
Yeast Mpd1p Reveals the Structural Diversity of the Protein Disulfide Isomerase Family
10
Yeast Mpd1p Reveals the Structural Diversity of the Protein Disulfide Isomerase Family Elvira Vitu 1 , Einav Gross 1 , Harry M. Greenblatt 1 , Carolyn S. Sevier 2 , Chris A. Kaiser 2 and Deborah Fass 1 ⁎ 1 Department of Structural Biology, Weizmann Institute of Science, Rehovot 76100, Israel 2 Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA Received 10 July 2008; received in revised form 2 September 2008; accepted 16 September 2008 Available online 27 September 2008 Oxidoreductases belonging to the protein disulfide isomerase (PDI) family promote proper disulfide bond formation in substrate proteins in the endoplasmic reticulum. In plants and metazoans, new family members continue to be identified and assigned to various functional niches. PDI-like proteins typically contain tandem thioredoxin-fold domains. The limited information available suggested that the relative orientations of these domains may be quite uniform across the family, and structural models based on this assumption are appearing. However, the X-ray crystal structure of the yeast PDI family protein Mpd1p, described here, demonstrates the radically different domain orientations and surface properties achievable with multiple copies of the thioredoxin fold. A comparison of Mpd1p with yeast Pdi1p expands our perspective on the contexts in which redox-active motifs are presented in the PDI family. © 2008 Elsevier Ltd. All rights reserved. Edited by R. Huber Keywords: disulfide bonds; oxidoreductase; protein disulfide isomerase; thioredoxin fold; endoplasmic reticulum Introduction Oxidative protein folding in the endoplasmic reti- culum (ER) depends on the Protein Disulfide Iso- merase (PDI) family of tandem thioredoxin (trx) fold proteins. The yeast ER has five known PDI variants, 1 and 19 have been identified so far in humans. 2 PDI- like proteins are highly divergent in amino acid sequence, in the number and types of domains they contain, and, for those that have been studied func- tionally, in their capacity to serve as oxidases, reduc- tases, isomerases, or chaperones. Currently, the extent to which structural and functional features can be derived directly from the sequences of PDI proteins is severely limited, despite the great depth to which the trx fold and its redox properties have been studied. One of the best characterized of the PDI family proteins is yeast Pdi1p. In the oxidative protein folding process, one function of Pdi1p is to act as a disulfide carrier from the sulfhydryl oxidase Ero1p to substrate proteins. 3 Pdi1p consists of four trx fold domains in the order a-b-b’-a’, with the two ter- minal domains (a and a’) containing the redox-active Cys-X-X-Cys motifs. These two di-cysteine motifs of Pdi1p have been proposed to have different and complementary roles in the introduction and iso- merization of substrate disulfides. 4 The structure of yeast Pdi1p has been determined by X-ray crystal- lography, revealing a contorted U-shaped molecule with the two redox-active motifs opposite one ano- ther across the opening. 5 An alternative way to view the protein, however, is as a loose cloverleaf (Fig. 1) composed of the first three trx domains (a-b-b’), with the fourth trx domain (a’) projecting at a 45° angle from the plane of the cloverleaf and making little contact with the rest of the structure. The cloverleaf structure observed for the a-b-b’ domains of Pdi1p was found originally in the protein calsequestrin (Fig. 1). 6 Calsequestrin contains three tandem trx domains but is not redox-active. Instead, this highly acidic protein functions in storage of calcium in the sacrcoplasmic reticulum, binding 40 – 50 calcium ions per protein molecule. 7 The pI, calc- ulated from amino acid sequence, 8 of rabbit skeletal muscle calsequestrin is 3.9, a consequence of acidic residues, namely aspartic and glutamic acid, con- stituting 30% of the protein. Notably, the calculated pI values of yeast Pdi1p and human PDI are also quite low, 4.4 and 4.8, respectively, and the majority *Corresponding author. E-mail address: [email protected]. Abbreviations used: ER, endoplasmic reticulum; PDI, protein disulfide isomerase; trx, thioredoxin. doi:10.1016/j.jmb.2008.09.052 J. Mol. Biol. (2008) 384, 631–640 Available online at www.sciencedirect.com 0022-2836/$ - see front matter © 2008 Elsevier Ltd. All rights reserved.
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Yeast Mpd1p Reveals the Structural Diversity of the Protein Disulfide Isomerase Family

doi:10.1016/j.jmb.2008.09.052 J. Mol. Biol. (2008) 384, 631–640
Available online at www.sciencedirect.com
Yeast Mpd1p Reveals the Structural Diversity of theProtein Disulfide Isomerase Family
Elvira Vitu1, Einav Gross1, Harry M. Greenblatt1, Carolyn S. Sevier2,Chris A. Kaiser2 and Deborah Fass1⁎
1Department of StructuralBiology, Weizmann Institute ofScience, Rehovot 76100, Israel2Department of Biology,Massachusetts Institute ofTechnology, Cambridge,MA 02139, USAReceived 10 July 2008;received in revised form2 September 2008;accepted 16 September 2008Available online27 September 2008
*Corresponding author. E-mail [email protected] used: ER, endoplas
protein disulfide isomerase; trx, thio
0022-2836/$ - see front matter © 2008 E
Oxidoreductases belonging to the protein disulfide isomerase (PDI) familypromote proper disulfide bond formation in substrate proteins in theendoplasmic reticulum. In plants and metazoans, new family memberscontinue to be identified and assigned to various functional niches. PDI-likeproteins typically contain tandem thioredoxin-fold domains. The limitedinformation available suggested that the relative orientations of thesedomains may be quite uniform across the family, and structural modelsbased on this assumption are appearing. However, the X-ray crystalstructure of the yeast PDI family protein Mpd1p, described here,demonstrates the radically different domain orientations and surfaceproperties achievable with multiple copies of the thioredoxin fold. Acomparison of Mpd1p with yeast Pdi1p expands our perspective on thecontexts in which redox-active motifs are presented in the PDI family.
© 2008 Elsevier Ltd. All rights reserved.
Keywords: disulfide bonds; oxidoreductase; protein disulfide isomerase;thioredoxin fold; endoplasmic reticulum
Edited by R. HuberIntroduction
Oxidative protein folding in the endoplasmic reti-culum (ER) depends on the Protein Disulfide Iso-merase (PDI) family of tandem thioredoxin (trx) foldproteins. The yeast ER has five known PDI variants,1
and 19 have been identified so far in humans.2 PDI-like proteins are highly divergent in amino acidsequence, in the number and types of domains theycontain, and, for those that have been studied func-tionally, in their capacity to serve as oxidases, reduc-tases, isomerases, or chaperones. Currently, theextent to which structural and functional featurescan be derived directly from the sequences of PDIproteins is severely limited, despite the great depthto which the trx fold and its redox properties havebeen studied.One of the best characterized of the PDI family
proteins is yeast Pdi1p. In the oxidative proteinfolding process, one function of Pdi1p is to act as adisulfide carrier from the sulfhydryl oxidase Ero1pto substrate proteins.3 Pdi1p consists of four trx fold
ess:
mic reticulum; PDI,redoxin.
lsevier Ltd. All rights reserve
domains in the order a-b-b’-a’, with the two ter-minal domains (a and a’) containing the redox-activeCys-X-X-Cys motifs. These two di-cysteine motifs ofPdi1p have been proposed to have different andcomplementary roles in the introduction and iso-merization of substrate disulfides.4 The structure ofyeast Pdi1p has been determined by X-ray crystal-lography, revealing a contorted U-shaped moleculewith the two redox-active motifs opposite one ano-ther across the opening.5 An alternative way to viewthe protein, however, is as a loose cloverleaf (Fig. 1)composed of the first three trx domains (a-b-b’),with the fourth trx domain (a’) projecting at a 45°angle from the plane of the cloverleaf and makinglittle contact with the rest of the structure.The cloverleaf structure observed for the a-b-b’
domains of Pdi1pwas found originally in the proteincalsequestrin (Fig. 1).6 Calsequestrin contains threetandem trx domains but is not redox-active. Instead,this highly acidic protein functions in storage ofcalcium in the sacrcoplasmic reticulum, binding 40 –50 calcium ions per protein molecule.7 The pI, calc-ulated from amino acid sequence,8 of rabbit skeletalmuscle calsequestrin is 3.9, a consequence of acidicresidues, namely aspartic and glutamic acid, con-stituting 30% of the protein. Notably, the calculatedpI values of yeast Pdi1p and human PDI are alsoquite low, 4.4 and 4.8, respectively, and the majority
d.
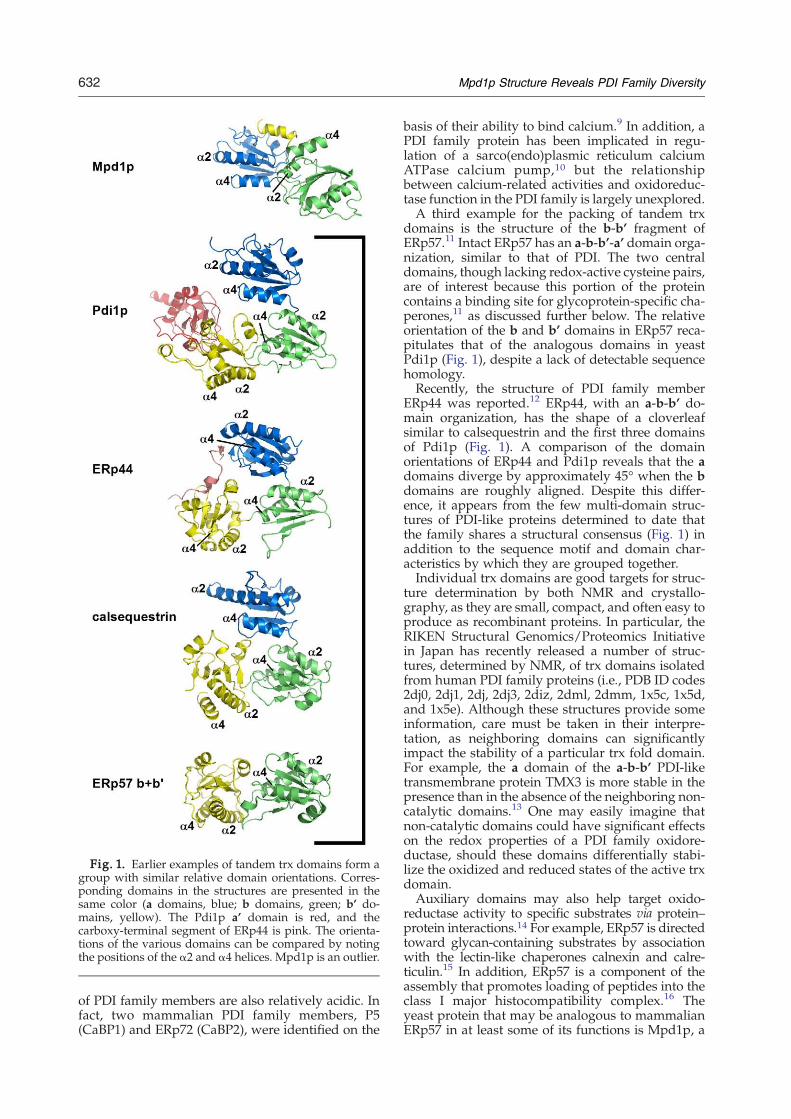
Fig. 1. Earlier examples of tandem trx domains form agroup with similar relative domain orientations. Corres-ponding domains in the structures are presented in thesame color (a domains, blue; b domains, green; b’ do-mains, yellow). The Pdi1p a’ domain is red, and thecarboxy-terminal segment of ERp44 is pink. The orienta-tions of the various domains can be compared by notingthe positions of the α2 and α4 helices. Mpd1p is an outlier.
632 Mpd1p Structure Reveals PDI Family Diversity
of PDI family members are also relatively acidic. Infact, two mammalian PDI family members, P5(CaBP1) and ERp72 (CaBP2), were identified on the
basis of their ability to bind calcium.9 In addition, aPDI family protein has been implicated in regu-lation of a sarco(endo)plasmic reticulum calciumATPase calcium pump,10 but the relationshipbetween calcium-related activities and oxidoreduc-tase function in the PDI family is largely unexplored.A third example for the packing of tandem trx
domains is the structure of the b-b’ fragment ofERp57.11 Intact ERp57 has an a-b-b’-a’ domain orga-nization, similar to that of PDI. The two centraldomains, though lacking redox-active cysteine pairs,are of interest because this portion of the proteincontains a binding site for glycoprotein-specific cha-perones,11 as discussed further below. The relativeorientation of the b and b’ domains in ERp57 reca-pitulates that of the analogous domains in yeastPdi1p (Fig. 1), despite a lack of detectable sequencehomology.Recently, the structure of PDI family member
ERp44 was reported.12 ERp44, with an a-b-b’ do-main organization, has the shape of a cloverleafsimilar to calsequestrin and the first three domainsof Pdi1p (Fig. 1). A comparison of the domainorientations of ERp44 and Pdi1p reveals that the adomains diverge by approximately 45° when the bdomains are roughly aligned. Despite this differ-ence, it appears from the few multi-domain struc-tures of PDI-like proteins determined to date thatthe family shares a structural consensus (Fig. 1) inaddition to the sequence motif and domain char-acteristics by which they are grouped together.Individual trx domains are good targets for struc-
ture determination by both NMR and crystallo-graphy, as they are small, compact, and often easy toproduce as recombinant proteins. In particular, theRIKEN Structural Genomics/Proteomics Initiativein Japan has recently released a number of struc-tures, determined by NMR, of trx domains isolatedfrom human PDI family proteins (i.e., PDB ID codes2dj0, 2dj1, 2dj, 2dj3, 2diz, 2dml, 2dmm, 1x5c, 1x5d,and 1x5e). Although these structures provide someinformation, care must be taken in their interpre-tation, as neighboring domains can significantlyimpact the stability of a particular trx fold domain.For example, the a domain of the a-b-b’ PDI-liketransmembrane protein TMX3 is more stable in thepresence than in the absence of the neighboring non-catalytic domains.13 One may easily imagine thatnon-catalytic domains could have significant effectson the redox properties of a PDI family oxidore-ductase, should these domains differentially stabi-lize the oxidized and reduced states of the active trxdomain.Auxiliary domains may also help target oxido-
reductase activity to specific substrates via protein–protein interactions.14 For example, ERp57 is directedtoward glycan-containing substrates by associationwith the lectin-like chaperones calnexin and calre-ticulin.15 In addition, ERp57 is a component of theassembly that promotes loading of peptides into theclass I major histocompatibility complex.16 Theyeast protein that may be analogous to mammalianERp57 in at least some of its functions is Mpd1p, a

633Mpd1p Structure Reveals PDI Family Diversity
PDI-like protein that associates with Cne1p, theyeast homolog of calnexin and calreticulin.17 Al-though all yeast PDI family members are multicopysuppressors of a pdi1 deletion,18–21 Mpd1p is theonly family member that, when placed under thecontrol of the PDI1 promoter, can rescue strainsdeleted of any or all other PDI homologs.1 This ob-servation suggests that, though Mpd1p may nor-mally be devoted to a specific substrate pool, it isstructurally and biochemically competent to carryout the general and essential functions of Pdi1p. Toobtain the complete structure of what is likely to be aglycoprotein-targeted PDI family oxidoreductase,and to provide a counterpoint to Pdi1p, we analyzedMpd1p using X-ray crystallography. The Mpd1pstructure reveals new ways in which individualthioredoxin domains can interact to produce novelstructures from a well-worn fold.
Results
Domain organization of Mpd1p
Mpd1p has two domains. The N-terminal domain,which shares approximately 35% sequence identitywith the Pdi1p a domain, contains the single Cys-X-X-Cys motif and is named the Mpd1p a domain.The second, redox-inactive Mpd1p domain (theMpd1p b domain) does not show detectable se-quence homology with any other protein, but it isidentified as a trx-like domain by fold recognitionalgorithms (data not shown).22 The mammalianprotein with the highest level of sequence homologyto Mpd1p is P5, a PDI family protein with an a-a’-bdomain organization. The a domains of Mpd1p andP5 have 44% amino acid sequence identity in theregion covered by the Mpd1p structure reported
Fig. 2. Amino acid sequence and structural features of Mpdsequence and labeled for each domain according to the canopolypeptide preceding helix α1 in each domain contains a stramake limited hydrogen bonds to the rest of the sheet and aresidues are highlighted in blue. The crystallized region, whichcarboxy terminal ER retention signal, is indicated between arrored triangle indicates the interdomain hinge residue (see Fig.
here. However, the sequence homology does notextend further into the two proteins.
Crystallization of Mpd1p
We expected that crystallization of Mpd1p wouldbe challenging because of the extraordinary over-representation of basic amino acids in the primarystructure (Fig. 2). Our over-expressed construct con-tains 50 (17.4%) basic amino acid residues, 38 ofwhich are lysine. Lysine is notoriously under-represented at crystal contacts,23,24 and mutation orreductive methylation of surface lysines is oftenconsidered as a strategy to increase the likelihood ofcrystallization of basic proteins.25,26 Nevertheless,we obtained crystals of wild-type Mpd1p withoutlysine modification. The major technical hurdle en-countered was the appearance of crystals of twodifferent space groups: a useful monoclinic form,which appeared rarely (∼2% of crystals), and acommon but poorly ordered orthorhombic formwith morphology indistinguishable from that of themonoclinic crystals. The two crystal forms hadsimilar unit cell dimensions; molecules related bynon-crystallographic symmetry in the monocliniccrystals were related by crystallographic symmetryin the orthorhombic crystals. Phaseswere obtained bymultiple anomalous dispersion,27 using the peak andremote wavelengths from a monoclinic crystal of theMpd1p mutant Leu65Met/Leu76Met containingselenomethionine (Table 1). The Mpd1p structurewas built into the resulting experimental electrondensity map and, after restoring leucine for methio-nine at the two mutated positions, refined to 2.0 Åresolution against data from wild-type Mpd1pcrystals. No significant difference was seen betweenthe two molecules in the crystal asymmetric unit,which had a main chain root-mean-square deviation(rmsd) of less than 0.2 Å. The active sites are buried by
1p. Secondary structural elements are indicated below thenical thioredoxin fold (β1-α1-β2-α2-β3-α3-β4-β5-α4). Thend-like segment corresponding to β1, but these segmentsre not recognized as strands by dssp.28 Basic amino aciddid not include the amino-terminal signal sequence or thews. Disulfide bonds are shown as linked yellow balls. The3).

Table 1. Summary of crystallographic data
A. Data collectionSpace group P21 native Semet peak Semet remoteWavelength (Å) 1.5418 0.9785 0.8855Unit cell parameters
a (Å) 48.16 47.94 47.95b (Å) 92.27 91.45 91.46c (Å) 79.34 79.17 79.18β (°) 92.23 92.44 92.43
Resolution a (Å) 50–2.0 (2.07–2.00) 50–2.16 (2.24–2.16) 50–1.96 (2.03–1.96)Completeness (%) 97.9 (94.7) 98.7 (89.4) 92.3 (63.8)Redundancy 5.8 (5.3) 8.3 (7.1) 4.0 (2.9)Rsym
b 0.064 (0.676) 0.151 (0.492) 0.051 (0.287)I/σ 17.0 (3.3) 14.9 (6.5) 11.9 (2.3)Overall figure of meritc 0.416, 0.667
B. Refinement statisticsReflections
Total 45,840Test set 3243
Rworkd 21.9
Rfreed 25.5
rmsd from idealityBond lengths (Å) 0.006Bond angles (°) 1.284
No. atomsProtein 4315Water 292Ethylene glycol, acetate 52a Values for the highest resolution shell are given in parentheses.b Rsym=∑hkl∑i|Ii(hkl)–bI(hkl)N|/∑hkl∑iIi(hkl), where Ii(hkl) is the observed intensity and bI(hkl)N is the average intensity for i
observations.c Figures of merit for data from 20 to 2.0 Å resolution obtained from SOLVE43, and after solvent flattening and non-crystallographic
symmetry averaging using DM.49d Rwork, Rfree=∑||Fobs|–|Fcalc||/∑|Fobs, where Fobs and Fcalc are the observed and calculated structure factors, respectively. A set of
reflections (7.1%) were excluded from the refinement and used to calculate Rfree.
634 Mpd1p Structure Reveals PDI Family Diversity
the packing of the molecules in the asymmetric unit,suggesting that the protein–protein interfaces seen inthe crystals are unlikely to be physiologically relevant.
Overall structure of Mpd1p
As anticipated, the Mpd1p structure is composedof two trx folds (Fig. 3). Both have the canonical β1-
Fig. 3. Structure of Mpd1p. A ribbon representation ofthe protein is shownwith the amino-terminal, redox activedomain in blue, the carboxy-terminal domain in green,and the carboxy-terminal extension that packs against theamino-terminal domain in yellow. The region betweenstrands β4 and β5 in the a domain that could not bemodeled due to lack of electron density is indicated by adotted line. Helices are numbered to correspond to Fig. 2,and cysteine side chains are shown as ball-and-stick. Thered triangle marks the position in Mpd1p at which thepolypeptide backbone diverges from that of Pdi1p,leading to a different relative domain orientation.
α1-β2-α2-β3-α3-β4-β5-α4 sequence of secondarystructural elements, although the segment corres-ponding to β1 of each domain makes few hydrogenbonds to the adjacent strand and is not rigorouslyclassified as a β-strand according to secondary struc-ture assignment algorithms.28 The rmsd for Cα
atoms between the N-terminal, redox-active adomain of Mpd1p and that of Pdi1p is 1.0 Å over102 residues, but between β-strands β4 and β5Mpd1p has a ∼15 amino acid insertion not found inPdi1p (Fig. 4a). Poor electron density was seen forthis region in experimental and 2Fo–Fc electrondensity maps, so the loop was not included in theMpd1p model. The b domains of Mpd1p and Pdi1pare also similar, with a Cα rmsd of 2.0 Å over 79residues. However, Mpd1p has an insertion in thisdomain as well: the∼30-residue loop downstream ofβ-strand β3, including helix α3, is not found in thePdi1p b domain and is present in truncated form(∼13 residues) in the Pdi1p b' domain (Fig. 4a).Preceding the last helix of the domain, Pdi1p b' hasan insertion of∼12 residues not found in the Pdi1p orMpd1p b domains.The relative orientations of the a and b domains
differ between Mpd1p and Pdi1p (Fig. 4b), placingMpd1p far outside the cluster of other tandem trxdomain proteins with known structure (Fig. 1).Whenthe a domains of Mpd1p and Pdi1p are superposed,the b domains are related by a rotation of nearly 180°(Fig. 4c). Ile153 of Mpd1p (Fig. 2), corresponding to
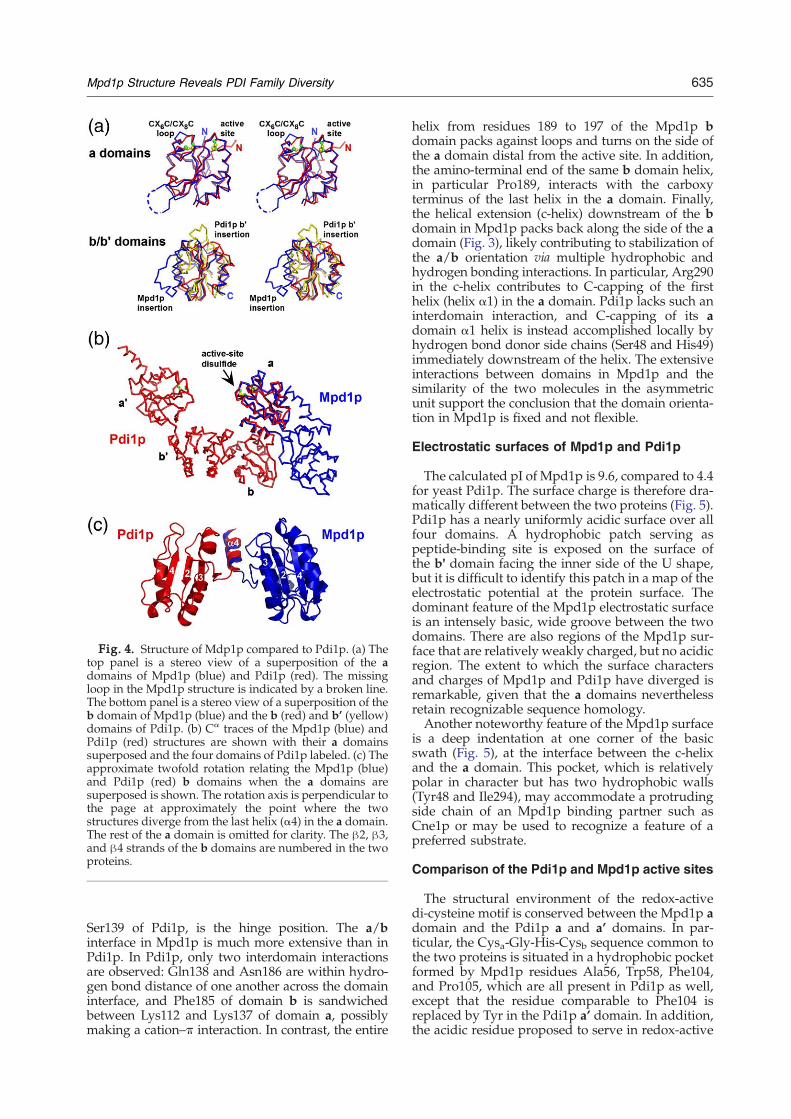
Fig. 4. Structure of Mdp1p compared to Pdi1p. (a) Thetop panel is a stereo view of a superposition of the adomains of Mpd1p (blue) and Pdi1p (red). The missingloop in the Mpd1p structure is indicated by a broken line.The bottom panel is a stereo view of a superposition of theb domain of Mpd1p (blue) and the b (red) and b’ (yellow)domains of Pdi1p. (b) Cα traces of the Mpd1p (blue) andPdi1p (red) structures are shown with their a domainssuperposed and the four domains of Pdi1p labeled. (c) Theapproximate twofold rotation relating the Mpd1p (blue)and Pdi1p (red) b domains when the a domains aresuperposed is shown. The rotation axis is perpendicular tothe page at approximately the point where the twostructures diverge from the last helix (α4) in the a domain.The rest of the a domain is omitted for clarity. The β2, β3,and β4 strands of the b domains are numbered in the twoproteins.
635Mpd1p Structure Reveals PDI Family Diversity
Ser139 of Pdi1p, is the hinge position. The a/binterface in Mpd1p is much more extensive than inPdi1p. In Pdi1p, only two interdomain interactionsare observed: Gln138 and Asn186 are within hydro-gen bond distance of one another across the domaininterface, and Phe185 of domain b is sandwichedbetween Lys112 and Lys137 of domain a, possiblymaking a cation–π interaction. In contrast, the entire
helix from residues 189 to 197 of the Mpd1p bdomain packs against loops and turns on the side ofthe a domain distal from the active site. In addition,the amino-terminal end of the same b domain helix,in particular Pro189, interacts with the carboxyterminus of the last helix in the a domain. Finally,the helical extension (c-helix) downstream of the bdomain in Mpd1p packs back along the side of the adomain (Fig. 3), likely contributing to stabilization ofthe a/b orientation via multiple hydrophobic andhydrogen bonding interactions. In particular, Arg290in the c-helix contributes to C-capping of the firsthelix (helix α1) in the a domain. Pdi1p lacks such aninterdomain interaction, and C-capping of its adomain α1 helix is instead accomplished locally byhydrogen bond donor side chains (Ser48 and His49)immediately downstream of the helix. The extensiveinteractions between domains in Mpd1p and thesimilarity of the two molecules in the asymmetricunit support the conclusion that the domain orienta-tion in Mpd1p is fixed and not flexible.
Electrostatic surfaces of Mpd1p and Pdi1p
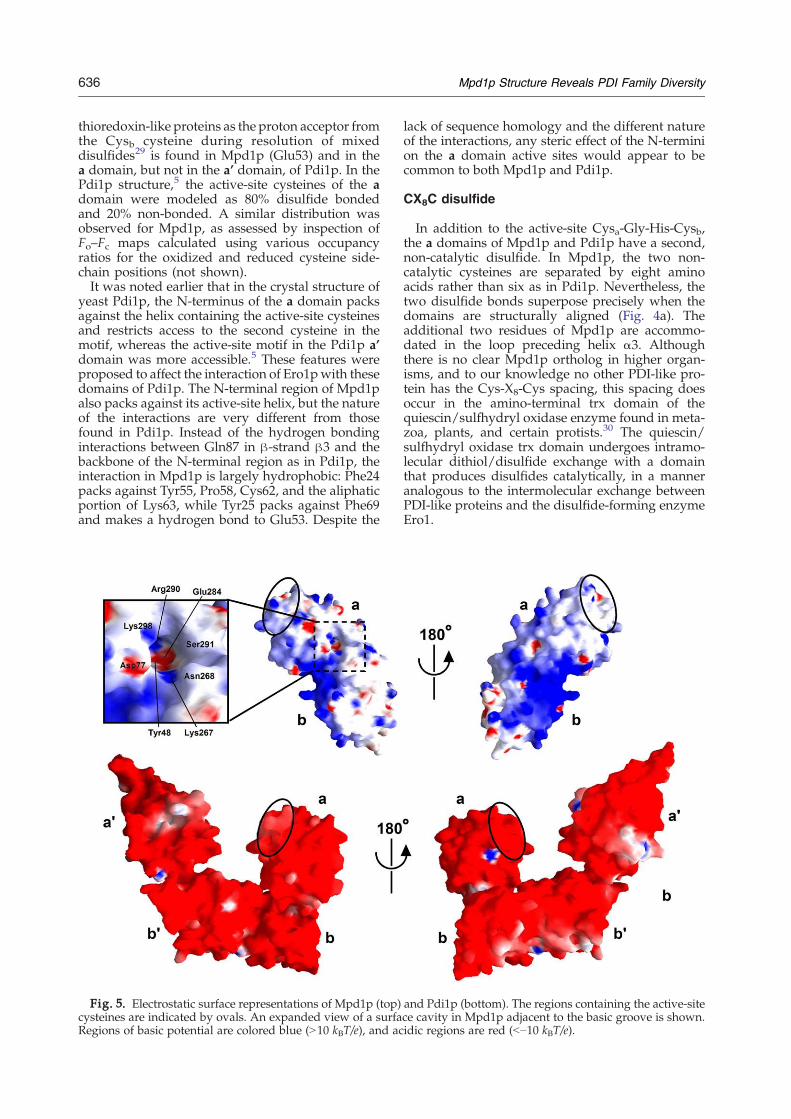
The calculated pI of Mpd1p is 9.6, compared to 4.4for yeast Pdi1p. The surface charge is therefore dra-matically different between the two proteins (Fig. 5).Pdi1p has a nearly uniformly acidic surface over allfour domains. A hydrophobic patch serving aspeptide-binding site is exposed on the surface ofthe b' domain facing the inner side of the U shape,but it is difficult to identify this patch in a map of theelectrostatic potential at the protein surface. Thedominant feature of the Mpd1p electrostatic surfaceis an intensely basic, wide groove between the twodomains. There are also regions of the Mpd1p sur-face that are relatively weakly charged, but no acidicregion. The extent to which the surface charactersand charges of Mpd1p and Pdi1p have diverged isremarkable, given that the a domains neverthelessretain recognizable sequence homology.Another noteworthy feature of the Mpd1p surface
is a deep indentation at one corner of the basicswath (Fig. 5), at the interface between the c-helixand the a domain. This pocket, which is relativelypolar in character but has two hydrophobic walls(Tyr48 and Ile294), may accommodate a protrudingside chain of an Mpd1p binding partner such asCne1p or may be used to recognize a feature of apreferred substrate.
Comparison of the Pdi1p and Mpd1p active sites
The structural environment of the redox-activedi-cysteine motif is conserved between the Mpd1p adomain and the Pdi1p a and a’ domains. In par-ticular, the Cysa-Gly-His-Cysb sequence common tothe two proteins is situated in a hydrophobic pocketformed by Mpd1p residues Ala56, Trp58, Phe104,and Pro105, which are all present in Pdi1p as well,except that the residue comparable to Phe104 isreplaced by Tyr in the Pdi1p a’ domain. In addition,the acidic residue proposed to serve in redox-active
636 Mpd1p Structure Reveals PDI Family Diversity
thioredoxin-like proteins as the proton acceptor fromthe Cysb cysteine during resolution of mixeddisulfides29 is found in Mpd1p (Glu53) and in thea domain, but not in the a’ domain, of Pdi1p. In thePdi1p structure,5 the active-site cysteines of the adomain were modeled as 80% disulfide bondedand 20% non-bonded. A similar distribution wasobserved for Mpd1p, as assessed by inspection ofFo–Fc maps calculated using various occupancyratios for the oxidized and reduced cysteine side-chain positions (not shown).It was noted earlier that in the crystal structure of
yeast Pdi1p, the N-terminus of the a domain packsagainst the helix containing the active-site cysteinesand restricts access to the second cysteine in themotif, whereas the active-site motif in the Pdi1p a’domain was more accessible.5 These features wereproposed to affect the interaction of Ero1pwith thesedomains of Pdi1p. The N-terminal region of Mpd1palso packs against its active-site helix, but the natureof the interactions are very different from thosefound in Pdi1p. Instead of the hydrogen bondinginteractions between Gln87 in β-strand β3 and thebackbone of the N-terminal region as in Pdi1p, theinteraction in Mpd1p is largely hydrophobic: Phe24packs against Tyr55, Pro58, Cys62, and the aliphaticportion of Lys63, while Tyr25 packs against Phe69and makes a hydrogen bond to Glu53. Despite the
Fig. 5. Electrostatic surface representations of Mpd1p (top)cysteines are indicated by ovals. An expanded view of a surfaRegions of basic potential are colored blue (N10 kBT/e), and ac
lack of sequence homology and the different natureof the interactions, any steric effect of the N-terminion the a domain active sites would appear to becommon to both Mpd1p and Pdi1p.
CX8C disulfide
In addition to the active-site Cysa-Gly-His-Cysb,the a domains of Mpd1p and Pdi1p have a second,non-catalytic disulfide. In Mpd1p, the two non-catalytic cysteines are separated by eight aminoacids rather than six as in Pdi1p. Nevertheless, thetwo disulfide bonds superpose precisely when thedomains are structurally aligned (Fig. 4a). Theadditional two residues of Mpd1p are accommo-dated in the loop preceding helix α3. Althoughthere is no clear Mpd1p ortholog in higher organ-isms, and to our knowledge no other PDI-like pro-tein has the Cys-X8-Cys spacing, this spacing doesoccur in the amino-terminal trx domain of thequiescin/sulfhydryl oxidase enzyme found in meta-zoa, plants, and certain protists.30 The quiescin/sulfhydryl oxidase trx domain undergoes intramo-lecular dithiol/disulfide exchange with a domainthat produces disulfides catalytically, in a manneranalogous to the intermolecular exchange betweenPDI-like proteins and the disulfide-forming enzymeEro1.
and Pdi1p (bottom). The regions containing the active-sitece cavity in Mpd1p adjacent to the basic groove is shown.idic regions are red (b−10 kBT/e).

637Mpd1p Structure Reveals PDI Family Diversity
Discussion
Molecular genetics experiments, homologysearches, and protein fold prediction have revealedthe presence in eukaryote genomes of numerousproteins containing tandem trx-like domains. Manyof these proteins are localized to the ER where theyfunction as either general or specific chaperones andcatalysts of disulfide bond formation and isomeri-zation.2,31 In addition, PDI family proteins arefound in chloroplasts32 and in mitochondria.33 Pro-tein fold and amino acid sequence analyses groupthe PDI-like proteins into a family, but it is be-coming clear that structural properties distinguishone member from another. These individual phy-sical characteristics are likely to profoundly impactphysiological activities and bestow functionaldiversity.Primary structure analysis does not provide infor-
mation about relative domain orientation or detailedsurface properties, both of which may contribute tosubstrate specificity, formation of stable complexeswith other proteins, and chaperone activity of PDIproteins. Although the Cys-X-X-Cys motif in thecontext of a predicted trx fold is a likely indicator of afunction in disulfide exchange, the tertiary andquaternary structural contexts of these motifs/foldsare critical. For example, in the network of oxido-reductases responsible for disulfide bond formationin the bacterial periplasm, dimerization of the iso-merase DsbC sterically prevents its trx-like domainsfrom being oxidized inappropriately by the disulfidebond-generating enzyme DsbB.34,35 In higher eukar-yotes, the plethora of PDI-like proteins probablyreflects a range of redox properties and targeting ofoxidoreductase activity to varied substrate pools, buthow differences in function and specificity are deter-mined is poorly understood.Global structural features are particularly impor-
tant in the activities of trx fold proteins due to theapparently limited extent to which the active sites ofthese domains dictate substrate specificity. Thio-redoxin was early on labeled a “male” enzyme,having a protruding active-site di-cysteine motif inmarked contrast to the substrate binding cavities ofother enzymes.36 In the latter, substrate specificitycan be set by the size of the binding pocket and thefunctional groups available for interaction with sub-strate. Although in trx-like proteins, the pKa valuesof the nucleophilic cysteines and the redox potentialsof the active-site Cys-X-X-Cys motifs vary,37 whichmay affect the rates and nature of reactions catalyzed(i.e., oxidation versus isomerization), these featuresprobably do not greatly affect substrate specificity orfunctional role per se. To illustrate this point, trx-likeproteins with vastly different redox potentials canfunctionally substitute for one another in vivo.38 It islikely that the particular activities of the many PDI-like proteins in the ER are determined less by active-site chemistry than by expression levels, localization,complex formation with other proteins, and perhapsby steric restrictions to prevent interactions that arenot physiologically beneficial.
Despite being a two-domain protein, yeast Mpd1pmay be functionally analogous to mammalianERp57, a four-domain ER protein that associateswith the glycan binding chaperones calnexin andcalreticulin.39 Mpd1p binds to Cne1p, the singlecalnexin/calreticulin homolog in yeast, with a dis-sociation constant of ∼200 nM.17 In this manner,ERp57 and Mpd1p oxidoreductase activity may berecruited to glycoprotein substrates. The structure ofthe ERp57 b and b’ domains, the two non-catalyticcentral domains of the protein, have been deter-mined by X-ray crystallography.11 The calnexinbinding site on the b’ domain was investigated byNMR and proposed to correspond to a conservedbasic patch on the domain surface.11 A positivelycharged binding site for calnexin in ERp57 wasexpected, based on the negative charge of the regionof calnexin that participates in the interaction.40 Thepositive potential of the Mpd1p surface (Fig. 5) ismuch more pronounced than that of ERp57, but thefunction of the surface charge of Mpd1p is notknown. The large basic patch of Mpd1p is centeredon the interdomain groove, rather than on the non-existent b’ domain, and therefore could not havebeen predicted without knowledge of the relativedomain orientation. One may speculate that themode of interaction of Cne1p with Mpd1p positionsthe Cne1p lectin domain and its cargo at theappropriate distance and orientation with respectto the Mpd1p active-site Cys-X-X-Cys motif.
Conclusions
PDI family proteins have been identified andclassified on the basis of sequence homology, theexistence of characteristic sequence motifs, andalgorithms that recognize the trx structural fold.Current interest lies in determining the structuraldifferences between PDI family members and inidentifying the roles and purposes for multiple PDIproteins in the ER lumen and in other organelles. Toreach this goal, it is essential to determine thestructural differences between PDI family membersas well as their unique redox properties, substratespecificities, and other distinguishing functionalfeatures. To date, research has focused primarily onPDI itself due to its status as a founding member ofthe PDI family and the essential nature of Pdi1p inyeast. Indeed, interpretation of structural and bio-chemical data on PDI family members has oftenrelied upon PDI as a reference. For example,to complement a study of the influence of redox-inactive domains on the stability and activity ofneighboring catalytic domains, the structure of thethree-domain lumenal region of TMX3 wasmodeled13 on the basis of the NMR structure of thea domain of human PDI41 and the X-ray crystalstructure of calsequestrin.6 The Mpd1p structureprovides an additional and highly divergent templatefor such modeling exercises. Nevertheless, a perhapsmore instructive lesson from elucidation of theMpd1p structure is the challenge in predicting the

638 Mpd1p Structure Reveals PDI Family Diversity
multidomain tertiary structures of PDI family pro-teins from primary structure alone. An amino acidsequence alignment of Pdi1p and Mpd1p at the a/binterdomain junction (Fig. 6) does not hint at thestructural divergence observed to occur in this region.
Fig. 6. The a/b interdomain linker sequences ofMpd1p(top) and Pdi1p (bottom) are aligned. The α4 helix is thelast secondary structure element in the a domain. The openarrowhead indicates the hinge residue as shown in red inFigs. 1 and 2. The filled arrowhead indicates a conservedburied hydrophobic residue that is superposed in astructural alignment of the b domains of the two proteins.This figure illustrates the limited number of amino acidsneeded to accommodate dramatic differences in domainorientation and the likelihood of not observing suchdifferences when inspecting sequence alignments.
†http://www.pymol.org
Materials and Methods
Mpd1p expression and purification
The sequence coding for yeast Mpd1p (residues 23–310)lacking the signal sequence and ER retention signal andcontaining a His6 tag at the amino terminus was insertedinto the pET-15b vector (Novagen) between the NcoI andBamHI restriction sites. This construct, which containedtwomethionine residues (theN-terminalMet andMet108),was mutagenized (Leu65Met/Leu76Met) to improve theanomalous signal for phase determination using multipleanomalous dispersion. The mutated positions are buriedbetween helix α2 and the β-sheet of the Mpd1p a domain,andmutagenesis did not affect the expression, purification,or structure of the protein significantly. Protein expressionwas done in the E. coli strain BL21 (DE3) plysS (Novagen).Cells containing the expression plasmid were grown to anA595nm of 0.4–0.6 at 37 °C, isopropyl-1-thio-β-D-galacto-pyranoside (IPTG) was added to a final concentration of1 mM, and the cultures were grown at 25 °C for a further12 h before harvesting. Cell pellets were suspended in50 mM sodium phosphate buffer (pH 8.0), 500 mM NaCl,and 10 mM imidazole, sonicated, and centrifuged for25 min at 15,000g to remove the cell debris. The super-natant was incubated with Ni-NTA agarose (Qiagen) for2 h at 4 °C. The beads were allowed to settle, thesupernatant was removed, and the beads were transferredto a column. The columnwas washed with 50 mM sodiumphosphate buffer (pH 8.0), 500 mM NaCl, and 20 mMimidazole. Mpd1p protein was eluted with a step gradientof imidazole in 50 mM sodium phosphate buffer (pH 8.0),300 mM NaCl and then dialyzed against 10 mM Tris(pH 8.0), 50 mM NaCl. Protein that precipitated duringdialysis was removed by centrifugation at 15,000g for10 min, and the remaining soluble fraction was furtherpurified by gel-filtration chromatography. Elution frac-tions containing monomeric and dimeric forms of Mpd1pwere pooled, and the combinedmaterial was concentratedto 24–28 mg/ml.
Crystallization, data collection, structure refinement,and analysis
Mpd1p was crystallized by the hanging-drop, vapor-diffusion method at 20 °C. The protein stock solution (2 μl)was mixed with well solution (2 μl) containing 30% PEG4000, 0.2M sodium acetate, and 100mM cacodylic acid (pH5.9). Needlelike crystals that appeared spontaneously wereused for streak-seeding hanging drops that had equili-brated overnight against a well solution of 17% PEG 4000,0.2 M sodium acetate, 100 mM cacodylic acid (pH 5.9), and6–10% ethylene glycol. Crystals of protein containingselenomethionine were grown by seeding over a wellsolution of 21–24% PEG 4000, 0.2 M sodium acetate,100mM cacodylic acid (pH 5.9), and 6–10% ethylene glycol.Crystals of monoclinic and orthorhombic space groupsappeared after about two days, but the orthorhombiccrystals suffered from high mosaicity and lattice disorders.The monoclinic crystals were selected for further study.
Crystals were transferred to 30% PEG 4000, 0.2 Msodium acetate, 100 mM cacodylic acid (pH 5.9), and 15%ethylene glycol before flash-freezing in a nitrogen streamat 120 K. Native data were collected using a RU-H3RRigaku rotating anode generator equipped with Osmicconfocal focusing mirrors and an R-AXIS IV++ detector.Multiple anomalous dispersion data were collected atbeamline BM14 of the European Synchrotron RadiationFacility (ESRF), Grenoble, France. Data were processedand scaled using the DENZO and SCALEPACK pro-grams.42 Selenium atoms were located and phases werecalculated using SOLVE.43 The structure was built usingO,44 and refinement was done with CNS.45 Molprobitywas used to analyze the structure and assess its quality.46The structure ranked in the 89th percentile amongstructures of comparable resolution (1.75 – 2.25 Å),98.3% of residues were in the most favored regions ofRamachandran space, and there was no outlier. Imageswere made using PyMOl†, except for electrostatic surfacerepresentations, which were generated using Grasp.47
The Pdi1p and Mpd1p a domains were compared bysuperposition over Pdi1p residues 31–48, 50–79, 82–92,93–113, and 118–139 and Mpd1p residues 28–45, 48–77,79–89, 92–112, and 132–153 using lsqkab.48 The b domainswere superposed using Pdi1p residues 142–149, 156–166,168–195, 199–208, 212–220, and 224–236 and Mpd1presidues 156–163, 171–181, 182–209, 244–253, 256–264,and 265–277.
Protein Data Bank accession code
Structure factors and coordinates have been depositedin the RCSB Protein Data Bank with accession code 3ED3.
Acknowledgements
The authors thank Chih-chen Wang and RobertoSitia for providing the ERp44 coordinates in advanceof their release. This work was supported by a grantfrom the U.S.-Israel Binational Science Foundation(to D.F. and C.A.K.) and by the Kimmelman Centerfor Macromolecular Assemblies (to D.F.).

639Mpd1p Structure Reveals PDI Family Diversity
References
1. Norgaard, P., Westphal, V., Tachibana, C., Alsoe, L.,Holst, B. &Winther, J. R. (2001). Functional differencesin yeast protein disulfide isomerases. J. Cell Biol. 152,553–562.
2. Appenzeller-Herzog, C. & Ellgaard, L. (2008). Thehuman PDI family: versatility packed into a singlefold. Biochim. Biophys. Acta, 1783, 535–548.
3. Frand, A. R. & Kaiser, C. A. (1999). Ero1p oxidizesprotein disulfide isomerase in a pathway for disulfidebond formation in the endoplasmic reticulum. Mol.Cell, 4, 469–477.
4. Kulp,M. S., Frickel, E.M., Ellgaard, L. &Weissman, J. S.(2006). Domain architecture of protein-disulfide isome-rase facilitates its dual role as an oxidase and an iso-merase in Ero1p-mediated disulfide formation. J. Biol.Chem, 281, 876–884.
5. Tian, G., Xiang, S., Noiva, R., Lennarz, W. J. &Schindelin, H. (2006). The crystal structure of yeastprotein disulfide isomerase suggests cooperativitybetween its active sites. Cell, 124, 61–73.
6. Wang, S., Trumble, W. R., Liao, H., Wesson, C. R.,Dunker, A. K. & Kang, C. H. (1998). Crystal structureof calsequestrin from rabbit skeletal muscle sarco-plasmic reticulum. Nat. Struct. Biol. 5, 476–483.
7. MacLennan, D. H., Abu-Abed, M. & Kang, C. H.(2002). Structure-function relationships in Ca2+ cyclingproteins. J. Mol. Cell. Cardiol. 34, 897–918.
8. Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S.,Wilkins, M. R., Appel, R. D. & Bairoch, A. (2005).Protein identification and analysis tools on theExPASy server. In The Proteomics Protocols Handbook(Walker, John M., ed), pp. 571–607, Humana Press,Totowa, NJ.
9. Van, P. N., Peter, F. & Söling, H. D. (1989). Fourintracisternal calcium-binding glycoproteins from ratliver microsomes with high affinity for calcium. Noindication for calsequestrin-like proteins in inositol1,4,5-trisphosphate-sensitive calcium sequestering ratliver vesicles. J. Biol. Chem. 264, 17494–17501.
10. Li, Y. & Camacho, P. (2004). Ca2+-dependent redoxmodulation of CERCA by ERp57. J. Cell Biol. 164,35–46.
11. Kozlov, G., Maattanen, P., Schrag, J. D., Pollock, S.,Cygler, M., Nagar, B. et al. (2006). Crystal structure ofthe bb' domains of the protein disulfide isomeraseERp57. Structure, 14, 1331–1339.
12. Wang, L., Wang, L., Vavassori, S., Li, S., Ke, H.,Anelli, T. et al. (2008). Crystal structure of humanERp44 shows a dynamic functional modulation byits carboxy-terminal tail. EMBO Rep. 9, 642–647.
13. Haugstetter, J., Maurer, M. A., Blicher, T., Pagac, M.,Wider, G. & Ellgaard, L. (2007). Structure-functionanalysis of the endoplasmic reticulum oxidoreduc-tase TMX3 reveals interdomain stabilization of theN-terminal redox-active domain. J. Biol. Chem. 282,33859–33867.
14. Ferrari, D. M. & Söling, H. D. (1999). The proteindisulphide-isomerase family: unravelling a string offolds. Biochem. J. 339, 1–10.
15. Jessop, C. E., Chakravarthi, S., Garbi, N., Hämmerling,G. J., Lovell, S. & Bulleid, N. J. (2007). ERp57 isessential for efficient folding of glycoproteins sharingcommon structural domains. EMBO J. 26, 28–40.
16. Cresswell, P., Ackerman, A. L., Giodini, A., Peaper,D. R. & Wearsch, P. A. (2005). Mechanisms of MHCclass I-restricted antigen processing and cross-presentation. Immunol. Rev. 207, 145–157.
17. Kimura, T., Hosada, T., Sato, Y., Kitamura, Y., Ikeda,T., Horibe, T. & Kikuchi, M. (2005). Interactions amongyeast protein-disulfide isomerase proteins and endo-plasmic reticulum chaperone proteins influence theiractivities. J. Biol. Chem. 280, 31438–31441.
18. Tachibana, C. & Stevens, T. H. (1992). The yeast EUG1gene encodes an endoplasmic reticulum protein that isfunctionally related to protein disulfide isomerase.Mol. Cell Biol. 12, 4601–4611.
19. Tachikawa, H., Funahashi, W., Takeuchi, Y., Nakanishi,H., Nishihara, R., Katoh, S. et al. (1997). Overproduc-tion of Mpd2p suppresses the lethality of proteindisulfide isomerase depletion in a CXXC sequencedependent manner. Biochem. Biophys. Res. Commun.239, 710–714.
20. Tachikawa, H., Takeuchi, Y., Funahashi, W., Miura, T.,Gao, X. D., Fujimoto, D. et al. (1995). Isolation andcharacterization of a yeast gene, MPD1, the over-expression of which suppresses inviability caused byprotein disulfide isomerase depletion. FEBS Lett. 369,212–216.
21. Wang, Q. Q. & Chang, A. (1999). Eps1, a novel PDI-related protein involved in ER quality control in yeast.EMBO J. 18, 5972–5982.
22. Kelley, L. A., MacCallum, R. M. & Sternberg, M. J. E.(2000). Enhanced genome annotation using structuralprofiles in the program 3D-PSSM. J. Mol. Biol. 299,499–520.
23. Dasgupta, S., Iyer, G. H., Bryant, S. H., Lawrence, C. E.& Bell, J. A. (1997). Extent and nature of contactsbetween protein molecules in crystal lattices and bet-ween subunits of protein oligomers. Proteins: Struct.Funct. Genet. 28, 494–514.
24. Iyer, G. H., Dasgupta, S. & Bell, J. A. (2000). Ionicstrength and intermolecular contacts in proteincrystals. J. Crystal Growth, 217, 429–440.
25. Walter, T. S., Meier, C., Assenberg, R., Au, K. F., Ren, J.,Verma, A. et al. (2006). Lysine methylation as a routinerescue strategy for protein crystallization. Structure,14, 1617–1622.
26. Goldschmidt, L., Cooper, D. R., Derewenda, Z. S. &Eisenberg, D. (2007). Toward rational protein crystal-lization: a web server for the design of crystallizableprotein variants. Protein Sci. 16, 1569–1576.
27. Hendrickson, W. A. (1991). Determination of macro-molecular structures from anomalous diffraction ofsynchrotron radiation. Science, 254, 51–58.
28. Kabsch, W. & Sander, C. (1983). Dictionary of proteinsecondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22,2577–2637.
29. LeMaster, D. M. (1996). Structural determinants of thecatalytic reactivity of the buried cysteine of Escher-ichia coli thioredoxin. Biochemistry, 35, 14876–14881.
30. Raje, S. & Thorpe, C. (2003). Inter-domain redoxcommunication in flavoenzymes of the quiescin/sulfhydryl oxidase family: role of a thioredoxindomain in disulfide bond formation. Biochemistry, 42,4560–4568.
31. Maattanen, P., Kozlov, G., Gehring, K. & Thomas,D. Y. (2006). ERp57 and PDI: multifunctional proteindisulfide isomerases with similar domain architec-tures but differing substrate-partner associations.Biochem. Cell Biol. 84, 881–889.
32. Trebitsh, T., Meiri, E., Ostersetzer, O., Adam, Z. &Danon, A. (2001). The protein disulfide isomerase-likeRB60 is partitioned between stroma and thylakoidsin Chlamydomonas reinhardtii chloroplasts. J. Biol.Chem, 276, 4564–4569.

640 Mpd1p Structure Reveals PDI Family Diversity
33. Kimura, T., Horibe, T., Sakamoto, C., Shitara, Y.,Fujiwara, F., Komiya, T. et al. (2008). Evidence formitochondrial localization of P5, a member of theprotein disulfide isomerase family. J. Biochem. 144,187–196.
34. Bader, M. W., Hiniker, A., Regeimbal, J., Goldstone,D., Haebel, P. W., Riemer, J. et al. (2001). Turning adisulfide isomerase into an oxidase: DsbC mutantsthat imitate DsbA. EMBO J. 20, 1555–1562.
35. Inaba, K., Murakami, S., Suzuki, M., Nakagawa, A.,Yamashita, E., Okada, K. & Ito, K. (2006). Crystal struc-ture of the DsbB-DsbA complex reveals a mechanismof disulfide bond generation. Cell, 127, 789–801.
36. Holmgren, A., Söderberg, B. O., Eklund, H. &Brändén, C. I. (1975). Three-dimensional structure ofEscherichia coli thioredoxin-S2 to 2.8 Å resolution.Proc. Natl Acad. Sci. USA, 72, 2305–2309.
37. Carvalho, A. P., Fernandez, P. A. & Ramos, M. J.(2006). Similarities and differences in the thioredoxinsuperfamily. Prog. Biophys. Mol. Biol. 91, 229–248.
38. Debarbieux, L. & Beckwith, J. (1998). The reductiveenzyme thioredoxin 1 acts as an oxidant when it isexported to the Escherichia coli periplasm. Proc. NatlAcad. Sci. USA, 95, 10751–10756.
39. Oliver, J. D., van der Wal, F. J., Bulleid, N. J. & High, S.(1997). Interaction of the thiol-dependent reductaseERp57 with nascent glycoproteins. Science, 275, 66–86.
40. Frickel, E.-M., Riek, R., Jelesarov, I., Helenius, A.,Wüthrich, K. & Ellgaard, L. (2002). TROSY-NMR revealsinteraction between ERp57 and the tip of the calreticulinP-domain. Proc. Natl Acad. Sci. USA, 99, 1954–1959.
41. Kemmink, J., Darby, N. J., Dijkstra, K., Nilges, M. &Creighton, T. E. (1996). Structure determination of the
N-terminal thioredoxin-like domain of protein dis-ulfide isomerase using multidimensional hetero-nuclear 13C/15N NMR spectroscopy. Biochemistry,35, 7684–7691.
42. Otwinowski, Z. & Minor, W. (1997). Processing ofX-ray diffraction data collected in oscillation mode.Methods Enzymol. 276, 307–326.
43. Terwilliger, T. & Berendzen, J. (1999). AutomatedMAD and MIR structure solution. Acta Crystallog. D,55, 849–861.
44. Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M.(1991). Improved methods for building proteinmodels in electron density maps and the location oferrors in these models. Acta Crystallog. A, 47, 110–119.
45. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano,W. L., Gros, P., Grosse-Kunstleve, R. W. et al. (1998).Crystallography and NMR system: a new softwaresuite for macromolecular structure determination.Acta Crystallog. D, 54, 905–921.
46. Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N.,Kapral, G. J., Wang, X. et al. (2007). MolProbity: all-atom contacts and structure validation for proteinsand nucleic acids. Nucleic Acids Res. 35, W375–W383.
47. Nicholls, A., Sharp, K. A. & Honig, B. (1991). Proteinfolding and association: insights from the interfacialand thermodynamic properties of hydrocarbons.Proteins: Struct. Funct. Genet. 11, 281–296.
48. Kabsch, W. (1976). A solution for the best rotation torelate two sets of vectors. Acta Crystallog. A, 32, 922–923.
49. Cowtan, K. (1994). 'dm': an automated procedure forphase improvement by density modification. JointCCP4 ESF-EACBM Newsletter. Protein Crystallogr. 31,34–38.




















