
Activating Stakeholders: An Approach by MNCs in Emerging Markets
Upload
independentCategory
view
0download
0
Platelet-Activating Factor and Bacteremia-InducedPulmonary Hypertension
Leonardo C. Clavijo, M.D.,* Mary B. Carter, M.D., Ph.D.,† Paul J. Matheson, Ph.D.,†,1
Lisa A. Wills-Frank, M.D.,‡ Mark A. Wilson, M.D., Ph.D.,† William B. Wead, Ph.D.,*and R. Neal Garrison, M.D.†
†Department of Surgery, ‡Department of Pathology, and *Department of Physiology and Biophysics, University of Louisville,and Louisville Veterans Administration Medical Center, Louisville, Kentucky 40206
Journal of Surgical Research 88, 173–180 (2000)doi:10.1006/jsre.1999.5748, available online at http://www.idealibrary.com on
lica
haSmmwoi
vnarti
rwtmcst
Submitted for pub
Background. Acute lung injury is a common compli-cation of gram-negative sepsis. Pulmonary hyperten-sion and increased lung vascular permeability arecentral features of lung injury following experimentalbacteremia. Platelet-activating factor is a prominentproinflammatory mediator during bacterial sepsis.Our previous studies have demonstrated that exoge-nous administration of platelet-activating factor(PAF) induces pulmonary edema without causing pul-monary hypertension. Interestingly, inhibition of PAFactivity during Escherichia coli bacteremia preventsthe development of both pulmonary hypertension andpulmonary edema. These data suggest that PAF con-tributes to pulmonary hypertension during sepsis, butthat this is unlikely to be a direct vascular effect ofPAF. The goal of the present study was to investigatethe mechanism by which acute E. coli bacteremia in-duces pulmonary injury and to define the role thatPAF plays in this injury. We hypothesized that theeffects of PAF on pulmonary hypertension during bac-teremia are due to the effects of PAF on other vascularmediators. Several studies suggest that PAF inducesthe expression of endothelin-1 (ET), a potent peptidevasoconstrictor. Further, our previous studies haveimplicated ET as a central mediator of systemic vaso-constriction during bacteremia. We therefore soughtto assess whether ET is modulated by PAF. E. coli hasalso been demonstrated to increase endothelial pro-duction of nitric oxide (NO), which contributes tomaintenance of basal vascular tone in the pulmonarycirculation. We hypothesized that PAF might increase
Presented at the 23rd Annual Meeting of the Association of Vet-erans Administration Surgeons, St. Louis, MO, May 2–4, 1999.
nt
1 To whom correspondence should be addressed at University ofLouisville—CAMR, 800 Zorn Avenue, Research Building 19, Louis-ville, KY 40206. E-mail: [email protected].
173
tion June 1, 1999
pulmonary vascular resistance during bacteremia byactivating neutrophils, increasing expression of ET,and decreasing the tonic release of NO. Furthermore,we hypothesized that hypoxic vasoconstriction didnot contribute to pulmonary vasoconstriction duringthe first 120 min of E. coli bacteremia.
Methods. Pulmonary artery pressure (PAP), bloodpressure (BP), heart rate (HR), and arterial bloodgases (ABG) were measured in anesthetized spontane-ously breathing adult male Sprague–Dawley rats. E.coli (109 CFU/100 g body wt) was injected at t 5 0, and
emodynamic data were obtained at 10-min intervalsnd ABG data at 30-min intervals for a total of 120 min.ham animals were treated equally but received nor-al saline in place of E. coli. In treatment groups, a 2.5g/kg dose of WEB 2086, a PAF receptor antagonist,as administered intravenously 15 min prior to thenset of sepsis or sham sepsis. The groups were (1)ntravenous E. coli (n 5 5); (2) intravenous WEB 2086
pretreatment 1 intravenous E. coli (n 5 5); (3) intra-enous WEB 2086 alone (n 5 5); and (4) intravenousormal saline (n 5 6). Nitric oxide metabolites (NOx)nd ET concentrations were assayed from arterial se-um samples obtained at the end of the protocol. Lungissue was harvested for measurement of myeloperox-dase (MPO) activity and pulmonary histology.
Results. E. coli bacteremia increased HR, PAP, andespiratory rate early during sepsis (within 20 min),hile hypoxemia, hypotension, and hemoconcentra-
ion were not manifest until the second hour. Pretreat-ent with WEB 2086 completely abrogated all of these
hanges. E. coli bacteremia increased the activity oferum ET, lung MPO, and neutrophil sequestration inhe lung parenchyma via a PAF-dependent mecha-
ism. However, the mechanism of increased produc-ion of NO appears to be PAF independent.Conclusions. These data support the hypothesis that0022-4804/00 $35.00Copyright © 2000 by Academic Press
All rights of reproduction in any form reserved.

tpsl
fLdwmpw
aacoMibnIv
H:
E. coli bacteremia rapidly induces pulmonary hyper-ension stimulated by PAF and mediated at least inart by endothelin-1 and neutrophil activation andequestration in the lung. Microvascular injury witheak is also mediated by PAF during E. coli bactere-
mia, but the time course of resultant hypoxemia andhemoconcentration is slower than that of pulmonaryhypertension. The contribution of hypoxic vasocon-striction in exacerbating pulmonary hypertension ingram-negative sepsis is probably a late (beyond 2 h)phenomenon. PAF receptor antagonism administeredearly in gram-negative sepsis might reduce pulmonaryhypertension and capillary leak. © 2000 Academic Press
Key Words: platelet-activating factor; gram-negativesepsis; Escherichia coli; lung injury; pulmonaryedema; pulmonary artery catheterization; rat; endo-thelin-1; nitric oxide; neutrophil.
INTRODUCTION
Acute respiratory distress syndrome (ARDS) repre-sents the pulmonary component of multisystem organfailure and occurs most often in the face of gram-negative sepsis. Despite advances in intensive caretherapies, the combination of sepsis and ARDS stillcarries a 50% mortality.
One of the most significant of the proinflammatorymediators during acute sepsis is platelet-activatingfactor (PAF). PAF is reported to contribute to hemody-namic instability and organ or cellular dysfunction in awide range of human [1–4], animal [5–8], and in vitro[9, 10] models of sepsis or systemic inflammation. Theorgan that seems to be most susceptible to the conse-quences of PAF is the lung.
In animal models, exogenously administered PAFcauses hemodynamic derangements and organ dys-function which in many ways mimics that seen inexperimental and clinical sepsis. It is reported thatexogenously administered PAF alters vascular reactiv-ity and increases microvascular permeability in thepulmonary system. The exact effects of PAF on thepulmonary vascular bed vary according to species andvessel, with responses on the arterial side differentfrom those on the venous side [11]. We [12] and others[13, 14] have shown that in the rat, exogenously ad-ministered PAF induces both systemic and pulmonaryhypotension, tachycardia, and acute lung injury. Theseeffects are all dose dependent and receptor mediated.
Gram-negative sepsis is a well-described investiga-tional model of systemic inflammation [15]. Interest-ingly, intravenous injection of Escherichia coli or en-dotoxin in the rat rapidly (,20 min) inducespulmonary hypertension and acute lung injury [12],while at the same time increases serum and lung tissue
174 JOURNAL OF SURGICAL RESEARC
concentrations of PAF [5]. Pretreatment with WEB2086, a PAF receptor antagonist, prevents the pulmo-nary vascular and parenchymal changes acutely dur-
ing E. coli bacteremia. Hence, although exogenouslyadministered PAF reduces pulmonary arterial pres-sure in the rat, the binding of PAF to its receptorsduring E. coli sepsis is prerequisite for the develop-ment of pulmonary vascular hypertension.
The present research addresses the following ques-tions: What is the mechanism by which E. coli acutelyinduces pulmonary hypertension in the rat, and whatrole does PAF play in this mechanism? Certainly acutelung injury during gram-negative bacteremia is a mul-tifactorial process during which PAF might shed itsinfluences either by direct or by indirect means. Hy-poxic vasoconstriction is another factor that might in-crease pulmonary artery pressure independent of PAFproduction. Our aim was to further elucidate PAF’seffects on neutrophil activation and on the balancebetween the endothelium-derived paracrine mediatorsnitric oxide (NO), a potent vasodilator, andendothelin-1 (ET), a peptide vasoconstrictor, during E.coli bacteremia in the rat. We also sought to quantitatearterial oxygen content using blood gas analysis toassess the contribution of hypoxic vasoconstriction toincreased pulmonary artery pressure in this model.Our hypothesis was that PAF might increase pulmo-nary vascular resistance during bacteremia by activat-ing neutrophils, by increasing release of ET, and bydecreasing the tonic release of NO. Furthermore, wehypothesized that hypoxic vasoconstriction would notcontribute significantly to pulmonary vasoconstrictionduring the first 120 min of E. coli bacteremia.
MATERIALS AND METHODS
Animal Preparation
All experimental protocols were approved by the VAMC AnimalStudies Subcommittee of the Research and Development Committee.Pathogen-free male Sprague–Dawley rats (284–341g body wt, mean305.7 6 3.2 g) were housed using reverse isolation in an animal careacility approved by the American Association for Accreditation ofaboratory Animal Care (AAALAC). Food but not water was with-rawn 12 h prior to experimentation. Animals were anesthetizedith pentobarbital (50 mg/kg) ip. Repeated doses of pentobarbital (17g/kg) were given ip as needed to maintain anesthesia. Body tem-
erature was measured with a rectal probe and maintained at 37°Cith a heating pad.Tracheostomy was performed with PE-240 polyethylene tubing,
nd animals breathed room air for the entire experiment. In somenimals (Experiment 1) the right femoral artery and vein wereannulated with stretched PE-90 and PE-50, respectively. The fem-ral artery was connected to a pressure transducer (TXD-310, Digi-ed, Louisville, KY) and a blood pressure analyzer (Digi-Med, Lou-
sville, KY). The right carotid artery was cannulated with PE-50 forlood gas determination. Normal saline (1 cc) was injected subcuta-eously on an hourly basis to compensate for insensible fluid losses.n other animals (Experiment 2) only the carotid artery and jugularein were cannulated with PE-50.
Closed Chest Pulmonary Artery Catheterization
VOL. 88, NO. 2, FEBRUARY 2000
We modified the closed-chest pulmonary artery (PA) catheteriza-tion technique originally described by Herget and Palecek [16].

2a
ba
swsfF
wpac
PU
Briefly, a 13-cm-long Silastic PA catheter (0.012 i.d., 0.025 o.d.) andintroducer (7.5-cm PE-90 tube with the tip turned anteriorly 30°)were passed via the right jugular vein into the right ventricle. TheSilastic catheter was advanced into the PA, and the introducer wasremoved from the atrium. The PA catheter was attached to a pres-sure transducer (TXD-310, Digi-Med) and low-pressure analyzer(LPA-2000 Digi-Med). A calibrated oscilloscope (Tektronix 2213,London, England) was used to assess the hemodynamic waveform.The catheter position was confirmed in vivo by the characteristicpulmonary artery wave profile and at autopsy by dissection of thepulmonary arteries.
Bacteremia Model
Acute bacteremia was induced by infusion of live E. coli [1 3 109
colony-forming units (CFU) per 100 g body wt over 5 min] whichconsistently produces a hyperdynamic state with elevated cardiacoutput and normal blood pressure [15].
Data Collection
Experiment 1
Hemodynamic variables. After 30 min of hemodynamic stabiliza-tion, two baseline measurements of mean arterial blood pressure(MAP), heart rate (HR), and mean pulmonary artery pressure (PAP)were obtained at 15-min intervals and averaged (BL-avg). MAP andHR were measured from the femoral arterial line, and PAP wasmeasured from the PA catheter.
Arterial blood gas and hemoglobin measurements. Arterial blood(0.2–0.3 cc) was drawn through the carotid artery cannula into aheparanized 1.0-cc syringe and blood gases were immediately ana-lyzed on a Radiometer ABL300 Acid–Base Laboratory (Copenhagen,Denmark). Arterial pH, PO2, PCO2, and hemoglobin were recorded,and the arterial line was flushed with 0.3 cc of dilute heparinizednormal saline (10 units/cc).
Experimental Groups
Experiment 1: Gram-Negative Bacteremia and PAFReceptor Inhibition: Hemodynamic Data andArterial Blood Gases
In the first experiment, animals were divided into four groups.Group 1 (E. coli, n 5 5) received E. coli (109 CFU/100 g body wt in1 cc normal saline) through the femoral vein over 5 min beginning att 5 0. Group 2 (Sham, n 5 6) received normal saline in place of E.coli at t 5 0. Group 3 (E. coli 1 WEB, n 5 5) received 2.5 mg/kgWEB 2086 iv (courtesy of Boehringer Ingelheim, Germany) dilutedin 1 cc of normal saline 15 min prior to infusion of E. coli. Group 4(Sham 1 WEB) received an equivalent dose of WEB 2086 15 minprior to t 5 0 and normal saline in place of E. coli. Hemodynamicmeasurements (MAP, HR, PAP) were obtained at 10-min intervals,and blood gas and hemoglobin measurements were taken at 30-minintervals for a total of 120 min. After the final hemodynamic andblood gas determination, animals were euthanized.
Experiment 2: Gram-Negative Bacteremia and PAFReceptor Inhibition: Pulmonary Nitric OxideMetabolites, Endothelin-1, Myeloperoxidase,and Histology
In the second experiment, animals were divided into three groupsof animals (n 5 7 in each group). Group 1 received E. coli (109
CFU/100 g body wt in 1 cc normal saline) through the jugular vein
CLAVIJO ET AL.: PAF AND
over 5 min beginning at t 5 0. Group 2 received normal saline inplace of E. coli at t 5 0. Group 3 (E. coli 1 WEB) received 2.5 mg/kgWEB 2086 iv diluted in 1 cc of normal saline 15 min prior to infusion
of E. coli. After 120 min, arterial blood from the carotid artery washarvested in a heparanized syringe and spun at 5000g for 7 min, andthe serum stored in aliquots at 280°C for nitric oxide metabolite andendothelin assay. Animals were then placed on a rodent ventilator(Harvard Apparatus, Model 683, South Natick, MA) and ventilatedat 70 breaths per minute with room air, tidal volume 2.0 cc, and 1-cmH2O positive end-expiratory pressure. After midline thoracotomy, a0-ml bolus of normal saline was injected into the right ventricle atpressure of 20 cm H2O to wash out the pulmonary intravascular
content. The right middle lobe was excised and placed in a 10%formaldehyde solution for histological analysis. The right lower lobewas excised, weighed, homogenized in 2 ml of cold homogenizationbuffer, centrifuged at 3900g for 10 min, resuspended in suspensionuffer, and stored at 220°C for myeloperoxidase (MPO) activityssay. Animals were euthanized.
Nitric oxide metabolite assay. Nitric oxide metabolites were mea-ured in serum by a fluorescent modified-Greiss reaction assay [17]hich has been previously described in detail [18]. Frozen serum
amples were thawed and filtered in 10,000-MW-cutoff microcentri-uge filter units (Millipore, Norwalk, CT) for 30 min at 10,000g.iltered serum samples were aliquoted (30 ml per well) into six wells
of an opaque 96-well microplate: two for serum blanks, two fornitrite, and two for total nitrite plus nitrate. Total nitrite plus nitratewas measured by enzymatic reduction of sample nitrate to nitritewith nitrate reductase (Aspergillus niger, 14.3 mU/well) in the pres-ence of NADPH (final well concentration, 48 mM) for 10 min at 20°Cwith gentle shaking. Standards were run in triplicate for nitrite andtotal nitrite plus nitrate. Fluorescence was measured on a Cam-bridge Technology Model 7600 (Cambridge, MA) fluorescent micro-plate reader (excitation, 365 nm; emission, 450 nm). First-orderregressions of the standard curves produced r 2 values of 0.9945 and0.9961 in these experiments, demonstrating a high degree of corre-lation between standard concentration and measured fluorescenceintensity (arbitrary fluorescent units).
Endothelin-1 assay. Endothelin-1 was measured in serum with acommercially available ELISA kit (Cayman Chemical Co., Ann Ar-bor, MI) according to the kit instructions. Frozen serum sampleswere thawed and filtered in 0.22-mm-pore-size Millipore microcen-trifuge filters at 10,000g for 30 min. Serum samples (100 ml per well)
ere assayed in quadruplicate with two serum blanks for each sam-le. Absorbance (405 nm with 620-nm internal blank) was read withBio-Rad automatic plate reader and endothelin concentration was
alculated from second-order regression of the standards (r 2 50.9999).
Myeloperoxidase assay. Myeloperoxidase was measured in tissuesamples by previously described methods [19, 20]. Briefly, homoge-nized tissue samples were thawed and sonicated for 10 s prior toheating the samples to 60°C for 2 h. Samples were refrozen (220°C)overnight prior to performing the assay. On the day of the assay,samples were thawed and centrifuged (5000g for 5 min), and super-natants were aliquoted into 12 3 75-mm glass tubes (100 ml/pertube). Samples were incubated with cocktail buffer containing tetra-methylbenzidine in dimethylformamide (Sigma Chemical Co., St.Louis, MO) and 15 mM hydrogen peroxide for 20 min at 37°C in ashaking water bath. Absorbance was measured at 655 nm for sam-ples and myeloperoxidase standards (first order, r 2 5 0.9990) andmyeloperoxidase activity was calculated for each sample.
Histology. Lung specimens in 10% formaldehyde were mountedin paraffin, sectioned into slices 4 mm thick, and stained with hema-toxylin and eosin. In a blinded fashion, histological examination wasperformed using light microscopy at 43, 103, 203, and 403 magni-
175LMONARY HYPERTENSION
fication. Specimens were graded on a scale of 1 to 10 for alveolar andcapillary wall integrity and on a scale of 1 to 5 for neutrophil infil-tration into the parenchymal tissue and alveolar space.
(dtamm
a(mpa
hmEa
Pt0
B
g
H:
Statistical Analysis
All data are expressed as means 6 SEM. To assess differencesbetween groups, one-way analysis of variance (ANOVA) withTukey’s test for honestly significant difference (HSD) was used. Toassess changes in a variable from baseline, one-way analysis ofvariance for repeated measures (REMANOVA) with Tukey’s HSDwas performed. The null hypothesis was rejected for P , 0.05.
RESULTS
Experiment 1
Hemodynamic Data
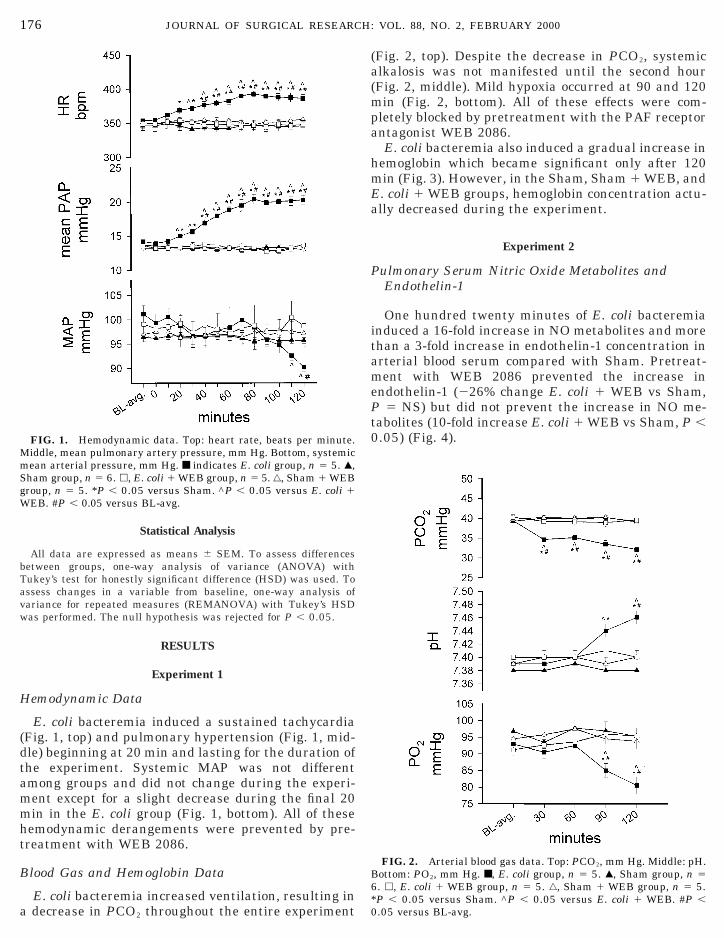
E. coli bacteremia induced a sustained tachycardiaFig. 1, top) and pulmonary hypertension (Fig. 1, mid-le) beginning at 20 min and lasting for the duration ofhe experiment. Systemic MAP was not differentmong groups and did not change during the experi-ent except for a slight decrease during the final 20in in the E. coli group (Fig. 1, bottom). All of these
hemodynamic derangements were prevented by pre-treatment with WEB 2086.
Blood Gas and Hemoglobin Data
FIG. 1. Hemodynamic data. Top: heart rate, beats per minute.Middle, mean pulmonary artery pressure, mm Hg. Bottom, systemicmean arterial pressure, mm Hg. ■ indicates E. coli group, n 5 5. Œ,Sham group, n 5 6. h, E. coli 1 WEB group, n 5 5. ‚, Sham 1 WEBroup, n 5 5. *P , 0.05 versus Sham. ^P , 0.05 versus E. coli 1
WEB. #P , 0.05 versus BL-avg.
176 JOURNAL OF SURGICAL RESEARC
E. coli bacteremia increased ventilation, resulting ina decrease in PCO2 throughout the entire experiment
*0
(Fig. 2, top). Despite the decrease in PCO2, systemiclkalosis was not manifested until the second hourFig. 2, middle). Mild hypoxia occurred at 90 and 120
in (Fig. 2, bottom). All of these effects were com-letely blocked by pretreatment with the PAF receptorntagonist WEB 2086.E. coli bacteremia also induced a gradual increase in
emoglobin which became significant only after 120in (Fig. 3). However, in the Sham, Sham 1 WEB, and. coli 1 WEB groups, hemoglobin concentration actu-lly decreased during the experiment.
Experiment 2
Pulmonary Serum Nitric Oxide Metabolites andEndothelin-1
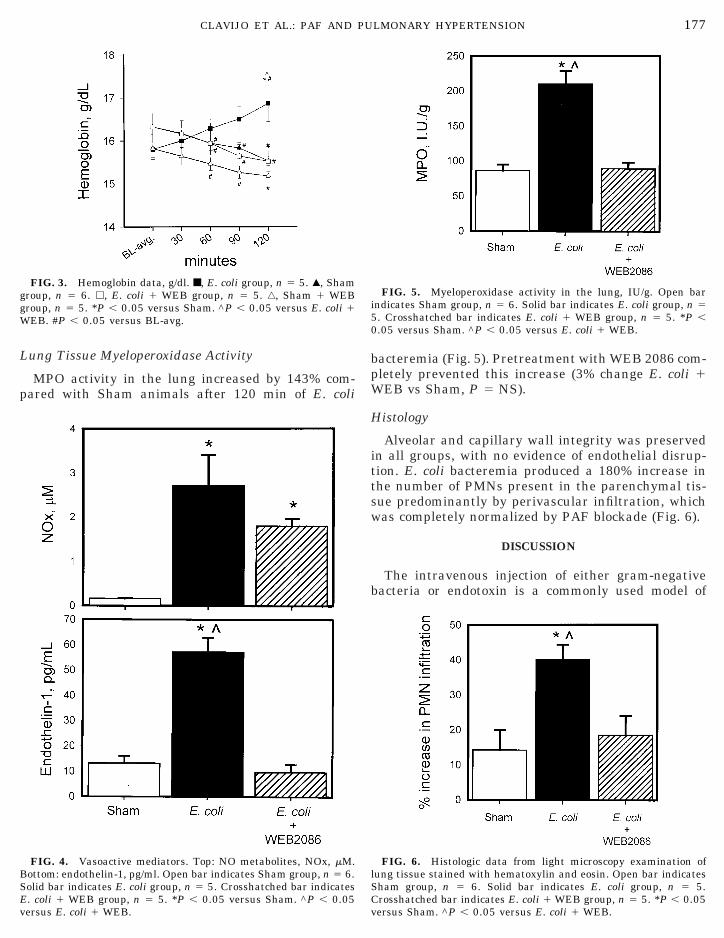
One hundred twenty minutes of E. coli bacteremiainduced a 16-fold increase in NO metabolites and morethan a 3-fold increase in endothelin-1 concentration inarterial blood serum compared with Sham. Pretreat-ment with WEB 2086 prevented the increase inendothelin-1 (226% change E. coli 1 WEB vs Sham,
5 NS) but did not prevent the increase in NO me-abolites (10-fold increase E. coli 1 WEB vs Sham, P ,.05) (Fig. 4).
FIG. 2. Arterial blood gas data. Top: PCO2, mm Hg. Middle: pH.ottom: PO2, mm Hg. ■, E. coli group, n 5 5. Œ, Sham group, n 5
VOL. 88, NO. 2, FEBRUARY 2000
6. h, E. coli 1 WEB group, n 5 5. ‚, Sham 1 WEB group, n 5 5.P , 0.05 versus Sham. ^P , 0.05 versus E. coli 1 WEB. #P ,.05 versus BL-avg.
BSE
ggW 5
0
PU
Lung Tissue Myeloperoxidase Activity
MPO activity in the lung increased by 143% com-pared with Sham animals after 120 min of E. coli
FIG. 4. Vasoactive mediators. Top: NO metabolites, NOx, mM.ottom: endothelin-1, pg/ml. Open bar indicates Sham group, n 5 6.
FIG. 3. Hemoglobin data, g/dl. ■, E. coli group, n 5 5. Œ, Shamroup, n 5 6. h, E. coli 1 WEB group, n 5 5. ‚, Sham 1 WEBroup, n 5 5. *P , 0.05 versus Sham. ^P , 0.05 versus E. coli 1EB. #P , 0.05 versus BL-avg.
CLAVIJO ET AL.: PAF AND
olid bar indicates E. coli group, n 5 5. Crosshatched bar indicates. coli 1 WEB group, n 5 5. *P , 0.05 versus Sham. ^P , 0.05
versus E. coli 1 WEB.C
bacteremia (Fig. 5). Pretreatment with WEB 2086 com-pletely prevented this increase (3% change E. coli 1WEB vs Sham, P 5 NS).
Histology
Alveolar and capillary wall integrity was preservedin all groups, with no evidence of endothelial disrup-tion. E. coli bacteremia produced a 180% increase inthe number of PMNs present in the parenchymal tis-sue predominantly by perivascular infiltration, whichwas completely normalized by PAF blockade (Fig. 6).
DISCUSSION
The intravenous injection of either gram-negativebacteria or endotoxin is a commonly used model of
FIG. 6. Histologic data from light microscopy examination oflung tissue stained with hematoxylin and eosin. Open bar indicates
FIG. 5. Myeloperoxidase activity in the lung, IU/g. Open barindicates Sham group, n 5 6. Solid bar indicates E. coli group, n 5
. Crosshatched bar indicates E. coli 1 WEB group, n 5 5. *P ,
.05 versus Sham. ^P , 0.05 versus E. coli 1 WEB.
177LMONARY HYPERTENSION
Sham group, n 5 6. Solid bar indicates E. coli group, n 5 5.rosshatched bar indicates E. coli 1 WEB group, n 5 5. *P , 0.05
versus Sham. ^P , 0.05 versus E. coli 1 WEB.

trt[mhe
c
t9gtcem
2hbcavsrb
setvsomrvssd(
vtN
aiotvoemp
1 H:
systemic inflammation. Many investigators use thesemodels, rather than cecal ligation and puncture, tostudy the pathophysiology of septic shock due to therapid induction of hemodynamic derangement and or-gan dysfunction, plus the ease in administering andcontrolling dose, which decreases variability and im-proves reproducibility. Gram-negative bacteremia hasbeen shown to produce pulmonary arterial hyperten-sion, hypoxia, increased lung MPO activity, and in-creased protein and neutrophil count in bronchoalveo-lar lavage fluid [21–24]. One of the clinical hallmarksof sepsis is a hyperdynamic phase characterized by anearly increase in cardiac output with decreased totalperipheral resistance, followed by a chronic hypody-namic phase with impaired cardiac performance. Inthis model, systemic mean arterial pressure and car-diac index biphasically increase acutely and decreasethereafter [22].
The hemodynamic responses to E. coli bacteremia inhe present study are consistent with those previouslyeported [15, 25], including a rapid onset of profoundachycardia, pulmonary hypertension, and tachypnea23]. Systemic hypotension did not manifest until 110in elapsed time, indicating that animals are in theyperdynamic phase of sepsis for the majority of thexperiment.Despite decreased PCO2, blood pH determinations in
septic animals did not demonstrate alkalosis until thesecond hour of the experiment. We interpret these datato indicate a metabolic acidosis was established lessthan 30 min after the onset of bacteremia. Hence, withthe injection of 109 CFU of E. coli per 100 g body wt,septic shock-induced conversion of cellular metabolismto anaerobic metabolism occurs rapidly in this model. Acompensatory tachypnea ensues which maintains theoverall pH at normal levels.
Although pulmonary vasoconstriction was evidentwithin 20 min of onset of bacteremia, arterial PO2 didnot decrease until 90 min elapsed time. We interpretthis to mean that in this model, pulmonary vasoactivesubstances are produced rapidly and exert their effectson the pulmonary vasculature early on. In contrast,pulmonary endothelial cell damage and basementmembrane disruption is a slower process, taking morethan an hour to accumulate sufficient interstitial fluidto cause decreased PO2 on room air. This is furthersupported by the slow increase in hemoglobin concen-tration seen in the E. coli group. It is likely that endo-thelial cell damage and microvascular leak occurredthroughout the peripheral as well as pulmonary vas-cular bed, causing extravasation of fluid out of thevascular space, resulting in hemoconcentration. How-ever, it took the entire 120 min of elapsed time for the
78 JOURNAL OF SURGICAL RESEARC
hemoconcentration to become statistically significant.The decrease in PO2, although statistically signifi-
ant, was probably not of physiological significance,
b
c
i.e., was not severe enough to result in hypoxic vaso-constriction. At a PO2 of 80.5 6 2.6 mm Hg (which washe lowest recorded in the E. coli group) greater than0% of hemoglobin molecules are saturated with oxy-en. Thus, it is highly likely that hypoxic vasoconstric-ion was not occurring in any significant amount toontribute to the increase in PAP observed in thesexperiments. Hypoxic vasoconstriction, therefore,ust be a “late” event if it occurs at all in this model.Pretreatment with the PAF receptor antagonist WEB
086 completely abrogated the tachycardia, pulmonaryypertension, systemic hypotension, tachypnea, meta-olic acidosis, and hemoconcentration. These data impli-ate PAF as a significant early mediator of cellular met-bolic dysfunction, microvascular leak, and pulmonaryascular constriction in the acute stages of gram-negativeepsis. Others [5, 26] report similar findings after PAFeceptor antagonism in rodent models of gram-negativeacteremia and endotoxemia.Gram-negative sepsis in both animal and human
tudies increases production of nitric oxide andndothelin-1 [27–29]. Nitric oxide is a potent vasodila-or which causes cyclic GMP-dependent relaxation inascular smooth muscle. Profound systemic hypoten-ion of clinical septic shock has been attributed toverproduction of NO [29]. Endothelin-1 is one of theost potent vasoconstrictors known, and ET might be
esponsible for the vasoconstriction reported in theascular beds of the kidney and intestine during septichock [30–32]. Hence, different vascular beds demon-trate differing vasoactive responses to systemic sepsisepending on the local balance between vasodilatorNO) and vasoconstrictor (ET) mediators.
To explore the mechanism by which PAF mediatesascular responses during sepsis, we measured concen-rations of ET and the stable NO metabolites NO2 andO3 in arterial blood. Two hours after the onset of E.
coli bacteremia, ET increased by 335% and NO metab-olites by 1600% compared with Sham animals. In thecase of ET, this increase was completely blocked byWEB 2086, but in the case of NO metabolites, PAFreceptor blockade showed only a trend toward an ef-fect. Hence, gram-negative sepsis induces productionof ET in a PAF-dependent mechanism, but E. coli prob-bly increases NO expression primarily by a PAF-ndependent mechanism. The fact that NO productionccurs by a mechanism independent of PAF might beeleologically advantageous. Others report that pre-ention of NO synthesis greatly worsens cellular andrgan dysfunction attributed to PAF during endotox-mia or bacteremia [33–35]. Especially in the lung, NOay exert a protective effect [34]. Perhaps the uncou-
ling of PAF from NO production provides a survival
VOL. 88, NO. 2, FEBRUARY 2000
enefit.The paradoxical findings that E. coli bacteremia
auses pulmonary hypertension which can be pre-

tpcgtmit
spvNtsrepppvn
mcaPrn
[Ptondrmtootnc“ftcbatams
tcdnbem
PU
vented by PAF receptor antagonism and that directintravenous PAF administration induces pulmonaryhypotension are not clearly understood [12]. This ob-servation might be explained by the idea that endo-toxin stimulates the production of many vasoactivecompounds (constrictors and dilators) by both PAF-and non-PAF-mediated pathways. Overall vasculartone is determined by the balance of constrictor anddilator forces present at any given time. Previous stud-ies demonstrate that PAF directly causes receptor-mediated systemic and pulmonary vasodilation [11–14]. The present study indicates that endotoxinstimulates ET production by a PAF-dependent butprobably indirect mechanism, but PAF’s dilatory ef-fects were not directly measured. PAF blockade earlyafter bacteremia appears to halt both direct PAF-mediated vascular effects and the complete progressionof the inflammatory cascade. In other words, PAF ap-pears to occupy a position early in the cascade and PAFblockade prevents direct PAF effects (vasodilation) aswell as indirect effects (production of other vasocon-strictor compounds such as ET) and eliminates theircontribution to vascular tone. Thus, during acute bac-teremia the direct effect of PAF on vascular tone mightbe dilatory, while the overall effect of bacteremia isinitially vasoconstriction and hypertension.
We further investigated the effects that E. coli bac-eremia had on pulmonary sequestration of neutro-hils. Lung myeloperoxidase activity and neutrophilounts both increased by 143 and 180% over Shamroups, respectively. In each case, pretreatment withhe PAF receptor antagonist WEB 2086 completelyitigated the response. Hence, the binding of PAF to
ts receptors is necessary for early neutrophil seques-ration in the lung in this model.
Neutrophil sequestration and activation has beenuggested to be one of the primary mechanisms of bothulmonary microvascular leak [36–38] and pulmonaryascular hypertension observed during sepsis [38].eutrophil-induced injury to the microvasculature in
he lung is shown to decrease vessels’ response to va-odilators [38, 39] and to increase permeability of mac-omolecules [36]. So, in this model, although exog-nously administered PAF is itself a vasodilator in theulmonary vascular bed [14], during sepsis PAF ap-ears to be prerequisite for the development of bothulmonary artery hypertension and pulmonary micro-ascular leak, mediated perhaps in part by activatedeutrophils and endothelin-1.One criticism of the present study is that pretreat-ent of animals with WEB 2086 does not mimic the
linical scenario where treatment is instituted onlyfter patients become septic. Although in this study
CLAVIJO ET AL.: PAF AND
AF receptor antagonism pretreatment completely ab-ogated many of the deleterious effects of gram-egative bacteremia, recent animal [40] and human
41] studies report no improvement in outcome whenAF receptor antagonism is used to treat already es-ablished septic shock. However, the fact that clinicalutcomes were not improved by PAF receptor antago-ist does not prove that PAF is not an important me-iator of clinical sepsis, and it does not mean that PAFeceptor antagonism has no role in the prevention oforbidity and mortality during septic shock. The sys-
emic inflammatory response is composed of a numberf enzymatic cascades which are both redundant andverlapping. Because of this redundancy, it is intuitivehat blockade of just one mediator of inflammation willot be sufficient to turn “off” the entire system. When itomes to systemic inflammatory response, there is nosilver bullet.” It is our hypothesis that clinical benefitrom manipulation of the inflammatory cascade inreatment of septic shock will not be manifest until theascade is blocked in several key points, with PAFeing only one such key point. Blockade of other medi-tors, such as complement, oxygen radicals, coagula-ion cascade, interleukins, leukotrienes, and thrombox-nes, in combination with PAF receptor blockade,ight be necessary to see clinical benefit and improved
urvival during septic shock.In summary, E. coli induces early pulmonary hyper-
ension, late hypoxemia, increased serum endothelin-1oncentration, and increased pulmonary myeloperoxi-ase and neutrophil sequestration, all of which areormalized with blockade of platelet-activating factory WEB 2086 pretreatment. E. coli also increases NOxpression, most probably by a PAF-independentechanism. E. coli-induced pulmonary hypertension
might be stimulated by PAF at least in part byendothelin-1 production and neutrophil activation. Hy-poxic vasoconstriction is probably not important withinthe first 2 h of bacteremia in this model.
REFERENCES
1. Bussolino, F., Porcellini, M. G., Varese, L., and Bosia, A. Intra-vascular release of platelet-activating factor in children withsepsis. Thromb. Res. 48: 619, 1987.
2. Graham, R. M., Stephens, C. J., Silvester, W., Leong, L. L.,Strum, M. J., and Taylor, R. R. Plasma degradation of platelet-activating factor in severely ill patients with clinical sepsis.Crit. Care Med. 22: 204, 1994.
3. Anderson, B. O., Bensard, D. D., and Harken, A. H. The role ofplatelet-activating factor and its antagonist in shock, sepsis andmultiple organ failure. Surgery 172: 415, 1991.
4. Fink, A., Geva, D., Zung, A., Konichezky, S., Eliraz, A., andBentwich, Z. Adult respiratory distress syndrome: Roles of leu-kotriene C4 and platelet-activating factor. Crit. Care Med. 18:905, 1990.
5. Chang, S., Fedderson, C. O., Henson, P. M., and Voekel, N. F.Platelet-activating factor mediates hemodynamic changes andlung injury in endotoxin-treated rats. J. Clin. Invest. 79: 1498,
179LMONARY HYPERTENSION
1987.6. Olson, N. C., Joyce, P. B., and Fleisher, L. N. Mono-
hydroxyeicosatetraenoic acids during porcine endotoxemia: Ef-

2
2
3
H:
fect of a platelet-activating factor receptor antagonist. Lab.Invest. 63: 221, 1989.
7. Carter, M. B., Wilson, M. A., Wead, W. B., and Garrison, R. N.Platelet-activating factor mediates pulmonary macromolecularleak following intestinal ischemia–reperfusion. J. Surg. Res. 60:403, 1996.
8. Filep, J., Herman, F., Braquet, P., and Mozes, T. Increased levelsof platelet-activating factor in blood following intestinal ischemiain the dog. Biochem. Biophys. Res. Commun. 158: 353, 1989.
9. Lewis, J. C., O’Flaherty, J. T., McCall, C. E., Wykle, R. L., andBond, M. G. Platelet-activating factor effects on pulmonaryultrastructure in rabbits. Exp. Mol. Pathol. 38: 100, 1983.
10. Bussolino, F., Camussi, G., Aglietta, M., Braquet, P., Bosia, A.,Pescarmona, G., Sanavio, F., D’Urso, N., and Marchisio, P. C.Human endothelial cells are target for platelet-activating fac-tor. J. Immunol. 139: 2439, 1987.
11. Gao, Y., Zhou, H., and Raj, U. PAF induces relaxation of pul-monary arteries but contraction of pulmonary veins in theferret. Am. Phys. 269: H704, 1985.
12. Clavijo, L. C., Carter, M. B., Matheson, P. J., Wilson, M. A.,Wead, W. B., and Garrison, R. N. Platelet-activating factorincreases pulmonary vascular permeability without increasingpulmonary artery pressure. Submitted for publication.
13. Laurindo, F. R. M., Goldstein, R. E., Davenport, N. J., Ezra, D.,and Feuerstein, G. Z. Mechanisms of hypotension produced byplatelet-activating factor. J. Appl. Physiol. 66: 2681, 1989.
14. McMurtry, I. F., and Morris, K. G. Platelet-activating factorcauses pulmonary vasodilation in the rat. Am. Rev. Respir. Dis.134: 757, 1986.
15. Spain, D. A., Wilson, M. A., and Garrison, R. N. The role ofnitric oxide in regulation of the rat small intestinal microcircu-lation during bacteremia. Shock 2: 41, 1994.
16. Herget, J., and Palecek, F. Pulmonary arterial blood pressurein closed chest rats: Changes after catecholamines, histamineand serotonin. Arch. Int. Pharmacodyn. 198: 107, 1972.
17. Misko, T. P., Schilling, R. J., Salvemini, D., Moore, W. M., andCurrie, M. G. A fluorometric assay for the measurement ofnitrite in biological samples. Anal. Biochem. 214: 11, 1993.
18. Matheson, P. J., Spain, D. A., Harris, P. D., Garrison, R. N., andWilson, M. A. Glucose and glutamine gavage increase portalvein nitric oxide metabolite levels via adenosine A2b activation.J. Surg. Res. 83: 105, 1999.
19. Grisham, M. B., Benoit, J. N., and Granger, D. N. Assessmentof leukocyte involvement during ischemia and reperfusion ofintestine. Methods Enzymol. 186: 729, 1990.
20. Schierwagen, C., Bylund-Fellenius, A.-C., and Lundberg, C.Improved method for quantitation of tissue PMN accumulationmeasured by myeloperoxidase activity. J. Pharmacol. Methods23: 179, 1990.
21. Bloomfield, G. L., Ridings, P. C., Blocher, C. R., Fisher, B. J.,Sugerman, H. J., Nagamoto, H., and Fowler, A. A. OPC-6535, asuperoxide anion production inhibitor, attenuates acute lunginjury. J. Surg. Res. 72: 70, 1997.
22. Ridings, P. C., Hollowway, S., Gbloomfield, G. L., Phillips,M. L., Fisher, B. J., Blocher, C. R., Sugarman, H. J., andFowler, A. A. Protective role of synthetic sialylated oligosaccha-ride in sepsis-induced acute lung injury. J. Appl. Physiol. 82:644, 1997.
23. Welty-Wolf, K. E., Carraway, M. S., Huang, Y. C. T., Simonson,S. G., Kantrow, S. P., and Piantadosi, C. A. Bacterial primingincreases lung injury in gram-negative sepsis. Am. J. Respir.Crit. Care Med. 158: 610, 1998.
180 JOURNAL OF SURGICAL RESEARC
24. Carraway, M. S., Welty-Wolf, K. E., Kantrow, S. P., Huang,Y. C. T., Simonson, S. G., Que, L. G., Kishimoto, T. K., andPiantadosi, C. A. Antibody to E- and L-selectin does not prevent
lung injury or mortality in septic baboons. Am. J. Respir. Crit.Care Med. 157: 938, 1998.
25. Bar-Natan, M. F., Wilson, M. A., Spain, D. A., and Garrison,R. N. Platelet-activating factor and sepsis-induced small intes-tinal microvascular hypoperfusion. J. Surg. Res. 58: 38, 1995.
26. Miotla, J. M., Jeffery, P. K., and Hellewell P. G. Platelet-activating factor plays a pivotal role in the induction of exper-imental lung injury. Am. J. Respir. Cell Mol. Biol. 18: 197, 1998.
27. Hom, G. J., Grant, S. K., Wolfe, G., Bach, T. J., Macintyre, D. E.,and Hutchinson N. I. Lipopolysaccharide-induced hypotensionand vascular hyporeactivity in the rat: Tissue analysis of nitricoxide synthase mRNA and protein expression in the presenceand absence of dexamethasone, NG-monomethyl-L-arginine orindomethacin. J. Pharmacol. Exp. Ther. 272: 452, 1995.
8. Buttery, L. D. K., Evans, T. J., Springall, D. R., Carpenter, A.,Cohen, J., and Polak, J. M. Immunochemical localization ofinducible nitric oxide synthase in endotoxin-treated rats. Lab.Invest. 71: 755, 1994.
9. Petros, A., Bennett, D., and Vallance P. Effect of nitric oxidesynthase inhibitors on hypotension in patients with septicshock. Lancet 338: 1557, 1991.
0. Chou, M. C., Wilson, M. A., Spain, D. A., Hadjiminas, D.,Anderson, G. L., Cheadle, W. G., and Garrison, R. N.Endothelin-1 expression in the small intestine during chronicperitonitis. Shock 4: 411, 1995.
31. Wilson, M. A., Steeb, G. D., and Garrison, R. N. Endothelinsmediate intestinal hypoperfusion during bacteremia. Shock 55:168, 1993.
32. Bloom I. T., Bentley, F. R., Wilson, M. A., and Garrison, R. N. Invivo effects of endothelin on the renal microcirculation. J. Surg.Res. 54: 274, 1993.
33. Spain, D. A., Wilson, M. A., Bar-Natan, M. F., and Garrison,R. N. Nitric oxide synthase inhibition aggravates intestinalmicrovascular vasoconstriction and hypoperfusion of bactere-mia. J. Trauma 36: 720, 1994.
34. Gryglewski, R. J., Wolkow, P. P., Uracz, W., Janowska, E.,Bartus, J. B., Balbatun, O., Patton, S., Brovkovych, V., andMalinski, T. Protective role of pulmonary nitric oxide in theacute phase of endotoxemia in rats. Circ. Res. 82: 819, 1998.
35. Meyer, J., Lentz, C. W., Stothert, J. C., Traber, L. D., Herndon,D. N., and Traber, D. L. Effects of nitric oxide synthesis inhibitionin hyperdynamic endotoxemia. Crit. Care Med. 22: 306, 1994.
36. Repine, J. E., Bowman, M., and Tate, R. M. Neutrophils andlung edema. Chest 81: 475, 1982.
37. Tate, R. M., and Repine, J. E. Neutrophils and the adult respi-ratory distress syndrome. Am. Rev. Respir. Dis. 128: 532, 1990.
38. Sheridan, B. C., McIntyre, R. C., Jr., Moore, E. E., Meldrum,D. R., Agrafojo, J., and Fullerton D. A. Neutrophils mediatepulmonary vasomotor dysfunction in endotoxin-induced acutelung injury. J. Trauma 42: 391, 1997.
39. Sheridan B. C., McIntyre, R. C., Agrafojo, J. Meldrum, D. R.,Meng, X., and Fullerton, D. A. Neutrophil depletion attenuatesendotoxin-induced dysfunction of cGMP-mediated pulmonaryvasorelaxation. Am. J. Phys. 271: L820, 1996.
40. Spapen, H., Zhang, H., Verhaeghe, V., Rogiers, P., Cabral, A.,and Vincent, J. L. Treatment with a platelet-activating factorantagonist has little protective effects during endotoxic shock inthe dog. Shock 8: 200, 1997.
41. Dhainaut, J.-F. A., Tenaillon, A., Tulzo, Y. L., Schlemmer, B. S.,Solet, J.-P., Wolff, M., Holzapfel, L., Zeni, F., Dreyfuss, D.,Mira, J.-P. Vathaire, F. D., Guinot, P., and the BN 52021 SepsisStudy Group. Platelet-activating factor receptor antagonist BN
VOL. 88, NO. 2, FEBRUARY 2000
52021 in the treatment of severe sepsis: A randomized, double-blind, placebo-controlled, multicenter clinical trial. Crit. CareMed. 22: 1720.
Copyright © 2022 FDOKUMEN




















