
Platelet-Activating Factor and Bacteremia-Induced Pulmonary Hypertension
Upload
independentCategory
view
1download
0
Protein disulphide isomerasemediates platelet aggregation and secretion
DAVID W. ESSE X AND MENGRU LI Department of Medicine, Division of Hematology/Oncologyand Department of Biochemistry, State University of New York, Health Science Center at Brooklyn,Brooklyn, New York, U.S.A.
Received 10 August 1998; accepted for publication 13 November 1998
Summary. Platelet surface thiols and disulphides play animportant role in platelet responses. Agents that reducedisulphide bonds expose the fibrinogen receptor in plateletsand activate the purified glycoprotein (GP) IIbIIIa receptor.Protein disulphide isomerase (PDI), an enzyme that re-arranges disulphides bonds, is found on the platelet surfacewhere it is catalytically active. We investigated the role of PDIin platelet responses using (1) rabbit anti-PDI IgG specific forPDI, (2) a competing substrate (scrambled ribonuclease A),and (3) the PDI inhibitor, bacitracin. Fab fragments of therabbit anti-PDI IgG inhibited platelet responses to theagonists tested (ADP and collagen), whereas Fab fragmentsprepared identically from normal rabbit IgG had noinhibitory effect. Scrambled ribonuclease A blocked plateletaggregation and secretion, but native ribonuclease A did not.
When biphasic platelet aggregation was examined usingplatelets in citrated plasma, the principle effect of bacitracinwas on second phase or irreversible aggregation responsesand the accompanying secretion. Using flow cytometry andan antibody specific for activated GPIIbIIIa (PAC-1), therabbit anti-PDI Fab fragments substantially inhibited activa-tion of GPIIbIIIa when added before, but not after, plateletactivation. In summary, we have demonstrated that proteindisulphide isomerase mediates platelet aggregation andsecretion, and that it activates GPIIbIIIa, suggesting thisreceptor as the target of the enzyme.
Keywords: platelet, aggregation, secretion, protein disul-phide isomerase, glycoprotein IIbIIIa.
A number of reports have demonstrated an important rolefor platelet surface thiol or sulphydryl groups and di-sulphides in platelet responses. Aledort et al (1968) foundthat the non-penetrating sulphydryl reactive reagent para-chloromercuribenzinesulphonate (PCMBS) inhibited plateletaggregation. MacIntyre and colleagues confirmed thisfinding and also demonstrated that concentrations of thereducing agent dithiothreitol (DTT) above 1 mM caused slowprogressive platelet aggregation after a lag period of 1–4 min, provided that fibrinogen was present (MacIntyre &Gordon, 1974a; MacIntyre et al, 1977). Unlike ADP-inducedaggregation, DTT-induced aggregation was not inhibited byprostaglandin E1 (PGE1) (MacIntyre & Gordon, 1974b;Zucker & Masiello, 1984), suggesting that reduction ofdisulphide bonds triggers a later step in the platelet response,specifically, the development of the ability to bind fibrinogen.Zucker & Masiello (1984) found that DTT and 2-mercapto-ethanol induced platelet aggregation in normal platelets butnot in platelets which lacked the GPIIbIIIa receptor (from
thrombasthenic patients). They also showed that normalplatelets stimulated with DTT bound 125I-labelled fibrinogen.Finally, Kouns et al (1993) have shown that DTT, by selectivedisulphide bond reduction, activates purified GPIIbIIIa to afibrinogen binding state.
Both GPIIb and GPIIIa contain disulphide bonds. GPIIbcontains 18 cysteine residues and nine disulphide bonds(Poncz et al, 1987), and GPIIIa contains 56 cysteineresidues, most of which are in a cysteine-rich region in theextracellular portion of the molecule. Each cysteine isinvolved in disulphide bond formation that stabilizes themature complex (Calvete et al, 1988, 1991; Beer & Coller,1989; Niewiarowski et al, 1989). The conversion ofGPIIbIIIa to a fibrinogen-binding conformation is a regulatedprocess, activated by cytoplasmic signalling (Naik & Parise,1997). Once fibrinogen binds to the receptor, furtherconformational changes in GPIIbIIIa lead to signalling intothe platelet. Much remains to be elucidated about theseprocesses, and the possibility exists that the same moleculesor pathways are involved in both processes.
Protein disulphide isomerase (PDI), an enzyme whichcatalyses disulphide exchange reactions (Noiva, 1994; Luz &Lennarz, 1996), has recently been localized to the external
British Journal of Haematology, 1999, 104, 448–454
448 q 1999 Blackwell Science Ltd
Correspondence: Dr D. W. Essex, Box 8, Department of Biochemistry,SUNY Health Science Center at Brooklyn, 450 Clarkson Avenue,Brooklyn, NY 11203, U.S.A.

449Protein Disulphide Isomerase mediates Platelet Aggregation
q 1999 Blackwell Science Ltd, British Journal of Haematology 104: 448–454
surface of mammalian cells and platelets where it iscatalytically active (Mandel et al, 1993; Chen et al, 1995;Essex et al, 1995). PDI has long been known as anendoplasmic reticulum protein involved in the arrangementof disulphide bonds in nascent polypeptide chains (Noiva,1994; Luz & Lennarz, 1996). PDI contains a peptide bindingsite (Klappa et al, 1998) that is necessary for catalyticactivity (Morjana & Gilbert, 1991; Darby & Creighton, 1995;Quan et al, 1995; Luz & Lennarz, 1996). It has twohomologous active sites, each with the amino acid sequenceCys-Gly-His-Cys. Ribonuclease A (RNase) that has beeninactivated by formation of randomly mismatched orscrambled disulphide bonds (scrambled RNase) is a well-known substrate of PDI (Noiva, 1994; Luz & Lennarz, 1996).PDI on the platelet surface has been shown to reactivatescrambled RNase, an activity inhibited by rabbit anti-PDI IgGand other inhibitors of PDI (Essex et al, 1995). Weinvestigated the effect of inhibitors of PDI on plateletaggregation and secretion and also examined the ability ofscrambled RNase as a competing substrate to inhibit plateletresponses.
MATERIALS AND METHODS
Materials. ATP, Chemo-Lumo reagents, ADP, collagen andepinephrine were purchased from Chronolog Corp. (Haver-town, Pa.). Collagen was also obtained from Bio Data Corp.(Horsham, Pa.). This collagen was made from calf skin (themanufacturers recommend using 190 mg/ml for its use inplatelet aggregation studies). Apyrase, heparin, bovineserum albumin, papain, aspirin, prostaglandin E1 (PGE1),protein A, dithiothreitol (DTT), bacitracin and normal rabbitIgG were purchased from Sigma Chemical Co. (St Louis,Mo.). Purified a- and g-thrombin were gifts of Dr JohnFenton II (Division of Laboratories and Research, New YorkState Department of Health, Albany, N.Y.). Highly purifiednative RNase from bovine pancreas was obtained from FlukaChemical Corp. (Milwaukee, Wis.).
PDI assay. Scrambled RNase was prepared as described(Hillson et al, 1984; Essex et al, 1995) from native RNase.The PDI assay was performed as described with PDI activitybeing detected by measuring the renaturation of scrambledRNase (Essex et al, 1995).
Antibodies. Rabbit anti-PDI antiserum was raised againsthuman platelet PDI and was passed through a humanalbumin–Sepharose affinity column and subsequentlythrough a protein A column as described (Chen et al,1995), from which IgG was eluted with 0·1 mol/l glycine atpH 3·0. The eluant was then adjusted to pH 7·4.Characterization of the IgG by immunoblotting of purifiedPDI, and platelet lysate following SDS-PAGE showed that thelysate contained the same immunoreactive PDI, seen as asingle band. The anti-PDI IgG, but not normal rabbit IgG,inhibited soluble PDI in the scrambled RNase assay. FITC-labelled PAC-1, which recognizes the activated plateletGPIIbIIIa receptor (Shattil et al, 1985) and binds to thefibrinogen binding site (Taub et al, 1989), was obtained fromBecton Dickinson (San Jose, Calif.).
Preparation of Fab fragments. The intact rabbit anti-PDI IgGinduced platelet aggregation which was blocked by themonoclonal antibody (IV.3) that blocks Fc receptor mediatedinteractions, indicating that this phenomena is similar tothat described for MoAbs which activate platelets. Thisunusual feature of a polyclonal rabbit antibody has rarelybeen reported (Hashimoto et al, 1992) and has beencharacterized (unpublished observations). Fab fragments ofthe rabbit anti-PDI antibody were prepared as describedelsewhere (Essex et al, 1995), with minor modifications, byincubation of 600 mg of immune or nonimmune IgG withpapain (10 mg) in 100 mM sodium acetate, pH 5·5, contain-ing 1 mM EDTA, at 378C. The reaction was stopped by theaddition of iodoacetamide to a final concentration of 75 mM
for 30 min at room temperature. The Fab fragments wereseparated from the Fc portion by affinity chromatographyover protein A. The Fab fragments were extensively dialysedagainst phosphate-buffered saline (PBS; 10 mM phosphateand 140 mM NaCl, pH 7·4) to remove the iodoacetamide andthen concentrated. Control Fab fragments from normalrabbit IgG were prepared in an identical manner. Theconcentration of the Fab was determined by spectrophoto-metry at 280 nm using an extinction coefficient of 15·0(Essex et al, 1995). The Fab fragments were 40 kD in size onSDS–(12%) PAGE and free of whole antibody. Two prepara-tions of anti-PDI Fab fragments were used in these studies.
Platelet preparation. Whole blood was collected in 3·8%sodium citrate (9:1, vol:vol) by venepuncture as described(Essex et al, 1995) from several different healthy male andfemale donors. Donors had been free of any medication for atleast 2 weeks before blood donation. Platelet-rich plasma(PRP) was obtained after centrifugation of whole blood at300 g for 20 min. Washed platelets were prepared fromwhole blood collected in acid citrate dextrose (Essex et al,1995). The aggregation and clotting inhibitors PGE1 (1 mM),apyrase (1 U/ml) and heparin (2 U/ml) were added to thewhole blood, and platelet-rich plasma was obtained follow-ing centrifugation of the whole blood at 300 g for 20 min at248C. Platelets were isolated by centrifugation of the PRP at1000 g for 20 min at 248C and washed three times using aTyrodes–albumin solution containing 137 mM NaCl, 2·7 mM
KCl, 1 mM MgCl2, 0·36 mM NaH2PO4, 12 mM NaHCO3, 2 mM
CaCl2, 5·5 mM glucose and 0·35% albumin at pH 7·35. Thefirst wash solution contained heparin, apyrase and PGE1; thesecond contained apyrase and PGE1, and the third containedonly PGE1. The final platelet pellets were re-suspended inTyrodes buffer (pH 7·35) in the absence of any inhibitors.Platelets were counted microscopically using a haemacyt-ometer.
Platelet aggregation and release studies. The experimentswere carried-out in a Chronolog Lumi-Aggregometer(Chronolog Corp., Havertown, Pa.). For experiments ofplatelet aggregation and secretion, 50 ml of chronologluciferin/luciferase was added to 0·45 ml of PRP or plateletssuspended in Tyrodes buffer. Washed platelet suspensionscontained 2–4 × 108/ml platelets to which fibrinogen(200 mg/ml) was added. Platelet activation was initiated byadding the agonists to platelets under constant stirringconditions at 378C. Bacitracin (which contains no salts), the
anti-PDI Fab fragments, or scrambled RNase were pre-incubated with the platelets for 10 min, although a 1 minpreincubation proved almost as effective. If we assume thereare 50 000 molecules of PDI per platelet, 100 mg/ml ofscrambled RNase would provide a 10-fold molar excess ofscrambled RNase to PDI. At the end of each experiment,platelet responses were retested without inhibitors to ensurecontinued responses.
Western blotting. Western blotting was performed asdescribed (Essex et al, 1995) using 7·5% SDS-PAGE underreducing conditions. After the membrane was incubatedwith the rabbit anti-PDI IgG, it was developed using goatanti-rabbit IgG conjugated with peroxidase and a chemi-luminescent substrate.
Flow cytometry analysis. For flow cytometry analysis citratedplatelet-rich plasma was diluted into an equal volume ofphosphate-buffered saline and incubated for 10 min with anti-PDI or normal rabbit IgG Fab fragments and then withcollagen (48 mg/ml; Bio Data) for 5 min without stirring atroom temperature. Bio Data collagen was used for thesestudies as it more consistently produced stronger PAC-1binding than Chronolog collagen. In some samples the Fabfragments were added after the collagen had been incubatedwith the platelets. 10 ml of FITC-conjugated PAC-1 was thenincubated with 10 ml of platelets for 30 min in the dark. Tenthousand platelets per sample were analysed by flowcytometry as described (Chen et al, 1995; Essex et al, 1995).
RESULTS
Effect of PDI inhibitors on platelet aggregationAnti-PDI Fab fragments prepared from rabbit anti-PDI IgGinhibited ADP and collagen-induced aggregation of washed
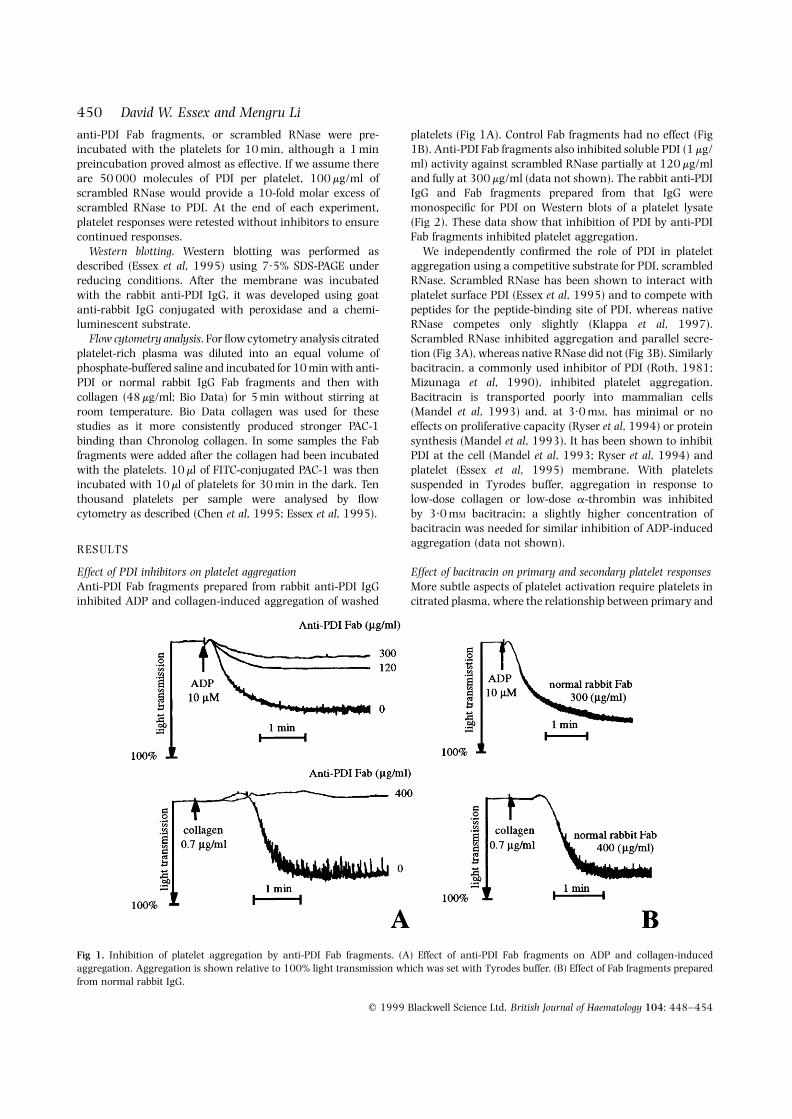
platelets (Fig 1A). Control Fab fragments had no effect (Fig1B). Anti-PDI Fab fragments also inhibited soluble PDI (1 mg/ml) activity against scrambled RNase partially at 120 mg/mland fully at 300 mg/ml (data not shown). The rabbit anti-PDIIgG and Fab fragments prepared from that IgG weremonospecific for PDI on Western blots of a platelet lysate(Fig 2). These data show that inhibition of PDI by anti-PDIFab fragments inhibited platelet aggregation.
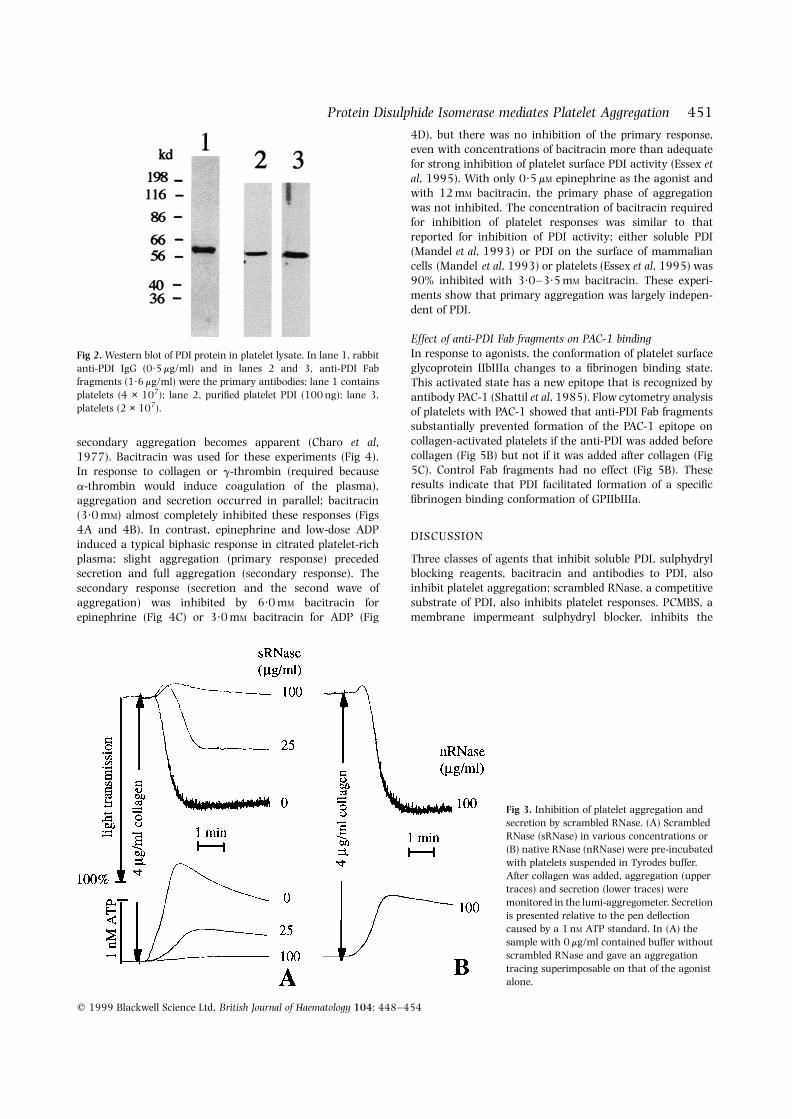
We independently confirmed the role of PDI in plateletaggregation using a competitive substrate for PDI, scrambledRNase. Scrambled RNase has been shown to interact withplatelet surface PDI (Essex et al, 1995) and to compete withpeptides for the peptide-binding site of PDI, whereas nativeRNase competes only slightly (Klappa et al, 1997).Scrambled RNase inhibited aggregation and parallel secre-tion (Fig 3A), whereas native RNase did not (Fig 3B). Similarlybacitracin, a commonly used inhibitor of PDI (Roth, 1981;Mizunaga et al, 1990), inhibited platelet aggregation.Bacitracin is transported poorly into mammalian cells(Mandel et al, 1993) and, at 3·0 mM, has minimal or noeffects on proliferative capacity (Ryser et al, 1994) or proteinsynthesis (Mandel et al, 1993). It has been shown to inhibitPDI at the cell (Mandel et al, 1993; Ryser et al, 1994) andplatelet (Essex et al, 1995) membrane. With plateletssuspended in Tyrodes buffer, aggregation in response tolow-dose collagen or low-dose a-thrombin was inhibitedby 3·0 mM bacitracin; a slightly higher concentration ofbacitracin was needed for similar inhibition of ADP-inducedaggregation (data not shown).
Effect of bacitracin on primary and secondary platelet responsesMore subtle aspects of platelet activation require platelets incitrated plasma, where the relationship between primary and
q 1999 Blackwell Science Ltd, British Journal of Haematology 104: 448–454
450 David W. Essex and Mengru Li
Fig 1. Inhibition of platelet aggregation by anti-PDI Fab fragments. (A) Effect of anti-PDI Fab fragments on ADP and collagen-inducedaggregation. Aggregation is shown relative to 100% light transmission which was set with Tyrodes buffer. (B) Effect of Fab fragments preparedfrom normal rabbit IgG.
451Protein Disulphide Isomerase mediates Platelet Aggregation
q 1999 Blackwell Science Ltd, British Journal of Haematology 104: 448–454
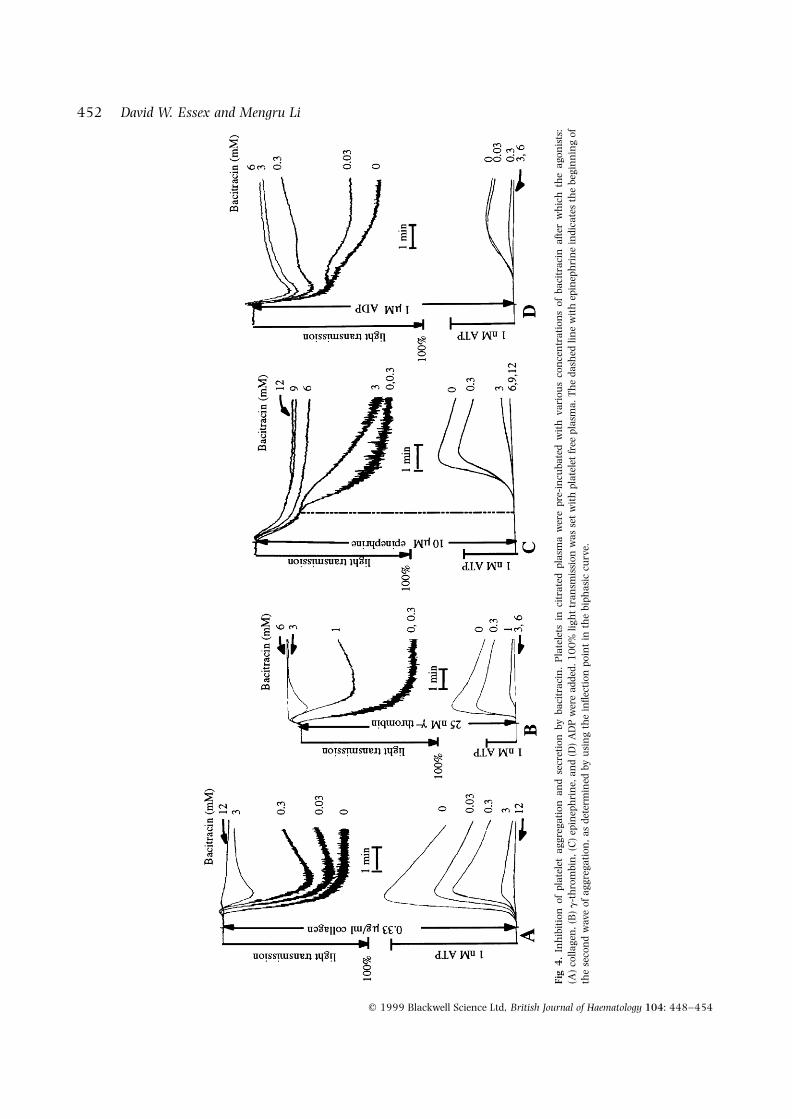
secondary aggregation becomes apparent (Charo et al,1977). Bacitracin was used for these experiments (Fig 4).In response to collagen or g-thrombin (required becausea-thrombin would induce coagulation of the plasma),aggregation and secretion occurred in parallel; bacitracin(3·0 mM) almost completely inhibited these responses (Figs4A and 4B). In contrast, epinephrine and low-dose ADPinduced a typical biphasic response in citrated platelet-richplasma; slight aggregation (primary response) precededsecretion and full aggregation (secondary response). Thesecondary response (secretion and the second wave ofaggregation) was inhibited by 6·0 mM bacitracin forepinephrine (Fig 4C) or 3·0 mM bacitracin for ADP (Fig
4D), but there was no inhibition of the primary response,even with concentrations of bacitracin more than adequatefor strong inhibition of platelet surface PDI activity (Essex etal, 1995). With only 0·5 mM epinephrine as the agonist andwith 12 mM bacitracin, the primary phase of aggregationwas not inhibited. The concentration of bacitracin requiredfor inhibition of platelet responses was similar to thatreported for inhibition of PDI activity; either soluble PDI(Mandel et al, 1993) or PDI on the surface of mammaliancells (Mandel et al, 1993) or platelets (Essex et al, 1995) was90% inhibited with 3·0–3·5 mM bacitracin. These experi-ments show that primary aggregation was largely indepen-dent of PDI.
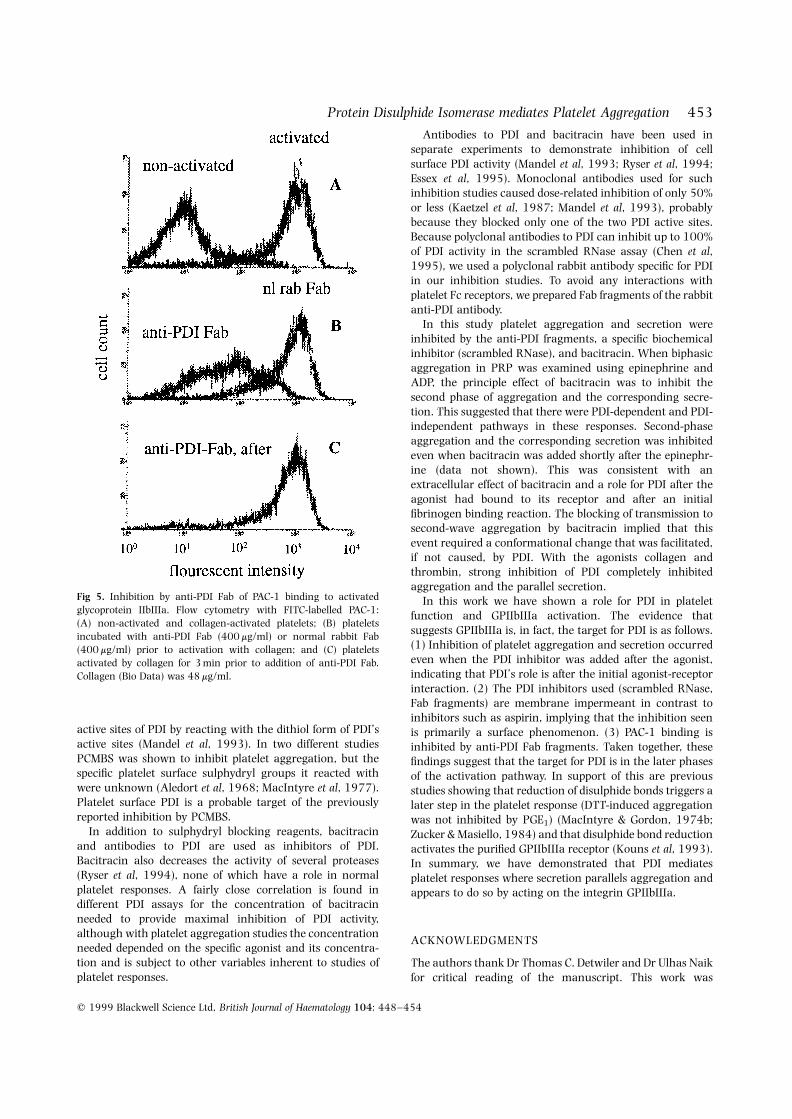
Effect of anti-PDI Fab fragments on PAC-1 bindingIn response to agonists, the conformation of platelet surfaceglycoprotein IIbIIIa changes to a fibrinogen binding state.This activated state has a new epitope that is recognized byantibody PAC-1 (Shattil et al, 1985). Flow cytometry analysisof platelets with PAC-1 showed that anti-PDI Fab fragmentssubstantially prevented formation of the PAC-1 epitope oncollagen-activated platelets if the anti-PDI was added beforecollagen (Fig 5B) but not if it was added after collagen (Fig5C). Control Fab fragments had no effect (Fig 5B). Theseresults indicate that PDI facilitated formation of a specificfibrinogen binding conformation of GPIIbIIIa.
DISCUSSION
Three classes of agents that inhibit soluble PDI, sulphydrylblocking reagents, bacitracin and antibodies to PDI, alsoinhibit platelet aggregation; scrambled RNase, a competitivesubstrate of PDI, also inhibits platelet responses. PCMBS, amembrane impermeant sulphydryl blocker, inhibits the
Fig 2. Western blot of PDI protein in platelet lysate. In lane 1, rabbitanti-PDI IgG (0·5 mg/ml) and in lanes 2 and 3, anti-PDI Fabfragments (1·6 mg/ml) were the primary antibodies; lane 1 containsplatelets (4 × 107); lane 2, purified platelet PDI (100 ng); lane 3,platelets (2 × 107).
Fig 3. Inhibition of platelet aggregation andsecretion by scrambled RNase. (A) ScrambledRNase (sRNase) in various concentrations or(B) native RNase (nRNase) were pre-incubatedwith platelets suspended in Tyrodes buffer.After collagen was added, aggregation (uppertraces) and secretion (lower traces) weremonitored in the lumi-aggregometer. Secretionis presented relative to the pen deflectioncaused by a 1 nM ATP standard. In (A) thesample with 0 mg/ml contained buffer withoutscrambled RNase and gave an aggregationtracing superimposable on that of the agonistalone.
q 1999 Blackwell Science Ltd, British Journal of Haematology 104: 448–454
452 David W. Essex and Mengru Li
Fig
4.
Inh
ibit
ion
ofpl
atel
etag
greg
atio
nan
dse
cret
ion
byba
citr
acin
.P
late
lets
inci
trat
edpl
asm
aw
ere
pre-
incu
bate
dw
ith
vari
ous
con
cen
trat
ions
ofba
citr
acin
afte
rw
hic
hth
eag
onis
ts:
(A)
colla
gen
,(B
)g
-th
rom
bin
,(C
)ep
inep
hri
ne,
and
(D)
AD
Pw
ere
adde
d.1
00
%lig
ht
tran
smis
sion
was
set
wit
hpl
atel
etfr
eepl
asm
a.T
he
dash
edlin
ew
ith
epin
eph
rin
ein
dica
tes
the
begi
nn
ing
ofth
ese
con
dw
ave
ofag
greg
atio
n,a
sde
term
ined
byu
sin
gth
ein
flect
ion
poin
tin
the
biph
asic
curv
e.
453Protein Disulphide Isomerase mediates Platelet Aggregation
q 1999 Blackwell Science Ltd, British Journal of Haematology 104: 448–454
active sites of PDI by reacting with the dithiol form of PDI’sactive sites (Mandel et al, 1993). In two different studiesPCMBS was shown to inhibit platelet aggregation, but thespecific platelet surface sulphydryl groups it reacted withwere unknown (Aledort et al, 1968; MacIntyre et al, 1977).Platelet surface PDI is a probable target of the previouslyreported inhibition by PCMBS.
In addition to sulphydryl blocking reagents, bacitracinand antibodies to PDI are used as inhibitors of PDI.Bacitracin also decreases the activity of several proteases(Ryser et al, 1994), none of which have a role in normalplatelet responses. A fairly close correlation is found indifferent PDI assays for the concentration of bacitracinneeded to provide maximal inhibition of PDI activity,although with platelet aggregation studies the concentrationneeded depended on the specific agonist and its concentra-tion and is subject to other variables inherent to studies ofplatelet responses.
Antibodies to PDI and bacitracin have been used inseparate experiments to demonstrate inhibition of cellsurface PDI activity (Mandel et al, 1993; Ryser et al, 1994;Essex et al, 1995). Monoclonal antibodies used for suchinhibition studies caused dose-related inhibition of only 50%or less (Kaetzel et al, 1987; Mandel et al, 1993), probablybecause they blocked only one of the two PDI active sites.Because polyclonal antibodies to PDI can inhibit up to 100%of PDI activity in the scrambled RNase assay (Chen et al,1995), we used a polyclonal rabbit antibody specific for PDIin our inhibition studies. To avoid any interactions withplatelet Fc receptors, we prepared Fab fragments of the rabbitanti-PDI antibody.
In this study platelet aggregation and secretion wereinhibited by the anti-PDI fragments, a specific biochemicalinhibitor (scrambled RNase), and bacitracin. When biphasicaggregation in PRP was examined using epinephrine andADP, the principle effect of bacitracin was to inhibit thesecond phase of aggregation and the corresponding secre-tion. This suggested that there were PDI-dependent and PDI-independent pathways in these responses. Second-phaseaggregation and the corresponding secretion was inhibitedeven when bacitracin was added shortly after the epinephr-ine (data not shown). This was consistent with anextracellular effect of bacitracin and a role for PDI after theagonist had bound to its receptor and after an initialfibrinogen binding reaction. The blocking of transmission tosecond-wave aggregation by bacitracin implied that thisevent required a conformational change that was facilitated,if not caused, by PDI. With the agonists collagen andthrombin, strong inhibition of PDI completely inhibitedaggregation and the parallel secretion.
In this work we have shown a role for PDI in plateletfunction and GPIIbIIIa activation. The evidence thatsuggests GPIIbIIIa is, in fact, the target for PDI is as follows.(1) Inhibition of platelet aggregation and secretion occurredeven when the PDI inhibitor was added after the agonist,indicating that PDI’s role is after the initial agonist-receptorinteraction. (2) The PDI inhibitors used (scrambled RNase,Fab fragments) are membrane impermeant in contrast toinhibitors such as aspirin, implying that the inhibition seenis primarily a surface phenomenon. (3) PAC-1 binding isinhibited by anti-PDI Fab fragments. Taken together, thesefindings suggest that the target for PDI is in the later phasesof the activation pathway. In support of this are previousstudies showing that reduction of disulphide bonds triggers alater step in the platelet response (DTT-induced aggregationwas not inhibited by PGE1) (MacIntyre & Gordon, 1974b;Zucker & Masiello, 1984) and that disulphide bond reductionactivates the purified GPIIbIIIa receptor (Kouns et al, 1993).In summary, we have demonstrated that PDI mediatesplatelet responses where secretion parallels aggregation andappears to do so by acting on the integrin GPIIbIIIa.
ACKNOWLEDGMENTS
The authors thank Dr Thomas C. Detwiler and Dr Ulhas Naikfor critical reading of the manuscript. This work was
Fig 5. Inhibition by anti-PDI Fab of PAC-1 binding to activatedglycoprotein IIbIIIa. Flow cytometry with FITC-labelled PAC-1:(A) non-activated and collagen-activated platelets; (B) plateletsincubated with anti-PDI Fab (400 mg/ml) or normal rabbit Fab(400 mg/ml) prior to activation with collagen; and (C) plateletsactivated by collagen for 3 min prior to addition of anti-PDI Fab.Collagen (Bio Data) was 48 mg/ml.

supported by a The New York Heart Association Grant-in-Aid for Research.
REFERENCES
Aledort, L.M., Troup, S.B. & Weed, R.I. (1968) Inhibition ofsulfhydryl-dependent platelet functions by penetrating and non-penetrating anologues of parachloromercuribenzene. Blood, 31,471–479.
Beer, J. & Coller, B.S. (1989) Evidence that platelet glycoproteinIIIa has a large disulfide bonded loop that is susceptible toproteolytic cleavage. Journal of Biological Chemistry, 264, 17564–17573.
Calvete, J.J., Henschen, A. & Gonzalez-Rodriguez, J. (1991) Assign-ment of disulfide bonds in human platelet GPIIIa: a disulfidepattern for the beta subunits of the integrin family. BiochemicalJournal, 274, 63–71.
Calvete, J.J., Rivas, G., Maruri, M., Alvarez, M.V., McGregor, J.L., Hew,C.L. & Gonzalez-Rodriguez, J. (1988) Tryptic digestion of GPIIIa.Isolation and biochemical characterization of the 23 kDaN-terminal glycopeptide carrying the antigenic determinant fora monoclonal antibody (P37) which inhibits platelet aggregation.Biochemical Journal, 250, 697–704.
Charo, I.F., Feinman, R.D. & Detwiler, T.C. (1977) Interrelations ofplatelet aggregation and secretion. Journal of Clinical Investigation,66, 866–873.
Chen, K., Detwiler, T.C. & Essex, D.W. (1995) Characterization ofprotein disulphide isomerase released from activated platelets.British Journal of Haematology, 90, 2168–2173.
Darby, N.J. & Creighton, T.E. (1995) Catalytic mechanism of DsbAand its comparison with that of protein disulfide isomerase.Biochemistry, 34, 3576–3587.
Essex, D.W., Chen, K. & Swiatkowska, M. (1995) Localization ofprotein disulfide isomerase to the external surface of the plateletplasma membrane. Blood, 86, 2168–2173.
Hashimoto, K., Sekiya, F., Takagi, J., Tsukada, T., Sato, F. & Saito, Y.(1992) Activation of phospholipases in platelets by polyclonalantibodies against a surface membrane protein. Biochimica etBiophysica Acta, 1165, 27–31.
Hillson, D.A., Lambert, N. & Freedman, R.B. (1984) Formation andisomerization of disulfide bonds in protein: protein disulfideisomerase. Methods in Enzymology, 107, 281.
Kaetzel, C.S., Rao, C.K. & Lamm, M.E. (1987) Protein disulfide-isomerase from human placenta and rat liver: purification andimmunological characterization with monoclonal antibodies.Biochemical Journal, 241, 39–47.
Klappa, P., Hawkins, H.C. & Freedman, R.B. (1997) Interactionsbetween protein disulphide isomerase and peptides. EuropeanJournal of Biochemistry, 248, 37–42.
Klappa, P., Ruddock, L.W., Darby, N.J. & Freedman, R.B. (1998) Theb0 domain provides the principal peptide-binding site of proteindisulfide isomerase but all domains contribute to binding ofmisfolded proteins. EMBO Journal, 17, 927–935.
Kouns, W.C., Jutzi, J., Jennings, L.K. & Steiner, B. (1993) Disulfidereduction ‘activates’ integrin aIIb3 (GPIIb–IIIa). (Abstract).Thrombosis and Haemostasis, 69, 785.
Luz, J.M. & Lennarz, W.J. (1996) Protein disulfide isomerase:a multifunctional protein of the endoplasmic reticulum.
Stress-inducible Cellular Responses (ed. by U. Feige, R. I. Morimoto,I. Yahara. and B. Polla), pp. 97–117. Verlag Basel, Switzerland.
MacIntyre, D.E. & Gordon, J.L. (1974a) Evidence for two populationsof disulphide bonds on blood platelets. Biochemical SocietyTransactions, 2, 873–875.
MacIntyre, D.E. & Gordon, J.L. (1974b) Prostaglandin receptor onblood platelets: effect of thiol reagents on inhibition of plateletaggregation by prostaglandin E1. Biochemical Society Transactions,2, 1265–1269.
MacIntyre, D.E., Grainge, C.A. Drummond, A.H. & Gordon, J.L.(1977) Effect of thiol reagents on platelet transport processes andresponses to stimuli. Biochemical Pharmacology, 26, 319–323.
Mandel, R., Ryser, H.J.-P., Ghani, F., Wu, M. & Peak, D. (1993)Inhibition of a reductive function of the plasma membrane bybacitracin and antibodies against protein disulfide-isomerase.Proceedings of the National Academy of Sciences of the United States ofAmerica, 90, 4112–4116.
Mizunaga, T., Katakura, Y., Miura, T. & Maruyama, Y. (1990)Purification and characterization of yeast protein disulfideisomerase. Journal of Biochemistry, 108, 846–851.
Morjana, N.A. & Gilbert, H.F. (1991) Effect of protein and peptideinhibitors on the activity of protein disulfide isomerase. Biochem-istry, 30, 4985–4990.
Naik, U.P. & Parise, L.V. (1997) Structure and function of plateletaIIb3. Current Opinion in Hematology, 4, 317–322.
Niewiarowski, S., Norton, K.J., Eckardt, A., Lukasiewics, H., Holt, J.C.& Kornecki, E. (1989) Structural and functional characterization ofmajor platelet membrane components derived by limited proteolysisof glycoprotein IIIa. Biochimica et Biophysica Acta, 983, 91–99.
Noiva, R. (1994) Enzymatic catalysis of disulfide formation. ProteinExpression and Purification, 5, 1–13.
Poncz, M., Eisman, R., Heidenreich, R., Silver, S.M., Vilaire, G.,Surrey, S., Schwartz, E. & Bennett, J.S. (1987) Structure of theplatelet membrane glycoprotein IIb: homology to the alphasubunits of the vitronectin and fibronectin membrane receptors.Journal of Biological Chemistry, 262, 8476–8482.
Quan, H., Fan, G. & Wang, C. (1995) Independence of thechaperone activity of protein disulfide isomerase from itsthioredoxin-like active site. Journal of Biological Chemistry, 270,17078–17080.
Roth, R.A. (1981) Bacitracin: an inhibitor of the insulin degradingactivity of glutathione-insulin transhydrogenase. Biochemical andBiophysical Research Communications, 98, 431–438.
Ryser, H.J.-P., Levy, E.M., Mandel, R. & DiSciullo, G.J. (1994)Inhibition of human immunodeficiency virus infection by agentsthat interfere with thiol-disulfide interchange upon virus receptorinteraction. Proceedings of the National Academy of Sciences of theUnited States of America, 91, 4559–4563.
Shattil, S.J., Hoxie, J.A., Cunningham, M. & Brass, L.F. (1985)Changes in the platelet membrane glycoprotein IIb–IIIa complexduring platelet activation. Journal of Biological Chemistry, 260,11107–11114.
Taub, R., Gould, R.J., Garsky, V.M., Ciccarone, T.M., Hoxie, J.,Friedman, P.A. & Shattil, S.J. (1989) A monoclonal antibodyagainst the platelet fibrinogen receptor contains a sequence thatmimics a receptor recognition domain in fibrinogen. Journal ofBiological Chemistry, 264, 259–265.
Zucker, M.B. & Masiello, N.C. (1984) Platelet aggregation caused bydithiothreitol. Thrombosis and Haemostasis, 51, 119–124.
q 1999 Blackwell Science Ltd, British Journal of Haematology 104: 448–454
454 David W. Essex and Mengru Li
Copyright © 2022 FDOKUMEN




















