
Hydrogen sulfide downregulates the aortic L-arginine/nitric oxide pathway in rats
34
1 Hydrogen Sulfide Downregulates the Aortic L-Arginine/Nitric Oxide Pathway in Rats Bin Geng 1,2* , Yuying Cui 1 , Jing Zhao 1,2 , Fang Yu 2 , Yi Zhu 2 , Geyang Xu 2 , Zhiwen Zhang 2 , Chaoshu Tang 1,2 , Junbao Du 3 1. Institute of Cardiovascular Research, First Hospital of Peking University. Beijing P.R. China 2. Department of Physiology, Peking University Health Science Center. Beijing P.R. China 3. Department of Pediatric, First Hospital of Peking University. Beijing P.R. China *Corresponding Author: Bin Geng. Institute of Cardiovascular Research, First Hospital of Peking University. Beijing xishuku street 8, 100034. E-mail: [email protected], Telephone: +86-010-82802183, Fax: +86-010-82802183. Page 1 of 34 Articles in PresS. Am J Physiol Regul Integr Comp Physiol (July 18, 2007). doi:10.1152/ajpregu.00207.2006 Copyright © 2007 by the American Physiological Society.
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Hydrogen sulfide downregulates the aortic L-arginine/nitric oxide pathway in rats

1
Hydrogen Sulfide Downregulates the Aortic L-Arginine/Nitric
Oxide Pathway in Rats
Bin Geng1,2*, Yuying Cui1, Jing Zhao1,2, Fang Yu2, Yi Zhu2, Geyang Xu2,
Zhiwen Zhang2, Chaoshu Tang1,2 , Junbao Du3
1. Institute of Cardiovascular Research, First Hospital of Peking University.
Beijing P.R. China
2. Department of Physiology, Peking University Health Science Center.
Beijing P.R. China
3. Department of Pediatric, First Hospital of Peking University. Beijing P.R.
China
*Corresponding Author: Bin Geng.
Institute of Cardiovascular Research, First Hospital of Peking University.
Beijing xishuku street 8, 100034. E-mail: [email protected],
Telephone: +86-010-82802183, Fax: +86-010-82802183.
Page 1 of 34Articles in PresS. Am J Physiol Regul Integr Comp Physiol (July 18, 2007). doi:10.1152/ajpregu.00207.2006
Copyright © 2007 by the American Physiological Society.

2
ABSTRACT
The aim of the present study was investigation the effect of H2S signaling by nitric
oxide (NO) in isolated rat aortas and cultured human umbilical vein endothelial cells
(HUVECs). Both administration of H2S and NaHS as well as endogenous H2S reduced
NO formation, eNOS activity, eNOS transcript abundance and L-Arg transport (all
P<0.01). The kinetics analysis of eNOS activity and L-Arg transport showed that H2S
reduced Vmax values (all P<0.01) without modifying Km parameters. Use of selective
NOS inhibitors verified that eNOS (versus iNOS and nNOS) was the specific target of
H2S regulation. H2S treatment (100 µmol/L) reduced Akt phosphorylation, and decreased
eNOS phosphorylation at Ser1177. H2S reduced L-Arg uptake by inhibition of a system
y+ transporter, and decreased the CAT-1 transcript. H2S treatment reduced protein
expression of eNOS, but not of nNOS and iNOS. Pinacidil (K ATP channel opener)
exhibited the similar inhibitory effects on the L-Arg/NOS/NO pathway. Glibenclamide
(KATP channel inhibitor) partly blocked the inhibitory effect of H2S and pinacidil. An in
vivo experiment revealed that H2S down-regulated the vascular L-Arg/eNOS/NO
pathway after intraperitoneal injection of NaHS (14 µmol/kg) in rats. Taken together, our
findings suggest that H2S downregulates the vascular L-Arg/NOS/NO pathway in vitro
and in vivo, and the KATP channel could be involved in the regulatory mechanism of H2S.
Keywords: Hydrogen sulfide, Nitric oxide, Nitric oxide synthase, L-Arg transport
Page 2 of 34

3
Hydrogen sulfide (H2S) is a well-known pungent gas, and its toxicology has been
extensively studied (40). Recently, H2S has been found to be endogenously generated in
various mammalian tissues by 2 pyridoxal-5’-phosphate-dependent enzymes such as
cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) (6), with L-cysteine
used as a main substrate. The gas may be a functional regulator in the nervous and
cardiovascular systems (21). In the cardiovascular system, CSE may be a key enzyme by
which H2S is generated (50). H2S causes a dose-dependent relaxation of the isolated rat
aorta (16, 50) and mesenteric arterioles (4); induces transient hypotension in anesthetized
rats in vivo (50), reduces proliferation of vascular smooth muscle cells (VSMCs) (8, 46),
and exerted a negative inotropic action of the heart in vivo and in vitro (13). Significantly,
there is evidence linking the endogenous CSE/H2S pathway to the pathogenesis of arterial
hypertension (45, 52), pulmonary hypertension induced by hypoxia (5), septic and
endotoxin shock (18), haemorrhagic shock (27), ischemic cardiac disease (2, 11),
ischemic brain damage (33) and hyperdynamic circulation in cirrhosis (10). Growing
evidence suggests that in addition to NO and CO (12), endogenous H2S may be a novel
cardiovascular gasotransmitter. As an endogenous ligand, H2S directly opens the KATP
channel in VSMCs and relaxes vascular smooth muscles in an endothelium-independent
manner (50), which is different from the effect of NO and CO.
Interestingly, the administration of sodium nitroprusside (SNP), an NO donor,
potentiates the vasorelaxation effects induced by H2S (16), and upregulats of H2S
production in rat vascular tissues in a concentration dependent manner (48). In contrast,
pretreatment with H2S attenuates the vasorelaxant effect induced by SNP in aortic rings
(49). In rats with septic and endotoxin shock, the plasma level of H2S is positively
correlated with that of NO (18) . In the brain, H2S acts as an endogenous peroxynitrite
scavenger (42). All these results suggest that NO and H2S signaling are irterrelated. Of
note, the expression of the H2S-generating enzyme CSE had been observed in VSMCs
(50), but not in the vascular endothelium, suggesting that H2S is generated in vascular
tissues mainly from VSMCs. Whether and how endogenous H2S from VSMCs affects the
endothelial NO production pathway is unknown. In the present study, we examined the
effects of H2S on NO production, endothelial NO synthase (eNOS) activity and
expression, and L-Arginine (L-Arg, substrate of NOS) transport in isolated rat aortas and
Page 3 of 34

4
cultured human umbilical vein endothelial cells (HUVECs) to explore the regulatory role
of H2S in the vascular L-Arg/NOS/NO pathway.
Page 4 of 34

5
MATERIALS AND METHODS
Animals
All animal care and experimental protocols complied with the Animal Management
Rules of the Ministry of Health of the People’s Republic of China (documentation
Number 55, 2001, Ministry of Health of P.R. China) and the guide for the Care and Use
of the Laboratory Animals of the First Hospital, Peking University. Male
Sprague-Dawley (SD) rats (250-300 g) were provided by the Animal Department, Health
Science Center of Peking University. All animals were maintained on normal rat chow
with unrestricted water and on a 12-h light/12 h dark cycle.
Materials
HUVECs (926 cell line) were purchased from the American Type Culture
Collection (ATCC, Manassas, USA). Tris-base, Dowex AG 50 w-x8 (Na+), NADPH,
tetrahydrobiopterin (BH4), calmodulin (CaM), flavin mononucleotide (FMN), flavin
adenine dinucleotide (FAD), L-Arg , L-canavanine, spermidine, pinacidil, glibenclamide,
N-ethylmaleimide (NEM), DL-propargylglycine (PAG), L-cysteine,
pyridoxal-5’-phosphate and sodium hydrosulfide (NaHS) were purchased from Sigma Co.
(St. Louis, MO, USA). L-[3H]-Arg (1.5 TBq/mmol) was from NEN Life Sci Products
(Boston, MA, USA); and eNOS antibody (SC-654), P-eNOS(Ser 1177)-R antibody
(SC-12972), iNOS antibody (SC-7271), nNOS antibody(SC-648),
phospho-Akt1/2/3(Ser473) antibody(Sc-7985R), Akt antibody(SC-8312) and β-tubulin
antibody (SC-5274) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA,
USA).
H2S-saturated solution (0.09 mol/L at room temperature) was made by bubbling
with pure H2S gas (offered by Beijing XianHeYu CO. LTD, China) and stored at -70°C.
The specific primers used for sample loading calibration were synthesized by SBS
Co (Beijing, China) and were as follows: eNOS-S, 5'GTTTGTCTGCGGCGATGT-3';
eNOS-A, 5'AAGAAACAGGAAGCGGGTG-3'; cationic amino acid transporter-1
(CAT-1)- S, 5'-TGAAGACAGGGAGTGAGGG-3'; CAT-1-A ,
5'-TAGACAAACGGTAGCAGT-3'; GAPDH- S,
Page 5 of 34

6
5'GGATTTGGTCGTATTGGG3';GAPDH-A, 5'GGAAGATGGTGATGGGATT3'.
Other chemicals and reagents were analytical grade.
Organ culture of aortas in vitro
At the selected time, recipient rats were anesthetized with pentobarbital sodium (30
mg/kg, given intraperitoneally) and then decapitated quickly. The entire
thoraco-abdominal aorta was quickly removed and placed in cold, sterile
phosphate-buffered saline (PBS) (4°C). The organ culture of aortic tissues was carried
out as previously described (20), with some modification. Briefly, aortas were cleaned of
adherent adipose tissues and collateral vessels under sterile conditions. The aortic tissues
were placed in ice-cold PBS, cut into pieces of approximately 1 × 1 mm2 and transferred
to 24-well plates containing 1 mL of culture medium (RPMI 1640 with 0.5% [w/v]
bovine serum albumin, BSA) per well covering the tissue slices completely. Tissues w ere
cultured in a shaking bath (60 beats/min) at 37°C, in an atmosphere of 95% O2 and 5%
CO2. NaHS, the donor of H2S, or H2S gas buffer were added to the wells at a final
concentration of 50 µmol/L, then incubated for 2, 4 and 6 h respectively. For other groups,
1, 10, 100 and 1000 µmol/L NaHS or H2S gas buffer with or without other drugs
(L-cysteine 10 mmol/L; pyridoxal-5’-phosphate 2 mmol/L; DL-propargylglycine 2
mmol/L; pinacidil, 10 µmol/L, and glibenclimide, 10 µmol/L) were added to the wells,
which were incubated for 2 h. At the end of incubation, the NO production (content of
nitrate and nitrite in medium), tissue NOS activity and [3H]-L-Arg transport were
measured. The activity of lactate dehydrogenase (LDH) in the culture medium was
measured using a LDH-cytotoxic test kit (44) to determine the tissue viability.
H2S assay by sulfide-sensitive electrode
Measurement of H2S concentration in culture medium involved use of a
sulfide-sensitive electrode (Model 9616; Orion Research; Beverly, MA). In brief, sulfide
antioxidant buffer (SAOB) was added into standard sulfide solution or blood samples at a
ratio of 1:1 and then stirred thoroughly. Electrodes were rinsed in distilled water, blotted
dry, and placed into standards or samples. When a stable reading was displayed, the
millivolt value was recorded. The H2S concentration was calculated against the
calibration curve of the standard S2- solution (50).
Page 6 of 34

7
HUVEC culture
Confluent HUVECs (926 cell line) were put into medium 199 containing 5 mmol/L
D-glucose, 10% fetal calf serum, 5 mmol/L glutamine, 100 u/mL penicillin-streptomycin
and 0.03 mg/mL gentamycin at 37°C in a 5% CO2 atmosphere (43).Cell viability was
determined by trypan blue exclusion assay and by measurement of the activity of LDH in
the culture medium (44). Cell viability remained at more than 95% throughout the
experiments.
H2S (50 µmol/L) was added after changing to serum-free medium; cells were
incubated for 2 h at 37°C in a 95% air-5% CO2 atmosphere, and eNOS enzyme kinetics
and L-Arg transport dynamics were measured. For other groups, 100 µmol/L of H2S was
added and cells were incubated for 4 h before gene expression of cationic amino acid
transporter (CAT-1) and eNOS were analyzed by RT-PCR. The protein expression of
cellular phospho-Akt and total Akt were detected by Western blotting after incubation
with H2S (500 µmol/L), pinacidil (10 µmol/L) or glibenclamide (10 µmol/L) for 0.5 h;
phospho-eNOS and eNOS protein expression was measured after 2-h incubation. H2S
(100, 500 and 1000 µmol/L), and/or pinacidil (10 µmol/L) and glibenclamide (10 µmol/L)
were added after incubation for 6 h, eNOS, then iNOS and nNOS protein expression was
analyzed.
Assay of nitrate plus nitrite
L-Arg (200 µmol/L) was added to the organ and cell culture medium, which was
incubated for different times according to the experiment protocol; the media was then
collected. The assay was performed in a standard flat-bottomed 96-well polystyrene
microtitre plate, with PBS used as a control. An amount of 50 µL of nitrate reductase and
β-NADPH was added to each well, for final concentrations of 300 U/L and 25 µmol/L,
respectively. The plate was incubated at room temperature for 3 h. Excess β-NADPH was
consumed by the addition of 50 µL of PBS containing L-glutamic dehydrogenase,
α-ketoglutaric acid, and NH4Cl, (for final concentrations of 500 U/L, 4 mmol/L and 100
mmol/L, respectively) followed by 10-min incubation at 37oC. The nitrite concentration
was then measured by the addition of 50 µL Griess reagents, and the absorbance was read
at 540 nm by use of a plate reader after 10-min incubation at room temperature (14).
Page 7 of 34

8
Assay of NOS activity and eNOS enzyme kinetics
Aortic samples were fast frozen and ground into a fine powder in liquid nitrogen by
use of a mortar and pestle before sonicating in 5 volumes of homogenization buffer (50
mmol/L Tris-HCl, pH 7.2, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, 0.1%
L-mercaptoethanol, 10 µg/mL leupeptin, 5 µg/mL pepstatin A, 1 mmol/L PMSF, 0.5
mmol/L dithiothreitol). NOS activity was measured by monitoring the conversion of
L-[3H]-Arg to L-[3H]-citrulline (23). For routine assay of total NOS (tNOS), 0.1 mg of
aortic tissue protein was incubated in a reaction buffer containing 1 mmol/L L-[3H]-Arg
(containing 1 mCi [3H]), 1 mmol/L NADPH, 300 IU/mL calmodulin, 5 µmol/L
tetrahydrobiopterin (BH4), 2 mmol/L CaCl2, 10 µmol/L flavine-adenine dinucleotide
(FAD) (31), and 10 µmol/L flavin mononucleotide (FMN), for 30 min at 37oC. For assay
of inducible NOS (iNOS) activity, the reaction buffer was Ca2+-free. The reaction was
stopped by adding 50 µL of ice cold Tris-HCl buffer (pH 5.5) containing 1 mmol/L
L-citrulline, 1 mmol/L EGTA and 1 mmol/L EDTA. The reaction substance was applied
to a 1 mL column of Dowex AG50WX-8 (Na+) to absorb L-[3H]-Arg. The NOS activity
was then quantified by determining the radioactivity on liquid scintillation spectroscopy
(Beckman, LS6500) and presented as nmol L-[3H]-citrulline/min/mg protein. eNOS
activity was calculated as the difference between total NOS activity and iNOS activity.
The measurement of eNOS enzyme kinetics in HUVECs was as previously
described (24), with minor modification. In brief, the cells were washed twice with
ice-cold buffer containing (in mmol/L) 100 NaCl, 25 NaH2PO4, and 80 Na2HPO4, pH 7.5.
The cells were pelleted and resuspended in ice-cold 50 mmol/L Tris-HCl buffer (pH 7.4)
containing l mmol/L EDTA, 5 mmol/L mercaptoethanol, 10 µg/mL pepstatin A, 10
µg/mL leupeptin, 90 µg/mL PMSF, and 1 µmol/L tetrahydrobiopterin. The cells were
disrupted by repeated freeze-thawing in liquid nitrogen. The cell lysate (50 µL) was
added to 50 µL of buffer as described above, except with different concentrations (0.1,
0.2, 0.4, 0.8, 1.6 and 3.2 mmol/L) of L-[3H]-Arg. The other reactions of eNOS activity
assay were further processed as described above.
Page 8 of 34

9
Assay of L-Arg uptake in aortic tissues and L-Arg transport kinetics in HUVECS
L-Arg uptake and transport kinetics analysis were performed by measured
intracellular incorporation L-[3H]-Arg. On completion of aortic incubation, the aortic
tissues were washed 3 times with ice-cold Krebs-Henseleit (KH) buffer (in mmol/L:
NaCl 131, KCl 5.6, NaHCO3 25, NaH2PO4 1, D-glucose 5, HEPES 20, CaCl2 2.5, MgCl2
1, pH 7.4). Aortic L-Arg uptake was measured by incubation with KH buffer containing
100 µmol/L L-Arg and 10 µCi/mL L-[3H]-Arg at 37oC for 15 min. Aortic tissues were
washed with ice-cold 10 mmol/L unlabeled L-Arg in PBS, then lysed with 200 µL
methanoic acid. Total radioactivity of L-[3H]-Arg was measured by liquid scintillation
counting (37).
Transport kinetics of L-Arg into endothelial cells was measured by the method of
Leoncini et al. (25), with minor modifications. Briefly, kinetic experiments were
performed in cells incubated for 15 min with KH buffer containing L-Arg (10 to 320
µmol/L) and 2 µCi/mL L-[3H]-Arg. Transport was terminated by removing the media and
washing the cells three times with ice-cold 10 mmol/L unlabeled L-Arg in PBS. Cells
were lysed with 0.5% Triton X-100 in 0.5 mol/L NaOH, and radioactive activity
[3H]-L-Arg taken by cells was assayed by liquid scintillation counting.
RT-PCR assay of gene expression of eNOS and CAT-1 in HUVECs
The mRNA levels of eNOS and CAT-1 were measured by RT-PCR, as described
previously by our lab (19). Total RNA from cells was extracted with Trizol reagent
(Applygen Technologies Inc. Beijing). RT-PCR was performed in a total volume of 25
µL. After denaturing at 95oC for 5 min, PCR was run at 94oC for 30 s, 61oC for 30 s, and
72oC for 40 s for 30 cycles. The PCR products were separated on a 1.5 % agarose gel and
stained with ethidium bromide. The optical density of the bands of eNOS cDNA (553 bp)
and CAT-1 cDNA (250 bp) was measured by use of the Gel Documentation System
(Bio-Rad, Hercules, CA). The PCR products were amplified again with the human
GAPDH primers at 94oC for 30 s, 55oC for 30 s, and 72oC for 30 s for 20 cycles, and the
optical density of the GAPDH band (205 bp) was measured. The ratio of eNOS or CAT-1
to GAPDH was considered as the relative amount of eNOS or CAT-1 gene expression.
Page 9 of 34

10
Western blot analysis
Protein isolation and Western blot analysis were carried out as previously described
(3). Briefly, a cellular lysate was prepared from the HUVECs and separated by
SDS-PAGE (on 7.5% gels). The proteins were transferred to nitrocellulose membranes
(Schleicher and Schuell; Dassel, Germany) by electroblotting (Bio-Rad). Proteins were
detected with polyclonal anti-eNOS, anti-P- eNOS, anti-iNOS, anti-nNOS, anti-Akt, and
anti-P- Akt antibodies and a monoclonal anti-tubulin antibody. After incubation with the
appropriate secondary antibodies conjugated to horseradish peroxidase, the membrane
was washed and color was developed by use of an enhanced chemiluminescence (7) kit
(Applygen Technologies Inc. Beijing).
The aortic alterations of L-Arg/NOS/NO pathway by injection H2S to in vivo rats
Rats were intraperitoneally injected with NaHS (3.5 µmol/kg, intraperitoneally each
30 min, for a total of 4 injections, total H2S 14 µmol/kg, n=6 rats) or normal saline (n=6
control rats). Two hours after the first injection, rats were anesthetized by intraperitoneal
injection with pentobarbital sodium (30 mg/kg). The blood was collected in heparinized
syringes from the abdominal aortas and transferred to tubes. The plasma was separated to
assay nitrate plus nitrite content. The aortic tissues were isolated, and its NOS activity
and L-Arg uptake were measured according to the above methods.
Statistical analysis
The results are expressed as means ± SD. Comparisons across more than 2 groups
were analyzed by one -way ANOVA followed by the Student-Newman-Keuls test. A
two-tailed P value<0.05 was considered statistically significant.
Page 10 of 34

11
RESULTS
H2S inhibited NO generation of aortic tissues
After L-Arg administration to cultured aortic tissues, the level of nitrate plus nitrite,
both stable products of NO, continuously accumulated in the medium up to 6 h (Fig. 1A).
After administration of H2S (50 µmol/L) and NaHS (50 µmol/L), the NO products in the
medium were decreased, by 63% and 47% (P<0.01) after 2 h, 48% and 30% (P<0.01)
after 4 h, and 32 % and 19% (P <0. 05) after 6 h, respectively, compared with that in
controls (Fig. 1A). According to the Fig. 1A results, we selected 2 h as a time of best
inhibitory effect, and observed the effects of various concentrations (from 1 to 1000
µmol/L) of H2S on NO release at this time. Administration of both H2S and NaHS
significantly reduced NO production in a concentration-dependent manner after a 2-h
incubation (Fig. 1B). The IC50 value was 19.5 µmol/L for H2S (95% confidence interval
[CI], 7.06-53.09 µmol/L) and 15.9 µmol/L for NaHS (95% CI, 7.99-31.59 µmol/L).
Although the IC50 of H2S was a little higher than that of NaHS, the inhibitory effect of
H2S was stronger than that of NaHS (Fig. 1C). The results showed that exogenous H2S
inhibits vascular NO production in vitro.
We felt it important to explore the effect of endogenously generated H2S.
Administration of L-cysteine (L-Cys, the substrate of CSE) and pyridoxal-5’-phosphate
(PLP, a co-factor of CSE) increased H2S release from incubated aortic tissues with H2S
concentration in the culture medium reaching 37.5±2.7 µmol/L (Fig.1D); baseline H2S
concentration in the culture medium was undetectable. Plasma H2S concentration was
48.96±10.59µmol/L according to the Zhao W reported (53). At the same time, L-Cys and
PLP treatment reduced NO production by 43% as compared with that in controls (P<0.01,
Fig. 1E). The effect of L-Cys and PLP treatment on NO production was blocked by
preincubation with an inhibitor of CSE, DL-propargylglycine (PAG) (Fig.1E), which
prevented endogenous H2S formation (the H2S concentration in culture medium could not
be detected by sensitive sulfur electrode, as shown in Fig.1D).
The KATP channel is an important molecular target of H2S in cardiovascular tissues
(4, 47, 50). To assess the role of KATP channel in H2S-induced decrease of NO production,
the KATP channel opener pinacidil and KATP channel inhibitor glibenclamide were used.
Pinacidil reduced NO production by 34% and H2S (100 µmol/L) level by 30% (P<0.01)
Page 11 of 34

12
(Fig. 1F). The activity of both agents was largely blocked by pretreatment with
glibenclamide (10 µmol/L). These results suggest that the KATP channel is involved in
inhibition of H2S-induced NO generation.
H2S downregulated aortic eNOS activity and changed its kinetics
eNOS is the principle form of NO synthase in normal arterial endothelium. On
incubation aortic tissue slices with H2S (50 µmol/L) and NaHS (50 µmol/L), reduced
eNOS activity by 56% and 47% (P<0.01) after 2 h, 39% and 27% (P<0.01) after 4 h, and
12% and 11% (P>0.05, Fig. 2A) after 6 h, respectively, compared with that in controls. In
addition, H2S and NaHS (from 1 to 1000 µmol/L) markedly decreased aortic eNOS
activity in a concentration-dependent manner after 2 h incubation (Fig. 2B). The IC50
values of H2S and NaHS for eNOS activity were 11.49 µmol/L (95% CI, 9.53-19.87
µmol/L) and 31.97 µmol/L (95% CI, 11.93-85.70 µmol/L), respectively. H2S was
stronger in inhibiting eNOS activity than NaHS (Fig. 2C). The results are similar to those
with NO generation, which suggests that the H2S reduced vascular NO generation at least
in part, by inhibiting eNOS activity.
The kinetics of eNOS regulation by H2S were studied in HUVEC cultures.
Incubation of HUVEC with NaHS (50 µmol/L, 2 h) inhibited the maximal activity of
eNOS (Vmax, 75.1±2.9 vs control 113.0±4.0 pmol/min/mg protein, P<0.01) without
modifying the Km value for L-Arg (33.6±4.4 vs control 39.3±3.0 µmol/L, P>0.05, Fig.
2D), thus significantly decreasing the enzyme activity efficienc y (ratio of Vmax to Km:
2.24±0.66 vs 2.88±1.33, P<0.01). We investigated the effect on aortic eNOS activity of
endogenously generated H2S by adding L-Cys and PLP to HUVECs. eNOS activity was
reduced by 48% (P<0.01; Fig. 2E), an effect that was attenuated by pretreatment with
PAG (a CSE inhibitor) (48). L-canavanine, an selective iNOS inhibitor (26) and
spermidine an nNOS inhibitor, alone or together, did not affect the inhibition effects of
H2S (100 µmol/L) on NOS activity (Fig. 2F), which indicates that H2S specifically
regulates eNOS activity. Like H2S, pinacidil inhibited eNOS activity, which was not
blocked by the iNOS or nNOS inhibitors used. However, glibenclamide blocked
inhibition of eNOS activity by H2S and pinacidil. Collectively, the results suggest that a
KATP channel is involved in the impact of H2S on eNOS activity.
Page 12 of 34

13
Both Ca2+-dependent and -independent pathways are involved in the regulation of
eNOS activity. There is evidence that H2S modulates Ca2+ influx in cardiovascular cells
(1) suggesting that H2S may inhibit eNOS activity through a Ca2+-dependent pathway.
Whether a Ca2+-independent pathway is involved in the inhibition of eNOS activity by
H2S is unknown. We detected phosphorylation of Ser1177, which activates eNOS by a
Ca2+-independent pathway (17), in eNOS protein. Incubation with H2S (500 µmol/L) for
2 h inhibited the phosphorylation of eNOS Ser1177 without changing eNOS expression
(Fig. 2G; P<0.01). Pinacidil also slightly inhibited eNOS phosphorylation. Pretreatment
with glibenclamide (10 µmol/L) partially blocked the effects of H2S and pinacidil on
eNOS phosphorylation (Fig. 2G). The results confirmed the role of a KATP channel in the
regulation of H2S in eNOS activity.
Akt (also named protein kinase B) mediates eNOS phosphorylation of Ser1177 in
vitro and in vivo, and plays a role in both calcium-dependent and -independent eNOS
activation (15). H2S treatment for 30 min significantly reduced Akt phosphorylation
without changing total Akt protein expression (Fig. 2H). Pinacidil also reduced Akt
phosphorylation, and glibenclamide blocked the effects of H2S and pinacidil. This
suggests that Akt signaling involves in the regulation of H2S to eNOS phosphorylation,
and which partially modulates by opening KATP channels.
H2S inhibited L-[3H]-Arg uptake by aortic tissues and changed L-[3H]-Arg
transport kinetics in HUVECs
To determine the impact of H2S on transcellular transport of L-Arg, we measured
L-[3H]-Arg uptake in aortic tissues. H2S (50 µmol/L) and NaHS (50 µmol/L) decreased
L-[3H]-Arg uptake by 58% and 50% (P<0.01) after 2 h, 39% and 30% (P<0.01) after 4 h,
and 12% and 8% (P>0.05) after 6 h, respectively (Fig. 3A). In addition, incubation with
H2S and NaHS for 2 h inhibited the L-[3H]-Arg uptake in a concentration-dependent
manner (1 to 1000 µmol/L; Fig. 3B). The IC50 values were 84.08 µmol/L (95% CI:
27.28-259.20 µmol/L) and 39.15 µmol/L (95% CI: 10.46-146.50 µmol/L) for H2S and
NaHS, respectively, the inhibitory effect of H2S being stronger than that of NaHS (Fig.
3C).
Page 13 of 34

14
To explore the regulation of L-Arg transport activity by H2S, we investigated the
kinetics of L-Arg transport in HUVECs. NaHS (50 µmol/L, 2 h) decreased the Vmax of
L-[3H]-Arg transport (17.5±1.2 vs control 20.6±1.1 pmol/105 cells/min, P<0.01) without
modifying the Km value (84.4±14.8 vs 73.0±10.4 µmol/L, P>0.05) and thus reduced
significantly the transport efficiency (Vmax / Km : 0.21±0.08 vs 0.28±0.11, P<0.01) (Fig.
3D).
In addition, administration of L-Cys and PLP inhibited L-Arg uptake, which was
blocked by PAG pretreatment, as shown in Fig. 3E. The results suggest that H2S, whether
exogenously administered or endogenously generated, inhibits L-Arg transport.
HUVECs transport arginine through two Na+-independent systems. System y+ is
sensitive to N-ethylmaleimide (NEM) and referable to the expression of CAT1 and
CAT2B. System y+L is referable to the expression of y+LAT2, y+LAT1, and 4F2hc (35).
To identify which type of transporter system acts as a main target of H2S, we investigated
L-Arg uptake with or without NEM (0.5 mmol/L, pretreatment 20 min). NEM reduced
L-Arg uptake by 32% compared with control (Fig.3F, P<0.01). NEM pretreatment plus
H2S (100 µmol/L) did not change the L-Arg influx compared with the effect of NEM
alone (Fig. 3F; P>0.05). The KATP channel opener pinacidil slightly reduced L-Arg
uptake (12% decrease compared to control) as did H2S, but NEM intensified the
inhibitory effects of pinacidil on L-Arg uptake compared with NEM treatment alone
(Fig.3F, P<0.01). Simultaneously, glibenclamide blocked in part the inhibitory effects of
both H2S and pinacidil.
H2S down-regulated CAT-1 and eNOS gene transcript level in HUVECs
To determine whether H2S modulates the transcript level of CAT-1, mRNA
expression of the L-Arg transporter and of eNOS was determined by RT-PCR in HUVEC
after incubation for 4 h with H2S (100 µmol/L). Transcript levels of CAT-1 and eNOS
were 40% and 43%, respectively (all P<0.01, Fig. 4) less than that in controls.
H2S inhibited eNOS but did not change iNOS and nNOS protein expression in
HUVECs
Page 14 of 34

15
To confirm whether H2S modulates eNOS, eNOS protein expression was assayed by
Western blots in HUVES after 6-h H2S incubation. H2S (100, 500 and 1000 µmol/L)
significantly inhibited eNOS protein expression, but not in a concentration-dependent
manner, and the inhibitory effect was partially blocked by glibenclamide preincubation
(10 µmol/L, Fig. 5A). Fig. 5B confirmed the above phenomena and showed
pinacidil-induced inhibition of eNOS protein expression and block of this by
glibenclamide.
Induction of iNOS protein expression by H2S was not observed (Fig. 5C). nNOS is
another constitutive NOS that is expressed in endothelial cells. Neither H2S nor pinacidil
altered nNOS expression (Fig. 5D).
NaHS inhibited L-Arg/NOS/NO pathway in rats in vivo
To demonstrate the in vivo effects of H2S on the vascular L-Arg/NOS/NO pathway,
we analyzed plasma levels of nitrite and nitrate after intraperitoneal administration of 14
µmol/kg NaHS in rats. At 2 h after NaHS injection, the plasma content of nitrate plus
nitrite was 21% (24.44±0.80 vs 31.00±2.66 µmol/L less than in controls, P<0.01, Fig.
6A). Aortic eNOS activity was reduced by 42% (23.06±5. 08 vs 39.50±8.67
pmol/min/mg protein, P<0.01, Fig. 6B), and L-Arg uptake decreased by 30%
(88.35±14.31 vs 125.40 ± 18.96 pmol/min/mg protein, P<0.01, Fig. 6C). iNOS activity
was not induced by H2S injection in the in vivo experiments (Fig. 6B).
Page 15 of 34

16
DISCUSSION
Recently, the novel endogenous gas H2S has been recognized as another
cardiovascular gasotransmitter that exerts important cardiovascular effects similar to
those of NO and CO. However, H2S, as opposed to NO and CO, is specifically released
from VSMCs but not endothelium, opens the KATP channel, induces cellular membrane
hyperpolarization and causes vessel dilation in an autocrine/paracrine manner (40, 41, 50),
which differs from the effect of NO and CO. Some studies have reported that NO
enhanced endogenous H2S production (48) and its vascular dilation effect (16), whereas
H2S inhibited vascular relaxation induced by NO (49). In the present study, we found that
endogenous H2S down-regulated the vascular L-Arg/NOS/NO pathway. This was
mediated in part through the KATP channel. Taken together these results suggest a delicate
crosstalk between endothelial and VSMCs by a complex interaction of the
gasotransmitters NO and H2S (Fig.7).
The physiological serum concentration of H2S in Sprague Dawley (SD) rats has
been reported to be 45.6+/-14.2 µmol/L (50). In the present study, an exogenously
administered physiological concentration of H2S or its donor, NaHS, inhibited NO
release from cultured aortic tissues. The inhibitory effect of H2S weakened with
incubation time but remained detectable for 6 h or more, consistent with anticipated
changes of H2S concentration in the medium (as a gasotransmitter, H2S could be
metabolized in the cell quickly (44). According to the concentration-dependent inhibitory
effects of H2S, the IC50 value of H2S was calculated as approximately half of the
physiological serum concentration. Endogenous H2S generation from aortic tissues is 3.6
nmol/min/g wet tissue (48). SNP (from 10 to 1000 nmol/L) increased aortic H2S
production in a concentration-dependent manner; 1 µmol/L SNP can increase H2S by 2
nmol/min/g wet tissue (48). One possibility is a negative feedback regulation in which
accumulated NO enhances H2S release from VSMCs during the short term and, over H2S
inhibits NO formation, achieving dynamic balance in the ultimate.
H2S endogenously generated by L-Cys plus PLP reduced NO production by 43%.
Pretreatment with PAG, a CSE inhibitor (51) and pinacidil, a non-selective KATP channel
opener, decreased aortic NO production, similar to the effect of H2S. Glibenclamide, a
non-selective inhibitor, blocked the inhibitory effects of H2S and pinacidil. Collectively,
Page 16 of 34

17
these data suggest that endogenously generated H2S downregulates vascular NO
production and opening of the KATP channel could be one of possible mechanisms.
NO is generated from the conversion of L-Arg to L-citrulline by the enzymatic
action of NADPH-dependent NO synthases (NOS). NOS exists in 3 distinct isoforms , a
constitutive neuronal NOS (NOS I or nNOS), an inducible NOS (NOS II or iNOS), and a
constitutive endothelial NOS (NOS III or eNOS) (1). In the vaculature, NO is produced
mainly in the endothelium by constitutively expressed eNOS. The present results suggest
that exogenous H2S or its donor NaHS reduced eNOS activity within 4 h in a
concentration (from 1 to 1000 µmol/L)-dependent manner. The IC50 values for H2S
(11.49 umol/L) were approximately 26% of the physiological serum H2S concentration,
and lower than that of H2S-inhibited NO production (19.5 umol/L). The kinetic study
showed that H2S did not modify the enzyme affinity to L-Arg (represented as Km value),
but decreased the maximum value of catalytic velocity (Vmax), indicating that H2S
reduces eNOS catalytic efficiency. H2S endogenously generated by administration of
L-Cys plus PLP also inhibited eNOS activity.
To verify the specificity of H2S-mediated inhibition of eNOS activity, we
investigated the effect of single or combined administration of the selective iNOS
inhibitor L-canavanine and nNOS inhibitor spermidine. These treatments did not change
the inhibitory effect of H2S on constitutive NOS activity. Although iNOS activity could
be induced by stimuli such as cytokines, reactive oxygen species or some drugs (1), we
detected very low iNOS activity in normal cultured aortic tissues, and neither H2S nor
NaHS incubation affected iNOS activity during the entire incubation period. These
results suggest that H2S is specifically inhibition eNOS activity. Additionally, H2S
inhibits eNOS transcript level and protein expression. These findings suggest that eNOS
is an important regulatory target of H2S-affected NO generation.
L-Arg transport is another key step to limiting NO generation besides NOS catalysis
(8). In the present study, exogenous administration of both H2S and NaHS significantly
reduced L-[3H]-Arg transporter activity within 4 h and returned to control levels after 6 h.
Inhibition was concentration-dependent in the 1 to 1000 umol/L range, and the inhibitory
effect of H2S (from 12.4% to 55.3%) was higher than that of NaHS (from 3.6% to 38.8%).
The IC50 values were 84 µmol/L for H2S, and approximated 182% of physiological serum
Page 17 of 34

18
concentrations. Intracellular levels of L-Arg are not generally considered rate-limiting for
eNOS catalysis (51). Consumption of intracellular L-Arg initiates and stimulates its
uptake by cells (30), at this time, NO generation is dependent on L-Arg uptake. These
may explain why the IC50 of H2S for L-Arg uptake was higher than that for NO
generation. Kinetic analysis of the L-Arg transport showed that H2S inhibited the Vmax of
L-[3H]-Arg uptake, but did not influence its Km value. Furthermore, administration of
L-Cys plus PLP increased endogenous H2S production and reduced L-Arg transport.
L-Arg enters mammalian cells through several membrane-bound cationic aminoacid
transporters: systems y+, b0, +, B0, +, and y+L (8). Previous studies have demonstrated
that Arg transport in the vascular endothelium is mainly attributed to system y+, and to
some extent to system y+L (35). NEM, a system y+ selective inhibitor, reduced L-Arg
influx per se. After pretreatment with NEM, H2S did not change L-Arg influx suggesting
that the NEM-sensitive system y+ transporter activity contributes to the regulation of
L-Arg uptake by H2S. In addition, the present study found that H2S also decreased the
CAT1 (encoding the system y+ transporter) transcript level by 34%. All the above results
suggest that endogenous H2S inhibits L-Arg transport under physiological conditions, and
that the system y+ transporter is the main pathway regulated by H2S.
The effect of injection of NaHS on the L-Arg/NOS/NO pathway was also examined
in vivo in rats.According to our previous work, injection of NaHS (14 µmol/kg) after 2 h
increased plasma H2S concentration by 67% over baseline (52). In the present study, the
plasma level of nitrate plus nitrite was decreased by 21%, and aortic eNOS activity by
42% and L-Arg by 30 % 2 h after injection of NaHS. These in vivo results were similar to
those of the in vitro experiments and support the notion that H2S down-regulates the
vascular L-Arg/NOS/NO pathway (i.e., inhibited L-Arg uptake, eNOS activity and NO
generation). Venous injection of H2S can directly induce vasodilation, and cause transient
hypotension (16, 50), and we have previously reported that intraperitoneal bolus injection
of NaHS reduces areterial blood pressure. Increasing plasma H2S level from 39µmol/L to
79 µmol/L reduced arterial blood pressure by 22 mmHg (from 134 mmHg to 112 mmHg);
but plasma H2S concentration increased from 79 µmol/L to 123 µmol/L, while arterial
blood pressure fell by 6 mmHg (from 112 mmHg to 106 mmHg) (52). The present in
Page 18 of 34

19
vivo studies showing inhibition of the L-Arg/NOS/NO pathway by H2S may explain in
part this phenomenon.
Our findings support the concept that eNOS is an important target molecule of H2S,
affecting its activity as well as gene and/or protein expression. Several mechanisms are
known to regulate eNOS activity, including the interaction of eNOS with caveolin-1, heat
shock protein 90 (Hsp90), or membrane phospholipids, and enzyme translocation and
phosphorylation (1). Ser1177 appears to be the most important site of eNOS
phosphorylation and is affected by most, if not all, of the diverse stimuli that promote
eNOS activation. Phosphorylation of eNOS-Ser1177 increases eNOS sensitivity to
Ca2+/calmodulin binding and leads to eNOS activation (28). Our study found that H2S
downregulated eNOS phosphorylation at Ser1177. This result implies that H2S inhibits
eNOS activity partly through inhibition eNOS phosphorylation at Ser 1177.
Akt-mediated-phosphorylation of eNOS-Ser1177 is a crucial step of eNOS activation
induced by estrogen, insulin, VEGF, statins and shear stress (15). H2S reduced Akt
phosphorylation without influencing total Akt protein, suggesting that the Akt pathway
may contribute to regulation of H2S by eNOS-Ser1177 phosphorylation. H2S is an
endogenous opener of the KATP channel in many cells types, e.g. (39, 47, 50). The KATP
channel inhibitor glibenclamide blocked the effects of H2S on eNOS activity (Fig.2G-H),
eNOS phosporylation and Akt phosphorylation. Glibenclamide also blocked inhibition of
eNOS phosphorylation by the non-selective KATP channel opener pinacidil. These data
imply that H2S may open KATP channels, followed by reduced Akt phosphorylation and
inhibition of eNOS phosphorylation, leading to downregulation of eNOS activity.
Our study found that H2S also decreased eNOS transcript abundance. Numerous
physiological and pathophysiological stimuli have been identified to modulate eNOS
expression. Reactive oxygen species, as a signal molecule, play an important role in
eNOS transcription and posttranslational regulation. Effects of H2O2 on eNOS
transcription and posttranslational modification were reported in bovine and human
endothelial cells. Chronic oxidative stress caused by excessive H2O2 production in vivo
evokes a compensatory response involving increased eNOS transcript (36). Our previous
work showed that H2S reduced oxidative radical release and scavenged H2O2 directly
(11). H2S may reduce the H2O2 signal in eNOS transcription and posttranslation, and
Page 19 of 34

20
thereby reduce eNOS transcription. Fig.5D revealed that KATP channel opening by H2S
and pinacidil inhibited eNOS protein expression, effects that were blocked by
glibenclamide. These data suggest that KATP channels are involved in the inhibition of
eNOS protein expression by H2S. However, the precise mechanisms of inhibition of
eNOS protein expression by H2S need to be further investigated.
L-Arg transporter could be additional target molecules of H2S regulation. L-Arg
transport from plasma into cells is mediated by several different classes of CATs. System
y+ activity, selective for cationic amino acids only, is encoded by 4 genes (CAT1, CAT2,
CAT3, and CAT4) representing different isoforms (35). Systems y+ (CAT-1) and y+L
(heavy chain subunit-4F2hc) have been detected in the endothelium (9). Transport of
cationic amino acids via CAT-1 is voltage dependent with alterations of membrane
potential affecting both Vmax and Km values for influx (22). L-Arg transport is sensitive
to the membrane potential; it is stimulated by drugs that cause membrane
hyperpolarization and inhibited by those that cause membrane depolarization (51). H2S
opens the KATP channel, and induces membrane hyperpolarization (50), which may
contribute to L-Arg transport activity, and furthermore, results in an accumulation of the
intracellular pool of L-Arg available for eNOS. However, opening KATP channels alone is
not enough to explain the mechanisms of H2S-mediated L-Arg influx. Administration of
pinacidil reduced L-Arg influx as did H2S, but NEM could not block the inhibitory
effects of pinacidil. Glibenclamide blocked the effect of pinacidil to a lesser extent than
H2S, suggesting that the mechanisms involved in the H2S regulation of L-Arg influx may
be more complex.
Administration of pinacidil reduced L-Arg influx as did H2S, but NEM could not
block the effects of pinacidil. Glibenclamide blocked almost completely the inhibition
effects of pinacidil, but only slightly blocked the effects of H2S, which suggests that the
mechanisms involved in the H2S regulation of L-Arg influx are complex. Opening KATP
channels alone is not enough to explain the mechanisms of H2S-mediated L-Arg influx.
Oh et al. found that H2S could inhibit NO production in LPS-stimulated macrophages
through a mechanism that involves heme oxygenase-1(HO-1)/carbon monoxide (CO)
(29). H2S increased pulmonary arterial HO-1 gene transcription and protein expression
(32). CO, derived from VSMCs and endothelium, has vasodilatory and antiproliferative
Page 20 of 34

21
effects and acts as a competitive antagonist for NO-mediated sGC activation or displaces
internal stores of NO. As a heme ligand, CO potentially inhibits NOS activity and
reduces NO production (34, 38).
Taken together, these results suggest that the gasotransmitter family, including NO,
CO and H2S, derived from vascular tissues in a paracrine/autocrine manner, interacts in
the regulation of vascular homeostasis. The interaction of these gasotransmitters in
distinct locations of the blood vessels could maintain a dynamic balance and form a
regulatory ″network″ (41). Imbalances of the network regulation may contribute to
pathogenesis of cardiovascular diseases. Research into the network regulation of
gasotransmitters should reveal a novel prospect for understanding the mechanism of
cardiovascular diseases and a novel target for prevention and treatment.
ACKNOWLEDGMENTS
This work was supported by The Major State Basic Research Development Program of
the People’s Republic of China (no. 2006CB503807) National Natural Science
Foundation of People’s Republic China (no. 30400151), National Natural Science
Foundation for Distinguished Young Scholars (no. 30425010) and Grant no.
20030001035 from the Ministry of Education, China.
Page 21 of 34

22
REFERENCES
1. Alderton WK, Cooper CE, and Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J 357: 593-615, 2001.2. Bian JS, Yong QC, Pan TT, Feng ZN, Ali MY, Zhou S, and Moore PK. Role of hydrogen sulfide in the cardioprotection caused by ischemic preconditioning in the rat heart and cardiac myocytes. J Pharmacol Exp Ther 316: 670-678, 2006.3. Bin G, Fen QY, Hua LX, Hong ZB, Zheng PY, and Shu TC. Dysfunction of myocardial sarcoplasmic reticulum in rats with myocardial calcification. Life Sci 77: 966-979, 2005.4. Cheng Y, Ndisang JF, Tang G, Cao K, and Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol287: H2316-2323, 2004.5. Chunyu Z, Junbao D, Dingfang B, Hui Y, Xiuying T, and Chaoshu T. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem Biophys Res Commun 302: 810-816, 2003.6. Cismasiu VB, Denes SA, Reilander H, Michel H, and Szedlacsek SE. The MAM (meprin/A5-protein/PTPmu) domain is a homophilic binding site promoting the lateral dimerization of receptor-like protein-tyrosine phosphatase mu. J Biol Chem 279: 26922-26931, 2004.7. Declercq JP, Evrard C, Clippe A, Stricht DV, Bernard A, and Knoops B. Crystal structure of human peroxiredoxin 5, a novel type of mammalian peroxiredoxin at 1.5 A resolution. J Mol Biol 311: 751-759, 2001.8. Du J, Hui Y, Cheung Y, Bin G, Jiang H, Chen X, and Tang C. The possible role of hydrogen sulfide as a smooth muscle cell proliferation inhibitor in rat cultured cells.Heart Vessels 19: 75-80, 2004.9. Dye JF, Vause S, Johnston T, Clark P, Firth JA, D'Souza SW, Sibley CP, and Glazier JD. Characterization of cationic amino acid transporters and expression of endothelial nitric oxide synthase in human placental microvascular endothelial cells. Faseb J 18: 125-127, 2004.10. Ebrahimkhani MR, Mani AR, and Moore K. Hydrogen sulphide and the hyperdynamic circulation in cirrhosis: a hypothesis. Gut 54: 1668-1671, 2005.11. Geng B, Chang L, Pan C, Qi Y, Zhao J, Pang Y, Du J, and Tang C. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochem Biophys Res Commun 318: 756-763, 2004.12. Geng B, Yan H, Zhong GZ, Zhang CY, Chen XB, Jiang HF, Tang CS, and Du JB. [Hydrogen sulfide: a novel cardiovascular functional regulatory gas factor]. Beijing Da Xue Xue Bao 36: 106, 2004.13. Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du J, and Tang C. H2S generated by heart in rat and its effects on cardiac function. Biochem Biophys Res Commun 313: 362-368, 2004.14. Giovannoni G, Land JM, Keir G, Thompson EJ, and Heales SJ. Adaptation of the nitrate reductase and Griess reaction methods for the measurement of serum nitrate plus nitrite levels. Ann Clin Biochem 34 ( Pt 2): 193-198, 1997.15. Govers R and Rabelink TJ. Cellular regulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol 280: F193-206, 2001.
Page 22 of 34

23
16. Hosoki R, Matsuki N, and Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun 237: 527-531, 1997.17. Hu H XM, Belayev LL, Zhang J, Block ER, Patel JM. Autoinhibitory domain fragment of endothelial NOS enhances pulmonary artery vasorelaxation by the NO-cGMP pathway. Am J Physiol Lung Cell Mol Physiol 286: L1066-1074., 2004.18. Hui Y, Du J, Tang C, Bin G, and Jiang H. Changes in arterial hydrogen sulfide (H(2)S) content during septic shock and endotoxin shock in rats. J Infect 47: 155-160, 2003.19. Jiang W, Cai DY, Pan CS, Qi YF, Jiang HF, Geng B, and Tang CS. Changes in production and metabolism of brain natriuretic peptide in rats with myocardial necrosis. Eur J Pharmacol 507: 153-162, 2005.20. Jiang W, Yang JH, Pan CS, Qi YF, Pang YZ, and Tang CS. Effects of adrenomedullin on cell proliferation in rat adventitia induced by aldosterone. J Hypertens22: 1953-1961, 2004.21. Kamoun P. Endogenous production of hydrogen sulfide in mammals. Amino Acids26: 243-254, 2004.22. Kavanaugh MP. Voltage dependence of facilitated arginine flux mediated by the system y+ basic amino acid transporter. Biochemistry 32: 5781-5785, 1993.23. Kullo IJ, Schwartz RS, Pompili VJ, Tsutsui M, Milstien S, Fitzpatrick LA, Katusic ZS, and O'Brien T. Expression and function of recombinant endothelial NO synthase in coronary artery smooth muscle cells. Arterioscler Thromb Vasc Biol 17: 2405-2412, 1997.24. Lantin-Hermoso RL, Rosenfeld CR, Yuhanna IS, German Z, Chen Z, and Shaul PW. Estrogen acutely stimulates nitric oxide synthase activity in fetal pulmonary artery endothelium. Am J Physiol 273: L119-126, 1997.25. Leoncini G, Pascale R, and Signorello MG. Effects of homocysteine on l-arginine transport and nitric oxide formation in human platelets. Eur J Clin Invest 33: 713-719, 2003.26. Liaudet L, Fishman D, Markert M, Perret C, and Feihl F. L-canavanine improves organ function and tissue adenosine triphosphate levels in rodent endotoxemia. American journal of respiratory and critical care medicine 155: 1643-1648, 1997.27. Mok YY, Atan MS, Yoke Ping C, Zhong Jing W, Bhatia M, Moochhala S, and Moore PK. Role of hydrogen sulphide in haemorrhagic shock in the rat: protective effect of inhibitors of hydrogen sulphide biosynthesis. Br J Pharmacol 143: 881-889, 2004.28. Mount PF, Kemp BE, and Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol, 2006.29. Oh GS, Pae HO, Lee BS, Kim BN, Kim JM, Kim HR, Jeon SB, Jeon WK, Chae HJ, and Chung HT. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic Biol Med 41: 106-119, 2006.30. Palacin M, Estevez R, Bertran J, and Zorzano A. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev 78: 969-1054, 1998.31. Poppe C, McFadden KA, and Demczuk WH. Drug resistance, plasmids, biotypes and susceptibility to bacteriophages of Salmonella isolated from poultry in Canada. Int J Food Microbiol 30: 325-344, 1996.
Page 23 of 34

24
32. Qingyou Z, Junbao D, Weijin Z, Hui Y, Chaoshu T, and Chunyu Z. Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem Biophys Res Commun 317: 30-37, 2004.33. Qu K, Chen CP, Halliwell B, Moore PK, and Wong PT. Hydrogen sulfide is a mediator of cerebral ischemic damage. Stroke 37: 889-893, 2006.34. Ryter SW and Otterbein LE. Carbon monoxide in biology and medicine. Bioessays26: 270-280, 2004.35. Sala R, Rotoli BM, Colla E, Visigalli R, Parolari A, Bussolati O, Gazzola GC, and Dall'Asta V. Two-way arginine transport in human endothelial cells: TNF-alpha stimulation is restricted to system y(+). American journal of physiology 282: C134-143, 2002.36. Searles CD. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. American journal of physiology 291: C803–C816, 2006.37. Sobrevia L, Nadal A, Yudilevich DL, and Mann GE. Activation of L-arginine transport (system y+) and nitric oxide synthase by elevated glucose and insulin in human endothelial cells. J Physiol 490 ( Pt 3): 775-781, 1996.38. Stevenson TH, Gutierrez AF, Alderton WK, Lian L, and Scrutton NS. Kinetics of CO binding to the haem domain of murine inducible nitric oxide synthase: differential effects of haem domain ligands. Biochem J 358: 201-208, 2001.39. Tang G WL, Liang W, Wang R. Direct stimulation of K(ATP) channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol Pharmacol 68: 1757-1764, 2005 40. Wang R. The gasotransmitter role of hydrogen sulfide. Antioxid Redox Signal 5: 493-501, 2003.41. Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? Faseb J 16: 1792-1798, 2002.42. Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, Cheung NS, Halliwell B, and Moore PK. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite 'scavenger'? J Neurochem 90: 765-768, 2004.43. Wu G, Mannam AP, Wu J, Kirbis S, Shie JL, Chen C, Laham RJ, Sellke FW, and Li J. Hypoxia induces myocyte-dependent COX-2 regulation in endothelial cells: role of VEGF. Am J Physiol Heart Circ Physiol 285: H2420-2429, 2003.44. Yamamoto C, Kaji T, Furuya M, Sakamoto M, Kozuka H, and Koizumi F.Basic fibroblast growth factor suppresses tissue plasminogen activator release from cultured human umbilical vein endothelial cells but enhances that from cultured human aortic endothelial cells. Thromb Res 73: 255-263, 1994.45. Yan H, Du J, and Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun 313: 22-27, 2004.46. Yang G, Cao K, Wu L, and Wang R. Cystathionine gamma-lyase overexpression inhibits cell proliferation via a H2S-dependent modulation of ERK1/2 phosphorylation and p21Cip/WAK-1. J Biol Chem 279: 49199-49205, 2004.47. Yang W YG, Jia X, Wu L, Wang R. Activation of KATP channels by H2S in rat insulin-secreting cells and the underlying mechanisms. J Physiol 569: 519-531, 2005.48. Zhao W, Ndisang JF, and Wang R. Modulation of endogenous production of H2S in rat tissues. Can J Physiol Pharmacol 81: 848-853, 2003.
Page 24 of 34

25
49. Zhao W and Wang R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol 283: H474-480, 2002.50. Zhao W, Zhang J, Lu Y, and Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. Embo J 20: 6008-6016, 2001.51. Zharikov SI. and Block ER. Characterization of l-arginine uptake by plasma membrane vesicles isolated from cultured pulmonary artery endothelial cells. Biochim Biophys Acta 1369: 173-183, 1998.52. Zhong G, Chen F, Cheng Y, Tang C, and Du J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J Hypertens 21: 1879-1885, 2003.
Page 25 of 34
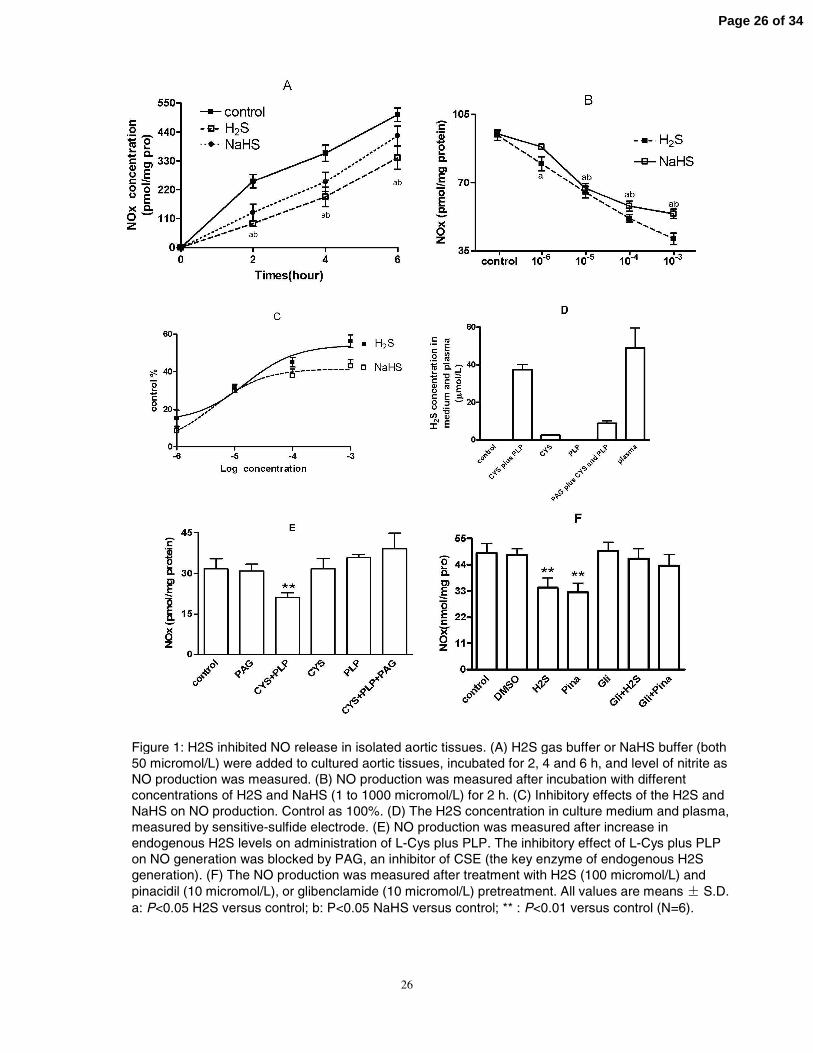
26
Figure 1: H2S inhibited NO release in isolated aortic tissues. (A) H2S gas buffer or NaHS buffer (both 50 micromol/L) were added to cultured aortic tissues, incubated for 2, 4 and 6 h, and level of nitrite as NO production was measured. (B) NO production was measured after incubation with different concentrations of H2S and NaHS (1 to 1000 micromol/L) for 2 h. (C) Inhibitory effects of the H2S and NaHS on NO production. Control as 100%. (D) The H2S concentration in culture medium and plasma, measured by sensitive-sulfide electrode. (E) NO production was measured after increase in endogenous H2S levels on administration of L-Cys plus PLP. The inhibitory effect of L-Cys plus PLP on NO generation was blocked by PAG, an inhibitor of CSE (the key enzyme of endogenous H2S generation). (F) The NO production was measured after treatment with H2S (100 micromol/L) and pinacidil (10 micromol/L), or glibenclamide (10 micromol/L) pretreatment. All values are means S.D. a: P<0.05 H2S versus control; b: P<0.05 NaHS versus control; ** : P<0.01 versus control (N=6).
Page 26 of 34

27
Page 27 of 34

28
• Figure 2: H2S downregulates eNOS activity without changing iNOS activity. (A) Alterations in aortic eNOS and iNOSactivities induced by H2S and NaHS (50 micromol/L) after 2, 4 and 6 h of incubation. (B) Effect of different concentrations of H2S and NaHS (1-1000 micromol/L) on aortic eNOS and iNOS activities after 2-h treatment. (C) Inhibitory effect of H2S and NaHS on eNOS activity. Control as 100%. (D) eNOS enzyme activity assay at various concentrations of L-Arg (0.1-3.2 mmol/L) in HUVECs with or without treatment with physiological concentrations of H2S (50 micromol/L). (E) Effect on eNOS and iNOS activities of endogenous H2S generated by treatment with L-Cys plus PLP. (F) The alteration of nNOS and iNOSactivity after treatment with H2S (100 micromol/L) and pinacidil (10 micromol/L), and the effects of the use of the selective iNOS inhibitor-L canavanine (2.5 mmol/L), the nNOS selective inhibitor spermidine (0.5 mmol/L) and the non-selective KATP channel inhibitor glibenclamide (10 micromol/L). (G) H2S (500micromol/L) and pinacidil (10 micromol/L) down-regulate eNOS phosphorylation at Ser1177, but do not change eNOS protein expression after 2-h incubation; glibenclamide(Gli) blocked the inhibitory effects in part (N=3). (H) H2S (500 micromol/L) and pinacidil (10 micromol/L) reduced phosphorylation of Akt and glibenclamide blocked the inhibitory effects. All values are means S.D. a: P<0.05 H2S versus control; b: P<0.05 NaHS versus control; *P<0.05 versus control; **: P<0.01 versus control; #:P<0.05 versus H2S treatment,&: P<0.05 versus pinacidil. $: P<0.01 versus control iNOS activity (N=5).
Page 28 of 34

29
Figure 3: H2S inhibited L-Arg transport. (A) After incubation with H2S and NaHS (50 micromol/L) for 2, 4 and 6 h, aortic L-Arg transport was measured by the isotope tracer method. (B) Aortic tissues were treated with various concentrations of H2S and NaHS (1-1000 micromol/L) for 2 h, and the aortic L-Arg uptake was assessed. (C) Correction statistics are given for concentrations of H2S and NaHS and inhibition proportion of L-Arg uptake. Control as 100%. (D) L-Arg transport activity assay at various concentrations of L-Arg (0.1-3.2 mmol/L) in HUVECs with or without H2S (50 micromol/L). (E) Alterations in L-Arg transport induced by endogenous H2S production. (F) The effect of the nonselective KATP channel opener pinacidil (10 micromol/L, Pina) on L-Arg uptake; and pretreatment with the system y+ transporter inhibitor NEM, the alteration of L-Arg uptake by H2S and pinacidil. All values are means S.D. a: P<0.05 H2S versus control; b: P<0.05 NaHS versus control; **: P<0.01 versus control, and #: P<0.05 versus H2S treatment. (N=5).
Page 29 of 34

30
Figure 4: H2S inhibited CAT-1 and eNOS gene expressions in HUVECs. (A) After 4 h of incubation with H2S (50micromol/L), cells underwent RT-PCR, separation on a 1.5 % agarose gel and staining with ethidium bromide; bands of eNOS mRNA (553 bp), CAT-1 mRNA (250 bp) and GAPDH mRNA (205 bp) are shown. M is the DNA marker. (B) The optical densities of the bands were measured by use of the Gel Documentation System. The mRNA ratio of eNOS and CAT-1 to GAPDH was considered the relative amount of eNOS and CAT-1 mRNA (N=3). **: P<0.01 versus control.
Page 30 of 34

31
Figure 5: H2S regulates eNOS, iNOS and nNOS protein expression. (A) eNOS protein expression was revealed by Western blotting after incubation with H2S (100, 500 and 1000 micromol/L) for 6 h. Glibenclamide (Gli), a KATP channel inhibitor blocked the inhibitory effects of H2S. Tubulin expression was detected as a protein control. (B)The effects of pinacidil on eNOS protein expression. (C) iNOS protein expression was analyzed after H2S treatment (100, 500 and 1000 micromol/L), and TNF (30 ng/ml) as a positive control. (D) nNOS protein expression was measured after treatment with H2S, pinacidil and glibenclamide. *: P<0.05 versus control, **: P<0.01 versus control; #: P<0.05 versus H2S treatment and $: P<0.05 versus pinacidil (N=3).
Page 31 of 34
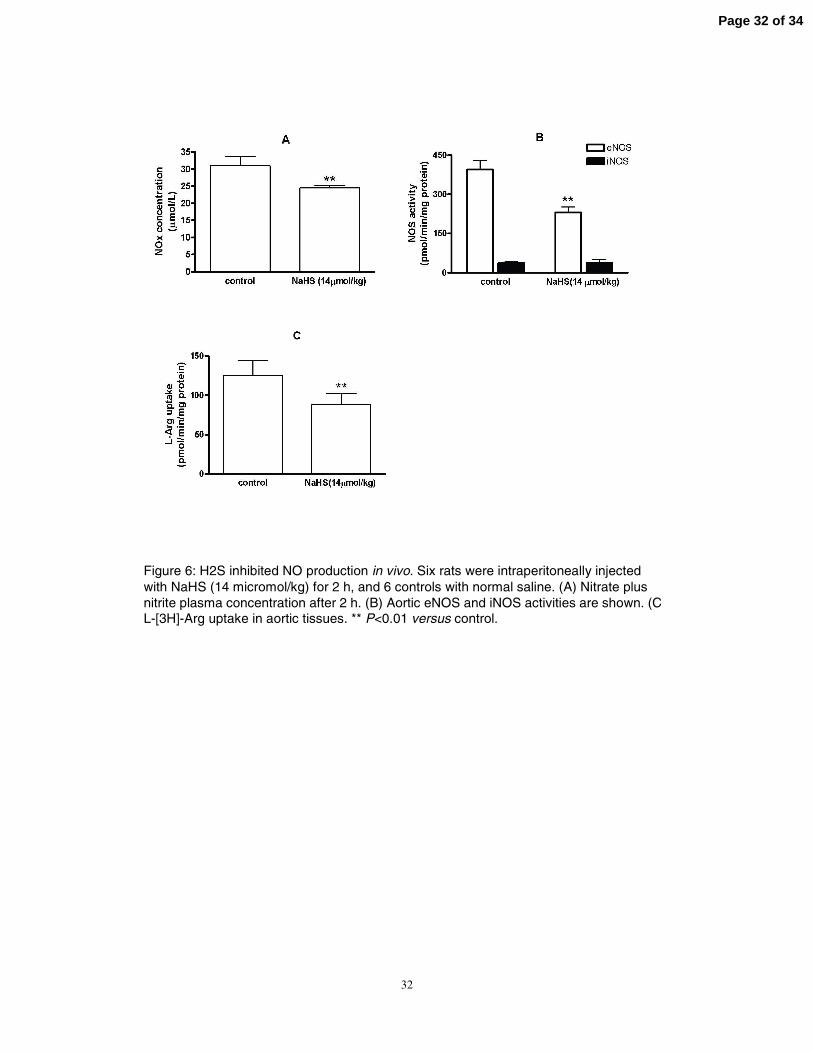
32
Figure 6: H2S inhibited NO production in vivo. Six rats were intraperitoneally injected with NaHS (14 micromol/kg) for 2 h, and 6 controls with normal saline. (A) Nitrate plus nitrite plasma concentration after 2 h. (B) Aortic eNOS and iNOS activities are shown. (C) L-[3H]-Arg uptake in aortic tissues. ** P<0.01 versus control.
Page 32 of 34

33
Figure 7: Summary of the interaction between H2S and NO in artery
Page 33 of 34

34
Page 34 of 34




















