
Reticulon1-C modulates protein disulphide isomerase function
Upload
independentCategory
view
1download
0
The crystal structure of human WD40 repeat-containingpeptidylprolyl isomerase (PPWD1)Tara L. Davis1,2, John R. Walker1, Hui Ouyang1, Farrell MacKenzie1, Christine Butler-Cole1,Elena M. Newman1, Elan Z. Eisenmesser3 and Sirano Dhe-Paganon1,2
1 Structural Genomics Consortium, Banting Institute, University of Toronto, Canada
2 Department of Physiology, University of Toronto, Canada
3 Department of Biochemistry and Molecular Genetics, University of Colorado, Denver and Health Sciences Center, Aurora, CO, USA
Cyclophilins (Cyps) are one of three subfamilies of the
peptidylprolyl isomerases (PPIases; E.C. 5.2.1.8),
together with the structurally unrelated FK506-binding
proteins and parvulins [1,2]. Biologists and clinicians
initially focused on the specific and high affinity of
PPIases for the immunosuppressants ciclosporin A,
FK506 and rapamycin [3–5]. These immunosuppressive
effects were eventually determined to be uncoupled
from the enzymatic function of the PPIases, which
involves the reversible cis–trans isomerization of Xaa–
Pro peptide bonds [2,6]. The evolutionary importance
of this fold and ⁄or function may be inferred from the
broad distribution of PPIases throughout Eukaryota,
Eubacteria, Archaea and, recently, a viral genome
[2,7,8]. However, there is very little direct evidence
to show that the enzymatic function encoded by the
PPIases is the only, or even the primary, physiological
role of these proteins in cells. There are a subset of
proteins whose association with Cyps is necessary for
correct folding and structural integrity: for instance,
maturation of steroid receptor complexes in conjunc-
tion with Hsp90 ⁄Hsc70 [9–11] and the function of Nin-
aA in Drosophila rhodopsin folding [12]. Cyp-catalyzed
isomerization can also play a part in host response
events, including the participation of host CypA in
binding the capsid protein Gag of HIV-1 during infec-
tion in humans and packaging into HIV virions, and
the association of a host CypB–viral polymerase com-
plex leading to viral replication during infection by
hepatitis C [13,14].
However, more recent studies of PPIases have
focused on their roles as signal transducers rather than
Keywords
crystal structure; cyclophilin; peptidyl-prolyl
isomerase; spliceosome; WD40
Correspondence
S. Dhe-Paganon, Structural Genomics
Consortium, Banting Institute, University of
Toronto, 100 College Street, Room 511,
Toronto, ON, Canada M5G 1L5
Fax: +1 416 946 0588
Tel: +1 416 946 3876
E-mail: [email protected]
(Received 18 January 2008, revised 3 March
2008, accepted 6 March 2008)
doi:10.1111/j.1742-4658.2008.06381.x
Cyclophilins comprise one of the three classes of peptidylprolyl isomerases
found in all eukaryotic and prokaryotic organisms, as well as viruses.
Many of the 17 annotated human cyclophilins contain the catalytic domain
in tandem with other domains, and many of the specific functions of a par-
ticular cyclophilin or its associated domains remain unknown. The struc-
ture of the isomerase domain from a spliceosome-associated cyclophilin,
PPWD1 (peptidylprolyl isomerase containing WD40 repeat), has been
solved to 1.65 A. In the crystal, the N-terminus of one isomerase domain is
bound in the active site of a neighboring isomerase molecule in a manner
analogous to substrate. NMR solution studies show that this sequence
binds to the active site of the cyclophilin, but cannot be turned over by the
enzyme. A pseudo-substrate immediately N-terminal to the cyclophilin
domain in PPWD1 could have wider implications for the function of this
cyclophilin in the spliceosome, where it is located in human cells.
Abbreviations
Cyp, cyclophilin; IC50, 50% inhibitory concentration; pNA, p-nitroaniline; PPIase, peptidylprolyl isomerase; PPWD1, peptidylprolyl isomerase
containing WD40 repeat; suc, succinyl.
FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS 2283

as chaperones or protein foldases. Specific examples
include the participation of CypD in the mitochondrial
permeability transition, leading to necrotic cell death
[15], the isomerase-dependent regulation of ligand
specificity for the SH2 domain of the non-receptor
tyrosine kinase Itk [16,17], and the role of multiple
Cyps in regulating transcriptional and spliceosomal
events [18–20]. In these cases, the substrate proteins
are already folded and performing a subset of their
normal functions; an isomerase interacts with this sub-
strate conformation, thereby allowing for a new set of
molecular interactions to occur with the product con-
former [1,17]. The molecular switch function of Cyps
may well have different sequence determinants than
their isomerization function, but very few studies have
been performed to probe the sequence specificity of
binding versus catalysis of Cyps. Early evidence indi-
cated that CypA is able to catalyze the isomerization
of a wide array of Xaa-Pro sequences with nearly iden-
tical catalytic efficiency [21]. In contrast, phage display
and subsequent amino acid substitution analysis to
identify CypA binding specificity identified a clear
preference for sequences N- and C-terminal to the
Xaa-Pro target culminating in a consensus sequence of
FG*PXLp. This work indicates that preferential bind-
ing of target proteins may be dictated by a select sub-
set of amino acids distinct from sequences that are
substrates for catalysis [22]. The relevance of this find-
ing is dependent on studies showing an in vivo context
for peptides capable of binding to PPIases without
being used as isomerization substrates; to date, no
such peptide sequence has been described in the liter-
ature.
Further complicating the field of Cyp biology is the
fact that many of the 17 Cyps annotated in the human
genome have not been thoroughly studied in terms of
either enzymatic or other functional significance. For
some of these Cyps, their location inside the cell
provides the only clue as to their function. The large
subset of Cyps found to be stably associated with
spliceosomes provides an example of the issues
involved in studying the complexity of Cyp function.
Spliceosomes are multi-megadalton complexes contain-
ing hundreds of proteins and five essential small
nuclear RNAs, whose function is to excise out non-
coding regions (introns) of translated pre-mRNAs
[23,24]. Recent technological advances in the purifica-
tion of spliceosomal complexes, coupled with advances
in mass spectrophotometric methodology, have led to
a massive increase in the identification of proteins
found to be associated with spliceosomal complexes
[25–28]. At least 11 of the 17 human PPIases have
been found to be associated with intermediate splice-
osomal subcomplexes [26,29]. One of the spliceosomal
Cyps was identified using yeast two-hybrid screens for
known spliceosomal components; another was recog-
nized as a spliceosomal component based on sequence
homology to an SR repeat containing splicing factors
[20,30]. For two other spliceosomal Cyps, functional
and structural studies eventually identified their cog-
nate spliceosomal binding partners: PPIL1 binds to the
SNW domain-containing protein 1 (SKIP), and CypH
binds to the small ribonucleoprotein Prp4 [18,31–33].
Interestingly, in both of these cases, the interaction
with spliceosomal components was found on surfaces
not involving the isomerase active site and did not
affect cis–trans isomerase activity, indicating that the
function of the isomerase in the spliceosomal complex
may involve simultaneous active site and second-site
interactions. The binding partners for the other seven
spliceosomal Cyps are undetermined. Moreover, the
physiological function of the spliceosomal Cyps
remains unclear, which perhaps is not surprising, con-
sidering that so many proteins encoding the same
enzymatic function are found simultaneously in splice-
osomal complexes. It is theorized that this high level
of complexity and seeming duplication of effort in the
spliceosomal complexes are a function of the exquisite
sophistication of dynamic networks needed to properly
regulate and proofread the splicing process [23,34].
PPWD1 (peptidylprolyl isomerase containing WD40
repeat) was cloned in 1994 [35] and later purified as
part of the catalytically competent form of the spliceo-
some C complex [26]. This polypeptide encodes an
N-terminal WD40 repeat domain and a C-terminal
domain homologous to Cyps. As part of an attempt to
structurally characterize the spliceosomal Cyps, we
have determined the high-resolution X-ray crystal
structure of the isomerase domain of PPWD1 and
monitored its activity via both UV kinetics and NMR
solution experiments. In this structure, PPWD1 forms
distinct intermolecular interactions within the asym-
metric unit with an internal peptide containing a
Gly–Pro sequence. Interestingly, the Pro residue is
found in trans, an unusual circumstance for a substrate
peptide. Further experiments have shown that PPWD1
is indeed a functional isomerase against a standard
substrate sequence, but that, surprisingly, a peptide
containing the internal sequence is able to bind, but is
not catalyzed by the isomerase, suggesting that it is
not a substrate. Both the intermolecular interaction
and lack of enzymatic turnover were confirmed using
NMR solution studies of the PPWD1 protein. This
work represents the first structural and biochemical
characterization of a WD40 repeat-containing spliceos-
omal Cyp, and the first instance of a Pro-containing
Structure of spliceosomal cyclophilin PPWD1 T. L. Davis et al.
2284 FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS

sequence that is sufficient to bind specifically to the
active site of a Cyp, but is not a substrate for cis–trans
isomerization.
Results
The crystal structure of the isomerase domain of
human PPWD1 (utilizing a construct encoding residues
473–646) was determined at 1.65 A resolution. The
final model of PPWD1 comprises three polypeptide
chains; each chain is disordered from residues 473–482,
such that the first interpretable density is for residue
Gln483. All other residues encoded by the PPWD1
construct are present in the final model. In addition,
the model contains a single glycerol molecule most
probably contributed from the cryoprotectant and 388
water molecules. With an architecture consisting of
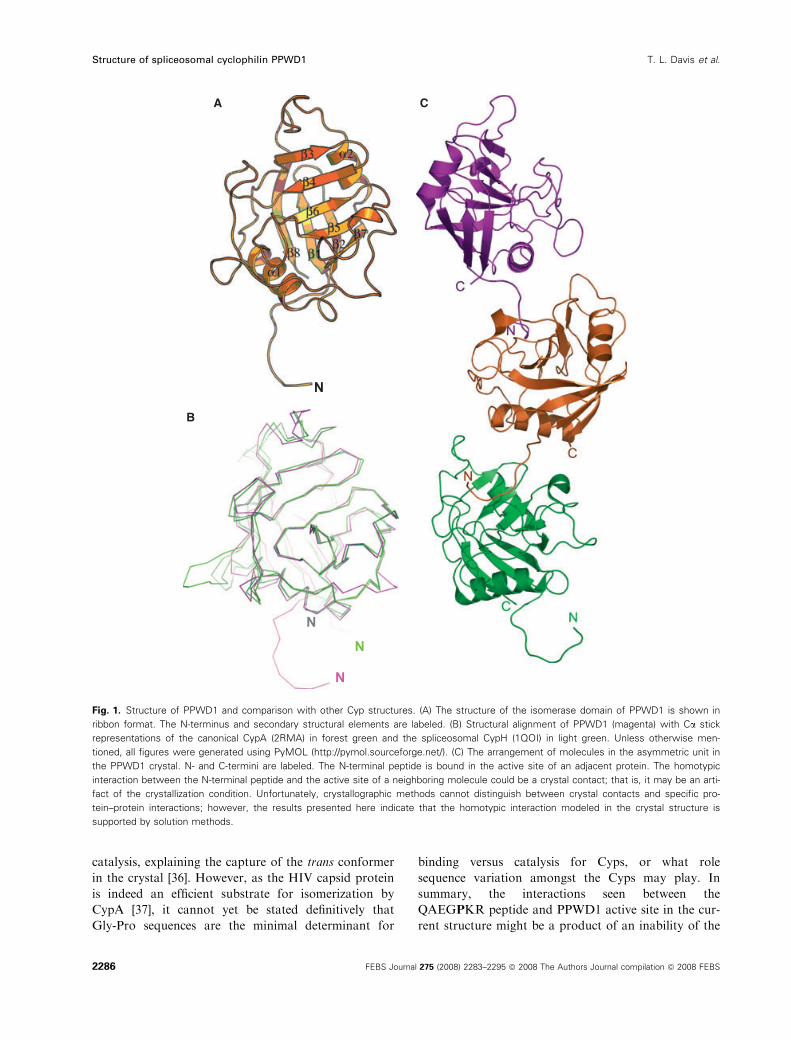
eight antiparallel b-sheets forming a closed b-barrelwith two a-helices packed against the core (Fig. 1A),
the structure of PPWD1 is similar to the structure of
the canonical CypA as well as to CypH and PPIL1,
two other spliceosomal Cyps (Fig. 1B). The Ca atoms
of CypA align to the isomerase domain of PPWD1
with an rmsd of 1.34 A over 124 atoms, corresponding
well to the 60% sequence similarity between the two
isomerase domains. The main conformational differ-
ences lie in the a2–b8, b1–b2 and b4–b5 loops. The
b1–b2 loop (corresponding to residues Thr498–
Asp502) is shorter by five residues in PPWD1 com-
pared with CypA. This deletion also occurs in six of
the 17 currently annotated human Cyps, including
PPIL1–PPIL4 (Fig. 2), but the significance of the resul-
tant shorter b1–b2 loop is not understood.
PPWD1 crystallized with three protein molecules in
the asymmetric unit. Unexpectedly, there was an inter-
molecular interaction, propagated throughout the crys-
tal, in which the active sites of all three molecules in
the asymmetric unit were bound to seven residues
(QAEGP487KR) of an adjacent molecule (Fig. 1C).
The three polypeptide chains in the asymmetric unit
are conformationally identical, with rmsd values of 0.4
and 0.2 A over all atoms in 176 residues. In addition,
the N-terminal peptide is oriented in very similar fash-
ion across all three molecules, also with rmsd values of
0.4 and 0.2 A over all atoms for the first seven resi-
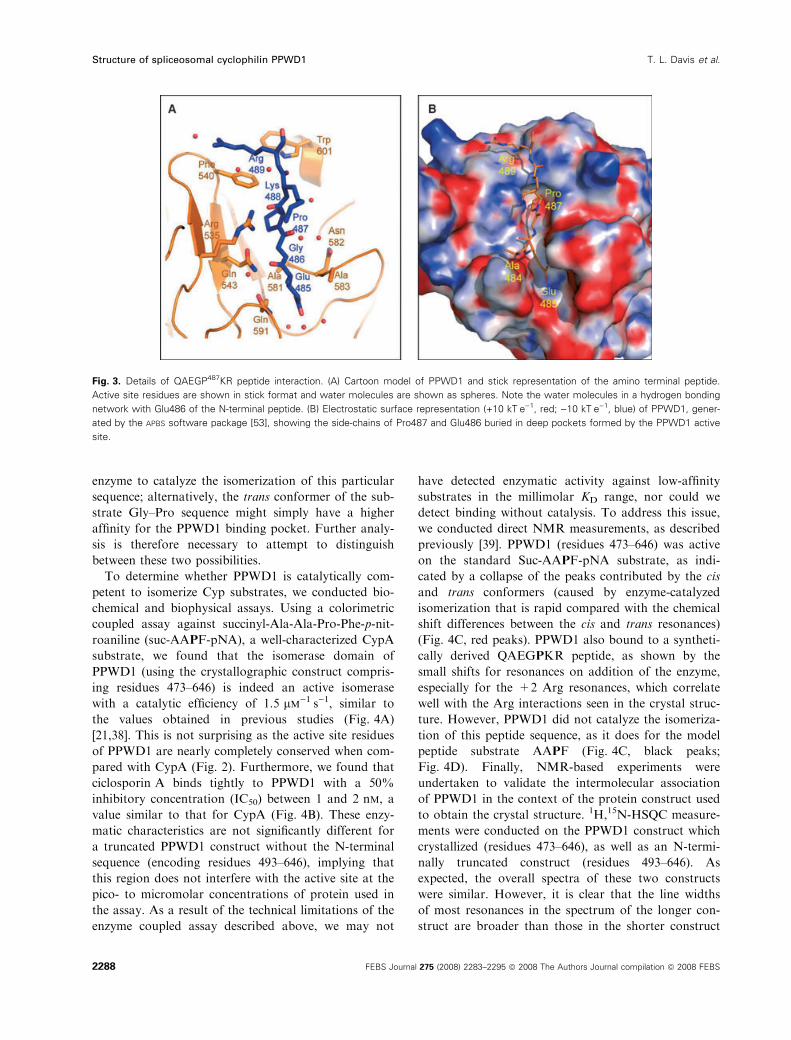
dues. Pro487 from one monomer is buried within the
active site of another, and is less than 3 A from the
side-chain of the conserved Arg535 (Arg55 in CypA)
(2.95 A for NH1 and 2.85 A for NH2). In addition,
Pro487 of the peptide is bound in a hydrophobic
pocket composed of Phe540, Phe593, Met541, Leu602
and His606, all of which are conserved between CypA
and PPWD1 (Figs 2 and 3). The Nd1 atom of the
conserved Trp602 (Trp121 in CypA) is coordinating
the backbone oxygen of Lys488 of the QAEGPKR
peptide (Fig. 3A), and Phe540 is coordinating Lys488
through a stacking interaction. The other interactions
between the side-chain of the QAEGPKR peptide in
the active site are centered about residue Glu485; in
addition to specific interactions between the backbone
nitrogen of Glu485 with the oxygen of Gly551, the
side-chain of Glu485 is nestled in a deep pocket of the
enzyme (Fig. 3B). An oxygen of the c-carboxyl groupforms hydrogen bonds with the backbone amide nitro-
gen of Asn582 and a water molecule buried in the
pocket; the other c-carboxyl oxygen is involved in a
network of water-mediated hydrogen bonds. Two resi-
dues in the PPWD1 active site, Gln543 and Gln591,
contribute to the complementary polar nature of this
pocket. The average B-factor for all three of the
QAEGPKR regions is 48 A2, and is comparable with
an all-atom B-factor of 45 A2 for all molecules in the
asymmetric unit. These observations indicate a stable
interaction, although PPWD1 runs as a monomer
using size exclusion chromatography, which suggests
that the interaction is either low affinity or has a high
off rate.
This mode of interaction and the trans conformation
of the peptide in the active site mimic the enzyme–sub-
strate interaction observed in the CypA–HIV-1 Gag
complex [36]. Pro487 is found to be bound to PPWD1
in the trans configuration, which is also the conformer
found in the complexes of the capsid protein Gag and
in some peptides derived from GAG protein with
CypA. For these complexes, the requirement for an
X-Gly-Pro (X „ Pro) sequence was proposed to
obtain stable binding of the trans isomer into the Cyp
active site; the PPWD1 structure confirms this observa-
tion and, indeed, any other amino acid at that position
would adopt /,w angles such that Cb would clash with
the catalytic Arg535 and destabilize the trans confor-
mation [36]. Although the backbone amide of the )2position has been shown to be involved in a configura-
tion-dependent hydrogen bond with the b4–b5 loop,
the function of the side-chain at this position has not
been exhaustively studied. As opposed to the mainly
hydrophobic residues found in the context of HIV-1
capsid variants (containing Ala, Val, or Met at posi-
tion )2), PPWD1 contains Glu at this position
(Glu485), which is pointing into a region of charge
complementarity in the PPWD1 active site that would
not be well accommodated by hydrophobic side-
chains. The relevance of finding a peptide bound in
trans in the active site of isomerases is ambiguous; in
the case of the HIV capsid, it has been proposed that
an X-Gly-Pro sequence represents a poor substrate for
T. L. Davis et al. Structure of spliceosomal cyclophilin PPWD1
FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS 2285
catalysis, explaining the capture of the trans conformer
in the crystal [36]. However, as the HIV capsid protein
is indeed an efficient substrate for isomerization by
CypA [37], it cannot yet be stated definitively that
Gly-Pro sequences are the minimal determinant for
binding versus catalysis for Cyps, or what role
sequence variation amongst the Cyps may play. In
summary, the interactions seen between the
QAEGPKR peptide and PPWD1 active site in the cur-
rent structure might be a product of an inability of the
Fig. 1. Structure of PPWD1 and comparison with other Cyp structures. (A) The structure of the isomerase domain of PPWD1 is shown in
ribbon format. The N-terminus and secondary structural elements are labeled. (B) Structural alignment of PPWD1 (magenta) with Ca stick
representations of the canonical CypA (2RMA) in forest green and the spliceosomal CypH (1QOI) in light green. Unless otherwise men-
tioned, all figures were generated using PyMOL (http://pymol.sourceforge.net/). (C) The arrangement of molecules in the asymmetric unit in
the PPWD1 crystal. N- and C-termini are labeled. The N-terminal peptide is bound in the active site of an adjacent protein. The homotypic
interaction between the N-terminal peptide and the active site of a neighboring molecule could be a crystal contact; that is, it may be an arti-
fact of the crystallization condition. Unfortunately, crystallographic methods cannot distinguish between crystal contacts and specific pro-
tein–protein interactions; however, the results presented here indicate that the homotypic interaction modeled in the crystal structure is
supported by solution methods.
Structure of spliceosomal cyclophilin PPWD1 T. L. Davis et al.
2286 FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS
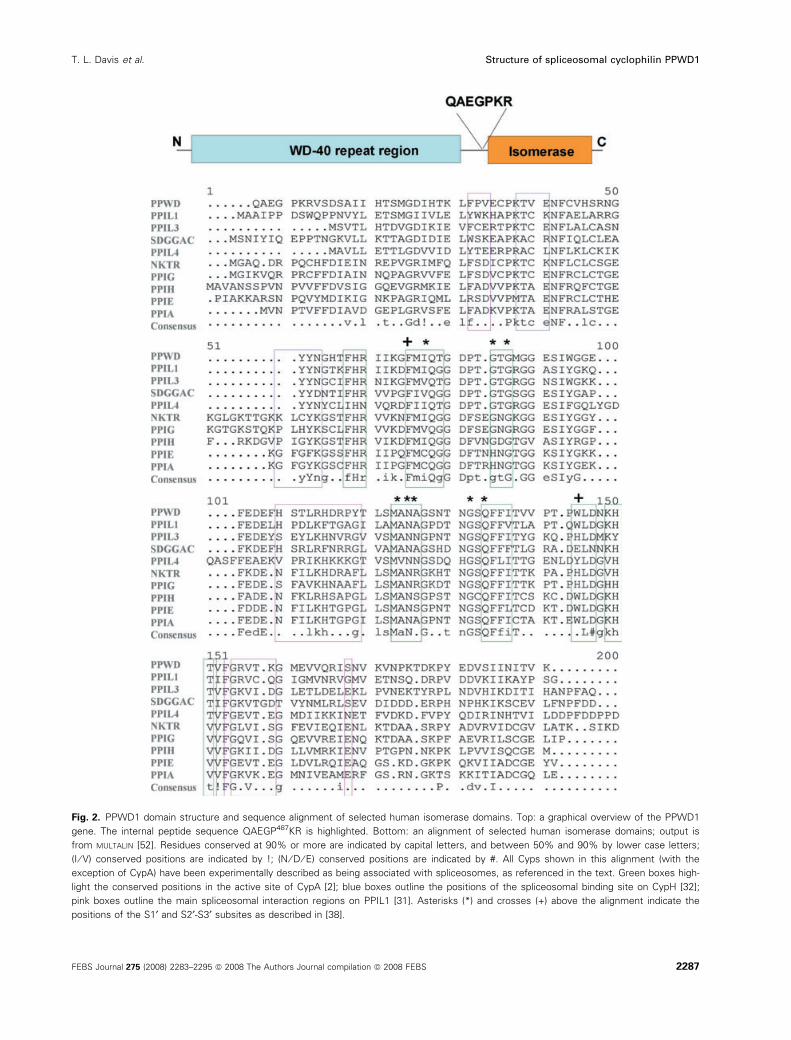
Fig. 2. PPWD1 domain structure and sequence alignment of selected human isomerase domains. Top: a graphical overview of the PPWD1
gene. The internal peptide sequence QAEGP487KR is highlighted. Bottom: an alignment of selected human isomerase domains; output is
from MULTALIN [52]. Residues conserved at 90% or more are indicated by capital letters, and between 50% and 90% by lower case letters;
(I ⁄ V) conserved positions are indicated by !; (N ⁄ D ⁄ E) conserved positions are indicated by #. All Cyps shown in this alignment (with the
exception of CypA) have been experimentally described as being associated with spliceosomes, as referenced in the text. Green boxes high-
light the conserved positions in the active site of CypA [2]; blue boxes outline the positions of the spliceosomal binding site on CypH [32];
pink boxes outline the main spliceosomal interaction regions on PPIL1 [31]. Asterisks (*) and crosses (+) above the alignment indicate the
positions of the S1¢ and S2¢-S3¢ subsites as described in [38].
T. L. Davis et al. Structure of spliceosomal cyclophilin PPWD1
FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS 2287
enzyme to catalyze the isomerization of this particular
sequence; alternatively, the trans conformer of the sub-
strate Gly–Pro sequence might simply have a higher
affinity for the PPWD1 binding pocket. Further analy-
sis is therefore necessary to attempt to distinguish
between these two possibilities.
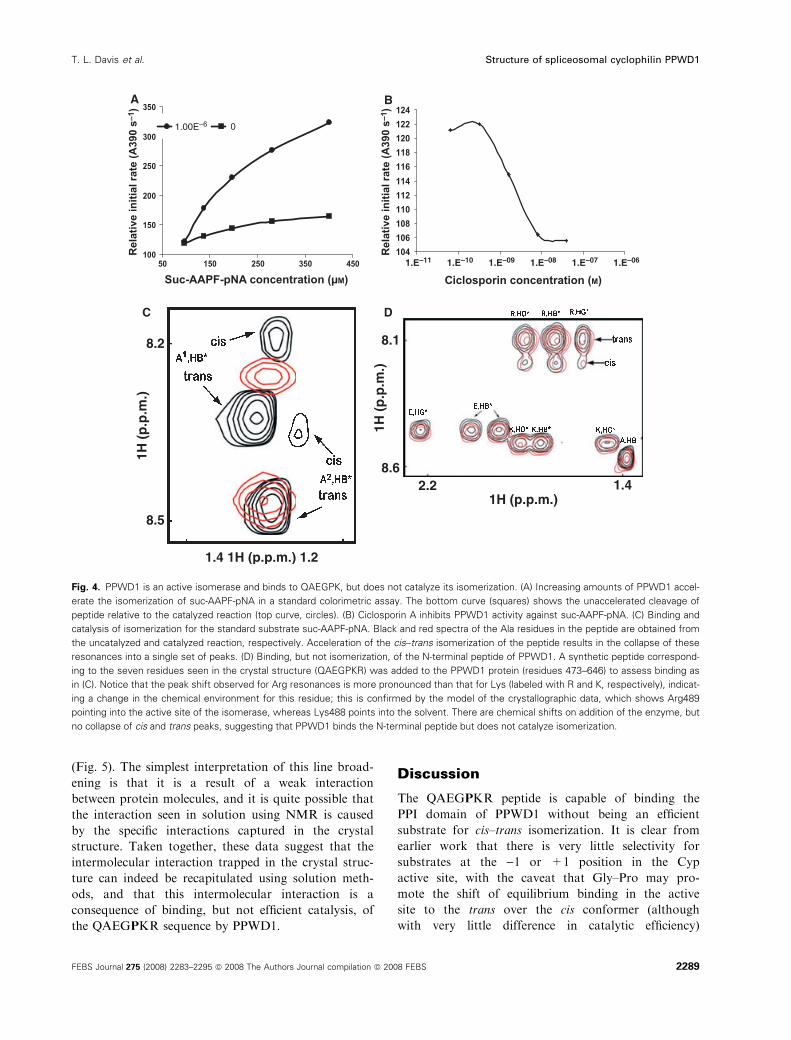
To determine whether PPWD1 is catalytically com-
petent to isomerize Cyp substrates, we conducted bio-
chemical and biophysical assays. Using a colorimetric
coupled assay against succinyl-Ala-Ala-Pro-Phe-p-nit-
roaniline (suc-AAPF-pNA), a well-characterized CypA
substrate, we found that the isomerase domain of
PPWD1 (using the crystallographic construct compris-
ing residues 473–646) is indeed an active isomerase
with a catalytic efficiency of 1.5 lm)1Æs)1, similar to
the values obtained in previous studies (Fig. 4A)
[21,38]. This is not surprising as the active site residues
of PPWD1 are nearly completely conserved when com-
pared with CypA (Fig. 2). Furthermore, we found that
ciclosporin A binds tightly to PPWD1 with a 50%
inhibitory concentration (IC50) between 1 and 2 nm, a
value similar to that for CypA (Fig. 4B). These enzy-
matic characteristics are not significantly different for
a truncated PPWD1 construct without the N-terminal
sequence (encoding residues 493–646), implying that
this region does not interfere with the active site at the
pico- to micromolar concentrations of protein used in
the assay. As a result of the technical limitations of the
enzyme coupled assay described above, we may not
have detected enzymatic activity against low-affinity
substrates in the millimolar KD range, nor could we
detect binding without catalysis. To address this issue,
we conducted direct NMR measurements, as described
previously [39]. PPWD1 (residues 473–646) was active
on the standard Suc-AAPF-pNA substrate, as indi-
cated by a collapse of the peaks contributed by the cis
and trans conformers (caused by enzyme-catalyzed
isomerization that is rapid compared with the chemical
shift differences between the cis and trans resonances)
(Fig. 4C, red peaks). PPWD1 also bound to a syntheti-
cally derived QAEGPKR peptide, as shown by the
small shifts for resonances on addition of the enzyme,
especially for the +2 Arg resonances, which correlate
well with the Arg interactions seen in the crystal struc-
ture. However, PPWD1 did not catalyze the isomeriza-
tion of this peptide sequence, as it does for the model
peptide substrate AAPF (Fig. 4C, black peaks;
Fig. 4D). Finally, NMR-based experiments were
undertaken to validate the intermolecular association
of PPWD1 in the context of the protein construct used
to obtain the crystal structure. 1H,15N-HSQC measure-
ments were conducted on the PPWD1 construct which
crystallized (residues 473–646), as well as an N-termi-
nally truncated construct (residues 493–646). As
expected, the overall spectra of these two constructs
were similar. However, it is clear that the line widths
of most resonances in the spectrum of the longer con-
struct are broader than those in the shorter construct
Fig. 3. Details of QAEGP487KR peptide interaction. (A) Cartoon model of PPWD1 and stick representation of the amino terminal peptide.
Active site residues are shown in stick format and water molecules are shown as spheres. Note the water molecules in a hydrogen bonding
network with Glu486 of the N-terminal peptide. (B) Electrostatic surface representation (+10 kTÆe)1, red; )10 kTÆe)1, blue) of PPWD1, gener-
ated by the APBS software package [53], showing the side-chains of Pro487 and Glu486 buried in deep pockets formed by the PPWD1 active
site.
Structure of spliceosomal cyclophilin PPWD1 T. L. Davis et al.
2288 FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS
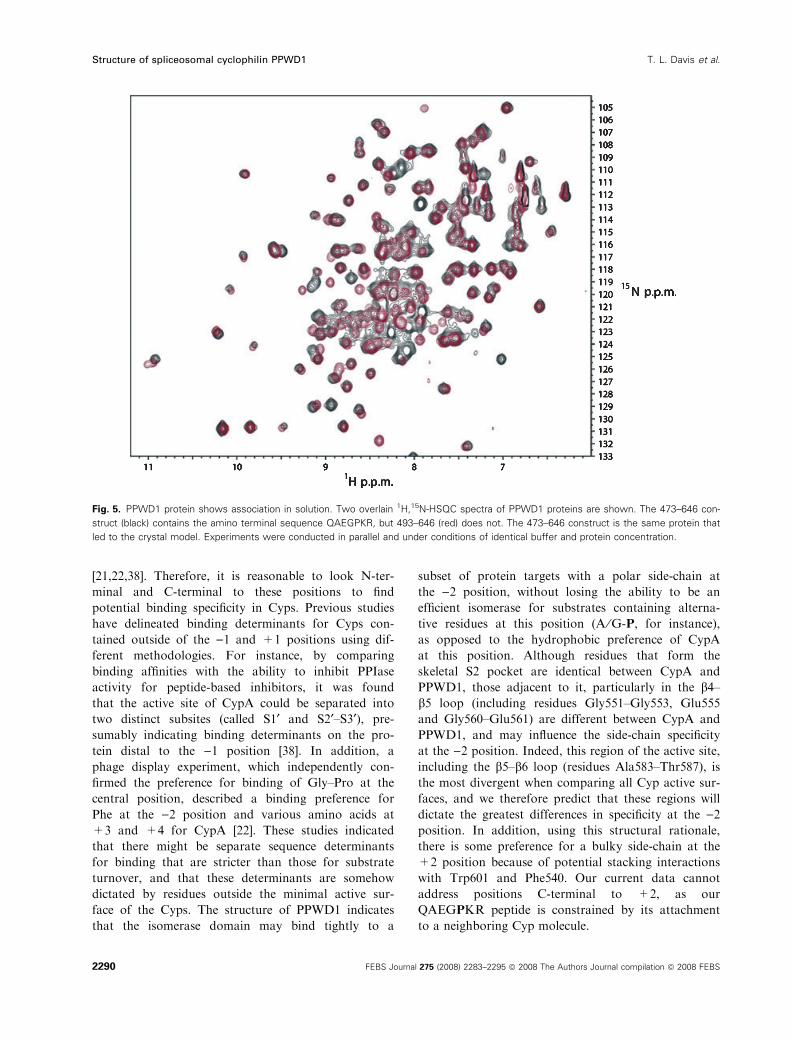
(Fig. 5). The simplest interpretation of this line broad-
ening is that it is a result of a weak interaction
between protein molecules, and it is quite possible that
the interaction seen in solution using NMR is caused
by the specific interactions captured in the crystal
structure. Taken together, these data suggest that the
intermolecular interaction trapped in the crystal struc-
ture can indeed be recapitulated using solution meth-
ods, and that this intermolecular interaction is a
consequence of binding, but not efficient catalysis, of
the QAEGPKR sequence by PPWD1.
Discussion
The QAEGPKR peptide is capable of binding the
PPI domain of PPWD1 without being an efficient
substrate for cis–trans isomerization. It is clear from
earlier work that there is very little selectivity for
substrates at the )1 or +1 position in the Cyp
active site, with the caveat that Gly–Pro may pro-
mote the shift of equilibrium binding in the active
site to the trans over the cis conformer (although
with very little difference in catalytic efficiency)
Fig. 4. PPWD1 is an active isomerase and binds to QAEGPK, but does not catalyze its isomerization. (A) Increasing amounts of PPWD1 accel-
erate the isomerization of suc-AAPF-pNA in a standard colorimetric assay. The bottom curve (squares) shows the unaccelerated cleavage of
peptide relative to the catalyzed reaction (top curve, circles). (B) Ciclosporin A inhibits PPWD1 activity against suc-AAPF-pNA. (C) Binding and
catalysis of isomerization for the standard substrate suc-AAPF-pNA. Black and red spectra of the Ala residues in the peptide are obtained from
the uncatalyzed and catalyzed reaction, respectively. Acceleration of the cis–trans isomerization of the peptide results in the collapse of these
resonances into a single set of peaks. (D) Binding, but not isomerization, of the N-terminal peptide of PPWD1. A synthetic peptide correspond-
ing to the seven residues seen in the crystal structure (QAEGPKR) was added to the PPWD1 protein (residues 473–646) to assess binding as
in (C). Notice that the peak shift observed for Arg resonances is more pronounced than that for Lys (labeled with R and K, respectively), indicat-
ing a change in the chemical environment for this residue; this is confirmed by the model of the crystallographic data, which shows Arg489
pointing into the active site of the isomerase, whereas Lys488 points into the solvent. There are chemical shifts on addition of the enzyme, but
no collapse of cis and trans peaks, suggesting that PPWD1 binds the N-terminal peptide but does not catalyze isomerization.
T. L. Davis et al. Structure of spliceosomal cyclophilin PPWD1
FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS 2289
[21,22,38]. Therefore, it is reasonable to look N-ter-
minal and C-terminal to these positions to find
potential binding specificity in Cyps. Previous studies
have delineated binding determinants for Cyps con-
tained outside of the )1 and +1 positions using dif-
ferent methodologies. For instance, by comparing
binding affinities with the ability to inhibit PPIase
activity for peptide-based inhibitors, it was found
that the active site of CypA could be separated into
two distinct subsites (called S1¢ and S2¢–S3¢), pre-
sumably indicating binding determinants on the pro-
tein distal to the )1 position [38]. In addition, a
phage display experiment, which independently con-
firmed the preference for binding of Gly–Pro at the
central position, described a binding preference for
Phe at the )2 position and various amino acids at
+3 and +4 for CypA [22]. These studies indicated
that there might be separate sequence determinants
for binding that are stricter than those for substrate
turnover, and that these determinants are somehow
dictated by residues outside the minimal active sur-
face of the Cyps. The structure of PPWD1 indicates
that the isomerase domain may bind tightly to a
subset of protein targets with a polar side-chain at
the )2 position, without losing the ability to be an
efficient isomerase for substrates containing alterna-
tive residues at this position (A ⁄G-P, for instance),
as opposed to the hydrophobic preference of CypA
at this position. Although residues that form the
skeletal S2 pocket are identical between CypA and
PPWD1, those adjacent to it, particularly in the b4–b5 loop (including residues Gly551–Gly553, Glu555
and Gly560–Glu561) are different between CypA and
PPWD1, and may influence the side-chain specificity
at the )2 position. Indeed, this region of the active site,
including the b5–b6 loop (residues Ala583–Thr587), is
the most divergent when comparing all Cyp active sur-
faces, and we therefore predict that these regions will
dictate the greatest differences in specificity at the )2position. In addition, using this structural rationale,
there is some preference for a bulky side-chain at the
+2 position because of potential stacking interactions
with Trp601 and Phe540. Our current data cannot
address positions C-terminal to +2, as our
QAEGPKR peptide is constrained by its attachment
to a neighboring Cyp molecule.
Fig. 5. PPWD1 protein shows association in solution. Two overlain 1H,15N-HSQC spectra of PPWD1 proteins are shown. The 473–646 con-
struct (black) contains the amino terminal sequence QAEGPKR, but 493–646 (red) does not. The 473–646 construct is the same protein that
led to the crystal model. Experiments were conducted in parallel and under conditions of identical buffer and protein concentration.
Structure of spliceosomal cyclophilin PPWD1 T. L. Davis et al.
2290 FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS

As mentioned earlier, many of the residues that
make up the distal subsites on PPWD1 are conserved
between the spliceosomal Cyps and CypA (Fig. 2).
Two exceptions are Ala583, which is variously changed
to Ser, Arg or Asn in spliceosomal Cyps, and Trp601
which is largely conserved amongst the non-spliceoso-
mal Cyps, but is variably changed to His, glutamate,
or Tyr in the spliceosomal subclass of Cyps (Fig. 2).
Interestingly Ala583 is part of the S1¢ subsite, and
Trp601 and Phe540 are part of the S2¢–S3¢ subsite
described previously [38]. It is reasonable to propose
that the ability of PPWD1 to bind to sequence deter-
minants that are not substrates for isomerization may
be quite relevant to the larger biological function of
Cyps if it is found to be a more general phenomenon.
The intermolecular interaction seen in the crystal
structure of PPWD1 may imply a new function within
the spliceosome, where it possibly plays a role in
spliceosomal assembly or activity. Homotypic interac-
tions are also observed in the crystal structure of
another spliceosomal Cyp, CypH (SnuCyp-20), where
isomerase domains interact with each other, again
through an extended loop region N-terminal to the
isomerase domain. PPWD1 and CypH have no
sequence similarity in this region and crystallize under
different conditions and with different spacegroups
[32]. It is interesting in the context of this dissimilarity
to observe that the two spliceosomal Cyps exhibit a
similar type of intermolecular association in structural
studies. Although the solution properties of the homo-
typic CypH interaction were not studied, the crystal
structure of CypH bound to a peptide derived from
the spliceosome shows that it binds a surface directly
opposite to the isomerase active site (Fig. 2), suggest-
ing that the homotypic interaction seen in the structure
would not be impaired by spliceosomal association
[32,33]. The overall sequence similarity between the
isomerase domain of CypH and the isomerase domain
of PPWD1 is reasonably high (55% over approxi-
mately 140 residues), but many of the residues that
form the spliceosomal binding site of CypH are not
conserved in PPWD1 (blue boxes, Fig. 2). In the case
of CypJ (PPIL3), another spliceosomal Cyp whose
interaction surface with the spliceosomal protein SKIP
has been probed using NMR, the interaction with the
spliceosomal component was again found to be distinct
from the active site (pink boxes, Fig. 2) [31]. Again,
the spliceosomal interacting region of CypJ is not well
conserved amongst spliceosomal Cyps, including
PPWD1. Although the spliceosomal target or interact-
ing region of PPWD1 has not been isolated, it is rea-
sonable to believe, based on the cases of CypH and
CypJ, that this region may lie well outside the active
site residues, and that these surfaces may be variable
in terms of sequence amongst the spliceosomal Cyps in
order to target isomerase binding to distinct spliceoso-
mal substituents. It may be that the bifunctional isom-
erase domains of spliceosomal Cyps may be directed
towards internal sequences in order to regulate the
activity of these enzymes or to serve as a signal trans-
duction element in addition to the isomerase function.
Indeed, it may be that isomerization must be prevented
in order for spliceosomal Cyps to perform these addi-
tional functions, and perhaps the homotypic interac-
tions seen in the spliceosomal Cyps are indicative of
peptide sequences that are binding determinants, but
not efficient substrates, as in the case of PPWD1.
Experimental procedures
Cloning, expression and purification
Full-length cDNA encoding human PPWD1 was obtained
from the Mammalian Gene Collection (BC015385). Con-
structs based around the predicted isomerase domain
boundaries were cloned into pET28a-LIC and transformed
into BL21 Gold (DE3) cells (Stratagene, La Jolla, CA,
USA). Cultures were grown in Terrific Broth medium
at 37 �C to D � 6, and induced at 15 �C with the addition
of 50 lm isopropyl thio-b-d-galactoside. Pellets were resus-
pended in 20 mL of lysis buffer (50 mm Tris, pH 8.0,
500 mm NaCl, 1 mm phenylmethanesulfonyl fluoride and
0.1 mL general protease inhibitor; Sigma P2714, St Louis,
MO, USA) and lysed by sonication; lysates were then cen-
trifuged for 20 min at 69 673 g. The supernatant was
loaded onto nickel nitrilotriacetic acid resin (Qiagen, Valen-
cia, CA, USA), washed with five column volumes of lysis
buffer and five column volumes of low imidazole buffer
(lysis buffer + 10 mm imidazole, pH 8), and eluted in
10 mL of elution buffer (lysis buffer + 250 mm imidazole,
pH 8, and 10% glycerol). One unit of thrombin (Sigma)
per milligram of protein was added to remove the tag
overnight at 4 �C. For gel filtration, an XK 16 · 65 column
(GE Healthcare, Piscataway, NJ, USA) packed with
HiLoad Superdex 200 resin was pre-equilibrated with gel
filtration buffer (lysis buffer + 5 mm b-mercaptoethanol
and 1 mm EDTA). Peak fractions were pooled and
concentrated using Amicon concentrators (10 000 molecular
mass cut-off; Millipore, Danvers, MA, USA). The protein
was used at 15 mgÆmL)1 for crystallization studies.
Crystallization, data collection and structure
solution
A construct of the PPWD1 isomerase domain containing
residues 473–646 crystallized in 1.7 m NH4SO4, 0.1 m
sodium cacodylate, pH 5.7, and 0.2 m sodium acetate.
T. L. Davis et al. Structure of spliceosomal cyclophilin PPWD1
FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS 2291

Crystals were harvested into mother liquor with 15% glyc-
erol and frozen in liquid nitrogen. Diffraction data were
collected on an FR-E SuperBright Cu rotating anode ⁄Raxis
IV++ detector (Rigaku Americas, The Woodlands, TX,
USA), and integrated and scaled using the hkl2000 pro-
gram package [40,41]. For structure solution and refine-
ment, the program phaser [42] was used as part of the
ccp4 suite [43] to find the molecular replacement solution
in the resolution range between 20 and 2.8 A, using PDB
ID 1XO7 as a search model. Following phaser, a round of
maximum-likelihood refinement and phase extension to
1.65 A was carried out with refmac5 [43,44], and the
phases were then input into arp ⁄warp [45] for automatic
model building and iterative refinement. The models were
completed using the graphics program o [46], and further
rounds of refinement using refmac5 resulted in an R-factor
of 19.9% (Rfree = 24.5%) for data from 44.32 to 1.65 A.
The model has excellent stereochemistry as judged by
procheck [47], with no residues in disallowed or unfavor-
able regions of Ramachandran space. The final model of
PPWD1 comprises three polypeptide chains corresponding
to the three molecules in the asymmetric unit; each chain is
disordered from residues 473–482, such that each model
reflects residues Gln483–Lys646. Data collection and refine-
ment statistics are provided in Table 1. Coordinates and
structure factors have been deposited in the Protein Data
Bank (PDB) with ID 2A2N.
PPIase colorimetric activity
PPIase activity was measured using a standard protease-cou-
pled assay [3,48] adapted to a 96-well format. The reaction
mixture contained 64 pm to 1 lm of PPIase and 200 nm chy-
motrypsin (Sigma) in reaction buffer (35 mm Hepes, pH 7.8,
368 mm trifluoroethanol, 50 mm NaCl2, 10 mm LiCl2, 5 mm
b-mercaptoethanol). The reaction was performed at 25 �Cusing 33–400 lm suc-AAPF-pNA (Bachem Americas, King
of Prussia, PA, USA), and read at 390 nm on a BioTek Syn-
ergy 2 plate reader using flat-bottomed well plates (Costar
3695). Initial velocities were plotted against the substrate
peptide concentrations to compare the uncatalyzed chymo-
trypsin rate with the isomerase-catalyzed reaction.
NMR experiments
Cells were inoculated into 20 mL of modified M9 medium
containing (15NH4)2SO4, trace elements and Kao & Mich-
ayluk vitamin solution (Sigma). Growth and induction were
performed as above, except that cells were induced at
D > � 3 with 100 lm isopropyl thio-b-d-galactoside. Pro-tein was purified as above. 15N-labeled proteins at 1 mm in
20 mm Hepes, pH 7.5, 100 mm NaCl, 10 mm dithiothreitol
and 10% D2O were pre-centrifuged at 35 000 g for 10 min,
and then subjected to NMR using a cryoprobed Bruker
AV500 spectrometer (Bruker, Milton, Canada). All spectra
were recorded at 25 �C. For 1H,15N-HSQC experiments, a
pulse sequence with ‘flip-back’ water suppression was used.
Typically, sweep widths of 8000 and 2000 Hz were used for
the F2 and F1 dimension, respectively. The data were pro-
cessed with Topspin [49] or NMRpipe [50] software.
All samples aimed at assessing PPWD1 binding and cataly-
sis of peptides were diluted to 300 lL with 5% D2O and
placed into a Shigemi microcell (Allison Park, PA, USA) in
50 mm Hepes, pH 7, 500 mm NaCl and 1 mm dithiothreitol.
Samples contained 3 mm peptide and either 0.5 mm suc-
AAPF-pNA (Bachem) or TQAEGPKR (Sigma-Genosys),
with and without 1 mm PPWD1-Cyp. Spectra were collected
at 10 �C on a Varian 600 or 900 MHz spectrometer (Palo
Alto, CA, USA). Spectra were acquired using standard Var-
ian BioPack sequences, processed using NMRpipe software
[50] and visualized using ccpn software [51].
Acknowledgements
Some of the NMR instrumentation used in the current
study belongs to the Ontario Center for Structural
Table 1. Data collection, phasing and refinement statistics [atomic
coordinates were deposited in the Protein Data Bank (PDB) (http://
www.rcsb.org): 2A2N].
Data set PPWD1
Space group C 1 2 1
Unit cell (A)
Unit cell (deg)
139.658, 39.893, 115.638
90.00, 122.33, 90.00
Beamline Rigaku FR-E
Wavelength (A) 1.54178
Resolution (A) 23.20–1.65
Unique reflections 59 800
Data redundancy (-fold) 3.4 (2.6)
Completeness (%)a 91.4 (63.1)
I ⁄ sigI 34.86 (2.0)
Rsymb 0.036
Refinement
Resolution (A) 23.20–1.65
Reflections used 56 773
All atoms {solvent} 4259 {338}
Rwork ⁄ Rfreec 0.199 ⁄ 0.245
rmsd bond length (A) 0.015
rmsd bond angle (deg) 1.368
Figure of merit 0.815
Average B-factor (A2) 46.22
Ramachandran plot
Favored (%) 94.46
Allowed (%) 5.54
Disallowed (%) 0
a Highest resolution shell is shown in parentheses. b Rsym = 100 ·sum(|I ) <I>|) ⁄ sum(<I>), where I is the observed intensity and <I>
is the average intensity from multiple observations of symmetry-
related reflections. c Rfree value was calculated with 5% of the
data.
Structure of spliceosomal cyclophilin PPWD1 T. L. Davis et al.
2292 FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS

Proteomics. The Structural Genomics Consortium is a
registered charity (number 1097737) that receives funds
from the Canadian Institutes for Health Research,
the Canadian Foundation for Innovation, Genome
Canada through the Ontario Genomics Institute, Glaxo-
SmithKline, Karolinska Institutet, the Knut and Alice
Wallenberg Foundation, the Ontario Innovation Trust,
the Ontario Ministry for Research and Innovation,
Merck & Co., Inc., the Novartis Research Foundation,
the Swedish Agency for Innovation Systems, the
Swedish Foundation for Strategic Research and the
Wellcome Trust.
References
1 Andreotti AH (2003) Native state proline isomerization:
An intrinsic molecular switch. Biochemistry 42, 9515–
9524.
2 Galat A & Metcalfe SM (1995) Peptidylproline cis ⁄ transisomerases. Prog Biophys Mol Biol 63, 67–118.
3 Fischer G, Wittmannliebold B, Lang K, Kiefhaber T &
Schmid FX (1989) Cyclophilin and peptidyl-prolyl cis–
trans isomerase are probably identical proteins. Nature
337, 476–478.
4 Takahashi N, Hayano T & Suzuki M (1989) Peptidyl-
prolyl cis–trans isomerase is the cyclosporin-A-binding
protein cyclophilin. Nature 337, 473–475.
5 Schreiber SL (1991) Chemistry and biology of the
immunophilins and their immunosuppressive ligands.
Science 251, 283–287.
6 Gething MJ & Sambrook J (1992) Protein folding in
the cell. Nature 355, 33–45.
7 Raoult D, Audic S, Robert C, Abergel C, Renesto P,
Ogata H, La Scola B, Suzan M & Claverie JM (2004)
The 1.2-megabase genome sequence of mimivirus.
Science 306, 1344–1350.
8 Pemberton TJ & Kay JE (2005) Identification and com-
parative analysis of the peptidyl-prolyl cis ⁄ trans isomer-
ase repertoires of H. sapiens, D. melanogaster,
C. elegans, S. cerevisiae and Sz. pombe. Comp Funct
Genomics 6, 277–300.
9 Kimmins S & MacRae TH (2000) Maturation of steroid
receptors: an example of functional cooperation among
molecular chaperones and their associated proteins. Cell
Stress Chaperones 5, 76–86.
10 Fanghanel J & Fischer G (2004) Insights into the cata-
lytic mechanism of peptidyl prolyl cis ⁄ trans isomerases.
Front Biosci 9, 3453–3478.
11 Schiene-Fischer C & Yu C (2001) Receptor accessory
folding helper enzymes: the functional role of
peptidyl prolyl cis ⁄ trans isomerases. FEBS Lett 495,
1–6.
12 Baker EK, Colley NJ & Zuker CS (1994) The cyclophi-
lin homolog NinaA functions as a chaperone, forming a
stable complex in-vivo with its protein target rhodopsin.
EMBO J 13, 4886–4895.
13 Scarlata S & Carter C (2003) Role of HIV-1 Gag
domains in viral assembly. Biochim Biophys Acta,
Biomembranes 1614, 62–72.
14 Watashi K, Ishii N, Hijikata M, Inoue D, Murata T,
Miyanari Y & Shimotohno K (2005) Cyclophilin B is a
functional regulator of hepatitis C virus RNA polymer-
ase. Mol Cell 19, 111–122.
15 Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska
H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb
RA, Dorn GW et al. (2005) Loss of cyclophilin D
reveals a critical role for mitochondrial permeability
transition in cell death. Nature 434, 658–662.
16 Brazin KN, Mallis RJ, Fulton DB & Andreotti AH
(2002) Regulation of the tyrosine kinase Itk by the
peptidyl-prolyl isomerase cyclophilin A. Proc Natl Acad
Sci U S A 99, 1899–1904.
17 Lu KP, Finn G, Lee TH & Nicholson LK (2007) Prolyl
cis–trans isomerization as a molecular timer. Nat Chem
Biol 3, 619–629.
18 Teigelkamp S, Achsel T, Mundt C, Gothel SF,
Cronshagen U, Lane WS, Marahiel M & Luhrmann
R (1998) The 20kD protein of human [U4 ⁄U6.U5]
tri-snRNPs is a novel cyclophilin that forms a
complex with the U4 ⁄UG-specific 60kD and 90kD
proteins. RNA-A Publication of the RNA Society 4,
127–141.
19 Horowitz DS, Lee EJ, Mabon SA & Misteli T (2002) A
cyclophilin functions in pre-mRNA splicing. EMBO J
21, 470–480.
20 Lin CL, Leu S, Lu MC & Ouyang P (2004) Over-
expression of SR-cyclophilin, an interaction partner of
nuclear pinin, releases SR family splicing factors from
nuclear speckles. Biochem Biophys Res Commun 321,
638–647.
21 Harrison RK & Stein RL (1990) Substrate specificities
of the peptidyl prolyl cis–trans isomerase activities of
cyclophilin and Fk-506 binding-protein – evidence for
the existence of a family of distinct enzymes. Biochemis-
try 29, 3813–3816.
22 Piotukh K, Gu W, Kofler M, Labudde D, Helms V &
Freund C (2005) Cyclophilin a binds to linear peptide
motifs containing a consensus that is present in many
human proteins. J Biol Chem 280, 23668–23674.
23 Valadkhan S (2007) The spliceosome: caught in a web
of shifting interactions. Curr Opin Struct Biol 17,
310–315.
24 Nilsen TW (2003) The spliceosome: the most complex
macromolecular machine in the cell? Bioessays 25,
1147–1149.
25 Jurica MS, Licklider LJ, Gygi SP, Grigorieff N &
Moore MJ (2002) Purification and characterization of
native spliceosomes suitable for three-dimensional
T. L. Davis et al. Structure of spliceosomal cyclophilin PPWD1
FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS 2293

structural analysis. RNA-A Publication of the RNA
Society 8, 426–439.
26 Jurica MS & Moore MJ (2003) Pre-mRNA splicing:
awash in a sea of proteins. Mol Cell 12, 5–14.
27 Chen YIG, Moore RE, Ge HY, Young MK, Lee TD &
Stevens SW (2007) Proteomic analysis of in vivo-assem-
bled pre-mRNA splicing complexes expands the catalog
of participating factors. Nucleic Acids Res 35, 3928–
3944.
28 Neubauer G (2005) The analysis of multiprotein com-
plexes: the yeast and the human spliceosome as case
studies. In Methods in Enzymology (Burlingame AL, ed)
405, 236–263.
29 Deckert J, Hartmuth M, Boehringer D, Behzadnia N,
Will CL, Kastner B, Stark H, Urlaub H & Luhrmann
R (2006) Protein composition and electron microscopy
structure of affinity-purified human spliceosomal B
complexes isolated under physiological conditions. Mol
Cell Biol 26, 5528–5543.
30 Mortillaro MJ & Berezney R (1998) Matrin CYP, an
SR-rich cyclophilin that associates with the nuclear
matrix and splicing factors. J Biol Chem 273, 8183–
8192.
31 Xu C, Zhang JH, Huang XJ, Sun JP, Xu YQ, Tang
YJ, Wu JH, Shi YY, Huang QH & Zhang QH (2006)
Solution structure of human peptidyl prolyl isomerase-
like protein 1 and insights into its interaction with
SKIP. J Biol Chem 281, 15900–15908.
32 Reidt U, Wahl MC, Fasshauer D, Horowitz DS,
Luhrmann R & Ficner R (2003) Crystal structure of a
complex between human spliceosomal cyclophilin H
and a U4 ⁄U6 snRNP-60K peptide. J Mol Biol 331,
45–56.
33 Ingelfinger D, Gothel SF, Marahiel MA, Reidt U,
Ficner R, Luhrmann R & Achsel T (2003) Two pro-
tein–protein interaction sites on the spliceosome-associ-
ated human cyclophilin CypH. Nucleic Acids Res 31,
4791–4796.
34 Stark H & Luhrmann R (2006) Cryo-electron micros-
copy of spliceosomal components. Annu Rev Biophys
Biomol Struct 35, 435–457.
35 Nomura N, Nagase T, Miyajima N, Sazuka T,
Tanaka A, Sato S, Seki N, Kawarabayasi Y,
Ishikawa Ki & Tabata S (1994) Prediction of the
coding sequences of unidentified human genes. II. The
coding sequences of 40 new genes (KIAA0041–
KIAA0080) deduced by analysis of cDNA clones
from human cell line KG-1 (Supplement). DNA Res
1, 251–262.
36 Howard BR, Vajdos FF, Li S, Sundquist WI & Hill CP
(2003) Structural insights into the catalytic mechanism
of cyclophilin A. Nat Struct Biol 10, 475–481.
37 Bosco DA, Eisenmesser EZ, Pochapsky S, Sundquist
WI & Kern D (2002) Catalysis of cis ⁄ trans
isomerization in native HIV-1 capsid by human
cyclophilin A. Proc Natl Acad Sci U S A 99, 5247–
5252.
38 Demange L, Moutiez M, Vaudry K & Dugave C (2001)
Interaction of human cyclophilin hCyp-18 with short
peptides suggests the existence of two functionally inde-
pendent subsites. FEBS Lett 505, 191–195.
39 Kern D, Kern G, Scherer G, Fischer G & Drakenberg
T (1995) Kinetic-analysis of cyclophilin-catalyzed prolyl
cis ⁄ trans isomerization by dynamic NMR-spectroscopy.
Biochemistry 34, 13594–13602.
40 Minor W, Cymborowski M & Otwinowski Z (2002)
Automatic system for crystallographic data collection
and analysis. Acta Phys Pol A 101, 613–619.
41 Otwinowski Z & Minor W (1997) Processing of X-ray
diffraction data collected in oscillation mode. Macromol
Crystallogr, Part A 276, 307–326.
42 Read RJ (2001) Pushing the boundaries of molecular
replacement with maximum likelihood. Acta Crystallogr
Sect D: Biol Crystallogr 57, 1373–1382.
43 Bailey S (1994) The Ccp4 suite – programs for protein
crystallography. Acta Crystallogr Sect D: Biol Crystal-
logr 50, 760–763.
44 Murshudov GN, Vagin AA & Dodson EJ (1997)
Refinement of macromolecular structures by the maxi-
mum-likelihood method. Acta Crystallogr Sect D: Biol
Crystallogr 53, 240–255.
45 Perrakis A, Morris R & Lamzin VS (1999) Automated
protein model building combined with iterative struc-
ture refinement. Nat Struct Biol 6, 458–463.
46 Jones TA, Zou JY, Cowan SW & Kjeldgaard M
(1991) Improved methods for building protein models
in electron density maps and the location of errors
in these models. Acta Crystallogr A 47 (Pt 2), 110–
119.
47 Laskowski RA, MacArthur MW, Moss DS & Thornton
JM (1993) Procheck – a program to check the stereo-
chemical quality of protein structures. J Appl Crystal-
logr 26, 283–291.
48 Kofron JL, Kuzmic P, Kishore V, Colonbonilla E &
Rich DH (1991) Determination of kinetic constants
for peptidyl prolyl cis–trans isomerases by an
improved spectrophotometric assay. Biochemistry 30,
6127–6134.
49 Zur Y (2004) An algorithm to calculate the NMR
signal of a multi spin-echo sequence with relaxation
and spin-diffusion. J Magn Reson 171, 97–106.
50 Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J
& Bax A (1995) Nmrpipe – a multidimensional spectral
processing system based on unix pipes. J Biomol NMR
6, 277–293.
51 Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon
A, Llinas P, Ulrich EL, Markley JL, Ionides J & Laue
ED (2005) The CCPN data model for NMR spectros-
Structure of spliceosomal cyclophilin PPWD1 T. L. Davis et al.
2294 FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS

copy: development of a software pipeline. Proteins-
Struct Funct Bioinformatics 59, 687–696.
52 Corpet F (1988) Multiple sequence alignment with
hierarchical-clustering. Nucleic Acids Res 16, 10881–
10890.
53 Baker NA, Sept D, Joseph S, Holst MJ & McCammon
JA (2001) Electrostatics of nanosystems: application
to microtubules and the ribosome. Proc Natl Acad Sci
U S A 98, 10037–10041.
T. L. Davis et al. Structure of spliceosomal cyclophilin PPWD1
FEBS Journal 275 (2008) 2283–2295 ª 2008 The Authors Journal compilation ª 2008 FEBS 2295
Copyright © 2022 FDOKUMEN




















