
Protein disulphide isomerase mediates platelet aggregation and secretion

PROTEIN AGGREGATION AND DISEASES
IntroductionEvolution has guided the design of amino acid sequences such
that globular proteins reliably assume a specific functional
native state, precisely bringing together residues to form,
for example, catalytic sites in enzymes or specific binding
site architectures for protein complexation and signaling.
The ability of the protein to find and maintain the native
state is therefore dependent on an amino acid sequence that
gives rise to a structural ensemble that is
thermodynamically stable at the physiological pressures and
temperatures and solution conditions in the normal cellular
or extracellular environment. Destabilizing sequence
mutations,1 chemical modification,2 or changes in protein
concentration and solution environment of the protein3 can
shift the equilibrium from the native state in favor of
aggregates, that is, misfolded states with interchain
contacts made with other proteins. These aggregates range
from structurally amorphous collections of misfolded
proteins often found in inclusion bodies when proteins are
overexpresssed in bacterial hosts4 to fibrils with regular
and repeating structure associated with a number of human
diseases.1 In order to change deleterious aggregation
outcomes, it is of critical importance to develop an
understanding of the molecular driving forces for early and

late aggregation events, which in turn might be reversed to
prevent disease proteins from nucleating thermodynamically
stable aggregate assemblies or to break up inclusion bodies
to recover functional protein.
Though the gross morphology of
large fibril aggregates can be investigated with current
biochemical or protein structural experimental techniques,1,5
these are more limited in application to early aggregation
events involving small and likely disordered oligomers at
very dilute concentration. Molecular simulations currently
offer great promise of directly observing the entire
aggregation process in molecular detail. In this project
report, we present the recent studies which show how
judicious use of coarse-grained models, validated against
appropriate experimental observables, can characterize the
aggregation thermodynamics and kinetic pathways at a level
of detail and insight not possible with experiment alone. We
use these models to quantify molecular mechanistic
differences in aggregation outcomes for nondisease proteins
L and G and the A - peptide indicted in Alzheimer’s
disease.
What is protein aggregation…………?Protein aggregation is a biological phenomenon in which
miss-folded protein aggregate(i.e; accumulate and clump
togather) either intra or extracellularly. These protein

aggregates are often toixic. Protein aggregates have
beenimplicated in a wide variety of diseases known
amyloidoses, including Alzheimer’s, Parkinson’ andPrion
diseases.
In this account we present the development and application
of computational models for theinvestigation of diseases
protein aggregatibn as illustrated for the proteins L and G
and the Alzheimers’s A beta systems, Parkinsons, huntingtons
and Prion disease’s
Common factors in neurodegenerative phenomena:-
Here we list …… Deposition of fibrillarproteinacious material in the
intracellular matrix.
Mitrochondrial dysfunction,increased oxidative stress
andproduction ROS(many mitrocondria,a lot of energy)
Increased apoptosis
Decreased proteasomal activity (ubiquitin is present in
all thelesions, plaques Lewy bodies, huntingtin
aggregates etc)
Decresed autophagy and lysosomal degradation of protein
Excitotoxicity

Alteration in the intrigrity of the cell membrane:
implication for alterd levels of intracellular
cholesterol
Protein misfolding, aggregation andneurodegeneration:-
Deposition of fibrillarproteinacious materialin the intracellular or extracellular matrix
Amyloid: amyloid fibrils are filamentous, hydrophobicstructures, width ~10nm, length between 0.1-10 mM. Ribbon-like -sheet motifs are formed by --strands and -turns.These kind of fibrils are common to differentneurodegenerative diseases, from Alzheimer’s to Huntington’sdisease. Conformation-specific antibodies (raised againstspecific proteins which form a specific amyloid formation)can recognize only the -sheet -polymeric conformation butnot the monomeric-soluble conformation of the proteinsubstrate. Thus, the β-sheet conformation and amyloidformation may be linked to neurodegenerative diseases.

Fig 1: beta sheet conformation of protein
Ref: ( Ross CA, Poirier MA. Nat Med. 2004 Jul: 10 Suppl:S 10-7. Review)
How proteins aggregate and form Amyloidinsoluble Fibrils…………?
Several factors may induce protein aggregation:-
That’s are………..
1-Protein oxidation (a-synuclein in PD)/ 2-Metal chelation (Prion disease
and AD) - 3-Specific protein cleavage (AD) 4-Inefficient protein degradation of b-sheet proteins/ubiquitin accumulates within the les (PD, AD, ALS, Prion Disease, HD) 5-Change in intracellular pH (AD)
Different evidences in the different neurodegenerative diseases :-AD: plaques, soluble Ab, correlate with progression of the
disease. However, Ab oligomers seem to be the more toxic
species.

PD: inclusion bodies do not follow the progression of the
disease.
HD: aggregates may be present ONLY in surviving neurons.
Fig 2 : Example for gain and loss of function mechanism
leading toneuronal dysfunction and cell death. (red-AD- blue
prion diseases.)
Representation of structural components of protein structures:
Primary Secondary Tertiary Quternary structure Structure structure structure

Fig 3 :Schematic representation of the structural components ofprotein . The order of the amino acids determines the primarystructure of the protein which can fold into secondary elementsalpha-helix, beta-sheet or random coils. The distribution ofsecondary elements ascertains tertiary structure that isstabilized by hydrogen bonds or ionic interactions. Finaly thequartenary structure describe the connection of poly peptidesubunits in multimeric proteins.
Ref: (Eszter Herczenik and Martijn F.B. G. Gebbink)
Diferent states of protein……….
Protein folding: a folding funnel to change the structure
and the energy of proteins.
Only folded, native proteins are functionally active.
Unfolded states: characterized by higher degree of
conformational entropy and free energy than native states.
This leads to “unstableness” of proteins when in the
unfolded state. As the folding funnel proceeds,
conformational entropy decreases as proteins have lower

number of conformational states, as well as the free energy
decreases. The minimum of the energy level of a protein is
reached when it’s in its native/folded state
Thermodynamics of proteinfolding/misfolding
Fig 4 : Energy state of protein folding under physiological andmisfolding condition. The shape of the graph shows energy stateof the protein conformations moving towards its native ormisfolded condition through multiple inter or intramolecularcontact arrangements.
(“The energy of the different conformations decreases withthe development of organized, native-like proteins”.)

Steps that lead to formation of aggregates:
Unfolding……
Nucleation: when proteins attach REVERSIBLY to a growing
core.
Aggregation: when proteins attach IRREVERSIBLY to the core
forming larger aggregates. Aggregation can be triggered by
hydrophobic residues in the sequence of the protein and by
b-sheet structure. Amyloid is one of the forms of protein
aggregates that occurs in nature, is very stable but its
formation can be still reversible. This is not true,
unfortunately, for most of the protein aggregates that are
responsible of neurodegenerative diseases.
Summary of protein folding diseases :

(Ref: Eszter Herczenik Martijn F. B. G. Gebbink)
Protein aggregation and neurodegenerative disease:Neurodegenerative diseases such as Alzheimer's disease (AD),
Parkinson's disease (PD), Huntington's disease (HD),
amyotrophic lateral sclerosis (ALS) and prion diseases are
increasingly being realized to have common cellular and
molecular mechanisms including protein aggregation and
inclusion body formation. The aggregates usually consist of
fibers containing misfolded protein with a -sheet
conformation, termed amyloid. There is partial but not
perfect overlap among the cells in which abnormal proteins

are deposited and the cells that degenerate. The most likely
explanation is that inclusions and other visible protein
aggregates represent an end stage of a molecular cascade of
several steps, and that earlier steps in the cascade may be
more directly tied to pathogenesis than the inclusions
themselves. For several diseases, genetic variants assist in
explaining the pathogenesis of the more common sporadic
forms and developing mouse and other models. There is now
increased understanding of the pathways involved in protein
aggregation, and some recent clues have emerged as to the
molecular mechanisms of cellular toxicity. These are leading
to approaches toward rational therapeutics.
Deposition of fibrillar proteinacious
material in Alzhimers disease:
Alzheimer's disease and tauopathies. AD is a late-onset
dementing illness, with progressive loss of memory, task
performance, speech, and recognition of people and objects.
There is degeneration of neurons (particularly in the basal
forebrain and hippocampus), but at least as important for
pathogenesis may be synaptic pathology and altered neuronal
connections AD involves two major kinds of protein
aggregates. Extracellular aggregates known as neuritic
plaques have as their major constituent the A peptide,
which is derived from proteolytic processing of the amyloid

precursor protein (APP). The A -containing aggregates have
-sheet structure and Congo red and thioflavin-T reactivity
characteristic of amyloid. There are also intracellular
aggregates of the microtubule-associated protein tau, called
neurofibrillary tangles. The pathogenesis of AD has been
greatly clarified by the identification of genetic mutations
responsible for rare familial forms of the disease. These
mutations are in APP itself and also in the
presenilins, which are involved with the cleavage of APP.
Fig 5: Plaques
Tangles Phospho-Tau
(Ref: Bossy-Wetzel E.et al. Nat Med.2004 Jul; 10 Suppl:S2-9. Review )
Deposition of fibrillar proteinacious material in Parkinsons disease: Parkinson's disease. PD is characterized by resting tremor,
rigidity, slow movements and other features such as postural
and autonomic instability. It is caused by degeneration of
dopaminergic neurons in the substantia nigra of the midbrain
and other monoaminergic neurons in the brain stem . The
discovery of several genes in which mutations cause early-
onset forms of PD has greatly accelerated research progress.
Point mutations or increased gene dosage of the -synuclein
gene cause autosomal dominant PD via a gain-of-function
mechanism. Recessive early-onset PD can be caused by
mutations in the genes encoding parkin, DJ-1 or PINK1,
presumably by a loss-of-function mechanism. The pathological
hallmark of adult-onset PD is the Lewy body, an inclusion
body found in the cytoplasm of neurons, often near the
nucleus. Lewy bodies are densest in the substantial nigra
but can also be present in monoaminergic, cerebral cortical
and other neurons. There are also aggregates in neurites,
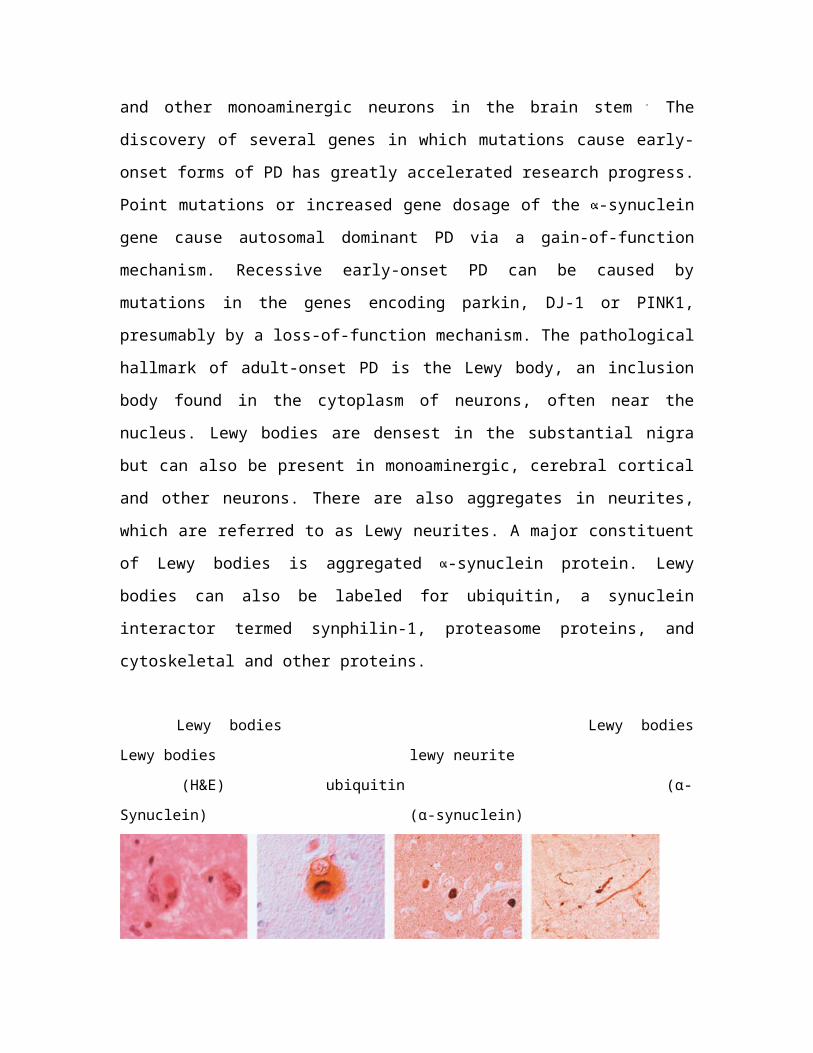
which are referred to as Lewy neurites. A major constituent
of Lewy bodies is aggregated -synuclein protein. Lewy
bodies can also be labeled for ubiquitin, a synuclein
interactor termed synphilin-1, proteasome proteins, and
cytoskeletal and other proteins.
Lewy bodies Lewy bodies
Lewy bodies lewy neurite
(H&E) ubiquitin (α-
Synuclein) (α-synuclein)

Fig 6 :Ref: (Bossy-wetzel E.et al.. Nat Med. 2004 Jul:s2-9 Revew )
Deposition of fibrillar proteinacious material in Hungtons disease:
Huntington's disease. HD is a progressive neurodegenerative
disorder caused by expansion of a CAG repeat coding for
polyglutamine in the N terminus of the huntingtin protein.
Because it is caused by a mutation in a single gene, HD has
emerged as a model for studying neurodegenerative disease
pathogenesis. There is a remarkable threshold effect, in
that polyglutamine stretches of 36 in huntingtin cause
disease, whereas 35 do not. Within the expanded range,
longer repeats cause earlier onset. There is a striking
correlation between the threshold for aggregation in vitro and
the threshold for disease in humans, consistent with the
idea that aggregation is related to pathogenesis. Inclusions
containing huntingtin are present in regions of the brain
that degenerate. However, the neurons with inclusions do not
correspond exactly to the neurons that degenerate. For
instance, inclusions are present in the striatum, which is
most affected, but they are more enriched in populations of
large interneurons, which are spared, than in medium spiny
projection neurons, which are selectively lost. There is a
good correlation, however, between the length of the CAG
repeat and the density of inclusions.
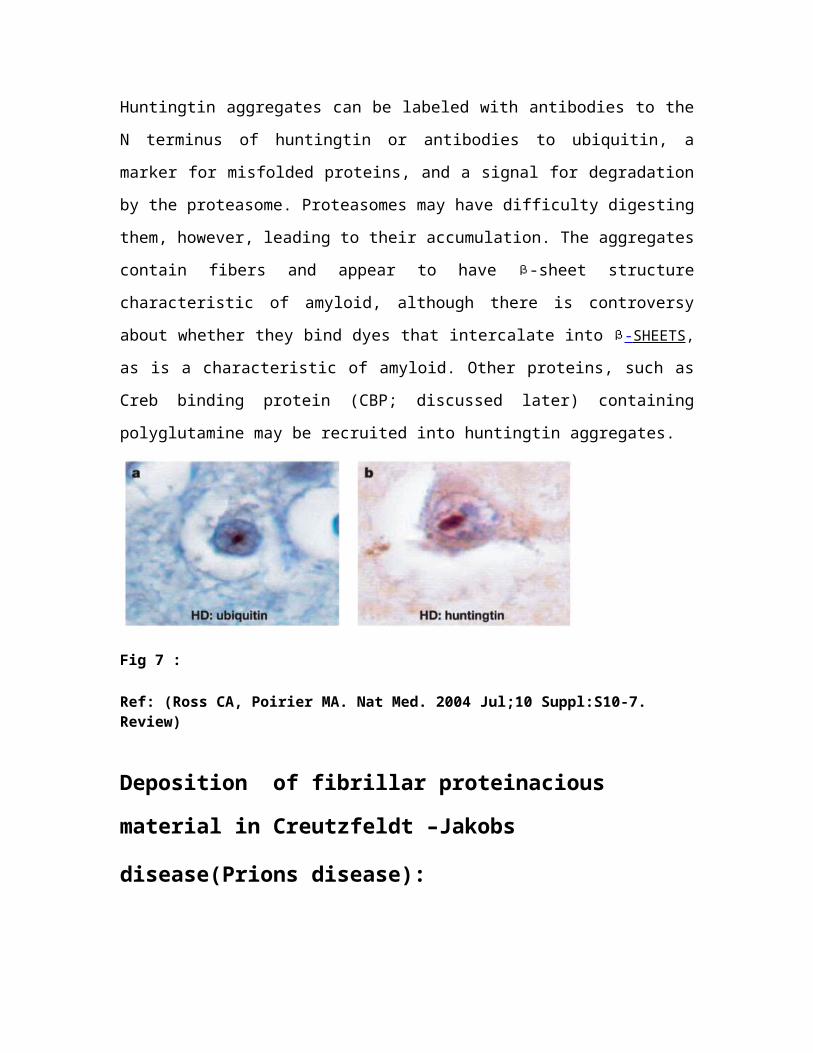
Huntingtin aggregates can be labeled with antibodies to the
N terminus of huntingtin or antibodies to ubiquitin, a
marker for misfolded proteins, and a signal for degradation
by the proteasome. Proteasomes may have difficulty digesting
them, however, leading to their accumulation. The aggregates
contain fibers and appear to have -sheet structure
characteristic of amyloid, although there is controversy
about whether they bind dyes that intercalate into - SHEETS ,
as is a characteristic of amyloid. Other proteins, such as
Creb binding protein (CBP; discussed later) containing
polyglutamine may be recruited into huntingtin aggregates.
Fig 7 :
Ref: (Ross CA, Poirier MA. Nat Med. 2004 Jul;10 Suppl:S10-7. Review)
Deposition of fibrillar proteinacious
material in Creutzfeldt –Jakobs
disease(Prions disease):
Neurodegenerative diseases caused by prions can be sporadic
or can be acquired either by environmental transmission or
via genetic mutations Environmental pathways include eating
prion particles derived from infected brain tissue or
surgical implantation via infected instruments. Prion
disease can also be caused by point mutations in the prion
gene, leading to alterations of the prion protein. Pathology
can include amyloid plaques that appear similar to those of
AD and that can be labeled with prion antibodies. Prion
disease is a prototypical protein conformation disease, in
that highly sophisticated studies have shown that it is
caused by abnormal protein structure and not an infective
viral agent. Mechanisms of prion disease have been
illuminated by the discovery of prionlike protein
conformational changes in yeast. In all cases, disease is
caused by abnormally folded prion proteins. Prion
aggregation can take place both extracellularly and
intracellularly.
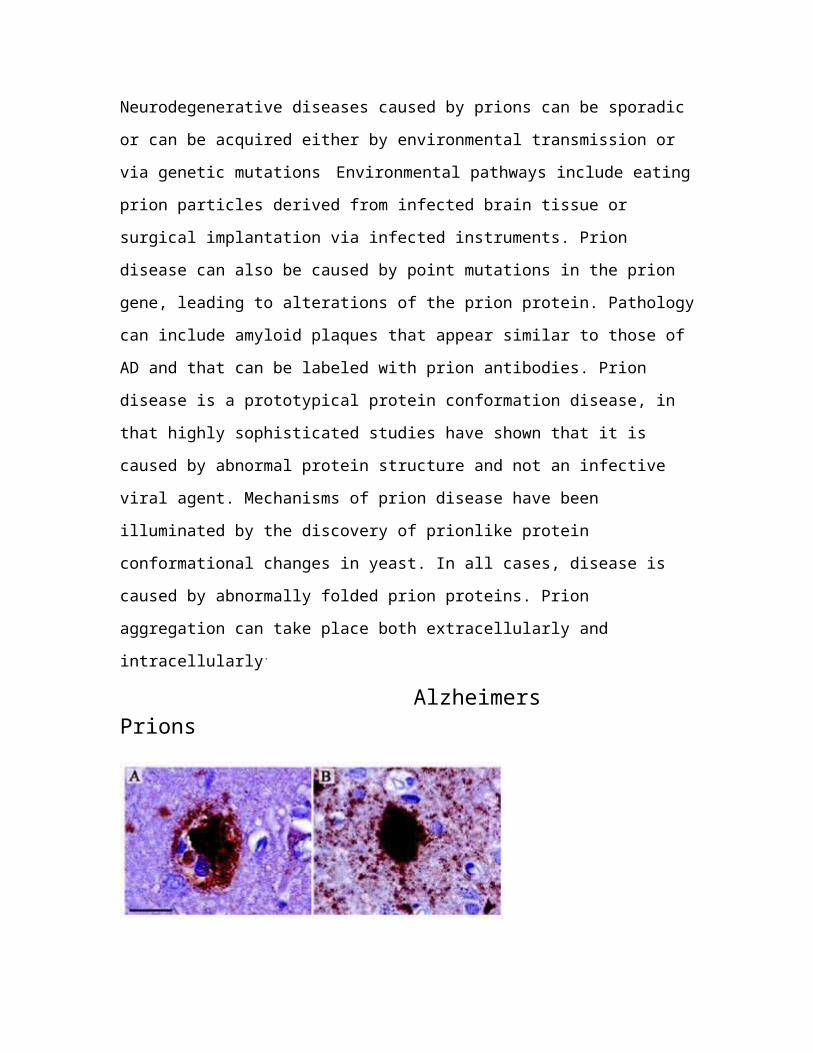
Alzheimers Prions

Fig 8 : Both Alzheimer’s and Prion diseases are characterized bythe deposition of pathological proteins in the brain, often inthe form of plaques. The brown colour is indicative ofimmunostained cortical deposit of A-beta peptide and of the prpsc
protein in brains of patients suffering from alzheimer’s disease(A) and prion disease (B) respectively
Deposition of fibrillar proteinacious
material in Cataract disease: A cataract is a clouding that develops in the crystallineiens of the eye or in its envelope (lens capsule), varying
in degree from slight to complete opacity and obstructing
the passage of light. Early in the development of age-
related cataract, the power of the lens may be increased,
causing near-sightedness (myopia), and the gradual yellowing
and opacification of the lens may reduce the perception of
blue colors. Cataracts typically progress slowly to cause
vision loss and are potentially blinding if untreated. The
condition usually affects both eyes, but almost always one
eye is affected earlier than the other
Fig 9: Cataract disease in human eye
Role of Water in ProteinAggregation andAmyloid Polymorphism:

Protein aggregation leading to amyloid fibril formation is
linked to a number of neurodegenerative diseases although in
some instances their formation is also beneficial.
Understanding how misfolded proteins polymerize into ordered
fibrils, which universallyhave a characteristic cross β-
structure, may be important in our ability to intervene and
prevent their formation. The physical basis of protein
aggregation involving a cascade of events that drive a
monomer to a fibrillar structure is complicated because of
interplay of a number of energy and time scales governing
amyloid formation. In addition, a number of other factors,
such as protein concentration, sequence of proteins, and
environmental conditions (pH, presence of osmolytes,
temperature) affect various kinetic steps in distinct ways,
thus making it difficult to describe even in vitro protein
aggregation in molecular terms. Despite these complexities,
significant advances have been made, especially in getting
structures of peptide amyloids and models for amyloid
fibrils on Aβ and fungal prion proteins. The availability of
structures have made it possible to undertake molecular
dynamics simulations, which have given insights into the
role water plays in oligomer formation as well as assembly
and growth of amyloid fibrils.
It has long been appreciated that water plays a major
role in the self-assembly of proteins5 in ensuring that
hydrophobic residues are (predominantly) sequestered in

protein interior. In contrast, the effects of water on
protein aggregation are poorly understood. Indeed, almost
all studies (experimental and computer simulations) on
amlyoid assembly mechanisms have been largely analyzed using
a protein centric perspective. The situation is further
exacerbated by experimental difficulties in directly
monitoring water activity during the growth process. Here,
we provide a perspective on the role water plays in protein
aggregation by synthesizing results from molecular dynamics
(MD) simulation studies. Briefly our goals are the
following: (i) Describe how water-mediated interactions
affect the energy landscape of monomers and drive oligomer
formation in Aβ peptides. (ii) The key role water plays in
late stages of fibril growth is described by large
variations in the sequence-dependent mechanism of self-
assembly to β-sheetrich amyloids. (iii) We use results of
recentMD simulations and concepts in protein crystallization
to provide scenarios for the role water plays in polymorphic
amyloid structures.
Fig 10 : Water absorb in protein

Water in protein aggregation and Amyloid polymorphis
Figure 11 : Schematic of protein aggregation mechanisms leadingto polymorphic fibrils. On the left are solvated peptides. Waterin the hydrationlayer is in red and the bulk water is in blue.Even isolated monomers sample aggregation-prone conformations,N*, which are coated with varyingnumbers of water molecules. Thepeptides with N* conformations aggregate to form disorderedprotein-rich droplets. A major driving force for aggregation isthe release of water molecules in the hydration layer into thebulk, which facilitates fibril formation being entropicallyfavorable. Thestructured protein aggregates nucleate from theprotein-rich droplet to form protofilaments, which further self-assemble to form a variety of matureamyloid fibrils. In some ofthe polymorphic structures, discrete numbers of water moleculesare confined in the fibril.
Conclusions:
Most of the neurodegenerative diseases are characterized byintracellular or extracellular deposition of insolublematerial. Whether this is a cause or a consequence of the diseases is
not known yet.
It is speculated that the early species in this process might be most toxic,
by being involved in abnormal interactions with other cellular proteins.

However, the fact that all these diseases are characterized
by the same common factors, and the observation that
inherited forms of these diseases cause a massive increase
in the production of b-sheet related proteins lead to
hypothesize that these b-sheet proteins and the subsequent
formation of the insoluble lesions may be upstream the
cascade of events that lead to neurodegeneration.

Department of Chemistry University of Kalyani
Kalyani, Nadia
Project report submitted for partial fulfillment of M.Sc . degree (PHYSICAL CHEMISTRY SPECIAL)
By

CHINMAY GHOSH MARCH 2012
Reference:
1 Dobson, C. M. Protein folding and misfolding. Nature 2003,426, 884–890.2 Goedert, M.; Spillantini, M. G. A century of Alzheimer’sdisease. Science 2006, 314,
777–781.Contrasting Disease and Nondisease Protein Aggregation Fawziet al. 1046 ACCUNTS OF CHEMICAL RESEARCH 1037-1047 August 2008
Vol. 41, No. 83 Cellmer, T.; Douma, R.; Huebner, A.; Prausnitz, J.; Blanch,H. Kinetic studies of
protein L aggregation and disaggregation. Biophys. Chem. 2007,125, 350–359.
4 Clark, E. D. Protein refolding for industrial processes.Curr. Opin. Biotechnol. 2001,
12, 202–207.5 Tycko, R.; Petkova, A.; Oyler, N.; Chan, C. C.; Balbach,J. Probing the molecular
structure of amyloid fibrils with solid-state NMR. Biophys. J.2002, 82, 187A.

6 King, J.; Haasepettingell, C.; Robinson, A. S.; Speed, M.;Mitraki, A. Thermolabile
folding intermediates-inclusion body precursors andchaperonin substrates. FASEBJ. 1996, 10, 57–66.
7 Uversky, V. N.; Li, J.; Fink, A. L. Evidence for apartially folded intermediate in alphasynuclein
fibril formation. J. Biol. Chem. 2001, 276, 10737–10744.8 Silow, M.; Tan, Y. J.; Fersht, A. R.; Oliveberg, M.Formation of short-lived protein
aggregates directly from the coil in two-state folding.Biochemistry 1999, 38,13006–13012.
9 Chiti, F.; Taddei, N.; Baroni, F.; Capanni, C.; Stefani,M.; Ramponi, G.; Dobson,
C. M. Kinetic partitioning of protein folding andaggregation. Nat. Struct. Biol. 2002,9, 137–143.
1 0 Kim, D. E.; Fisher, C.; Baker, D. A breakdown of symmetryin the folding transition
state of protein L. J. Mol. Biol. 2000, 298, 971–984.11 Park, S. H.; Shastry, M. C. R.; Roder, H. Folding dynamics
of the B1-domain ofprotein G explored by ultrarapid mixing. Nat. Struct. Biol. 1999,6, 943–947.
12 Fawzi, N. L.; Chubukov, V.; Clark, L. A.; Brown, S.; Head-Gordon, T. Influence ofdenatured and intermediate states of folding on proteinaggregation. Protein Sci.2005, 14, 993–1003.
13 Head-Gordon, T.; Brown, S. Minimalist models for proteinfolding and design. Curr.Opin. Struct. Biol. 2003, 13, 160–167.
14 Brown, S.; Fawzi, N.; Head-Gordon, T. Coarse-grainedsequences for protein foldingand design. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 10712–10717.

15 Yap, E. H.; Fawzi, N. L.; Head-Gordon, T. A coarse-grainedalpha-carbon proteinmodel with anisotropic hydrogen-bonding. Proteins 2008, 70,626–638.
16 Thirumalai, D.; Klimov, D. K. Deciphering the timescalesand mechanisms of proteinfolding using minimal off-lattice models. Curr. Opin. Struct. Biol.1999, 9, 197–207. Wikstrom, M.; Drakenberg, T.; Forsen, S.; Sjobring, U.;Bjorck, L. Three-dimensionalsolution structure of an immunoglobulin light chain-bindingdomain of protein-L.Comparison with the IgG-binding domains of protein G.Biochemistry 1994, 33,14011–14017.
17 Gronenborn, A. M.; Filpula, D. R.; Essig, N. Z.; Achari,A.; Whitlow, M.; Wingfield,P. T.; Clore, G. M. A novel, highly stable fold of theimmunoglobulin binding domainof streptococcal protein-G. Science 1991, 253, 657–661. Brown, S.; Head-Gordon, T. Intermediates and the folding ofproteins L and G.Protein Sci. 2004, 13, 958–970.
18 Klyubin, I.; Walsh, D. M.; Cullen, W. K.; Fadeeva, J. V.;Anwyl, R.; Selkoe, D. J.;
19 Rowan, M. J. Soluble Arctic amyloid beta protein inhibitshippocampal long-term
20 potentiation in vivo. Eur. J. Neurosci. 2004, 19, 2839–2846.2121 Taylor, J.P., Hardy, J. & Fischbeck, K.H. Toxic proteinsin neurodegenerative disease. Science 296, 1991−1995(2002). | Article |
22 Bates, G. Huntingtin aggregation and toxicity inHuntington's disease. Lancet 361, 1642−1644(2003). | Article | 23 Caughey, B. & Lansbury, P.T. Protofibrils, pores,fibrils, and neurodegeneration: separating the responsible

protein aggregates from the innocent bystanders. Annu. Rev. Neurosci.26, 267−298 (2003). | Article | 24 Berke, S.J. & Paulson, H.L. Protein aggregation and theubiquitin proteasome pathway: gaining the UPPer hand onneurodegeneration. Curr. Opin. Genet. Dev. 13, 253−261(2003). | Article | 25 Ross, C.A. & Pickart, C. The ubiquitin-proteasome pathway inParkinson's and other neurodegenerative diseases. Trends Cell Biol.(2004). 26 Nussbaum, R.L. & Ellis, C.E. Alzheimer's disease andParkinson's disease. N. Engl. J. Med. 348, 1356−1364(2003). | Article | 27 Wong, P.C., Cai, H., Borchelt, D.R. & Price, D.L. Geneticallyengineered mouse models of neurodegenerative diseases. Nat. Neurosci.5, 633−639 (2002). | Article | 28 Ross, C.A. When more is less: pathogenesis of glutaminerepeat neurodegenerative diseases. Neuron 15, 493−496(1995). | Article | 29 Selkoe, D.J. Folding proteins in fatal ways. Nature 426,900−904 (2003). | Article | 30 Davies, S.W. et al. Formation of neuronal intranuclearinclusions underlies the neurological dysfunction in micetransgenic for the HD mutation. Cell 90, 537−548(1997). | Article | 31 Scherzinger, E. et al. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils:implications for Huntington's disease pathology. Proc. Natl. Acad. Sci.USA 96, 4604−4609 (1999). | Article | 32 Vonsattel, J.P. et al. Neuropathological classification ofHuntington's disease. J. Neuropathol. Exp. Neurol. 44, 559−577 (1985). 34 Kuemmerle, S. et al. Huntington aggregates may not predictneuronal death in Huntington's disease. Ann. Neurol. 46, 842−849(1999). | Article | 35 Gutekunst, C.A. et al. Nuclear and neuropil aggregates inHuntington's disease: relationship to neuropathology. J. Neurosci.19, 2522−2534 (1999).

36 Becher, M.W. et al. Intranuclear neuronal inclusions inHuntington's disease and dentatorubral and pallidoluysianatrophy: correlation between the density of inclusions and IT15CAG triplet repeat length. Neurobiol. Dis. 4, 387−397(1998). | Article | 37 Myers, R.H. et al. Clinical and neuropathologic assessment ofseverity in Huntington disease. Neurology 38, 341−347 (1988). 38 Venkatraman, P., Wetzel, R., Tanaka, M., Nukina, N. &Goldberg, A.L. Eukaryotic proteasomes cannot digest polyglutaminesequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 14, 95−104 (2004). | Article |
39 Shankar, G. M.; Li, S.; Mehta, T. H.; Garcia-Munoz, A.;Shepardson, N. E.; Smith, I.; Brett,
F. M.; Farrell, M. A.; Rowan, M. J.; Lemere, C. A.; Regan,C. M.; Walsh, D. M.; Sabatini,B. L.; Selkoe, D. J. Amyloid-beta protein dimers isolateddirectly from Alzheimer'sbrains impair synaptic plasticity and memory. Nat. Med.2008,14, 837–842.
40 Aguzzi, A. Cell biology: Beyond the prion principle.Nature2009, 459, 924–925.
41 Maji, S. K.; Perrin, M. H.; Sawaya, M. R.; Jessberger,S.; Vadodaria, K.; Rissman, R. A.;
Singru, P. S.; Nilsson, K. P. R.; Simon, R.; Schubert, D.;Eisenberg, D.; Rivier, J.;Sawchenko, P.; Vale, W.; Riek, R. Functional amyloids asnatural storage of peptidehormones in pituitary secretory granules. Science2009, 325,328–332.
42 Tycko, R. Insights into the amyloid folding problem fromsolid-state NMR. Biochemistry
2003, 42, 3151–3159.43 Dill, K. Dominant forces in protein folding.
Biochemistry1990, 29, 7133–7155.44 Thirumalai, D.; Klimov, D. K.; Dima, R. I. Emerging ideason the molecular basis of protein

and peptide aggregation. Curr. Opin. Struct. Biol.2003, 13,146–159.
45 Tarus, B.; Straub, J. E.; Thirumalai, D. Dynamics ofAsp23_Lys28 salt-bridge formation in
Aβ10_35 monomers. J. Am. Chem. Soc.2006, 128, 16159–16168.46 Li, M. S.; Klimov, D. K.; Straub, J. E.; Thirumalai, D.Probing the mechanisms of fibril
formation using lattice models. J. Chem. Phys.2008, 129, No.175101.
47 Li, M. S.; Co, N. T.; Reddy, G.; Hu, C.-K.; Straub, J. E.;Thirumalai, D. Factors
governing fibrillogenesis of polypeptide chains revealed bylattice models.Phys. Rev. Lett.2010, 105, No. 218101.
48 Bellesia, G.; Shea, J.-E. What determines the structure andstability of KFFE monomers,
dimers, and protofibrils? Biophys. J.2009, 96, 875–886.49 Massi, F.; Straub, J. Energy landscape theory forAlzheimer's amyloid beta-peptide fibril
elongation. Proteins2001, 42, 217–229.50 Straub, J. E.; Thirumalai, D. Principles governing oligomerformation in amyloidogenic
peptides. Curr. Opin. Struct. Biol.2010, 20, 187–195.51 Dima, R.; Thirumalai, D. Exploring the propensities ofhelices in PrPc to form beta sheet
using NMR structures and sequence alignments. Biophys.J.2002, 83, 1268–1280.
52 Dima, R.; Thirumalai, D. Probing the instabilities in thedynamics of helical fragments from
mouse PrPc. Proc. Natl. Acad. Sci. U.S.A.2004, 101, 15335–15340.
ACKNOWLEDGEMENT

Let me take the opportunity toexpress my deepest sense ofgratitude to my supervisor Prof.Nilashis Nandi for his patientguidance and teaching through courseof my project paper.
My sincerely gratitude goes toour H.O.D. Prof. Mitali Sarkar andall the faculties to give me theirvaluable suggestion and to completemy project paper.
I am also very thankful toSindrila Duttabanik , Md. Najib Alam, Kumarjyoti Roy (Resarch ScholarsDepartment of Chemistry, K.U.) fortheir valuable time and continuoushelp throughout course of myassigned paper.
Finally I gratefully thank tomy parents for inspiring me.

CHINMAY GHOSH

CONTENTS
1 - Acknowledgement
2 - Introduction
3 - What is proteinaggregation…?
4 - Protein missfolding,aggregation and Neurodegeneration.
5 - How protein aggregates andform amyloid Insoluble fibrils…..?

6 - Steps that lead to formationof aggregate. 7 - Protein aggregation and Neurodegenerative disease
8 - Role of water in proteinaggregation and Amyloid polymorphism.
9 - Conclusion
10 - References

Copyright © 2022 FDOKUMEN




















