
Hepatocyte growth factor ameliorates progression of interstitial fibrosis in rats with established...
11
Kidney International, Vol. 65 (2004), pp. 409–419 Hepatocyte growth factor ameliorates progression of interstitial fibrosis in rats with established renal injury LANCE D. DWORKIN,RUJUN GONG,EVELYN T OLBERT,J ASON CENTRACCHIO,NAHIRO Y ANO, ABDUL R. ZANABLI,ALFREDO ESPARZA, and ABDALLA RIFAI Brown Medical School, Providence, Rhode Island; and Rhode Island Hospital, Providence, Rhode Island Hepatocyte growth factor ameliorates progression of interstitial fibrosis in rats with established renal injury. Background. Hepatocyte growth factor (HGF) has been re- ported to prevent injury in several models of renal disease; how- ever, whether HGF can also retard progression of established renal disease is not known. Methods. The aim of the present study was to examine the effects of HGF on progression of chronic renal disease in rats with remnant kidneys and established injury. Studies were per- formed in rats that underwent subtotal nephrectomy, were ob- served for two weeks without therapy, and then randomized to receive HGF or vehicle by continuous infusion for an additional two weeks. Results. HGF administration was associated with a reduc- tion in morphologic evidence of interstitial, but not glomerular injury. The beneficial effects of HGF were not associated with reductions in the expression of transforming growth factor-beta (TGF-b ), or in the extent epithelial cell apoptosis or transdif- ferentiation. Rather, HGF appeared to induce fibrinolytic path- ways by increasing expression of metalloproteinase-9 (MMP-9) and decreasing levels of plasminogen activator inhibitor-1 (PAI-1) and tissue inhibitor of metalloproteinase-1 (TIMP-2). HGF administration was also associated with an apparent in- crease in renal endothelin production and a significant reduc- tion in glomerular capillary pressure. Conclusion. These findings suggest that HGF can retard pro- gression of chronic renal disease even after injury is already established, primarily by promoting matrix degradation. Following subtotal nephrectomy, rats develop a syn- drome of hypertension, proteinuria, and renal dysfunc- tion that eventually leads to progressive glomerular sclerosis, interstitial fibrosis, and end-stage renal failure [1]. A number of factors have been proposed to ex- plain progression in this model, including alterations in glomerular hemodynamics [2], and/or in the production Key words: glomerular hemodynamics, metalloproteinase, PAI-1, TIMP-2, remnant kidney. Received for publication January 3, 2003 and in revised form June 9, 2003, and July 23, 2003 Accepted for publication September 22, 2003 C 2004 by the International Society of Nephrology of various growth factors and cytokines that regulate matrix production and degradation and, thereby, renal scarring [3, 4]. However, exactly which factors are truly important modulators of chronic renal disease progres- sion are still uncertain. Originally characterized as a potent mitogen for liver cells, hepatocyte growth factor (HGF) has multiple ef- fects on renal tubular epithelial cells, including stimula- tion of cell proliferation, motility, and differentiation [5]. We reported that the HGF/c-met system is also activated in remnant kidneys and that blockade of endogenous HGF by administration of a specific antibody markedly worsens kidney function and morphologic evidence of renal injury in this model [6]. In that and other studies, HGF was shown to have antifibrotic effects, including blockade of epithelial to fibroblast transdifferentiation and stimulation matrix degradative pathways [7–9]. Of note, although HGF has now been reported to amelio- rate injury in several models of renal disease [6–11], in each instance, HGF was initiated either before or coin- cident with the initial onset of injury. As a result, these studies demonstrate that HGF can partially prevent re- nal damage, but not that HGF can retard progression of established renal disease. The purpose of this study was to examine the effects of HGF on progression of chronic renal disease in rats with remnant kidneys and established injury. We found that administration of ex- ogenous HGF not only reduced morphologic evidence of interstitial injury but also had unsuspected effects on glomerular hemodynamics. We also examined the effects of HGF on a number of other parameters associated with progression of chronic renal disease, including glomeru- lar and tubular cell apoptosis, as well as the expression of various cytokines, growth factors, and matrix-regulating molecules. METHODS Design of the animal studies All studies were approved by the Rhode Island Hos- pital Institutional Animal Care and Use Committee and 409
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Hepatocyte growth factor ameliorates progression of interstitial fibrosis in rats with established...

Kidney International, Vol. 65 (2004), pp. 409–419
Hepatocyte growth factor ameliorates progression of interstitialfibrosis in rats with established renal injury
LANCE D. DWORKIN, RUJUN GONG, EVELYN TOLBERT, JASON CENTRACCHIO, NAHIRO YANO,ABDUL R. ZANABLI, ALFREDO ESPARZA, and ABDALLA RIFAI
Brown Medical School, Providence, Rhode Island; and Rhode Island Hospital, Providence, Rhode Island
Hepatocyte growth factor ameliorates progression of interstitialfibrosis in rats with established renal injury.
Background. Hepatocyte growth factor (HGF) has been re-ported to prevent injury in several models of renal disease; how-ever, whether HGF can also retard progression of establishedrenal disease is not known.
Methods. The aim of the present study was to examine theeffects of HGF on progression of chronic renal disease in ratswith remnant kidneys and established injury. Studies were per-formed in rats that underwent subtotal nephrectomy, were ob-served for two weeks without therapy, and then randomized toreceive HGF or vehicle by continuous infusion for an additionaltwo weeks.
Results. HGF administration was associated with a reduc-tion in morphologic evidence of interstitial, but not glomerularinjury. The beneficial effects of HGF were not associated withreductions in the expression of transforming growth factor-beta(TGF-b), or in the extent epithelial cell apoptosis or transdif-ferentiation. Rather, HGF appeared to induce fibrinolytic path-ways by increasing expression of metalloproteinase-9 (MMP-9)and decreasing levels of plasminogen activator inhibitor-1(PAI-1) and tissue inhibitor of metalloproteinase-1 (TIMP-2).HGF administration was also associated with an apparent in-crease in renal endothelin production and a significant reduc-tion in glomerular capillary pressure.
Conclusion. These findings suggest that HGF can retard pro-gression of chronic renal disease even after injury is alreadyestablished, primarily by promoting matrix degradation.
Following subtotal nephrectomy, rats develop a syn-drome of hypertension, proteinuria, and renal dysfunc-tion that eventually leads to progressive glomerularsclerosis, interstitial fibrosis, and end-stage renal failure[1]. A number of factors have been proposed to ex-plain progression in this model, including alterations inglomerular hemodynamics [2], and/or in the production
Key words: glomerular hemodynamics, metalloproteinase, PAI-1,TIMP-2, remnant kidney.
Received for publication January 3, 2003and in revised form June 9, 2003, and July 23, 2003Accepted for publication September 22, 2003
C© 2004 by the International Society of Nephrology
of various growth factors and cytokines that regulatematrix production and degradation and, thereby, renalscarring [3, 4]. However, exactly which factors are trulyimportant modulators of chronic renal disease progres-sion are still uncertain.
Originally characterized as a potent mitogen for livercells, hepatocyte growth factor (HGF) has multiple ef-fects on renal tubular epithelial cells, including stimula-tion of cell proliferation, motility, and differentiation [5].We reported that the HGF/c-met system is also activatedin remnant kidneys and that blockade of endogenousHGF by administration of a specific antibody markedlyworsens kidney function and morphologic evidence ofrenal injury in this model [6]. In that and other studies,HGF was shown to have antifibrotic effects, includingblockade of epithelial to fibroblast transdifferentiationand stimulation matrix degradative pathways [7–9]. Ofnote, although HGF has now been reported to amelio-rate injury in several models of renal disease [6–11], ineach instance, HGF was initiated either before or coin-cident with the initial onset of injury. As a result, thesestudies demonstrate that HGF can partially prevent re-nal damage, but not that HGF can retard progressionof established renal disease. The purpose of this studywas to examine the effects of HGF on progression ofchronic renal disease in rats with remnant kidneys andestablished injury. We found that administration of ex-ogenous HGF not only reduced morphologic evidenceof interstitial injury but also had unsuspected effects onglomerular hemodynamics. We also examined the effectsof HGF on a number of other parameters associated withprogression of chronic renal disease, including glomeru-lar and tubular cell apoptosis, as well as the expression ofvarious cytokines, growth factors, and matrix-regulatingmolecules.
METHODS
Design of the animal studies
All studies were approved by the Rhode Island Hos-pital Institutional Animal Care and Use Committee and
409

410 Dworkin et al: HGF and interstitial fibrosis
performed in accordance with published guidelines. Stud-ies were performed in two groups (N = 9 per group) ofmale Sprague-Dawley rats with initial weights of 180–240 g. To produce the remnant kidney model, rats un-derwent right nephrectomy and segmental infarction ofapproximately 2/3 of the left kidney by ligation of two tothree branches of the left main renal artery as previouslydescribed [6]. To select rats with near uniform reductionsin renal mass, serum creatinine was measured three daysafter ablation and only rats with a creatinine between 0.8and 1.2 mg/dL were included. Rats were then followed fortwo weeks without therapy, by which time hypertensionand proteinuria are present and morphologic evidence ofinjury is already apparent [6].
Beginning two weeks after ablation, HGF or vehicle(control) was continuously infused intravenously for twoweeks by implantation of an osmotic minipump. Ratswere anesthetized; a small incision was made over the leftjugular vein, which was then cannulated with polyethy-lene tubing (PE50) connected to an osmotic pump (Alzet,Cupertino, CA, USA) that delivered its contents at con-stant rate of 0.5 lL/hr. Pumps were filled with recombi-nant human HGF (Genentech, South San Francisco, CA,USA) in saline in a concentration calculated to result inan infusion rate of 20 lg HGF per day. Control rats re-ceived pumps containing saline alone.
Micropuncture studies
Four weeks after ablation and two weeks after admin-istration of HGF or vehicle, rats were anesthetized withthiobutabarbital (100 mg/kg, intraperitoneally), placedon a heated table, and prepared for micropuncture deter-mination of the pressures, flows, and resistances govern-ing glomerular ultrafiltration as described previously [12],with minor modifications. Briefly, polyethylene catheterswere inserted into the femoral artery, left and rightjugular veins, and the left ureter for infusion of solu-tions and collection of samples. A tracheostomy was per-formed. Arterial pressure was continuously measured bya pressure transducer connected to a computer runningWINDAS software (DATAQ Instruments, Inc., Akron,OH, USA). All rats received isoncotic rat plasma at arate of 0.1 mL/min for a total of 10 mL/kg body weight,followed by a sustained infusion of plasma at a rate of0.5 mL/hr to compensate for surgical losses and spec-imen collection. Rats also received a 0.5 mL bolusand a sustained infusion (0.5 mL/hr) of [3H] inulin insaline. Individual tubules were punctured using sharp-ened glass pipettes. Tubular fluid, urine, and blood sam-ples were collected for determination of single nephronand whole kidney GFR by the measurement of 3H in-ulin activity. Pressures were measured in cortical tubulesand efferent arterioles using the servo-null, micropipettetechnique. Glomerular capillary pressure was estimatedusing the stop-flow technique. Whole kidney renal plasma
flow (RPF) was measured after the conclusion of the mi-cropuncture studies using an ultrasonic flow probe (Tran-sonic Systems, Inc., Ithaca, NY, USA). In this technique,the left renal artery was dissected away from the per-inephritic fat and a flow probe placed around the renalartery near the hilum of the kidney. The area around theprobe was infused with a viscous gel. The probe is con-nected via a flow meter to a computer and renal bloodflow (RBF) recorded. Whole kidney filtration fraction(FF) was calculated using the standard formula. Initialglomerular capillary plasma flow rate was estimated us-ing the measured values for single nephron glomerularfiltration rate (SNGFR) and whole kidney FF.
Determination of plasma HGF levels by ELISA
The plasma concentration of HGF was measured usinga specific sandwich enzyme-linked immunosorbent assay(ELISA) kit (R&D Systems, Minneapolis, MN, USA) ac-cording to the manufacturer’s instructions.
Morphologic techniques
At the conclusion of the hemodynamic studies, kid-neys were perfused with iced saline and a portion of thekidney fixed by immersion in formalyn. Coronal slices ofeach kidney were stained with hematoxylin/eosin (HE).Microscopic abnormalities were assessed in a blindedfashion by a single observer. Glomerular and tubulointer-stitial changes were individually assessed. For glomerularchanges, all glomeruli in a section (∼60–80) were exam-ined. Each tuft received a score of 0–4 based on how manyquadrants were abnormal (0 = none; 4 = all). A compos-ite glomerular score was obtained for each rat by addingthe scores for each glomerulus and dividing the total bythe number of glomeruli that were examined. Tubuloint-erstitial damage was quantified in a similar fashion. Thirtyrandomly selected fields were examined and graded 0–4based on the percentage of the field that was abnormal(0 = none, 1 = 25%, 2 = 50%, 3 = 75%, 4 = 100%).The scores were added and divided by the number offields examined to produce a composite interstitial injuryscore.
Immunohistochemistry
Alpha-smooth muscle actin (a-SMA), metalmetalloproteinase-9 (MMP-9), tissue inhibitor ofmetalloproteinase-2 (TIMP-2), endothelin (ET-1),and plasminogen activator inhibitor-1 (PAI-1) proteinlevels were assessed in kidney sections by immuno-histochemistry. Remnant kidneys were embedded inparaffin, sliced, and mounted on glass slides. Staining fora-SMA was performed using a monoclonal mouse anti-a-SMA antibody (Cat. #A2547; Sigma Chemical Co.,St. Louis, MO, USA) in appropriate buffer accordingto Vectastain� (Vector Laboratories, Burlingame, CA,USA) anti-mouse ABC kit instructions. The following

Dworkin et al: HGF and interstitial fibrosis 411
polyclonal antibodies were used to detect their specificligands: rabbit anti-MMP-9 antibody (Cat. #M-5302;Sigma Chemical Co.), rabbit anti-TIMP-2 antibody (Cat.#T-8062; Sigma Chemical Co.), rabbit anti-ET-1 antibody(Cat. #PC266; Oncogene Research Products, Cambridge,MA, USA), rabbit anti-PAI-1 antibody (Cat. #sc-8979;Santa Cruz Biotechnology, Santa Cruz, CA, USA), alsousing the Vectastain� ABC Kit. Slides were stained withdiaminobenzidine tetrahydro chloride (DAB) and coun-terstained with methyl green or hematoxylin, cleared,and mounted using Vectamount� (Vector Laboratories)permanent mounting medium. The extent of a-SMAexpression was assessed in a blinded fashion by a singleinvestigator. Fifteen fields per slide were examined,scored from 0 to 3, and the field scores averaged togenerate a composite score for each animal. Expressionof other proteins was assessed semiquantatively andresults of these studies were presented as representativeimages.
Determination of apoptosis score by TUNEL staining
Remnant kidneys embedded in paraffin were sec-tioned onto glass slides. Dehydration was performed us-ing xylene and ethanol in series as recommended bythe kit protocol. TUNEL staining was performed us-ing the TdT-FragELTM DNA Fragmentation DetectionKit (Cat. #QIA33; Oncogene Research Products). Detec-tion of apoptotic cells and counter stain of sections wereproduced using DAB (12 minutes) and methyl green(3 minutes), respectively. The slides were mounted witha coverglass using VectamountTM permanent mountingmedium. Glomerular and interstitial apoptosis scoreswere individually assessed in a blinded fashion under 40×magnification. The number of positive apoptotic bodiesin 20 randomly selected glomeruli from each section wasdetermined and an average glomerular apoptosis scorecalculated for each rat. An interstitial apoptosis score wascalculated by counting the number of positive cells in 10randomly selected interstitial fields per specimen.
Preparation of cDNA probe from kidney
A cortical section of the remnant kidney from each ratwas snap frozen in liquid nitrogen and stored at −80◦C.Total RNA was extracted from homogenate of the renalcortex using TRIzol (Invitrogen, Carlsbad, CA, USA),and then treated with DNase I (Promega, Madison, WI,USA) to eliminate potential contamination with resid-ual genomic DNA. The cDNA was prepared from 2 lgof the total RNA using SuperScript II RNase H-reversetranscriptase (Invitrogen), followed by ethanol precipi-tation with ammonium acetate to remove surplus dNTP.The purified cDNA preparations from three rats in thesame group were pooled and the mixture was labeledwith 25 lCi [a-33P]dCTP (NEN Life Science Products,Boston, MA, USA) using Random Primers DNA Label-
ing System (Invitrogen), followed by purification throughSephadex G-50 (Amersham Biosciences, Piscataway, NJ)spin column.
Dot-blot analysis
Genes of glyceraldehyde 3-phosphate dehydrogenase(GAPDH), connective tissue growth factor (CTGF), vas-cular alpha-smooth muscle actin (a-SMA), fibronectin,HGF, hepatocyte growth factor receptor (c-met),and TGF-b1 were amplified by reverse transcription-polymerase chain reaction (RT-PCR) using specificprimers listed in Table 1. The PCR products were pu-rified by precipitation with ethanol and ammonium ac-etate. Each heat-denatured PCR product (25 ng/spot) wasapplied in duplicate to positively charged nylon mem-brane (Nytran Plus; Schleiger & Schuell, Keene, NH,USA) using a vacuum dot blotting apparatus followedby ultraviolet cross-linking. Strips of the dot blots werepreincubated in 100 mL hybridization bottles with 5 mLof hybridization solution (0.5 mol/L NaHPO4, pH 7.2,1 mmol/L EDTA, 7% SDS). After two hours of prehy-bridization, 1 × 107 cpm of each 33P-labeled cDNA probewas added to each membrane strip. After 16 hours of hy-bridization with rotation at 17 rpm, the membrane stripswere rinsed with washing solution (40 mmol/L Na2HPO4,1% SDS) and washed three times at 65◦C for 20 minutesper wash. The membrane strips were plastic-wrapped andexposed to BAS-MS2040 imaging plate (Fuji Photo FilmCo., Kanagawa, Japan) for 48 hours, and then the im-age was processed by a BAS-2500 phosphoimager (FujiPhoto Film Co.). Hybridization signal intensity of eachspot measured as photo-stimulated luminescence (PSL)developed on the imaging plates were processed forquantitative analysis using Array Vision imaging soft-ware (IMAGING Research, Inc., St. Catharines, Ontario,Canada).
Cell culture studies
To further examine the effects of HGF on matrix de-grading pathways in kidney cells, human proximal tubularcells were serum deprived for 24 hours, and then exposedto HGF or control media in the presence of absence ofvarying concentrations of angiotensin II. After 24 hoursof incubation, RNA was isolated from approximately 2 ×106 human proximal tubular epithelial cell (HKC) cellsusing TRIzol solution according to the instructions spec-ified by the manufacturer. Briefly, cells grown in 6-wellplates were harvested in TRIzol and 200 lL chloroformwas added. After centrifugation for 15 minutes at 12,000g,the aqueous phase was mixed with an equal volume ofisopropylalcohol. After 12 hours at −20◦C, the RNA waspelleted for 15 minutes at 10,000g, washed twice with 70%ethanol, dried, and dissolved in H2O. RNA was quantifiedby measuring ultraviolet absorbance at 260 nm, its puritydocumented by comparison of the ultraviolet absorbance

412 Dworkin et al: HGF and interstitial fibrosis
Table 1. List of PCR primers
PCR productGene (Dot blot) 5′-Primer 3′-Primer size (bp)
Glyceraldehyde-3-phosphate- AACTTTGGCATCGTGGAAGG CCACCACCCTGTTGCTGTAG 478dehydrogenase (GAPDH)
Transforming growth factor, beta 1 CGCGTGCTAATGGTGGAAAC CGGTAGTGAACCCGTTGATG 400Connective tissue growth factor GGAGTGGGTGTGTGATGAGC TGTAATGGCAGGCACAGGTC 497Vascular alpha-smooth muscle actin GAAGCGCAGAGCAAGAGAGG CCAGAGCGACATAGCACAGC 496Fibronectin CGCTTTGACTTCACCACCAG GCCGTTTCAGGAAGGTTGAG 503Hepatocyte growth factor AGGGCTTTCCATTCACTTGC CAAGAATTTGTGCCGGTGTG 506c-met CCGAGGTTCACTGCATGTTC TACTGACATACGCGGCTTGG 507Gene (RT-PCR)MMP-9 5′-GAC TCG GTC TTT GAG GAG CC-3′(sense)
5′-GAA CTC ACG CGC CAG TAG AA-3′(antisense)TIMP-2 5′-GTTTTGCAATGCAGATGTAG-3′(sense)
5′-ATGTCGAGAAACTCCTGCTT-3′(antisense)PAI-1 5′-ACTACGACCGCGACAAGA-3′(sense)
5′-AGGTCAATGTCAGGAGAGGC-3′(antisense)GAPDH 5′-GAAGGTGAAGGTCGGAGTC-3′(sense)
5′-GAAGATGGTGATGGGATTTC-3′(antisense)
readings at 260 and 280 nm, and its integrity assured bydocumentation of intact ribosomal RNA bands on ethid-ium bromide–stained agarose gels.
cDNA generation and RT-PCR
Total RNA harvested from HKC cells was diluted to0.2 lg/lL in H2O and first-strand cDNA was preparedusing 5 lg of RNA, Superscript RT reverse transcrip-tase and oligo(dT) primer according to the instructionsof the manufacturer. Reverse transcriptase was then in-activated by heating at 70◦C for 15 minutes, and RNasewas added to remove the template RNA. Aliquots ofthe cDNA were then amplified using specific primers(Table 1). The housekeeping gene GAPDH served as aninternal control. PCR products were electrophoresed in1.5% agarose gels containing 0.5 lg/mLof ethidium bro-mide. Resolved gels were photographed under UV lightwith Polaroid negative film (Polaroid, Cambridge, MA,USA).
Statistics
Statistical analyses were performed using a personalcomputer running SigmaStat 2.0 software (SPSS, Inc.,Chicago, IL, USA). The Student t test was employedto compare normally distributed data from control andHGF-infused rats and the Mann-Whitney rank sum testwas used for nonparametric data. Data are presented asmean ± SE.
RESULTS
Whole animal data
The average serum creatinine at day three after ab-lation was 0.99 ± 0.03 for control rats and 0.97 ± 0.04in HGF-treated rats. These values were not significantlydifferent. On average, rats gained weight from time of
ablation to time of sacrifice. At 4 weeks after ablation thevalues for mean weight were identical, 271 ± 13 g in bothgroups. ELISA determination of plasma HGF concentra-tion after 2 weeks of constant infusion and immediatelybefore sacrifice demonstrated that rats receiving HGFby minipump had about a threefold elevation in plasmaHGF level to 660 ± 147 pg/mL as compared to 231 ±10 pg/mL in saline-infused, control rats (P < 0.05).
Micropuncture data
Results of the micropuncture studies are summarizedin Table 2. Although mean arterial pressure was not sig-nificantly changed, infusion of HGF was associated witha significant reduction in the glomerular capillary hy-draulic pressure (PGC) of 14 mm Hg on average. Of note,this magnitude of reduction in PGC is similar to that pro-duced in this model by dietary protein restriction [13] orby the administration of renin-angiotensin system block-ing drugs [14], and has been associated with ameliorationof glomerular injury in these settings. There were no sta-tistically significant changes in any other hemodynamicparameter, including whole kidney or single nephronplasma flow or GFR, or in the calculated values for affer-ent or efferent arteriolar resistance.
Morphologic data
Representative light micrographs from control andHGF-infused rats are shown in Figure 1. Typical glomeru-lar and tubulointerstitial changes in the remnant kidneymodel have been previously described [13] and include,within the glomerulus, hypertrophy, microaneurysm, andmicrothrombus formation, mesangial expansion, seg-mental and global sclerosis. Interstitial changes typicallycorrelate with glomerular lesions and include tubular di-latation, cast formation, interstitial collagen deposition,and fibrosis. The severities of glomerular and interstitial

Dworkin et al: HGF and interstitial fibrosis 413
Table 2. Results of the micropuncture study
mL/min mm HgMAP FF CA
Group mm Hg GFR RPF % PGC PT �P g/dL
Control N = 12 169 ± 8 0.51 ± 0.10 3.4 ± 1.2 0.18 ± 0.02 71 ± 3 15 ± 1 56 ± 3 5.8 ± 0.2HGF N = 9 151 ± 12 0.47 ± 0.13 2.9 ± 0.3 0.15 ± 0.03 57 ± 3 13 ± 1 44 ± 3 5.5 ± 0.1
NS NS NS NS <0.01 NS <0.02 NS
dyne∗s∗cm−5 × 1010SNGFR KfnL/min QA nL/smm Hg RA RE RA/RE
113 ± 7 677 ± 108 0.059 ± 0.005 0.51 ± 0.09 0.29 ± 0.05 1.79 ± 0.1292 ± 8 831 ± 207 0.069 ± 0.008 0.48 ± 0.13 0.22 ± .0.07 2.45 ± 0.24
= 0.07 NS NS NS NS <0.02
N, number per group. Values are mean ± SE. Abbreviations are: NS, not significantly different; MAP, mean arterial pressure; GFR, glomerular filtration rate; RPF,renal plasma flow; FF, filtration fraction; PGC, glomerular capillary pressure; PT, proximal tubular pressure; �P, glomerular transcapillary hydraulic pressure difference;CA, plasma protein concentration; SNGFR, single nephron glomerular filtration rate; QA, initial glomerular capillary plasma flow rate; Kf, ultrafiltration coefficient;RA, afferent arteriolar resistance; RE, efferent arteriolar resistance.
A B
Fig. 1. Representative light micrographs ofkidney cortex from a remnant kidney froma control rat that received vehicle (A) ascompared to a rat that received intravenoushepatocyte growth factor (HGF) (B) for twoweeks. The kidney in (A) shows the typicalfindings in remnant kidneys at 4 weeks afterablation including glomerular sclerosis, inter-stitial inflammation, tubular atrophy, and castformation. Although some interstitial inflam-mation is observed, glomerular and tubulararchitecture are better preserved in the HGF-treated rat (B). (PAS, × 40).
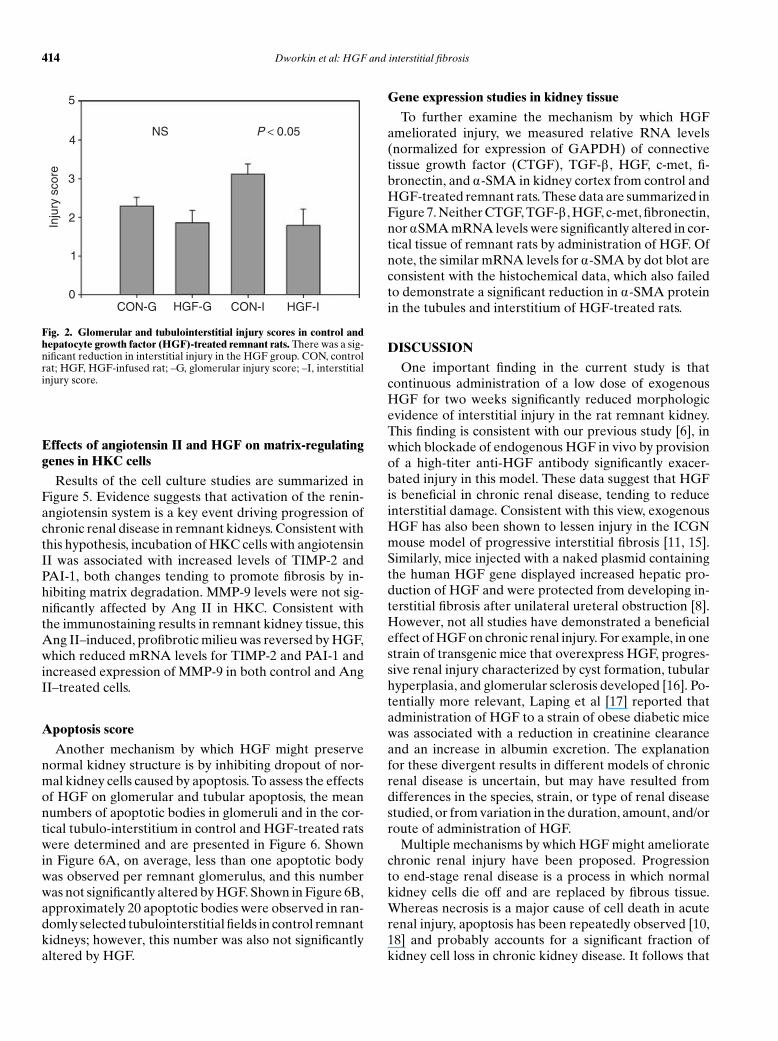
changes were scored separately and are presented inFigure 2. Glomerular lesions tended to be less extensivein HGF-treated rats, although the difference was not sig-nificantly different. However, the interstitial injury scorewas significantly reduced by approximately 50% by HGFadministration.
Immunohistochemistry
One purported mechanism by which HGF reduces in-terstitial fibrosis is by inhibiting transdifferentiation ofrenal epithelial cells to a fibroblastic phenotype. Theseinterstitial fibroblasts are hypothesized to be major pro-ducers of interstitial matrix material, thereby contribut-ing to progression of interstitial fibrosis. To determinethe extent of epithelial transdifferention, remnant kid-neys were stained with a specific anti-a-SMA antibody.The results of these studies are summarized in Figure 3.In the normal kidney, anti-a-SMA staining is essen-tially limited to vascular profiles and Bowman’s capsulewith little or no protein detected in renal tubular cells(Fig. 3A). In contrast and shown in Figure 3B, much
more extensive staining is detected in remnant kidneysand not only vessel walls, but also many tubule cells arepositive. However, when sections from control and HGF-infused rats were scored and compared, no reduction inanti-a-SMA staining was observed in the HGF-treatedgroup.
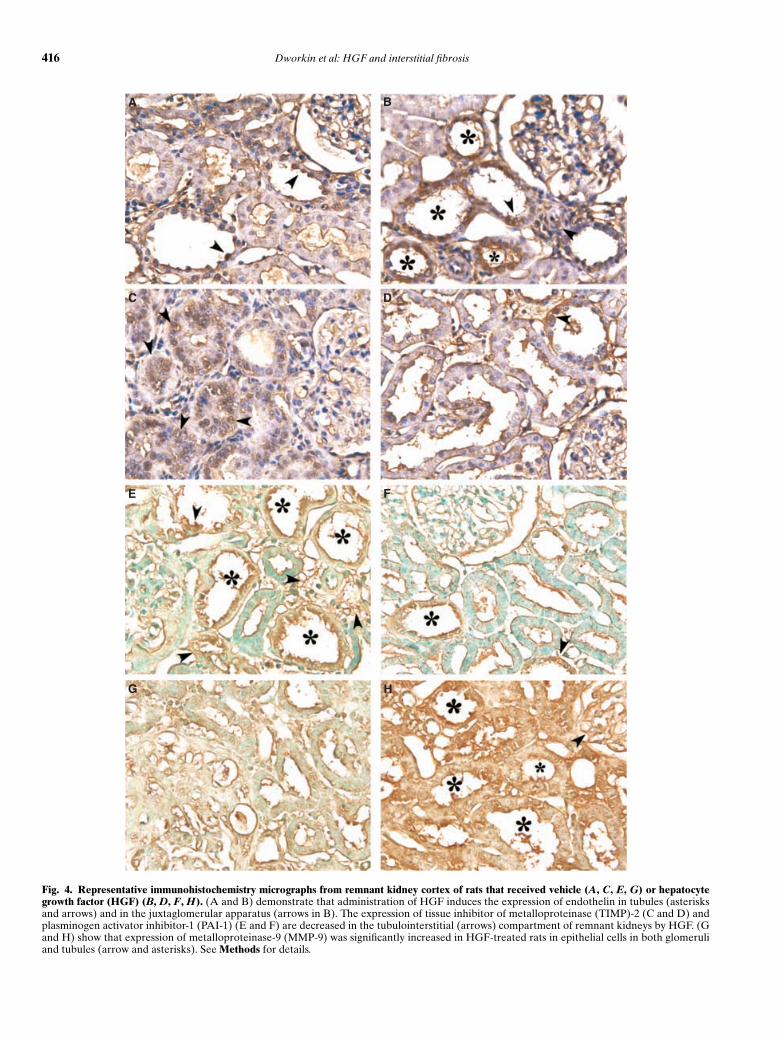
Representative micrographs from the immunohisto-chemical assessment of renal cortical endothelin, TIMP-2, PAI-1, and MMP-9 expression are presented inFigure 4. Administration of HGF was associated withinduction of endothelinf expression in distal tubule pro-files and particularly in those adjacent to the glomerulusin the region of the juxtaglomerular apparatus (Fig. 4B).Endothelin was also present in these segments in rem-nant kidneys from rats not given HGF, but staining wasless intense. Shown in Figure 4 D and F, respectively, ex-pression of TIMP-2 and PAI-1 was suppressed in HGF-treated rats, as compared to control remnant kidneys (Fig.4 C and E). In contrast, expression of the matrix degrad-ing metalloproteinase MMP-9 was markedly induced byHGF (Fig. 4H) as compared to untreated rats (Fig. 4G).
414 Dworkin et al: HGF and interstitial fibrosis
0
1
2
3
4
5
Inju
ry s
core
CON-G CON-IHGF-G HGF-I
NS P < 0.05
Fig. 2. Glomerular and tubulointerstitial injury scores in control andhepatocyte growth factor (HGF)-treated remnant rats. There was a sig-nificant reduction in interstitial injury in the HGF group. CON, controlrat; HGF, HGF-infused rat; –G, glomerular injury score; –I, interstitialinjury score.
Effects of angiotensin II and HGF on matrix-regulatinggenes in HKC cells
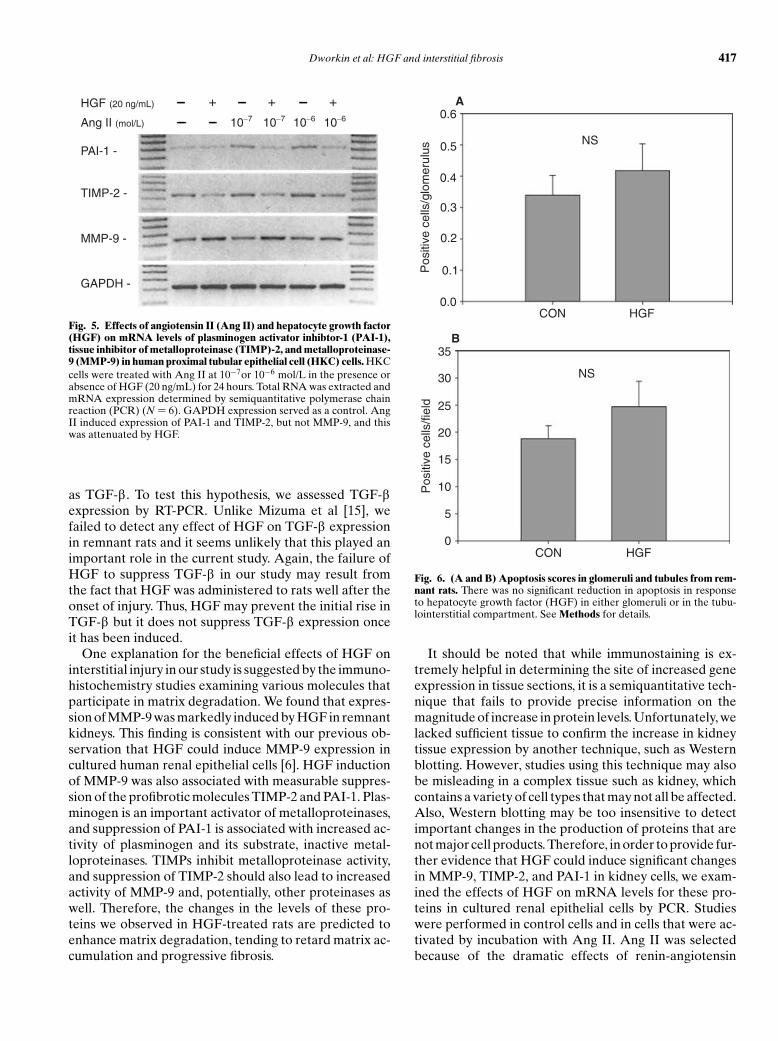
Results of the cell culture studies are summarized inFigure 5. Evidence suggests that activation of the renin-angiotensin system is a key event driving progression ofchronic renal disease in remnant kidneys. Consistent withthis hypothesis, incubation of HKC cells with angiotensinII was associated with increased levels of TIMP-2 andPAI-1, both changes tending to promote fibrosis by in-hibiting matrix degradation. MMP-9 levels were not sig-nificantly affected by Ang II in HKC. Consistent withthe immunostaining results in remnant kidney tissue, thisAng II–induced, profibrotic milieu was reversed by HGF,which reduced mRNA levels for TIMP-2 and PAI-1 andincreased expression of MMP-9 in both control and AngII–treated cells.
Apoptosis score
Another mechanism by which HGF might preservenormal kidney structure is by inhibiting dropout of nor-mal kidney cells caused by apoptosis. To assess the effectsof HGF on glomerular and tubular apoptosis, the meannumbers of apoptotic bodies in glomeruli and in the cor-tical tubulo-interstitium in control and HGF-treated ratswere determined and are presented in Figure 6. Shownin Figure 6A, on average, less than one apoptotic bodywas observed per remnant glomerulus, and this numberwas not significantly altered by HGF. Shown in Figure 6B,approximately 20 apoptotic bodies were observed in ran-domly selected tubulointerstitial fields in control remnantkidneys; however, this number was also not significantlyaltered by HGF.
Gene expression studies in kidney tissue
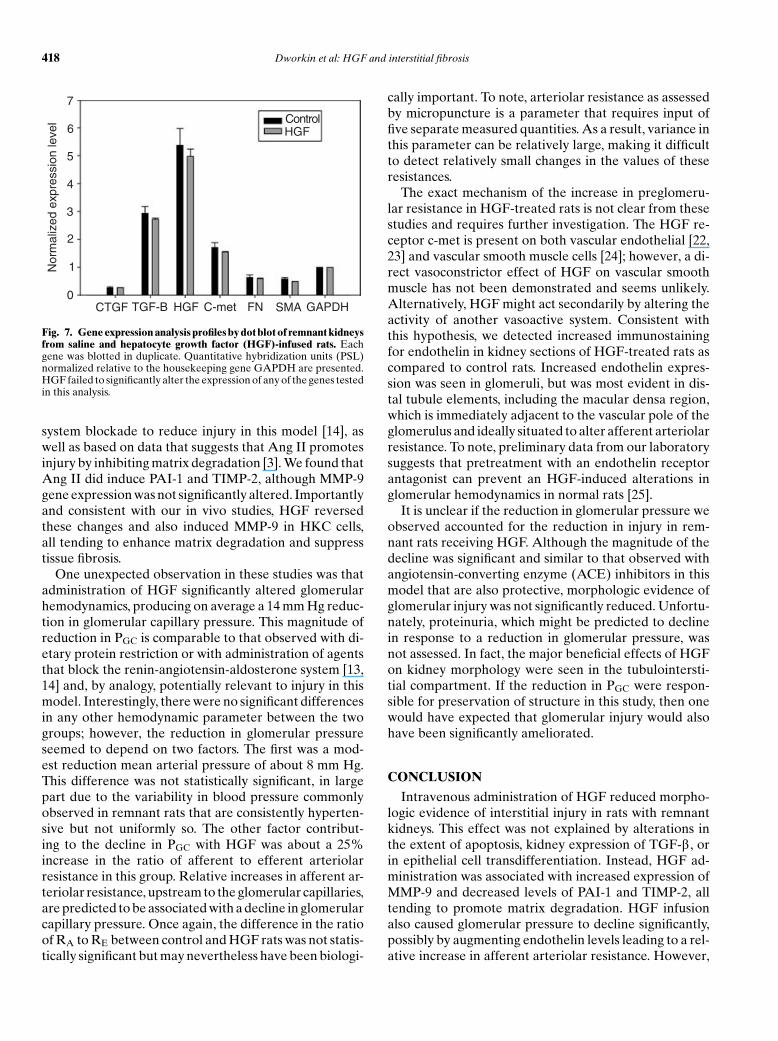
To further examine the mechanism by which HGFameliorated injury, we measured relative RNA levels(normalized for expression of GAPDH) of connectivetissue growth factor (CTGF), TGF-b , HGF, c-met, fi-bronectin, and a-SMA in kidney cortex from control andHGF-treated remnant rats. These data are summarized inFigure 7. Neither CTGF, TGF-b , HGF, c-met, fibronectin,nor aSMA mRNA levels were significantly altered in cor-tical tissue of remnant rats by administration of HGF. Ofnote, the similar mRNA levels for a-SMA by dot blot areconsistent with the histochemical data, which also failedto demonstrate a significant reduction in a-SMA proteinin the tubules and interstitium of HGF-treated rats.
DISCUSSION
One important finding in the current study is thatcontinuous administration of a low dose of exogenousHGF for two weeks significantly reduced morphologicevidence of interstitial injury in the rat remnant kidney.This finding is consistent with our previous study [6], inwhich blockade of endogenous HGF in vivo by provisionof a high-titer anti-HGF antibody significantly exacer-bated injury in this model. These data suggest that HGFis beneficial in chronic renal disease, tending to reduceinterstitial damage. Consistent with this view, exogenousHGF has also been shown to lessen injury in the ICGNmouse model of progressive interstitial fibrosis [11, 15].Similarly, mice injected with a naked plasmid containingthe human HGF gene displayed increased hepatic pro-duction of HGF and were protected from developing in-terstitial fibrosis after unilateral ureteral obstruction [8].However, not all studies have demonstrated a beneficialeffect of HGF on chronic renal injury. For example, in onestrain of transgenic mice that overexpress HGF, progres-sive renal injury characterized by cyst formation, tubularhyperplasia, and glomerular sclerosis developed [16]. Po-tentially more relevant, Laping et al [17] reported thatadministration of HGF to a strain of obese diabetic micewas associated with a reduction in creatinine clearanceand an increase in albumin excretion. The explanationfor these divergent results in different models of chronicrenal disease is uncertain, but may have resulted fromdifferences in the species, strain, or type of renal diseasestudied, or from variation in the duration, amount, and/orroute of administration of HGF.
Multiple mechanisms by which HGF might amelioratechronic renal injury have been proposed. Progressionto end-stage renal disease is a process in which normalkidney cells die off and are replaced by fibrous tissue.Whereas necrosis is a major cause of cell death in acuterenal injury, apoptosis has been repeatedly observed [10,18] and probably accounts for a significant fraction ofkidney cell loss in chronic kidney disease. It follows that

Dworkin et al: HGF and interstitial fibrosis 415
BA
Fig. 3. Alpha-smooth muscle actin (a -SMA)expression in normal (A) and remnant (B)kidney cortex. In normal kidney, a-SMA ex-pression is limited to vascular profiles. In con-trast, in the remnant kidney, a-SMA expres-sion is observed not only in the vascular tissue,but in tubular cells as well (asterisks) and inthe peritubular interstitium. See Methods fordetails.
one mechanism by which HGF might ameliorate injuryis by inhibiting apoptosis of tubular cells. Consistent withthis view, HGF prevents renal epithelial cells from under-going apoptotic cell death following exposure to varioustoxins [19, 20]. However, in the current study, we wereunable to demonstrate any reduction in apoptosis in thekidneys of rats given HGF by TUNEL staining. This sug-gests that the beneficial effect of HGF on progression ofchronic disease does not depend primarily on enhancedcell survival.
Alternatively, we have suggested that HGF may slowprogression of chronic renal disease by reducing accu-mulation of extracellular matrix [6]. Against this hypoth-esis, HGF directly stimulates production of individualmatrix proteins such as fibronectin by epithelial cells[5]. However, matrix accumulation leading to tissue fi-brosis is a complex process involving multiple cell typesand alterations in both matrix synthesis and degradation.Potentially relevant to progressive interstitial fibrosis inchronic kidney disease is the process of transdifferention,by which renal epithelial cells convert to a more fibroblas-tic phenotype. Epithelially derived interstitial fibroblastsare thought to be the major source of the excess matrixmaterial that accumulates during progressive interstitialfibrosis [21]. Transdifferentiation in vivo can be trackedby examining the expression of a-SMA, a protein not pro-duced by renal epithelial cells under normal conditions,but abundantly expressed in epithelial cells undergoingtransdifferentiation into fibroblasts. To investigate the ef-fects of HGF on transdifferentiation in the current study,a-SMA expression was examined in control and HGF-treated remnant rats by RT-PCR and by immunohisto-chemistry. As previously reported, tubular expression ofa-SMA was readily detected in remnant kidneys; how-ever, this was not reduced by HGF.
The failure of HGF to suppress epithelial transdiffer-entiation in the current study is surprising and appearsinconsistent with previous reports [9, 21]. One possi-ble explanation for these divergent results relates to themarked induction of endogenous HGF observed in thissetting. HGF levels increase significantly in remnant ascompared to control rats, possibly resulting in full recep-tor occupancy and near maximal activation of the HGF/c-met axis at some sites [6]. If so, then further elevation incirculating HGF levels by provision of exogenous HGFmight have little effect. It should be noted, however, thatthe dose we administered was sufficient to suppress inter-stitial injury, even though extent of transdifferentiationwas unaffected. It seems more likely that the failure ofHGF to suppress transdifferentiation relates to the tim-ing of administration of the growth factor. In previousstudies of the effects of HGF in models of chronic renalinjury, HGF therapy has been initiated before, or at thevery latest, concurrent with, the onset of injury. There-fore, in these studies, epithelial transdifferentiation wasprevented but not reversed. In contrast, in the currentstudy, injury was allowed to progress for two to threeweeks before the onset of HGF administration. In thissetting, a significant number of epithelial cells may havealready undergone transdifferentiation and HGF mightbe unable to reverse this process. If this hypothesis is cor-rect, then the clinical relevance of this particular action ofHGF is likely to be marginal, as chronic renal disease isalways well established before the initiation of any ther-apy to slow progression. In any event, it seems apparentthat the reduction in interstitial injury we observed inHGF-treated remnant rats did not depend on inhibitionof transdifferentiation.
Mizuma et al [15] suggested that HGF prevents fibrosisby suppressing production of fibrogenic cytokines such
416 Dworkin et al: HGF and interstitial fibrosis
A B
C D
E F
G H
Fig. 4. Representative immunohistochemistry micrographs from remnant kidney cortex of rats that received vehicle (A, C, E, G) or hepatocytegrowth factor (HGF) (B, D, F, H). (A and B) demonstrate that administration of HGF induces the expression of endothelin in tubules (asterisksand arrows) and in the juxtaglomerular apparatus (arrows in B). The expression of tissue inhibitor of metalloproteinase (TIMP)-2 (C and D) andplasminogen activator inhibitor-1 (PAI-1) (E and F) are decreased in the tubulointerstitial (arrows) compartment of remnant kidneys by HGF. (Gand H) show that expression of metalloproteinase-9 (MMP-9) was significantly increased in HGF-treated rats in epithelial cells in both glomeruliand tubules (arrow and asterisks). See Methods for details.
Dworkin et al: HGF and interstitial fibrosis 417
TIMP-2 -
PAI-1 -
MMP-9 -
GAPDH -
Ang II (mol/L)
HGF (20 ng/mL)
10−7 10−7 10−6 10−6
+ + +
Fig. 5. Effects of angiotensin II (Ang II) and hepatocyte growth factor(HGF) on mRNA levels of plasminogen activator inhibtor-1 (PAI-1),tissue inhibitor of metalloproteinase (TIMP)-2, and metalloproteinase-9 (MMP-9) in human proximal tubular epithelial cell (HKC) cells. HKCcells were treated with Ang II at 10−7or 10−6 mol/L in the presence orabsence of HGF (20 ng/mL) for 24 hours. Total RNA was extracted andmRNA expression determined by semiquantitative polymerase chainreaction (PCR) (N = 6). GAPDH expression served as a control. AngII induced expression of PAI-1 and TIMP-2, but not MMP-9, and thiswas attenuated by HGF.
as TGF-b . To test this hypothesis, we assessed TGF-bexpression by RT-PCR. Unlike Mizuma et al [15], wefailed to detect any effect of HGF on TGF-b expressionin remnant rats and it seems unlikely that this played animportant role in the current study. Again, the failure ofHGF to suppress TGF-b in our study may result fromthe fact that HGF was administered to rats well after theonset of injury. Thus, HGF may prevent the initial rise inTGF-b but it does not suppress TGF-b expression onceit has been induced.
One explanation for the beneficial effects of HGF oninterstitial injury in our study is suggested by the immuno-histochemistry studies examining various molecules thatparticipate in matrix degradation. We found that expres-sion of MMP-9 was markedly induced by HGF in remnantkidneys. This finding is consistent with our previous ob-servation that HGF could induce MMP-9 expression incultured human renal epithelial cells [6]. HGF inductionof MMP-9 was also associated with measurable suppres-sion of the profibrotic molecules TIMP-2 and PAI-1. Plas-minogen is an important activator of metalloproteinases,and suppression of PAI-1 is associated with increased ac-tivity of plasminogen and its substrate, inactive metal-loproteinases. TIMPs inhibit metalloproteinase activity,and suppression of TIMP-2 should also lead to increasedactivity of MMP-9 and, potentially, other proteinases aswell. Therefore, the changes in the levels of these pro-teins we observed in HGF-treated rats are predicted toenhance matrix degradation, tending to retard matrix ac-cumulation and progressive fibrosis.
0.0
0.1
0.2
0.3
0.4
0.5
0.6
Pos
itive
cel
ls/g
lom
erul
us
CON HGF
A
NS
0
5
10
15
20
25
30
35
Pos
itive
cel
ls/fi
eld
CON HGF
B
NS
Fig. 6. (A and B) Apoptosis scores in glomeruli and tubules from rem-nant rats. There was no significant reduction in apoptosis in responseto hepatocyte growth factor (HGF) in either glomeruli or in the tubu-lointerstitial compartment. See Methods for details.
It should be noted that while immunostaining is ex-tremely helpful in determining the site of increased geneexpression in tissue sections, it is a semiquantitative tech-nique that fails to provide precise information on themagnitude of increase in protein levels. Unfortunately, welacked sufficient tissue to confirm the increase in kidneytissue expression by another technique, such as Westernblotting. However, studies using this technique may alsobe misleading in a complex tissue such as kidney, whichcontains a variety of cell types that may not all be affected.Also, Western blotting may be too insensitive to detectimportant changes in the production of proteins that arenot major cell products. Therefore, in order to provide fur-ther evidence that HGF could induce significant changesin MMP-9, TIMP-2, and PAI-1 in kidney cells, we exam-ined the effects of HGF on mRNA levels for these pro-teins in cultured renal epithelial cells by PCR. Studieswere performed in control cells and in cells that were ac-tivated by incubation with Ang II. Ang II was selectedbecause of the dramatic effects of renin-angiotensin
418 Dworkin et al: HGF and interstitial fibrosis
0
1
2
3
4
5
6
7
Nor
mal
ized
exp
ress
ion
leve
l
CTGF TGF-B HGF C-met FN SMA GAPDH
ControlHGF
Fig. 7. Gene expression analysis profiles by dot blot of remnant kidneysfrom saline and hepatocyte growth factor (HGF)-infused rats. Eachgene was blotted in duplicate. Quantitative hybridization units (PSL)normalized relative to the housekeeping gene GAPDH are presented.HGF failed to significantly alter the expression of any of the genes testedin this analysis.
system blockade to reduce injury in this model [14], aswell as based on data that suggests that Ang II promotesinjury by inhibiting matrix degradation [3]. We found thatAng II did induce PAI-1 and TIMP-2, although MMP-9gene expression was not significantly altered. Importantlyand consistent with our in vivo studies, HGF reversedthese changes and also induced MMP-9 in HKC cells,all tending to enhance matrix degradation and suppresstissue fibrosis.
One unexpected observation in these studies was thatadministration of HGF significantly altered glomerularhemodynamics, producing on average a 14 mm Hg reduc-tion in glomerular capillary pressure. This magnitude ofreduction in PGC is comparable to that observed with di-etary protein restriction or with administration of agentsthat block the renin-angiotensin-aldosterone system [13,14] and, by analogy, potentially relevant to injury in thismodel. Interestingly, there were no significant differencesin any other hemodynamic parameter between the twogroups; however, the reduction in glomerular pressureseemed to depend on two factors. The first was a mod-est reduction mean arterial pressure of about 8 mm Hg.This difference was not statistically significant, in largepart due to the variability in blood pressure commonlyobserved in remnant rats that are consistently hyperten-sive but not uniformly so. The other factor contribut-ing to the decline in PGC with HGF was about a 25%increase in the ratio of afferent to efferent arteriolarresistance in this group. Relative increases in afferent ar-teriolar resistance, upstream to the glomerular capillaries,are predicted to be associated with a decline in glomerularcapillary pressure. Once again, the difference in the ratioof RA to RE between control and HGF rats was not statis-tically significant but may nevertheless have been biologi-
cally important. To note, arteriolar resistance as assessedby micropuncture is a parameter that requires input offive separate measured quantities. As a result, variance inthis parameter can be relatively large, making it difficultto detect relatively small changes in the values of theseresistances.
The exact mechanism of the increase in preglomeru-lar resistance in HGF-treated rats is not clear from thesestudies and requires further investigation. The HGF re-ceptor c-met is present on both vascular endothelial [22,23] and vascular smooth muscle cells [24]; however, a di-rect vasoconstrictor effect of HGF on vascular smoothmuscle has not been demonstrated and seems unlikely.Alternatively, HGF might act secondarily by altering theactivity of another vasoactive system. Consistent withthis hypothesis, we detected increased immunostainingfor endothelin in kidney sections of HGF-treated rats ascompared to control rats. Increased endothelin expres-sion was seen in glomeruli, but was most evident in dis-tal tubule elements, including the macular densa region,which is immediately adjacent to the vascular pole of theglomerulus and ideally situated to alter afferent arteriolarresistance. To note, preliminary data from our laboratorysuggests that pretreatment with an endothelin receptorantagonist can prevent an HGF-induced alterations inglomerular hemodynamics in normal rats [25].
It is unclear if the reduction in glomerular pressure weobserved accounted for the reduction in injury in rem-nant rats receiving HGF. Although the magnitude of thedecline was significant and similar to that observed withangiotensin-converting enzyme (ACE) inhibitors in thismodel that are also protective, morphologic evidence ofglomerular injury was not significantly reduced. Unfortu-nately, proteinuria, which might be predicted to declinein response to a reduction in glomerular pressure, wasnot assessed. In fact, the major beneficial effects of HGFon kidney morphology were seen in the tubulointersti-tial compartment. If the reduction in PGC were respon-sible for preservation of structure in this study, then onewould have expected that glomerular injury would alsohave been significantly ameliorated.
CONCLUSION
Intravenous administration of HGF reduced morpho-logic evidence of interstitial injury in rats with remnantkidneys. This effect was not explained by alterations inthe extent of apoptosis, kidney expression of TGF-b , orin epithelial cell transdifferentiation. Instead, HGF ad-ministration was associated with increased expression ofMMP-9 and decreased levels of PAI-1 and TIMP-2, alltending to promote matrix degradation. HGF infusionalso caused glomerular pressure to decline significantly,possibly by augmenting endothelin levels leading to a rel-ative increase in afferent arteriolar resistance. However,

Dworkin et al: HGF and interstitial fibrosis 419
further studies are needed to determine the mechanismby which HGF alters renal hemodynamics in this setting.
ACKNOWLEDGMENT
These studies were supported by a National Institutes of Health RO1grant DK52314.
Reprint requests to Lance D. Dworkin, M.D., Rhode Island Hospital,593 Eddy Street, Providence, RI 02903.Email: [email protected]
REFERENCES
1. PURKERSON ML, JOIST JH, GREENBERG JM, et al: Pathogenesis of theglomerulopathy associated with renal infarction in rats. Kidney Int22:407–417, 1976
2. HOSTETTER TH, OLSON JL, RENNKE HG, et al: Hyperfiltration inremnant nephrons: A potentially adverse response to renal ablation.Am J Physiol 241:F85–F93, 1981
3. FOGO AB: Progression and potential regression of glomeruloscle-rosis. Kidney Int 59:804–819, 2001
4. TAIPALE J, KESKI-OJA J: Growth factors in the extracellular matrix.FASEB J 11:51–59, 1997
5. LIU Y, CENTRACCHIO JN, LIN L, et al: Constitutive expressionof HGF modulates renal epithelial cell phenotype and inducesc-met and fibronectin expression. Exp Cell Res 242: 174–185,1998
6. LIU Y, RAJUR K, TOLBERT E, DWORKIN LD: Endogenous hepatocytegrowth factor ameliorates chronic renal injury by activating matrixdegradation pathways. Kidney Int 58:2028–2043, 2000
7. TANAK T, ICHIMARU N, TAKAHARA S, et al: In vivo gene transfer ofhepatocyte growth factor to skeletal muscle prevents changes inrat kidneys after 5/6 nephrectomy. Am J Transplantation 2:828–836,2002
8. YANG J, DAI C, LIU Y: Hepatocyte growth factor gene therapy andangiotensin II blockade synergistically attenuate renal interstitialfibrosis in mice. J Am Soc Nephrol 13:2464–2477, 2002
9. YANG J, LIU Y: Blockage of tubular epithelial to myofibroblast tran-sition by hepatocyte growth factor prevents renal interstitial fibrosis.J Am Soc Nephrol 13:96–107, 2002
10. MIZUNO S, MATSUMOTO K, NAKAMURA T, et al: Hepatocyte growthfactor suppresses interstitial fibrosis in a mouse model of obstructivenephropathy. Kidney Int 59:1304–1314, 2001
11. MIZUNO S, KUROSAWA T, MATSUMOTO K, et al: Hepatocyte growth
factor prevents renal fibrosis and dysfunction in a mouse model ofchronic renal disease. J Clin Invest 101:1827–1834, 1998
12. DWORKIN LD, BENSTEIN JA, TOLBERT E, FEINER HD: Salt restrictioninhibits renal growth and stabilizes injury in rats with establishedrenal disease. J Am Soc Nephrol 7:437–442, 1996
13. HOSTETTER TH, OLSON JL, RENNKE HG, et al: Hyperfiltration inremnant nephrons: a potentially adverse response to renal ablation.Am J Physiol 241:F85–93 1981
14. ANDERSON S, MEYER T, RENNKE HG, BRENNER BM: Control ofglomerular hypertension limits glomerular injury in rats with re-duced renal mass. J Clin Invest 76:612–619, 1985
15. MIZUNO S, MATSUMOTO K, KUROSAWA T, et al: Reciprocal balanceof hepatocyte growth factor and transforming growth factor-beta 1in renal fibrosis in mice. Kidney Int 57:937–948, 2000
16. TAKAYAMA H, LAROCHELLE W, SABNIS SG, et al: Renal tubular hy-perplasia, polycystic disease, and glomerulosclerosis in transgenicmice overexpressing hepatocyte growth factor/scatter factor. LabInvest 77:131–138, 1997
17. LAPING NJ, OLSON BA, HO T, et al: Hepatocyte growth factor: Aregulator of extracellular matrix genes in mouse mesangial cells.Biochem Pharmacology 59:847–853, 2000
18. SCHELLING JR, CLEVELAND RP: Involvement of Fas-dependentapoptosis in renal tubular epithelial cell deletion in chronic renalfailure. Kidney Int 56:1313–1316, 1999
19. FORNONI A, LI H, FOSCHI A, et al: Hepatocyte growth factor, but notinsulin-like growth factor I, protects podocytes against cyclosporinA-induced apoptosis. Am J Pathol 158:275–280, 2001
20. LIU Y, SUN AM, DWORKIN LD: Hepatocyte growth factor protectsrenal epithelial cells from apoptotic cell death. Biochem BiophysRes Commun 246:821–826, 1998
21. YANG J, LIU Y: Dissection of key events in tubular epithelial to my-ofibroblast transition and its implications in renal interstitial fibrosis.Am J Pathol 159:1465–1475, 2001
22. NAKANO N, MORISHITA R, MORIGUCHI A, et al: Negative regulationof local hepatocyte growth factor expression by angiotensin II andtransforming growth factor-beta in blood vessels: Potential role ofHGF in cardiovascular disease. Hypertension 32:444–451, 1998
23. MORISHITA R, NAKAMURA S, HAYASHI S, et al: Contribution of avascular modulator, hepatocyte growth factor (HGF), to the patho-genesis of cardiovascular disease. J Atheroscler Thromb 4:128–134,1998
24. NAKAMURA Y, MORISHITA R, HIGAKI J, et al: Expression of local hep-atocyte growth factor system in vascular tissues. Biochem BiophysRes Commun 215:483–488, 1995
25. ROY A, TOLBERT E, CENTRACCHIO J, et al: HGF induces anendothelin-mediated decline in renal plasma flow and GFR in rat.J Am Soc Nephrol 12:A2996, 2001




















