
Protein refolding in reversed micelles: Interactions of the protein with micelle components
10
Protein Refolding in Reversed Micelles: Interactions of the Protein with Micelle Components Anna J. Hagen, T. Alan Hatton, and Daniel 1. C. Wang* Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02 139 Accepted for publication September 29, 1989 A novel process has been developed to improve the re- folding yield of denatured proteins. It uses reversed mi- celles to isolate denatured protein molecules from each other and thus, upon refolding, reduces the intermolec- ular interactions which lead to aggregation. The feasibility of this process was first demonstrated with Ribonucle- ase A as a model protein. In the present work, we ex- panded the scope of this study to better understand both the general mechanisms of protein refolding in re- versed micelles and the biotechnological applicability of the process. First, we investigated the interactions be- tween the individual components of the reversed micellar system (the protein molecule, the denaturant guanidine hydrochloride (GuHCI), and the surfactant (AOT)) during the refolding process. We then extended our studies to a more hydrophobic protein, y-interferon, which aggre- gates upon refolding in aqueous solution. However, it was also found to aggregate in our reversed micelle process during the extraction step. Since y-interferon is a much more hydrophobic protein than RNase, we hy- pothesize that interactions between hydrophobic amino acids and the surfactant layer may interfere with refold- ing. This hypothesis was tested by studying the refold- ing of chemically modified RNase. The substitution of 55% of the surface lysine residues with hydrophobic caproyl groups caused a significant decrease in the re- folding yield of RNase in the reversed micellar system without affecting aqueous solution renaturation. In addi- tion, the extraction efficiency of the enzyme from re- versed micelles back into aqueous solution was severely reduced and resulted in aggregation. These exper- iments indicate that unfolded hydrophobic proteins in- teract with the surfactant molecules, which limits their ability to refold in reversed micelles. INTRODUCTION The.study of protein refolding in reversed micelles was un- dertaken to reduce the problem of aggregation, which causes a severe decrease in yield during the recovery of re- combinant proteins from bacterial inclusion bodies. The reversed micelles isolate the protein molecules from each other during refolding, and thus reduce the intermolecular * To whom all correspondence should be addressed Biotechnology and Bioengineering, Vol. 35, Pp. 966-975 (1990) 0 1990 John Wiley & Sons, Inc. interactions which lead to aggregation. The feasibility of this process was first demonstrated with RNase.' Our ob- jective is now to understand the general mechanisms of protein refolding in reversed micelles in order to determine the important parameters affecting this process. We would then like to extend this study to other proteins and deter- mine the types of proteins and surfactants for which this process can be used. The interior of a reversed micelle system consists of several interacting components: protein, surfactant, water and guanidine hydrochloride (GuHCl). In order to under- stand the folding process within reversed micelles, we must analyze these interactions, which can have significant effects on protein conformation and folding. First, the guanidinium ion concentration could be a very important variable in the reversed micelle system since it is concen- trated in the interior of the micelle' and is known to act as a protein denaturant.2 In aqueous solution, at low concen- trations of GuHCl (up to 1.2M) the refolding of reduced and denatured chymotrypsinogen is enhanced. In con- trast, above 2M GuHCl, refolding is completely inhibited.3 Similar behavior has been observed with carbonic anhy- dra~e,~ and with BSA in the presence of low concentration of urea.5 Interactions of the unfolded protein with the surfactant could have deleterious influences since many detergents are also strong protein denaturants, producing conforma- tional changes at very low concentrations upon binding.* In contrast, some nondenaturing detergents have been shown to protect exposed hydrophobic groups from aggre- gation during refolding.6 All of these interactions are very specific to the protein-denaturant combination. The un- folded protein could also come into contact with the sol- vent, since organic solvents preferentially solubilize the hydrophobic groups normally present on the interior of the protein, which are already exposed in this case.2 However, if a monolayer of water is present around the molecule, it can retain its native conformation and display activity in an organic solvent.' We therefore do not know a priori, how these different components will influence protein refold- CCC 0006-3592/901010 0966- 10$04.00
Transcript of Protein refolding in reversed micelles: Interactions of the protein with micelle components

Protein Refolding in Reversed Micelles: Interactions of the Protein with Micelle Components
Anna J. Hagen, T. Alan Hatton, and Daniel 1. C. Wang* Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02 139
Accepted for publication September 29, 1989
A novel process has been developed to improve the re- folding yield of denatured proteins. It uses reversed mi- celles to isolate denatured protein molecules from each other and thus, upon refolding, reduces the intermolec- ular interactions which lead to aggregation. The feasibility of this process was first demonstrated with Ribonucle- ase A as a model protein. In the present work, we ex- panded the scope of this study to better understand both the general mechanisms of protein refolding in re- versed micelles and the biotechnological applicability of the process. First, we investigated the interactions be- tween the individual components of the reversed micellar system (the protein molecule, the denaturant guanidine hydrochloride (GuHCI), and the surfactant (AOT)) during the refolding process. We then extended our studies to a more hydrophobic protein, y-interferon, which aggre- gates upon refolding in aqueous solution. However, it was also found to aggregate in our reversed micelle process during the extraction step. Since y-interferon is a much more hydrophobic protein than RNase, we hy- pothesize that interactions between hydrophobic amino acids and the surfactant layer may interfere with refold- ing. This hypothesis was tested by studying the refold- ing of chemically modified RNase. The substitution of 55% of the surface lysine residues with hydrophobic caproyl groups caused a significant decrease in the re- folding yield of RNase in the reversed micellar system without affecting aqueous solution renaturation. In addi- tion, the extraction efficiency of the enzyme from re- versed micelles back into aqueous solution was severely reduced and resulted in aggregation. These exper- iments indicate that unfolded hydrophobic proteins in- teract with the surfactant molecules, which limits their ability to refold in reversed micelles.
INTRODUCTION
The.study of protein refolding in reversed micelles was un- dertaken to reduce the problem of aggregation, which causes a severe decrease in yield during the recovery of re- combinant proteins from bacterial inclusion bodies. The reversed micelles isolate the protein molecules from each other during refolding, and thus reduce the intermolecular
* To whom all correspondence should be addressed
Biotechnology and Bioengineering, Vol. 35, Pp. 966-975 (1990) 0 1990 John Wiley & Sons, Inc.
interactions which lead to aggregation. The feasibility of this process was first demonstrated with RNase.' Our ob- jective is now to understand the general mechanisms of protein refolding in reversed micelles in order to determine the important parameters affecting this process. We would then like to extend this study to other proteins and deter- mine the types of proteins and surfactants for which this process can be used.
The interior of a reversed micelle system consists of several interacting components: protein, surfactant, water and guanidine hydrochloride (GuHCl). In order to under- stand the folding process within reversed micelles, we must analyze these interactions, which can have significant effects on protein conformation and folding. First, the guanidinium ion concentration could be a very important variable in the reversed micelle system since it is concen- trated in the interior of the micelle' and is known to act as a protein denaturant.2 In aqueous solution, at low concen- trations of GuHCl (up to 1.2M) the refolding of reduced and denatured chymotrypsinogen is enhanced. In con- trast, above 2M GuHCl, refolding is completely inhibited.3 Similar behavior has been observed with carbonic anhy- d r a ~ e , ~ and with BSA in the presence of low concentration of urea.5
Interactions of the unfolded protein with the surfactant could have deleterious influences since many detergents are also strong protein denaturants, producing conforma- tional changes at very low concentrations upon binding.* In contrast, some nondenaturing detergents have been shown to protect exposed hydrophobic groups from aggre- gation during refolding.6 All of these interactions are very specific to the protein-denaturant combination. The un- folded protein could also come into contact with the sol- vent, since organic solvents preferentially solubilize the hydrophobic groups normally present on the interior of the protein, which are already exposed in this case.2 However, if a monolayer of water is present around the molecule, it can retain its native conformation and display activity in an organic solvent.' We therefore do not know a priori, how these different components will influence protein refold-
CCC 0006-3592/901010 0966- 10$04.00

ing. It will be important to determine to what extent the yields attainable in our system will be affected by these in- teractions, and whether they are comparable to those ob- tained in aqueous solution.
The location and conformation of unfolded proteins in reversed micelles represents critical information for under- standing the mechanism of protein refolding in this system. There has been very little research previously conducted in this area, but literature is available for other guest molecules in reversed micelles. For the case of AOT in iso-octane, several hydrophilic enzymes were found to retain activity and native conformation after incorporation into reversed micelles.’ Petit et al. showed that electrostatic attractions between the protein and the negative surfactant heads cause association of the protein with the interface.’ Incor- poration of the hormones glucagon and adrenocoticotropin [ACTH( 1-24)] into AOT reversed micelles induces sec- ondary structure (absent in aqueous solution) in these pep- tides by interaction with the structured water or the surfactant interface. I” A similar result has been seen with the membrane proteins, Myelin Basic Protein (MBP) and the Folch-Pi proteolipid, where electrostatic interactions and strong binding of interfacial water cause the induced a-helicity. “ . I 2
For the case of small molecules, hydrophobicity is the most important parameter determining their interactions with the water, solvent and surfactant.I3 The behavior of amino acids in an AOT-iso-octane system has been investi- gated by Leotidis and H a t t ~ n . ’ ~ They found that hydropho- bic amino acids interact very strongly with the micellar interface, acting as cosurfactants.
It is clear that specific regions in the unfolded protein have the potential to interact with the micelle components and interfere with refolding. One approach to study this problem and identify the interfering reactions is to examine the effect of protein modifications on refolding a reversed micellar environment. In aqueous solution, covalent modi- fications of proteins have been carried out to identify which amino acids are important in stability and refold- ing. Chemical modifications have also been used to determine the location of a protein within the reversed mi- celles. l7 We will combine these two methods of analysis in our studies in order to better understand the interactions between protein and micelle components, and to determine which of the reversed micelle system parameters are most important during the refolding of denatured proteins.
The reversed micelle process was designed to isolate de- natured protein molecules from each other during refolding and thus reduce the intermolecular interactions which cause aggregation. It consists of four stages, as shown schemati- cally in Figure 1. The denatured protein molecules are first transferred to the reversed micelles at a concentration of less than one molecule per micelle. The level of denaturant within the micelle can then be reduced by a series of con- tacting stages with an aqueous phase containing no dena- turant. Addition of a redox reagent then causes reoxidation
AGGREGATED PROTEIN
‘1 DENATURANT
DENATURED PROTEIN
SURFACTANT
UNFOLDED PROTEIN IN REVERSED MICE L L E
REDOX REAGENT DENATURANT %IH@
PROTEININ
SURFACTANT HI@ REFOLDED PROTEIN
SOLUTION
+ SOLVENT
INAQUEOUS
Figure 1. Proposed process for protein folding in reversed micelles: (1) Denatured protein is transfened to reversed micelles. ( 2 ) Denaturant is removed and (3) redox reagent is added which allows for refolding. (4) Refolded protein is extracted from reversed micelles back into aque- ous solution.
of disulfide bonds and refolding of the denatured enzyme into the biologically active conformation within the re- versed micelle. Finally, the renatured protein can be ex- tracted from the organic phase back into aqueous solution, and retains biological activity. The feasibility of this pro- cess was first demonstrated with ribonuclease A as a model system.
EXPERIMENTAL
Materials
RNase A was obtained from Worthington Biochemicals (Freehold, NJ). ?-Interferon was supplied in both native and denatured forms by Biogen, Inc. (Cambridge, MA). The radioimmunoassay kit for the y-interferon was pur- chased from Centocor (Malvern, PA). AOT was initially purchased from Pfalz and Bauer (Waterbury, CT), but for the work with hydrophobic modifications, it was obtained from Sigma (St. Louis, MO). Riedel-de-Haen Hydranal titrant and solvent for Karl Fischer titration were pur- chased from Crescent Chemical Company (Hauppauge, NY). All other chemicals were purchased from Sigma (St. Louis, MO).
HAGEN, HATTON, AND WANG: PROTEIN REFOLDING 967

Experimental Methods Desalting of Denatured RNase and Capro yl-RNase Solution
A desalting column (prepared by filling a 50-mL glass BioRad column with Sephadex (3-25) was equilibrated with 1M GuHCl + 1 mM EDTA in O.1M tris, pH 8.7. A sample (to 2 mL) of denatured RNase in 6M GuHCl was applied to the column, and a buffer flow rate of about 1 mL/min was used to remove P-mercaptoethanol and reduce the GuHCl concentration to that of the running buffer. The final protein concentration was 3-4 mg/mL in the elution buffer. For renaturation of RNase in high GuHCl concentrations, the desalting column was equili- brated with up to 3M GuHCl in the same buffer. The samples were then diluted to the appropriate GuHCl con- centration prior to glutathione addition. All sulfhydryl groups were reduced after this step as measured by the DTNB assay.20
Hydrophobic Modifications of RNase
Caproylation of the &-amino group of the lysines was based on the procedure of Epstein and Goldberger.” Two hundred milligrams of RNase were suspended in 8 mL of 50% saturated sodium acetate in O.1M Na,PO,, and al- lowed to dissolve for l h. The pH was adjusted to 8.6 with IN NaOH and the sample was cooled to 0°C. Pure caproic anhydride was then added in aliquots of 100 to 400 p L every 10 min for 20 to 30 min (total volumes of 0, 300, 800, and 1600 pL), depending on the degree of substitu- tion desired. The reaction temperature was maintained at 0°C and the pH was maintained at 8.6 during the reaction by addition of 1N NaOH as required. The sample was then transferred to dialysis tubing (with about twice the sample volume available for expansion). After exhaustive dialysis against water, any remaining precipitate was removed by centrifugation and filtration, and the clear supernatant was lyophilized.
The degree of modification was assayed by the TNBS method of Fields using N-acetyl lysine as a standard. We carried out three levels of modification and one control which was treated in the same way as the samples, but without addition of caproic anhydride. We obtained 6.9, 6.7, 5.2, and 3.1 free amino groups, corresponding to 0, 3, 25, and 55% substitution, respectively. Ribonuclease contains a total of eleven free amino groups (10 lysines plus the N-terminal amino acid). Seven of these eleven lysine residues are accessible in the folded enzyme. Titra- tion of free amino groups in reduced and carboxymeth- ylated RNase’’ gave a value of 10.9 free amino groups per RNase molecule. The 0, 3, and 25% substitution retained approximately their original activities in both aqueous so- lution and upon injection (100 pL/mL of 400 mM AOT) into reversed micelles. However, the 55% modified RNase showed only 66% of its original activity in aqueous solu- tion and 40% in the reversed micelles. We also observed an increase in the refolding time of the enzyme following the modification process (48 h instead of 24 h for untreated RNase’) and obtained a reduced refolding yield (80% in- stead of 100% for the untreated RNase). These effects could be due to the treatment itself, which involves high ionic strength, dialysis and lyophilization and could have had a negative effect on protein stability and refolding.
Denaturation of RNase and Capro yl-RNase
RNase A at concentrations between 20 and 30 mg/mL was dissolved in 6M GuHCl in 0.025M phosphate buffer, pH 7.5. The P-mercaptoethanol was added to give a concen- tration of 0.3M. The solution was incubated at room tem- perature overnight in a sealed tube under nitrogen. Total volume was 1-3 mL.
Assay for Free SH Groups
The test reagent was 10 mM DTNB [5,5‘-dithiobis-(2-nitro benzoic acid)] in O.1M phosphate buffer + 1mM EDTA, pH 7. A 100 p L volume of this solution was added to 3 mL of sample, allowed to sit 10 min, then the absorbance was measured at 412 nm. Concentrations were determined from a standard curve with cysteine in the sample buffer with concentrations from 0 to 200 p M . This method is described by Riddles et a1.”
Reoxidation of RNase and Capro yl-RNase in Aqueous Solution
Reduced and oxidized glutathione ( 5 mM each) were added to the sample containing denatured and reduced RNase (after the desalting column). The time of addition was considered as time = 0 for renaturation.
Transfer of Denatured RNase and Capro yl-RNase to Reversed Micelies
Equal volumes (typically 4-5 mL) of denatured RNase so- lution (containing 0.5-3 mg/mL RNase and 0.5M-1M GuHCl) and 400 mM AOT in iso-octane were combined in a 20-mL scintillation vial and stirred magnetically for 15 min at room temperature. The cloudy mixture was transferred to a capped centrifuge tube and centrifuged for 15 min, resulting in the formation of two clear phases, which were separated by careful pipetting. A blank sample containing no protein was treated in parallel. A thin layer of white precipitate was sometimes seen at the interface and was discarded. It probably consisted of surfactant, since very little protein was lost, and it was also observed in the absence of protein.
For the experiment at high RNase concentrations (Fig. 2), the reversed micelle solutions containing RNase
968 BIOTECHNOLOGY AND BIOENGINEERING, VOL. 35, APRIL 1990

100
6 80
0 6 8 60 > U
40 6 ; 20
s?
4..
X., Aqueous Solution
'A W0=7.2 W0=5 7 wo=9.9
L
1 2 3 4 5
RNase (mg/rnl overall)
Figure 2. Reversed micelle solutions were prepared with denatured and reduced RNase in 0.5M GuHCl. After dilution and glutathione addition, final pro- tein concentrations were as indicated on the graph. Final W, values were 9.9, 7.2, and 5 . 7 . Data for aqueous solution renaturation of RNase are from ref. 26.
Refolding efficiency of RNase in reversed micelles
were prepared as above. Then two dilutions (1 : 2 and 1 : 5) were carried out with AOT in iso-octane (containing no reversed micelles) to reduce the water content of the re- versed micelles as well as the overall enzyme concentra- tion. Glutathione was then added to these samples as described below.
Removal of Guanidine from Reversed Micelles
Subsequent contacting stages to remove GuHCl involved combining 3-5 mL of the reversed micelle phase with an equal volume of an aqueous solution consisting of O.1M KCl in O.1M tris + 1mM EDTA, pH 8.7, mixing for 15 min, then centrifuging and separating phases as above. This step was repeated up to ten times with the resulting organic phase and a fresh aqueous solution to remove addi- tional denaturant.
Glu ta thion e A ddition
Stock solutions of reduced (GSH) and oxidized (GSSG) glutathione were made up separately at concentrations of 40 mM each in 0.1M tris + 1 mM EDTA, pH 8.7. Prior to use, they were mixed (typically at a 3 : 1 or 4: 1 ratio of the reduced to oxidized forms), and an aliquot of 50 to 100 p L was injected into 1 mL of 400 mM AOT solution (i.e., dripped in slowly while vortexing). One volume of glu- tathione reversed micelle solution (0.5 to 1 mL) was added to two volumes of sample solution (1-2mL), vortexed, and the tubes were capped. The time of addition of glu- tathione was considered as time = 0 for the reoxidation experiments, and enzyme activity was measured over time in the reversed micelles.
Extraction of Capro yl-RNase from Reversed Micelles
Equal volumes of a reversed micelle solution containing RNase (3-5 mL) and an aqueous solution of 1M KCI in 0.1M tris buffer, pH 8.7, were combined in a 20-mL scin- tillation vial with 100 pL ethyl acetate/mL. The mixture was stirred magnetically for 15 min at room temperature, then transferred to a capped centrifuge tube and cen- trifuged for 15 min. The phases were then separated.
Water Content of Reversed Micelles
Water content was determined by Karl Fisher Titration in an automated Mettler DL-18 Karl Fisher titrimeter.2' Sample volumes of 100 to 300 p L were used. Parameter W,, was determined as the molar ratio of water to surfac- tant. Samples were assayed in duplicate or triplicate, and the average value was used.
RNase and Capro yl-RNase Activity Assay
In aqueous solution. The substrate solution was 0.1 mg/ mL cytidine 2' : 3'-cyclic monophosphate in 0.025M phos- phate buffer, pH 7.5. A 1.4-mL volume of substrate solu- tion was pipetted into each of two cuvettes, and the spectrophotometer was set to zero at 286 nm. A 100-pL volume of sample was added to one cuvette and 100 p L of buffer to the other, and the contents were mixed by invert- ing several times. The change in absorbance at 286 nm was recorded over time for the difference between the two cu- vettes, and the initial rate was determined. Concentrations of active RNase were determined from a standard curve for native RNase in the sample buffer which was linear over the concentration range of 0 to 5 mg/mL. This assay is described by Crook et a1.22 This assay and all others re- quiring this equipment were performed with a Hewlett Packard diode array spectrophotometer linked to a HP Vec- tra ES/12 computer.
In Reversed Micelles. The substrate solution was 10 mg/ mL cytidine 2' : 3'-cyclic monophosphate in 0.05M borate buffer, pH 9.5. A 150-pL volume of this solution was in- jected into 9 mL of 50 mM AOT in iso-octane. One ml of substrate solution in reversed micelles was pipetted into each of two cuvettes, and the spectrophotometer was set to zero at 286 nm. Initially, 200pL of the sample were added to one cuvette, and 200pL of 400 mM AOT were added to the other, and the contents were mixed by inverting several times. For high salt concentrations in particular, we ob- tained better results with smaller sample volumes such as 100 p L , which was used for the later experiments. The change in OD reading was recorded over time for the dif- ference between the two cuvettes, and the initial rate was determined. Concentrations of active RNase were deter- mined from a standard curve for native RNase injected into reversed micelles. It was linear over a range of 0.05 to 0.91 mg/mL (prepared from 0 to 10 mg/mL RNase in
HAGEN, HATTON, AND WANG: PROTEIN REFOLDING 969

O.1M tris + ImM EDTA, pH 8.7 (no GuHCI), injected into 400 mM AOT at 100 pL/mL AOT solution). This assay was adapted from Wolf and Lu i~ i .*~
Enzymatic Activities. The results for both of these assays were also expressed as enzymatic activities in units of moles substrate converted to product per unit enzyme per unit time. They were calculated from the kinetic data by dividing the initial rate by the RNase concentration and miltiplying by a constant as follows:
Spec Act in units o f
moles S - p mole RNase x At
x AOD,,,/At C
cone RNase (mg/ml) - -
where C is 8.9 for the aqueous assay and 14 for the re- versed micelle assay (using 100 p1 sample).
Determination of RNase and Capro yl-RNase Concentration
Enzyme concentration was measured by ODzB0, which is a standard method for determining protein concentrations. In addition, the value of the absorbance at 310 nm was sub- tracted in order to reduce the error due to solution turbid- it^.'^ Sample concentrations were obtained from standard curves which, in the case of reversed micelles, were pre- pared by injection of aqueous solutions of known concen- trations. The following formulae were used to calculate concentrations (in mg/mL):
native (aqueous soln): C = (OD,,, - OD,,,)/0.60 denatured (aqueous soln): C = (OD,,, - OD,,,)/0.60 native (RM): C = (OD,,, - OD,,,)/0.65 denatured (RM): C = (OD,,, - OD,,,)/0.65
During phase transfer experiments, Lowry assays were performed to determine protein concentrations in the aque- ous phase before and after transfer. The difference agreed, within experimental error, with the concentrations obtained in the reversed micelle phase from absorbance measure- ments. The Lowry assay was mainly used to verify the rotein concentrations obtained by absorbance measure-
ments. This is the standard assay as described by Schleif and Wensink. 25
Gamma-Interferon Sample Preparation
Denatured y-interferon was obtained from Biogen in the form of inclusion bodies suspended in phosphate buffered saline (PBS) and was stored in 50 mL aliquots at -40°C. Prior to use, the required amount (usually 100-200 mL) was thawed overnight at 4°C. The sample, still in aliquots of 50 mL, was then centrifuged, the supernatant removed, and the pellet (about 5 mL) resuspended in 50 mL of PBS with 10 mM dithiothreitol (DTT), followed by centrifuga- tion. This washing step was repeated three times. The final pellet (about 2 mL) was then resuspended in 10 mL of 4M
GuHCl with 10 mM DTT on a rotary shaker stirring at slow speed for I h. After centrifugation, the supernatant was retained, and contained typically 20 mg/mL protein by Biorad assay. This product was characterized by gel fil- tration on Superose 12 and by gel electrophoresis, and was found to contain about 60% y-interferon. It was used with- out further purification. Native y-interferon has an activity of 3.45 U/mg, as measured by cytopathic effect assay (Biogen). The composition of PBS is as follows: 0.137M NaCI, 1 mM KH,PO,, 10 mM Na,HPO,, and 10 mM EDTA, pH 7.
Determination of y-Interferon Concentration
Most of this work was performed with the denatured pro- tein, as described above. Since it does not represent pure y-interferon, our extinction coefficients are specific to this mixture. The same batch of protein was used throughout all the experiments, and so we do not expect any variation.
Aqueous concentrations were determined by Lowry as- say, and these were correlated with absorbance measure- ments. In the case of the reversed micelle solution, gel electrophoresis of the aqueous solution before and after transfer showed no difference in the extent of transfer be- tween the protein components. We therefore used values from the aqueous Lowry assay before and after transfer to calculate the extinction coefficients.
For the denatured protein:
Concentration (aqueous) in mg/mL = (OD,,, - OD,,, - blank)/0.74 Concentration (RM) in mg/mL = (OD,,, - OD310 - blank)/0.72
Transfer of Denatured y-Interferon into Reversed Micelles
A solution of denatured y-interferon was prepared by mix- ing 0.1-5 mL of our 20 mg/mL stock solution in 4M GuHCl with an additional 0.5-15 mL of 4M GuHCl to achieve the desired protein concentration (0.5-5 mg/mL). Simultaneous additions of PBS containing 10 mM DTT (to reduce the GuHCl concentration within the range of 1M- 1.5M and protein to 0.2-2 mg/mL) and of 400 mM AOT (volume equal to total aqueous volume) were carried out, and mixing was started immediately. The total volume of one phase ranged from 5 mL in a 20-mL scintillation vial (for transfer studies) to 50 mL in a 200-mL centrifuge bottle (for experiments that involve many subsequent contacting steps for GuHCl removal). After 15 min, the samples were transferred into capped tubes and centrifuged for 20 min. The phases were then separated, and the absorbances at 280 and 3 10 nm were measured to determine protein concen- trations. An additional centrifugation step was occasionally necessary to clarify the organic phase, in particular at the high GuHCl concentrations.
970 BIOTECHNOLOGY AND BIOENGINEERING, VOL. 35, APRIL 1990

Removal of Guanidine Hydrochloride from the Reversed Micelles
Subsequent contacting stages involved combining the re- versed micelle solution prepared as above (5-50 mL) with an equal volume of an aqueous solution containing no de- naturant, typically 0 . lM KCI with 10 mM DTT in 0.025M phosphate buffer, pH 7 . After mixing magnetically for 15 min in a 20-mL scintillation vial, the samples were transferred to a capped centrifuge tube and centrifuged for 10-20 min. The phases were then separated, and this con- tacting stage was repeated with the resulting organic phase up to ten times. The volumes decreased with each separa- tion because of the difficulty of removing all of the organic phase without disturbing the interface. About 30% of the organic volume was lost over ten contacting stages.
Extraction of y-Interferon from Reversed Micelles
Equal volumes, typically 3-5 mL each of reversed micelle solution and aqueous extractant were mixed for 15 min, centrifuged, and the phases separated as above. A variety of compositions of the aqueous extractant solution were used containing different types [NaCI, KCI, GuHCl, and (NH,),SO,J and concentrations (0 .1M-3M) of salts as well as additives and cosurfactants.
RESULTS AND DISCUSSION
Protein-Denaturant-Surfactant Interactions in Reversed Micelles
Our first objective was to determine the extent of protein- reversed micelle interactions by attempting to refold RNase without aggregation at higher protein concentra- tions than in aqueous solution. If the yields obtained were lower than in aqueous solution, then interactions must have occurred between the protein molecule and the re- versed micelle which limited its refolding. We started with 5 mg/mL RNase reduced and denatured in 6M GuHCl. At this protein concentration, RNase usually aggregates in aqueous solution upon complete denaturant removal .26
Since the presence of GuHCl interferes with the transfer of proteins into reversed micelles, the lowest denaturant concentration must be chosen which maintains the protein soluble and unfolded.’ Therefore, in order to maximize protein transfer to reversed micelles, we reduced the GuHCl concentration to 0.5M prior to transfer. We then contacted the protein solutions (at varying initial concen- trations up to 5.1 mg/mL) with 400 mM AOT in iso- octane to incorporate the protein molecules into reversed micelles. During these steps, the protein remained soluble and unfolded. In order to initiate renaturation, the redox reagent glutathione was added to reoxidize disulfide bonds. These manipulations (see the Materials and Methods sec- tion for details) resulted in three final levels of water con- tent during the refolding step (W,, of 5.7, 7.2, and 9.9). Recovery of activity was measured over time in the reversed
micellar phase, and the results are shown in Figure 2. It can be seen that, as we increased the RNase concentration at a constant water content, the refolding yield in reversed micelles decreased, as is generally observed in aqueous so- lution. We also see that the renaturation yield was very de- pendent on the water content of the micelles. For example, at 2 mg/mL RNase, the refolding efficiency increased from 10 to 75% as the W, value is increased from 7.2 to 9.9. This result indicates that we were able to improve the refolding yields of denatured proteins in reversed micelles by using higher water contents in the rnicelles.
We can compare our results in the reversed micelles to those obtained by Anfinsen and HaberZ6 for refolding of RNase in aqueous solution (dotted line on Fig. 2). As the RNase concentration was reduced at a constant water con- tent, the reactivation yield approached that obtained in aqueous solution. In addition, we can see that, if the W , were increased even greater than 9.9, the reactivation pro- file would probably coincide with the aqueous solution results. It therefore appears that the yields obtained in aqueous solution also represent the maximum level of re- activation for RNase in reversed micelles. It remains to be seen whether this maximum is protein dependent, or whether it could also be attained for other proteins.
The size of the reversed micelles can be determined from the water content, based on the correlation of Eicke and Rehak,27 as used by Leotidis and Hattm.” It varied significantly between W, of 5.7 and 9.9 (Table I), with the volume increasing by a factor of 3.7 (column 3 ) , while the number of micelles decreased by a factor of 2.1 (column 4). At the highest W,, we therefore had a fewer number of larger micelles, and the proportion of micelles containing a protein molecule also increased. However, this proportion of filled micelles was not in itself a deter- mining variable. As an illustration, the two samples at 2 mg/mL overall RNase concentration (at W, values of 7.2 and 9.9) both had the same proportion of filled micelles (about 2.6%), yet we can see that the recovery yields were very different ( 10% vs. 70%, respectively). Therefore, other factors such as water content (and thus micelle size) or guanidinium ion concentration inside the micelle must play a crucial role in determining the reactivation yield.
In order to address the effect of GuHCl on the refolding of RNase in reversed micelles. we need to know the
Table I. Effect of W , on micelle size and concentration.
Micelle volume Micelle radius (A’) MicelleslmL
Water content (W,) (A, ( X 10-31 ( X 10-’7
5.7 13 8.4 4.9 7.2 1s 15 3.5 9.9 19 31 2.3
Water content (column 1) was determined by Karl Fischer Titration, and micelle size and concentrations were determined based on the corre- lation of ref. 27.
971 HAGEN, HATTON, A N D WANG: PROTEIN REFOLDING
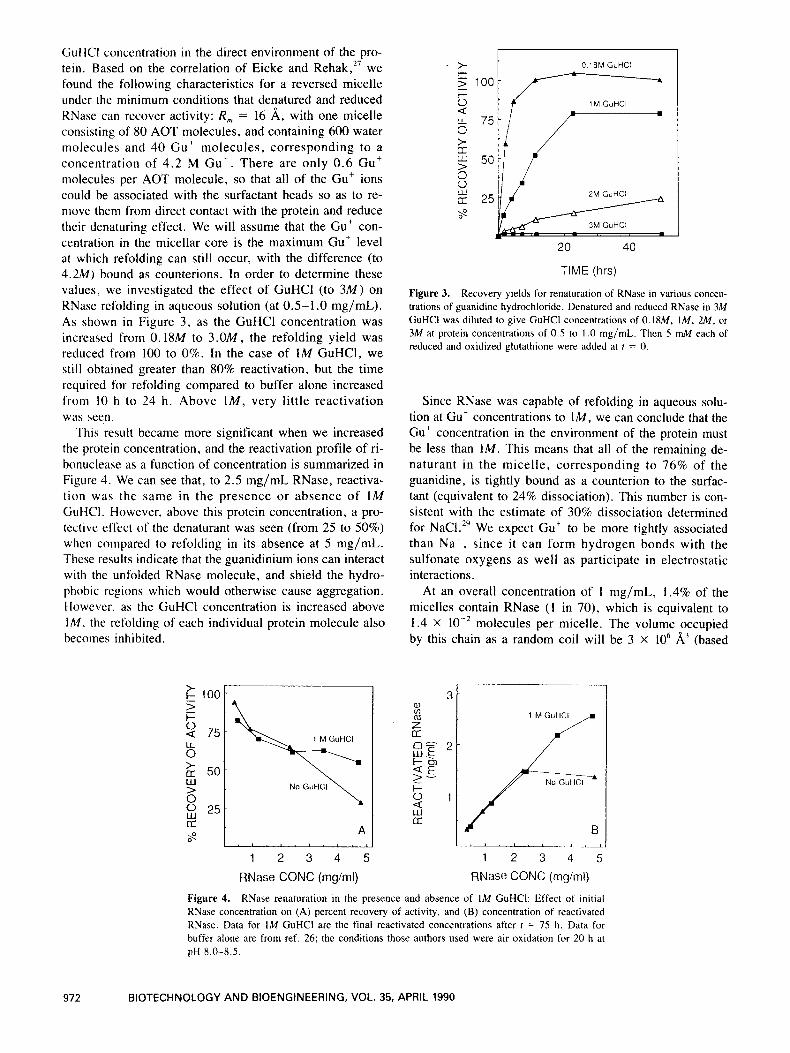
CiuHCl concentration in the direct environment of the pro- tein. Based on the correlation of Eicke and Rehak,27 we found the following characteristics for a reversed micelle under the minimum conditions that denatured and reduced KNase can recover activity: R , = 16 A, with one micelle consisting of 80 AOT molecules, and containing 600 water molecules and 40 Gut molecules, corresponding to a concentration of 4.2 M Gut . There are only 0 .6 Gut molecules per AOT molecule, so that all of the Gu’ ions could be associated with the surfactant heads so as to re- move them from direct contact with the protein and reduce their denaturing effect. We will assume that the Gu’ con- centration in the micellar core is the maximum Gut level at which refolding can still occur, with the difference (to 4.2M) bound as counterions. In order to determine these values, we investigated the effect of GuHCl (to 3 M ) on RNase refolding in aqueous solution (at 0.5-1.0 mg/mL). As shown in Figure 3 , as the GuHCl concentration was increased from 0. ISM to 3 . 0 M , the refolding yield was reduced from 100 to 0%. In the case of 1M GuHCl, we still obtained greater than 80% reactivation, but the time required for refolding compared to buffer alone increased from 10 h to 24 h . Above lM, very little reactivation was seen.
This result became more significant when we increased the protein concentration, and the reactivation profile of ri- bonuclease as a function of concentration is summarized in Figure 4. We can see that, to 2.5 mg/mL RNase, reactiva- tion was the same in the presence or absence of 1M GuHCI. However, above this protein concentration, a pro- tective effect of the denaturant was seen (from 25 to 50%) when compared to refolding in its absence at 5 mg/mL. These results indicate that the guanidinium ions can interact with the unfolded RNase molecule, and shield the hydro- phobic regions which would otherwise cause aggregation. However, as the GuHCl concentration is increased above IM, the refolding of each individual protein molecule also becomes inhibited.
b 1 MGuHCI
1 2 3 4 5
RNase CONC (mg/ml)
0 18M GuHCl
50
25
20 40
TIME (hrs)
Figure 3. Recovery yields for renaturation of RNase in various concen- trations of guanidine hydrochloride. Denatured and reduced RNase in 3M GuHCI was diluted to give GuHCl concentrations of 0. IXM, IM, 2M, or 3M at protein concentrations of 0.5 to 1 .0 mg/mL. Then 5 mM each of reduced and oxidized glutathione were added at I = 0.
Since RNase was capable of refolding in aqueous solu- tion at Gut concentrations to lM, we can conclude that the Gut concentration in the environment of the protein must be less than 1M. This means that all of the remaining de- naturant i n the micelle, corresponding to 76% of the guanidine, is tightly bound as a counterion to the surfac- tant (equivalent to 24% dissociation). This number is con- sistent with the estimate of 30% dissociation determined for NaC1.” We expect Cut to be more tightly associated than Na’, since i t can form hydrogen bonds with the sulfonate oxygens as well as participate in electrostatic interactions.
At an overall concentration of 1 mg/mL, 1.4% of the micelles contain RNase ( I in 70), which is equivalent to 1.4 X molecules per micelle. The volume occupied by this chain as a random coil will be 3 X lo6 A3 (based
I -lr-
1 2 3 4 5 RNase CONC (rng/rnl)
Figure 4. RNase renaturation in the presence and absence of 1M GuHCI: Effect of initial RNase concentration on (A) percent recovery of activity, and (B) concentration of reactivated RNase. Data for 1M GuHCl are the final reactivated concentrations after t = 75 h. Data for buffer alone are from ref. 26; the conditions those authors used were air oxidation for 20 h at pH 8.0-8.5.
972 BIOTECHNOLOGY AND BIOENGINEERING, VOL. 35, APRIL 1990

on Lapanje'), which is much larger than the volume of a micelle (3 X lo4 A3). This leads us to conclude that the protein molecule is constrained by the micellar environ- ment, possibly, for RNase, into a conformation close to the native.
Effect of Protein Hydrophobicity on Refolding in Reversed Micelles
We have now studied some of the important interactions occurring between the protein and the micellar components during refolding of RNase in reversed micelles. However, in order to determine the role of protein hydrophobicity, as well as the applicability of this system to protein folding in general, we extended our studies to a hydrophobic protein, y-interferon. Although both proteins under study are small and monomeric, unlike RNase, y-interferon aggregates in aqueous solution upon removal of denat~rant.~'
The folding process in reversed micelles was carried out with y-interferon as previously described for RNase (see Fig. I ) . Transfer of denatured y-interferon from a GuHCl solution to reversed micelles occurred very readily, as did GuHCl removal from the organic phase. However, upon attempting extraction of the protein from the organic phase to the aqueous, no protein was recovered in aqueous solu- tion; it either remained in the reversed micelle phase or ag- gregated at the interface. We would expect this type of severe aggregation behavior if the y-interferon was still unfolded i n the reversed micelles. This occurs during extraction where a high concentration of hydrophobic unfolded molecules would be suddenly present at the inter- face between the two phases and would therefore aggre- gate. We would not encounter this problem in dilute aqueous solution since y-interferon was capable of refold- ing from a denatured state with a 40% yield at 0.01 mg/mL. Therefore, the individual molecules are able to recover ac- tivity, but there must be interactions within the reversed micelle system that prevent refolding. However, in the case of active, correctly folded y-interferon, extraction from the reversed micellar phase also resulted in aggrega- tion at the interface. The reversed micellar environment apparently has a denaturing effect on the native protein which could also account for the inability of the denatured protein to refold.
Our approach so far has been to consider the micelles as simple compartments which isolate the denatured protein molecules from each other during refolding. However, since we have now encountered problems with aggrega- tion, we must take into account the interactions between the micelle and the protein, specifically its amino acid side chains. Ionic linkages between the protein and the surfac- tant head groups play a very important role in the location and the conformation of native proteins and peptides. How- ever, for denatured proteins, as for membrane proteins, hy- drophobic interactions may occur with the surfactant tails. y-Interferon contains a much higher proportion of those
amino acids which have a strong preference for the surfac- tant layer when compared to RNase; in addition, they are arranged into regions of high hydr~phobicity.~' We can hy- pothesize that these hydrophobic amino acid regions can anchor the protein into the surfactant layer and stabilize the molecule in an unfolded form. This will also greatly reduce the driving force for refolding. However, RNase contains hydrophobic amino acids, although fewer than y-interferon, and the enzyme was still capable of refold- ing. Therefore, we can envision an intermediate level of hydrophobicity above which a denatured protein can no longer refold due to competing surfactant interactions. In order to study the effect of hydrophobicity on refolding, we used covalent modifications of RNase so that the hy- drophobic variants could be easily assayed in reversed mi- celles and then compared under the same conditions.
The requirements for a suitable type of modification are first, that the enzyme retains its catalytic activity and sec- ond, that it still be capable of refolding from a denatured state in aqueous solution. The caproyl group (5 methylenes and an amide) was selected, since it fulfills these two crite- ria. Is We carried out three different degrees of modifica- tion ranging from 0 to 55% substitution of the lysines. These derivatives retained enzymatic activity in aqueous as well as reversed micellar solutions, with only the most hy- drophobic modification showing reduced activity in both systems (see the Materials and Methods section).
We first determined that these modified RNases were still capable of refolding in aqueous solution. The proteins were denatured and reduced in 6 M GuHCl and 0 .3M P-mercaptoethanol, then passed through a desalting column to reduce the concentration of denaturant. It was then di- luted to give 0.8M GuHCl and 0.2 mg/mL protein. Glu- tathione (5 mM each of reduced and oxidized forms) was added, and enzymatic activity was measured over time. We found that all of the modified RNases were capable of recovering their full activity, as had been reported previ- 0us1y.'~ A study of the refolding of these proteins in re- versed micelles will therefore provide information on the interactions of the added caproyl groups with the micellar environment.
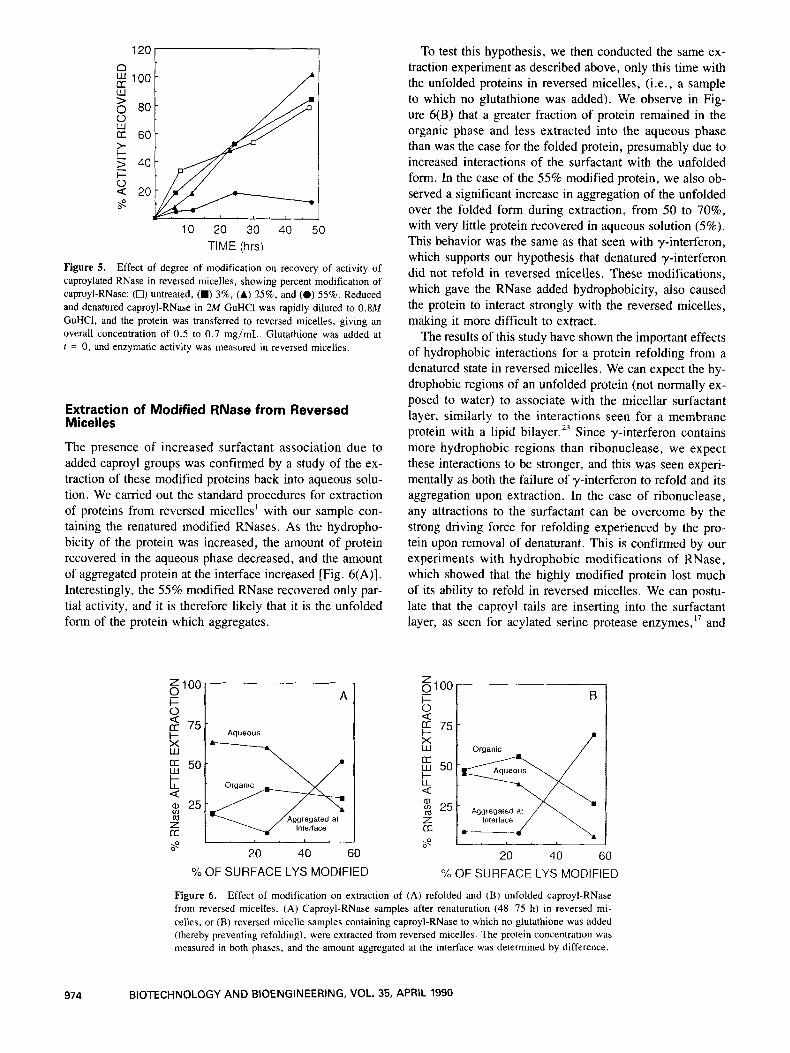
The steps of the reversed micelle process were then car- ried out as discussed previously for native RNase (see Fig. 1). The transfer efficiency to reversed micelles was not significantly affected by the added hydrophobicity. In contrast, the level of modification was a very important parameter in the reoxidation and recovery of activity of the modified RNases. Glutathione in reversed micelles was added to the micellar solution of denatured protein and ac- tivity in the micellar phase was determined over time. As shown in Figure 5 , with 0, 3, and 25% modifications, a high degree of enzymatic activity was recovered (80- 100%). However, at 55% modification, we saw a very sig- nificant reduction in the ability of the protein to refold. There must therefore be unfavorable interactions between the added caproyl groups and the micelle.
HAGEN, HAlTON, AND WANG: PROTEIN REFOLDING 973
120 I I
!d n i 100 U I / I
10 20 30 40 50 TIME (hrs)
Figure 5. Effect of degree of modification on recovery of activity of caproylated RNase in reversed micelles, showing percent modification of caproyl-RNase: (0) untreated, (W) 3%, (A) 25%. and (0) 55%. Reduced and denatured caproyl-RNase in 2M GuHCl was rapidly diluted to 0.8M GuHCI, and the protein was transferred to reversed micelles, giving an overall concentration of 0.5 to 0.7 mg/mL. Glutathione was added at f = 0, and enzymatic activity was measured in reversed micelles.
Extraction of Modified RNase from Reversed Micelles
The presence of increased surfactant association due to added caproyl groups was confirmed by a study of the ex- traction of these modified proteins back into aqueous solu- tion. We carried out the standard procedures for extraction of proteins from reversed micelles' with our sample con- taining the renatured modified RNases. As the hydropho- bicity of the protein was increased, the amount of protein recovered in the aqueous phase decreased, and the amount of aggregated protein at the interface increased [Fig. 6(A)]. Interestingly, the 55% modified RNase recovered only par- tial activity, and it is therefore likely that it is the unfolded form of the protein which aggregates.
To test this hypothesis, we then conducted the same ex- traction experiment as described above, only this time with the unfolded proteins in reversed micelles, (i.e., a sample to which no glutathione was added). We observe in Fig- ure 6(B) that a greater fraction of protein remained in the organic phase and less extracted into the aqueous phase than was the case for the folded protein, presumably due to increased interactions of the surfactant with the unfolded form. In the case of the 55% modified protein, we also ob- served a significant increase in aggregation of the unfolded over the folded form during extraction, from 50 to 70%, with very little protein recovered in aqueous solution (5%). This behavior was the same as that seen with y-interferon, which supports our hypothesis that denatured y-interferon did not refold in reversed micelles. These modifications, which gave the RNase added hydrophobicity, also caused the protein to interact strongly with the reversed micelles, making it more difficult to extract.
The results of this study have shown the important effects of hydrophobic interactions for a protein refolding from a denatured state in reversed micelles. We can expect the hy- drophobic regions of an unfolded protein (not normally ex- posed to water) to associate with the micellar surfactant layer, similarly to the interactions seen for a membrane protein with a lipid bilayer. 23 Since y-interferon contains more hydrophobic regions than ribonuclease, we expect these interactions to be stronger, and this was seen experi- mentally as both the failure of y-interferon to refold and its aggregation upon extraction. In the case of ribonuclease, any attractions to the surfactant can be overcome by the strong driving force for refolding experienced by the pro- tein upon removal of denaturant. This is confirmed by our experiments with hydrophobic modifications of RNase, which showed that the highly modified protein lost much of its ability to refold in reversed micelles. We can postu- late that the caproyl tails are inserting into the surfactant layer, as seen for acylated serine protease enzymes,17 and
= 100 0 l- 0
75
3
t 50
Q a, 25 m Z [r
ffl
s
A / Aqueous I
20 40 60
%OF SURFACE LYS MODIFIED 20 40 60
Yo OF SURFACE LYS MODIFIED
Figure 6. Effect of modification on extraction of (A) refolded and (B) unfolded caproyl-RNase from reversed micelles. (A) Caproyl-RNase samples after renaturation (48-75 h) in reversed mi- celles, or (B) reversed micelle samples containing caproyl-RNase to which no glutathione was added (thereby preventing refolding), were extracted from reversed micelles. The protein concentration was measured in both phases, and the amount aggregated at the interface was determined by difference.
974 BIOTECHNOLOGY AND BIOENGINEERING, VOL. 35, APRIL 1990

stabilizing the unfolded protein molecule. By chemically modifying RNase enough to significantly increase the hy- drophobicity, we were able to counteract the driving force for refolding and reduce the extraction efficiency. For y- interferon, we can postulate that this stabilizing effect was so strong that it completely prevented refolding, and also inhibited extraction. These results seem to indicate that only hydrophilic or moderately hydrophobic proteins will be able to refold in AOT reversed micelles.
Since all of our experiments were carried out with only one surfactant system (AOT in iso-octane), we cannot gen- eralize our results to other reversed micelle systems until alternative surfactants and solvents have been investigated. In conjunction with this analysis, the feasibility studies should be continued with other proteins to determine the general applicability of the reversed micelle system for re- ducing aggregation and improving refolding yields during the refolding of denatured proteins.
The authors acknowledge the financial support from the National Science Foundation under the Engineering Research Center Initia- tive to the MIT Biotechnology Process Engineering Center (CDR- 88-03014) and the fellowship to A. J . Hagen from the Natural Sciences and Engineering Research Council of Canada.
References
I .
2.
3 .
4.
5 . 6.
7 .
A. 1. Hagen, T. A. Hatton, and D. I . C. Wang, Biotech. Eioeng. Submitted for Publication (1989). S. Lapanie, "Physicochemical Aspects of Protein Denaturation". John Wiley and Sons, Publ. (1978). G . Ortini and M . E. Goldberg, J . Biol. Chem., 253(10), 3453 (1978). U. Carlsson, L. E. Henderson, and S. Lindskog, Biochim. Biophys. Acra. 310, 376 (1973). S . Damodaran, Eiochim. Biophys. Acta, 914, 114 (1987). P. Horowitz and N. L. Criscimagna, J . Biol. Chem., 261, 15652 (1986) A. M. Klibanov, Trends Biochem. Sci.. 14, 141 (1989).
8.
9.
10.
1 1
12.
13.
14.
15. 16. 17.
18. 19.
20.
21
22.
23.
24.
25.
26. 27. 28. 29.
30.
31.
32.
L. Luisi and B. Steinmann-Hofmann, Methods Enzym., 136, 188 (1987). C. Petit, P. Brochette, and M. P. Pileni, J . Phys. Chem., 90, 6517 (1986). J . Gallay, M. Vincent, C. Nicot, and M. Waks, Biochemistry, 26, 5738 (1987). C. Nicot, M. Vacher, M. Vincent, J . Gallay, and M. Waks, Eio- chemistry, 24, 7024 (1985). A. Delahodde, M. Vacher, C. Nicot, and M. Waks, FEES Lett., 172, 343 (1984). P. L. Luisi, M. Giomini, M. P. Pileni, and B. H. Robinson, Biochim. Biophys. Acra., 947, 209 (1988). E. B. Leotidis and T. A. Hatton, in Slructure and Reactivity in Re- versed Micelles, M. P. Pileni, Ed. (Elsevier, New York, 1989). C. Epstein and R. F. Goldberger, J . Biol. Chem., 239, 1087 (1964). A. Light, BioTechniques, 3(4), 298 (1985). A. V. Kabanov, A. V. Levashov, and K. Martinek, V. Mosk. Univ. Khim., 41(6), 591 (1986). F. Fields, Methods Enzymol., 25, 464 (1972). A. M. Crestfeld, F. Moire, and W. H. Stein, J . Biol. Chem., 238, 622 (1963). P. W. Riddles, R. L. Blakely, and B. Zerner, in Methods in Enzymol- ogy, Volume 91 (Academic, New York, 1983), pp. 49-61. H. A . Laitman and W. E. Harris, Chemical Analysis, 2nd ed. McGraw-Hill, New York, (1975), pp. 361-363. E. M. Crook, A. P. Mathias, and B. R. Robin, Biochem J . , 74, 234 ( 1960). R. Wolf and P. L. Luisi, Biochem. Biophys. Res. Commun.. 89, 209 (1979). J. M. Woll, T. A. Hatton, and M. L. Yarmush, Biorerhnol. Prog., 5(2), 57 (1989). R. F. Schleif, and P. C. Wensink, Practical Methods in Molecular Bi- ology (Springer-Verlag. New York, 1981), p. 75. C. B. Anfinsen and E. Haber, J . BioL. Chem., 236, 1361 (1961). H-F. Eicke and J. Rehak, Helv. Chim. Acra., 59(306), 2883 (1976). E. B. Leotidis and T. A. Hatton. Langmuir, 5, 741 (1989). K. F. Thompson and L. M. Gierasch, J . Am. Chem. Soc.. 106, 3648 (1984). T. Arakawa, N. K . Alton, and Y-R. Hsu, J . Biol. Chem., 260, 14435 (1985). Protein Sequence Query Program from the Protein Identification Re- source, National Biomedical Research Foundation, Washington, DC, ( 1985). J-L. Popot, S-E. Gerchman, and D. L. Engelman. J . Mol. Biol., 198, 655 (1987).
HAGEN, HATTON, AND WANG: PROTEIN REFOLDING 975




















