
Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease
12
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
-
Upload
herzzentrum-goettingen -
Category
Documents
-
view
2 -
download
0
Transcript of Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IVcollagen disease
Oliver Gross a,b,⁎, Rainer Girgert a, Bogdan Beirowski b, Matthias Kretzler c, Hee Gyung Kang c,h,Jenny Kruegel d, Nicolai Miosge d, Ann-Christin Busse e, Stephan Segerer f, Wolfgang F. Vogel g,Gerhard-Anton Müller a, Manfred Weber b
a Department of Nephrology & Rheumatology, University Medicine Goettingen Robert-Koch Str. 40, 37075 Goettingen, Germanyb Cologne General Hospital, Med. Clinic I, University Witten-Merdecke, Cologne, Germanyc Division of Nephrology, Department of Internal Medicine, Center for Computation in Medicine and Biology, University of Michigan, Ann Arbor, MI, USAd Department of Prosthodontics, Tissue regeneration work group, Georg-August-University Goettingen, Goettingen, Germanye Department of Diagnostic Radiology, Georg-August-University Goettingen, Goettingen, Germanyf Clinic for Nephrology, USZ and Institute of Anatomy, University of Zurich, Switzerlandg Department of Laboratory Medicine and Pathobiology, Faculty of Medicine, University of Toronto, Toronto, Canadah Pediatric Nephrology, Seoul National University Children's Hospital, Seoul, South Korea
a b s t r a c ta r t i c l e i n f o
Article history:Received 10 January 2010Received in revised form 1 March 2010Accepted 11 March 2010
Keywords:Type IV collagenCollagen receptorsAlport syndromeRenal fibrosisHereditary kidney disease
Alport syndrome is a hereditary type IV collagen disease leading to progressive renal fibrosis, hearing lossand ocular changes. End stage renal failure usually develops during adolescence. COL4A3−/− mice serve asan animal model for progressive renal scarring in Alport syndrome. The present study evaluates the role ofDiscoidin Domain Receptor 1 (DDR1) in cell–matrix interaction involved in pathogenesis of Alport syndromeincluding renal inflammation and fibrosis.DDR1/COL4A3 Double-knockouts were compared to COL4A3−/− mice with 50% or 100% expression ofDDR1, wildtype controls and to DDR1−/− COL4A3+/+ controls for over 6 years. Double-knockouts lived47% longer, mice with 50% DDR1 lived 29% longer and showed improved renal function (reduction inproteinuria and blood urea nitrogen) compared to animals with 100% DDR1 expression. Loss of DDR1reduced proinflammtory, profibrotic cells via signaling of TGFβ, CTGF, NFκB and IL-6 and decreaseddeposition of extracellular matrix. Immunogold-staining and in-situ hybridisation identified podocytes asmajor players in DDR1-mediated fibrosis and inflammation within the kidney.In summary, glomerular epithelial cells (podocytes) express DDR1. Loss of DDR1-expression in the kidneydelayed renal fibrosis and inflammation in hereditary type IV collagen disease. This supports our hypothesisthat podocyte–matrix interaction via collagen receptors plays an important part in progression of renalfibrosis in Alport disease. The blockade of collagen-receptor DDR1 might serve as an important newtherapeutic concept in progressive fibrotic and inflammatory diseases in the future.
© 2010 Elsevier B.V. All rights reserved.
1. Introduction
Basement membranes are important barriers for filtration ofmacromolecules. This function isparticularly apparent in theglomerularbasement membrane (GBM). The GBM is a fusion product of twobasement membranes built by endothelial and epithelial cells (podo-cytes) (Abrahamson et al., 2009). Fenestrations within the endothelialcell layer allow direct access of plasma proteins to the GBM (Kriz 2008).The intricate network of foot processes reaching out from the podocytesfurther contributes to the filtration unit. The podocytes' foot processes
are linked by a slit-diaphragm, originally described as a zipper likestructure by Rodewald and Karnovsky (1974). Since this early workmuch has been learned about the molecules that generate its uniquepermselectivity andmechanical stability of the filtration unit within thekidney (Kriz 2008). Onemajor constituent of the GBM is the network oftype IV collagen, large molecules with interrupted triple-helicalstretches, aggregating into a highly ordered network via their N-terminal andC-terminal globular domains (Hudsonet al., 2003). Type IVcollagen plays a part in a variety of protein–protein interactions,including laminins, proteoglycans, nidogens, as well as in cellattachment via α1β1 integrin (Cosgrove et al., 2000). In mammalsincluding humans and mice, six genetically distinct type IV collagen α-chains α1–α6 have been identified, encoded by six separate genesCOL4A1–COL4A6 (Hudson et al., 2003). While the immature GBMconsists of α1/α1/α2(IV) chains, α3/α4/α5(IV)-heterotrimers are
Matrix Biology 29 (2010) 346–356
⁎ Corresponding author. Department of Nephrology & Rheumatology, UniversityMedicine Goettingen Robert-Koch Str. 40, 37075 Goettingen, Germany. Tel.: +49 551396331; fax: +49 551 398906.
E-mail address: [email protected] (O. Gross).
0945-053X/$ – see front matter © 2010 Elsevier B.V. All rights reserved.doi:10.1016/j.matbio.2010.03.002
Contents lists available at ScienceDirect
Matrix Biology
j ourna l homepage: www.e lsev ie r.com/ locate /matb io

Author's personal copy
found in the mature GBM (Miner and Sanes, 1994; Borza et al., 2002;Hudson et al., 2003). Mature GBM contains numerous disulfide-bondsproviding advanced mechanical strength needed for filtration of about150 l of primary urine per day.
The α3/α4/α5(IV)-chains of mature GBM are built exclusively bypodocytes (not by endothelial cells) (Abrahamson et al., 2009),placing the podocytes into the focus of GBM-diseases such as Alportsyndrome. Alport syndrome (AS) is caused by mutations in theCOL4A3, 4 or 5 genes coding for the α3/α4/α5-chains of type IV
collagen. AS is characterized by hematuria and proteinuria, progres-sive renal failure, sensorineural deafness, and typical ocular changes(Hudson et al., 2003; Kashtan 2004). AS affects about 1:10,000children and usually leads to end stage renal failure duringadolescence. While the loss of the α3/α4/α5-chains in AS leads tocharacteristic ultra-structural changes within the GBM, the chain ofmolecular events leading from mutation in a type IV collagen gene toprogressive renal scarring and loss of kidney function has yet to bedetermined. α β integrin plays a distinct role in pathogenesis of AS
Fig. 1. Nephroprotective effect of loss of DDR1 on renal failure (lifespan): A: box-blot of lifespan in DDR1+/+ COL4A3−/− animals (left) compared to DDR1+/−COL4A3−/−animals (middle) and Double-knockouts (right). 50% loss of DDR1-expression prolonged survival until death from progressing renal fibrosis by 29%, complete loss of DDR1-expression by 47%. B: Blood urea levels in wildtype littermates compared to DDR1+/+ COL4A3−/−, DDR1+/− COL4A3−/−, and Double-knockout animals (each pointrepresenting 3–4 different animals). C: Amount of proteinuria in wildtype littermates compared to DDR1+/+ COL4A3−/−, DDR1+/− COL4A3−/−, and Double-knockout animals(each point representing 5 different animals). D: Contrast-enhanced CT-scans of living animals comparing kidney size as a parameter of nephron loss and progressive fibroticshrinkage. Left: wildtype animal; middle: DDR1+/+ COL4A3−/− animal; right: Double-knockout mouse.
347O. Gross et al. / Matrix Biology 29 (2010) 346–356
Author's personal copy
(Cosgrove et al., 2000). The role of further collagen receptors (otherthan integrins) such as DDR1 has not been addressed previously.
DDR1 and DDR2 are receptor tyrosine kinases with a characteristicextracellular discoidin element (Vogel et al., 2000). DDR1 is activatedby triple-helical collagens such as type I and IV independently fromintegrins (Vogel et al., 2000). In adherent cells, highest DDR1phosphorylation is reached after several hours of collagen stimulation(L'hote et al., 2002). DDR1 mRNA expression is predominantly seen inepithelial cells, particularly within the kidney, lung, gastrointestinaltract, and brain; however, little is known about its precise cellulardistribution (Vogel et al., 2006).
Deletion of DDR1 in themouse germ line resulted in viable animalswith hearing loss (Meyer zum Gottesberge et al., 2007), females showdefect blastocyst-implantation and mammary gland development(Vogel et al., 2006). Vascular smooth muscle cells from DDR1-nullmice show decreased proliferation, collagen-attachment, and migra-tion, pointing to a role of DDR1 in atherosclerosis (Franco et al., 2008).DDR1 controls growth and adhesion of renal mesangial cells (Curatand Vogel, 2002). Further, deletion of DDR1 protects – to a certainextent – from hypertension-induced renal disease (Flamant et al.,2006) as well as from bleomycin-induced pulmonary fibrosis byinfluencing leukocyte differentiation, collagen production, myofibro-blast expansion, apoptosis and NFκB-activation (Avivi-Green et al.,2006). DDR1−/− mice develop mild proteinuria due to altered GBMmorphology, but no progressive renal disease or loss of renal function(Gross et al., 2004b).
These data indicate a possible general role of DDR1 in chronicinflammatory and fibrotic renal diseases (Ronco and Chatziantoniou,
2008), which is investigated in the present study. Additionally,previous data in COL4A3−/− mice pointed to a possible role of DDR1in Alport disease (Gross et al., 2004a). The present study demon-strates that DDR1 is expressed in glomerular epithelial cells(podocytes). Loss of DDR1 in Alport-mice delays renal fibrosis andinflammation. Podocyte–matrix interaction via DDR1 plays a role inAS and might have a universal role in common human inflammatoryand fibrotic diseases. Therefore, the blockade of DDR1 might becomean important new therapeutic concept in progressive fibrosis andinflammation in the future.
2. Results
2.1. Loss of DDR1 in COL4A3−/−mice prolongs lifespan until death fromprogressive renal fibrosis
Lifespan was continuously documented over a period of 6 years(Fig. 1). No animals were lost due to infections or adverse events.Cross breeding of Double-heterozygous DDR+/− COL4A3+/− miceresulted in expected numbers of different genotypes, including ∼6%Double-knockouts. Lifespan of DDR1+/+ COL4A3−/− animalswas 64.3 days (mean; standard deviation +/−8.0; n=16) (Fig. 1A).DDR1+/− COL4A3−/− mice with 50% expression of DDR1 lived82.5 days (+/−7.3; n=27) which was an increase of 29% overDDR1+/+ COL4A3−/− mice (pb0.01). Lifespan of Double-knock-outs without DDR1 expression improved to 94.2 days (+/−9.7;n=17), an increase of 47% compared to DDR1+/+ COL4A3−/−mice (pb0.001; no significant gender differences). Further, lifespan
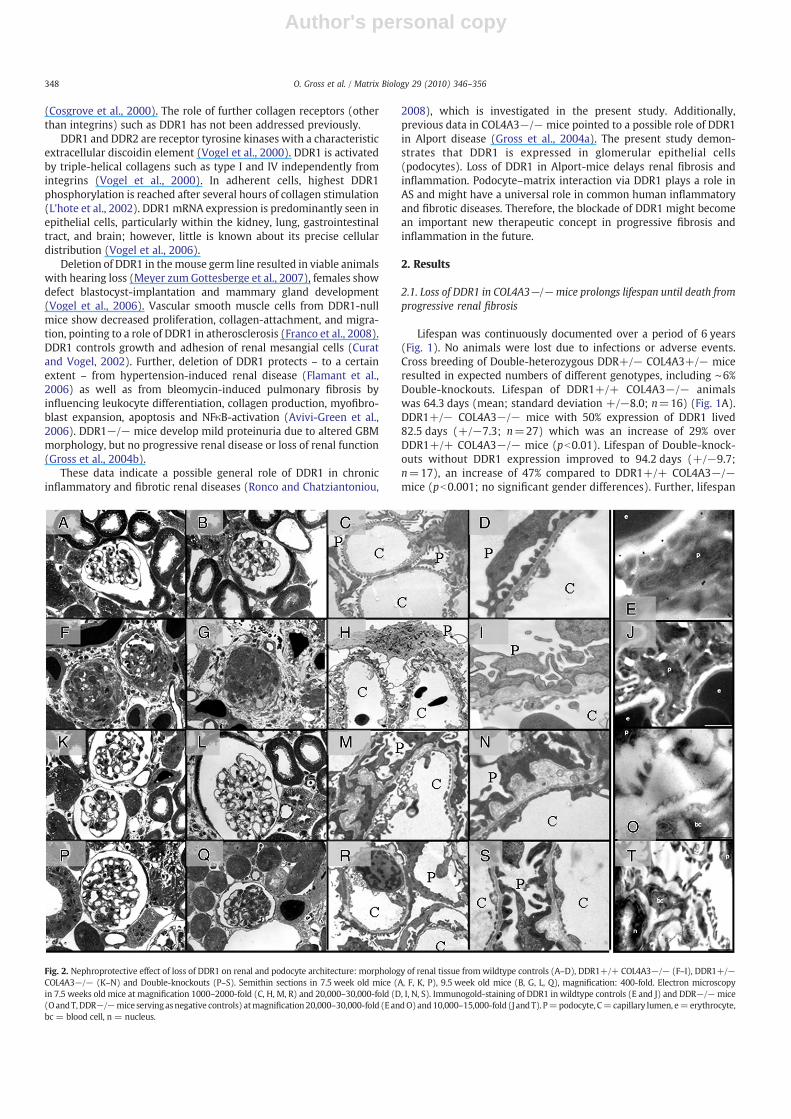
Fig. 2. Nephroprotective effect of loss of DDR1 on renal and podocyte architecture: morphology of renal tissue from wildtype controls (A–D), DDR1+/+ COL4A3−/− (F–I), DDR1+/−COL4A3−/− (K–N) and Double-knockouts (P–S). Semithin sections in 7.5 week old mice (A, F, K, P), 9.5 week old mice (B, G, L, Q), magnification: 400-fold. Electron microscopyin 7.5 weeks old mice at magnification 1000–2000-fold (C, H, M, R) and 20,000–30,000-fold (D, I, N, S). Immunogold-staining of DDR1 in wildtype controls (E and J) and DDR−/−mice(OandT,DDR−/−mice servingasnegative controls) atmagnification20,000–30,000-fold (EandO)and10,000–15,000-fold (J andT). P=podocyte, C=capillary lumen, e=erythrocyte,bc = blood cell, n = nucleus.
348 O. Gross et al. / Matrix Biology 29 (2010) 346–356
Author's personal copy
of Double-knockouts increased significantly compared to DDR1+/−COL4A3−/− (pb0.05). DDR1−/− COL4A3+/+served as an additionalcontrol with a mild renal phenotype and normal life-expectancy asdescribed earlier (Gross et al., 2004b).
2.2. Loss of DDR1 delays uremia, reduces proteinuria, and preserveskidney size
Blood urea began to rise slightly after 7.5 weeks in all COL4A3−/−mice irrespectiveof theirDDR1status (Fig. 1B). By9.5 weeks of age, bloodurea had increased to 246.7+/−27.2 mg/dl in DDR1+/+ COL4A3−/−animals. Serum-urea concentrations of COL4A3−/− mice with 50%DDR1 expression were significantly lower (165.3+/−38.1 mg/dl) andurea of Double-knockouts decreased further to 85.2+/−18.4 mg/dl
(pb0.05). Proteinuria increased to 5.0+/−1.8 g/l at 7.5 weeks and to12.1+/−3.3 g/l at 9.5 weeks in DDR1+/+ COL4A3−/− animals(Fig. 1C). In contrast, proteinuria of COL4A3−/− mice with 50%DDR1 expression was significantly lower (2.7+/−1.2 g/l at 7.5 weeksand 7.5+/−2.7 g/l at 9.5 weeks) and proteinuria of Double-knockoutsdecreased further to 0.9+/−0.5 g/l at 7.5 weeks, 2.2+/−1.2 g/l at9.5 weeksand5.6+/−2.8 g/l at 13 weeks (pb0.05compared towildtypeand to DDR1+/+ COL4A3−/−). CT-scans in living animals mirroredthese findings: kidneys of 9.5 weeks old DDR1+/+ COL4A3−/− micewere 33% smaller than kidneys from Double-knockouts (indicatingfibrosis and loss of nephrons/ kidney function) (Fig. 1D). Compared towildtype littermates, kidneys of DDR1+/+ COL4A3−/− mice were55.6% smaller, in contrast, kidneys from Double-knockouts shrunk by33.8% “only”.
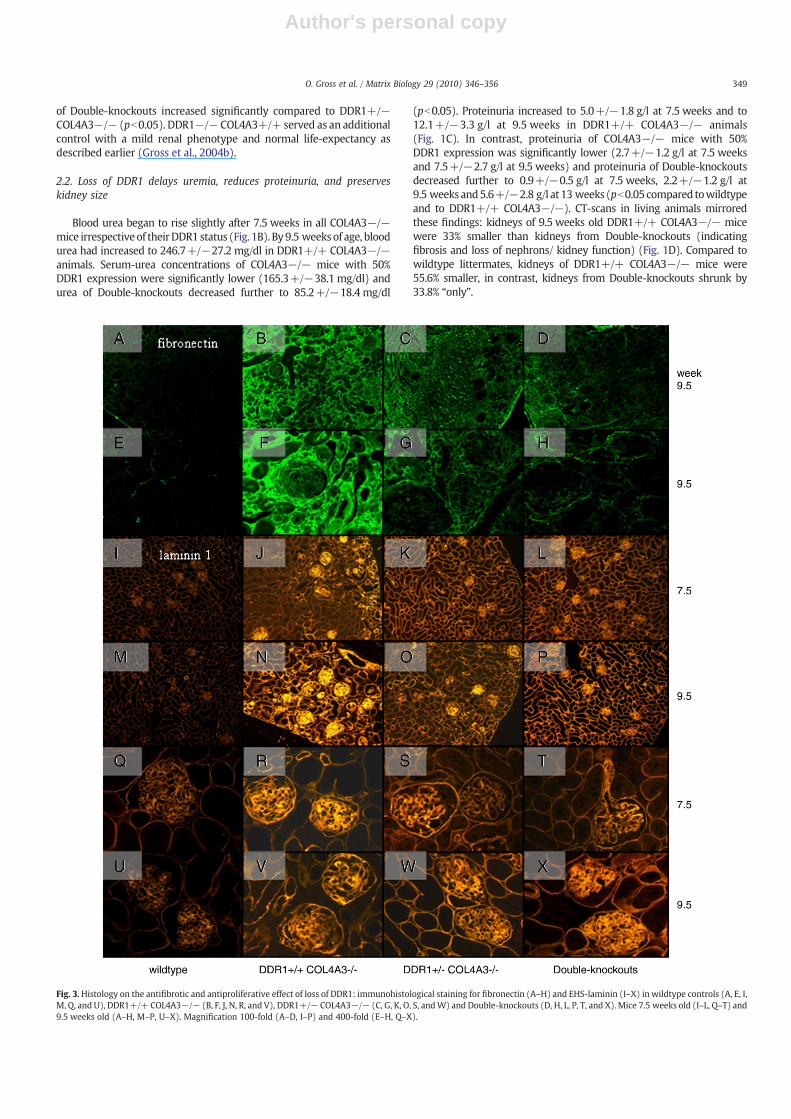
Fig. 3.Histology on the antifibrotic and antiproliferative effect of loss of DDR1: immunohistological staining for fibronectin (A–H) and EHS-laminin (I–X) in wildtype controls (A, E, I,M, Q, and U), DDR1+/+ COL4A3−/− (B, F, J, N, R, and V), DDR1+/− COL4A3−/− (C, G, K, O, S, andW) and Double-knockouts (D, H, L, P, T, and X). Mice 7.5 weeks old (I–L, Q–T) and9.5 weeks old (A–H, M–P, U–X). Magnification 100-fold (A–D, I–P) and 400-fold (E–H, Q–X).
349O. Gross et al. / Matrix Biology 29 (2010) 346–356
Author's personal copy
2.3. Loss of DDR1 preserves podocyte architecture, and reduces glomerularand tubulointerstitial matrix deposition and fibrosis via downregulation ofTGF-β and CTGF
Light microscopy of semithin kidney sections demonstrated severeglomerular, periglomerular, tubulointerstitial matrix deposition in 7.5and 9.5 week old DDR1+/+ COL4A3−/− mice (Fig. 2F and G)compared to wildtype controls (Fig. 2A and B). In contrast, DDR1+/−COL4A3−/− mice (Fig. 2K and L) and even more Double-knockouts(Fig. 2P and Q) showed less severe glomerular, periglomerular andtubulointerstitial fibrosis. Similarly, electron microscopy demonstratedthickening and splitting of the glomerular basement membrane (GBM)typical for Alport syndrome and loss and effacement of podocytes' footprocesses (Fig. 2H and I, wildtype control Fig. 2C and D). DDR1+/−COL4A3−/− mice showed less severe GBM-splitting (Fig. 2M and N).GBM-appearance improved further inDouble-knockouts paralleled by apreserved podocyte architecture including foot processes with lesspodocyte effacement (Fig. 2R and S).
In order to further elucidate the antifibrotic effects of loss of DDR1we immunostained fibronectin (representing scar-tissue, Fig. 3A–H)and EHS-laminin (representing extracellular matrix deposition,Fig. 3I–X): compared to wildtype controls (Fig. 3 first column) severeglomerular and tubulointerstitial scarring (Fig. 3B and F) and matrix
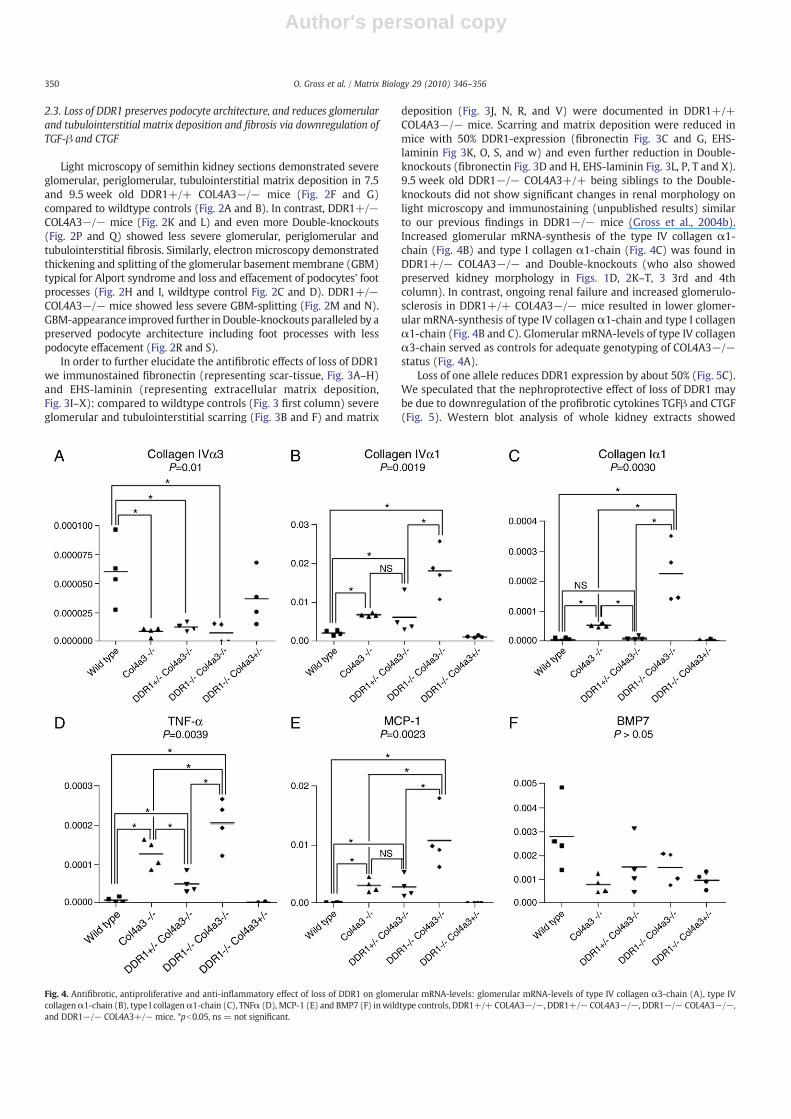
deposition (Fig. 3J, N, R, and V) were documented in DDR1+/+COL4A3−/− mice. Scarring and matrix deposition were reduced inmice with 50% DDR1-expression (fibronectin Fig. 3C and G, EHS-laminin Fig 3K, O, S, and w) and even further reduction in Double-knockouts (fibronectin Fig. 3D and H, EHS-laminin Fig. 3L, P, T and X).9.5 week old DDR1−/− COL4A3+/+ being siblings to the Double-knockouts did not show significant changes in renal morphology onlight microscopy and immunostaining (unpublished results) similarto our previous findings in DDR1−/− mice (Gross et al., 2004b).Increased glomerular mRNA-synthesis of the type IV collagen α1-chain (Fig. 4B) and type I collagen α1-chain (Fig. 4C) was found inDDR1+/− COL4A3−/− and Double-knockouts (who also showedpreserved kidney morphology in Figs. 1D, 2K–T, 3 3rd and 4thcolumn). In contrast, ongoing renal failure and increased glomerulo-sclerosis in DDR1+/+ COL4A3−/− mice resulted in lower glomer-ular mRNA-synthesis of type IV collagen α1-chain and type I collagenα1-chain (Fig. 4B and C). Glomerular mRNA-levels of type IV collagenα3-chain served as controls for adequate genotyping of COL4A3−/−status (Fig. 4A).
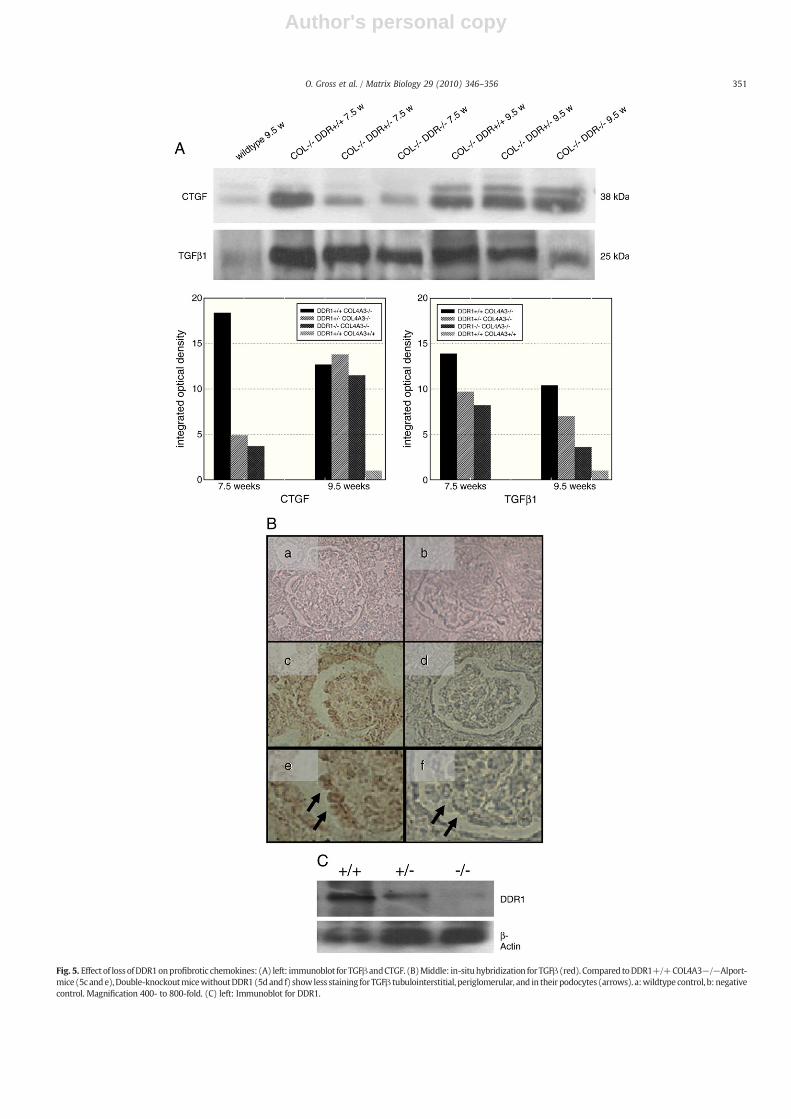
Loss of one allele reduces DDR1 expression by about 50% (Fig. 5C).We speculated that the nephroprotective effect of loss of DDR1 maybe due to downregulation of the profibrotic cytokines TGFβ and CTGF(Fig. 5). Western blot analysis of whole kidney extracts showed
Fig. 4. Antifibrotic, antiproliferative and anti-inflammatory effect of loss of DDR1 on glomerular mRNA-levels: glomerular mRNA-levels of type IV collagen α3-chain (A), type IVcollagenα1-chain (B), type I collagenα1-chain (C), TNFα (D), MCP-1 (E) and BMP7 (F) inwildtype controls, DDR1+/+ COL4A3−/−, DDR1+/− COL4A3−/−, DDR1−/− COL4A3−/−,and DDR1−/− COL4A3+/− mice. *pb0.05, ns = not significant.
350 O. Gross et al. / Matrix Biology 29 (2010) 346–356
Author's personal copy
Fig. 5. Effect of lossofDDR1onprofibrotic chemokines: (A) left: immunoblot forTGFβandCTGF. (B)Middle: in-situhybridization for TGFβ (red). Compared toDDR1+/+COL4A3−/−Alport-mice (5c ande),Double-knockoutmicewithoutDDR1(5dand f) show less staining forTGFβ tubulointerstitial, periglomerular, and in their podocytes (arrows). a:wildtype control, b: negativecontrol. Magnification 400- to 800-fold. (C) left: Immunoblot for DDR1.
351O. Gross et al. / Matrix Biology 29 (2010) 346–356
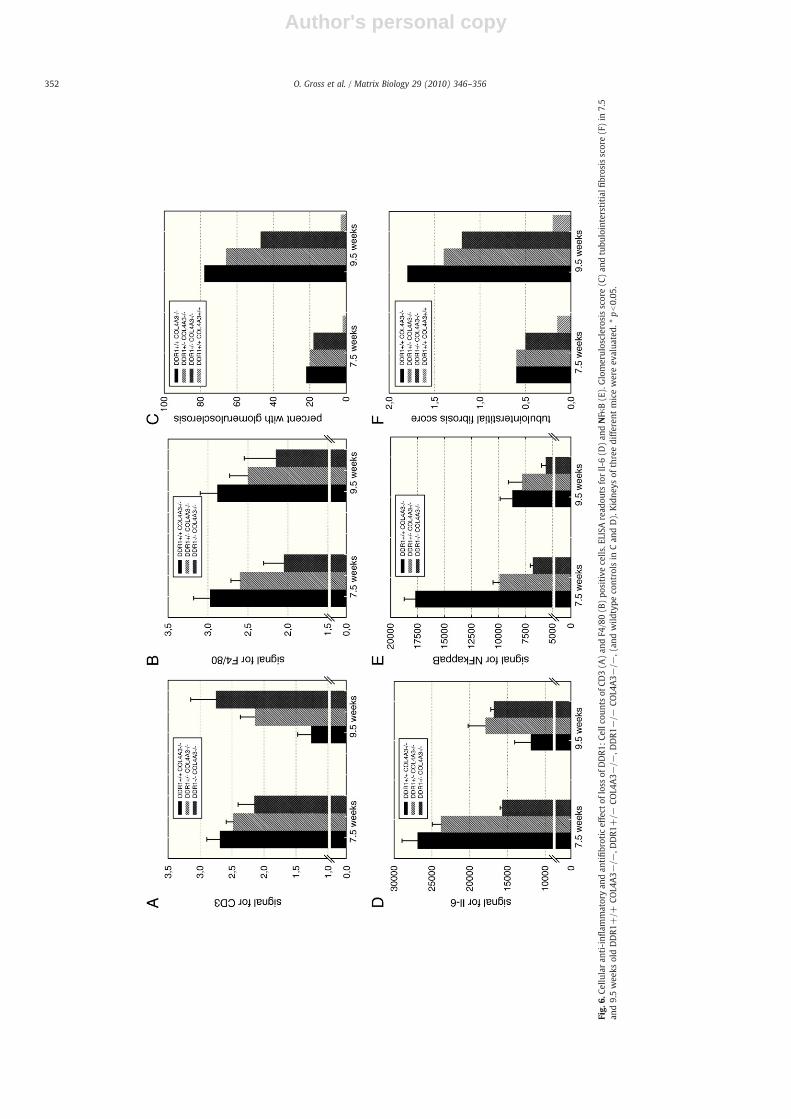
Author's personal copy
Fig.
6.Ce
llularan
ti-infl
ammatoryan
dan
tifibrotic
effect
ofloss
ofDDR1
:Cellc
ountsof
CD3(A
)an
dF4
/80(B
)po
sitive
cells
.ELISA
read
outs
forIl-6(D
)an
dNFκ
B(E
).Glomerulosclerosisscore(C
)an
dtubu
lointerstitial
fibrosisscore(F)in
7.5
and9.5wee
ksoldDDR1
+/+
COL4
A3−
/−,D
DR1
+/−
COL4
A3−
/−,D
DR1
−/−
COL4
A3−
/−,(an
dwild
type
controls
inCan
dD).Kidne
ysof
threedifferen
tmicewereev
alua
ted.
*pb0.05
.
352 O. Gross et al. / Matrix Biology 29 (2010) 346–356

Author's personal copy
marked upregulation of the profibrotic cytokines TGFβ and connectivetissue growth factor (CTGF) in DDR+/+ COL4A3−/− mice (Fig. 5,lanes 2 and 5) compared to wildtype controls (Fig. 5, lane 1). 50% lossof DDR1 (Fig. 5, lanes 3 and 6) and even further complete loss of DDR1(Fig. 5, lanes 4 and 7) resulted in a marked downregulation of TGFβand CTGF.
Tubulointerstitial fibrosis scores of DDR1+/+ COL4A3−/− micewere0.6 at 7.5 weeks and1.8at 9.5 weeks (Fig. 6F). This tubulointerstitialfibrosis score improved to 0.6 and 1.4 in DDR1+/− COL4A3−/− miceand to 0.5 and 1.2 in Double-knockouts (Fig. 6F). In parallel, glomerulo-sclerosis indexes of DDR1+/+COL4A3−/− of 22% at 7.5 weeks and 78%at 9.5 weeks improved to 20% and 64% in DDR1+/− COL4A3−/− miceand to 18% and 45% in Double-knockouts (Fig. 6C).
2.4. Loss of DDR1 influences glomerular inflammation and reducestubulointerstitial infiltration of T-lymphocytes and macrophages
In order to further elucidate the role of DDR1 in chronic in-flammation we stained and quantified T-lymphocytes (Fig. 6A) andmacrophages (Fig. 6B): numbers of CD3- and F4/80-positive cellswere reduced in 7.5 week old Col4A3−/−mice with 50% loss of DDR1and even more with the complete loss of DDR1. F4/80 positive cellsfurther decreased in 9.5 week old Double-knockouts compared toDDR1+/+ COL4A3−/− mice (pointing to an important role of DDR1in activation of macrophages). Numbers of CD3-positive cells in-creased in 9.5 weeks old Double-knockouts compared to DDR1+/+COL4A3−/− mice.
The anti-inflammatory effect of loss of DDR1 was confirmed intotal kidney extracts by ELISA of Il-6 (Fig. 6D) and NFκB (Fig. 6E). Inparallel to the CD3- and F4/80-counts, signaling of Il-6 and NFκB in7.5 week old Double-knockouts was significantly reduced comparedto DDR1+/+Col4A3−/−mice. Downregulation of NFκB continued inweek 9.5, whereas the Il-6 signal increased by week 9.5 in Double-knockouts similarly to the effects seen for CD3-positive cells.
In order to further evaluate the role of glomerular (podocyte)damage and signaling in chronic renal fibrosis and inflammation weanalysed glomerular mRNA-levels of TNFα (Fig. 4D) and MCP1(Fig. 4E). Interestingly, both signals increased in Double-knockoutscompared to DDR1+/+COL4A3−/−mice. Again, these datamight beinterpreted by focusing on glomerular damage: while Double-knock-outs had a better glomeruloscerosis score in weeks 7.5 and 9.5 (andtherefore more intact cells left to produce chemokines), DDR1+/+COL4A3−/− showed more severe damage and fibrosis (reducing thenumber of cells being able to produce proinflammatory chemokines).DDR1+/− COL4A3−/− and Double-knockouts only showed a ten-dency towards higher mRNA BMP7-levels compared to DDR1+/+COL4A3−/− (data not significant, Fig. 4F).
2.5. Immunogold electron microscopy and in-situ hybridization identifypodocytes as possible key-players in DDR1-mediated fibrosis andinflammation
DDR1 has been localized as a tyrosine kinase receptor on severalcell-types. However, the role of DDR1 within the kidney, especiallywithin the glomerulus is less clear. We hypothesized that podocyte–matrix interaction via collagen receptors such as DDR1 plays a keyrole in progression of fibrosis and inflammation. In order to verify ourhypothesis we performed immunogold-localisation of DDR1 in cellsnext to the GBM (as Alport syndrome is a GBM-disease): DDR1 couldbe found at the basal side of podocytes' foot processes right betweenthe GBM and the slit-diaphragm. DDR1 could always be stained on thesurface of podocytes, but never on the surface of endothelial cells. In-situ hybridization for TGFβ confirmed these results (Fig. 5B): Loss ofDDR1 in the Double-KOs led to reduced TGFβ-staining tubulointer-stitial, periglomerular and in podocytes compared to COL4A3−/−DDR+/+ mice. In summary, these data point to a key role of
podocyte-GBM interaction via DDR1 in chronic fibrotic and inflam-matory renal diseases.
3. Discussion
The present study investigates a possible general role of DDR1 inchronic inflammatory and fibrotic diseases. As the major finding, ourstudy demonstrates that loss of DDR1-expression in the kidney setsback renal fibrosis and inflammation. This mechanism markedlyprolongs lifespan until death from progressive renal fibrosis inCOL4A3−/− mice, pointing to a general important new therapeuticconcept in progressive fibrotic and inflammatory diseases.
DDR−/− mice develop a mild renal phenotype without renalfailure and without severe proteinuria (Gross et al. 2004b). Their localdefect of the GBM affected less than 2% of the total GBM-area only.Further, their mild renal phenotype showed up later in life (6 to9 months of age) compared to the two to three month old Double-knockout mice used in the present study. Our findings on thecollagen-receptor DDR1 are in accordance to a previous study byCosgrove and coworkers about the collagen-receptor α1/β1 integrin(Cosgrove et al., 2000). The present study further reinforces the role ofthe collagen-receptor DDR1 in chronic renal fibrosis by improving thehard end point lifespan until death from renal failure. Additionally,the localization of the collagen-receptor DDR1 at the lateral base ofpodocytes points to an important role of DDR1 in matrix–cellinteraction between GBM and podocyte. Activation of DDR1 requiresa transmembrane leucine zipper (Noordeen et al., 2006) and fourneighboring, surface-exposed collagen-loops (Abdulhussein et al.,2008). These collagen-loops can be altered by glycinemutations in AS.AS patients with a 3′ glycine mutation develop a different phenotypethan patients with 5′ glycine mutations (Gross et al., 2002). Differentmutated collagen-loops might result in different binding affinity tocollagen receptors such as DDR1. Therefore, DDR1 seems to play animportant role in pathogenesis of AS.
Abrahamson and coworkers (2009) recently demonstrated adistinct role of the podocyte in preservation and production ofmature GBM-components such as α3/4/5(IV)-chains. This groupfurther verified that collagen receptors (integrins) are linked to thecytoskeleton of podocytes. In accordance to this data, our study pointsto a similar function of DDR1 in maintenance of podocyte'scytoskeleton and architecture (Fig. 2). This function of DDR1 seemsnot to rely on integrin-signaling, as DDR1 is activated independentlyfrom integrins (Vogel et al., 2000).
3.1. Collagen receptors play an important role in pathogenesis of chronicfibrosis
In chronic renal fibrotic diseases such as Alport syndrome,glomerular and tubulointerstitial matrix deposition and scarring areregulated by TGFβ and CTGF. The present study demonstrates thatdownregulation or loss of the collagen-receptor DDR1 reduce totalamounts of TGFβ and CTGF within the kidney (Fig. 5A). Down-regulation of TGFβ by ACE-inhibition delayed fibrosis and progressiverenal failure in Alport-mice (Gross et al., 2003). Glomerular down-regulation of TGFβwithin the podocytes (Fig. 5B) further strengthensour hypothesis that the podocytes do not only produce mature GBM-components (α3/4/5(IV) collagen), but react on altered GBM-composition via their collagen receptors such as DDR1 by upregulat-ing TGFβ finally leading to glomerular (and tubulointerstitial) fibrosis.Activation of DDR1 influences smooth muscle cell migration andmatrix metalloproteinase expression (Hou et al., 2002), bothmechanisms have been shown to influence Alport pathogenesis(Cosgrove et al., 1996, 2000; Gross et al., 2003; Hudson et al., 2003;Ronco and Chatziantoniou, 2008). Interaction between podocytes,their actin cytoskeleton and the slit-diaphragm is important forpodocyte foot process formation (Mundel and Shankland, 2002;
353O. Gross et al. / Matrix Biology 29 (2010) 346–356

Author's personal copy
Saleem et al., 2002). According to our data, interaction betweenpodocytes, their collagen receptors and the GBM play a major role inmaintenance of podocyte- and GBM-architecture in AS, but inglomerular fibrosis (and inflammation) as well. These rules mightnot only apply for patients with hereditary GBM-alternations such asAlport syndrome but in acquired GBM-changes such as in hyperten-sive nephropathy or lupus nephritis.
3.2. Collagen receptors play an important role in pathogenesis of chronicinflammation (Franco et al., 2008; Vogel et al., 2006)
Activation of DDR1 by collagens upregulates chemokine produc-tion in macrophages (Matsuyama et al., 2004), known to play a majorrole in pathogenesis of renal fibrosis in AS similar to lymphocytes(LeBleu et al., 2008; Rao et al., 2006; Rodgers et al., 2003). These dataare strengthened by our findings of reduced numbers of F4/80 andCD3 positive cells in Alport-mice without DDR1. However, due to thepathogenesis of Alport disease, our data on inflammatory cells cannotprove if this antiinflammtory effect is a direct consequence of loss ofDDR1 or of reduction of proteinuria, fibrosis or proinflammatorysignaling. Similarly, reduced amounts of total kidney IL-6, NFκB, andglomerular MCP1 and TNFα in Alport-mice without DDR1 support ourhypothesis that DDR1 plays a general role in human chronicinflammatory diseases. This hypothesis is supported by humanbiopsies with different glomerular injury, glomerular inflammationand fibrosis: DDR1 mRNA-levels from the European renal biopsy databank were compared in Affymetrix based gene expression profiling inglomeruli from human patients with hypertensive nephropathy,diabetic nephropathy, membranous glomerulonephritis, IgA-ne-phropathy, lupusnephritis and focal segmental glomerulosclerosis(FSGS) (Cohen et al., 2002). DDR1mRNAwas found to be significantlyregulated in lupus nephritis and hypertensive nephropathy ascompared to normal controls (data supplied in Supplementary data).
Future experiments should focus on the role of collagen receptorssuch as DDR1 on podocyte's architecture and cytoskeleton. Further,interaction in between the GBM, the podocytes and the slit membranemight become increasingly important in pathogenesis of chronicinflammatory, chronic fibrotic renal diseases and pathogenesis (andtherapy) of proteinuria.
In conclusion, podocyte–matrix interaction via collagen receptorssuch as DDR1 plays an important part in fibrosis. Loss of DDR1-expression in the kidney sets back renal fibrosis and inflammation.Therefore, our data suggest inhibition of the collagen-receptor DDR1to become an important new therapeutic concept in progressivefibrotic and inflammatory diseases.
4. Experimental procedures
4.1. Knockout mice
The generation and PCR based genotyping of DDR1 and COL4A3-null mice has been described earlier (Cosgrove et al., 1996; Grosset al., 2004b). All DDR1mice used originated from amixed 129/Sv andICR background (Curat and Vogel 2002) and backcrossed to the 129/Sv background from 10/2002 to 9/2003, all COL4A3 mice (JacksonImmunoresearch Laboratories, Westgrove, PA, USA) were from aclean 129/Sv background and free of pathogens. Double-heterozygousmice were crossbred for more than 20 generations between 9/2003and 2009 (see Supplementary data). Control mice were wild type orheterozygous littermates. For this study, a total of 47 Double-knockouts were used from both sexes. All animal procedures werein accordance with the Declaration of Helsinki and the Guide for theCare and Use of Laboratory Animals (NIH). No animals were lost due toinfections during monitoring.
4.2. Clinical chemistry and urine analyses
Proteinuria was measured using a gradient polyacrylamide gel,stained with Coomassie Blue and analysed by densitometry, asemiquantitative method described before (Gross et al., 2004a).Serum-urea was determined on a Hitachi 917 Automatic analyzer(Boehringer Mannheim, Germany) that was validated on a daily basis.
4.3. Reconstruction of kidneys by flat-panel volume computedtomography
Mice (littermates with the same age and weight) were imagedwith flat-panel volume computed tomography (fpVCT) (GeneralElectric Global Research, Niskayuna, NY, USA). The fpVCT-prototypeconsists of a modified circular CT gantry and two amorphous siliconflat-panel X-ray detectors each of 20.5×20.5 cm2 with a matrix of1024×1024 detector elements and a resolution of 200 µm. It operatesin a step-and-shoot acquisition mode. The standard z-coverage of onestep is 4.21 cm. Mice were anaesthetized with vaporized isoflurane(0.8–1%, Forene®, Abbott AG, Baar, Switzerland) during the wholeimaging session. For enhanced contrast 150 µl of Isovist 300 (Bayer-Schering, Berlin, Germany), an iodine containing contrast agent, wasapplied intravenously in the tail vein approximately 30 s before scan.Mice were centered on the fpVCT gantry axis of rotation and placedperpendicularly to the z-axis, so that the whole mouse could bescanned with one rotation. All data sets were acquired with the sameimaging parameters: 1000 views per rotation, 8-s rotation time, 360used detector rows and 80 kVp and 100 mA. A modified Feldkampalgorithm was used for image reconstruction resulting in isotropichigh resolution volume data sets (512×512 matrix, resolution about150 µm). Images were analysed with a special imaging software(Voxtools 3.0.64Z, General Electric Global Research, Niskayuna, NY,USA) at an Advantage Workstation (AW 4.2, General Electric GlobalResearch, Niskayuna, NY, USA).
4.4. Light and electron microscopy
Mice were sacrificed, transcardially perfused with a solution ofparaformaldehyde and glutaraldehyde and kidneys were immersion-fixed as described previously (Gross et al., 2004a,b). Semithin and thinsections were taken on a Reichert Ultracut UCT ultramicrotome. AZeiss Axiophot and a Zeiss EM 902 microscope (Göttingen, Germany)were used for histological documentation. For Immunogold-staining,kidney samples of 1 mm3 were fixed and embedded in a hydrophilicresin (LR-Gold; London Resin Company, Reading, UK) as describedpreviously (Miosge et al., 1999). Ultrathin sections were blocked with1% BSA in PBS for 20 min at room temperature. The primary antibodyrabbit-anti-mouse DDR-1 (ab37838-100, abcam, Cambridge, UK) wasincubated 1:50 in PBS for 1 h at room temperature. A secondary goat-anti-rabbit IgG (Medac, Hamburg, Germany) was coupled to 16-nmcolloidal gold particles according to standard protocols (Kruegel et al.,2008) and then incubated 1:300 in PBS for 20 min at roomtemperature. Sections were contrasted with 1% uranyl acetate andlead citrate, each for 10 min at room temperature. Negative controlexperiments were performed with the aid of DDR-1−/− kidneys, aswell as by treating the sections with 1% BSA instead of the primaryantibody. In order to exclude binding of uncoupled gold particles totissue structures, sections were incubated with pure colloidal goldsolution.
4.5. Immunohistochemistry of tissue sections, cell count and scoring offibrosis
For paraffin embedding, fresh kidneys were fixed in 4% parafor-maldehyde in 0.1 mol/l phosphate-buffered saline (PBS) and vacuum-embedded in a Shandon Citadel 1000 (Pittsburg, PA, USA). Sections
354 O. Gross et al. / Matrix Biology 29 (2010) 346–356

Author's personal copy
were cut at 3 μm were rehydrated in graded alcohol, treated with 1%Trypsin for 7 min and blocked with 5% BSA at room temperature.Endogenous peroxidases were blocked in 3% hydrogen peroxide andendogenous biotin using the Avidin/Biotin blocking Kit (Vector,Burlingame, CA). Antigen retrieval was performed in an autoclaveoven (CD3) or by protease digestion (for F4/80) (Segerer et al., 2008).Tissues were incubated by a secondary biotinylated anti ratimmunoglobulin G antibody (Vector). For F4/80 staining an additionalbiotinylated antibody against the secondary reagent was used forsignal amplification. The ABC reagent (Vector) was applied, for signaldevelopment DAB (combined with nickel chloride to produce a blackcolored product) was used. For the counterstain we used a briefincubation in methylgreen. Primary antibodies were incubatedovernight at 4 °C (rabbit-anti-mouse EHS-laminin and rabbit-anti-mouse fibronectin, 1:1000, gift from M. Paulsson, Cologne, Germany).Goat-anti-rabbit Cy3 (Jackson ImmunoResearch Laboratories Inc.,West Grove, PA, USA) served as a secondary antibody. Sections wereanalysed on a Zeiss Axiophot microscope (Göttingen, Germany).Glomeruloscerosis was defined as loss of more than 50% of glomerularlumen due to extracellular matrix accumulation. Tubulointerstitialfibrosis was evaluated in a similar matter by grading 12 kidneysections each of three animals per experimental group into zero, 1+and 2+ accumulations of extracellular matrix by a blinded observer.
4.6. RNA-isolation and RT-PCR
Total RNA from glomeruli was isolated and purified using theRNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to themanufacturer's instructions. The integrity of the purified RNA wasassessed on an Agilent 2100 Bioanalyzer using a RNA 6000 NanoLabChip kit (Agilent Technologies, Santa Clara, CA, USA). Changes inexpression of selected genes identified by microarray analysis wereconfirmed by real-time RT-PCR using an Applied Biosystems 7900HTFast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA).Suitable primers and FAM-labelled probes were designed usingversion 1.5 of the Primer Express software (Applied Biosystems) orused as commercially available assays. Reverse transcription of 2 µgtotal kidney cortex RNA with random hexamers and PCR of theresulting cDNA was essentially done as described before (Cohen et al.,2002). Control reactions without addition of reverse transcriptasewere performed in parallel to exclude amplification of contaminatinggenomic DNA. Relative expression values were calculated for eachRNA sample by dividing the raw expression value of the target gene bythe corresponding value of the 18S RNA. The following primer pairsand probes and assays were used:
Col4a3 procollagen, type IV, alpha 3 Mm00483656_m1Col4a1 procollagen, type IV, alpha 1 Mm00802377_m1BMP7 bone morphogenetic protein 7 Mm00432102_m1Collagen 1 alpha 1Forward primer: 5′-TGCTTTCTGCCCGGAAGA-3′Reverse primer: 5´-GGGATGCCATCTCGTCCA-3´Probe: 5´-CCAGGGTCTCCCTTGGGTCCTACATCT-3´MCP-1: PDAR (pre-developed assay reagent) 4329581FTNF-alpha: PDAR (pre-developed assay reagent) 4329596F
4.7. Immunoblotting, in-situ hybridisation and ELISA
3–4 kidneys from different animals were pooled to minimiseindividual differences. Protein was extracted in a TBS solution, usingprotease inhibitors phenylmethylsulfonyl-fluoride (PMSF) and n-ethylmaleimide (NEM). Aliquots with 30 μg protein as shown by BCAprotein assay were dissolved in SDS-sample buffer, separated byelectrophoresis on a 4–12% NuPage Novex Bis–Tris Gel (Invitrogen,Carlsbad, USA), transferred to a polyvinylidenfluorid-membrane, and
blocked with 5% BSA and milk powder. Mouse anti-TGFβ1 (1:100,R&D Systems, Minneapolis, USA) and rabbit-anti-CTGF (1:5000,Abcam Limited, Cambridge, United Kingdom) were incubated for60 min. The membrane was then incubated with the secondaryantibody conjugated with HRP (Dako, Hamburg, Germany). The blotwas developed using chemoluminescence. Protein expression wasanalysed by densitometry using a gel-pro analyser software (MediaCybernetics, USA), density readings were corrected for loadinginequalities, immunoblots were repeated three times.
Paraffin-embedded sections (5 µm) of kidneys from knockoutmice of various genotypes were dewaxed and rehydrated indecreasing ethanol concentrations according to standard histologicalprotocols. After two washes in PBS sections were partially digested by6 µg/ml proteinase K (Qiagen, Hilden, Germany) for 30 min. Sectionswere post-fixed by 4% formaldehyde for 5 min before hybridizationwith a Digoxigenin-labelled RNA-probe specific either for TGFβmRNAor CTGF mRNA.
Probes for in-situ hybridization were labelled with Digoxigenin byin vitro transcription in the presence of DIG-UTP using RT-PCRproducts of TGFβ and CTGF as templates linked to promoters of SP6 orT7 RNA polymerase by molecular cloning. DIG-labelled probes werehybridized to the sections in hybridization solution at 42 °C overnight. Subsequently, sectionswerewashed twice in 2× SSC and in PBS.After inactivation of endogenous peroxidises by incubation in 3%H2O2 bound probes were detected by sequential incubation withrabbit-anti-DIG-HRP (Roche, Penzberg, Germany) and goat-anti-rabbit HRP (Envision, DAKO, Hamburg, Germany). Bound probeswere visualized using the HRP substrate AEC (DAKO, Hamburg,Germany).
Kidneys were homogenized in K2HPO4/MgCl2/EDTA buffer andpurified by centrifugation. Mouse Anti IL-6 ELISA (eBioscience, SanDiego, CA, USA) was performed according to the instructions of themanufacturer. In brief, 100 µl of each supernatant were added to thewells of a microwell plate pre-coated with a capture antibody andincubated at room temperature. A biotin-labelled detection antibodywas added before 100 µl Avidin-HRPwas added.Wells were washed 7times before 100 µl of substrate solution (tetramethylbenzidine) wasadded. After 15min the color reaction was stopped by addition of50 µl stop-solution (1 M H3PO4/H2SO4) and optical density mea-sured at 450 nm/ 570 nm. Values were compared to values of aparallel standard curve ranging from 4 pg/ml to 500 pg/ml.
4.8. Statistics
Data were analysed by Log Rank statistic (survival analysis) andone-way analysis of variance (ANOVA). Comparison of gene expres-sion level was analysed by non-parametric t-test, MannWhitney test.All results are given as mean and standard deviation.
Acknowledgements
The authors want to thank the staff of the animal facilities at theCenter of Molecular Medicine Cologne, at the MPI ExperimentalMedicine Goettingen and the Medical Faculty of the University ofGoettingen for animal care. We thank Mrs. Andrea Bernhard and Mrs.Oya Goektas for excellent technical assistance. Parts of this work werepresented as abstracts at the annual meetings of the German and theAmerican Societies of Nephrology. This work was supported by grantsfrom the Kidney Foundation of Canada (W.F.V.), the Cologne FortuneProgram (O.G.) and is supported by the Deutsche Forschungsge-meinschaft DFG GR 1852/4-1 and 4-2 (O.G.).
The authors would like to dedicate this paper to the memory oftheir friend and colleague,Wolfgang Vogel, who sadly died during thisstudy. His work in the field of matrix biology will always be linkedwith Discoidin Domain Receptors.
355O. Gross et al. / Matrix Biology 29 (2010) 346–356

Author's personal copy
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.matbio.2010.03.002.
References
Abdulhussein, R., Koo, D.H., Vogel, W.F., 2008. Identification of disulfide-linked dimersof the receptor tyrosine kinase DDR1. J. Biol. Chem. 2 283 (18), 12026–12033.
Abrahamson, D.R., Hudson, B.G., Stroganova, L., Borza, D.B., St John, P.L., 2009. Cellularorigins of glomerular basement membrane type IV collagen networks. J. Am. Soc.Nephrol. 20 (7), 1471–1479 Jul, Electronic publication 2009 May 7.
Avivi-Green, C., Singal, M., Vogel, W.F., 2006. Discoidin domain receptor 1-deficientmice are resistant to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care 15174 (4), 420–427.
Borza, D.B., Bondar, O., Todd, P., Sundaramoorthy, M., Sado, Y., Ninomiya, Y., Hudson, B.G.,2002. Quaternary organization of the goodpasture autoantigen, the alpha 3(IV)collagen chain. Sequestration of two cryptic autoepitopes by intrapromoter interac-tions with the alpha4 and alpha5 NC1 domains. J. Biol. Chem. 277, 40075–40083.
Cohen, C.D., Frach, K., Schlöndorff, D., Kretzler, M., 2002. Quantitative gene expressionanalysis in renal biopsies: a novel protocol for a high-throughput multicenterapplication. Kidney Int. 61, 133–140.
Cosgrove, D., Meehan, D.T., Grunkemeyer, J.A., Kornak, J.M., Sayers, R., Hunter, W.J.,Samuelson, G.C., 1996. Collagen COL4A3 knockout: a mouse model for autosomalAlport syndrome. Genes Dev. 10, 2981–2992.
Cosgrove, D., Rodgers, K., Meehan, D., Miller, C., Bovard, K., Gilroy, A., Gardner, H.,Kotelianski, V., Gotwals, P., Amatucci, A., Kalluri, R., 2000. Integrin alpha1beta1 andtransforming growth factor-beta1 play distinct roles in alport glomerularpathogenesis and serve as dual targets for metabolic therapy. Am. J. Pathol. 157,1649–1659.
Curat, C.A., Vogel, W.F., 2002. Discoidin domain receptor 1 controls growth andadhesion of mesangial cells. J. Am. Soc. Nephrol. 13, 2648–2656.
Flamant, M., Placier, S., Rodenas, A., Curat, C.A., Vogel, W.F., Chatziantoniou, C.,Dussaule, J.C., 2006. Discoidin domain receptor 1 null mice are protected againsthypertension-induced renal disease. J. Am. Soc. Nephrol. 17 (12), 3374–3381.
Franco, C., Hou, G., Ahmad, P.J., Fu, E.Y., Koh, L., Vogel, W.F., Bendeck, M.P., 2008.Discoidin domain receptor 1 (ddr1) deletion decreases atherosclerosis byaccelerating matrix accumulation and reducing inflammation in low-densitylipoprotein receptor-deficient mice. Circ. Res. 23 102 (10), 1202–1211.
Gross, O., Netzer, K.-O., Lambrecht, R., Jung, C., Seibold, S., Leinonen, A., Weber, M., 2002.Meta-analysis of genotype–phenotype correlation in X-linked Alport Syndrome:impact on genetic counseling. Nephrol. Dial. Transpl. 17, 1218–1227.
Gross, O., Beirowski, B., Koepke, M.-L., Kuck, J., Reiner, M., Addicks, K., Smyth, N.,Schulze-Lohoff, E., Weber, M., 2003. Preemptive ramipril therapy delays renalfailure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome.Kidney Int. 63, 438–446.
Gross, O., Schulze-Lohoff, E., Koepke, M.-L., Beirowski, B., Addicks, K., Bloch, W., Smyth,N., Weber, M., 2004a. Antifibrotic, nephroprotective potential of ACE-inhibitor vsAT1 antagonist in a murine model of renal fibrosis. Nephrol. Dial. Transplant. 19,1716–1723.
Gross, O., Beirowski, B., Harvey, S.J., McFadden, C., Chen, D., Tam, S., Thorner, P.S., Smyth,N., Addicks, K., Bloch, W., Ninomiya, Y., Sado, Y., Weber, M., Vogel, W., 2004b.DDR1-deficient mice show localized subepithelial GBM thickening with focal lossof slit diaphragms and proteinuria. Kidney Int. 66, 102–111.
Hou, G., Vogel, W.F., Bendeck, M.P., 2002. Tyrosine kinase activity of discoidin domainreceptor 1 is necessary for smooth muscle cell migration and matrix metallopro-teinase expression. Circ. Res. 90, 1147–1149.
Hudson, B.G., Tryggvason, K., Sundaramoorthy, M., Neilson, E.G., 2003. Alport'ssyndrome, Goodpasture's syndrome, and type IV collagen. N. Engl. J. Med. 348,2543–2556.
Kashtan, C.E., 2004. Familial hematuria due to type IV collagen mutations: alportsyndrome and thin basement membrane nephropathy. Curr. Opin. Pediatr. 16 (2),177–181.
Kriz, W., 2008. Fenestrated glomerular capillaries are unique. J. Am. Soc. Nephrol. 19,1439–1440.
Kruegel, J., Sadowski, B., Miosge, N., 2008. Nidogen-1 and nidogen-2 in healthy humancartilage and in late-stage osteoarthritis cartilage. Arthritis Rheum. 58 (5),1422–1432.
Lebleu, V.S., Sugimoto, H., Miller, C.A., Gattone 2nd, V.H., Kalluri, R., 2008. Lymphocytesare dispensable for glomerulonephritis but required for renal interstitial fibrosis inmatrix defect-induced Alport renal disease. Lab. Invest. 88 (3), 284–292.
L'hote, C.G., Thomas, P.H., Ganesan, T.S., 2002. Functional analysis of discoidin domainreceptor 1: effect of adhesion on DDR1 phosphorylation. FASEB J. 16, 234–236.
Matsuyama, W., Wang, L., Farrar, W.L., Faure, M., Yoshimura, T., 2004. Activation ofdiscoidin domain receptor 1 isoform b with collagen up-regulates chemokineproduction in human macrophages: role of p38 mitogen-activated protein kinaseand NF-kappa B. J. Immunol. 15 172 (4), 2332–2340.
Meyer zum Gottesberge, A.M., Gross, O., Becker-Lendzian, U., Massing, T., Vogel, W.F.,2007. Inner ear defects and hearing loss inmice lacking the collagen receptor DDR1.Lab. Invest. 88 (1), 27–37.
Miner, J.H., Sanes, J.R., 1994. Collagen IV alpha 3, alpha 4, and alpha 5 chains in rodentbasal laminae: sequence, distribution, association with laminins, and developmen-tal switches. J. Cell Biol. 127, 879–891.
Miosge, N., Sasaki, T., Timpl, R., 1999. Angiogenesis inhibitor endostatin is a distinctcomponent of elastic fibers in vessel walls. FASEB J. 13 (13), 1743–1750.
Mundel, P., Shankland, S.J., 2002. Podocyte biology and response to injury. J. Am. Soc.Nephrol. 13, 3005–3015.
Noordeen, N.A., Carafoli, F., Hohenester, E., Horton, M.A., Leitinger, B., 2006. Atransmembrane leucine zipper is required for activation of the dimeric receptortyrosine kinase DDR1. J. Biol. Chem. 11 281 (32), 22744–22751.
Rao, V.H., Meehan, D.T., Delimont, D., Nakajima, M., Wada, T., Gratton, M.A., Cosgrove,D., 2006. Role for macrophage metalloelastase in glomerular basement membranedamage associated with alport syndrome. Am. J. Pathol. 169 (1), 32–46.
Rodewald, R., Karnovsky, M.J., 1974. Porous substructure of the glomerular slitdiaphragm in the rat and mouse. J. Cell. Biol. 60, 423–433.
Rodgers, K.D., Rao, V., Meehan, D.T., Fager, N., Gotwals, P., Ryan, S.T., Koteliansky, V.,Nemori, R., Cosgrove, D., 2003. Monocytes may promote myofibroblast accumu-lation and apoptosis in Alport renal fibrosis. Kidney Int. 63 (4), 1338–1355.
Ronco, P., Chatziantoniou, C., 2008. Matrix metalloproteinases and matrix receptors inprogression and reversal of kidney disease: therapeutic perspectives. Kidney Int. 74(7), 873–878.
Saleem, M.A., Ni, L., Witherden, I., Tryggvason, K., Ruotsalainen, V., Mundel, P.,Mathieson, P.W., 2002. Co-localization of nephrin, podocin, and the actincytoskeleton: evidence for a role in podocyte foot process formation. Am. J. Pathol.161, 1459–1466.
Segerer, S., Heller, F., Lindenmeyer, M.T., Schmid, H., Cohen, C.D., Draganovici, D.,Mandelbaum, J., Nelson, P.J., Gröne, H.J., Gröne, E.F., Figel, A.M., Nössner, E.,Schlöndorff, D., 2008. Compartment specific expression of dendritic cell markers inhuman glomerulonephritis. Kidney Int. 74, 37–46.
Vogel, W., Brakebusch, C., Fässler, R., Alves, F., Ruggiero, F., Pawson, T., 2000. Discoidindomain receptor 1 is activated independently of beta1 integrin. J. Biol. Chem. 275(8), 5779–5784.
Vogel, W.F., Abdulhussein, R., Ford, C.E., 2006. Sensing extracellular matrix: an updateon discoidin domain receptor function. Cell. Signal. 18 (8), 1108–1116.
356 O. Gross et al. / Matrix Biology 29 (2010) 346–356




















