
Interaction of a “nido”-ruthenium terpyridylamine complex with charged elongated micellar...
7
Colloids and Surfaces B: Biointerfaces 88 (2011) 641–647 Contents lists available at ScienceDirect Colloids and Surfaces B: Biointerfaces jou rn al h om epage: www.elsevier.com/locate/colsurfb Interaction of a “nido”-ruthenium terpyridylamine complex with charged elongated micellar scaffolds Arnab Maity, Prasun Ghosh, Tarasankar Das, Soumik Mandal, Parna Gupta ∗ , Pradipta Purkayastha ∗ Department of Chemical Sciences, Indian Institute of Science Education and Research, Kolkata, Mohanpur Campus, Mohanpur 741252, WB, India a r t i c l e i n f o Article history: Received 14 June 2011 Received in revised form 22 July 2011 Accepted 26 July 2011 Available online 4 August 2011 Keywords: Ionic surfactant Ruthenium(II) complex Elongated scaffold Binding interaction a b s t r a c t Influence on ionic surfactants by a specially designed terpyridylamine ligand and the ruthenium(II) com- plex formed with it has been studied in aqueous solution. The ligand coordinates to Ru 2+ into an octahedral geometry in such a way that the final form takes a “nido” or nest-like structure. The substitution on the pyridinyl moiety is kept at the ortho position to acquire the specified geometry. Similar complexes have been reported to have anti-tumor properties and thus the ruthenium complexes can effectively replace platinum complexes that serve the same purpose but with certain drawbacks. The “nido” geometry was chosen to minimize the cytotoxicity that creeps in when para substituents of the pyridinyl moiety are used. The latter variety forms a dendridic scaffold. In presence of both the ligand and the complex, ionic surfactants form elongated aggregates. The surface charge of those aggregates decides the nature of interaction of the ligand and the complex formed therefore. Interaction with anionic surfactant scaffold is found to be stronger than the cationic one. © 2011 Elsevier B.V. All rights reserved. 1. Introduction The considerable importance of transition metal complexes as potential chemotherapeutics and in diagnostic medicine has defined their wide use as antitumor and antiviral agents [1,2]. Platinum metal based antitumor agents were primarily discovered and found their vivid applications in medicinal chemistry [3,4]. Later findings provoked the problems associated with platinum- based chemotherapies, which were due to the side-effects of such complexes [4]. Thus, other coordination complexes having tran- sition metal centers have been established [5–8]. From these studies ruthenium compounds have been shown to display less general toxicity than their platinum counterparts are also able to interact with DNA and proteins [9–11] Some typical chemical properties, such as rate of ligand exchange, range of accessible oxi- dation states, and ability of ruthenium to mimic iron in binding to certain biological molecules, make the ruthenium compounds promising candidates for medicinal chemistry [12]. Ruthenium(II) compounds exhibit a low general toxicity and since cancer cells can also become oxidized at certain stages of their growth cycle, oxidation of the ruthenium cannot be excluded [13]. The potential of arene ruthenium(II) complexes as anticancer agents stemmed from the stabilization of ruthenium in its 2+ oxidation state by ∗ Corresponding authors. Fax: +91 33 25873020. E-mail addresses: [email protected] (P. Gupta), [email protected] (P. Purkayastha). -coordinated aromatic hydrocarbons [14,15]. The ruthenium(II) arene fragment coordination with multidrug resistance modulator modeled ligand has shown profound improvement on cytotoxi- city [16]. Research is on to explore whether the combination of an arene ruthenium motif with some potent biologically active ligands would result in novel efficient chemotherapeutic agents against human cancer [17]. As cancer cells overexpress transferrin receptors to satisfy their increased demand for iron, ruthenium- based drugs (containing the iron homologue of ruthenium) may be delivered more efficiently to cancer cells [13,18]. Moreover, the amphiphilic properties of the arene ruthenium unit, provided by the hydrophobic arene ligand counterbalanced by the hydrophilic metal center, and the synthetic diversity of the arene ligand pro- vides an excellent scaffold for the coupling of organic segments for targeted chemotherapy [19,20]. The tetra- and octanuclear arene ruthenium complexes con- taining the first or second generation of a diaminobutane-based dendrimer were found to be moderately cytotoxic without any pronounced size effect [21]. With the established anticancer activ- ity of arene ruthenium complexes and dendrimers as interesting ligands for biological applications, synthesis of multinuclear ruthe- nium complexes based on a poly(propyleneimine) scaffold were investigated to monitor cytotoxicity against A2780 human ovar- ian cancer cell line [21]. One issue in these works is the sizes of the dendrimers that may or may not be beneficial to the tissues as far as their propagation through the blood vessels are concerned. Thus, while keeping similar arene moiety but eliminating the tox- icity due to size effect, we have designed a ruthenium complex 0927-7765/$ – see front matter © 2011 Elsevier B.V. All rights reserved. doi:10.1016/j.colsurfb.2011.07.057
Transcript of Interaction of a “nido”-ruthenium terpyridylamine complex with charged elongated micellar...

Ie
AD
a
ARRAA
KIREB
1
adPaLbcssgtpdtpccoof
(
0d
Colloids and Surfaces B: Biointerfaces 88 (2011) 641– 647
Contents lists available at ScienceDirect
Colloids and Surfaces B: Biointerfaces
jou rn al h om epage: www.elsev ier .com/ locate /co lsur fb
nteraction of a “nido”-ruthenium terpyridylamine complex with chargedlongated micellar scaffolds
rnab Maity, Prasun Ghosh, Tarasankar Das, Soumik Mandal, Parna Gupta ∗, Pradipta Purkayastha ∗
epartment of Chemical Sciences, Indian Institute of Science Education and Research, Kolkata, Mohanpur Campus, Mohanpur 741252, WB, India
r t i c l e i n f o
rticle history:eceived 14 June 2011eceived in revised form 22 July 2011ccepted 26 July 2011vailable online 4 August 2011
a b s t r a c t
Influence on ionic surfactants by a specially designed terpyridylamine ligand and the ruthenium(II) com-plex formed with it has been studied in aqueous solution. The ligand coordinates to Ru2+ into an octahedralgeometry in such a way that the final form takes a “nido” or nest-like structure. The substitution on thepyridinyl moiety is kept at the ortho position to acquire the specified geometry. Similar complexes havebeen reported to have anti-tumor properties and thus the ruthenium complexes can effectively replace
eywords:onic surfactantuthenium(II) complexlongated scaffoldinding interaction
platinum complexes that serve the same purpose but with certain drawbacks. The “nido” geometry waschosen to minimize the cytotoxicity that creeps in when para substituents of the pyridinyl moiety areused. The latter variety forms a dendridic scaffold. In presence of both the ligand and the complex, ionicsurfactants form elongated aggregates. The surface charge of those aggregates decides the nature ofinteraction of the ligand and the complex formed therefore. Interaction with anionic surfactant scaffoldis found to be stronger than the cationic one.
. Introduction
The considerable importance of transition metal complexess potential chemotherapeutics and in diagnostic medicine hasefined their wide use as antitumor and antiviral agents [1,2].latinum metal based antitumor agents were primarily discoverednd found their vivid applications in medicinal chemistry [3,4].ater findings provoked the problems associated with platinum-ased chemotherapies, which were due to the side-effects of suchomplexes [4]. Thus, other coordination complexes having tran-ition metal centers have been established [5–8]. From thesetudies ruthenium compounds have been shown to display lesseneral toxicity than their platinum counterparts are also ableo interact with DNA and proteins [9–11] Some typical chemicalroperties, such as rate of ligand exchange, range of accessible oxi-ation states, and ability of ruthenium to mimic iron in bindingo certain biological molecules, make the ruthenium compoundsromising candidates for medicinal chemistry [12]. Ruthenium(II)ompounds exhibit a low general toxicity and since cancer cellsan also become oxidized at certain stages of their growth cycle,
xidation of the ruthenium cannot be excluded [13]. The potentialf arene ruthenium(II) complexes as anticancer agents stemmedrom the stabilization of ruthenium in its 2+ oxidation state by∗ Corresponding authors. Fax: +91 33 25873020.E-mail addresses: [email protected] (P. Gupta), [email protected]
P. Purkayastha).
927-7765/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.colsurfb.2011.07.057
© 2011 Elsevier B.V. All rights reserved.
�-coordinated aromatic hydrocarbons [14,15]. The ruthenium(II)arene fragment coordination with multidrug resistance modulatormodeled ligand has shown profound improvement on cytotoxi-city [16]. Research is on to explore whether the combination ofan arene ruthenium motif with some potent biologically activeligands would result in novel efficient chemotherapeutic agentsagainst human cancer [17]. As cancer cells overexpress transferrinreceptors to satisfy their increased demand for iron, ruthenium-based drugs (containing the iron homologue of ruthenium) maybe delivered more efficiently to cancer cells [13,18]. Moreover, theamphiphilic properties of the arene ruthenium unit, provided bythe hydrophobic arene ligand counterbalanced by the hydrophilicmetal center, and the synthetic diversity of the arene ligand pro-vides an excellent scaffold for the coupling of organic segments fortargeted chemotherapy [19,20].
The tetra- and octanuclear arene ruthenium complexes con-taining the first or second generation of a diaminobutane-baseddendrimer were found to be moderately cytotoxic without anypronounced size effect [21]. With the established anticancer activ-ity of arene ruthenium complexes and dendrimers as interestingligands for biological applications, synthesis of multinuclear ruthe-nium complexes based on a poly(propyleneimine) scaffold wereinvestigated to monitor cytotoxicity against A2780 human ovar-ian cancer cell line [21]. One issue in these works is the sizes of
the dendrimers that may or may not be beneficial to the tissues asfar as their propagation through the blood vessels are concerned.Thus, while keeping similar arene moiety but eliminating the tox-icity due to size effect, we have designed a ruthenium complex
642 A. Maity et al. / Colloids and Surfaces B: Biointerfaces 88 (2011) 641– 647
N
N N
N
N
N
N
N
N
N
N
NN
N
Ru2+
-alde
tflcwowafpnita
2
wTS
2
2(
(aipe(m2m(((
2
[hEsmt
(I)
Scheme 1. Non-optimized representative structures of (I) Tris[(pyridine-2
hat has a “nido” type of structural motif. The ligand has inherentuorescence that acts as a signal for our photophysical studies toharacterize the system in aqueous medium. In the present studye have used micellar scaffolds to observe the effect of the complex
n the rearrangement of the surfactant molecules and interactionith them. The ligand that we have synthesized is Tris[(pyridine-2-
ldene)ethyl]amine (L2) and the ruthenium(II) complex resultingrom that ligand is designated as A as shown in Scheme 1. Therincipal motivation behind the work is to characterize the ruthe-ium(II) complex that could have a potential anti-cancer activity
n presence of charged surface scaffolds as mimicked by surfac-ants. We have chosen cetyltrimethylammonium bromide (CTAB)nd sodium dodecyl sulfate (SDS) as the model surfactants.
. Materials and methods
All the chemicals were procured from Sigma–Aldrich, Mil-aukee, WI, USA and were used without further purification.
he solvents were procured either from Spectrochem, India origma–Aldrich and were of UV spectroscopy grade.
.1. Syntheses
.1.1. Synthesis of Tris[((pyridine-2-aldene)amino)ethyl]amineL2)
To a solution of pyridine-2-aldehyde (1 g, 9.34 mmol) in ethanol20 mL), Tren (0.46 g, 3.11 mmol) was added in dry toluene (50 mL)nd the mixture was refluxed and stirred for 24 h. The result-ng liquid evaporated to dryness affording an oily substance. Theure ligand is obtained upon washing the oily mass with coldthanol. Yield: 0.64 g (50%); ESI-MS (m/z): 414.83 (M+H)+, 147.94NCH2CH2N CHPy)+. 1H NMR: 10.05 (–CH, 3H) 8.57 (3H, d, aro-
atic ring), 7.6–8.35 (9H, aromatic ring), 3.76 (6H, t, NCH2CH2),.96 (6H, t, NCH2CH2); 13C NMR: 162.64 (N C), 154.38 (aro-atic ring), 149.24 (aromatic ring), 136.44 (aromatic ring), 124.52
aromatic ring), 121.18 (aromatic ring), 59.73 (NCH2CH2), 55.22NCH2CH2). IR (cm−1, KBr pellet): 3435–2850 (�̄C–H), 2850, 1651�̄C H), 1587, 1469, 1436, 774.
.1.2. Synthesis of [RuL2](BPh4)2To a hot solution of L2 (0.043 g, 0.10 mmol) in toluene (40 mL)
Ru(PPh3)2Cl2] (100 mg, 0.10 mmol) was added. The mixture waseated to reflux for 24 h resulting in a dark reddish-brown solution.
vaporation of this solution under vacuum gave a reddish-brownolid, which was dissolved in methanol and NaBPh4 was added. Theixture was again stirred for 30 min. The resulting solution is fil-ered, and the residue was washed with a small amount of methanol
(II)
ne)ethyl]amine (L2) and (II) the coordination complex of L2 with Ru2+ (A).
to yield a dark brown colored crystalline [RuL2](BPh4)2. Yield:0.032 g, 50%; ESI-MS (m/z): 607.7 (M+); 1H NMR: 3.35–4.5 (com-plex), 6.0–8.0 (complex), 10.08; IR (cm−1, KBr pellet): 3792–2920,1614, 1478, 1265, 1093, 755.
2.2. Preparation of solutions
Solutions of the ligand and the complex were prepared by ini-tially dissolving them in acetonitrile. The aqueous solutions of thesystems were made by mixing 0.2% of the mother solution to 99.8%triple distilled water. There was no separation of the solute underthis condition and acetonitrile acted as a good co-solvent. Differ-ent concentrations of surfactant solutions were prepared in tripledistilled water and either the ligand or the complex was added toa final concentration of 2 × 10−6 M.
2.3. Methods
The characterizations of the ligand and the complex were doneusing a Bruker 500 MHz NMR spectrometer and a PerkinElmerSpectrum RX I FTIR spectrometer. The absorption spectra weretaken in a Varian Cary 300 Bio spectrophotometer, the fluorescencemeasurements were done with a PerkinElmer LS 55 spectrofluo-rimeter and the atomic force microscopy (AFM) data were collectedusing an NT-MDT NTEGRA instrument.
3. Results and discussion
3.1. Absorption spectral studies
Fig. 1 shows the absorption spectrum of the ligand L2 thatdemonstrates a structured peak at 260 nm and a shoulder at∼300 nm. The absorbance gets enhanced on addition of CTAB tothe solution of L2. Similar observation is recorded for the complex[RuL2](BPh4)2 (A) in presence of CTAB. The absorption band in thiscase is unstructured. Enhancement in absorbance for both the casesdoes not go through the customary nature that reflects the for-mation of spherical micelles at the critical micellar concentration(CMC). Instead we observe a regular increase in the absorbance asindicated by the plot of relative absorbance against the surfactantconcentration in Fig. 2. Normally on the occurrence of spheri-cal micelles a distinct break in the changing physical parameter
appears at the CMC. Non-obtaining of the break point probably indi-cates the non-occurrence of spherical micelles of CTAB in presenceof L2 and the complex A. However, the regular enhancement in theabsorbance of the species shows evidence for mutual interaction.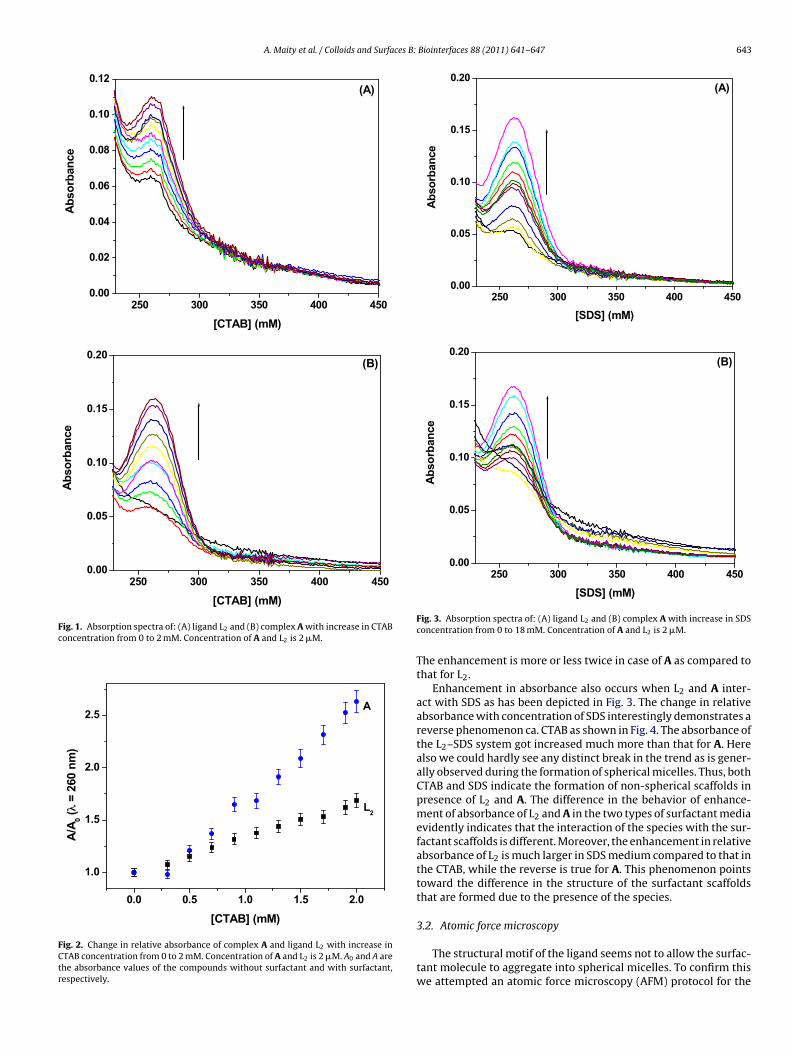
A. Maity et al. / Colloids and Surfaces B: Biointerfaces 88 (2011) 641– 647 643
250 300 350 400 4500.00
0.02
0.04
0.06
0.08
0.10
0.12(A)
Abso
rban
ce
[CTAB] (mM)
250 300 350 400 4500.00
0.05
0.10
0.15
0.20(B)
Abso
rban
ce
[CTAB] (mM)
Fig. 1. Absorption spectra of: (A) ligand L2 and (B) complex A with increase in CTABconcentration from 0 to 2 mM. Concentration of A and L2 is 2 �M.
0.0 0.5 1.0 1.5 2.0
1.0
1.5
2.0
2.5A
L2
A/A 0 (
λ =
260
nm)
[CTAB] (mM)
Fig. 2. Change in relative absorbance of complex A and ligand L2 with increase inCTAB concentration from 0 to 2 mM. Concentration of A and L2 is 2 �M. A0 and A arethe absorbance values of the compounds without surfactant and with surfactant,respectively.
250 30 0 35 0 40 0 45 00.00
0.05
0.10
0.15
0.20(A)
Abso
rban
ce
[SDS] (mM)
250 300 35 0 40 0 45 00.00
0.05
0.10
0.15
0.20(B)
Abso
rban
ce
[SDS] (mM)
Fig. 3. Absorption spectra of: (A) ligand L2 and (B) complex A with increase in SDSconcentration from 0 to 18 mM. Concentration of A and L2 is 2 �M.
The enhancement is more or less twice in case of A as compared tothat for L2.
Enhancement in absorbance also occurs when L2 and A inter-act with SDS as has been depicted in Fig. 3. The change in relativeabsorbance with concentration of SDS interestingly demonstrates areverse phenomenon ca. CTAB as shown in Fig. 4. The absorbance ofthe L2–SDS system got increased much more than that for A. Herealso we could hardly see any distinct break in the trend as is gener-ally observed during the formation of spherical micelles. Thus, bothCTAB and SDS indicate the formation of non-spherical scaffolds inpresence of L2 and A. The difference in the behavior of enhance-ment of absorbance of L2 and A in the two types of surfactant mediaevidently indicates that the interaction of the species with the sur-factant scaffolds is different. Moreover, the enhancement in relativeabsorbance of L2 is much larger in SDS medium compared to that inthe CTAB, while the reverse is true for A. This phenomenon pointstoward the difference in the structure of the surfactant scaffoldsthat are formed due to the presence of the species.
3.2. Atomic force microscopy
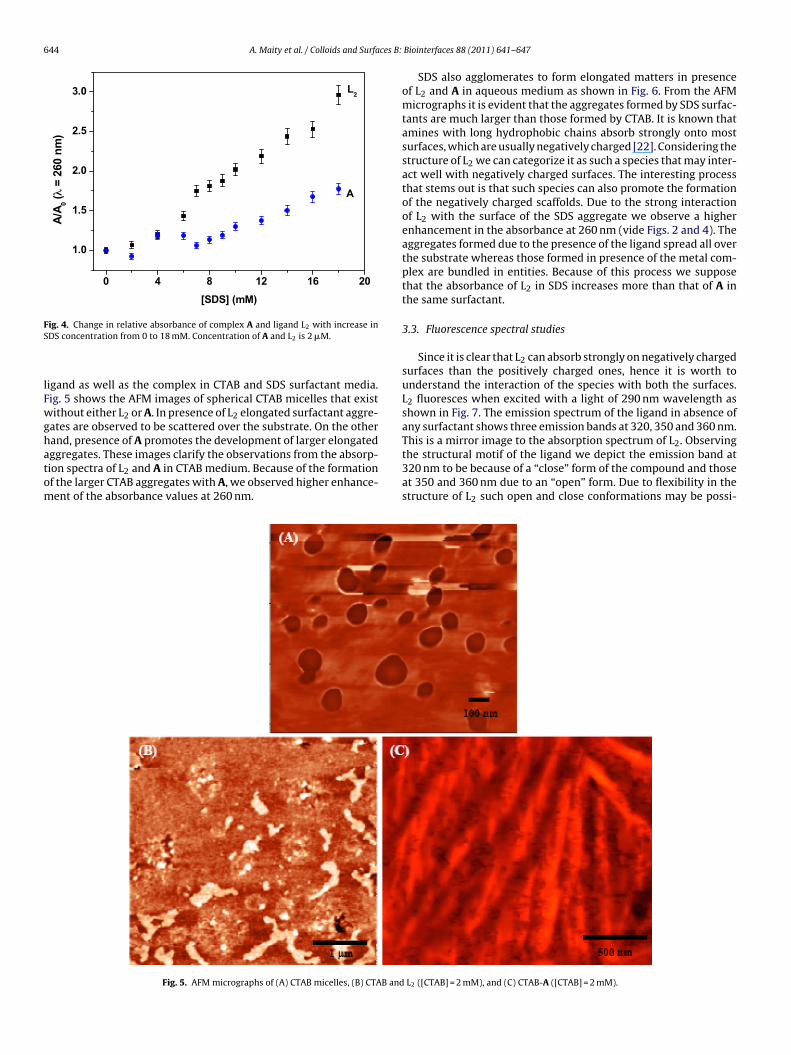
The structural motif of the ligand seems not to allow the surfac-tant molecule to aggregate into spherical micelles. To confirm thiswe attempted an atomic force microscopy (AFM) protocol for the
644 A. Maity et al. / Colloids and Surfaces B:
0 4 8 12 16 20
1.0
1.5
2.0
2.5
3.0
A
L2
A/A 0 (
λ =
260
nm)
[SDS] (mM)
Fig. 4. Change in relative absorbance of complex A and ligand L2 with increase inS
lFwghatom
the structural motif of the ligand we depict the emission band at
DS concentration from 0 to 18 mM. Concentration of A and L2 is 2 �M.
igand as well as the complex in CTAB and SDS surfactant media.ig. 5 shows the AFM images of spherical CTAB micelles that existithout either L2 or A. In presence of L2 elongated surfactant aggre-
ates are observed to be scattered over the substrate. On the otherand, presence of A promotes the development of larger elongatedggregates. These images clarify the observations from the absorp-
ion spectra of L2 and A in CTAB medium. Because of the formationf the larger CTAB aggregates with A, we observed higher enhance-ent of the absorbance values at 260 nm.Fig. 5. AFM micrographs of (A) CTAB micelles, (B) CTAB and
Biointerfaces 88 (2011) 641– 647
SDS also agglomerates to form elongated matters in presenceof L2 and A in aqueous medium as shown in Fig. 6. From the AFMmicrographs it is evident that the aggregates formed by SDS surfac-tants are much larger than those formed by CTAB. It is known thatamines with long hydrophobic chains absorb strongly onto mostsurfaces, which are usually negatively charged [22]. Considering thestructure of L2 we can categorize it as such a species that may inter-act well with negatively charged surfaces. The interesting processthat stems out is that such species can also promote the formationof the negatively charged scaffolds. Due to the strong interactionof L2 with the surface of the SDS aggregate we observe a higherenhancement in the absorbance at 260 nm (vide Figs. 2 and 4). Theaggregates formed due to the presence of the ligand spread all overthe substrate whereas those formed in presence of the metal com-plex are bundled in entities. Because of this process we supposethat the absorbance of L2 in SDS increases more than that of A inthe same surfactant.
3.3. Fluorescence spectral studies
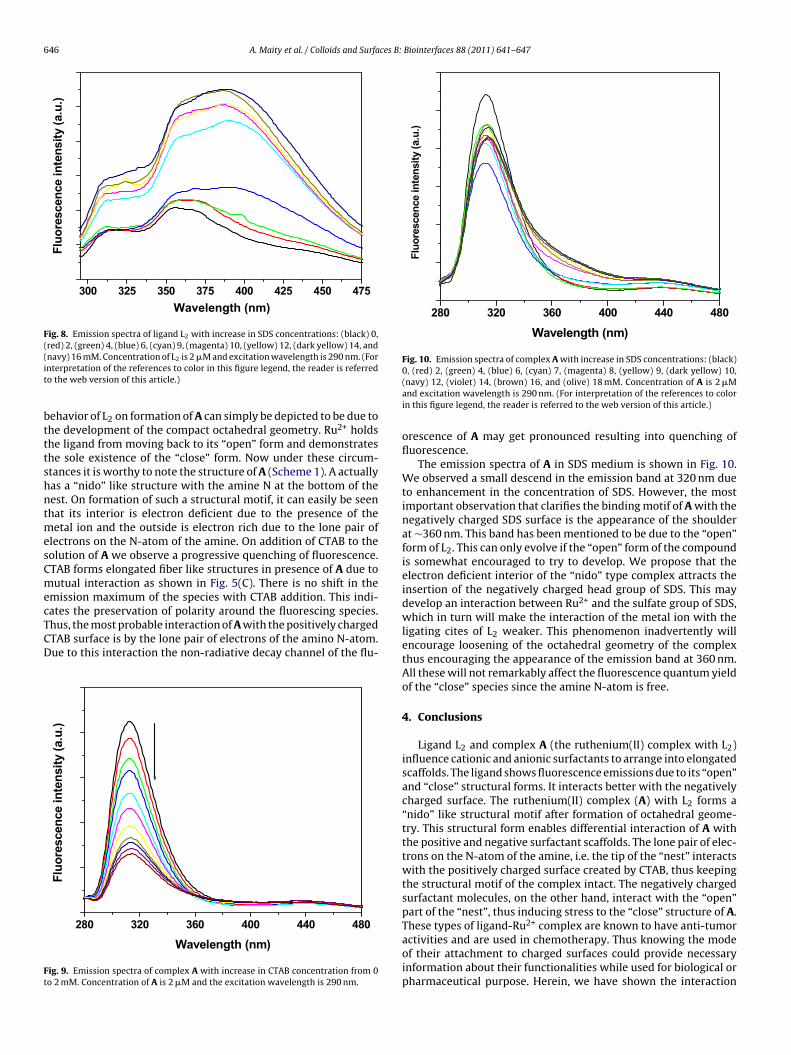
Since it is clear that L2 can absorb strongly on negatively chargedsurfaces than the positively charged ones, hence it is worth tounderstand the interaction of the species with both the surfaces.L2 fluoresces when excited with a light of 290 nm wavelength asshown in Fig. 7. The emission spectrum of the ligand in absence ofany surfactant shows three emission bands at 320, 350 and 360 nm.This is a mirror image to the absorption spectrum of L2. Observing
320 nm to be because of a “close” form of the compound and thoseat 350 and 360 nm due to an “open” form. Due to flexibility in thestructure of L2 such open and close conformations may be possi-
L2 ([CTAB] = 2 mM), and (C) CTAB-A ([CTAB] = 2 mM).
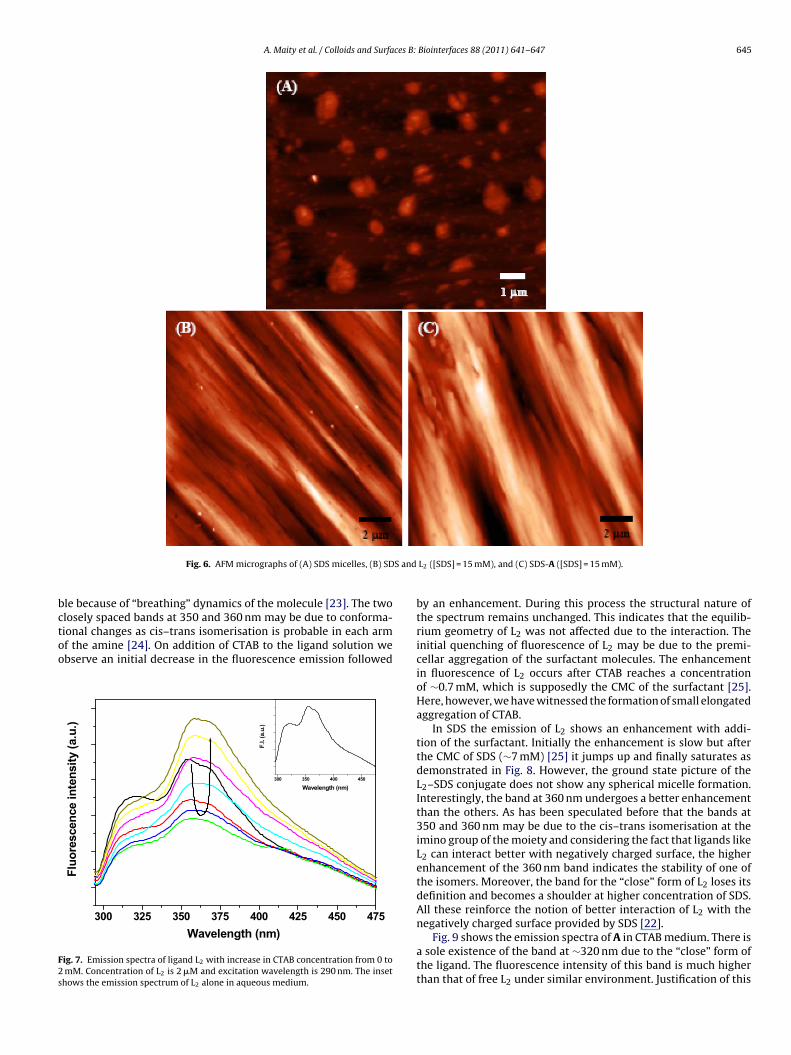
A. Maity et al. / Colloids and Surfaces B: Biointerfaces 88 (2011) 641– 647 645
S and
bctoo
F2s
Fig. 6. AFM micrographs of (A) SDS micelles, (B) SD
le because of “breathing” dynamics of the molecule [23]. The twolosely spaced bands at 350 and 360 nm may be due to conforma-
ional changes as cis–trans isomerisation is probable in each armf the amine [24]. On addition of CTAB to the ligand solution webserve an initial decrease in the fluorescence emission followed300 325 35 0 375 400 425 450 475
F.I.
(a.u
.)
Wavelength (nm)
Fluo
resc
ence
inte
nsity
(a.u
.)
Waveleng th (nm)
ig. 7. Emission spectra of ligand L2 with increase in CTAB concentration from 0 to mM. Concentration of L2 is 2 �M and excitation wavelength is 290 nm. The insethows the emission spectrum of L2 alone in aqueous medium.
L2 ([SDS] = 15 mM), and (C) SDS-A ([SDS] = 15 mM).
by an enhancement. During this process the structural nature ofthe spectrum remains unchanged. This indicates that the equilib-rium geometry of L2 was not affected due to the interaction. Theinitial quenching of fluorescence of L2 may be due to the premi-cellar aggregation of the surfactant molecules. The enhancementin fluorescence of L2 occurs after CTAB reaches a concentrationof ∼0.7 mM, which is supposedly the CMC of the surfactant [25].Here, however, we have witnessed the formation of small elongatedaggregation of CTAB.
In SDS the emission of L2 shows an enhancement with addi-tion of the surfactant. Initially the enhancement is slow but afterthe CMC of SDS (∼7 mM) [25] it jumps up and finally saturates asdemonstrated in Fig. 8. However, the ground state picture of theL2–SDS conjugate does not show any spherical micelle formation.Interestingly, the band at 360 nm undergoes a better enhancementthan the others. As has been speculated before that the bands at350 and 360 nm may be due to the cis–trans isomerisation at theimino group of the moiety and considering the fact that ligands likeL2 can interact better with negatively charged surface, the higherenhancement of the 360 nm band indicates the stability of one ofthe isomers. Moreover, the band for the “close” form of L2 loses itsdefinition and becomes a shoulder at higher concentration of SDS.All these reinforce the notion of better interaction of L2 with thenegatively charged surface provided by SDS [22].
Fig. 9 shows the emission spectra of A in CTAB medium. There isa sole existence of the band at ∼320 nm due to the “close” form ofthe ligand. The fluorescence intensity of this band is much higherthan that of free L2 under similar environment. Justification of this
646 A. Maity et al. / Colloids and Surfaces B: Biointerfaces 88 (2011) 641– 647
300 325 350 375 40 0 42 5 45 0 475
Fluo
resc
ence
inte
nsity
(a.u
.)
Wavelength (nm)
Fig. 8. Emission spectra of ligand L2 with increase in SDS concentrations: (black) 0,(red) 2, (green) 4, (blue) 6, (cyan) 9, (magenta) 10, (yellow) 12, (dark yellow) 14, and(it
btttshntmesCmecTCD
Ft
280 32 0 360 400 44 0 480
Fluo
resc
ence
inte
nsity
(a.u
.)
Waveleng th (nm)
Fig. 10. Emission spectra of complex A with increase in SDS concentrations: (black)0, (red) 2, (green) 4, (blue) 6, (cyan) 7, (magenta) 8, (yellow) 9, (dark yellow) 10,
navy) 16 mM. Concentration of L2 is 2 �M and excitation wavelength is 290 nm. (Fornterpretation of the references to color in this figure legend, the reader is referredo the web version of this article.)
ehavior of L2 on formation of A can simply be depicted to be due tohe development of the compact octahedral geometry. Ru2+ holdshe ligand from moving back to its “open” form and demonstrateshe sole existence of the “close” form. Now under these circum-tances it is worthy to note the structure of A (Scheme 1). A actuallyas a “nido” like structure with the amine N at the bottom of theest. On formation of such a structural motif, it can easily be seenhat its interior is electron deficient due to the presence of the
etal ion and the outside is electron rich due to the lone pair oflectrons on the N-atom of the amine. On addition of CTAB to theolution of A we observe a progressive quenching of fluorescence.TAB forms elongated fiber like structures in presence of A due toutual interaction as shown in Fig. 5(C). There is no shift in the
mission maximum of the species with CTAB addition. This indi-ates the preservation of polarity around the fluorescing species.
hus, the most probable interaction of A with the positively chargedTAB surface is by the lone pair of electrons of the amino N-atom.ue to this interaction the non-radiative decay channel of the flu-280 320 360 400 440 480
Fluo
resc
ence
inte
nsity
(a.u
.)
Wavelength (n m)
ig. 9. Emission spectra of complex A with increase in CTAB concentration from 0o 2 mM. Concentration of A is 2 �M and the excitation wavelength is 290 nm.
(navy) 12, (violet) 14, (brown) 16, and (olive) 18 mM. Concentration of A is 2 �Mand excitation wavelength is 290 nm. (For interpretation of the references to colorin this figure legend, the reader is referred to the web version of this article.)
orescence of A may get pronounced resulting into quenching offluorescence.
The emission spectra of A in SDS medium is shown in Fig. 10.We observed a small descend in the emission band at 320 nm dueto enhancement in the concentration of SDS. However, the mostimportant observation that clarifies the binding motif of A with thenegatively charged SDS surface is the appearance of the shoulderat ∼360 nm. This band has been mentioned to be due to the “open”form of L2. This can only evolve if the “open” form of the compoundis somewhat encouraged to try to develop. We propose that theelectron deficient interior of the “nido” type complex attracts theinsertion of the negatively charged head group of SDS. This maydevelop an interaction between Ru2+ and the sulfate group of SDS,which in turn will make the interaction of the metal ion with theligating cites of L2 weaker. This phenomenon inadvertently willencourage loosening of the octahedral geometry of the complexthus encouraging the appearance of the emission band at 360 nm.All these will not remarkably affect the fluorescence quantum yieldof the “close” species since the amine N-atom is free.
4. Conclusions
Ligand L2 and complex A (the ruthenium(II) complex with L2)influence cationic and anionic surfactants to arrange into elongatedscaffolds. The ligand shows fluorescence emissions due to its “open”and “close” structural forms. It interacts better with the negativelycharged surface. The ruthenium(II) complex (A) with L2 forms a“nido” like structural motif after formation of octahedral geome-try. This structural form enables differential interaction of A withthe positive and negative surfactant scaffolds. The lone pair of elec-trons on the N-atom of the amine, i.e. the tip of the “nest” interactswith the positively charged surface created by CTAB, thus keepingthe structural motif of the complex intact. The negatively chargedsurfactant molecules, on the other hand, interact with the “open”part of the “nest”, thus inducing stress to the “close” structure of A.These types of ligand-Ru2+ complex are known to have anti-tumor
activities and are used in chemotherapy. Thus knowing the modeof their attachment to charged surfaces could provide necessaryinformation about their functionalities while used for biological orpharmaceutical purpose. Herein, we have shown the interaction
ces B:
obb
A
gaAa
R
[
[
[[[
[
[
[
[
[[[
[
A. Maity et al. / Colloids and Surfa
f the ligand and the complex from photophysical aspect usingiomimicking scaffolds. Further studies on biological systems wille performed to understand their impacts.
cknowledgements
Financial support by CSIR, New Delhi [01(2261)/08-EMR-II] isratefully acknowledged. PG acknowledges Department of Sciencend Technology, India, for research grant no. SR/FT/CS-057/2009.M and PG, SM, and TD acknowledge UGC, Government of Indiand CSIR, New Delhi, respectively for their fellowships.
eferences
[1] L. Ronconi, P.J. Sadler, Coord. Chem. Rev. 251 (2007) 1633.[2] C. Orvig, M.J. Abrams, Chem. Rev. 99 (1999) 2201.[3] B. Lippert (Ed.), Cisplatin Chemistry and Biochemistry of a Leading Anticancer
Drug, VCHA & Wiley-VCH, Zurich, 1999.[4] M. Galanski, M.A. Jakupec, B.K. Keppler, Curr. Med. Chem. 12 (2005) 2075.[5] C.V. Christodoulou, D.R. Ferry, D.W. Fyfe, A. Young, J. Doran, T.M. Sheehan, A.
Eliopoulos, K. Hale, J. Baumgart, G. Sass, D.J. Kerr, J. Clin. Oncol. 16 (1998) 2761.[6] F. Caruso, M. Rossi, J. Tanski, C. Pettinari, F. Marchetti, J. Med. Chem. 46 (2003)
1737.[7] C.G. Hartinger, S. Zorbas-Seifried, M.A. Jakupec, B. Kynast, H. Zorbas, B.K. Kep-
pler, J. Inorg. Biochem. 100 (2006) 891.
[[
[
Biointerfaces 88 (2011) 641– 647 647
[8] W.H. Ang, P.J. Dyson, Eur. J. Inorg. Chem. (2006) 4003.[9] M.J. Clarke, Chem. Rev. 99 (1999) 2511.10] F. Wang, J. Xu, A. Habtemariam, J. Bella, P.J. Sadler, J. Am. Chem. Soc. 127 (2005)
17734.11] A.R. Timerbaev, C.G. Hartinger, S.S. Aleksenko, B.K. Keppler, Chem. Rev. 106
(2006) 2224.12] V. Brabec, O. Nováková, Drug Resist. Updat. 9 (2006) 111.13] P.J. Dyson, G. Sava, Dalton Trans. (2006) 1929.14] C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T.J.
Geldbach, G. Sava, P.J. Dyson, J. Med. Chem. 48 (2005) 4161.15] T. Burgarcic, A. Habtemariam, J. Stepankova, P. Heringova, J. Kasparkova, R.J.
Deeth, R.D.L. Johnstone, A. Prescimone, A. Parkin, S. Parsons, V. Brabec, P.J.Sadler, Inorg. Chem. 47 (2008) 11470.
16] C.A. Vock, W.H. Ang, C. Scolaro, A.D. Phillips, L. Lagopoulos, L. Juillerat-Jeanneret,G. Sava, R. Scopelliti, P.J. Dyson, J. Med. Chem. 50 (2007) 2166.
17] C.-H. Wu, D.-H. Wu, X. Liu, G. Guoyiqibayi, D.-D. Guo, G. Lv, X.-M. Wang, H. Yan,H. Jiang, Z.-H. Lu, Inorg. Chem. 48 (2009) 2352.
18] C.S. Allardyce, A. Dorcier, C. Scolaro, P.J. Dyson, Appl. Organomet. Chem. 19(2005) 1.
19] P.J. Dyson, Chimia 61 (2007) 698.20] G. Süss-Fink, Dalton Trans. 39 (2010) 1673.21] P. Govender, N.C. Antonels, J. Mattsson, A.K. Renfrew, P.J. Dyson, J.R. Moss, B.
Therrien, G.S. Smith, J. Organomet. Chem. 694 (2009) 3470.22] M.J. Rosen, Surfactants and Interfacial Phenomena, 3rd ed., Wiley-Interscience,
New Jersey, USA, 2004, p 17.
23] J. Wu, J. Prausnitz, Fluid Phase Equilib. 194–197 (2002) 689.24] M. Amati, C. Bonini, M. D’Auria, M. Funicello, F. Lelj, R. Racioppi, J. Org. Chem.71 (2006) 7165.25] S.S. Jaffer, M. Sowmiya, S.K. Saha, P. Purkayastha, J. Colloid Interface Sci. 325
(2008) 236.
















 and electrochemical behaviour](https://static.fdokumen.com/doc/165x107/6333101f576b626f850db0e4/ligand-substitution-and-growth-reactions-with-ru-3-se-2-nido-clusters-coordination.jpg)



