
Forensic electrochemistry: sensing the molecule of murder atropine
Upload
independentCategory
view
3download
0
Send Orders of Reprints at [email protected]
Current Topics in Medicinal Chemistry, 2013, 13, 000-000 1
1568-0266/13 $58.00+.00 © 2013 Bentham Science Publishers
Protein Interface Pharmacophore Mapping Tools for Small Molecule Protein: Protein Interaction Inhibitor Discovery
Arnout Voet1,*, Eleanor F. Banwell2, Kamlesh K. Sahu1, Jonathan G. Heddle2 and Kam Y. J. Zhang1
1Zhang Initiative Research Unit, and
2Heddle Initiative Research Unit, Advanced Science Institute, RIKEN, 2-1 Hiro-
sawa, Wako, Saitama 351-0198, Japan
Abstract: Protein:protein interactions are becoming increasingly significant as potential drug targets; however, the ra-tional identification of small molecule inhibitors of such interactions remains a challenge. Pharmacophore modelling is a popular tool for virtual screening of compound libraries, and has previously been successfully applied to the discovery of enzymatic inhibitors. However, the application of pharmacophore modelling in the field of protein:protein interaction in-hibitors has historically been considered more of a challenge and remains limited. In this review, we explore the interac-tion mimicry by known inhibitors that originate from in vitro screening, demonstrating the validity of pharmacophore mapping in the generation of queries for virtual screening. We discuss the pharmacophore mapping methods that have been successfully employed in the discovery of first-in-class inhibitors. These successful cases demonstrate the usefulness of a “tool kit” of diverse strategies for application across a range of situations depending on the available structural infor-mation.
Keywords: Drug discovery, pharmacophore modelling, protein:protein interaction (PPI), small molecule protein:protein inter-action inhibitor (SMPPII), virtual screening.
1. INTRODUCTION
Proteins are essential for virtually every biological proc-ess; they function via interactions with other molecules, and the complexes they form are often extensive and themselves only small parts of vast interaction networks. The exact number of protein:protein interactions (PPIs) in humans is unknown, but has been estimated at between 130,000 and 650,000 [1, 2]. All diseases are departures from normal bio-logical processes and, as these are often controlled by PPIs, disruption of unwanted interactions through targeting with Small Molecule PPI Inhibitors (SMPPIIs) is an attractive therapeutic strategy [3].
The pharmaceutical industry has extensively explored the interactions between enzymes and their substrates and nu-merous small molecule enzyme inhibitors have been de-signed to fit active sites, influence ion channels and trans-porters, or modulate receptor activities. While the interac-tions involved are similar to those found in enzyme inhibi-tors, the design of SMPPIIs is a more complex problem. En-zyme substrates are typically small and bind in clear cavi-ties/clefts where they are shielded from the solvent. They interact with their receptor via highly orientation specific hydrophilic contacts that serve as excellent templates for drug designers to mimic [4, 5]. However, PPIs generally comprise large shallow surfaces with areas in the range 1500-3000 Å2, as compared to the 300-1000 Å2 that is typi-cal for interactions between small molecules and proteins [6, 7]. These interactions are usually predominantly hydro-
*Address correspondence to this author at the Zhang IRU, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan; Tel: +81-48-4679209; Fax:+81-48-4678790; Email: [email protected]
phobic in character and composed of non-contiguous se-quences of amino acids, meaning interactions do not depend on the precise placement of particular functional groups ar-rayed linearly and cannot simply be grafted onto small pep-tides [5]. As a result of these differences, SMPPIIs have been considered “high-hanging fruits” [8, 9]. However, this para-digm has recently started to change, and the number of novel SMPPIIs being reported has increased dramatically [5, 10-15].
Due to their importance and ubiquity, PPIs are of great interest as potential targets for drug discovery. However, as described above, they are not easily amenable to the enzy-matic inhibitor design approach. Consequently, most re-ported SMPPIIs originate from in vitro (including fragment based) screening techniques [16-18]. In vitro screening is expensive and time consuming and virtual screening (VS) alternatives are highly desirable. VS is a computer-aided drug-discovery (CADD) method comprising the in silico screening of a library of chemical compounds in order to identify those that are most likely to bind to a specified tar-get. Through VS, a large number of molecules likely to be inactive can be filtered out of the search before in vitro evaluation, speeding up the process and reducing the costs [19].
One of the most commonly applied VS methods is mo-lecular docking, where drug-like compounds are placed into their receptor structure virtually, and are scored for potency using computational simulations [20]. Typically, several different putative binding modes are identified per com-pound, each with different scores, and it is often observed that the true binding mode is ranked lower than false binding modes [21]. Molecular docking is of interest in the design of

2 Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 Voet et al.
SMPPIIs but, as with in vitro screening, compound libraries are large and docking and scoring them all is a laborious task where the vast majority of the molecules screened will turn out to be unsuitable. Any method for searching libraries and detecting only those molecules with a reasonable likelihood of interacting will further speed up the discovery of new drugs [22]. Chemo-informational analysis of previously re-ported SMPPIIs provides insights into their chemical and spatial properties. This information can be used to inform the design of high-throughput screening libraries and to improve VS via post filtering [13, 15, 23-25].
A pharmacophore is defined as the set of features neces-sary for molecular recognition of a ligand by a receptor and to trigger a biological event, including inhibition [26]. In CADD, a pharmacophore model is the abstract 3D represen-tation of the chemical functionalities considered to be essen-tial for this process. Pharmacophore activity is independent of the scaffold, and this explains why similar biological events can be triggered by chemically divergent molecules [27]. Pharmacophore models can be used as the query with which compound libraries can be searched in order to pull out molecules of interest for later VS and are also of interest in the chemo-informatics field [26]. They comprise sphere-like features representing molecular functionalities including “hydrophobic”, “aromatic”, “hydrogen bond acceptor”, “hy-drogen bond donor”, “cation”, or “anion”, and any logical combination of these (Fig. 1). Where a lot of structural in-formation about an interaction exists, a pharmacophore model can be built by mapping the key interactions between the ligand and the protein [28]. This can be further refined by superimposing multiple ligands in order to discover the common features [29]. Where less information is available, the information in the receptor structure alone can be used to produce the inverse map and to deduce the pharmacophore model best able to bind to it. All these methods have been shown to be valuable for speeding up the drug-discovery process; however, as discussed above, the differences be-tween SMPPIIs and enzyme inhibitors make constructing pharmacophore queries for the former more of a challenge.
For the purpose of this review, we have performed a lit-erature search in Pubmed using the search term “small mole-cule protein interaction inhibitor”. Here we review the struc-tural information of all published SMPPII and PPI com-plexes that reveal mimicry of the PPI interface by SMPPIIs in order to demonstrate the validity of pharmacophore map-ping for SMPPII discovery. We also explore the various methods that have been developed to assist in the construc-tion of pharmacophore models for SMPPIIs in order to lower these high-hanging but valuable fruits to within reach. As this field is in its infancy, the number of publications is small enough that we were able to review all the cases published as of November 2012.
2. MIMICRY OF THE PPI INTERFACE BY KNOWN SMPPIIS
It is reasonable to assume that, much like enzyme inhibi-tors, the majority of successful SMPPIIs are likely to work by binding directly to the interface and blocking access by the native ligand [30]. Although there will be exceptions, intuitively the best SMPPIIs should be those that most
closely resemble the molecular function and geometry of the ligands. However, it is important to test this assumption. Fortunately, for the few examples of SMPPIIs discovered by traditional in vitro screens where we have the relevant crystal structures for both the protein:protein complex and pro-tein:inhibitor complex, there is substantial evidence of such a relationship. These have become the archetypal examples for the SMPPII drug-design community and are summarized in (Table 1) and graphically depicted in (Fig. 2).
Fig. (1). (A) Pharmacophore models are composed of functional features, represented as spheres. These features are typically hydro-phobic, aromatic, cationic or anionic moieties and hydrogen bond donors or acceptors. (B) A kinase-inhibitor pharmacophore model, showing various features mapped on top of a known inhibitor (sticks) sitting in the binding pockets of the CDK2 kinase protein (cartoon) (PDB 1KE5). This visualization of pharmacophore fea-tures and coloring scheme will be employed throughout this manu-script according to the Molecular Operating Environment (MOE) software package [108]. The features at the bottom right of the molecule do not take part in the interaction with the protein and are not included in the pharmacophore model.
One example of clear PPI mimicry by an inhibitor dis-covered by in vitro screening is the inhibitor of Bcl2 and “bad” (or bax) discovered by Hajduk, et al. in 2006 [17]. In B-cell lymphoma, the pro-apoptotic “bad” is inhibited in cancerous cells by over expression of B-cell lymphoma 2

Protein Interface Pharmacophore Mapping Tools Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 3
Table 1. Summary of the Archetypal Cases of PPI Mimicry by SMPPIIs. For Each Example, the PDB Codes for the Pro-
tein:protein and Protein: Inhibitor Structures are Given, Along with the Pharmacophore Feature Count and the Relevant
Amino Acids on which the Features are Mapped.
Complex PDB Codes Pharmacophore Feature Count
Interaction Figure PPI SMPPII Hydrophobic Polar Total Amino Acids to Map Features on
Bcl2:bad 2A 2BZW 2O2M 3 1 4 hydrophobic: Leu109, Phe116, and Phe10; polar: Glu117
HDM2:p53 2B 1YCR 3TU1 3 1 4 hydrophobic: Phe19, Trp23 and Leu26; polar: Trp23
XIAP:Smac 2C 1G73 3MUP 3 0 7 hydrophobic: Val2, Pro3 and Ile4 polar: Ala1, Val2,
Pro3 and Ile4
ZipA:FtsZ 2D 1F47 1Y2F 4 4 4 hydrophobic: Phe377, Leu372, Leu378 and Ile374
Integrin:Fibrinogen 2E 2VDO 2VDM 2 2 5 hydrophobic: Ala408 and Lys406; polar
Lys406 and Asp410(2)
Integrase:LEDGF/p75 2F 2B4J 3LPU 2 3 4 hydrophobic: Ile365 and Ile368; polar: Asp366(2)
VHL:HIF1 2G 1LQB 3ZRC 3 3 6 hydrophobic: Leu562, Hyp564, Ile566 polar:
Leu 562, Hyp564(2)
IL2:IL2R 2H 1Z92 1PY2 2 2 4 hydrophobic: Leu2 and Met25; polar: Arg36 (2)
(Bcl2). This prevents amplification of the apoptotic cascade and stops cell death [31]. Interrupting this interaction could restore the apoptotic cascade and cause cell death, making SMPPIIs of Bcl2 attractive anti-cancer drug targets [32, 33]. The first Bcl2 targeting SMPPII were generated by SAR by NMR (Structure Activity Relationship by Nuclear magnetic Resonance: a fragment-based drug design method in which molecular fragment activity is determined by NMR spectros-copy), and structural analysis of the superposition of the PPI complex and protein:inhibitor complex indicates a great deal of functional overlap (Fig. 2A) [17]. Regulation of apoptosis is a popular SMPPII drug target and similar PPI interface mimicry by SMPPIIs has also been demonstrated for the HDM2:p53 and XIAP:Smac inhibitors (Fig. 2B & C respec-tively) [30, 34-39].
Protein:SMPPII structures that are not related to apopto-sis, but exhibit mimicry also exist for:
1) ZipA:FtsZ, an essential PPI required for bacterial cell division and therefore of interest as a possible antibiotic target (Fig. 2D) [40-44].
2) Integrin:Fibrinogen, a PPI involved in blood clot forma-tion and therefore of interest to prevent thrombosis (Fig. 2E) [45, 46].
3) Integrase:LEDGF/p75, a PPI between a cellular and an HIV enzyme and therefore of interest for anti-retroviral therapy (Fig. 2F) [47-52].
4) VHL:HIF1, an ubiquitination related PPI which is impor-tant for a wide variety of diseases (Fig. 2G) [53-55].
5) IL2:IL2R, a PPI important for the immune response (Fig. 2H) [56-58].
From these examples, and the summary of the type of in-teractions in (Table 1), it is apparent the majority of interac-
tions are formed by hydrophobic contacts, which is in agreement with the hydrophobic nature of PPI interfaces. However, polar interactions are also present, especially in the case of the XIAP:Smac interaction where the inhibitor binds by exploiting the -strand backbone interactions (Fig. 2C).
From these observations, it seems probable that building a pharmacophore query for selection of SMPPIIs is possible [59]. However, it should be noted that these interactions are highly complex. For example, although mimicry exists in the case of the IL2:IL2R interaction (Fig. 2H), binding occurs after induced fit. Thus, while pharmacophore models can be derived by mapping the features of the amino acids that make up the PPI interface, this technique is not a panacea.
Mimicry is particularly clear in the cases where the PPI interface is formed by a contiguous stretch of interacting amino acids and the receptor has a rigid surface — that is, where interactions are similar in character to those seen in enzyme inhibition. The PPI interactions of the Bcl2:bad, HDM2:p53 and ZipA:FtsZ are all formed by a single -helix, while the XIAP:Smac interaction consists of single -strand. In the case of the Integrin:Fibrinogen or the VHL:HIF interaction, the interface comprises a single loop.
There are examples of SMPPIIs where mimicry has not been observed (for example, inhibitors of the HPV E1:E2 interaction and the TNF trimer), or where they exhibit sig-nificant induced fit (the IL2:IL2R interaction), but in these PPI interfaces, the major interactions are discontinuous stretches of amino acids [60, 61].
These insights can be taken into account to assess the level of challenge a PPI interface poses before starting a drug design project.
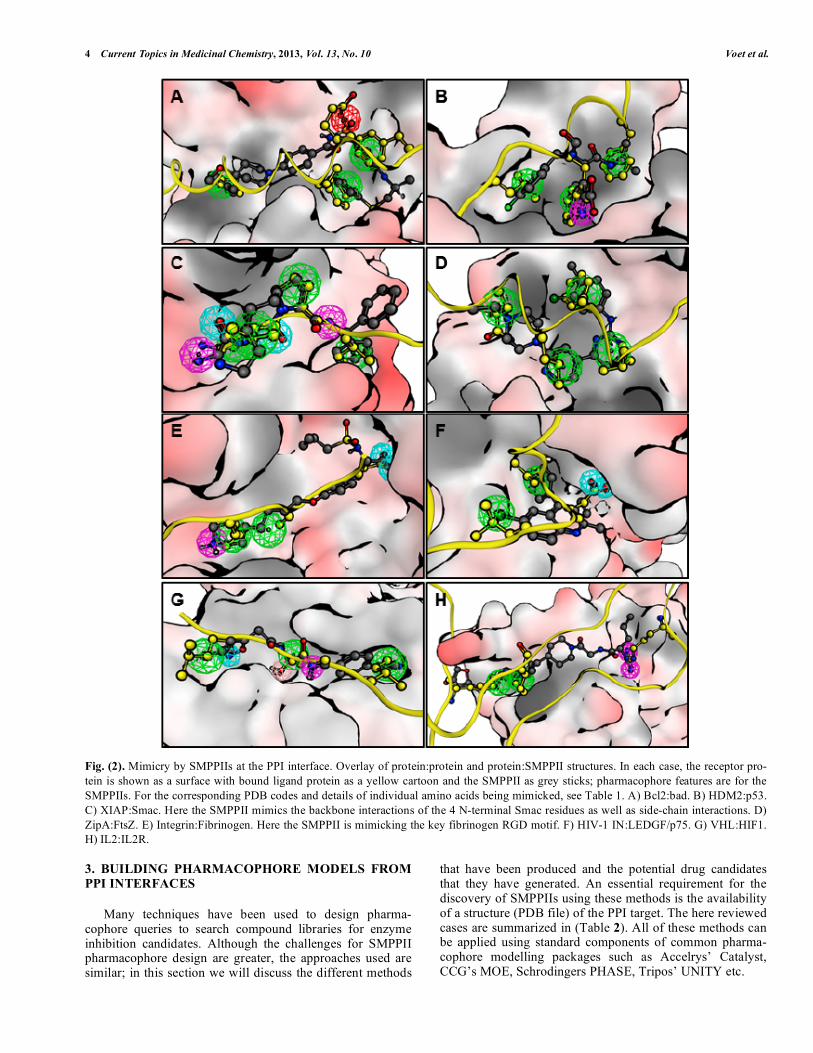
4 Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 Voet et al.
Fig. (2). Mimicry by SMPPIIs at the PPI interface. Overlay of protein:protein and protein:SMPPII structures. In each case, the receptor pro-tein is shown as a surface with bound ligand protein as a yellow cartoon and the SMPPII as grey sticks; pharmacophore features are for the SMPPIIs. For the corresponding PDB codes and details of individual amino acids being mimicked, see Table 1. A) Bcl2:bad. B) HDM2:p53. C) XIAP:Smac. Here the SMPPII mimics the backbone interactions of the 4 N-terminal Smac residues as well as side-chain interactions. D) ZipA:FtsZ. E) Integrin:Fibrinogen. Here the SMPPII is mimicking the key fibrinogen RGD motif. F) HIV-1 IN:LEDGF/p75. G) VHL:HIF1. H) IL2:IL2R. 3. BUILDING PHARMACOPHORE MODELS FROM
PPI INTERFACES
Many techniques have been used to design pharma-cophore queries to search compound libraries for enzyme inhibition candidates. Although the challenges for SMPPII pharmacophore design are greater, the approaches used are similar; in this section we will discuss the different methods
that have been produced and the potential drug candidates that they have generated. An essential requirement for the discovery of SMPPIIs using these methods is the availability of a structure (PDB file) of the PPI target. The here reviewed cases are summarized in (Table 2). All of these methods can be applied using standard components of common pharma-cophore modelling packages such as Accelrys’ Catalyst, CCG’s MOE, Schrodingers PHASE, Tripos’ UNITY etc.

Protein Interface Pharmacophore Mapping Tools Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 5
Table 2. Examples of SMPPII Discovery Using Pharmacophore Mapping. For Each Example, the PDB Codes for the Pro-
tein:inhibitor Structure is Given, Along with the Type of Method and the Key Reference.
Target name Complex pdb Code Method Type Reference
AnnexinA2:S100A11 1BT6 manual Kerry, et al. 2012
Integrase:LEDGF/p75 2B4J automatic + manual De Luca, et al. 2010
Integrase:LEDGF/p75 2B4J, 1HYV,1BI4,1BL3 consensus Christ, et al. 2010
Bcr-Abl:14-3-3 1YWT automatic + MD Corradi, et al. 2011
PUMA:Bcl2 2VOF,2ROC consensus Mustata, et al. 2010
HIV-1 RT p55:p66 1RTH MIF Grohmann, et al. 2008
Lysozyme:PliC 3F6Z manual + MIF Voet, et al. 2011
3.1. Manual Pharmacophore Model Design
The simplest method for designing pharmacophore mod-els is to examine the PPI manually and map the pharma-cophore query onto the PPI using personal insights and pref-erences. SMPPII design is still a young field, and manual model design allows the most interesting drug targets to be investigated without waiting for automated tools to be devel-oped.
An example of SMPPIIs discovered using a manually created pharmacophore query are the compounds that inhibit the S100A10:AnnexinA2 interaction. The S100A10 protein is an important component in cellular structural scaffolding. Recent studies have indicated that, together with its binding partner AnnexinA2, it is also involved in angiogenesis [62, 63], making it a possible cancer drug target. Reddy, et al. used the Tripos UNITY module, to map a pharmacophore query onto the crystal structure of the PPI complex [64]. S100A10 was considered the receptor as AnnexinA2 inter-acts with a cleft-like area of S100A10 via a single -helix at its N-terminal end. Pharmacophore features were positioned over manually selected and highly buried amino acids in the PPI interface. The model consisted of 2 hydrophobic, 2 hy-drogen bond donors and 5 hydrogen bond acceptor features and atoms from S100A10 were considered as forbidden vol-ume features (Fig. 3A). A query was produced such that both hydrophobic features and at least 1 (up to a maximum of 5) hydrogen-bonding features were required. This was then used to screen a commercial library of around 700,000 mole-cules, delivering 568 hit compounds, which were subse-quently docked into the S100A10 protein using the GOLD algorithm. The final selection of 190 hits was based on the combination of a good GOLD score and a good simulated binding mode according to the pharmacophore query. From the 190 ordered compounds, 7 compounds showed inhibitory activity in vitro with a maximal IC50 potency of 24 M.
3.2. Automation
Although manual pharmacophore model design can be started without waiting for new computational tools to be written, it depends entirely on the skill of the designer and is extremely labor intensive. In contrast, automated methods build a pharmacophore query using a computational algo-
rithm that gives more consistent results and facilitates a more rational model design process. One such tool is LigandScout [65], which detects interactions and uses them to determine the placement of pharmacophore features. Our literature search revealed 2 different examples where an automated approach was used to map a pharmacophore query on the PPI interface: the first describes the discovery of an SMPPII targeting the HIV-1 Integrase:LEDGF/p75 interaction (Fig. 3B, top), and the second targets the Bcr-Abl:14-3-3 interac-tion (Fig. 3C).
3.2.1. HIV-1 Integrase:LEDGF/p75
The replication of the Human Immunodeficiency Virus (HIV) is a complex cycle of several essential consecutive steps. One of these is the integration of viral cDNA into the human genome, which is mediated by the viral integrase (IN). To carry out its role in HIV replication, IN participates in a number of interactions of interest as drug targets [66], including binding to human LEDGF/p75 [67, 68]. In 2005, Cherepanov, et al. published the first crystal structure of the interaction of the IBD domain of LEDGF/p75 with a dimer of HIV-1 IN Catalytic Core Domain (CCD) revealing a drug-like cleft in the interface, and providing a platform for structure based drug design for this interaction (PDB 2B4J) [69].
For the discovery of the first SMPPII targeting this inter-action, De Luca, et al manually selected residues Ile365 and Asp366 from LEDGF/p75, and then used LigandScout to automatically map their pharmacophore features (Fig. 3B, top) [70]. The model consists of 1 hydrophobic interaction, 2 hydrogen bond acceptor interactions, and a hydrogen bond donor interaction. Additionally, 9 excluded volumes repre-senting the protein pocket wall were added by LigandScout. The query was used against the in-house CHIME database and the selected molecules are evaluated using the Alpha-screen assay, resulting in the discovery of CHIBA-3002 that has a reported activity of 46% inhibition at 100 μM concen-tration. These inhibitors were later optimized to reach a maximum inhibitory potency (IC50) of 35 μM [52]. It should however be noted that this class of molecules is also reported to be inhibitors of the enzymatic strand transfer step, obscur-ing the evaluation of the inhibition of viral replication in vivo.

6 Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 Voet et al.
3.2.2. Bcr-Abl:14-3-3
LigandScout is a valuable tool for pharmacophore map-ping, however, it was originally developed to create pharma-cophore models for small drug-like molecules. When used on PPIs, LigandScout returns pharmacophore models that are far too complex to use as queries. De Luca, et al. resolved this problem by manually selecting the residues to be mapped, however, this method can really only be considered semi-automated, as it still relies on considerable insight from the operator to identify the residues most important for the interaction. Molecular Dynamics (MD) simulations calculate the dynamic behavior of molecules on the atomistic scale, detecting the most frequent interactions and delivering in-sights into the relative importance of these within the PPI. Combining the use of LigandScout with MD provides a way to trim the number of pharmacophore features returned with-out relying on operator knowledge, resulting in a fully auto-mated method. However, MD is highly computationally in-tensive.
The usefulness of combining LigandScout with MD was demonstrated by Corradi, et al. in their report on the discov-ery of the first drug-like inhibitors of the Bcr-Abl:14-3-3 interaction [71]. 14-3-3 proteins are important for the sub-cellular redistribution of proteins in chronic myelogenous leukemia. LigandScout was used to produce a pharma-cophore model from the structure of 14-3-3 with its inter-acting motif, the MARSH-phosphoS-YPA nonapeptide (PDB 1YWT) [65, 72]. Due to the large number of intermo-lecular interactions, the resulting pharmacophore model con-tained too many features and was too complex for VS. A molecular dynamics (MD) simulation of the complex was employed and the most important interactions identified [73]. Less significant interactions were removed from the pharmacophore model in order to generate a query consisting of only 5 features (Fig. 3C). The query was used to screen a collection of 200,000 molecules resulting in 99 that were further filtered for drug likeness according to the Lipinski’s rule of 5 [74]. The 87 remaining compounds were docked into the 14-3-3 receptor, and the best conformations were compared to the pharmacophore model a second time in or-der to evaluate their similarity after docking and parsed ac-cordingly. Eventually, 24 molecules were biologically evalu-ated, leading to the identification of BV02. Experiments in-dicated that BV02 has a dose-dependent impact on the re-productive integrity of both wild-type Ba/F3 cells and mu-tants that have previously developed resistance to existing cancer drugs, with LD50 activities of 1.04 μM and 1.47 μM respectively. Further evaluation of BV02 indicated that the cytotoxic effect originated from inducing apoptosis, validat-ing the proposed mechanism of action of the inhibitors for the Bcr-Abl:14-3-3 interaction [75].
3.3. Consensus-Based Pharmacophore Models
All of the examples covered so far base their pharma-cophore model on a single interacting pair of molecules. However, overlaying multiple structures reveals consensus interactions at the receptor interface and thus provides an alternative to MD for identifying which interactions are the most important. Our literature search again revealed 2 differ-ent cases where consensus-based pharmacophores led to the
discovery of SMPPII. The first targets the HIV-1 Inte-grase:LEDGF/p75 interaction (Fig. 3B, bottom), while the second targets the PUMA interactions.
3.3.1. HIV-1 Integrase:LEDGF/p75
The LEDGINS are a group of SMPPIIs discovered by Christ, et al. in 2010 using consensus-based pharmacophore modelling [76]. Like CHIBA-3002, discovered by De Luca, et al. (section 3.2.1), LEDGINS inhibit the HIV-1 IN:LEDGF/p75 complex. The consensus model was created from a superposition of 4 different crystal structures: the HIV-1 IN:LEDGF/p75 complex (PDB 2B4J) )[69]; 2 struc-tures of HIV-1 IN CCD dimers with crystal packing contacts in the IBD binding cleft (PDBs 1BI4 and 1BL3) [77] and a structure of the CCD dimer with bound tetraphenylarsonium and a chlorine ion (PDB 1HYV) [78]. The model consisted of 6 features and a forbidden volume representing IN (Fig. 3B, bottom). This technique provides a way to distill out the most important molecular interactions without restricting the area under consideration, a fact that is evident in contrast to the model generated by De Luca, et al. to target the same interaction (see section 3.2.1), which does not include inter-actions outside the main binding cleft (Fig. 3B). The phar-macophore model, in combination with a chemo-informatics SMPPII-like filter was used as the query to search a library of 200,000 molecules. Hits were docked into the 1HYV re-ceptor structure and then filtered again using similarity to the pharmacophore model. The top scoring and most divergent 25 molecules were selected for biological evaluation using the Alpha-screen assay and one of the compounds exhibited 36% inhibitory activity at 100 μM. After testing the deriva-tives of the first hit compounds, a superior molecule was identified that exhibited in vitro as well as in vivo activity (IC50 = 12.2 μM). Further optimization led to greater im-provements, the best of which (CX04328) delivers low mi-cromolar activity (IC50 = 1.37 μM in vitro according to the Alpha-screen method and IC50 = 2.35 μM and CC50 = 58.9 μM according to the MTT/MT-4 assay viral replication as-say) [79]. Two X-ray structures with soaked inhibitors have been obtained (PDBs 3LPU and 3LPT). Overlapping these structures with the crystal structure of LEDGF/p75 IBD and HIV-1 IN CCD (PDB 2B4J) also indicates effective PPI mimicry by the SMPPII and validates the pharmacophore query employed for the discovery of these compounds (Fig. 2F).
This class of compounds — the LEDGINS (LEDGF/p75 Integrase inhibitors) — is the first class of inhibitors target-ing the HIV-1 IN:LEDGF/p75 with demonstrated viral repli-cation inhibitory activity. Following further validation and a cross-resistance profile against known drug-resistant HIV strains, it was shown that full activity was retained for all strains tested, including integrase strand-transfer inhibitor resistant strains. This new class of inhibitors have demon-strated the value of the HIV-1 IN:LEDGF/p75 as a promis-ing new target for antiretroviral therapy, and are themselves promising as new antiretroviral agents. To this end, this class of compounds has been licensed to Pfizer for further devel-opment [80].
In the above example, HIV-1 IN was the targeted recep-tor, but the same research group has also examined the in-verse case, using LEDGF/p75 as the receptor and designing

Protein Interface Pharmacophore Mapping Tools Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 7
Fig. (3). Pharmacophore mapping on the PPI interface allows for the discovery of novel SMPPIIs. In each case, the left image is the PPI complex with the receptor protein shown as a white cartoon and the ligand protein in yellow. The box shows an enlargement of the binding site, with the ligand now rendered as yellow sticks and the mapped pharmacophore model overlaid. A) AnnexinA2:S100A10. B) Inte-grase:LEDGF/p75 according to De Luca, et al. (top query) and Christ, et al.. (bottom query). C) Bcr-Abl:14-3-3. D) Lysozyme:PliC. SMPPIIs based around features on HIV-1 IN. These inhibi-tors were designed de novo, using a pharmacophore-guided scaffold replacement methodology, where the key interacting amino acids of HIV-1 IN were transferred onto a small molecule scaffold with an IC50 of 30 μM [47]. Although this is an example of rational design rather than pharmacophore-model VS, this is the first time that a PPI has proven to be disruptable by SMPPIIs targeting both protein partners.
These inhibitors also inhibit the interaction of LEDGF/p75 with its natural binding partner JPO2 (IC50 = 37 μM). This case where both proteins of the same PPI can be targeted by SMPPII raises the future possibility of “tool kits” of drugs where the decision over which partner in the interface should be targeted depends on the other interactions the proteins participate in.

8 Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 Voet et al.
3.3.2. PUMA
Mustata, et al. have also applied consensus-based phar-macophore mapping to discover a putative radio-protectant that targets PUMA (p53 upregulated modulator of apoptosis) [81]. PUMA is a key mediator of radiation-induced apopto-sis in the gastrointestinal and hematopoietic system. Upon radiation, PUMA is expressed and binds any of the 5 known anti-apoptotic Bcl-2 family members, releasing bak or bax (both pro-apoptotic factors) and inducing apoptosis [82-84]. SMPPIIs that target PUMA should prevent the release of bak/bax from the anti-apoptotic Bcl-2 proteins and suppress radiation-induced apoptosis in healthy cells.
In order to create the pharmacophore model, the struc-tures of PUMA in complex with A-1 protein (PDB 2VOF) [85] and MC1-1 protein (PDB 2ROC) [86] were aligned. Then, 4 features (2 hydrophobic, an anionic and a cationic) were mapped onto the consensus interaction in the aligned PPI interfaces. Using this pharmacophore, the ZINC data-base, a centralized collection of all organic/drug-like com-mercially available small molecules, was screened [87]. Af-ter a chemoinformatics-based drug-likeness evaluation, 13 molecules were acquired for testing. In an assay for PUMA induced apoptosis, 10 out of the 13 compounds showed in-hibitory activity. The structures of these molecules have not been published, but the most potent hit molecule was found to significantly suppress radiosensitivity of the cells upon irradiation.
3.4. Molecular Interaction Field
The consensus PPI pharmacophore mapping method is effective for maximizing efficacy where multiple structures exist, however, for the majority of PPIs this is not the case. Furthermore, there are many interesting examples where we only have the structure of one of the proteins involved. In order to continue with SMPPII design, methods that are able to extract the important features from only one side of an interaction are necessary. Molecular Interaction Field (MIF) based strategies such as GRID [88] or DrugScore [89, 90] are able to quantify and visualize “hotspots” — features of particular binding importance — from only one side of the interface. Using MIF methods, the non-bonded interaction energy (GRID) or the interaction score (DrugScore) for a certain chemical probe is mapped onto a grid, which covers the area of interest. Certain points on this grid have a better score (local optima) and these putative interaction hotspots are highlighted on the returned structure in the form of a contour map. These areas can thus be considered centers on which to manually map a pharmacophore feature with the corresponding (inverse) interaction property. MIF-based methods only require one side of the interface and are very fast, however the larger the interface the greater the number of identified hotspots and the greater the requirement for manual picking of features. Once again 2 different successful SMPPII discovery cases were identified in the literature. The first one targets the HIV-1-RT heterodimer PPI, while the second case describes inhibitors of the lysozyme:PLIC inter-action.
3.4.1. HIV-1-RT Heterodimer
Grohmann, et al. employed GRID as the MIF generating algorithm for the discovery of an SMPPII targeting the HIV-RT heterodimer (consisting of p55 and p66) [91]. Structural analysis of the p66 subunit (extracted from PDB 1RTH) re-vealed a drug-like cleft. MD simulations were performed to generate an ensemble of 6 different possible site conforma-tions. For each one of these conformations, an MIF analysis for hydrophobic interactions, hydrogen bond donor interac-tions and hydrogen bond acceptor interactions was per-formed on the target site. The coordinates agreeing with the minima of the MIF were replaced by pharmacophore fea-tures representing their respective type of interaction. Six pharmacophore models were derived, and later reduced to yield 3 unique models. These were used to query the AsinexGold library, resulting in 114 compounds that fitted any one of the 3 models and conformed to the Lipinski rule of 5. The ligands were subsequently docked into a receptor conformation corresponding to the pharmacophore model they were targeting. In the end 10 ligands were commercially acquired and evaluated in an RT redimerization assay. One compound was able to inhibit RT dimer formation (IC50 = 150 μM) as well as the RT polymerase (IC50 = 155 μM) and RNaseH activity (IC50 = 111 μM) in vitro [92].
3.4.2. Lysozyme:Plic
MIF-based methods allow for the creation of pharma-cophore models using only the receptor structure informa-tion, but, in combination with homology modelling, they can also be useful where structural information is completely absent. Lysozymes (EC3.2.1.17) are enzymes from the in-nate immune system that are present in most mammals, in-cluding humans. The defensive role of lysozyme against pathogenic bacteria is generally accepted and supported by lysozymal knock-out and over-expression experiments [93-95]. Nevertheless bacteria can successfully survive lysozyme rich environments via the production of proteinaceous lysozyme inhibitors (PliCs) (for a recent review see Calle-waert, et al.) [96]. The disruption of the interaction between human lysozyme and bacterial PliC may be a useful sensitiz-ing agent to boost the host’s ability to resist infection by an-tibiotic-resistant bacterial strains. Many bacteria produce lysozyme inhibitory proteins [96], however, PliC sequences vary from species to species and we lack structures with which to design inhibitors for the vast majority. In order to solve this problem, Voet, et al. produced a pharmacophore model for S. typhimurium PliC inhibitory protein (for which no structural information exists) by first creating a homology model based on the P. aeruginosa MliC lysozyme complex (3F6Z) [82][97, 98]. The majority of the interactions in the interface were conserved, delivering a highly accurate model suitable for further pharmacophore analysis. Inspection re-vealed a binding cleft in the Salmonella PliC, posited to be the receptor for a protruding lysozyme loop. On this basis, a pharmacophore model taking into account prominent fea-tures of both sides of the interface was created. Hydrophobic features picked up by the MIF analysis of the cleft were combined with interactions of the protruding loop to produce a composite feature map (Fig. 3D). The model was used, following the usual protocols, to search a library for potential active compounds, followed by the standard molecular dock-

Protein Interface Pharmacophore Mapping Tools Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 9
ing and second-pass pharmacophore filtering. The resulting 28 molecules were tested in vitro resulting in 4 hit com-pounds that are suitable for further optimization.
3.5. Other Computational Methods for Future Pharma-cophore Mapping
With the rise in interest in targeting PPIs for drug discov-ery, new structural analysis tools have been developed. While their application for the discovery of novel SMPPII is not yet demonstrated, they have been validated by retrospec-tively analyzing the structural information of existing SMPPIIs and are of interest for the future.
3.5.1. Free Energy of Binding Decomposition
MM/GBSA (Molecular Mechanics/Generalized Born Surface Area) is a “free energy of binding” calculation method, usually computed with the AMBER force field package, that uses the non-bonded interaction terms of a Mo-lecular Mechanics (MM) force field and the Generalized Born (GB) electrostatics model combined with the molecular Surface Area (SA) to estimate the energy of solvation [99, 100]. To account for the dynamic nature of molecules, mul-tiple structures are tested, and the given energy is the result of averaging. Typically this method is combined with MD simulations to create an ensemble of different receptor con-formations, although other conformational sampling methods such as the Monte Carlo method can also be used. The MM/GBSA method facilitates decomposition of the ener-getic contributions at the amino acid level in order to give insights into the importance/energetics of the interactions in the PPI complex. Thus, amino acids that significantly con-tribute to the binding energy of the PPI interface can be con-sidered hotspots for mediating interface recognition and binding [101-104]. These amino acids could serve as the anchor residues on which to map the pharmacophore fea-tures. The usefulness of this kind of simulation was demon-strated by Zhao, et al. and Tintori, et al., who could success-fully identified the key interactions mimicked by the previ-ously mentioned LEDGINS [103, 104]. Since MM/GBSA uses MD as the sampling method it is slow and highly com-putationally intensive, limiting its use as a first choice for pharmacophore mapping.
3.5.2. Solvent Wrapping Analysis
Another new and promising technique for designing SMPPIIs is analysis of solvent wrapping. In a recent paper, Accordino, et al. analyzed the structural information of sev-eral SMPPIIs, and demonstrated the importance of intermo-lecular wrapping at PPI interface hotspots, using an in-house developed tool. Molecular wrapping is the way in which the nonpolar groups of a particular molecule can promote the dehydration of hydrophobic groups on a partner, facilitating binding [105]. Their results show that the successful SMPPIIs closely mimic the wrapping behavior of the protein they replace. Solvent-wrapping analysis of PPIs may also provide a way to identify the amino acids most important for binding.
3.5.3. Small-Molecule Inhibitor Starting Points
Finally, “Small-Molecule Inhibitor Starting Points” (SMISPs)” is a Support Vector Machine (SVM) based
method proposed by Koes, et al. [106]. SMISPs is a compu-tational artificial intelligence method able to identify clusters of PPI interface residues from a 3D PPI complex structure, in order to provide a starting point for inhibitor design. The SVM was trained using the available structural SMPPII in-formation, and therefore learned the optimal receptor residue interaction properties that enable good mimicry. Using their SVM method, they scanned the PDB database and their re-sults suggest that half of all PPIs present may possess these features and be susceptible to small-molecule inhibition, giving hope for the SMPPII drug discovery field. Recently, they reported an online interface for their method called PocketQuery (http://pocketquery.csb.pitt.edu) [107]. This web interface of their SVM method exports the residue clus-ters that are the “chemical starting points”. Like solvent wrapping analysis, SMISPs was developed too recently to have been reported as a successful tool for the identification of novel SMPPIIs. It is promising for the future; however, as with many artificial intelligence-based methods, it is impor-tant to bear in mind that black-box methods deliver results based on rules that cannot be interpreted by the operator.
CONCLUSION
Despite the fact that the research field of pharmacophore-based VS is relatively young, there have been enough suc-cesses to indicate that these approaches are likely to be valu-able in the search for novel SMPPIIs. Not only are they be-ing reported in increasing numbers, but chemo-informatical analysis of the reported SMPPIIs provides further informa-tion that can be fed back to create VS filters that, in conjunc-tion with pharmacophores, improve the design process. There are now many available tools for PPI mapping and pharmacophore modelling. As the different cases reviewed in this paper indicate, this variety of tools has been used with success, and supply researchers with an excellent choice of strategies to apply, depending on the circumstances under which they are working. While this choice is valuable, it is important to thoroughly analyze the available structural in-formation in order to use the best method or methods with which to generate the most useful consensus model. Prelimi-nary analysis of the putative receptor sites is likely to be im-proved by checking whether the PPI interface area is rigid, whether induced fit regions can be avoided, and whether there is a site that can accommodate a drug-like molecule. When examining the structural information for the previ-ously successful SMPPII discovery cases, it is apparent that the most druggable PPI interfaces are much like enzymatic interactions and amenable to the techniques used in classical inhibitor design.
Pharmacophores derived from PPI interfaces should not be considered the Holy Grail for the discovery of SMPPIIs. They can only be applied if a 3D structure of the PPI com-plex is available and there is, as yet, no method for designing inhibitors capable of acting through more complex mecha-nisms such as induced fit or allosterically. However, the re-ported successes indicate that pharmacophore modelling and VS are valuable tools for the discovery of novel inhibitors of PPIs, giving hope that PPIs may become more accessible pharmaceutical targets in the future and that many of their SMPPIIs can be rationally identified using CADD strategies.

10 Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 Voet et al.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no con-flicts of interest.
ACKNOWLEDGEMENT
Declared none.
LIST OF ABBREVIATIONS
3D = 3-Dimensional
Abl = Abelson murine leukemia viral oncogene homolog
Bcl2 = B-cell lymphoma protein 2
BCR = Breakpoint Cluster Region
CADD = Computer-Aided Drug Design
CCD = Catalytic Core Domain (of HIV-1 inte-grase)
CML = Chronic Myelogenous Leukemia
FtsZ = Filamenting temperature-sensitive mutant Z
HIF = Hypoxia Inducible Factor
HIV = Human Immunodeficiency Virus
HDM2 = Human double minute 2
HPV E1 = Human Papilloma Virus Early protein 1
HPV E2 = Human Papilloma Virus Early protein 2
IBD = Integrase Binding Domain
ICAM = Intercellular Adhesion Molecule
IL2 = Interleukin 2
IL2R = Interleukin 2 Receptor
IN = Integrase
iNOS = inducible Nitric Oxyde Synthase
LFA = Leukocyte Function Associated Antigen
MD = Molecular Dynamics
MIF = Molecular interaction Fields
MOE = Molecular Operating Environment
NMR = Nuclear Magnetic Resonance
PDB = Protein Data Bank entry code
PliC/MliC = Periplasmatic/Membrane-bound lysozyme inhibitor of c-type lysozyme
PPI = Protein:Protein Interaction
PUMA = p53 Upregulated Modulator of Apoptosis
RMSD = Root Mean Square Deviation
RT = Reverse Transcriptase
SAR = Structure Activity Relationship
SMISP = Small Molecule Inhibitor Starting Point
SMPPII = Small Molecule Protein:Protein Interaction Inhibitor
SVM = Support Vector Machine
TNF = Tumor Necrosis Factor
VHL = Von Hippel Lindau protein
VS = Virtual Screening
XIAP = X-linked Inhibitor of Apoptosis
ZipA = Z-interacting protein A
REFERENCES
[1] Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh KI, Yildirim MA, Simonis N, Heinzmann K, Gebreab F, Sahalie JM, Cevik S, Simon C, de Smet AS, Dann E, Smolyar A, Vinayagam A, Yu H, Szeto D, Borick H, Dricot A, Klitgord N, Murray RR, Lin C, Lalowski M, Timm J, Rau K, Boone C, Braun P, Cusick ME, Roth FP, Hill DE, Tavernier J, Wanker EE, Barabasi AL, Vidal M. An empirical framework for binary interactome mapping. Nat Methods, 2009; 6: 83-90.
[2] Stumpf MP, Thorne T, de Silva E, Stewart R, An HJ, Lappe M, Wiuf C. Estimating the size of the human interactome. Proc Natl Acad Sci U S A, 2008; 105: 6959-64.
[3] Sperandio O. Editorial: Toward the design of drugs on protein-protein interactions. Curr Pharm Des, 2012; 18: 4585.
[4] Zhou P, Huang J, Tian F. Specific noncovalent interactions at protein-ligand interface: implications for rational drug design. Curr Med Chem, 2012; 19: 226-38.
[5] Yin H, Hamilton AD. Strategies for targeting protein-protein interactions with synthetic agents. Angew Chem Int Ed Engl, 2005; 44: 4130-63.
[6] Marshall G. Three-dimensional structure of peptide—protein complexes: implications for recognition. Current Opinion in Structural Biology, 1992; 2: 904-919
[7] Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol, 1999; 285: 2177-98.
[8] Verdine GL, Walensky LD. The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin Cancer Res, 2007; 13: 7264-70.
[9] Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature, 2007; 450: 1001-9.
[10] Berg T. Small-molecule inhibitors of protein-protein interactions. Curr Opin Drug Discov Devel, 2008; 11: 666-74.
[11] Rechfeld F, Gruber P, Hofmann J, Kirchmair J. Modulators of protein-protein interactions: novel approaches in targeting protein kinases and other pharmaceutically relevant biomolecules. Curr Top Med Chem, 2011; 11: 1305-19.
[12] Meireles LM, Mustata G. Discovery of Modulators of Protein-Protein Interactions: Current Approaches and Limitations. Curr Top Med Chem, 2010.
[13] Pagliaro L, Felding J, Audouze K, Nielsen SJ, Terry RB, Krog-Jensen C, Butcher S. Emerging classes of protein-protein interaction inhibitors and new tools for their development. Curr Opin Chem Biol, 2004; 8: 442-9.
[14] Wilson AJ. Inhibition of protein-protein interactions using designed molecules. Chem Soc Rev, 2009; 38: 3289-300.
[15] Morelli X, Bourgeas R, Roche P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2P2I). Curr Opin Chem Biol, 2011; 15: 475-81.
[16] Fletcher S, Hamilton AD. Protein-protein interaction inhibitors: small molecules from screening techniques. Curr Top Med Chem, 2007; 7: 922-7.
[17] Hajduk PJ. SAR by NMR: putting the pieces together. Mol Interv, 2006; 6: 266-72.
[18] Kumar A, Voet A, Zhang KY. Fragment based drug design: from experimental to computational approaches. Curr Med Chem, 2012.
[19] Zhang S. Computer-aided drug discovery and development. Methods Mol Biol, 2011; 716: 23-38.
[20] Bottegoni G. Protein-ligand docking. Front Biosci; 17: 2289-306.

Protein Interface Pharmacophore Mapping Tools Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 11
[21] Ha S, Andreani R, Robbins A, Muegge I. Evaluation of docking/scoring approaches: a comparative study based on MMP3 inhibitors. J Comput Aided Mol Des, 2000; 14: 435-48.
[22] Kellenberger E, Foata N, Rognan D. Ranking targets in structure-based virtual screening of three-dimensional protein libraries: methods and problems. J Chem Inf Model, 2008; 48: 1014-25.
[23] Reynes C, Host H, Camproux AC, Laconde G, Leroux F, Mazars A, Deprez B, Fahraeus R, Villoutreix BO, Sperandio O. Designing focused chemical libraries enriched in protein-protein interaction inhibitors using machine-learning methods. PLoS Comput Biol; 6: e1000695.
[24] Sperandio O, Reynes CH, Camproux AC, Villoutreix BO. Rationalizing the chemical space of protein-protein interaction inhibitors. Drug Discov Today; 15: 220-9.
[25] Neugebauer A, Hartmann RW, Klein CD. Prediction of protein-protein interaction inhibitors by chemoinformatics and machine learning methods. J Med Chem, 2007; 50: 4665-8.
[26] Raimund Mannhold HK, Gerd Folkers. Pharmacophores and Pharmacophore Searches. John Wiley & Sons 2006.
[27] Gao Q, Yang L, Zhu Y. Pharmacophore based drug design approach as a practical process in drug discovery. Curr Comput Aided Drug Des, 2010; 6: 37-49.
[28] Tan L, Batista J, Bajorath J. Computational methodologies for compound database searching that utilize experimental protein-ligand interaction information. Chem Biol Drug Des, 2010; 76: 191-200.
[29] Horvath D. Pharmacophore-based virtual screening. Methods Mol Biol, 2011; 672: 261-98.
[30] Fry DC. "Small-Molecule Inhibitors of Protein-Protein Interactions: How to Mimic a Protein Partner". Curr Pharm Des, 2012.
[31] Teijido O, Dejean L. Upregulation of Bcl2 inhibits apoptosis-driven BAX insertion but favors BAX relocalization in mitochondria. FEBS Lett, 2010; 584: 3305-10.
[32] Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov, 2008; 7: 989-1000.
[33] Andersen MH, Reker S, Kvistborg P, Becker JC, thor Straten P. Spontaneous immunity against Bcl-xL in cancer patients. J Immunol, 2005; 175: 2709-14.
[34] Momand J, Wu HH, Dasgupta G. MDM2--master regulator of the p53 tumor suppressor protein. Gene, 2000; 242: 15-29.
[35] Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov, 2008; 7: 979-87.
[36] Jones SN, Hancock AR, Vogel H, Donehower LA, Bradley A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc Natl Acad Sci U S A, 1998; 95: 15608-12.
[37] Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science, 1996; 274: 948-53.
[38] Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature, 1997; 388: 300-4.
[39] Oost TK, Sun C, Armstrong RC, Al-Assaad AS, Betz SF, Deckwerth TL, Ding H, Elmore SW, Meadows RP, Olejniczak ET, Oleksijew A, Oltersdorf T, Rosenberg SH, Shoemaker AR, Tomaselli KJ, Zou H, Fesik SW. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem, 2004; 47: 4417-26.
[40] Haney SA, Glasfeld E, Hale C, Keeney D, He Z, de Boer P. Genetic analysis of the Escherichia coli FtsZ.ZipA interaction in the yeast two-hybrid system. Characterization of FtsZ residues essential for the interactions with ZipA and with FtsA. J Biol Chem, 2001; 276: 11980-7.
[41] Fry DC. Drug-like inhibitors of protein-protein interactions: a structural examination of effective protein mimicry. Curr Protein Pept Sci, 2008; 9: 240-7.
[42] Jennings LD, Foreman KW, Rush TS, 3rd, Tsao DH, Mosyak L, Kincaid SL, Sukhdeo MN, Sutherland AG, Ding W, Kenny CH, Sabus CL, Liu H, Dushin EG, Moghazeh SL, Labthavikul P, Petersen PJ, Tuckman M, Haney SA, Ruzin AV. Combinatorial synthesis of substituted 3-(2-indolyl)piperidines and 2-phenyl indoles as inhibitors of ZipA-FtsZ interaction. Bioorg Med Chem, 2004; 12: 5115-31.
[43] Jennings LD, Foreman KW, Rush TS, 3rd, Tsao DH, Mosyak L, Li Y, Sukhdeo MN, Ding W, Dushin EG, Kenny CH, Moghazeh SL, Petersen PJ, Ruzin AV, Tuckman M, Sutherland AG. Design and synthesis of indolo[2,3-a]quinolizin-7-one inhibitors of the ZipA-FtsZ interaction. Bioorg Med Chem Lett, 2004; 14: 1427-31.
[44] Tsao DH, Sutherland AG, Jennings LD, Li Y, Rush TS, 3rd, Alvarez JC, Ding W, Dushin EG, Dushin RG, Haney SA, Kenny CH, Malakian AK, Nilakantan R, Mosyak L. Discovery of novel inhibitors of the ZipA/FtsZ complex by NMR fragment screening coupled with structure-based design. Bioorg Med Chem, 2006; 14: 7953-61.
[45] Hartman GD, Egbertson MS, Halczenko W, Laswell WL, Duggan ME, Smith RL, Naylor AM, Manno PD, Lynch RJ, Zhang G, et al. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J Med Chem, 1992; 35: 4640-2.
[46] Springer TA, Zhu J, Xiao T. Structural basis for distinctive recognition of fibrinogen gammaC peptide by the platelet integrin alphaIIbbeta3. J Cell Biol, 2008; 182: 791-800.
[47] Cavalluzzo C, Voet A, Christ F, Singh BK, Sharma A, Debyser Z, Maeyer MD, Eycken EVd. De novo design of small molecule inhibitors targeting the LEDGF/p75-HIV integrase interaction. RSC Advances, 2012; 2: 974.
[48] Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol, 2010; 6: 442-8.
[49] De Luca L, Ferro S, Morreale F, Christ F, Debyser Z, Chimirri A, Gitto R. Fragment hopping approach directed at design of HIV IN-LEDGF/p75 interaction inhibitors. J Enzyme Inhib Med Chem, 2012.
[50] De Luca L, Ferro S, Morreale F, Chimirri A. Inhibition of the interaction between HIV-1 integrase and its cofactor LEDGF/p75: a promising approach in anti-retroviral therapy. Mini Rev Med Chem, 2011; 11: 714-27.
[51] De Luca L, Ferro S, Morreale F, De Grazia S, Chimirri A. Inhibitors of the interactions between HIV-1 IN and the cofactor LEDGF/p75. ChemMedChem, 2011; 6: 1184-91.
[52] De Luca L, Ferro S, Gitto R, Barreca ML, Agnello S, Christ F, Debyser Z, Chimirri A. Small molecules targeting the interaction between HIV-1 integrase and LEDGF/p75 cofactor. Bioorg Med Chem, 2010; 18: 7515-21.
[53] Garber K. Missing the target: ubiquitin ligase drugs stall. J Natl Cancer Inst, 2005; 97: 166-7.
[54] Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, Jorgensen WL, Ciulli A, Crews CM. Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1alpha. Angew Chem Int Ed Engl, 2012.
[55] Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc, 2012; 134: 4465-8.
[56] Grigorian A, Mkhikian H, Demetriou M. Interleukin-2, Interleukin-7, T cell-mediated autoimmunity, and N-glycosylation. Ann N Y Acad Sci, 2012; 1253: 49-57.
[57] Wilson CG, Arkin MR. Small-molecule inhibitors of IL-2/IL-2R: lessons learned and applied. Curr Top Microbiol Immunol, 2010; 348: 25-59.
[58] Braisted AC, Oslob JD, Delano WL, Hyde J, McDowell RS, Waal N, Yu C, Arkin MR, Raimundo BC. Discovery of a potent small molecule IL-2 inhibitor through fragment assembly. J Am Chem Soc, 2003; 125: 3714-5.
[59] Voet A, Zhang KY. Pharmacophore Modelling as a Virtual Screening Tool for the Discovery of Small Molecule Protein-protein Interaction Inhibitors. Curr Pharm Des, 2012; 18: 4586-98.
[60] He MM, Smith AS, Oslob JD, Flanagan WM, Braisted AC, Whitty A, Cancilla MT, Wang J, Lugovskoy AA, Yoburn JC, Fung AD, Farrington G, Eldredge JK, Day ES, Cruz LA, Cachero TG, Miller SK, Friedman JE, Choong IC, Cunningham BC. Small-molecule inhibition of TNF-alpha. Science, 2005; 310: 1022-5.
[61] Goudreau N, Cameron DR, Deziel R, Hache B, Jakalian A, Malenfant E, Naud J, Ogilvie WW, O'Meara J, White PW, Yoakim C. Optimization and determination of the absolute configuration of a series of potent inhibitors of human papillomavirus type-11 E1-E2 protein-protein interaction: a combined medicinal chemistry,

12 Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 Voet et al.
NMR and computational chemistry approach. Bioorg Med Chem, 2007; 15: 2690-700.
[62] Ling Q, Jacovina AT, Deora A, Febbraio M, Simantov R, Silverstein RL, Hempstead B, Mark WH, Hajjar KA. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J Clin Invest, 2004; 113: 38-48.
[63] Surette AP, Madureira PA, Phipps KD, Miller VA, Svenningsson P, Waisman DM. Regulation of fibrinolysis by S100A10 in vivo. Blood, 2011; 118: 3172-81.
[64] Reddy TR, Li C, Fischer PM, Dekker LV. Three-dimensional pharmacophore design and biochemical screening identifies substituted 1,2,4-triazoles as inhibitors of the annexin A2-S100A10 protein interaction. ChemMedChem, 2012; 7: 1435-46.
[65] Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model, 2005; 45: 160-9.
[66] Voet AR, Maeyer MD, Debyser Z, Christ F. In search of second-generation HIV integrase inhibitors: targeting integration beyond strand transfer. Future Med Chem, 2009; 1: 1259-74.
[67] Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J Biol Chem, 2003; 278: 372-81.
[68] De Rijck J, Vandekerckhove L, Gijsbers R, Hombrouck A, Hendrix J, Vercammen J, Engelborghs Y, Christ F, Debyser Z. Overexpression of the lens epithelium-derived growth factor/p75 integrase binding domain inhibits human immunodeficiency virus replication. J Virol, 2006; 80: 11498-509.
[69] Cherepanov P, Ambrosio AL, Rahman S, Ellenberger T, Engelman A. Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc Natl Acad Sci U S A, 2005; 102: 17308-13.
[70] De Luca L, Barreca ML, Ferro S, Christ F, Iraci N, Gitto R, Monforte AM, Debyser Z, Chimirri A. Pharmacophore-based discovery of small-molecule inhibitors of protein-protein interactions between HIV-1 integrase and cellular cofactor LEDGF/p75. ChemMedChem, 2009; 4: 1311-6.
[71] Corradi V, Mancini M, Manetti F, Petta S, Santucci MA, Botta M. Identification of the first non-peptidic small molecule inhibitor of the c-Abl/14-3-3 protein-protein interactions able to drive sensitive and Imatinib-resistant leukemia cells to apoptosis. Bioorg Med Chem Lett, 2010; 20: 6133-7.
[72] Wilker EW, Grant RA, Artim SC, Yaffe MB. A structural basis for 14-3-3sigma functional specificity. J Biol Chem, 2005; 280: 18891-8.
[73] Cui Y. Using molecular simulations to probe pharmaceutical materials. J Pharm Sci, 2011; 100: 2000-19.
[74] Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev, 2001; 46: 3-26.
[75] Mancini M, Corradi V, Petta S, Barbieri E, Manetti F, Botta M, Santucci MA. A new nonpeptidic inhibitor of 14-3-3 induces apoptotic cell death in chronic myeloid leukemia sensitive or resistant to imatinib. J Pharmacol Exp Ther, 2010; 336: 596-604.
[76] Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol, 2010; 6: 442-448.
[77] Maignan S, Guilloteau JP, Zhou-Liu Q, Clement-Mella C, Mikol V. Crystal structures of the catalytic domain of HIV-1 integrase free and complexed with its metal cofactor: high level of similarity of the active site with other viral integrases. J Mol Biol, 1998; 282: 359-68.
[78] Molteni V, Greenwald J, Rhodes D, Hwang Y, Kwiatkowski W, Bushman FD, Siegel JS, Choe S. Identification of a small-molecule binding site at the dimer interface of the HIV integrase catalytic domain. Acta Crystallogr D Biol Crystallogr, 2001; 57: 536-44.
[79] Pannecouque C, Daelemans D, De Clercq E. Tetrazolium-based colorimetric assay for the detection of HIV replication inhibitors: revisited 20 years later. Nat Protoc, 2008; 3: 427-34.
[80] K.U.Leuven. K.U.Leuven enters into license agreement with Pfizer In: ed.^eds., 2010.
[81] Mustata G, Li M, Zevola N, Bakan A, Zhang L, Epperly M, Greenberger JS, Yu J, Bahar I. Development of Small-Molecule
PUMA Inhibitors for Mitigating Radiation-Induced Cell Death. Curr Top Med Chem, 2010.
[82] Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, Look AT. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell, 2005; 123: 641-53.
[83] Qiu W, Carson-Walter EB, Liu H, Epperly M, Greenberger JS, Zambetti GP, Zhang L, Yu J. PUMA regulates intestinal progenitor cell radiosensitivity and gastrointestinal syndrome. Cell Stem Cell, 2008; 2: 576-83.
[84] Potten CS. Radiation, the ideal cytotoxic agent for studying the cell biology of tissues such as the small intestine. Radiat Res, 2004; 161: 123-36.
[85] Smits C, Czabotar PE, Hinds MG, Day CL. Structural plasticity underpins promiscuous binding of the prosurvival protein A1. Structure, 2008; 16: 818-29.
[86] Day CL, Smits C, Fan FC, Lee EF, Fairlie WD, Hinds MG. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J Mol Biol, 2008; 380: 958-71.
[87] Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model, 2005; 45: 177-82.
[88] Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem, 1985; 28: 849-57.
[89] Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol, 2000; 295: 337-56.
[90] Neudert G, Klebe G. DSX: a knowledge-based scoring function for the assessment of protein-ligand complexes. J Chem Inf Model, 2011; 51: 2731-45.
[91] Grohmann D, Corradi V, Elbasyouny M, Baude A, Horenkamp F, Laufer SD, Manetti F, Botta M, Restle T. Small molecule inhibitors targeting HIV-1 reverse transcriptase dimerization. Chembiochem, 2008; 9: 916-22.
[92] Tintori C, Corradi V, Magnani M, Manetti F, Botta M. Targets looking for drugs: a multistep computational protocol for the development of structure-based pharmacophores and their applications for hit discovery. J Chem Inf Model, 2008; 48: 2166-79.
[93] Markart P, Korfhagen TR, Weaver TE, Akinbi HT. Mouse lysozyme M is important in pulmonary host defense against Klebsiella pneumoniae infection. Am J Respir Crit Care Med, 2004; 169: 454-8.
[94] Akinbi HT, Epaud R, Bhatt H, Weaver TE. Bacterial killing is enhanced by expression of lysozyme in the lungs of transgenic mice. J Immunol, 2000; 165: 5760-6.
[95] Cole AM, Thapa DR, Gabayan V, Liao HI, Liu L, Ganz T. Decreased clearance of Pseudomonas aeruginosa from airways of mice deficient in lysozyme M. J Leukoc Biol, 2005; 78: 1081-5.
[96] Callewaert L, Van Herreweghe JM, Vanderkelen L, Leysen S, Voet A, Michiels CW. Guards of the great wall: bacterial lysozyme inhibitors. Trends Microbiol, 2012; 20: 501-10.
[97] Voet A, Callewaert L, Ulens T, Vanderkelen L, Vanherreweghe JM, Michiels CW, De Maeyer M. Structure based discovery of small molecule suppressors targeting bacterial lysozyme inhibitors. Biochem Biophys Res Commun, 2011; 405: 527-32.
[98] Yum S, Kim MJ, Xu Y, Jin XL, Yoo HY, Park JW, Gong JH, Choe KM, Lee BL, Ha NC. Structural basis for the recognition of lysozyme by MliC, a periplasmic lysozyme inhibitor in Gram-negative bacteria. Biochem Biophys Res Commun, 2009; 378: 244-8.
[99] Srinivasan J, Miller J, Kollman PA, Case DA. Continuum solvent studies of the stability of RNA hairpin loops and helices. J Biomol Struct Dyn, 1998; 16: 671-82.
[100] Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE, 3rd. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res, 2000; 33: 889-97.
[101] Venken T, Krnavek D, Munch J, Kirchhoff F, Henklein P, De Maeyer M, Voet A. An optimized MM/PBSA virtual screening approach applied to an HIV-1 gp41 fusion peptide inhibitor. Proteins, 2011; 79: 3221-35.

Protein Interface Pharmacophore Mapping Tools Current Topics in Medicinal Chemistry, 2013, Vol. 13, No. 10 13
[102] Venken T, Daelemans D, De Maeyer M, Voet A. Computational investigation of the HIV-1 Rev multimerization using molecular dynamics simulations and binding free energy calculations. Proteins, 2012; 80: 1633-46.
[103] Zhao Y, Li W, Zeng J, Liu G, Tang Y. Insights into the interactions between HIV-1 integrase and human LEDGF/p75 by molecular dynamics simulation and free energy calculation. Proteins, 2008; 72: 635-45.
[104] Tintori C, Veljkovic N, Veljkovic V, Botta M. Computational studies of the interaction between the HIV-1 integrase tetramer and the cofactor LEDGF/p75: insights from molecular dynamics simulations and the informational spectrum method. Proteins, 2010; 78: 3396-408.
[105] Accordino SR, Morini MA, Sierra MB, Fris JA, Appignanesi GA, Fernandez A. Wrapping mimicking in drug-like small molecules disruptive of protein-protein interfaces. Proteins, 2012; 80: 1755-65.
[106] Koes DR, Camacho CJ. Small-molecule inhibitor starting points learned from protein-protein interaction inhibitor structure. Bioinformatics, 2012; 28: 784-91.
[107] Koes DR, Camacho CJ. PocketQuery: protein-protein interaction inhibitor starting points from protein-protein interaction structure. Nucleic Acids Res, 2012; 40: W387-92.
[108] Chemical Computing Group Inc. SSW, Suite #910, Montreal, QC, Canada, H3A 2R7, 2011. Molecular Operating Environment (MOE), 2011.10. In: ed.^eds., 2011.
Received: ???????????????? Revised: ???????????????? Accepted: ????????????????
Copyright © 2022 FDOKUMEN




















