
Protein aggregation as a paradigm of aging
17
Review Protein aggregation as a paradigm of aging Ariel B. Lindner ⁎, Alice Demarez INSERM U571, Paris, F-75015, France Paris Descartes University, Paris, F-75015, France abstract article info Article history: Received 2 February 2009 Received in revised form 8 June 2009 Accepted 9 June 2009 Available online 13 June 2009 Keywords: Chaperone Protein misfolding Protein aggregation Asymmetry Proteostasis Aging network The process of physiological decline leading to death of the individual is driven by the deteriorating capacity to withstand extrinsic and intrinsic hazards, resulting in damage accumulation with age. The dynamic changes with time of the network governing the outcome of misfolded proteins, exemplifying as intrinsic hazards, is considered here as a paradigm of aging. The main features of the network, namely, the non-linear increase of damage and the presence of amplifying feedback loops within the system are presented through a survey of the different components of the network and related cellular processes in aging and disease. © 2009 Elsevier B.V. All rights reserved. 1. Introduction Aging is a fundamental characteristic of all living organisms down to bacteria. Yet while we commonly apprehend aging intuitively, arguably its concise global scientific understanding is still elusive. Aging can be generally defined and understood in the dynamic context of diminished fitness of the individual, recapitulated in a decrease of reproduction rate and an eventual exponential increase in mortality with time at the population level. This can be accounted for by time-dependent deterioration of physiological parameters of the organism. While aging is an overall deterministic phenomenon that can be viewed as the surrender of the living system to thermodynamic equilibration with its environment, it is a process driven by stochastic hazards of extrinsic (e.g., accidents, predation, weather, pollution, infections…) and intrinsic (e.g., reactive oxygen species (ROS), off- pathway toxic metabolites, replication, transcription and translation errors, misfolded proteins…) nature. Indeed, aging prevails even when minimizing the extrinsic contribution, as in controlled labora- tory settings. Inherent to the hazards implicated in the physiology of aging is their potential capacity to self- and-cross amplify their own frequencies as well as the deleterious consequences of their ensued damages (Fig. 1). According to this view, the non-linear accumulation of different damages results in the aging of the ‘host’ organism. This stochasti- cally-initiated non-linearity may account for the large variability in aging phenotypes and life-span, independent of environmental and clonal differences. It follows that the internal environment (intracel- lular, tissue, organ, body) modulates the probability and extent of hazard and damage. In particular, this lays ground for evolution to devise molecular systems to keep hazards and damages at bay until at least reproduction is assured by avoidance (fidelity) and maintenance (repair). This however does not come without a cost — given limited resources, investment into minimizing damages is on account of growth and reproduction. Thus, understanding aging amounts to the quantitative study of the time evolution of the global network [1–3], consisting of hazards, damages, quality control and regulation systems (Fig. 1). This review focuses on a subset of this network that captures many of the essential features of the global network and governs one of the main molecular phenotypes associated with aging: the time-dependent accumulation of protein aggregates. We describe the network consisting of proteins and their intrinsic potential propensity to misfold, aggregate and damage, as well as the imperfect, aggravated with age, cellular strategies to avoid, repair and eliminate this damage within the context of the cellular and external environment. Though aging and longevity studies mainly focused on signaling out the contribution of individual physiological traits to the aging process (e.g., dietary restriction, oxidative stress, DNA damage, protein aggregation), these cannot be easily disentangled. Given the emergent complexity due to their non-linear co-dependence, an integrated framework should be sought for and the understanding of the interplay between protein aggregation and other aging/longevity traits is crucial. Indeed, recent work has shed light to the intricate synergy between protein homeostasis and the caloric restriction- related insulin/insulin-like growth factor-1 [4,5] and sirtuin [6] Biochimica et Biophysica Acta 1790 (2009) 980–996 ⁎ Corresponding author. Laboratoire de Genetique Moleculaire Evolutive et Medicale, INSERM U571, Faculté de Médecine Univ. Paris Descartes,156, Rue de Vaugirard, 75730 Paris cedex 15, France. Tel.: +33 1 4061 5348; fax: +33 1 4061 5322. E-mail address: [email protected] (A.B. Lindner). 0304-4165/$ – see front matter © 2009 Elsevier B.V. All rights reserved. doi:10.1016/j.bbagen.2009.06.005 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbagen
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Protein aggregation as a paradigm of aging

Biochimica et Biophysica Acta 1790 (2009) 980–996
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r.com/ locate /bbagen
Review
Protein aggregation as a paradigm of aging
Ariel B. Lindner ⁎, Alice DemarezINSERM U571, Paris, F-75015, FranceParis Descartes University, Paris, F-75015, France
⁎ Corresponding author. Laboratoire de Genetique MoINSERM U571, Faculté de Médecine Univ. Paris DescartesParis cedex 15, France. Tel.: +33 1 4061 5348; fax: +33
E-mail address: [email protected] (A.B. Lindne
0304-4165/$ – see front matter © 2009 Elsevier B.V. Adoi:10.1016/j.bbagen.2009.06.005
a b s t r a c t
a r t i c l e i n f oArticle history:Received 2 February 2009Received in revised form 8 June 2009Accepted 9 June 2009Available online 13 June 2009
Keywords:ChaperoneProtein misfoldingProtein aggregationAsymmetryProteostasisAging network
The process of physiological decline leading to death of the individual is driven by the deteriorating capacityto withstand extrinsic and intrinsic hazards, resulting in damage accumulation with age. The dynamicchanges with time of the network governing the outcome of misfolded proteins, exemplifying as intrinsichazards, is considered here as a paradigm of aging. The main features of the network, namely, the non-linearincrease of damage and the presence of amplifying feedback loops within the system are presented through asurvey of the different components of the network and related cellular processes in aging and disease.
© 2009 Elsevier B.V. All rights reserved.
1. Introduction
Aging is a fundamental characteristic of all living organisms downto bacteria. Yet while we commonly apprehend aging intuitively,arguably its concise global scientific understanding is still elusive.Aging can be generally defined and understood in the dynamiccontext of diminished fitness of the individual, recapitulated in adecrease of reproduction rate and an eventual exponential increase inmortality with time at the population level. This can be accounted forby time-dependent deterioration of physiological parameters of theorganism. While aging is an overall deterministic phenomenon thatcan be viewed as the surrender of the living system to thermodynamicequilibration with its environment, it is a process driven by stochastichazards of extrinsic (e.g., accidents, predation, weather, pollution,infections…) and intrinsic (e.g., reactive oxygen species (ROS), off-pathway toxic metabolites, replication, transcription and translationerrors, misfolded proteins…) nature. Indeed, aging prevails evenwhen minimizing the extrinsic contribution, as in controlled labora-tory settings. Inherent to the hazards implicated in the physiology ofaging is their potential capacity to self- and-cross amplify their ownfrequencies as well as the deleterious consequences of their ensueddamages (Fig. 1).
According to this view, the non-linear accumulation of differentdamages results in the aging of the ‘host’ organism. This stochasti-cally-initiated non-linearity may account for the large variability in
leculaire Evolutive et Medicale,, 156, Rue de Vaugirard, 757301 4061 5322.r).
ll rights reserved.
aging phenotypes and life-span, independent of environmental andclonal differences. It follows that the internal environment (intracel-lular, tissue, organ, body) modulates the probability and extent ofhazard and damage. In particular, this lays ground for evolution todevise molecular systems to keep hazards and damages at bay until atleast reproduction is assured by avoidance (fidelity) and maintenance(repair). This however does not come without a cost — given limitedresources, investment into minimizing damages is on account ofgrowth and reproduction. Thus, understanding aging amounts to thequantitative study of the time evolution of the global network [1–3],consisting of hazards, damages, quality control and regulationsystems (Fig. 1). This review focuses on a subset of this networkthat captures many of the essential features of the global network andgoverns one of the main molecular phenotypes associated with aging:the time-dependent accumulation of protein aggregates. We describethe network consisting of proteins and their intrinsic potentialpropensity to misfold, aggregate and damage, as well as theimperfect, aggravated with age, cellular strategies to avoid, repairand eliminate this damage within the context of the cellular andexternal environment.
Though aging and longevity studies mainly focused on signalingout the contribution of individual physiological traits to the agingprocess (e.g., dietary restriction, oxidative stress, DNA damage, proteinaggregation), these cannot be easily disentangled. Given the emergentcomplexity due to their non-linear co-dependence, an integratedframework should be sought for and the understanding of theinterplay between protein aggregation and other aging/longevitytraits is crucial. Indeed, recent work has shed light to the intricatesynergy between protein homeostasis and the caloric restriction-related insulin/insulin-like growth factor-1 [4,5] and sirtuin [6]

Fig. 1. Schematic depiction of cellular network governing aging. External and internalhazards lead to the accumulation of damagewith time, resulting in fitness decrease anddeath. Key characteristics of the network include intra- and inter-connectivity ofhazards and control mechanisms, potential positive feedback of hazards as well ascross-amplification and cross-inhibition between control mechanisms and internalhazards.
981A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
pathways in longevity and protein aggregation disease models asreviewed elsewhere [4,7].
1.1. Multiple paths to folding and misfolding
Proteins' function relies on their 3D structure, yet for a nascentpolypeptide to achieve its correct fold, coded in its sequence [8], isoften not trivial. A unified framework has converged in the past fewyears from a great body of works to close the gap between the initialview of describing protein folding as an inevitable context-indepen-dent path to its final least energy unique form and the computationalparadox of the astronomically large number of possible conformers fora given polypeptide that can be scanned only within an astronomicaltimescale [9]. Early works focused on in vitro folding of small (up to100 amino acids) globular proteins in dilute solutions, where a singlerate-limiting transition state governs folding [10]. These works werekey to present the folding problem in the context of transition statekinetics, leading to experimental and theoretical work addressinglarger proteins with multiple partially folded intermediates andpotential folding pathways [11]. Evolution has thus encoded in thegenotype not only the final structure but also the pathways leading toit within what can be represented by a free energy landscape ofrugged nature, delineated by ensembles of conformations aroundintermediate and ground states [12,13]. A major effort in the field is tocharacterize such landscapes experimentally and computationally[14]. This landscape is punctuated by transition states representingenergy barriers that determine the life-time, e.g., the kinetic stability,of the flanked intermediates. The energy differences between thedifferent conformational species is often rather small (5–10 kcal),suggesting that the unfolded polypeptide chain travels within thislandscape, driven by energy minimization through stochastic sam-pling of different native and non-native intra-molecular interactionsby structural fluctuations. Few, key interactions (mostly of hydro-phobic nature) may govern the structure of the intermediate steps,followed in a cooperative manner by many small contributions of
spatially neighboring side-chains and backbone interactions [15–17].Thus, rather than an invariant resolution towards the folded ‘native’state (through multiple possible pathways [18]), proteins may betrapped in alternative pathways, resolving into a non-functional ‘mis-folded’ conformation, often resulting in insolubility.
1.2. Multiple paths from misfolding to aggregation
The above-described ‘ideal’ landscape for a given isolated proteinis further complexified when considering that in non-dilutedsolutions as in the context of the highly concentrated crowdedintracellular milieu [19]. A competition there occurs between thefolding-driving intra-protein and the inter-protein interactions withsurrounding polypeptides' folding intermediates that may lead tooligomerization that may collapse into insoluble, aggregated forms;their thermodynamic stability is often higher than that of the nativefolded form [17]. Indeed, folded proteins when left long enough insolution may eventually aggregate, as many experimentalists oftenencountered in their aging test-tubes. Thus, the aggregation propen-sity of a protein relies intrinsically on its folding kinetics and lifetimeof intermediates as well as on its concentration and environment. Theconcentration dependence of aggregation is exemplified by recentresult demonstrating that aggregation propensity of a battery ofhuman proteins, as modeled in silico and measured in vitro, isnegatively correlated with their in vivo expression levels, suggestingan evolutionary tuning of protein stability with respect to theircellular concentration. A further implication is that the cellularprotein quality control system (see below) has evolved to provide onlya limited capacity to buffer decreasing solubility [20]. Indeed, overexpression of α-synuclein as a result of the gene's loci genomicamplification is sufficient to lead to its aggregation and Parkinson'sdisease [21]. The environmental component is exemplified in vitro bysensitivity to pH, heat, pressure [22] and presence of co-solvents [15]that differentially shape the landscape kinetically and thermodyna-mically (e.g. the relative stabilities of the native, misfolded states).The interactions governing both correct folding and aggregation areof similar nature, namely, hydrophobicity and hydrogen-bonding asin secondary structure β-sheets of amyloids (see below). Theevolutionary correlate is evident from structural analysis, suggestinga strong selection pressure on the protein sequence to stabilize theirnative folding pathway conformations in order to avoid misfoldingkinetic traps. This is achieved through the presence of chargedresidues [23], dispersion and covering aggregation-prone β-sheetstretches within stable α-helices, flanking aggregation-prone frag-ments [24–27] by ‘gate-keeping’, flexible residues (e.g., glycine) [28]and conformation-breaking residues (e.g., proline) [17,29]. Whetherkinetic intermediates are shared between the least-energy pathwaysleading to the native and the misfolded ground states or distinct fromeach other is currently unresolved and may be protein specific[30,31]. It is clear though that the energy landscape of individualprotein (mis)folding is extended and modified by presence of otherpolypeptides to include the aggregation outcome [15–17].
2. The amyloids
Rather than a direct collapse, aggregation follows key intermediatesteps that can be kinetically resolved [32,33], leading to aggregates thatmay be classed into two groups: amorphous [34] or rather orderedfibrillar amyloids of heterogeneous forms [35–39]. Scarce knowledge isavailable concerning the physiological outcome from amorphousaggregates from the aging perspective, though arguably they areformed and may accumulate in all cells. Most studies were focused onunicellular over-expression systems that often result in such inclusionbodies [40]. Though the effect of ‘amorphous’ aggregates on aging isnot as spectacular as of amyloids, they were associated to aging in theE. coli bacteria [41] and yeast [42–44] (see below). In contrast, much
982 A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
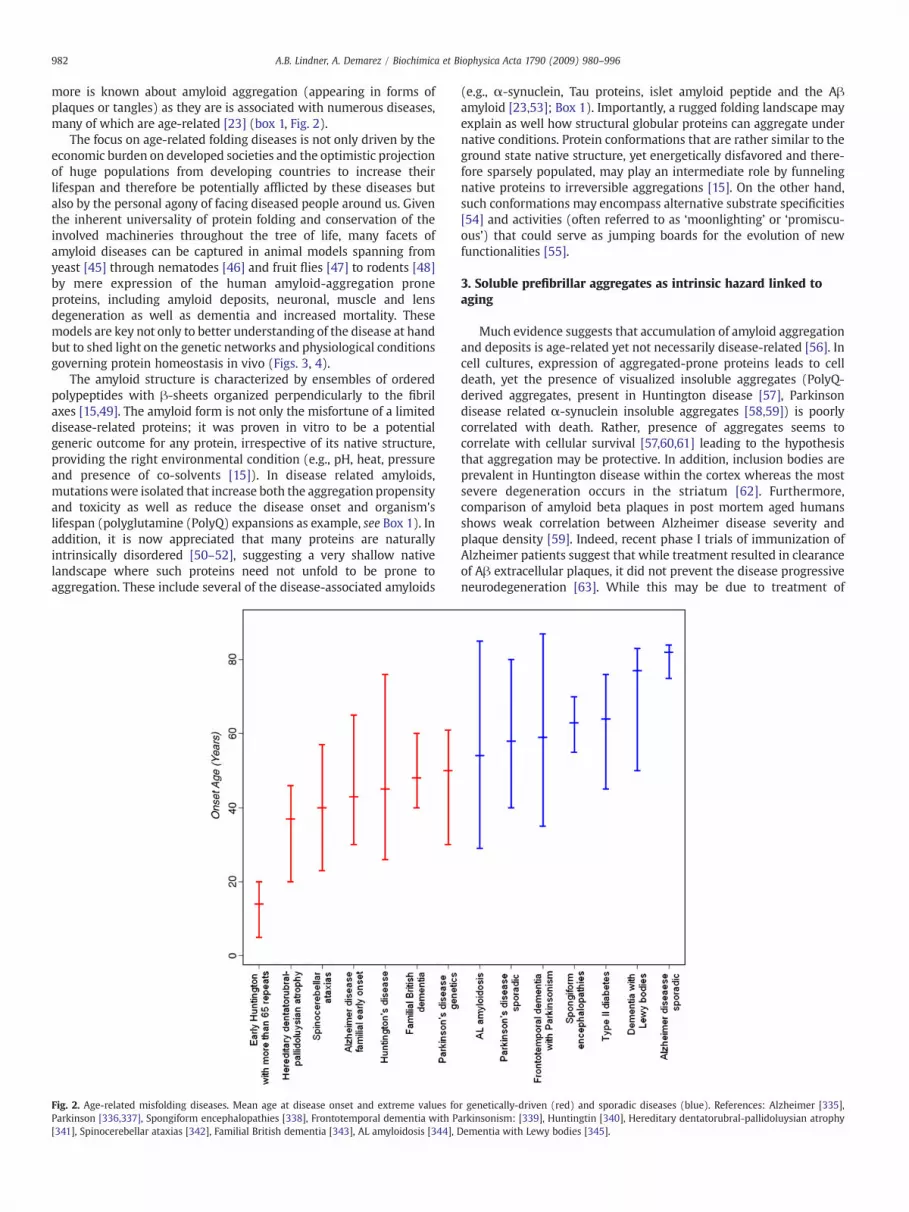
more is known about amyloid aggregation (appearing in forms ofplaques or tangles) as they are is associated with numerous diseases,many of which are age-related [23] (box 1, Fig. 2).
The focus on age-related folding diseases is not only driven by theeconomic burden on developed societies and the optimistic projectionof huge populations from developing countries to increase theirlifespan and therefore be potentially afflicted by these diseases butalso by the personal agony of facing diseased people around us. Giventhe inherent universality of protein folding and conservation of theinvolved machineries throughout the tree of life, many facets ofamyloid diseases can be captured in animal models spanning fromyeast [45] through nematodes [46] and fruit flies [47] to rodents [48]by mere expression of the human amyloid-aggregation proneproteins, including amyloid deposits, neuronal, muscle and lensdegeneration as well as dementia and increased mortality. Thesemodels are key not only to better understanding of the disease at handbut to shed light on the genetic networks and physiological conditionsgoverning protein homeostasis in vivo (Figs. 3, 4).
The amyloid structure is characterized by ensembles of orderedpolypeptides with β-sheets organized perpendicularly to the fibrilaxes [15,49]. The amyloid form is not only the misfortune of a limiteddisease-related proteins; it was proven in vitro to be a potentialgeneric outcome for any protein, irrespective of its native structure,providing the right environmental condition (e.g., pH, heat, pressureand presence of co-solvents [15]). In disease related amyloids,mutationswere isolated that increase both the aggregation propensityand toxicity as well as reduce the disease onset and organism'slifespan (polyglutamine (PolyQ) expansions as example, see Box 1). Inaddition, it is now appreciated that many proteins are naturallyintrinsically disordered [50–52], suggesting a very shallow nativelandscape where such proteins need not unfold to be prone toaggregation. These include several of the disease-associated amyloids
Fig. 2. Age-related misfolding diseases. Mean age at disease onset and extreme values foParkinson [336,337], Spongiform encephalopathies [338], Frontotemporal dementia with P[341], Spinocerebellar ataxias [342], Familial British dementia [343], AL amyloidosis [344], D
(e.g., α-synuclein, Tau proteins, islet amyloid peptide and the Aβamyloid [23,53]; Box 1). Importantly, a rugged folding landscape mayexplain as well how structural globular proteins can aggregate undernative conditions. Protein conformations that are rather similar to theground state native structure, yet energetically disfavored and there-fore sparsely populated, may play an intermediate role by funnelingnative proteins to irreversible aggregations [15]. On the other hand,such conformations may encompass alternative substrate specificities[54] and activities (often referred to as ‘moonlighting’ or ‘promiscu-ous’) that could serve as jumping boards for the evolution of newfunctionalities [55].
3. Soluble prefibrillar aggregates as intrinsic hazard linked toaging
Much evidence suggests that accumulation of amyloid aggregationand deposits is age-related yet not necessarily disease-related [56]. Incell cultures, expression of aggregated-prone proteins leads to celldeath, yet the presence of visualized insoluble aggregates (PolyQ-derived aggregates, present in Huntington disease [57], Parkinsondisease related α-synuclein insoluble aggregates [58,59]) is poorlycorrelated with death. Rather, presence of aggregates seems tocorrelate with cellular survival [57,60,61] leading to the hypothesisthat aggregation may be protective. In addition, inclusion bodies areprevalent in Huntington disease within the cortex whereas the mostsevere degeneration occurs in the striatum [62]. Furthermore,comparison of amyloid beta plaques in post mortem aged humansshows weak correlation between Alzheimer disease severity andplaque density [59]. Indeed, recent phase I trials of immunization ofAlzheimer patients suggest that while treatment resulted in clearanceof Aβ extracellular plaques, it did not prevent the disease progressiveneurodegeneration [63]. While this may be due to treatment of
r genetically-driven (red) and sporadic diseases (blue). References: Alzheimer [335],arkinsonism: [339], Huntingtin [340], Hereditary dentatorubral-pallidoluysian atrophyementia with Lewy bodies [345].

Fig. 3. Protein network in aging and aggregation. This protein interaction map was realized on the basis of automated semantic data-mining and graphic representation (Ali Babafreeware, Humboldt-Universität, Berlin), querying the 200 most recent research articles in PubMed database with “aging AND (aggregation OR misfolding)”. Isolated and generic(e.g., ‘kinase’) terms were manually deleted, relevant missing edges were added (b5% of links), followed by isolating and color coding the emerged groups. The resulted networkrepresents well current knowledge in the area. Given the exponential expansion of scientific literature, advancements in semantic data-mining are key to our scientific apprehension.
983A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
advanced disease [64], it may also indicate that the plaques aredownstream of the cause of disease. This initial frustration had leadresearchers to probe in detail the amyloid formation pathway,suggesting fibrillation follows distinct intermediate oligomeric states[23,32]. In particular, small soluble oligomeric spherical prefibrillarassemblies of 2–10 monomers were identified, their assembly intoprotofibrils may be the potential source of aggregate toxicity [65].
Fig. 4. Contribution of model organisms to the understanding of age-related amyloid diseaHuman subjects and derived cell-lines (pink), Mammalian (blue) and Other models (e.g., in(manual PubMed data-mining). A feedback emerges lending credence to pursue biomedical rby extensive research in model organisms, resulting in the identification of new factors. PT
Such soluble oligomeric intermediates can be detected both in vitro[15], in vivo within model organisms [66] as well as in diseasedpatients [67]. Structural studies suggest that these intermediates arepartially unstructured yet present some cross β-sheet motifs andshow different assembly structures (as beads, string of beads or inannular form probably by folding of the string) and pathways[33,39,68–71]. These protofibrils serve as nucleation centers for the
ses. Different components of proteostasis associated to the diseases as emerged fromvitro, yeast, vertebrates; green), ordered within each block by relative publication dateesearch with model systems: preliminary phenotype identification in patients followedM — post-transcriptional modifications.

984 A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
formation of mature protofilaments that constitute the amyloid fibrils[15,72] in a dynamical manner where unit molecules dissociate andre-associate frequently [73]. Such nucleation process is reminiscent tocrystallization nucleation [74] and follows similar kinetics, namelylong lag followed by fast auto-catalytic increase of fibril size [23,75].The discovery of protofibril-specific, sequence-independent antibo-dies that inhibit further aggregation and toxicity [76–78] suggest thatthese intermediates, sharing no apparent sequence, charge nor aminoacid profile, share common detailed structure. Indeed, cellular toxicitycan be induced by prefibrillar but not mature fibrils from disease-unrelated polypeptides (e.g., bacterial HipF and SH3 domain frag-ments) in tissue culture and animal models [79,80]. Thus, whilefolding and to a large extent misfolding is largely sequence-determined, the aggregation process and associated toxicity arepartially blind to the detailed sequence [81]. Thus, misfolded proteinsmay induce the co-aggregation of other proteins, not only into‘amorphous’ aggregated bodies but to some extent into structuredfibrils in vitro (down to 30% homology within immunoglobulindomains [82]) as well as in vivo [83]. Indeed, numerous cases wererecorded of seizing heterologous proteins within amyloid aggregates(e.g., Ataxin and Huntingtin [84] Synuclein and Tau, Synphilin-1 [85],Huntingtin and mTor [86]). Such amplification may result in co-occurrence of two distinct diseases as Alzheimer and ALS [87] orParkinson [88,89].
The hypothesis that protofibril intermediates, rather than themature fibrils, are the main toxic element leading to downstreamcellular degeneration. is well-supported by numerous data asreviewed for Alzheimer's and Parkinson's diseases [90]. For instance,a series of papers demonstrated that dimer to tetrameric prefibrillarintermediates, isolated from human cerebral cortex and cerebrospinalfluid [91–93] as well synthetic Aβ protofibrils [94] (or Aβ diffusibleligands [95]) can recapture key disease manifestations in modelanimals. Furthermore, in silico predictions of protofibrillar aggregationpropensities of Aβ42 synthetic mutants, meet their measuredaggregation in vitro and correlate with their pathogenic effect whenexpressed inDrosophila, in terms of decreased locomotion and lifespan[96]. Similar indications concerning other cases (e.g. Parkinson'sdisease [97–99], Huntington's disease [60,100], type II diabetes [101],model amyloid — bacterial HypF [102]) resulted in the dogma shift ofpresenting kinetic intermediates of the fibril formation as key tounderstanding aggregation toxicity. Interestingly, as the formation ofprefibrillar or protofibrillar intermediates may be reversible, they maybe involved in other diseases where matured aggregation in forms ofinclusion bodies, plaques or fibrils are not detected.
4. Age-related damage by protein aggregates
Toxic aggregate intermediate hazards may lead to the propagationof damage in either a direct or indirect manner. The latter includessequestering other proteins to the aggregate, eliminating key cellularfunctions (e.g., the cAMP response element binding (CREB)-bindingprotein (CBP) by polyQ [84]) or by inhibiting the cell's protein qualitycontrol network members (see below). In an early work, solubleaggregates of Aβ amyloids in a mouse mode correlated withneuroendocrine dysfunction, impairment of stress response andregulation of blood glucose levels [103], suggesting disruption ofsignaling pathways and cross-talk between multiple aging pheno-types. In another damaging venue, the association of polyQHuntingtinfragments to mitochondria increases with age in knock-in mice,disrupting mitochondria transport and resulting in decreased ATPproduction [104] and increased oxidative stress (as shown withnumerous amyloidoses in tissue culture [105]. Evidence to directtoxicity includes membrane integration and depolarization [106],damaging neuronal signaling and triggering apoptosis [107]. Impor-tantly, permeabilizing mitochondria leads to burst of reactiveoxidative species (ROS) [105] that can lead to damage amplification
by destabilizing proteins and increase their aggregation (see below).The permeabilization mechanism is still under debate; formation ofchannels was evoked [108,109] though often challenged [107]. Specificdamage to neuronal cells in the different diseases includes inductionof hippocampal synapse requiring NMDA-type glutamate receptoractivity by Aβ dimer and trimer oligomers [66,67,92], depressingsynaptic function and integrity [110], and inhibition of hippocampallong-term potentiation [111,112]. In yet another example of neuronaldamage, Synuclein soluble accumulation correlates with gradual lossof dopamine caused by a decrease of its synthesis rate-limitingtyrosine hydroxylase levels. The dopamine loss is accelerated whenSynuclein levels pass a threshold and aggregate due to induceddamaged lysosomal activity [113].
5. The protein quality control network
Within the cellular physiological context, protein folding is rarelyachieved autonomously. Not only does the nascent polypeptide chainarising from the ribosome needs to be held by auxiliary proteins [114]to reach its translation end, many proteins (20%–30% [115]) may neverreach their folding state without assistance, resulting mostly in rapiddegradation [116]. An adaptive network of universally conservedfunctions, commonly referred to as the protein quality control (PQC)[117], has evolved to maintain the cellular protein homeostasis [118](or ‘proteostasis’ [119]), through maximizing cellular protein foldingcapacity (by the chaperone system),minimizing intrinsic and extrinsicprotein assaults and degradation of misfolded proteins (by proteases,the ubiquitin–proteosome system (UPS), and lysosome) (Fig. 5).
6. Chaperones: controlling protein folding fidelity and repair
Chaperones [120,121] evolved to assist in folding proteins and theirmaintenance in the native form. The chaperones do not serve astemplate or code for their substrates' structures but help to avoid orreverse unproductive, non-functional conformations [122]. They con-stitute a significant fraction of cellular proteins yet can also be inducedby different proteotoxic stress conditions [123] ofwhich heat-shock (forwhich they are named ‘heat shock proteins’ or ‘hsp’) serves as aparadigm [124]. The chaperone expression is controlled, both inprokaryotes and eukaryotes by specific transcription factors (TF)(RpoH [125] and HSF (heat shock factor) variants [126], respectively),whereby under native conditions the TF are held as monomers bychaperones away from their chromosomal targets, resulting in basaltranscription level of their downstream targets. In the presence ofmisfolding stress, the heat-shock response (or unfolded proteinresponse (UPR) in the ER), is induced by the rapid presence of unfoldedproteins that titrate the chaperones, releasing the TF to trimerize,translocate to the nucleus and induce chaperones expression [127].Chaperones overlap in their substrate profile and act together and insynergy within the chaperone network [128] and with other PQCsystems as the proteosome and lysosomal degradation pathways (seebelow). Further cross-talk with other cellular networks (as the insulin-like signaling (ILS) and sirtuin pathways) provide further control withinthe context of the organism's well-being [4,7]. Importantly, there isdifference in chaperone profile within cellular organelles and amongcellular lineages, resulting in differences in handling misfolded proteinstress. For example, extended PolyQ polypeptides form readily amyloidaggregates in eukaryotic cytoplasm yet fail to do so in mitochondria orthe endoplasmic reticulum [129]. Furthermore, within the brain,chaperone expression differs between cell types [123].
7. The HSP chaperones
The different chaperones are divided into different families,classified both by size and partially differentiated yet concertedactions ([130–133]):

Fig. 5. Protein folding and aggregation cellular network. Schematic depiction of the interactions between the protein components of the PQC and the folding/misfolding of cellularproteins. Hatched — rate modulation; Solid — direct concentration modulation.
985A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
The small hsp (sHSP) of molecular mass b43 kDa have the capacityto maintain the solubility of partially unfolded proteins throughbinding by protecting their exposed hydrophobic surface [134,135].They can be found bound either to individual polypeptides as well aswithin formed misfolded oligomers and aggregates (hence namedinclusion body proteins or Ibp in prokaryotes [136]). Evidencesuggests that sHSP destabilizes the aggregates, facilitating theirsolubilisation and refolding (or degradation), mediated by Hsp70/40and Hsp104 chaperones [137–139]. The sHSP are organized withinlarge dynamic oligomeric structures, maintained through a conservedα-crystalline domain which function is not yet clarified. It washypothesized that the oligomeric state is a ‘resting state’ providingsHSP stabilization (themselves prone to aggregation) and that thisstructure is dismantled with heat, providing active subunits, possiblyas dimers [140]. Within the sHSP, the Hsp33 chaperones provide anexample for the interplay between oxidative and misfolding stresses.It is only in the presence of the two stresses that the chaperone isactivated [141].
The hsp60 chaperones are constructed from two stacked 7-merrings, facilitating protein folding in an isolated cavity (referred to asthe ‘Anfinsen cage’ [122]). Structural studies revealed that the innersurface of the Hsp60 (together with its cap protein Hsp10) cavity ishydrophobic in the substrate binding state and changes to ahydrophilic surface when the folded product is released through anallosteric conformational change mediated by ATP hydrolysis [142]. Ineukaryotes such chaperones are present in the mitochondria, whereasa second, cytosolic class has no identified Hsp10 partner [143].
The Hsp70 chaperones [144,145] are cytosolic and, in eukaryotes,exist as well within organelles as the endoplasmic reticulum andmitochondria. Together with numerous co-factors (N40 in humans),Hsp70s take part in multiple tasks including folding of nascentpolypeptides and their assembly, refolding of misfolded proteins,membrane translocation of proteins targeted for organelles andsecretion as well as controlling the function and life-time of regulatoryand signaling proteins. These multiple goals are achieved throughATP-driven cycles of substrate binding and release mediated by abattery of co-chaperones (e.g. Hsp40 family [146]) and factorseffecting its nucleotide exchange rate (e.g., GrpE [147] and Bag-1family proteins [148]). Co-factors also mediate the delivery ofmisfolded proteins after hsp70 unfolding to proteosome degradation(e.g. Bag-1 and ubiquitin ligase CHIP [149]; see below). Other factors
(e.g. Hop) may couple Hsp70 to the Hsp90 family and modulate theiractivity [150]. Rather than caging their substrate, the Hsp70 proteinsinteract mainly with short, around 7 amino-acid, hydrophobic regionsthat are statistically well-represented within proteins [143].
The ATP-dependent Hsp90 family [151], shares similar functions toHsp70 to prevent non-specific aggregation of generic proteins. Hsp90can mediate conformational changes of folded proteins, leading notonly to their stabilization but also to their activation or degradation.Furthermore, together with it numerous co-factors, Hsp90 preferen-tially interacts more specifically with ‘client’ proteins, many of whichin higher eukaryotes are involved in signal transduction (e.g. tyrosinekinases and hormone receptors), linking it in mammals to play a rolein carcinogenesis [152]. However Hsp90 has many pleiotropic effects,as emerge through systematic screening approaches, includingparticipation in cell cycle, meiosis, transport, secretion and chromatinremodeling, epigenetic gene regulation and viral replication [153–155]. Moreover, Hsp90 (and to some extent Hsp70) is considered agenetic capacitor by buffering sequence variability through stabiliza-tion of native structure. Upon stress, or in absence of the capacitor,new phenotypes emerge that can, in high frequency, yield selectiveevolutionary advantages [156–158].
Finally, unlike the latter chaperone families, the Hsp104 proteins,members of the AAA+ (ATPase Associated with diverse Activities)family, are not abundant yet are highly induced upon externalstress and confer strong resistance to it [159]. Furthermore, ratherthan prevention of misfolding, its main function may be disag-gregation [160]. Hsp104 is sought to bind disaggregated oligomersand in synergy with Hsp70/Hsp40 and, at least in some cases sHSP,restore native structure and function or direct the disaggregatedproteins to degradation [137,138]. Curiously, hsp104 was lost inrecent evolution and it remains to be seen whether other proteinsconfer this unique function in metazoans. The Hsp104 mechanismhas not been completely deciphered yet it involves ATP-dependentpassage of the polypeptide through a pore within the Hsp104 two-tiered ring-shape hexamer [161]. Earlier results showing bacterial(ClpB) and yeast Hsp104 mediated disaggregation has beenrecently coupled to observations suggesting that Hsp104 canmediate the disaggregation of amyloids by preferentially actingon aggregation intermediates in vitro (Aβ Amyloid prefibrillaroligomers, protofibrils [162]) as well as in vivo in rat model ofParkinson disease [163].

986 A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
8. Chaperone network age-linked phenotypes
Numerous studies recorded a general decrease in stress respon-siveness of the chaperone system as well as diminished activity ofmembers of the chaperone system with age [164]. Studies of agedhepatocytes and fibroblasts suggest that the HSF1 transcriptionactivity but not its protein levels decrease with age [164–166]. Inaddition, decreased heat-shock responsiveness in brain tissue withagewas observed (rabbits [167] rats [168]). Importantly, a direct effectof HSF1 levels on longevity was shown in nematodes where decreasedexpression resulted in faster mortality whereas its over-expressionyielded a significant increase in longevity [169], acting synergisticallywith the daf-16 longevity pathway, and accompanied by delay inpolyQ aggregation onset [5]. The mechanism governing the decreasein HSF1 activity with age is still unknown though clues may arise fromrecent findings on novel factors that control its activity [168,170].
Measurement of relative specific chaperone activity in elders ascompared to young individuals suggest a decrease of Hsp70chaperone activity in retina (human and rhesus monkeys, [171])and in myocardial tissue after exercise [172] and, along with Hsp90, inhepatocytes of rats [173,174]. Decreased Hsp70 activity was alsoobserved in lymphoblasts of aged humans but not of centenariansubjects [175]. Other examples include the amyloid aggregation andconcomitant decrease of activity of the sHSP α-crystallin in agedcataractous eye lens [176–178] and of αB-crystallin in mouseAlzheimer model [179]. The decreased chaperone activity may beassigned to their overload by increasing levels of misfolded proteinswith age [3] — an amplifying loop where initial causality cannot beeasily discerned. While it was shown by fluorescent microscopy thatHsp70-YFP fusion is not sequestered by CFP-tagged polyQ aggregatesin tissue culture model system [180] though it does interact withdiffusible polyQ polypeptides, it may be that chaperones arekinetically sequestered away from their native substrates. Furtherreasoning includes inhibitory post-translational modifications asoxidation, glycation (see below), rendering the chaperones ‘sick’with time [164]. Chaperone activity decrease may have pronouncedeffects on cellular networks' robustness and impair the organism'sadaptation capacity [3].
A correlate suggests that over-expression of chaperones willdiminish the accumulation and toxicity of protein aggregates andmay increase longevity. Indeed, numerous studies in neurodegenera-tive disease models of yeast [181,182], worms [183], fruit flies [184]and mice [185] demonstrate suppression of disease upon increasingchaperone levels [186]. The direct association of chaperones to aging isdiscerned from experimental systems permitting the tuning of theirexpression levels either bymild stress or over-expression. As example,ubiquitous or targeted expression of HSP22 in motor neuronsmitochondria, increases the lifespan and the resistance to oxidativestress and heat shock in D.melanogaster [187]; inversely decreasedlifespan is observed in absence of this sHSP [188]. However, increase inhsp70 copy number, though having a protective effect against heatstress did not effect significantly the fruit fly's lifespan [189] Earlierresults of over-expression of Hsp70 in D.melanogaster derived cellularcultures [190] and animals [191] suggest a deleterious effect anddecreased lifespan, possibly as a result of accumulation of Hsp70 asinsoluble aggregates. These results and the fact that chaperonesgovern the function and lifetime numerous physiologically essentialproteins suggest that chaperone homeostasis is critical to theorganism's well being. Yet, tuning the stress response by “hormesis”—mild punctual [192,193] or repeated heat shock [194,195] appears tobe beneficial both in providing stress tolerance and increasedlongevity in many experimental systems including nematodes, fruitflies, mice and human-derived cell-lines [196]. Similar hormesis wasrecorded using other stresses as dietary restriction [197], in support ofthe interplay between proteostasis and lifespan controlling networks[198]. Indeed, potentially, hormesis inducing drugs, as well as such
that can upregulate the QPC are currently validated in animal andhuman trials [119].
9. Peptidyl-Prolyl Isomerases (PPI) and Protein DisulfideIsomerase (PDI) role in age-related protein aggregation
On top of the HSP chaperones, two classes of enzymes exist tocatalyze key rate-limiting steps that ensure proteins' proper foldingand stability: prolyl isomerization by the Peptidyl-Prolyl Isomerases(PPI) and cystine formation by Protein Disulfide Isomerase (PDI).Prolyl residues in proteins may exist with either trans or cis peptidebond conformations of similar energetic stability. Specific prolylresidues within native protein structures adopt a specific isomer andas the intrinsic isomerization rate is slow, enzymes (PPI) were evolvedfor its amplification. Moreover, specific PPI can interact with alreadyfolded proteins, leading to possible control of their conformation andtherefore function [199]. For the Pin1 PPI, specific Ser/Thr-prophosphorylation pattern is recognized on the substrate. As the activityof Ser/Thr kinases is highly dependent on the prolyl isomer, Pin1 playsa direct role in controlling large spectra of activities, including stressresponses, cellular development and growth regulation, immuneresponse, neuronal differentiation and survival [199]. Its anti-aggregation activity is exemplified in its protective role in Alzheimer[200,201]. Indeed, Pin1 knock-out mice develop Alzheimer-likecognitive, motor and aggregation pathological symptoms [202].
Disulfide bonds, covalently linking cysteine thiol side-chainswithin proteins through their oxidation, play a major role in thelatter's folding and stabilization [203]. Such oxidative folding takesplace in confined organelles (e.g., the endoplasmic reticulum (ER),bacterial periplasm), protected from the cytoplasmic reducingenvironment. It is catalyzed by the PDI enzymes that ensure correctcysteine coupling and phasing of their formation with the foldingprocess [204]. PDI, together with the hsp class chaperones are inducedupon unfolding stress as part of the UPR [205]. Perturbation of theredox potential in the ER as well as negative modulation of the UPRresults therefore in accumulation of misfolded, aggregated proteins.As example, PDI plays a role in attenuation of neuronal cell deathtriggered by misfolded proteins and its inhibition by nitrosylation (seebelow) results in higher neurotoxicity, as observed in sporadicParkinson and Alzheimer diseased brains [206].
10. Proteolytic clearance of misfolded proteins
Despite the elaborative work of the chaperone system, manyproteins never reach their native stable form and are degraded [116].Cellular protein catabolic systems evolved not only to carefully tunethe life-time of cellular proteins, exerting control on all essentialcellular functions (e.g., transcription, cell cycle and apoptosis, antigenprocessing [207,208]) but also to ensure the proteostasis quality byelimination of non-functional, potentially toxic proteins [209]. Twomajor pathways, the ubiquitin–proteosome system and autophagy–lysosome systems that govern protein degradation in eukaryotes aredescribed below. A third system of proteolysis, the calpain Ca2+-activated cystein proteases is implicated in aging, with reports ofincreased activity [210–212] as well as decreased activity [213] withage, suggesting that deregulation of calpains is deleterious tomaintenance of proteostasis and that the effects may be context,tissue dependent.
11. The ubiquitin–proteosome system (UPS)
The (UPS) provides the cell with selective degradation of proteins,governing their lifetime and disposing of damaged proteins. Indeed,aggregation is enhanced when the UPS is impaired or inhibited[209,214]. The UPS is not limited to cytosolic proteins, but may handlecompartmentalized proteins as ER-misfolded proteins through the

987A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
ability to export such proteins back to cytosol (ER associateddegradation (ERAD) [215]). The 26S proteosome is made of 50 proteinsubunits arranged to form a core, barrel-shaped 20S particle,encompassing a hollow channel where proteolysis takes place.Different subunit compositions of the proteosome exist, modulatingspecific functions. In the human proteosome, two active sites withdistinct substrate specificities (e.g., chymotrypsin-, trypsin-like,activities) reside within the channel to digest unfolded proteins intosmall, 9–12mer peptides. The associated 19S particle facilitates,through extensive ATPase activity, the substrate recognition and‘holding’ and translocation of the unfolded proteins into the 20S core[216]. An intricate control system drives proteins selectively toproteosome degradation by ATP-driven covalent attachment of multi-ple ubiquitin (a small 9 kDa peptide) units, mediated by numerousubiquitin-activating (E1) and -carrier proteins (E2) as well as -ligases(E3) [217]. Energy consuming proteolysis, though not evident from athermodynamic viewpoint, occurs universally also in absence of UPSwithin prokaryotes and mitochondria, their proteases' activity andstructure often resembles that of chaperones (e.g., the Lon, ClpP, FtsHand HslUV proteases [218]), suggesting a common ancestor and aselective pressure to efficiently control and degrade aberrant proteins.
Essentially, the function of different UPS factors is directly linked toproteotoxicity and lifespan (as shown for the 19S subunit homologue[219] and E3 ligase [220] in nematode studies). A strong link existsbetween the chaperone and the UPS systems [221]. The UPS relies onthe ability tomaintainmisfolded proteins in a soluble form to facilitatetheir degradation and thus depends at least in part on the chaperonemachinery. As example, CHIP functions both as an Hsp70 co-factor andas ubiquitin-ligase, demonstrating the tight interaction between thefolding and degradation pathways [149]. Indeed, CHIP knock-out miceexhibit accumulation of toxic oligomeric aggregates and decreasedlongevity [222]. The orchestrated build-up of the proteosomemachinery is chaperone-dependent [223], revealing another level ofnetworking within the PQC. A close link exists as well between theUPS and the lysosomal degradation system (see below).
The crucial role of the UPS in aging is reflected by the identificationof specific neurodegenerative-prone mutations [224]. In addition,many studies correlated decreased proteosomal activity with age andrelated diseases [225–229]. Examples include cultured cells (senes-cence [230]), eye lens (cataract [231]), muscle (sporadic inclusion-body myositis [232]), epidermis (‘photoaging’ [233]), heart tissue(aging, [234], arteriosclerosis [235]) spinal cord [236] and the brain(neurodegenerative diseases [237]). This age-related proteosomalactivity reduction with frequent concurrent accumulation of ubiqui-nylated and SUMOilated proteins may be due to accumulation of post-translational damaged targeted proteins lending them difficult todigest (see below). Furthermore, proteosome reactivity towards somepolypeptides as the Huntington-related long polyQ stretches may beimpaired [238,239], leading to their age-related accumulation as toxicaggregates [240] due to entrapment of essential ERAD proteins by theHuntingtin polyQ aggregation [241]. Indeed, proteosomes' activitydecline may be mainly due to their sequestration by the accumulatedburden of misfolded protein compounds [242] as was observed inmany age-related diseases (e.g., Alzheimer's [243] and Parkinson's[244] diseases). A complementary hypothesis of a ‘misfolding trap’was recently put forward, suggesting that, transient binding ofamyloid oligomers to folding intermediates of ‘normal’ newlysynthesized proteins may lead to their ubiquitinylation and degrada-tion, disrupting cellular proteostasis by loss of control of protein's lifetime [245].
12. The autophagy–lysosome system
As discussed above, misfolded proteins can collapse into aninsoluble form and co-aggregate, either due to faster kinetics orfailure of the chaperone system. Another line of defense is provided by
lysosomal autophagy [246,247]. In parallel to its role in regulation ofcell-death and in elimination of dysfunctional organelles (e.g.,mitochondria, peroxisomes) and pathogens, autophagy contributesto proteostasis through multiple pathways [248,249]. Macroauto-phagy, coordinated by multiple autophagy-related gene (ATG),eliminates bulk accumulation of aggregates (aggrosomes; see below)through engulfment and fusion with the resulting vacuoles. Further-more, through stress-triggered chaperone-mediated autophagy(CMA), specific individual proteins containing a consensus amino-acid motif are delivered to the lysosome for degradation [250].Lysosomes have in addition the capacity of engulfing small cytosolicportions to uptake and degrade proteins (microautophagy [251]).Ubiquitinylation plays a role in autophagy where E1, E2 and E3-likeproteins are part of the Atg proteins that mediate membraneexpansion and engulfment, suggesting a close cross-talk betweenthe UPS and lysosomal degradation pathways [249,252]. As in theabove described proteostasis-maintaining systems, the lysosomalsystem exhibits a general decrease of reactivity with age [246]. Aswas shown recently, this diminution can be reversed transgenically inrodents by modulating the expression of the LAMP-2A CMA receptor[253]. Lysosomal autophagy failure results in aggregate accumulationlinked to age diseases as manifested in the case of Parkinson's [254],Alzheimer's diseases [255], polyQ aggregates [256] and neurodegen-eration in mice models [257,258]. A further link between UPS andautophagy has been made recently by demonstrating that Parkin (anE3 phosphorylation-modulated ubiquitin ligase of which loss-of-function mutations are causally related to Parkinson's disease)associates to damaged mitochondria and mediates their removal bylysosomal autophagy. Under disease (or perhaps aging) conditions,this may lead to accumulation of defected mitochondria, resulting infurther amplification of damage by spread of reactive oxygen species(see below) [259].
13. Intrinsic hazards
Within the global scenario of proteostasis: protein folding andaggregation landscapes modulated by the quality control system asdetailed above, lay the details of individual proteins, each with itsspecific folding/aggregation landscape shaped by its genotype(sequence) and environment. The former may be subjected tomutations that can result in destabilization of the native form andpromote aggregation. Such aggregation-prone mutations are closelylinked to age-related misfolding diseases (as examples [260–264]).Moreover, limited by their fidelity, transcriptional [265–267] andtranslational [268,269] error rates (10−5–10−6 and 10−3–10−4 percodon, respectively) suggest that up to 20% of the synthesizedproteins may be mistranslated [270] and be prone to aggregation[271]. Even silent mutations (where nucleotide replacement does notchange the codon amino acid coupling), may influence protein'sfolding, probably by affecting its balanced translation and folding rates[272,273]. Importantly, proteins' natural amino-acids, some of whichare inherently unstable (e.g., aspartyl and arginyl residues) mayundergo spontaneous isomerization [274], racemization (e.g., aspar-tyl; debatable role in Aβ amyloid aggregation [275,276] as well asseryl, threonyl, and tyrosyl residues) and aspargine and glutaminedeamidation (e.g., triggering cataract-associated crystallin aggrega-tion [277]). These modifications further amplify the effect of inherent,replication, transcription and translation mutations. Moreover in vivo,proteins encounter molecular, covalent assaults that change as welltheir surface topology and charge that influence their misfoldingpropensity. These effects are exacerbated by the fact that foldingintermediates by nature exposemore residues to the bulk and are thusmore sensitive to modifications that in turn can canalize the modifiedpolypeptide to misfolding pathways by stabilization of the aggregatedground state, the intermediates leading to it or by destabilizing thenative ground state. Yet again, mutations at all levels that result in

988 A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
destabilizing the folded state or increase the life time of intermediateswill increase the chances of further post-modification and therefore ofmisfolding [278], accumulation of toxic intermediates and groundstate aggregate inclusions. Indeed, the sensitivity of folding inter-mediates to modification can be harnessed by researchers to studyfolding dynamics by use of highly reactive modifiers and massspectroscopy ([279]).
Post-translation modifications that increase misfolding propensityand aggregation accumulationwith age (and related diseases) includeoxidative adducts generated by reactive oxygen and nitrosyl species(ROS [280] and RNS [281]) that escape the cellular capacity toeradicate them as well as by metabolic, e.g. advanced glycation end(AGE) products, lipid aldehydes (advanced lipid peroxidation end(ALE) products, and cholesterol abducts as examples [282–284])generated by ROS. AGE-related cross-linked proteins, further modifiedby free radicals may be the source of the 19th century describedaccumulation of age pigments (also referred to as lipofuscin or ceroid[228]). Given its abundance and facilitated detection by derivation andantibody detection, protein carbonylation by metal-mediated oxida-tion by hydroxy radicals (Fenton reaction [285]) is a hallmark of theinterplay between oxidative damage, protein aggregation and damageaccumulation with age as is widely documented in all studied speciesfrom bacteria [286] and yeast [42,44] to humans where carbonylationand aggregation was documented for the SOD1 major antioxidantenzyme, further amplifying the damage [287].
Only some of these modifications can be reversed by specializedenzymes, linking their function to age-related aggregation anddisease, as methionine sulfoxide reductase (yeast aging [288],Parkinson's disease [289]) and iso-aspartate methyltransferase (Alz-heimer's disease [274]). Indeed methionine, one of the mostoxidation-sensitive amino-acid, is oxidized into both R- and S-stereoisomers of methionine sulfoxide (MetO). Methionine sulfoxidereductase A and B (MsrA/B) are reducing those sulfoxides andtherefore serve as ROS scavengers. Numerous studies show an effect ofthe MsrA/B expression on age relative oxidative stress and longevity;an over expression of MsrA in flies leads to an increase in longevity[290] whereas knock-out mice show a decrease in longevity [291].Moreover it was shown that MsrA is involved in protection ofdopaminergic cells against Parkinson's disease insults [292]. Accu-mulation of damaged proteins increases the burden on the qualitycontrol system and lowers its capacity to maintain proteostasis innative and stressed conditions. As example, cross-linked AGE proteinsresist proteosome degradation [226] and impair autophagy [293],leading to accumulation of corrupt mitochondria that can furtherensue ROS-mediated aggregation and damage. Furthermore, QPCmembers themselves are prone to such modifications [43] (andperhaps even more sensitive to them due to their exposed activity-specific residues surfaces [118]). This in turn results in a viciousdamage accumulation cascade and disease. As example, S-nitrosyla-tion of PDI [206], Parkin (E3 ligase) and of Uch-L1(PARK5; Ubiquitinterminal hydrolase L1) are associated with Parkinson's disease [281].In another intertwined example, in Alzheimer's disease, oxidativestress promotes kinase-mediated hyper-phosphorylation of the Taoprotein and formation of tangled amyloids. Indeed, a kinase inhibitorprevents both Tao hyper-phosphorylation and motor impairment inmouse model [294]. In turn, Tao tangles result in increase of oxidativestress leading to an auto-catalytic process [295,296].
14. Extrinsic hazards
Environmental factors may as well promote aggregation and age-related neurodegenerative disease [262]. Reports include the role ofheavy metals as iron and zinc (either through their catalytic role inoxidation or directly in promoting misfolding [297]) and prenatalexposure to lead effecting disease onset age [298]. The possibleassociative role of inorganic mercury, present in dental cavity
amalgam, in Alzheimer's disease is under controversy [299]. Pesticides(rotenone and paraquat [300]) and inhalation of illegal drugscontaminant (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)[301] are associated with Parkinson's disease. Perhaps the moststriking evidence for an environmental involvement in misfoldingdisease is exemplified by a recent study of an Iceland-specifichereditary amyloid angiopathy, caused by a single mutation in thecystatin C gene, leading to its amyloid deposition, intracerebralhemorrhage, dementia and differing levels of paralysis. Noteworthy,wild-type cystatin C is found within Aβ amyloid Alzheimer's plaquesand its locus is associated to susceptibility to the disease's sporadicform. Tracing back the mutation-carriers' lineages to the 19th centurysuggests that this disease could have been considered as an aging-related amyloid disease, with mean lifespan of 65 years. Albeit atpresent, the mean lifespan is reduced to 31 [302], unexplainable byaccumulation of other mutations within two generations. This adds tothe notion of age-related misfolding disease as “cultural diseases”,that prevail in modern, medically-developed societies where thepopulation reaches abundantly the diseases' onset age [303].
15. Localisation and sequestration of misfolded and aggregatedproteins
As was discussed earlier, while some oligomeric aggregationintermediates can be toxic, their retention to large, stable aggregatesmay be protective and may mediate their clearance (as in autophagy).Microscopy studies identified protein inclusions within eukaryoticcells as autophagy-related vacuoles [304], aggrosomes [34], ER-associated compartments [305] and as co-existence of porous anddense inclusions [306], to name a few. Yet, how they are formed,regulated and whether these different observations relate to similarcompartments, to different kinetic steps in aggregate accumulationand processing or rather point to functional disparity has remainedlargely unclear. A recent study, addressing the localisation andoutcome of different aggregate-prone proteins in yeast and mamma-lian cells, sheds new light to this matter [307]. Two compartments,distinct in their cellular localisation, content and function, wereidentified. The first, ‘juxtanuclear quality control’ (‘JUNQ’) compart-ment retains mostly cytosolic ubiquinylated misfolded proteins thatare at least partially soluble (as concluded by the possibility ofproteins to diffuse/exit away) and is characterized by strong presenceof proteosomes. The second perivacular compartment, designated as‘insoluble protein deposit’ (or IPOD), appears after prolongedexpression of non-amyloid aggregated proteins or upon expressionof amyloid-prone proteins (yeast prion Ure2 and polyQ expandedHuntingtin protein), consists of non-diffusible, mostly aggregatedproteins. As in previous observations, the formation of thesecompartments is microtubules-dependent; their disruption by aspecific drug results in multiple small foci throughout the cytosol. Itis therefore tempting to assume that JUNQ compartment deals withreversible refolding and degradation by chaperones and proteosomeswhereas the IPOD structure sequesters aggregated proteins in aprotective manner, possibly further processed by membrane engulf-ment and lysosomal autophagy. Further studies should shed light onthe link between these observations and previously describedinclusions, on whether differential chaperone content, sortingmachinery exist, on the activity of microtubules in their assembly,on what maintains the boundary of the compartments, and on theirpresence and in vivo significance to aggregate processing in healthand disease.
16. Asymmetric division of aggregates and aging
Most of the data linking protein aggregation to aging are derivedfrom observations in post-mitotic, non-dividing cells as in neuronsand mature multi-cellular organisms as the fruit-fly and nematode. In

Fig. 6. Aging as time-dependent change of global network dynamics.
989A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
dividing cells, protein aggregates are diluted through cellular growthand division and thus their aging-related phenotype may depend onthe balance between their accumulation and dilution. Furthermore,the accumulation of misfolded proteins into small number ofinclusions, suggests that their asymmetric division within progenywill result at the rejuvenation of part of the progeny, devoid ofaggregates, at the expense of aging of an aggregate-carrying fractionof the progeny. In this context, unicellular organisms are relevantexperimental models to understand the role of protein aggregation inaging of dividing cells. Indeed, recent studies in yeast [42–44] andbacteria [41] demonstrated that protein inclusions are not onlydivided asymmetrically but do so in a deterministic fashion, wherethe mother cell retains the aggregates to give rise to aggregate-free,rejuvenated daughter cells.
The asymmetrically dividing yeast, Saccharomyces cerevisiae, is thefirst unicellular organism where aging was observed as mother cellsgive rise to limited number of small daughter buds before senescing[308]. A well-documented molecular asymmetry underlying yeastaging is the accumulation of extrachromosomal rDNA circles inmother cells [309]. More recently, protein aggregates (identifiedimmunocytochemically by targeting protein carbonylation and aggre-gate-associated Hsp104-gfp fusion), were shown to accumulatepreferentially in mother cells though ROS levels were similar inprogenitor and daughter cells [42,43]. An active mechanism wasimplied, dependent on the actin cytoskeleton and the lifespan-associated Sir2p (NAD (nicotinamide adenine dinucleotide)-depen-dent histone deacetylase)). Sir2p deletion resulted in elevatedcarbonylation levels (with preference to chaperones), disruption ofasymmetry and decreased lifespan. These phenotypes could bereversed by over-expression of Hsp104, suggesting that chaperonesplay a role in aggregate assembly and asymmetric segregation that isin turn crucial to minimize aging [43]. Further work with the Schizo-saccharomyces pombe yeast demonstrated that uneven accumulationof carbonylated aggregates, results in cells with high damage contentassociated with longer generation time and accelerated aging [44].Interestingly, in the asymmetrically dividing bacteria, Caulobactercrescentus, the abundance of heat-shock proteins involved indisaggregation (e.g., Lon and DnaK) is biased towards the aging[310] stalked cell [311].
Aging has been observed in apparent symmetric division bacteriaas Escherichia coli [312] and Bacillus subtilis [313]. In these bacteria, thetwo apparently identical offspring cells can be distinguished by theage (in divisions) of their poles. Recording individual growth rate ofeach individual cell within a microcolony in homogenous andcontrolled environment demonstrated that cells inheriting the oldpole exhibit diminished growth rates, decreased offspring production,and increased incidence of death [312]. Importantly, asymmetricsegregation of protein aggregates (detected in vivo through theassociation of fluorescent protein-tagged sHSP chaperone (IbpA[136])) was observed, resulting in their accumulation within the oldpole volume of aged bacteria. The measurement of cellular fitness(e.g., their growth rate) and, in parallel, of aggregation levels lead tothe conclusion that up to 40% of the aging phenotype can be attributedto the presence of protein aggregates [41]. A passive process may driveE. coli's misfolded protein accumulation into inclusions, suggestingthat this primordial route of segregation was harnessed to evolve theactive mechanism present in eukaryotes. Indeed, modelingapproaches generally suggest that asymmetric segregation of damageis potentially beneficial and can thus be evolved [44,314–316]. Anopen question remains, whether the aggregate load per se isresponsible to the associated aging or whether the asymmetricpresence of such insoluble aggregates reflects a concurrent bias ofsoluble toxic aggregates or a linkage to yet another unknownphenotype. While bacterial aggregates (inclusion bodies) were oftenconsidered ‘amorphous’, recent results suggest that at least whenover-expressing heterologous amyloid-prone peptides [317,318] or
globular proteins [319], amyloids are formed. It remains to be seenwhether under unstressed ‘native’ conditions, bacterial aggregates,over-represented in senescent (non cultivable) bacterial cells [320]are of amyloid nature, yet the above results support the importance ofstudying aging and protein aggregation in bacteria.
Evidence to asymmetry in damaged proteins segregation wasestablished in higher eukaryotic dividing cells when expressingexpanded Huntingtin in human embryonic kidney or hamster cell-lines cell lines. In addition, the polarized asymmetric inheritance ofaggrosomes in Drosophila melanogaster neuronal precursor cells aswell as in epithelial crypts of ataxia type 3 patients was reported. Inthe latter, no polyQ aggregates where found in putative stem cellsalthough numbers of aggregates was strikingly high in the tissue,suggesting that stem cells are protected against aggregation. Thisresult is compatible with the hypothesis that aggregate proteins areasymmetrically segregated in order to protect stem cells fromdamages proteins [321]. Interestingly, in mitotic division, thought toresult in two identical cells, proteins targeted for degradation arepreferentially inherited by one of the resulting somatic cells [322].These accumulating results suggest that asymmetry may play a majorrole in proteostasis, in correspondence to its manifestations in manyother cellular processes [323].
17. Converging abstraction, modeling and quantification
The main features of the proteostasis network documented in thisreview (Fig. 5), namely the non-linear increase of misfolded proteinhazard probability, the presence of amplification loops within thesystem (as the capacity of misfolded proteins to self-amplify, inhibitthe control systems and induce damage) can be further abstracted as arepresentative paradigm of the aging process (Fig. 6). In thisframework, perceiving aging is synonymous to the understanding ofthe dynamic changes through time of such basic network, leadingfrom a life supporting system to increased hazard, damage accumula-tion and death. Given the high variability in expression levels of thePQC members and stochastic nature of the aging process severalpredictions weremadewithin the network framework. For instance, itwas hypothesized that variability in chaperone expression can lead tobistability, underlying the non-linear transition to fast damageaccumulation state [324]. In another venue, it was suggested thatthe pleiotropic nature of chaperone low affinity connectivity

990 A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
(common feature of proteins associated with senescence [325]), mayplay a critical role whereby their susceptibility to hazard anddiminished activity with aging increases cellular ‘noise’ and deterio-rates the coupling within the network, leading to increased damageand system's disintegration (“weak link theory of aging” [3]).Computational modeling of both abstract and specific networkdynamics [324,326] in context of cellular dynamics (as asymmetricinheritance of damage upon division)may yield predictions that couldthen be tested experimentally.
Indeed, the acquired qualitative molecular knowledge of theproteostasis network components and interactions forms the neces-sary basis to pursue the understanding of aging. To this end, the in vivoquantification of the network interactions kinetics and their change intime remains is critical and defines themajor challenge that lies ahead.This amounts to parameterize the arrows (as those in Fig. 5) anddeciphering the rules governing their change with time (Fig. 6).Recent advances in quantitative platforms as microscopy, microflui-dics and proteomics can now support such “in vivo Biochemistry”approach. Studies of short-lived organisms amenable to fast geneticmanipulations, can now couple controlled perturbations withindefined environment with quantitative measures of network mem-bers' concentration and activities through time. Whether proteinmisfolding will emerge as a major causative factor of aging remains tobe seen yet through such studies, key contributors to aging can bediscerned [327]. The ground is set for merging predictive modelingand quantitative experimentation, suggesting near future break-throughs in our understanding of aging.
Box-1. Age-related misfolding diseases
Perhaps the most pronounced role of protein aggregations in agingis manifested by their association to cellular degeneracy in manyage-related diseases [23] (Fig. 2). Key examples, mostly afflictingthe central nervous systems, but also muscular tissues and eyes'lens are briefly described below.Alzheimer's disease (AD) is the most prevalent neurodegenerativedisease with around seventeen million patients worldwide [328].The disease symptoms include memory loss and/or dementia.Physiologically, AD is characterized by extracellular amyloidplaques deposition of the proteolitically derived short (40 and 42amino acid forms) amyloid-beta (Aβ) protein in neurites andcerebral blood vessels. The 42-long isomer has a much higheraggregation propensity and its presence is correlated to disease.The other physiological marker of AD is presence of intracellularhallmark neurofibrillary tangles in neuron and glial cells. Theseinclusions are composed of hyperphosphorylated tau proteins (amicrotubule-associated protein), assembled in paired helicalfilaments, twisted ribbons or straight filament. Sporadic PDrepresents 95% of the cases, but mutations leading to developmentof the disease are described and lead to a decrease in the mean ageat onset of 77 years in familial AD and 82 years in sporadic cases[260,329].Parkinson's disease (PD) is the second most common neurodegen-erative disease. PD is the general name given to diseases having thesame symptoms; loss of motor control associated with tremors,muscle rigidity, bradykinesia (slowness in movement), cognitiveimpairment and dementia. These syndromes are correlatedwith thepresence of amyloid Lewy bodies and Lewy neurites. The majorprotein forming these abnormal amyloid fibrils is α-synuclein, ahighly conserved protein expressed mainly in nervous tissues and
particularly in presynaptic terminals. Its function is still uncertainbut it may play a role in synaptic dynamics and particularly inregulation of dopamine transmission and vesicular recycling [90].Many mutations as α-synuclein duplication or triplication or in theparkin gene coding for an E3 ubiquitin ligase, have been identifiedto lead to hereditary PD with autosomal dominant or recessiveinheritance [261].Huntington's disease (HD) is an autosomal dominant neurodegen-erative disorder affecting the brain. HD is characterized bymovement disorder, chorea, and behavioral changes. This diseaseis caused by the extension of the polyQ domain at the N-Terminalof the Huntingtin protein (Htt), leading to its amyloid stableaggregation. The numbers of CAG repeat range from 11 to 35 inhealthy people and from 30 to N70 repeats in patients [330]. Theonset age of the disease is strongly correlated with the number ofrepeats but most onsets take place between the ages of 30 and40 years [331]. HD represents one of the known polyQ diseases;others include spinobulbar muscular atrophy and spinocerebellarataxia.Creutzfeldt–Jakob disease (CDJ) is a rare and fatal neurodegenera-tive disease causing degeneration in the brain and spinal cord,leading to psychiatric disorders like depression, schizophrenia anddementia. CJD is a prion protein disease characterized by atransmissible spongiform encephalopathy caused by an accumula-tion and amyloid aggregation of abnormal isoforms of prionprotein (PrP), a membrane glycosylphosphatidylinositol-anchoredglycoprotein. Sporadic cases of CDJ represent the majority ofoccurrences, joint by familial (mutation in the prion protein gene),and transmissible (cannibalism, ingestion of contaminated animal,or medical errors) cases [332].Aged lens: cataract and presbyopia. Decrease in sight ability is oneof the common characteristics of aging. Presbyopia affects almostall humans by the age of 50. An increase in the stiffness of aginglenses has been observed and correlated with a decrease in freesoluble α-crystallin. α-crystallin is the most abundant protein inthe lens, around 40% of the protein content, and belongs to thefamily of the small heat-shock chaperones [333]. A cataract is adecrease in transparency of the cornea due to formation of stableamyloid fibrils by the crystalline [231]. Cataracts can be caused bymutations, environmental factors, as exposure to UV, or in relationto other diseases (e.g., diabetes and hypertension).Sporadic inclusion-body myositis (s-IMB) the most abundantmuscle disease associated with aging. S-IMB is an inflammatorymyopathy. This disease is characterized by a T-cell inflammatoryinfiltration, cytotoxic necrosis and the presence of congophilicinclusions containing among others, Aβ and phosphorylated tau aswell as other proteins [334].
Acknowledgements
We wish to thank the former and present members of our groupand in particular G. Paul, M. Ni, E.J. Stewart, M. Radman, and F.Taddei for numerous and enriching discussions that provided theframework to this review. Our work on aging is funded by grantsfrom the French National Research Agency (ANR), HFSP and the AxaFoundation.
References
[1] T.B. Kirkwood, A. Kowald, Network theory of aging, Exp. Gerontol. 32 (1997)395–399.

991A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
[2] T.B. Kirkwood, Understanding the odd science of aging, Cell 120 (2005) 437–447.[3] C. Soti, P. Csermely, Aging cellular networks: chaperones as major participants,
Exp. Gerontol. 42 (2007) 113–119.[4] E. Cohen, A. Dillin, The insulin paradox: aging, proteotoxicity and neurodegen-
eration, Nat. Rev. Neurosci. 9 (2008) 759–767.[5] A.L. Hsu, C.T. Murphy, C. Kenyon, Regulation of aging and age-related disease by
DAF-16 and heat-shock factor, Science 300 (2003) 1142–1145.[6] D. Chen, A.D. Steele, G. Hutter, et al., The role of calorie restriction and SIRT1 in
prion-mediated neurodegeneration, Exp. Gerontol. 43 (2008) 1086–1093.[7] L. Gan, L. Mucke, Paths of convergence: sirtuins in aging and neurodegeneration,
Neuron 58 (2008) 10–14.[8] C.B. Anfinsen, Principles that govern folding of protein chains, Science 181 (1973)
223–230.[9] K.A. Dill, S.B. Ozkan, M.S. Shell, T.R. Weikl, The protein folding problem, Annu.
Rev. Biophys. 37 (2008) 289–316.[10] A. Matouschek, J.T. Kellis, L. Serrano, A.R. Fersht, Mapping the transition-state
and pathway of protein folding by protein engineering, Nature 340 (1989)122–126.
[11] A.R. Fersht, From the first protein structures to our current knowledge of proteinfolding: delights and scepticisms, Nat. Rev. Mol. Cell. Biol. 9 (2008) 650–654.
[12] P.G. Wolynes, J.N. Onuchic, D. Thirumalai, Navigating the folding routes, Science267 (1995) 1619–1620.
[13] C.M. Dobson, A. Sali, M. Karplus, Protein folding: a perspective from theory andexperiment, Angew. Chem., Int. Ed. 37 (1998) 868–893.
[14] S. Matysiak, C. Clementi, Mapping folding energy landscapes with theory andexperiment, Arch. Biochem. Biophys. 469 (2008) 29–33.
[15] F. Chiti, C.M. Dobson, Amyloid formation by globular proteins under nativeconditions, Nat. Chem. Biol. 5 (2009) 15–22.
[16] P.L. Clark, Protein folding in the cell: reshaping the folding funnel, TrendsBiochem. Sci. 29 (2004) 527–534.
[17] T.R. Jahn, S.E. Radford, The Yin and Yang of protein folding, FEBS J. 272 (2005)5962–5970.
[18] J.N. Onuchic, Z. Luthey-Schulten, P.G. Wolynes, Theory of protein folding: theenergy landscape perspective, Annu. Rev. Phys. Chem. 48 (1997) 545–600.
[19] R.J. Ellis, A.P. Minton, Protein aggregation in crowded environments, Biol. Chem.387 (2006) 485–497.
[20] G.G. Tartaglia, S. Pechmann, C.M. Dobson, M. Vendruscolo, Life on the edge: a linkbetween gene expression levels and aggregation rates of human proteins, TrendsBiochem. Sci. 32 (2007) 204–206.
[21] A.B. Singleton, M. Farrer, J. Johnson, et al., alpha-Synuclein locus triplicationcauses Parkinson's disease, Science 302 (2003) 841.
[22] D. Foguel, J.L. Silva, New insights into the mechanisms of protein misfolding andaggregation in amyloidogenic diseases derived frompressure studies, Biochemistry43 (2004) 11361–11370.
[23] F. Chiti, C.M. Dobson, Protein misfolding, functional amyloid, and human disease,Annu. Rev. Biochem. 75 (2006) 333–366.
[24] F. Chiti, N. Taddei, F. Baroni, et al., Kinetic partitioning of protein folding andaggregation, Nat. Struct. Biol. 9 (2002) 137–143.
[25] F. Rousseau, J. Schymkowitz, L. Serrano, Protein aggregation and amyloidosis:confusion of the kinds? Curr. Opin. Struct. Biol. 16 (2006) 118–126.
[26] F. Rousseau, L. Serrano, J.W. Schymkowitz, How evolutionary pressure againstprotein aggregation shaped chaperone specificity, J. Mol. Biol. 355 (2006)1037–1047.
[27] J.M. Bui, A. Cavalli, J. Gsponer, Identification of aggregation-prone elements byusing interaction-energy matrices, Angew. Chem. Int. Ed. Engl. 47 (2008)7267–7269.
[28] D.E. Otzen, M. Oliveberg, Salt-induced detour through compact regions ofthe protein folding landscape, Proc. Natl. Acad. Sci. U. S. A. 96 (1999)11746–11751.
[29] E. Monsellier, F. Chiti, Prevention of amyloid-like aggregation as a driving force ofprotein evolution, EMBO Rep. 8 (2007) 737–742.
[30] Y.J. Bollen, I.E. Sanchez, C.P. van Mierlo, Formation of on- and off-pathwayintermediates in the folding kinetics of Azotobacter vinelandii apoflavodoxin,Biochemistry 43 (2004) 10475–10489.
[31] M. Vendruscolo, E. Paci, M. Karplus, C.M. Dobson, Structures and relative freeenergies of partially folded states of proteins, Proc. Natl. Acad. Sci. U. S. A. 100(2003) 14817–14821.
[32] C.J. Roberts, Non-native protein aggregation kinetics, Biotechnol. Bioeng. 98(2007) 927–938.
[33] S. Kumar, J.B. Udgaonkar, Conformational conversion may precede or followaggregate elongation on alternative pathways of amyloid protofibril formation,J. Mol. Biol. 385 (2009) 1266–1276.
[34] R.R. Kopito, Aggresomes, inclusion bodies and protein aggregation, Trends CellBiol. 10 (2000) 524–530.
[35] H. Heise, W. Hoyer, S. Becker, O.C. Andronesi, D. Riedel, M. Baldus, Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR, Proc. Natl. Acad. Sci. U. S. A. 102(2005) 15871–15876.
[36] L.C. Serpell, C.C. Blake, P.E. Fraser, Molecular structure of a fibrillar Alzheimer's Abeta fragment, Biochemistry 39 (2000) 13269–13275.
[37] S. Hess, S.L. Lindquist, T. Scheibel, Alternative assembly pathways of theamyloidogenic yeast prion determinant Sup35-NM, EMBO Rep. 8 (2007)1196–1201.
[38] A.T. Petkova, R.D. Leapman, Z.H. Guo, W.M. Yau, M.P. Mattson, R. Tycko, Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils,Science 307 (2005) 262–265.
[39] C. Goldsbury, P. Frey, V. Olivieri, U. Aebi, S.A. Muller, Multiple assembly pathwaysunderlie amyloid-beta fibril polymorphisms, J. Molec. Biol. 352 (2005) 282–298.
[40] F. Baneyx, M. Mujacic, Recombinant protein folding andmisfolding in Escherichiacoli, Nat. Biotechnol. 22 (2004) 1399–1408.
[41] A.B. Lindner, R. Madden, A. Demarez, E.J. Stewart, F. Taddei, Asymmetricsegregation of protein aggregates is associated with cellular aging andrejuvenation, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 3076–3081.
[42] H. Aguilaniu, L. Gustafsson, M. Rigoulet, T. Nystrom, Asymmetric inheritanceof oxidatively damaged proteins during cytokinesis, Science 299 (2003)1751–1753.
[43] N. Erjavec, L. Larsson, J. Grantham, T. Nystrom, Accelerated aging and failure tosegregate damaged proteins in Sir2 mutants can be suppressed by over-producing the protein aggregation-remodeling factor Hsp104p, Genes Dev. 21(2007) 2410–2421.
[44] N. Erjavec, M. Cvijovic, E. Klipp, T. Nystrom, Selective benefits of damagepartitioning in unicellular systems and its effects on aging, Proc. Natl. Acad.Sci. U. S. A. 105 (2008) 18764–18769.
[45] S. Willingham, T.F. Outeiro, M.J. DeVit, S.L. Lindquist, P.J. Muchowski, Yeast genesthat enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein,Science 302 (2003) 1769–1772.
[46] S. Hamamichi, R.N. Rivas, A.L. Knight, S. Cao, K.A. Caldwell, G.A. Caldwell,Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkin-son's disease model, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 728–733.
[47] K. Iijima, K. Iijima-Ando, Drosophila models of Alzheimer's amyloidosis: thechallenge of dissecting the complex mechanisms of toxicity of amyloid-beta42, J. Alzheimers. Dis. 15 (2008) 523–540.
[48] E. Rockenstein, L. Crews, E. Masliah, Transgenic animal models of neurodegen-erative diseases and their application to treatment development, Adv. Drug.Deliv. Rev. 59 (2007) 1093–1102.
[49] M. Sunde, C. Blake, The structure of amyloid fibrils by electron microscopy andX-ray diffraction, Adv. Protein. Chem. 50 (1997) 123–159.
[50] A.L. Fink, Natively unfolded proteins, Curr. Opin. Struct. Biol. 15 (2005) 35–41.[51] H.J. Dyson, P.E. Wright, Intrinsically unstructured proteins and their functions,
Nat. Rev. Mol. Cell. Biol. 6 (2005) 197–208.[52] J. Gsponer, M.E. Futschik, S.A. Teichmann, M.M. Babu, Tight regulation of
unstructured proteins: from transcript synthesis to protein degradation, Science322 (2008) 1365–1368.
[53] J.W. Kelly, Structural biology: proteins downhill all the way, Nature 442 (2006)255–256.
[54] A.B. Lindner, Z. Eshhar, D.S. Tawfik, Conformational changes affect binding andcatalysis by ester-hydrolysing antibodies, J. Mol. Biol. 285 (1999) 421–430.
[55] N. Tokuriki, D.S. Tawfik, Protein dynamism and evolvability, Science 324 (2009)203–207.
[56] C.A. Ross, M.A. Poirier, Opinion: what is the role of protein aggregation inneurodegeneration? Nat. Rev. Mol. Cell. Biol. 6 (2005) 891–898.
[57] F. Saudou, S. Finkbeiner, D. Devys, M.E. Greenberg, Huntingtin acts in the nucleusto induce apoptosis but death does not correlate with the formation ofintranuclear inclusions, Cell 95 (1998) 55–66.
[58] L. Stefanis, K.E. Larsen, H.J. Rideout, D. Sulzer, L.A. Greene, Expression of A53Tmutant but notwild-type alpha-synuclein in PC12 cells induces alterations of theubiquitin-dependent degradation system, loss of dopamine release, andautophagic cell death, J. Neurosci. 21 (2001) 9549–9560.
[59] R.D. Terry, E. Masliah, D.P. Salmon, et al., Physical basis of cognitive alterations inAlzheimer's disease: synapse loss is the major correlate of cognitive impairment,Ann. Neurol. 30 (1991) 572–580.
[60] M. Arrasate, S. Mitra, E.S. Schweitzer, M.R. Segal, S. Finkbeiner, Inclusion bodyformation reduces levels of mutant huntingtin and the risk of neuronal death,Nature 431 (2004) 805–810.
[61] S. Engelender, Z. Kaminsky, X. Guo, et al., Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions, Nat. Genet. 22(1999) 110–114.
[62] C.A. Gutekunst, S.H. Li, H. Yi, et al., Nuclear and neuropil aggregates in Huntington'sdisease: relationship to neuropathology, J. Neurosci. 19 (1999) 2522–2534.
[63] C. Holmes, D. Boche, D. Wilkinson, et al., Long-term effects of Abeta42immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial, Lancet 372 (2008) 216–223.
[64] A. Abbott, Neuroscience: the plaque plan, Nature 456 (2008) 161–164.[65] B. Caughey, P.T. Lansbury, Protofibrils, pores, fibrils, and neurodegeneration:
separating the responsible protein aggregates from the innocent bystanders,Annu. Rev. Neurosci. 26 (2003) 267–298.
[66] G.M. Shankar, B.L. Bloodgood, M. Townsend, D.M.Walsh, D.J. Selkoe, B.L. Sabatini,Natural oligomers of the Alzheimer amyloid-beta protein induce reversiblesynapse loss by modulating an NMDA-type glutamate receptor-dependentsignaling pathway, J. Neurosci. 27 (2007) 2866–2875.
[67] G.M. Shankar, S. Li, T.H. Mehta, et al., Amyloid-beta protein dimers isolateddirectly from Alzheimer's brains impair synaptic plasticity and memory, Nat.Med. 14 (2008) 837–842.
[68] A.R. Hurshman, J.T. White, E.T. Powers, J.W. Kelly, Transthyretin aggregationunder partially denaturing conditions is a downhill polymerization, Biochem-istry 43 (2004) 7365–7381.
[69] A.J. Modler, K. Gast, G. Lutsch, G. Damaschun, Assembly of amyloid protofibrilsvia critical oligomers—a novel pathway of amyloid formation, J. Mol. Biol. 325(2003) 135–148.
[70] T.R. Serio, A.G. Cashikar, A.S. Kowal, et al., Nucleated conformational conversionand the replication of conformational information by a prion determinant,Science 289 (2000) 1317–1321.

992 A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
[71] A.M. Smith, T.R. Jahn, A.E. Ashcroft, S.E. Radford, Direct observation of oligomericspecies formed in the early stages of amyloid fibril formation using electrosprayionisation mass spectrometry, J. Mol. Biol. 364 (2006) 9–19.
[72] R. Pellarin, E. Guarnera, A. Caflisch, Pathways and intermediates of amyloid fibrilformation, J. Mol. Biol. 374 (2007) 917–924.
[73] N. Carulla, G.L. Caddy, D.R. Hall, et al., Molecular recycling within amyloid fibrils,Nature 436 (2005) 554–558.
[74] D. Thirumalai, D.K. Klimov, R.I. Dima, Emerging ideas on the molecular basis ofprotein and peptide aggregation, Curr. Opin. Struct. Biol. 13 (2003) 146–159.
[75] A.M. Morris, M.A. Watzky, J.N. Agar, R.G. Finke, Fitting neurological proteinaggregation kinetic data via a 2-step, minimal/“Ockham's razor” model: theFinke–Watzky mechanism of nucleation followed by autocatalytic surfacegrowth, Biochemistry 47 (2008) 2413–2427.
[76] R. Kayed, E. Head, J.L. Thompson, et al., Common structure of soluble amyloidoligomers implies common mechanism of pathogenesis, Science 300 (2003)486–489.
[77] D.G. Georganopoulou, L. Chang, J.M. Nam, et al., Nanoparticle-based detection incerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer's disease,Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 2273–2276.
[78] X.P. Wang, J.H. Zhang, Y.J. Wang, et al., Conformation-dependent single-chainvariable fragment antibodies specifically recognize beta-amyloid oligomers,FEBS Lett. (2009).
[79] M. Bucciantini, E. Giannoni, F. Chiti, et al., Inherent toxicity of aggregates impliesa common mechanism for protein misfolding diseases, Nature 416 (2002)507–511.
[80] S. Baglioni, F. Casamenti, M. Bucciantini, et al., Prefibrillar amyloid aggregatescould be generic toxins in higher organisms, J. Neurosci. 26 (2006) 8160–8167.
[81] M.P. Hinault, A. Ben-Zvi, P. Goloubinoff, Chaperones and proteases — cellularfold-controlling factors of proteins in Neurodegenerative diseases and aging,J. Mol. Neurosci. 30 (2006) 249–265.
[82] C.F. Wright, S.A. Teichmann, J. Clarke, C.M. Dobson, The importance of sequencediversity in the aggregation and evolution of proteins, Nature 438 (2005)878–881.
[83] T. Gidalevitz, A. Ben-Zvi, K.H. Ho, H.R. Brignull, R.I. Morimoto, Progressivedisruption of cellular protein folding in models of polyglutamine diseases,Science 311 (2006) 1471–1474.
[84] Y. Chai, J. Shao, V.M. Miller, A. Williams, H.L. Paulson, Live-cell imaging revealsdivergent intracellular dynamics of polyglutamine disease proteins and supportsa sequestration model of pathogenesis, Proc. Natl. Acad. Sci. U. S. A. 99 (2002)9310–9315.
[85] K.K. Chung, Y. Zhang, K.L. Lim, et al., Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation inParkinson disease, Nat. Med. 7 (2001) 1144–1150.
[86] B. Ravikumar, C. Vacher, Z. Berger, et al., Inhibition of mTOR induces autophagyand reduces toxicity of polyglutamine expansions in fly and mouse models ofHuntington disease, Nat. Genet. 36 (2004) 585–595.
[87] R.L. Hamilton, R. Bowser, Alzheimer disease pathology in amyotrophic lateralsclerosis, Acta Neuropathol. 107 (2004) 515–522.
[88] M.S. Forman, M.L. Schmidt, S. Kasturi, D.P. Perl, M. Lee, J.Q. Trojanowski, Tau andalpha-synuclein pathology in amygdala of Parkinsonism-dementia complexpatients of Guam, Am. J. Pathol. 160 (2002) 1725–1731.
[89] I.F. Tsigelny, L. Crews, P. Desplats, Mechanisms of hybrid oligomer formation inthe pathogenesis of combined Alzheimer's and Parkinson's diseases, PLoS ONE 3(2008) e3135.
[90] G.B. Irvine, O.M. El-Agnaf, G.M. Shankar, D.M. Walsh, Protein aggregation in thebrain: the molecular basis for Alzheimer's and Parkinson's diseases, Mol. Med. 14(2008) 451–464.
[91] D.M. Walsh, I. Klyubin, J.V. Fadeeva, et al., Naturally secreted oligomers ofamyloid beta protein potently inhibit hippocampal long-term potentiation invivo, Nature 416 (2002) 535–539.
[92] J.P. Cleary, D.M. Walsh, J.J. Hofmeister, et al., Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function, Nat. Neurosci. 8 (2005)79–84.
[93] B. Calabrese, G.M. Shaked, I.V. Tabarean, J. Braga, E.H. Koo, S. Halpain, Rapid,concurrent alterations in pre- and postsynaptic structure induced by naturally-secreted amyloid-beta protein, Mol. Cell. Neurosci. 35 (2007) 183–193.
[94] D.M. Hartley, D.M. Walsh, C.P. Ye, et al., Protofibrillar intermediates of amyloidbeta-protein induce acute electrophysiological changes and progressive neuro-toxicity in cortical neurons, J. Neurosci. 19 (1999) 8876–8884.
[95] P.N. Lacor, M.C. Buniel, P.W. Furlow, et al., Abeta oligomer-induced aberrations insynapse composition, shape, and density provide a molecular basis for loss ofconnectivity in Alzheimer's disease, J. Neurosci. 27 (2007) 796–807.
[96] L.M. Luheshi, G.G. Tartaglia, A.C. Brorsson, Systematic in vivo analysis of theintrinsic determinants of amyloid Beta pathogenicity, PLoS Biol. 5 (2007) e290.
[97] M.S. Goldberg, P.T. Lansbury, Is there a cause-and-effect relationship betweenalpha-synuclein fibrillization and Parkinson's disease? Nat. Cell. Biol. 2 (2000)E115–119.
[98] M.J. Volles, P.T. Lansbury, Zeroing in on the pathogenic form of alpha-synucleinand its mechanism of neurotoxicity in Parkinson's disease, Biochemistry 42(2003) 7871–7878.
[99] D.P. Smith, D.J. Tew, A.F. Hill, et al., Formation of a high affinity lipid-bindingintermediate during the early aggregation phase of alpha-synuclein, Biochemistry47 (2008) 1425–1434.
[100] J.P. Taylor, F. Tanaka, J. Robitschek, et al., Aggresomes protect cells by enhancingthe degradation of toxic polyglutamine-containing protein, Hum. Mol. Genet. 12(2003) 749–757.
[101] M. Anguiano, R.J. Nowak, P.T. Lansbury, Protofibrillar islet amyloid polypeptidepermeabilizes synthetic vesicles by a pore-like mechanism that may be relevantto type II diabetes, Biochemistry 41 (2002) 11338–11343.
[102] M. Bucciantini, G. Calloni, F. Chiti, et al., Prefibrillar amyloid proteinaggregates share common features of cytotoxicity, J. Biol. Chem. 279 (2004)31374–31382.
[103] W.A. Pedersen, C. Culmsee, D. Ziegler, J.P. Herman, M.P. Mattson, Aberrant stressresponse associated with severe hypoglycemia in a transgenic mouse model ofAlzheimer's disease, J. Mol. Neurosci. 13 (1999) 159–165.
[104] A.L. Orr, S. Li, C.E. Wang, et al., N-terminal mutant huntingtin associates withmitochondria and impairs mitochondrial trafficking, J. Neurosci. 28 (2008)2783–2792.
[105] D. Schubert, C. Behl, R. Lesley, et al., Amyloid peptides are toxic via a commonoxidative mechanism, Proc. Natl. Acad. Sci. U. S. A. 92 (1995) 1989–1993.
[106] A. Relini, S. Torrassa, R. Rolandi, et al., Monitoring the process of HypFfibrillization and liposome permeabilization by protofibrils, J. Mol. Biol. 338(2004) 943–957.
[107] C.G. Glabe, Common mechanisms of amyloid oligomer pathogenesis indegenerative disease, Neurobiol. Aging. 27 (2006) 570–575.
[108] T.A. Mirzabekov, M.C. Lin, B.L. Kagan, Pore formation by the cytotoxic isletamyloid peptide amylin, J. Biol. Chem. 271 (1996) 1988–1992.
[109] B.L. Kagan, Amyloidosis and protein folding, Science 307 (2005) 42–43 authorreply 42–43.
[110] F. Kamenetz, T. Tomita, H. Hsieh, et al., APP processing and synaptic function,Neuron 37 (2003) 925–937.
[111] M. Townsend, G.M. Shankar, T. Mehta, D.M. Walsh, D.J. Selkoe, Effects of secretedoligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potentrole for trimers, J. Physiol. 572 (2006) 477–492.
[112] C. Haass, D.J. Selkoe, Soluble protein oligomers in neurodegeneration: lessonsfrom the Alzheimer's amyloid beta-peptide, Nat. Rev. Mol. Cell. Biol. 8 (2007)101–112.
[113] Y. Chu, J.H. Kordower, Age-associated increases of alpha-synuclein in monkeysand humans are associated with nigrostriatal dopamine depletion: is this thetarget for Parkinson's disease? Neurobiol. Dis. 25 (2007) 134–149.
[114] T. Maier, L. Ferbitz, E. Deuerling, N. Ban, A cradle for new proteins: trigger factorat the ribosome, Curr. Opin. Struct. Biol. 15 (2005) 204–212.
[115] S. Wickner, M.R. Maurizi, S. Gottesman, Posttranslational quality control: folding,refolding, and degrading proteins, Science 286 (1999) 1888–1893.
[116] U. Schubert, L.C. Anton, J. Gibbs, C.C. Norbury, J.W. Yewdell, J.R. Bennink, Rapiddegradation of a large fraction of newly synthesized proteins by proteasomes,Nature 404 (2000) 770–774.
[117] S. Gottesman, S. Wickner, M.R. Maurizi, Protein quality control: triage bychaperones and proteases, Genes Dev. 11 (1997) 815–823.
[118] C. Soti, P. Csermely, Aging and molecular chaperones, Exp. Gerontol. 38 (2003)1037–1040.
[119] W.E. Balch, R.I. Morimoto, A. Dillin, J.W. Kelly, Adapting proteostasis for diseaseintervention, Science 319 (2008) 916–919.
[120] M.Morange,What history tells us II. The discovery of chaperone function, J. Biosci.30 (2005) 461–464.
[121] R.J. Ellis, Proteins as molecular chaperones, Nature 328 (1987) 378–379.[122] R.J. Ellis, Molecular chaperones. Opening and closing the Anfinsen cage, Curr.
Biol. 4 (1994) 633–635.[123] R.I. Morimoto, Proteotoxic stress and inducible chaperone networks in
neurodegenerative disease and aging, Genes Dev. 22 (2008) 1427–1438.[124] S. Lindquist, The heat-shock response, Annu. Rev. Biochem. 55 (1986) 1151–1191.[125] A.D. Grossman, J.W. Erickson, C.A. Gross, The htpR gene product of E. coli is a
sigma factor for heat-shock promoters, Cell 38 (1984) 383–390.[126] L. Pirkkala, P. Nykanen, L. Sistonen, Roles of the heat shock transcription factors
in regulation of the heat shock response and beyond, FASEB J. 15 (2001)1118–1131.
[127] J. Anckar, L. Sistonen, Heat shock factor 1 as a coordinator of stress anddevelopmental pathways, Adv. Exp. Med. Biol. 594 (2007) 78–88.
[128] P. Csermely, T. Korcsmaros, I. Kovacs, M. Szalay, C. Soti, Systems biology ofmolecular chaperone networks, Novartis Found. Symp. 291 (2008) 54–58.
[129] E. Rousseau, B. Dehay, L. Ben-Haiem, Y. Trottier, M. Morange, A. Bertolotti,Targeting expression of expanded polyglutamine proteins to the endoplasmicreticulumormitochondria prevents their aggregation, Proc. Natl. Acad. Sci. U. S. A.101 (2004) 9648–9653.
[130] S. Walter, J. Buchner, Molecular chaperones—cellular machines for proteinfolding, Angew. Chem. Int. Ed. Engl. 41 (2002) 1098–1113.
[131] P.J. Muchowski, J.L. Wacker, Modulation of neurodegeneration by molecularchaperones, Nat. Rev. Neurosci. 6 (2005) 11–22.
[132] K. Liberek, A. Lewandowska, S. Zietkiewicz, Chaperones in control of proteindisaggregation, EMBO J. 27 (2008) 328–335.
[133] F.U. Hartl, M. Hayer-Hartl, Molecular chaperones in the cytosol: from nascentchain to folded protein, Science 295 (2002) 1852–1858.
[134] M.J. Welsh, M. Gaestel, Small heat-shock protein family: function in health anddisease, Ann. N.Y. Acad. Sci. 851 (1998) 28–35.
[135] U. Jakob, M. Gaestel, K. Engel, J. Buchner, Small heat shock proteins are molecularchaperones, J. Biol. Chem. 268 (1993) 1517–1520.
[136] S.P. Allen, J.O. Polazzi, J.K. Gierse, A.M. Easton, Two novel heat shock genesencoding proteins produced in response to heterologous protein expression inEscherichia coli, J. Bacteriol. 174 (1992) 6938–6947.
[137] A. Mogk, E. Deuerling, S. Vorderwulbecke, E. Vierling, B. Bukau, Small heat shockproteins, ClpB and the DnaK system form a functional triade in reversing proteinaggregation, Mol. Microbiol. 50 (2003) 585–595.

993A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
[138] A.G. Cashikar, M. Duennwald, S.L. Lindquist, A chaperone pathway in proteindisaggregation. Hsp26 alters the nature of protein aggregates to facilitatereactivation by Hsp104, J. Biol. Chem. 280 (2005) 23869–23875.
[139] E. Ratajczak, S. Zietkiewicz, K. Liberek, Distinct activities of Escherichia colismall heat shock proteins IbpA and IbpB promote efficient proteindisaggregation, J. Mol. Biol. (2008).
[140] Y. Sun, T.H. MacRae, Small heat shock proteins: molecular structure andchaperone function, Cell. Mol. Life Sci. 62 (2005) 2460–2476.
[141] M. Ilbert, J. Horst, S. Ahrens, et al., The redox-switch domain of Hsp33 functionsas dual stress sensor, Nat. Struct. Mol. Biol. 14 (2007) 556–563.
[142] A. Horovitz, Structural aspects of GroEL function, Curr. Opin. Struct. Biol. 8 (1998)93–100.
[143] B. Bukau, A.L. Horwich, The Hsp70 and Hsp60 chaperone machines, Cell 92(1998) 351–366.
[144] M.P. Mayer, B. Bukau, Hsp70 chaperones: cellular functions and molecularmechanism, Cell. Mol. Life Sci. 62 (2005) 670–684.
[145] P. Goloubinoff, P. De Los Rios, The mechanism of Hsp70 chaperones: (entropic)pulling the models together, Trends Biochem. Sci. 32 (2007) 372–380.
[146] J.C. Bardwell, K. Tilly, E. Craig, J. King, M. Zylicz, C. Georgopoulos, The nucleotidesequence of the Escherichia coli K12 dnaJ+ gene. A gene that encodes a heatshock protein, J. Biol. Chem. 261 (1986) 1782–1785.
[147] C. Harrison, GrpE, a nucleotide exchange factor for DnaK, Cell Stress Chaperones8 (2003) 218–224.
[148] S. Alberti, C. Esser, J. Hohfeld, BAG-1—a nucleotide exchange factor of Hsc70 withmultiple cellular functions, Cell Stress Chaperones 8 (2003) 225–231.
[149] V. Arndt, C. Rogon, J. Hohfeld, To be, or not to be—molecular chaperones inprotein degradation, Cell. Mol. Life Sci. 64 (2007) 2525–2541.
[150] O.O. Odunuga, V.M. Longshaw, G.L. Blatch, Hop: more than an Hsp70/Hsp90adaptor protein, Bioessays 26 (2004) 1058–1068.
[151] M.P. Mayer, C. Prodromou, J. Frydman, The Hsp90mosaic: a picture emerges, Nat.Struct. Mol. Biol. 16 (2009) 2–6.
[152] L. Whitesell, S.L. Lindquist, HSP90 and the chaperoning of cancer, Nat. Rev.Cancer 5 (2005) 761–772.
[153] A.J. McClellan, Y. Xia, A.M. Deutschbauer, R.W. Davis, M. Gerstein, J. Frydman,Diverse cellular functions of the Hsp90 molecular chaperone uncovered usingsystems approaches, Cell 131 (2007) 121–135.
[154] R. Zhao, M. Davey, Y.C. Hsu, et al., Navigating the chaperone network: anintegrative map of physical and genetic interactions mediated by the hsp90chaperone, Cell 120 (2005) 715–727.
[155] L.H. Pearl, C. Prodromou, P. Workman, The Hsp90 molecular chaperone: an openand shut case for treatment, Biochem. J. 410 (2008) 439–453.
[156] S. Rutherford, Y. Hirate, B.J. Swalla, The Hsp90 capacitor, developmentalremodeling, and evolution: the robustness of gene networks and the curiousevolvability of metamorphosis, Crit. Rev. Biochem. Mol. Biol. 42 (2007) 355–372.
[157] S.L. Rutherford, S. Lindquist, Hsp90 as a capacitor for morphological evolution,Nature 396 (1998) 336–342.
[158] T.A. Sangster, N. Salathia, S. Undurraga, et al., HSP90 affects the expression ofgenetic variation and developmental stability in quantitative traits, Proc. Natl.Acad. Sci. U. S. A. 105 (2008) 2963–2968.
[159] B. Bosl, V. Grimminger, S. Walter, The molecular chaperone Hsp104—a molecularmachine for protein disaggregation, J. Struct. Biol. 156 (2006) 139–148.
[160] J.R. Glover, S. Lindquist, Hsp104, Hsp70, and Hsp40: a novel chaperone systemthat rescues previously aggregated proteins, Cell 94 (1998) 73–82.
[161] P. Tessarz, A. Mogk, B. Bukau, Substrate threading through the central pore of theHsp104 chaperone as a common mechanism for protein disaggregation andprion propagation, Mol. Microbiol. 68 (2008) 87–97.
[162] M. Arimon, V. Grimminger, F. Sanz, H.A. Lashuel, Hsp104 targets multipleintermediates on the amyloid pathway and suppresses the seeding capacity ofAbeta fibrils and protofibrils, J. Mol. Biol. 384 (2008) 1157–1173.
[163] C. Lo Bianco, J. Shorter, E. Regulier, et al., Hsp104 antagonizes alpha-synucleinaggregation and reduces dopaminergic degeneration in a rat model of Parkinsondisease, J. Clin. Invest. 118 (2008) 3087–3097.
[164] A.J. Macario, E. Conway de Macario, Sick chaperones, cellular stress, and disease,N. Engl. J. Med. 353 (2005) 1489–1501.
[165] A. Gutsmann-Conrad, A.R. Heydari, S. You, A. Richardson, The expression of heatshock protein 70 decreases with cellular senescence in vitro and in cells derivedfrom young and old human subjects, Exp. Cell. Res. 241 (1998) 404–413.
[166] R. Singh, S. Kolvraa, P. Bross, et al., Reduced heat shock response in humanmononuclear cells during aging and its association with polymorphisms inHSP70 genes, Cell Stress Chaperones 11 (2006) 208–215.
[167] G.K. Sprang, R. Brown, Selective induction of a heat shock gene in fibre tracts andcerebellar neurons of the rabbit brain detected by in situ hybridization, Brain Res.427 (1987) 89–93.
[168] I. Shamovsky, D. Gershon, Novel regulatory factors of HSF-1 activation: facts andperspectives regarding their involvement in the age-associated attenuation ofthe heat shock response, Mech. Ageing. Dev. 125 (2004) 767–775.
[169] J.F. Morley, R.I. Morimoto, Regulation of longevity in Caenorhabditis elegans byheat shock factor and molecular chaperones, Mol. Biol. Cell. 15 (2004) 657–664.
[170] I. Shamovsky, M. Ivannikov, E.S. Kandel, D. Gershon, E. Nudler, RNA-mediatedresponse to heat shock in mammalian cells, Nature 440 (2006) 556–560.
[171] S.L. Bernstein, A.M. Liu, B.C. Hansen, R.I. Somiari, Heat shock cognate-70 geneexpression declines during normal aging of the primate retina, Invest.Ophthalmol. Vis. Sci. 41 (2000) 2857–2862.
[172] J.W. Starnes, A.M. Choilawala, R.P. Taylor, M.J. Nelson, M.D. Delp, Myocardial heatshock protein 70 expression in young and old rats after identical exerciseprograms, J. Gerontol. A Biol. Sci. Med. Sci. 60 (2005) 963–969.
[173] G. Nardai, P. Csermely, C. Soti, Chaperone function and chaperone overload in theaged. A preliminary analysis, Exp. Gerontol. 37 (2002) 1257–1262.
[174] R.R. Erickson, L.M. Dunning, J.L. Holtzman, The effect of aging on the chaperoneconcentrations in the hepatic, endoplasmic reticulum of male rats: the possiblerole of protein misfolding due to the loss of chaperones in the decline inphysiological function seen with age, J. Gerontol. A Biol. Sci. Med. Sci. 61 (2006)435–443.
[175] R. Ambra, E. Mocchegiani, R. Giacconi, et al., Characterization of the hsp70response in lymphoblasts from aged and centenarian subjects and differ-ential effects of in vitro zinc supplementation, Exp. Gerontol. 39 (2004)1475–1484.
[176] B.K. Derham, J.J. Harding, The effects of ageing on the chaperone-like function ofrabbit alpha-crystallin, comparing three methods of assay, Biochim. Biophys.Acta 1336 (1997) 187–194.
[177] R.J. Kapphahn, C.M. Ethen, E.A. Peters, L. Higgins, D.A. Ferrington, Modified alphaA crystallin in the retina: altered expression and truncation with aging,Biochemistry 42 (2003) 15310–15325.
[178] J. Horwitz, T. Emmons, L. Takemoto, The ability of lens alpha crystallin to protectagainst heat-induced aggregation is age-dependent, Curr. Eye Res. 11 (1992)817–822.
[179] C. Zabel, D.C. Chamrad, J. Priller, et al., Alterations in the mouse and humanproteome caused by Huntington's disease,Mol. Cell. Proteomics 1 (2002) 366–375.
[180] S. Kim, E.A. Nollen, K. Kitagawa, V.P. Bindokas, R.I. Morimoto, Polyglutamineprotein aggregates are dynamic, Nat. Cell. Biol. 4 (2002) 826–831.
[181] S. Bagriantsev, S. Liebman, Modulation of Abeta42 low-n oligomerization using anovel yeast reporter system, BMC Biol. 4 (2006) 32.
[182] T. von der Haar, L. Josse, P. Wright, J. Zenthon, M.F. Tuite, Development of a novelyeast cell-based system for studying the aggregation of Alzheimer's disease-associated Abeta peptides in vivo, Neurodegener. Dis. 4 (2007) 136–147.
[183] V. Fonte, D.R. Kipp, J. Yerg, et al., Suppression of in vivo beta-amyloid peptidetoxicity by overexpression of the HSP-16.2 small chaperone protein, J. Biol. Chem.283 (2008) 784–791.
[184] J.A. McLear, D. Lebrecht, A. Messer, W.J. Wolfgang, Combinational approach ofintrabody with enhanced Hsp70 expression addresses multiple pathologies in afly model of Huntington's disease, FASEB J. 22 (2008) 2003–2011.
[185] C.J. Cummings, Y. Sun, P. Opal, et al., Over-expression of inducible HSP70chaperone suppresses neuropathology and improves motor function in SCA1mice, Hum. Mol. Genet. 10 (2001) 1511–1518.
[186] P.J. Muchowski, Protein misfolding, amyloid formation, and neurodegeneration:a critical role for molecular chaperones? Neuron 35 (2002) 9–12.
[187] G. Morrow, M. Samson, S. Michaud, R.M. Tanguay, Overexpression of the smallmitochondrial Hsp22 extends Drosophila life span and increases resistance tooxidative stress, FASEB J. 18 (2004) 598–599.
[188] G. Morrow, S. Battistini, P. Zhang, R.M. Tanguay, Decreased lifespan in theabsence of expression of the mitochondrial small heat shock protein Hsp22 inDrosophila, J. Biol. Chem. 279 (2004) 43382–43385.
[189] N. Minois, S. Vaynberg, Fecundity and life span in transgenic Drosophilamelanogaster overexpressing hsp70, Biogerontology 3 (2002) 301–306.
[190] J.H. Feder, J.M. Rossi, J. Solomon, N. Solomon, S. Lindquist, The consequences ofexpressing hsp70 in Drosophila cells at normal temperatures, Genes Dev. 6(1992) 1402–1413.
[191] R.A. Krebs, M.E. Feder, Deleterious consequences of Hsp70 overexpression inDrosophila melanogaster larvae, Cell Stress Chaperones 2 (1997) 60–71.
[192] M. Tatar, A.A. Khazaeli, J.W. Curtsinger, Chaperoning extended life, Nature 390(1997) 30.
[193] S.L. Rea, D. Wu, J.R. Cypser, J.W. Vaupel, T.E. Johnson, A stress-sensitive reporterpredicts longevity in isogenic populations of Caenorhabditis elegans, Nat. Genet.37 (2005) 894–898.
[194] G.J. Lithgow, T.M. White, S. Melov, T.E. Johnson, Thermotolerance and extendedlife-span conferred by single-gene mutations and induced by thermal stress,Proc. Natl. Acad. Sci. U. S. A. 92 (1995) 7540–7544.
[195] M.J. Hercus, V. Loeschcke, S.I. Rattan, Lifespan extension of Drosophilamelanogaster through hormesis by repeated mild heat stress, Biogerontology 4(2003) 149–156.
[196] Rattan-SIS., Hormetic modulation of aging and longevity by mild heat stress,Dose-Response 3 (2005) 533–546.
[197] E.J. Masoro, The role of hormesis in life extension by dietary restriction,Interdiscip. Top. Gerontol. 35 (2007) 1–17.
[198] D. Gems, L. Partridge, Stress–response hormesis and aging: “that which does notkill us makes us stronger”, Cell. Metabolism. 7 (2008) 200–203.
[199] K.P. Lu, G. Finn, T.H. Lee, L.K. Nicholson, Prolyl cis-trans isomerization as amolecular timer, Nat. Chem. Biol. 3 (2007) 619–629.
[200] P.J. Lu, G.Wulf, X.Z. Zhou, P. Davies, K.P. Lu, The prolyl isomerase Pin1 restores thefunction of Alzheimer-associated phosphorylated tau protein, Nature 399 (1999)784–788.
[201] D.A. Butterfield, H.M. Abdul, W. Opii, et al., Pin1 in Alzheimer's disease,J. Neurochem. 98 (2006) 1697–1706.
[202] L. Pastorino, A. Sun, P.J. Lu, et al., The prolyl isomerase Pin1 regulates amyloidprecursor protein processing and amyloid-beta production, Nature 440 (2006)528–534.
[203] B.S. Mamathambika, J.C. Bardwell, Disulfide-linked protein folding pathways,Annu. Rev. Cell. Dev. Biol. 24 (2008) 211–235.
[204] B. Wilkinson, H.F. Gilbert, Protein disulfide isomerase, Biochim. Biophys. Acta1699 (2004) 35–44.
[205] C. Christis, N.H. Lubsen, I. Braakman, Protein folding includes oligomerization —
examples from the endoplasmic reticulum and cytosol, Febs J. 275 (2008) 4700–4727.

994 A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
[206] T. Uehara, T. Nakamura, D. Yao, et al., S-nitrosylated protein-disulphideisomerase links protein misfolding to neurodegeneration, Nature 441 (2006)513–517.
[207] A. Hershko, The ubiquitin system for protein degradation and some of its roles inthe control of the cell-division cycle (Nobel lecture), Angew. Chem. Int. Ed. Engl.44 (2005) 5932–5943.
[208] M.H. Glickman, A. Ciechanover, The ubiquitin–proteasome proteolytic pathway:destruction for the sake of construction, Physiol. Rev. 82 (2002) 373–428.
[209] A.L. Goldberg, Protein degradation and protection against misfolded or damagedproteins, Nature 426 (2003) 895–899.
[210] K. Saito, J.S. Elce, J.E. Hamos, R.A. Nixon, Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: apotential molecular basis for neuronal degeneration, Proc. Natl. Acad. Sci. U. S. A.90 (1993) 2628–2632.
[211] H. Manya, M. Inomata, T. Fujimori, et al., Klotho protein deficiency leads tooveractivation of mu-calpain, J. Biol. Chem. 277 (2002) 35503–35508.
[212] M. Benuck, M. Banay-Schwartz, T. DeGuzman, A. Lajtha, Changes in brainprotease activity in aging, J. Neurochem. 67 (1996) 2019–2029.
[213] M.D. Covington, D.D. Arrington, R.G. Schnellmann, Calpain 10 is required for cellviability and is decreased in the aging kidney, Am. J. Physiol. Renal. Physiol. (2009).
[214] S. Paul, Dysfunction of the ubiquitin–proteasome system in multiple diseaseconditions: therapeutic approaches, Bioessays 30 (2008) 1172–1184.
[215] S.S. Vembar, J.L. Brodsky, One step at a time: endoplasmic reticulum-associateddegradation, Nat. Rev. Mol. Cell. Biol. 9 (2008) 944–957.
[216] D.H. Wolf, W. Hilt, The proteasome: a proteolytic nanomachine of cell regulationand waste disposal, Biochim. Biophys. Acta 1695 (2004) 19–31.
[217] A. Hershko, A. Ciechanover, The ubiquitin system, Annu. Rev. Biochem. 67 (1998)425–479.
[218] C. Herman, R. D'Ari, Proteolysis and chaperones: the destruction/reconstructiondilemma, Curr. Opin. Microbiol. 1 (1998) 204–209.
[219] C. Yun, A. Stanhill, Y. Yang, et al., Proteasomal adaptation to environmental stresslinks resistance to proteotoxicity with longevity in Caenorhabditis elegans, Proc.Natl. Acad. Sci. U. S. A. 105 (2008) 7094–7099.
[220] W. Li, B. Gao, S.M. Lee, K. Bennett, D. Fang, RLE-1, an E3 ubiquitin ligase, regulatesC. elegans aging by catalyzing DAF-16 polyubiquitination, Dev. Cell 12 (2007)235–246.
[221] A.J. McClellan, S. Tam, D. Kaganovich, J. Frydman, Protein quality control:chaperones culling corrupt conformations, Nat. Cell. Biol. 7 (2005) 736–741.
[222] J.N. Min, R.A. Whaley, N.E. Sharpless, P. Lockyer, A.L. Portbury, C. Patterson,CHIP deficiency decreases longevity, with accelerated aging phenotypesaccompanied by altered protein quality control, Mol. Cell. Biol. 28 (2008)4018–4025.
[223] R. Rosenzweig, M.H. Glickman, Chaperone-driven proteasome assembly, Bio-chem. Soc. Trans. 36 (2008) 807–812.
[224] A. Ciechanover, The ubiquitin proteolytic system: from a vague idea, throughbasic mechanisms, and onto human diseases and drug targeting, Neurology 66(2006) S7–19.
[225] G. Carrard, A.L. Bulteau, I. Petropoulos, B. Friguet, Impairment of proteasomestructure and function in aging, Int. J. Biochem. Cell. Biol. 34 (2002) 1461–1474.
[226] N. Chondrogianni, E.S. Gonos, Proteasome dysfunction in mammalian aging:steps and factors involved, Exp. Gerontol. 40 (2005) 931–938.
[227] B. Dahlmann, Role of proteasomes in disease, BMC Biochem. 8 (Suppl. 1) (2007)S3.
[228] T. Grune, T. Jung, K. Merker, K.J. Davies, Decreased proteolysis caused by proteinaggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ duringoxidative stress, aging, and disease, Int. J. Biochem. Cell Biol. 36 (2004)2519–2530.
[229] V.A. Vernace, T. Schmidt-Glenewinkel, M.E. Figueiredo-Pereira, Aging andregulated protein degradation: who has the UPPer hand? Aging Cell 6 (2007)599–606.
[230] N. Sitte, K. Merker, T. Von Zglinicki, T. Grune, K.J. Davies, Protein oxidation anddegradation during cellular senescence of human BJ fibroblasts: part I—effects ofproliferative senescence, FASEB J. 14 (2000) 2495–2502.
[231] M. Zetterberg, A. Petersen, J. Sjostrand, J. Karlsson, Proteasome activity in humanlens nuclei and correlation with age, gender and severity of cataract, Curr. EyeRes. 27 (2003) 45–53.
[232] V. Askanas, W.K. Engel, Inclusion-body myositis: a myodegenerative conforma-tional disorder associated with Abeta, protein misfolding, and proteasomeinhibition, Neurology 66 (2006) S39–48.
[233] A.L. Bulteau, M. Moreau, C. Nizard, B. Friguet, Proteasome and photoaging: theeffects of UV irradiation, Ann. N.Y. Acad. Sci. 1100 (2007) 280–290.
[234] F. Li, L. Zhang, J. Craddock, et al., Aging and dietary restriction effects onubiquitination, sumoylation, and the proteasome in the heart, Mech. Ageing.Dev. 129 (2008) 515–521.
[235] J. Herrmann, S.M. Soares, L.O. Lerman, A. Lerman, Potential role of the ubiquitin–proteasome system in atherosclerosis: aspects of a protein quality disease, J. Am.Coll. Cardiol. 51 (2008) 2003–2010.
[236] J.N. Keller, F.F. Huang, W.R. Markesbery, Decreased levels of proteasome activityand proteasome expression in aging spinal cord, Neuroscience 98 (2000)149–156.
[237] A. Ciechanover, P. Brundin, The ubiquitin proteasome system in neurodegen-erative diseases: sometimes the chicken, sometimes the egg, Neuron 40 (2003)427–446.
[238] C.I. Holmberg, K.E. Staniszewski, K.N. Mensah, A. Matouschek, R.I. Morimoto,Inefficient degradation of truncated polyglutamine proteins by the proteasome,EMBO J. 23 (2004) 4307–4318.
[239] P. Venkatraman, R. Wetzel, M. Tanaka, N. Nukina, A.L. Goldberg, Eukaryoticproteasomes cannot digest polyglutamine sequences and release them duringdegradation of polyglutamine-containing proteins, Mol. Cell. 14 (2004) 95–104.
[240] E.J. Bennett, T.A. Shaler, B. Woodman, et al., Global changes to the ubiquitinsystem in Huntington's disease, Nature 448 (2007) 704–708.
[241] M.L. Duennwald, S. Lindquist, Impaired ERAD and ER stress are early and specificevents in polyglutamine toxicity, Genes Dev. 22 (2008) 3308–3319.
[242] N.F. Bence, R.M. Sampat, R.R. Kopito, Impairment of the ubiquitin–proteasomesystem by protein aggregation, Science 292 (2001) 1552–1555.
[243] Y.A. Lam, C.M. Pickart, A. Alban, et al., Inhibition of the ubiquitin–proteasomesystem in Alzheimer's disease, Proc. Natl. Acad. Sci. U. S. A. 97 (2000) 9902–9906.
[244] H. Snyder, K. Mensah, C. Theisler, J. Lee, A. Matouschek, B. Wolozin, Aggregatedand monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibitproteasomal function, J. Biol. Chem. 278 (2003) 11753–11759.
[245] J.M. Gruschus, Do amyloid oligomers act as traps for misfolded proteins? Ahypothesis, Amyloid 15 (2008) 160–165.
[246] A.M. Cuervo, J.F. Dice, How do intracellular proteolytic systems change with age?Front Biosci. 3 (1998) d25–d43.
[247] E.L. Eskelinen, New insights into the mechanisms of macroautophagy inmammalian cells, Int. Rev. Cell. Mol. Biol. 266 (2008) 207–247.
[248] M. Kundu, C.B. Thompson, Autophagy: basic principles and relevance to disease,Annu. Rev. Pathol. 3 (2008) 427–455.
[249] N.B. Nedelsky, P.K. Todd, J.P. Taylor, Autophagy and the ubiquitin–proteasomesystem: collaborators in neuroprotection, Biochim. Biophys. Acta 1782 (2008)691–699.
[250] J.F. Dice, Chaperone-mediated autophagy, Autophagy 3 (2007) 295–299.[251] J. Ahlberg, L. Marzella, H. Glaumann, Uptake and degradation of proteins by
isolated rat liver lysosomes. Suggestion of a microautophagic pathway ofproteolysis, Lab. Invest. 47 (1982) 523–532.
[252] S.E. Lenk, W.A. Dunn, J.S. Trausch, A. Ciechanover, A.L. Schwartz, Ubiquitin-activating enzyme, E1, is associated with maturation of autophagic vacuoles,J. Cell. Biol. 118 (1992) 301–308.
[253] C. Zhang, A.M. Cuervo, Restoration of chaperone-mediated autophagy in agingliver improves cellular maintenance and hepatic function, Nat. Med. 14 (2008)959–965.
[254] A.M. Cuervo, L. Stefanis, R. Fredenburg, P.T. Lansbury, D. Sulzer, Impaireddegradation of mutant alpha-synuclein by chaperone-mediated autophagy,Science 305 (2004) 1292–1295.
[255] W.H. Yu, A.M. Cuervo, A. Kumar, et al., Macroautophagy—a novel Beta-amyloidpeptide-generating pathway activated in Alzheimer's disease, J. Cell. Biol. 171(2005) 87–98.
[256] B. Ravikumar, A. Acevedo-Arozena, S. Imarisio, et al., Dynein mutations impairautophagic clearance of aggregate-prone proteins, Nat. Genet. 37 (2005)771–776.
[257] T. Hara, K. Nakamura, M. Matsui, et al., Suppression of basal autophagy in neuralcells causes neurodegenerative disease in mice, Nature 441 (2006) 885–889.
[258] M. Komatsu, S. Waguri, T. Chiba, et al., Loss of autophagy in the central nervoussystem causes neurodegeneration in mice, Nature 441 (2006) 880–884.
[259] D. Narendra, A. Tanaka, D.F. Suen, R.J. Youle, Parkin is recruited selectively toimpaired mitochondria and promotes their autophagy, J. Cell. Biol. 183 (2008)795–803.
[260] L. Bertram, R.E. Tanzi, Thirty years of Alzheimer's disease genetics: theimplications of systematic meta-analyses, Nat. Rev. Neurosci. 9 (2008) 768–778.
[261] S. Biskup, M. Gerlach, A. Kupsch, et al., Genes associated with Parkinsonsyndrome, J. Neurol. 255 (Suppl. 5) (2008) 8–17.
[262] F. Coppede, M. Mancuso, G. Siciliano, L. Migliore, L. Murri, Genes and theenvironment in neurodegeneration, Biosci. Rep. 26 (2006) 341–367.
[263] M. Katsuno, H. Banno, K. Suzuki, et al., Molecular genetics and biomarkers ofpolyglutamine diseases, Curr. Mol. Med. 8 (2008) 221–234.
[264] V.N. Uversky, Alpha-synuclein misfolding and neurodegenerative diseases, Curr.Protein Pept. Sci. 9 (2008) 507–540.
[265] D.A. Erie, O. Hajiseyedjavadi, M.C. Young, P.H. von Hippel, Multiple RNApolymerase conformations and GreA: control of the fidelity of transcription,Science 262 (1993) 867–873.
[266] N. Alic, N. Ayoub, E. Landrieux, et al., Selectivity and proofreading both contributesignificantly to the fidelity of RNA polymerase III transcription, Proc. Natl. Acad.Sci. U. S. A. 104 (2007) 10400–10405.
[267] N.K. Nesser, D.O. Peterson, D.K. Hawley, RNA polymerase II subunit Rpb9 isimportant for transcriptional fidelity in vivo, Proc. Natl. Acad. Sci. U. S. A. 103(2006) 3268–3273.
[268] P. Edelmann, J. Gallant, Mistranslation in E. coli, Cell 10 (1977) 131–137.[269] H.S. Zaher, R. Green, Quality control by the ribosome following peptide bond
formation, Nature 457 (2009) 161–166.[270] J.M. Ogle, V. Ramakrishnan, Structural insights into translational fidelity, Annu.
Rev. Biochem. 74 (2005) 129–177.[271] D.A. Drummond, C.O. Wilke, Mistranslation-induced protein misfolding as a
dominant constraint on coding-sequence evolution, Cell 134 (2008) 341–352.[272] P. Cortazzo, C. Cervenansky, M. Marin, C. Reiss, R. Ehrlich, A. Deana, Silent
mutations affect in vivo protein folding in Escherichia coli, Biochem. Biophys. Res.Commun. 293 (2002) 537–541.
[273] C. Kimchi-Sarfaty, J.M. Oh, W. Kim, et al., A “silent” polymorphism in the MDR1gene changes substrate specificity, Science 315 (2007) 525–528.
[274] T. Shimizu, Y. Matsuoka, T. Shirasawa, Biological significance of isoaspartate andits repair system, Biol. Pharm. Bull. 28 (2005) 1590–1596.
[275] K. Murakami, M. Uno, Y. Masuda, T. Shimizu, T. Shirasawa, K. Irie, Isomerizationand/or racemization at Asp23 of Abeta42 do not increase its aggregative ability,

995A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
neurotoxicity, and radical productivity in vitro, Biochem. Biophys. Res. Commun.366 (2008) 745–751.
[276] J. Orpiszewski, N. Schormann, B. Kluve-Beckerman, J.J. Liepnieks, M.D.Benson, Protein aging hypothesis of Alzheimer disease, FASEB J. 14 (2000)1255–1263.
[277] T. Takata, J.T. Oxford, B. Demeler, K.J. Lampi, Deamidation destabilizes andtriggers aggregation of a lens protein, betaA3-crystallin, Protein Sci. 17 (2008)1565–1575.
[278] S. Dukan, A. Farewell, M. Ballesteros, F. Taddei, M. Radman, T. Nystrom, Proteinoxidation in response to increased transcriptional or translational errors, Proc.Natl. Acad. Sci. U. S. A. 97 (2000) 5746–5749.
[279] B.B. Stocks, L. Konermann, Structural characterization of short-lived proteinunfolding intermediates by laser-induced oxidative labeling and mass spectro-metry, Anal. Chem. 81 (2009) 20–27.
[280] T.C. Squier, Oxidative stress and protein aggregation during biological aging, Exp.Gerontol. 36 (2001) 1539–1550.
[281] T. Nakamura, S.A. Lipton, Emerging roles of S-nitrosylation in protein misfoldingand neurodegenerative diseases, Antioxid. Redox. Signal. 10 (2008) 87–101.
[282] Q. Zhang, E.T. Powers, J. Nieva, et al., Metabolite-initiated protein misfoldingmay trigger Alzheimer's disease, Proc. Natl. Acad. Sci. U. S. A. 101 (2004)4752–4757.
[283] A.R. Hipkiss, Accumulation of altered proteins and ageing: causes and effects,Exp. Gerontol. 41 (2006) 464–473.
[284] V. Soskic, K. Groebe, A. Schrattenholz, Nonenzymatic posttranslational proteinmodifications in ageing, Exp. Gerontol. 43 (2008) 247–257.
[285] C.C. Winterbourn, Toxicity of iron and hydrogen peroxide: the Fenton reaction,Toxicol. Lett. 82–83 (1995) 969–974.
[286] E. Maisonneuve, B. Ezraty, S. Dukan, Protein aggregates: an aging factor involvedin cell death, J. Bacteriol. 190 (2008) 6070–6075.
[287] J. Choi, H.D. Rees, S.T. Weintraub, A.I. Levey, L.S. Chin, L. Li, Oxidativemodifications and aggregation of Cu,Zn-superoxide dismutase associatedwith Alzheimer and Parkinson diseases, J. Biol. Chem. 280 (2005)11648–11655.
[288] A. Koc, A.P. Gasch, J.C. Rutherford, H.Y. Kim, V.N. Gladyshev, Methionine sulfoxidereductase regulation of yeast lifespan reveals reactive oxygen species-dependentand -independent components of aging, Proc. Natl. Acad. Sci. U. S. A. 101 (2004)7999–8004.
[289] C.B. Glaser, G. Yamin, V.N. Uversky, A.L. Fink, Methionine oxidation, alpha-synuclein and Parkinson's disease, Biochim. Biophys. Acta 1703 (2005)157–169.
[290] H. Ruan, X.D. Tang, M.L. Chen, et al., High-quality life extension by the enzymepeptide methionine sulfoxide reductase, Proc. Natl. Acad. Sci. U. S. A. 99 (2002)2748–2753.
[291] J. Moskovitz, S. Bar-Noy, W.M. Williams, J. Requena, B.S. Berlett, E.R.Stadtman, Methionine sulfoxide reductase (MsrA) is a regulator of antioxidantdefense and lifespan in mammals, Proc. Natl. Acad. Sci. U. S. A. 98 (2001)12920–12925.
[292] F. Liu, J. Hindupur, J.L. Nguyen, et al., Methionine sulfoxide reductase A protectsdopaminergic cells from Parkinson's disease-related insults, Free Radic. Biol.Med. 45 (2008) 242–255.
[293] U.T. Brunk, A. Terman, The mitochondrial–lysosomal axis theory of aging:accumulation of damaged mitochondria as a result of imperfect autophagocy-tosis, Eur. J. Biochem. 269 (2002) 1996–2002.
[294] S. Le Corre, H.W. Klafki, N. Plesnila, et al., An inhibitor of tau hyperpho-sphorylation prevents severe motor impairments in tau transgenic mice, Proc.Natl. Acad. Sci. U. S. A. 103 (2006) 9673–9678.
[295] D. Dias-Santagata, T.A. Fulga, A. Duttaroy, M.B. Feany, Oxidative stress mediatestau-induced neurodegeneration in Drosophila, J. Clin. Invest. 117 (2007)236–245.
[296] C.W. Wittmann, M.F. Wszolek, J.M. Shulman, et al., Tauopathy in Drosophila:neurodegeneration without neurofibrillary tangles, Science 293 (2001)711–714.
[297] N. Uversky, J. Li, K. Bower, A.L. Fink, Synergistic effects of pesticides and metalson the fibrillation of alpha-synuclein: implications for Parkinson's disease,Neurotoxicology 23 (2002) 527–536.
[298] M.R. Basha, W. Wei, S.A. Bakheet, et al., The fetal basis of amyloidogenesis:exposure to lead and latent overexpression of amyloid precursor protein andbeta-amyloid in the aging brain, J. Neurosci. 25 (2005) 823–829.
[299] J. Mutter, J. Naumann, C. Sadaghiani, R. Schneider, H. Walach, Alzheimer disease:mercury as pathogenetic factor and apolipoprotein E as a moderator, NeuroEndocrinol. Lett. 25 (2004) 331–339.
[300] A.L. McCormack, M. Thiruchelvam, A.B. Manning-Bog, et al., Environmentalrisk factors and Parkinson's disease: selective degeneration of nigraldopaminergic neurons caused by the herbicide paraquat, Neurobiol. Dis. 10(2002) 119–127.
[301] J.W. Langston, P. Ballard, J.W. Tetrud, I. Irwin, Chronic Parkinsonism inhumans due to a product of meperidine-analog synthesis, Science 219 (1983)979–980.
[302] A. Palsdottir, A. Helgason, S. Palsson, A drastic reduction in the life span ofcystatin C L68Q carriers due to life-style changes during the last two centuries,PLoS Genet. 4 (2008) e1000099.
[303] P. Csermely, Chaperone overload is a possible contributor to ‘civilizationdiseases’, Trends Genet. 17 (2001) 701–704.
[304] A. Iwata, J.C. Christianson, M. Bucci, et al., Increased susceptibility of cytoplasmicover nuclear polyglutamine aggregates to autophagic degradation, Proc. Natl.Acad. Sci. U. S. A. 102 (2005) 13135–13140.
[305] S. Kamhi-Nesher, M. Shenkman, S. Tolchinsky, S.V. Fromm, R. Ehrlich, G.Z.Lederkremer, A novel quality control compartment derived from the endoplas-mic reticulum, Mol. Biol. Cell. 12 (2001) 1711–1723.
[306] G. Matsumoto, S. Kim, R.I. Morimoto, Huntingtin and mutant SOD1 formaggregate structures with distinct molecular properties in human cells, J. Biol.Chem. 281 (2006) 4477–4485.
[307] D. Kaganovich, R. Kopito, J. Frydman, Misfolded proteins partition between twodistinct quality control compartments, Nature 454 (2008) 1088–1095.
[308] R.K. Mortimer, J.R. Johnston, Life span of individual yeast cells, Nature 183 (1959)1751–1752.
[309] D.A. Sinclair, K. Mills, L. Guarente, Molecular mechanisms of yeast aging, TrendsBiochem. Sci. 23 (1998) 131–134.
[310] M. Ackermann, S.C. Stearns, U. Jenal, Senescence in a bacteriumwith asymmetricdivision, Science 300 (2003) 1920.
[311] S.H. Reuter, L. Shapiro, Asymmetric segregation of heat-shock proteins upon celldivision in Caulobacter crescentus, J. Mol. Biol. 194 (1987) 653–662.
[312] E.J. Stewart, R. Madden, G. Paul, F. Taddei, Aging and death in an organismthat reproduces by morphologically symmetric division, PLoS Biol. 3 (2005)e45.
[313] J.W. Veening, E.J. Stewart, T.W. Berngruber, F. Taddei, O.P. Kuipers, L.W. Hamoen,Bet-hedging and epigenetic inheritance in bacterial cell development, Proc. Natl.Acad. Sci. U. S. A. 105 (2008) 4393–4398.
[314] M. Ackermann, L. Chao, C.T. Bergstrom, M. Doebeli, On the evolutionary origin ofaging, Aging Cell 6 (2007) 235–244.
[315] S.N. Evans, D. Steinsaltz, Damage segregation at fissioning may increase growthrates: a superprocess model, Theor. Popul. Biol. 71 (2007) 473–490.
[316] M. Watve, S. Parab, P. Jogdand, S. Keni, Aging may be a conditional strategicchoice and not an inevitable outcome for bacteria, Proc. Natl. Acad. Sci. U. S. A.103 (2006) 14831–14835.
[317] M. Carrio, N. Gonzalez-Montalban, A. Vera, A. Villaverde, S. Ventura, Amyloid-like properties of bacterial inclusion bodies, J. Mol. Biol. 347 (2005)1025–1037.
[318] M. Morell, R. Bravo, A. Espargaro, et al., Inclusion bodies: specificity in theiraggregation process and amyloid-like structure, Biochim. Biophys. Acta 1783(2008) 1815–1825.
[319] L. Wang, S.K. Maji, M.R. Sawaya, D. Eisenberg, R. Riek, Bacterial inclusion bodiescontain amyloid-like structure, PLoS Biol. 6 (2008) e195.
[320] E. Maisonneuve, L. Fraysse, D. Moinier, S. Dukan, Existence of abnormal proteinaggregates in healthy Escherichia coli cells, J. Bacteriol. 190 (2008) 887–893.
[321] M.A. Rujano, F. Bosveld, F.A. Salomons, Polarised asymmetric inheritance ofaccumulated protein damage in higher eukaryotes, PLoS Biol. 4 (2006)e417.
[322] L.C. Fuentealba, E. Eivers, D. Geissert, V. Taelman, E.M. De Robertis, Asymmetricmitosis: unequal segregation of proteins destined for degradation, Proc. Natl.Acad. Sci. U. S. A. 105 (2008) 7732–7737.
[323] I.G. Macara, S. Mili, Polarity and differential inheritance—universal attributes oflife? Cell 135 (2008) 801–812.
[324] T.R. Rieger, R.I. Morimoto, V. Hatzimanikatis, Mathematical modeling of theeukaryotic heat-shock response: dynamics of the hsp70 promoter, Biophys. J. 88(2005) 1646–1658.
[325] D.E. Promislow, Protein networks, pleiotropy and the evolution of senescence,Proc. Biol. Sci. 271 (2004) 1225–1234.
[326] C.J. Proctor, C. Soti, R.J. Boys, et al., Modelling the actions of chaperones and theirrole in ageing, Mech. Ageing. Dev. 126 (2005) 119–131.
[327] M.P. Murphy, L. Partridge, Toward a control theory analysis of aging, Annu. Rev.Biochem. 77 (2008) 777–798.
[328] C. Qiu, D. De Ronchi, L. Fratiglioni, The epidemiology of the dementias: an update,Curr. Opin. Psychiatr. 20 (2007) 380–385.
[329] R. Mayeux, Genetic epidemiology of Alzheimer disease, Alzheimer Dis. Assoc.Disord. 20 (2006) S58–62.
[330] R.H. Myers, M.E. MacDonald, W.J. Koroshetz, et al., De novo expansion of a (CAG)n repeat in sporadic Huntington's disease, Nat. Genet. 5 (1993) 168–173.
[331] T. Foroud, J. Gray, J. Ivashina, P.M. Conneally, Differences in duration ofHuntington's disease based on age at onset, J. Neurol. Neurosurg. Psychiatry 66(1999) 52–56.
[332] G. Mallucci, J. Collinge, Update on Creutzfeldt–Jakob disease, Curr. Opin. Neurol.17 (2004) 641–647.
[333] J. Horwitz, Alpha-crystallin can function as a molecular chaperone, Proc. Natl.Acad. Sci. U. S. A. 89 (1992) 10449–10453.
[334] M. Needham, F.L. Mastaglia, Sporadic inclusion body myositis: a continuingpuzzle, Neuromuscul. Disord. 18 (2008) 6–16.
[335] E.H. Corder, A.M. Saunders, N.J. Risch, et al., Protective effect of apolipoprotein Etype 2 allele for late onset Alzheimer disease, Nat. Genet. 7 (1994) 180–184.
[336] S. Lesage, A. Durr, M. Tazir, et al., LRRK2 G2019S as a cause of Parkinson's diseasein North African Arabs, N. Engl. J. Med. 354 (2006) 422–423.
[337] W.C. Shyu, S.Z. Lin, M.F. Chiang, et al., Early-onset Parkinson's disease in a Chinesepopulation: 99mTc-TRODAT-1 SPECT, Parkin gene analysis and clinical study,Parkinsonism. Relat. Disord. 11 (2005) 173–180.
[338] Leeds University, Spongiform encephalopathies — age onset, http://www.bmb.leeds.ac.uk/mbiology/ug/ugteach/micr3290/bse_table_3.html 2006.
[339] A.C. Bruni, P. Momeni, L. Bernardi, Heterogeneity within a large kindred withfrontotemporal dementia: a novel progranulin mutation, Neurology 69 (2007)140–147.
[340] F. Squitieri, G. Sabbadini, P. Mandich, et al., Family and molecular data for a fineanalysis of age at onset in Huntington disease, Am. J. Med. Genet. 95 (2000)366–373.

996 A.B. Lindner, A. Demarez / Biochimica et Biophysica Acta 1790 (2009) 980–996
[341] M. Wardle, E. Majounie, N.M. Williams, A.E. Rosser, H.R. Morris, N.P. Robertson,Dentatorubral pallidoluysian atrophy in South Wales, J. Neurol. Neurosurg.Psychiatry 79 (2008) 804–807.
[342] R. Rengaraj, M. Dhanaraj, T. Arulmozhi, B. Chattopadhyay, N.P.Battacharyya, High prevalence of spinocerebellar ataxia type 1 in anethnic Tamil community in India, Neurol. India 53 (2005) 308–310discussion 311.
[343] S. Mead, M. James-Galton, T. Revesz, et al., Familial British dementia withamyloid angiopathy: early clinical, neuropsychological and imaging findings,Brain 123 (Pt 5) (2000) 975–991.
[344] S.W. Dubrey, K. Cha, M. Skinner, M. LaValley, R.H. Falk, Familial and primary (AL)cardiac amyloidosis: echocardiographically similar diseases with distinctlydifferent clinical outcomes, Heart 78 (1997) 74–82.
[345] I.G. McKeith, Dementia with Lewy bodies, Br. J. Psychiatry 180 (2002) 144–147.




















