
Regulation of Renal Fibrosis by Smad3 Thr388 Phosphorylation
9
SHORT COMMUNICATION Regulation of Renal Fibrosis by Smad3 Thr388 Phosphorylation Xinli Qu,* Xueling Li, y Yaowu Zheng, z Yi Ren, x Victor G. Puelles,* Georgina Caruana,* David J. Nikolic-Paterson, { and Jinhua Li* From the Department of Anatomy and Developmental Biology,* Monash University, Clayton, Australia; the Key Laboratory of Mammalian Reproductive Biology and Biotechnology, y Ministry of Education, Inner Mongolia University, Hohhot, China; the Transgenic Research Center, z School of Life Sciences, Northeast Normal University, Changchun, China; the Department of Biomedical Sciences, x Florida State University College of Medicine, Tallahassee, Florida; and the Department of Nephrology, Monash Health and the Department of Medicine, { Monash University, Clayton, Australia Accepted for publication December 19, 2013. Address correspondence to Jinhua Li, Ph.D., Department of Anatomy and Developmental Biology, Monash University, Clayton, VIC 3800, Australia. E-mail: [email protected]. Transforming growth factor-b (TGF-b) promotes tissue fibrosis via receptor-mediated phosphorylation of the receptor-activated Smad2/3, together with Smad4. Of these, Smad3 plays a major profibrotic role in mouse models of tissue fibrosis. Transcriptional activity of the Smad3 protein is regulated by phosphorylation of residues in the C-terminal domain and the linker region. Herein, we examined the role of a novel phosphorylation site within the MH2 domain (T388) in the regulation of Smad3 activity. Confocal microscopy using an Smad3 phosphorylated T388-specific antibody identified phosphorylation of Smad3 T388 in myofibroblasts and tubular epithelial cells in human focal and segmental glomerulosclerosis and mouse models of unilateral ureteric obstruction and diabetic ne- phropathy, whereas phosphorylated T388 was largely absent in normal kidney. In vitro, TGF-b1 induced phosphorylation of Smad3 T388 in a biphasic pattern. A point mutation of T388/V in an Smad3 construct demonstrated that phosphorylation of T388 promotes Smad3 binding to Smad4 and CDK8, but was not necessary for nuclear translocation. Furthermore, T388 phosphorylation was required for TGF-beinduced collagen I gene promoter activity and extracellular matrix production in cultured fibroblasts. In conclusion, our study identifies phosphorylation of T388 in the Smad3 MH2 domain as an important mechanism that regulates the profibrotic TGF-b/Smad3 signaling pathway, which has direct relevance to human and experimental fibrotic kidney disease. (Am J Pathol 2014, 184: 944e952; http://dx.doi.org/10.1016/j.ajpath.2013.12.003) Transforming growth factor-b1 (TGF-b1) is a secreted cytokine that regulates a diverse range of cellular responses, including the control of cell proliferation, differentiation, and apoptosis. 1 TGF-b1 also plays a key role in the development and progression of tissue fibrosis. 2e5 Administration of an antieTGF-b1 neutralizing antibody attenuates renal fibrosis in experimental kidney disease. 6 TGF-b1 acts directly on fibroblasts to induce expression of a-smooth muscle actin (a- SMA) and transition into collagen Ieproducing myofibro- blasts. The major biological effects of TGF-b operate via the TGF-b type II and type I receptors and the receptor-activated Smad family (Smads 2, 3, and 4). 7,8 On binding TGF-b, type II and type I receptors are activated by phosphorylation. The active TGF-b type I receptor then phosphorylates serine amino acid side chains in the SSXS motif in the C-terminal region of Smad2 and Smad3. Phosphorylated Smad2/3 oli- gomerizes with Smad4, and this complex translocates to the nucleus and regulates targeted gene transcription. 7,8 Conditional deletion of Smad2 from kidney tubular epithe- lial cells exacerbates renal interstitial fibrosis in the mouse unilateral ureteral obstruction (UUO) model, demonstrating an antifibrotic role for Smad2. 9 However, mice with global Smad3 gene deletion are resistant to tissue fibrosis in experimental models of kidney, lung, and cardiac disease. 10e12 In addition, Supported by the Monash University Accelerating Programme (J.L.), the National Health and Medical Research Council (NHMRC) of Australia (J.L.), and an NHMRC Career Development Award (J.L.). X.Q. and X.L. contributed equally to this work. Disclosures: None declared. Copyright ª 2014 Published by Elsevier Inc. on behalf of American Society for Investigative Pathology. http://dx.doi.org/10.1016/j.ajpath.2013.12.003 ajp.amjpathol.org The American Journal of Pathology, Vol. 184, No. 4, April 2014
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Regulation of Renal Fibrosis by Smad3 Thr388 Phosphorylation

The American Journal of Pathology, Vol. 184, No. 4, April 2014
ajp.amjpathol.org
SHORT COMMUNICATIONRegulation of Renal Fibrosis by Smad3 Thr388PhosphorylationXinli Qu,* Xueling Li,y Yaowu Zheng,z Yi Ren,x Victor G. Puelles,* Georgina Caruana,* David J. Nikolic-Paterson,{ andJinhua Li*
From the Department of Anatomy and Developmental Biology,* Monash University, Clayton, Australia; the Key Laboratory of Mammalian ReproductiveBiology and Biotechnology,y Ministry of Education, Inner Mongolia University, Hohhot, China; the Transgenic Research Center,z School of Life Sciences,Northeast Normal University, Changchun, China; the Department of Biomedical Sciences,x Florida State University College of Medicine, Tallahassee,Florida; and the Department of Nephrology, Monash Health and the Department of Medicine,{ Monash University, Clayton, Australia
Accepted for publication
C
A
h
December 19, 2013.
Address correspondence toJinhua Li, Ph.D., Department ofAnatomy and DevelopmentalBiology, Monash University,Clayton, VIC 3800, Australia.E-mail: [email protected].
opyright ª 2014 Published by Elsevier Inc.
merican Society for Investigative Pathology.
ttp://dx.doi.org/10.1016/j.ajpath.2013.12.003
Transforming growth factor-b (TGF-b) promotes tissue fibrosis via receptor-mediated phosphorylationof the receptor-activated Smad2/3, together with Smad4. Of these, Smad3 plays a major profibroticrole in mouse models of tissue fibrosis. Transcriptional activity of the Smad3 protein is regulated byphosphorylation of residues in the C-terminal domain and the linker region. Herein, we examined therole of a novel phosphorylation site within the MH2 domain (T388) in the regulation of Smad3activity. Confocal microscopy using an Smad3 phosphorylated T388-specific antibody identifiedphosphorylation of Smad3 T388 in myofibroblasts and tubular epithelial cells in human focal andsegmental glomerulosclerosis and mouse models of unilateral ureteric obstruction and diabetic ne-phropathy, whereas phosphorylated T388 was largely absent in normal kidney. In vitro, TGF-b1induced phosphorylation of Smad3 T388 in a biphasic pattern. A point mutation of T388/V in anSmad3 construct demonstrated that phosphorylation of T388 promotes Smad3 binding to Smad4 andCDK8, but was not necessary for nuclear translocation. Furthermore, T388 phosphorylation wasrequired for TGF-beinduced collagen I gene promoter activity and extracellular matrix production incultured fibroblasts. In conclusion, our study identifies phosphorylation of T388 in the Smad3 MH2domain as an important mechanism that regulates the profibrotic TGF-b/Smad3 signaling pathway,which has direct relevance to human and experimental fibrotic kidney disease. (Am J Pathol 2014,184: 944e952; http://dx.doi.org/10.1016/j.ajpath.2013.12.003)
Supported by the Monash University Accelerating Programme (J.L.), theNational Health and Medical Research Council (NHMRC) of Australia(J.L.), and an NHMRC Career Development Award (J.L.).X.Q. and X.L. contributed equally to this work.Disclosures: None declared.
Transforming growth factor-b1 (TGF-b1) is a secretedcytokine that regulates a diverse range of cellular responses,including the control of cell proliferation, differentiation, andapoptosis.1 TGF-b1 also plays a key role in the developmentand progression of tissue fibrosis.2e5 Administration of anantieTGF-b1 neutralizing antibody attenuates renal fibrosisin experimental kidney disease.6 TGF-b1 acts directly onfibroblasts to induce expression of a-smooth muscle actin (a-SMA) and transition into collagen Ieproducing myofibro-blasts. The major biological effects of TGF-b operate via theTGF-b type II and type I receptors and the receptor-activatedSmad family (Smads 2, 3, and 4).7,8 On binding TGF-b, typeII and type I receptors are activated by phosphorylation. Theactive TGF-b type I receptor then phosphorylates serineamino acid side chains in the SSXS motif in the C-terminal
on behalf of
region of Smad2 and Smad3. Phosphorylated Smad2/3 oli-gomerizes with Smad4, and this complex translocates to thenucleus and regulates targeted gene transcription.7,8
Conditional deletion of Smad2 from kidney tubular epithe-lial cells exacerbates renal interstitial fibrosis in the mouseunilateral ureteral obstruction (UUO)model, demonstrating anantifibrotic role for Smad2.9However,micewith global Smad3gene deletion are resistant to tissue fibrosis in experimentalmodels of kidney, lung, and cardiac disease.10e12 In addition,

Smad3 T388 Phosphorylation in Fibrosis
Smad3-deficient fibroblasts fail to autoinduce TGF-b1expression.13 It is generally accepted that phosphorylationof Smad3 in the C-terminal SSXS motif by the type ITGF-b receptor kinase plays an essential role in mediatingthe TGF-b response.14 In addition, a range of protein ki-nases, including mitogen-activated protein kinases [p38,extracellular signaleregulated kinase (ERK), and c-Junkinase], protein kinase B (Akt), and CDKs (CDK4 andCDK8), can phosphorylate the linker region of Smad3(T179, S204, S208, and S213), which positively ornegatively regulates Smad3 function.15e22 A recent studyhas identified a novel phosphorylation site (T388) withinthe MH2 domain of Smad3, which has the potential toregulate Smad3 function.23 Herein, we demonstrate thatSmad3 T388 is phosphorylated in human and experimentalkidney fibrosis and identify a functional role for Smad3T388 phosphorylation in the TGF-b1einduced profibroticresponse of renal fibroblasts.
Materials and Methods
Experimental Animals
C57BL6/J mice were purchased from Monash AnimalServices, Monash University (Clayton, Australia). Breedingpairs of Smad3�/� and Nos3�/� mice were purchased fromJackson Laboratories (Bar Harbor, ME) and maintained atMonash Animal Services. All experiments were approvedby the Monash University Animal Ethics Committee andadhered to the Australian Code of Practice for the Care andUse of Animals for Scientific Purposes.
UUO surgery was performed on groups of six male wild-type (WT) or Smad3�/� C57Bl6/J mice under isofluraneanesthesia. The left ureter was visualized after a flankincision and ligated with a vascular clamp. Sham mice un-derwent the same procedure, except that the ureter was notligated. Mice were sacrificed 4 days after UUO surgery, andkidney tissue was processed for analysis.
Diabetes was induced in Nos3�/� mice (n Z 10) at 8weeks of age by i.p. administration of 50 mg/g streptozo-tocin (Sigma-Aldrich, St. Louis, MO) for 5 consecutivedays. Control Nos3�/� mice (n Z 6) received daily i.p.injections of normal saline for 5 days. Mice were sacrificed3 months after the onset of diabetes. Kidney tissues werecollected for analysis.
Human Renal Biopsy Specimens
Studies using human tissue were approved by the HumanEthics Committee of Monash Medical Centre, and writteninformed consent was obtained from the patients. Cryostatsections of snap frozen tissue, excess to that required fordiagnosis, were examined in four cases of focal andsegmental glomerulosclerosis. Normal kidney tissue wasobtained from the noninvolved pole of nephrectomies per-formed as the result of renal carcinoma.
The American Journal of Pathology - ajp.amjpathol.org
Polyclonal Antibody to Smad3 Phosphorylated T388
A polyclonal antibody was generated by repeated immuniza-tion of a rabbit with a linear Smad3 peptide containing phos-phorylated T388 (GWGAEYRRQT388VTSTPCWIELHL),which is absolutely conserved between human and mouse. Theantibody was affinity purified using the p-T388 peptide andthen absorbed against the non-phosphorylation peptide.
Cell Culture
NRK49F rat kidney fibroblasts and NRK52E rat proximaltubular epithelial cells were cultured as previously de-scribed.24,25 For experiments, cells were cultured in Dul-becco’s modified Eagle’s medium containing 0.1% fetalbovine serum, with or without 0 to 4 ng/mL human TGF-b1(Cat. No. T1654; Sigma-Aldrich). Each experiment wasrepeated at least three times. In the blocking studies, NRK49Fcells were pretreated with 4 mg/mL mouse antieTGF-b1neutralizing antibody (R&D Systems, Minneapolis, MN) orcontrol mouse IgG, 100 nmol/L TGF-b receptor I inhibitor(ALK5I; Calbiochem, La Jolla, CA), or 20 mmol/L ERK1/2inhibitor, PD98059 (Calbiochem), for 30 minutes, followedby addition of TGF-b1 for different periods.
Confocal Microscopy
Cryostat tissue sections and cultured cells in chamber slideswere fixed in 4% paraformaldehyde (Sigma-Aldrich), blockedwith 2% bovine serum albumin in PBS, and incubated withone or more of rabbit anti-phosphorylated Smad3 T388 anti-body, followed by goat anti-rabbit Alexa Fluor 488 (Invi-trogen, Mount Waverley, Australia), Cy3-conjugated mouseantiea-SMA antibody (Sigma-Aldrich), or fluoresceinisothiocyanateeconjugated mouse anti-Flag antibody (Sigma-Aldrich). Sections were counterstained with DAPI (Sigma-Aldrich) to visualize nuclei and analyzed with an OlympusFluoview 1000 confocal microscope (Olympus, Tokyo,Japan) under an oil UPLFL 60� objective (numerical aperture,1.25; Olympus) using FV10-ASW software version 1.3c(Olympus). The numbers of p-T388þ cells and dual-stainedp-T388þa-SMAþ cells were counted in blinded slides.
Immunoprecipitation and Western Blot Analysis
Cultured cells were lysed in 0.4 mL radioimmunoprecipitationassay lysis buffer and sonicated. Cell lysates (1 mg) wereimmunoprecipitated with mouse antibodies to Smad3, Smad2,or Flag (Sigma-Aldrich) and protein A/G agarose beads (SantaCruz Biotechnology, Dallas, TX). Proteins were separated by10%SDS-PAGE and transferred to a polyvinylidene difluoridemembrane. After blocking for 30 minutes at 4�C in 5% bovineserum albumen in PBS with 0.1% Tween 20 (Sigma-Aldrich),the membrane was incubated overnight with rabbit antiep-Smad3 T388 or commercial rabbit antibodies to Smad2/3,p-Smad3 C-terminus, Smad4, or CDK8 (Cell Signaling
945

Qu et al
Technology, San Francisco, CA). Antibody binding wasdetected using an electrochemiluminescence kit (AmershamPharmacia Biotech, Arlington, IL) and the Kodak 4000MMimage station (Waltham, MA). Band density was quantifiedusing ImageJ software version 1.46r (NIH, Bethesda, MD).Rabbit anti-collagen I (Biorbyt Ltd, Cambridge, UK), mouseantiea-SMA antibody (Sigma-Aldrich), and rabbit anti-fibronectin (Santa Cruz Biotechnology) were also used inWestern blot analysis. a-Tubulin, glyceraldehyde-3-phosphatedehydrogenase, and Smad3 were used as internal controls.
Plasmid Transfection and Real-Time RT-PCR
Primary kidney fibroblasts, isolated from newborn Smad3�/�
mice, were transfected using Lipofectamine 2000 (Invi-trogen) with the following plasmids expressing Flag-taggedWT or mutated Smad3 plus enhanced green fluorescenceprotein (EGFP): pMSCV-Flag-Smad3 WT-IRES-EGFP,pMSCV-Flag-Smad3 T388/V-IRES-EGFP, and pMSCV-Flag-Smad3 3S/A-IRES-EGFP. Transfected cells were iso-lated 24 hours later by fluorescence-activated cell sorting(FACS). The EGFP-positive cells were then cultured with orwithout TGF-b1 for 24 hours. Cells were fixed for confocalmicroscopy or lysed for Western blot analysis, as previouslydescribed. In addition, cells were lysed for RNA extractionand real-time RT-PCR analysis of a-SMA, collagen I, andfibronectin mRNA levels, as previously described.25
Luciferase Reporter Assay
HEK293T cells were transfected with various Flag-Smad3-IRES-EGFP constructs. Twenty-four hours later, the trans-fected cells were purified by FACS and then EGFP-positivecells were transfected with a luciferase reporter constructunder control of the �3200 to 54 fragment of the a2(I)collagen promoter and the control PRL-TK plasmid usingLipofectamine 2000 and a maximum of 1.0 mg total DNAper well, as described previously.26 Twenty-four hours aftertransfection, the transfected cells were stimulated with orwithout TGF-b1 for 18 hours and then examined using theDual-Luciferase assay kit (Promega, Madison, WI).
Statistical Analysis
Data are shown as means � SD, with statistical analysesperformed using one-way analysis of variance, with posthoc analysis with Tukey’s multiple comparison test usingGraphPad Prism version 6.0 (Graph-Pad Software, SanDiego, CA).
Results
Characterization of p-T388 Smad3 Antibody
Immunoprecipitation/Western blot (IP/WB) analysis showedthat the rabbit antibody to p-T388 bound to Smad3, but notSmad2, in TGF-b1 stimulated NRK49F renal fibroblasts
946
(Figure 1A). In addition, transfection of Smad3�/� renal fi-broblasts with various Smad3 constructs demonstrated thatmutation of T388 to valine abolished detection of Smad3 inTGF-b1e stimulated cells, whereas mutation of the SSXSmotif in the C-terminal domain had no effect on detection ofTGF-b1einduced T388 phosphorylation (Figure 1B). Con-trol NRK49F renal fibroblasts and NRK52E tubular epithelialcells were negative for anti-pT388 staining, but distinct nu-clear staining was evident 15 minutes after TGF-b1 stimula-tion in both cell types (Figure 1C).
Phosphorylation of Smad3 T388 in Kidney Fibrosis
Only occasional cells exhibited p-T388 immunostaining innormal mouse kidney. In contrast, numerous cells exhibitednuclear p-T388 staining in the mouse UUO model of renalfibrosis, being most prominent in tubular epithelial cells and a-SMAþmyofibroblasts (Figure 2, A, B, and I). This stainingwasabolished using the immunizing p-T388 peptide (Figure 2C)and was absent in the obstructed kidney of Smad3�/� mice(Figure 2D). Compared with saline controls, Nos3�/� micewith streptozotocin-induced diabetic nephropathy showed asignificant increase in the number of p-T388þ cells in bothsclerotic glomeruli and areas of interstitial fibrosis, including p-T388 staining ina-SMAþmyofibroblasts (Figure 2,E, F, and J).Few p-T388þ cells were seen in normal human kidney from theuninvolved pole of carcinoma nephrectomy samples. However,numerous tubular cells and a-SMAþ myofibroblasts exhibitedp-T388 staining in biopsy specimens of focal and segmentalglomerulosclerosis (Figure 2, G and H).
Phosphorylation of Smad3 T388 Is Not Required forNuclear Translocation
The kinetics of TGF-b1einduced phosphorylation of Smad3T388 were examined in NRK49F kidney fibroblasts. Tran-sient induction of Smad3 T388 phosphorylation was evidentover 5 to 15 minutes after TGF-b1 stimulation, and a secondphase of T388 phosphorylation was seen over 1 to 5 hours(Figure 1D). Immunostaining identified that Smad3, phos-phorylated at T388, translocates to the cell nucleus within 15minutes of TGF-b1 stimulation (Figure 1C). To determinethe functional role of T388 phosphorylation in this response,Smad3�/� renal fibroblasts were transfected with variousFlag-tagged Smad3 constructs. WT Smad3 showed nucleartranslocation 15 minutes after TGF-b1 stimulation, whichhad gone by 2 hours (Figure 1E). However, blocking phos-phorylation at T388 (T388/V) or the C-terminal (3S/A) didnot affect Smad3 nuclear translocation (Figure 1, F and G).
Phosphorylation of Smad3 T388 Depends on TGF-bType I Receptor and Facilitates Interaction with Smad4and Other Kinases
Blockade of the kinase activity of the TGF-b type I receptorinhibited TGF-b1einduced phosphorylation of Smad3
ajp.amjpathol.org - The American Journal of Pathology
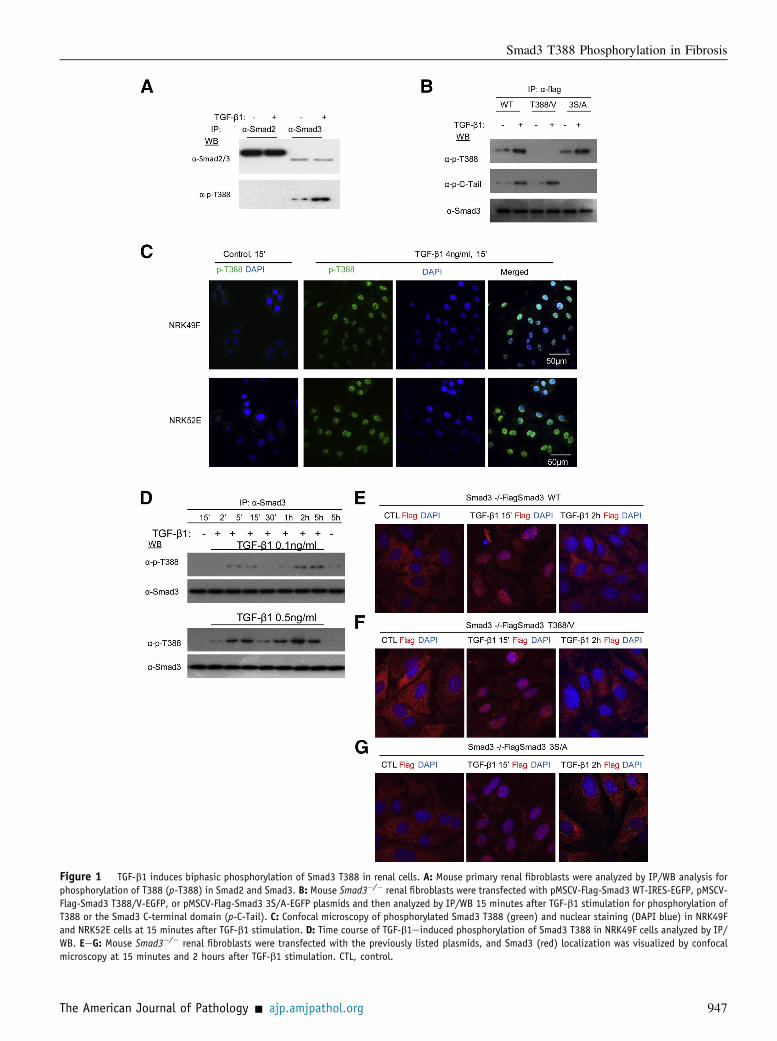
Figure 1 TGF-b1 induces biphasic phosphorylation of Smad3 T388 in renal cells. A: Mouse primary renal fibroblasts were analyzed by IP/WB analysis forphosphorylation of T388 (p-T388) in Smad2 and Smad3. B: Mouse Smad3�/� renal fibroblasts were transfected with pMSCV-Flag-Smad3 WT-IRES-EGFP, pMSCV-Flag-Smad3 T388/V-EGFP, or pMSCV-Flag-Smad3 3S/A-EGFP plasmids and then analyzed by IP/WB 15 minutes after TGF-b1 stimulation for phosphorylation ofT388 or the Smad3 C-terminal domain (p-C-Tail). C: Confocal microscopy of phosphorylated Smad3 T388 (green) and nuclear staining (DAPI blue) in NRK49Fand NRK52E cells at 15 minutes after TGF-b1 stimulation. D: Time course of TGF-b1einduced phosphorylation of Smad3 T388 in NRK49F cells analyzed by IP/WB. EeG: Mouse Smad3�/� renal fibroblasts were transfected with the previously listed plasmids, and Smad3 (red) localization was visualized by confocalmicroscopy at 15 minutes and 2 hours after TGF-b1 stimulation. CTL, control.
Smad3 T388 Phosphorylation in Fibrosis
The American Journal of Pathology - ajp.amjpathol.org 947
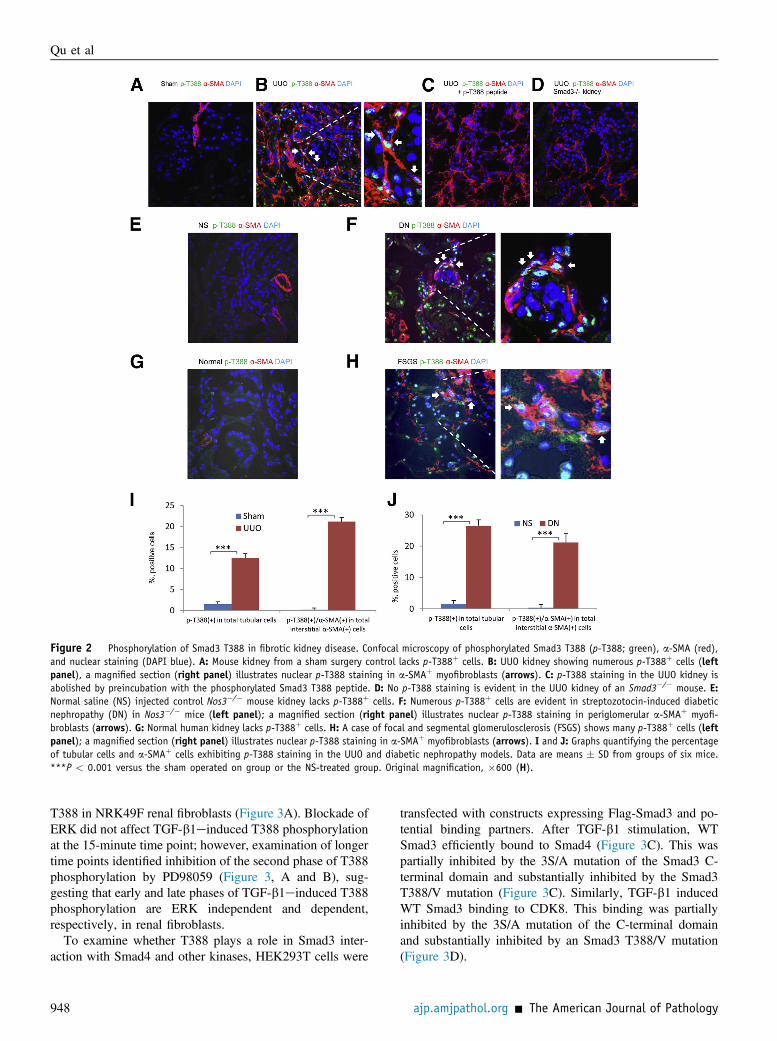
Figure 2 Phosphorylation of Smad3 T388 in fibrotic kidney disease. Confocal microscopy of phosphorylated Smad3 T388 (p-T388; green), a-SMA (red),and nuclear staining (DAPI blue). A: Mouse kidney from a sham surgery control lacks p-T388þ cells. B: UUO kidney showing numerous p-T388þ cells (leftpanel), a magnified section (right panel) illustrates nuclear p-T388 staining in a-SMAþ myofibroblasts (arrows). C: p-T388 staining in the UUO kidney isabolished by preincubation with the phosphorylated Smad3 T388 peptide. D: No p-T388 staining is evident in the UUO kidney of an Smad3�/� mouse. E:Normal saline (NS) injected control Nos3�/� mouse kidney lacks p-T388þ cells. F: Numerous p-T388þ cells are evident in streptozotocin-induced diabeticnephropathy (DN) in Nos3�/� mice (left panel); a magnified section (right panel) illustrates nuclear p-T388 staining in periglomerular a-SMAþ myofi-broblasts (arrows). G: Normal human kidney lacks p-T388þ cells. H: A case of focal and segmental glomerulosclerosis (FSGS) shows many p-T388þ cells (leftpanel); a magnified section (right panel) illustrates nuclear p-T388 staining in a-SMAþ myofibroblasts (arrows). I and J: Graphs quantifying the percentageof tubular cells and a-SMAþ cells exhibiting p-T388 staining in the UUO and diabetic nephropathy models. Data are means � SD from groups of six mice.***P < 0.001 versus the sham operated on group or the NS-treated group. Original magnification, �600 (H).
Qu et al
T388 in NRK49F renal fibroblasts (Figure 3A). Blockade ofERK did not affect TGF-b1einduced T388 phosphorylationat the 15-minute time point; however, examination of longertime points identified inhibition of the second phase of T388phosphorylation by PD98059 (Figure 3, A and B), sug-gesting that early and late phases of TGF-b1einduced T388phosphorylation are ERK independent and dependent,respectively, in renal fibroblasts.
To examine whether T388 plays a role in Smad3 inter-action with Smad4 and other kinases, HEK293T cells were
948
transfected with constructs expressing Flag-Smad3 and po-tential binding partners. After TGF-b1 stimulation, WTSmad3 efficiently bound to Smad4 (Figure 3C). This waspartially inhibited by the 3S/A mutation of the Smad3 C-terminal domain and substantially inhibited by the Smad3T388/V mutation (Figure 3C). Similarly, TGF-b1 inducedWT Smad3 binding to CDK8. This binding was partiallyinhibited by the 3S/A mutation of the C-terminal domainand substantially inhibited by an Smad3 T388/V mutation(Figure 3D).
ajp.amjpathol.org - The American Journal of Pathology

Figure 3 Analysis of Smad3 T388 phosphorylation and the role of T388 in Smad3 complex formation with Smad4 and CDK8. A: NRK49F renal fibroblastswere pretreated with a neutralizing antieTGF-b1 antibody, control antibody, TGF-b receptor 1 inhibitor (TGF-b1 RI), dimethyl sulfoxide (DMSO) vehicle, or theERK1/2 inhibitor, PD98059, for 30 minutes before 15-minute stimulation with TGF-b1. Cells were analyzed by IP/WB analysis for phosphorylation of T388(p-T388) or the Smad3 C-terminal domain (p-C-Tail; A). Graphs provide quantification of the results. B: Time course study of ERK inhibition in TGF-b1einducedSmad3 T388 phosphorylation in NRK49F renal fibroblasts. HEK293T cells were transfected with plasmids expressing WT Smad3 or Smad3 with point mutations(T388/V or 3S/A) and then harvested 15 minutes after TGF-b1 stimulation. C: IP/WB identifies complex formation between Smad3 and Smad4; graph shows thequantification. D: HEK293T cells transfected with Smad3 plasmids were analyzed by IP/WB for TGF-b1einduced complex formation between Smad3 and CDK8;graph shows the quantification. Data are means � SD. ***P < 0.001 versus control; yyyP < 0.001 versus FlagSmad3 WT; zzzP < 0.001 versus WT or FlagSmad3T388/V (T388/V).
Smad3 T388 Phosphorylation in Fibrosis
Phosphorylation of Smad3 T388 Is Necessary for theTGF-b1eInduced Fibrotic Response in RenalFibroblasts
The functional role of Smad3 T388 phosphorylation in theTGF-b1einduced profibrotic response was examined in twodifferent settings. First, HEK293T cells were transfected withFlag-Smad3-IRES-EGFP constructs. The transfected cellswere purified using flow cytometry, and then EGFP-positivecells were analyzed for collagen I gene transcription activityusing a collagen I promoter luciferase reporter assay. WT-Smad3 facilitated robust TGF-b1einduced collagen I genetranscription (Figure 4A). This was partially attenuated by the3S/A mutation of the Smad3 C-terminal domain and almostabolished by the T388/V mutation (Figure 4A). Second, theSmad3�/� kidney fibroblasts of mice were transfected withvarious Flag-Smad3-IRES-EGFP constructs. Transfected
The American Journal of Pathology - ajp.amjpathol.org
cells were purified 24 hours later and then stimulated with orwithout TGF-b1. Real-time RT-PCR and Western blot ana-lyses demonstrated that TGF-b1 induced robust mRNA andprotein expression of a-SMA, collagen I, and fibronectin incells transfected with WT Smad3 (Figure 4, BeD). The 3S/Amutation of the C-terminal domain caused a partial reductionin the TGF-b1einduced profibrotic response, whereas theSmad3 T388 mutation substantially reduced this response(Figure 4, BeD).
Discussion
To our knowledge, this is the first study to identify Smad3T388 phosphorylation in human or experimental tissuefibrosis. Smad3 T388 phosphorylation within a-SMAþ
myofibroblasts, the main cell type producing collagen I in
949
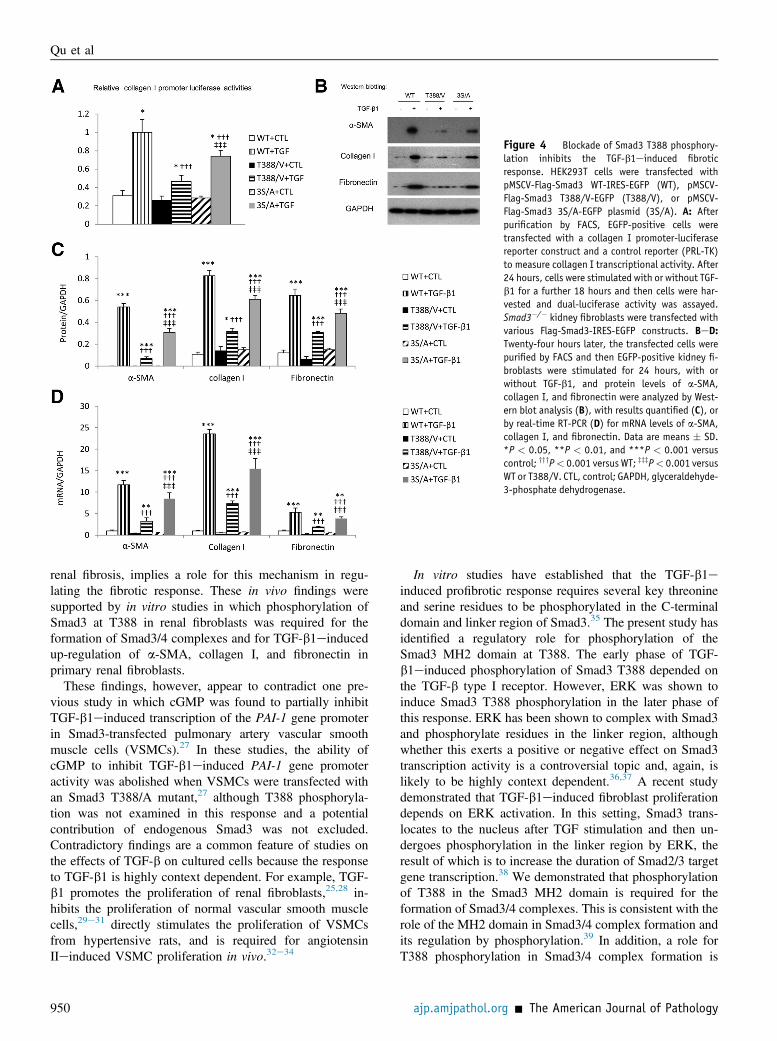
Figure 4 Blockade of Smad3 T388 phosphory-lation inhibits the TGF-b1einduced fibroticresponse. HEK293T cells were transfected withpMSCV-Flag-Smad3 WT-IRES-EGFP (WT), pMSCV-Flag-Smad3 T388/V-EGFP (T388/V), or pMSCV-Flag-Smad3 3S/A-EGFP plasmid (3S/A). A: Afterpurification by FACS, EGFP-positive cells weretransfected with a collagen I promoter-luciferasereporter construct and a control reporter (PRL-TK)to measure collagen I transcriptional activity. After24 hours, cells were stimulated with or without TGF-b1 for a further 18 hours and then cells were har-vested and dual-luciferase activity was assayed.Smad3�/� kidney fibroblasts were transfected withvarious Flag-Smad3-IRES-EGFP constructs. BeD:Twenty-four hours later, the transfected cells werepurified by FACS and then EGFP-positive kidney fi-broblasts were stimulated for 24 hours, with orwithout TGF-b1, and protein levels of a-SMA,collagen I, and fibronectin were analyzed by West-ern blot analysis (B), with results quantified (C), orby real-time RT-PCR (D) for mRNA levels of a-SMA,collagen I, and fibronectin. Data are means � SD.*P < 0.05, **P < 0.01, and ***P < 0.001 versuscontrol; yyyP< 0.001 versus WT; zzzP< 0.001 versusWT or T388/V. CTL, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Qu et al
renal fibrosis, implies a role for this mechanism in regu-lating the fibrotic response. These in vivo findings weresupported by in vitro studies in which phosphorylation ofSmad3 at T388 in renal fibroblasts was required for theformation of Smad3/4 complexes and for TGF-b1einducedup-regulation of a-SMA, collagen I, and fibronectin inprimary renal fibroblasts.
These findings, however, appear to contradict one pre-vious study in which cGMP was found to partially inhibitTGF-b1einduced transcription of the PAI-1 gene promoterin Smad3-transfected pulmonary artery vascular smoothmuscle cells (VSMCs).27 In these studies, the ability ofcGMP to inhibit TGF-b1einduced PAI-1 gene promoteractivity was abolished when VSMCs were transfected withan Smad3 T388/A mutant,27 although T388 phosphoryla-tion was not examined in this response and a potentialcontribution of endogenous Smad3 was not excluded.Contradictory findings are a common feature of studies onthe effects of TGF-b on cultured cells because the responseto TGF-b1 is highly context dependent. For example, TGF-b1 promotes the proliferation of renal fibroblasts,25,28 in-hibits the proliferation of normal vascular smooth musclecells,29e31 directly stimulates the proliferation of VSMCsfrom hypertensive rats, and is required for angiotensinIIeinduced VSMC proliferation in vivo.32e34
950
In vitro studies have established that the TGF-b1einduced profibrotic response requires several key threonineand serine residues to be phosphorylated in the C-terminaldomain and linker region of Smad3.35 The present study hasidentified a regulatory role for phosphorylation of theSmad3 MH2 domain at T388. The early phase of TGF-b1einduced phosphorylation of Smad3 T388 depended onthe TGF-b type I receptor. However, ERK was shown toinduce Smad3 T388 phosphorylation in the later phase ofthis response. ERK has been shown to complex with Smad3and phosphorylate residues in the linker region, althoughwhether this exerts a positive or negative effect on Smad3transcription activity is a controversial topic and, again, islikely to be highly context dependent.36,37 A recent studydemonstrated that TGF-b1einduced fibroblast proliferationdepends on ERK activation. In this setting, Smad3 trans-locates to the nucleus after TGF stimulation and then un-dergoes phosphorylation in the linker region by ERK, theresult of which is to increase the duration of Smad2/3 targetgene transcription.38 We demonstrated that phosphorylationof T388 in the Smad3 MH2 domain is required for theformation of Smad3/4 complexes. This is consistent with therole of the MH2 domain in Smad3/4 complex formation andits regulation by phosphorylation.39 In addition, a role forT388 phosphorylation in Smad3/4 complex formation is
ajp.amjpathol.org - The American Journal of Pathology

Smad3 T388 Phosphorylation in Fibrosis
consistent with the substantial reduction in Smad3 tran-scription activity seen on mutation of this amino acid intransfected renal fibroblasts. Furthermore, nuclear CDK8can phosphorylate the linker region of Smad3 complexesand, thus, promote Smad transcriptional activity and theturnover of activated Smad proteins.22 Thus, blockade ofSmad3 T388 phosphorylation decreases the interaction be-tween Smad3 and CDK8, which, via decreased linkerphosphorylation, will result in decreased Smad3 targetedgene transcription.
It is generally assumed that TGF-b type I receptoremediated phosphorylation of the SSXS motif in the C-terminal domain of Smad2 and Smad3 is required for theirassociation with Smad4 and subsequent nuclear trans-location.7 However, Smad2/3 nuclear translocation is acomplex process involving several mechanisms. Indeed, it isargued that the TGF-b signal does not influence the Smadnuclear import and export process itself, but rather modifiesthe affinity of these Smads with their binding partners,which retains them in either the nucleus or the cytoplasm. Itis the change in affinity for retention factors on both sides ofthe nuclear envelope that leads to enhanced nuclear locali-zation of Smads 2, 3, and 4 in response to TGF-b.40 Forexample, mutation of the SSMS motif in the C-terminaldomain was unable to prevent nuclear translocation ofSmad2,41 indicating that Smad2 has intrinsic nuclear importactivity without TGF-b receptoremediated phosphoryla-tion. In addition, TGF-b receptoremediated phosphoryla-tion of Smad2 significantly decreases its affinity for Smadanchor for receptor activation (SARA) and thereby pro-motes nuclear accumulation of Smad2.42 Also, expressionof the MH2 domain of Smad3 is sufficient to enable nucleartranslocation, thereby eliminating the requirement forC-terminal domain phosphorylation by the TGF-b type Ireceptor.40 This is consistent with our results showing noeffect of mutation of the SSXS motif in the C-terminaldomain on TGF-beinduced Smad3 nuclear translocation.Indeed, a KKLKK motif in the MH1 domain of Smad3 canbind directly to importin-b and trigger nuclear trans-location.43 Also, on TGF-b1 stimulation, Smad3 losesinteraction with SARA,44 suggesting that TGF-b1 inducesSmad3 nuclear translocation partly through dissociation ofSmad3 from cytoplasmic retention factors, such as SARA.Furthermore, a recent study has implicated an SPS domainwithin the MH2 domain as being critical for TGF-beinduced nuclear translocation of Smad3.45 Thus, phos-phorylation of the SPS domain and dissociation fromfactors, such as SARA, may be sufficient to allow TGF-beinduced Smad3 nuclear translocation in renal fibroblasts inthe absence of Smad3 C-terminal or T388 phosphorylation.The exact mechanism by which phosphorylation of T388regulates Smad3 function requires further investigation.However, one possibility is that T388 phosphorylation fa-cilitates the formation of Smad3/Smad4 heterotrimers. Thisis based on in vitro immunoprecipitation experimentsshowing reduced Smad4 binding to mutated Smad3 T388V.
The American Journal of Pathology - ajp.amjpathol.org
In addition, crystallographic studies of the MH2 domain inSmad3/Smad4 heterotrimers identified the T388 residue atthe Smad3/Smad4 interface.46 Alternatively, phosphoryla-tion of Smad3 T388 may promote either the binding oftranscriptional cofactors or the disassociation of Smad3repressors to maximize Smad3 transcriptional activity.
In conclusion, we have identified that Smad3 T388phosphorylation occurs in a-SMAþ myofibroblasts duringthe pathogenesis of human and experimental renal fibrosis.Smad3 T388 phosphorylation is required for the formationof Smad3/4 complexes and for CDK8 binding to Smad3.Although Smad3 T388 phosphorylation did not affect nu-clear translocation, T388 phosphorylation was shown tobe a major mechanism promoting TGF-b1einduced up-regulation of a-SMA, collagen I, and fibronectin in pri-mary renal fibroblasts. Studies of transgenic mice withmutated Smad3 are needed to define the regulatory role ofSmad3 phosphorylation in experimental models of tissuefibrosis and provide target validation for therapeutic strate-gies to modify Smad3 phosphorylation.
Acknowledgment
Confocal imaging was performed at the Monash MicroImaging Facility at Monash University (Clayton, Australia).
References
1. Massagué J: TGFb signalling in context. Nat Rev Mol Cell Biol 2012,13:616e630
2. Bottinger EP: TGF-beta in renal injury and disease. Semin Nephrol2007, 27:309e320
3. Rosenkranz S: TGF-b1 and angiotensin networking in cardiacremodeling. Cardiovasc Res 2004, 63:423e432
4. Border WA, Noble NA: TGF-beta in kidney fibrosis: a target for genetherapy. Kidney Int 1997, 51:1388e1396
5. Liu Y: Renal fibrosis: new insights into the pathogenesis and thera-peutics. Kidney Int 2006, 69:213e217
6. Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti EA:Suppression of experimental glomerulonephritis by antiserum againsttransforming growth factor beta 1. Nature 1990, 346:371e374
7. Kretzschmar M, Massague J: Smads: mediators and regulators ofTGF-b signaling. Curr Opin Genet Dev 1998, 8:103e111
8. Zimmerman CM, Padgett RW: Transforming growth factor Bsignaling mediators and regulators. Gene 2000, 249:17e30
9. Meng XM, Huang XR, Chung AC, Qin W, Shao X, Igarashi P, Ju W,Bottinger EP, Lan HY: Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol 2010, 21:1477e1487
10. Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A: Targeteddisruption of TGF-beta1/Smad3 signaling protects against renal tubu-lointerstitial fibrosis induced by unilateral ureteral obstruction. J ClinInvest 2003, 112:1486e1494
11. Zhao J, Shi W, Wang YL, Chen H, Bringas P Jr., Datto MB,Frederick JP, Wang XF, Warburton D: Smad3 deficiency attenuatesbleomycin-induced pulmonary fibrosis in mice. Am J Physiol LungCell Mol Physiol 2002, 282:L585eL593
12. Huang XR, Chung AC, Yang F, Yue W, Deng C, Lau CP, Tse HF,Lan HY: Smad3 mediates cardiac inflammation and fibrosis inangiotensin II-induced hypertensive cardiac remodeling. Hypertension2010, 55:1165e1171
951

Qu et al
13. Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL,Weinstein M, Deng C, Kucherlapati R, Bottinger EP, Roberts AB:Functional characterization of transforming growth factor betasignaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem2001, 276:19945e19953
14. ten Dijke P, Miyazono K, Heldin CH: Signaling inputs converge onnuclear effectors in TGF-beta signaling. Trends Biochem Sci 2000, 25:64e70
15. Velden JL, Alcorn JF, Guala AS, Badura EC, Janssen-Heininger YM:c-Jun N-terminal kinase 1 promotes transforming growth factor-beta1-induced epithelial-to-mesenchymal transition via control oflinker phosphorylation and transcriptional activity of Smad3. Am JRespir Cell Mol Biol 2011, 44:571e581
16. Liu Q, Zhang Y, Mao H, Chen W, Luo N, Zhou Q, Chen W, Yu X: Acrosstalk between the Smad and JNK signaling in the TGF-beta-induced epithelial-mesenchymal transition in rat peritoneal mesothe-lial cells. PLoS One 2012, 7:e32009
17. Lan HY: Diverse roles of TGF-beta/Smads in renal fibrosis andinflammation. Int J Biol Sci 2011, 7:1056e1067
18. Wang G, Matsuura I, He D, Liu F: Transforming growth factor-{beta}-inducible phosphorylation of Smad3. J Biol Chem 2009, 284:9663e9673
19. Kretzschmar M, Doody J, Massague J: Opposing BMP and EGFsignalling pathways converge on the TGF-beta family mediatorSmad1. Nature 1997, 389:618e622
20. Kretzschmar M, Doody J, Timokhina I, Massague J: A mechanism ofrepression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev1999, 13:804e816
21. Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F: Cyclin-dependent kinases regulate the antiproliferative function of Smads.Nature 2004, 430:226e231
22. Alarcón C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, Barlas A,Miller AN, Manova-Todorova K, Macias MJ, Sapkota G, Pan D,Massagué J: Nuclear CDKs drive Smad transcriptional activation andturnover in BMP and TGF-beta pathways. Cell 2009, 139:757e769
23. Li P, Wang D, Lucas J, Oparil S, Xing D, Cao X, Novak L,Renfrow MB, Chen YF: Atrial natriuretic peptide inhibits transforminggrowth factor beta-induced Smad signaling and myofibroblast trans-formation in mouse cardiac fibroblasts. Circ Res 2008, 102:185e192
24. Li J, Qu X, Ricardo SD, Bertram JF, Nikolic-Paterson DJ: Resveratrolinhibits renal fibrosis in the obstructed kidney: potential role indeacetylation of Smad3. Am J Pathol 2010, 177:1065e1071
25. Qu X, Zhang X, Yao J, Song J, Nikolic-Paterson DJ, Li J: ResolvinsE1 and D1 inhibit interstitial fibrosis in the obstructed kidney via in-hibition of local fibroblast proliferation. J Pathol 2012, 228:506e519
26. McGaha TL, Kodera T, Spiera H, Stan AC, Pines M, Bona CA:Halofuginone inhibition of COL1A2 promoter activity via a c-Jun-dependent mechanism. Arthritis Rheum 2002, 46:2748e2761
27. Gong K, Xing D, Li P, Hilgers RH, Hage FG, Oparil S, Chen YF:cGMP inhibits TGF-beta signaling by sequestering Smad3 with cyto-solic beta2-tubulin in pulmonary artery smooth muscle cells. MolEndocrinol 2011, 25:1794e1803
28. Johnson DW, Saunders HJ, Baxter RC, Field MJ, Pollock CA: Para-crine stimulation of human renal fibroblasts by proximal tubule cells.Kidney Int 1998, 54:747e757
952
29. Owens GK, Geisterfer AA, Yang YW, Komoriya A: Transforminggrowth factor-beta-induced growth inhibition and cellular hypertrophyin cultured vascular smoothmuscle cells. J Cell Biol 1988, 107:771e780
30. Kirschenlohr HL, Metcalfe JC, Weissberg PL, Grainger DJ: Prolifer-ation of human aortic vascular smooth muscle cells in culture ismodulated by active TGF beta. Cardiovasc Res 1995, 29:848e855
31. Seay U, Sedding D, Krick S, Hecker M, Seeger W, Eickelberg O:Transforming growth factor-beta-dependent growth inhibition in pri-mary vascular smooth muscle cells is p38-dependent. J Pharmacol ExpTher 2005, 315:1005e1012
32. Saltis J, Agrotis A, Kanellakis P, Bobik A: Developmentally regulatedtransforming growth factor-beta 1 action on vascular smooth musclegrowth in the SHR. Clin Exp Pharmacol Physiol 1994, 21:149e152
33. Sato M, Ohsaki Y, Tobise K: Transforming growth factor-beta 1proliferated vascular smooth muscle cells from spontaneously hyper-tensive rats. Am J Hypertens 1995, 8:160e166
34. Yurovsky VV, Gerzanich V, Ivanova S, Dong Y, Tsymbalyuk O,Simard JM: Autocrine TGF-beta1 mediates angiotensin II-inducedproliferative response of cerebral vessels in vivo. Am J Hypertens2007, 20:950e956
35. Matsuzaki K: Smad phosphoisoform signals in acute and chronic liverinjury: similarities and differences between epithelial and mesen-chymal cells. Cell Tissue Res 2012, 347:225e243
36. Kretzschmar M, Doody J, Timokhina I, Massagué J: A mechanism ofrepression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev1999, 13:804e816
37. Hayashida T, Decaestecker M, Schnaper HW: Cross-talk betweenERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. FASEB J 2003, 17:1576e1578
38. Hough C, Radu M, Doré JJ: TGF-beta induced Erk phosphorylation ofSmad linker region regulates Smad signaling. PLoS One 2012, 7:e42513
39. ten Dijke P, Hill CS: New insights into TGF-beta-Smad signalling.Trends Biochem Sci 2004, 29:265e273
40. Xu L, Alarcon C, Col S, Massagué J: Distinct domain utilization bySmad3 and Smad4 for nucleoporin interaction and nuclear import.J Biol Chem 2003, 278:42569e42577
41. Xu L, Chen YG, Massagué J: The nuclear import function of Smad2 ismasked by SARA and unmasked by TGFbeta-dependent phosphory-lation. Nat Cell Biol 2000, 2:559e562
42. Xu L, Kang Y, Cöl S, Massagué J: Smad2 nucleocytoplasmic shuttlingby nucleoporins CAN/Nup214 and Nup153 feeds TGFbeta signalingcomplexes in the cytoplasm and nucleus. Mol Cell 2002, 10:271e282
43. Xiao Z, Liu X, Lodish HF: Importin beta mediates nuclear trans-location of Smad 3. J Biol Chem 2000, 275:23425e23428
44. Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL: SARA, aFYVE domain protein that recruits Smad2 to the TGFbeta receptor.Cell 1998, 95:779e791
45. Chuderland D, Konson A, Seger R: Identification and characterizationof a general nuclear translocation signal in signaling proteins. Mol Cell2008, 31:850e861
46. Chacko BM, Qin BY, Tiwari A, Shi G, Lam S, Hayward LJ, DeCaestecker M, Lin K: Structural basis of heteromeric Smad proteinassembly in TGF-b signaling. Mol Cell 2004, 15:813e823
ajp.amjpathol.org - The American Journal of Pathology




















