
Catalytic and interfacial aspects of enzymatic polymer synthesis in reversed micellar systems
10
Catalytic and Interfacial Aspects of Enzymatic Polymer Synthesis in Reversed Micellar Systems A. Madhusudhan Rao, Vijay T. John,* and Richard D. Gonzalez Department of Chemical Engineering, Tulane University, New Orleans, Louisiana 70 1 18 Joseph A. Akkara and David L. Kaplan Department of the Army, Natick Research, Development and Engineering Center, Natick, Massachusetts 01 760 Received April 7, 1992/Accepted October 13, 1992 The enzyme horseradish peroxidase, when encapsulated in reversed micelles, is capable of catalyzing the syn- thesis of phenolic and aromatic amine polymers. The synthesis of polyethylphenol is specifically considered in this article and is found to be extremely feasible in the micellar system. Polymer chain growth can be controlled to some degree by manipulating the ability of the solvent to sustain chain solubility; this is effectively done by adjusting the surfactant concentration. This results in a degree of control of polymer molecular weight. The synthesized polymer drops out of solution and can be easily recovered. 0 1993 John Wiley & Sons, Inc. Key words: horseradish peroxidase reversed micelles phenolic polymers enzyme kinetics INTRODUCTION Phenolic and aromatic amine-based polymers have a variety of applications in making resins for coatings, laminates, etc." In recent years, the synthesis of such polymers through enzyme catalysis has been intensively examined.'r3 This is due to the possibility that such synthesis carried out at ambient conditions may have advantages in comparison to the conventional formaldehyde-based high-temperature process, where undesirable side reactions lead to poor con- trol of polymer structure and molecular weight. In addition, concern over the toxicity of formaldehyde" necessitates the study of alternative technologies to produce such polymers. The enzyme horseradish peroxidase catalyzes the oxida- tive coupling of a variety of substrates including phenols and aromatic amines'8,22 through activation by hydrogen peroxide. It has been used as the enzyme of choice in recent studies which describe the polymerization of such sub~trates.'?~ In aqueous media, the reaction is not very effective due to poor substrate solubility, and the polymeri- zation does not proceed much further than low molecular weight oligomer formation before precipitation limits fur- ther growth." There is therefore an advantage to carrying out the reaction in organic media in order to sustain the growing chain in solution. The ability of horseradish peroxidase to function in a variety of organic solvents and the reported feasibility of polymerization in such media are * To whom all correspondence should be addressed. direct evidence of the application potential of nonaque- ous enzymology.' Indeed, molecular weights as high as 4 X 16 have been reported in the polymerization of p- phenylphenol using a solvent containing 85% dioxane and 15% water.' In this article, we examine horseradish peroxidase cata- lyzed polymerization in the microstructured media of water- in-oil microemulsions traditionally known as reversed mi- celles, schematically shown in Figure l. Reversed micelles have been of much research interest recently due to their remarkable ability to solubilized macromolecular poly- electrolytes such as proteins without destroying catalytic activity. In addition, the fact that protein occupancy per micelle is seldom greater than unity indicates that the catalyst dispersion is extremely high, leading to efficient utilization. The observation that enzyme activity is retained in reversed micelles has led to a variety of synthetic applications.8 Of special interest are reactions in which the presence of excess water leads to equilibrium limitations in yield. Our rationale for conducting polymerization in reversed micelles is based on much more than the exploratory objec- tive of determining whether the reaction is feasible in media other than monophasic organic solvents. Reversed micelles are characterized by extremely large oil-water interfacial areas. A rough calculation for AOT- (dioctyl sodium sulfosuccinate-) water-isooctane systems indicates an in- terfacial area of 18 m2/mL of solution when the surfactant concentration is kept at 50 mM and the water-to-surfactant molar ratio wo is 15. Since the monomeric species do possess amphiphilic characteristics, especially with hy- drocarbon moieties attached to the aromatic ring, it is possible that the monomeric species may align themselves at the oil-water interface with the hydroxyl (or amine) groups directed at the microaqueous core. This leads to the possibility that the initial alignment of monomeric species prior to reaction may result in the synthesis of oriented polymers. Figure 2 illustrates the reaction mechanisms with initial activation at the hydroxyl position' and migration of the free radical to the resonance-stabilized aromatic ring, following which coupling between monomeric units occurs. Biotechnology and Bioengineering, Vol. 41, Pp. 531-540 (1993) 0 1993 John Wiley & Sons, Inc. CCC 0006-3592/93/050531- 10
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Catalytic and interfacial aspects of enzymatic polymer synthesis in reversed micellar systems

Catalytic and Interfacial Aspects of Enzymatic Polymer Synthesis in Reversed Micellar Systems A. Madhusudhan Rao, Vijay T. John,* and Richard D. Gonzalez Department of Chemical Engineering, Tulane University, New Orleans, Louisiana 70 1 18
Joseph A. Akkara and David L. Kaplan Department of the Army, Natick Research, Development and Engineering Center, Natick, Massachusetts 01 760
Received April 7, 1992/Accepted October 13, 1992
The enzyme horseradish peroxidase, when encapsulated in reversed micelles, is capable of catalyzing the syn- thesis of phenolic and aromatic amine polymers. The synthesis of polyethylphenol is specifically considered in this article and is found to be extremely feasible in the micellar system. Polymer chain growth can be controlled to some degree by manipulating the ability of the solvent to sustain chain solubility; this is effectively done by adjusting the surfactant concentration. This results in a degree of control of polymer molecular weight. The synthesized polymer drops out of solution and can be easily recovered. 0 1993 John Wiley & Sons, Inc. Key words: horseradish peroxidase reversed micelles phenolic polymers enzyme kinetics
INTRODUCTION
Phenolic and aromatic amine-based polymers have a variety of applications in making resins for coatings, laminates, etc." In recent years, the synthesis of such polymers through enzyme catalysis has been intensively examined.'r3 This is due to the possibility that such synthesis carried out at ambient conditions may have advantages in comparison to the conventional formaldehyde-based high-temperature process, where undesirable side reactions lead to poor con- trol of polymer structure and molecular weight. In addition, concern over the toxicity of formaldehyde" necessitates the study of alternative technologies to produce such polymers.
The enzyme horseradish peroxidase catalyzes the oxida- tive coupling of a variety of substrates including phenols and aromatic amines'8,22 through activation by hydrogen peroxide. It has been used as the enzyme of choice in recent studies which describe the polymerization of such sub~trates.'?~ In aqueous media, the reaction is not very effective due to poor substrate solubility, and the polymeri- zation does not proceed much further than low molecular weight oligomer formation before precipitation limits fur- ther growth." There is therefore an advantage to carrying out the reaction in organic media in order to sustain the growing chain in solution. The ability of horseradish peroxidase to function in a variety of organic solvents and the reported feasibility of polymerization in such media are
* To whom all correspondence should be addressed.
direct evidence of the application potential of nonaque- ous enzymology.' Indeed, molecular weights as high as 4 X 16 have been reported in the polymerization of p- phenylphenol using a solvent containing 85% dioxane and 15% water.'
In this article, we examine horseradish peroxidase cata- lyzed polymerization in the microstructured media of water- in-oil microemulsions traditionally known as reversed mi- celles, schematically shown in Figure l. Reversed micelles have been of much research interest recently due to their remarkable ability to solubilized macromolecular poly- electrolytes such as proteins without destroying catalytic activity. In addition, the fact that protein occupancy per micelle is seldom greater than unity indicates that the catalyst dispersion is extremely high, leading to efficient utilization. The observation that enzyme activity is retained in reversed micelles has led to a variety of synthetic applications.8 Of special interest are reactions in which the presence of excess water leads to equilibrium limitations in yield.
Our rationale for conducting polymerization in reversed micelles is based on much more than the exploratory objec- tive of determining whether the reaction is feasible in media other than monophasic organic solvents. Reversed micelles are characterized by extremely large oil-water interfacial areas. A rough calculation for AOT- (dioctyl sodium sulfosuccinate-) water-isooctane systems indicates an in- terfacial area of 18 m2/mL of solution when the surfactant concentration is kept at 50 mM and the water-to-surfactant molar ratio wo is 15. Since the monomeric species do possess amphiphilic characteristics, especially with hy- drocarbon moieties attached to the aromatic ring, it is possible that the monomeric species may align themselves at the oil-water interface with the hydroxyl (or amine) groups directed at the microaqueous core. This leads to the possibility that the initial alignment of monomeric species prior to reaction may result in the synthesis of oriented polymers. Figure 2 illustrates the reaction mechanisms with initial activation at the hydroxyl position' and migration of the free radical to the resonance-stabilized aromatic ring, following which coupling between monomeric units occurs.
Biotechnology and Bioengineering, Vol. 41, Pp. 531 -540 (1993) 0 1993 John Wiley & Sons, Inc. CCC 0006-3592/93/050531- 10
\ 0
i II 0
Figure 1. Schematic of an enzyme in a reversed micelle. The surfactant shown is the double-tailed anionic, bis(2-ethylhexyl) sodium sulfosuccinate.
I . . .
H 0
. A
Figure 2. Reaction scheme for polyethylphenol synthesis.
The idea, therefore, is to determine if initial alignment of the monomeric species at the interface would result in optimal coupling and chain growth, perhaps with all chain linkages at positions which are ortho to the hydroxyl functional group. Such polymers with aligned functional groups may possess novel properties such as the ability to chelate with metals. Even more important, such conjugated polymers exhibit optical n~nlinearity,'~ and it is one of our continuing objectives to determine if synthesis in reversed micelles improves the second and third order nonlinear susceptibilities (x2 and x3 values) with applications to photonics.
This article deals with the polymerization of a model phenolic compound, p-ethylphenol. The objective here is
not so much to examine the application potential of the polymer, but rather to quantify the feasibility of the reaction in terms of kinetics and monomer conversions and to measure gross characteristics of the polymer, especially its molecular weight distribution. We report comparisons of polymerization in different media, in particular the monophasic organic solvent system of 85% (v/v) dioxane in water. The role of the reversed micellar environment in sustaining polymerization is an aspect we have studied. Finally, an attempt is made to understand the physiochemi- cal characteristics of enzyme-containing reversed micelles to determine the retention of micellar structural integrity and modifications to micellar structure introduced by the addition of monomer and due to subsequent reaction.
532 BIOTECHNOLOGY AND BIOENGINEERING, VOL. 41, NO. 5, MARCH 5, 1993

EXPERIMENTAL
Materials
Reversed micelles were made with the anionic surfac- tant bis(2-ethylhexyl) sodium sulfosuccinate (AOT), ob- tained from Aldrich Chemicals, Milwaukee, WI (>99% purity). The solvent, isooctane, was also purchased from Aldrich Chemicals, [high-performance liquid chromatog- raphy (HPLC) grade] and used as supplied. Peroxidase (type 11: from horseradish, molecular weight 40,000, activ- ity 200 units/mg), hydrogen peroxide (30%), and HEPES buffer were purchased from Sigma Chemical Company (St. Louis, MO). Acetonitrile used in the HPLC mobile phase was supplied by Baxter Chemicals (Muskegon, MI). Deionized and doubly distilled water was used in all preparations.
Methods
On the preparative scale, a typical reaction was carried out as follows. First 0.15 M AOT was dissolved in isooctane. As a representative starting reaction volume, we used 10 mL of this solution. To this solution we directly added HEPES buffer to a concentration of 1.69 M. We also made up a stock solution of 1.25 mM enzyme in HEPES buffer (assuming an enzyme molecular weight of 40,000 and full enzyme content in the material supplied by Sigma Chemical Company); an aliquot of this enzyme-in-buffer solution was added to the reaction mixture to a concentration of 0.56 M. The total amount of buffer added (2.25 M) results in a reversed micellar solution of wg (water/AOT molar ratio) of 15 and with final overall enzyme concentration of 12.5 p M . The substrate, p-ethylphenol, was then introduced at a concentration of 0.15 M. After dissolving the monomer, the reaction was initiated by the addition of 0.2 M of 30% H202, a concentration in excess of the substrate concentra- tion. Hydrogen peroxide is an inhibitor of peroxidase; we have determined that in the reversed micellar system, H202- to-enzyme molar ratios greater than 1.6 X lo4 completely inhibit enzyme activity. At the concentrations used, this ratio is 1.6 X lo4 and the H202 has minimal effect on enzyme activity. Nevertheless, we added H202 in three batches in fairly rapid succession to further minimize any inhibitory effects.
We have found that the reaction in reversed micelles is extremely rapid and occurs with the formation of a dark precipitate within a minute of reaction initiation. A faint yellow color observed in the supernatant is due to oligomers that remain suspended in solution. Accordingly, the precipitate was isolated from the supernatant, washed with water to remove any traces of enzyme if present, and washed repeatedly with isooctane to remove any residual surfactant. The final precipitate was dried free of the solvent and water at 60°C and stored for subsequent polymer characterization. The water content of the polymer as
measured by Karl-Fischer titration (Mettler DL-18) was less than 1% by weight.
To measure the kinetics of the reaction, it was nec- essary to drastically reduce enzyme and substrate con- centrations. For kinetic analysis, the concentrations of the reaction mixture constituents were as follows: 50 mM AOT, wo 15, 0.17 p M enzyme, 5 mM H202, and 10-50 mM p-ethylphenol. During the early stages of the reaction, a gradual color change in the medium was observed, with some precipitation seen after about 1 h. The observed reaction rates were calculated from the linear portion of the conversion-vs.-time plots. The extent of conversion and the reaction progress were monitored by HPLC analysis (Perkin-Elmer chromatograph with an LC 235 diode array detector and a model 250 LC binary pump) using two 3-cm long, 0.46-diameter C-18 reverse phase columns in series. The mobile phase was a 56/44 acetonitrile/water mixture used under isocratic conditions at a flow rate of 1.5 mL/min. The phenol concentration was monitored at 280 nm. Monomer elution time was about 0.95 min, and there was good separation between the monomer and soluble oligomers. Reaction mixture samples were diluted 40-fold with acetonitrile prior to injection. Finally, the polymer yields were calculated using the chromatographic data for monomer conversions and the final dry weights of the polymer.
Polymer molecular weight distributions were measured using gel permeation chromatography (Toyopearl TSK- GEL column). A 56-cm column was gravity packed using TSK-GEL Toyopearl packing (HW-55) from Supelco (Bellefonte, PA). The eluant was a 4 : 1 mixture of dimethyl formamide (DMF) and methanol (MeOH), and the column was operated under the eluant head pressure adjusted to maintain a flow rate of 0.35 mL/min. The dry polymer was dissolved in the 4/1 DMF/MeOH solvent at a concentration of 0.1% (w/v), and 1 mL of this solution was introduced into the column. One-milliliter fractions were collected automatically at equal intervals and analyzed on a Shimadzu UV-160 spectrometer at 270 nm. Polystyrene samples in the molecular weight range of 687-410,000 (Supelco) were used as calibration standards.
The reversed micellar system was characterized through quasi-elastic light scattering (QELS) techniques and through Fourier transform infrared spectroscopy (FTIR). The QELS system described previ~usly'~ consisted of a Coherent Innova 90-5 argon ion laser operating at a wavelength (A) of 488 nm, a Thorn EM1 head-on phototube for single photon counting, and a Brookhaven BI 2030 autocorrelator. All measurements were carried out at an observation angle (0) of 90", and autocorrelation decay curves were analyzed by standard cumulant methods." The light scattering data gives results for the equivalent hydrodynamic diameter DH, obtained from the Stokes-Einstein equation DH =
kt/3?rqD. The micelle diffusion coefficient D is (27q2)-', with 7 being the exponential decay time obtained from the autocorrelation decay curve and q = (4~n/A)sin(0/2) being the scattering vector. The index of refraction n and
533 RAO ET AL.: SYNTHESIS OF POLYMERS IN REVERSED MICELLES

the solution viscosity q were taken as 1.39 and 0.491 cP, respectively, for isooctane. The FTIR analysis was done using a Mattson 6021 spectrometer. The CaFz windows were used in a liquid sample cell (Spectra-Tech) with path length 0.3 mm.
RESULTS AND DISCUSSION
Our first observation is that the reaction is feasible in the reversed micellar system. When preparative concentrations are used, the reaction is extremely rapid, leading to the formation of a polymer precipitate. However, prior to quantitation of the reaction in reversed micelles, it is instructive to compare the reaction to reaction in other modes of substrate-enzyme contact. Table I lists qualitative results of polymerization in three other systems: (i) the monophasic organic solvent systems of dioxane + water, (ii) the monophasic organic solvent system of isooctane, and (iii) the aqueous solvent system. The distinction be- tween polymer and oligomer is simply that between the product that stays in solution and that of the insoluble product. There are clear distinctions between the different reaction media, and it is evident that the reversed micel- lar system and the dioxane + water system are the most favorable systems and therefore merit further study. The significant difference between polymerization in reversed micelles and in the monophasic isooctane system needs to be pointed out briefly. The high conversion levels and polymer yields in the reversed micellar case indicates that the addition of surfactant (and water) to isooctane has a significant effect on keeping the growing chain in solution
and in promoting continued polymerization. In addition to providing a high degree of dispersion to the enzyme, the surfactant does appear to interact with the monomer and with polymeric species, promoting the reaction. This is a theme that will be elaborated on in subsequent discussion.
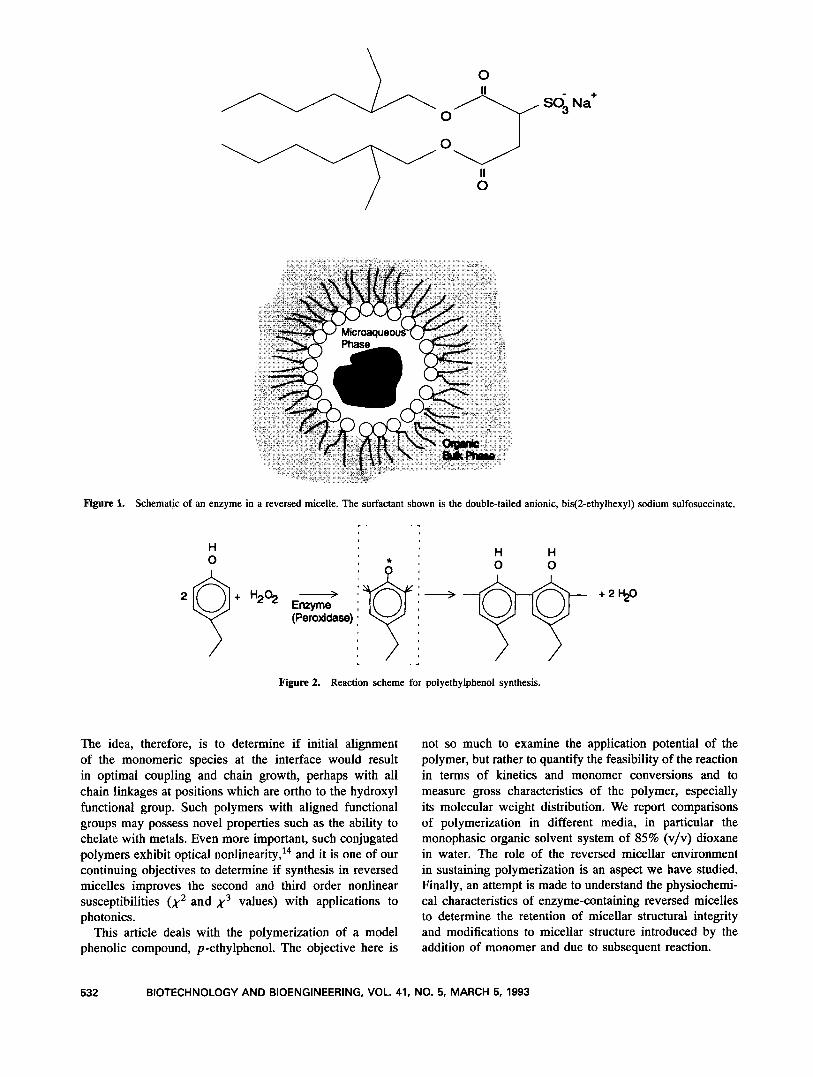
Based on the qualitative distinctions between the dif- ferent systems for polymerization, the reversed micellar system and the dioxane/water system are further charac- terized. As shown in Table I, the two systems differ in the oligomer-to-polymer ratio (soluble-to-insoluble-product ratio), although we emphasize that the two systems have different solvating abilities and hence may sustain the growing chain in solution to differing extents. Figures 3a and 3b illustrate the liquid chromatograms of the two systems at about 95% monomer conversion. The monomer peak elutes at 0.95 min, and the subsequent peaks are due to oligomeric species in solution. It is evident that the oligomeric products are much more predominant in the dioxide/water system than in the micellar case.
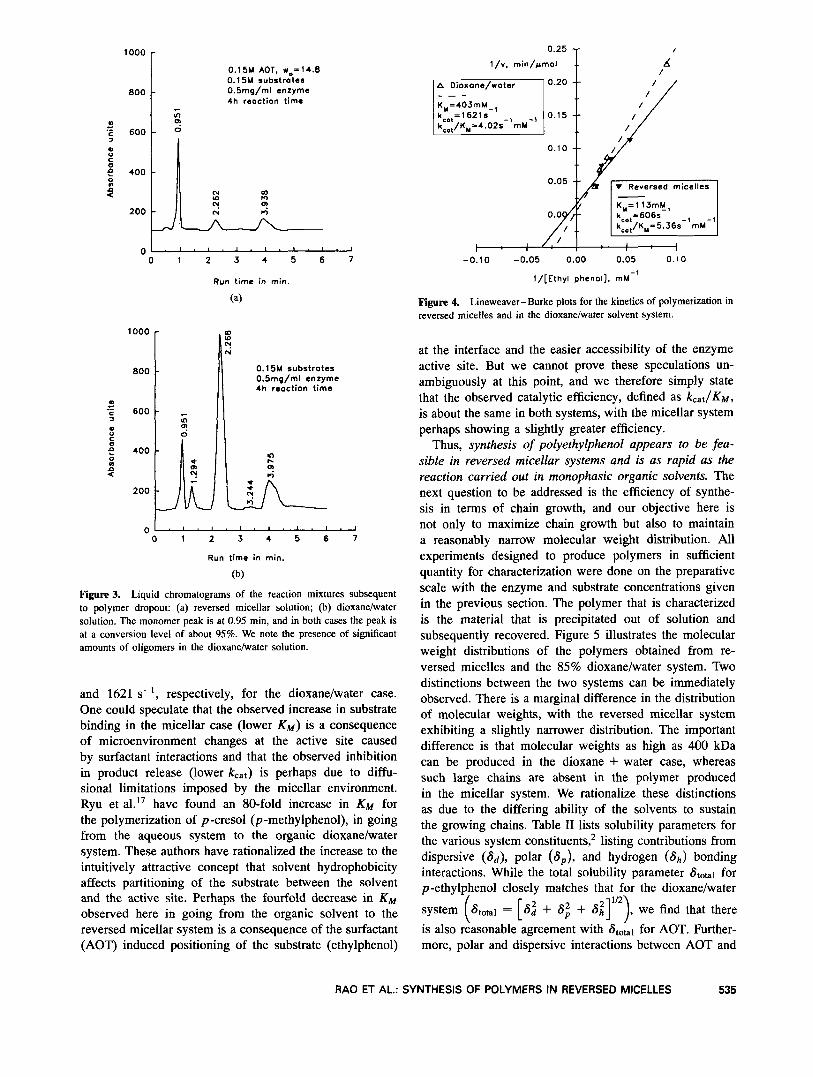
Further characterization of the two systems is based on kinetic studies of the relative rates of reaction using the species concentrations described in the preceding section. Lineweaver-Burke plots for the two systems are shown in Figure 4, where the conversions and reaction rates were monitored through liquid chromatography. Although this is a bisubstrate reaction, we have considered the reaction as pseudo-monosubstrate, keeping the H202 concentration fixed and varying the ethylphenol concentration, following the procedure of Ryu et a1.l’ From the plots, the calcu- lated values of KM and k,,, are 0.113 M and 606 s-l, respectively, for the reversed micellar case and 0.403 M
Table I. Qualitative aspects of polyethylphenol synthesized in various media.
Monomer Polymer yield, Reaction Conversion Polymer producedl medium (after 2 h) monomer converted Comments
-95% monomer soluble; 1. Reversed 295% micelles high enzyme dispersion wo = 15; (solution clear); [AOT] = 0.15 M minimal oligomer formation;
polymer precipitates
2. Isooctane
3. Dioxane (85%) in water
4. Water
-6%
95%
35%
95100%
=20%
155%
monomer soluble; poor enzyme dispersion (in insoluble aggregates); little oligomer formation; polymer at the air/isooctane interface
monomer soluble; fairly high enzyme dispersion (enzyme in suspension, cloudy solution); significant oligomer formation; polymer precipitates
poor monomer solubility; high enzyme dispersion (clearly soluble); some oligomer formation; polymer precipitates
534 BIOTECHNOLOGY AND BIOENGINEERING, VOL. 41, NO. 5, MARCH 5, 1993
1000 r
n
E 3
u 0 c 0
- .-
c n n I
n C c .- a a
n
n
0
O
0
<
L
800
600
400
200
0.15Y AOT. we-14.8 0.15M substrates 0.5mg/ml enzyme 4h reaction time -
ul t : m c) N 10
1000
800
600
400
200
L m (D
N 1,
0.15M substrates 0.5mg/ml enzyme 4h reaction time
m
v) t i- m 4 I 0.15M substrates
0.5mg/ml enzyme 4h reaction time
v) i-
4
0 1 2 3 4 5 6 7
Run time in min.
(b)
Figure 3. Liquid chromatograms of the reaction mixtures subsequent to polymer dropout: (a) reversed micellar solution; (b) dioxane/water solution. The monomer peak is at 0.95 min, and in both cases the peak is at a conversion level of about 95%. We note the presence of significant amounts of oligomers in the dioxane/water solution.
and 1621 s-l, respectively, for the dioxanelwater case. One could speculate that the observed increase in substrate binding in the micellar case (lower K M ) is a consequence of microenvironment changes at the active site caused by surfactant interactions and that the observed inhibition in product release (lower kcat) is perhaps due to diffu- sional limitations imposed by the micellar environment. Ryu et al." have found an 80-fold increase in KM for the polymerization of p -cresol ( p -methylphenol), in going from the aqueous system to the organic dioxane/water system. These authors have rationalized the increase to the intuitively attractive concept that solvent hydrophobicity affects partitioning of the substrate between the solvent and the active site. Perhaps the fourfold decrease in KM observed here in going from the organic solvent to the reversed micellar system is a consequence of the surfactant (AOT) induced positioning of the substrate (ethylphenol)
-0.10 -0.05 0.00 0.05 0.10
l/[Ethyl phenol], mM-'
Figure 4. Lineweaver-Burke plots for the kinetics of polymerization in reversed micelles and in the dioxane/water solvent system.
at the interface and the easier accessibility of the enzyme active site. But we cannot prove these speculations un- ambiguously at this point, and we therefore simply state that the observed catalytic efficiency, defined as k,at/KM, is about the same in both systems, with the micellar system perhaps showing a slightly greater efficiency.
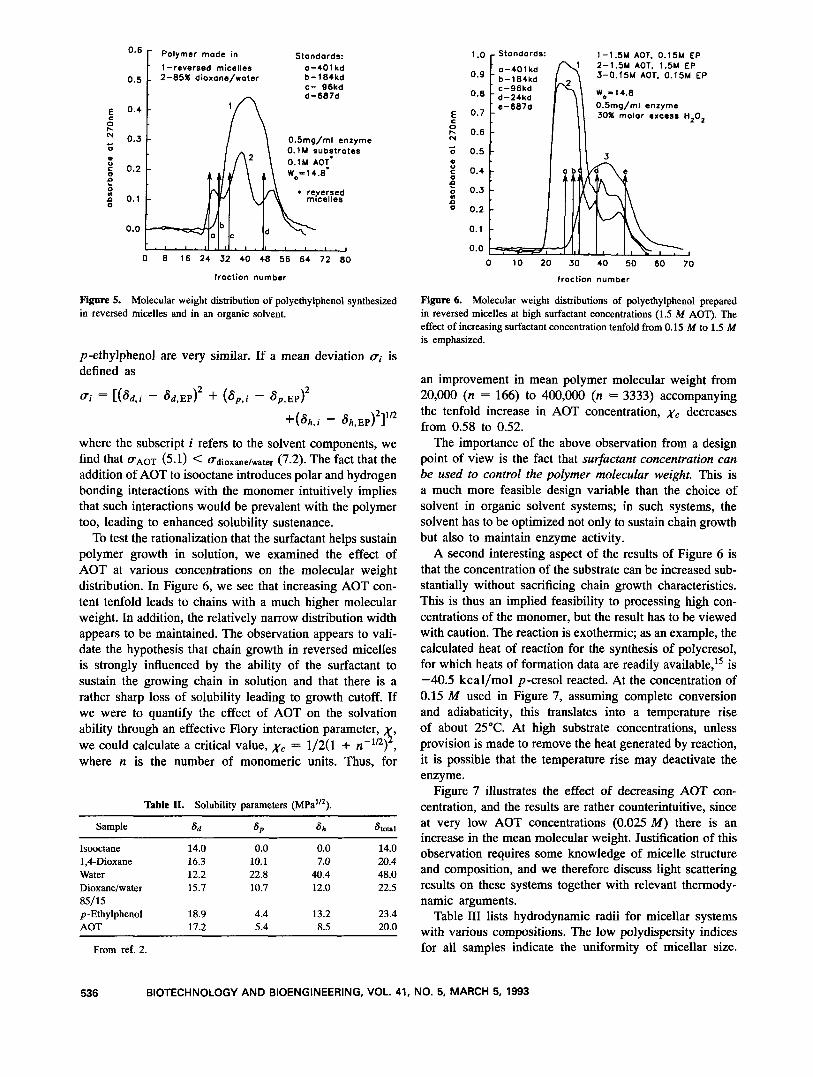
Thus, synthesis of polyethylphenol appears to be fea- sible in reversed micellar systems and is as rapid as the reaction carried out in monophasic organic solvents. The next question to be addressed is the efficiency of synthe- sis in terms of chain growth, and our objective here is not only to maximize chain growth but also to maintain a reasonably narrow molecular weight distribution. All experiments designed to produce polymers in sufficient quantity for characterization were done on the preparative scale with the enzyme and substrate concentrations given in the previous section. The polymer that is characterized is the material that is precipitated out of solution and subsequently recovered. Figure 5 illustrates the molecular weight distributions of the polymers obtained from re- versed micelles and the 85% dioxanelwater system. Two distinctions between the two systems can be immediately observed. There is a marginal difference in the distribution of molecular weights, with the reversed micellar system exhibiting a slightly narrower distribution. The important difference is that molecular weights as high as 400 kDa can be produced in the dioxane + water case, whereas such large chains are absent in the polymer produced in the micellar system. We rationalize these distinctions as due to the differing ability of the solvents to sustain the growing chains. Table I1 lists solubility parameters for the various system constituents? listing contributions from dispersive (ad), polar (ap ) , and hydrogen (ah) bonding interactions. While the total solubility parameter Stotal for p -ethylphenol closely matches that for the dioxanelwater
system (atota1 = [a: + 8; + &]ln), we find that there is also reasonable agreement with Gtotal for AOT. Further- more, polar and dispersive interactions between AOT and
RAO ET AL.: SYNTHESIS OF POLYMERS IN REVERSED MICELLES 535
Polymer mode in
1 -reversed micelles 2-85% dioxone/watcr
a C O
f n n 0
0.2
0.1
0.0
Standards: a -40 lkd b- 184kd C- 96kd d-687d
O.Smg/ml enzyme 0.1M subftrotes O . l M AOT W0=14.8'
0 8 16 2 4 32 4 0 48 5 6 64 72 80
fraction number
Figure 5. Molecular weight distribution of polyethylphenol synthesized in reversed micelles and in an organic solvent.
p-ethylphenol are very similar. If a mean deviation ui is defined as
gi = L(6d.i - 6d,EP)2 + ( 6 p . i - 6p.EP)'
+(Sh,i - s ~ , E P ) ~ I ' "
where the subscript i refers to the solvent components, we find that UAOT (5.1) < gdioxane/mtm (7.2). The fact that the addition of AOT to isooctane introduces polar and hydrogen bonding interactions with the monomer intuitively implies that such interactions would be prevalent with the polymer too, leading to enhanced solubility sustenance.
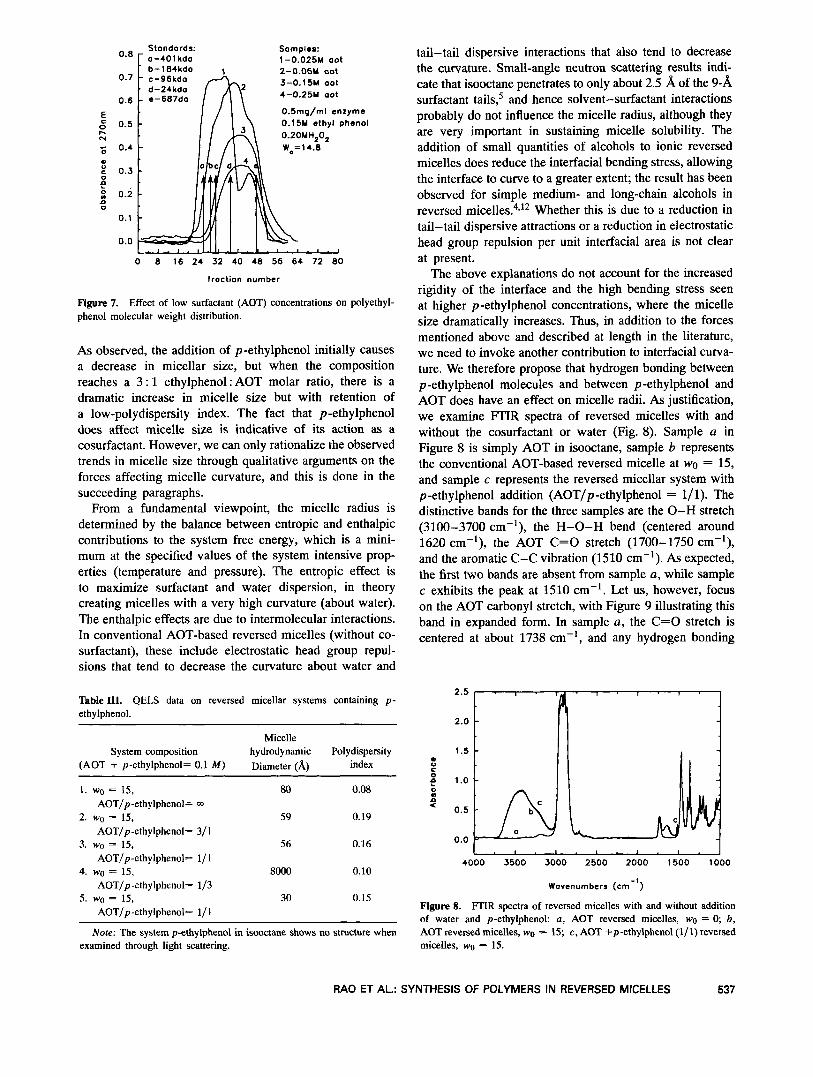
To test the rationalization that the surfactant helps sustain polymer growth in solution, we examined the effect of AOT at various concentrations on the molecular weight distribution. In Figure 6, we see that increasing AOT con- tent tenfold leads to chains with a much higher molecular weight. In addition, the relatively narrow distribution width appears to be maintained. The observation appears to vali- date the hypothesis that chain growth in reversed micelles is strongly influenced by the ability of the surfactant to sustain the growing chain in solution and that there is a rather sharp loss of solubility leading to growth cutoff. If we were to quantify the effect of AOT on the solvation ability through an effective Flory interaction parameter, we could calculate a critical value, xC = 1/2( 1 + n-ll2) , where n is the number of monomeric units. Thus, for
r7
Table II. Solubility parameters (MPa'").
Sample s d SP s h total
Isooctane 14.0 0.0 0.0 14.0 1,4-Dioxane 16.3 10.1 7.0 20.4 Water 12.2 22.8 40.4 48.0 Dioxane/water 15.7 10.7 12.0 22.5 85/15 p-Ethylphenol 18.9 4.4 13.2 23.4 AOT 17.2 5.4 8.5 20.0
From ref. 2.
1 .o Standards:
0-401 kd b-184kd 0.9 1
0.6
0.5
1-1.5M AOT. 0.15M EP 2-1.5M AOT, 1.5M EP 3-0.15M AOT. 0.15M EP
Wo= 14.8 0.5mg/ml enzyme 30% molar excess H202
3
0 10 20 30 4 0 50 6 0 70
fraction number
Figure 6. Molecular weight distributions of polyethylphenol prepared in reversed micelles at high surfactant concentrations (1.5 M AOT). The effect of increasing surfactant concentration tenfold from 0.15 M to 1.5 M is emphasized.
an improvement in mean polymer molecular weight from 20,000 (n = 166) to 400,000 (n = 3333) accompanying the tenfold increase in AOT concentration, xc decreases from 0.58 to 0.52.
The importance of the above observation from a design point of view is the fact that surfactant Concentration can be used to control the polymer molecular weight. This is a much more feasible design variable than the choice of solvent in organic solvent systems; in such systems, the solvent has to be optimized not only to sustain chain growth but also to maintain enzyme activity.
A second interesting aspect of the results of Figure 6 is that the concentration of the substrate can be increased sub- stantially without sacrificing chain growth characteristics. This is thus an implied feasibility to processing high con- centrations of the monomer, but the result has to be viewed with caution. The reaction is exothermic; as an example, the calculated heat of reaction for the synthesis of polycresol, for which heats of formation data are readily a~ailable,'~ is -40.5 kcal/mol p-cresol reacted. At the concentration of 0.15 M used in Figure 7, assuming complete conversion and adiabaticity, this translates into a temperature rise of about 25°C. At high substrate concentrations, unless provision is made to remove the heat generated by reaction, it is possible that the temperature rise may deactivate the enzyme.
Figure 7 illustrates the effect of decreasing AOT con- centration, and the results are rather counterintuitive, since at very low AOT concentrations (0.025 M) there is an increase in the mean molecular weight. Justification of this observation requires some knowledge of micelle structure and composition, and we therefore discuss light scattering results on these systems together with relevant thermody- namic arguments.
Table 111 lists hydrodynamic radii for micellar systems with various compositions. The low polydispersity indices for all samples indicate the uniformity of micellar size.
536 BIOTECHNOLOGY AND BIOENGINEERING, VOL. 41, NO. 5, MARCH 5, 1993
Standards: Samples: o-8 r a-40lkdo 1-0.025M aot
b-184kdO 2 - 0 . 0 5 ~ O O ~ 0.7 1 c-96kdo h2 3-0.15M oat
4-0.25Y aot d-24kda 0.6 e-687da
0.5
;; 0.4
c" 0.3
e
I- cy
u
:: 0.2 n 0
0.1
0.0
0.5mg/ml enzyme 0.15Y ethyl phenol
0 8 16 24 32 40 48 56 64 72 80
froction number
Figure 7. phenol molecular weight distribution.
Effect of low surfactant (AOT) concentrations on polyethyl-
As observed, the addition of p-ethylphenol initially causes a decrease in micellar size, but when the composition reaches a 3 : 1 ethylpheno1:AOT molar ratio, there is a dramatic increase in micelle size but with retention of a low-polydispersity index. The fact that p-ethylphenol does affect micelle size is indicative of its action as a cosurfactant. However, we can only rationalize the observed trends in micelle size through qualitative arguments on the forces affecting micelle curvature, and this is done in the succeeding paragraphs.
From a fundamental viewpoint, the micelle radius is determined by the balance between entropic and enthalpic contributions to the system free energy, which is a mini- mum at the specified values of the system intensive prop- erties (temperature and pressure). The entropic effect is to maximize surfactant and water dispersion, in theory creating micelles with a very high curvature (about water). The enthalpic effects are due to intermolecular interactions. In conventional AOT-based reversed micelles (without co- surfactant), these include electrostatic head group repul- sions that tend to decrease the curvature about water and
Table 111. QELS data on reversed micellar systems containing p - ethylphenol.
Micelle System composition hydrodynamic Polydispersity
(AOT + p-ethylphenol= 0.1 M) Diameter (A) index ~~ ~
1. wo = 15, 80 0.08 AOT/p-ethylphenol= m
AOT/p-ethylphenol= 3/1
AOT/p-ethylphenol= 1/1
AOT/p-ethylphenol= 1/3
AOT/p-ethylphenol= 1/1
2. WO = 15, 59 0.19
3. wo = 15, 56 0.16
4. wo = 15, 8000 0.10
5. wo = 15, 30 0.15
Note: The system p-ethylphenol in isooctane shows no structure when examined through light scattering.
tail-tail dispersive interactions that also tend to decrease the curvature. Small-angle neutron scattering results indi- cate that isooctane penetrates to only about 2.5 A of the 9-A surfactant tails: and hence solvent-surfactant interactions probably do not influence the micelle radius, although they are very important in sustaining micelle solubility. The addition of small quantities of alcohols to ionic reversed micelles does reduce the interfacial bending stress, allowing the interface to curve to a greater extent; the result has been observed for simple medium- and long-chain alcohols in reversed micel le~.~, '~ Whether this is due to a reduction in tail-tail dispersive attractions or a reduction in electrostatic head group repulsion per unit interfacial area is not clear at present.
The above explanations do not account for the increased rigidity of the interface and the high bending stress seen at higher p-ethylphenol concentrations, where the micelle size dramatically increases. Thus, in addition to the forces mentioned above and described at length in the literature, we need to invoke another contribution to interfacial curva- ture. We therefore propose that hydrogen bonding between p-ethylphenol molecules and between p -ethylphenol and AOT does have an effect on micelle radii. As justification, we examine FTIR spectra of reversed micelles with and without the cosurfactant or water (Fig. 8). Sample a in Figure 8 is simply AOT in isooctane, sample b represents the conventional AOT-based reversed micelle at wo = 15, and sample c represents the reversed micellar system with p-ethylphenol addition (AOT/p-ethylphenol = 1/1). The distinctive bands for the three samples are the 0-H stretch (3100-3700 cm-'), the H-0-H bend (centered around 1620 cm-'), the AOT C=O stretch (1700-1750 cm-'), and the aromatic C-C vibration (1510 cm-'). As expected, the first two bands are absent from sample a, while sample c exhibits the peak at 1510 cm-'. Let us, however, focus on the AOT carbonyl stretch, with Figure 9 illustrating this band in expanded form. In sample a, the C=O stretch is centered at about 1738 cm-', and any hydrogen bonding
2.5
2.0
1.5
1 .o
0.5
0.0
4000 3500 3000 2500 2000 1500 1000
Wovenumbera (cm-')
Figure 8. FTIR spectra of reversed micelles with and without addition of water and p-ethylphenol: a, AOT reversed micelles, wo = 0; b, AOT reversed micelles, wo = 15; c, AOT +p-ethylphenol (1/1) reversed micelles, wo = 15.
RAO ET AL.: SYNTHESIS OF POLYMERS IN REVERSED MICELLES 537
0.4 0.5 f-----7
1755 1740 1725 1710 1695 1680
Wovenumbers (cm-')
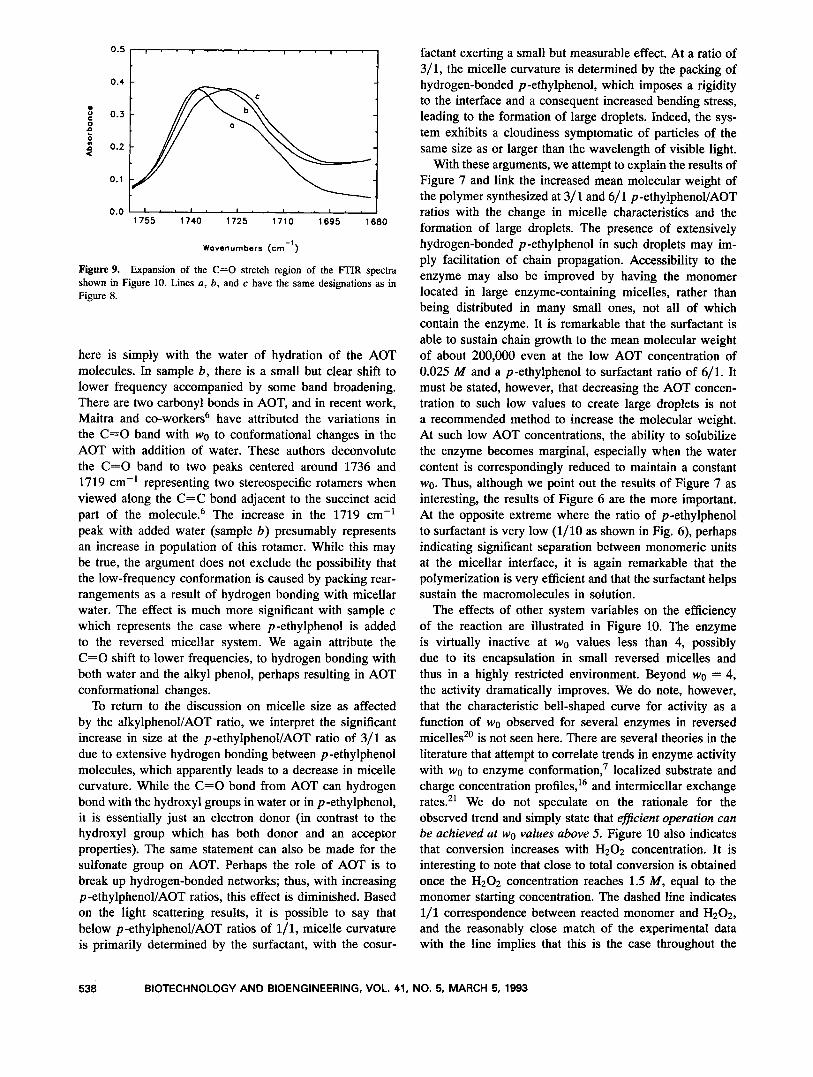
Figure 9. Expansion of the C=O stretch region of the ITIR spectra shown in Figure 10. Lines a, b, and c have the same designations as in Figure 8.
here is simply with the water of hydration of the AOT molecules. In sample b, there is a small but clear shift to lower frequency accompanied by some band broadening. There are two carbonyl bonds in AOT, and in recent work, Maitra and co-worker&' have attributed the variations in the C=O band with wo to conformational changes in the AOT with addition of water. These authors deconvolute the C=O band to two peaks centered around 1736 and 1719 cm-' representing two stereospecific rotamers when viewed along the C=C bond adjacent to the succinct acid part of the molecule.6 The increase in the 1719 cm-' peak with added water (sample b) presumably represents an increase in population of this rotamer. While this may be true, the argument does not exclude the possibility that the low-frequency conformation is caused by packing rear- rangements as a result of hydrogen bonding with micellar water. The effect is much more significant with sample c which represents the case where p-ethylphenol is added to the reversed micellar system. We again attribute the C=O shift to lower frequencies, to hydrogen bonding with both water and the alkyl phenol, perhaps resulting in AOT conformational changes.
To return to the discussion on micelle size as affected by the alkylphenol/AOT ratio, we interpret the significant increase in size at the p-ethylphenollAOT ratio of 3/1 as due to extensive hydrogen bonding between p-ethylphenol molecules, which apparently leads to a decrease in micelle curvature. While the C=O bond from AOT can hydrogen bond with the hydroxyl groups in water or in p-ethylphenol, it is essentially just an electron donor (in contrast to the hydroxyl group which has both donor and an acceptor properties). The same statement can also be made for the sulfonate group on AOT. Perhaps the role of AOT is to break up hydrogen-bonded networks; thus, with increasing p -ethylphenol/AOT ratios, this effect is diminished. Based on the light scattering results, it is possible to say that below p -ethylphenol/AOT ratios of 1/1, micelle curvature is primarily determined by the surfactant, with the cosur-
factant exerting a small but measurable effect. At a ratio of 3/1, the micelle curvature is determined by the packing of hydrogen-bonded p-ethylphenol, which imposes a rigidity to the interface and a consequent increased bending stress, leading to the formation of large droplets. Indeed, the sys- tem exhibits a cloudiness symptomatic of particles of the same size as or larger than the wavelength of visible light.
With these arguments, we attempt to explain the results of Figure 7 and link the increased mean molecular weight of the polymer synthesized at 3/1 and 6/1 p-ethylphenoVAOT ratios with the change in micelle characteristics and the formation of large droplets. The presence of extensively hydrogen-bonded p -ethylphenol in such droplets may im- ply facilitation of chain propagation. Accessibility to the enzyme may also be improved by having the monomer located in large enzyme-containing micelles, rather than being distributed in many small ones, not all of which contain the enzyme. It is remarkable that the surfactant is able to sustain chain growth to the mean molecular weight of about 200,000 even at the low AOT concentration of 0.025 M and a p-ethylphenol to surfactant ratio of 6/1. It must be stated, however, that decreasing the AOT concen- tration to such low values to create large droplets is not a recommended method to increase the molecular weight. At such low AOT concentrations, the ability to solubilize the enzyme becomes marginal, especially when the water content is correspondingly reduced to maintain a constant WO. Thus, although we point out the results of Figure 7 as interesting, the results of Figure 6 are the more important. At the opposite extreme where the ratio of p-ethylphenol to surfactant is very low (1/10 as shown in Fig. 6), perhaps indicating significant separation between monomeric units at the micellar interface, it is again remarkable that the polymerization is very efficient and that the surfactant helps sustain the macromolecules in solution.
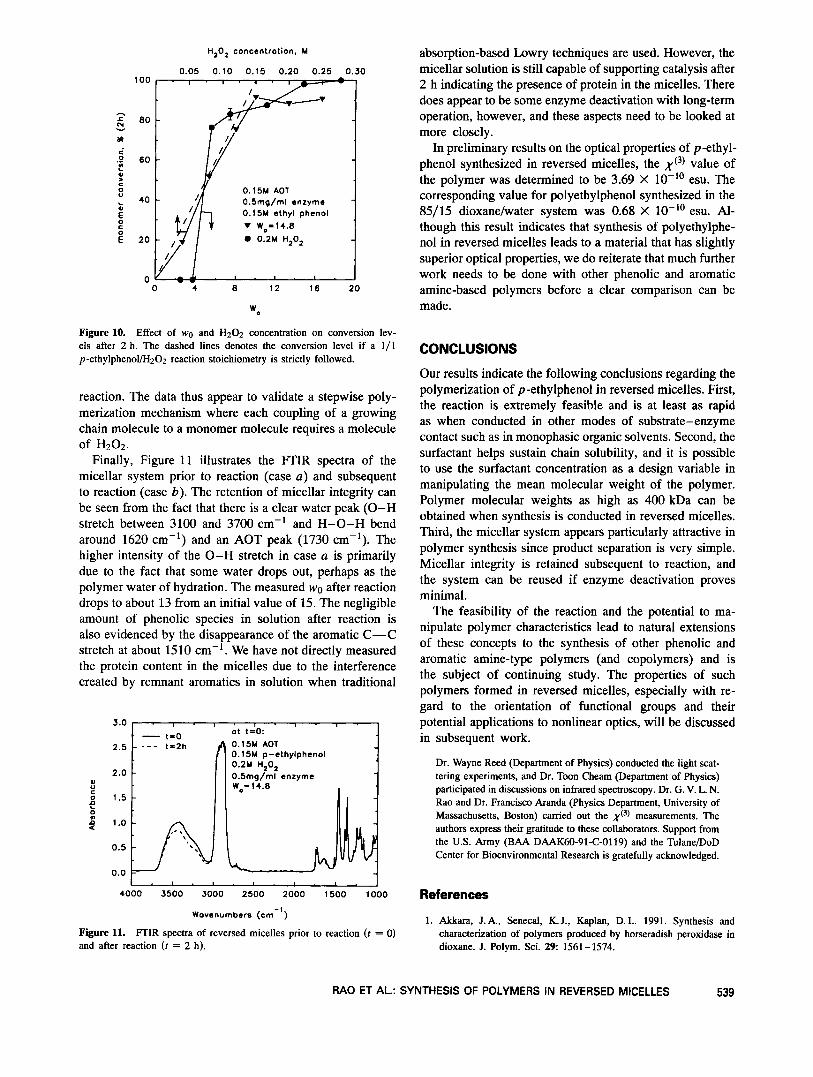
The effects of other system variables on the efficiency of the reaction are illustrated in Figure 10. The enzyme is virtually inactive at wo values less than 4, possibly due to its encapsulation in small reversed micelles and thus in a highly restricted environment. Beyond wo = 4, the activity dramatically improves. We do note, however, that the characteristic bell-shaped curve for activity as a function of wo observed for several enzymes in reversed micelles2' is not seen here. There are several theories in the literature that attempt to correlate trends in enzyme activity with wo to enzyme c~nformation,~ localized substrate and charge concentration profiles,16 and intermicellar exchange rates.21 We do not speculate on the rationale for the observed trend and simply state that eficient operation can be achieved at wo values above 5. Figure 10 also indicates that conversion increases with H202 concentration. It is interesting to note that close to total conversion is obtained once the H202 concentration reaches 1.5 M, equal to the monomer starting concentration. The dashed line indicates 1/1 correspondence between reacted monomer and H202, and the reasonably close match of the experimental data with the line implies that this is the case throughout the
538 BIOTECHNOLOGY AND BIOENGINEERING, VOL. 41, NO. 5, MARCH 5, 1993
HzOZ concentration. M
, ' , . , . , . I . at t=o:
.-- t=2h 0.15M AOT 0.1% p-ethylphenol 0.2M H,O, 0.5mg/ml enzyme Wo= 14.8
t -0 . -
0.05 0.10 0.15 0.20 0.25 0.30
& 80 v
6p
0.05 0.10 0.15 0.20 0.25 0.30 loor . 9 . , . , 9 ,
i c .- > C
0 0
u L
; E
60 -
4 0 -
20 -
0.15M AOT 0.5mg/ml enzyme 0.1% ethyl phenol
0 O.2M H,O,
v W0=14.8
0 I . , . , . - - 0 4 8 12 16 20
wo
Figure 10. Effect of wo and H202 concentration on conversion lev- els after 2 h. The dashed lines denotes the conversion level if a 1/1 p-ethylphenollH202 reaction stoichiometry is strictly followed.
reaction. The data thus appear to validate a stepwise poly- merization mechanism where each coupling of a growing chain molecule to a monomer molecule requires a molecule
Finally, Figure 11 illustrates the FTIR spectra of the micellar system prior to reaction (case a ) and subsequent to reaction (case b). The retention of micellar integrity can be seen from the fact that there is a clear water peak (0-H stretch between 3100 and 3700 cm-' and H-0-H bend around 1620 cm-') and an AOT peak (1730 cm-'). The higher intensity of the 0-H stretch in case a is primarily due to the fact that some water drops out, perhaps as the polymer water of hydration. The measured wo after reaction drops to about 13 from an initial value of 15. The negligible amount of phenolic species in solution after reaction is also evidenced by the disappearance of the aromatic C-C stretch at about 1510 cm-'. We have not directly measured the protein content in the micelles due to the interference created by remnant aromatics in solution when traditional
of H202.
u
C 0 ; n <
3.0
2.5
2.0
1.5
1 .o
0.5
0.0 l . ~ . I . l . I . 8 . I
4 0 0 0 3500 3000 2500 2000 1500 1000
Wovenumberr (cm-')
Figure 11. and after reaction (t = 2 h).
FTIR spectra of reversed micelles prior to reaction (t = 0)
absorption-based Lowry techniques are used. However, the micellar solution is still capable of supporting catalysis after 2 h indicating the presence of protein in the micelles. There does appear to be some enzyme deactivation with long-term operation, however, and these aspects need to be looked at more closely.
In preliminary results on the optical properties of p-ethyl- phenol synthesized in reversed micelles, the ,y(3) value of the polymer was determined to be 3.69 X lo-'* esu. The corresponding value for polyethylphenol synthesized in the 85/15 dioxane/water system was 0.68 X lo-'' esu. Al- though this result indicates that synthesis of polyethylphe- no1 in reversed micelles leads to a material that has slightly superior optical properties, we do reiterate that much further work needs to be done with other phenolic and aromatic amine-based polymers before a clear comparison can be made.
CONCLUSIONS
Our results indicate the following conclusions regarding the polymerization of p-ethylphenol in reversed micelles. First, the reaction is extremely feasible and is at least as rapid as when conducted in other modes of substrate-enzyme contact such as in monophasic organic solvents. Second, the surfactant helps sustain chain solubility, and it is possible to use the surfactant concentration as a design variable in manipulating the mean molecular weight of the polymer. Polymer molecular weights as high as 400 kDa can be obtained when synthesis is conducted in reversed micelles. Third, the micellar system appears particularly attractive in polymer synthesis since product separation is very simple. Micellar integrity is retained subsequent to reaction, and the system can be reused if enzyme deactivation proves minimal.
The feasibility of the reaction and the potential to ma- nipulate polymer characteristics lead to natural extensions of these concepts to the synthesis of other phenolic and aromatic amine-type polymers (and copolymers) and is the subject of continuing study. The properties of such polymers formed in reversed micelles, especially with re- gard to the orientation of functional groups and their potential applications to nonlinear optics, will be discussed in subsequent work.
Dr. Wayne Reed (Department of Physics) conducted the light scat- tering experiments, and Dr. Toon Cheam (Department of Physics) participated in discussions on infrared spectroscopy. Dr. G . V. L. N. Rao and Dr. Francisco Aranda (Physics Department, University of Massachusetts, Boston) carried out the ,yf3) measurements. The authors express their gratitude to these collaborators. Support from the US. Army (BAA DAAK60-91-C-0119) and the Tulane/DoD Center for Bioenvironmental Research is gratefully acknowledged.
References
1. Akkara, J.A., Senecal, K.J., Kaplan, D.L. 1991. Synthesis and characterization of polymers produced by horseradish peroxidase in dioxane. J. Polym. Sci. 2 9 1561-1574.
RAO ET AL.: SYNTHESIS OF POLYMERS IN REVERSED MICELLES 539

2. Barton, A.F.M. 1983. CRC handbook of solubility parameters and other cohesion parameters. CRC Press, Boca Raton, FL.
3. Dordick, N., Marletta, M. A., Klibanov, A.M. 1987. Polymerization of phenols catalyzed by peroxidase in nonaqueous media. Biotechnol. Bioeng. 30: 31-36.
4. Hou, M.-J., Shah, D. 0. 1987. Effects of the molecular structure of the interface and continuous phase on solubilization of water in watedoil microemulsions. Langmuir 3: 1086-1096.
5. Huang, J. S., Safran, S. A., Kim, M. W., Grest, G. S., Kotlarchyk, M., Quirke, N. 1984. Attractive interactions in micelles and microemul- sions. Phys. Rev. Lett. 53: 592-594.
6. Jain, T.K., Varshney, M., Maitra, A. 1989. Structural studies of aerosol OT reverse micellar aggregates by FT-IR spectroscopy. J. Phys. Chem. 93: 7409-7416.
7. Kahanov, A.V., Levashov, A.V., Klyachko, N.L., Pshezhetesky, A. V., Martinek, K. 1988. Enzymes entrapped in reversed micelles of surfactants in organic solvents: A theoretical treatment of the catalytic activity regulation. J. Theor. Biol. 133: 327-343.
8. Khmelnitsky, Yu.L., Kabanov, A.V., Klyachko, N.L., Levashov, A. V., Martinek, K. 1989. Enzymatic catalysis in reversed micelles. pp. 230-261 In: M. P. Pileni (ed.), Structure and reactivity in reversed micelles. Elsevier, New York.
9. Klibanov, A.M. 1986. Enzymes that work in organic solvents. Chemtech. 16: 354-359.
10. Kopf, P.W. 1985. Phenolic resins. In: Encyclopedia of polymer science and engineering, vol. 11. John Wiley & Sons, New York.
11. Koppel, D. E. 1972. Analysis of macromolecular polydispersity in intensity correlation spectroscopy: The methods of cumulants. J. Chem. Phys. 57: 4814-4820.
12. Lang, J., Lalem, N., Zana, R. 1991. Quaternary water in oil mi- croemulsions. l. Effect of alcohol chain length and concentration on droplet size and exchange of material between droplets. J. Phys. Chem. 95: 9533-9541.
13. Nguyen, H., John, V.T., Reed, W.F. 1991. Characteristics of protein- containing reversed micelles subjected to clathrate hydrate formation conditions. J. Phys. Chem. 95: 1467-1471.
14. Prasad, P. N., Williams, D. J. 1991. Introduction to nonlinear optical effects in molecules and polymers. John Wiley & Sons, New York.
15. Reid, R. C., Prausnitz, J. M., Sherwood, T. K. 1977. The properties of gases and liquids. McGraw-Hill, New York.
16. Ruckenstein, E., Karpe, P. 1990. On the enzymatic superactivity in ionic reverse micelles. J. COIL Inted Sci. 139: 408-436.
17. Ryu, K., Stafford, D.R., Dordick, J.S. 1989. Peroxidase catalyzed polymerization of phenols: Kinetics of p-cresol oxidation in organic media. ACS Symp. Ser. 389: 141-157.
18. Saunders, B. C., Holmes-Siedle, A. G., Stark, B. P. 1964. Peroxidase. Butterworth Publishers, London.
19. Schwartz, R. D., Hutchinson, D. B. 1981. Microbial and enzymic pro- duction of 4,4’-dihydroxybiphenyl via phenol coupling. Enz. Microb. Technol. 3: 361-364.
20. Shield, J. W., Ferguson, H. D., Bommarius, A. S., Hatton, T. A. 1986. Enzymes in reversed micelles as catalysts for organic phase synthesis reactions. Ind. Eng. Chem. Fund. 25: 603-612.
21. Verhaert, R. M. D., Hilhorst, R., Vermue, M., Schaafsma, T. J., Veeger, C. 1990. Description of enzyme kinetics in reversed micelles. Eur. J. Biochem. 187: 59-72.
22. Westerfield, W.W., Lowe, C. 1942. The oxidation of p-cresol by peroxidase. J. Biol. Chem. 145: 463-470.
540 BIOTECHNOLOGY AND BIOENGINEERING, VOL. 41, NO. 5, MARCH 5, 1993




















