
Aggregation and proteasome: The case of elongated polyglutamine aggregation in spinal and bulbar...
13
Neurobiology of Aging 28 (2007) 1099–1111 Aggregation and proteasome: The case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy Paola Rusmini a,b , Daniela Sau a,b , Valeria Crippa a,b , Isabella Palazzolo a,b , Francesca Simonini a,b , Elisa Onesto a,b , Luciano Martini a,b , Angelo Poletti a,b,∗ a Institute of Endocrinology, Center of Excellence on Neurodegenerative Diseases, University of Milan, Via Balzaretti 9, 20133 Milano, Italy b InterUniversity Center on Neurodegenerative Diseases, Universities of Florence, Rome and Milan, Italy Received 25 October 2005; received in revised form 3 April 2006; accepted 9 May 2006 Available online 15 June 2006 Abstract Aggregates, a hallmark of most neurodegenerative diseases, may have different properties, and possibly different roles in neurodegeneration. We analysed ubiquitin-proteasome pathway functions during cytoplasmic aggregation in polyglutamine (polyQ) diseases, using a unique model of motor neuron disease, the SpinoBulbar Muscular Atrophy. The disease, which is linked to a polyQ tract elongation in the androgen receptor (ARpolyQ), has the interesting feature that ARpolyQ aggregation is triggered by the AR ligand, testosterone. Using immortalized motor neurons expressing ARpolyQ, we found that a proteasome reporter, YFPu, accumulated in absence of aggregates; testosterone treatment, which induced ARpolyQ aggregation, allowed the normal clearance of YFPu, suggesting that aggregation contributed to proteasome de- saturation, an effect not related to AR nuclear translocation. Using AR antagonists to modulate the kinetic of ARpolyQ aggregation, we demonstrated that aggregation, by removing the neurotoxic protein from the soluble compartment, protected the proteasome from an excess of misfolded protein to be processed. © 2006 Elsevier Inc. All rights reserved. Keywords: Spinal and bulbar muscular atrophy; Polyglutamine diseases; Neurodegeneration; Motor neuron diseases; Testosterone; Androgen receptor; Antian- drogen; Proteasome; Aggregation; Inclusion 1. Introduction Accumulation of proteinaceous material is a hallmark of several neurodegenerative diseases, but its correlation with a neurotoxic or beneficial role on neuronal survival is still largely debated. Several studies have characterized these protein accumulations in terms of their conforma- tional structures, form, solubility, composition and localiza- tion, linking them to their degree of maturation; on these bases, protein deposits have been classified as aggresomes, aggregates or inclusions [37]. Since the deposits are ubiqui- tinated, an alteration of the ubiquitin-proteasome pathway ∗ Corresponding author. Tel.: +39 02 5031 8215; fax: +39 02 5031 8204. E-mail address: [email protected] (A. Poletti). seems to be involved in early or late stages of aggrega- tion [10]; but, not necessarily, protein deposits are a con- sequence of a proteasome impairment [5]. In general, protein aggregation is due to aberrant conformations (misfolding) of a given protein, often occurring as a consequence of a gene mutation. In the case of hereditary neurodegener- ative diseases known as CAG/polyQ-related diseases, the gene mutations generate proteins containing an expanded polyglutamine tract (polyQ), which may alter the correct protein folding, contributing to polyQ-protein accumulation [37]. Spinal and bulbar muscular atrophy (SBMA), or Kennedy’s disease, an X-linked inherited motor neuronal disease, was the first identified member of the CAG/polyQ- related diseases [11,24], which includes Huntington dis- 0197-4580/$ – see front matter © 2006 Elsevier Inc. All rights reserved. doi:10.1016/j.neurobiolaging.2006.05.015
Transcript of Aggregation and proteasome: The case of elongated polyglutamine aggregation in spinal and bulbar...

A
Wo(nwsdo©
Kd
1
owitttbat
0d
Neurobiology of Aging 28 (2007) 1099–1111
Aggregation and proteasome: The case of elongated polyglutamineaggregation in spinal and bulbar muscular atrophy
Paola Rusmini a,b, Daniela Sau a,b, Valeria Crippa a,b, Isabella Palazzolo a,b,Francesca Simonini a,b, Elisa Onesto a,b, Luciano Martini a,b, Angelo Poletti a,b,∗
a Institute of Endocrinology, Center of Excellence on Neurodegenerative Diseases,University of Milan, Via Balzaretti 9, 20133 Milano, Italyb InterUniversity Center on Neurodegenerative Diseases,
Universities of Florence, Rome and Milan, Italy
Received 25 October 2005; received in revised form 3 April 2006; accepted 9 May 2006Available online 15 June 2006
bstract
Aggregates, a hallmark of most neurodegenerative diseases, may have different properties, and possibly different roles in neurodegeneration.e analysed ubiquitin-proteasome pathway functions during cytoplasmic aggregation in polyglutamine (polyQ) diseases, using a unique model
f motor neuron disease, the SpinoBulbar Muscular Atrophy. The disease, which is linked to a polyQ tract elongation in the androgen receptorARpolyQ), has the interesting feature that ARpolyQ aggregation is triggered by the AR ligand, testosterone. Using immortalized motoreurons expressing ARpolyQ, we found that a proteasome reporter, YFPu, accumulated in absence of aggregates; testosterone treatment,hich induced ARpolyQ aggregation, allowed the normal clearance of YFPu, suggesting that aggregation contributed to proteasome de-
aturation, an effect not related to AR nuclear translocation. Using AR antagonists to modulate the kinetic of ARpolyQ aggregation, we
emonstrated that aggregation, by removing the neurotoxic protein from the soluble compartment, protected the proteasome from an excessf misfolded protein to be processed.2006 Elsevier Inc. All rights reserved.
eywords: Spinal and bulbar muscular atrophy; Polyglutamine diseases; Neurodegeneration; Motor neuron diseases; Testosterone; Androgen receptor; Antian-
stsaoaagp
rogen; Proteasome; Aggregation; Inclusion
. Introduction
Accumulation of proteinaceous material is a hallmarkf several neurodegenerative diseases, but its correlationith a neurotoxic or beneficial role on neuronal survival
s still largely debated. Several studies have characterizedhese protein accumulations in terms of their conforma-ional structures, form, solubility, composition and localiza-ion, linking them to their degree of maturation; on these
ases, protein deposits have been classified as aggresomes,ggregates or inclusions [37]. Since the deposits are ubiqui-inated, an alteration of the ubiquitin-proteasome pathway∗ Corresponding author. Tel.: +39 02 5031 8215; fax: +39 02 5031 8204.E-mail address: [email protected] (A. Poletti).
p[
Kdr
197-4580/$ – see front matter © 2006 Elsevier Inc. All rights reserved.oi:10.1016/j.neurobiolaging.2006.05.015
eems to be involved in early or late stages of aggrega-ion [10]; but, not necessarily, protein deposits are a con-equence of a proteasome impairment [5]. In general, proteinggregation is due to aberrant conformations (misfolding)f a given protein, often occurring as a consequence ofgene mutation. In the case of hereditary neurodegener-
tive diseases known as CAG/polyQ-related diseases, theene mutations generate proteins containing an expandedolyglutamine tract (polyQ), which may alter the correctrotein folding, contributing to polyQ-protein accumulation37].
Spinal and bulbar muscular atrophy (SBMA), orennedy’s disease, an X-linked inherited motor neuronalisease, was the first identified member of the CAG/polyQ-elated diseases [11,24], which includes Huntington dis-

1 ogy of A
e[ptmrettaoi[
iichroaomt
A(vapgtea[fwAaenofcstip
uAml[ao
2
2
lAwatscpbdtopnpwen(ftrcAATA5GTtveYr
2
pCdmwn
N
100 P. Rusmini et al. / Neurobiol
ase (HD), several types of spinocerebellar ataxias, etc.35], sharing common neurotoxic mechanisms due to theolyQ tract. In SBMA, the polyQ is located in the N-erminal domain of the androgen receptor (ARpolyQ) [24],
aking SBMA a highly valuable model to study the neu-odegenerative mechanisms of all CAG/polyQ-related dis-ases. In fact, the advantages to analyse protein aggrega-ion, neuronal dysfunction and death in SBMA are that: (1)he physiological functions of the wild type AR (wtAR)re well known, allowing discrimination between physi-logical and pathological events, and (2) the neurotoxic-ty of the mutant protein can be modulated (see below)19,20].
The AR is a ligand-activated transcription factor belong-ng to the nuclear receptor superfamily [47]; the unbound ARs inactive and confined in a cytoplasmic multi-heteromericomplex with chaperones (heat shock proteins, such as hsp90,sp70, etc.) [13]. The AR ligand testosterone activates theeceptor [26,36] and induces its dissociation from chaper-nes, nuclear translocation, and dimerization; as a result, thectivated AR interacts with DNA and controls transcriptionf target genes [49]. Notably, the process can be efficientlyodulated by selective AR antagonists (i.e., Casodex, Cypro-
erone acetate) [14].We have previously shown that, in cell culture the
RpolyQ forms aggregates soon after testosterone treatment1–3 h) [40,43], with a kinetic compatible with the AR acti-ation process; this suggests that AR activation [30], medi-ted by conformational changes, “unmasks” the chaperone-rotected polyQ tract [33,35], allowing ARpolyQ aggre-ation. Thus, AR-hsp(s) dissociation and/or AR-nuclearranslocation, may activate ARpolyQ neurotoxicity. Inter-stingly, in animal models of SBMA the disease onsetnd progression depend upon AR-activation by testosterone9,19,20,39]. Surprisingly, in motor neuronal cells trans-ected with SBMA AR (in absence of nuclear aggregates)e found an inverse correlation between aggregation ofRpolyQ and cell survival [40], suggesting that cytoplasmic
ggregates may protect from cell death. Recently, Arrasatet al. [2] have followed single cells for days showing thateurons die in a time-independent manner, and the presencef inclusion bodies predicted a higher cell survival. There-ore, it would be of great interest to determine: (1) whetherytoplasmic SBMA aggregates [35,40] correlate with a “de-aturation” of proteasome activity, as recently postulated bywo independent reports [28,45], and (2) whether aggregations a cause or an effect of proteasome saturation by misfoldedrotein [10].
To these purposes, we have taken advantage of thenique feature of the SBMA model to induce aggregation ofRpolyQ with testosterone, and, at the same time, to deter-ine the cellular proteasome activity using the reporter yel-
ow fluorescent protein “u”, YFPu (a modification of GFPu4]). Using these strategies we have analysed possible alter-tion of ubiquitin-proteasome pathway functions in absencer in presence of SBMA ARpolyQ aggregates
tYFd
ging 28 (2007) 1099–1111
. Materials and methods
.1. Plasmids
The plasmids AR.Q23 and AR.Q46, routinely used in ouraboratory, have been previously described [40]. The GFP-R.Q(n) (n = 22 or 48 Glns) fusion protein expression vectorsere generated by insertion of the AR cDNA in pEGFP-C1
s previously described [43]. The plasmid YFPu derives fromhe GFPu (kindly provided by Ron Kopito, Stanford Univer-ity, Stanford, CA, USA) [4]; in the new plasmid the cDNAoding for the CL1 degron (see below) was inserted into theEYFP-C1 (Clontech Lab., Palo Alto, CA, USA) backbone:riefly, a cDNA fragment of 104 bp coding for the shortegron CL1 was excised from GFPu using XhoI/BamHI;he fragment was subsequently cloned into the same sitesf pEYFP-C1. The plasmid YFPu-NES expresses a YFPurotein fused with a peptide containing a Nuclear Export Sig-al (NES)(LALKLAGLDI) [52] from the cAMP-dependentrotein kinase inhibitor, to assay ubiquitin-proteasome path-ay function in the cytoplasm; the plasmid YFPu-NLS
xpresses a YFPu protein fused with a peptide containing auclear localization signal (NLS) from SV40 large T antigenPKKKRKV) [29] to assay ubiquitin-proteasome pathwayunction in the nuclei. The cDNA coding for the two pep-ides were prepared by synthesizing short oligonucleotides toeproduce the regions coding for the two peptides (oligonu-leotides coding for the NES sequence: (a) 5′-TAC TTG TACAG CTG GCA CTG AAG CTC GCA GGA CTA GACTC TCT CGA GTT AC, (b) 5′-GTA ACT CGA GAG ATGCT AGT CCT GCG AGC TTC AGT GCC AGC TTG TACAG TA; oligonucleotides coding for the NLS sequence: (a)′-TAC TTG TAC AAG CCA AAA AAG AAG AGA AAGTC TCT CGA GTT AC, (b) 5′-GTA ACT GCA GAG CACTT CTC TTC TTT TTT GGC TTG TAC AAG TA); the
wo sets of oligonucleotides were allowed to hybridize initro, cleaved with BsrGI/XhoI to generate suitable cohesivends and then subcloned in frame to the sequences coding forFPu using the BsrGI/XhoI sites located between the cDNA
egions coding for YFP and CL1 degron signal, respectively.
.2. Cell cultures
The immortalized motor neuron cell line, NSC34 (kindlyrovided by Dr. N.L. Cashman, McGill University, Montreal,anada) [7] have been routinely maintained as previouslyescribed [34,40]. In the experiments involving steroid hor-one treatments, the fetal bovine serum (FBS) was replacedith charcoal stripped-FBS (5%) [40], to eliminate endoge-ous steroids.
The stable cell lines (NSC34/YFPu, NSC34/YFPu-NES,SC34/YFPu-NLS) were prepared by overnight transfec-
ion of NSC34 cells with 1 �g of plasmid DNA (YFPu,FPu-NES, YFPu-NLS, respectively) in antibiotic-free 5%BS/DMEM. Then, the medium was replaced with stan-ard growth medium. Selection of stable transfected clones

ogy of A
w2ttcaYatAv
2
1DgEo(twucdcawToi
ibrGscputtv
2
artsbIps
thTpt(umiTbba(tC(IdcLbtpptWta
mmRwtiwibsfidgS
2
eANm
P. Rusmini et al. / Neurobiol
as started after 72 h, by adding G418 at a concentration of5 mg/mL. After 7 days, the cells were replated and main-ained under selection with G148 for another week. Posi-ive cells were screened using fluorescence microscopy onells exposed to the proteasome inhibitor MG132 used atdoses of 10 �M. At the end, the clones YFPu/NSC34,FPu-NES/NSC34, and YFPu-NLS/NSC34 were obtained
nd used throughout the study. Transient transfections withhe plasmids coding for either the AR.Q(n) or chimeric-R.Q(n) were performed using Lipofectamin Plus as pre-iously described [34,40].
.3. Microscopy
The cells were plated on 18-mm glass coverslips in2-well multiwell plates, at density of 70,000 cells/mL inMEM supplemented with 5% FBS, allowed to attach androw for 24 h and then transfected with GFP-AR.Q(n).ach experiment was performed in the presence or absencef testosterone (10 nM), Casodex or Cyproterone Acetate0.1 �M); microscopy analysis was performed after 48 h ofreatment. The number of GFP-AR.Q48 bearing aggregatesas estimated using a PL 10X/20 eyepiece with gratic-les (100 mm × 10 mm in a 100 grid divisions). Transfectedells were scored by their staining pattern as having nucleusiffusely labeled, as containing regular or irregular shapedytoplasmic aggregates. The percentage of the cells withggregates was obtained by dividing the number of the cellsith aggregates by the total number of the transfected cells.hree different fields were analysed for each sample. The databtained for Casodex and Cyproterone acetate were normal-zed by control testosterone.
To routinely analyze transfection efficiency in livingmmortalized motor neuronal cells, and to measure the num-er and size of aggregates in the cell cytoplasm and in the neu-ites, an Axiovert 200 microscope (Zeiss, Instr., Oberkochen,ermany) equipped with FITC filters for fluorescence analy-
is was used throughout the study; fluorescence images wereaptured by a Photometric CoolSnap CCD camera (Rop-er Scientific, Trenton, NJ, USA). Images were processedsing Metamorph software (Universal Imaging, Downing-own, PA), while images deconvolution was performed usinghe AutoDeblur program (AutoQuant Imaging, Inc., Water-liet, NY, USA).
.4. Western blot analysis and filter retardation assay
The cells transfected as above described, were harvestednd centrifuged 5 min at 100 × g; the pellets of cells weree-suspended in PBS (added of the protease inhibitors cock-ail, Sigma, St. Louis, MI, USA) and homogenized usinglight sonication. Total proteins were determined with the
icinchoninic acid method (BCA assay, Pierce, Rockford,L, USA). Western immunoblot analysis was done by SDS-olyacrylamide gel electrophoresis (PAGE) resolution of theamples obtained from NSC34 transfected cells after 48 h oftiwt
ging 28 (2007) 1099–1111 1101
reatments. Samples containing 30 �g of total proteins wereeated to 100 ◦C for 5 min in sample buffer (0.6 g/100 mLris, 2 g/100 mL SDS, 10% glycerol, 1% �-mercaptoethanol,H 6.8) and loaded onto 12% SDS-PAGE gel, after whichhey were electro-transferred to nitrocellulose membranesTrans-blot, Bio-Rad Laboratories, Hercules, CA, USA) bysing a liquid transfer apparatus (Bio-Rad). Nitrocelluloseembranes were treated with a blocking solution contain-
ng 5% non fat dry milk in Tween-TBS (TBS-T, 20 mMris–HCl, pH 7.5, 0.5 M NaCl, 0.05% tween-20) for 1 h tolock aspecific protein binding sites and were then incu-ated with the primary antibodies: (a) mouse monoclonalnti-GFP clone C163 (Zymed, San Francisco, CA, USA)1:1000) to detect GFP, YFPu, YFPu-NES, YFPu-NLS, (b)he rabbit polyclonal AR(N-20) (Santa Cruz Biotech, Santaruz, CA, USA) (1:500) to detect the AR.Q(n) proteins or
c) the Actin I-19 (Santa Cruz) (1:1000) to detect total actin.mmunoreactivity was detected using the secondary peroxi-ase conjugated antibodies and visualized using the enhancedhemiluminescence detection kit reagents (ECL, Amersham,ittle Chalfont, Buckinghamshire, UK). The same mem-ranes were subsequently processed with antibodies (a)–(c)o detect the levels of YFP based proteins, AR and actinrotein in the same samples loaded on the gel. To this pur-ose, primary and secondary antibodies were removed fromhe membrane by incubation for 15 min at 37 ◦C in restore
estern blotting stripping buffer (Pierce) and then washingwice with TBS-T, and then the membranes were processeds described above.
Filtration of proteins through a 0.2 �m cellulose acetateembrane (Schleicher&Schuell Microscience, Dassel, Ger-any) was performed using a slot–blot apparatus (Bio-ad). The membranes were treated with 20% methanol inater and then washed with PBS buffer. Samples of pro-
ein (1,5–3–6 �g) were prepared in a final volume of 50 �Ln PBS, loaded, and gently vacuumed. Membranes wereashed twice with PBS, and then rinsed with 20% methanol
n water. Slot–blots were probed as described for Westernlots. To compare SDS versus PBS solubility of the AR.Q(n)pecies formed in NSC34 after transfection, the samples forlter retardation assay were prepared following the methodescribed by Sittler, et al. [41] to detect SDS-insoluble aggre-ates, or as described for western blot samples, to detectDS-soluble, PBS-insoluble aggregates.
.5. Real time-polymerase chain reaction
To evaluate testosterone-dependent changes in thexpression of YFPu mRNA in NSC34/YFPu expressingR.Q(n), we have performed real time PCR analysis. TheSC34/YFPu were plated at 80,000 cells/mL in six-wellultiwell plates in DMEM plus 5% FBS and subsequently
ransfected with AR.Q(n). Cells were transfected and treatedn triplicate with testosterone (10 nM), and control cellsere treated with vehicle (ethanol). Following 48 h of
reatment, cells were lysed and total RNA extracted using
1 ogy of A
aCmaS
C
102 P. Rusmini et al. / Neurobiol
n RNeasy Mini Kit with DNase 1 (Qiagen, Valencia,
A, USA). Primers for real time RT-PCR of the YFPuRNA were designed in accordance with recommendationsccompanying the ABI Prism 7700 Sequence Detectionystem (Applied Biosystem, Foster City, CA, USA) on
PpCp
ging 28 (2007) 1099–1111
-G base content and 5′ content and using the program
rimer Express 1.5. Forward and reverse primers for YFPuroduce a product of 83 bases: YFPu upstream primer, 5′-ACATGAAGCAGCACGACTT-3′ and YFPu downstreamrimer, 5′-CCGTCGTCCTTGAAGAAGAT-3′. GAPDH
ogy of A
hpCTiRa(tbmrgtrewrflYPacen
2
SsC
3
3S
tc
ftayctabbuslawafrwamhtfiiAtAslotohs
gl
FovetStmch(uA
P. Rusmini et al. / Neurobiol
ousekeeping gene was used as control, with the followingrimers: upstream primer 5′-CCAGAACATCATCCCTG-AT-3′, downstream primer 5′-CAGTGAGCTTCCCGT-CAG-3′. iTaq SYBR Green Supermix with ROX (contain-
ng SYBR Green enzyme, the internal fluorescent controlOX and all components required for PCR, excluding cDNAnd primers; Bio-Rad) was used in real time PCR analysisApplied Biosystems). Preliminary assays were performedo optimize the primer concentration and avoid non-specificinding; these assays were performed according to theanufacturer’s recommendations using two sets of primers
ecognizing YFPu and the GAPDH mRNA housekeepingene. The resulting amplification plot was used to selecthe optimal primer concentrations and ensure similareaction efficiencies for both primers. Relative YFPu mRNAxpression levels in NSC34-YFPu cells were assessedith real time RT-PCR according to the manufacturer’s
ecommendations using optimized primer levels. A setuorescence level was selected at which samples amplifyingFPu and GAPDH were both in a point of exponentialCR amplification. Treatments were performed in triplicatend then each sample read in duplicate and mean cyclesorresponding with the selected fluorescence level forach duplicate sample assessed; YFPu values were thenormalized to those of GAPDH.
.6. Statistical analysis
Statistical analysis has been performed using an unpairedtudent’s t-test (two-tailed P-value) by utilizing the PRISMoftware (version 4.0b, GraphPad Software, Inc., San Diego,A, USA).
. Results and discussion
.1. Testosterone-dependent aggregate formation inBMA
We have initially extended our earlier observations on theestosterone dependence of ARpolyQ aggregation using twoomplementary approaches.
Acwc
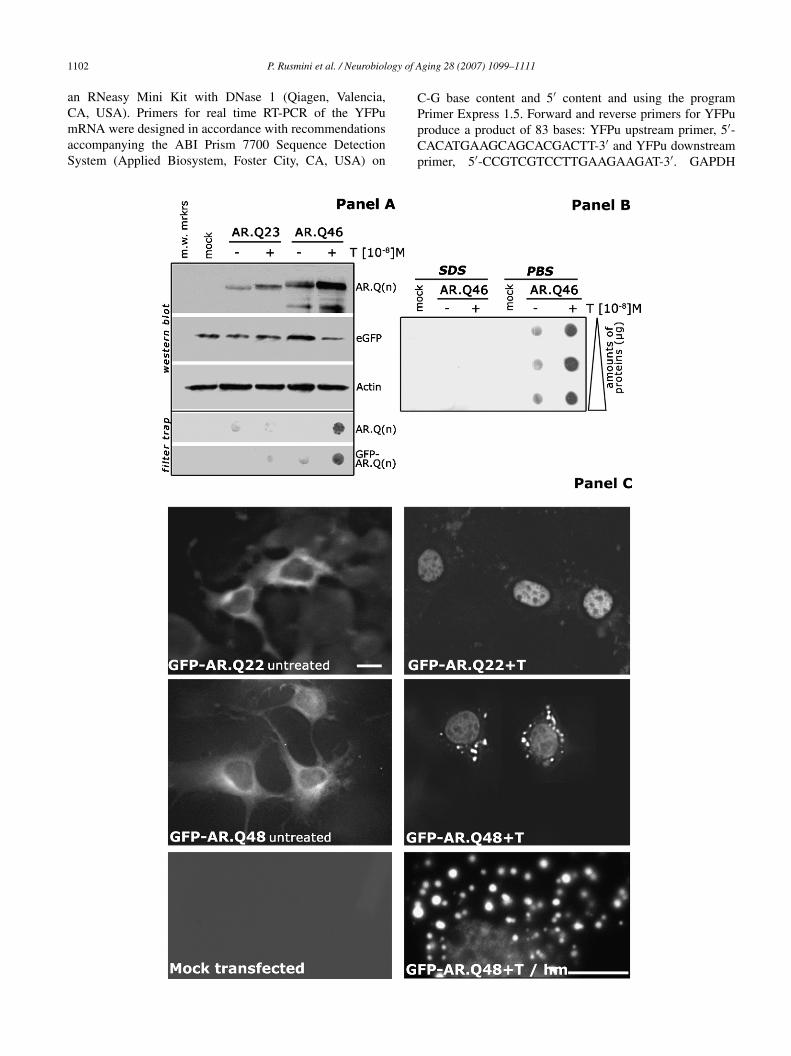
ig. 1. Effects of testosterone on solubility and aggregation of wt or SBMA mutanf NSC34 transfected with wt (AR.Q23) or SBMA mutant (AR.Q46) ARs in abseisualized using the AR(N-20) antibody. Samples were normalized either using anvaluated with co-transfection of the pEGFP-N1 vector or with anti-actin antibodyrap) performed on cell lysates of NSC34 transfected with either wt or SBMA mBMA AR.Q(n), n = 46; lower inset, GFP tagged ARs: wt GFP-AR.Q(n), n = 22; S
estosterone for 48 h. The insoluble AR protein was visualized using the AR(N-20utant AR.Q46 in NSC34, evaluted in filter retardation assay on cellulose acetat
ontaining 1% SDS; PBS = cell lysates obtained by harvesting the cells in PBS. Inigh resolution fluorescent miscroscopy analysis on NSC34 cells transfected withuntreated) or in presence (+T) of 10 nM testosterone for 48 h. Mock transfected insing a 40× objective; GFP-AR.Q48 + T/hm = higher magnification, the image wautoDeblur. Scale bars = 10 �m.
ging 28 (2007) 1099–1111 1103
Immortalized motor neurons (NSC34) have been trans-ected with wt (AR.Q23) or mutant (AR.Q46) AR, andhe effects of testosterone on AR properties were evalu-ted [14]. Fig. 1, panel A, shows the western blot anal-sis and the filter retardation assay performed on NSC34ells expressing wt or mutant ARs. It appears that testos-erone induces the typical “up-shift” of the AR bands [15]nd increases the AR levels; the higher AR levels haveeen previously associated to the stabilization of the ligand-ound AR protein [55]; interestingly, the effect is partic-larly robust in the case of SBMA ARpolyQ [34,35,40],uggesting that wt and mutant ARs differ in their intracel-ular relative steady state levels. Fig. 1, panel A also shows
filter retardation assay performed on NSC34 expressingt or mutant AR.Q(n) or GFP tagged AR [GFP-AR.Q(n)]
nd demonstrates that in both cases testosterone induces theormation of large amounts of insoluble ARpolyQ speciesetained by the cellulose acetate membrane; in the case oftARs (either unactivated or testosterone treated), negible
mounts of immunoreactive material were present on theembrane, excluding the formation of significant amounts of
igh molecular weight (MW) insoluble AR species. Testos-erone untreated (unbound) ARpolyQ also fully crossed thelter, proving that in NSC34 cells the ARpolyQ is soluble
n its inactive status. On the other hand, a large amount ofR(N-20) immunoreactive insoluble protein was present in
estosterone-treated NSC34 cells expressing the ARpolyQ.s shown in Fig. 1, panel B, the insoluble ARpolyQ
pecies induced by testosterone were easily detectable inysates of NSC34 cells obtained using PBS, but not in thosebtained using a SDS-containing PBS; this suggests thathe insoluble ARpolyQ species are not SDS-resistant, anbservation which is further supported by the absence ofigh MW species of ARpolyQ in the stacking gels (nothown).
Fig. 1, panel C shows the process of ARpolyQ aggre-ation in motor neuron; the analysis was performed iniving NSC34 cells expressing GFP tagged wtAR (GFP-
R.Q22) or mutant SBMA ARpolyQ (GFP-AR.Q48). Itlearly appears that the unactivated (testosterone untreated)tAR (GFP-AR.Q22 untreated) is diffusely localized in the
ell cytoplasm; testosterone (GFP-AR.Q22 + T) induced a
t ARs. Panel A, western analysis (Western blot) performed on cell lysatesnce (−T) or in presence (+T) of 10 nM testosterone for 48 h. The AR wasti GFP antibody (anti GFP clone C163), to measure transfection efficiencyto evaluate protein loading on SDS-PAGE. Filter retardation assay (Filterutant ARs (upper inset, untagged full length ARs: wt AR.Q(n), n = 23;
BMA GFP-AR.Q(n), n = 48) in absence (−T) or in presence (+T) of 10 nM) antibody. Panel B, effects of different buffers on the solubility of SBMAe membranes. SDS = cell lysates obtained by harvesting the cells in PBSsoluble AR protein was visualized using the AR(N-20) antibody. Panel C,GFP tagged wt (GFP-AR.Q22) or mutant (GFP-AR.Q48) ARs in absencedicates NSC34 transfected with the empty vector. The images were takens taken using a 63× objective. The images have been deconvoluted using
1 ogy of A
rTAtgtomadptb[abA
cfo“uibMbbAmt
Fpbbttvsvottag
104 P. Rusmini et al. / Neurobiol
apid (2–3 h) and complete nuclear translocation of the wtAR.he distribution of the unbound, unactivated ARpolyQ (GFP-R.Q48 untreated) was very similar to that of the wtAR, with
he protein diffused in the cytoplasm and no detectable aggre-ates; testosterone (GFP-AR.Q48 + T) induced the nuclearranslocation of ARpolyQ, but also a massive aggregationf the ARpolyQ in the cytoplasm (see also inset at higheragnification) in about 40% of transfected cells. In the
dopted experimental conditions, no nuclear aggregates wereetectable; in fact, polyQ longer that 90 Gln (never found inatients) are required to generate AR nuclear aggregates inransfected cells [50]. Since both types of aggregates haveeen found in spinal cord motor neurons of SBMA patients1], cytoplasmic and nuclear aggregates may reflect different
spects of the SBMA disease. The data of Fig. 1, panel C haveeen confirmed using the AR(N-20) antibody on untaggedR(s) (not shown) [34,40,43].in[
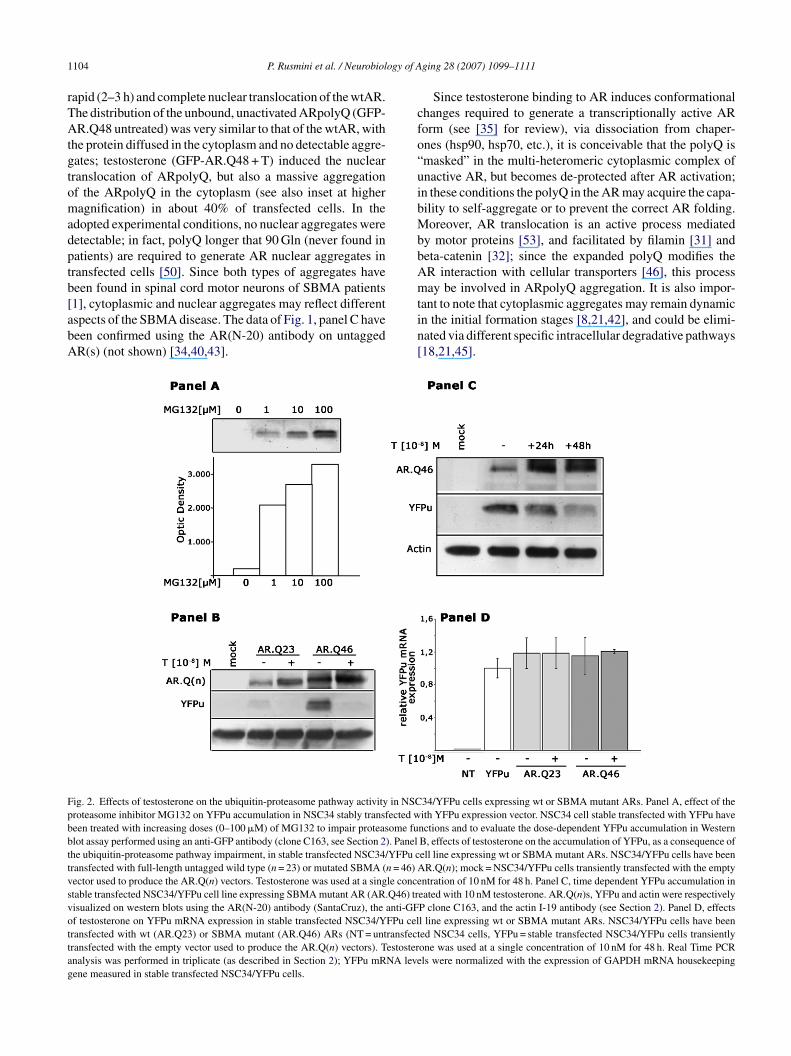
ig. 2. Effects of testosterone on the ubiquitin-proteasome pathway activity in NSCroteasome inhibitor MG132 on YFPu accumulation in NSC34 stably transfected ween treated with increasing doses (0–100 �M) of MG132 to impair proteasome fulot assay performed using an anti-GFP antibody (clone C163, see Section 2). Panelhe ubiquitin-proteasome pathway impairment, in stable transfected NSC34/YFPu cransfected with full-length untagged wild type (n = 23) or mutated SBMA (n = 46) Aector used to produce the AR.Q(n) vectors. Testosterone was used at a single concetable transfected NSC34/YFPu cell line expressing SBMA mutant AR (AR.Q46) trisualized on western blots using the AR(N-20) antibody (SantaCruz), the anti-GFf testosterone on YFPu mRNA expression in stable transfected NSC34/YFPu celransfected with wt (AR.Q23) or SBMA mutant (AR.Q46) ARs (NT = untransfectransfected with the empty vector used to produce the AR.Q(n) vectors). Testosternalysis was performed in triplicate (as described in Section 2); YFPu mRNA leveene measured in stable transfected NSC34/YFPu cells.
ging 28 (2007) 1099–1111
Since testosterone binding to AR induces conformationalhanges required to generate a transcriptionally active ARorm (see [35] for review), via dissociation from chaper-nes (hsp90, hsp70, etc.), it is conceivable that the polyQ ismasked” in the multi-heteromeric cytoplasmic complex ofnactive AR, but becomes de-protected after AR activation;n these conditions the polyQ in the AR may acquire the capa-ility to self-aggregate or to prevent the correct AR folding.oreover, AR translocation is an active process mediated
y motor proteins [53], and facilitated by filamin [31] andeta-catenin [32]; since the expanded polyQ modifies theR interaction with cellular transporters [46], this processay be involved in ARpolyQ aggregation. It is also impor-
ant to note that cytoplasmic aggregates may remain dynamic
n the initial formation stages [8,21,42], and could be elimi-ated via different specific intracellular degradative pathways18,21,45].34/YFPu cells expressing wt or SBMA mutant ARs. Panel A, effect of theith YFPu expression vector. NSC34 cell stable transfected with YFPu havenctions and to evaluate the dose-dependent YFPu accumulation in WesternB, effects of testosterone on the accumulation of YFPu, as a consequence ofell line expressing wt or SBMA mutant ARs. NSC34/YFPu cells have beenR.Q(n); mock = NSC34/YFPu cells transiently transfected with the empty
ntration of 10 nM for 48 h. Panel C, time dependent YFPu accumulation ineated with 10 nM testosterone. AR.Q(n)s, YFPu and actin were respectivelyP clone C163, and the actin I-19 antibody (see Section 2). Panel D, effectsl line expressing wt or SBMA mutant ARs. NSC34/YFPu cells have beened NSC34 cells, YFPu = stable transfected NSC34/YFPu cells transientlyone was used at a single concentration of 10 nM for 48 h. Real Time PCRls were normalized with the expression of GAPDH mRNA housekeeping

ogy of A
3a
tuatfTdfdt
tYbdAeitllA[mfom
ofofbdYito[muulwutacdeso
tta[
atYmctsbtifSA)antarthcwTitacsbsr
gstmrwhavticaa
P. Rusmini et al. / Neurobiol
.2. Effects of ARpolyQ aggregation on proteasomectivity
To analyse the effects of polyQ proteins aggregation onhe ubiquitin-proteasome pathway, we took advantage of thenique opportunity to trigger ARpolyQ aggregation. We havessayed the effects of soluble (unactivated), insoluble (testos-erone activated) and aggregated ARpolyQ on proteasomeunctions using the GFPu (or YFPu) reporter system [4,5].he YFPu derives from the GFPu and consists of a shortegron (CL1) signal for the ubiquitin-proteasome pathway,used to the YFP C-terminus [4]. YFPu is normally rapidlyegraded by the proteasome, but proteasome impairment leado fluorescence accumulation within the cells.
To limit assay variability, we have produced a stableransfected cell line based on NSC34 [7] expressing theFPu. YFPu efficiency to detect proteasome impairment haseen tested on NSC34/YFPu cells treated with increasingoses of the proteasome inhibitor, MG132; Fig. 2, panel
shows the dose dependent response of the YFPu lev-ls in NSC34/YFPu as a function of proteasome inhibition,ndicating that YFPu accumulation increases proportionallyo ubiquitin-proteasome pathway impairment. Since in pre-iminary experiments we observed that AR over-expressioned to high variation of the assays performed (because ofR retention into structures similar to the Golgi apparatus
17]), we avoided AR over-expression, by carefully deter-ining the amount of AR cDNA used in transient trans-
ection of NSC34/YFPu; all the data presented were thusbtained limiting to 1 �g of DNA the transfection in a six-wellultiwells.Fig. 2, panels B, shows the results of the study performed
n NSC34/YFPu transiently transfected with different ARorms (wt and polyQ). In both cases, testosterone (48 hf treatment at a dose of 10−8 M) stabilizes the two ARorms considered (see also Fig. 1, panel A); in the samelots, only negligible amounts of the YFPu proteins wereetectable in NSC34/YFPu expressing wtAR, while a robustFPu accumulation was present in motor neurons express-
ng the ARpolyQ in its inactive (testosterone untreated) sta-us. In these conditions, no insoluble or aggregated formsf unactivated ARpolyQ were detectable (see Fig. 1 and34,35,40,43]); it is conceivable that proteasome impair-ent occurs as a consequence of the fast turnover of the
nactivated AR, which is normally rapidly degraded by thebiquitin-proteasome pathway [6,54], a physiological regu-ator of its steady state level in basal conditions. Therefore,hile the unactivated wtAR is efficiently processed by thebiquitin-proteasome pathway (and YFPu rapidly degraded),he ARpolyQ, in absence of androgens (inactive status),bnormally interacts with the ubiquitin-proteasome pathway,ompeting with YFPu for degradation; the effect has to be
ue to the abnormally long polyQ size that might not befficiently cleaved by the enzymatic activity of the protea-ome system [16,21]. Interestingly, impairment or saturationf the proteasome activity does not cause protein aggrega-masi
ging 28 (2007) 1099–1111 1105
ion in the relatively short time of observation adopted (upo 48–72 h) (see Fig. 1), suggesting that the two processesre not directly linked, as previously suggested by others5].
Fig. 2, panels B, also shows the effect of AR-ligand inter-ction on proteasome function. Surprisingly, testosteronereatment, which activated the ARpolyQ, resulted in theFPu disappearance in Western blot, suggesting a nor-al clearance of this proteasome reporter protein in these
onditions. The AR.Q46 stabilization induced by testos-erone seems to correlate with the degree of proteasome de-aturation (Fig. 2, panel C). It must be noted that testosterone,y triggering ARpolyQ aggregation, induces AR seques-ration into a specific subcellular compartment preventingts intracellular degradation. Since cytoplasmic aggregatesormed in NSC34 cells in our experimental condition areDS-soluble/PBS insoluble (Fig. 1, panel B), the amounts ofRpolyQ detected in SDS-PAGE must also include the (PBS-
insoluble AR fraction sequestered into physically definedggregates, and not degraded by the proteasome; this phe-omenon explains the large accumulation in western blot (athe expected size of 110 kDa) of full length AR.Q46 measuredfter testosterone treatment. As shown in Fig. 2, panel D, theeduction of the YFPu level was not due to modifications ofhe YFPu mRNA expression, since real time PCR analysisas excluded variations of the YFPu mRNA levels in NSC34ells transfected with AR.Q(n), either untreated or treatedith the same dose (and for the same time) of testosterone.herefore, as mentioned above, the fact that we found a large
ncrease of insoluble and aggregated forms of testosteronereated ARpolyQ (Fig. 1) along with a reduction of YFPuccumulation, indicates that the subcellular segregation intoytoplasmic ARpolyQ aggregates, might contribute to de-aturate the ubiquitin-proteasome pathway activity, alteredy the presence of soluble (untreated) ARpolyQ. The dataupport the hypothesis that aggregates may be a protective,ather than a pathogenic, response [5].
We previously shown that ARpolyQ cytoplasmic aggre-ation also correlates with an increase of motor neuronalurvival [40]; thus, it is conceivable that protein aggrega-ion helps to protect from the neurotoxicity of a misfolded
utant protein. In this line of evidences, Arrasate et al. haveecently demonstrated that huntingtin aggregation correlatesith an improved survival and a decreased level of mutantuntingtin in neurons, protecting the cell from death [2]. Ingreement with our [40] and other [22,38,44] early obser-ations, it has been recently proposed that polyQ aggrega-ion in the cytoplasm might act as a “sink” that temporar-ly removes the toxic proteins from the soluble neuronalompartments [28,45], waiting for degradation through theutophagosome–lysosome fusion pathway [18,51]. Clearly,ggregate neurotoxicity may depend on the stage of their
aturation, since they may remain dynamic (soluble) soonfter formation (see Fig. 1, panel B), and only at latertages they may evolve to an insoluble status referred to asnclusions.
1 ogy of A
3n
acotcntfsfopcfitas
rbNwpi(meNaoliTsia
FAetoae(toS
106 P. Rusmini et al. / Neurobiol
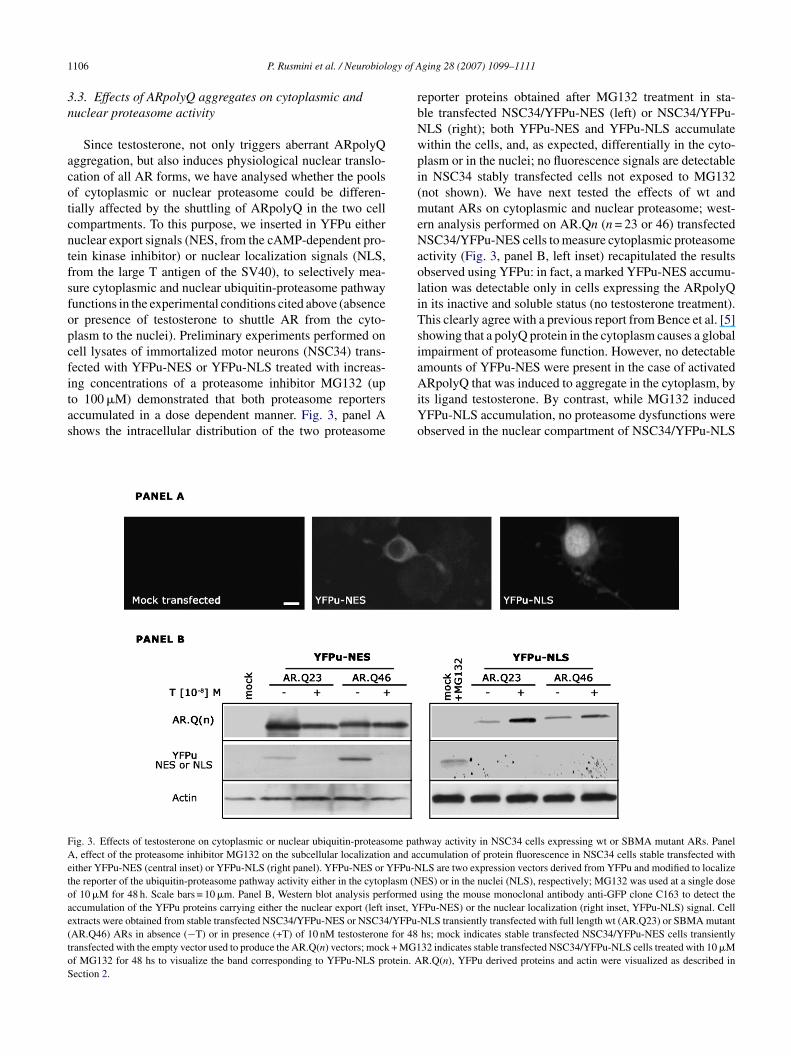
.3. Effects of ARpolyQ aggregates on cytoplasmic anduclear proteasome activity
Since testosterone, not only triggers aberrant ARpolyQggregation, but also induces physiological nuclear translo-ation of all AR forms, we have analysed whether the poolsf cytoplasmic or nuclear proteasome could be differen-ially affected by the shuttling of ARpolyQ in the two cellompartments. To this purpose, we inserted in YFPu eitheruclear export signals (NES, from the cAMP-dependent pro-ein kinase inhibitor) or nuclear localization signals (NLS,rom the large T antigen of the SV40), to selectively mea-ure cytoplasmic and nuclear ubiquitin-proteasome pathwayunctions in the experimental conditions cited above (absencer presence of testosterone to shuttle AR from the cyto-lasm to the nuclei). Preliminary experiments performed onell lysates of immortalized motor neurons (NSC34) trans-ected with YFPu-NES or YFPu-NLS treated with increas-
ng concentrations of a proteasome inhibitor MG132 (upo 100 �M) demonstrated that both proteasome reportersccumulated in a dose dependent manner. Fig. 3, panel Ahows the intracellular distribution of the two proteasomeAiYo
ig. 3. Effects of testosterone on cytoplasmic or nuclear ubiquitin-proteasome pat, effect of the proteasome inhibitor MG132 on the subcellular localization and ac
ither YFPu-NES (central inset) or YFPu-NLS (right panel). YFPu-NES or YFPu-Nhe reporter of the ubiquitin-proteasome pathway activity either in the cytoplasm (Nf 10 �M for 48 h. Scale bars = 10 �m. Panel B, Western blot analysis performedccumulation of the YFPu proteins carrying either the nuclear export (left inset, Yxtracts were obtained from stable transfected NSC34/YFPu-NES or NSC34/YFPu-AR.Q46) ARs in absence (−T) or in presence (+T) of 10 nM testosterone for 48ransfected with the empty vector used to produce the AR.Q(n) vectors; mock + MG1f MG132 for 48 hs to visualize the band corresponding to YFPu-NLS protein. Aection 2.
ging 28 (2007) 1099–1111
eporter proteins obtained after MG132 treatment in sta-le transfected NSC34/YFPu-NES (left) or NSC34/YFPu-LS (right); both YFPu-NES and YFPu-NLS accumulateithin the cells, and, as expected, differentially in the cyto-lasm or in the nuclei; no fluorescence signals are detectablen NSC34 stably transfected cells not exposed to MG132not shown). We have next tested the effects of wt andutant ARs on cytoplasmic and nuclear proteasome; west-
rn analysis performed on AR.Qn (n = 23 or 46) transfectedSC34/YFPu-NES cells to measure cytoplasmic proteasome
ctivity (Fig. 3, panel B, left inset) recapitulated the resultsbserved using YFPu: in fact, a marked YFPu-NES accumu-ation was detectable only in cells expressing the ARpolyQn its inactive and soluble status (no testosterone treatment).his clearly agree with a previous report from Bence et al. [5]howing that a polyQ protein in the cytoplasm causes a globalmpairment of proteasome function. However, no detectablemounts of YFPu-NES were present in the case of activated
RpolyQ that was induced to aggregate in the cytoplasm, byts ligand testosterone. By contrast, while MG132 inducedFPu-NLS accumulation, no proteasome dysfunctions werebserved in the nuclear compartment of NSC34/YFPu-NLS
hway activity in NSC34 cells expressing wt or SBMA mutant ARs. Panelcumulation of protein fluorescence in NSC34 cells stable transfected withLS are two expression vectors derived from YFPu and modified to localizeES) or in the nuclei (NLS), respectively; MG132 was used at a single doseusing the mouse monoclonal antibody anti-GFP clone C163 to detect theFPu-NES) or the nuclear localization (right inset, YFPu-NLS) signal. CellNLS transiently transfected with full length wt (AR.Q23) or SBMA mutanths; mock indicates stable transfected NSC34/YFPu-NES cells transiently32 indicates stable transfected NSC34/YFPu-NLS cells treated with 10 �MR.Q(n), YFPu derived proteins and actin were visualized as described in
ogy of A
e(infA
3a
To further analyse the negative correlation between pro-
FtaraAptaou
P. Rusmini et al. / Neurobiol
xpressing soluble (unactivated) ARpolyQ in the cytoplasmFig. 3, panel B, right inset); furthermore, while testosterone-nduced nuclear translocation of ARpolyQ, it did not sig-ificantly modify the accumulation of YFPu-NLS; there-
ore nuclear proteasome activity is not affected by activatedRpolyQ.ta
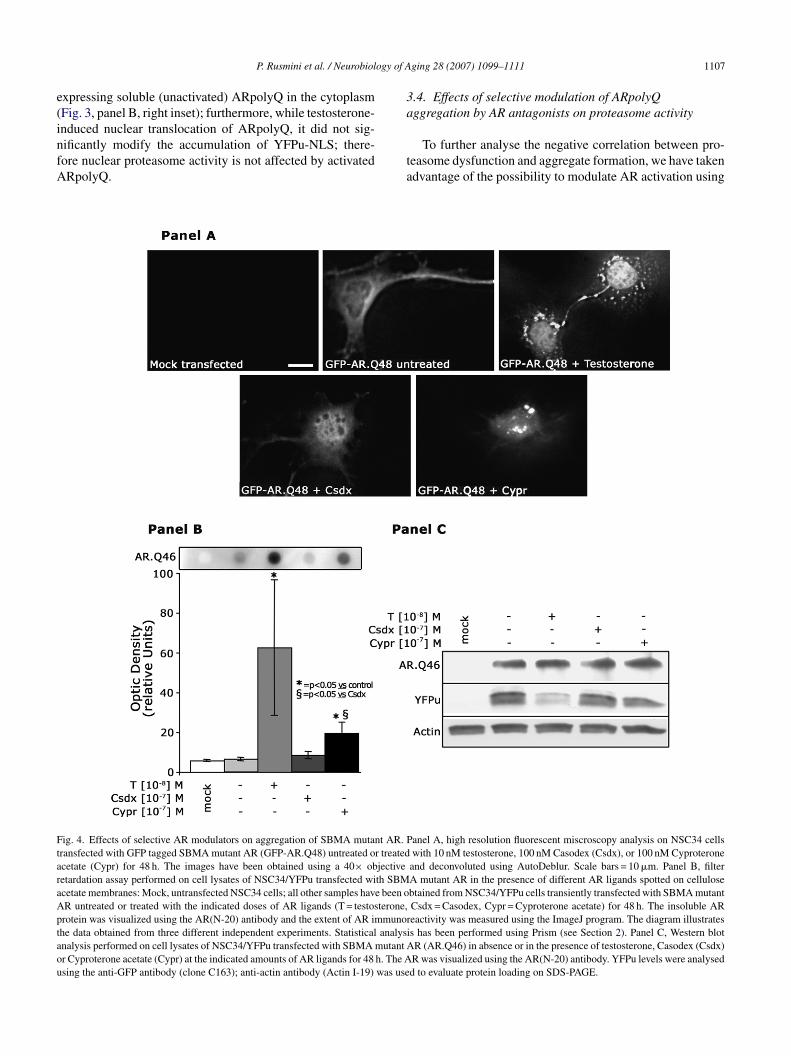
ig. 4. Effects of selective AR modulators on aggregation of SBMA mutant AR. Pransfected with GFP tagged SBMA mutant AR (GFP-AR.Q48) untreated or treatedcetate (Cypr) for 48 h. The images have been obtained using a 40× objectiveetardation assay performed on cell lysates of NSC34/YFPu transfected with SBMcetate membranes: Mock, untransfected NSC34 cells; all other samples have been oR untreated or treated with the indicated doses of AR ligands (T = testosterone,rotein was visualized using the AR(N-20) antibody and the extent of AR immunorhe data obtained from three different independent experiments. Statistical analysinalysis performed on cell lysates of NSC34/YFPu transfected with SBMA mutant Ar Cyproterone acetate (Cypr) at the indicated amounts of AR ligands for 48 h. The Asing the anti-GFP antibody (clone C163); anti-actin antibody (Actin I-19) was use
ging 28 (2007) 1099–1111 1107
.4. Effects of selective modulation of ARpolyQggregation by AR antagonists on proteasome activity
easome dysfunction and aggregate formation, we have takendvantage of the possibility to modulate AR activation using
anel A, high resolution fluorescent miscroscopy analysis on NSC34 cellswith 10 nM testosterone, 100 nM Casodex (Csdx), or 100 nM Cyproteroneand deconvoluted using AutoDeblur. Scale bars = 10 �m. Panel B, filterA mutant AR in the presence of different AR ligands spotted on cellulosebtained from NSC34/YFPu cells transiently transfected with SBMA mutantCsdx = Casodex, Cypr = Cyproterone acetate) for 48 h. The insoluble AReactivity was measured using the ImageJ program. The diagram illustratess has been performed using Prism (see Section 2). Panel C, Western blotR (AR.Q46) in absence or in the presence of testosterone, Casodex (Csdx)R was visualized using the AR(N-20) antibody. YFPu levels were analysedd to evaluate protein loading on SDS-PAGE.

1 ogy of A
taitcthbtAtACCdete
aptttrgsbnatsswotp(tblieAthai
aAatn(c
isowurt
pmelilAcpgpsaitef
snanoawa
ebsob
rarattgSntc
108 P. Rusmini et al. / Neurobiol
he selective antiandrogens, like Casodex and Cyproteronecetate. These two compounds interact with the AR, induc-ng conformational changes, the nuclear translocation andhe AR interaction with DNA, but block the recruitment ofo-activators required for transcription. It has been shownhat the antagonists decrease the dissociation rate from thesp90, slow down the AR nuclear transfer rate [14], andlock the trans-conformational change of AR preventinghe acquisition of the active conformation [14]. DifferentR antagonists induce different effects [23,48]; i.e., Cypro-
erone acetate is less efficient than testosterone in promotingR nuclear translocation, but is much more efficient thanasodex [3,25]. Since previous reports have also shown thatyproterone acetate induces AR aggregation, while Casodexoes not trigger aggregate formation [3], we used this pow-rful system to modulate AR aggregation and to determinehe effects of antiandrogens on proteasome activity in cellsxpressing mutant SBMA ARpolyQ.
Fig. 4, panel A shows the effects of the different AR lig-nds on GFP-AR.Q48 aggregation. In all the examined sam-les, Casodex was unable to induce AR aggregation, whileestosterone and Cyproterone acetate induced AR aggrega-ion, even if several differences were observed: (1) whileestosterone aggregates were generally characterized by aegular spherical shape, Cyproterone acetate induced aggre-ates that often presented irregular shapes; (2) the aggregateize was generally lower when aggregation was inducedy Cyproterone acetate. We have statistically analysed theumber and type of aggregates induced by the two AR lig-nds: testosterone-induced aggregates in about 47% of totalransfected cells; of these, about 87% presented a regularpherical shape (the remaining 13% presented an irregularhape). In the case of Cyproterone acetate, visible aggregatesere present in about 41% of total transfected cells; of these,nly 34% presented the regular spherical shape observed inhe case of the testosterone induction, while almost 64%resented aggregates characterized by an irregular shapespherical aggregates induced by testosterone versus Cypro-erone acetate p < 0.05; irregular shaped aggregates inducedy testosterone versus Cyproterone acetate p < 0.01). The bio-ogical meaning of these observations is still obscure, but its conceivable that these differences may reflect the differ-nt kinetics of activation induced by the two AR ligands.s stated above, Cyproterone acetate is less efficient than
estosterone to promote AR dissociation from the chaperonessp(s) and the AR nuclear translocation; Casodex is even lessctive than Cyproterone acetate and this may correlate withts inability to induce ARpolyQ aggregation [3,14].
Fig. 4, panel B shows the results of the filter retardationssay performed on cell lysates of motor neurons expressingR.Q46 and treated with different AR ligands; the results
re the means (±S.D.) of three different assays performed in
he same experimental conditions in duplicate. As expected,o signals were present in untransfected YFPu/NSC34 cellsmock) and in untreated NSC34/AR.Q46 cells (second spot)onfirming that unactivated AR is soluble; again, testosteronepapw
ging 28 (2007) 1099–1111
nduced the formation of a high amounts of insoluble ARpecies retained by the cellulose acetate membrane. Casodexnly marginally modified the solubility of the AR proteins,hile Cyproterone acetate significantly decreased the AR sol-bility (p < 0.05), since a large amount of insoluble AR wasetained by the cellulose acetate membrane after binding tohis antagonist.
Fig. 4, panel C shows the results of the Western bloterformed, using the same samples utilized in panel B, toeasure the level of YFPu accumulation in motor neurons
xpressing the ARpolyQ after treatment with different ARigands. Again, untreated cytoplasmic and soluble ARpolyQmpaired proteasome activity since YFPu protein accumu-ates in these samples; testosterone treatment, which inducesR aggregation, allows the normal clearance of the YFPu,
onfirming that AR aggregation contributes to de-saturate theroteasome; Casodex, which is not able to trigger AR aggre-ation and only marginally affects AR solubility, does notrevent YFPu accumulation, suggesting that the ARpolyQoluble forms are responsible for proteasome dysfunction; ingreement with this hypothesis, Cyproterone acetate, whichncreases the level of insoluble forms and triggers aggrega-ion of AR, also prevents YFPu accumulation, mimicking theffect of testosterone, and thus preventing proteasome dys-unctions.
Interestingly, Furutani et al. [12] have recently demon-trated, by monitoring neurodegeneration as a rough eye phe-otype in a Drosophila SBMA model, that both testosteronend several known AR antagonists induced a rough eye phe-otype, considered as marker of polyQ toxicity, leading to thenset of neurodegeneration. Notably, Casodex (also knowns Bicalutamide), which does not induce AR aggregation,as able to damage the fly eyes, suggesting that aggregation
nd neurodegeneration may not be directly linked.The data here presented suggest that the use of differ-
nt AR ligands might have different impact on the disease,ecause of their selective effects on aggregation and protea-ome impairment. Clearly, these AR ligands need to be testedn the SBMA animal models to characterize their possibleeneficial effects against motor neuronal loss.
Therefore, cytoplasmic aggregates may play a beneficialole to motor neuron cell functions, and since cytoplasmic ARggregates have been described in motor and sensory neu-ons of SBMA patients [1], the mechanism may be relevantlso in human patients. However, it is unclear how testos-erone, which, using the mechanism just proposed, seemso protect from motor neuron cell damage, might also trig-er ARpolyQ neurotoxicity, as shown in animal models ofBMA [19,20]. We have proposed that testosterone-inducedeuropil aggregates by altering the kinesin-mediated axonalransport [33–35] may deprive the synapses from essentialomponents for motor neuron function. Other groups have
roposed the involvement of testosterone-dependent nuclearggregates in neurotoxicity [11,20,27]. Therefore, severalrocesses might all be activated by the AR ligand(s) ande must consider that different types of aggregates may be
P. Rusmini et al. / Neurobiology of Aging 28 (2007) 1099–1111 1109
Fig. 5. Effect of testosterone on SBMA mutant AR in immortalized motor neuronal cells. The SBMA mutant AR, in its unactive status, is entrapped into acytoplasmic multi-heteromeric complex composed of several chaperones proteins (hsp90, hsp70, etc.); its turnover is regulated by the ubiquitin-proteasomepathway. After the activation of the SBMA mutant AR, induced by its ligand testosterone, the receptor protein must dissociate from the hsp(s) changing itsconformation to reach the “active status” by exposing a nuclear localization signal present in its DNA binding domain. This step is absolutely required forthe AR nuclear translocation, and its interaction with the genome to activate transcription of androgen responsive genes. However, the AR/hsp(s) complexdissociation might be a crucial step to allow testosterone-dependent SBMA AR aggregation; in fact, the release of the hsp(s) may “unmask” the elongated polyQtract located in the N-terminus of the mutant AR, allowing either (a) its physical interaction with other AR polyQs, thus triggering the aggregation process,or (b) the generation of aberrant conformational changes (misfolding) in the AR during its transition to the “active status” leading to aggregate formation.In both cases, since the aggregation seems to precede the interaction with the ubiquitin-proteasome pathway, the polyQ is segregated into physically definedsubcellular compartments, the aggregates, preventing its intracellular degradation. In this case the aggregates act as “sinks” temporarily removing the polyQf n helpsi ome fus
ftlnr
4
Apepmcapmtppanss
ei
ms
A
#sMtDa(vo9
rom the soluble compartment; it is thus conceivable that protein aggregatiots degradation through alternative pathways (i.e.: the autophagosome-lysos
ormed during disease, and (depending upon polyQ size, cellypes and/or experimental or environmental conditions) beocated in selected cell compartments (cytoplasm, neurites,ucleus, see [35] for review), playing different role in neu-odegeneration.
. Conclusions
The data presented in this paper suggest that (a) mutantR, and possibly wtAR, is processed by the ubiquitin-roteasome pathway, and this activity is impaired by the pres-nce of the elongated polyQ of AR; (b) ubiquitin-proteasomeathway is normally functioning in presence of cytoplas-ic aggregates; (c) aggregation process in the cytoplasm
ould be an intrinsic property of the mutant protein and notconsequence of the saturation of the ubiquitin-proteasomeathway; (d) the aggregation process induced by testosteroneay have a beneficial effect, by sequestering the mutant pro-
ein into the insoluble fraction and preserving the ubiquitin-roteasome pathway from an excess of soluble misfoldedrotein to be degraded. It is also possible that the mech-
nisms of protein partitioning (or sequestration) into theuclei, may help to de-saturate the cytoplasmic proteasomeystem (decreased YFPu levels) and that the nuclear protea-ome system may be less sensitive to the same protein in ourR
to protect from the neurotoxicity of a misfolded mutant protein, waiting forion pathway?).
xperimental conditions. An explanatory diagram is reportedn Fig. 5.
It remains to be determined whether ARpolyQ aggregatesay then be processed through other intracellular degradative
ystems, such as the autophagosome-lysosome pathway [18].
cknowledgements
The financial support of Telethon – Italy (Grants no.1283, and GP0222Y01), the Italian Ministry of Univer-ity and Research (MIUR-FIRB (# RBAU01NXFP) and
IUR-Cofin (2003054414 003 and 2005057598 002)),he Italian Ministry of Health convenzione no. 93, FON-AZIONE CARIPLO, the University of Milan are gratefullycknowledged. The authors wish to thank Dr. N.R. CashmanUniversity of British Columbia, Canada) for having pro-ided the NSC34 cells, and Prof. Ron K. Kopito (Departmentf Biological Sciences, Stanford University, Stanford, CA4305, USA) for having provided the GFPu vector.
eferences
[1] Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, NakagomiY, et al. Widespread nuclear and cytoplasmic accumulation of mutantandrogen receptor in SBMA patients. Brain Res 2005;128:659–70.

1 ogy of A
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
110 P. Rusmini et al. / Neurobiol
[2] Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusionbody formation reduces levels of mutant huntingtin and the risk ofneuronal death. Nature 2004;431:805–10.
[3] Becker M, Martin E, Schneikert J, Krug HF, Cato AC. Cytoplasmiclocalization and the choice of ligand determine aggregate formationby androgen receptor with amplified polyglutamine stretch. J Cell Biol2000;149:255–62.
[4] Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001;292:1552–5.
[5] Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impair-ment of the ubiquitin-proteasome system by nuclear or cytoplas-mic protein aggregates precedes inclusion body formation. Mol Cell2005;17:351–65.
[6] Cardozo CP, Michaud C, Ost MC, Fliss AE, Yang E, Patterson C, etal. C-terminal hsp-interacting protein slows androgen receptor syn-thesis and reduces its rate of degradation. Arch Biochem Biophys2003;410:134–40.
[7] Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, etal. Neuroblastoma × spinal cord (nsc) hybrid cell lines resemble devel-oping motor neurons. Devel Dyn 1992;194:209–21.
[8] Chai Y, Shao J, Miller VM, Williams A, Paulson HL. Live-cell imag-ing reveals divergent intracellular dynamics of polyglutamine diseaseproteins and supports a sequestration model of pathogenesis. Proc NatlAcad Sci USA 2002;99:9310–5.
[9] Chevalier-Larsen ES, O’Brien CJ, Wang H, Jenkins SC, HolderL, Lieberman AP, et al. Castration restores function and neurofila-ment alterations of aged symptomatic males in a transgenic mousemodel of spinal and bulbar muscular atrophy. J Neurosci 2004;24:4778–86.
10] Ciechanover A, Brundin P. The ubiquitin proteasome system in neu-rodegenerative diseases: Sometimes the chicken, sometimes the egg.Neuron 2003;40:427–46.
11] Fischbeck KH. Polyglutamine expansion neurodegenerative disease.Brain Res Bull 2001;56:161–3.
12] Furutani T, Takeyama KI, Tanabe M, Koutoku H, Ito S, Taniguchi N,et al. Human expanded polyq androgen receptor mutants in neurode-generation as a novel ligand target. J Pharmacol Exp Ther 2005.
13] Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol2002;20:3001–15.
14] Georget V, Terouanne B, Nicolas JC, Sultan C. Mechanism ofantiandrogen action: Key role of hsp90 in conformational changeand transcriptional activity of the androgen receptor. Biochemistry2002;41:11824–31.
15] Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD,et al. Androgen receptor phosphorylation. Regulation and identificationof the phosphorylation sites. J Biol Chem 2002;277:29304–14.
16] Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, MorimotoRI. Inefficient degradation of truncated polyglutamine proteins by theproteasome. EMBO J 2004;23:4307–18.
17] Huynh DP, Yang HT, Vakharia H, Nguyen D, Pulst SM. Expan-sion of the polyq repeat in ataxin-2 alters its golgi localization,disrupts the golgi complex and causes cell death. Hum Mol Genet2003;12:1485–96.
18] Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, etal. Increased susceptibility of cytoplasmic over nuclear polyglutamineaggregates to autophagic degradation. Proc Natl Acad Sci USA 2005.
19] Katsuno M, Adachi H, Doyu M, Minamiyama M, Sang C, KobayashiY, et al. Leuprorelin rescues polyglutamine-dependent phenotypes ina transgenic mouse model of spinal and bulbar muscular atrophy. NatMed 2003;9:768–73.
20] Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, et
al. Testosterone reduction prevents phenotypic expression in a trans-genic mouse model of spinal and bulbar muscular atrophy. Neuron2002;35:843–54.21] Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglu-tamine protein aggregates are dynamic. Nat Cell Biol 2002;4:826–31.
[
ging 28 (2007) 1099–1111
22] Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, ClarkHB, et al. Ataxin-1 nuclear localization and aggregation—role inpolyglutamine-induced disease in sca1 transgenic mice. Cell 1998;95:41–53.
23] Kuil CW, Berrevoets CA, Mulder E. Ligand-induced conformationalalterations of the androgen receptor analyzed by limited trypsiniza-tion. Studies on the mechanism of antiandrogen action. J Biol Chem1995;270:27569–76.
24] La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH.Androgen receptor gene mutations in x-linked spinal and bulbar mus-cular atrophy. Nature 1991;352:77–9.
25] Marcelli M, Stenoien DL, Szafran AT, Simeoni S, Agoulnik IU, WeigelNL, et al. Quantifying effects of ligands on androgen receptor nucleartranslocation, intranuclear dynamics, and solubility. J Cell Biochem2006;98:770–88.
26] Marron TU, Guerini V, Rusmini P, Sau D, Brevini TAL, Martini L, et al.Androgen-induced neurite outgrowth is mediated by neuritin in motorneurones. J Neurochem 2005;92:10–20.
27] McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J,et al. Creb-binding protein sequestration by expanded polyglutamine.Hum Mol Genet 2000;9:2197–202.
28] Muchowski PJ, Ning K, D’Souza-Schorey C, Fields S. Requirement ofan intact microtubule cytoskeleton for aggregation and inclusion bodyformation by a mutant huntingtin fragment. Proc Natl Acad Sci USA2002;99:727–32.
29] Newmeyer DD, Forbes DJ. Nuclear import can be separated intodistinct steps in vitro: nuclear pore binding and translocation. Cell1988;52:641–53.
30] Ohara-Nemoto Y, Nemoto T, Sato N, Ota M. Characterization of thenontransformed and transformed androgen receptor and heat shockprotein 90 with high-performance hydrophobic-interaction chromatog-raphy. J Steroid Biochem 1988;31:295–304.
31] Ozanne DM, Brady ME, Cook S, Gaughan L, Neal DE, Rob-son CN. Androgen receptor nuclear translocation is facilitated bythe f-actin cross-linking protein filamin. Mol Endocrinol 2000;14:1618–26.
32] Pawlowski JE, Ertel JR, Allen MP, Xu M, Butler C, Wilson EM, etal. Liganded androgen receptor interaction with beta-catenin: nuclearco-localization and modulation of transcriptional activity in neuronalcells. J Biol Chem 2002;277:20702–10.
33] Piccioni F, Simeoni S, Andriola I, Armatura E, Bassanini S, PozziP, et al. Polyglutamine tract expansion of the androgen receptor in amotoneuronal model of spinal and bulbar muscular atrophy. Brain ResBull 2001;56:215–20.
34] Piccioni F, Pinton P, Simeoni S, Pozzi P, Fascio U, Vismara G, et al.Androgen receptor with elongated polyglutamine tract forms aggre-gates that alter axonal trafficking and mitochondrial distribution inmotor neuronal processes. FASEB J 2002;160:1418–20.
35] Poletti A. The polyglutamine tract of androgen receptor: Fromfunctions to dysfunctions in motor neurons. Front Neuroendocrinol2004;25:1–26.
36] Pozzi P, Bendotti C, Simeoni S, Piccioni F, Guerini V, Marron TU, etal. Androgen 5-alpha-reductase type 2 is highly expressed and activein rat spinal cord motor neurones. J Neuroendocrinol 2003;15:882–7.
37] Ross CA, Poirier MA. Protein aggregation and neurodegenerative dis-ease. Nat Med 2004;10(Suppl):S10–7.
38] Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts inthe nucleus to induce apoptosis but death does not correlate with theformation of intranuclear inclusions. Cell 1998;95:55–66.
39] Schmidt BJ, Greenberg CR, Allingham-Hawkins DJ, Spriggs El.Expression of x-linked bulbospinal muscular atrophy (kennedy disease)in two homozygous women. Neurology 2002;59:770–2.
40] Simeoni S, Mancini MA, Stenoien DL, Marcelli M, WeigelNL, Zanisi M, et al. Motoneuronal cell death is not corre-lated with aggregate formation of androgen receptors containingan elongated polyglutamine tract. Hum Mol Genet 2000;9:133–44.

ogy of A
[
[
[
[
[
[
[
[
[
[
[
[
[
[
degradation pathway. Endocrinology 2003;144:4841–50.
P. Rusmini et al. / Neurobiol
41] Sittler A, Walter S, Wedemeyer N, Hasenbank R, Scherzinge E, EickhoffH, et al. Sh3gl3 associates with the huntingtin exon 1 protein andpromotes the formation of polygln-containing protein aggregates. MolCell 1998;2:427–36.
42] Stenoien DL, Mielke M, Mancini MA. Intranuclear ataxin1 inclusionscontain both fast- and slow-exchanging components. Nat Cell Biol2002;10:806–10.
43] Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K,DeMartino G, et al. Polyglutamine-expanded androgen receptors formaggregates that sequester heat shock proteins, proteasome componentsand src-1, and are suppressed by the hdj-2 chaperone. Hum Mol Genet1999;8:731–41.
44] Tagawa K, Hoshino M, Okuda T, Ueda H, Hayashi H, Engemann S, etal. Distinct aggregation and cell death patterns among different types ofprimary neurons induced by mutant huntingtin protein. J Neurochem2004;89:974–87.
45] Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, et al. Aggresomes protect cells by enhancing the degra-dation of toxic polyglutamine-containing protein. Hum Mol Genet2003;12:749–57.
46] Thomas M, Dadgar N, Aphale A, Harrell JM, Kunkel R, Pratt WB, et al.Androgen receptor acetylation site mutations cause trafficking defects,misfolding, and aggregation similar to expanded glutamine tracts. J
Biol Chem 2004;279:8389–95.47] Trapman J, Klaassen P, Kuiper GGJM, van der Korput JAGM, FaberPW, van Rooij HCJ, et al. Cloning, structure and expression of a CDNAencoding the human androgen receptor. Biochem Biophys Res Com-mun 1988;153:241–8.
[
ging 28 (2007) 1099–1111 1111
48] Veldscholte J, Berrevoets CA, Brinkmann AO, Grootegoed JA, MulderE. Anti-androgens and the mutated androgen receptor of lncap cells:differential effects on binding affinity, heat-shock protein interaction,and transcription activation. Biochemistry 1992;31:2393–9.
49] Verrijdt G, Haelens A, Claessens F. Selective DNA recognition by theandrogen receptor as a mechanism for hormone-specific regulation ofgene expression. Mol Genet Metab 2003;78:175–85.
50] Walcott JL, Merry DE. Ligand promotes intranuclear inclusions in anovel cell model of spinal and bulbar muscular atrophy. J Biol Chem2002;277:50855–9.
51] Webb JL, Ravikumar B, Rubinsztein DC. Microtubule disruptioninhibits autophagosome-lysosome fusion: implications for studying theroles of aggresomes in polyglutamine diseases. Int J Biochem Cell Biol2004;36:2541–50.
52] Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signalfor rapid export of proteins from the nucleus. Cell 1995;82:463–73.
53] Whitaker HC, Hanrahan S, Totty N, Gamble SC, Waxman J, Cato AC, etal. Androgen receptor is targeted to distinct subcellular compartmentsin response to different therapeutic antiandrogens. Clin Cancer Res2004;10:7392–401.
54] Woodham C, Birch L, Prins GS. Neonatal estrogen down-regulatesprostatic androgen receptor through a proteosome-mediated protein
55] Zhou ZX, Lane MV, Kemppainen JA, French FS, Wilson EM. Speci-ficity of ligand-dependent androgen receptor stabilization: receptordomain interactions influence ligand dissociation and receptor. MolEndocrinol 1995;9:208–18.




















