
Cytomegalovirus Downregulates IRE1 to Repress the Unfolded Protein Response
Upload
independentCategory
view
1download
0
Cancer Cell
Article
JAK2V617F-Mediated Phosphorylation of PRMT5Downregulates Its Methyltransferase Activityand Promotes MyeloproliferationFan Liu,1,4 Xinyang Zhao,1,4 Fabiana Perna,1 Lan Wang,1 Priya Koppikar,2 Omar Abdel-Wahab,2 Michael W. Harr,1
Ross L. Levine,2 Hao Xu,1 Ayalew Tefferi,3 Anthony Deblasio,1 Megan Hatlen,1 Silvia Menendez,1 and Stephen D. Nimer1,*1Molecular Pharmacology and Chemistry Program, Sloan-Kettering Institute2Human Oncology and Pathogenesis ProgramMemorial Sloan-Kettering Cancer Center, New York, NY 10065, USA3Division of Hematology, Mayo Clinic, Rochester, MN 55905, USA4These authors contributed equally to this work
*Correspondence: [email protected] 10.1016/j.ccr.2010.12.020
SUMMARY
The JAK2V617F constitutively activated tyrosine kinase is found in most patients with myeloproliferativeneoplasms. While examining the interaction between JAK2 and PRMT5, an arginine methyltransferase orig-inally identified as JAK-binding protein 1, we found that JAK2V617F (and JAK2K539L) bound PRMT5 morestrongly than did wild-type JAK2. These oncogenic kinases also acquired the ability to phosphorylatePRMT5, greatly impairing its ability to methylate its histone substrates, and representing a specific gain-of-function that allows them to regulate chromatin modifications. We readily detected PRMT5 phosphoryla-tion in JAK2V617F-positive patient samples, and when we knocked down PRMT5 in human CD34+ cellsusing shRNA, we observed increased colony formation and erythroid differentiation. These results indicatethat phosphorylation of PRMT5 contributes to the mutant JAK2-induced myeloproliferative phenotype.
INTRODUCTION
The myeloproliferative neoplasms (MPNs) are stem cell disor-
ders whose proliferation is thought to be driven by activating
tyrosine kinase gene mutations, such as the BCR-ABL fusion
gene in chronicmyelogenous leukemia (CML) (De Keersmaecker
and Cools, 2006). The JAK2 kinase V617F mutation is found in
most patients with non-CML MPN (Baxter et al., 2005; James
et al., 2005; Kralovics et al., 2005; Levine et al., 2005). It is
a constitutively active kinase that can phosphorylate STAT5 in
the absence of upstream signals, confer cytokine-independent
growth to Ba/F3 cells, and induce a myeloproliferative disease
in mouse models (Akada et al., 2010; James et al., 2005; Marty
et al., 2010; Mullally et al., 2010; Xing et al., 2008). In addition
to V617F, mutations within the exon 12 region of JAK2, such
as K539L, have been observed in patients with MPN, although
they are much rarer (Pikman and Levine, 2007; Scott et al.,
Significance
The JAK2V617F mutation has been found in most cases of MPkinases acquire the ability to phosphorylate and downregulatand erythroid differentiation promoting effects of JAK2V617Fbetween oncogenic kinases and histone arginine methylationhematopoietic stem/progenitor cells. These findings provide igies for the MPNs.
C
2007). Like the V617F mutation, these mutations disrupt the
negative regulation of JAK2 and lead to constitutive kinase
activity.
The JAK2 protein associates with the cytoplasmic domains of
a number of cytokine receptors and is crucial for mediating
signals triggered by several hematopoietic growth factors,
including erythropoietin (Epo), thrombopoietin (Tpo), and granu-
locyte colony-stimulating factor (G-CSF). Murine JAK2 knockout
embryos die of severe anemia on days 11–13 in utero, demon-
strating the importance of JAK2 in hematopoietic cytokine
signaling (Neubauer et al., 1998; Parganas et al., 1998). It has
been reported by several groups that the transforming effects
of JAK2V617F require an intact FERM domain, which binds to
homodimeric type I cytokine receptors (Lu et al., 2005; Wernig
et al., 2008a). This suggests that interactions between JAK2
and cytokine receptors remain capable of regulating the biolog-
ical function of JAK2V617F.
Ns, and in this study we show that oncogenic JAK2 mutante PRMT5 activity. This contributes to the myeloproliferationand represents a gain-of-function that leads to crosstalk, altering the gene expression profile and the behavior ofnsight into the pathogenesis and possible treatment strate-
ancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc. 283

Cancer Cell
JAK2V617F Regulates PRMT5 Activity
Upon activation, the receptor-bound JAK2 phosphorylates
specific tyrosine residues of its downstream targets, activating
cell survival/proliferation-promoting signaling pathways (Ihle
and Gilliland, 2007). Several kinase cascades are activated by
JAK2V617F, including the STAT5/BCL-XL, PI3K/AKT, and
ERK/MAPK pathways (James et al., 2005; Wang et al., 2009);
however, they may not completely account for the MPN
phenotype.
The type II arginine methyltransferase PRMT5 was first identi-
fied as JAK-binding protein 1 (JBP1) in a yeast two-hybrid assay
(Pollack et al., 1999). It mediates the symmetrical dimethylation
of arginine residues within histones H2A, H3, and H4 (Ancelin
et al., 2006; Branscombe et al., 2001; Pal et al., 2004), and meth-
ylates other cellular proteins as well, such as p53, SPT5, and
MBD2 (Jansson et al., 2008; Kwak et al., 2003; Tan and Nakielny,
2006). Together with the WD40-repeat containing MEP50
protein andwith pICln, PRMT5 forms a large 20S protein arginine
methyltransferase complex, termed the ‘‘methylosome.’’ This
complex functions in RNA processing by methylating Sm
proteins and affecting snRNP biogenesis (Chari et al., 2008;
Friesen et al., 2001, 2002; Meister and Fischer, 2002). PRMT5
has been also found in the hSWI/SNF and NURD chromatin-
remodeling complexes (Le Guezennec et al., 2006; Pal et al.,
2004), where it can exert transcriptional control on target gene
expression.
Although first identified as JAK2-binding protein, there are no
functional data linking PRMT5 with JAK2. To gain insight into
JAK2V617F-induced MPN, we investigated the in vivo interac-
tion between PRMT5 and the oncogenic mutant JAK2 kinases
(JAK2V617F and JAK2K539L), and determined how this interac-
tion contributes to the myeloproliferative phenotype that they
induce.
RESULTS
PRMT5 Interacts with JAK2V617F and JAK2K539LMore Strongly than Wild-Type JAK2First, we examined whether PRMT5 interacts with JAK2 and if
the V617F (and K539L) activating mutations in JAK2 affect this
interaction. We coexpressed FLAG-PRMT5 with HA-tagged
wild-type JAK2 and JAK2V617F, or HA-PRMT5 with nontagged
versions of the wild-type JAK2, JAK2V617F, and JAK2K539L
proteins in 293T cells, and found that whereas the wild-type
JAK2 interacts with PRMT5, both the JAK2V617F and
JAK2K539L mutants bound PRMT5 more strongly than wild-
type JAK2 (Figures 1A and 1B), demonstrating that both consti-
tutively activated forms of JAK2 have increased affinity for
PRMT5. Next, to determine whether the endogenous
JAK2V617F and PRMT5 proteins interact in leukemia cells, we
performed co-immunoprecipitation (Co-IP) assays using two
different anti-JAK2 antibodies and the JAK2V617F-positive
HEL cell line. The interaction of JAK2V617F with PRMT5 was
readily detected using either antibody (Figure 1C). Because
none of the commercially available anti-PRMT5 antibodies effi-
ciently immunoprecipitates PRMT5, we also utilized a HEL cell
line that we engineered to stably express HA-tagged PRMT5.
Using an anti-HA antibody, we could detect a robust interaction
between PRMT5 and the mutant JAK2 (Figure 1D). We
confirmed that the interaction between PRMT5 and JAK2V617F
284 Cancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc.
is stronger than the interaction between PRMT5 and wild-type
JAK2 in hematopoietic cells using Ba/F3 cell lines that stably
express the wild-type or V617F mutant JAK2 proteins. Although
these cell lines express endogenous JAK2 protein (see and
compare lanes 2 and 3 to lane 1 in Figure S1A. available online),
using an anti-JAK2 antibody for the IP, we see that the mutant
JAK2V617F pulls down significantly more endogenous PRMT5
protein than does wild-type JAK2. We also determined the
subcellular localization of the JAK2-PRMT5 interaction by
performing Co-IP experiments using cytoplasmic and nuclear
fractions of HEL cells that stably express HA-tagged PRMT5
(Figure S1C). The interaction between JAK2 and PRMT5 can
be detected in both the cytoplasmic and nuclear fractions of
these hematopoietic cells. This reflects their normal localization
in the cell because the endogenous JAK2 and PRMT5 proteins
are found in both the cytoplasmic and nuclear fractions of HEL
cells (Figure S1B).
To map the region(s) in JAK2 that interacts with PRMT5,
we constructed a series of N-terminal deletion mutants of HA-
tagged JAK2 with or without the V617F substitution, and
expressed these proteins with FLAG-tagged PRMT5 in 293T
cells (Figure 1E). Co-IP experiments showed that deletion of
the first 382 amino acids (which contain the receptor-binding
FERM domain) from JAK2V617F greatly reduced its interaction
with PRMT5. Given that the C terminus of JAK2 (808-1132)
does not bind PRMT5, this indicates that the N-terminal portion
of JAK2 is responsible for binding PRMT5. The JAK2D382WT
deletion mutant protein also binds weakly to PRMT5, suggesting
that loss of the FERMdomainmay expose other epitopes in wild-
type JAK2 that can bind PRMT5.
Oncogenic JAK2 Kinases Phosphorylate PRMT5 In VivoTo determine if JAK2 kinase can directly phosphorylate PRMT5,
we performed an in vitro kinase assay using bacterially purified
GST-PRMT5 as the substrate (Figure 2A). JAK2-dependent
phosphorylation of GST-PRMT5 was readily detected because
a JAK2 inhibitor (JAK Inhibitor I) (1 mM) completely abrogated
the phosphorylation (lane 8). We next determined if JAK2 phos-
phorylates PRMT5 in vivo, by coexpressing HA-PRMT5 with
wild-type JAK2 or the JAK2 mutants in 293T cells, and using
anti-HA immunoprecipitation followed by anti-phosphotyrosine
immunoblotting. We found that PRMT5 was phosphorylated by
the mutant JAK2V617F and JAK2K539L kinases, but not the
wild-type JAK2 kinase (Figures 2B and 2C). The phosphorylation
of PRMT5 by JAK2V617F was further confirmed using an anti-
phosphotyrosine antibody to pull down proteins from 293T cells
coexpressing JAK2V617F and FLAG-tagged PRMT5 (Figure S2).
To determine whether wild-type JAK2 can phosphorylate
PRMT5 when it is activated by signaling through the erythropoi-
etin receptor (EpoR), we transfected 293T cells with JAK2 wild-
type or V617F mutant with or without the EpoR, and added
20 U/ml of Epo to the cells for 20 min (Figure 2D). Wild-type
JAK2 was activated by the presence of Epo and its receptor
(as shown by JAK2 autophosphorylation and phosphorylation
of HA-STAT5, lane 10). However, unlike JAK2V617F, the acti-
vated wild-type JAK2 kinase did not detectably phosphorylate
PRMT5, indicating that PRMT5 phosphorylation is indeed an
acquired function of themutant JAK2 kinase. Interestingly, phos-
phorylation of PRMT5 by JAK2V617F was reduced in cells

FL.PRMT5JAK2WT.HA
JAK2V617F.HA +
+
FLAG
JAK2
FLAGTubulin
IP: HA
Input
JAK2HA-PRMT5
--+ + +-- --
WT WT V617F K539L
Input
IP: HA
JAK2
JAK2
PRMT5
-- + + +-- -- ---- -- --
Input IgG JAK2IP
JAK2
PRMT5
Input
IP: HA
HA
FLAG
FLAG
HA
JAK2 -- -- WTFLVFFL
∆N382W
T
∆N382V
F
∆N807
∆N499V
F
∆N499W
T
+ FL.PRMT51 2 3 4 5 6 7 8 9
1 2 3 41 2 3 4 5
WTFLVFFL
ΔN382WT
ΔN382VFΔN499WTΔN499VFΔN807
1 1132
383
500
808
V617F
JAK2
A B
C
E
IP Input
IP: IgG HA
JAK2
PRMT5
JAK2
PRMT5
D
(125kDa)
JAK2HA
(70kDa)
IgG HA
Figure 1. The Oncogenic JAK2 Mutants Interact More Strongly with PRMT5 than Wild-Type JAK2 and Gain the Ability to Phosphorylate
PRMT5
(A) The V617Fmutation enhances the interaction between JAK2 and PRMT5. 293T cells were transiently transfected with vectors expressing FLAG-PRMT5 alone
(lane 2) or with HA-tagged wild-type (lane 3), or V617F (lane 4) JAK2 proteins. Immunoprecipitation was performed using an anti-HA antibody and immunoblotting
with anti-FLAG or anti-JAK2 antibodies.
(B) Both the V617F and K539Lmutations enhance the PRMT5/JAK2 association. HA-tagged PRMT5was coexpressed in 293T cells with wild-type (lane 3), V617F
(lane 4), or K539L (lane 5) JAK2 proteins. Proteins were precipitated by anti-HA immunoprecipitation, and the blots were probed with antibodies specific for HA,
JAK2, or PRMT5.
(C) Endogenous interaction between PRMT5 and JAK2V617F is detected in HEL cells. Proteins were precipitated from HEL cell extracts using two different anti-
JAK2 antibodies; normal rabbit IgG was used as a control for the immunoprecipitation.
(D) Interaction between HA-PRMT5 and endogenous JAK2V617F is detected in HEL cells. Co-IP was performed using a HEL cell line stably expressing
HA-PRMT5. Proteins were precipitated by either a normal mouse IgG or anti-HA antibody. Immunoblotting was performed using antibodies specific for JAK2
and PRMT5.
(E) The N-terminal region of JAK2 interacts with PRMT5. The left diagram is of full-length JAK2 and various amino terminal-deleted JAK2 proteins with or without
the V617F mutation. Numbers indicate the relevant amino acids. Right panel shows that HA-tagged JAK2 full-length or various amino terminal-deletion mutants
were coexpressed in 293T cells with FLAG-PRMT5. JAK2 and any associated PRMT5 were pulled down using anti-HA immunoprecipitation. Membranes were
immunoblotted with an anti-HA antibody to detect JAK2 and an anti-FLAG antibody to detect PRMT5. WTFL, wild-type full-length JAK2 protein; VFFL,
JAK2V617F full-length protein.
See also Figure S1.
Cancer Cell
JAK2V617F Regulates PRMT5 Activity
overexpressing the EpoR, suggesting that the level of EpoR
expression can affect the ability of JAK2V617F to phosphorylate
PRMT5.
We next determined whether phosphorylation of endogenous
PRMT5 by JAK2V617F occurs in JAK2V617F-positive HEL
leukemia cells. HEL cells were treated with either DMSO or
JAK Inhibitor I for 16 hr, and the phosphorylated proteins were
C
immunoprecipitated using an anti-phosphotyrosine antibody
(Figure 2E). Although PRMT5 (and STAT5) is phosphorylated
in DMSO-treated HEL cells, and the phosphorylation of
both proteins is greatly reduced by the JAK2 inhibitor, PRMT5
is not phosphorylated in the TF-1 hematopoietic cells (which
express wild-type JAK2), even when the JAK2 kinase is
activated by Epo (or GM-CSF), which clearly triggers STAT5
ancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc. 285

PRMT5
MBP
JAK2 kinase(ng)GST-PRMT5(ug)
MBP(ug)JAK inhibitor I
-- 1.25 1.251.25 0.5
--0.5 2.5
-- 2.5 -- -- -- -- ---- -- -- -- -- -- --
0.5 1.251.252.5 -- -- 2.5 0.5
B
JAK2 -- -- WT V617FHA-PRMT5 -- + + +
Input
IP: HA
JAK2
P-PRMT5
PRMT5
2.5
Input
IP: HA
JAK2
P-JAK2
P-PRMT5P-STAT5
PRMT5STAT5 (95kDa)
JAK2WTJAK2VF
EpoR
JAK2
P-PRMT5
JAK2 -- -- WT VF KL
HA-PRMT5 -- + + + + + + +
PRMT5
-- -- + -- + -- -- + -- + ---- -- +-- -- +-- +---- +-- -- -- -- + + -- -- -- + +
1 2 3 4 5 6 7 8 9 10 11HA-PRMT5 HA-STAT5
InputIP
PY99 IgG
TubulinSTAT5PRMT5
D JI1 D JI1 DSTAT5
PRMT5
Input
IP: PY99
-- Epo GM-CSF
Epo+J
I1
STAT5
PRMT5
STAT5
PRMT5
Cytokine
1 2 3 4 5 6 7 8
A
C D
E F
+
Figure 2. The Constitutively Active JAK2 Mutants Phosphorylate PRMT5 In Vivo
(A) JAK2 phosphorylates PRMT5 in vitro. In vitro kinase assays were performed using bacterial-purified GST-PRMT5 and the active JAK2 kinase. The amount of
protein in the reaction is indicated. Myelin basic protein (MBP) was used as a positive control (lane 3).
(B) PRMT5 is phosphorylated by JAK2V617F in vivo. HA-PRMT5 was cotransfected into 293T cells with an empty vector or with vectors expressing wild-type or
V617F mutant JAK2 protein. PRMT5 was precipitated by anti-HA immunoprecipitation, and phosphorylation of PRMT5 was detected using a phosphotyrosine-
specific antibody (PY350).
(C) Both JAK2V617F and JAK2K539L phosphorylate PRMT5 in vivo. 293T cells were transiently transfected with HA-PRMT5 alone or cotransfected with an
increasing concentration of JAK2 wild-type, JAK2V617F- or JAK2K539L-expressing vector. HA-PRMT5was purified by anti-HA immunoprecipitation, and phos-
phorylation of PRMT5 was detected by immunoblotting with the PY350 antibody.
(D) Activated wild-type JAK2 is unable to phosphorylate PRMT5 in vivo. 293T cells were transfected with vectors expressing the proteins indicated in the figure.
Proteins were purified by anti-HA immunoprecipitation, and phosphorylation of PRMT5 and STAT5 was detected using an anti-phosphotyrosine antibody. Input
was from 5% of the total lysate. JAK2 phosphorylation was detected using an antibody specific for phospho-JAK2.
(E) Endogenous PRMT5 is phosphorylated in HEL cells. HEL cells were treated with either DMSO or 2 mM JAK Inhibitor I for 16 hr. Phosphorylated proteins were
purified using the PY99 anti-phosphotyrosine antibody. Normal mouse IgG was used as a control for the immunoprecipitation. The precipitated STAT5 and
PRMT5 proteins were detected using anti-STAT5 and anti-PRMT5 antibodies, respectively. JI1, JAK Inhibitor I.
(F) Phosphorylated PRMT5 is not detected in TF-1 cells. TF-1 cells cultured in growth medium supplemented with 2 ng/ml of recombinant human IL-3 were
deprived of cytokine for 16 hr. The cells (5 3 106) were then treated with GM-CSF (25 ng/ml), Epo (20 U/ml), or Epo plus 2 mM of JAK Inhibitor I (JI1) for
20 min. Phosphorylated STAT5 and PRMT5 proteins were immunoprecipitated using the PY99 antibody.
See also Figure S2.
Cancer Cell
JAK2V617F Regulates PRMT5 Activity
phosphorylation (Figure 2F). These results identify PRMT5 as
a bona fide in vivo substrate of JAK2V617F, but not activated
wild-type JAK2.
Phosphorylation of PRMT5 by JAK2V617F GreatlyImpairs Its Methyltransferase ActivityPRMT5 has been shown to methylate histones H2A, H3, and H4
in vitro and in vivo. To determine whether phosphorylation of
PRMT5 affects its enzymatic activity, we purified HA-tagged
PRMT5 protein from 293T cells engineered to express either
wild-type or mutant JAK2 and HA-PRMT5. After we confirmed
286 Cancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc.
the phosphorylation of PRMT5 by JAK2V617F, we incubated
the purified PRMT5 with [3H] S-adenosylmethionine and
recombinant histone H4 (Figure 3A) or histone H2A (Figure 3B)
in an in vitro methylation assay. Although coexpression of wild-
type JAK2 had little effect on PRMT5 methyltransferase activity
(Figure 3A, lanes 8–10), JAK2V617F significantly impaired
the ability of PRMT5 to methylate histone H4 (lanes 11–13). As
expected, coexpressionofMEP50withPRMT5greatly enhanced
its enzymatic activity (lanes 2–4). Similar results were seen for
JAK2V617F (and JAK2K539L) on histone H2A methylation (Fig-
ure 3B; data not shown).

AMethylated H4
PRMT5
H4
--
1 2 3 4 5 6 7 8 9 10 11 12 13MEP50 EV JAK2WT JAK2VF
HA-PRMT5 -- --
1 2 3 4 5 6 7 8 9
MEP50
JAK2WTJAK2VF
-- -- -- -- -- -- -- -- +-- -- -- -- -- -- + + ---- -- -- -- + + -- -- --
Methylated H2A
PRMT5
H2A(14kDa)
H2A/H4R3me
H3(18kDa)
JAK2 -- WT V617F K539L
D
B
C
Genes regulated by PRMT5Genes regulated by JAK2
E
Down:353
Up:34842
Up:528
Down:23748
+
Hours 0 1 2 4
H2AR3me
H3
P-STAT5
Tubulin
F
G
DMSOJI1 CEP70
1
H3
H2AR3me
P-PRMT5-747
PRMT5
(11kDa)
SS Starve
d
DMSOJI1 CEP70
1
P-STAT5
H2AR3me
H3
HA-PRMT5 Figure 3. Phosphorylation of PRMT5 by
JAK2V617F Impairs Its HistoneMethyltrans-
ferase Activity
(A) Coexpression of JAK2V617F, but not the wild-
type JAK2, impairs the ability of PRMT5 to meth-
ylate histone H4 in vitro. An in vitro methylation
assay was performed using HA-PRMT5 purified
from 293T cells transfected with HA-PRMT5 alone
(lanes 5–7) or cotransfected with MEP50 (lanes
2–4), wild-type JAK (lanes 8–10), or JAK2V617F
(lanes 11–13). Increasing amount of proteins (10,
15, or 20 ml) were added to each methylation
reaction with 2.5 mg recombinant H4 and 1 mCi of3H-SAM. Even loading of the proteins (PRMT5
and histone H4) was visualized using Coomassie
blue staining. EV, empty vector.
(B) In vitro methylation assays were performed
with recombinant H2A and similarly purified
HA-PRMT5. Increasing amount of purified protein
(10 or 20 ml) was added to the reaction.
(C) Overexpression of the mutant JAK2 proteins,
but not the wild-type JAK2 protein, downregulates
H2A/H4 R3 methylation. The core histones were
purified from 293T cells transfected with either
an empty vector or with vectors expressing the
JAK2 proteins indicated in the figure. Methylation
of H2A/H4 R3 was detected using a rabbit poly-
clonal antibody specific for H2A/H4 R3 symmetric
dimethylation (H2A/H4R3me2s). An anti-histone
H3 antibody was used to show the equal loading.
(D) Inhibition of JAK2 kinase activity increases H2A
R3methylation in HEL cells. HEL cells were treated
with DMSO or with the JAK2 inhibitors JAK Inhib-
itor I (1 mM) or CEP701 (0.2 mM) for 2 hr. Histone
methylation was detected in cell lysates using an
antibody specific for symmetric dimethylation of
H2A/H4 R3. JI1, JAK Inhibitor I.
(E) Inhibition of JAK2 rapidly upregulates H2A R3
methylation in HEL cells. HEL cells were treated
with TG101348 (3 mM) for 0, 1, 2, and 4 hr. JAK2
kinase inhibition was monitored by measuring
STAT5 phosphorylation using an anti-phospho-
STAT5 antibody. Phosphorylation of PRMT5 was
detected with the phospho-specific PRMT5
antibody (P-PRMT5-747). Histones were purified
by acidic extraction, and H2A R3 methylation
was detected using an anti-H2A/H4R3me2s anti-
body. The levels of histone H3 and tubulin are
shown as loading controls.
(F) JAK2 inhibitors do not change the level of H2A R3 methylation in TF-1 cells. TF-1 cells growing in medium supplemented with recombinant human IL-3 were
treated with DMSO or JAK2 inhibitors (as indicated in the figure) for 2 hr. Histone methylation was detected using an anti-H2A/H4R3me2s antibody and STAT5
phosphorylation detected using a phospho-STAT5 antibody. SS, steady state. Starved cells were deprived of IL-3 for 2 hr.
(G) Gene expression profiles were generated using Affymetrix HG133 GeneChips and CEP701 (0.5 mM)-treated or shPRMT5-treated HEL cells, versus controls
(DMSO or a lentivirus expressing a scrambled shRNA, respectively). Genes reciprocally up- or downregulated R1.5-fold in both duplicate samples from drug-
treated samples, and the shPRMT5-treated samples are compared. The top shows the number of genes that are downregulated by CEP701 treatment and
upregulated by knockdown of PRMT5. The bottom illustrates the number of genes that are upregulated by CEP701 treatment and downregulated by PRMT5
knockdown.
See also Table S1.
Cancer Cell
JAK2V617F Regulates PRMT5 Activity
We next examined whether JAK2V617F and JAK2K539L
expression affects global H2A/H4 R3 symmetric dimethylation
levels in vivo using 293T cells transiently expressing JAK2
wild-type, JAK2V617F, or JAK2K539L. Wild-type JAK2 had no
effect on the global level of H2A/H4 R3 methylation; however,
both oncogenic JAK2 kinases nearly abolished H2A/H4 R3
methylation (Figure 3C). We next assessed histone arginine
C
methylation levels in HEL cells, in the presence or absence of
JAK Inhibitor I, CEP701, which is a JAK2 (and FLT3) inhibitor
(Hexner et al., 2008) and TG101348 (Lasho et al., 2008; Wernig
et al., 2008b), the most specific JAK2 inhibitor tested. Treatment
with all three JAK2 inhibitors markedly increased H2A R3
symmetric dimethylation in the cell (Figures 3D and 3E) (H4 R3
symmetric dimethylation is not found in HEL cells in the presence
ancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc. 287

Cancer Cell
JAK2V617F Regulates PRMT5 Activity
or absence of the JAK2 inhibitor). To determine how rapidly JAK2
inhibition affects the level of H2A/H4 R3 methylation, we
performed a time course experiment using HEL cells treated
with TG101348 (Figure 3E). An increase in H2A/H4 R3 methyla-
tion was seen within 1 hr, which peaked at the 2-hr time point.
At 4 hr, the effect on H2A/H4 R3 methylation began to reverse,
likely due to instability of the inhibitor because both STAT5 and
PRMT5 phosphorylation began to increase at the 4-hr time point.
In contrast, these JAK2 inhibitors had minimal effect on H2A/H4
R3 methylation levels in TF-1 cells, even though they blocked
STAT5 phosphorylation (Figure 3F). Thus, the oncogenic JAK2
proteins gain the ability to regulate global H2A/H4 R3 symmetric
dimethylation levels, presumably via phosphorylation of PRMT5.
Although JAK2 regulates gene transcription through the
canonical JAK2-STAT5 pathway, as well as other pathways,
our data suggest that the oncogenic JAK2 kinases (like V617F
and K539L) can also regulate gene expression via repression of
PRMT5 activity and possibly changes in the methylation of the
histone H2A and H4 tails. We performed gene expression profile
analysis onDMSO-treated versusCEP701-treatedHEL cells and
on PRMT5-directed shRNA-expressing HEL cells versus control
shRNA-expressing cells using Affymetrix HG133GeneChips.We
found 881 genes whose mRNA levels changed more than
1.5-fold in both duplicate samples of the CEP701-treated cells,
and 585 genes whose expression reproducibly changed R1.5-
fold in cellswherePRMT5wasknockeddown.Because inhibition
of JAK2 activity derepresses PRMT5 activity, we hypothesize
that genes that are reciprocally regulated between the inhibitor-
treated samples and the shPRMT5-treated samples will be regu-
lated by JAK2-induced PRMT5 phosphorylation. Indeed, we
found 90 such genes (42 upregulated by the inhibitor and down-
regulated by the shRNA, and 48 downregulated by the inhibitor
and upregulated by the shRNA), including genes involved in ribo-
somal biogenesis and autophagy (Figure 3G; Table S1).
The Major Phosphorylation Sites in PRMT5 Mapto Its N-Terminal RegionTo map the site(s) in PRMT5 phosphorylated by JAK2V617F, we
performed in vitro kinase assays and found that PRMT5 is
phosphorylated at multiple sites located between amino acids
268 and 320 (data not shown). There are six tyrosine residues
in this region (at position 280, 283, 286, 297, 304, and 307),
and we mutated the first three (M3), first four (M4), and all six
tyrosine residues (M6) to phenylalanine, and examined the
extent of PRMT5 phosphorylation in transfected 293T cells (Fig-
ure 4A). Compared to wild-type PRMT5, the M6 form of PRMT5
had markedly reduced phosphorylation when coexpressed with
JAK2V617F. The residual phosphorylation could be due to
dimerization with endogenous wild-type PRMT5, or to the pres-
ence of other phosphorylation sites within the protein. In contrast
the M3 form of PRMT5 had a similar degree of phosphorylation
as wild-type PRMT5, suggesting that the last three tyrosines
are the major sites of JAK2 phosphorylation in PRMT5.
To demonstrate that PRMT5 is phosphorylated at these tyro-
sine residues, we generated a phospho-specific anti-PRMT5
antibody (P-PRMT5-747) using a mixture of peptides containing
all combinations of phosphorylated Y297, Y304, and Y307
tyrosine residues. A dot blot assay confirmed that this antibody
recognizes the phosphorylated and not the unphosphorylated
288 Cancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc.
peptides (data not shown). To confirm that endogenous
PRMT5 is phosphorylated by JAK2V617F in HEL cells, we
treated the cells with the JAK inhibitor I, CEP701 (Figure 4B),
and TG101348 (Figure 3F) and performed several immunoblots.
These JAK2 inhibitors significantly reduced the phosphorylation
of PRMT5 and STAT5, demonstrating that endogenous PRMT5
is phosphorylated within these three tyrosine residues by
JAK2V617F. These three tyrosine residues are highly conserved
in PRMT5, from Xenopus to human (Figure S3A).
Phosphorylation of PRMT5 by JAK2V617F DisruptsIts Association with MEP50Because MEP50 markedly enhances the enzymatic activity of
PRMT5 (Figures 3A and 3B), we examined whether JAK2V617F
impairs PRMT5 activity by disrupting the PRMT5/MEP50
complex. Using 293T cells that transiently express HA-PRMT5,
and either the wild-type JAK2 or the V617F mutant protein, we
could readily co-immunoprecipitate endogenous MEP50 protein
from cells expressing wild-type JAK2 with an anti-HA antibody
(Figure 4C). However, coexpression of JAK2V617F significantly
reduced the interaction between PRMT5 andMEP50. The kinase
activity of JAK2V617F is required to disrupt the PRMT5/MEP50
association because treating the cells with JAK Inhibitor I
blocked the phosphorylation of PRMT5 and restored the interac-
tion between PRMT5 and MEP50 (Figure 4D). Expression of
JAK2V617F also disrupted the PRMT5/MEP50 interaction in
HeLa cells and U2OS cells (Figures S3B and S3C).
To determine whether phosphorylation of PRMT5 affects the
PRMT5/MEP50 complex in JAK2V617F-positive HEL cells, we
established a stable HEL cell line that expresses myc-tagged
MEP50 and purified the PRMT5/MEP50 complex using an anti-
myc antibody (Figure 4E). Although the unphosphorylated form
of PRMT5 bound to MEP50, the phosphorylated form of
PRMT5 was exclusively in the flowthrough following the immu-
noprecipitation. Thus, phosphorylation of PRMT5 by JAK2V617F
blocks its association with MEP50 in hematopoietic cells.
PRMT5 Negatively Regulates HematopoieticStem/Progenitor Cell Expansion and ErythroidDifferentiationWe examined whether decreased PRMT5 activity promotes
myeloproliferation and/or erythroid differentiation, by knocking
down PRMT5 expression in human CD34+ CB cells using
shRNA. We achieved only 60%–70% knockdown of PRMT5
mRNA but still observed a 2-fold increase in CFUs (Figure 5A).
We then overexpressed HA-tagged wild-type PRMT5, or the
M6 mutant form of PRMT5 (PRMT5M6), in CD34+ CB cells and
performed CFU assays. Consistent with the knockdown experi-
ments, PRMT5 overexpression significantly decreased colony
formation (Figure 5B), indicating that PRMT5 negatively regu-
lates progenitor cell proliferation and expansion.
We also examined whether PRMT5 activity regulates erythroid
differentiation, using in vitro liquid culture assays. We knocked
down PRMT5 or overexpressed wild-type PRMT5 or the
PRMT5M6 mutant in CD34+ CB cells and cultured the GFP+
cells in medium supporting erythroid differentiation for 1 or
2weeks. Although PRMT5 knockdown cells showed a significant
increase in CD71/Ter-119-positive cells, overexpression of
either the wild-type or the M6 mutant form of PRMT5 blocked
Input
IP: HA
JAK2
P-PRMT5
PRMT5
HA-PRMT5 -- WT M3 WT M3 M6M4M4 M6JAK2VF -- -- -- -- -- + + + +
1 2 3 4 5 6 7 8 9
A B
P-PRMT5-747
P-STAT5
PRMT5
DMSOJI1 CEP70
1
Input
IP: HA
JAK2
MEP50(50kDa)
P-PRMT5
MEP50
PRMT5
JAK2HA-PRMT5
-- -- WT V617F-- + + +
Input
IP: HA
JAK2
MEP50
MEP50
P-PRMT5
PRMT5
DMSO JI1
HA-PRMT5JAK2
-- + + + -- + + +-- -- WT VF -- -- WT VF
1 2 3 4 5 6 7 8
D
IP-M
yc-M
EP50Flo
w-thro
ugh
PRMT5
MEP50
E
C
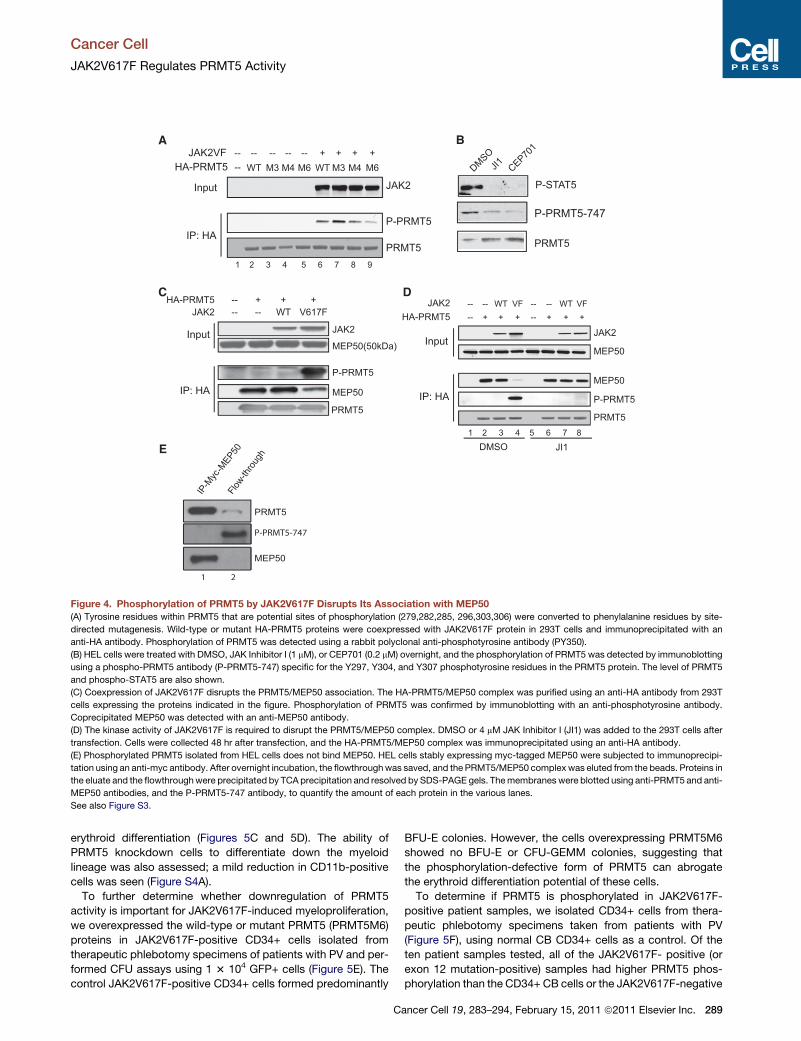
Figure 4. Phosphorylation of PRMT5 by JAK2V617F Disrupts Its Association with MEP50
(A) Tyrosine residues within PRMT5 that are potential sites of phosphorylation (279,282,285, 296,303,306) were converted to phenylalanine residues by site-
directed mutagenesis. Wild-type or mutant HA-PRMT5 proteins were coexpressed with JAK2V617F protein in 293T cells and immunoprecipitated with an
anti-HA antibody. Phosphorylation of PRMT5 was detected using a rabbit polyclonal anti-phosphotyrosine antibody (PY350).
(B) HEL cells were treated with DMSO, JAK Inhibitor I (1 mM), or CEP701 (0.2 mM) overnight, and the phosphorylation of PRMT5 was detected by immunoblotting
using a phospho-PRMT5 antibody (P-PRMT5-747) specific for the Y297, Y304, and Y307 phosphotyrosine residues in the PRMT5 protein. The level of PRMT5
and phospho-STAT5 are also shown.
(C) Coexpression of JAK2V617F disrupts the PRMT5/MEP50 association. The HA-PRMT5/MEP50 complex was purified using an anti-HA antibody from 293T
cells expressing the proteins indicated in the figure. Phosphorylation of PRMT5 was confirmed by immunoblotting with an anti-phosphotyrosine antibody.
Coprecipitated MEP50 was detected with an anti-MEP50 antibody.
(D) The kinase activity of JAK2V617F is required to disrupt the PRMT5/MEP50 complex. DMSO or 4 mM JAK Inhibitor I (JI1) was added to the 293T cells after
transfection. Cells were collected 48 hr after transfection, and the HA-PRMT5/MEP50 complex was immunoprecipitated using an anti-HA antibody.
(E) Phosphorylated PRMT5 isolated from HEL cells does not bind MEP50. HEL cells stably expressing myc-tagged MEP50 were subjected to immunoprecipi-
tation using an anti-myc antibody. After overnight incubation, the flowthroughwas saved, and the PRMT5/MEP50 complex was eluted from the beads. Proteins in
the eluate and the flowthroughwere precipitated by TCA precipitation and resolved by SDS-PAGE gels. Themembranes were blotted using anti-PRMT5 and anti-
MEP50 antibodies, and the P-PRMT5-747 antibody, to quantify the amount of each protein in the various lanes.
See also Figure S3.
Cancer Cell
JAK2V617F Regulates PRMT5 Activity
erythroid differentiation (Figures 5C and 5D). The ability of
PRMT5 knockdown cells to differentiate down the myeloid
lineage was also assessed; a mild reduction in CD11b-positive
cells was seen (Figure S4A).
To further determine whether downregulation of PRMT5
activity is important for JAK2V617F-induced myeloproliferation,
we overexpressed the wild-type or mutant PRMT5 (PRMT5M6)
proteins in JAK2V617F-positive CD34+ cells isolated from
therapeutic phlebotomy specimens of patients with PV and per-
formed CFU assays using 1 3 104 GFP+ cells (Figure 5E). The
control JAK2V617F-positive CD34+ cells formed predominantly
C
BFU-E colonies. However, the cells overexpressing PRMT5M6
showed no BFU-E or CFU-GEMM colonies, suggesting that
the phosphorylation-defective form of PRMT5 can abrogate
the erythroid differentiation potential of these cells.
To determine if PRMT5 is phosphorylated in JAK2V617F-
positive patient samples, we isolated CD34+ cells from thera-
peutic phlebotomy specimens taken from patients with PV
(Figure 5F), using normal CB CD34+ cells as a control. Of the
ten patient samples tested, all of the JAK2V617F- positive (or
exon 12 mutation-positive) samples had higher PRMT5 phos-
phorylation than the CD34+ CB cells or the JAK2V617F-negative
ancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc. 289

CB PV(#3)JA
K2VF+
PV(#4)JA
K2VF-
CLKO shPRMT5
pBGJR HA-PRMT5WT HA-PRMT5M6
ET(#1)
PV(#2)
HEL
JAK2V617F+
1 2 3 4 5 6
F
D
P-PRMT5-747
PRMT5
JAK2VF (exon12)+ JAK2VF-
7 8 9 10 11 12
A
E
PV(#9)
PV(#10)
PV(#8)
PV(#7)
PV(#6)
PV(#5)
pBGJR HA-PRMT5WT HA-PRMT5M6
Colon
ynum
berp
er1x
104
cells
BFUGEMMGM
pBGJR HA-PRMT5WT HA-PRMT5M6
Col
ony
num
berp
er2x
104
cells
BFUGEMMGM
LKO shPRMT5
Col
ony
num
ber p
er1x
104
BFUGEMMGM
LKO shPRMT5
Fold
chan
gevs
.con
trol
B
Figure 5. PRMT5 Activity Regulates Progenitor Cell Expansion/Differentiation in Human CD34+ Cells
(A) Knockdown of PRMT5 promotes CFU formation. Isolated human CBCD34+ cells were transduced with lentiviruses expressing a scrambled shRNA or shRNA
specific to PRMT5. GFP-positive cells were sorted 2 days after infection. Left panel shows that PRMT5 mRNA expression was detected by real-time PCR. Right
Cancer Cell
JAK2V617F Regulates PRMT5 Activity
290 Cancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc.

Cancer Cell
JAK2V617F Regulates PRMT5 Activity
CD34+ cells. We found less phospho-PRMT5 in two of the three
JAK2V617F-negative patient samples analyzed, compared to
the JAK2V617F+ MPN patient samples (Figure 5F, lane 6 and
11). The clinical information on these patients (including their
JAK2 mutant allele status) is summarized in Table S2. The
weak positivity in CB CD34+ cells and the high level of phos-
pho-PRMT5 in the one JAK2 wild-type patient sample suggest
that other tyrosine kinases can also phosphorylate PRMT5 in
hematopoietic cells. Computer-based programs, such as GPS
2.1 (Xue et al., 2008), indicate that the Y297, Y304, and Y307
tyrosine residues in PRMT5 are potentially phosphorylatable by
ABL1, FGFR, Src, and JAK1. In addition to examining CD34+
cells, we also examined granulocytes isolated from normal CB,
or from the phlebotomy specimens of patients with MPN, and
detected PRMT5 phosphorylation (Figure S4B).
DISCUSSION
We have determined that oncogenic mutations within the JAK2
tyrosine kinase (V617F and K539L) enhance its interaction with
PRMT5, leading to PRMT5 phosphorylation in vivo. Although
both the wild-type and mutant forms of JAK2 proteins interact
with PRMT5, phosphorylation of PRMT5 is a ‘‘gain-of-function’’
of the mutant JAK2 kinases, which reduces PRMT5methyltrans-
ferase activity and decreases global histone H2A/H4 R3
methylation.
The PRMT5-interacting region in the JAK2 protein maps to its
N terminus, making it unlikely that the mutated residues in the
JH2 domain play a critical role in this interaction. Furthermore,
it appears that the active conformation of the mutant JAK2
proteins, and not necessarily their kinase activity, allows them
to bind PRMT5 more strongly because the increased binding
persists in the presence of JAK2 kinase inhibitors (data not
shown).
We find that PRMT5 that is phosphorylated by JAK2V617F no
longer binds MEP50, even though we can detect an interaction
of MEP50 with unphosphorylated PRMT5 in HEL cells. It is
possible that much of the PRMT5:MEP50 complex that contains
unphosphorylated PRMT5 is cytoplasmic, rather than nuclear,
because we and others have shown that both proteins are found
in the nucleus and the cytoplasm, and PRMT5 is known to
panel illustrates that approximately 13 104 GFP-positive CD34+ cells were plated
tion of CFU-GM, CFU-GEMM, and BFU-E. Colonies were scored 2 weeks after th
iments with error bars indicating ±SD.
(B) Overexpression of PRMT5 inhibits colony formation. Isolated human CB CD34
together with HA-tagged wild-type PRMT5 or an HA-tagged M6 mutant form of
assays. The average results of two independent experiments are shown here. Th
(C) Knockdown of PRMT5 promotes erythroid differentiation. GFP+ CD34+ cells
erythroid differentiation for 14 days. CD71-positive/Ter-115-positive cells were d
(D) PRMT5 overexpression inhibits erythroid differentiation. HA-PRMT5 or HA
erythroid-promoting medium for 7 days; erythroid differentiation was determined
refers to the control lentiviral vector.
(E) PRMT5 overexpression inhibits colony formation in JAK2V617F-positive CD3
botomy units of patients with PV and transduced by control lentivirus or lentiviruse
in methylcellulose with cytokines supporting GM, GEMM, and BFU colony forma
(F) Phosphorylation of PRMT5 was detected in CD34+ cells isolated from patients
or from ten phlebotomy units taken from patients with MPN (lanes 2–3 and 5–12)
was detected using the P-PRMT5-747 antibody. The total amount of PRMT5 wa
cythemia vera; HEL, HEL leukemia cells.
See also Figure S4 and Table S2.
C
complex with MEP50 in both locations (Liang et al., 2007). Given
the enhanced proliferation seen following knockdown of PRMT5
in normal CD34+ cells, it would seem ‘‘easier’’ for the
JAK2V617F+ mutant cells to simply degrade, or not express
PRMT5. However, PRMT5 preferentially promotes p53-depen-
dent cell cycle arrest at the expense of p53-dependent
apoptosis (Jansson et al., 2008), and indeed, we find that knock-
ing down PRMT5 in HEL cells triggers cell death (data not
shown). Thus, JAK2V617F-expressing hematopoietic cells may
require that some PRMT5 protein be present, to complex with
MEP50, and perhaps to promote dimerization of the JAK2V617F
protein, similar to the role that the EpoR has been shown to play
(Lu et al., 2005; Wernig et al., 2008a). Given our confirmation of
the recent demonstrations that JAK2 can be found in the nucleus
(Dawson et al., 2009; Rinaldi et al., 2010), PRMT5 phosphoryla-
tion may be differentially regulated in the nucleus versus the
cytoplasm of the cell, and in hematopoietic cells versus other
cell types.
The kinase-dependent regulation of global H2A/H4 R3
symmetricmethylation reveals a link between an oncogenic tyro-
sine kinase and this particular chromatin modification. We have
also shown the dynamic nature of this regulation. Symmetric
dimethylation of histone H2A/H4 R3 has been shown to nega-
tively, as well as positively, regulate gene expression (Fabbrizio
et al., 2002; Richard et al., 2005). To gain insight into how
JAK2V617F could regulate gene expression through phosphor-
ylation of PRMT5, we performed gene expression profiling
comparing the JAK2-regulated genes (defined using a JAK2
inhibitor) with the PRMT5-regulated genes (defined using shRNA
directed against PRMT5) and found 90 genes that were recipro-
cally regulated. The recently reported interplay between histone
arginine methylation and DNA methylation (Zhao et al., 2009)
suggests that the oncogenic JAK2 kinases may not only regulate
histone arginine methylation but also DNA methylation, thereby
‘‘maintaining’’ a myeloproliferative epigenetic signature that
could be established by activation of the JAK/STAT pathway.
Recent work has shown an important role for arginine methyl-
ation in regulating hematopoiesis and leukemogenesis. PRMT1
(a type I arginine methyltransferase) has been shown to be an
essential component of MLL fusion protein-driven leukemogen-
esis (Cheung et al., 2007), and to regulate AML1(Runx1) function
in methylcellulose culture supplemented with cytokines to support the forma-
e plating. The results shown here are the averages of two independent exper-
+ cells were transduced with lentiviruses expressing either GFP alone, or GFP
PRMT5. GFP-positive cells were sorted, and 2 3 104 cells were used for CFU
e error bars indicate ±SD.
were cultured in serum-free medium supplemented with cytokines supporting
etermined by FACS analysis.
-PRMT5M6 was overexpressed in CB CD34+ cells, which were cultured in
by FACS analysis, staining for CD71 (y axis) and Glycophorin A (x axis). pBGJR
4+ cells isolated from patients with PV. CD34+ cells were isolated from phle-
s expressing HA-PRMT5 or HA-PRMT5M6. The 13 104 GFP+ cells were plated
tion. Colonies were scored 14 days after plating.
with MPN. CD34+ cells were isolated from human umbilical cord blood (lane 1)
. HEL cells were used as a positive control (lane 4). Phosphorylation of PRMT5
s also assessed by immunoblotting. ET, essential thrombocythemia; PV, poly-
ancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc. 291

Cancer Cell
JAK2V617F Regulates PRMT5 Activity
(Zhao et al., 2008). PRMT5 has been shown to repress gamma-
globin gene expression by recruiting DNMT3A and inducing
additional repressive epigenetic marks, such as H4S1ph and
H4K20me3 (Rank et al., 2010; Zhao et al., 2009); this suggests
a role for PRMT5 in erythropoiesis (which we have demon-
strated) and hematopoiesis in general. In contrast, we found
that downregulation of PRMT5 activity (a type II arginine methyl-
transferase) promotes progenitor cell expansion and accelerates
erythroid differentiation. Forced expression of JAK2V617F
induces a PV-like disease in mouse models (Akada et al.,
2010; Marty et al., 2010; Mullally et al., 2010), and our studies
suggest that in addition to activating the STAT5 pathway, this
mutant JAK2 kinase can induce myeloproliferation and erythro-
cytosis by abrogating PRMT5 activity. Although knockdown of
PRMT5 in many cell lines, such as HEL and K562, led to
apoptosis and/or growth arrest, its downregulation in normal
CB CD34+ cells provides a proliferative signal. We too find that
PRMT5 regulates globin gene expression in HEL, CB CD34+
cells as well as in K562 cells (data not shown). However, the
distinct effects of PRMT5 knockdown on gene expression in
HEL versus CD34+ cells (data not shown) demonstrate the
importance of cell context on PRMT5 function.
Although PRMT5 is most highly phosphorylated in
JAK2V617F-positive MPN patient CD34+ cells (and in granulo-
cytes), it is also phosphorylated to some degree in JAK2V617F-
negative MPN and normal CB CD34+ cells. Overexpressing
PRMT5 (wild-type and, in particular, the M6 mutant form of
PRMT5) in JAK2V617F-positive CD34+ patient cells resulted in
a block in cell expansion and erythroid differentiation, providing
further evidence that phosphorylation and abrogation of
PRMT5 activity are important functions of this oncogenic kinase.
Nonetheless, further experiments are needed to assess the rela-
tive contribution of PRMT5phosphorylation to the various clinical
syndromes associated with the JAK2V617F mutation.
In conclusion, we have identified a ‘‘gain of function’’ for the
constitutively activated forms of JAK2 kinase (JAK2V617F and
JAK2K539L), namely phosphorylation of PRMT5, which allows
them to control transcription by regulating histone H2A and H4
arginine methylation. Given the diverse functions of PRMT5 in
the cell, further studies of the proteins methylated by PRMT5
and the pathways affected in JAK2V617F-expressing cells will
shed additional light on the molecular pathogenesis of the
MPNs.
EXPERIMENTAL PROCEDURES
Cell Culture, Transfection, and Plasmids
The human leukemia cell lines TF-1, HEL, and Ba/F3 were grown in RPMI
Media 1640 with 10% FBS (Invitrogen), supplemented with recombinant
human IL-3 (2 ng/ml) for TF-1 and Ba/F3 cells. To inhibit JAK2 activity in
HEL and TF-1 cells, cells were treated with three distinct JAK2 inhibitors:
JAK Inhibitor I (Calbiochem), CEP701 (LC Laboratories), and TG101348 at
the indicated concentrations (DMSO was used as the diluent control). In
the time course experiment, HEL cells were treated with 3 mM TG101348 for
0, 1, 2, and 4 hr. To activate JAK2 in TF-1 cells, the cells were deprived of
cytokine for 12–16 hr, and thenGM-CSF (25 ng/ml) or Epo (20U/ml) was added
to the growth medium for 20 min. Transient transfection of 293T cells was
performed using the PolyFect Transfection Reagent (QIAGEN) per the manu-
facturer’s instructions. To express the mutant or wild-type JAK2 proteins in
293T cells, the wild-type JAK2, JAK2V617F, and JAK2K539L cDNAs were
subcloned into the CMV promoter-driven pCDNA3 vector (Invitrogen) using
292 Cancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc.
EcoRV and Not1 sites. FLAG-tagged or HA-tagged PRMT5 and HA-tagged
STAT5 cDNAs were also subcloned into the pCDNA3 vector through appro-
priate restriction sites. For in vitro kinase assays, the full-length PRMT5
open reading frame was subcloned into the bacterial expression vector
pSBET, which contains a glutathione S-transferase (GST) tag and a 63 Histi-
dine tag at the N and C termini of the inserted sequence, respectively.
In Vitro Kinase Assay
In vitro kinase assays were performed using commercially available JAK2
kinase (Upstate), which contains only the kinase domain (from amino acid
800 to C-terminal end), and bacterial-purified GST-PRMT5. For each reaction,
0.5 or 2.5 mg of GST-PRMT5, 0.5 or 1.25 ng of JAK2, and 5 mCi of [g-32p] ATP
were added to the kinase buffer (8 mM MOPS [pH 7.0], 0.2 mM EDTA, 10 mM
MgAc, and 0.1 mM cold ATP). Reactions were incubated at 30�C for 20 min,
and proteins resolved by gel electrophoresis.
In Vitro Methylation Assay
The methylation assay was done as described (Zhao et al., 2008). HA-PRMT5
was purified from transfected 293T cells by anti- HA immunoprecipitation. The
immobilized proteins were then incubated with 25 ml of HMT buffer (20 mMTris
[pH 8.8], 4 mM EDTA, 1 mM PMSF, 0.5 mM DTT) supplemented with 2.5 mg of
recombinant histone H4 or H2A (New England Biolabs) and 1 mCi 3H-SAM
(Amersham) at 30�C for 4 hr. The reaction was stopped by adding SDS loading
buffer, and the proteins were resolved on SDS-PAGE gels.
Microarray Analysis
RNA was isolated from HEL cells expressing control shRNA or PRMT5-
directed shRNA using the RNAeasy Plus Kit (QIAGEN), transcribed into
cDNA using random hexamer priming and Superscriptase (Invitrogen), and
then hybridized to the Affymetrix HG-U133 GeneChips. The data were
analyzed using Partek Genomic Suites, version 6.5, using a false discovery
rate (FDR) of 1% to filter our data and identify differentially expressed genes.
Duplicate, independent samples were prepared for each condition.
Isolation of Granulocytes and CD34+ Cells from Patient
Samples and/or Cord Blood
Granulocytes and mononuclear cells were isolated from human umbilical cord
blood by Ficoll-Hypaque Plus (Sigma Chemical) density centrifugation. CD34+
cells were then purified from the mononuclear cells by positive selection using
a Midi MACS separator, LS+ column, and CD34 Micro Bead Kit (Miltenyi
Biotec). For CFU and cell differentiation assays, CD34+ cells were cultured
in IMDM (Cellgro) containing 20% BIT 9500 (Stem Cell Technology) for 24 hr
before infection. The media were also supplemented with SCF (100 ng/ml),
FLT-3 (10 ng/ml), IL-6 (20 ng/ml), and TPO (100 ng/ml) (all purchased from
PeproTech).
For the analysis of patient samples, CD34+ cells and granulocytes were iso-
lated from phlebotomy units taken from patients with MPN treated at MSKCC,
with appropriate IRB-approved informed consent. All samples were obtained
as discarded samples during the routine clinical care of the patients, and they
were de-identified prior to analysis. To obtain enough cells for western blot-
ting, the CD34+ cells were cultured in medium supplemented with cytokines
(including SCF, IL3, IL6, and Epo) for 1 week before being subjected to
analysis. Phosphorylation of PRMT5 was detected using the P-PRMT5-747
antibody. The same membrane was stripped and re-probed to quantify the
total amount of PRMT5.
Generation of Lentiviruses and Infection of Primary
Hematopoietic CD34+ Cells
shRNA targeting PRMT5 or a scrambled shRNA was cloned into the
pLKO.1-GFP lentiviral vector. HA-PRMT5 WT or M6 cDNAs were cloned into
the lentiviral vector pBGJR (modified by replacing the CMV promoter with
the EF1a promoter). Viruses were produced by transfecting pLKO.1-GFP or
pBGJR vector with helper plasmids into 293T cells, according to standard
protocols. After 24 hr of growth, CD34+ cells were infected with high-titer
concentrated lentiviral suspensions in the presence of 8 mg/ml Polybrene
(Aldrich). GFP-positive cells were sorted 2 days after infection.

Cancer Cell
JAK2V617F Regulates PRMT5 Activity
CFU Assay
The 1 3 104 or 2 3 104 transduced GFP-positive CD34+ cells isolated from
patients with ET or PV were plated (in duplicate) in methylcellulose supple-
mented with Epo (5 IU/ml), SCF (50 ng/ml), IL-3 (20 ng/ml), IL-6 (20 ng/ml),
G-CSF (20 ng/ml), andGM-CSF (20 ng/ml). BFU-E, CFU-GM, andCFU-GEMM
colonies were scored 14 days after seeding.
In Vitro Differentiation Assay
GFP-positive CD34+ CB cells were cultured in 20% BIT in IMDM medium
with different cytokines that support erythroid or myeloid cell differentiation
for 7 or 14 days. The erythroid culture was supplemented with Epo (6 IU/ml)
and SCF (100 ng/ml), and the myeloid culture with SCF (100 ng/ml), FLT-3
(10 ng/ml), IL-3 (20 ng/ml), IL-6 (20 ng/ml), GM-CSF (20 ng/ml), and G-CSF
(20 ng/ml). Cells were collected and stained with CD71-APC (BD PharMingen)
andGlycophorin A-PE (Invitrogen) tomonitor erythroid differentiation, and with
CD11b-APC (BD PharMingen) to monitor myeloid differentiation.
ACCESSION NUMBERS
Coordinates have been deposited in the NCBI GEO with accession code
GSE25725.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures,
four figures, and two tables and can be found with this article online at
doi:10.1016/j.ccr.2010.12.020.
ACKNOWLEDGMENTS
We thankMinkui Luo and themembers of the S.D.N. laboratory for reading this
manuscript and providing thoughtful suggestions and comments. We thank
Erica Chuang for assistance in preparing this manuscript. This work was
supported by a Leukemia Lymphoma Society SCOR Grant (S.D.N.), a Starr
Foundation Award (S.D.N.), a Clinical Scholar Grant (awarded to F.L.) and
R01-CA151949 (to R.L.L.). The authors have no conflicts of interest or financial
disclosures to declare.
Received: August 3, 2010
Revised: November 10, 2010
Accepted: December 4, 2010
Published: February 14, 2011
REFERENCES
Akada, H., Yan, D., Zou, H., Fiering, S., Hutchison, R.E., andMohi,M.G. (2010).
Conditional expression of heterozygous or homozygous Jak2V617F from its
endogenous promoter induces a polycythemia vera-like disease. Blood 115,
3589–3597.
Ancelin,K., Lange,U.C.,Hajkova, P., Schneider, R., Bannister, A.J.,Kouzarides,
T., and Surani, M.A. (2006). Blimp1 associates with Prmt5 and directs histone
arginine methylation in mouse germ cells. Nat. Cell Biol. 8, 623–630.
Baxter, E.J., Scott, L.M., Campbell, P.J., East, C., Fourouclas, N., Swanton, S.,
Vassiliou, G.S., Bench, A.J., Boyd, E.M., Curtin, N., et al. (2005). Acquired
mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders.
Lancet 365, 1054–1061.
Branscombe, T.L., Frankel, A., Lee, J.H., Cook, J.R., Yang, Z., Pestka, S., and
Clarke, S. (2001). PRMT5 (Janus kinase-binding protein 1) catalyzes the forma-
tion of symmetric dimethylarginine residues in proteins. J. Biol. Chem. 276,
32971–32976.
Chari, A., Golas, M.M., Klingenhager, M., Neuenkirchen, N., Sander, B.,
Englbrecht, C., Sickmann, A., Stark, H., and Fischer, U. (2008). A. assembly
chaperone collaborates with the SMN complex to generate spliceosomal
SnRNPs. Cell 135, 497–509.
Cheung, N., Chan, L.C., Thompson, A., Cleary, M.L., and So, C.W. (2007).
Protein arginine-methyltransferase-dependent oncogenesis. Nat. Cell Biol.
9, 1208–1215.
C
Dawson, M.A., Bannister, A.J., Gottgens, B., Foster, S.D., Bartke, T., Green,
A.R., and Kouzarides, T. (2009). JAK2 phosphorylates histone H3Y41 and
excludes HP1alpha from chromatin. Nature 461, 819–822.
De Keersmaecker, K., and Cools, J. (2006). Chronic myeloproliferative disor-
ders: a tyrosine kinase tale. Leukemia 20, 200–205.
Fabbrizio, E., El Messaoudi, S., Polanowska, J., Paul, C., Cook, J.R., Lee, J.H.,
Negre, V., Rousset, M., Pestka, S., Le Cam, A., and Sardet, C. (2002). Negative
regulation of transcription by the type II arginine methyltransferase PRMT5.
EMBO Rep. 3, 641–645.
Friesen, W.J., Paushkin, S., Wyce, A., Massenet, S., Pesiridis, G.S., Van
Duyne, G., Rappsilber, J., Mann, M., and Dreyfuss, G. (2001). The methylo-
some, a 20S complex containing JBP1 and pICln, produces dimethylargi-
nine-modified Sm proteins. Mol. Cell. Biol. 21, 8289–8300.
Friesen, W.J., Wyce, A., Paushkin, S., Abel, L., Rappsilber, J., Mann, M., and
Dreyfuss, G. (2002). A novel WD repeat protein component of the methylo-
some binds Sm proteins. J. Biol. Chem. 277, 8243–8247.
Hexner, E.O., Serdikoff, C., Jan, M., Swider, C.R., Robinson, C., Yang, S.,
Angeles, T., Emerson, S.G., Carroll, M., Ruggeri, B., and Dobrzanski, P.
(2008). Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/
STAT5 signaling and the proliferation of primary erythroid cells from patients
with myeloproliferative disorders. Blood 111, 5663–5671.
Ihle, J.N., and Gilliland, D.G. (2007). Jak2: normal function and role in hemato-
poietic disorders. Curr. Opin. Genet. Dev. 17, 8–14.
James, C., Ugo, V., Le Couedic, J.P., Staerk, J., Delhommeau, F., Lacout, C.,
Garcon, L., Raslova, H., Berger, R., Bennaceur-Griscelli, A., et al. (2005).
A unique clonal JAK2 mutation leading to constitutive signalling causes poly-
cythaemia vera. Nature 434, 1144–1148.
Jansson, M., Durant, S.T., Cho, E.C., Sheahan, S., Edelmann, M., Kessler, B.,
and La Thangue, N.B. (2008). Argininemethylation regulates the p53 response.
Nat. Cell Biol. 10, 1431–1439.
Kralovics, R., Passamonti, F., Buser, A.S., Teo, S.S., Tiedt, R., Passweg, J.R.,
Tichelli, A., Cazzola, M., and Skoda, R.C. (2005). A gain-of-functionmutation of
JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790.
Kwak, Y.T., Guo, J., Prajapati, S., Park, K.J., Surabhi, R.M., Miller, B., Gehrig,
P., and Gaynor, R.B. (2003). Methylation of SPT5 regulates its interaction with
RNA polymerase II and transcriptional elongation properties. Mol. Cell 11,
1055–1066.
Lasho, T.L., Tefferi, A., Hood, J.D., Verstovsek, S., Gilliland, D.G., and
Pardanani, A. (2008). TG101348, a JAK2-selective antagonist, inhibits primary
hematopoietic cells derived from myeloproliferative disorder patients with
JAK2V617F,MPLW515K or JAK2 exon 12mutations aswell asmutation nega-
tive patients. Leukemia 22, 1790–1792.
Le Guezennec, X., Vermeulen, M., Brinkman, A.B., Hoeijmakers, W.A., Cohen,
A., Lasonder, E., and Stunnenberg, H.G. (2006). MBD2/NuRD and MBD3/
NuRD, two distinct complexes with different biochemical and functional prop-
erties. Mol. Cell. Biol. 26, 843–851.
Levine, R.L., Wadleigh, M., Cools, J., Ebert, B.L., Wernig, G., Huntly, B.J.,
Boggon, T.J., Wlodarska, I., Clark, J.J., Moore, S., et al. (2005). Activating
mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombo-
cythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7, 387–397.
Liang, J.J., Wang, Z., Chiriboga, L., Greco, M.A., Shapiro, E., Huang, H., Yang,
X.J., Huang, J., Peng, Y., Melamed, J., et al. (2007). The expression and func-
tion of androgen receptor coactivator p44 and protein arginine methyltransfer-
ase 5 in the developing testis and testicular tumors. J. Urol. 177, 1918–1922.
Lu, X., Levine, R., Tong, W., Wernig, G., Pikman, Y., Zarnegar, S., Gilliland,
D.G., and Lodish, H. (2005). Expression of a homodimeric type I cytokine
receptor is required for JAK2V617F-mediated transformation. Proc. Natl.
Acad. Sci. USA 102, 18962–18967.
Marty, C., Lacout, C., Martin, A., Hasan, S., Jacquot, S., Birling, M.C.,
Vainchenker, W., and Villeval, J.L. (2010). Myeloproliferative neoplasm
induced by constitutive expression of JAK2V617F in knock-in mice. Blood
116, 783–787.
ancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc. 293

Cancer Cell
JAK2V617F Regulates PRMT5 Activity
Meister, G., and Fischer, U. (2002). Assisted RNP assembly: SMN and PRMT5
complexes cooperate in the formation of spliceosomal UsnRNPs. EMBO J. 21,
5853–5863.
Mullally, A., Lane, S.W., Ball, B., Megerdichian, C., Okabe, R., Al-Shahrour, F.,
Paktinat, M., Haydu, J.E., Housman, E., Lord, A.M., et al. (2010). Physiological
Jak2V617F expression causes a lethal myeloproliferative neoplasm with
differential effects on hematopoietic stem and progenitor cells. Cancer Cell
17, 584–596.
Neubauer, H., Cumano, A., Muller, M., Wu, H., Huffstadt, U., and Pfeffer, K.
(1998). Jak2 deficiency defines an essential developmental checkpoint in
definitive hematopoiesis. Cell 93, 397–409.
Pal, S., Vishwanath, S.N., Erdjument-Bromage, H., Tempst, P., and Sif, S.
(2004). Human SWI/SNF-associated PRMT5 methylates histone H3 arginine
8 and negatively regulates expression of ST7 and NM23 tumor suppressor
genes. Mol. Cell. Biol. 24, 9630–9645.
Parganas, E., Wang, D., Stravopodis, D., Topham, D.J., Marine, J.C., Teglund,
S., Vanin, E.F., Bodner, S., Colamonici, O.R., van Deursen, J.M., et al. (1998).
Jak2 is essential for signaling through a variety of cytokine receptors. Cell 93,
385–395.
Pikman, Y., and Levine, R.L. (2007). Advances in the molecular characteriza-
tion of Philadelphia-negative chronic myeloproliferative disorders. Curr.
Opin. Oncol. 19, 628–634.
Pollack, B.P., Kotenko, S.V., He, W., Izotova, L.S., Barnoski, B.L., and Pestka,
S. (1999). The human homologue of the yeast proteins Skb1 and Hsl7p inter-
acts with Jak kinases and contains protein methyltransferase activity. J. Biol.
Chem. 274, 31531–31542.
Rank, G., Cerruti, L., Simpson, R.J., Moritz, R.L., Jane, S.M., and Zhao, Q.
(2010). Identification of a PRMT5-dependent repressor complex linked to
silencing of human fetal globin gene expression. Blood 116, 1585–1592.
Richard, S., Morel, M., and Cleroux, P. (2005). Arginine methylation regulates
IL-2 gene expression: a role for protein arginine methyltransferase 5 (PRMT5).
Biochem. J. 388, 379–386.
Rinaldi, C.R., Rinaldi, P., Alagia, A., Gemei, M., Esposito, N., Formiggini, F.,
Martinelli, V., Senyuk, V., Nucifora, G., and Pane, F. (2010). Preferential nuclear
accumulation of JAK2V617F in CD34+ but not in granulocytic, megakaryocytic
or erythroid cells of patients with Philadelphia-negative myeloproliferative
neoplasia. Blood 116, 6023–6026.
294 Cancer Cell 19, 283–294, February 15, 2011 ª2011 Elsevier Inc.
Scott, L.M., Tong, W., Levine, R.L., Scott, M.A., Beer, P.A., Stratton, M.R.,
Futreal, P.A., Erber, W.N., McMullin, M.F., Harrison, C.N., et al. (2007). JAK2
exon 12 mutations in polycythemia vera and idiopathic erythrocytosis.
N. Engl. J. Med. 356, 459–468.
Tan, C.P., and Nakielny, S. (2006). Control of the DNA methylation system
component MBD2 by protein arginine methylation. Mol. Cell. Biol. 26, 7224–
7235.
Wang, Y., Fiskus, W., Chong, D.G., Buckley, K.M., Natarajan, K., Rao, R.,
Joshi, A., Balusu, R., Koul, S., Chen, J., et al. (2009). Cotreatment with pano-
binostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and
signaling and exerts synergistic cytotoxic effects against humanmyeloprolifer-
ative neoplastic cells. Blood 114, 5024–5033.
Wernig, G., Gonneville, J.R., Crowley, B.J., Rodrigues, M.S., Reddy, M.M.,
Hudon, H.E., Walz, C., Reiter, A., Podar, K., Royer, Y., et al. (2008a). The
Jak2V617F oncogene associated with myeloproliferative diseases requires
a functional FERM domain for transformation and for expression of the Myc
and Pim proto-oncogenes. Blood 111, 3751–3759.
Wernig, G., Kharas, M.G., Okabe, R., Moore, S.A., Leeman, D.S., Cullen, D.E.,
Gozo, M., McDowell, E.P., Levine, R.L., Doukas, J., et al. (2008b). Efficacy of
TG101348, a selective JAK2 inhibitor, in treatment of a murine model of
JAK2V617F-induced polycythemia vera. Cancer Cell 13, 311–320.
Xing, S., Wanting, T.H., Zhao, W., Ma, J., Wang, S., Xu, X., Li, Q., Fu, X., Xu, M.,
and Zhao, Z.J. (2008). Transgenic expression of JAK2V617F causesmyelopro-
liferative disorders in mice. Blood 111, 5109–5117.
Xue, Y., Ren, J., Gao, X., Jin, C., Wen, L., and Yao, X. (2008). GPS 2.0, a tool to
predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell.
Proteomics 7, 1598–1608.
Zhao, Q., Rank, G., Tan, Y.T., Li, H., Moritz, R.L., Simpson, R.J., Cerruti, L.,
Curtis, D.J., Patel, D.J., Allis, C.D., et al. (2009). PRMT5-mediated methylation
of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in
gene silencing. Nat. Struct. Mol. Biol. 16, 304–311.
Zhao, X., Jankovic, V., Gural, A., Huang, G., Pardanani, A., Menendez, S.,
Zhang, J., Dunne, R., Xiao, A., Erdjument-Bromage, H., et al. (2008).
Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates
its transcriptional activity. Genes Dev. 22, 640–653.
Copyright © 2022 FDOKUMEN




















