
Characterization of mutations in ATP8B1 associated with hereditary cholestasis
12
LIVER FAILURE AND LIVER DISEASE Characterization of Mutations in ATP8B1 Associated With Hereditary Cholestasis Leo W. J. Klomp, 1,2 Julie C. Vargas, 3 Saskia W. C. van Mil, 2 Ludmila Pawlikowska, 3,4,5 Sandra S. Strautnieks, 6 Michiel J. T. van Eijk, 2 Jenneke A. Juijn, 1 Carlos Pab ´ on-Pe ˜ na, 5 Lauren B. Smith, 5 Joseph A. DeYoung, 5 Jane A. Byrne, 6 Justijn Gombert, 1 Gerda van der Brugge, 1 Ruud Berger, 1 Irena Jankowska, 7 Joanna Pawlowska, 7 Erica Villa, 8 A. S. Knisely, 6 Richard J. Thompson, 6 Nelson B. Freimer, 5,9 Roderick H. J. Houwen, 2 and Laura N. Bull 3 Progressive familial intrahepatic cholestasis (PFIC) and benign recurrent intrahepatic cholestasis (BRIC) are clinically distinct hereditary disorders. PFIC patients suffer from chronic cholestasis and develop liver fibrosis. BRIC patients experience intermittent attacks of cholestasis that resolve spontaneously. Mutations in ATP8B1 (previously FIC1) may result in PFIC or BRIC. We report the genomic organization of ATP8B1 and mutation analyses of 180 families with PFIC or BRIC that identified 54 distinct disease mutations, including 10 mutations predicted to disrupt splicing, 6 nonsense mutations, 11 small insertion or deletion mutations predicted to induce frameshifts, 1 large genomic deletion, 2 small inframe deletions, and 24 missense mutations. Most mutations are rare, occurring in 1–3 families, or are limited to specific populations. Many patients are compound heterozygous for 2 mutations. Mutation type or location correlates overall with clinical severity: missense mutations are more common in BRIC (58% vs. 38% in PFIC), while nonsense, frameshifting, and large deletion mutations are more common in PFIC (41% vs. 16% in BRIC). Some mutations, however, lead to a wide range of phenotypes, from PFIC to BRIC or even no clinical disease. ATP8B1 mutations were detected in 30% and 41%, respectively, of the PFIC and BRIC patients screened. Supplementary material for this article can be found on the HEPATOLOGY website (http://interscience.wiley.com/jpages/0270-9139/sup- pmat/index.html) and at www.atp8b1-primers.nl (HEPATOLOGY 2004;40:27–38.) P rogressive familial intrahepatic cholestasis (PFIC) is a chronic autosomal recessive disorder causing hepatic fibrosis and end-stage liver disease. 1–3 Be- nign recurrent intrahepatic cholestasis (BRIC), with both autosomal recessive and autosomal dominant forms, is characterized by intermittent cholestasis without liver scarring. 4–6 Normal serum concentrations of cholesterol and -glutamyltranspeptidase (-GT) activity despite conjugated hyperbilirubinemia (“low–-GT cholestasis”) distinguish PFIC and BRIC from most cholestatic disor- ders. Defects in bile acid secretion and/or absorption, causing hepatic and systemic accumulation of bile acids with reduced enteric bile acid availability, underlie BRIC and PFIC. 7–9 Studies of autosomal recessive low–-GT PFIC and BRIC mapped 2 disease loci, at 18q21 (BRIC and progressive familial intrahepatic cholestasis 1 [PFIC1]) and 2q24 (PFIC2). 10 –15 Mutations in ATP8B1 (formerly FIC1) were identified in patients with PFIC1, BRIC, and Greenland familial cholestasis. 16 –20 We term these disorders collectively FIC1 disease, for FIC1 (famil- ial intrahepatic cholestasis 1), the protein encoded by ATP8B1. FIC1, a P4-subfamily P-type adenosine triphosphatase (ATPase), 16,21 is a putative aminophos- pholipid translocase. 22 Mutations were identified in ABCB11 (formerly BSEP) in PFIC2 patients. 18,23,24 ABCB11 encodes bile salt export pump, an ATPase trans- Abbreviations: PFIC, progressive familial intrahepatic cholestasis; BRIC, benign recurrent intrahepatic cholestasis; -GT, -glutamyltranspeptidase; PFIC1, pro- gressive familial intrahepatic cholestasis 1; ATPase, adenosine triphosphatase; FIC1, familial intrahepatic cholestasis 1; DHPLC, denaturing high performance liquid chromatography; cDNA, complementary DNA; PCR, polymerase chain re- action; SSCP, single-stranded conformation polymorphism. From the Departments of 1 Metabolic and Endocrine Diseases and 2 Pediatric Gastroenterology, University Medical Center, Utrecht, The Netherlands; the 3 UCSF Liver Center Laboratory and Department of Medicine, San Francisco General Hospital, San Francisco, CA; the 4 Program in Biomedical Sciences and 5 Neurogenetics Labor- atory, Department of Psychiatry and Liver Center, University of California, San Fran- cisco, San Francisco, CA; the 6 Institute of Liver Studies, King’s College Hospital, Denmark Hill, London, United Kingdom; the 7 Department of Gastroenterology, Hepatology, and Immunology, The Children’s Memorial Health Institute, Warsaw, Poland; the 8 Department of Internal Medicine/Gastroenterology, University of Modena; e reggio Emilia Medical School, Modena, Italy; and the 9 Department of Psychiatry and Human Genetics, University of California, Los Angeles, Los Angeles, CA. Received September 23, 2003; accepted March 24, 2004. This work was supported by grant WS98-12 of the Dutch Digestive Disease Foundation (to L. W. J. K.) and R01 grant DK50697 from the National Institutes of Health (to N. B. F. and L. N. B.). The Pharmacogenetics of Membrane Trans- porters study is supported by grant GM61390. Address reprint requests to: Dr. Laura N. Bull, University of California, San Francisco Liver Center Laboratory, San Francisco General Hospital, 1001 Potrero Avenue, Building 40, Room 4102, San Francisco, CA 94110. E-mail: [email protected]; fax: 415-641-0517; or Dr. Leo W.J. Klomp, Department of Metabolic and Endocrine Diseases, University Medical Center Utrecht, Wil- helmina Children’s Hospital, Room KC.02.069.1, Lundlaan 6, 3584 EA Utrecht, The Netherlands. E-mail: [email protected]; fax: 31-30-2504295. Copyright © 2004 by the American Association for the Study of Liver Diseases. Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/hep.20285 27
Transcript of Characterization of mutations in ATP8B1 associated with hereditary cholestasis

LIVER FAILURE AND LIVER DISEASE
Characterization of Mutations in ATP8B1 AssociatedWith Hereditary Cholestasis
Leo W. J. Klomp,1,2 Julie C. Vargas,3 Saskia W. C. van Mil,2 Ludmila Pawlikowska,3,4,5 Sandra S. Strautnieks,6
Michiel J. T. van Eijk,2 Jenneke A. Juijn,1 Carlos Pabon-Pena,5 Lauren B. Smith,5 Joseph A. DeYoung,5 Jane A. Byrne,6
Justijn Gombert,1 Gerda van der Brugge,1 Ruud Berger,1 Irena Jankowska,7 Joanna Pawlowska,7 Erica Villa,8 A. S. Knisely,6
Richard J. Thompson,6 Nelson B. Freimer,5,9 Roderick H. J. Houwen,2 and Laura N. Bull3
Progressive familial intrahepatic cholestasis (PFIC) and benign recurrent intrahepatic cholestasis(BRIC) are clinically distinct hereditary disorders. PFIC patients suffer from chronic cholestasisand develop liver fibrosis. BRIC patients experience intermittent attacks of cholestasis thatresolve spontaneously. Mutations in ATP8B1 (previously FIC1) may result in PFIC or BRIC. Wereport the genomic organization of ATP8B1 and mutation analyses of 180 families with PFIC orBRIC that identified 54 distinct disease mutations, including 10 mutations predicted to disruptsplicing, 6 nonsense mutations, 11 small insertion or deletion mutations predicted to induceframeshifts, 1 large genomic deletion, 2 small inframe deletions, and 24 missense mutations.Most mutations are rare, occurring in 1–3 families, or are limited to specific populations. Manypatients are compound heterozygous for 2 mutations. Mutation type or location correlatesoverall with clinical severity: missense mutations are more common in BRIC (58% vs. 38% inPFIC), while nonsense, frameshifting, and large deletion mutations are more common in PFIC(41% vs. 16% in BRIC). Some mutations, however, lead to a wide range of phenotypes, fromPFIC to BRIC or even no clinical disease. ATP8B1 mutations were detected in 30% and 41%,respectively, of the PFIC and BRIC patients screened. Supplementary material for this articlecan be found on the HEPATOLOGY website (http://interscience.wiley.com/jpages/0270-9139/sup-pmat/index.html) and at www.atp8b1-primers.nl (HEPATOLOGY 2004;40:27–38.)
Progressive familial intrahepatic cholestasis (PFIC)is a chronic autosomal recessive disorder causinghepatic fibrosis and end-stage liver disease.1–3 Be-
nign recurrent intrahepatic cholestasis (BRIC), with bothautosomal recessive and autosomal dominant forms, ischaracterized by intermittent cholestasis without liverscarring.4–6 Normal serum concentrations of cholesteroland �-glutamyltranspeptidase (�-GT) activity despiteconjugated hyperbilirubinemia (“low–�-GT cholestasis”)distinguish PFIC and BRIC from most cholestatic disor-ders. Defects in bile acid secretion and/or absorption,causing hepatic and systemic accumulation of bile acidswith reduced enteric bile acid availability, underlie BRICand PFIC.7–9 Studies of autosomal recessive low–�-GTPFIC and BRIC mapped 2 disease loci, at 18q21 (BRICand progressive familial intrahepatic cholestasis 1[PFIC1]) and 2q24 (PFIC2).10–15 Mutations in ATP8B1(formerly FIC1) were identified in patients with PFIC1,BRIC, and Greenland familial cholestasis.16–20 We termthese disorders collectively FIC1 disease, for FIC1 (famil-ial intrahepatic cholestasis 1), the protein encoded byATP8B1. FIC1, a P4-subfamily P-type adenosinetriphosphatase (ATPase),16,21 is a putative aminophos-pholipid translocase.22 Mutations were identified inABCB11 (formerly BSEP) in PFIC2 patients.18,23,24
ABCB11 encodes bile salt export pump, an ATPase trans-
Abbreviations: PFIC, progressive familial intrahepatic cholestasis; BRIC, benignrecurrent intrahepatic cholestasis; �-GT, �-glutamyltranspeptidase; PFIC1, pro-gressive familial intrahepatic cholestasis 1; ATPase, adenosine triphosphatase;FIC1, familial intrahepatic cholestasis 1; DHPLC, denaturing high performanceliquid chromatography; cDNA, complementary DNA; PCR, polymerase chain re-action; SSCP, single-stranded conformation polymorphism.
From the Departments of 1Metabolic and Endocrine Diseases and 2PediatricGastroenterology, University Medical Center, Utrecht, The Netherlands; the 3UCSFLiver Center Laboratory and Department of Medicine, San Francisco General Hospital,San Francisco, CA; the 4Program in Biomedical Sciences and 5Neurogenetics Labor-atory, Department of Psychiatry and Liver Center, University of California, San Fran-cisco, San Francisco, CA; the 6Institute of Liver Studies, King’s College Hospital,Denmark Hill, London, United Kingdom; the 7Department of Gastroenterology,Hepatology, and Immunology, The Children’s Memorial Health Institute, Warsaw,Poland; the 8Department of Internal Medicine/Gastroenterology, University ofModena; e reggio Emilia Medical School, Modena, Italy; and the 9Department ofPsychiatry and Human Genetics, University of California, Los Angeles, Los Angeles, CA.
Received September 23, 2003; accepted March 24, 2004.This work was supported by grant WS98-12 of the Dutch Digestive Disease
Foundation (to L. W. J. K.) and R01 grant DK50697 from the National Institutesof Health (to N. B. F. and L. N. B.). The Pharmacogenetics of Membrane Trans-porters study is supported by grant GM61390.
Address reprint requests to: Dr. Laura N. Bull, University of California, SanFrancisco Liver Center Laboratory, San Francisco General Hospital, 1001 PotreroAvenue, Building 40, Room 4102, San Francisco, CA 94110. E-mail:[email protected]; fax: 415-641-0517; or Dr. Leo W.J. Klomp, Departmentof Metabolic and Endocrine Diseases, University Medical Center Utrecht, Wil-helmina Children’s Hospital, Room KC.02.069.1, Lundlaan 6, 3584 EA Utrecht,The Netherlands. E-mail: [email protected]; fax: 31-30-2504295.
Copyright © 2004 by the American Association for the Study of Liver Diseases.Published online in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/hep.20285
27

ferring bile salts from hepatocyte into canalicu-lus.14,23,25–27 We present a spectrum of mutations inATP8B1 responsible for FIC1 disease with features dis-tinguishing mutations associated with PFIC1 from thoseassociated with BRIC.
Patients and MethodsPatients and Controls
Study protocols were approved by the Children’sStudy Committee of the University Medical CenterUtrecht, the Committee on Human Research of the Uni-versity of California, San Francisco, and the Ethics Com-mittees of the Children’s Memorial Health Institute,Warsaw, and King’s College Hospital, London. Patientsof diverse ancestry were diagnosed with PFIC or BRICbased on clinical and routine laboratory findings. BRICwas diagnosed in patients with at least 2 bouts of cholesta-sis whose symptoms resolved completely between attacks.Patients diagnosed with PFIC had nonremitting pruritusmanifest in infancy, poor growth, and icterus, with con-jugated hyperbilirubinemia. Biliary tracts were normal onimaging. No patient had elevated serum concentrations of�-GT activity. Urine screening, performed in most cases,excluded primary disorders of bile acid synthesis.
Genomic DNA of peripheral blood leukocytes was ex-tracted routinely. Reference control DNA was derivedfrom Centre d’Etude du Polymorphisme Humain (Paris,France) individuals 1331-1, 1331-2, or 1347-2. Controldata were from a screen for ATP8B1 variation in 247ethnically diverse normal individuals (hence 494 controlchromosomes; human variation panels, Coriell Institutefor Medical Research, Camden, NJ) using denaturinghigh performance liquid chromatography (DHPLC) andDNA sequencing (http://www.pharmgkb.org).28 Fiftyhealthy Dutch females (100 control chromosomes) alsowere screened for some mutations.
Determination of ATP8B1 StructureTo determine ATP8B1 exon–intron junctions, we
used the overlapping P1 artificial chromosome clones233B12 and 63L4, which together contain ATP8B1.16
Duplicate minilibrary filters were hybridized with 32P-labeled FIC1 complementary DNA (cDNA) probes. Plas-mids isolated from colonies with probe-marked replicateswere sequenced. To identify additional exons, portions ofclones 233B12 and 63L4 were sequenced directly usingATP8B1 cDNA-derived primers and a [32P] dATP-basedadaptation of the cycle sequencing protocol (Roche Mo-lecular Systems, Branchburg, NJ).29
Determination of Intron SizesEach intron was amplified using 2 independent primer
pairs. Both 1331-2 genomic DNA and clones 233B12 or
63L4 were used as templates. Polymerase chain reaction(PCR) was performed using the TaKaRa LA Long PCRkit (Takara Shiuzo, Otsu, Shiga, Japan). Intron sizes wereestimated after separation of PCR products on 0.8% aga-rose gels. Sequencing verified exon–intron boundaries.
Mutation Screening
General Strategy. DNA samples from some familieswere genotyped using markers spanning and flankingATP8B1.15 We developed primer sets for amplification ofall ATP8B1 coding sequences, including intervening ex-on–intron boundaries (http://www.atp8b1-primers.nl).Samples were screened via single-stranded conformationpolymorphism (SSCP) analysis (Utrecht) or DHPLC(San Francisco) and sequencing. In several families, pa-tient DNA was unavailable, so parental DNA wasscreened. All mutations were confirmed with indepen-dent PCR reamplification and sequencing—except forI661T, which generally was identified using a mutation-specific PCR restriction fragment length polymorphismassay. When DNA from patients’ families was available,segregation of disease-associated mutations was investi-gated; data uniformly were consistent with autosomal re-cessive inheritance.
PCR. Oligonucleotide primers were designed to am-plify all exons containing coding sequence, with flankingexon–intron boundaries (SSCP and DHPLC primers,http://www.atp8b1-primers.nl [Supplementary Table 1];DHPLC primers are identical to those described at http://pharmacogenetics.ucsf.edu, with one exception). PCRproducts were generated using AmpliTaq or AmpliTaqGold DNA polymerase (Roche Molecular Systems) for35 cycles. For DHPLC, primers were purified by way ofion exchange (Operon, Alameda, CA).
SSCP Analysis. Formamide was added to PCR prod-ucts at a final concentration of 50%. The samples weredenatured at 95°C for 5 minutes and quickly chilled onice. Aliquots were applied to 12.5% polyacrylamide Excelgels (Amersham Pharmacia Biotech, Piscataway, NJ) andelectrophoresed using the FBE-3000 apparatus at 600 Vand 15°C. Gels were then silver-stained (Amersham Phar-macia Biotech). Sixteen exons were screened using SSCPanalysis. Eleven exons, including those with insufficientlyspecific and reproducible SSCP analysis, were sequenceddirectly.
Sequencing. PCR products were purified on Qia-quick spin columns (Qiagen, Valencia, CA) or PerformaDTR Gel filtration cartridges (Edge Biosystems, Gaith-ersburg, MD) and sequenced using the BigDye Termina-tor Cycle Sequencing kit and an ABI prism 377 sequencer
28 KLOMP ET AL. HEPATOLOGY, July 2004

or 3700 DNA analyzer (Applied Biosystems, Warrington,UK).
DHPLC. For each exon, PCR products were pooled(3 samples per well), denatured at 95°C for 8 minutes,then cooled to 25°C, decreasing the temperature 0.5°Cevery 19 seconds. Melting profiles of each exon weregenerated (http://insertion.stanford.edu/melt.html),and DHPLC was performed at multiple temperatures,using either a Wave (Transgenomic, Tokyo, Japan) orHelix (Varian, Palo Alto, CA) apparatus (2 �L ofpooled sample injected per temperature). We per-formed DHPLC on all 27 coding exons; for 13 exons,DHPLC results were unclear or identified many vari-ant-bearing samples (usually due to a common poly-morphism within the PCR product screened), so PCRproducts were sequenced.
Restriction Fragment Length Polymorphism. PCRamplification of genomic DNA with forward primer 5�-CTATTCTTTGCATTGGTGGATTT and reverseprimer 5�-TTCTGTAAATTCTTTTTCTGCA yieldeda 112-bp product. The mismatch in the reverse primer(underlined) introduces a PstI restriction site when cyto-sine, but not thymidine, lies at position 1982. Amplifiedproducts were digested with PstI (Roche Molecular Sys-tems) for 4 hours at 37°C and electrophoresed on a 6%polyacrylamide gel.
ATP8B1 Transcript Analysis. Peripheral leukocytetotal RNA (10 �g) from the index patient from familyB16 was reverse-transcribed using oligo(dT) and Super-script reverse transcriptase (Invitrogen, Leek, The Neth-erlands). PCR was performed using nested primerslocated in exons 21 and 25. PCR products were TA-cloned and sequenced.
Results
Organization of ATP8B1. We initially identified 27exons from 44 bp to 2.3 kb long, containing the complete
coding sequence and the 3� untranslated region ofATP8B1, and 22 bp of the 5� untranslated region. Wesearched the expressed sequence tag database with the 5�untranslated region sequence, using the Basic LocalAlignment Search Tool (http://www.ncbi.nlm.nih.gov/BLAST), and found a corresponding 538-bp clone (ac-cession number AW997038). PCR amplification ofcDNA obtained from human intestinal RNA revealedthat the expressed sequence tag contained 472 bp of novelsequence information contiguous with the 5� end ofATP8B1 cDNA. This expressed sequence tag encom-passed 2 exons (exons 1 and 2) and mapped to P1 artificialchromosome 63L4, a clone known to harbor the 5� por-tion of ATP8B1.16 The genomic sequence upstream ofthe novel ATP8B1 cDNA was determined. Reverse-tran-scriptase PCR experiments revealed that exon 1 contains437 additional bp beyond the 5� end of the sequencecontained within expressed sequence tag AW997038.Exon 1 of ATP8B1 is thus at least 909 bp long and con-tains only the 5� untranslated region. The exact transcrip-tion start site has not been experimentally determined.These data reveal that ATP8B1 comprises 28 exons (Fig.1). The translation initiation codon lies in exon 2 and thestop codon lies in exon 28. The complete ATP8B1 cDNAis 6805 bp long, in accord with results of Northern blot-ting (7 kb).16 All splice junctions keep the ag-gt rule. Thecomplete sequences of small introns (introns 5, 7, 9, and25) were determined experimentally. Lengths of otherintrons were estimated via long PCR (http://www.atp8b1-primers.nl) (Supplementary Table 2). Thelargest intron, intron 15, is almost 10 kb long. ATP8B1spans at least 77 kb on chromosome 18q21 (see Fig. 1).
ATP8B1 Mutations: Types and Intragenic Distri-bution. We screened all protein-coding sequences fornucleotide differences from ATP8B1 reference sequencesin 180 families (130 PFIC, 50 BRIC), with these excep-tions: PFIC patients from 5 families were not further
Fig. 1. ATP8B1: genomic organizationand location of splice site mutations. Sche-matic representation of the exon–intronstructure of ATP8B1; exons are numbered1–28. Exons and introns are depicted atdifferent scales as indicated. Arrows indi-cate splice site mutations detected in BRIC(top) and PFIC1 patients (bottom); red ar-rows in both sections indicate a mutationdetected in both BRIC and PFIC patients.UTR, untranslated region.
HEPATOLOGY, Vol. 40, No. 1, 2004 KLOMP ET AL. 29

screened after being found to carry mutations previouslyobserved in families from similar ethnic backgrounds.BRIC patients were initially screened for the commonI661T mutation; I661T homozygotes were not furtherscreened.
Figures 1–3 illustrate the 54 distinct ATP8B1 mu-tations that we have identified, including 9 that wehave previously reported.16,17 Seven of the 54 havebeen independently detected by others.19,20 Ten are
predicted to interfere with splicing (see Fig. 1; Table1A). One of these (279 G�A) is a synonymous exonicchange; the affected guanine is the last base of exon 3,resulting in a predicted change in messenger RNAsplicing. Six nonsense mutations have been identified,as well as 11 small insertions or deletions predicted tocause frameshifts (see Fig. 2; Table 1B). These clusterin the 3� half of the gene. All 5 insertion mutationsinvolve duplication of neighboring sequence. One
Fig. 2. Location of nonsense, frameshift, and large deletion mutations within the coding region of ATP8B1. Exons, drawn to scale, are shown asnumbered boxes; dashed lines indicate the start (atg) and stop (tga) codons. Below the ATP8B1 messenger RNA, FIC1 is schematized; N and Cindicate amino- and carboxy-termini, respectively. Transmembrane domains are depicted in orange, blue designates functional P-type ATPaseconsensus domains, and green designates P4 P-type ATPase-specific consensus domains.32–34 Arrows indicate mutations detected in BRIC (top) andPFIC1 patients (bottom); red arrows in both sections identify a mutation detected in both BRIC and PFIC patients. All mutations are labeled by theresidue at which amino acid sequence is predicted to change. Scale bar: 100 codons (amino acid residues).
Fig. 3. Location of missense and smallin-frame deletion mutations in the codingregion of ATP8B1. ATP8B1 messenger RNA,FIC1 protein structure, and mutations areschematically presented as in Fig. 2. Mis-sense and deletion mutations are pre-sented as the predicted amino acidsubstitutions or deletions. Scale bar: 100codons (amino acid residues). N, amino-terminus; C, carboxy-terminus.
30 KLOMP ET AL. HEPATOLOGY, July 2004

large genomic deletion, Ex1_Ex13del, is also shown(see Fig. 2; Table 1B).16 We identified 2 small in-framedeletions and 24 missense mutations (see Fig. 3; Table1C). These mutations lie throughout the ATP8B1 se-quence. The in-frame deletion, 1587_1589delCTT,deletes one of 3 contiguous CTT repeats and was iden-tified in 2 PFIC patients (PFIC19, of Japanese ances-try,19 and PFIC33, of Caucasian–African Americanancestry; Table 2).
Mutations in PFIC Patients. ATP8B1 mutationswere identified in 39 (30%) of the 130 PFIC families ofdiverse ethnic origin screened in the current study (Table2; see Figs. 1–3). In 28 families, patients were homozy-gous for an ATP8B1 mutation (Table 2A); in 9 families,patients were compound heterozygotes (Table 2B); and in2 families, we detected a mutation on only one allele(Table 2C). None of these mutations was identified incontrol samples. In total, 36 distinct mutations were de-tected (Tables 1 and 2). No mutations common to abroad cross-section of populations were identified, and 25of the 36 mutations were detected in only one family. Themost frequently identified mutation in this sample wasE665X, which was detected in 5 Dominican families. TheD554N mutation17 previously identified in Inuit patientswas detected in 1 additional Inuit family, but also in afamily from the neighboring yet ethnically distinct Atha-bascan population. The IVS18�2 T�C mutation, previ-ously identified in 2 families,16,20 was detected in 2additional families in this study. Five other novel muta-tions identified in this study occur in patients from 2families (Tables 1 and 2).
Mutations in BRIC Patients. ATP8B1 mutationswere identified in 20 (40%) of the 50 BRIC families(most of European descent) screened in the current study(see Table 3). We previously identified the I661T muta-tion as frequent in European BRIC patients, occurring inapproximately 45% of families screened.15,16,30 We havenow identified 3 additional families with I661T-homozy-gous patients, or 16 families total (Table 3A).16,30 Weidentified 5 additional families with I661T-heterozygouspatients, or 11 such families total.16 We detected a secondheterozygous mutation in patients from 8 of these families(Table 3A).
Patients from 36 BRIC families did not carry I661T;ATP8B1 sequence variations undetected in controls werefound in only 6 of these families (17%). The probandswere homozygous in 2 families (families BRIC15 andBRIC16; Table 3B) and compound heterozygous in 4families (families BRIC17–BRIC20; Table 3C).
In total, I661T, G892R (homozygous in a PFIC pa-tient16 and detected in compound heterozygote patient
PFIC31), and 15 distinct novel mutations associated withBRIC were detected (see Tables 1 and 3, Figs. 1–3).
Intermediate Phenotype. A genome screen of the first3 identified patients in family BRIC16 initially mappedBRIC to 18q21.10 We have now detected a mutation,IVS23-3C�A, in homozygous form in all 6 affected in-dividuals of this family. To determine the effect of thismutation on the ATP8B1 transcript, we evaluated leuko-cyte RNA from 1 patient and found complete in-frameskipping of exon 24 (results not shown). The correspond-ing transcript is predicted to encode FIC1 lacking thecomplete sixth transmembrane domain and 7 flankingamino acid residues. Consistent with the presumed majoreffect of this structural change on protein function, dis-ease in 2 patients from this family became progressive,representing a form of FIC1 disease intermediate in sever-ity between BRIC and PFIC.31 Why this mutation doesnot lead invariably to PFIC is unknown.
Genotype–Phenotype Correlation. Mutations likelyto alter structure and/or function of FIC1 severely, such asframeshift, nonsense, and large genomic deletion muta-tions, compose a greater proportion of distinct mutationsdetected in PFIC patients than in BRIC patients, whereasthe proportion of distinct missense mutations is greater inBRIC patients than in PFIC patients (Fig. 4). In the 2BRIC families with nonsense mutations (familiesBRIC10 and BRIC18; see Tables 3A and 3C), these occurin compound heterozygous form with a missense muta-tion. In the single BRIC family (BRIC16) with a homozy-gous frameshift mutation (G1241fs; see Table 3B), themutation lies only 11 amino acid residues from the pro-tein C-terminus. Patients who are homozygous for frame-shift mutations nearer the N-terminus of the protein allhave PFIC (Table 2A). For example, homozygosity forA1208fs produces PFIC (family PFIC11), although thisframeshift occurs only 33 residues farther from the pro-tein C-terminus than does the G1241fs mutation. Theprecise effect on transcript structure of only 1 of 10 mu-tations likely to affect splicing could be determinedthrough RNA studies. Nevertheless, the effects of most ofthese mutations are likely to be severe; six alter invariantsplice junction residues. Those detected in BRIC patientsoccured in combination with I661T (families BRIC4 andBRIC11) and/or in highly conserved, but not invariant,residues implicated in splicing (see Table 1A).16 Thesedata indicate that mutation type or location correlatesoverall with disease severity.
Most missense mutations seen in PFIC or BRIC pa-tients occur in highly conserved residues, cause noncon-servative amino acid changes, and are predicted to affectactivity of FIC1 substantially (see Table 1C, Fig. 4). Forexample, the S453Y, D454G, and T456M missense mu-
HEPATOLOGY, Vol. 40, No. 1, 2004 KLOMP ET AL. 31

Table 1. Characteristics of ATP8B1 Mutations
A. Mutations Predicted to Interfere With Splicing
Nucleotide Change Disease Number of Families Ethnicity References
279 G�A (A93A) P, B 2 Belgian This studyIVS3�1_�3delGTG P 1 Bolivian This studyIVS3-2 A�G P 1 Caucasian/African American This studyIVS8�1 G�T P 1 Saudi This studyIVS17-1 G�A P 1 South Asian This studyIVS18�2 T�C P 4 1 Polish, 3 unknown 16, 20; this studyIVS20-4 CT�AA B 1 Dutch 16IVS21�5 G�A P 2 Polish This studyIVS23-3 C�A B, I 1 (extended) Dutch This studyIVS26�2 T�A B 1 Unknown This study
B. Nonsense, Frameshift, and Large Deletion Mutations
Nucleotide Change Predicted Effect Disease Number of Families Ethnicity References
Ex1_Ex13del No transcript P 1 Unknown 16614_615insA N205fs P 1 English This study1804 C�T R602X P 1 Asian/Hispanic/Caucasian This study1993 G�T E665X P 5 Dominican This study2016delG K672fs P 1 Caucasian This study2124_2125insGAGCTACAGCTATTGAAGGC K709fs P 1 Japanese 19; this study1208 C�A P792fs P 1 Japanese 19; this study2543_2556dupAGCGGCAGAAAAAC F853fs P 1 Hungarian This study2596_2599dupTGCC R867fs P 1 South Asian This study2788 C�T R930X P 2 French Canadian, Caucasian This study2854 C�T R952X P, B 3 European, English, German This study2873delA N958fs P 1 Northern European This study3040 C�T R1014X P 1 Belgian This study3069_3070delAA Q1023fs P 1 Northern European This study3140delT L1047fs P 1 (extended) Finnish This study3490 C�T R1164X B 1 Italian This study3622_3628delGCCTACG A1208fs P 1 (extended) Irish This study3722delG G1241fs B 1 Pakistani This study
C. Missense and Small Inframe Deletion Mutations
Nucleotide ChangePredicted
Effect
Amino Acid Residue Conservation in Human ATP8B1 Homologues
Disease
NumberofFamilies Ethnicity References8A1 8A2 8B2 8B3 8B4 9A 9B 10B 10C 10D 11A 11B 11C
208 G�A D70N — — D N D — — — — — — — — B 1 Unknown This study380 T�C L127P I I L I L L I L I L I I I P 1 Japanese 19; this study863 T�C L288S L L L L L L L F L L L L L P 1 Polish 16923 G�T G308V G G G G G G G G G G G G G P 1 (large
kindred)Amish 15, 16
923 G�A G308D G G G G G G G G G G G G G B 1 German This study1030 A�T I344F L L I I I L L C C C Y Y Y B 1 Unknown This study1208 C�A S403Y S S S S S S S S S S S S S P 1 Japanese 19; this study1235 G�C R412P K K R Y R K K K K K K K K P 1 Japanese 19; this study1358 C�A S453Y S S S S S T T S S S T T T B 1 Unknown This study1361 A�G D454G D D D D D D D D D D D D D B 1 French This study1367 C�T T456M T T T T T T T T T T T T T P 1 Italian This study1498 T�C Y500H — — L F Q — — — — — — — — P 1 Hispanic This study1587_1589delCTT F529del T T R R R K K L I I R K R P 2 Japanese, Caucasian/
African American19; this study
1604 A�T H535L H H H H H H H N N N H H H P 1 Iranian This study1660 G�A D554N D D D D D D D D D D D D D P 10 9 Inuit, 1 Athabascan/
Norwegian17; this study
1798 C�T R600W R R R R R S S R R R R R R B 1 Dutch This study1799 G�A R600Q R R R R R S S R R R R R R B 3 Dutch, unknown This study1882 C�T R628W R R R R K I I L L L R K R B 1 German This study1982 T�C I661T I L L V L L L V L M L F I B,P 29 �all Western
European15, 16, 30;
this study2063 A�G D688G E E A Q A A A M F L A A E P 1 Unknown This study2081 T�C I694T I I V M I L L L L L I I I B 1 French This study2197 G�A G733R G G G G G G G G G G G G G P 1 Caucasian This study2384_2392delGAAACCGTG 795delGNR — — — EPR — — — — — — — — — B 1 Dutch 162558 T�C F853S F F F F L F F F F F F F F P 1 Caucasian This study2674 G�A G892R G G G G G G G G G G G G G P,B 3 Italian, Caucasian,
unknown16; this study
3118 G�A G1040R G A G G G S S A A A G G A P 2 Saudi This study
NOTE. Mutations are listed in the order in which they occur in ATP8B1. Amino acid conservation is indicated as follows: human ATP8B1 homologues are listed by their class, type, andwhere applicable, member number designation (e.g., 8A1 for ATP8A1). Dark shading indicates amino acid residues that are identical to the amino acid present in ATP8B1 at the indicatedposition in the alignment of these protein sequences. Light shading indicates residues at which a conservative substitution between ATP8B1 and the indicated homologue is present.Unshaded residues represent nonconservative substitutions between ATP8B1 and the homologue. A dash (—) indicates residues not present in the homologue, or regions of the homologuethat did not align with ATP8B1. Sequences were aligned using MultAlin42 with manual editing. Amino acid substitutions were classified as conservative using the blosum62 substitutionmatrix,43 with a cutoff of ��1.Abbreviations: B, BRIC; P, PFIC1; I, intermediate severity disease.

tations affect the phosphorylation domain of FIC1,highly conserved in all P-type ATPases. This region con-tains an aspartic acid residue (D454) transiently phos-phorylated for catalytic activity.32 G733R lies in a regionimplicated in ATP binding and G892R lies in the hingeregion; both regions are highly conserved in all P-typeATPases.32–34 D554N is located in a sequence motif spe-cifically conserved in the P4 subfamily of P-type ATPases,to which FIC1 belongs.17,21 Three missense mutations(L127P, S403Y, G1040R) occur in highly conserved res-idues in predicted transmembrane domains. In BRIC pa-tients, mutations in conserved functional domains wereinvariably detected in compound heterozygous form (seeTable 3).
Four mutations were detected in both PFIC and BRICpatients, but the combinations of mutations in BRIC pa-tients differed from those in PFIC patients (see Tables1–3). Also, at R600 and G308, 2 distinct mutations weredetected. G308D, found in heterozygous form in theBRIC patient from family BRIC5, alters the same aminoacid residue mutated in Amish PFIC1 patients, who arehomozygous for G308V.16 In the BRIC patient, theG308D mutation occurs in compound heterozygousform with the common I661T mutation.
I661T in PFIC. The I661T mutation previously re-ported only in BRIC patients16 is present in most BRICpatients with detected ATP8B1 mutations. This muta-tion is also one of the four detected in both BRIC andPFIC patients; this mutation occurs in compound het-erozygous form with a nonsense mutation (R602X, fam-ily PFIC29; R952X, family PFIC30). The I661Tmutation, in heterozygous form with another mutation,thus is associated with either episodic or chronic progres-sive cholestasis.
Reduced Penetrance. In a large Italian pedigree inwhich we previously identified presence of I661T (familyBRIC21),16 we detected 4 homozygous I661T individu-als, 1 male (deceased at �77 years of age) and 3 females(born in 1964, 1959, and 1949) who were symptom-freetheir entire lives (Fig. 5). The male (II-3) had three chil-dren with BRIC. Another unaffected man (born in 1962)homozygous for I661T has a younger sibling with BRIC(family BRIC1). The proband in family BRIC17 is com-pound heterozygous for 2 missense mutations (D454Gand I694T). This patient has an older, clinically unaf-fected brother (born in 1956) who carries the same mu-tations. These results suggest gender-independentreduced penetrance of FIC1 disease.
Rare Variants. We identified 3 rare sequence variantsin ATP8B1 for which association with disease is equivocal(Table 4). Two patients (PFIC41 and BRIC25) were het-erozygous for synonymous variants (G457G and T177T,
respectively) not detected in control samples. In neitherpatient was an ATP8B1 mutation detected on the otherallele, nor were any ABCB11 mutations identified inPFIC41 (Strautnieks et al., unpublished data). A thirdvariant, E429A, was present on one of approximately 600control chromosomes. In screening of approximately 360potential PFIC or BRIC chromosomes, this variant wasfound in heterozygous form in 1 PFIC patient, 2 BRICpatients, and a parent of a third BRIC patient (DNA fromthis patient was not available) (see Table 4). In the 3patients with this variant, no disease mutation was de-tected on the other allele; however, one patient (PFIC40)was compound heterozygous for 2 mutations in ABCB11(Strautnieks et al., unpublished data). Whether this rareallele contributes to disease in these individuals is uncer-tain.
DiscussionGenes encoding P-type ATPases often extend over a
large genomic region and are divided into many ex-ons.35,36 We have determined that ATP8B1 contains 28exons and spans at least 77 kb. We found no evidence foradditional exons or alternatively spliced transcripts. Thetranscription start site of ATP8B1 must be establishedexperimentally to define the upstream elements that reg-ulate ATP8B1 expression.
Determination of the genomic structure of ATP8B1allowed the first large-scale screening for ATP8B1 muta-tions associated with BRIC and PFIC. In conjunctionwith previous small series of ATP8B1 mutations,16–20 thiswork describes a spectrum of mutations causing FIC1disease. With one exception (D7ON, present on 0.5% ofcontrol chromosomes), none of these mutations was iden-tified in control samples, providing strong genetic evi-dence of association with disease. Future RNA-based andfunctional studies could further support the identity ofsplice site and missense mutations as disease-causing—rather than rare normal—variants. Nevertheless, the loca-tion of the splice site mutations in highly conserved orinvariant splicing consensus residues, the high levels ofconservation of most mutated amino acid residues, andthe nonconservative nature of the amino acid substitu-tions, provide further evidence that these changes are dis-ease mutations. Disease-associated mutations in ATP8B1are diverse; only the I661T mutation is common in mul-tiple populations. Other mutations are relatively preva-lent in particular populations due to founder effects.Mutations do not cluster in a specific gene region, al-though missense mutations concentrate in highly con-served residues, and there is a relative lack of frameshiftand nonsense mutations in the gene’s 5� half.
HEPATOLOGY, Vol. 40, No. 1, 2004 KLOMP ET AL. 33
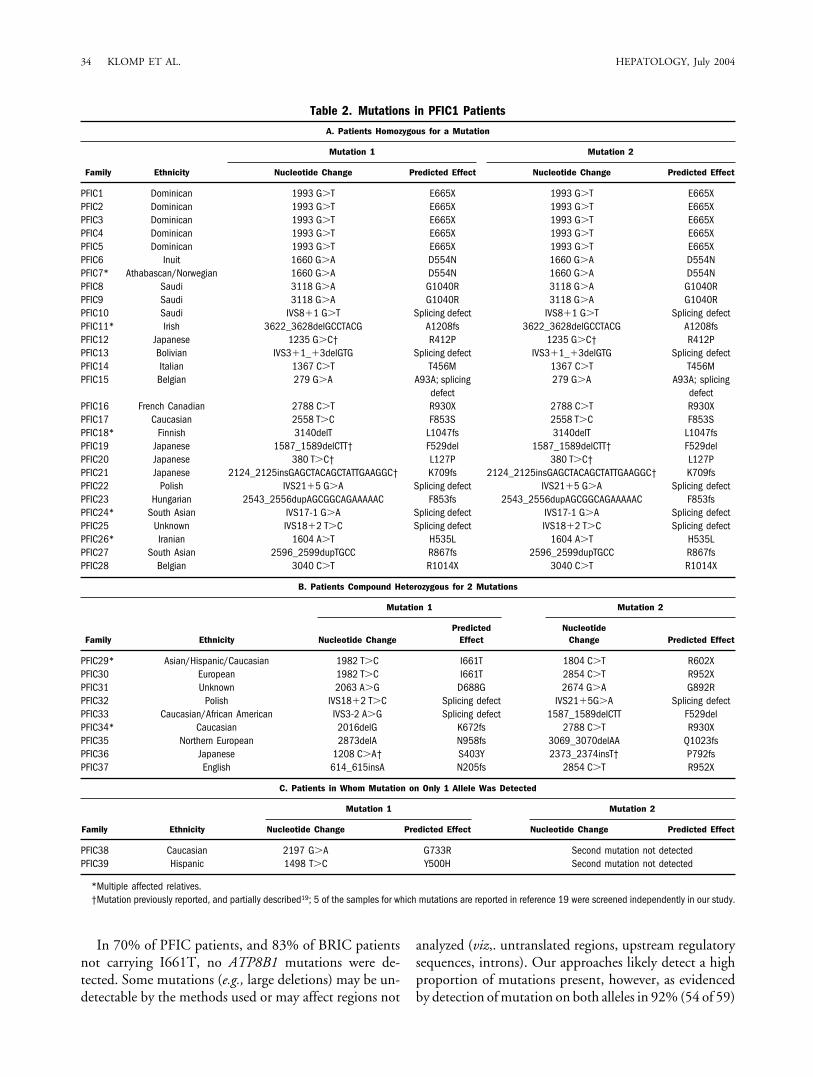
In 70% of PFIC patients, and 83% of BRIC patientsnot carrying I661T, no ATP8B1 mutations were de-tected. Some mutations (e.g., large deletions) may be un-detectable by the methods used or may affect regions not
analyzed (viz,. untranslated regions, upstream regulatorysequences, introns). Our approaches likely detect a highproportion of mutations present, however, as evidencedby detection of mutation on both alleles in 92% (54 of 59)
Table 2. Mutations in PFIC1 Patients
A. Patients Homozygous for a Mutation
Family Ethnicity
Mutation 1 Mutation 2
Nucleotide Change Predicted Effect Nucleotide Change Predicted Effect
PFIC1 Dominican 1993 G�T E665X 1993 G�T E665XPFIC2 Dominican 1993 G�T E665X 1993 G�T E665XPFIC3 Dominican 1993 G�T E665X 1993 G�T E665XPFIC4 Dominican 1993 G�T E665X 1993 G�T E665XPFIC5 Dominican 1993 G�T E665X 1993 G�T E665XPFIC6 Inuit 1660 G�A D554N 1660 G�A D554NPFIC7* Athabascan/Norwegian 1660 G�A D554N 1660 G�A D554NPFIC8 Saudi 3118 G�A G1040R 3118 G�A G1040RPFIC9 Saudi 3118 G�A G1040R 3118 G�A G1040RPFIC10 Saudi IVS8�1 G�T Splicing defect IVS8�1 G�T Splicing defectPFIC11* Irish 3622_3628delGCCTACG A1208fs 3622_3628delGCCTACG A1208fsPFIC12 Japanese 1235 G�C† R412P 1235 G�C† R412PPFIC13 Bolivian IVS3�1_�3delGTG Splicing defect IVS3�1_�3delGTG Splicing defectPFIC14 Italian 1367 C�T T456M 1367 C�T T456MPFIC15 Belgian 279 G�A A93A; splicing
defect279 G�A A93A; splicing
defectPFIC16 French Canadian 2788 C�T R930X 2788 C�T R930XPFIC17 Caucasian 2558 T�C F853S 2558 T�C F853SPFIC18* Finnish 3140delT L1047fs 3140delT L1047fsPFIC19 Japanese 1587_1589delCTT† F529del 1587_1589delCTT† F529delPFIC20 Japanese 380 T�C† L127P 380 T�C† L127PPFIC21 Japanese 2124_2125insGAGCTACAGCTATTGAAGGC† K709fs 2124_2125insGAGCTACAGCTATTGAAGGC† K709fsPFIC22 Polish IVS21�5 G�A Splicing defect IVS21�5 G�A Splicing defectPFIC23 Hungarian 2543_2556dupAGCGGCAGAAAAAC F853fs 2543_2556dupAGCGGCAGAAAAAC F853fsPFIC24* South Asian IVS17-1 G�A Splicing defect IVS17-1 G�A Splicing defectPFIC25 Unknown IVS18�2 T�C Splicing defect IVS18�2 T�C Splicing defectPFIC26* Iranian 1604 A�T H535L 1604 A�T H535LPFIC27 South Asian 2596_2599dupTGCC R867fs 2596_2599dupTGCC R867fsPFIC28 Belgian 3040 C�T R1014X 3040 C�T R1014X
B. Patients Compound Heterozygous for 2 Mutations
Family Ethnicity
Mutation 1 Mutation 2
Nucleotide ChangePredicted
EffectNucleotide
Change Predicted Effect
PFIC29* Asian/Hispanic/Caucasian 1982 T�C I661T 1804 C�T R602XPFIC30 European 1982 T�C I661T 2854 C�T R952XPFIC31 Unknown 2063 A�G D688G 2674 G�A G892RPFIC32 Polish IVS18�2 T�C Splicing defect IVS21�5G�A Splicing defectPFIC33 Caucasian/African American IVS3-2 A�G Splicing defect 1587_1589delCTT F529delPFIC34* Caucasian 2016delG K672fs 2788 C�T R930XPFIC35 Northern European 2873delA N958fs 3069_3070delAA Q1023fsPFIC36 Japanese 1208 C�A† S403Y 2373_2374insT† P792fsPFIC37 English 614_615insA N205fs 2854 C�T R952X
C. Patients in Whom Mutation on Only 1 Allele Was Detected
Family Ethnicity
Mutation 1 Mutation 2
Nucleotide Change Predicted Effect Nucleotide Change Predicted Effect
PFIC38 Caucasian 2197 G�A G733R Second mutation not detectedPFIC39 Hispanic 1498 T�C Y500H Second mutation not detected
*Multiple affected relatives.†Mutation previously reported, and partially described19; 5 of the samples for which mutations are reported in reference 19 were screened independently in our study.
34 KLOMP ET AL. HEPATOLOGY, July 2004

of the BRIC or PFIC patients in whom any ATP8B1mutation was found. If a high proportion of mutationswas undetectable as a result of methodological limita-tions, the proportion of patients should be larger in whommutation on only one allele was identified. We infer thatmany PFIC and BRIC patients do not carry mutations inATP8B1. Although some PFIC patients carry mutationsin ABCB11,18,23,24 in others, linkage to both ATP8B1 andABCB11 has been excluded.37 Over 30 of the PFIC pa-tients in our study have been screened for mutations inboth ATP8B1 and ABCB11 with none found (Strautniekset al., unpublished data). These data strongly suggest anunidentified locus for autosomal recessive BRIC andPFIC. Another study indicates an additional, unmappedlocus for autosomal dominant BRIC.6
Screened patients lacking mutations in both ATP8B1and ABCB11 are good candidates for studies to identifyadditional loci mutated in low–�-GT PFIC. Screening ofTJP2 and BAAT may also be merited, as these genes are
mutated in some cases of familial hypercholanemia, thepresentation of which may overlap clinically with PFIC.38
Although several ATP8B1 mutations can be associatedwith both PFIC and BRIC, PFIC patients more oftencarry homozygous nonsense and frameshift mutationspredicted to affect protein expression or function severely.BRIC patients are more often compound heterozygousand usually carry at least one mutation that may affectprotein function only moderately (notably I661T). De-velopment of an in vitro assay for FIC1 function wouldpermit further characterization of genotype–phenotyperelationships in FIC1 disease. However, phenotypic se-verity can range widely (from normal health to PFIC) fora single mutation. Most notably, I661T demonstrates in-complete penetrance in homozygous form, while in com-pound heterozygous form it can even cause PFIC1.Among 6 patients homozygous for the IVS23-3C�Amutation, all initially presenting with episodic cholestasis(viz. BRIC), two have subsequently developed chronic
Table 3. Mutations in BRIC Patients
A. Patients Homozygous or Compound Heterozygous for I661T (1982 T>C)
Family Ethnicity
Mutation 1 Mutation 2
Nucleotide Change Predicted Effect Nucleotide Change Predicted Effect
BRIC1 Belgian 1982 T�C I661T 1982 T�C I661TBRIC2 Dutch 1982 T�C I661T 1982 T�C I661TBRIC3 Portuguese 1982 T�C I661T 1982 T�C I661T
BRIC4 Belgian 1982 T�C I661T 279 G�A Splicing defectBRIC5 German 1982 T�C I661T 923 G�A G308DBRIC6 Dutch 1982 T�C I661T 1798 C�T R600WBRIC7 Dutch 1982 T�C I661T 1799 G�A R600QBRIC8 Dutch 1982 T�C I661T 1799 G�A R600QBRIC9 Italian 1982 T�C I661T 2674 G�A G892RBRIC10 Italian 1982 T�C I661T 3490 C�T R1164XBRIC11 Unknown 1982 T�C I661T IVS26�2 T�A Splicing defectBRIC12 Dutch 1982 T�C I661T Second mutation not yet foundBRIC13 German 1982 T�C I661T Second mutation not yet foundBRIC14 Dutch 1982 T�C I661T Second mutation not yet found
B. Patients Homozygous for Non-I661T Mutations
Family Ethnicity
Mutation 1 Mutation 2
Nucleotide Change Predicted Effect Nucleotide Change Predicted Effect
BRIC15* Dutch IVS23-3 C�A Skip exon 24† IVS23-3 C�A Skip exon 24†BRIC16 Pakistani 3722delG G1241fs 3722delG G1241fs
C. Patients Compound Heterozygous for Non-I661T Mutations
Family Ethnicity
Mutation 1 Mutation 2
Nucleotide Change Predicted Effect Nucleotide Change Predicted Effect
BRIC17 French 1361 A�G D454G 2081 T�C I694TBRIC18 German 1882 C�T R628W 2854 C�T R952XBRIC19 Unknown 1030 A�T I344F 1358 C�A S453YBRIC20 German 208 G�A D70N 1799 G�A R600Q
*Disease in 2 of 6 patients in this family has since worsened, indicating that they have disease of intermediate severity between PFIC and BRIC.31
†Determined experimentally.
HEPATOLOGY, Vol. 40, No. 1, 2004 KLOMP ET AL. 35

cholestasis, exhibiting a phenotype intermediate betweenBRIC and PFIC (family BRIC15).31,39 FIC1 disease thusranges along a clinicopathological continuum, rather thansegregating clearly between mild (BRIC) and severe(PFIC1). The large phenotypic differences among pa-
tients with the same mutation implicate additional factorsin disease. If these can be identified, the biology of FIC1may be better understood and treatment of FIC1 diseasemay improve.
How mutations in ATP8B1 cause cholestasis is un-known. FIC1 is located at the brush border of enterocytes,the canalicular membrane of hepatocytes, and the apicalmembrane of cholangiocytes, as well as in other tis-sues.16,22,40 It may be an aminophospholipid translo-case.22 These data are consistent with an indirect role ofFIC1 in bile salt secretion, and in enterohepatic andcholangiohepatic recirculation of bile salts. Mice homozy-gous for the FIC1 G308V mutation display no decrease inbiliary bile salt secretion, but have dramatically increasedserum bile salt concentrations when fed cholate. Thesefindings suggest that FIC1 dysfunction perturbs bile saltresorption.41
Our work affects carrier detection, genetic counseling,and prenatal diagnosis. Because 79% (27 of 34 combiningthis and our previous studies15,16,30) of BRIC patientswith detected ATP8B1 mutations carried at least one copyof I661T, screening for I661T should identify most BRIC
Fig. 4. Categories of mutations identified in FIC1 disease. Red indicates categories of mutations likely to prevent production of structurally normalprotein (nonsense, frameshift, and large deletion mutations). Green indicates categories of mutation with the potential to yield structurally grosslynormal but functionally deficient protein (missense and small inframe deletion mutations). Splice site mutations are represented by green and redstriped areas to indicate the uncertain severity of their effects on the transcript, in the absence of RNA studies. PFIC, progressive familial intrahepaticcholestasis; BRIC, benign recurrent intrahepatic cholestasis.
Fig. 5. Segregation analysis of the I661T substitution in familyBRIC21. Exon 18–specific ATP8B1 fragments were amplified by PCR,using a mismatch primer, from genomic DNA of the individuals indicatedin the pedigree and of indicated control individuals. After incubation withPstI, samples were analyzed via polyacrylamide gel electrophoresis andstained with ethidium bromide. In individuals without the mutation, theintroduced PstI restriction site allows PCR-product cleavage into a largefragment and a small one (the latter not resolved). Molecular sizemarkers: bp. All unaffected individuals are homozygous for I661T.
Table 4. Rare ATP8B1 Variants Detected in Patients
Patient Ethnicity Variant 1 Predicted Effect Variant 2
PFIC40 Caucasian 1286 A�C E429A None detectedPFIC41 Unknown 1371 G�A G457G None detectedBRIC22 Unknown 1286 A�C E429A None detectedBRIC23 Dutch 1286 A�C E429A None detectedBRIC24*
(parent) Unknown 1286 A�C E429ABRIC25 Asian 531 G�A T177T None detected
*Insufficient DNA was available from this individual to permit screening of allATP8B1 coding exons, so we do not know if other sequence changes might alsobe present.
36 KLOMP ET AL. HEPATOLOGY, July 2004

patients of Western European descent with FIC1 disease.Of wider value is that phenotypic criteria were not used toenrich our sample for patients more likely to carryATP8B1 rather than ABCB11 mutations. Comparison ofthese patients’ phenotypes, having FIC1 disease geneti-cally confirmed without selection bias, with those of sim-ilarly identified patients having genetically confirmed bilesalt export pump disease, should permit definition of dif-ferences in presentation, histopathology, biochemistry,and treatment outcomes between these disorders, facili-tating better diagnosis and treatment. Such comparison isunderway. The data presented also lay the foundation foranalysis of FIC1 disease pathogenesis. Studies of expres-sion and function will address the effects of the mutationsdescribed on the localization and molecular role of FIC1.
Acknowledgment: We thank families for their partic-ipation and many clinicians for case referral. We acknowl-edge Serieta Mohkamsing and Eyun Song for experttechnical assistance, and Patricia Babbitt for helpful dis-cussion.
References1. Clayton RJ, Iber FL, Ruebner BH, McKusick VA. Byler disease. Fatal
familial intrahepatic cholestasis in an Amish kindred. Am J Dis Child1969;117:112–124.
2. Linarelli LG, Williams CN, Phillips MJ. Byler’s disease: fatal intrahepaticcholestasis. J Pediatr 1972;81:484–492.
3. Bourke B, Goggin N, Walsh D, Kennedy S, Setchell KD, Drumm B.Byler-like familial cholestasis in an extended kindred. Arch Dis Child1996;75:223–227.
4. Summerskill WHJ. The syndrome of benign recurrent cholestasis. Am JMed 1965;38:298–305.
5. De Koning TJ, Sandkuijl LA, De Schryver JE, Hennekam EA, Beemer FA,Houwen RH. Autosomal-recessive inheritance of benign recurrent intra-hepatic cholestasis. Am J Med Genet 1995;57:479–482.
6. Floreani A, Molaro M, Mottes M, Sangalli A, Baragiotta A, Roda A, et al.Autosomal dominant benign recurrent intrahepatic cholestasis (BRIC) un-linked to 18q21 and 2q24. Am J Med Genet 2000;95:450–453.
7. Knisely AS. Progressive familial intrahepatic cholestasis: a personal per-spective. Pediatr Dev Pathol 2000;3:113–125.
8. van Mil SWC, Klomp LWJ, Bull LN, Houwen RH. FIC1 disease: aspectrum of intrahepatic cholestatic disorders. Semin Liver Dis 2001;21:535–544.
9. Oude Elferink RP, Groen AK. Genetic defects in hepatobiliary transport.Biochim Biophys Acta 2002;1586:129–145.
10. Houwen RHJ, Baharloo S, Blankenship K, Raeymaekers P, Juyn J, Sand-kuijl LA, et al. Genome screening by searching for shared segments: map-ping a gene for benign recurrent intrahepatic cholestasis. Nat Genet 1994;8:380–386.
11. Carlton VEH, Knisely AS, Freimer NB. Mapping of a locus for progressivefamilial intrahepatic cholestasis (Byler disease) to 18q21-q22, the benignrecurrent intrahepatic cholestasis region. Hum Mol Genet 1995;4:1049–1053.
12. Strautnieks SS, Kagalwalla AF, Tanner MS, Gardiner RM, Thompson RJ.Locus heterogeneity in progressive familial intrahepatic cholestasis. J MedGenet 1996;33:833–836.
13. Bull LN, Carlton VEH, Stricker NL, Baharloo S, DeYoung JA, FreimerNB, et al. Genetic and morphological findings in progressive familial in-trahepatic cholestasis (Byler disease [PFIC-1] and Byler syndrome): evi-dence for heterogeneity. HEPATOLOGY 1997;26:155–164.
14. Strautnieks SS, Kagalwalla AF, Tanner MS, Knisely AS, Bull L, Freimer N,et al. Identification of a locus for progressive familial intrahepatic cholesta-sis PFIC2 on chromosome 2q24. Am J Hum Genet 1997;61:630–633.
15. Bull LN, Juijn JA, Liao M, van Eijk MJ, Sinke RJ, Stricker NL, et al.Fine-resolution mapping by haplotype evaluation: the examples of PFIC1and BRIC. Hum Genet 1999;104:241–248.
16. Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, etal. A gene encoding a P-type ATPase mutated in two forms of hereditarycholestasis. Nat Genet 1998;18:219–224.
17. Klomp LW, Bull LN, Knisely AS, van Der Doelen MA, Juijn JA, Berger R,et al. A missense mutation in FIC1 is associated with Greenland familialcholestasis. HEPATOLOGY 2000;32:1337–1341.
18. Chen HL, Chang PS, Hsu HC, Ni YH, Hsu HY, Lee JH, et al. FIC1 andBSEP defects in Taiwanese patients with chronic intrahepatic cholestasiswith low alpha-glutamyltranspeptidase levels. J Pediatr 2002;140:119–124.
19. Egawa H, Yorifuji T, Sumazaki R, Kimura A, Hasegawa M, Tanaka K.Intractable diarrhea after liver transplantation for Byler’s disease: successfultreatment with bile adsorptive resin. Liver Transpl 2002;8:714–716.
20. Lykavieris P, van Mil S, Cresteil D, Fabre M, Hadchouel M, Klomp L, etal. Progressive familial intrahepatic cholestasis type 1 and extrahepaticfeatures: no catch-up of stature growth, exacerbation of diarrhea, and ap-pearance of liver steatosis after liver transplantation. J Hepatol 2003;39:447–452.
21. Palmgren MG, Axelson KB. Evolution of P-type ATPases. Biochim Bio-phys Acta 1998;1365:37–45.
22. Ujhazy P, Ortiz D, Misra S, Li S, Moseley J, Jones H, et al. Familialintrahepatic cholestasis 1: studies of localization and function. HEPATOL-OGY 2001;34:768–775.
23. Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, et al.A gene encoding a liver-specific ABC transporter is mutated in progressivefamilial intrahepatic cholestasis. Nat Genet 1998;20:233–238.
24. Jansen PLM, Strautnieks SS, Jacquemin E, Hadchouel M, Sokal E, Hoo-iveld GJ, et al. Hepatocanalicular bile salt export pump deficiency in pa-tients with progressive familial intrahepatic cholestasis. Gastroenterology1999;117:1370–1379.
25. Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, et al.The sister of P-glycoprotein represents the canalicular bile salt exportpump of mammalian liver. J Biol Chem 1998;273:10046–10050.
26. Byrne JA, Strautnieks SS, Mieli-Vergani G, Higgins CF, Linton KJ,Thompson RJ. The human bile salt export pump: characterization of sub-strate specificity and identification of inhibitors. Gastroenterology 2002;123:1649–1658.
27. Noe J, Stieger B, Meier PJ. Functional expression of the canalicular bile saltexport pump of human liver. Gastroenterology 2002;123:1659–1666.
28. Leabman MK, Huang CC, DeYoung J, Carlson EJ, Taylor TR, de la CruzM, et al. Natural variation in human membrane transporter genes revealsevolutionary and functional constraints. Proc Natl Acad Sci U S A 2003;100:5896–5901.
29. Vredendaal PJ, van den Berg IE, Stroobants AK, van der AD, MalingreHE, Berger R. Structural organization of the human short-chain L-3-hydroxyacyl-CoA dehydrogenase gene. Mamm Genome 1998;9:763–768.
30. Tygstrup N, Steig BA, Juijn JA, Bull LN, Houwen RHJ. Recurrent familialintrahepatic cholestasis in the Faeroe Islands. Phenotypic heterogeneitybut genetic homogeneity. HEPATOLOGY 1999;29:506–508.
31. van Ooteghem NAM, Klomp LW, van Berge Henegouwen GP, HouwenRH. Benign recurrent intrahepatic cholestasis progressing to progressivefamilial intrahepatic cholestasis: low GGT cholestasis is a clinical contin-uum. J Hepatol 2002;36:439–443.
32. Lutsenko S, Kaplan JH. Organization of P-type ATPases: significance ofstructural diversity. Biochemistry 1995;34:15607–51613.
33. Catty P, de Kerchove d’Exaerde A, Goffeau A. The complete inventory ofthe yeast Saccharomyces cerevisiae P-type transport ATPases. FEBS Lett1997;409:325–332.
HEPATOLOGY, Vol. 40, No. 1, 2004 KLOMP ET AL. 37

34. Halleck MS, Pradhan D, Blackman C, Berkes C, Williamson P, SchlegelRA. Multiple members of a third subfamily of P-type ATPases identifiedby genomic sequences and ESTs. Genome Res 1998;8:354–361.
35. Petrukhin K, Lutsenko S, Chernov I, Ross BM, Kaplan JH, GilliamTC. Characterization of the Wilson disease gene encoding a P-typecopper transporting ATPase: genomic organization, alternative splic-ing, and structure/function predictions. Hum Mol Genet 1994;3:1647–1656.
36. Dierick HA, Ambrosini L, Spencer J, Glover TW, Mercer JF. Molecularstructure of the Menkes disease gene (ATP7A). Genomics 1995;28:462–469.
37. Strautnieks S, Byrne J, Knisely AS, Bull LN, Sokal E, Lacaille F, et al. Theremust be a third locus for low GGT PFIC [Abstract]. HEPATOLOGY 2001;34:240A.
38. Carlton VEH, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Rob-inson DL, et al. Complex inheritance of familial hypercholanemia withassociated mutations in TJP2 and BAAT. Nat Genet 2003;34:91–96.
39. Berman L, Markowitz J, Kahn E, Kocoshis SA, Agostini RM Jr, HouwenRH, et al. Benign recurrent intrahepatic cholestasis and progressive familialintrahepatic cholestasis linked to PFIC1: points on a continuum [Ab-stract]. HEPATOLOGY 1997;26:384A.
40. Eppens EF, van Mil SWC, de Vree JM, Mok KS, Juijn JA, Oude ElferinkRP, et al. FIC1, the protein affected in two forms of hereditary cholestasis,is localized in the cholangiocyte and the canalicular membrane of thehepatocyte. J Hepatol 2001;35:436–443.
41. Pawlikowska L, Groen A, Eppens EF, Kunne C, Ottenhoff R, Looije N, etal. A mouse genetic model for familial cholestasis caused by ATP8B1mutations reveals perturbed bile salt homeostasis but no impairment in bilesecretion. Hum Mol Genet 2004;13:881–892.
42. Corpet F. Multiple sequence alignment with hierarchical clustering. Nu-cleic Acids Res 1988;16:10881–10890.
43. Henikoff S, Henikoff JG. Amino acid substitution matrices from proteinblocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919.
38 KLOMP ET AL. HEPATOLOGY, July 2004




















