
Protein arginine-methyltransferase-dependent oncogenesis
8
LETTERS Protein arginine-methyltransferase-dependent oncogenesis Ngai Cheung 1 , Li Chong Chan 2 , Alex Thompson 3 , Michael L Cleary 4 and Chi Wai Eric So 1,5 Enzymes that mediate reversible epigenetic modifications have not only been recognized as key in regulating gene expression 1 and oncogenesis 2,3 , but also provide potential targets for molecular therapy 4 . Although the methylation of arginine 3 of histone 4 (H4R3) by protein arginine methyltransferase 1 (PRMT1) is a critical modification for active chromatin 5,6 and prevention of heterochromatin spread 7 , there has been no direct evidence of any role of PRMTs in cancer. Here, we show that PRMT1 is an essential component of a novel Mixed Lineage Leukaemia (MLL) oncogenic transcriptional complex with both histone acetylation and H4R3 methylation activities, which also correlate with the expression of critical MLL downstream targets. Direct fusion of MLL with PRMT1 or Sam68, a bridging molecule in the complex for PRMT1 interaction, could enhance self-renewal of primary haematopoietic cells. Conversely, specific knockdown of PRMT1 or Sam68 expression suppressed MLL-mediated transformation. This study not only functionally dissects the oncogenic transcriptional machinery associated with an MLL fusion complex, but also uncovers — for the first time — an essential function of PRMTs in oncogenesis and reveals their potential as novel therapeutic targets in human cancer. Acquisition of aberrant transcription properties is a key feature for many oncogenic transcription factors involved in human malignancies 2,8 . The Mixed Lineage Leukaemia (MLL) gene, which encodes a lysine methyl- transferase, is rearranged by chromosomal translocation to form chi- meric fusion proteins. The carboxy-terminal SET domain of wild-type MLL, which associates with three highly conserved core components (RbBP5, Ash2L and WDR5) and mediates H3K4 trimethylation 9,10 , is replaced by sequences from fusion partner genes 11 . Although deletion of the SET domain would indicate a loss of transactivation function, the resultant MLL fusion proteins act as dominant-positive transcriptional regulators that can bind DNA and aberrantly activate, via unknown mechanisms, the expression of MLL downstream targets such as Hox genes 11,12 . The resultant oncogenic MLL fusion proteins can transform both haematopoietic stem-cell and early myeloid progenitors 13,14 , in which they resurrect the self-renewal transcriptional programmes in the leukaemic stem cells 15 . To gain further insights into underly- ing mechanisms and epigenetic networks being deregulated by MLL fusions, a retroviral transduction and transplantation assay (RTTA) 14 was used to assess the oncogenic properties of MLL–EEN, the found- ing member of MLL SH3-domain-containing fusion proteins 16 , which constitutes a major family of MLL fusion proteins 11 . In contrast to cells transduced with truncated MLL 5’, truncated EEN 3’ or the vector con- trols that exhausted their proliferative ability within the first round of plating, murine primary haematopoietic cells that have been transduced with MLL–EEN oncoprotein exhibited enhanced self-renewal ability and could form compact CFU-GEMM-like colonies in the third and subsequent rounds of plating (Fig. 1a). Immunophenotypes of trans- formed cells were consistent with early myeloid precursors, which were positive for c-Kit, Mac-1 and Gr-1 (Fig. 1b, and data not shown). When MLL–EEN-transduced cells were transplanted into sub-lethally irradiated syngeneic mice, all the mice succumbed to acute leukaemia, with a median disease latency of 9 months (Fig. 1c). Histological and immunophenotypic studies on autopsy samples were consistent with acute myeloid leukaemia (AML) (Fig. 1d–e, and data not shown) that closely recapitulated the corresponding human disease associated with this fusion oncoprotein 16 . These results confirm the leukaemogenic properties of MLL–EEN and validate the usefulness of this approach for subsequent functional studies. Although aberrant self-association has been proposed as an oncogenic mechanism for various leukaemia-associated transcription factors 17 , MLL–EEN mutants, with deletion of both the EEN coiled-coil domain and the putative SH3 binding sites upstream of the MLL breakpoint, could still efficiently transform primary haematopoietic cells (Fig. 1f– g). This indicates that self-association plays a minimal, if any, role in MLL–EEN-mediated transformation. In contrast, deletion of the EEN SH3 domain abolished transformation, indicating that the SH3 domain is the minimal transformation domain that is necessary and sufficient 1 Haemato-Oncology Section, The Institute of Cancer Research, Sutton, Greater London SM2 5NG, UK; 2 Department of Pathology, The University of Hong Kong, Hong Kong; 3 Department of Haematology, Queen’s University, Belfast BT9 7AB, Northern Ireland; 4 Department of Pathology, Stanford University Medical Center, Stanford, CA 93405, USA 5 Correspondence should be addressed to C.W.E.S. ([email protected]) Received 2 March 2007; accepted 24 July 2007; published online 23 September 2007; DOI: 10.1038/ncb1642 1208 NATURE CELL BIOLOGY VOLUME9|NUMBER10|OCTOBER2007 © 2007 Nature Publishing Group
Transcript of Protein arginine-methyltransferase-dependent oncogenesis

l e t t e r s
Protein arginine-methyltransferase-dependent oncogenesisNgai Cheung1, Li Chong Chan2, Alex Thompson3, Michael L Cleary4 and Chi Wai Eric So1,5
Enzymes that mediate reversible epigenetic modifications have not only been recognized as key in regulating gene expression1 and oncogenesis2,3, but also provide potential targets for molecular therapy4. Although the methylation of arginine 3 of histone 4 (H4R3) by protein arginine methyltransferase 1 (PRMT1) is a critical modification for active chromatin5,6 and prevention of heterochromatin spread7, there has been no direct evidence of any role of PRMTs in cancer. Here, we show that PRMT1 is an essential component of a novel Mixed Lineage Leukaemia (MLL) oncogenic transcriptional complex with both histone acetylation and H4R3 methylation activities, which also correlate with the expression of critical MLL downstream targets. Direct fusion of MLL with PRMT1 or Sam68, a bridging molecule in the complex for PRMT1 interaction, could enhance self-renewal of primary haematopoietic cells. Conversely, specific knockdown of PRMT1 or Sam68 expression suppressed MLL-mediated transformation. This study not only functionally dissects the oncogenic transcriptional machinery associated with an MLL fusion complex, but also uncovers — for the first time — an essential function of PRMTs in oncogenesis and reveals their potential as novel therapeutic targets in human cancer.
Acquisition of aberrant transcription properties is a key feature for many oncogenic transcription factors involved in human malignancies2,8. The Mixed Lineage Leukaemia (MLL) gene, which encodes a lysine methyl-transferase, is rearranged by chromosomal translocation to form chi-meric fusion proteins. The carboxy-terminal SET domain of wild-type MLL, which associates with three highly conserved core components (RbBP5, Ash2L and WDR5) and mediates H3K4 trimethylation9,10, is replaced by sequences from fusion partner genes11. Although deletion of the SET domain would indicate a loss of transactivation function, the resultant MLL fusion proteins act as dominant-positive transcriptional regulators that can bind DNA and aberrantly activate, via unknown mechanisms, the expression of MLL downstream targets such as Hox
genes11,12. The resultant oncogenic MLL fusion proteins can transform both haematopoietic stem-cell and early myeloid progenitors13,14, in which they resurrect the self-renewal transcriptional programmes in the leukaemic stem cells15. To gain further insights into underly-ing mechanisms and epigenetic networks being deregulated by MLL fusions, a retroviral transduction and transplantation assay (RTTA)14 was used to assess the oncogenic properties of MLL–EEN, the found-ing member of MLL SH3-domain-containing fusion proteins16, which constitutes a major family of MLL fusion proteins11. In contrast to cells transduced with truncated MLL 5’, truncated EEN 3’ or the vector con-trols that exhausted their proliferative ability within the first round of plating, murine primary haematopoietic cells that have been transduced with MLL–EEN oncoprotein exhibited enhanced self-renewal ability and could form compact CFU-GEMM-like colonies in the third and subsequent rounds of plating (Fig. 1a). Immunophenotypes of trans-formed cells were consistent with early myeloid precursors, which were positive for c-Kit, Mac-1 and Gr-1 (Fig. 1b, and data not shown). When MLL–EEN-transduced cells were transplanted into sub-lethally irradiated syngeneic mice, all the mice succumbed to acute leukaemia, with a median disease latency of 9 months (Fig. 1c). Histological and immunophenotypic studies on autopsy samples were consistent with acute myeloid leukaemia (AML) (Fig. 1d–e, and data not shown) that closely recapitulated the corresponding human disease associated with this fusion oncoprotein16. These results confirm the leukaemogenic properties of MLL–EEN and validate the usefulness of this approach for subsequent functional studies.
Although aberrant self-association has been proposed as an oncogenic mechanism for various leukaemia-associated transcription factors17, MLL–EEN mutants, with deletion of both the EEN coiled-coil domain and the putative SH3 binding sites upstream of the MLL breakpoint, could still efficiently transform primary haematopoietic cells (Fig. 1f–g). This indicates that self-association plays a minimal, if any, role in MLL–EEN-mediated transformation. In contrast, deletion of the EEN SH3 domain abolished transformation, indicating that the SH3 domain is the minimal transformation domain that is necessary and sufficient
1Haemato-Oncology Section, The Institute of Cancer Research, Sutton, Greater London SM2 5NG, UK; 2Department of Pathology, The University of Hong Kong, Hong Kong; 3Department of Haematology, Queen’s University, Belfast BT9 7AB, Northern Ireland; 4Department of Pathology, Stanford University Medical Center, Stanford, CA 93405, USA5Correspondence should be addressed to C.W.E.S. ([email protected])
Received 2 March 2007; accepted 24 July 2007; published online 23 September 2007; DOI: 10.1038/ncb1642
1208 � nature cell biology �volume�9�|�number�10�|�oCTober�2007
© 2007 Nature Publishing Group

l e t t e r s
for MLL–EEN-mediated oncogenesis (Fig. 1f–g). To confirm the in vivo leukaemogenic properties of MLL–EEN SH3-domain mutants, cells transduced with MLL–EEN∆N302 were injected into sub-lethally irra-diated mice for disease development. All injected mice developed AML
with very similar latency and phenotype as the full-length MLL–EEN (Fig. 1c and data not shown). Taken together, our results reveal that the aberrant acquisition of the EEN SH3 domain is the major driving force and a novel mechanism for oncogenic activation of MLL.
0 100 200 300 400 500
Number of colonies
First platingSecond platingThird plating
c-kit
0 50 100 150 200 250 300 350
Third plating
0 3 6 9 12 150
25
50
75
100
Month
ab
d
f
g
AT-hook
AT-hook
MTase
AT-hook MTase
AT-hook MTase
AT-hook MTase
AT-hook MTase
AT-hook MTase
AT-hook MTase
MTase AcyIT
AcyIT
CC
CC
H
H
SH3
SH3
AcyIT
AcyIT
CC
CC
CC
H
H
H
SH3
SH3
SH3
AcyIT
AcyIT
CC
CC
H SH3
MLL-5′
3′-EEN
Vector control
MLL–EEN16
1250
1250 1362
1362
125 290 368
c
Mock controlMLL–EEN∆N302MLL–EEN
MLL–EEN
Normal
PB Liver Spleen
e
11 10 100 1000 10000
1 10 100 1000 10000
104
103
102
101
1
10000
1000
100
10
Gr1
11 10 100 1000 10000
104
103
102
101
1 10 100 1000 100001
10000
1000
100
10
M–EEN∆C237
MLL–EEN
M–EEN∆C303
M–EEN∆PRD
M–EEN∆N237
M–EEN∆N302
Number of colonies
WB: α-MLL
Mr(K)
Mac-1
Mac-1
Mac-1
MLL EEN
1
16 125 290
237
303
237 368
368302
1250
1250
1250 1362
368
PRD
PRD
PRD
MLL
-5′
M–E
EN
M–E
EN
∆C23
7
M–E
EN
∆C30
3
M–E
EN
∆N23
7
M–E
EN
∆N30
2
M–E
EN
∆PR
D
M–S
am68
203
c-kit
Gr1Mac-1
Sur
viva
l (p
erce
ntag
e)
PRD
PRD
Figure 1 The EEN-SH3 domain is necessary and sufficient for MLL–EEN-mediated transformation. (a) A schematic diagram indicating the retroviral constructs used in the retroviral transduction and transplantation assay (RTTA; left). The bar chart (right) represents the corresponding number of colonies after each round of plating in methylcellulose (average number of three independent assays). Typical morphology of the compact third round colony generated by MLL–EEN-transduced cells (insert). Scale bar, 50 µm. (b) Phenotypic analysis of MLL–EEN-transformed cells by fluorescence-activated cell sorting (FACS) using indicated antibodies. (c) Survival curves for a cohort (n = 10) of sub-lethally irradiated C56BL/6 mice injected with
indicated MLL–EEN (FL or ∆N302)-transduced cells, or mock control. (d) Representative histology is shown for control and MLL–EEN mice, in which leukaemic cells were present in the peripheral blood (1200× magnification) and infiltrated into the spleen and liver (40× magnification). (e) FACS analysis of MLL–EEN leukaemic blasts. (f) Schematic diagram (left) shows the deletion mutants of MLL–-EEN used for RTTA. Bar graph (right) represents the number of colonies generated by the respective constructs in the third plating (mean ± s.d., n = 3). (g) Detection of expression of the various MLL fusion constructs used in RTTA by western blotting (WB) with MLL antibody. Relative molecular mass in thousands (K). Error bars indicate standard derivations (n = 3).
nature cell biology �volume�9�|�number�10�|�oCTober�2007� 1209
© 2007 Nature Publishing Group

l e t t e r s
Next, we sought to affinity purify the critical protein complex recruited by the EEN SH3 domain to the MLL fusion that leads to enhanced self-renewal and oncogenic transformation. Three major and distinctive bands with apparent relative molecular masses (Mr) of 62,000 (62 K), 68,000 (68 K) and 110,000 (110 K) were observed in the complex pulled down by GST–EEN-SH3 but not with the GST control (Fig. 2a). By mass spectrometry, these proteins were identified as nonO, Sam68 and PSF, respectively, all of which are involved in transcriptional regulation and/or RNA processing. As epigenetic de-regulation has long been speculated as the oncogenic mechanism mediated by MLL fusion proteins11,17,18, we decided to focus on Sam68, which has been shown to independently recruit the histone methyltransferase PRMT1 and the histone acetyl-transferase CBP19, and promote RNA splicing together with RNA PolII and Brm20. The identity of Sam68 was first confirmed by western blotting (Fig. 2b), and its in vivo interaction with EEN was further demonstrated by co-immunoprecipitation assay, in which Sam68 and EEN could recip-rocally precipitate each other (Fig. 2c). Further deletion analyses revealed the interaction interface between the SH3 domain of EEN and a region spanning 263–331 aa of Sam68 that contains the polyproline motif 3 (P3) (Fig. 2c). This indicates that a direct interaction occurs, which was further confirmed by a GST pulldown assay using in vitro translated and recombinant proteins (see Supplementary Information, Fig. 1). The in vivo interaction between Sam68 and MLL–EEN oncoprotein was also demonstrated by both co-immunoprecipitation assay (Fig. 2d) and immunofluorescent staining (Fig. 2e), which showed sub-nuclear
co-localization between MLL–EEN and Sam68. Taken together, these results validate Sam68 as an in vivo interacting partner for MLL–EEN.
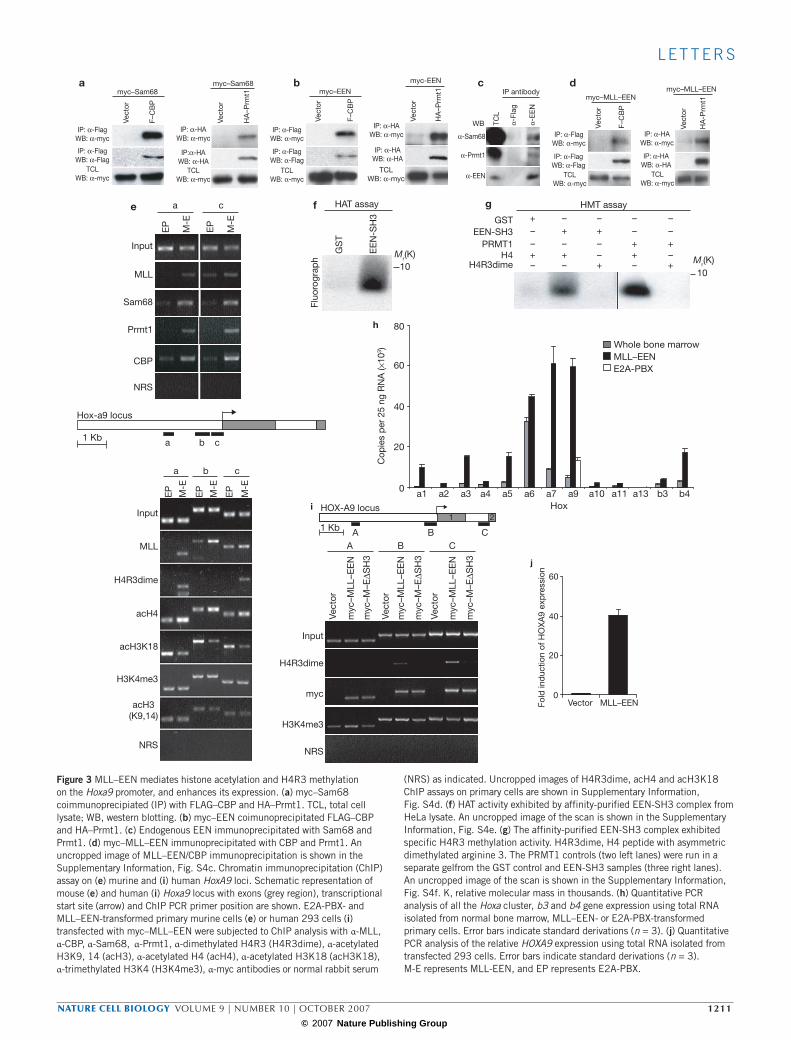
To confirm the presence of PRMT1 and CBP in the EEN complex, we performed western blotting on the affinity-purified EEN-SH3 complex and were able to detect specific bands corresponding to PRMT1 and CBP, respectively (Fig. 2b). Consistently, co-immunoprecipitation vali-dated their in vivo interaction via Sam68 (Fig. 3a). To demonstrate that CBP and PRMT1 associated with EEN in vivo, we further showed that EEN could indeed interact and immunoprecipitate CBP and PRMT1 in 293 cells (Fig. 3b). In addition, the in vivo interaction among endogenous EEN, Sam68 and PRMT1 were confirmed by immunoprecipitation assays in which EEN antibody could specifically precipitate endogenous Sam68 and PRMT1 (Fig. 3c). Finally, the equivalent interaction between the oncogenic MLL–EEN fusion and PRMT1–CBP was demonstrated and confirmed using co-immunopreciptiation assays (Fig. 3d) and chro-matin immunoprecipitation (ChIP) assays in transformed primary cells, showing in vivo recruitment of these endogenous histone modification enzymes by MLL–EEN to downstream targets (that is, Hoxa9 gene) (Fig. 3e, upper panel).
To assess the histone modification ability of the EEN-SH3 complex, the affinity-purified complex was incubated with the histone H4 tail in the presence of 3H-labelled acetyl-coenzyme A. Histone acetyla-tion could be detected in the presence of the EEN-SH3 complex but not the control (Fig. 3f). For the in vitro methylation assay, the EEN-SH3 complex was incubated with the histone H4 tail in the presence of
myc–Sam68
Merge
IP: α-FlagWB: α-myc
TCLWB: α-myc
myc–Sam68
IP: α-FlagWB: α-myc
TCLWB: α-myc
-PSF/SFPQ
-Sam68
-NonO
50-
60-
70-
90-80-
100-
75
50
75
50
50
75
353025
50
75
250
35
250
75
75
120- GST
GST
–SH
3
Vect
or
F-E
EN
F-E
EN
∆SH
3
*
Vect
or
1–44
3
50–4
43
263–
443
1–33
1
1–26
3
1–19
5
myc–EEN
5% In
put
GST
GST
–EEN
-SH
3
Vect
or
F-M
-EE
NWB
α-Sam68
WBα-CBP
WBα-PRMT1
IP: α-FlagWB: α-myc
IP: α-FlagWB: α-Flag
TCLWB: α-myc
b d
ca
e
IP: α-FlagWB: α-Flag IP: α-Flag
WB: α-Flag
Mr(K)
Mr(K)Mr(K)
Mr(K)
Mr(K) myc–MLL–EEN Flag–Sam68 DAPI
Figure 2 Identification of the protein complexes associated with the EEN-SH3 domain. (a) Coomassie blue staining of the proteins bound to GST–EEN-SH3 or GST control from HeLa cell lysate. (b) Immunodetection of Sam68, CBP and Prmt1 in the affinity-purified EEN-SH3 complex with the respective antibodies. (c) In vivo interaction was demonstrated by co-immunoprecipitation (IP) with anti-FLAG and immunodetected with anti-myc antibody on 293 cells cotransfected with (left) myc–Sam68, together with FLAG-tagged EEN proteins; or (right) myc–EEN, together with FLAG-tagged Sam68 proteins.
TCL, total cell lysate. Asterisk indicates Ig heavy chain. An uncropped image for the coimmunoprecipitation of FLAG–Sam68/myc–EEN is shown in the Supplementary Information, Fig. S4a. WB, western blotting. (d) FLAG–MLL–EEN coimmunoprecipitated with myc–Sam68. An uncropped image is shown in the Supplementary Information, Fig. S4b. (e) Immunofluorescence image of double staining for anti-FLAG and anti-myc antibody in COS7 cells co-expressing myc–MLL–EEN and FLAG–Sam68 with DNA counterstained with DAPI. Scale bars, 10 µm. K, relative molecular mass in thousands.
1210 � nature cell biology �volume�9�|�number�10�|�oCTober�2007
© 2007 Nature Publishing Group
l e t t e r s
IP: α-FlagWB: α-myc
IP: α-FlagWB: α-Flag
TCLWB: α-myc
IP: α-HAWB: α-myc
IP: α-HAWB: α-HA
TCLWB: α-myc
IP: α-HAWB: α-myc
IP: α-HAWB: α-HA
IP: α-FlagWB: α-myc
IP: α-FlagWB: α-Flag
IP: α-HAWB: α-myc
IP:α-HAWB: α-HA
1 Kb a b c
Hox-a9 locus
HMT assayHAT assay
a
CA B
HOX-A9 locus1 2
A B C
NRS
Input
myc
H4R3dime
H3K4me3
a1 a2 a4a3 a5 a6 b4b3a13a7 a9 a10 a11Hox
20
40
60
80
0
IP antibody
EP
M-E
EP
M-E
a c
b c
++–
–
––––
––––
– ––
––
++ +
+ + +++
1 Kb
WB
MLL–EENE2A-PBX
1010
myc–Sam68myc–Sam68 myc–EEN
myc-EEN
e
a b c d
f g
EEN-SH3GST
PRMT1H4
H4R3dime
Input
MLL
Sam68
Prmt1
CBP
NRS
h
Input
MLL
H4R3dime
acH4
acH3K18
H3K4me3
acH3(K9,14)
NRS
Fluo
rogr
aph
Vect
or
F–C
BP
Vect
or
HA
–Prm
t1
Vect
or
F–C
BP
Vect
or
F–C
BP
Vect
or
HA
–Prm
t1
Vect
or
HA
-Prm
t1
TCL
α-Fl
ag
α-E
EN
TCLWB: α-myc
TCLWB: α-myc
TCLWB: α-myc
GS
T
EE
N-S
H3
Mr(K)Mr(K)
myc–MLL–EENmyc–MLL–EEN
j
i
MLL–EENVectorFold
ind
uctio
n of
HO
XA
9 ex
pre
ssio
n
0
20
40
60
IP: α-FlagWB: α-myc
IP: α-FlagWB: α-Flag
TCLWB: α-myc
α-Sam68
α-Prmt1
α-EEN
Cop
ies
per
25 n
g R
NA
(×10
3 ) Whole bone marrow
EP
M-E
EP
M-E
EP
M-E
Vect
or
myc
–MLL
–EE
N
myc
–M–E
∆SH
3
Vect
or
myc
–MLL
–EE
N
myc
–M–E
∆SH
3
Vect
or
myc
–MLL
–EE
N
myc
–M–E
∆SH
3
Figure 3 MLL–EEN mediates histone acetylation and H4R3 methylation on the Hoxa9 promoter, and enhances its expression. (a) myc–Sam68 coimmunoprecipiated (IP) with FLAG–CBP and HA–Prmt1. TCL, total cell lysate; WB, western blotting. (b) myc–EEN coimunoprecipitated FLAG–CBP and HA–Prmt1. (c) Endogenous EEN immunoprecipitated with Sam68 and Prmt1. (d) myc–MLL–EEN immunoprecipitated with CBP and Prmt1. An uncropped image of MLL–EEN/CBP immunoprecipitation is shown in the Supplementary Information, Fig. S4c. Chromatin immunoprecipitation (ChIP) assay on (e) murine and (i) human HoxA9 loci. Schematic representation of mouse (e) and human (i) Hoxa9 locus with exons (grey region), transcriptional start site (arrow) and ChIP PCR primer position are shown. E2A-PBX- and MLL–EEN-transformed primary murine cells (e) or human 293 cells (i) transfected with myc–MLL–EEN were subjected to ChIP analysis with α-MLL, α-CBP, α-Sam68, α-Prmt1, α-dimethylated H4R3 (H4R3dime), α-acetylated H3K9, 14 (acH3), α-acetylated H4 (acH4), α-acetylated H3K18 (acH3K18), α-trimethylated H3K4 (H3K4me3), α-myc antibodies or normal rabbit serum
(NRS) as indicated. Uncropped images of H4R3dime, acH4 and acH3K18 ChIP assays on primary cells are shown in Supplementary Information, Fig. S4d. (f) HAT activity exhibited by affinity-purified EEN-SH3 complex from HeLa lysate. An uncropped image of the scan is shown in the Supplementary Information, Fig. S4e. (g) The affinity-purified EEN-SH3 complex exhibited specific H4R3 methylation activity. H4R3dime, H4 peptide with asymmetric dimethylated arginine 3. The PRMT1 controls (two left lanes) were run in a separate gelfrom the GST control and EEN-SH3 samples (three right lanes). An uncropped image of the scan is shown in the Supplementary Information, Fig. S4f. K, relative molecular mass in thousands. (h) Quantitative PCR analysis of all the Hoxa cluster, b3 and b4 gene expression using total RNA isolated from normal bone marrow, MLL–EEN- or E2A-PBX-transformed primary cells. Error bars indicate standard derivations (n = 3). (j) Quantitative PCR analysis of the relative HOXA9 expression using total RNA isolated from transfected 293 cells. Error bars indicate standard derivations (n = 3). M-E represents MLL-EEN, and EP represents E2A-PBX.
nature cell biology �volume�9�|�number�10�|�oCTober�2007� 1211
© 2007 Nature Publishing Group

l e t t e r s
3H-labelled S-adenosylmethionine. Histone methylation was detected in the presence of either purified PRMT1 or GST–EEN-SH3 complex but not GST, indicating specific de novo histone methyltransferase activity associated with the EEN-SH3 complex (Fig. 3g). As PRMT1 is the only known enzyme that mediates asymmetric dimethylation of H4R3, puri-fied PRMT1 and the GST–EEN-SH3 complex were separately incubated with histone H4 tail modified by asymmetric dimethylated arginine 3. As a result, methylation by PRMT1 or the EEN-SH3 complex was com-pletely abolished (Fig. 3g), confirming that H4R3 is the specific meth-ylation site and the activity was not due to contamination with other methyltransferases during the purification process.
To further demonstrate the enzymatic activities of the MLL–EEN complex in vivo, we assessed the effect of MLL–EEN on histone mod-ification of critical downstream target genes such as Hoxa9 (refs 21, 22). As PRMT1 can mediate specific H4R3 asymmetric dimethylation that promotes histone acetylation by CBP/p300 for gene expression23, recruitment of PRMT1 and CBP via MLL–EEN can potentially main-tain or even activate the expression of Hox genes. To this end, we first confirmed, by quantitative RT-PCR, the overexpression of Hox genes (including Hoxa9) in MLL–EEN-transformed primary haematopoietic cells (Fig. 3h). Further ChIP analysis revealed specific H4R3 asymmetric dimethylation and an increased global H4 acetylation on the Hoxa9 promoter in MLL–EEN but not E2A-PBX-transformed cells (Fig. 3e, lower panel) — although the Hoxa9 gene was also expressed in the lat-ter (Fig. 3h). In contrast, the Hoxa9 promoter in E2A-PBX-transformed cells had a higher level of H3K18 acetylation (Fig. 3e, lower panel). These data strongly indicate that H4R3 methylation and global acetylation of
H4 were specific epigenetic modifications that were mediated by MLL–EEN. To further demonstrate that these epigenetic marks were not due to random association with the differentiation status of the transformed cells or culturing artefacts, ChIP assays were also carried out on human 293 cells that were transiently transfected with myc-tagged MLL–EEN. Consistently, we detected specific H4R3 asymmetric dimethylation marks on the HOXA9 gene (Fig. 3i), which were significantly upregu-lated following ectopic expression of MLL–EEN (Fig. 3j). Finally, we also confirmed the essential role of the SH3 domain in H4R3 methylation using a MLL–EEN ∆SH3 mutant, which was incapable of mediating H4R3 asymmetric dimethylation on the HOXA9 promoter (Fig. 3i). Together with the in vitro enzymatic data, these results confirm the de novo H4R3 asymmetric dimethylation and the H4 acetylation activities of the MLL–EEN oncogenic complex.
We hypothesized that if the recruitment of histone-modifying enzymes by Sam68 is a critical step for MLL–EEN oncogenicity, it would be expected that Sam68 may be able to substitute for EEN in transformation. To this end, a synthetic MLL–Sam68 construct was made and subjected to RTTA (Fig. 4a). In contrast to cells transduced with truncated MLL or Sam68 retroviruses that exhausted their proliferative capability in the sec-ond round of plating, MLL–Sam68-transduced cells could be replated to the third round and produced compact CFU-GEMM colonies (Fig. 4a). Immunophenotypic analysis revealed their phenotypes as early myeloid precursors, reminiscent of MLL–EEN-transformed cells (Fig. 4b, 1b). Next, we assessed the functional requirement of Sam68 for MLL–EEN-mediated transformation using RTTA in combination with short hairpin RNAs (shRNAs). We developed and validated two independent shRNAs
Figure 4 The role of Sam68 in MLL–EEN-mediated transformation of primary haematopoietic cells. (a) Schematic diagram of MLL–Sam68 retroviral constructs used in the retroviral transduction and transplantation assay (left). The bar chart (right) represents the corresponding number of third-round colonies (average number of three independent assays). Inserts are typical third-round colonies generated from MLL–Sam68-transduced cells. Scale bar, 50 µm. (b) Phenotypic analysis of MLL–Sam68-transformed cells by fluorescence-activated cell sorting using indicated antibodies. (c) Western blot (WB) analysis of primary haematopoietic cells cotransduced with MLL–EEN, pSUPER control or Sam68-shRNA
(short-hairpin RNA) retroviruses with indicated antibodies. K, relative molecular mass in thousands. (d) ChIP analysis of murine Hoxa9 promotor on MLL–EEN-transformed primary cells with Sam68 knockdown. (e) Co-transduction of haematopoietic progenitor cells with MLL–EEN or E2A-PBX in combination with pSUPER control or Sam68-shRNA retroviruses resulted in specific and significant suppression of MLL-mediated transformation by Sam68 shRNA. Bar diagram represents the number of third-round colonies relative to pSUPER control in percentage (mean ± s.d., n = 3). Both Sam68-sh4 and Sam68-sh6 behaved similarly in all the assays tested. Only the results from Sam68-sh4 are shown.
c d
a
1 1250MLL Sam68
1 443
443
P1 P2 P3 P4 P5
MLL–Sam68
MLL-5′
b Mac-1
Gr-1Mac-1
c-kitUnstained controlMLL–Sam68
WBα-Sam68
pS
UP
ER
Sam
68-s
h4
WBα-actin
Number of third-round colonies
Relative number of third-round colonies (percentage)
0 20 40 60 80 100 120 140
MLL–EEN
E2A-PBX
pSUPERSam68-sh4
0 20 40 60 80
P1 P2 P3 P4 P5
Sam68
NRS
MLL
H4R3dime
Input
pSUPERSam68-sh4
MLL–EEN + + + ++ +
+ +– –
––
e
75
50
Ty-rich
Ty-rich
KH
KH
RGG
1 RGG
AT-hook PRD
1362
AT-hook PRD
MTase
MTase
Hoxa9a
Primerc
MLL–Sam68
1 10 102 103 104
1 10 102 103 104
1
10
102
103
104
1
10
102
103
104
Mr(K)
1212 � nature cell biology �volume�9�|�number�10�|�oCTober�2007
© 2007 Nature Publishing Group
l e t t e r s
that specifically inhibited the expression of Sam68 in primary haemat-opoietic cells (Fig. 4c, and data not shown). Consistently, Sam68, but not control or scramble shRNAs, could specifically inhibit H4R3 asymmetric dimethylation by MLL–EEN, confirming the functional significance of Sam68 for MLL–EEN-mediated H4R3 methylation (Fig. 4d, and data not shown). In contrast to cells co-transduced with MLL–EEN and shRNA vector control, significant growth inhibition was observed in MLL–EEN-transformed cells that were co-transduced with Sam68 shRNAs (Fig. 4e, and see Supplementary Information, Fig. S2). To eliminate the possibility that this growth inhibition was due to general toxicity of the shRNAs, we further demonstrated that the transformation of primary haematopoietic cells by a non-MLL oncoprotein (E2A-PBX) was inert to the treatment (Fig. 4e, and see Supplementary Information, Fig. S2). These results con-sistently indicate a critical and essential function of Sam68 as a bridging molecule in the oncogenic MLL fusion complex for transformation.
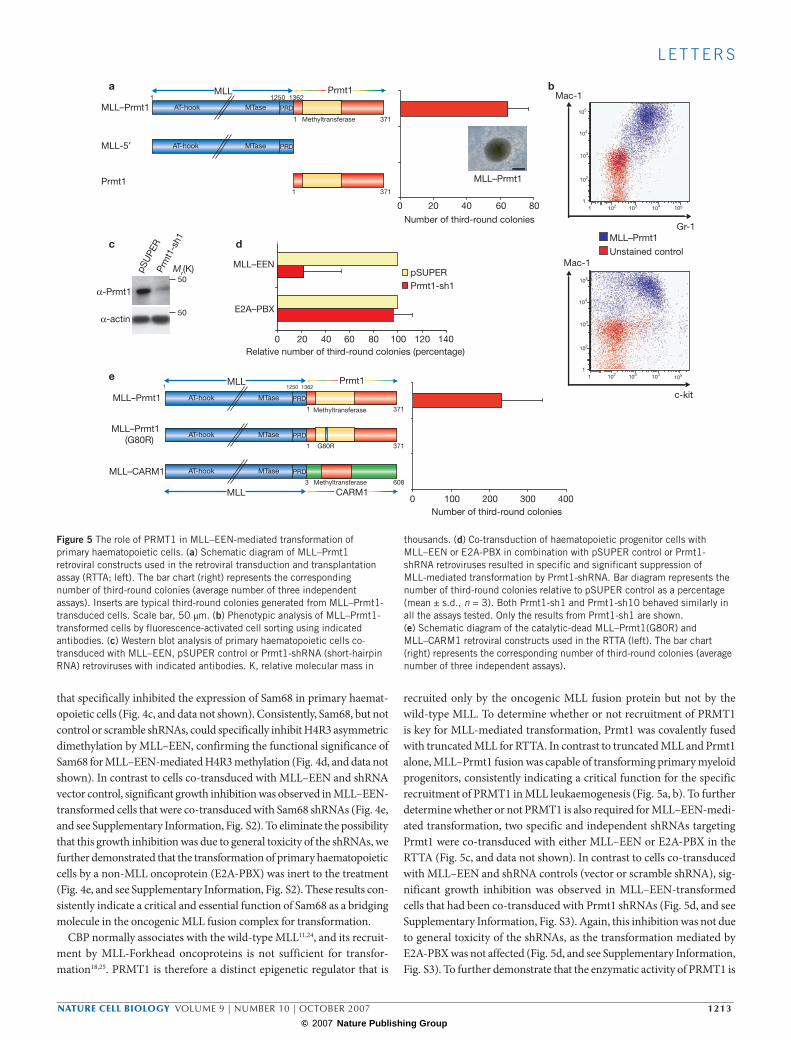
CBP normally associates with the wild-type MLL11,24, and its recruit-ment by MLL-Forkhead oncoproteins is not sufficient for transfor-mation18,25. PRMT1 is therefore a distinct epigenetic regulator that is
recruited only by the oncogenic MLL fusion protein but not by the wild-type MLL. To determine whether or not recruitment of PRMT1 is key for MLL-mediated transformation, Prmt1 was covalently fused with truncated MLL for RTTA. In contrast to truncated MLL and Prmt1 alone, MLL–Prmt1 fusion was capable of transforming primary myeloid progenitors, consistently indicating a critical function for the specific recruitment of PRMT1 in MLL leukaemogenesis (Fig. 5a, b). To further determine whether or not PRMT1 is also required for MLL–EEN-medi-ated transformation, two specific and independent shRNAs targeting Prmt1 were co-transduced with either MLL–EEN or E2A-PBX in the RTTA (Fig. 5c, and data not shown). In contrast to cells co-transduced with MLL–EEN and shRNA controls (vector or scramble shRNA), sig-nificant growth inhibition was observed in MLL–EEN-transformed cells that had been co-transduced with Prmt1 shRNAs (Fig. 5d, and see Supplementary Information, Fig. S3). Again, this inhibition was not due to general toxicity of the shRNAs, as the transformation mediated by E2A-PBX was not affected (Fig. 5d, and see Supplementary Information, Fig. S3). To further demonstrate that the enzymatic activity of PRMT1 is
c d
Relative number of third-round colonies (percentage)
MLL–EEN
E2A–PBX
0 20 40 60 80 100 120 140
pSUPERPrmt1-sh1
a
AT-hook MTase PRDMLL-5′
MLL–Prmt1
Number of third-round colonies
b
Mac-1
102 103 1041051
102
103
104
105
1
c-kit
Mac-1
Gr-1
102 103 104 1051
102
103
104
105
1
Unstained control
MLL–Prmt1
α-Prmt1
α-actin
MLL–Prmt1
1 1362MLL Prmt1
1 371
AT-hook MTase PRD
1 371Prmt1
0 20 40 60 80
MLL–Prmt11 1250 1362
MLL Prmt1
1 371
AT-hook MTase PRD
MLL–CARM1
MLL3 608
AT-hook MTase PRD
AT-hook MTase PRD
1 371
MLL–Prmt1 (G80R)
Number of third-round colonies0 100 200 300 400
e
50
50
Methyltransferase
Methyltransferase
Methyltransferase
pSU
PER
Prm
t1-s
h1
Mr(K)
G80R
CARM1
1250
Figure 5 The role of PRMT1 in MLL–EEN-mediated transformation of primary haematopoietic cells. (a) Schematic diagram of MLL–Prmt1 retroviral constructs used in the retroviral transduction and transplantation assay (RTTA; left). The bar chart (right) represents the corresponding number of third-round colonies (average number of three independent assays). Inserts are typical third-round colonies generated from MLL–Prmt1-transduced cells. Scale bar, 50 µm. (b) Phenotypic analysis of MLL–Prmt1-transformed cells by fluorescence-activated cell sorting using indicated antibodies. (c) Western blot analysis of primary haematopoietic cells co-transduced with MLL–EEN, pSUPER control or Prmt1-shRNA (short-hairpin RNA) retroviruses with indicated antibodies. K, relative molecular mass in
thousands. (d) Co-transduction of haematopoietic progenitor cells with MLL–EEN or E2A-PBX in combination with pSUPER control or Prmt1-shRNA retroviruses resulted in specific and significant suppression of MLL-mediated transformation by Prmt1-shRNA. Bar diagram represents the number of third-round colonies relative to pSUPER control as a percentage (mean ± s.d., n = 3). Both Prmt1-sh1 and Prmt1-sh10 behaved similarly in all the assays tested. Only the results from Prmt1-sh1 are shown. (e) Schematic diagram of the catalytic-dead MLL–Prmt1(G80R) and MLL–CARM1 retroviral constructs used in the RTTA (left). The bar chart (right) represents the corresponding number of third-round colonies (average number of three independent assays).
nature cell biology �volume�9�|�number�10�|�oCTober�2007� 1213
© 2007 Nature Publishing Group

l e t t e r s
essential for the MLL-mediated transformation, a point mutation (G80R) was introduced into the catalytic domain of the MLL–Prmt1 construct, which results in a catalytic-dead mutant23. In contrast to MLL–Prmt1, MLL–Prmt1 (G80R) completely lost its transforming capacity (Fig. 5e), confirming a critical function of H4R3 asymmetric dimethylation in MLL–EEN transformation. Although PRMT1 is the only known arginine methyltransferase that confers H4R3 asymmetric dimethylation marks, there are multiple PRMT family members with slightly different enzymatic activities. Among them, PRMT4 (CARM1) has been better characterized and possesses H3R17 methylation activity5,6. To deter-mine whether other PRMTs can also activate the oncogenic property of MLL, full-length CARM1 was fused to truncated MLL and subjected to transformation assay. In contrast to MLL–Prmt1, MLL–CARM1 failed to transform primary haematopoietic cells (Fig. 5e). Taken together, this study discovers a specific and essential role of PRMT1 in a novel protein arginine-methyltransferase-dependent oncogenic pathway.
Epigenetic therapy has given renewed optimism for targeting onco-genic transcriptional processes that may otherwise not be targeted by small molecules. However, other than DNA methyltransferase and his-tone deacetylases (HDACs) that are aberrantly recruited by some onco-genic transcription factors4, the precise chromatin-modifying enzymes that are involved in aberrant transcriptional deregulation in most malig-nancies remain unknown. Arginine methylation plays a critical role in the regulation of protein function, gene expression and is an integral part of the histone code5. Recently, arginine methylation of histones has been implicated as a major determinant that regulates pluripotency in early mouse embryo26 and epigenetic reprogramming of germ-cell lineage27. Here, we have identified an arginine methyltransferase as an essential component of MLL oncogenic complexes and have unveiled a novel func-tion of this class of epigenetic-modifying enzymes in human cancer.
Recent functional analyses of oncogenic RARα fusion complexes have provided important mechanistic insights and novel molecular targets for acute promyelocytic leukaemia28–32. In contrast to RXR29,31, HDACs4 and PRC232 that are essential for transcriptional repression and trans-formation mediated by RARα fusions, PRMT1 confers an aberrant tran-scriptional activation property to MLL fusion complexes that is critical for the induction of leukaemia. These findings further strengthen our hypothesis for the existence of two functionally distinctive subgroups of oncogenic transcription factors with aberrant repression and activa-tion activities, respectively17. It has been shown that PRMT1 cooperates with p300 and CARM1 to maintain an active chromatin structure for a maximal transcription33. Thus, recruitment of both PRMT1 and CBP by an oncogenic MLL fusion complex may mediate step-wise histone modifications that efficiently facilitate an open chromatin structure for active gene expression. Although alteration of epigenetic codes on his-tones by MLL fusion complexes is an attractive model, and consistent with the recent findings that hDot1L with H3K79 methyltransferase activity is recruited by MLL–AF10 (ref. 34), it is important to note that most of the histone-modifying enzymes (including PRMTs) have multiple substrates that may also be critical for oncogenesis1,5. Notably, PRMT1 can methylate and regulate the activities of the components of transcriptional machinery, including transcription factors (for exam-ple, STAT1), elongation factors (for example, SPT5) and RNA-process-ing factors (for example, Sam68)5. Although the current study links PRMT1 to leukaemia, PRMTs may also play a role in other forms of cancer. PRMT1 and CARM1 are co-activators for nuclear receptors23 and
β-catenin6, which are frequent targets for both haematological and solid malignancies. PRMT5 is recruited to the promoters and suppresses the expression of tumour suppressors ST7 and NM23 (refs 5, 6). Therefore, future work elucidating the oncogenic pathways mediated by PRMTs and their prevalence in leukaemia and other cancers will provide important insights not only into specific targeting of MLL leukaemic stem cells, but also into the role that these proteins play in human malignancies and facilitate the development of optimal combinatorial strategies for epigenetic therapy.
METHoDSConstructs. MLL–EEN was constructed identically to the reported patient sequence16 into an MSCV-neo retroviral vector. FLAG or myc-tagged constructs were in pFLAG–CMV2 or pCS2+MT vectors, respectively. shRNA constructs were made by subcloning shRNA oligonucleotides into pSUPER.retro.puro according to the manufacturer’s instructions (OligoEngine, Seattle, WA). The 19-bp shRNA target sequences for Sam68-sh4, Sam68-sh6, Prmt1-sh1 and Prmt1-sh10 were: 5’-CAAGGAGGAAGAGTTGCGC-3’, 5’-AAGATGACGAGGAGAATTA-3’, 5’-GACATGACATCCAAAGACT-3’ and 5’-CAAGTGAAGAGGAACGACT-3’, respectively. HA-CARM1, HA-Prmt1, FLAG-CBP and Sam68 constructs were kind gifts from Dong Shuo, Michael Stallcup, Tso-Pang Yao and Flossie Wong-Staal, respectively.RTTA assay. RTTA assay was performed as previously described14. Briefly, ret-roviral supernatants were collected 3 d after transfection of Phoenix cells and used to infect haematopoietic progenitors and stem cells (harvested from the bone marrow of 4–10-week-old C57BL/6 mice) that were positively selected for c-Kit expression by magnetic-activated cell sorting. After spinoculation by centrifugation at 500 g for 2 h at 32 oC, transduced cells were cultured overnight in RPMI supplemented with 10% fetal calf serum, 20 ng ml–1 stem cell factor, plus 10 ng ml–1 each of interleukin (IL)-3 and IL-6 (R&D Systems, Minneapolis, MN). Transduced cells were then plated in 1% methylcellulose (Stem Cell Technologies, Vancouver, BC, Canada) supplemented with the same cytokines plus 10 ng ml–1 GM-CSF (R&D Systems) in the presence or absence of 1 mg ml–1 G418. For co-transduction experiments with shRNA constructs, the cells were co-selected with 1 µg ml–1 puromycin. After 7 d culture, colonies were counted to calculate the transduction efficiency. Single-cell suspensions (104 cells) of antibiotic-resistant colonies were then replated in methylcellulose media supplemented with the same growth factors without antibiotics. Plating was repeated every 7 d. For tumorigenicity assays, 106 immortalized cells were injected via the tail vein into 6-week-old syngeneic C57BL/6 mice that had received a sub-lethal dose of 5.25 Gy total body irradiation. Mice were main-tained on antibiotic water and monitored for the development of leukaemia by complete blood count, blood smear and fluorescence-activated cell-sorting analysis. Tissues were fixed in buffered formalin, sectioned and stained with hematoxylin and eosin for histological analysis. All the animal works were per-formed according to the guidelines and regulations of the Animal (Scientific Procedures) Act 1986.
GST-pulldown affinity purification and mass spectrometry. A total of 100 µg of GST fusion protein was incubated with 2 mg HeLa cell lysate in NP-40 lysis buffer (150 mM NaCl, 50 mM Tris (pH 8), 5 mM EDTA, Complete Protease Inhibitor (Roche, Mannheim, Germany), 1% NP-40) for 2 h at 4 °C, washed with lysis buffer, eluted with SDS sample buffer and finally resolved in 10% SDS–PAGE. Identity of the proteins was determined by MALDI-TOF-MS in reflector mode and the peptide masses from the samples were used for database searches with Profound (Genomic Solutions, Ann Arbor, MI) against the NCBInr database (TopLab GmbH, Martinsreid, Germany). GST-pulldown of radiolabelled and recombinant protein was performed as described previously25.
Immunoblotting and immunofluorescence. Antibodies used for immunoblot-ting were obtained from Santa Cruz Biotechnology (Santa Cruz, CA; anti-CBP; anti-myc, A14; anti-Sam68; anti-EEN), Sigma-Aldrich (Dorset, UK); anti-FLAG, M2; and anti-actin); Upstate Biotech (Billerica, MA; anti-Prmt1; and N4.4 anti-MLL); and Roche (anti-HA). For immunocolocalization, COS7 cells were cotrans-fected using Fugene 6 transfection reagent (Roche), according to the protocol, 24 h prior to fixation. Immunofluorescence was performed as described previously35
1214 � nature cell biology �volume�9�|�number�10�|�oCTober�2007
© 2007 Nature Publishing Group

l e t t e r s
using rabbit polyclonal anti-myc (A14) plus monoclonal anti-FLAG (M2), and subsequently immunodetected with FITC and Texas-Red-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA), respectively.
Transfection and immunoprecipitation. Subconfluent 293 cells transfected using calcium precipitation were harvested after 36–48 h. For generic immuno-precipitation, transfected cells were lysed in NP-40 lysis buffer (0.5% NP-40 for Sam68 and EEN, 0.1% for Prmt1) for 30 min on ice, incubated with the respective antibody overnight and then precipitated with protein-G sepharose (Amersham, Bucks, UK) at 4 °C for 4 h. Eluted proteins were resolved by SDS–PAGE and detected with the corresponding antibody. For CBP immunoprecipitation, NP-40 lysis buffer with 300 mM NaCl and 0.1% NP-40 was used.
In vitro acetylation and methylation assay. The histone acetyltransferase com-plex associated with the GST–EEN-SH3 was assayed in a final volume of 30 µl reaction containing 20 mM Tris (pH 8), 2% glycerol, 5 mM EDTA, 1 mM DTT, 50 mM sodium butyrate, 4 µg histone H4 peptide (aa 1–29) and 1 µCi 3H-acetyl-coenzyme A. For the histone methyltransferase assay, 20 mM Tris (pH 8), 5 mM EDTA, 0.5 mM DTT, 1 mM PMSF, 1 µCi 3H-S-adenosyl-l-methionine, 4 µg histone H4 peptide (aa 1–29) or H4R3dime peptide (a kind gift from Suming Huang) were used. The reactions were incubated for 1 h at 30 °C, terminated by heating in sample buffer and then separated by 15% SDS–PAGE gel. Gels were fixed, immersed into Amplify solution (Amersham) for 30 min before drying, then exposed overnight.
Quantitative RT-PCR. The quantity and quality of total RNAs extracted using Trizol (Invitrogen, Carlsbad, CA) followed by DNase treatment (Ambion, Austin, TX) were determined by Bioanalyzer (Agilent RNA 6000 nano). Primer sequences and conditions for Q-PCRs are available on request.
Chromatin immunoprecipitation. The ChIP assay kit was used according to the manufacturer’s protocol (Upstate Biotech) with some modifications. Briefly, cells were fixed with 1% formaldedyde for 15 min at room temperature and then quenched with 0.125 M glycine for 5 min. To generate DNA fragments of 0.2–1 kb, 1 × 106 cells were sonicated by Bioruptor (Diagenode, Liege, Belgium) on maximum power for 15 min with an on–off interval of 30 s. Chromatin frag-ments were incubated with the antibody overnight and collected in protein-A agarose (Upstate Biotech). All antibodies against modified histones were from Upstate except anti-MLL (Bethyl Laboratories, Montgomery, TX); anti-H3K4me3 and anti-Prmt1 (Abcam, Cambridge, UK). Cross-linked products were reversed by heating overnight at 65 °C and then treated with proteinase K at 45 °C for 1 h. Eluted DNA was purified using QIAquick PCR purification kit (Qiagen, West Sussex, UK) and resuspended in elution buffer containing 50 ng ml–1 tRNA (Invitrogen). PCRs were performed using Platinum Taq (Invitrogen) according to the manufacturer’s protocol. Primer sequences for human and murine Hoxa9 are available on request ([email protected]).
Note: Supplementary Information is available on the Nature Cell Biology website.
ACkNoWLEdgEMENTSWe thank M. Greaves, P. Workman, A. Ashworth, T. Lappin, S. Armstrong, A. Zelent, S. Huang and D. Shuo for useful discussion; J. Yam, G. McGonigle, B. Zeisig, W. Yue, A. Wilson, M. Boix-Chornet for excellent technical help; A. Thornhill, F. Darling, BSU staff for husbandry of mice; and P. Tse for professional artwork. This work is supported by the Association for International Cancer Research (AICR), and CWE So is an AICR research fellow.
AuThor CoNTribuTioNSN.C. performed most of the experiments; A.T. did the Q–RT–QPCR; L.C.C. and M.L.C. provided critical advice; C.W.E.S. designed and oversaw the project and wrote the manuscript.
Published online at http://www.nature.com/naturecellbiology/ reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
1. Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007).
2. Lund, A. H. & van Lohuizen, M. Epigenetics and cancer. Genes Dev. 18, 2315–2335 (2004).
3. Schneider, R., Bannister, A. J. & Kouzarides, T. Unsafe SETs: histone lysine methyl-transferases and cancer. Trends Biochem. Sci. 27, 396–402 (2002).
4. Minucci, S. & Pelicci, P. G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature Rev. Cancer 6, 38–51 (2006).
5. Bedford, M. T. & Richard, S. Arginine methylation an emerging regulator of protein function. Mol. Cell 18, 263–272 (2005).
6. Lee, D. Y., Teyssier, C., Strahl, B. D. & Stallcup, M. R. Role of protein methylation in regulation of transcription. Endocr. Rev. 26, 147–170 (2005).
7. Huang, S., Litt, M. & Felsenfeld, G. Methylation of histone H4 by arginine methyltrans-ferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev. 19, 1885–1893 (2005).
8. Look, A. T. Oncogenic transcription factors in the human acute leukemias. Science 278, 1059–1064 (1997).
9. Dou, Y. et al. Regulation of MLL1 H3K4 methyltransferase activity by its core compo-nents. Nature Struct. Mol. Biol. 13, 713–719 (2006).
10. Wysocka, J. et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121, 859–872 (2005).
11. Daser, A. & Rabbitts, T. H. The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin. Cancer Biol. 15, 175–188 (2005).
12. Milne, T. A. et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 10, 1107–1117 (2002).
13. Cozzio, A. et al. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 17, 3029–3035 (2003).
14. So, C. W. et al. MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell 3, 161–171 (2003).
15. Krivtsov, A. V. et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822 (2006).
16. So, C. W. et al. EEN encodes for a member of a new family of proteins containing an Src homology 3 domain and is the third gene located on chromosome 19p13 that fuses to MLL in human leukemia. Proc. Natl Acad. Sci. USA 94, 2563–2568 (1997).
17. So, C. W. & Cleary, M. L. Dimerization: a versatile switch for oncogenesis. Blood 104, 919–922 (2004).
18. So, C. W. & Cleary, M. L. Common mechanism for oncogenic activation of MLL by forkhead family proteins. Blood 101, 633–639 (2003).
19. Lukong, K. E. & Richard, S. Sam68, the KH domain-containing superSTAR. Biochim. Biophys. Acta 1653, 73–86 (2003).
20. Batsche, E., Yaniv, M. & Muchardt, C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nature Struct. Mol. Biol. 13, 22–29 (2006).
21. So, C. W., Karsunky, H., Wong, P., Weissman, I. L. & Cleary, M. L. Leukemic transfor-mation of hematopietic progenitors by MLL-GAS7 in the absence of Hoxa7 or Hoxa9. Blood 103, 3192–3199 (2004).
22. Ayton, P. M. & Cleary, M. L. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 17, 2298–2307 (2003).
23. Wang, H. et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 293, 853–857 (2001).
24. Ernst, P., Wang, J., Huang, M., Goodman, R. H. & Korsmeyer, S. J. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol. Cell Biol. 21, 2249–2258 (2001).
25. So, C. W. & Cleary, M. L. MLL-AFX requires the transcriptional effector domains of AFX to transform myeloid progenitors and transdominantly interfere with forkhead protein function. Mol. Cell Biol. 22, 6542–6552 (2002).
26. Torres-Padilla, M. E., Parfitt, D. E., Kouzarides, T. & Zernicka-Goetz, M. Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature 445, 214–218 (2007).
27. Ancelin, K. et al. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nature Cell Biol. 8, 623–630 (2006).
28. Kwok, C., Zeisig, B. B., Dong, S. & So, C. W. Forced homo-oligomerization of RARα leads to transformation of primary hematopoietic cells. Cancer Cell 9, 95–108 (2006).
29. Zeisig, B. B. et al. Recruitment of RXR by homotetrameric RARα fusion proteins is essential for transformation. Cancer Cell 12, 36–51 (2007).
30. Sternsdorf, T. et al. Forced retinoic acid receptor alpha homodimers prime mice for APL-like leukemia. Cancer Cell 9, 81–94 (2006).
31. Zhu, J. et al. RXR is an essential component of the oncogenic PML/RARA complex in vivo. Cancer Cell 12, 23–35 (2007).
32. Villa, R. et al. Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer Cell 11, 513–525 (2007).
33. An, W., Kim, J. & Roeder, R. G. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 117, 735–748 (2004).
34. Okada, Y. et al. hDOT1L links histone methylation to leukemogenesis. Cell 121, 167–178 (2005).
35. Cheung, N. et al. Subcellular localization of EEN/endophilin A2, a fusion partner gene in leukemia. Biochem. J. 383, 27–35 (2004).
nature cell biology �volume�9�|�number�10�|�oCTober�2007� 1215
© 2007 Nature Publishing Group




















