
Determination of Maximal Lactate Steady State in Healthy Adults
Upload
unesco-iheCategory
view
7download
0
267
Clinica Chimica Acta, 108 (1980) 267-276 @ Elsevier/North-Holland Biomedical Press
CCA 1561
HEREDITARY DEFICIENCY OF LACTATE DEHYDROGENASE M-SUBUNIT
TAKASHI KANNO a**, KAYOKO SUDO a, IZUMI TAKEUCHI a, SHINJI KANDA a, NISHIO HONDA b, YOSHIRO NISHIMURA b and KUNIO OYAMA b
a Department of Laboratory Medicine and b First Department of Medicine, Hamamatsu University, School of Medicine, Handa-cho 3600, Hamamatsu 431-31 (Japan)
(Received April 18th, 3 980)
Summary
A family with a complete deficiency of lactate dehydrogenase M-subunit was investigated. The propositus was an l&year-old male who complained of exer- tional pigmenturia and easy fatigue. Marked discrepancy was observed in the ratio between creatine kinase and lactate dehydrogenase (CK/LDH). Electro- phoretic analysis of serum LDH isoenzymes of the propositus demonstrated only one activity band of LDH H4. A complete lack of the LDH M-subunit was similarly demonstrated in erythrocytes, leukocytes and in the intermediate vastus muscle. LDH levels in the muscle specimen were markedly decreased in the patient, whereas CK and aspartate aminotransferase were almost the same as in a control subject. LDH isoenzymes of erythrocytes were analyzed in 5 siblings and in the parents. This demonstrated a complete lack of LDH M-sub- unit in 3 siblings. The ratio between H-subunit and M-subunit (H/M) in erythro- cyte LDH suggested a partial absence of the M-subunit in two siblings and in the parents.
An abortive increase of blood lactate and a marked increase in blood pyruvate were observed immediately after ischemic work of the forearm, accompanied by an increase in serum creatine kinase and myoglobinuria. The present case represents a newly described form of genetically determined myopathy.
Introduction
A complete deficiency of lactate dehydrogenase H-subunit was reported by Kitamura et al. [l] in a patient with moderately impaired glucose tolerance. However, a lack of M-subunit production as a result of genetic abnormality has
* To whom correspondence should be addressed.

268
never been reported. In this study, a genetic abnormality of the M-subunit of lactate dehydrogenase was investigated in a patient with exertional pigmenturia and easy fatigue. The relationship between M-subunit deficiency and muscle disorders is discussed.
Materials and methods
Erythrocytes and leukocytes were prepared by the dextran centrifugation method described by Skoog and Beck [2]. Preparation of the hemolysate was performed according to the method of Beutler et al. [3]. The collected leuko- cytes were re-suspended in 67 mmol/l phosphate buffer containing 2 mmol/l of magnesium chloride, and sonicated with a microtop Ultra-Sonicator (Model UR-BOP, Tomy. Seiko Co. Ltd., Japan) in an ice-cold container for 4 X 15 s at 20 kHz. The erythrocyte hemolysates and ultra-sonicated leukocytes were used for the experiments.
Lactate dehydrogenase (LDH) activity was measured by a spectrophotometric continuous monitoring method using a JEOL 6 channel reaction rate analyser (Model JEOL H6R, Japan Electron Optics Laboratory Co. Ltd.). The assay conditions were mainly based on the method described by Wroblewski and LaDue [4]. Final reagent concentrations of the assay mixture are as follows: pyruvate, 0.68 mmol/l; NADH, 0.17 mmol/l; sodium-sodium phosohate pH 7.4, 100 mmol/l, respectively. The activity of the enzyme is expressed in U/l and that of erythrocytes and leukocytes are expressed in U/10’ cells at 37°C. The normal range of serum LDH is 160-320 U/l. Creatine kinase (CK) activity was measured by the method described by Szasz et al. [5] using a kit from Boehringer, Auto-Pat CK/NAC. The normal ranges of serum CK activity for adult males are 20-110 and for adult females 20-80 U/l, respectively.
Isoenzymes of LDH were electrophoretically separated by the method described by Shioya et al. [6] using Cellogel membrane. Activity bands were visualized using lactate as a substrate, NAD and tetrazolium salt (MTT) as the final hydrogen acceptor. The final concentrations of reagents were as follows; D,L-lactate, 0.13 mol/l; NAD, 1.0 mmol/l; MTT, 0.4 mmol/l; PMS, 0.1 mmol/l; Tris-HCl pH 7.4, 60 mmol/l.
Blood pyruvate and lactate were enzymatically measured by the slightly modified methods described by Bucher et al. [ 71 and Hohorst [ 81, respectively.
The muscle specimen was obtained by biopsy of the intermediate vastus muscle under local anesthesia. A biopsy sample of the same muscle during surgical procedure was obtained as a control. The muscle specimen was homo- genized by 10 ~01s. of 67 mmol/l phosphate buffer, pH 7.4, containing 2 mmol/l of MgCl*. The supernatant resulting from refrigerated centrifugation 10 000 X g was used for the experiments.
Case report An l&year-old male who had complained for 2 years of brownish discolora-
tion of the urine and easy fatigue lo-12 h after hard exercise was referred to our hospital for further examination. The propositus (111-5) had 5 siblings and in one of them (III-l, the eldest brother), a high serum level of aspartate amino- transferase had been found 2 years previously. The elevation of the aminotrans-

269
ferase was observed transiently after hard work and decreased by bed rest. Examination of the serum enzyme levels of the propositus was performed
during the episode of exertional pigmenturia. Aspartate aminotransferase showed 390 U/l (normal ranges of aspartate aminotransferase were 5-25 U/l), alanine aminotransferase, 81 U/l (3-17 U/l), creatine kinase, 26 290 U/l, and lactate dehydrogenase, 443 U/l. The brownish substance in the urine was iden- tified as myoglobin. Other laboratory data of the propositus were normal.
Results
Ratio of CK and LDH (CWLDH) in muscle disorders and in the propositus In subjects without muscle disorders, a correlation between CK and LDH
was not usually demonstrated. However, higher correlations between these two enzymes were observed in patients with muscle disorders involving heart injury. Moreover, the ratio between these two enzymes (CK/LDH) increased in this group. As shown in Table I, a group of patients with hereditary skeletal muscle disorders (6 cases of Duchenne type muscular dystrophy, and one congenital) and a group with myocardial infarction (studied within 3 days after the onset) show almost the same ranges of CK/LDH. Patients with non-hereditary muscle injury (cf. arteriosclerosis obliterans, carbon monoxide toxicosis) had higher ratios, and a remarkably high ratio was demonstrated in the propositus with symptomatic myoglobinuria. The aspartate aminotransferase activity in the patient was increased in parallel with CK activity. Hence, incomplete release of LDH from the skeletal muscles was suspected (Table I).
Electrophoretic analysis of LDH isoenzymes Electrophoretic separation of LDH isoenzymes was performed to study the
low serum LDH levels of the propositus. Isoenzyme patterns of the serum, erythrocytes, leukocytes and the intermediate vastus muscle are shown in Fig. 1. As compared with the normal 5 isoenzyme bands, the isoenzyme pat- terns of the propositus show only one isoenzyme band of H4 (LDH I), which migrates fastest toward the anode.
This single isoenzyme pattern is also observed in LDH of erythrocytes, leukocytes and that of the muscle specimen. A lack of the M-subunit of LDH was demonstrated in all specimens from the patient. A similar analysis of LDH isoenzyme was performed on the serum, erythrocytes and leukocytes of the eldest brother of the propositus and the results are shown in the middle frame
TABLE I
RATIO BETWEEN CREATINE KINASE AND LACTATE DEHYDROGENASE IN PATIENTS WITH MUSCLE DISORDERS
Muscle disorders n Range of ratio
Patients with hereditary skeletal muscle disorders Patients with non-hereditary skeletal muscle disorders Patients with myocardial infarction Patients without muscle disorders Propositus (stage of myoglobinuria)
I 0.9- 4.9 39 2.4-11.2 28 O.S- 4.8
273 <0.5 1 59.3

H4
Fig. 1. Zymograms of lactate dehydrogenase isoenzymes obtained from the serum, erythrocytes. leuko- cytes and the intermediate vastus muscle. As compared with the normal control, only one activity band of H4 which has the fastest mobility to the anodic side is demonstrated in the propositus (111-5) and one of the siblings (III-l). These results anticipated a lack of M-subunit production in all of the specimens.

271
of Fig. 1. The results are identical to those of the propositus. A genetic disorder was suspected from these results.
Levels of LDH activities in erythrocytes and leukocytes were analyzed in the propositus. As summarized in Table II, LDH activities in both these types of blood cell are decreased as compared to those of normal subjects. As described above, a deficiency of M-subunit production was found on electrophoretic analysis of LDH isoenzymes, and the decrease of specific activities of LDH ob- served in both erythrocytes and leukocytes is explained by the absence of pro- duction of this subunit.
Enzyme teds in the muscle specimen LDH, CK and aspartate aminotransferase levels of a muscle specimen were
analyzed, and are summarized in Table III. Aspartate aminotransferase and CK levels were almost the same in both the control and in the patient, however, LDH activity was markedly decreased in the patient. Thus, the ratio between CK and LDH is markedly increased. As demonstrated in Fig. 1, the lack of LDH M-subunit was confirmed electrophoretically. The decreased activity of LDH in the muscle of the patient was presumed to be the result of the lack of M-subunit.
Isckemic work During exercise, glycolysis markedly accelerates in the skeletal muscle, and
lactate formed from pyruvate by the enzyme action of LDH is actively dis- charged into the blood circulation. In this particular case, the lack of LDH M-subunit was suspected of leading to a retardation in the conversion of
TABLE II
LACTATE DEHYDROGENASE IN BLOOD CELLS
Subjects Red blood cell White blood cells
LDH activity LDH isoenzymes LDH activity LDH &enzymes
w *> (U *I
ProPosltus 5.25 H4 105.8 H4 Normal control 6.5-8.2 H4, HJMI, H2M2 150-300 H4. H3Mx. %Mz. HIM3, M4
* LDH activity is expressed in U/lo9 cells.
TABLE III
ENZYME LEVELS IN THE MUSCLE SPECIMEN OBTAINED FROM THE PATIENT
Enzymes
Lactate dehydrogenase Aspartate aminotransferase Creatine kinase CKILDH
Control Patient
(U/g of wet weight) (U/g of wet weight)
226 11
52 58
2332 2964
10.3 269.4
0 p<o.o5 l p,o.o5
035 10 20 30 minutes
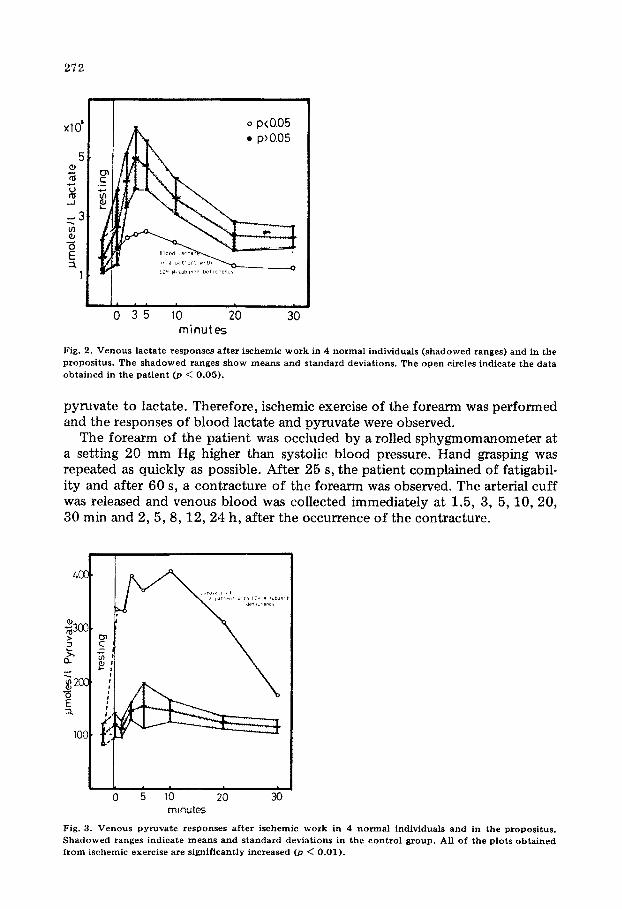
Fig. 2. Venous lactate responses after ischemic work in 4 normal individuals (shadowed ranges) and in the propositus. The shadowed ranges show means and standard deviations. The open circles indicate the data obtained in the patient @ < 0.05).
pyruvate to lactate, Therefore, ischemic exercise of the forearm was performed and the responses of blood lactate and pyruvate were observed.
The forearm of the patient was occluded by a rolled sphy~om~ome~r at a setting 20 mm Hg higher than systolic blood pressure. Hand grasping was repeated as quickly as possible. After 25 s, the patient complained of fatigabil- ity and after 60 s, a contracture of the forearm was observed. The arterial cuff was released and venous blood was collected immediately at 1.5, 3, 5, 10, 20, 30 min and 2,5,8,12,24 h, after the occurrence of the contracture.
L
0 5 10 20 30 minutes
Fig. 3. Venous pyruvate responses after ischemic work in 4 normal individuals and in the propositus. Shadowed ranges indicate means and standard deviations in the control group. All of the plots obtained from ischemic exercise are significantly increased @ < 0.01).
273
=?
RO 35 10 20
hours
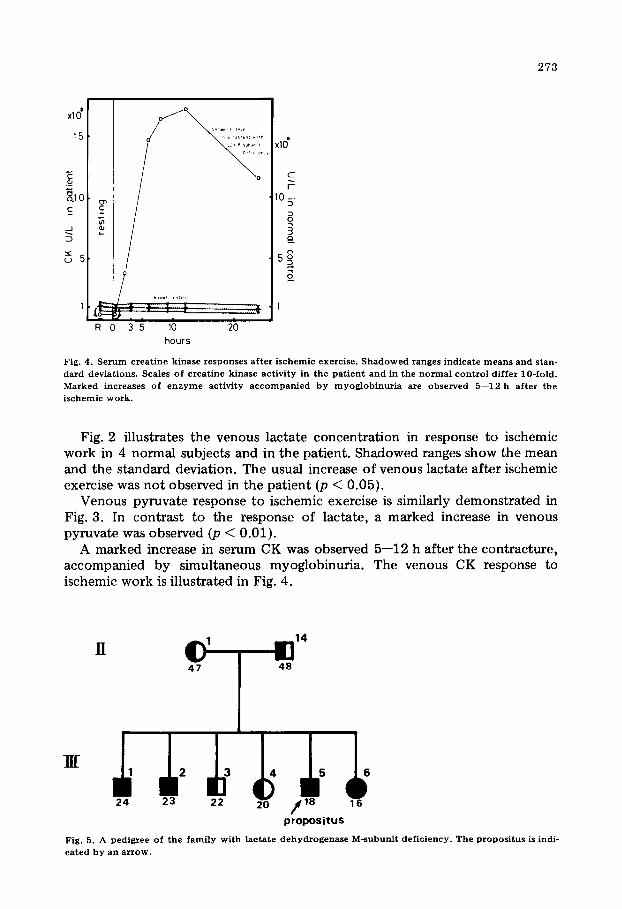
Fig. 4. Serum creatine kinase responses after ischemic exercise. Shadowed ranges indicate means and stan- dard deviations. Scales of creatine kinase activity in the patient and in the normal control differ lo-fold. Marked increases of enzyme activity accompanied by myoglobinuria are observed 5-12 h after the ischemic work.
Fig. 2 illustrates the venous lactate concentration in response to ischemic work in 4 normal subjects and in the patient. Shadowed ranges show the mean and the standard deviation. The usual increase of venous lactate after ischemic exercise was not observed in the patient (p < 0.05).
Venous pyruvate response to ischemic exercise is similarly demonstrated in Fig. 3. In contrast to the response of lactate, a marked increase in venous pyruvate was observed (p < 0.01).
A marked increase in serum CK was observed 5-12 h after the contracture, accompanied by simultaneous myoglobinuria. The venous CK response to ischemic work is illustrated in Fig. 4.
14
p;opositUS
Fig. 5. A pedigree of the family with lactate dehydrogenase M-subunit deficiency. The propositus is indi-
cated by an arrow.
TA
BLE
IV
DIS
TR
IBU
TIO
N
OF
LA
CT
AT
E
DE
HY
DR
OG
EN
ASE
IS
OE
NZY
ME
S
OF
T
HE
FA
MIL
Y
Subj
ects
Ser
um
R
BC
I II
II
I IV
V
H
IM
I II
II
I IV
v
H/M
LD
H
%
56
%
%
w
%
I 90
%
I u*
III-
5
pro
posi
tus
100
0 0
0 0
III-
1 br
oth
er
100
0 0
0
0 -
HI-
Z br
oth
er
100
0 0
0 0
III-
3 br
oth
er
37.9
35.4
18.2
4.6
4.0
3.0
H
I-4 si
ster
34.6
37.3
19.2
5.2
3.2
2.8
II
I-6 si
ster
100
0
0
0
0
II-1
m
oth
er
40.1
37.8
18.3
2.9
1.4
3.5
II
-14
fath
er
32.8
40.9
20.0
4.2
1.8
3.0
100
0 0
0 0
- 5.2
5
100
0 0
0 0
- 5.4
4
100
0 0
0 0
- 4.8
9
56.0
26.8
17.0
5.4
5.7
1
53.6
33.7
12.7
5.8
6.6
4
100
0
0
0
0
- 5.0
5
55.8
30.5
13.7
5.9
N
T
55.6
26.8
17.3
5.4
7.7
5
Con
trol
25-3
0
3540
24-2
8
2-7
2-5
1.9
-2.7
38-4
3
30-3
5
25-3
0
- -
2.7
-4.3
* LD
H
acti
vity
is
expre
ssed
in U
/lo9
cell
s.

215
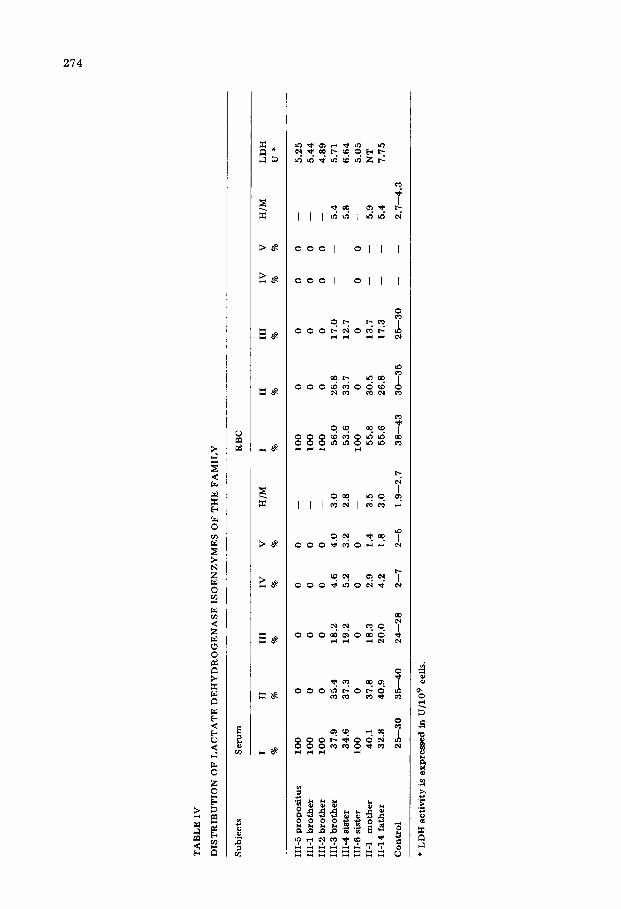
Family studies and detection of heterozygote The results of similar LDH analyses performed on 5 siblings and both parents
are summarized in Table IV. It is shown that only one isoenzyme band, iso- enzyme I (H4), is observed in the serum and erythrocytes of 3 siblings. The levels of erythrocyte LDH activities in family members are not so significantly different from those of normal subjects. However, similar isoenzyme patterns of LDH in erythrocytes were found in two siblings and in the parents. As com- pared to the patterns of normal subjects, isoenzyme II (H3M1) and isoenzyme III (H,M,) of this group are correspondingly diminished. The ratio between the H- and M-subunit is calculated from the RBC isoenzyme patterns and a signifi- cant increase of the ratio between H- and M-subunits is observed in these two siblings and in the parents. A pedigree chart of this family is demonstrated in Fig. 5.
Discussion
A complete deficiency of lactate dehydrogenase H-subunit was reported by Kitamura et al. in 1971 [l] and the erythrocyte metabolism in this disorder was studied by Miwa et al. [ 91. In general, a hereditary abnormality of a glyco- lytic enzyme of erythrocytes is accompanied by obvious evidence of hemolytic anemia. However, neither reticulocytosis nor hemolytic anemia were present in this patient with LDH H-subunit deficiency. On the other hand, a marked increase of the ratio between CK and LDH was observed in a patient who com- plained of exertional pigmenturia and easy fatigue. A complete absence of LDH M-subunit was demonstrated in the serum of the patient. The absence of M-sub- unit was confirmed electrophoretically in the blood cells and in the muscle specimen. The LDH content of muscle was markedly decreased, whereas levels of the other enzymes, CK and aspartate aminotransferase, closely resembled the control specimen. Furthermore, a complete lack of LDH M-subunit was found in three siblings. A significant increase of the ratio between H- and M-subunit in RBC LDH was observed in two other siblings and in the parents. These increases are probably based on the partial lack of M-subunit production of this group. Thus, the two siblings and the parents can be regarded as the heterozygotes of the LDH M-subunit deficiency. The detection of this hetero- zygote state can probably be most effectively accomplished by the analysis of the H/M ratio of the erythrocytes.
Localization of the human LDH M-subunit locus has been established on chromosome 11 [lo]. Thus, the disorder is considered to be a single autosomal recessive trait. A lack of LDH M-subunit as a result of genetic abnormality has never previously been reported. Moreover, our patient complained of exertional nyoglobinuria and easy fatigue compared with the symptomless propositus of the previously reported LDH H-subunit deficiency [ 91.
An abortive increase of venous lactate and a marked increase of venous pyruvate were observed immediately after ischemic exercise of the forearm. These abnormal responses can best be explained by the decrease in LDH levels in the skeletal muscle as the result of M-subunit deficiency. Because of this, pyruvate is released directly into the blood stream without prior conversion to lactate. This abnormal response of pyruvate is different from that observed in

276
patients with McArdle’s disease and in other glycogenolytic disorders [ 11,121. There are many reports of exertional myoglobinuria of unknown origin [ 3-51. The hazards of acute tubular necrosis are well known and can lead to fatal attacks in patients with myoglobinuria. The present case represents a newly described form of a genetically determined myopathy. Further studies must be done in attempt to explain the relationship between decreased levels of LDH and myopathy.
References
1 Kitamura, M.. Iilima. N., Hashimoto, F. and Hiratsuka. A. (1971) Hereditary deficiency of subunit H of lactate dehydrogenase. Clin. Chim. Acta 34,419-423
2 Skoog. W.A. and Beck, W.S. (1956) Studies on the fibrinogen, dextran and phytohemagglutinin meth- od of isolating leukocytes. Blood 11, 436-454
3 Beutler, E., Blume, K.G., Kaplan, J.C., Lohr, G.W., Ramot, B. and Valentine, W.N. (1977) Interna- tional Committee for Standardization in Haematology: Recommended methods for red-cell enzyme analysis. Br. J. Haematol. 35, 331-340
4 Wroblewski. F. and LaDue, J.S. (1955) Lactic dehydrogenase activity in blood. Proc. Sot. EXP. Biol. Med. 90,210-213
5 Szasz, G., Gruber. W. and Bernt, E. (1976) Creatine kinase in serum: 1. Determination of optimum reactmn conditions. Clin. Chem. 22, 650-656
6 Shioya. M., Yanagisawa, M., Kawamura, T. and Kanno. T. (1971) Separation of lactate dehydrogenase isoenzymes on cellulose acetate (Cellogel). Jap. J. Clin. Path. (Rinsho-Byori) 19, 469-472
7 Bucher, T., Czok, R., Lamprecht, W. and Latzko. E. (1963) Pyruvate. in Methods of Enzymatic Analysis (Bergmeyer. H.U.. ed.). pp. 253-259. Academic Press, New York
8 Hohorst, H.J. (1963) L(+)-Lactate, determination with lactic dehydrogenase and DPN. In: Methods of Enzymatic Analysis (Bergmeyer, H.U., ed.), PP. 266-270, Academic Press, New York
9 Miwa, S.. Nishina, T., Kanehashi, Y., Kitamura, M.. Hiratsuka, A. and Shizume, K. (1971) Studies on erythrocyte metabolism in a case with hereditary deficiency of H-subunit of lactate dehydrogenase.
Acta Haematol. Jap. 34. 228-232 10 Boone, C.M.. Chen. T.R. and Ruddle, F.H. (1972) Assignment of LDH-A locus in man to chromo-
some c-11 using cell hybrids. Proc. Natl. Acad. Sci. U.S.A. 69. 510-514 11 Rowland, L.P., Lovelace. R.E.. Schotland. D.L., Araki. S. and Carmel. P. (1966) The clinical diagnosis
of McArdle’s disease. Neurology 16.93-100 12 Schmid. R. and Mahler, R. (1959) Chronic progressive myopathy with myoglobinuria; demonstration
of a glycogenolytic defect in the muscle. J. Clin. Invest. 38. 2044-2058 13 Kontos, H.A., Harley, E.L., Wasserman, A.J., Kelly, J.J. and Magee. J.H. (1963) Exertional idiopathic
paroxysmal myoglobinuria, Amer. J. Med. 35, 283-292 14 Rowland, L.P., Fahn, S., Hirschberg, E. and Harter, D.H. (1964) Myoglobinuria. Arch. Neural. 10,
537-562 15 Rowland. L.P. and Penn, A.S. (1972) Myoglobinuria, Med. Clin. North amer. 56,1233-1256
Copyright © 2022 FDOKUMEN




















