
Cellular Toxicity of Polyglutamine Expansion Proteins
11
Molecular Cell, Vol. 15, 95–105, July 2, 2004, Copyright 2004 by Cell Press Cellular Toxicity of Polyglutamine Expansion Proteins: Mechanism of Transcription Factor Deactivation plasm of affected neurons. How the formation of these aggregates is mechanistically linked to cellular toxicity is not yet understood. The pathogenic length of the polyQ stretch in HD and other polyQ diseases is typically 37 residues or Gregor Schaffar, 1 Peter Breuer, 1 Raina Boteva, 2,3 Christian Behrends, 1,3 Nikolay Tzvetkov, 1 Nadine Strippel, 1 Hideki Sakahira, 1,4 Katja Siegers, 1 Manajit Hayer-Hartl, 1 and F. Ulrich Hartl 1, * greater, and this correlates with the ability of such se- 1 Department of Cellular Biochemistry quences to self-associate into fibrillar aggregates (Da- Max-Planck Institute of Biochemistry vies et al., 1997; DiFiglia et al., 1997; Scherzinger et al., Am Klopferspitz 18 1997). In these fibrils the polyQ repeats are proposed D-82152 Martinsried to form a curved, parallel sheet with 20 glutamines Germany per turn (Perutz et al., 2002; Wetzel, 2002). The hydrogen 2 Radiobiology Department bonded polypeptide chain runs perpendicular to the fi- National Center of Radiobiology and Radiation ber axis, and the glutamines form hydrogen bonded Protection “zippers,” conferring a high degree of structural stability. 1756 Sofia HD is caused by a polyQ extension in the N-terminal Bulgaria exon1 segment of huntingtin (Htt), an 350 kDa protein of unclear function expressed in all neurons. Whereas normal Htt is mainly located in the cytoplasm, mutant Summary Htt is increasingly found in the nucleus, a primary site of polyQ toxicity (e.g., Saudou et al., 1998). Aggregation- The expression of polyglutamine-expanded mutant competent N-terminal fragments of mutant Htt encom- proteins in Huntington’s disease and other neurode- passing the polyQ tract are produced by proteolysis and generative disorders is associated with the formation are thought to be critical in nuclear toxicity (DiFiglia et of intraneural inclusions. These aggregates could po- al., 1997; Lunkes et al., 2002). tentially cause cellular toxicity by sequestering essen- Mainly two nonexclusive ideas have been put forward tial proteins possessing normal polyQ repeats, includ- to explain how polyQ expansion proteins may cause ing the transcription factors TBP and CBP. We show, cellular dysfunction (reviewed in Sakahira et al., 2002). in vitro and in cells, that monomers or small soluble In one model, expanded polyQ proteins engage the ubi- oligomers of huntingtin exon1 accumulate in the nu- quitin-proteasome system in a nonproductive manner, cleus and inhibit the function of TBP in a polyQ-depen- interfering with proteasome-dependent cell regulation dent manner. FRET experiments indicate that these (Bence et al., 2001). In the second model, neurotoxicity toxic forms are generated through a conformational originates from the ability of the polyQ proteins to induce rearrangement in huntingtin. Interaction of toxic hun- the coaggregation of other proteins essential for cell tingtin with the benign polyQ repeat of TBP structurally viability, among them several transcription components destabilizes the transcription factor, independent of which possess nonpathogenic polyQ repeats. These the formation of insoluble coaggregates. Hsp70/Hsp40 factors include the TATA box binding protein (TBP), a chaperones interfere with the conformational change central component of the general transcription initiation in mutant huntingtin and inhibit the deactivation of complex, and the transcriptional coactivator CREB TBP. These results outline a molecular mechanism of binding protein (CBP). TBP, CBP, and other transcription cellular toxicity in polyQ disease and can explain the factors have been reported to colocalize with the aggre- beneficial effects of molecular chaperones. gates of disease-related polyQ proteins (e.g., McCamp- bell et al., 2000; Perez et al., 1998), and this may result Introduction in their sequestration and loss of function (Chai et al., 2002; Kim et al., 2002; Rajan et al., 2001). Indeed, transcriptional dysregulation has recently Expanded CAG repeats coding for polyglutamine (polyQ) been implicated in several polyQ diseases (reviewed in tracts in otherwise unrelated proteins are the cause of Sugars and Rubinsztein, 2003); however, the underlying at least nine different neurodegenerative diseases, in- mechanism is far from clear. PolyQ-induced transcrip- cluding Huntington’s disease (HD) (Ross, 2002; Zoghbi tional dysregulation in neurons can manifest itself before and Orr, 2000). These maladies are dominantly inherited cellular aggregates are detectable (Sugars and Ru- and typically show late onset with progressive neuronal binsztein, 2003), and it has been suggested that aggre- dysfunction and eventual loss of neurons in specific gate formation may even be a protective mechanism brain regions. A characteristic feature of their cytopa- (Cummings et al., 1999; Saudou et al., 1998). The in- thology is the formation of insoluble aggregates (inclu- creased expression of molecular chaperones such as sions) of the disease protein in the nucleus and cyto- Hsp70 and Hsp40 can suppress the neurotoxicity of polyQ-expanded proteins without preventing the forma- *Correspondence: [email protected] tion of polyQ inclusions (Cummings et al., 2001; Warrick 3 These authors contributed equally to this work. et al., 1999), although the chaperones do modulate the 4 Present address: The Institute of Medical Science, The University aggregation process (Chan et al., 2000; Krobitsch and of Tokyo, Advanced Clinical Research Center, Division of Infectious Diseases, 4-6-1 Shirokanedai Minato-ku, Tokyo 108-8639, Japan. Lindquist, 2000; Muchowski et al., 2000). These findings
-
Upload
biochem-mpg -
Category
Documents
-
view
0 -
download
0
Transcript of Cellular Toxicity of Polyglutamine Expansion Proteins

Molecular Cell, Vol. 15, 95–105, July 2, 2004, Copyright 2004 by Cell Press
Cellular Toxicity of PolyglutamineExpansion Proteins: Mechanism ofTranscription Factor Deactivation
plasm of affected neurons. How the formation of theseaggregates is mechanistically linked to cellular toxicityis not yet understood.
The pathogenic length of the polyQ stretch in HD andother polyQ diseases is typically �37 residues or
Gregor Schaffar,1 Peter Breuer,1 Raina Boteva,2,3
Christian Behrends,1,3 Nikolay Tzvetkov,1
Nadine Strippel,1 Hideki Sakahira,1,4
Katja Siegers,1 Manajit Hayer-Hartl,1
and F. Ulrich Hartl1,*greater, and this correlates with the ability of such se-1Department of Cellular Biochemistryquences to self-associate into fibrillar aggregates (Da-Max-Planck Institute of Biochemistryvies et al., 1997; DiFiglia et al., 1997; Scherzinger et al.,Am Klopferspitz 181997). In these fibrils the polyQ repeats are proposedD-82152 Martinsriedto form a curved, parallel � sheet with 20 glutaminesGermanyper turn (Perutz et al., 2002; Wetzel, 2002). The hydrogen2 Radiobiology Departmentbonded polypeptide chain runs perpendicular to the fi-National Center of Radiobiology and Radiationber axis, and the glutamines form hydrogen bondedProtection“zippers,” conferring a high degree of structural stability.1756 SofiaHD is caused by a polyQ extension in the N-terminalBulgariaexon1 segment of huntingtin (Htt), an �350 kDa proteinof unclear function expressed in all neurons. Whereasnormal Htt is mainly located in the cytoplasm, mutantSummaryHtt is increasingly found in the nucleus, a primary siteof polyQ toxicity (e.g., Saudou et al., 1998). Aggregation-The expression of polyglutamine-expanded mutantcompetent N-terminal fragments of mutant Htt encom-proteins in Huntington’s disease and other neurode-passing the polyQ tract are produced by proteolysis andgenerative disorders is associated with the formationare thought to be critical in nuclear toxicity (DiFiglia etof intraneural inclusions. These aggregates could po-al., 1997; Lunkes et al., 2002).tentially cause cellular toxicity by sequestering essen-
Mainly two nonexclusive ideas have been put forwardtial proteins possessing normal polyQ repeats, includ-to explain how polyQ expansion proteins may causeing the transcription factors TBP and CBP. We show,cellular dysfunction (reviewed in Sakahira et al., 2002).in vitro and in cells, that monomers or small solubleIn one model, expanded polyQ proteins engage the ubi-oligomers of huntingtin exon1 accumulate in the nu-quitin-proteasome system in a nonproductive manner,cleus and inhibit the function of TBP in a polyQ-depen-interfering with proteasome-dependent cell regulation
dent manner. FRET experiments indicate that these(Bence et al., 2001). In the second model, neurotoxicity
toxic forms are generated through a conformational originates from the ability of the polyQ proteins to inducerearrangement in huntingtin. Interaction of toxic hun- the coaggregation of other proteins essential for celltingtin with the benign polyQ repeat of TBP structurally viability, among them several transcription componentsdestabilizes the transcription factor, independent of which possess nonpathogenic polyQ repeats. Thesethe formation of insoluble coaggregates. Hsp70/Hsp40 factors include the TATA box binding protein (TBP), achaperones interfere with the conformational change central component of the general transcription initiationin mutant huntingtin and inhibit the deactivation of complex, and the transcriptional coactivator CREBTBP. These results outline a molecular mechanism of binding protein (CBP). TBP, CBP, and other transcriptioncellular toxicity in polyQ disease and can explain the factors have been reported to colocalize with the aggre-beneficial effects of molecular chaperones. gates of disease-related polyQ proteins (e.g., McCamp-
bell et al., 2000; Perez et al., 1998), and this may resultIntroduction in their sequestration and loss of function (Chai et al.,
2002; Kim et al., 2002; Rajan et al., 2001).Indeed, transcriptional dysregulation has recentlyExpanded CAG repeats coding for polyglutamine (polyQ)
been implicated in several polyQ diseases (reviewed intracts in otherwise unrelated proteins are the cause ofSugars and Rubinsztein, 2003); however, the underlyingat least nine different neurodegenerative diseases, in-mechanism is far from clear. PolyQ-induced transcrip-cluding Huntington’s disease (HD) (Ross, 2002; Zoghbitional dysregulation in neurons can manifest itself beforeand Orr, 2000). These maladies are dominantly inheritedcellular aggregates are detectable (Sugars and Ru-and typically show late onset with progressive neuronalbinsztein, 2003), and it has been suggested that aggre-dysfunction and eventual loss of neurons in specificgate formation may even be a protective mechanismbrain regions. A characteristic feature of their cytopa-(Cummings et al., 1999; Saudou et al., 1998). The in-thology is the formation of insoluble aggregates (inclu-creased expression of molecular chaperones such assions) of the disease protein in the nucleus and cyto-Hsp70 and Hsp40 can suppress the neurotoxicity ofpolyQ-expanded proteins without preventing the forma-
*Correspondence: [email protected] of polyQ inclusions (Cummings et al., 2001; Warrick3 These authors contributed equally to this work.et al., 1999), although the chaperones do modulate the4 Present address: The Institute of Medical Science, The Universityaggregation process (Chan et al., 2000; Krobitsch andof Tokyo, Advanced Clinical Research Center, Division of Infectious
Diseases, 4-6-1 Shirokanedai Minato-ku, Tokyo 108-8639, Japan. Lindquist, 2000; Muchowski et al., 2000). These findings
Molecular Cell96
would question the general significance of the seques-tration model for the disruption of transcriptional path-ways in polyQ diseases.
We have investigated the interaction of polyQ-expandedHtt exon1 constructs with TBP and CBP in vitro as wellas in neuroblastoma cells and in a yeast model. Ourresults show that monomers and small soluble oligo-mers of mutant Htt deactivate the transcription factorsby a polyQ-mediated interaction, independent of theformation of insoluble coaggregates. These solubletoxic forms are generated through a conformational re-arrangement in mutant Htt and accumulate to high levelswhen the polyQ-expanded protein is targeted to thenucleus. The Hsp70/Hsp40 chaperones modulate thetoxic intramolecular transition in mutant Htt, therebypreventing its aberrant interaction with the transcriptionfactor proteins.
Results
Efficient Recruitment of Nuclear Factors to HttAggregates Requires Ongoing Synthesisof Mutant HttWe confirmed the recruitment of CBP and TBP to polyQprotein aggregates in mouse neuroblastoma (N2a) cellsusing the exon1 fragment of Htt as a model diseaseprotein (see Supplemental Figure S1 at http://www.molecule.org/cgi/content/full/15/1/95/DC1). Upontransient expression, normal TBP with 38 Q, a mutantform of TBP lacking the polyQ tract (TBP�Q), and CBPwith 18 Q were located in the nucleus by immunofluores-cence. Mutant Htt with 96 Q (Htt96Q) formed large cyto-plasmic inclusions, whereas normal Htt with 20 Q was
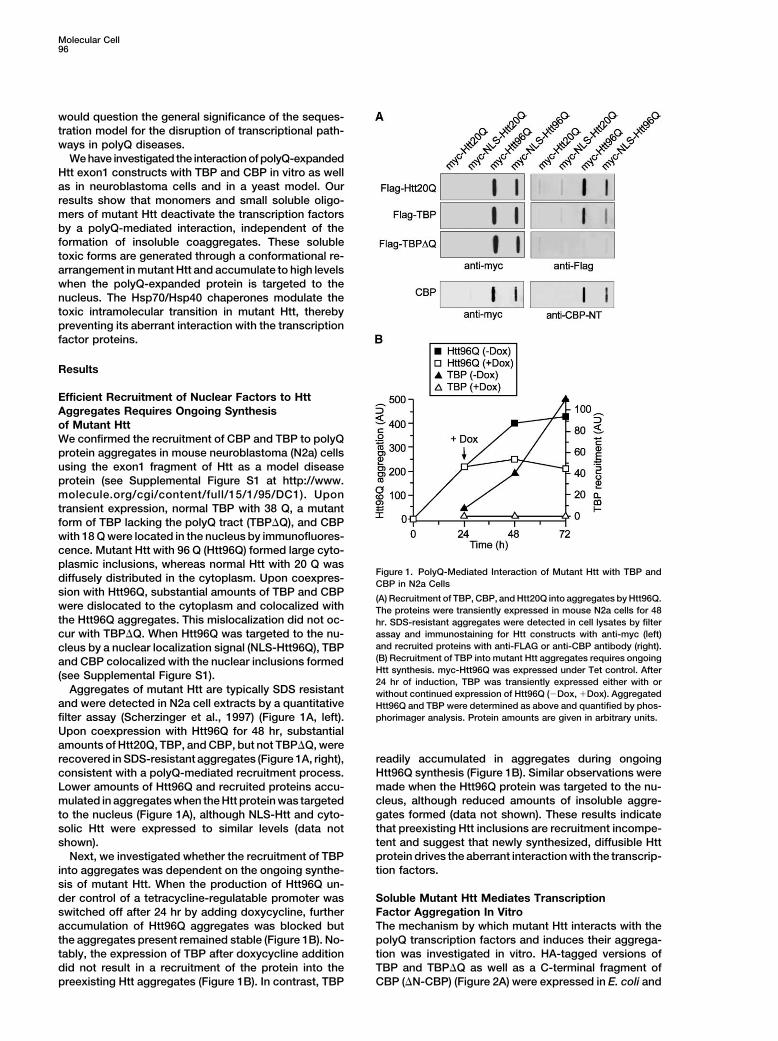
Figure 1. PolyQ-Mediated Interaction of Mutant Htt with TBP anddiffusely distributed in the cytoplasm. Upon coexpres- CBP in N2a Cellssion with Htt96Q, substantial amounts of TBP and CBP
(A) Recruitment of TBP, CBP, and Htt20Q into aggregates by Htt96Q.were dislocated to the cytoplasm and colocalized with The proteins were transiently expressed in mouse N2a cells for 48the Htt96Q aggregates. This mislocalization did not oc- hr. SDS-resistant aggregates were detected in cell lysates by filter
assay and immunostaining for Htt constructs with anti-myc (left)cur with TBP�Q. When Htt96Q was targeted to the nu-and recruited proteins with anti-FLAG or anti-CBP antibody (right).cleus by a nuclear localization signal (NLS-Htt96Q), TBP(B) Recruitment of TBP into mutant Htt aggregates requires ongoingand CBP colocalized with the nuclear inclusions formedHtt synthesis. myc-Htt96Q was expressed under Tet control. After(see Supplemental Figure S1).24 hr of induction, TBP was transiently expressed either with or
Aggregates of mutant Htt are typically SDS resistant without continued expression of Htt96Q (�Dox, �Dox). Aggregatedand were detected in N2a cell extracts by a quantitative Htt96Q and TBP were determined as above and quantified by phos-
phorimager analysis. Protein amounts are given in arbitrary units.filter assay (Scherzinger et al., 1997) (Figure 1A, left).Upon coexpression with Htt96Q for 48 hr, substantialamounts of Htt20Q, TBP, and CBP, but not TBP�Q, were
readily accumulated in aggregates during ongoingrecovered in SDS-resistant aggregates (Figure 1A, right),Htt96Q synthesis (Figure 1B). Similar observations wereconsistent with a polyQ-mediated recruitment process.made when the Htt96Q protein was targeted to the nu-Lower amounts of Htt96Q and recruited proteins accu-cleus, although reduced amounts of insoluble aggre-mulated in aggregates when the Htt protein was targetedgates formed (data not shown). These results indicateto the nucleus (Figure 1A), although NLS-Htt and cyto-that preexisting Htt inclusions are recruitment incompe-solic Htt were expressed to similar levels (data nottent and suggest that newly synthesized, diffusible Httshown).protein drives the aberrant interaction with the transcrip-Next, we investigated whether the recruitment of TBPtion factors.into aggregates was dependent on the ongoing synthe-
sis of mutant Htt. When the production of Htt96Q un-der control of a tetracycline-regulatable promoter was Soluble Mutant Htt Mediates Transcription
Factor Aggregation In Vitroswitched off after 24 hr by adding doxycycline, furtheraccumulation of Htt96Q aggregates was blocked but The mechanism by which mutant Htt interacts with the
polyQ transcription factors and induces their aggrega-the aggregates present remained stable (Figure 1B). No-tably, the expression of TBP after doxycycline addition tion was investigated in vitro. HA-tagged versions of
TBP and TBP�Q as well as a C-terminal fragment ofdid not result in a recruitment of the protein into thepreexisting Htt aggregates (Figure 1B). In contrast, TBP CBP (�N-CBP) (Figure 2A) were expressed in E. coli and
Toxicity of PolyQ Expansion Sequences97
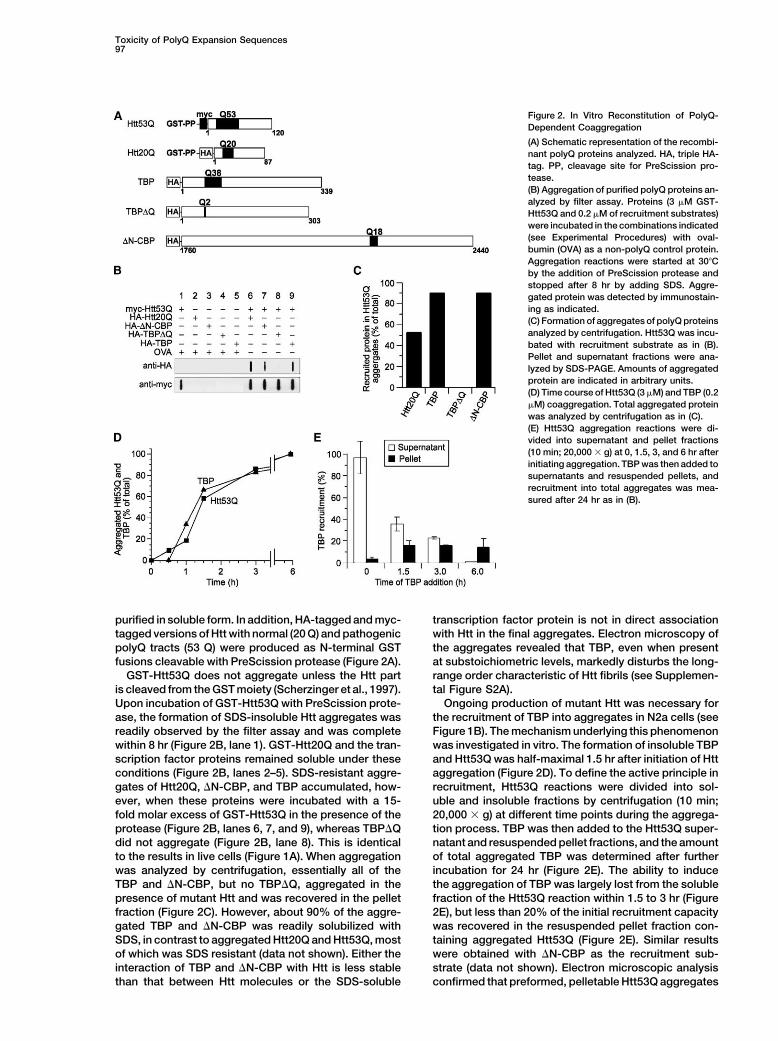
Figure 2. In Vitro Reconstitution of PolyQ-Dependent Coaggregation
(A) Schematic representation of the recombi-nant polyQ proteins analyzed. HA, triple HA-tag. PP, cleavage site for PreScission pro-tease.(B) Aggregation of purified polyQ proteins an-alyzed by filter assay. Proteins (3 �M GST-Htt53Q and 0.2 �M of recruitment substrates)were incubated in the combinations indicated(see Experimental Procedures) with oval-bumin (OVA) as a non-polyQ control protein.Aggregation reactions were started at 30�Cby the addition of PreScission protease andstopped after 8 hr by adding SDS. Aggre-gated protein was detected by immunostain-ing as indicated.(C) Formation of aggregates of polyQ proteinsanalyzed by centrifugation. Htt53Q was incu-bated with recruitment substrate as in (B).Pellet and supernatant fractions were ana-lyzed by SDS-PAGE. Amounts of aggregatedprotein are indicated in arbitrary units.(D) Time course of Htt53Q (3 �M) and TBP (0.2�M) coaggregation. Total aggregated proteinwas analyzed by centrifugation as in (C).(E) Htt53Q aggregation reactions were di-vided into supernatant and pellet fractions(10 min; 20,000 � g) at 0, 1.5, 3, and 6 hr afterinitiating aggregation. TBP was then added tosupernatants and resuspended pellets, andrecruitment into total aggregates was mea-sured after 24 hr as in (B).
purified in soluble form. In addition, HA-tagged and myc- transcription factor protein is not in direct associationwith Htt in the final aggregates. Electron microscopy oftagged versions of Htt with normal (20 Q) and pathogenic
polyQ tracts (53 Q) were produced as N-terminal GST the aggregates revealed that TBP, even when presentat substoichiometric levels, markedly disturbs the long-fusions cleavable with PreScission protease (Figure 2A).
GST-Htt53Q does not aggregate unless the Htt part range order characteristic of Htt fibrils (see Supplemen-tal Figure S2A).is cleaved from the GST moiety (Scherzinger et al., 1997).
Upon incubation of GST-Htt53Q with PreScission prote- Ongoing production of mutant Htt was necessary forthe recruitment of TBP into aggregates in N2a cells (seease, the formation of SDS-insoluble Htt aggregates was
readily observed by the filter assay and was complete Figure 1B). The mechanism underlying this phenomenonwas investigated in vitro. The formation of insoluble TBPwithin 8 hr (Figure 2B, lane 1). GST-Htt20Q and the tran-
scription factor proteins remained soluble under these and Htt53Q was half-maximal 1.5 hr after initiation of Httaggregation (Figure 2D). To define the active principle inconditions (Figure 2B, lanes 2–5). SDS-resistant aggre-
gates of Htt20Q, �N-CBP, and TBP accumulated, how- recruitment, Htt53Q reactions were divided into sol-uble and insoluble fractions by centrifugation (10 min;ever, when these proteins were incubated with a 15-
fold molar excess of GST-Htt53Q in the presence of the 20,000 � g) at different time points during the aggrega-tion process. TBP was then added to the Htt53Q super-protease (Figure 2B, lanes 6, 7, and 9), whereas TBP�Q
did not aggregate (Figure 2B, lane 8). This is identical natant and resuspended pellet fractions, and the amountof total aggregated TBP was determined after furtherto the results in live cells (Figure 1A). When aggregation
was analyzed by centrifugation, essentially all of the incubation for 24 hr (Figure 2E). The ability to inducethe aggregation of TBP was largely lost from the solubleTBP and �N-CBP, but no TBP�Q, aggregated in the
presence of mutant Htt and was recovered in the pellet fraction of the Htt53Q reaction within 1.5 to 3 hr (Figure2E), but less than 20% of the initial recruitment capacityfraction (Figure 2C). However, about 90% of the aggre-
gated TBP and �N-CBP was readily solubilized with was recovered in the resuspended pellet fraction con-taining aggregated Htt53Q (Figure 2E). Similar resultsSDS, in contrast to aggregated Htt20Q and Htt53Q, most
of which was SDS resistant (data not shown). Either the were obtained with �N-CBP as the recruitment sub-strate (data not shown). Electron microscopic analysisinteraction of TBP and �N-CBP with Htt is less stable
than that between Htt molecules or the SDS-soluble confirmed that preformed, pelletable Htt53Q aggregates

Molecular Cell98
are recruitment incompetent (see Supplemental Figure Thus, it seemed likely that the release from GST triggersa conformational rearrangement in Htt53Q that facili-S2B). On the basis of these findings, an interaction with
soluble Htt may be sufficient to affect the functional tates the nucleation of oligomers.To monitor conformational changes in mutant Htt, weproperties of the transcription factors, with sequestra-
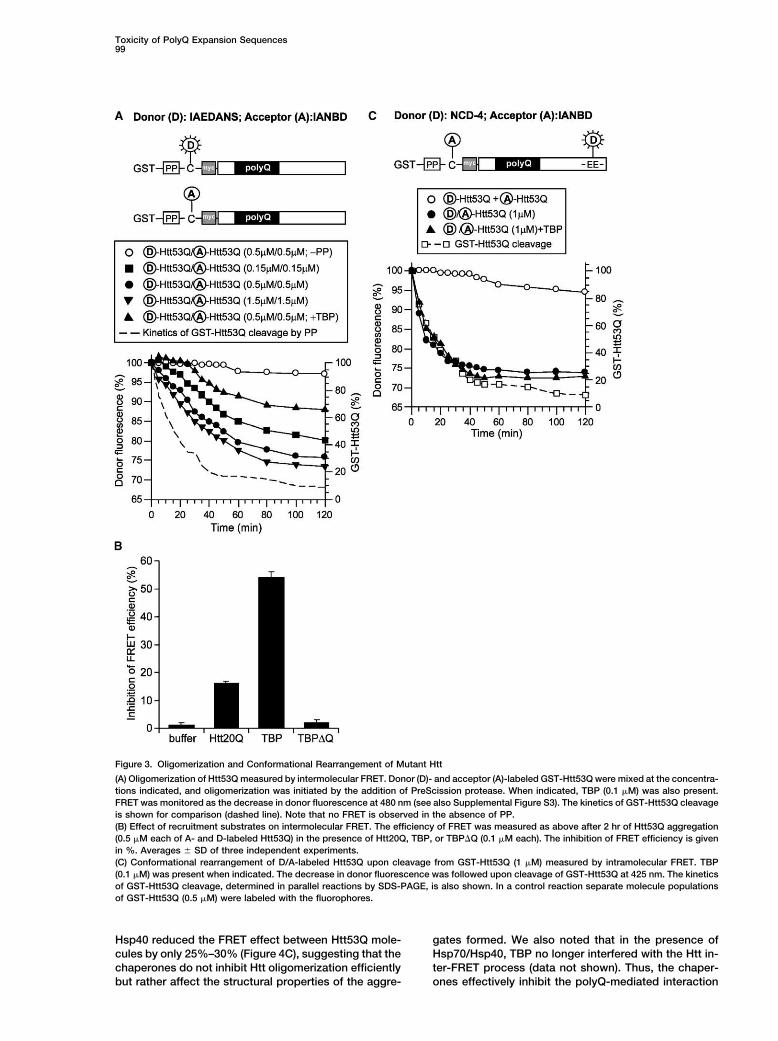
tion in coaggregates being a secondary phenomenon. performed intramolecular FRET experiments with NCDand IANBD as a fluorescence donor-acceptor pair forshort-range interactions up to 27–30 A. Htt53Q was la-TBP Interacts with Mutant Htt Very Earlybeled with IANBD at the unique cysteine preceding theduring OligomerizationpolyQ tract and with NCD at one of two adjacent gluta-Fluorescence Resonance Energy Transfer (FRET) mea-mates (residues 115 and 116) near the C terminus of thesurements were performed in vitro to characterize theprotein (Figure 3C). Upon cleavage of the labeled Htt53Qearly events during Htt53Q oligomerization and the inter-from the GST moiety, a decrease in NCD fluorescenceaction of Htt53Q with TBP. Intermolecular FRET wasresulting from energy transfer to IANBD occurred (Figurefollowed with 1.5-IAEDANS and IANBD as a donor and3C and Supplemental Figure S3). This effect representedacceptor pair able to interact over distances up to 70 A.intramolecular FRET because it was not observed uponCovalent labeling of Htt53Q was performed at a uniquemixing separate Htt preparations, one labeled with NCDcysteine (Figure 3A). When donor- and acceptor-labeledand the other with IANBD (Figure 3C). The intramolecularGST-Htt53Q were mixed at a 1:1 ratio and aggregationFRET process occurred essentially with the same kinet-was initiated by the addition of PreScission protease, aics as the cleavage of GST-Htt53Q (Figure 3C) and thusrobust, time-dependent decrease of the donor fluores-was considerably faster than Htt oligomerization mea-cence and an increase in acceptor fluorescence wassured by intermolecular FRET (Figure 3A). A critical dis-observed due to FRET (Figure 3A and Supplementaltance R0 of 17 A and an average distance r of 19.0 AFigure S3). This effect was kinetically dependent on thewas calculated for the intramolecular NCD-IANBD do-Htt53Q concentration, reaching a maximal rate at 1–3nor-acceptor pair. As no FRET was detectable with un-�M total Htt but was significantly slower than the pro-cleaved GST-Htt53Q, the average donor-acceptor dis-duction of Htt53Q by proteolytic cleavage from the GSTtance in Htt prior to cleavage from GST would be greaterfusion protein (Figure 3A and Supplemental Figure S3).than �30 A. NCD-IANBD-labeled Htt20Q showed onlyNo FRET was observed prior to cleavage, confirminga weak FRET signal upon cleavage from GST (data notthe inability of the GST fusion proteins to aggregate.shown), resulting in a greater average distance betweenThe oligomerization of Htt measured by FRET reachedthe NCD and IANBD groups of 26 A, as compared toa plateau at 1.5–2 hr, indicating that at this time most19 A for Htt53Q. Importantly, TBP did not interfere withmolecules have formed at least small oligomers (up tothe intramolecular FRET process in Htt53Q (Figure 3C),tetramers), the species most efficiently detected byin contrast to the effect of TBP on intermolecular FRETFRET (Figure 3A). Assuming a random orientation of the(Figures 3A and 3B). Moreover, no interaction was de-chromophores, we calculated a critical distance R0 oftected between TBP and uncleaved GST-Htt53Q.32 A and an average distance r of 38 A between the
These results indicate that Htt53Q undergoes a rapiddonor and acceptor chromophores in the Htt53Q oligo-structural rearrangement upon its cleavage from GST,mers, suggesting a head-to-tail oligomerization of Htt53Qcompatible with a compaction due to the formation ofmolecules. Some intermolecular FRET was also ob-� sheets in the mutant polyQ segment. This conforma-served with Htt20Q, but it occurred with significantlytional change likely represents the potentiating step forslower kinetics and lower efficiency (data not shown),the self-association of Htt53Q and its interaction withconsistent with a more transient interaction.other polyQ repeat proteins in this system.Next, we tested the effect of TBP addition on the
efficiency of FRET between Htt53Q molecules. We rea-soned that a physical interaction between TBP and Hsp70/Hsp40 Interfere with the ConformationalHtt53Q occurring early during Htt oligomerization should Change in Mutant Httinterfere with the FRET process. Indeed, addition of TBP Hsp70 and Hsp40 molecular chaperones have beenmarkedly reduced the FRET efficiency, whereas TBP�Q shown to suppress the neurotoxicity of polyQ-expandedwas without effect (Figures 3A and 3B). TBP affected the proteins, typically without preventing the formation ofHtt53Q inter-FRET process more strongly than Htt20Q visible polyQ inclusions in neurons (see Introduction).(Figure 3B) and interacted most efficiently with Htt53Q We found that mammalian Hsp70 and its cofactor Hsp40immediately upon cleavage of the GST-Htt53Q fusion (Hdj-1) are potent inhibitors of the Htt53Q-induced ag-protein (Figure 3A), consistent with ongoing Htt produc- gregation of TBP and �N-CBP in vitro (Figure 4A), al-tion being required for recruitment in vivo (Figure 1B). though they do not block Htt aggregation per se but
rather prevent the formation of Htt fibrils in favor ofdisordered aggregates (Muchowski et al., 2000). ThisA Conformational Rearrangement in Htt53Q
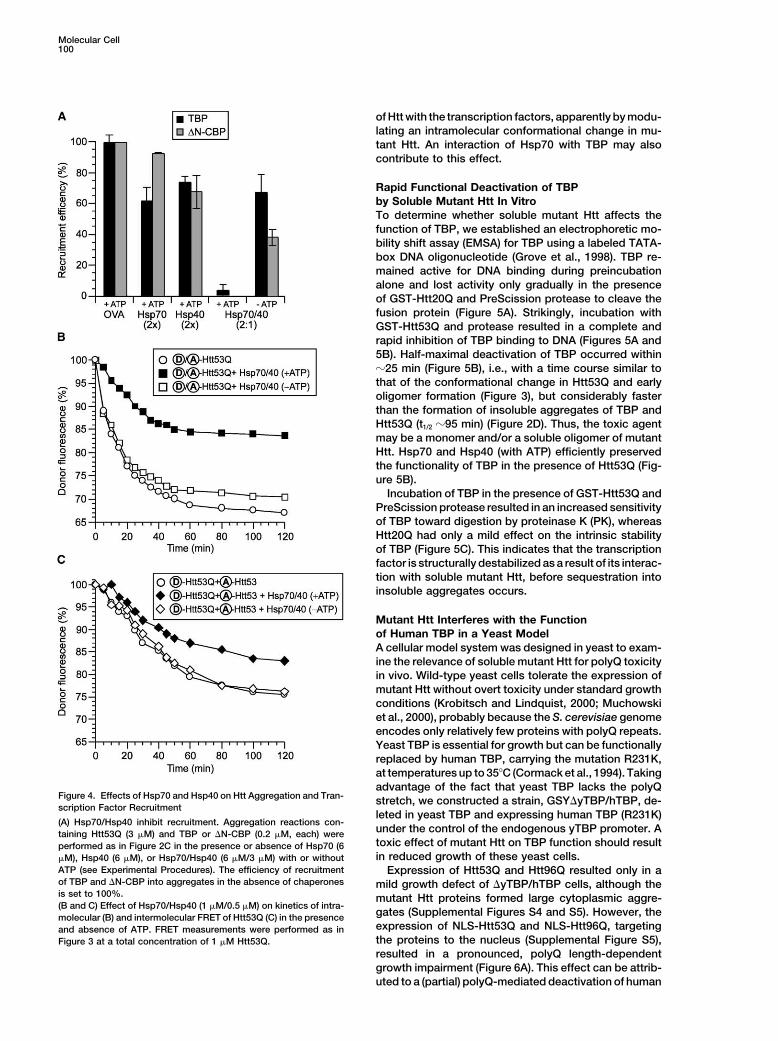
Precedes Oligomerization effect was ATP dependent and required both Hsp70 andHsp40 (Figure 4A).PolyQ repeats up to at least 53 Q are in an unstructured
conformation when attached to GST (Masino et al., 2002; FRET experiments with labeled Htt53Q showed thatthe chaperones interfered most efficiently with the intra-G.S., unpublished data). We established that the polyQ
segment in GST-Htt53Q is solvent exposed based on molecular rearrangement in Htt53Q occurring uponcleavage from GST (Figure 4B). In the presence ofits accessibility to protease and the observation that
uncleaved GST-Htt53Q, but not GST alone, is efficiently Hsp70, Hsp40, and ATP, the efficiency of the intra-FRETprocess was reduced by up to 60%. In contrast, Hsp70/recruited into aggregates by Htt53Q (data not shown).
Toxicity of PolyQ Expansion Sequences99
Figure 3. Oligomerization and Conformational Rearrangement of Mutant Htt
(A) Oligomerization of Htt53Q measured by intermolecular FRET. Donor (D)- and acceptor (A)-labeled GST-Htt53Q were mixed at the concentra-tions indicated, and oligomerization was initiated by the addition of PreScission protease. When indicated, TBP (0.1 �M) was also present.FRET was monitored as the decrease in donor fluorescence at 480 nm (see also Supplemental Figure S3). The kinetics of GST-Htt53Q cleavageis shown for comparison (dashed line). Note that no FRET is observed in the absence of PP.(B) Effect of recruitment substrates on intermolecular FRET. The efficiency of FRET was measured as above after 2 hr of Htt53Q aggregation(0.5 �M each of A- and D-labeled Htt53Q) in the presence of Htt20Q, TBP, or TBP�Q (0.1 �M each). The inhibition of FRET efficiency is givenin %. Averages � SD of three independent experiments.(C) Conformational rearrangement of D/A-labeled Htt53Q upon cleavage from GST-Htt53Q (1 �M) measured by intramolecular FRET. TBP(0.1 �M) was present when indicated. The decrease in donor fluorescence was followed upon cleavage of GST-Htt53Q at 425 nm. The kineticsof GST-Htt53Q cleavage, determined in parallel reactions by SDS-PAGE, is also shown. In a control reaction separate molecule populationsof GST-Htt53Q (0.5 �M) were labeled with the fluorophores.
Hsp40 reduced the FRET effect between Htt53Q mole- gates formed. We also noted that in the presence ofHsp70/Hsp40, TBP no longer interfered with the Htt in-cules by only 25%–30% (Figure 4C), suggesting that the
chaperones do not inhibit Htt oligomerization efficiently ter-FRET process (data not shown). Thus, the chaper-ones effectively inhibit the polyQ-mediated interactionbut rather affect the structural properties of the aggre-
Molecular Cell100
of Htt with the transcription factors, apparently by modu-lating an intramolecular conformational change in mu-tant Htt. An interaction of Hsp70 with TBP may alsocontribute to this effect.
Rapid Functional Deactivation of TBPby Soluble Mutant Htt In VitroTo determine whether soluble mutant Htt affects thefunction of TBP, we established an electrophoretic mo-bility shift assay (EMSA) for TBP using a labeled TATA-box DNA oligonucleotide (Grove et al., 1998). TBP re-mained active for DNA binding during preincubationalone and lost activity only gradually in the presenceof GST-Htt20Q and PreScission protease to cleave thefusion protein (Figure 5A). Strikingly, incubation withGST-Htt53Q and protease resulted in a complete andrapid inhibition of TBP binding to DNA (Figures 5A and5B). Half-maximal deactivation of TBP occurred within�25 min (Figure 5B), i.e., with a time course similar tothat of the conformational change in Htt53Q and earlyoligomer formation (Figure 3), but considerably fasterthan the formation of insoluble aggregates of TBP andHtt53Q (t1/2 �95 min) (Figure 2D). Thus, the toxic agentmay be a monomer and/or a soluble oligomer of mutantHtt. Hsp70 and Hsp40 (with ATP) efficiently preservedthe functionality of TBP in the presence of Htt53Q (Fig-ure 5B).
Incubation of TBP in the presence of GST-Htt53Q andPreScission protease resulted in an increased sensitivityof TBP toward digestion by proteinase K (PK), whereasHtt20Q had only a mild effect on the intrinsic stabilityof TBP (Figure 5C). This indicates that the transcriptionfactor is structurally destabilized as a result of its interac-tion with soluble mutant Htt, before sequestration intoinsoluble aggregates occurs.
Mutant Htt Interferes with the Functionof Human TBP in a Yeast ModelA cellular model system was designed in yeast to exam-ine the relevance of soluble mutant Htt for polyQ toxicityin vivo. Wild-type yeast cells tolerate the expression ofmutant Htt without overt toxicity under standard growthconditions (Krobitsch and Lindquist, 2000; Muchowskiet al., 2000), probably because the S. cerevisiae genomeencodes only relatively few proteins with polyQ repeats.Yeast TBP is essential for growth but can be functionallyreplaced by human TBP, carrying the mutation R231K,at temperatures up to 35�C (Cormack et al., 1994). Takingadvantage of the fact that yeast TBP lacks the polyQ
Figure 4. Effects of Hsp70 and Hsp40 on Htt Aggregation and Tran- stretch, we constructed a strain, GSY�yTBP/hTBP, de-scription Factor Recruitment
leted in yeast TBP and expressing human TBP (R231K)(A) Hsp70/Hsp40 inhibit recruitment. Aggregation reactions con-
under the control of the endogenous yTBP promoter. Ataining Htt53Q (3 �M) and TBP or �N-CBP (0.2 �M, each) weretoxic effect of mutant Htt on TBP function should resultperformed as in Figure 2C in the presence or absence of Hsp70 (6in reduced growth of these yeast cells.�M), Hsp40 (6 �M), or Hsp70/Hsp40 (6 �M/3 �M) with or without
ATP (see Experimental Procedures). The efficiency of recruitment Expression of Htt53Q and Htt96Q resulted only in aof TBP and �N-CBP into aggregates in the absence of chaperones mild growth defect of �yTBP/hTBP cells, although theis set to 100%. mutant Htt proteins formed large cytoplasmic aggre-(B and C) Effect of Hsp70/Hsp40 (1 �M/0.5 �M) on kinetics of intra-
gates (Supplemental Figures S4 and S5). However, themolecular (B) and intermolecular FRET of Htt53Q (C) in the presenceexpression of NLS-Htt53Q and NLS-Htt96Q, targetingand absence of ATP. FRET measurements were performed as inthe proteins to the nucleus (Supplemental Figure S5),Figure 3 at a total concentration of 1 �M Htt53Q.resulted in a pronounced, polyQ length-dependentgrowth impairment (Figure 6A). This effect can be attrib-uted to a (partial) polyQ-mediated deactivation of human
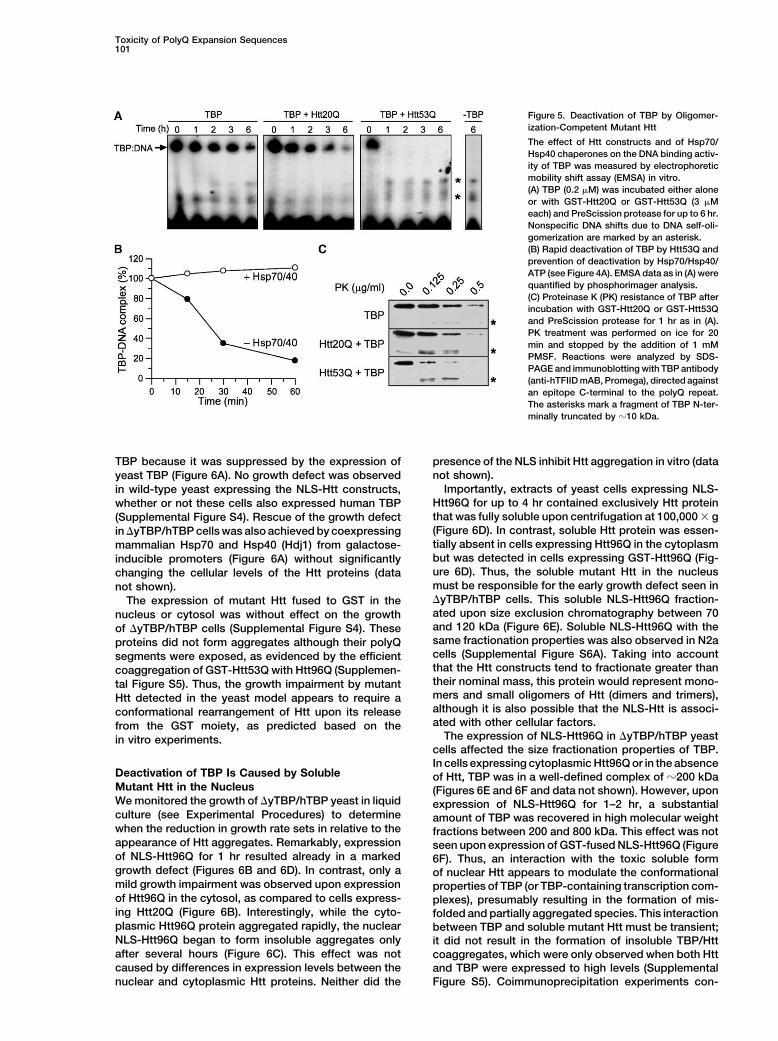
Toxicity of PolyQ Expansion Sequences101
Figure 5. Deactivation of TBP by Oligomer-ization-Competent Mutant Htt
The effect of Htt constructs and of Hsp70/Hsp40 chaperones on the DNA binding activ-ity of TBP was measured by electrophoreticmobility shift assay (EMSA) in vitro.(A) TBP (0.2 �M) was incubated either aloneor with GST-Htt20Q or GST-Htt53Q (3 �Meach) and PreScission protease for up to 6 hr.Nonspecific DNA shifts due to DNA self-oli-gomerization are marked by an asterisk.(B) Rapid deactivation of TBP by Htt53Q andprevention of deactivation by Hsp70/Hsp40/ATP (see Figure 4A). EMSA data as in (A) werequantified by phosphorimager analysis.(C) Proteinase K (PK) resistance of TBP afterincubation with GST-Htt20Q or GST-Htt53Qand PreScission protease for 1 hr as in (A).PK treatment was performed on ice for 20min and stopped by the addition of 1 mMPMSF. Reactions were analyzed by SDS-PAGE and immunoblotting with TBP antibody(anti-hTFIID mAB, Promega), directed againstan epitope C-terminal to the polyQ repeat.The asterisks mark a fragment of TBP N-ter-minally truncated by �10 kDa.
TBP because it was suppressed by the expression of presence of the NLS inhibit Htt aggregation in vitro (datanot shown).yeast TBP (Figure 6A). No growth defect was observed
Importantly, extracts of yeast cells expressing NLS-in wild-type yeast expressing the NLS-Htt constructs,Htt96Q for up to 4 hr contained exclusively Htt proteinwhether or not these cells also expressed human TBPthat was fully soluble upon centrifugation at 100,000 � g(Supplemental Figure S4). Rescue of the growth defect(Figure 6D). In contrast, soluble Htt protein was essen-in �yTBP/hTBP cells was also achieved by coexpressingtially absent in cells expressing Htt96Q in the cytoplasmmammalian Hsp70 and Hsp40 (Hdj1) from galactose-but was detected in cells expressing GST-Htt96Q (Fig-inducible promoters (Figure 6A) without significantlyure 6D). Thus, the soluble mutant Htt in the nucleuschanging the cellular levels of the Htt proteins (datamust be responsible for the early growth defect seen innot shown).�yTBP/hTBP cells. This soluble NLS-Htt96Q fraction-The expression of mutant Htt fused to GST in theated upon size exclusion chromatography between 70nucleus or cytosol was without effect on the growthand 120 kDa (Figure 6E). Soluble NLS-Htt96Q with theof �yTBP/hTBP cells (Supplemental Figure S4). Thesesame fractionation properties was also observed in N2aproteins did not form aggregates although their polyQcells (Supplemental Figure S6A). Taking into accountsegments were exposed, as evidenced by the efficientthat the Htt constructs tend to fractionate greater thancoaggregation of GST-Htt53Q with Htt96Q (Supplemen-their nominal mass, this protein would represent mono-tal Figure S5). Thus, the growth impairment by mutantmers and small oligomers of Htt (dimers and trimers),Htt detected in the yeast model appears to require aalthough it is also possible that the NLS-Htt is associ-conformational rearrangement of Htt upon its releaseated with other cellular factors.from the GST moiety, as predicted based on the
The expression of NLS-Htt96Q in �yTBP/hTBP yeastin vitro experiments.cells affected the size fractionation properties of TBP.In cells expressing cytoplasmic Htt96Q or in the absence
Deactivation of TBP Is Caused by Soluble of Htt, TBP was in a well-defined complex of �200 kDaMutant Htt in the Nucleus (Figures 6E and 6F and data not shown). However, uponWe monitored the growth of �yTBP/hTBP yeast in liquid expression of NLS-Htt96Q for 1–2 hr, a substantialculture (see Experimental Procedures) to determine amount of TBP was recovered in high molecular weightwhen the reduction in growth rate sets in relative to the fractions between 200 and 800 kDa. This effect was notappearance of Htt aggregates. Remarkably, expression seen upon expression of GST-fused NLS-Htt96Q (Figureof NLS-Htt96Q for 1 hr resulted already in a marked 6F). Thus, an interaction with the toxic soluble formgrowth defect (Figures 6B and 6D). In contrast, only a of nuclear Htt appears to modulate the conformationalmild growth impairment was observed upon expression properties of TBP (or TBP-containing transcription com-of Htt96Q in the cytosol, as compared to cells express- plexes), presumably resulting in the formation of mis-ing Htt20Q (Figure 6B). Interestingly, while the cyto- folded and partially aggregated species. This interactionplasmic Htt96Q protein aggregated rapidly, the nuclear between TBP and soluble mutant Htt must be transient;NLS-Htt96Q began to form insoluble aggregates only it did not result in the formation of insoluble TBP/Httafter several hours (Figure 6C). This effect was not coaggregates, which were only observed when both Httcaused by differences in expression levels between the and TBP were expressed to high levels (Supplemental
Figure S5). Coimmunoprecipitation experiments con-nuclear and cytoplasmic Htt proteins. Neither did the
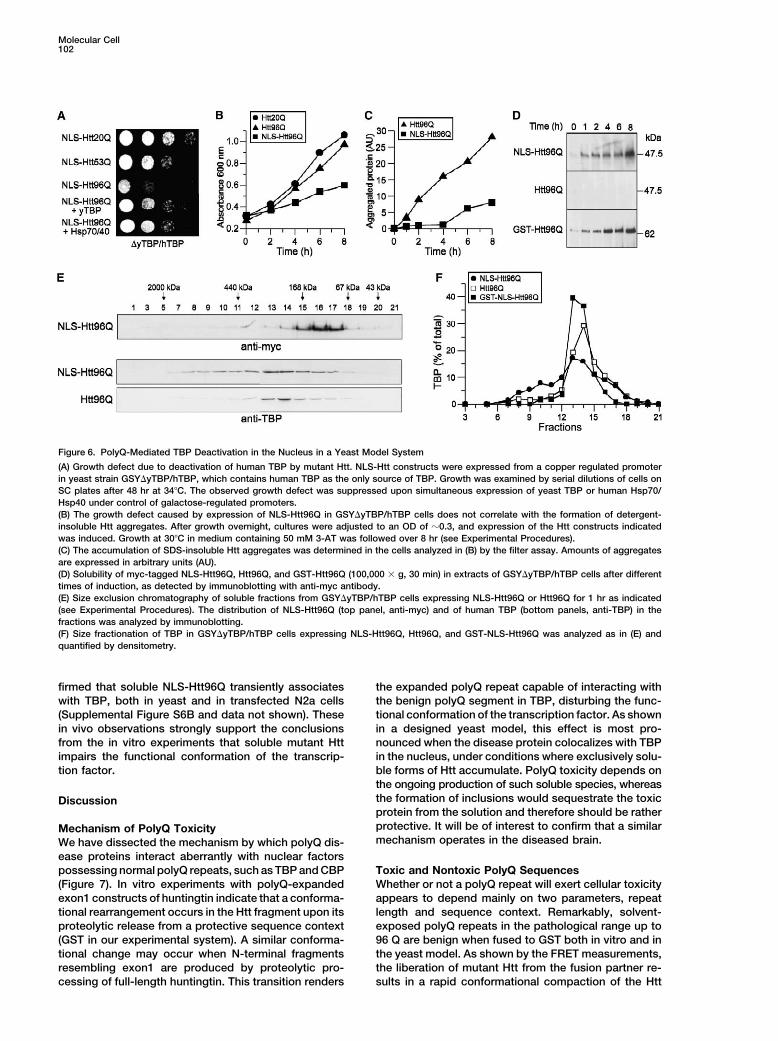
Molecular Cell102
Figure 6. PolyQ-Mediated TBP Deactivation in the Nucleus in a Yeast Model System
(A) Growth defect due to deactivation of human TBP by mutant Htt. NLS-Htt constructs were expressed from a copper regulated promoterin yeast strain GSY�yTBP/hTBP, which contains human TBP as the only source of TBP. Growth was examined by serial dilutions of cells onSC plates after 48 hr at 34�C. The observed growth defect was suppressed upon simultaneous expression of yeast TBP or human Hsp70/Hsp40 under control of galactose-regulated promoters.(B) The growth defect caused by expression of NLS-Htt96Q in GSY�yTBP/hTBP cells does not correlate with the formation of detergent-insoluble Htt aggregates. After growth overnight, cultures were adjusted to an OD of �0.3, and expression of the Htt constructs indicatedwas induced. Growth at 30�C in medium containing 50 mM 3-AT was followed over 8 hr (see Experimental Procedures).(C) The accumulation of SDS-insoluble Htt aggregates was determined in the cells analyzed in (B) by the filter assay. Amounts of aggregatesare expressed in arbitrary units (AU).(D) Solubility of myc-tagged NLS-Htt96Q, Htt96Q, and GST-Htt96Q (100,000 � g, 30 min) in extracts of GSY�yTBP/hTBP cells after differenttimes of induction, as detected by immunoblotting with anti-myc antibody.(E) Size exclusion chromatography of soluble fractions from GSY�yTBP/hTBP cells expressing NLS-Htt96Q or Htt96Q for 1 hr as indicated(see Experimental Procedures). The distribution of NLS-Htt96Q (top panel, anti-myc) and of human TBP (bottom panels, anti-TBP) in thefractions was analyzed by immunoblotting.(F) Size fractionation of TBP in GSY�yTBP/hTBP cells expressing NLS-Htt96Q, Htt96Q, and GST-NLS-Htt96Q was analyzed as in (E) andquantified by densitometry.
firmed that soluble NLS-Htt96Q transiently associates the expanded polyQ repeat capable of interacting withthe benign polyQ segment in TBP, disturbing the func-with TBP, both in yeast and in transfected N2a cells
(Supplemental Figure S6B and data not shown). These tional conformation of the transcription factor. As shownin a designed yeast model, this effect is most pro-in vivo observations strongly support the conclusions
from the in vitro experiments that soluble mutant Htt nounced when the disease protein colocalizes with TBPin the nucleus, under conditions where exclusively solu-impairs the functional conformation of the transcrip-
tion factor. ble forms of Htt accumulate. PolyQ toxicity depends onthe ongoing production of such soluble species, whereasthe formation of inclusions would sequestrate the toxicDiscussionprotein from the solution and therefore should be ratherprotective. It will be of interest to confirm that a similarMechanism of PolyQ Toxicitymechanism operates in the diseased brain.We have dissected the mechanism by which polyQ dis-
ease proteins interact aberrantly with nuclear factorspossessing normal polyQ repeats, such as TBP and CBP Toxic and Nontoxic PolyQ Sequences
Whether or not a polyQ repeat will exert cellular toxicity(Figure 7). In vitro experiments with polyQ-expandedexon1 constructs of huntingtin indicate that a conforma- appears to depend mainly on two parameters, repeat
length and sequence context. Remarkably, solvent-tional rearrangement occurs in the Htt fragment upon itsproteolytic release from a protective sequence context exposed polyQ repeats in the pathological range up to
96 Q are benign when fused to GST both in vitro and in(GST in our experimental system). A similar conforma-tional change may occur when N-terminal fragments the yeast model. As shown by the FRET measurements,
the liberation of mutant Htt from the fusion partner re-resembling exon1 are produced by proteolytic pro-cessing of full-length huntingtin. This transition renders sults in a rapid conformational compaction of the Htt

Toxicity of PolyQ Expansion Sequences103
Figure 7. Working Model for the Deactivationof TBP by a PolyQ Disease Protein
Toxic species of a polyQ-expanded diseaseprotein (monomers and small oligomers) aregenerated as a result of a conformationalchange in the polyQ segment, shown sche-matically as the formation of an intramolecu-lar, cylindrical � sheet (Perutz et al., 2002).See Discussion for details.
exon1 fragment. This effect correlates with the acquisi- apparently because the formation of insoluble Htt aggre-gates is retarded in the nuclear environment relative totion of toxicity and is compatible with the formation ofthe cytoplasm, with small soluble oligomers reachingintramolecular � sheet structure in the mutant polyQhigh local concentrations close to the sites of transcrip-segment. How the GST domain stabilizes the polyQ se-tion. While the attachment of a strong NLS to Htt exon1quence of Htt exon1 in a nontoxic conformation remainsmay contribute to this effect, other factors may play ato be established, but it is an attractive possibility thatrole as well, including a modification of mutant Htt inthe context of the authentic full-length huntingtin pro-the nucleus and the absence of a microtubule-mediatedvides a similar protective effect. This could explain, inprocess that concentrates microaggregates into largepart, why HD typically remains asymptomatic for manyaggresome-type inclusions in the cytoplasm (Muchow-years, although the disease protein is synthesizedski et al., 2002; Waelter et al., 2001). Thus, the toxicitythroughout life span.of polyQ expansion proteins may generally be enhancedOur results indicate that the toxic property of an ex-in the nuclear environment. Soluble nuclear oligomerspanded polyQ sequence, i.e., to interact aberrantly withof mutant Htt were also detected in N2a cells (Supple-the benign polyQ repeats of other proteins, is structur-mental Figure S6), but their existence in nondividingally linked with its ability to nucleate self-oligomeriza-neuronal cells remains to be demonstrated.tion. In the presence of a permissive sequence context,
Transcription factor molecules functionally compro-such as Htt exon1, this property requires a thresholdmised by the aberrant polyQ-polyQ interaction may pop-length of �37 Q (Scherzinger et al., 1997), where intra-ulate misfolded and partially aggregated states. Alter-molecular � sheet formation is thought to be sufficientlynatively, the conformationally altered protein may befavorable (Perutz et al., 2002). Because toxicity and oli-degraded or may coaggregate with the polyQ diseasegomerization competence are shared properties, �protein (Figure 7). However, the efficiency of coaggrega-sheet reactive agents such as Congo red may exert theirtion is limited by the low natural concentrations of theseprotective effect in polyQ disease models by blockingproteins, at least in dividing yeast cells.toxic interactions with benign polyQ proteins rather than
by inhibiting the formation of polyQ inclusions (SanchezFunction of Hsp70/Hsp40 in Suppressinget al., 2003). Benign polyQ repeats, on the other hand,PolyQ Toxicitywould be characterized by a low propensity to formHsp70 and its cochaperone Hsp40 interfere early withintramolecular � sheet structure because they are eitherpolyQ toxicity by retarding the conformational compac-below the pathological threshold length (as in CBP) ortion of the polyQ disease protein and presumably also byare embedded in a protective sequence context (as intransiently shielding benign polyQ sequences in targetTBP and GST-fused Htt96Q, and presumably full-proteins such as TBP (G.S., unpublished data) (Figure 7).length huntingtin).Although Hsp70 interacts preferentially with extendedsequences enriched in hydrophobic residues, glutamine
Significance of Soluble PolyQ Intermediates is not excluded from binding (Sakahira et al., 2002).Deactivation of TBP in vitro and in the yeast model Through their conformational effect on the polyQ pro-results from a destabilization of the protein induced by tein, the chaperones may either facilitate the degrada-a transient, polyQ-mediated interaction with soluble mo- tion of the potentially toxic agent or mediate its disposalnomers or small oligomers of mutant Htt. This is consis- in the form of nonfibrillar inclusions (Figure 7). The exis-tent with observations of polyQ-mediated interference tence of similar defense mechanisms in neurons wouldwith transcription in the apparent absence of inclusions help to explain why these cells have the capacity to deal(Sugars and Rubinsztein, 2003). Notably, this mecha- effectively with polyQ disease proteins for decades, untilnism of toxicity was most effective when the polyQ- the aging process diminishes the cell’s capacity to han-
dle these toxic molecules.expanded model protein was targeted to the nucleus,

Molecular Cell104
Experimental Procedures normalized to the same absorbance at 360 nm. The quantum yieldof the standard was set to 0.55.
FRET was initiated by PreScission protease cleavage of GST-HttExpression ConstructsGST-Htt exon1 constructs were as described (Muchowski et al., proteins at 30�C. FRET efficiency was calculated from the decrease
of the donor quantum yield (QD) in the presence of the acceptor2000). Htt sequences were N-terminally tagged with c-myc or tripleHA-epitopes. TBP was amplified from a human liver cDNA library (QDA). The average r and the critical distance R0 of the donor-acceptor
pair were calculated (Forster, 1948).(Clontech) and cloned with an N-terminal triple HA-tag into pProEX-Hta (Life Technologies). A mutant version with 2 Q (TBP�Q) wasgenerated by PCR mutagenesis. The C-terminal region of human Electrophoretic Mobility Shift AssayCBP (aa 1760–2440; �N-CBP) was amplified from pRSV-CBP (a kind In vitro aggregation reactions containing GST-Htt (3 �M) and TBPgift of T. Papamarcaki) and cloned into pProEX-Hta. (0.2 �M) as above were initiated with PreScission protease and
For tetracycline-inducible expression in N2a cells, c-myc tagged stopped by freezing in liquid nitrogen. Aliquots were thawed on iceHtt exon1 constructs were subcloned into pTREhygro (Clontech). and diluted 20-fold into EMSA buffer (20 mM HEPES-KOH [pH 8.0],Htt20Q, TBP, and TBP�Q were subcloned into pEF-BOSEX (Mizus- 150 mM KCl, 4 mM MgCl2, 1 mM DTT, 5% glycerol) at 28�C withhima and Nagata, 1990) with an N-terminal Flag-epitope. Non- 2 pmol 32P-labeled TA-30-B6 DNA containing a looped TBP bindingtagged, full-length CBP was expressed from pRSV-CBP. When indi- site (Grove et al., 1998) and analyzed immediately on 6% polyacryl-cated, a nuclear localization sequence, DPKKKRKV, from SV40 was amide gels.introduced N-terminally followed by an ELGTA-linker.
Plasmids YEp105-Htt20, YEp105-Htt53, and YEp105-Htt96Q (2 �,Htt Aggregation and PolyQ Recruitment In Vivo
TRP1) were used for copper-inducible expression of c-myc andN2a Cells
GST-c-myc-Htt constructs in yeast (with or without NLS as above).Htt exon1 constructs were transiently expressed from pTREhygro
Yeast TBP (yTBP, SPT15) was cloned into pYES2 expression vectorTet response plasmids in cells stably transfected with the Tet-Off
(2 �, URA3, Invitrogene). The genes encoding human Hsp40 (HDJ-1)regulator plasmid (Clontech). Doxycycline (1 �g/ml) was added to
and Hsp70 were cloned into p426Gal1 (2 �, URA3) (Sikorski andsuppress Htt expression. Recruitment substrates were transiently
Hieter, 1989).expressed. For biochemical analysis cells were lysed on ice in 1�
TBS, 0.5% Triton X-100, 0.5% deoxycholate, and Complete proteaseProtein Purification inhibitors (Roche). Htt96Q was detected with anti-myc, Htt20Q andGST-Htt20Q, GST-Htt53Q, bovine brain Hsp70, and human His6- TBP with anti-FLAG (M2, Sigma) and CBP with polyclonal antibodiestagged Hdj-1 were purified as described (Muchowski et al., 2000). against the N-terminal domain (Upstate Biotechnology).TBP, TBP�Q, and �N-CBP were expressed in E. coli BL21 (DE3) S. cerevisiaeand purified by Ni-NTA chromatography. A haploid yeast strain chromosomally deleted in yTBP (SPT15) and
carrying the gene for human TBP containing the mutation R231KIn Vitro Aggregation (Cormack et al., 1994) was constructed. The endogenous yTBP geneAggregation of GST-Htt proteins (3 �M) was initiated by the addition was replaced by a His marker (Sp HIS5) gene in YPH499 (MATaof 2.5 U PreScission protease (Amersham) in buffer A (50 mM Tris- ura3-52 lys2-801 ade2-101 trp1�63 his3�200 leu2�1 ) (Sikorski andHCl [pH 7.5], 150 mM KCl, 1 mM DTT) at 30�C. When indicated, Hieter, 1989) cells expressing hTBP (R231K) on a pRS415 plasmid2 mM ATP/5 mM MgCl2 and an ATP-regenerating system was added. under control of the yeast TBP promoter. The resulting GSY�yTBP/Substrates for recruitment were typically added at 0.2 �M and hTBP strain is temperature sensitive for growth above 34�C. TheHsp70/Hsp40 at 6 �M/3 �M. To detect SDS-insoluble aggregates, effect on growth of Htt exon1 expression from a copper controllablereactions were stopped by an equal volume of 4% SDS/100 mM promoter (Muchowski et al., 2000) was determined by serial dilutionsDTT and heating (95�C, 5 min), followed by filtration through a nitro- of cells on selective media plates at 34�C. Growth curves werecellulose-acetate membrane and immunodetection (Muchowski et recorded in liquid media (SC-Leu/His/Trp) in the presence of 50 mMal., 2000). To determine total pelletable aggregates, reactions with- 3-AT at 30�C. 3-AT is an inhibitor of HIS5, expression of which isout prior SDS treatment were subjected to centrifugation (20,000 � regulated by two TATA box elements.g, 30 min at 4�C), followed by SDS-PAGE or filter assay. Yeast cell extracts were prepared by glass bead lysis in 25 mM
Tris-HCl (pH 7.5), 50 mM KCl, 10 mM Mg2Cl, 1 mM EDTA, 5% glyc-erol, 1% Triton-X-100 and Complete protease inhibitors (Roche),FRET Analysisfollowed by incubation with benzonase (Merck) for 1 hr at 4�C. GelIntermolecular FRET was measured with the thiol-reactive probesfiltration of soluble fractions (100,000 � g, 30 min) was carried outIAEDANS and IANBD ester (Molecular Probes) as donor-acceptoron a Superdex 200 HR column (Amersham).pair. GST-Htt fusion proteins and GST (60 �M) were labeled for 3 hr
at 4�C in buffer C (50 mM Tris-HCl [pH 7.5], 150 mM KCl, 10%glycerol, 0.1 mM DTT) with a 30-fold molar excess of the dyes Acknowledgmentsadded from DMF. Free dye was removed by gel filtration (BioSpin30, BioRad). Labeling was quantified using the molar extinction The authors thank M. Meisterernst and G. Stelzer for valuable advicecoefficients of GST-Htt53Q and GST at 280 nm (4.3 � 104 M�1cm�1 with the TBP assays, T. Papamarcaki for providing the CBP expres-and 4.1 � 104 M�1cm�1), of IAEDANS at 336 nm (5.7 � 103 M�1cm�1) sion construct, and the Hereditary Disease Foundation for reagents.and of IANBD at 472 nm (23 � 103 M�1cm�1). Two molecules of R.B. was the recipient of a fellowship from the Humboldt Foundation.IAEDANS or IANBD dyes bound per molecule of GST-Htt: one to a This work was supported by the Deutsche Forschungsgemein-cysteine of GST and the other to the unique cysteine in front of the schaft.c-myc epitope of Htt (see Figure 3). All FRET experiments wereperformed with labeled GST-Htt53Q and GST, and the contribution Received: April 6, 2004of the marker in GST was subtracted (see Supplemental Figure S3 Revised: April 28, 2004and legend for additional controls [at http://www.molecule.org/cgi/ Accepted: May 6, 2004content/full/15/1/95/DC1]). Intramolecular FRET was measured with Published: July 1, 2004NCD-4 (Molecular Probes) and IANBD ester as donor-acceptor. La-beling with NCD-4 was in 50 mM potassium phosphate (pH 5.5).
ReferencesTryptic digests and sequence analysis of fluorescent peptides iden-tified one of two adjacent glutamates (E 115 and 116) of Htt as
Bence, N.F., Sampat, R.M., and Kopito, R.R. (2001). Impairment ofthe NCD attachment site (see Figure 4). NCD-labeled protein wasthe ubiquitin-proteasome system by protein aggregation. Scienceincubated with IANBD ester in buffer C. Relative quantum yields292, 1552–1555.of the bound donors (IAEDANS and NCD-4) were determined by
comparing their integrated emission spectra (excitation at 360 nm) Chai, Y., Shao, J., Miller, V.M., Williams, A., and Paulson, H.L. (2002).Live-cell imaging reveals divergent intracellular dynamics of poly-with that of the reference quinine bisulfate in 0.1 M H2SO4 and

Toxicity of PolyQ Expansion Sequences105
glutamine disease proteins and supports a sequestration model of Rajan, R.S., Illing, M.E., Bence, N.F., and Kopito, R.R. (2001). Speci-ficity in intracellular protein aggregation and inclusion body forma-pathogenesis. Proc. Natl. Acad. Sci. USA 99, 9310–9315.tion. Proc. Natl. Acad. Sci. USA 98, 13060–13065.Chan, H.Y.E., Warrick, J.M., Gray-Board, G.L., Paulson, H.L., andRoss, C.A. (2002). Polyglutamine pathogenesis: emergence of unify-Bonini, N.M. (2000). Mechanisms of chaperone suppression of poly-ing mechanisms for Huntington’s disease and related disorders.glutamine disease: selectivity, synergy and modulation of proteinNeuron 35, 819–822.solubility in Drosophila. Hum. Mol. Genet. 9, 2811–2820.
Sakahira, H., Breuer, P., Hayer-Hartl, M.K., and Hartl, F.U. (2002).Cormack, B.P., Strubin, M., Stargell, L.A., and Struhl, K. (1994).Molecular chaperones as modulators of polyglutamine protein ag-Conserved and nonconserved functions of the yeast and humangregation and toxicity. Proc. Natl. Acad. Sci. USA 99, 16412–16418.TATA-binding proteins. Genes Dev. 8, 1335–1343.
Sanchez, I., Mahlke, C., and Yuan, J. (2003). Pivotal role of oligomeri-Cummings, C.J., Reinstein, E., Sun, Y., Antalffy, B., Jiang, Y., Ciecha-zation in expanded polyglutamine neurodegenerative disorders.nover, A., Orr, H.T., Beaudet, A.L., and Zoghbi, H.Y. (1999). MutationNature 421, 373–379.of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency
while accelerating polyglutamine-induced pathology in SCA1 mice. Saudou, F., Finkbeiner, S., Devys, D., and Greenberg, M.E. (1998).Neuron 24, 879–892. Huntingtin acts in the nucleus to induce apoptosis but death does
not correlate with the formation of intranuclear inclusions. Cell 95,Cummings, C.J., Sun, Y.L., Opal, P., Antalffy, B., Mestril, R., Orr,55–66.H.T., Dillmann, W.H., and Zoghbi, H.Y. (2001). Over-expression of
inducible Hsp70 chaperone suppresses neuropathology and im- Scherzinger, E., Lurz, R., Turmaine, M., Mangiarini, L., Hollenbach,proves motor function in SCA1 mice. Hum. Mol. Genet. 10, 1511– B., Hasenbank, R., Bates, G.P., Davies, S.W., Lehrach, H., and1518. Wanker, E.E. (1997). Huntingtin-encoded polyglutamine expansions
form amyloid-like protein aggregates in vitro and in vivo. Cell 90,Davies, S.W., Turmaine, M., Cozens, B.A., DiFiglia, M., Sharp, A.H.,549–558.Ross, C.A., Scherzinger, E., Wanker, E.E., Mangiarini, L., and Bates,
G.P. (1997). Formation of neuronal intranuclear inclusions underlies Sikorski, R.S., and Hieter, P. (1989). A system of shuttle vectors andthe neurological dysfunction in mice transgenic for the HD mutation. yeast host strains designed for efficient manipulation of DNA inCell 90, 537–548. Saccharomyces cerevisiae. Genetics 122, 19–27.
DiFiglia, M., Sapp, E., Chase, K.O., Davies, S.W., Bates, G.P., Vonsat- Sugars, K.L., and Rubinsztein, D.C. (2003). Transcriptional abnor-tel, J.P., and Aronin, N. (1997). Aggregation of huntingtin in neuronal malities in Huntington disease. Trends Genet. 19, 233–238.intranuclear inclusions and dystrophic neurites in brain. Science Waelter, S., Boeddrich, A., Lurz, R., Scherzinger, E., Lueder, G.,277, 1990–1993. Lehrach, H., and Wanker, E.E. (2001). Accumulation of mutant hun-
tingtin fragments in aggresome-like inclusion bodies as a result ofForster, T. (1948). Intermolecular energy migration and fluorescence.insufficient protein degradation. Mol. Biol. Cell 12, 1393–1407.Ann. Phys. 2, 55–75.
Warrick, J.M., Chan, H.Y.E., Gray-Board, G.L., Chai, Y.H., Paulson,Grove, A., Galeone, A., Yu, E., Mayol, L., and Geiduschek, E.P. (1998).H.L., and Bonini, N.M. (1999). Suppression of polyglutamine-medi-Affinity, stability and polarity of binding of the TATA binding proteinated neurodegeneration in Drosophila by the molecular chaperonegoverned by flexure at the TATA Box. J. Mol. Biol. 282, 731–739.Hsp70. Nat. Genet. 23, 425–428.Kim, S., Nollen, E.A., Kitagawa, K., Bindokas, V.P., and Morimoto,Wetzel, R. (2002). Ideas of order for amyloid fibril structure. StructureR.I. (2002). Polyglutamine protein aggregates are dynamic. Nat. Cell10, 1031–1036.Biol. 4, 826–831.
Zoghbi, H.Y., and Orr, H.T. (2000). Glutamine repeats and neurode-Krobitsch, S., and Lindquist, S. (2000). Aggregation of huntingtin ingeneration. Annu. Rev. Neurosci. 23, 217–247.yeast varies with the length of the polyglutamine expansion and
the expression of chaperone proteins. Proc. Natl. Acad. Sci. USA97, 1589–1594.
Lunkes, A., Lindenberg, K.S., Ben-Haiem, L., Weber, C., Devys, D.,Landwehrmeyer, G.B., Mandel, J.L., and Trottier, Y. (2002). Prote-ases acting on mutant huntingtin generate cleaved products thatdifferentially build up cytoplasmic and nuclear inclusions. Mol. Cell10, 259–269.
Masino, L., Kelly, G., Leonard, K., Trottier, Y., and Pastore, A. (2002).Solution structure of polyglutamine tracts in GST-polyglutamine fu-sion proteins. FEBS Lett. 513, 267–272.
McCampbell, A., Taylor, J.P., Taye, A.A., Robitschek, J., Li, M., Wal-cott, J., Merry, D., Chai, Y., Paulson, H., Sobue, G., and Fischbeck,K.H. (2000). CREB-binding protein sequestration by expanded po-lyglutamine. Hum. Mol. Genet. 9, 2197–2202.
Mizushima, S., and Nagata, S. (1990). pEF-BOS, a powerful mamma-lian expression vector. Nucleic Acids Res. 18, 5322.
Muchowski, P.J., Schaffar, G., Sittler, A., Wanker, E.E., Hayer-Hartl,M.K., and Hartl, F.U. (2000). Hsp70 and Hsp40 chaperones caninhibit self-assembly of polyglutamine proteins into amyloid-like fi-brils. Proc. Natl. Acad. Sci. USA 97, 7841–7846.
Muchowski, P.J., Ning, K., D’Souza-Schorey, C., and Fields, S.(2002). Requirement of an intact microtubule cytoskeleton for aggre-gation and inclusion body formation by a mutant huntingtin frag-ment. Proc. Natl. Acad. Sci. USA 99, 727–732.
Perez, M.K., Paulson, H.L., Pendse, S.J., Saionz, S.J., Bonini, N.M.,and Pittman, R.N. (1998). Recruitment and the role of nuclear local-ization in polyglutamine-mediated aggregation. J. Cell Biol. 143,1457–1470.
Perutz, M.F., Finch, J.T., Berriman, J., and Lesk, A. (2002). Amyloidfibers are water-filled nanotubes. Proc. Natl. Acad. Sci. USA 99,5591–5595.




















