
A Chemical Theory for Ion Distribution Equilibria in Reverse Micellar Systems. New Experimental Data...
8
1162 Langmuir 1995,11, 1162-1169 A Chemical Theory for Ion Distribution Equilibria in Reverse Micellar Systems. New Experimental Data for Aerosol-OT-Isooctane-Water-Salt Systems Hamid R. Rabie and Juan H. Vera* Department of Chemical Engineering, McGill University, Montreal, Quebec, Canada H3A 2A7 Received October 31, 1994. In Final Form: January 12, 1995@ Aerosol-OT and some other surfactant molecules form reverse micelles in an organic phase in contact with an aqueous salt solution. Experimental data of the distribution of cations in Aerosol-OT-isooctane- water-salt systems have been measured at 23 "C for different chloride salts. The effect of different variables on the equilibriumion distribution between the organic phase, containingreverse micelles, and an aqueous phase, containingdifferentsalts, has been formulated in terms of dimensionless groups. These variables are (i) the initial surfactant concentration, (ii) the nature and initial salt concentrationof each salt, and (iii) the initial volume ratio of the two phases. The model distinguishes between different ions via the equilibriumconstants of their ion exchange reactions with the surfactant counterion. It accurately predicts the distribution of single charge and multicharge ions between the two phases of the system. A model formulated in dimensionless form, with few independent dimensionless groups rather than many independent variables, has definite advantages for the treatment of complex systems. In this work, the predictions of the model are compared with experimental results and with the predictions of other phenomenological models published in the literature for reverse micellar systems. Introduction Reverse micelles have attracted much interest over the past few decades because of their extensive technological and biological importance for oil recovery, lubrication, detergency, catalysis, metal and biomolecule extraction, or use for probes in immunological processes and drug de1ivery.l Electrolytes are important components in the two-phase systems under consideration. It has been long established that a high salt concentration is a prerequisite for the formation of a Winsor I1 ~ ystem,~,~ which is a water- in-oil (W/O)microemulsion in equilibrium with an excess aqueous phase. An understanding of the behavior of the system with different electrolytes, coupled with a knowl- edge of the ion distribution in the two-phase system, is desirable if the solubilization characteristics of solutes (metals, biomolecules,etc.) are to be interpreted correctly. The effect of electrolytes on reverse micelles has been studied by titration experiments which give a different perspective than that of the present study. The effects of temperature, salt type, and concentration on the maximum water uptake by the surfactant Aerosol-OT (AOT)reverse micellar phases before any excess aqueous phase is formed, was investigated by Kon-No and Kitahara.4-8 A similar study was undertaken by Chou and Shah.g In more recent years, Kunieda and Shinodalo-l2 and Gosh and Miller13 focused on the influence of salinity on the phase behavior of AOT/water/oil systems. Tosch et al.,14 Fletcher,15and Aveyard et a1.16 have measured the distribution of sodium * To whom correspondence should be addressed. @ Abstract published inAduameACSAbstracts, March 15,1995. (1) Luzar, A,; Bratko, D. J. Chem. Phys. 1990,92, 642. (2) Akoum, F.; Parodi, 0. J. Phys. (Les Uis, Fr.) 1986, 46, 1675. (3) Lampert, A.; Martinelli, R. U. Chem. Phys. 1984, 88, 399. (4) Kitahara, A.; Watanabe, K.; Kon-No, K.; Ishikawa, T. J. Colloid (5) Kon-No, K.; Kitahara, A. J. Colloid Interface Sci. 1971,35,403. (6) Kon-No, K.; Kitahara, A. J. Colloid Interface Sci. 1971,35,636. (7) Kon-No, K.; Kitahara, A. J. Colloid Interface Sci. 1971,37,469. (8) Kon-No, K.; Kitahara, A. J. Colloid Interface Sci. 1972,41, 47. (9) Chou, S. I.; Shah, D. 0. J. Colloid Interface Sci. 1981, 80, 311. (10) Kunieda, H.; Shinoda, K. J. Colloid Interface Sci. 1979,70,577. (11) Kunieda, H.; Shinoda, K. J. ColloidInterface Sci. 1980,75,601. (12) Kunieda, H.; Shinoda, K. J. Colloid Interface Sci. 1987, 118, (13) Ghosh, 0.; Miller, C. A. J. Phys. Chem. 1987,91, 4528. Interface Sci. 1969, 29, 48. 586. salts between an aqueous electrolyte solution and a W/O microemulsion in equilibrium. They also measured the water uptake by the reverse micelles and reported the AOT distribution between the two phases, the size of the reverse micelles, and the values of the interfacial tension. Studies more relevant to this work have been presented by Leodidis and Hatton," Vijayalakshmi et al.,18J9 and Plucinski et a1.20 A phenomenological model for the selective solubilization of cations in the reverse micelles ofAOT surfactant was presented by Leodidis and Hatton.l' Their primary goal was to model the large differences in water uptake and the ion distribution. They used the modified Poisson-Boltzmann theory with the assumption that the chemical potential for the ions can be expressed as a linear combination of a series of different interaction terms, while neglecting the ion-ion terms. Their model distinguished the different cations through their charge, hydrated size, and electrostatic free energy of hydration. Vijayalakshmi et a1.18 and Vijayalakshmi and Gularilg proposed a model which assumed that the adsorption of counterions onto the surfactant surface of the reverse micelle is described by the Stern double layer model. Their model is simpler than the Leodidis-Hatton model, but it cannot distinguish between different ions with the same charge. Plucinski et a1.20 studied the specific ion effect of the ion exchange in the AOT reverse micellar system in equilibrium as well as in kinetic experiments. They used the Graham model of metal ion adsorptionz1 to express the adsorption ofmetal ions in the inner Helmholtz plane. We present here a theoretical model in which the specific character of each exchangeable counterion (hydrated size, free energy of hydration and electronic properties) is (14) Tosch, W. C.; Jones, S. C.; Adamson, A. W. J. Colloid Interface (15)Fletcher, P. D. I. J. Chem. Soc., Faraday Trans. 1 1986, 82, (16)Aveyard, R.; Binks, B. P.; Clark, S.; Mead, J. J. Chem. Soc., (17) Leodidis, E. B.; Hatton, T. A. Langmuir 1989, 5, 741. (18) Vijayalakshmi, C. S.; Annapragada, A. V.; Gulari, E. Sep. Sci. (19) Vijayalakshmi, C. S.; Gulari, E. Sep. Sci. Technol. 1991,26,291. (20) Plucinski, P.; Nitsch, W. Langmuir 1994, 10, 371. (21) Hunter, R. J. Foundations ofcolloid Science; Clarendon Press: Sci. 1989, 31, 297. 2651. Faraahy Trans. 1 1986,82, 125. Technol. 1990,25, 711. Oxford, 1989. 0743-7463/95/2411-1162$09.00/0 0 1995 American Chemical Society
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of A Chemical Theory for Ion Distribution Equilibria in Reverse Micellar Systems. New Experimental Data...

1162 Langmuir 1995,11, 1162-1169
A Chemical Theory for Ion Distribution Equilibria in Reverse Micellar Systems. New Experimental Data for
Aerosol-OT-Isooctane-Water-Salt Systems Hamid R. Rabie and Juan H. Vera*
Department of Chemical Engineering, McGill University, Montreal, Quebec, Canada H3A 2A7
Received October 31, 1994. In Final Form: January 12, 1995@
Aerosol-OT and some other surfactant molecules form reverse micelles in an organic phase in contact with an aqueous salt solution. Experimental data of the distribution of cations in Aerosol-OT-isooctane- water-salt systems have been measured at 23 "C for different chloride salts. The effect of different variables on the equilibrium ion distribution between the organic phase, containing reverse micelles, and an aqueous phase, containing different salts, has been formulated in terms of dimensionless groups. These variables are (i) the initial surfactant concentration, (ii) the nature and initial salt concentration of each salt, and (iii) the initial volume ratio of the two phases. The model distinguishes between different ions via the equilibrium constants of their ion exchange reactions with the surfactant counterion. It accurately predicts the distribution of single charge and multicharge ions between the two phases of the system. A model formulated in dimensionless form, with few independent dimensionless groups rather than many independent variables, has definite advantages for the treatment of complex systems. In this work, the predictions of the model are compared with experimental results and with the predictions of other phenomenological models published in the literature for reverse micellar systems.
Introduction
Reverse micelles have attracted much interest over the past few decades because of their extensive technological and biological importance for oil recovery, lubrication, detergency, catalysis, metal and biomolecule extraction, or use for probes in immunological processes and drug de1ivery.l Electrolytes are important components in the two-phase systems under consideration. It has been long established that a high salt concentration is a prerequisite for the formation of a Winsor I1 ~ y s t e m , ~ , ~ which is a water- in-oil (W/O) microemulsion in equilibrium with an excess aqueous phase. An understanding of the behavior of the system with different electrolytes, coupled with a knowl- edge of the ion distribution in the two-phase system, is desirable if the solubilization characteristics of solutes (metals, biomolecules, etc.) are to be interpreted correctly.
The effect of electrolytes on reverse micelles has been studied by titration experiments which give a different perspective than that of the present study. The effects of temperature, salt type, and concentration on the maximum water uptake by the surfactant Aerosol-OT (AOT) reverse micellar phases before any excess aqueous phase is formed, was investigated by Kon-No and Kitahara.4-8 A similar study was undertaken by Chou and Shah.g In more recent years, Kunieda and Shinodalo-l2 and Gosh and Miller13 focused on the influence of salinity on the phase behavior of AOT/water/oil systems. Tosch et al.,14 Fletcher,15 and Aveyard et a1.16 have measured the distribution of sodium
* To whom correspondence should be addressed. @ Abstract published inAduameACSAbstracts, March 15,1995. (1) Luzar, A,; Bratko, D. J. Chem. Phys. 1990,92, 642. (2) Akoum, F.; Parodi, 0. J. Phys. (Les Uis, Fr.) 1986, 46, 1675. (3) Lampert, A.; Martinelli, R. U. Chem. Phys. 1984, 88, 399. (4) Kitahara, A.; Watanabe, K.; Kon-No, K.; Ishikawa, T. J. Colloid
( 5 ) Kon-No, K.; Kitahara, A. J. Colloid Interface Sci. 1971,35,403. (6) Kon-No, K.; Kitahara, A. J. Colloid Interface Sci. 1971,35,636. (7) Kon-No, K.; Kitahara, A. J. Colloid Interface Sci. 1971,37,469. (8) Kon-No, K.; Kitahara, A. J. Colloid Interface Sci. 1972,41, 47. (9) Chou, S. I.; Shah, D. 0. J. Colloid Interface Sci. 1981, 80, 311. (10) Kunieda, H.; Shinoda, K. J. Colloid Interface Sci. 1979,70,577. (11) Kunieda, H.; Shinoda, K. J. ColloidInterface Sci. 1980,75,601. (12) Kunieda, H.; Shinoda, K. J. Colloid Interface Sci. 1987, 118,
(13) Ghosh, 0.; Miller, C. A. J. Phys. Chem. 1987,91, 4528.
Interface Sci. 1969, 29, 48.
586.
salts between an aqueous electrolyte solution and a W/O microemulsion in equilibrium. They also measured the water uptake by the reverse micelles and reported the AOT distribution between the two phases, the size of the reverse micelles, and the values of the interfacial tension.
Studies more relevant to this work have been presented by Leodidis and Hatton," Vijayalakshmi et al.,18J9 and Plucinski et a1.20 A phenomenological model for the selective solubilization of cations in the reverse micelles ofAOT surfactant was presented by Leodidis and Hatton.l' Their primary goal was to model the large differences in water uptake and the ion distribution. They used the modified Poisson-Boltzmann theory with the assumption that the chemical potential for the ions can be expressed as a linear combination of a series of different interaction terms, while neglecting the ion-ion terms. Their model distinguished the different cations through their charge, hydrated size, and electrostatic free energy of hydration. Vijayalakshmi et a1.18 and Vijayalakshmi and Gularilg proposed a model which assumed that the adsorption of counterions onto the surfactant surface of the reverse micelle is described by the Stern double layer model. Their model is simpler than the Leodidis-Hatton model, but it cannot distinguish between different ions with the same charge. Plucinski et a1.20 studied the specific ion effect of the ion exchange in the AOT reverse micellar system in equilibrium as well as in kinetic experiments. They used the Graham model of metal ion adsorptionz1 to express the adsorption ofmetal ions in the inner Helmholtz plane.
We present here a theoretical model in which the specific character of each exchangeable counterion (hydrated size, free energy of hydration and electronic properties) is
(14) Tosch, W. C.; Jones, S. C.; Adamson, A. W. J. Colloid Interface
(15)Fletcher, P. D. I. J. Chem. Soc., Faraday Trans. 1 1986, 82,
(16)Aveyard, R.; Binks, B. P.; Clark, S.; Mead, J. J. Chem. Soc.,
(17) Leodidis, E. B.; Hatton, T. A. Langmuir 1989, 5, 741. (18) Vijayalakshmi, C. S.; Annapragada, A. V.; Gulari, E. Sep. Sci.
(19) Vijayalakshmi, C. S.; Gulari, E. Sep. Sci. Technol. 1991,26,291. (20) Plucinski, P.; Nitsch, W. Langmuir 1994, 10, 371. (21) Hunter, R. J. Foundations ofcolloid Science; Clarendon Press:
Sci. 1989, 31, 297.
2651.
Faraahy Trans. 1 1986,82, 125.
Technol. 1990,25, 711.
Oxford, 1989.
0743-7463/95/2411-1162$09.00/0 0 1995 American Chemical Society
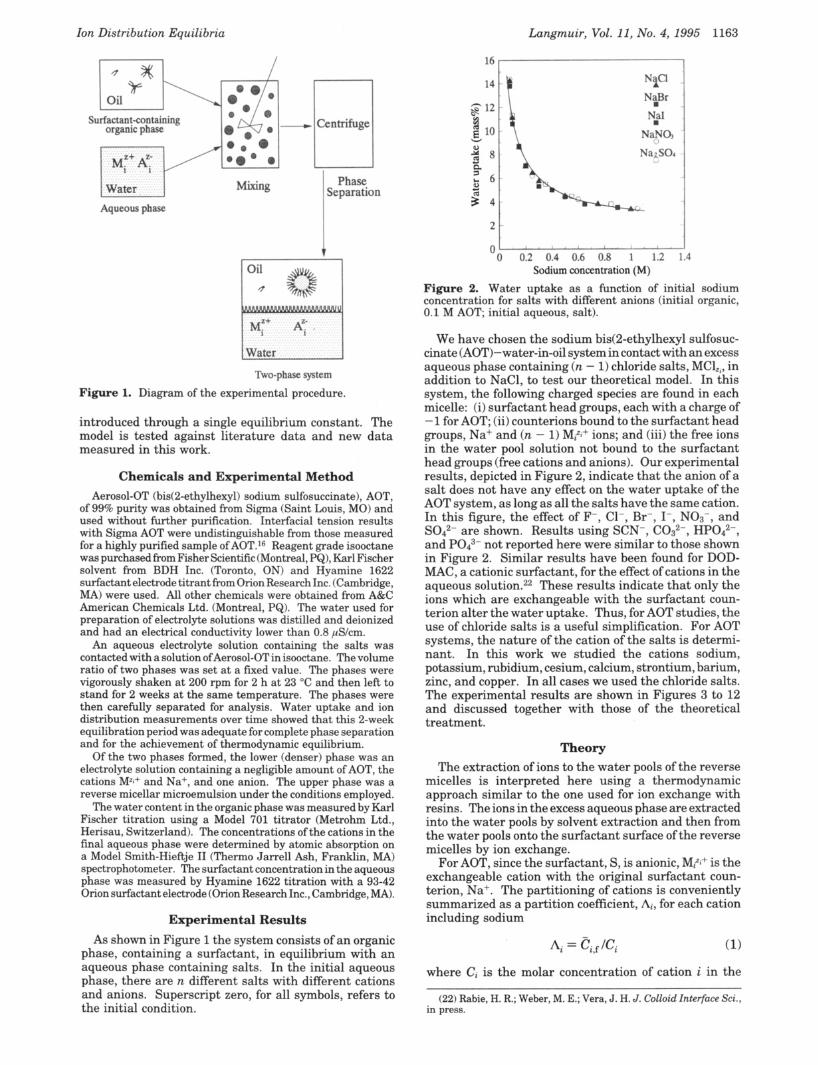
Ion Distribution Equilibria
Phase Separation
Langmuir, Vol. 11, No. 4, 1995 1163
[water. Mixing Aqueous phase
I
Centrifuge
I Water I Two-phase system
Figure 1. Diagram of the experimental procedure.
introduced through a single equilibrium constant. The model is tested against literature data and new data measured in this work.
Chemicals and Experimental Method Aerosol-OT (bis(2-ethylhexyl) sodium sulfosuccinate), AOT,
of 99% purity was obtained from Sigma (Saint Louis, MO) and used without further purification. Interfacial tension results with Sigma AOT were undistinguishable from those measured for a highly purified sample of AOT.16 Reagent grade isooctane was purchased from Fisher Scientific (Montreal, PQ), Karl Fischer solvent from BDH Inc. (Toronto, ON) and Hyamine 1622 surfactant electrode titrant from Orion Research Inc. (Cambridge, MA) were used. All other chemicals were obtained from A&C American Chemicals Ltd. (Montreal, PQ). The water used for preparation of electrolyte solutions was distilled and deionized and had an electrical conductivity lower than 0.8 ,uS/cm.
An aqueous electrolyte solution containing the salts was contacted with a solution ofAerosol-OT in isooctane. The volume ratio of two phases was set a t a fixed value. The phases were vigorously shaken at 200 rpm for 2 h a t 23 "C and then left to stand for 2 weeks at the same temperature. The phases were then carefully separated for analysis. Water uptake and ion distribution measurements over time showed that this 2-week equilibration period was adequate for complete phase separation and for the achievement of thermodynamic equilibrium.
Of the two phases formed, the lower (denser) phase was an electrolyte solution containing a negligible amount of AOT, the cations Mzl+ and Na+, and one anion. The upper phase was a reverse micellar microemulsion under the conditions employed.
The water content in the organic phase was measured by Karl Fischer titration using a Model 701 titrator (Metrohm Ltd., Herisau, Switzerland). The concentrations of the cations in the final aqueous phase were determined by atomic absorption on a Model Smith-Hieftje I1 (Therm0 Jarrell Ash, Franklin, MA) spectrophotometer. The surfactant concentration in the aqueous phase was measured by Hyamine 1622 titration with a 93-42 Orion surfactant electrode (Orion Research Inc., Cambridge, MA).
Experimental Results As shown in Figure 1 the system consists of an organic
phase, containing a surfactant, in equilibrium with an aqueous phase containing salts. In the initial aqueous phase, there are n different salts with different cations and anions. Superscript zero, for all symbols, refers to the initial condition.
16
14
g 12
10 W
2 8 c.
$ 4
a 2 6 c
2
n
h NiBr N$
-0 0.2 0.4 0.6 0.8 1 1.2 1.4 Sodium concentration (M)
Figure 2. Water uptake as a function of initial sodium concentration for salts with different anions (initial organic, 0.1 M AOT; initial aqueous, salt).
We have chosen the sodium bis(2-ethylhexyl sulfosuc- cinate (A0T)-water-in-oil system in contact with an excess aqueous phase containing (n - 1) chloride salts, MCl,,, in addition to NaCl, to test our theoretical model. In this system, the following charged species are found in each micelle: (i) surfactant head groups, each with a charge of - 1 for AOT; (ii) counterions bound to the surfactant head groups, Na+ and (n - 1) Mp+ ions; and (iii) the free ions in the water pool solution not bound to the surfactant head groups (free cations and anions). Our experimental results, depicted in Figure 2, indicate that the anion of a salt does not have any effect on the water uptake of the AOT system, as long as all the salts have the same cation. In this figure, the effect of F-, C1-, Br-, I-, NO3-, and
are shown. Results using SCN-, Cos2-, HP042-, and Po43- not reported here were similar to those shown in Figure 2. Similar results have been found for DOD- MAC, a cationic surfactant, for the effect of cations in the aqueous solution.22 These results indicate that only the ions which are exchangeable with the surfactant coun- terion alter the water uptake. Thus, for AOT studies, the use of chloride salts is a useful simplification. For AOT systems, the nature of the cation of the salts is determi- nant. In this work we studied the cations sodium, potassium, rubidium, cesium, calcium, strontium, barium, zinc, and copper. In all cases we used the chloride salts. The experimental results are shown in Figures 3 to 12 and discussed together with those of the theoretical treatment.
Theory The extraction of ions to the water pools of the reverse
micelles is interpreted here using a thermodynamic approach similar to the one used for ion exchange with resins. The ions in the excess aqueous phase are extracted into the water pools by solvent extraction and then from the water pools onto the surfactant surface of the reverse micelles by ion exchange.
For AOT, since the surfactant, S, is anionic, Mp+ is the exchangeable cation with the original surfactant coun- terion, Na+. The partitioning of cations is conveniently summarized as a partition coefficient, Ai, for each cation including sodium
Ai = ci,f/Ci
where Ci is the molar concentration of cation i in the
(22) Rabie, H. R.; Weber, M. E.; Vera, J. H. J. Colloid Interface Sei., in press.

1164 Langmuir, Vol. 11, No. 4, 1995
aqueous phase, and ci,f is the molar concentration of the free cation i in the water pools. The free cations in the water pools are assumed to undergo a reaction of ion exchange with the cations bound to the surfactant head groups. Following Shallcross et al.23 and Allen and Addison,24 the ion exchange for an anionic surfactant is represented by a reversible reaction of the form
(2)
where the free ion M i , f in the water pools replaces the surfactant bound counterion Na+ in the reverse micelles. In eq 2, zi is the charge number of ion M i and subscript "f' refers to the free ions inside the water pools. The equilibrium constant KN:, in terms of concentrations, for this reaction is
zi SNa + Mi,?+ == SzMi + ziNaf+
Rabie and Vera
(3)
where c i , b and c N a , b are the molar concentrations ofcation i and sodium, respectively, bound to the surfactant head group in the reverse micellar phase. Equations 2 and 3 are written for an anionic surfactant; however, they can be easily applied for cationic surfactants as well. In a ternary surfactant counterion system, the three equilib- rium constants must satisfy a so-called triangular rela- t i ~ n ~ ~ , of the form
(Kj)zr(K$i(<K,')zj = 1 (4)
Combining eqs 1 and 3 results in
ei,b cNa *' = (C)(G)
Equation 5 describes an ion exchange reaction between the cations in the excess aqueous phase and the sodium ofthe surfactant in the organic phase with the equilibrium constant, K s ~ 2 , defined by the following equation:
(6)
In this work, we have retained the basic assumptions of ele~troneutrality'~ and mon~dispersity'~ of the reverse micelles. For the mass balance on surfactant and cations the experimental situation is described as follows. In the initial state, an aqueous phase of volume VO containing HzO, n different cations with concentrations e, includ- ing Na+, and chloride anion with concentr_ation is contacted with an organic phase of volume VO containing isooctane and the surfactant with concentration c. In the final state (at equilibrium), the aqueous phase has a volume Vand it contains surfactant, cations, and chloride anion at concentrations C,, Ci, and Ccl-, respectively. The organic phase of vclume V contains isopctane, surfactant (C,), free cations (Ci,f), bound cations (Ci,b), chloride anion (Ccl-), and HzO in reverse micelles.
The concentrations of free ions in the organic phase are defined as moles of ion per unit volume ofwater pool while the concentrations of the surfactant and the bound ions in organic phase are defined as moles of surfactant per
(23) Shallcross, D. C.; Hermann, C. C.; McCoy, B. J. Chem. Eng. Sei. 1988.43. 279.
(24)Alen, R. M.; Addison, P. A. Chem. Eng. J. 1990, 44, 113. (25) Bajpai, R. K.; Gupta, A. K.; Rao, M. G. J. Phys. Chem. 1973,77,
1288.
unit volume oforganic phase. The mass balance of sodium is then formulated as:
(7)
where w is the volume ratio of water in the organic phase at equilibrium and it is only a function of final ion concentrations at equilibrium. For each of the other cations present in the initial aqueous phase we write:
Note that in eq 7, the surfactant solubilized in the aqueous phase is assumed to be completely dissociated. Assuming that in the reverse micelles all the surfactant sites are filled with either sodium or other cations, we write
(n-1)
- i=l
(9)
Equation 9 assumes that all the surfactant in the oil phase is found in the reverse micellar interfaces. In light of the results ofAveyard et a1.,16 this is a reasonable assumption for the AOT surfactant at the salt concentrations used in this work.
The partition coefficient ofAOT between the oil and the water phases was measured with a surfactant electrode, as described above. In no case the fraction of AOT in the bulk water was greater than 1%. Similar results have been found by other worker^.'^,^^ Thus, we assume that the concentration of the AOT surfactant in the aqueous phase, at equilibrium, is zero (C, = 0). Since the organic solvents are insoluble in the aqueous phase, the only source of organic or aqueous phase volume change is the solubilized water in the reverse micelles. Thus, the relation between the initial and final volumes ofeach phase is
Combining eqs 7-10 provides a useful relation between the initial and equilibrium concentrations of ions in the aqueous solution.
n n
(11) i=l i=l
The term Xi in eq 11 is defined by the following equation:
A'i = 1 + K ( A i - 1) l - w (12)
with "r" defined as the initial volume ratio of the organic to the aqueous phase
r = P P (13)
The value of A'< is unity either when the water uptake is close to zero or when the value of Ai is unity.
Following Leodidis and Hatton,17 we use the following ratios of equivalents to describe the final and initial conditions in the system.
(26) Bruno, P.; Caselli, M.; Luisi, P. L.; Maestro, M.; Traini, A. J . Phys. Chem. 1990,94, 5908.

Ion Distribution Equilibria Langmuir, Vol. 11, No. 4, 1995 1165
1 . Binary Counterwn Systems. For a binary counterion system, the solution can be written as
i=l
It is important to observe that when no NaCl is added to the initial aqueous phase, RP and (116) are equal to zero. However, the value of Ri will be different from zero since sodium is the original counterion of the surfactant and it will exchange with other cations present in the initial aqueous phase.
If the values ofRi for all cations are known, the following equations, obtained from combining eq 11 and the mass balance of a particular ion, can be used to determine its concentration in aqueous and organic phases at equilib- rium
(16)
Equation 16 stands for any cation including sodium. For all cations, excluding sodium, the concentration of bound cation in organic phase can be written as:
(17)
The concentration of bound sodium in organic phase can then be calculated from the following equation
The concentrations of free cations in the organic phase are found from eqs 1 and 16.
Leodidis and Hatton17 experimentally found that in a system of AOT and MCl,, the ratio 6 was the most satisfactory choice for monitoring the behavior of the cation liquid-liquid extraction. In their system, since no NaCl was initially present in the aqueous phase, the Ro parameter was zero. When they plotted Ri, the ratio of sodium to cation concentration in the final aqueous solution against the parameter 6 , the data of three different experiments collapsed onto a single plot. They did not provide any explanation for such an interesting finding. As explained below, the solution of eqs 1, 5, and 7-10, provides a simple relation between the initial and the equilibrium conditions of the system which explains the findings of Leodidis and Hatton.17 For the cases of a binary counterion system, e.g., NaCl and one other electrolyte MCl,, for AOT, and of a ternary counterion system, e.g., NaCl and two other electrolytes MCl,, and MCl,, for AOT, the solution ofthe above equations is analytical. The final expressions are given below. It is important to note that these expressions are written for AOT reverse micellar system. However, they are identical for all other anionic or cationic surfactants if the sodium, other cations added to the aqueous phase, and the anion are replaced by the original surfactant counterion, the other surfactant coun- terions added to the aqueous phase, and by the surfactant co-ion, respectively.
(19)
where K s ~ 2 has been replaced by K,’ for simplicity. The relationship between Ri and 6 for two limits of 6 , infinity and zero, can be written as:
and
Equation 19 together with another equation for the water uptake as a function of equilibrium concentrations of the ions can be used to find both the distribution of ions and the water uptake. However, in most of the cases, the water uptake is small enough so the change in the volumes of the organic and the aqueous phases, between the initial and the equilibrium states, can be neglected. Thus, the volumes of aqueous and organic phases can be assumed to be constant and denoted by Vand V, respectively. Since the volume of organic phase is assumed to be constant, the concentration of the surfactant in the organic phase remains at its initial value, denoted by e. As discussed in eq 12 the assumption of small w gives A’i close to unity. For monovalent cations under the above assumptions, eq 19 can be solved for Ri to give
It is important to note that eq 21, for monovalent salts, is valid for any value of water uptake if the values of Ai for sodium and the other cation are equal.
Equation 21 shows that the ratio of the equilibrium equivalents of sodium to another cation in the final aqueous solution is only a function of the RP and 6 parameters. As long as the RP and 6 parameters are fixed, no matter whether the experiments are performed by changing the initial concentration of salts in the aqueous solution, changing the surfactant concentration in the organic phase, or changing the volume ratio of the two phases in contact, the same value of Ri ratio is obtained.
It is interesting to note that for such systems, the effect of four independent variables (initial concentrations of surfactant, NaCl and MCl,, and the volume ratio of the two phases) can be described by only two dimensionless groups and one constant. Setting the initial concentration of NaCl at zero reduces the number of parameters to one, 6 . This is exactly the case studied by Leodidis and Hatton.17
The above analysis also shows that the value of equilibrium constant can then be obtained from a single data point. For a binary system containing MCl,, and
1166 Langmuir, Vol. 11, No. 4, 1995
having zero initial concentration of NaC1, i.e., RP = 0, when the values of 6 and volume ratio, r, are unity, the equilibrium constant of cation Mzl+ with sodium is simply:
~ , i = ( ~ ~ ) ( * l + l ) at 6 = 1; r = 1; R P = 0
(22)
Thus, the experimental measurement of R, for one point provides with the value ofK,” valid for all other conditions.
2. Ternary Counterion Systems. For a ternary system, containing three cations when no NaCl is initially added to the aqueous phase, two equations have to be solved simultaneously to find the distribution of ions in the final aqueous solution. These two equations relate the R ratios of two cations as follows:
Rabie and Vera
6(R1A’, + RzAt1 + R RIR2
(23)
K,2 = f (R1K2 + RzAf1 + R R A‘ Na ) - A’2)
4- RZA‘, 4- R,R2A”,) R1(1+ f l
RlR2
22
-
i (24)
where the subscripts or superscripts 1 and 2 refer to the cations 1 and 2, respectively, and f is defined by the following equation:
f = z2c; /zlc; (25)
Both Rz and R1 are zero when 6 is zero but their ratio, RZIR1= ClICz, is equal to 1 If. At large values of 6 this ratio can be written as:
It is interesting to note, when both cations have the same charge number this ratio is independent of 6, r, and of water uptake.
When the water uptake is negligible, the values of A’ are close to unity. The values of the equilibrium constants can be obtained from data for binary counterion systems, as described by eq 21, and then these same values can be used to predict the ion distributions for different conditions of a ternary system.
Results and Discussion The predictions of the model proposed in this work are
compared with experimental results for the solubilization of different monovalent and divalent cations, in binary and ternary counterion aqueous solutions, under varying concentrations of salts and surfactant and a t different volume ratios of the two phases.
1. Binary Counterion Systems. In these experi- ments the value of 6 was changed in the following ways: (i) by changing the initial concentration of salt while
Uniform distribution NNC ......
s
8 , ’I
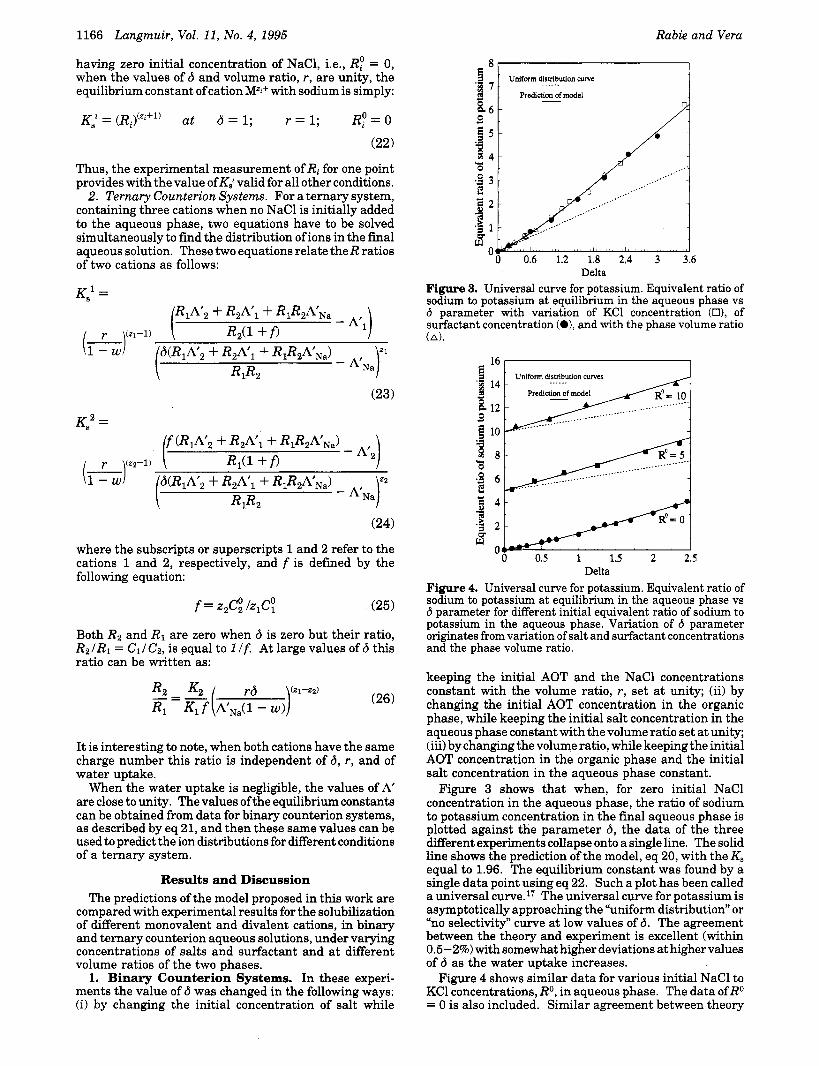
Delta Figure 3. Universal curve for potassium. Equivalent ratio of sodium to potassium at equilibrium in the aqueous phase vs 6 parameter with variation of KC1 concentration (O), of surfactant concentration (O), and with the phase volume ratio (A).
16 I Uniform distribution ...... N ~ V ~ S
Delta Figure 4. Universal curve for potassium. Equivalent ratio of sodium to potassium at equilibrium in the aqueous phase vs 6 parameter for different initial equivalent ratio of sodium to potassium in the aqueous phase. Variation of 6 parameter originates from variation of salt and surfactant concentrations and the phase volume ratio.
keeping the initial AOT and the NaCl concentrations constant with the volume ratio, r, set at unity; (ii) by changing the initial AOT concentration in the organic phase, while keeping the initial salt concentration in the aqueous phase constant with the volume ratio set at unity; (iii) by changing the volume ratio, while keeping the initial AOT concentration in the organic phase and the initial salt concentration in the aqueous phase constant.
Figure 3 shows that when, for zero initial NaCl concentration in the aqueous phase, the ratio of sodium to potassium concentration in the final aqueous phase is plotted against the parameter 6, the data of the three different experiments collapse onto a single line. The solid line shows the prediction of the model, eq 20, with the K, equal to 1.96. The equilibrium constant was found by a single data point using eq 22. Such a plot has been called a universal cu17re.l’ The universal curve for potassium is asymptotically approaching the “uniform distribution” or “no selectivity” curve at low values of 6. The agreement between the theory and experiment is excellent (within 0.5-2%) with somewhat higher deviations at higher values of 6 as the water uptake increases.
Figure 4 shows similar data for various initial NaCl to KC1 concentrations, RO, in aqueous phase. The data ofRo = 0 is also included. Similar agreement between theory
Ion Distribution Equilibria
10
Lungmuir, Vol. 11, No. 4, 1995 1167
Prcscg data
Literature data'"
PrescEmodel
Electrical double layer model"
Uniform distribution .....
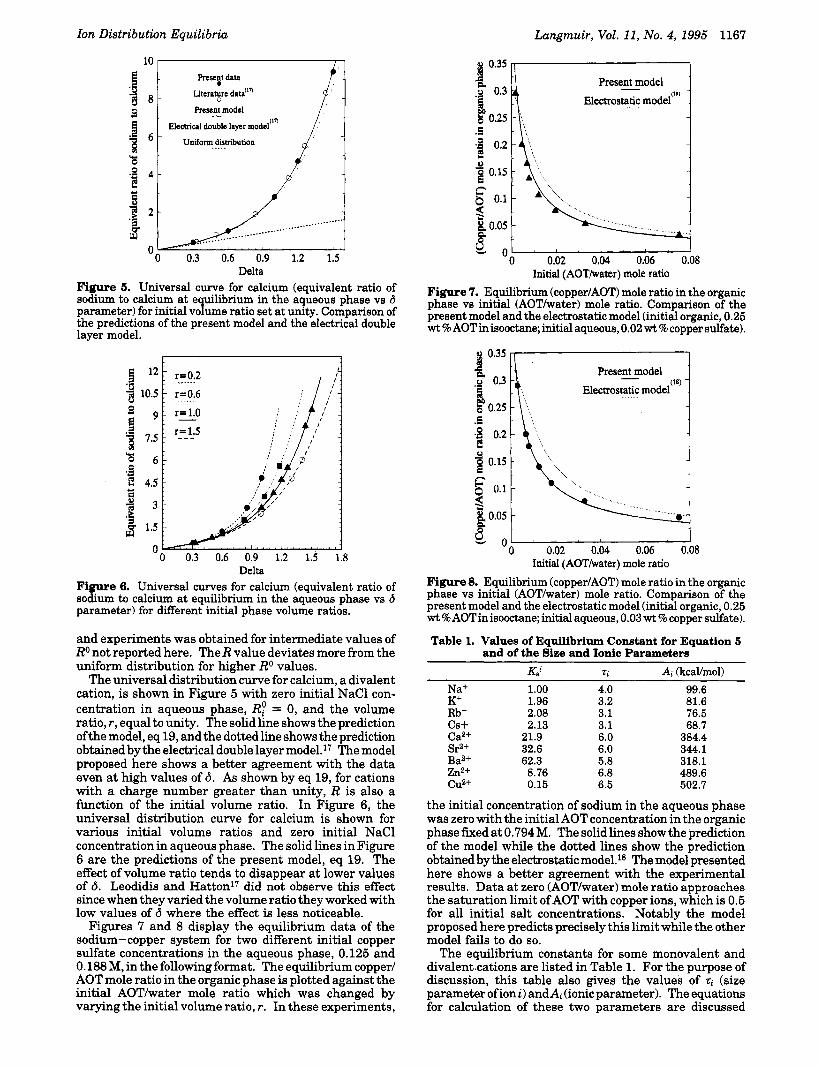
Delta Figure 5. Universal curve for calcium (equivalent ratio of sodium to calcium at equilibrium in the aqueous phase vs 6 parameter) for initial volume ratio set at unity. Comparison of the predictions of the present model and the electrical double layer model.
r
...... ' 5 / :4
Delta Figure 6. Universal curves for calcium (equivalent ratio of sodium to calcium at equilibrium in the aqueous phase vs 6 parameter) for different initial phase volume ratios.
and experiments was obtained for intermediate values of Ro not reported here. The R value deviates more from the uniform distribution for higher Ro values.
The universal distribution curve for calcium, a divalent cation, is shown in Figure 5 with zero initial NaCl con- centration in aqueous phase, RP = 0, and the volume ratio, r, equal to unity. The solid line shows the prediction ofthe model, eq 19, and the dotted line shows the prediction obtained by the electrical double layer mode1.l' The model proposed here shows a better agreement with the data even a t high values of 6. As shown by eq 19, for cations with a charge number greater than unity, R is also a function of the initial volume ratio. In Figure 6, the universal distribution curve for calcium is shown for various initial volume ratios and zero initial NaCl concentration in aqueous phase. The solid lines in Figure 6 are the predictions of the present model, eq 19. The effect of volume ratio tends to disappear at lower values of 6. Leodidis and Hattonl' did not observe this effect since when they varied the volume ratio they worked with low values of 6 where the effect is less noticeable.
Figures 7 and 8 display the equilibrium data of the sodium-copper system for two different initial copper sulfate concentrations in the aqueous phase, 0.125 and 0.188 M, in the following format. The equilibrium copper/ AOT mole ratio in the organic phase is plotted against the initial AOT/water mole ratio which was changed by varying the initial volume ratio, r. In these experiments,
p 0.35 -1 Presecmodel
Electrostatic modelo 0.25
.5 s 0.2 s 0 = 0.15 6
& 0.1 d & 0.05 -9 P
I 0.02 0.04 0.06 0.08
0; ' I ' ' ' '
Initial (AOTkater) mole ratio
Figure 7. Equilibrium (copper/AOT) mole ratio in the organic phase vs initial (AOT/water) mole ratio. Comparison of the present model and the electrostatic model (initial organic, 0.25 wt % AOTinisoodane; initial aqueous, 0.02 w t % copper sulfate).
p 0.35 r\ I
Presecmodel
Electrostatic model
Y 8 0.15 O S 2 l i
2 . 0 ; ' 0.02 ' ' 0.04 ' ' 0.06 ' ' 0.08 ' Figure 8. Equilibrium (copper/AOT) mole ratio in the organic phase vs initial (AOT/water) mole ratio. Comparison of the present model and the electrostatic model (initial organic, 0.25 wt%AOTinisooctane;initialaqueous, 0.03wt%copper sulfate).
Table 1. Values of Equilibrium Constant for Equation 5 and of the Size and Ionic Parametera
Initial (AOTkater) mole ratio
K.' Ti Ai (kcdmol) ~~
Na+ 1.00 4.0 99.6 K+ 1.96 3.2 81.6 Rb+ 2.08 3.1 76.5 cs+ 2.13 3.1 68.7 Ca2+ 21.9 6.0 384.4 Sr2+ 32.6 6.0 344.1 Ba2+ 62.3 5.8 318.1 Zn2+ 8.76 6.8 489.6 cu2+ 0.15 6.5 502.7
the initial concentration of sodium in the aqueous phase was zero with the initial AOT concentration in the organic phase fixed at 0.794 M. The solid lines show the prediction of the model while the dotted lines show the prediction obtained by the electrostatic model.la The model presented here shows a better agreement with the experimental results. Data at zero (AOT/water) mole ratio approaches the saturation limit ofAOT with copper ions, which is 0.5 for all initial salt concentrations. Notably the model proposed here predicts precisely this limit while the other model fails to do so.
The equilibrium constants for some monovalent and divalent cations are listed in Table 1. For the purpose of discussion, this table also gives the values of zi (size parameter of ion i ) andAi (ionic parameter). The equations for calculation of these two parameters are discussed
1168 Langmuir, Vol. 11, No. 4, 1995 Rabie and Vera
35 I I 1
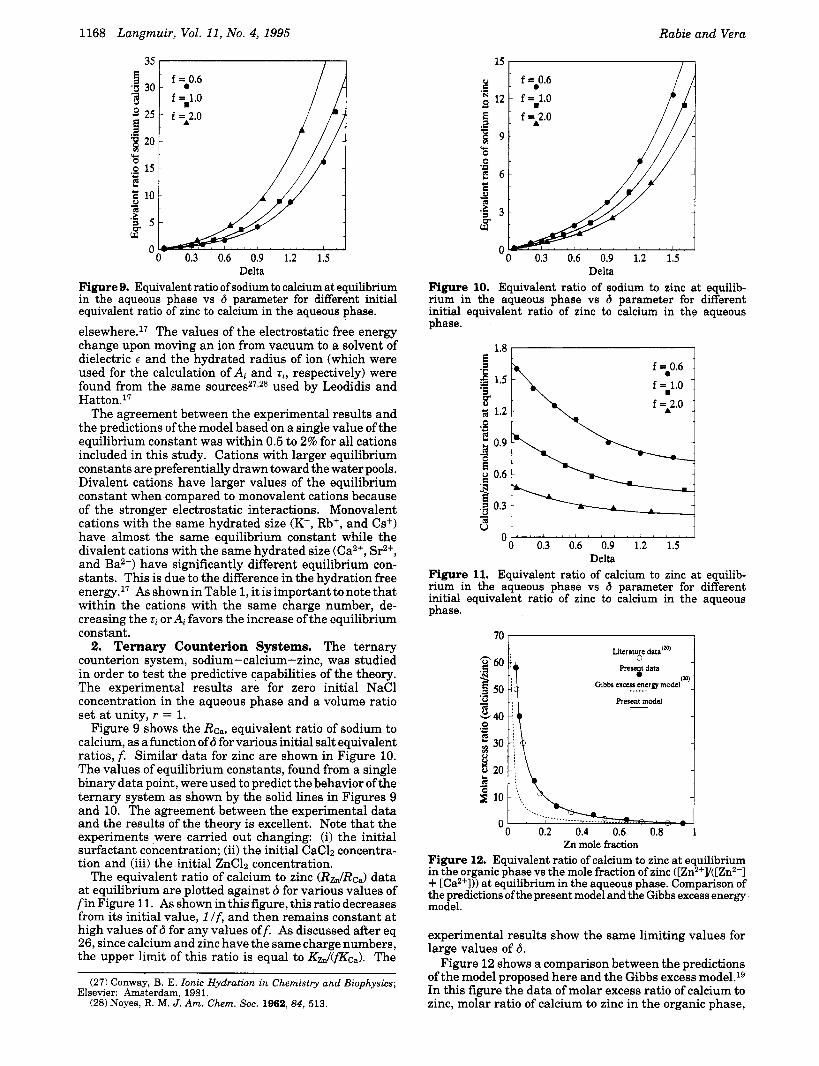
Delta Figure 9. Equivalent ratio of sodium to calcium at equilibrium in the aqueous phase vs 6 parameter for different initial equivalent ratio of zinc to calcium in the aqueous phase. e1~ewhere.l~ The values of the electrostatic free energy change upon moving an ion from vacuum to a solvent of dielectric E and the hydrated radius of ion (which were used for the calculation of A, and tr, respectively) were found from the same source^^^,^^ used by Leodidis and Hatton.17
The agreement between the experimental results and the predictions of the model based on a single value of the equilibrium constant was within 0.5 to 2% for all cations included in this study. Cations with larger equilibrium constants are preferentially drawn toward the water pools. Divalent cations have larger values of the equilibrium constant when compared to monovalent cations because of the stronger electrostatic interactions. Monovalent cations with the same hydrated size (K+, Rb+, and Cs+) have almost the same equilibrium constant while the divalent cations with the same hydrated size (Ca2+, Sr2+, and Ba2+) have significantly different equilibrium con- stants. This is due to the difference in the hydration free energy.17 As shown in Table 1, it is important to note that within the cations with the same charge number, de- creasing the zi or Ai favors the increase of the equilibrium constant.
2. Ternary Counterion Systems. The ternary counterion system, sodium-calcium-zinc, was studied in order to test the predictive capabilities of the theory. The experimental results are for zero initial NaCl concentration in the aqueous phase and a volume ratio set at unity, r = 1.
Figure 9 shows the Rea, equivalent ratio of sodium to calcium, as a function of 6 for various initial salt equivalent ratios, f. Similar data for zinc are shown in Figure 10. The values of equilibrium constants, found from a single binary data point, were used to predict the behavior ofthe ternary system as shown by the solid lines in Figures 9 and 10. The agreement between the experimental data and the results of the theory is excellent. Note that the experiments were carried out changing: (i) the initial surfactant concentration; (ii) the initial CaCl2 concentra- tion and (iii) the initial ZnClz concentration.
The equivalent ratio of calcium to zinc (RzdRc,) data at equilibrium are plotted against 6 for various values of fin Figure 11. As shown in this figure, this ratio decreases from its initial value, 1 If, and then remains constant at high values of 6 for any values off. As discussed after eq 26, since calcium and zinc have the same charge numbers, the upper limit of this ratio is equal to Kzd(flic,). The
(27) Conway, B. E. Ionic Hydration in Chemistry and Biophysics;
(28)Noyes, R. M. J. Am. Chem. SOC. 1962, 84, 513. Elsevier: Amsterdam, 1981.
15 I I 1
Delta Figure 10. Equivalent ratio of sodium to zinc at equilib- rium in the aqueous phase vs 6 parameter for different initial equivalent ratio of zinc to calcium in the aqueous phase.
1.8 [-I 5
'El 4 1.5 .I
% p 1.2 .z
0.9
0.6
- 'R .j 0.3 3
3
f =,0.6 f =.LO :'.... f =2.0 i
-
O b ' ' Ol3 ' ' Ol6 ' ' Old ' '1.2 ' 1;s Delta
Figure 11. Equivalent ratio of calcium to zinc at equilib- rium in the aqueous phase vs 6 parameter for different initial equivalent ratio of zinc to calcium in the aqueous phase.
70 r I
Literature data'" 0
Present data
Gibbs excus energy model
Present model
e - (w ...... - 'E l
Zn mole fraction Figure 12. Equivalent ratio of calcium to zinc at equilibrium in the organic phase vs the mole fraction of zinc ([Zn2+y([Zn2+] + [Ca2+])) at equilibrium in the aqueous phase. Comparison of the predictions ofthe present model and the Gibbs excess energy model.
experimental results show the same limiting values for large values of 6.
Figure 12 shows a comparison between the predictions of the model proposed here and the Gibbs excess m0de1.l~ In this figure the data of molar excess ratio of calcium to zinc, molar ratio of calcium to zinc in the organic phase,

Ion Distribution Equilibria
are shown as a function of the mole fraction of zinc in the aqueous phase at equilibrium. The predictions of the model proposed in this work agree well with experimental results while the Gibbs excess model underpredicts the data.
Conclusions A chemical model which accurately represents the
distribution of cations between a reverse micellar solution of AOT in isooctane and an excess aqueous phase has been developed. This model uses one single parameter for each cation. This parameter, which depends only on the nature of the cation (charge, hydrated size, and electrostatic free energy of hydration), is the equilibrium constant of the ion exchange reaction between the cation in aqueous phase and the cation bound to the surfactant head group. The parameter can be evaluated from one single data point from a binary system of sodium and the
Langmuir, Vol. 11, No. 4, 1995 1169
cation. The equilibrium constant found from a single data point in a binary counterion system can be used for the prediction of multicomponent systems.
The predictions of the model are in excellent agreement with experimental results. This model gives better correlation and predictions than other phenomenological models published in the literature. Dimensionless groups have been defined to regroup several independent vari- ables. The dimensionless form of the model, with few dimensionless groups rather than many independent variables, has definite advantages for the treatment of complex systems.
Acknowledgment. The authors are grateful to the Natural Sciences and Engineering Research Council of Canada for financial support and to Dr. M. E. Weber for helpful discussions and comments. LA940854V




















