasi radiologi tidak dapat dipercaya pada
16-24 jam pertama kehidupan. Kadang, beberapa
tanda klinis yang telah dijelaskan sebelumnya
tidak muncul setelah 24 jam observasi, maka
evaluasi radiologi terindikasikan.6
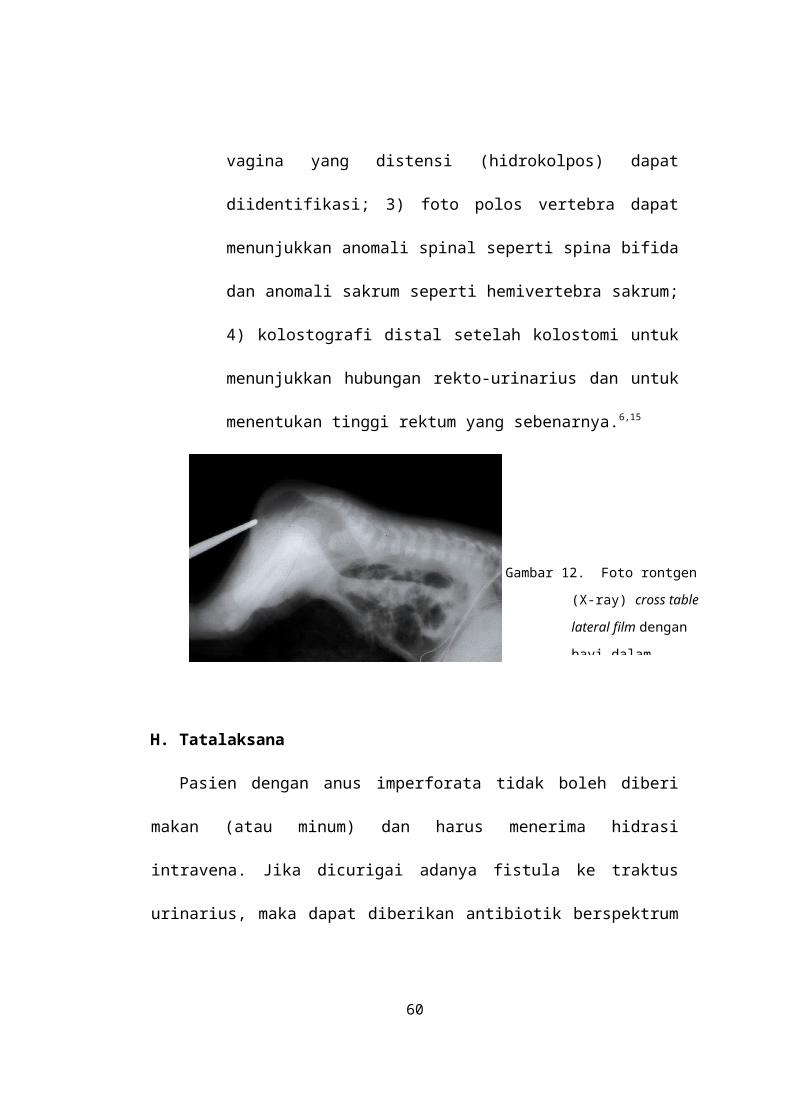
Pemeriksaan radiologi pasien dengan anus
imperforata antara lain: 1) cross table lateral film
dengan pasien dalam posisi pronasi diambil
dalam 16-24 jam kehidupan, penting untuk
menentukan posisi kantung rektum (Gambar 12);
2) USG abdomen, untuk mengevaluasi adanya
anomali urologi dan pada kasus anomali kloaka,59
vagina yang distensi (hidrokolpos) dapat
diidentifikasi; 3) foto polos vertebra dapat
menunjukkan anomali spinal seperti spina bifida
dan anomali sakrum seperti hemivertebra sakrum;
4) kolostografi distal setelah kolostomi untuk
menunjukkan hubungan rekto-urinarius dan untuk
menentukan tinggi rektum yang sebenarnya.6,15
H. Tatalaksana
Pasien dengan anus imperforata tidak boleh diberi
makan (atau minum) dan harus menerima hidrasi
intravena. Jika dicurigai adanya fistula ke traktus
urinarius, maka dapat diberikan antibiotik berspektrum
60
Gambar 12. Foto rontgen
(X-ray) cross table
lateral film dengan
bayi dalam

luas, meskipun antibiotik untuk bakteri anaerob belum
diperlukan dalam 48 jam pertama kehidupan.14
Penanganan lebih awal untuk bayi baru lahir dengan
malformasi anorektal sangat penting, dan ada 2
pertanyaan yang harus dijawab dalam 24-48 jam pertama
kehidupan. Yang pertama, adakah anomali yang
berhubungan yang mengancam nyawa dan harus diatasi
segera? Yang kedua, apakah bayi harus menjalani
prosedur primer tanpa kolostomi protektif, ataukah
kolostomi protektif dan perbaikan definitif nantinya?
Untuk bayi yang lahir dengan kloaka, dokter bedah harus
menentukan apakan ada dilatasi vagina dan apakah harus
didrainase, juga menentukan apakah pengalihan berkemih
dibutuhkan. Manuver ini ditujukan untuk mencegah sepsis
atau asidosis metabolik. Keputusan untuk melakukan
anoplasti pada periode neonatus atau untuk menunda
perbaikan dan untuk melakukan kolostomi didasarkan pada
pemeriksaan fisik bayi, tampilan perineum dan berbagai
perubahan yang terjadi dalam 24 jam pertama
kehidupan.15
61

Distensi abdomen tidak muncul dalam beberapa jam
pertama kehidupan dan distensi ini dibutuhkan untuk
mendorong mekonium keluar lewat fistula rektoperineal
atau fistula rektouretra. Hal ini dikarenakan bagian
paling distal dari rektum pada bayi-bayi ini
dikelilingi oleh struktur otot volunter yang mirip
terowongan, yang menjaga bagian rektum tersebut kolaps
dan kosong. Tekanan intraabdomen harus cukup tinggi
untuk ‘mengatasi’ tonus otot yang mengelilingi rektum.
Karena itu, keputusan untuk melakukan kolostomi ataukah
anoplasti harus menunggu 16-24 jam ini, ketika dokter
bedah mengawasi bukti klinis mengenai anomali anorektal
yang ada pada bayi.15
Pada inspeksi bokong, bila ditemukan perineum atau
dasar yang rata, merupakan bukti kurangnya lipatan
garis tengah gluteal, dan tidak adanya lekukan anal
mengindikasikan bahwa pasien memiliki sangat sedikit
otot pada perineum. Penemuan ini berhubungan dengan
malformasi letak tinggi dan karenanya harus dilakukan
kolostomi. Tanda pada perineum yang ditemukan pada62

pasien dengan malformasi letak rendah antara lain
adanya mekonium pada perineum, adanya tonjolan kulit
pada lekukan anal yang merupakan tempat lewatnya feses,
dan membran anus.15
Untuk bayi laki-laki dengan fistula rektoperineal
tidak membutuhkan kolostomi. Bayi-bayi tersebut dapat
menjalani anoplasti posterior sagital sedangkan bayi
dengan fistula rektourinarius harus menjalani pemisahan
feses dengan kolostomi. Untuk bayi perempuan dengan
fistula rektovestibuler, dilakukan kolostomi. Kadang
fistula pada bayi perempuan cukup besar untuk
dekompresi traktus gastrointestinal, dan dapat
didilatasi untuk memfasilitasi drainase feses sampai
usia bayi lebih tua dan dapat dilakukan perbaikan
definitif. Perbaikan definitif meliputi pendekatan
posterior sagital. Hal yang paling sulit dari operasi
ini adalah pemisahan rektum dan vagina yang membagi
dinding yang sama. Bayi perempuan dengan fistula
rektoperineal dilakukan anoplasti neonatus.15
63

Untuk bayi perempuan dengan kloaka, jika salurannya
< 3 cm, dapat dilakukan pendekatan posterior sagital.
Jika salurannya > 3 cm, makan biasanya dibutuhkan
laparotomi, dimana kadang vagina dan traktus urinarius
harus dipisahkan dan uretra harus direkonstruksi.15
I. Outcome dan Prognosis
Hal yang perlu diperhatikan setelah operasi adalah
konstipasi dan kontinensia. Konstipasi merupakan hal
yang harus dicegah setelah operasi, terutama pada
pasien perempuan dengan fistula rektoperineal atau
rektovestibular dan untuk pasien laki-laki dengan
fistula rektoperineal dan anus imperforata tanpa
fistula. Untuk mencapai kontinensia, dapat dilakukan
dengan pergerakan usus.15
Semua pasien dengan malformasi anorektal tanpa
komorbiditas yang mengancam nyawa memiliki prognosis
baik.14
64

65

BAB III
KESIMPULAN
1. DSD dan atresia ani merupakan kelainan bawaan yang
dapat ditemukan pada 1 dari 5000 dan 1 dari 4500 –
5500 kelahiran hidup.
2. Etiologi DSD beragam, dan dapat diklasifikasikan
menjadi DSD kromosom seks, 46,XX DSD dan 46,XY DSD;
sedangkan untuk atresia ani diduga multifaktorial.
3. DSD dan atresia ani dapat didiagnosis melalui
anamnesis, pemeriksaan fisik dan pemeriksaan
penunjang, serta dapat didiagnosis sebelum kelahiran
(prenatal).
4. Tatalaksana DSD membutuhkan tim multidispliner,
sedangkan kasus atresia ani bergantung pada jenis
anomalinya dan terapi suportif berupa hidrasi dan
nutrisi intravena dan antibiotik spektrum luas.
66

Daftar Pustaka
1. Arensman RM, Bambini DA, Almons PS. Pediatric
surgery. Texas: Landes Bioscience; 2000.
2. Ikatan dokter anak Indonesia UKK Perinatologi. Buku
ajar neonatologi. Jakarta: IDAI; 2010.
3. Batubara JRL, Tridjaja B, Pulungan AB, editor. Buku
ajar endokrinologi. Jakarta: IDAI; 2010.
4. Ikatan dokter anak Indonesia. Pedoman pelayanan
medis IDAI edisi II. Jakarta: IDAI; 2011.
5. Lee PA, Houk CP, Ahmed F, Hughes IA. Consensus
statement on management of intersex disorders.
Pediatrics 2006;118;e488 DOI: 10.1542/peds.2006-0738.
67

6. Kliegman RM, Behrman RE, Jenson HB, Stanton BF.
Nelson textbook of pediatrics, 18th ed. Washington
DC: Saunders Elsivier; 2007.
7. Paranton H. Embriologi sistem alat-alat urogenital.
Dalam: Buku Ilmu Kandungan, edisi ke-3. Jakarta: PT
Bina Pustaka Sarwono Prawirohardjo; 2011. hal.39-43.
8. Sadler TW. Langman’s medical embryology, 10th ed.
USA: Lippincott Williams & Wilkins; 2006.
9. Hutcheson J, Cendron M. Ambigous genitalia and
intersexuality. [internet]. 2012 Jan 11 [cited 2014
July 02];[7 screens]. Available from:
http://emedicine.medscape.com/article/1015520-
overview#showall
10. Nabhan ZM, Lee PA. Disorder of sex development.
Curr Opin Obstet Gynecol 19:440–445. © 2007
Lippincott Williams & Wilkins.
11. Chitayat D, Glanc P. Diagnostic approach in
prenatally detected genital abnormalities.
Ultrasound Obstet Gynecol 2010; 35: 637–646.
68

12. Siregar CD. Pendekatan diagnostik interseksualitas
pada anak. Cermin Dunia Kedokteran No. 126, 2000.
hal.32.
13. Lee PA, Houk CP. Long-term outcome and adjusment
among patients with DSD born with testicular
differentiation and masculinized external genitalia
[abstract]. Pediatr Endocrinol Rev 2012
Nov;10(1):140-51.
14. Rosen NG, Cuffari C. [internet]. Pediatric
imperforate anus. 2012 Nov 13 [cited 2014 June 29];
[5 screens]. Available from:
http://emedicine.medscape.com/article/929904-
overview
15. Levitt MA, Pena A. Anorectal malformations.
Orphanet Journal of Rare Diseases 2007, 2:33
doi:10.1186/1750-1172-2-33.
16. Brantberg A, Blaas HGK, Haugen SE, Isaksen CV,
Eiknes SH. Imperforate anus: a relatively common
anomaly rarely diagnosed prenatally. Ultrasound
Obstet Gynecol 2006; 28: 904–910.69

70




















