
Preparation and characterization of Pt/TiO 2 nanotubes catalyst for methanol electro-oxidation
7
Applied Catalysis B: Environmental 106 (2011) 609–615 Contents lists available at ScienceDirect Applied Catalysis B: Environmental jo ur n al homepage: www.elsevier.com/locate/apcatb Preparation and characterization of Pt/TiO 2 nanotubes catalyst for methanol electro-oxidation Bochra Abida a,∗ , Lotfi Chirchi a , Stève Baranton b , Teko Wilhelmin Napporn b , Hafedh Kochkar a , Jean-Michel Léger b , Abdelhamid Ghorbel a a Laboratoire de Chimie des Matériaux et Catalyse, Faculté des Sciences de Tunis, Campus Universitaire El-Manar 2092, El-Manar, Tunisia b Laboratoire de Catalyse en Chimie Organique, Equipe Electrocatalyse, UMR-CNRS 6503, Universite de Poitiers, 40 avenue du Recteur Pineau, 86022 Poitiers Cedex, France a r t i c l e i n f o Article history: Received 6 April 2011 Received in revised form 14 June 2011 Accepted 17 June 2011 Available online 24 June 2011 Keywords: Electrocatalysts Hydrothermal process Platinum nanoparticles Titanium dioxide nanotubes Methanol electro-oxidation a b s t r a c t Titanium dioxide nanotubes were prepared via a hydrothermal treatment of TiO 2 powder (Degussa P25). Obtained samples were analyzed by various techniques, such as transmission electron microscopy (TEM) and X-ray diffraction (XRD), which revealed that the crystal structure of the obtained materials was similar to that of H 2 Ti 2 O 5 ·H 2 O nanotubes, and were about 50 nm in length and 20 nm in diameter. Nitrogen adsorption–desorption isotherms indicated that synthesized solids are mesoporous materials with a multi-walled nanotubular structure and high specific surface area. The methanol oxidation reaction was investigated on platinum nanoparticles supported TiO 2 nanotubes (XC72). The electrocatalytic activity of the catalyst was measured by cyclic voltammetry. CO stripping voltammetry in acidic solutions was investigated to study the reaction of the catalysts towards poisoning by carbonyl compounds. The results demonstrated that Pt/TiO 2 nanotubes catalyst exhibits the best activity for methanol oxidation and were favorable for improving the tolerance to poisoning species. © 2011 Elsevier B.V. All rights reserved. 1. Introduction Direct methanol fuel cells have attracted considerable attention for their low weight, high power density, low operating tem- perature and low pollutant emission. In the near future, they represent an alternative power generation system especially for mobile and portable applications [1–5]. Platinum is a suitable elec- trocatalyst for the electro-oxidation of methanol. However pure platinum is a poor anode catalyst for methanol electro-oxidation at room temperature, because CO is generated as an interme- diate during methanol oxidation reaction and strongly adsorbed on platinum active sites [6–9]. The electrocatalytic activity of Pt nanoparticles for methanol reaction is dependent on various fac- tors including the size and dispersion of the particles, preparation methods, supporting materials, and their surface conditions. Gen- erally, a high dispersion of Pt based catalysts on a support is very critical for its electrocatalytic activity [10,11]. Recently, various works have been focused on Pt/transition metal oxide compos- ites systems, such as Pt/TiO 2 , Pt/SnO 2 , Pt/CeO 2 [12,13]. The metal oxide stabilizes Pt particles dispersion to favor the increase of active surface per unit weight of the catalyst. The aim of our studies is to investigate the effect of the substrate composition on the electrocatalytic activity of pure platinum nanoparticles. ∗ Corresponding author. Tel.: +216 71 88 34 24. E-mail address: [email protected] (B. Abida). Especially, titanium nanotubes prepared from TiO 2 (Degussa P25) powder by hydrothermal treatment are an attractive substrate for this study. This treatment was discovered by Kasuga et al. [14] treated TiO 2 in the 10 mol/L NaOH aqueous solution for 20 h at 383 K and nanotubes with 8 nm in diameter and 100 nm in length were obtained. This synthesis method was optimized by differ- ent studies by using different concentrations of NaOH aqueous solution as well using several raw materials and hydrothermal syn- thetic conditions [15]. Apart from the crystalline structure of TiO 2 anatase or a mixture of anatase and rutile some nanostructures of nanotubes were obtained with the formation of hydrogenoti- tanate H 2 Ti 2 O 5 ·H 2 O, H 2 Ti 3 O 7 , H 2 Ti 4 O 9 ·H 2 O [16]. Their tubular structure showed a larger surface area and a higher degree of porosity than TiO 2 powder, and an improvement of their photocat- alytic activity was evidenced [16]. In this work we are interested particularly to titanium nanotubes (H 2 Ti 2 O 5 ·H 2 O) [17,18], which present a high specific surface area compared with TiO 2 powders, caused by the nanotubular morphology [19]. They also exhibit a high ion-exchange capacity for cations of different metals [20], open mesoporous morphology [21], pronounced proton conduc- tivity and relatively good stability at elevated temperatures [22]. Furthermore, a strong interaction has been reported in the case of platinum catalysts dispersed on TiO 2 nanotubes leading to an improvement of the electrocatalytic activity of platinum when compared with platinum dispersed on other substrates. For ethanol oxidation, the results demonstrated that the titanium dioxide nanotubes can greatly enhance the catalytic activity of Pt and 0926-3373/$ – see front matter © 2011 Elsevier B.V. All rights reserved. doi:10.1016/j.apcatb.2011.06.022
Transcript of Preparation and characterization of Pt/TiO 2 nanotubes catalyst for methanol electro-oxidation

Pe
BHa
b
a
ARRAA
KEHPTM
1
fprmtpadontmecwioaso
0d
Applied Catalysis B: Environmental 106 (2011) 609– 615
Contents lists available at ScienceDirect
Applied Catalysis B: Environmental
jo ur n al homepage: www.elsev ier .com/ locate /apcatb
reparation and characterization of Pt/TiO2 nanotubes catalyst for methanollectro-oxidation
ochra Abidaa,∗, Lotfi Chirchia, Stève Barantonb, Teko Wilhelmin Nappornb,afedh Kochkara, Jean-Michel Légerb, Abdelhamid Ghorbela
Laboratoire de Chimie des Matériaux et Catalyse, Faculté des Sciences de Tunis, Campus Universitaire El-Manar 2092, El-Manar, TunisiaLaboratoire de Catalyse en Chimie Organique, Equipe Electrocatalyse, UMR-CNRS 6503, Universite de Poitiers, 40 avenue du Recteur Pineau, 86022 Poitiers Cedex, France
r t i c l e i n f o
rticle history:eceived 6 April 2011eceived in revised form 14 June 2011ccepted 17 June 2011vailable online 24 June 2011
a b s t r a c t
Titanium dioxide nanotubes were prepared via a hydrothermal treatment of TiO2 powder (Degussa P25).Obtained samples were analyzed by various techniques, such as transmission electron microscopy (TEM)and X-ray diffraction (XRD), which revealed that the crystal structure of the obtained materials was similarto that of H2Ti2O5·H2O nanotubes, and were about 50 nm in length and 20 nm in diameter. Nitrogenadsorption–desorption isotherms indicated that synthesized solids are mesoporous materials with a
eywords:lectrocatalystsydrothermal processlatinum nanoparticlesitanium dioxide nanotubes
multi-walled nanotubular structure and high specific surface area. The methanol oxidation reaction wasinvestigated on platinum nanoparticles supported TiO2 nanotubes (XC72). The electrocatalytic activityof the catalyst was measured by cyclic voltammetry. CO stripping voltammetry in acidic solutions wasinvestigated to study the reaction of the catalysts towards poisoning by carbonyl compounds. The resultsdemonstrated that Pt/TiO2 nanotubes catalyst exhibits the best activity for methanol oxidation and were
he to
ethanol electro-oxidation favorable for improving t. Introduction
Direct methanol fuel cells have attracted considerable attentionor their low weight, high power density, low operating tem-erature and low pollutant emission. In the near future, theyepresent an alternative power generation system especially forobile and portable applications [1–5]. Platinum is a suitable elec-
rocatalyst for the electro-oxidation of methanol. However purelatinum is a poor anode catalyst for methanol electro-oxidationt room temperature, because CO is generated as an interme-iate during methanol oxidation reaction and strongly adsorbedn platinum active sites [6–9]. The electrocatalytic activity of Ptanoparticles for methanol reaction is dependent on various fac-ors including the size and dispersion of the particles, preparation
ethods, supporting materials, and their surface conditions. Gen-rally, a high dispersion of Pt based catalysts on a support is veryritical for its electrocatalytic activity [10,11]. Recently, variousorks have been focused on Pt/transition metal oxide compos-
tes systems, such as Pt/TiO2, Pt/SnO2, Pt/CeO2 [12,13]. The metalxide stabilizes Pt particles dispersion to favor the increase of
ctive surface per unit weight of the catalyst. The aim of ourtudies is to investigate the effect of the substrate compositionn the electrocatalytic activity of pure platinum nanoparticles.∗ Corresponding author. Tel.: +216 71 88 34 24.E-mail address: [email protected] (B. Abida).
926-3373/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.apcatb.2011.06.022
lerance to poisoning species.© 2011 Elsevier B.V. All rights reserved.
Especially, titanium nanotubes prepared from TiO2 (Degussa P25)powder by hydrothermal treatment are an attractive substrate forthis study. This treatment was discovered by Kasuga et al. [14]treated TiO2 in the 10 mol/L NaOH aqueous solution for 20 h at383 K and nanotubes with 8 nm in diameter and 100 nm in lengthwere obtained. This synthesis method was optimized by differ-ent studies by using different concentrations of NaOH aqueoussolution as well using several raw materials and hydrothermal syn-thetic conditions [15]. Apart from the crystalline structure of TiO2anatase or a mixture of anatase and rutile some nanostructuresof nanotubes were obtained with the formation of hydrogenoti-tanate H2Ti2O5·H2O, H2Ti3O7, H2Ti4O9·H2O [16]. Their tubularstructure showed a larger surface area and a higher degree ofporosity than TiO2 powder, and an improvement of their photocat-alytic activity was evidenced [16]. In this work we are interestedparticularly to titanium nanotubes (H2Ti2O5·H2O) [17,18], whichpresent a high specific surface area compared with TiO2 powders,caused by the nanotubular morphology [19]. They also exhibit ahigh ion-exchange capacity for cations of different metals [20],open mesoporous morphology [21], pronounced proton conduc-tivity and relatively good stability at elevated temperatures [22].Furthermore, a strong interaction has been reported in the caseof platinum catalysts dispersed on TiO2 nanotubes leading to an
improvement of the electrocatalytic activity of platinum whencompared with platinum dispersed on other substrates. For ethanoloxidation, the results demonstrated that the titanium dioxidenanotubes can greatly enhance the catalytic activity of Pt and
6 B: Env
iont[eaTo
hplg(w
2
2
to1s2fit(0fiw
2
iaTsaawd
2
teeaoaXirwtQpsc7
10 B. Abida et al. / Applied Catalysis
ncrease the utilization rate of platinum [23,24]. Studies carriedut on commercial Pt/C catalysts mixed with titanium dioxideanotubes for direct alcohol fuel cells (DAFC) have shown thatitanium dioxide nanotubes provided better catalytic performance25]. Recently, titanium dioxide nanotubes were synthesized bylectrochemical anodization as Pt support. The catalyst was testeds electrode for electrochemical catalysis of methanol oxidation.he results exhibit an improvement of catalytic activity of methanolxidation [26].
In this paper, titanium dioxide nanotubes were synthesized byydrothermal treatment and mixed with H2PtCl6 solution to obtainlatinum nanoparticles by the impregnation method. The cata-
ysts were characterized by X-ray diffraction (XRD), the thermalravimetric analysis (TGA) and Transmission electron microscopyTEM), and their electrocatalytic activities for methanol oxidationere investigated by cyclic voltammetry.
. Experimental
.1. Preparation of TiO2 nanotubes by hydrothermal process
TiO2 nanotubes were prepared by hydrothermal method similaro that described by Kasuga et al. [14]. In a typical procedure, 0.5 gf TiO2 (Degussa P25) were dispersed in an aqueous solution of1.25 mol/L NaOH (15 mL) and placed in a Teflon-lined stainless-teel autoclave. The autoclave was statically heated at 130 ◦C for0 h. After the hydrothermal treatment, one proceeded to the firstltration with 1 L of distilled water (at approximately 80 ◦C) ando a neutralization with 0.1 mol/L of a hydrochloric acid solutionHCl) until pH 7. After this neutralization, a second washing with.5 L ebullient water was carried out, followed by a filtration andnally by a drying at 80 ◦C for 24 h. Titanium dioxide nanotubesere obtained.
.2. Preparation of Pt nanoparticles
The platinum nanoparticles where synthesized using thempregnation method. In order to produce 10 wt% Pt/substrate cat-lysts, 0.5 g of substrate (carbon Vulcan XC72, TiO2 Degussa P25 oriO2 nanotubes) was mixed with 6.25 mL of hexachloroplatinic acidolution (20 g/L). The mixture was ultrasonically treated for 30 minnd stirred for 2 h. Then 200 mL of 0.4 mol/L NaBH4 at 80 ◦C wasdded to the mixture under vigorous stirring. The catalytic powderas then filtered and washed with a large amount of water andried under vacuum at 110 ◦C for 4 h.
.3. Characterization of catalysts
The morphology of the prepared catalyst was investigated byransmission electron microscopy. The sample specimens for TEMxperiment were prepared by dispersing the catalyst powder inthanol by ultrasonic treatment for several minutes, dropping onto
holey carbon film supported by a copper grid. TEM images werebtained using instrument a JEOL JEM-2001 LaB6 microscope withn accelerating voltage of 200 kV and a resolution of ca. 0.19 nm.-ray diffraction analysis was performed using a “Philips analyt-
cal” X-ray diffractometer with Cu K� radiation. The 2� angularegions between 2◦ and 70◦ were explored at a scan rate of 6◦ min−1
ith step of 0.02◦. The thermal gravimetric analysis and differentialhermal analysis (DTA) were performed using TA Instruments “SDT
600” instrument, within the temperature range of room tem-
erature to 900 ◦C with the temperature rate of 10 ◦C min−1. BETurface area and BJH pore distribution of the TiO2 nanotubes werealculated from nitrogen adsorption and desorption isotherms at7 K using a “Micromeritics ASAP 2000” instrument. 100 mg of TiO2ironmental 106 (2011) 609– 615
nanotubes sample was sealed in the reactor equipped with a tem-perature control unit. The adsorption isotherm was measured at25 ◦C using a standard sequence, with a 30 min time out for eachpoint.
Atomic absorption spectroscopy was performed to determinethe platinum loading on titanium oxide based substrates. An AAnalyst 200 atomic absorption Spectrometer (Perkin Elmer) withplatinum hollow cathode lamp (� = 265 nm) was used. The catalyticpowder was previously mineralized in a hydrofluoric, hydrochlo-ric and nitric acid solution under microwave activation for 40 min.The excess of hydrofluoric acid is then removed by the addition ofH3BO3.
The loading of platinum on substrate for Pt/C Vulcan XC72sample was determined by TGA (Pt represent 12.4 wt% of thePt/C catalytic powder). For Pt/TiO2 and Pt/TiO2 nanotubes sam-ples, the platinum loading on the substrate was determined byatomic absorption spectroscopy (Pt represent 1.8 wt% and 9.5 wt%of the catalytic powder for Pt/TiO2 and Pt/TiO2 nanotubes samples,respectively).
The cyclic voltammetry and CO stripping measurements wereobtained using an Autolab potentiostat–galvanostat. All theelectrochemical measurements were carried out at 25 ◦C in athermostated three-compartment electrochemical cell. All thepotentials in this work were related to a reference saturatedcalomel electrode (SCE) in a 0.5 mol/L H2SO4 Suprapur sulphuricacid (Merck) was used for preparing the electrolytic solutions and1 mol/L CH3OH + 0.5 mol/L H2SO4 solution for methanol oxidationactivity test. The working electrode was a glassy carbon disk witha 3 mm diameter (geometric surface area, 0.071 cm2). It was pol-ished with Al2O3 powder and washed carefully before catalystdeposition. Platinum based working electrodes were prepared from12 wt% Pt/C (Vulcan XC72), 1.83 wt% Pt/TiO2 or 10 wt% Pt/TiO2nanotubes catalysts. An ink was prepared by ultrasonically dis-persing 5 mg catalytic powder with 5 mg of carbon Vulcan XC72in 0.5 mL 5 wt%. Nafion® solution (from Aldrich). The 5 mg of car-bon Vulcan XC72 were added in order to ensure a good electronicconductivity of the catalytic layer, especially in the case of platinumdispersed on TiO2 based substrates. A drop of 4.0 �L catalyst ink wasdeposited onto the working electrode surface. The platinum loadingobtained on the working electrode is 71 �gPt cm−2, 10 �gPt cm−2
and 54 �gPt cm−2 for Pt/C, Pt/TiO2 and Pt/TiO2 nanotubes catalysts,respectively. Glassy carbon and home-made reference saturatedcalomel electrodes were used as the counter and the reference elec-trode, respectively. The reference electrode was separated from theworking electrode compartment by an electrolyte bridge. Beforethe cyclic voltammetry measurements, nitrogen was bubbled in thesupporting electrolyte for 30 min at room temperature. Recordedat 50 mV s−1 from −0.2 V to 1.1 V, 6 cycles of voltammetry werenecessary to stabilize the current–potential signal.
3. Results and discussion
3.1. TEM analysis of Pt/TiO2 nanoparticles and Pt/TiO2 nanotubes
Fig. 1a shows the TEM image of TiO2 (Degussa P25), it can be seenthat the orientation of the crystal planes is random, and the generalparticle morphology can be assumed to be spherical, with an aver-age size between 15 and 50 nm.TEM images of Pt/TiO2 (DegussaP25) catalyst are shown in Fig. 1b and c which clearly demonstratehomogenous structure with small platinum particles dispersed ontitanium dioxide. Metal particle sizes were about 4–10 nm.
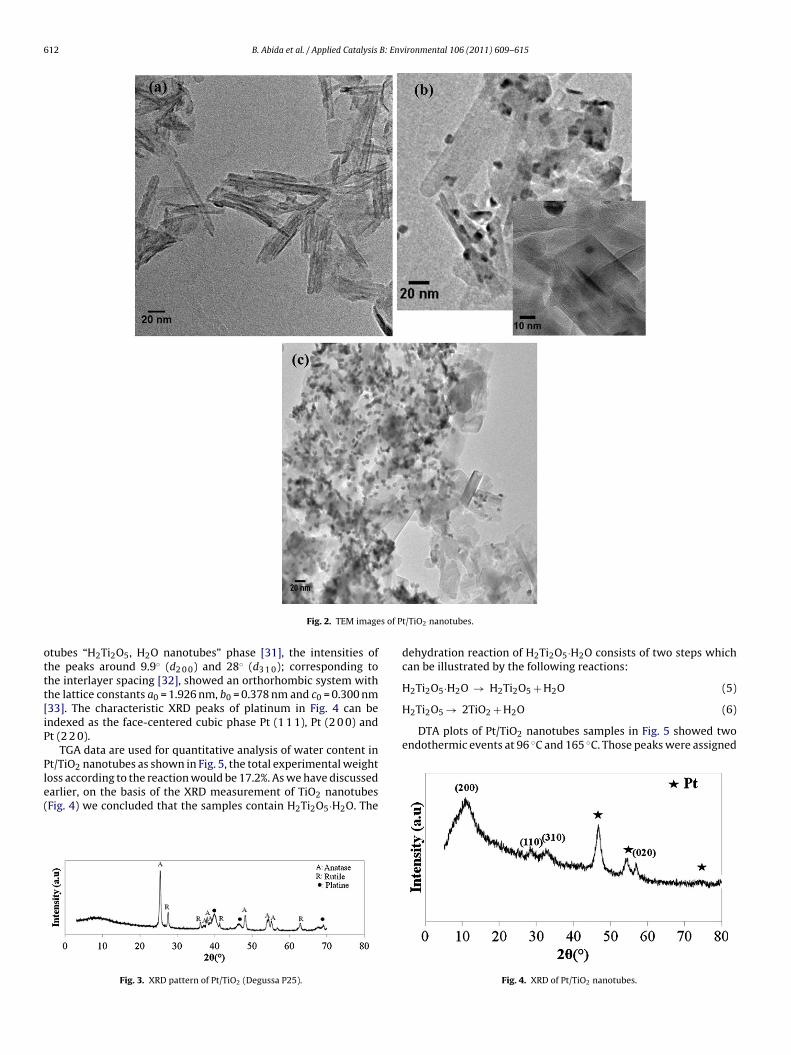
TiO2 powders have been hydrothermally transformed into TiO2nanotubes at 130 ◦C for 20 h. TEM image Fig. 2a shows a typicalstructure of nanotubes, which are about 25–50 nm in length and10–20 nm in diameter also they present a multi-layered structure

B. Abida et al. / Applied Catalysis B: Environmental 106 (2011) 609– 615 611
of Pt/T
(0is
3P
raasf
x
war7
Fig. 1. TEM images
2–10 layers), the interlamellar space ranges between 0.25 and.8 nm (inset Fig. 2b). The dispersion of Pt on the titanium diox-
de nanotubes is uniform as depicted in Fig. 2b and c. Metal particleizes were about 3–9 nm.
.2. Morphology and structure of Pt/TiO2 nanoparticles andt/TiO2 nanotubes
The XRD pattern of Pt/TiO2 (Degussa P25) powder in Fig. 3eveals the peaks of anatase (1 0 1) and rutile (1 1 0) at 2� = 25.38◦
nd 2� = 27.47◦ respectively, indicating that the TiO2 composition is mixture of rutile and anatase. Based on the respective peak inten-ities, the rutile content in the powder of TiO2 can be calculatedrom the following equation:
=(
1 + 0.8IAIR
)−1(1)
here x is the weight fraction of rutile in the powders, while IAnd IR are the X-ray intensities of the anatase and the rutile peaksespectively. The results indicate that there are 28.4% rutile and1.6% anatase.
iO2 (Degussa P25).
The crystallite size can be determined from the broadening ofcorresponding X-ray spectral peaks of anatase phase by Scherrerformula:
L = K�
cos �(2)
where L is the crystallite size, � is the wavelength of the X-ray radi-ation, K is usually taken as 0.94, is the line width at half maximumheight, and the crystallite size is about 25 nm [27].
The characteristic XRD peaks of platinum in Fig. 3 can be indexedas the face-centered cubic phase Pt (1 1 1), Pt (2 0 0) and Pt (2 2 0).
Fig. 4 shows the XRD pattern of the Pt/TiO2 nanotubes sam-ple, the different TiO2 phases (anatase, rutile or brookite) werenot detected. In fact, XRD pattern revealed that the crystalstructure of the materials obtained during the hydrothermal treat-ment was similar to that of hydrogenotitanate, H2Ti2O5·H2O (orto H2−xNaxTi2O5·H2O) obtained through the following reaction[28–30]:
2TiO2 + 2NaOH → Na2Ti2O5·H2O (3)
Ionic exchange could also take place in the following step duringthe acid washing procedure:
Na2Ti2O5·H2O + xH+ → HxNa2−xTi2O5·H2O + xNa+(0 ≤ x ≤ 2) (4)
All peaks in the XRD pattern (Fig. 4) at 2� = 9.9◦, 25◦, 28◦
and 48◦ which reveal the presence of hydrogenotitanate nan-
612 B. Abida et al. / Applied Catalysis B: Environmental 106 (2011) 609– 615
s of P
ottt[iP
Ple(
H2Ti2O5 → 2TiO2 + H2O (6)
DTA plots of Pt/TiO2 nanotubes samples in Fig. 5 showed twoendothermic events at 96 ◦C and 165 ◦C. Those peaks were assigned
Fig. 2. TEM image
tubes “H2Ti2O5, H2O nanotubes” phase [31], the intensities ofhe peaks around 9.9◦ (d2 0 0) and 28◦ (d3 1 0); corresponding tohe interlayer spacing [32], showed an orthorhombic system withhe lattice constants a0 = 1.926 nm, b0 = 0.378 nm and c0 = 0.300 nm33]. The characteristic XRD peaks of platinum in Fig. 4 can bendexed as the face-centered cubic phase Pt (1 1 1), Pt (2 0 0) andt (2 2 0).
TGA data are used for quantitative analysis of water content int/TiO2 nanotubes as shown in Fig. 5, the total experimental weightoss according to the reaction would be 17.2%. As we have discussed
arlier, on the basis of the XRD measurement of TiO2 nanotubesFig. 4) we concluded that the samples contain H2Ti2O5·H2O. TheFig. 3. XRD pattern of Pt/TiO2 (Degussa P25).
t/TiO2 nanotubes.
dehydration reaction of H2Ti2O5·H2O consists of two steps whichcan be illustrated by the following reactions:
H2Ti2O5·H2O → H2Ti2O5 + H2O (5)
Fig. 4. XRD of Pt/TiO2 nanotubes.

B. Abida et al. / Applied Catalysis B: Environmental 106 (2011) 609– 615 613
Fig. 5. TGA–DTA plots of Pt/TiO2 nanotubes.
F(
te3m
3
tTItmwltots
dbav
TT
ig. 6. Adsorption–desorption isotherms of nitrogen at 77 K for (a) TiO2 nanotubes;b) TiO2.
o the loss of humidity and crystalline water, respectively. Twoxothermic events were also observed with maxima at 240 and50 ◦C. These events are probably related to anatase phase’s for-ation respectively.
.3. Textural properties and elemental analysis
The adsorption–desorption isotherm of nitrogen at 77 K foritanium dioxide nanotubes (Fig. 6) is of Type IV isotherms withype H3 hysteresis loops, according to the classification of theUPAC [34], the initial part of the Type IV isotherm is attributedo monolayer–multilayer adsorption. Hysteresis appearing in the
ultilayer range of physisorption isotherms is usually associatedith capillary condensation in mesopore structures. The Type H3
oop is observed with aggregates of plate-like particles giving riseo slit-shaped pores. Taking into consideration that titanate nan-tubes are multiwalled scrolls which are believed to be formedhrough sheet folding or wrapping mechanism it seems to be rea-onable that they exhibit the H3 loop.
Textural parameters derived from nitrogen adsorption–
esorption isotherm data are summarized in Table 1. As cane noted, the transformation of TiO2 into TiO2 nanotubes wasccompanied by a marked increase in surface area and poreolume.able 1exture of the different TiO2 based support.
Samples SBET (m2/g) Pore volume(cm3/g)
Average pore size(nm)
Titanium dioxidenanotubes
246 0.54 8.9
Titanium dioxide(Degussa P25)
43 0.1 –
Fig. 7. (a) Cyclic voltammogram of the Pt/TiO2 nanotubes electrode in 0.5 mol/LH2SO4 and (b) cyclic voltammogram of Pt/TiO2 nanotubes electrode in 1 mol/LCH3OH + 0.5 mol/L H2SO4. Room temperature, scan rate: 50 mV s−1.
The lowest BET surface area was observed for sample TiO2(43 m2/g), whereas the highest surface area was measured in caseof TiO2 nanotubes (246 m2/g). The difference of the specific sur-face areas between the two types of TiO2 may be due to themutilayer-type structure of the TiO2 nanotubes. Similarly, the totalpore volume was increasing from 0.10 to 0.54 cm3/g for TiO2–TiO2nanotubes.
3.4. Electrochemical performance of catalysts
Cyclic voltammogram of Pt/TiO2 nanotubes electrocatalyst innitrogen-purged 0.5 mol/L H2SO4 is presented in Fig. 7a. Thevoltammetric features of Pt/TiO2 nanotubes electrocatalyst arecharacteristic of Pt metal presence, with Pt oxide formation inthe range between 0.6 and 0.9 V, which is due to the adsorptionon Pt oxide/hydroxides [35], and a well-defined peak at around0.5 V attributable to the reduction of the oxide/hydrous oxidelayer. Furthermore, hydrogen adsorption–desorption peaks areseen between 0 and −0.2 V. This result suggests that the Pt particlesare in good electronic contact with the TiO2 nanotubes surface.
The values of the electrochemically active surface areas of plat-inum (PtEASA) are obtained by measuring the charge from thehydrogen desorption peaks after subtracting the charge from thedouble-layer region and with the assumption that the smooth Ptelectrode gives the hydrogen adsorption charge of 210 �C cm−2
represents the quantity of charge corresponding to the adsorptionof a monolayer of hydrogen on Pt [36]. The active surface area of thePt/TiO2 nanotubes catalyst, which is determined by the magnitudeof the hydrogen adsorption–desorption peaks, in turn, dependsmainly on the size of the Pt particles and the presence of differentcrystallites of Pt, Pt(1 1 1), Pt(1 1 0) and Pt(1 0 0) facets. The active
surface areas of catalysts (PtEASA) were calculated with the for-mula PtEASA = QH/0.21. The electrochemically active surface areasof the Pt/TiO2 nanotubes, Pt/TiO2 and Pt/C electrodes are listed inTable 2. All methanol oxidation current densities (j) represented
614 B. Abida et al. / Applied Catalysis B: Environmental 106 (2011) 609– 615
in(
j
tbisitatweto
a
C
trc
oPbwta
saisvsDC
TCa
results in important variations of the platinum specific surface.The improvement of the synthesis method could lead to a bettercorrelation between COads oxidation experiment and methanoloxidation.
Fig. 8. Reaction pathways of methanol oxidation at a Pt surface.
n cyclic voltammograms are expressed as the current recorded (i)ormalized by the platinum electrochemically active surface areaPtEASA):
= i
PtEASA(7)
Formation of platinum oxide whose peak was observed duringhe scan around 0.5 V leads to the inhibition of methanol oxidationut reactivation of the surface, as the oxide, which is stable cathod-
cally, leads to another large anodic peak as the former in Fig. 7bhows the cyclic voltammograms for Pt/TiO2 nanotubes electroden 1 mol/L CH3OH + 0.5 mol/L H2SO4. On the first scan starting at 0 V,he onset potential of the methanol oxidation was around 0.3 V and
large methanol oxidation peak was observed at 0.62 V on the posi-ive irreversible scan and another acute peak of methanol oxidationas seen at 0.45 V on the reverse scan. This peak is usually consid-
red as being associated with the oxidation of methanol and withhe removal of the carbonaceous species generated via incompletexidation of alcohol in the forward scan [9,20] as shown in Fig. 8.
These carbonaceous species included absorbed HCHO, HCOOHnd absorbed CO species in methanol oxidation.
The complete electro-oxidation of methanol is expressed as:
H3OH + H2O → 6Haq+ + 6e− + CO2 (8)
The data for the three catalysts are listed in Table 2 hencehe ratio of the forward oxidation peak current density (jf) to theeverse peak current density (jr), jf/jr, can be used to describe theatalyst tolerance to the poisoning species.
In Fig. 9, the higher jf/jr ratio indicates more effective removalf the poisoning species on the catalyst surface, the jf/jr ratios oft/TiO2 nanotubes and Pt/TiO2 were 1.04 and 1.68 respectively,oth higher than that of Pt/C (0.80), illustrating better tolerance ofater-containing catalysts to carbonaceous species. However, the
hree catalysts present similar activity in terms of onset potentialnd current densities.
The oxidation of pre-adsorbed carbon monoxide (CO) was mea-ured by CO stripping voltammetry in 0.5 mol/L H2SO4 solution at
scan rate of 20 mV s−1. As the CO species are the main poisoningntermediate during methanol electro-oxidation, a good catalysthould possess excellent CO electro-oxidation ability, which can be
erified by CO stripping measurements. In Fig. 10 are presented COtripping curves of Pt/TiO2 nanotubes, Pt/TiO2 and Pt/C catalysts.ifferent catalytic behavior can be observed. As can be seen, theO oxidation potential of Pt/TiO2 nanotubes catalyst was betweenable 2haracterization of platinum based electrodes (0.071 cm2 geometric surface area)nd performance for methanol oxidation reaction.
Electrodes Pt/C XC72 Pt/TiO2 nanotubes Pt/TiO2
Platinum loading on substrate(wt.%)
12.4 9.52 1.83
Platinum loading on theelectrode (�gPt cm−2
geo)71 54 10
PtEASA (cm2) 2.23 1.041 0.2777Specific surface area (m2/g−1) 45 27 38E peak (V/SCE) 0.64 0.62 0.59jf/jr 0.80 1.04 1.68
Fig. 9. Cyclic voltammograms in 1 mol/L CH3OH + 0.5 mol/L H2SO4 of catalysts: Pt/C;(b) Pt/TiO2 nanotubes; (c) Pt/TiO2. Room temperature, scan rate: 50 mV s−1.
0.040 and 0.57 V versus SCE, which is much lower than that ofPt/TiO2 and Pt/C catalysts.
These results demonstrated that Pt/TiO2nanotubes catalystcan oxidize CO more easily at lower potential than Pt/TiO2 andPt/C catalysts, which is helpful for releasing the active sites ofPt at low potential for further oxidation of methanol in directmethanol fuel cell (DMFC) [37,38]. This difference of behaviorbetween COads oxidation and methanol oxidation could be dueto the high difference in electrochemically active surface area.Indeed, the platinum nanoparticle synthesis method (impreg-nation) is very dependent on the nature of the substrate, and
Fig. 10. CO stripping voltammograms in 0.5 mol/L H2SO4 solutions. Room temper-ature, scan rate: 20 mV s−1.

B: Env
4
cbiwaumspiofinsfpp
R
[[
[
[[
[[
[[[
[[[[[
[
[
[[
[
[[
[
[
[pratique de caractérisation, Technip, Paris, 2001.
B. Abida et al. / Applied Catalysis
. Conclusions
The hydrothermal treatment of TiO2 powder (P25) by a con-entrated NaOH aqueous solution allowed synthesizing titaniumased nanotubes (H2Ti2O5·H2O nanotubes). Studies of adsorption
sotherms showed a higher specific surface area for TiO2 nanotubeshich is suitable for a good dispersion of electro-catalysts. The TEM
nd XRD characterizations evidenced that TiO2 nanotubes exhibitniform distribution in length and diameter. The cyclic voltam-etry demonstrated that the Pt/TiO2 nanotubes catalyst exhibits
imilar catalytic activity for methanol electro-oxidation when com-ared with classical Pt/TiO2 and Pt/C catalyst. The TGA–DTA results
llustrated that the amount of water contained in the TiO2 nan-tubes was higher than that of TiO2 Degussa P25, validating theormation of titanium nanotubes. Furthermore, the water contentn the nanotubes could improve the oxidation of COads on platinumanoparticles compared with carbon substrate or TiO2 (P25) sub-trate. Hence, the use of hydrogenotitanate nanotube as a substrateor platinum catalyst improves considerably the COads oxidation onlatinum but the methanol oxidation reaction still occurs at highotential.
eferences
[1] M.B. Ji, Z.D. Wei, S.G. Chen, X.Q. Qi, L. Li, Q. Zhang, C. Liao, R. Tang, Int. J. HydrogenEnergy 34 (2009) 2765–2770.
[2] C. Grolleau, C. Coutanceau, F. Pierre, J.-M. Leger, Electrochim. Acta 53 (2008)7157–7165.
[3] H.J. Kim, D. Kim, H. Han, Y. Shul, J. Power Sources 159 (2006) 484–490.[4] G. Wu, T. Chen, X. Zong, H. Yan, G. Ma, X. Wang, Q. Xu, D. Wang, Z. Lei, C. Li, J.
Catal. 253 (2008) 225–227.[5] V. Baglio, A.S. Arico, A. Di Blasia, V. Antonucci, P.L. Antonucci, S. Licoccia, E.
Traversa, F. Serraino Fiory, Electrochim. Acta 50 (2005) 1241–1246.
[6] J.M. Leger, S. Rousseau, C. Coutanceau, F. Hahn, C. Lamy, Electrochim. Acta 50(2005) 5118–5125.[7] I. Ávila-García, C. Ramírez, J.M. Hallen López, E.M. Arce Estrada, J. Alloys Compd.
495 (2010) 462–465.[8] V. Selvaraj, M. Alagar, Electrochem. Commun. 9 (2007) 1145–1153.
[[[[
ironmental 106 (2011) 609– 615 615
[9] S.L. Gojkovié, J. Electroanal. Chem. 573 (2004) 271–276.10] W. Vogel, J. Phys. Chem. C 112 (2008) 13475–13482.11] H. Ren, M.P. Humbert, C.A. Menning, J.G. Chen, Y. Shu, U.G. Singh, W. Cheng,
Appl. Catal. A 375 (2010) 303–309.12] N. Rajalakshmi, N. Lakshmi, K.S. Dhathathreyan, Int. J. Hydrogen Energy 33
(2008) 7521–7526.13] H. Yuana, D. Guo, X. Qiu, W. Zhu, L. Chen, J. Power Sources 188 (2009) 8–13.14] T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino, K. Niihara, Adv. Mater. 11 (1999)
1307–1311.15] C. Tsai, H. Teng, Chem. Mater. 18 (2006) 367–373.16] C. Lee, C. Wang, M. Lyu, L. Juang, S. Liu, S. Hung, J. Colloid Interface Sci. 316
(2007) 562–569.17] Y. Xu, X. Fang, J. Xiong, Z. Zhang, Mater. Res. Bull. 45 (2010) 799–804.18] H. Ou, S. Lo, Sep. Purif. Technol. 58 (2007) 179–191.19] Z.Q. Song, H.Y. Xu, K.W. Li, H. Wang, H. Yan, J. Mol. Catal. A: Chem. 239 (2005)
87–91.20] D. He, L. Yang, S. Kuang, Q. Cai, Electrochem. Commun. 9 (2007) 2467–2472.21] C.-C. Tsai, J.-N. Nian, H. Teng, Appl. Surf. Sci. 253 (2006) 1898–1902.22] B. Lei, J. Xue, D. Jin, S. Ni, H. Sun, Rare Met. 27 (2008) 445–450.23] H. Song, X. Qiu, X. Li, F. Li, W. Zhu, Chen, J. Power Sources 170 (2007) 50–54.24] L. Timperman, Y.J. Feng, W. Vogel, N. Alonso-Vante, Electrochim. Acta 55 (2010)
7558–7563.25] P. Xiao, H. Songa, X. Qiua, W. Zhua, L. Chen, U. Stimmingc, P. Belec, Appl. Catal.
B: Environ. 97 (2010) 204–212.26] F. Hassan, H. Nanjo, S. Venkatachalam, M. Kanakubo, T. Ebina, J. Power Sources
195 (2010) 5889–5895.27] J.F. Porter, Y.G. Li, C.K. Chan, J. Mater. Sci. 34 (1999) 1523–1531.28] H. Kochkar, A. Turki, L. Bergaoui, G. Berhault, A. Ghorbel, J. Colloid Interface Sci.
331 (2009) 27–31.29] H. Kochkar, N. Lakhdhar, G. Berhault, M. Bausach, A. Ghorbel, J. Phys. Chem. C
113 (2009) 1672–1679.30] K. Jabou, H. Kochkar, G. Berhault, A. Ghorbel, J. Mater. Sci. 44 (2009) 6677–6682.31] G.-S. Guo, C.-N. He, Z.-H. Wang, F.-B. Gu, D.-M. Han, Talanta 72 (2007)
1687–1692.32] M. Qamar, C.R. Yoon, H.J. Oh, N.H. Lee, K. Park, D.H. Kim, K.S. Lee, W.J. Lee, S.J.
Kim, Catal. Today 131 (2008) 3–14.33] J. Yang, Z. Jin, X. Wang, W. Li, J. Zhang, S. Zhang, X. Guo, Z. Zhang, Dalton Trans.
(2003) 3898–3901.34] J. Lynch (Ed.), Analyse physico-chimique des catalyseurs industriels, Manuel
35] C. Naour, Thesis, Université de Paris XI, 1994.36] X.S. Peng, K. Koczkur, S. Nigro, A.C. Chen, Chem. Commun. 24 (2004) 2872–2873.37] S. Thomas, X. Ren, S.J. Gottesfeld, J. Electrochem. Soc. 146 (1999) 4354–4359.38] M. Wang, D. Guo, H. Li, J. Solid State Chem. 178 (2005) 1996–2000.




















